
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards polymetallic lanthanide complexes as dual contrast agents for magnetic resonance and optical imaging
Elke
Debroye
and
Tatjana N.
Parac-Vogt
*
Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: tatjana.vogt@chem.kuleuven.be; Fax: +32 16 327992; Tel: +32 16 327612
First published on 11th September 2014
Abstract
Magnetic resonance imaging (MRI) is a popular imaging technique in medical diagnostics. With the development of contrast agents, interest in its applications has grown tremendously. Significant effort has been made in order to identify the most important parameters that enhance the relaxation efficiency of MRI probes. Taking into account the requirements for an optimal magnetic performance, different contrast agents have been synthesized and studied. Moreover, novel bimodal probes have been developed in order to exploit the high sensitivity and resolution of optical microscopy with the ability of MRI to image opaque samples. Employing this strategy enables the simultaneous visualization of the same biological structures at different resolutions and depths. Throughout this review, different approaches used to improve relaxivity, especially by increasing the molecular volume and hence the rotational tumbling time of the agent, are highlighted. Several ways to obtain bimodal contrast agents are discussed in detail. Finally, lanthanide complexes incorporating an aromatic unit permitting efficient sensitization of lanthanide luminescence in combination with the relaxometric properties of gadolinium analogues are listed.
 Elke Debroye | Dr Elke Debroye completed her master in Chemistry in 2009 at the KU Leuven. In 2013 she obtained her PhD during which she worked on developing novel bimodal MRI/optical contrast agents in the laboratory of Prof. Tatjana N. Parac-Vogt. She is currently a post-doctoral fellow of the Research Foundation Flanders (FWO) as member of the group of Prof. Johan Hofkens at KU Leuven where she is studying the photocatalytic activity of nanomaterials at the single molecule level using electron microscopy and super-resolution fluorescence microscopy. Her research interests cover areas of organic synthesis and molecular spectroscopy. |
 Tatjana N. Parac-Vogt | Professor Tatjana N. Parac-Vogt studied chemistry at the University of Belgrade, former Yugoslavia. After obtaining her PhD at Iowa State University in Ames, USA, she has performed a post-doctoral research at the University of California at Berkeley in the group of Prof. Ken Raymond. She later moved to Germany as Alexander von Humboldt Fellow and has been at KU Leuven since 2000, where she is currently a full professor of Chemistry and the head of the Laboratory of Bioinorganic Chemistry. Her research interests span various areas of coordination chemistry, including development of functional complexes for potential applications in medicine and biochemistry. |
1 Introduction
Magnetic resonance imaging (MRI) has become an important diagnostic technique in clinical medicine. It provides high quality three-dimensional images of soft tissue with sub-millimeter spatial resolution and no depth limit. Contrary to other diagnostic techniques, like X-ray imaging or positron emission tomography (PET), no harmful ionizing radiation is needed for MR imaging since it is based on the principles of nuclear magnetic resonance (NMR) spectroscopy. The obtained signal intensity in MRI depends on the relaxation rate of protons of water molecules, which are on average 65% of a person's weight. By making use of three-dimensional magnetic field gradients, anatomical information can be acquired from every selected region in the human body. Differences among tissues can be detected due to variations in water density, proton relaxation times or water diffusion rates and the contrast can be improved by the administration of a paramagnetic contrast agent (CA), which enhances the rate of relaxation. In 1988, a first contrast agent containing gadolinium(III) was approved for clinical usage and nowadays, gadolinium-based complexes remain the most commonly employed probes in MRI investigations, comprising over 40% of all MRI scans in 2012.1 Gd(III) is a paramagnetic metal ion with seven unpaired electrons resulting in a high magnetic moment (7.94 μB) and its symmetric 8S7/2 ground state provides relatively long electron relaxation times. These features make Gd(III) a very attractive component for imaging applications. Since relatively high doses of gadolinium of 0.1–0.3 mmol per kg body weight are needed2 and free lanthanide(III) ions are known to be toxic (LD50 = 0.2 mmol kg−1 in mice), Gd(III) cannot be administered to a patient in its aqueous form. For clinical examinations, the metal ion is bound by a strongly coordinating ligand exhibiting high thermodynamic stability and high kinetic resistance towards acid catalyzed dissociation or transmetallation. The approved commercial contrast agents contain acyclic or macrocyclic polyaminocarboxylates to which gadolinium is eight-coordinated by nitrogen and oxygen donor atoms ensuring the stability. Across the lanthanide series, the coordination number (CN) decreases from nine for the light, to eight for the heavy trivalent metal ions, with CN for Gd(III) = 9. As a consequence, one Gd(III) coordination site remains available for the binding of a water molecule, which is beneficial for the water proton relaxation efficiency. The two most known complexes are the acyclic Gd(III)-diethylene triamine pentaacetic acid (Gd-DTPA, Magnevist®, Bayer Schering Pharma AG) and the twelve-membered ring structure Gd(III)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (Gd-DOTA, Dotarem®, Guerbet).Important physicochemical factors influencing the proton relaxivity of a gadolinium(III) complex will be discussed in the first section. In order to improve the contrast of a MR image, much effort has been made to increase the proton relaxation rate of MRI contrast agents. In general, appropriate modulation of the water exchange rate and the rotational correlation time of the complex can significantly enhance the overall relaxivity. A rigid macromolecule formed by covalent or non-covalent interactions displaying an increased rotational correlation time should be accompanied by a corresponding optimal water residence time and within this search, no loss of the thermodynamic stability or kinetic inertness of the complex may occur.
Despite the high spatial resolution and tissue penetration of MRI, this technique suffers from a low sensitivity. Much effort has been made to increase the sensitivity and to improve the relaxivity, also at higher magnetic field strengths. However, in certain cases it is necessary to validate imaging experiments by more than one approach. As the optical imaging technique is much more sensitive than MRI, the combination of these two techniques may result in images which reveal more details than when the two techniques are used separately. However, simultaneous administration of two different kinds of contrast agents needed for two imaging techniques may be problematic, as they often may not exhibit the same pharmacokinetics. Recently, the trend has shifted toward the development of bimodal contrast agents in order to investigate samples in exquisite detail.3 This review reports the most recent developments in the field of combined MRI/optical probes and indicates new areas that remain to be explored.
2 Design of efficient MRI contrast agents
2.1 Physicochemical considerations
The efficacy of contrast agents with regard to their application in MRI is defined by their relaxivity. This property describes the ability of a 1 mM solution of CA to enhance the relaxation rate of solvent nuclei in close vicinity of the paramagnetic entity, which increases signal intensity in a magnetic resonance image. Already in the period between 1948 and 1966, Solomon, Bloembergen and Morgan among others explored in detail the relaxation phenomena leading to the establishment of the Solomon, Bloembergen and Morgan (SBM) theory.4–6 In the presence of a paramagnetic compound, both the longitudinal 1/T1 and transverse relaxation rate 1/T2 of solvent (water) nuclei will be increased. The observed solvent relaxation rate is defined by eqn (1) consisting of a diamagnetic term (1/Ti,d) corresponding to the relaxation rate in the absence of a contrast agent and a paramagnetic term (1/Ti,p) representing the contribution of the paramagnetic entity.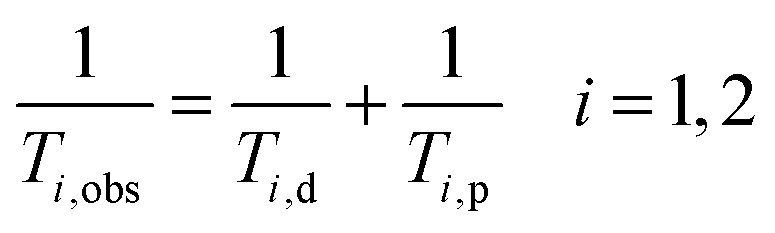 | (1) |
The paramagnetic contribution is directly proportional to the added gadolinium concentration:
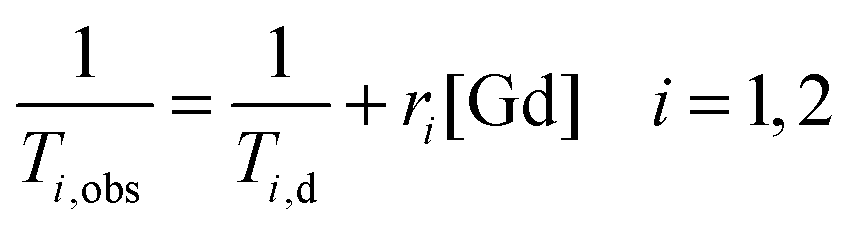 | (2) |
The paramagnetic relaxation of water protons is due to dipole–dipole interactions between the proton nuclear spin and the fluctuating local magnetic field generated by the unpaired electron spins of the paramagnetic substance.7 This effect can be separated into two components representing the inner and outer sphere interactions:
 | (3) |
In this equation, the superscript IS refers to the increased relaxation rate of water molecules directly coordinated to Gd(III) and OS stands for the paramagnetic influence on bulk water molecules diffusing in the near environment of the complex. In some cases, a second sphere contribution is taken into account arising from water molecules, which are hydrogen bonded to the chelating unit, so eqn (3) can also be written as follows:
| ri = rISi + r2ndi + rOSi i = 1, 2 | (4) |
The observable NMR signal reports on the proton longitudinal relaxivity of bulk water protons which are in chemical exchange with Gd(III)-bound water nuclei according to the following equation:8
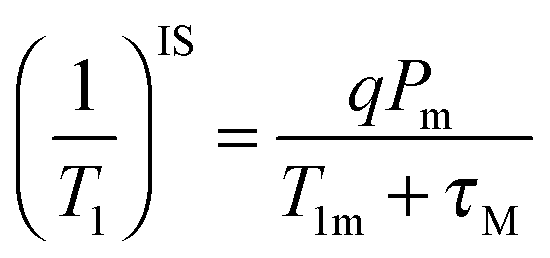 | (5) |
P m is the mole fraction of bound solvent nuclei, q is the number of water molecules in the first coordination sphere of the complex (also known as the hydration number), τM stands for the residence time of the solvent nuclei in the inner sphere, with kex = 1/τM, and T1m is the longitudinal relaxation rate of the coordinated water protons. In the case of fast water exchange (τM ≪ T1m), the relaxivity enhancement experienced by the bulk nuclei is equal to that for the bound nuclei.
According to the Solomon–Bloembergen–Morgan equations, the relaxation mechanism operating on the inner sphere water protons can be subdivided into the dipole–dipole (DD) and scalar or contact (SC) interactions.
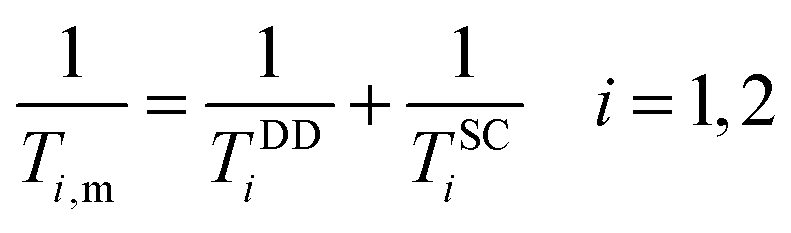 | (6) |
The dipole–dipole mechanism (1/TDDi) is governed by the reorientation of the nuclear spin–electron spin vector, by electron spin relaxation and by the water exchange rate, while the scalar term (1/TSCi) does not depend on the reorientation of the molecule, but only on electron spin relaxation and water exchange. The scalar contribution to the longitudinal relaxation rate is usually very small, since the bond formation in Gd(III) complexes is ionic and the water proton is separated from the metal ion by two bonds. Consequently, this term can be neglected. On the other hand, the important dipole–dipole interactions are influenced by a set of parameters, related to each other as follows:
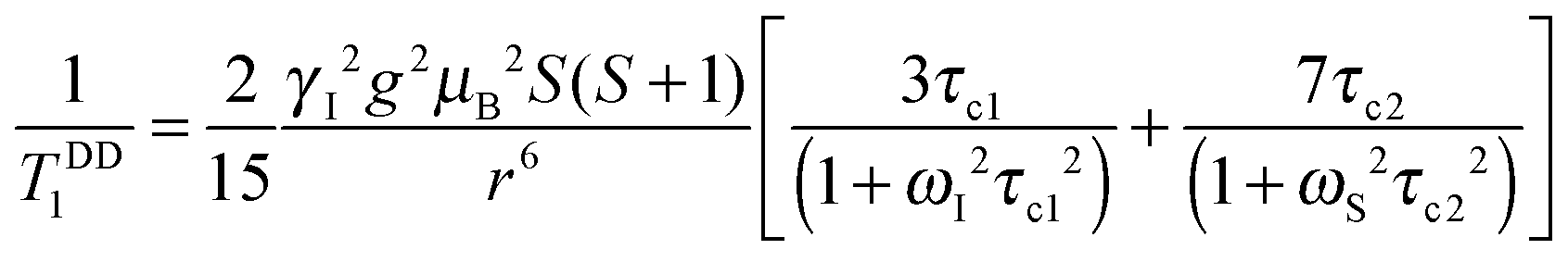 | (7) |
 | (8) |
Referring to eqn (5), we can conclude that the longitudinal relaxation rate (1/T1) will be enhanced if the overall correlation time (τc) increases. If the water residence time is rather long (T1m ≪ τM), then it will be the main factor limiting proton relaxivity. In contrast, in the case of fast water exchange (τM ≪ T1m), the relaxivity efficiency will depend on the relaxation rate of the coordinated protons (T1m), which in turn is determined by τR, τM and Tie. Maximum relaxivity is achieved when the inverse correlation time 1/τci approaches the proton Larmor frequency ωI. The remaining challenge exists in the fact that τR, τM and Tie have to be simultaneously optimized in order to obtain highly efficient contrast agents. Fig. 1 illustrates the key factors affecting proton relaxivity of a gadolinium(III) complex.
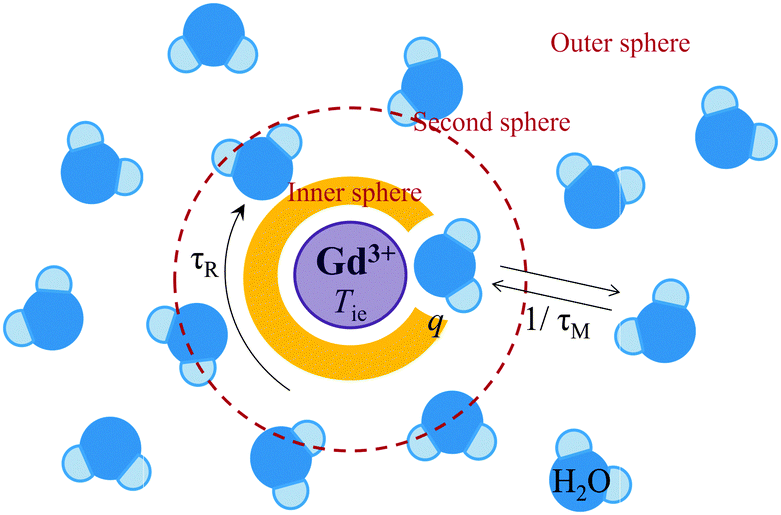 | ||
| Fig. 1 Physical parameters influencing the relaxation efficiency of MRI contrast agents at the molecular level. | ||
Thanks to extensive studies on a broad variety of gadolinium complexes based on DTPA or DOTA scaffolds and their derivatives, a relatively good insight into the essential requirements for designing new contrast agents has been achieved. Numerous papers and reviews have been published, describing not only the determining factors for obtaining highly efficient contrast agents, but also noting the difficulties that need to be taken into account.1,9–15 In the next section, a short overview of the most important tunable parameter and a discussion of corresponding recent results are given.
2.2 Molecular tumbling
One of the most critical variables affecting relaxation at the currently applied field strength (1.5 Tesla, 64 MHz) is the rotational correlation time τR.1 It was quickly found that the effective correlation time τc (eqn (8)) was dominated by this parameter. A higher τR value means slower molecular tumbling of the Gd(III) chelate, which is favourable for the overall proton relaxivity. A rough estimation of τR for spherical molecules can be obtained from the Debye–Stokes equation: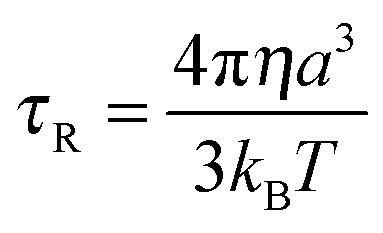 | (9) |
The rotational correlation time of small monomeric gadolinium complexes used in clinical imaging is in the order of 0.1 ns, resulting in a rather inefficient relaxation of approximately 4–5 s−1 mM−1 at low magnetic fields and a significant decrease in relaxivity at higher field strengths. However, the trend in MR imaging is toward higher fields (7 T, 300 MHz) since this results in a greater signal to noise ratio and increased resolution. During the last couple of decades, different strategies have been applied in order to slow down the molecular tumbling of contrast agents achieving better magnetic properties, even at increasing field strengths.14,17,18
The formation of supramolecular structures containing several paramagnetic ions has been explored in our research group. Copper[15]-metallacrown-5 gadolinium complexes derived from alpha-aminohydroxamic acids have been studied, for which a linear relationship between the relaxivity and the molecular mass of the metallacrown complex was found.19 A different strategy comprises the formation of a high-molecular weight tetra-metallic complex upon self-assembly of three phenantroline-substituted DTPA units around one iron(II) ion improving relaxivity to 9.5 s−1 mM−1 at 20 MHz. In vivo evaluation indicated potent contrast enhancement in organs including the liver.20
Monomeric Gd(III) chelates have been conjugated to macromolecular carriers, such as polymers leading to a notable increase of τR. It has been found that the rotational dynamic rate in hyperbranched polymers lies in between the values obtained for linear (fast rotation) and star polymers (slow rotation). However, the hyperbranched structures possess the shortest τM value since these are not too densely packed, resulting in better relaxivity r1.21 Numerous variations of dendrimer-based contrast agents for site-specific MR imaging have been synthesized.16,22 The hydrophobic structures display a preference for accumulation in liver and kidney, while functionalisation with polyethylene glycol allows imaging of the lymphatic system (Fig. 2).
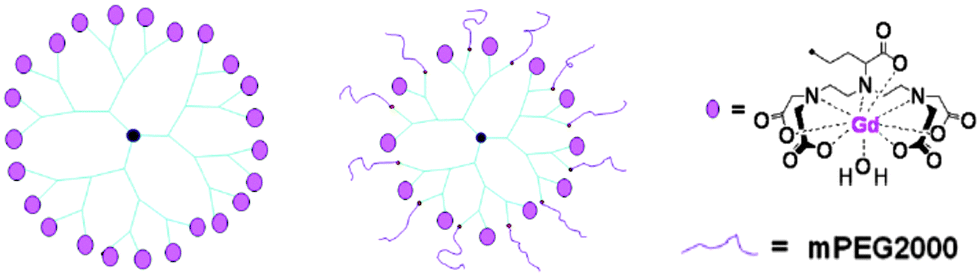 | ||
| Fig. 2 Examples of a hydrophobic G3-24Gd-DTPA (left) and hydrophilic G3-12Gd-DTPA-12mPEG (centre) dendrimer conjugated with Gd(III)-DTPA chelates.16 | ||
Likewise, conjugates of Gd(III)-based paramagnetic centres attached to monosaccharide sugars23 achieving a molecular weight of 3500 g mol−1 or of polysaccharide inulin24 of which each monosaccharide unit was attached to one Gd(III) chelate, have been studied. The average molecular weight of the latter conjugate equalled 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110 g mol−1 and the average number of Gd(III) ions per molecule was 24. For both systems, strong enhancements of the relaxation rates, up to 23 s−1 mM−1 at 20 MHz, were obtained.
110 g mol−1 and the average number of Gd(III) ions per molecule was 24. For both systems, strong enhancements of the relaxation rates, up to 23 s−1 mM−1 at 20 MHz, were obtained.
The incorporation of amphiphilic Gd(III) complexes into slowly tumbling micelles or liposomes also led to higher proton relaxivities.12,25 The aggregates are formed by hydrophobic interactions between the lipid tails while the hydrophilic moieties of the molecules face water. The formation of a micellar monolayer structure or a liposomal bilayer encapsulating an aqueous core depends on the nature and the relative sizes of the hydrophobic and hydrophilic parts. Very often, cholesterol or phospholipids are added in order to acquire more stable and monodisperse aggregates. During the last decade, many paramagnetic micelles of about 20–30 nm have been synthesized, achieving relaxivities of 18–25 s−1 mM−1 at 20 MHz and 37 °C.27–30 The liposomal vesicles as shown in Fig. 3 benefit from high observed relaxation rates from 10–15 s−1 mM−1 up to 30 s−1 mM−1, under the same conditions induced by the Gd(III) complexes entrapped in the inner aqueous cavity, in addition to the chelates at the external periphery.26,31–33
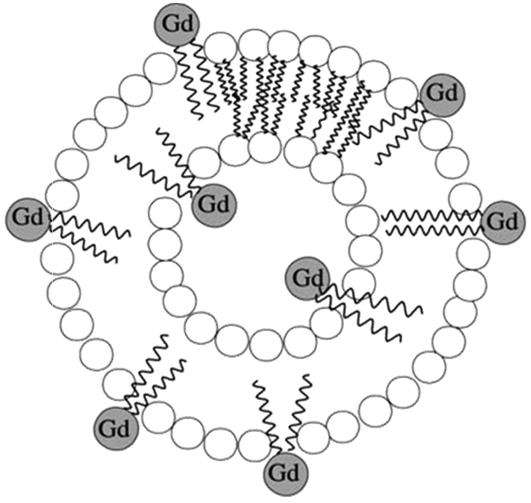 | ||
| Fig. 3 Illustration of a paramagnetic liposome loaded with amphiphilic Gd(III) complexes.26 | ||
A relatively accessible approach to restrict rotational motion involves non-covalent binding of the chelating ligand to proteins, such as human serum albumin (HSA) – the most abundant protein in blood. The reported albumin-targeted angiography agents generally display improved relaxivity (8–1534,35 to even 4036 s−1 mM−1 at 37 °C and 20 MHz) in the presence of 4% HSA. However, after elucidating the molecular mechanism, it was found that the protein itself can diminish hydration of the metal complex and/or the rate of water exchange due to limited access to the water binding site.10,37
Very high relaxivity values were obtained by inserting Gd(III) ions into nano-sized particles due to the fixation of numerous paramagnetic centres in a compact volume.2 The surface of gold nanocrystals has been coated with thiol derivatives of DTPA38,39 or DTTA40 leading to 2–3 nm particles with approximately 150 chelating units able to bind a gadolinium ion (Fig. 4). The overall relaxation enhancement per mM nanoparticle was calculated to be 600 and 3000 s−1 respectively. Immobilizing Gd(III) ions inside GdNaY-zeolite cavities showed increased proton relaxivity, but the efficiency was limited to 33 s−1 mM−1 by slow diffusion of water molecules from the interior of the silica-based particle (60–100 nm) to the bulk water.41 This limitation could be overcome by increasing the volume of the inner cages, improving the paramagnetic relaxation enhancement to 45 s−1 mM−1.42 Another family of silica nanoparticles consists of nanocontainers43 and mesoporous nanoparticles.44 The cylindrically shaped systems expose about 370 Gd(III)-DOTA units per zeolite crystal exhibiting an overall relaxation rate of 11000 s−1 per mM nanoparticle. One more example comprises ultrasmall (5–10 nm) Gd2O3 nanocrystals capped with diethylene glycol chains.45,46 The relaxation enhancement per particle reaches 39800 s−1 mM−1.
 | ||
| Fig. 4 Covalent binding of Gd(III)-DTTA to a gold nanoparticle.2,40 | ||
Despite the significant efforts in order to increase the molecular tumbling time of the MRI contrast agent, the theoretical maximum relaxation enhancement per Gd(III) ion (∼100 mM−1 s−1) has not yet been reached.1 Unfortunately, the rigidity of the formed macromolecular compound is very often accompanied by local motion of the gadolinium complex. Careful attention should be given to the flexibility of the linker group during the design of new high-molecular weight contrast agents. The total rotational dynamics can be moderated by considering the construction of polymer conjugates or via multilocus binding of the complex resulting in better fixation at the target micelles, liposomes33 or proteins.47
3 Design of MRI/optical contrast agents
Optical imaging is characterized by a low detection limit in the sub-micromolar scale versus millimolar for commercial T1 contrast agents. Optical imaging offers good resolution as well, but it is limited by a low tissue penetration. Light situated in the visible region (400–700 nm) cannot propagate more than 5 mm in tissue whereas light of higher wavelengths is less absorbed by physiological chromophores such as hemoglobin and melanin.48 Numerous reports affirm that NIR light of 700–900 nm can cross a few centimetres of tissue by multiple scattering, enabling in vivo fluorescence imaging.49,50 Luminescent/MRI bimodal agents have been developed exploiting the emissive properties of organic dyes, transition metal complexes or lanthanide ions.3,51–53 By this approach, the high resolution of magnetic resonance imaging and the high sensitivity of optical imaging are combined in one single probe displaying the same biodistribution for each diagnostic technique.3.1 Attachment of a MRI contrast agent to an organic dye
Conjugate peptides containing a Cy5.5-like dye and a Gd-DTPA chelate54 or fluorescein and Gd-DOTA (Fig. 5)55 have been reported. Good in vivo fluorescence properties and an average four-fold increase of the longitudinal relaxation rate compared to the parent gadolinium chelates were obtained. Another approach consists of the synthesis of Gd(III) hydroxycarbonate nanoparticles coated with a silica shell embedded with rhodamine B (RhB).56 These particles behave as moderate T1 or T2 relaxing contrast agents. They are clearly visible via the characteristic red emission of RhB with a low degree of photobleaching due to their incorporation inside the silica matrix. Furthermore, BODIPY-DOTA derivatives have been designed57 and can also be employed as a dual MR/optical imaging reporter characterized by considerably high quantum yields (20–50% in water). Intracellular delivered star polymers containing a blue fluorescent fluorene core were modified in order to chelate gadolinium(III) or europium(III). Rapid water exchange in the highly hydrated star polymer provided large ionic relaxivities up to 84 s−1 mM−1 at 20 MHz and 310 K.58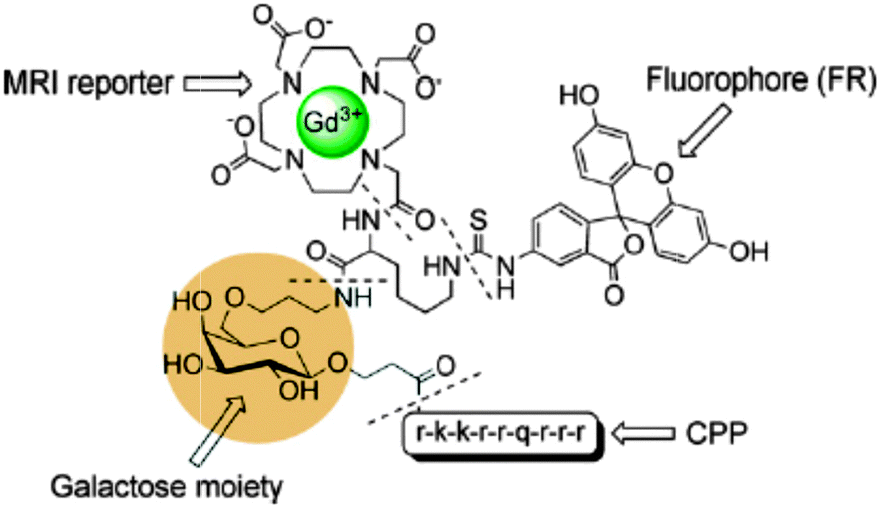 | ||
| Fig. 5 A cell penetrating peptide, functionalized with a galactose moiety to report on the β-galactosidase enzyme, that incorporates two imaging moieties Gd-DOTA and fluorescein.55 | ||
3.2 Heteropolymetallic complexes
The synthesis of a ditopic ligand containing a gadolinium coordinating unit and a moiety with affinity for luminescent transition metals allows the formation of a self-assembling bimodal heterometallic complex. In 2008, Faulkner et al. linked Gd(III)-DOTA to a rhenium(I) complex exhibiting red luminescence59 (Fig. 6a). A relatively long emissive lifetime (τLMCT) of 0.24 μs is observed for Gd3Re(Bpy)(CO)3, since there is no competitive quenching pathway through the lanthanide ion. The high relaxivity (8.6 s−1 mM−1 at 500 MHz) in aqueous solution is in line with the existence of a diaqua complex which tumbles slowly in solution. Later, six Gd(III)-DTTA complexes have been assembled around ruthenium(II) via three bipyridine coordinating units (Fig. 6b). The NMRD profile of Ru(Gd2bpy-DTTA2)3 showed a relaxivity hump between 10 and 100 MHz characteristic of slowly rotating molecules. The quantum yield of Ru(bpy)3 compounds in aqueous solutions is modest, lies in the range 0.001–0.1%, depending on pH and temperature, and can be accompanied by formation of singlet oxygen.60 Our group has designed novel metallostar complexes in which the longitudinal relaxivity of Gd(III)-DTPA was combined with a green-emissive titanium(IV) catecholate61 or aluminum(III) 8-hydroxyquinoline62 core (Fig. 6c and d). The complex (Gd4)3Ti(H2O)3 exhibited green broad-band emission with a maximum of 490 nm but with a relatively low absolute quantum yield of 0.054%. The decreased tumbling rate of the supramolecular complex led to an enhanced r1 relaxivity up to 12.3 s−1 mM−1 per Gd(III) ion at 20 MHz and 310 K that corresponds to 36.9 s−1 mM−1 per metallostar molecule. Formation of the metallostar compound (Gd5)3Al(H2O)3 resulted in a r1-value of 10.9 s−1 mM−1 per Gd(III) ion, corresponding to 32.7 s−1 mM−1 per heteropolymetallic complex. In addition to the high relaxivity values, (Gd5)3Al(H2O)3 exhibited green broad-band emission luminescence upon excitation at 367 nm. Heteropolymetallic complexes based on a ruthenium(II) 1,10-phenantroline centre were also synthesized and are depicted in Fig. 6e and f.63,64 The r1 relaxivities of [Ru(bpy)2]2GdDTPA(ph-phen)2 and (Gd-DTPA-ph-phen)3Ru per Gd(III) ion at 20 MHz and 310 K were equal to 7.0 and 12.0 s−1 mM−1, respectively. Both compounds exhibited bright-red luminescence centred at 610–620 nm and the quantum yields were found to be 4.7% and 4.8%. The luminescence lifetime of [Ru(bpy)2]2GdDTPA(ph-phen)2 equals 0.54 μs which is long enough to permit reasonable gating of any fluorescent background (for which lifetimes of <10 ns are typical) in applications such as time resolved microscopy. Unfortunately, in general, the broad emission bands and relatively short luminescence lifetimes (<1 μs) of transition metal chelates are less favourable for in vivo measurements since the observed emission can be hardly differentiated from background autofluorescence. Relaxation values and photophysical details of the discussed f–d heteropolymetallic complexes are summarized in Table 1.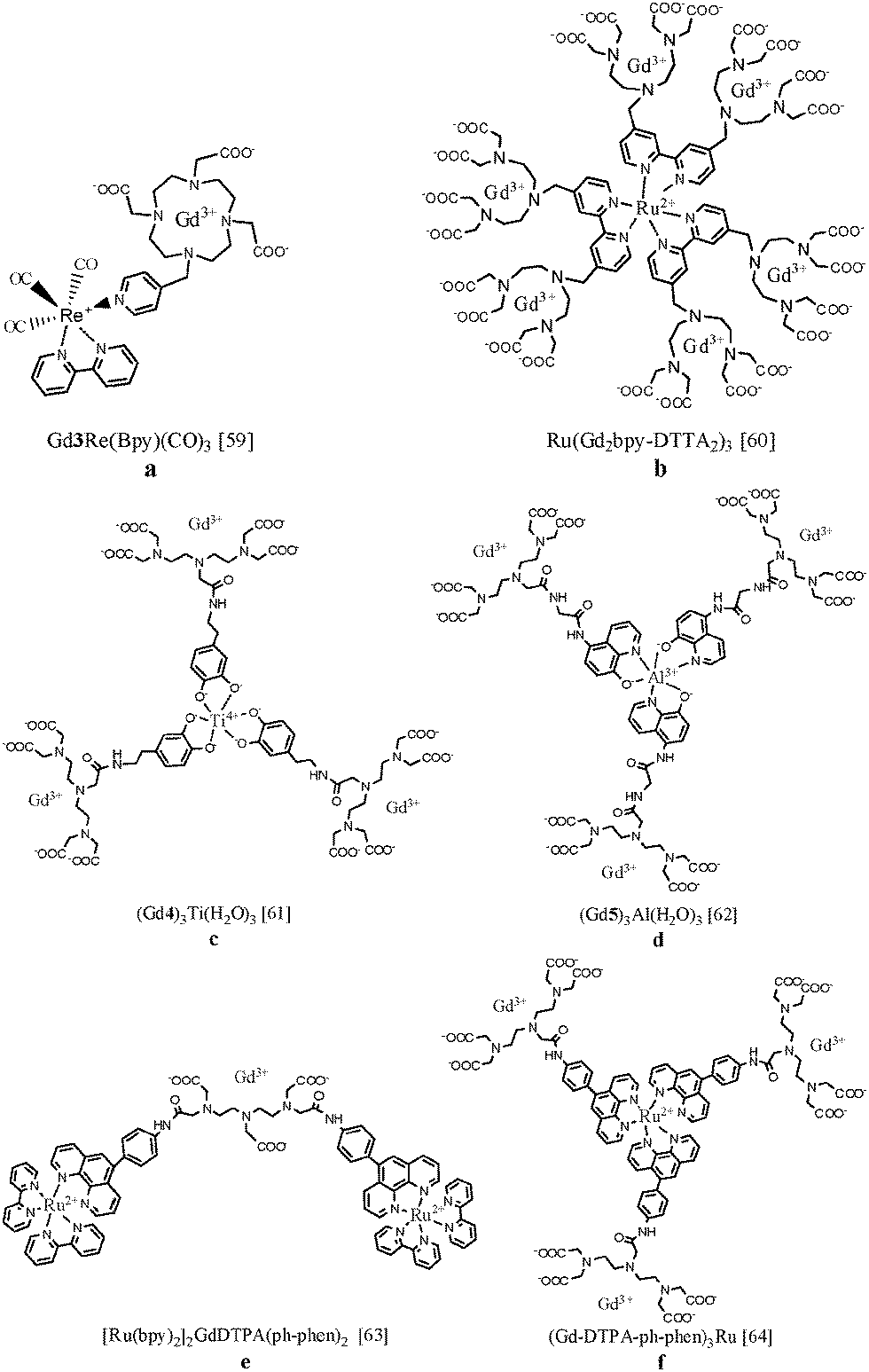 | ||
| Fig. 6 Chemical structures of lanthanide-transition metal heteropolymetallic complexes for potential bimodal MR/optical imaging.59–64 | ||
| Relaxometric Ln(III) | Luminescent metal ion |
r
1a (s−1 mM−1) |
λ exc (nm) | Quantum yield Φb (%) | |
|---|---|---|---|---|---|
| a Relaxivity r1 per millimolar Gd(III) at 20 MHz and 310 K unless stated otherwise. b Quantum yield relative to a standard solution. | |||||
| Gd3Re(Bpy)(CO)359 | GdIII | ReI | 8.6 (500 MHz) | 337 | — |
| Ru(Gd2bpy-DTTA2)360 | GdIII | RuII | 23.0 | 293 | 0.1 |
| (Gd4)3Ti(H2O)361 | GdIII | TiIV | 12.3 | 380 | 0.05 |
| (Gd5)3Al(H2O)362 | GdIII | AlIII | 10.9 | 367 | 0.52 |
| [Ru(bpy)2]2GdDTPA(ph-phen)263 | GdIII | RuII | 7.0 | 440 | 4.7 |
| (Gd-DTPA-ph-phen)3Ru64 | GdIII | RuII | 12.0 | 450 | 4.8 |
3.3 Bimodal lipophilic aggregates
The two diagnostic features were also combined into supramolecular aggregates such as micelles or liposomes. The surface of liposomes encapsulating several magnetite cores has been covered with Gd(III) complexes resulting in a very high r2/r1 ratio. The high flexibility of the lipid bilayer and the ease of preparation enable the composition to be fine-tuned and permit fluorescent moieties to be incorporated in the conjugates.65 Likewise, iron oxide nanoparticles and hydrophobic luminescent polymers were encapsulated in micelles. They emit across the visible spectrum with a relatively low quantum yield of 1.2%, which is typically a problem with fluorescent-magnetic nanocomposites. In MRI studies, they exhibited a shortening effect on the T2 relaxation time.66 Liposomes containing Gd-DTPA directly attached to bis(stearyl) and fluorescent lipids could be detected via in vitro and in vivo optical imaging and were also tested for MRI. The relaxivity per millimolar nanoparticle has been estimated to be 2000 s−1 mM−1.67 In order to avoid the use of two separate signaling compounds, more robust bimodal PEGylated liposomes were made out of a lipid molecule covalently bound via a Gly–Lys linker to Gd-DOTA and rhodamine. In vitro labeling of cells could be monitored by fluorescence microscopy and resulted in an over six-fold tumour to muscle signal enhancement.68 A key concern is to ensure that the Gd-based probes remain in the aqueous compartment in order to have access to water, while it has to be taken into account that the environment can suppress the emission intensity of the fluorophores.3.4 Nanoparticles
Nanoparticles for dual imaging are typically formed starting from existing particles that already possess one imaging functionality. An extra modality is then introduced in a spherical way around the core material. Numerous bimodal systems based on core–shell quantum dots or iron oxides have been comprehensively described in several reviews.3,51,69 Quantum dots expose a large amount of free amines over the entire surface, so that gadolinium chelates can easily be attached. Hydrophobic QDs coated with the tripeptide glutathione (Fig. 7)70 or with a PEGylated silica shell71 were labeled with Gd-DOTA. The Gd-ion relaxivities r1 and r2 at 60 MHz are up to 23 s−1 mM−1 and 54 s−1 mM−1 respectively, while NIR efficiency of 8.4% resulting from the synthesized nanomaterial was obtained. Besides the coupling of an iron oxide to organic fluorophores such as rhodamine,72 AlexaFluor,73 oligothiophene74 or Cy5.5,75 the T2 superparamagnetic particles can also be encapsulated in a shell, including the most popular silica shell. Porous silica layers enable good photon-transparency, water solubility and a large load of a variety of molecules like the organic dyes, fluorescein isothiocyanate76 and rhodamine77 or inorganic fluorophores such as terbium complexes.78 Alternatively, it is possible to have both imaging probes in the same matrix material by inserting the desired components into a nanocrystal. An anti-Stokes type emission has been observed with (Gd,Yb,Tb)PO4 nanocrystals, which have been synthesized via a hydrothermal method. Relaxometric measurements reveal that they are efficient T2-weighted contrast agents while strong green luminescence of terbium(III) is generated by two-photon infra-red excitation.79 Another approach involves hybrid silica nanospheres containing a luminescent ruthenium-bipyridine core and a paramagnetic coating of silylated gadolinium complexes. At 60 MHz, they exhibit a longitudinal relaxivity of 19.7 s−1 and a transverse relaxivity of 60.0 s−1 per millimolar Gd(III).80 Because of the high payload of Gd-DTTA chelates, the nanoparticles display r1 and r2 values of 2.0 × 105 and 6.1 × 105 s−1, respectively, on a per millimolar particle basis.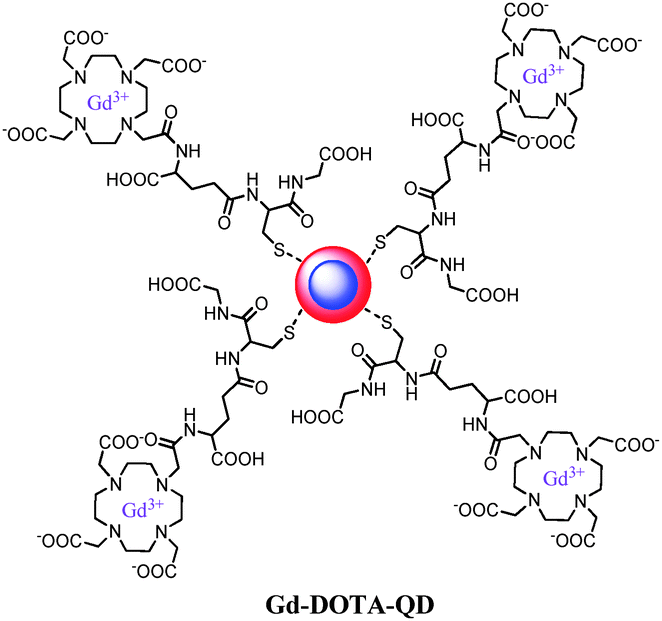 | ||
| Fig. 7 Gd(III)-DOTA functionalized cadmium-based quantum dots with a glutathione coating.70 | ||
4 Lanthanide-based bimodal systems
Although significant improvements concerning the utility of dual imaging reporters have been achieved, practical applications of the aforementioned conjugates are limited due to short luminescence lifetimes (100–300 ns), a small Stokes shift and poor resistance to photobleaching. During the last decade, lanthanide-based systems combining magnetic and optical properties have been attracting increasing attention.88 Besides the excited state lifetimes in the range of milliseconds (allowing filtering from short lifetime autofluorescence), lanthanides display a sharply spiked emission spectrum and a large energy difference between their emission bands and the absorption bands of the coordinated ligands, resulting in an exquisite luminescence. One must take into account that the f–f transitions of Ln(III) ions are characterized by low molar absorption coefficients since they are Laporte forbidden. For this reason, it is necessary to integrate an appropriate chromophore (aromatic unit) into the chelating ligand to trigger a process called ‘sensitization’. Upon absorption of light of a certain wavelength, the ligand is excited to the singlet excited state. The captured energy can be transferred to the lanthanide ion (singlet energy transfer), to the triplet state (intersystem crossing), or can be seen as fluorescence through an immediate decay to the ground state. The triplet state can subsequently pass the energy to the lanthanide ion or decays to its ground state by phosphorescence. Generally, although there are some exceptions, energy is transferred via the triplet state because the intersystem crossing is forced by the neighbouring paramagnetic lanthanide ion. In addition, energy transfer via the singlet state is not as fast as fluorescence or intersystem crossing. The emission intensity of lanthanide luminescence can significantly be reduced by a non-radiative energy dissipation via the vibronic modes of solvent molecules. Typically, this occurs by harmonic oscillators in the inner coordination sphere or in the near vicinity of the metal complex. The most common and efficient quencher is the O–H oscillator. In order to minimize this non-radiative decay effect, the lanthanide ion should be effectively shielded from the solvent by using chelating ligands that firmly bind and encapsulate the metal ion. It is not surprising that, apart from the well-known gadolinium-based MRI contrast agents, a selection of lanthanide compounds is used as luminescent probes in the visible and near infrared region for bioassay and live-cell microscopy.89,904.1 Mixed lanthanide systems
Mixtures of complexes in which the ligand is coordinated to paramagnetic gadolinium(III) or to lanthanide(III) ions emitting in the visible or NIR region have been intensively studied for their magnetic resonance and optical abilities.A pyridine-based DTPA substrate (Fig. 8a) allowing heptadentate coordination to lanthanide ions and bishydration of the complex has been synthesized. Improved MRI properties were obtained for the corresponding Gd(III) complexes (6.21 s−1 mM−1) and also NIR luminescence could be observed after complexation with neodymium(III). A luminescence quantum yield of 0.01% indicated that the presence of two water molecules bound to the Ln(III), beneficial for MRI applications, is not an absolute limitation for the development of NIR luminescent probes.81 The same ligand was attached to the surface of silica nanoparticles which was subsequently loaded with Gd(III) as well as Eu(III)/Tb(III) for relaxometry and visible luminescence, respectively.82 The aromatic backbone onto the silica surface enhanced the emission quantum yield of the Eu(III)-containing nanoparticles fivefold (0.05%) compared to similar systems without aromatic antennae. The Gd(III) paramagnetic relaxation at 20 MHz and 310 K equaled 7.95 s−1 mM−1. Derivatives of the pyridine-based DTPA ligand with an extra methoxy or triazole ring system (Fig. 8b–d) led to similar quantum yields of 0.01–0.02% for the NIR-emitting Nd(III) and Yb(III) complexes. Especially the synthon with a conjugation-expanding phenyl group displayed a somewhat increased sensitivity due to the higher extinction coefficient. Furthermore, the shift of the excitation wavelengths to higher values was beneficial for biological applications.83In vivo toxicity studies revealed that the bis-hydrated complexes were non-toxic and that they could be safely used for both magnetic resonance (MR) and optical imaging applications.
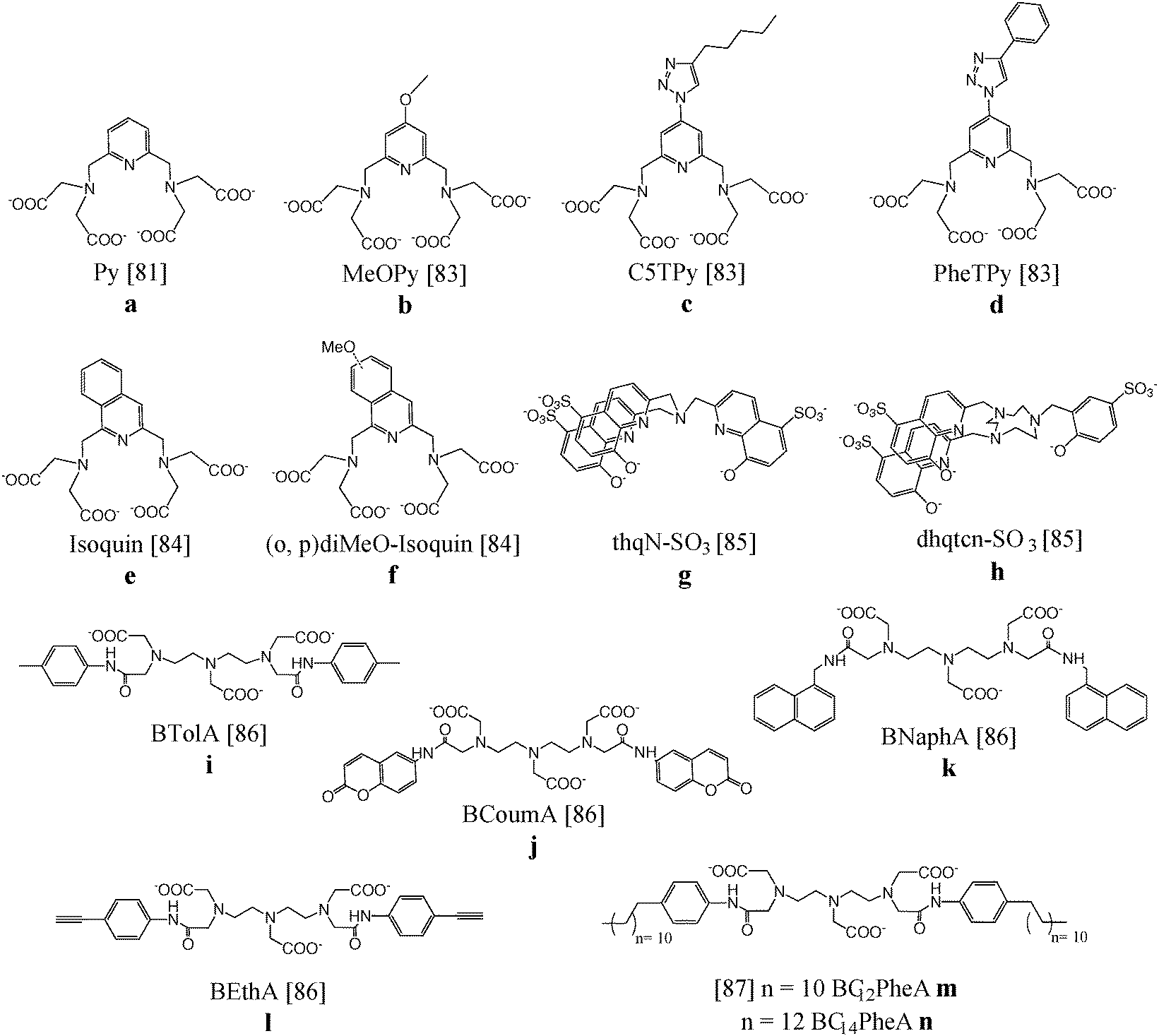 | ||
| Fig. 8 Chemical structures of chelating ligands for dual imaging applications.81–87 | ||
The luminescent properties were further optimized by the substitution of the pyridine core by isoquinoline (Fig. 8e and f). The excitation wavelength shifted over 100 nm toward lower energy in comparison to the pyridine-based analogue providing more adapted NIR emitting complexes for biological studies. The luminescence quantum yields of the Nd(III) (0.013–0.016%) and Yb(III) chelates (0.028–0.040%) are in the range of non-hydrated complexes, despite the presence of two inner-sphere water molecules. Nevertheless, good stability was obtained and promising r1 values of 8.5 s−1 mM−1 for the corresponding gadolinium(III) compounds were acquired.84
Despite the coordination of two water molecules, NIR emission could also be observed after complexation of the tripodal hydroxyquinolinate ligand, depicted in Fig. 8g, to Nd(III) or Yb(III). Unfortunately, a slow water exchange limited the expected high relaxivity for this bishydrate complex. On the other hand, the octadentate ligand based on triazacyclononane and 8-hydroxyquinolinate/phenolate binding units (Fig. 8h) was found to lead to gadolinium complexes with a relaxivity of 9.1 s−1 mM−1 at 20 MHz as a result of a long rotational correlation time, fast water exchange and slow electronic relaxation. The ligand was also a good sensitizer for near-IR emission of neodymium and ytterbium ions, displaying luminescence quantum yields up to 0.02%.85 Ln(III) complexes (Ln = Gd, Eu, Tb) of the ligands DTPA-bis-p-toluidine-amide, DTPA-bis-6-coumarin-amide, DTPA-bis-1-naphthylmethyl-amide and DTPA-bis-4-ethynylphenyl-amide (Fig. 8i–l) were prepared and studied for their bimodal MRI/optical properties.86 Eu(III) and Tb(III) derivatives in aqueous solutions exhibit characteristic red and green emission, respectively, with quantum yields of 0.73% for Eu(III)–DTPA-BNaphA and 2.5% for Tb(III)–DTPA-BEthA. The relaxivity of the Gd(III) complexes was improved compared to Gd-DTPA (±5.5 s−1 mM−1versus 3.8 s−1 mM−1 at 20 MHz and 310 K) due to a general increase of the rotational tumbling time, τR. Moreover, interaction studies in 4% HSA solutions revealed a further increase of relaxivity to values of 19 s−1 mM−1 caused by a further decrease of molecular motions. Gd(III) and Eu(III) complexes of two DTPA bisamide derivatives functionalized with p-dodecylaniline and p-tetradecylaniline (Fig. 8m and n) have been assembled with phospholipid DPPC and surfactant Tween 80® forming mixed micelles. Taking into account the sensitivity difference between magnetic resonance and optical imaging techniques, several ratios of Gd and Eu complexes were combined in one single micelle and their optical and relaxometric properties have been characterized in detail. The Eu(III) micelles exhibited quantum yields in the range of 1.0% and the relaxivity per Gd(III) ion reaches a maximum value of 16.0 s−1 mM−1 for the Gd-DTPA-BC14PheA assemblies, due to large τR values of 6.6 ns. The concentration ratio of 20:1 Gd/Eu compounds in the micelles provided the optimal required bimodal performance which can be useful in the search for other potential bimodal assemblies.87 The most relevant data regarding relaxometric and photophysical features of the aforementioned lanthanide complexes are listed in Table 2.
| Ligand | Relaxometric Ln(III) | Luminescent Ln(III) |
r
1a (s−1 mM−1) at 20 MHz |
r
2b (s−1 mM−1) at 500 MHz |
λ exc (nm) | Quantum yield Φc (%) |
|---|---|---|---|---|---|---|
| a Relaxivity r1 per millimolar Gd(III). b Relaxivity r2 per millimolar of Ln(III) ion. c Quantum yield relative to a standard solution. | ||||||
| Py81 | GdIII | NdIII/YbIII | 6.2 | — | 267 | 0.01/0.02 |
| SiO2@APS/PMN82 | GdIII | EuIII, TbIII | 7.9 | 56.4 | 270 | 0.05 |
| MeOPy83 | GdIII | NdIII/YbIII | — | — | 250 | 0.01/0.02 |
| C5TPy83 | GdIII | NdIII/YbIII | — | — | 290 | 0.01/0.02 |
| PheTPy83 | GdIII | NdIII, YbIII | — | — | 320 | 0.01 |
| Isoquin84 | GdIII | NdIII/YbIII | 8.5 | — | 320 | 0.02/0.04 |
| diMeO-isoquin84 | GdIII | NdIII/YbIII | — | — | 360 | 0.02/0.04 |
| thqN-SO385 | GdIII | NdIII, YbIII | 5.7 | — | 375 | 0.01 |
| dhqtcn-SO385 | GdIII | NdIII, YbIII | 9.1 | — | 375 | 0.02 |
| BTolA86 | GdIII | EuIII/TbIII | 4.1 | — | 280 | 0.3/0.6 |
| BCoumA86 | GdIII | EuIII/TbIII | 5.1 | — | 330 | 0.6/1.2 |
| BNaphA86 | GdIII | EuIII/TbIII | 6.4 | — | 292 | 0.7/1.0 |
| BEthA86 | GdIII | EuIII/TbIII | 5.7 | — | 290 | 0.6/2.5 |
| BC12PheA Mic87 | GdIII | EuIII | 14.2 | — | 290 | 1.0 |
| BC14PheA Mic87 | GdIII | EuIII | 16.0 | — | 290 | 1.1 |
| BTolA86 | DyIII | DyIII | 0.2 | 19.0 | 280 | 0.3 |
| BCoumA86 | DyIII | DyIII | 0.1 | 19.5 | 330 | 0.4 |
| BNaphA86 | DyIII | DyIII | 0.1 | 13.2 | 292 | 0.3 |
| BEthA86 | DyIII | DyIII | 0.1 | 27.4 | 290 | 0.5 |
| BC12PheA Mic87 | DyIII | DyIII | 0.2 | 36.0 | 290 | 1.0 |
| BC14PheA Mic87 | DyIII | DyIII | 0.2 | 33.4 | 290 | 1.0 |
Gd(III), Eu(III) and Tb(III) tris(amide) derivatives of DOTA in which one of the macrocyclic backbone nitrogen atoms has been replaced by a pyridine nitrogen (Fig. 9a), have been examined as potential contrast agents for imaging applications. The gadolinium chelates displayed high proton relaxivities of 7.9 s−1 mM−1 at 20 MHz and 310 K due to rapid water exchange and a large outer-sphere contribution, while the europium and terbium complexes revealed clearly visible luminescence for the q = 2 species.91 An aryl-phosphonate moiety was attached to DOTA since it can directly coordinate to the lanthanide ion maintaining a low hydration number and it acts as an effective antenna sensitizing green Tb(III) luminescence, obtaining quantum yields of 0.2–0.3% (Fig. 9b). Despite the eight-fold coordination to Gd(III), moderate relaxation properties around 5.0 s−1 mM−1 including second sphere contributions were obtained. Structural variations of the ligand, such as the alkyl chain length or the nature of the phenolic para substituent, enable further fine tuning possibilities.92 A somewhat different approach starts from dinuclear Gd(III), Eu(III) or Tb(III) complexes comprising DO3A units linked by a 4,4′-dimethyl-2,2′-bipyridine or a 2,6-bis(1H-pyrazol-1-yl)pyridine spacer (Fig. 9c and d). NMRD profiles characteristic of slowly tumbling compounds with high relaxivities between 10 and 60 MHz were observed due to the formation of nanosized aggregates. The relaxivity recorded at 20 MHz and 298 K equaled 13.7 s−1 mM−1 per molecule with two coordinated Gd(III) ions. The overall luminescence quantum yields were determined to be up to 0.08% for Eu(III) and 0.50% for Tb(III), pointing to an effective sensitization of the metal ion, especially for the compounds exhibiting terbium-centred emission.93 Relaxivity values and luminescence details of the DOTA derivatives are collected in Table 3.
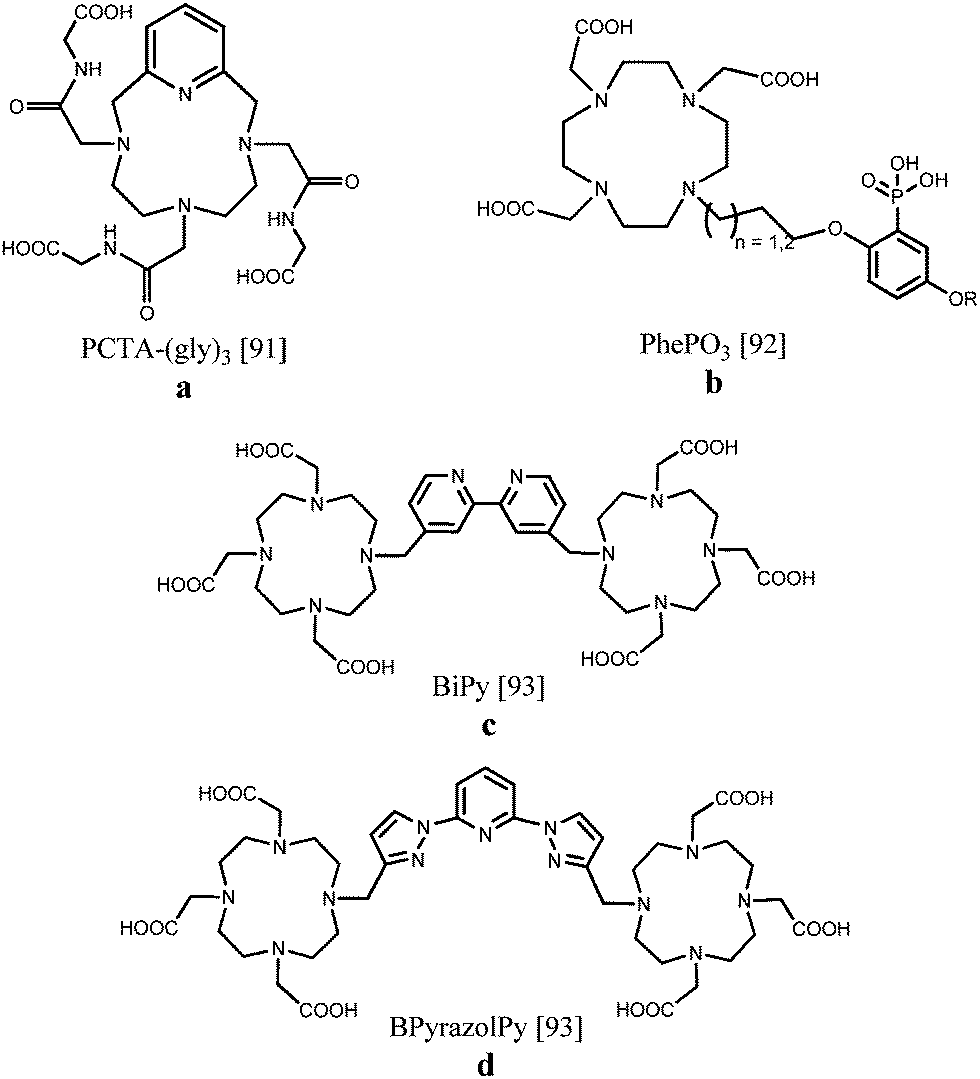 | ||
| Fig. 9 DOTA-based scaffolds allowing bimodal MR/optical imaging.91–93 | ||
| Ligand | Relaxometric Ln(III) | Luminescent Ln(III) |
r
1a (s−1 mM−1) at 20 MHz |
λ exc (nm) | Quantum yield Φb (%) |
|---|---|---|---|---|---|
| a Relaxivity r1 per millimolar Gd(III). b Quantum yield relative to a standard solution. | |||||
| PCTA-(gly)391 | GdIII | EuIII, TbIII | 9.9 | 284 | 0.05 |
| PhePO392 | GdIII | TbIII | 5.0 | 300 | 0.2–0.3 |
| BiPy93 | GdIII | EuIII/TbIII | 5.1 | 287 | 0.08/0.25 |
| BPyrazolPy93 | GdIII | EuIII/TbIII | 6.9 | 287 | 0.01/0.50 |
NIR and magnetic resonance studies have been performed exploiting the properties of the dysprosium(III) and ytterbium(III) ions. These lanthanides were chelated with a hexadentate ligand and subsequently conjugated to an esteramide dendrimer in order to improve bioavailability and solubility. At 60 MHz and 310 K, the Dy-containing macromolecules had the largest r1 relaxivity, 7.60 s−1 mM−1, while the Yb-analogues apparently displayed the largest r2 relaxivity, 23.0 s−1 mM−1. Furthermore, a Yb(III) NIR quantum yield of 0.2% was measured, thereby establishing an ytterbium-based bimodal NIR/T2 contrast agent.94 The six DTPA bisamide derivatives (Fig. 8i–n) have also been coordinated to dysprosium(III) and the magnetic and optical properties of the corresponding complexes and micelles were examined in detail.95 The complexes displayed characteristic Dy(III) emission with quantum yields of 0.3–0.5% and quantum yields up to 1% were obtained for the micelles. The transverse relaxivity r2 per Dy(III) ion at 500 MHz and 310 K reaches a maximum value of 27.4 s−1 mM−1 for Dy-DTPA-BEthA and 36.0 s−1 mM−1 for the Dy-DTPA-BC12PheA assemblies which is about 40-fold higher compared with a value of 0.8 s−1 mM−1 for Dy-DTPA. The efficient T2 relaxation is sustained by the high magnetic moment of the dysprosium ion, the coordination of water molecules with slow water exchange kinetics and long rotational correlation times. These findings open the way to the further development of bimodal optical and magnetic resonance imaging probes starting from single lanthanide compounds.
4.2 Heterometallic lanthanide complexes
The controlled site-selective synthesis of heterometallic lanthanide complexes remains a challenge due to the very similar coordination behaviour across the lanthanide series. For this reason it is important to highlight a few known approaches to successful preparation of this type of compounds. In a first example, two Tb(III)-DOTA chelates bearing a benzyl function have been linked by a DTPA binding site. Subsequently, Yb(III) was incorporated into the DTPA unit, yielding a trinuclear complex containing two different luminescent ions. After direct excitation of terbium at 488 nm, near-IR sensitized emission of ytterbium at 980 nm could be observed, inferring energy transfer between the two lanthanide metals due to their close proximity96 (Fig. 10a). On the other hand, europium(III) and terbium(III) have been consecutively integrated into a branched tetrapeptide-based DOTA/DTPA bis-chelate. The luminescence properties were compared in a series of alcohol solvents, and the Eu(III) emission band increased at the expense of Tb(III) emission with decreasing solvent polarity, offering potential applications in time-resolved solvent polarity sensing97 (Fig. 10b). Orthogonal protection of equivalent binding sites in a given ligand has been reported to allow selective lanthanide coordination. Fig. 10c depicts the discretely synthesized Tb(III)–Yb(III) bismetallic complex with a 4-aminoaniline bridge between two DOTA sites. The compound displayed sensitized luminescence in the visible and near IR region from both the Tb(III) and Yb(III) centres, respectively.98 Another strategy concerns the linkage of related bismetallic complexes containing Yb(III) or Nd(III) using diazotation reactions. Upon formation of the tetrametallic compound, an azo-dye is simultaneously generated which ensures efficient lanthanide sensitization leading to emission in the near IR region of the spectrum99 (Fig. 10d). Binuclear lanthanide complexes as presented in Fig. 10e have been obtained through “click”-chemistry.100 Copper catalyzed cycloaddition reactions with alkyne-functionalized Ln(III)-DO3A and benzyl azides lead to the formation of linked lanthanide complexes incorporating triazole units. It has been found that coordination of one of the triazole nitrogen atoms to the metal centre is able to switch on the luminescence or to change the spectroscopic properties by a change in the coordination environment. As an alternative, the Ugi four-component reaction has been applied to synthesize ditopic chelators by reacting different aldehydes, isocyanides and DOTA monoamides as amino and acid components (Fig. 10f). Relaxivity studies on the Gd2-complexes with R = octadecyl revealed enhanced T1 relaxation values of 20.0 s−1 mM−1 at 20 MHz and 310 K due to aggregation of the lipophilic structures forming micelles or even 38.1 s−1 mM−1via non-covalent interactions in the presence of HSA.101 The introduced “click”- and Ugi-reactions have been combined in a novel strategy creating tetranuclear lanthanide complexes containing two dysprosium ions, a terbium and a europium ion102 (Fig. 10g). The emission spectrum of the Dy2TbEu compound exhibits the luminescence transitions from the three different lanthanide ions. In such systems, the overall emission spectrum can be tuned by varying the building blocks and their relative distances.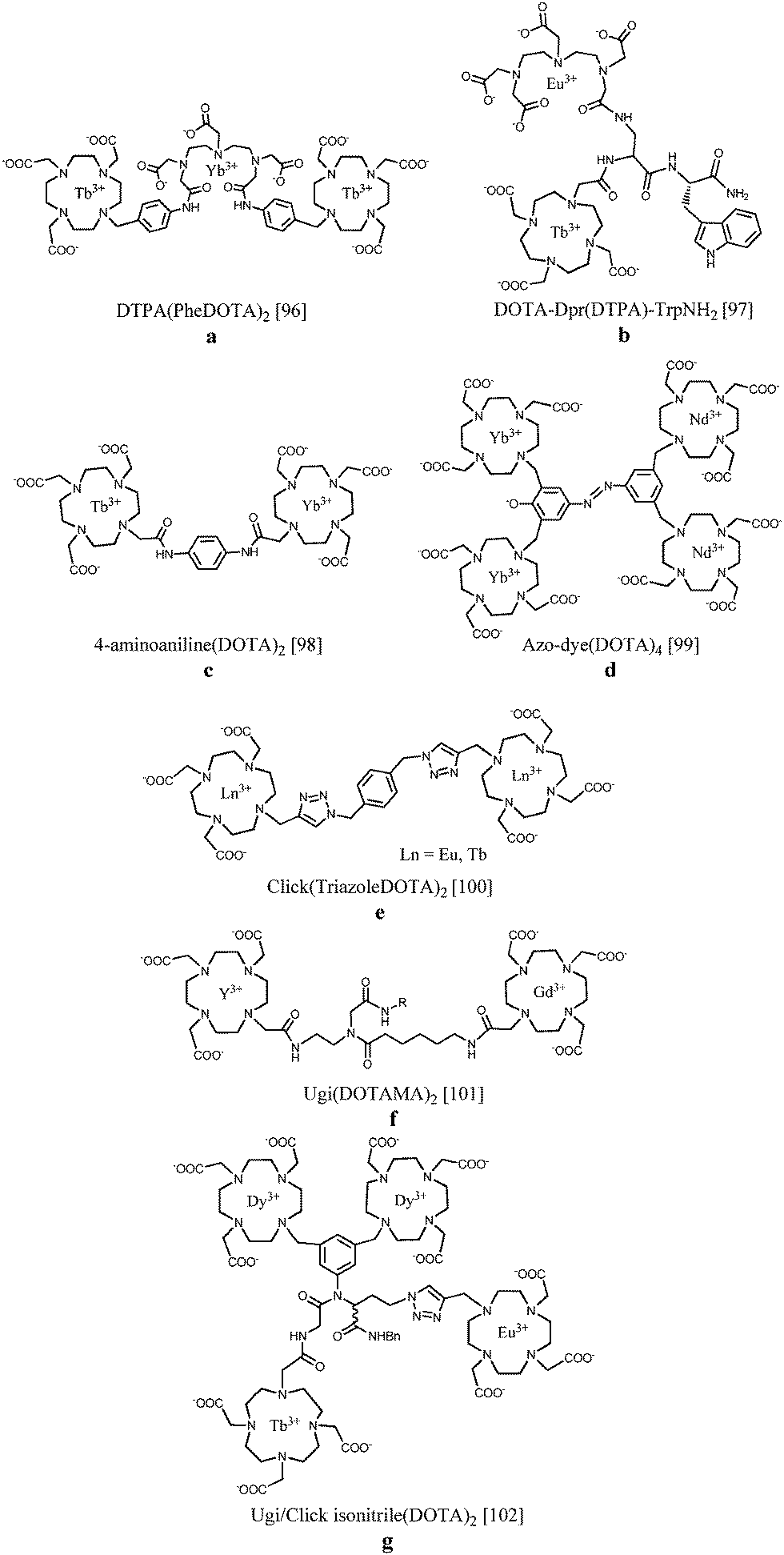 | ||
| Fig. 10 Sequentially metallated heteronuclear lanthanide complexes.96–102 The coordination bonds have been omitted for clarity. | ||
Several routes towards dual MR/optical imaging lanthanide-based systems have been explored. An aryl-group, functioning as the antenna, has been directly integrated into an acyclic polyaminocarboxylate chelator which was attached to a DOTA ring structure (Fig. 11a). The longitudinal relaxivity of the compound incorporating Gd(III) in the DOTA unit was calculated to be 5.48 s−1 mM−1. The antenna was able to sensitize both Eu(III) as well as Tb(III) when coordinated to the ligand. Compared to DOTA, the acyclic moiety is less kinetically and thermodynamically stable, however, the required emission and relaxometric characteristics have been demonstrated.103 As an alternative, two DOTA units bearing Gd(III) and Nd(III), Er(III) or Yb(III) were linked together by a diaminoanthraquinone (AQ) chromophore (Fig. 11b). The 1,4-diamino-substituted AQ has been compared with the 1,5-substitution pattern concerning relaxometric and optical properties.104 DNA intercalation studies showed that stronger binding interactions were obtained with 1,4-diamino-substituted AQ, promoting a relaxivity of 5.8 s−1 per mM Gd(III) at 20 MHz and 310 K when bound to the macromolecular DNA duplex due to slower rotational rates and the immobilization of the Gd(III) unit. Characteristic lanthanide-based emission in the near IR region could be observed for both chromophores, however, the 1,4-substituted AQ species possessed longer wavelength visible absorption. In 2011, Lewis et al.105 demonstrated the importance of the proximity of the lanthanide centres during the design of multimodal probes. In their work, two DTPA-bis-p-thiophenol amide derivatives were assembled via two disulphide bonds (Fig. 11c). The macrocyclic compound in which Gd(III) and Eu(III) were integrated displayed a 15% reduction in europium emission intensity compared to the non-macrocyclic Eu(III) and Gd(III) complexes. This observation has been ascribed to the increased rate of non-radiative emission pathways induced by the presence of internal gadolinium. Nevertheless, regarding the imaging sensitivities, the proposed structures provide interesting scaffolds for bimodal MR/optical applications. A novel synthetic strategy towards a heteropolymetallic lanthanide complex with selectively incorporated gadolinium and europium ions has been reported. For this purpose, a ditopic ligand able to coordinate with two different lanthanide ions was used.106 A DTPA-based moiety taking care of gadolinium(III) chelation is linked via an amide bond to a pyridine-2,6-dicarboxylate derivative ensuring self-assembly around europium(III) (Fig. 11d). Due to the easy accessibility of water to the three paramagnetic components per molecule, a relaxivity of up to 31.0 s−1 mM−1 is achieved. On the other hand, the exclusion of water from the first coordination sphere of Eu(III) results in a bright emissive compound exhibiting a quantum yield up to 10%. Stability studies revealed relatively high binding constants, paving a way for further research towards heteropolymetallic f–f assemblies offering interesting applications in the field of dual imaging. Table 4 displays a summary of the relaxation rate values and luminescence quantum yields of the selected complexes.
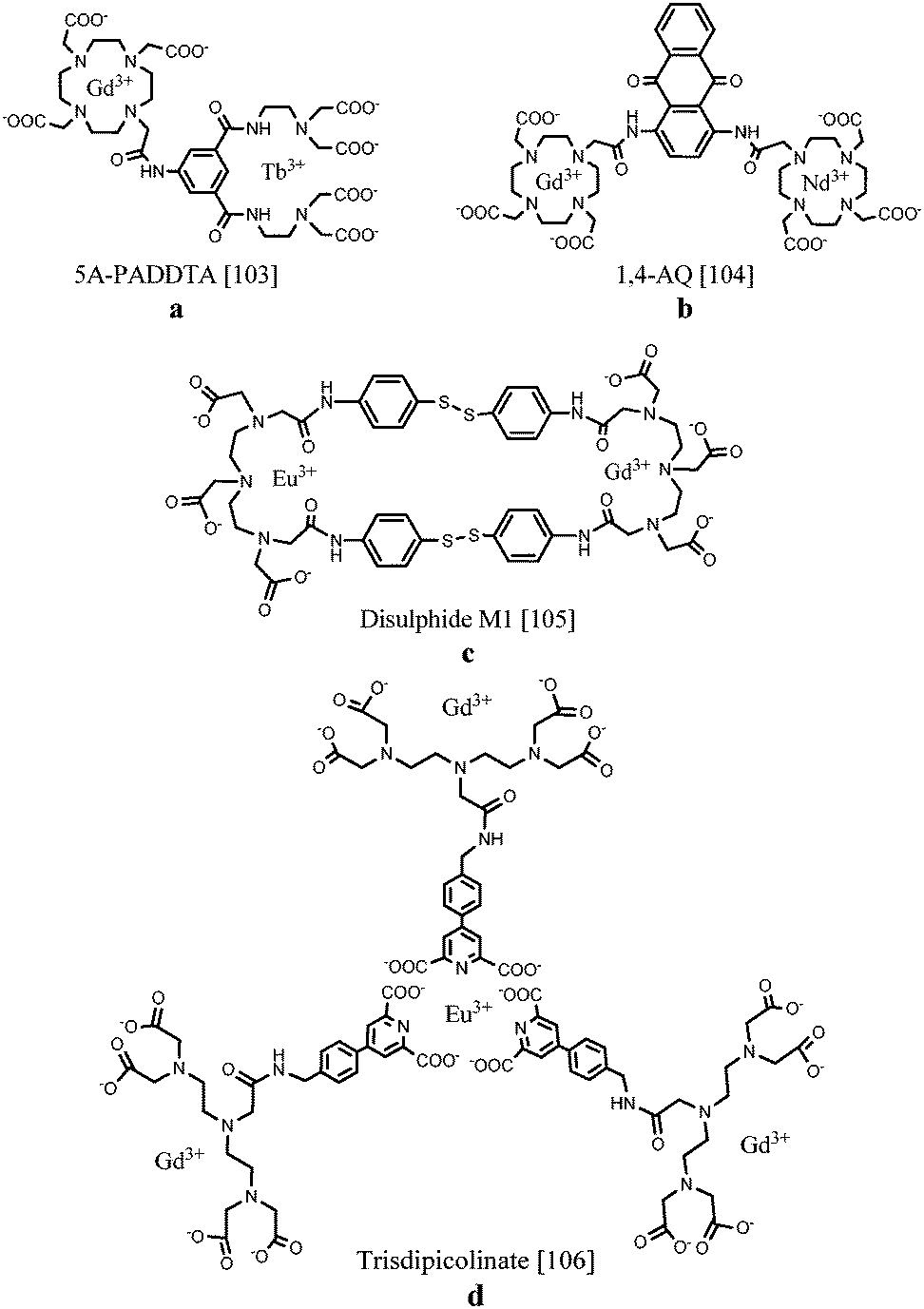 | ||
| Fig. 11 Chemical structures of selectively coordinated lanthanide-based bimodal systems.103–106 The metal coordination and Ln–water bonds have been omitted for clarity. | ||
| Relaxometric Ln(III) | Luminescent Ln(III) |
r
1a (s−1 mM−1) at 20 MHz |
λ exc (nm) | Quantum yield Φb (%) | |
|---|---|---|---|---|---|
| a Relaxivity r1 per millimolar Gd(III). b Quantum yield relative to a standard solution. | |||||
| 5A-PADDTA103 | GdIII | EuIII, TbIII | 5.5 | 297 | 0.1 |
| 1,4-AQ104 | GdIII | NdIII, ErIII, YbIII | 5.8 | 440 | 0.1 |
| Disulphide M1105 | GdIII | EuIII | — | 266 | — |
| Trisdipicolinate106 | GdIII | EuIII | 10.3 | 290 | 10.0 |
5 Conclusions and outlook
In the search towards efficient MRI contrast agents, one of the promising approaches involves the formation of supramolecular structures, preferably containing several gadolinium(III) ions. The slow molecular tumbling time of such higher MW systems offers the advantage of high proton relaxation rate values at the magnetic fields of interest, and in addition, the concentration of several lanthanide ions into a small volume reinforces this effect. In order to complement the low sensitivity of MRI, probes suitable for optical imaging with a low detection limit have been introduced into MRI contrast agents. Dual imaging contrast agents could provide diagnostic information at the early stages of the clinical follow up and avoid invasive procedures. So far, a number of different synthetic procedures have been reported in order to create bimodal magnetic resonance/optical imaging agents. MRI agents have been coupled to organic fluorophores or both probes have been incorporated in lipophilic aggregates or nanosized particles. For these compounds, broad emission bands and relatively short luminescence lifetimes are observed, which makes it difficult to distinguish their emission from background autofluorescence. The novel trend involving the combination of multiple lanthanide ions in one molecule has been a significant breakthrough, since these compounds offer excellent relaxivity and beneficial luminescence quantum yields with long lifetimes, even under physiological conditions. However, it is important to note that the limited tissue penetration of visible/near infrared light and substantial luminescence quenching in an aqueous environment will restrict in vivo applications of dual agents to imaging of tissue sections or as markers in surgical operations. Despite this limitation, molecular imaging plays an essential role in obtaining more detailed insights into cellular processes and for discovering diseases in an early stage. Therefore, the search for optimal imaging probes remains an issue of high priority.Acknowledgements
This work by E.D. and T.N.P.V. on the synthesis and characterization of newly designed contrast agents has been supported by the IWT Flanders and the FWO Flanders (G.0412.09). We would like to thank Svetlana V. Eliseeva, Luce Vander Elst, Sophie Laurent and Robert N. Muller for their help with the optical and NMRD measurements as part of our work reported in this paper.Notes and references
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- H. Lothar and T. Eva, Gadolinium-Based Bionanomaterials for Magnetic Resonance Imaging, 2011 Search PubMed.
- A. Louie, Chem. Rev., 2010, 110, 3146 CrossRef CAS PubMed.
- I. Solomon, Phys. Rev., 1955, 99, 559 CrossRef CAS.
- N. Bloembergen, J. Chem. Phys., 1957, 27, 572 CrossRef CAS PubMed.
- N. Bloembergen and L. O. Morgan, J. Chem. Phys., 1961, 34, 842 CrossRef CAS PubMed.
- V. C. Pierre, M. Botta, S. Aime and K. N. Raymond, Inorg. Chem., 2006, 45, 8355 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed.
- É. Tóth, L. Helm and A. E. Merbach, in Contrast Agents I. Magnetic Resonance Imaging, ed. W. Krause, Springer Verlag, 2002 Search PubMed.
- P. Caravan, Acc. Chem. Res., 2009, 42, 851 CrossRef CAS PubMed.
- S. Aime, S. G. Crich, E. Gianolio, G. B. Giovenzana, L. Tei and E. Terreno, Coord. Chem. Rev., 2006, 250, 1562 CrossRef CAS PubMed.
- S. Aime, D. D. Castelli, S. G. Crich, E. Gianolio and E. Terreno, Acc. Chem. Res., 2009, 42, 822 CrossRef CAS PubMed.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019 CrossRef CAS PubMed.
- P. Hermann, J. Kotek, V. Kubicek and I. Lukes, Dalton Trans., 2008, 3027 RSC.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568 CrossRef CAS PubMed.
- K. Luo, G. Liu, W. She, Q. Wang, G. Wang, B. He, H. Ai, Q. Gong, B. Song and Z. Gu, Biomaterials, 2011, 32, 7951 CrossRef CAS PubMed.
- A. Accardo, D. Tesauro, L. Aloj, C. Pedone and G. Morelli, Coord. Chem. Rev., 2009, 253, 2193 CrossRef CAS PubMed.
- A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921 CrossRef CAS PubMed.
- T. N. Parac-Vogt, A. Pacco, P. Nockemann, S. Laurent, R. N. Muller, M. Wickleder, G. Meyer, L. Vander Elst and K. Binnemans, Chem. – Eur. J., 2006, 12, 204 CrossRef CAS PubMed.
- T. N. Parac-Vogt, L. Vander Elst, K. Kimpe, S. Laurent, C. Burtea, F. Chen, R. Van Deun, Y. C. Ni, R. N. Muller and K. Binnemans, Contrast Media Mol. Imaging, 2006, 1, 267 CrossRef CAS PubMed.
- Y. Li, M. Beija, S. Laurent, L. Vander Elst, R. N. Muller, H. T. T. Duong, A. B. Lowe, T. P. Davis and C. Boyer, Macromolecules, 2012, 45, 4196 CrossRef CAS.
- H. Kobayashi and M. W. Brechbiel, Curr. Pharm. Biotechnol., 2004, 5, 539 CAS.
- D. A. Fulton, E. M. Elemento, S. Aime, L. Chaabane, M. Botta and D. Parker, Chem. Commun., 2006, 1064 RSC.
- P. Lebdušková, J. Kotek, P. Hermann, L. Vander Elst, R. N. Muller, I. Lukeš and J. A. Peters, Bioconjugate Chem., 2004, 15, 881 CrossRef PubMed.
- D. Delli Castelli, E. Gianolio, S. Geninatti Crich, E. Terreno and S. Aime, Coord. Chem. Rev., 2008, 252, 2424 CrossRef CAS PubMed.
- T. N. Parac-Vogt, K. Kimpe, S. Laurent, C. Piérart, L. Vander Elst, R. N. Muller and K. Binnemans, Eur. Biophys. J., 2006, 35, 136 CrossRef CAS PubMed.
- T. N. Parac-Vogt, K. Kimpe, S. Laurent, C. Piérart, L. Vander Elst, R. N. Muller and K. Binnemans, Eur. J. Inorg. Chem., 2004, 3538 CrossRef CAS PubMed.
- S. Torres, J. A. Martins, J. P. André, C. F. G. C. Geraldes, A. E. Merbach and É. Tóth, Chem. – Eur. J., 2006, 12, 940 CrossRef CAS PubMed.
- E. Gianolio, G. B. Giovenzana, D. Longo, I. Longo, I. Menegotto and S. Aime, Chemistry, 2007, 13, 5785 CrossRef CAS PubMed.
- C. Vanasschen, N. Bouslimani, D. Thonon and J. F. Desreux, Inorg. Chem., 2011, 50, 8946 CrossRef CAS PubMed.
- E. Terreno, D. Delli Castelli, C. Cabella, W. Dastrù, A. Sanino, J. Stancanello, L. Tei and S. Aime, Chem. Biodiversity, 2008, 5, 1901 CAS.
- S. Hak, H. M. H. F. Sanders, P. Agrawal, S. Langereis, H. Grüll, H. M. Keizer, F. Arena, E. Terreno, G. J. Strijkers and K. Nicolay, Eur. J. Pharm. Biopharm., 2009, 72, 397 CrossRef CAS PubMed.
- F. Kielar, L. Tei, E. Terreno and M. Botta, J. Am. Chem. Soc., 2010, 132, 7836 CrossRef CAS PubMed.
- T. N. Parac-Vogt, K. Kimpe, S. Laurent, L. Vander Elst, C. Burtea, F. Chen, R. N. Muller, Y. Ni, A. Verbruggen and K. Binnemans, Chem. – Eur. J., 2005, 11, 3077 CrossRef CAS PubMed.
- C. Henoumont, V. Henrotte, S. Laurent, L. Vander Elst and R. N. Muller, J. Inorg. Biochem., 2008, 102, 721 CrossRef CAS PubMed.
- P. Caravan, G. Parigi, J. M. Chasse, N. J. Cloutier, J. J. Ellison, R. B. Lauffer, C. Luchinat, S. A. McDermid, M. Spiller and T. J. McMurry, Inorg. Chem., 2007, 46, 6632 CrossRef CAS PubMed.
- S. G. Zech, H. B. Eldredge, M. P. Lowe and P. Caravan, Inorg. Chem., 2007, 46, 3576 CrossRef CAS PubMed.
- P. J. Debouttière, S. Roux, F. Vocanson, C. Billotey, O. Beuf, A. Favre-Réguillon, Y. Lin, S. Pellet-Rostaing, R. Lamartine, P. Perriat and O. Tillement, Adv. Funct. Mater., 2006, 16, 2330 CrossRef PubMed.
- C. Alric, J. Taleb, G. L. Duc, C. Mandon, C. Billotey, A. L. Meur-Herland, T. Brochard, F. Vocanson, M. Janier, P. Perriat, S. Roux and O. Tillement, J. Am. Chem. Soc., 2008, 130, 5908 CrossRef CAS PubMed.
- L. C. C. Moriggi, C. Cannizzo, E. Dumas, C. D. R. Mayer, A. Ulianov and L. Helm, J. Am. Chem. Soc., 2009, 131, 10828 CrossRef CAS PubMed.
- C. Platas-Iglesias, L. Vander Elst, W. Zhou, R. N. Muller, C. F. G. C. Geraldes, T. Maschmeyer and J. A. Peters, Chem. – Eur. J., 2002, 8, 5121 CrossRef CAS.
- É. Csajbók, I. Bányai, L. Vander Elst, R. N. Muller, W. Zhou and J. A. Peters, Chem. – Eur. J., 2005, 11, 4799 CrossRef PubMed.
- M. Tsotsalas, M. Busby, E. Gianolio, S. Aime and L. De Cola, Chem. Mater., 2008, 20, 5888 CrossRef CAS.
- K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154 CrossRef CAS PubMed.
- M.-A. Fortin, R. M. Petoral, Jr., F. Söderlind, A. Klasson, M. Engström, T. Veres, P.-O. Käll and K. Uvdal, Nanotechnology, 2007, 18, 395501 CrossRef.
- J.-L. Bridot, A.-C. Faure, S. Laurent, C. Rivière, C. Billotey, B. Hiba, M. Janier, V. Josserand, J.-L. Coll, L. Vander Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS PubMed.
- Z. Zhang, M. T. Greenfield, M. Spiller, T. J. McMurry, R. B. Lauffer and P. Caravan, Angew. Chem., Int. Ed., 2005, 44, 6766 CrossRef CAS PubMed.
- E. M. Sevick-Muraca, J. P. Houston and M. Gurfinkel, Curr. Opin. Chem. Biol., 2002, 6, 642 CrossRef CAS.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17 CrossRef CAS PubMed.
- E. Cassette, M. Helle, L. Bezdetnaya, F. Marchal, B. Dubertret and T. Pons, Adv. Drug Delivery Rev., 2013, 65, 719 CrossRef CAS PubMed.
- L. Frullano and T. J. Meade, JBIC, J. Biol. Inorg. Chem., 2007, 12, 939 CrossRef CAS PubMed.
- D. Jańczewski, Y. Zhang, G. K. Das, D. K. Yi, P. Padmanabhan, K. K. Bhakoo, T. T. Y. Tan and S. T. Selvan, Microsc. Res. Tech., 2011, 74, 563 CrossRef PubMed.
- L. E. Jennings and N. J. Long, Chem. Commun., 2009, 3511 RSC.
- J. Kuil, T. Buckle, H. Yuan, N. S. van den Berg, S. Oishi, N. Fujii, L. Josephson and F. W. B. van Leeuwen, Bioconjugate Chem., 2011, 22, 859 CrossRef CAS PubMed.
- A. Keliris, T. Ziegler, R. Mishra, R. Pohmann, M. G. Sauer, K. Ugurbil and J. Engelmann, Bioorg. Med. Chem., 2011, 19, 2529 CrossRef CAS PubMed.
- W. Di, S. K. P. Velu, A. Lascialfari, C. Liu, N. Pinna, P. Arosio, Y. Sakka and W. Qin, J. Mater. Chem., 2012, 22, 20641 RSC.
- C. Bernhard, C. Goze, Y. Rousselin and F. Denat, Chem. Commun., 2010, 46, 8267 RSC.
- C. T. Adkins, J. N. Dobish, C. S. Brown, B. Mayrsohn, S. K. Hamilton, F. Udoji, K. Radford, T. E. Yankeelov, J. C. Gore and E. Harth, Polym. Chem., 2012, 3, 390 RSC.
- T. Koullourou, L. Natrajan, H. Bhavsar, S. J. A. Pope, J. Feng, R. Kauppinen, J. Narvainen, R. Shaw, E. Scales, A. Kenwright and S. Faulkner, J. Am. Chem. Soc., 2008, 130, 2178 CrossRef CAS PubMed.
- L. Moriggi, A. Aebischer, C. Cannizzo, A. Sour, A. Borel, J.-C. G. Bunzli and L. Helm, Dalton Trans., 2009, 2088 RSC.
- G. Dehaen, S. V. Eliseeva, K. Kimpe, S. Laurent, L. VanderElst, R. N. Muller, W. Dehaen, K. Binnemans and T. N. Parac-Vogt, Chem. – Eur. J., 2012, 18, 293 CrossRef CAS PubMed.
- E. Debroye, G. Dehaen, S. V. Eliseeva, S. Laurent, L. Vander Elst, R. N. Muller, K. Binnemans and T. N. Parac-Vogt, Dalton Trans., 2012, 41, 10549 RSC.
- G. Dehaen, P. Verwilst, S. V. Eliseeva, S. Laurent, L. Vander Elst, R. N. Muller, W. M. De Borggraeve, K. Binnemans and T. N. Parac-Vogt, Inorg. Chem., 2011, 50, 10005 CrossRef CAS PubMed.
- G. Dehaen, S. V. Eliseeva, P. Verwilst, S. Laurent, L. Vander Elst, R. N. Muller, W. De Borggraeve, K. Binnemans and T. N. Parac-Vogt, Inorg. Chem., 2012, 51, 8775 CrossRef CAS PubMed.
- S. J. Soenen, G. V. Velde, A. Ketkar-Atre, U. Himmelreich and M. De Cuyper, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 197 CrossRef CAS PubMed.
- P. Howes, M. Green, A. Bowers, D. Parker, G. Varma, M. Kallumadil, M. Hughes, A. Warley, A. Brain and R. Botnar, J. Am. Chem. Soc., 2010, 132, 9833 CrossRef CAS PubMed.
- W. J. M. Mulder, G. J. Strijkers, G. A. F. van Tilborg, D. P. Cormode, Z. A. Fayad and K. Nicolay, Acc. Chem. Res., 2009, 42, 904 CrossRef CAS PubMed.
- N. Kamaly, T. Kalber, G. Kenny, J. Bell, M. Jorgensen and A. Miller, Org. Biomol. Chem., 2010, 8, 201 CAS.
- J. T. Rosenberg, J. M. Kogot, D. D. Lovingood, G. F. Strouse and S. C. Grant, Magn. Reson. Med., 2010, 64, 871 CrossRef CAS PubMed.
- T. Jin, Y. Yoshioka, F. Fujii, Y. Komai, J. Seki and A. Seiyama, Chem. Commun., 2008, 5764 RSC.
- D. Gerion, J. Herberg, R. Bok, E. Gjersing, E. Ramon, R. Maxwell, J. Kurhanewicz, T. F. Budinger, J. W. Gray, M. A. Shuman and F. F. Chen, J. Phys. Chem. C, 2007, 111, 12542 CAS.
- J. J. Gallagher, R. Tekoriute, J.-A. O'Reilly, C. Kerskens, Y. K. Gun'ko and M. Lynch, J. Mater. Chem., 2009, 19, 4081 RSC.
- J. Gunn, H. Wallen, O. Veiseh, C. Sun, C. Fang, J. Cao, C. Yee and M. Zhang, Small, 2008, 4, 712 CrossRef CAS PubMed.
- A. Quarta, R. Di Corato, L. Manna, S. Argentiere, R. Cingolani, G. Barbarella and T. Pellegrino, J. Am. Chem. Soc., 2008, 130, 10545 CrossRef CAS PubMed.
- M. J. Pittet, F. K. Swirski, F. Reynolds, L. Josephson and R. Weissleder, Nat. Protoc., 2006, 1, 73 CrossRef CAS PubMed.
- S. Ronchi, M. Colombo, P. Verderio, S. Mazzucchelli, F. Corsi, C. De Palma, R. Allevi, E. Clementi and D. Prosperi, AIP Conf. Proc., 2010, 1275, 102 CrossRef CAS PubMed.
- K. Lee, H.-Y. Moon, C. Park, O. R. Kim, E. Ahn, S. Y. Lee, H. E. Park, S.-H. Ihm, K.-B. Seung, K. Chang, T.-J. Yoon, C. Lee, C. Cheong and K. S. Hong, Curr. Appl. Phys., 2009, 9, S15 CrossRef PubMed.
- Z. Ma, D. Dosev, M. Nichkova, R. K. Dumas, S. J. Gee, B. D. Hammock, K. Liu and I. M. Kennedy, J. Magn. Magn. Mater., 2009, 321, 1368 CrossRef CAS PubMed.
- M. L. Debasu, D. Ananias, S. L. C. Pinho, C. F. G. C. Geraldes, L. D. Carlos and J. Rocha, Nanoscale, 2012, 4, 5154 RSC.
- W. J. Rieter, J. S. Kim, K. M. L. Taylor, H. An, W. Lin, T. Tarrant and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 3680 CrossRef CAS PubMed.
- L. Pellegatti, J. Zhang, B. Drahos, S. Villette, F. Suzenet, G. Guillaumet, S. Petoud and E. Toth, Chem. Commun., 2008, 6591 RSC.
- S. L. C. Pinho, H. Faneca, C. F. G. C. Geraldes, J. Rocha, L. D. Carlos and M.-H. Delville, Eur. J. Inorg. Chem., 2012, 2828 CrossRef CAS PubMed.
- C. S. Bonnet, F. Buron, F. Caillé, C. M. Shade, B. Drahoš, L. Pellegatti, J. Zhang, S. Villette, L. Helm, C. Pichon, F. Suzenet, S. Petoud and É. Tóth, Chem. – Eur. J., 2012, 18, 1419 CrossRef CAS PubMed.
- F. Caillé, C. S. Bonnet, F. Buron, S. Villette, L. Helm, S. Petoud, F. Suzenet and É. Tóth, Inorg. Chem., 2012, 51, 2522 CrossRef PubMed.
- G. Tallec, P. H. Fries, D. Imbert and M. Mazzanti, Inorg. Chem., 2011, 50, 7943 CrossRef CAS PubMed.
- E. Debroye, S. V. Eliseeva, S. Laurent, L. Vander Elst, S. Petoud, R. N. Muller and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2013, 2629 CrossRef CAS PubMed.
- E. Debroye, S. V. Eliseeva, S. Laurent, L. Vander Elst, R. N. Muller and T. N. Parac-Vogt, Dalton Trans., 2014, 43, 3589 RSC.
- C. S. Bonnet and É. Tóth, C. R. Chim., 2010, 13, 700 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750 RSC.
- F. A. Rojas-Quijano, E. T. Benyó, G. Tircsó, F. K. Kálmán, Z. Baranyai, S. Aime, A. D. Sherry and Z. Kovács, Chem. – Eur. J., 2009, 15, 13188 CrossRef CAS PubMed.
- M. P. Placidi, J. Engelmann, L. S. Natrajan, N. K. Logothetis and G. Angelovski, Chem. Commun., 2011, 47, 11534 RSC.
- M. Regueiro-Figueroa, A. Nonat, G. A. Rolla, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, L. J. Charbonnière, M. Botta and C. Platas-Iglesias, Chem. – Eur. J., 2013, 11696 CrossRef CAS PubMed.
- C. M. Andolina, P. J. Klemm, W. C. Floyd, J. M. J. Fréchet and K. N. Raymond, Macromolecules, 2012, 45, 8982 CrossRef CAS PubMed.
- E. Debroye, S. Laurent, L. Vander Elst, R. N. Muller and T. N. Parac-Vogt, Chem. – Eur. J., 2013, 19, 16019 CrossRef CAS PubMed.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526 CrossRef CAS PubMed.
- M. S. Tremblay and D. Sames, Chem. Commun., 2006, 4116 RSC.
- L. S. Natrajan, A. J. L. Villaraza, A. M. Kenwright and S. Faulkner, Chem. Commun., 2009, 6020 RSC.
- M. P. Placidi, A. J. L. Villaraza, L. S. Natrajan, D. Sykes, A. M. Kenwright and S. Faulkner, J. Am. Chem. Soc., 2009, 131, 9916 CrossRef CAS PubMed.
- M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283 RSC.
- L. Tei, G. Gugliotta, S. Avedano, G. B. Giovenzana and M. Botta, Org. Biomol. Chem., 2009, 7, 4406 CAS.
- T. J. Sorensen, M. Tropiano, O. A. Blackburn, J. A. Tilney, A. M. Kenwright and S. Faulkner, Chem. Commun., 2013, 49, 783 RSC.
- I. Mamedov, T. N. Parac-Vogt, N. K. Logothetis and G. Angelovski, Dalton Trans., 2010, 39, 5721 RSC.
- J. E. Jones, A. J. Amoroso, I. M. Dorin, G. Parigi, B. D. Ward, N. J. Buurma and S. J. A. Pope, Chem. Commun., 2011, 47, 3374 RSC.
- D. Lewis, Supramol. Chem., 2012, 24, 135 CrossRef CAS.
- E. Debroye, M. Ceulemans, L. Vander Elst, S. Laurent, R. N. Muller and T. N. Parac-Vogt, Inorg. Chem., 2014, 53, 1257 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |