
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological metals and metal-targeting compounds in major neurodegenerative diseases
Kevin J.
Barnham
abc and
Ashley I.
Bush
 *ade
*ade
aFlorey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, 3010, Australia
bBio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, 3010, Australia
cDepartment of Pharmacology and Therapeutics, The University of Melbourne, Parkville, 3010, Australia
dDepartment of Pathology, The University of Melbourne, Parkville, 3010, Australia
eCooperative Research Center for Mental Health, Carlton South, Victoria, Australia. E-mail: ashley.bush@florey.edu.au
First published on 7th August 2014
Abstract
Multiple abnormalities occur in the homeostasis of essential endogenous brain biometals in age-related neurodegenerative disorders, Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis. As a result, metals both accumulate in microscopic proteinopathies, and can be deficient in cells or cellular compartments. Therefore, bulk measurement of metal content in brain tissue samples reveal only the “tip of the iceberg”, with most of the important changes occurring on a microscopic and biochemical level. Each of the major proteins implicated in these disorders interacts with biological transition metals. Tau and the amyloid protein precursor have important roles in normal neuronal iron homeostasis. Changes in metal distribution, cellular deficiencies, or sequestration in proteinopathies all present abnormalities that can be corrected in animal models by small molecules. These biochemical targets are more complex than the simple excess of metals that are targeted by chelators. In this review we illustrate some of the richness in the science that has developed in the study of metals in neurodegeneration, and explore its novel pharmacology.
 Kevin J. Barnham | Kevin Barnham received his BSc in Chemistry from the University of Queensland in 1986 and his PhD in 1993. His doctoral worked focused on NMR spectroscopy to study the interactions of Pt anticancer drugs with amino acids and nucleobases under the supervision of Trevor Appleton. His postdoctoral work (1992–1995) under the supervision of Peter Sadler at The University of London expanded on this work. In 1995, he joined the Biomolecular Research Institute, The University of Melbourne, where he used NMR spectroscopy to determine the structures of proteins as potential drug targets. Since 2001, his research has focused on developing new therapeutic strategies for neurodegenerative diseases. |
 Ashley I. Bush | Ashley Bush heads the Oxidation Biology Unit at the Florey Institute of Neuroscience & Mental Health, is Professor of Neuroscience at The University of Melbourne, NHMRC Australia Fellow, co-director of biomarker development for the Australian Imaging Biomarker Lifestyle Study, Chief Scientific Officer of the Cooperative Research Center for Mental Health, and has staff appointments at the Massachusetts General Hospital. He has received numerous awards including the Potamkin Prize and the Beeson Award. He has authored >300 publications, with >22 |
Introduction
Metal ions play a major role in biological processes, whether it be structural (e.g. stabilizing various protein or nucleic acid configurations) or functional (e.g. being second messengers, or being the active site of metalloenzymes). Of particular interest to the concepts described within this review is the biology of the late first row transition metals Fe, Cu and Zn. We will not be reviewing toxicological heavy metals (e.g. Hg, Pb, Cd), non-biological metals (e.g. Al), or biologically important metals of lower abundance (e.g. Mn).Fe and Cu have the ability to exist in multiple valence states and this attribute is essential for enabling biological systems to capture, store, transport and utilize O2. At the quantum level, the O2 in our atmosphere is in a triplet spin state. As such, interactions with most organic molecules are spin-forbidden (i.e. energetically unfavorable) and this intrinsic feature of O2 must be overcome before organisms utilize O2. Fe and Cu have the ability to coordinate O2, and by cycling through oxidation states, are able to activate O2 by converting the relatively inactive triplet state into a highly reactive single state. By generating singlet state oxygen, organisms not only create a powerful driving force for life but also a molecule that has significant destructive capacity through its ability to cause oxidative stress. Therefore organisms that rely on O2 have developed a complex array of checks and balances to limit the destructive potential of the unregulated activation of oxygen. Not only do these checks and balances include the molecules such as superoxide dismutase 1 (SOD1) and catalase that detoxify reactive oxygen species, but the metabolism of the metal ions responsible for the activation of O2 is tightly regulated so that there is little, if any, freely exchangeable Fe and Cu present in biological systems (reviewed in ref. 1). These regulatory processes are energy-dependent and their efficiency degrades with age leading to increased oxidative stress giving rise to the oxidative stress hypothesis as a cause of aging.2,3
Age is the greatest risk factor for neurodegenerative diseases. We have for many years theorized that age-dependent deterioration of the metal homeostatic system of the brain drives neurodegenerative diseases. The evidence in support of this hypothesis is outlined in this review, as are the potential therapeutic strategies that arise from it.
Neurotransmission and second messenger roles of Zn and Cu
Zinc and copper are both essential metal ions, and are highly concentrated in grey matter.4,5 Zn2+ transporter proteins are classified into two families, Slc30a (known as ZnT) and Slc39a (termed Zrt-Irt like proteins, ZIPs). The ZIP transporters traffic Zn2+ into the cytoplasm, and the ZnT traffic Zn2+ out of the cytoplasm (Fig. 1). The importance and need for precision in Zn2+ trafficking is highlighted by the observation that, in the human and mouse genome, there are 14 ZIP and 9 ZnT known transporters reviewed in ref. 6. Of these, the ZnT3 transporter is selectively expressed in the cortex, and responsible for the concentration of chemically exchangeable Zn2+ (sometimes referred to as Labile Zn2+) in glutamatergic vesicles.7 This pool of vesicular Zn2+ is characteristically identified by Timm's staining and represents 20–30% of brain zinc.8 Co-release of Zn2+ with glutamate has been observed in hippocampal mossy fiber synapses, critical for memory, where endogenous Zn2+ was demonstrated to modulate postsynaptic excitability at NMDA and GABA receptors.9 The concentration peak of exchangeable Zn2+ in these synapses is estimated to be 100–300 μM.10–12 It is likely that Zn2+ is the counter-ion to glutamate in some boutons, since it forms a complex that slowly dissociates upon release.13 This is reminiscent of Zn2+ loading of insulin in the secretory granules of pancreatic β-cells. In this sense the zinc transporters involved in loading Zn2+ into the secretory or neurotransmitter vesicles have functional homology; ZnT8 for pancreatic β-cells and ZnT3 for glutamatergic vesicles, are both located in the vesicular membrane, and predominantly expressed in the pancreas and brain, respectively.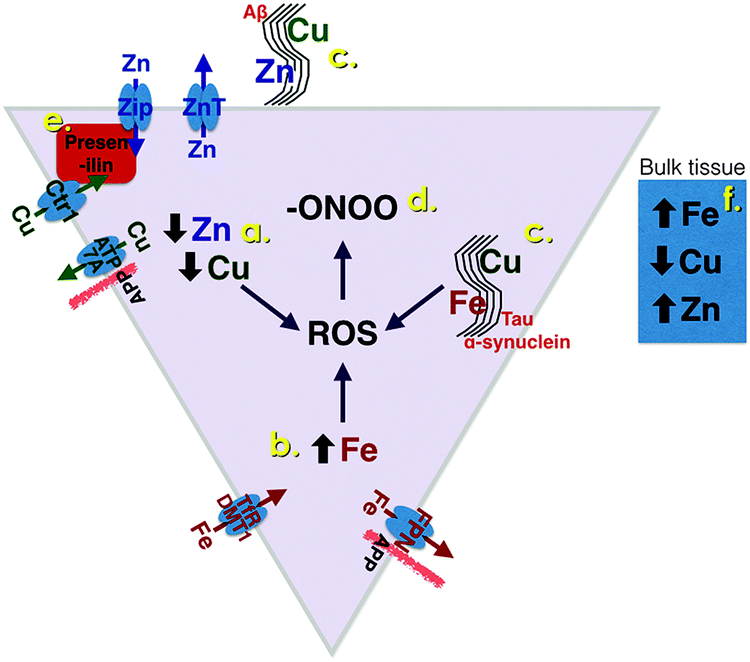 | ||
| Fig. 1 Redistribution of biometals in neurodegenerative diseases: targets for potential therapeutics. A notional AD-affected cortical neuron is shown, prototypic of other proteinopathies. Novel metal-associated small molecules that target biochemical and subcellular pathological changes include, (a) ionophores (e.g. clioquinol, PBT2) remove adventitial metals from being trapped within extracellular Aβ aggregates in AD and facilitate their uptake into the deficient intracellular compartment. (b) Iron chelators (e.g. deferiprone, clioquinol, M30) lower elevated intraneuronal iron concentrations, which are a source of ROS in AD, PD, possibly HD and ALS. (c) MPACs (e.g. PBT2) selectively target adventitial transition metal binding sites on proteinopathies (neurofibrillary tangles in AD and PD, HTT aggregates in HD), and suppress ROS generation. (d) CuIIBTSCs are catalytic peroxynitrite scavengers. The major classes of metal ion transporters are shown at the surface of the cell. For simplicity, their presence on endosomal membranes is not shown (FPN: ferroportin, TfR: transferrin receptor, DMT: divalent metal ion transporter 1, ATP7A: Cu transporter ATPase 7a). APP directly interacts with FPN to promote iron export. APP also promotes copper export, but the mechanism is uncertain. (e) Presenilins play a major role is promoting the uptake of Cu and Zn, but their interaction with Cu and Zn transporters is still not clear. (f) When total metals are measured in a section of affected grey matter (“Bulk tissue”: a mixture of neuronal and non-neuronal cells), the changes seen include elevated iron, decreased copper and, in advanced AD, elevated zinc. The typical locations of Aβ (AD), tau (AD and PD) and α-synuclein (PD) proteopathies are shown. | ||
A steadily growing literature has shown that Zn2+ released on neurotransmission plays a prominent role in post-synaptic and presynaptic responses, reviewed in ref. 14 and 15. Arrays of post-synaptic targets for Zn2+ have been reported, as well us uptake into the neurites with consequent signalling events. This has led investigators to propose that Zn2+ may qualify both as a neurotransmitter and second messenger.
Synaptic Zn2+ has been shown to inhibit the NMDA receptor through low and high affinity binding sites.16,17 Recent work has demonstrated that presynaptic vesicular Zn2+ release is needed for presynaptic plasticity that causes mossy-fiber long term potentiation (LTP),18 a classical form of LTP considered to underlie learning and memory. In that study, pleiotropic effects of synaptic Zn2+ release were observed since post-synaptic mossy fiber LTP was inhibited in the hippocampal CA3 region by Zn2+.18
Transactivation targets for synaptically-released Zn2+ have been identified, and include TrkB independently of brain-derived neurotrophic factor (BDNF).19 In addition, GPR39 has been characterized as a metabotropic Zn2+-sensing receptor (mZnR) with post-synaptic neuronal activity in the hippocampus20 and the dorsal cochlear nucleus.21 Zn2+ can also affect chloride ion homeostasis, as a recent study reported that elevated intracellular Zn2+ inhibits the activity of the K+/Cl− co-transporter-2 (KCC2), so modulating GABAergic neurotransmission.22 Additionally, caspase-3 activation and consequent apoptosis is inhibited by cytosolic Zn2+, as well as by the X-linked inhibitor of apoptosis protein (XIAP), whose activation is inhibited by Cu2+.23 On the other hand, neuronal death induced by oxidative stress is mediated in part by cytosolic Zn2+ release, liberated from metallothionein.24,25 Oxidant exposure leads to the simultaneous release of Ca2+, with consequent activation of CaMKII.26 Meanwhile, the liberated Zn2+ promotes phosphorylation of p38 via 12-lipoxygenase associated activation of ASK-1.25 These parallel pathways then converge to facilitate the association of syntaxin with Kv2.1, leading to injurious, Kv2.1-dependent, K+ current increase in neurons.26 This type of mechanism of neuronal death mediated by cytosolic Zn2+ increase may feature in Alzheimer's disease where there is a decrease in metallothionein III (MT-III),27,28 and inadequate buffering of zinc.29
The reuptake of Zn2+ into neurons still requires characterization, but is important since delayed reuptake may lead to extracellular pooling that can both interfere with NMDA receptor behavior as well as engender Aβ aggregation (see below). Voltage-gated calcium channels and the NMDA receptor are routes of clearance for synaptic Zn2+,30 but other mechanisms are likely to contribute, such as the participation of ZIPs. One recent report attributed about 50% of the uptake of Zn2+ and Cu2+ to the presenilins,31 mutations within these proteins can cause familial Alzheimer's disease. In addition, the cellular prion protein (PrPc) has also been reported to enhance the uptake of zinc into neuronal cells through a mechanism that involves AMPA receptors, but does not require endocytosis.32 Zinc-sensitive intracellular tyrosine phosphatase activity was decreased in cells expressing prion protein and increased in the brains of PrPc-null mice. Importantly, Zn2+ uptake was attenuated in cells expressing familial prionosis-associated PrP mutants and in prion-infected cells.32
Synaptic Zn2+ also plays critical roles in the assembly of the post-synaptic density (through Shank3), which is rich in zinc,33,34 and in sustaining the health of the synapse with aging. ZnT3 knockout mice develop marked cognitive loss by 6 months of age, accompanied by decreases in glutamate receptors, BDNF, TrkB, and PSD-95.35 The ability of Aβ to trap Zn2+ (ref. 36) outside of the neurite may contribute synaptic Shank3 scaffold deficiencies seen in Alzheimer's disease.37
Copper
Less is known about the neurophysiology of brain copper, although the importance of copper is underscored by the severe neurodevelopmental impairment that occurs in Menkes disease, caused by the loss of function mutation of the copper transporter ATP7a that moves copper out of the cell (Fig. 1), and from the gut into the blood, reviewed in ref. 38. Loss of ATP7a function in MoBr mice has established the importance of copper in axon outgrowth and synaptogenesis.39 Copper exists in health in the Cu+ oxidation state in the cytoplasm, and is extensively trafficked by a system of intracellular chaperones that prevent any free ionic copper from appearing in the cytoplasm,40 in order to prevent this reactive metal ion species from generating radicals and reactive oxygen.Activation of neurons causes a marked redistribution of cellular Cu+ towards the processes in a Ca2+-dependent manner.41 Activated synaptosomes release ionic copper,42–44 and ATP7a acts to concentrate Cu2+ into post-synaptic vesicles that are transcytosed and released upon NMDA receptor activation,45,46 to achieve transient synaptic concentrations of ≈100 μM reviewed in ref. 47. It is curious that the ionic copper is in the Cu2+ oxidation state when released by the neurite, since the uptake of copper and its cytoplasmic trafficking are mediated as the Cu+ species by Ctr1 (Fig. 1). The glutamatergic synapse is also the only known location in the body where freely exchangeable ionic Cu2+ appears.
Several papers have reported acute inhibitory effects of exogenously applied Cu2+ on the responses of NMDA, AMPA, glycine and GABA receptors,48–50 as well as voltage-dependent transient outward K+ currents and delayed rectifier K+ currents51,52 in neurons in culture, and in inhibiting purinergic receptors (P2X2, P2X4, P2X7).53–56 Cu2+ (1 μM) has been reported to suppress long-term potentiation in rat hippocampal slices.57 Removing endogenous Cu2+ released from hippocampal slices abolishes desensitization to prolonged NMDA exposure (which causes calcium overload), indicating that extracellular Cu2+ release has a protective role.58 This is mediated by Cu2+ interaction with PrPc, which then in turn interacts with the NMDA receptor complex to allosterically decrease the affinity of glycine for the receptor. More prolonged (3 h) exposure of primary rat hippocampal neurons to Cu2+ (10 μM) actually promotes activity through the AMPA receptor (GluR1 subunit) by markedly increasing PSD-95, indicating that extracellular Cu2+ may have biphasic effects on neuronal activity.59
The transsynaptic effects of Zn2+ and Cu2+ signaling were also demonstrated by the effects in cell culture of PBT2, a Zn2+/Cu2+ ionophore being developed for the treatment of Alzheimer's disease (vide infra).60 PBT2 induced inhibitory phosphorylation of the α- and β-isoforms of glycogen synthase kinase 3 by translocating extracellular Zn2+ and Cu2+ into cells. This was contributed to by Zn2+ inhibition of the phosphatase, calcineurin.
The effects of aging on brain metal homeostasis
Both normal development and aging markedly affect the distribution and concentrations of transition metals in brain tissue. Since aging is the major risk factor for AD and PD, we suspect that the timing of an elevation of iron, decrease in copper and fatigue of extracellular zinc clearance within the cerebral cortex in post-reproductive life may combine to create a change in metal milieu that induces proteostasis and neurotoxicity in these disorders. This is an area where more research is needed, but the existing data strongly encourage the exploration of aging as being a key factor in causing a pro-oxidant chemical environment in brain tissue mediated by the accumulation or redistribution of redox-active metals (mainly Fe and Cu). Robust elevations of brain copper and iron commence soon after weaning in mice,61 with genetic background strain and gender having a considerable impact.62 A marked elevation of copper in brain capillaries isolated from aged mice has been described, which may adversely impact on the clearance of Aβ (see below).63 A similar marked elevation of copper has been found in the subventricular zone of aged rats, associated with decreased neurogenesis.64 However, with advanced aging in humans (after the age of 50), levels of brain cortical copper decline markedly,65 potentially promoting Aβ amyloidogenesis,66–68 as discussed below.Several studies of post-mortem human brain tissue have also found age-dependent elevation of iron in several regions ref. 69 and references within. Book-keeping studies in rats have demonstrated that there is a net influx of iron trafficking across the blood–brain barrier, leading to gradual accumulation of iron.70 This elevation in brain iron is also detectable in humans by MRI and influenced by gender and alleles of iron regulatory genes, HFE and transferrin C2.71–73 While the cause of this elevation is uncertain, caloric restriction suppresses the age-dependent rise in iron (measured by MRI) in the globus pallidus, nigra, red nucleus, and temporal cortex in rhesus monkeys, while concomitantly sparing the age-dependent decline in their motor performance.74 This finding indicates that iron elevation with aging is a product of metabolism, and causal in the deteriorating function of motor nuclei.
In addition, zinc trafficking in the neocortex fatigues with aging (reviewed in ref. 75), which could lead to abnormal pooling of Zn2+ in compartments that lead to the excessive aggregation of Aβ species and consequent amyloid pathology (vide infra). This is an area of emphasis for future research.
The breakdown in metal homeostasis in Alzheimer's disease
Alzheimer's disease brain tissue is characterized by two hallmark pathological features, extracellular amyloid plaques, the major component of which is the amyloid-β peptide (Aβ), and intracellular neurofibrillary tangles composed of hyper-phosphorylated forms of the microtubule associated protein, tau. As described in this section, the Aβ proteopathy co-enriches with copper and zinc, whereas the tau proteopathy coenriches with iron (Fig. 1). There are also changes in cellular levels and compartmentalization of these metals in AD-affected brain tissue.29,65,76,77 In addition, the behaviour, trafficking or processing of each of the proteins implicated in AD pathogenesis in sensitive to copper, zinc and/or iron, and some (APP, presenilins and tau) play an established role in metal homeostasis. Taken together, AD emerges as a disorder of metal homeostasis with a proteopathic admixture of metals due to the participant proteins' normally close biochemical association with metal ions. We are of the view that AD may be a disorder as much of abnormal metal homeostasis (a metallopathy) as it is a proteopathy, but the proteopathic elements have attracted more attention for approaches to therapeutic intervention. However, strategies that target the underlying metallopathy are now being tested in the clinic and show promise.Physiological interactions of APP, presenilin and tau with biological metals
APP has an important functional role in cellular copper and iron export, as indicated by its transcription being promoted by copper,78 and its translation being promoted by iron through a 5′UTR iron regulatory element interacting with Iron Regulatory Protein 1.79,80 Aβ is generated by the action of two proteases (β- and γ-secretase) on the amyloid precursor protein (APP). APP is a multi-domain single pass transmembrane protein, with many different functional activities attributed to it, including a role in maintaining metal homeostasis. One of the ectodomains of APP has been termed the copper binding domain and this domain has been demonstrated to coordinate and reduce Cu2+ at moderate affinities,81 to modulate APP conformation,82 and copper efflux from cells78,83 (Fig. 1). Treatment of neuronal cells with copper increases the level of APP at the cell surface by a phosphorylation-dependent process that involves increased exocytosis and diminished endocytosis.84,85Decreased intracellular copper promotes amyloidogenic processing of APP.68,86 This is important because of reports of decreased copper levels in the human brain with old age,65 and a further decrease in cortical copper levels in AD.76,87 Amyloidogenic processing of APP occurs in lipid rafts, membrane microdomains enriched in cholesterol. β- and γ-secretases, as well as Aβ have been identified in lipid rafts from cells, human and rodent brains. Under conditions of copper deficiency, copper becomes paradoxically enriched in lipid rafts where it co-enriches with Aβ,88 potentially leading to hypermetallation of the peptide (discussed below) and the catalytic oxidation of cholesterol to produce H2O2.89–91
Yet AD does not appear to be caused by nutritional copper deficiency since a randomized, placebo controlled clinical trial of copper orotate (8 mg) over 12 months was safely tolerated but failed to impact disease progression.92 That is possibly because very little of this copper would have actually reached the brain, since ionic copper is a minute fraction of plasma, and very little penetrates the blood brain barrier. Hence the need for chaperones such as PBT2 and Cu(gtsm), discussed below. Indeed, excess copper exposure has been implicated in AD. Rabbits maintained on an elevated cholesterol and copper diet demonstrated accelerated plaque formation and increased oxidative stress in the brain,93 results that are supported by epidemiological data showing that diet high copper and fat increases the risk of AD.94 Copper exposure (90 days) of APP transgenic mice has been reported to increase brain Aβ levels, and, unlike normal mice, raises brain Cu levels and impairs neurocognition.63
APP has been recently shown to promote the efflux of iron from neurons and other cells by interaction with ferroportin29,95 (Fig. 1), so that APP knockout mice develop exaggerated iron elevation in brain and other tissues with aging.29,96 Despite the accumulation of insoluble tau in neurofibrillary tangles, soluble tau levels actually decrease in affected cortex in AD, and since tau promotes iron efflux by trafficking APP to the cell surface, tau knockout mice develop brain atrophy with cognitive loss and a parkinsonian phenotype from 12 months of age.97 Hence, two of the major proteins implicated in AD pathogenesis have major roles in iron efflux that may be compromised in the disease, and may contribute to the accumulation of iron in the disorder.
The presenilins, whose mutations cause familial AD, have been shown to play a major role in the uptake and turnover of Cu and Zn in several cell types (Fig. 1). Loss of presenilin expression markedly decreases CuZn superoxide dismutase (SOD1) activity by lowering cellular levels of the copper chaperone of SOD (CCS1),31 which can increase Aβ production98 potentially because CCS1 binds to the intracellular domain of BACE1 and may deliver copper to it.99 Hence, cytoplasmic copper may link presenilin, needed for γ-secretase cleavage, with BACE1 β-cleavage. SOD1 may contribute to pathogenesis since partial and full knock out in an APP-overexpressing mouse model accelerated Aβ accumulation, oligomerization and behavioral abnormalities, which preceded oxidative damage.100
The expression of low density lipoprotein receptor-related protein 1 (LRP1) in the brain microvasculature, and associated clearance of Aβ from the brain, has been recently reported to be very sensitive to copper.63 Treatment with low levels of Cu in drinking water for 90 days was sufficient to selectively reduce LRP1 and Aβ clearance. Indeed, treatment with very low levels of Cu2+ (200 nM) suppressed LRP1 levels in cultured primary mouse brain endothelial cells through oxidative stress, and the same levels of Al3+, Zn2+, or Fe2+ had no effect.63 This association is especially intriguing since Cu2+ induces the endocytosis of PrPc through a LRP1-dependent mechanism,101 and Aβ neurotoxicity has been described to be mediated through an interaction between Cu2+, PrPc and the NMDA receptor.58
Taken together, these studies support a sensitive interaction between the proteins implicated in the pathogenesis of AD and brain metal concentrations. In the instances of APP, presenilin and tau, the argument for the protein playing a constitutive role in metal homeostasis is quite strong (Fig. 1), and supports a loss of function in metal homeostasis as being upstream in AD pathogenesis.
Changes in metal levels, distribution and homeostasis in AD
The amyloid plaques that define AD have been shown to be metal sinks, with microParticle-Induced-X day Emission (PIXE) analysis showing high levels of Zn (1055 μM), Fe (940 μM) and Cu (390 μM) present. This analysis also demonstrated a 3–5 fold increase in the cortical and accessory basal nuclei of the amygdala in Zn, Cu and Fe in the neuropil of AD patients, as compared to age-matched controls.102 Iron is enriched within ferritin in the dystrophic neurites adjacent to amyloid plaques.103 This is also visualized in the plaques of APP transgenic mice by MRI104 and histochemistry.105 While Aβ directly coordinates and reduces Fe3+,106 to date iron has not been identified as coordinating Aβ within amyloid pathology, while Cu and Zn have been found to directly coordinate the histidine residues of Aβ by Raman spectroscopy,107 and to co-purify with Aβ from post-mortem human tissue90 (vide infra). The zinc within plaques and congophilic amyloid angiopathy (CAA) can be vividly stained by a modification of the Timm's stain.8,108–110 The density of amyloid plaques per unit volume is greatest in the layers of the cortex that contain the highest concentrations of exchangeable zinc, supporting the likelihood that the plaques are formed from the condensation of Zn2+ from the nearby tissue reacting with Aβ.110While there is an emerging consensus of an increase in metals ions in the plaques and CAA, in both AD108,111 and transgenic mouse models of AD (which contain lower metal concentrations than human plaques),112,113 total cellular copper levels are decreased in AD-affected cortex65,76,87 (Fig. 1). Despite this, there is evidence that the remaining tissue Cu is poorly ligated, causing an elevation of labile, redox-active Cu2+ in the brain to be associated with the increased oxidative stress present in the brains of AD subjects.76 The emergence of Cu2+ in the cytoplasm is possibly always a pathological event, and has been tied to the promotion of apoptosis through the inhibition of XIAP,23 or MAPK/ERK kinase 1 (MEK1) interaction with Extracellular signal-regulated kinase (ERK).114
Increased oxidative stress levels in AD are also associated with the production of 4-hydroxynonenal, which has the capacity to inhibit zinc export.115 This may contribute to the increased zinc levels in AD-affected brain, which rise late in the disorder.65,116 Interestingly, rodent studies have shown that zinc deficiency can increase zinc retention in the brain as the expression levels of the intracellular Zn export promoter ZnT1 are suppressed.117,118 This may contribute to the enlargement of amyloid plaques in the brains of APP/PS1 transgenic mice with dietary zinc deficiency.110 Levels of a number of zinc transporters including of ZnT1, ZnT4, ZnT6,119 and ZnT3,120 are altered in AD brain tissue. ZnT1 levels have been reported as altered in mild cognitive impairment,121 suggesting metal dyshomeostasis in early stages of the disease. Estrogen suppresses ZnT3 expression as well as synaptic zinc levels, yet ovariectomy causes a rebound elevation in synaptic zinc and ZnT3 expression.122 This is significant because women are at higher risk of developing AD and female APP transgenic mice have heavier amyloid burdens.123 ZnT3 knockout mice have been described as a phenocopy for the synaptic and memory deficits of Alzheimer's disease,120 consistent with amyloid pathology trapping zinc and inducing what is, in effect, a physiological ZnT3 knockout.
A more detailed fractionation study of post-mortem hippocampal tissue revealed an elevation of Zn2+ in both soluble and synaptic vesicle fractions in AD,124 paralleled by (as previously observed)35 a decreased expression of ZnT3. In cases of non-demented individuals with a comparable amyloid burden, levels of Zn2+ in soluble fractions were significantly lower than in AD, whereas in synaptic vesicles the levels of Zn2+ were similar to AD, with no change in levels of ZnT3. Aβ oligomers were markedly increased in the post-synaptic density fractions of the AD cases, but not the non-demented amyloid-bearing controls, arguing that the elevation of zinc facilitates the attachment of Aβ oligomers to post-synaptic targets, such as the NMDA receptor, as previously reported.125 Therefore, the buffering of free Zn2+ in the tissue would be expected to play an important role in preventing such pathological consequences. The major Zn2+ buffers in tissue are the metallothioneins (MTs). MT levels altered in AD with MT-1/II increased and MT-III decreased.27,28 MT-III released from astrocytes may play a role in preventing zinc-induced Aβ aggregation.126
Iron levels have been measured in AD post-mortem brain samples in several series. An emerging consensus is that iron levels are elevated in several cortical regions in AD.29,87,127–129 This is important since excess iron is a source of oxidative stress. As with mice, MRI can assay tissue iron in humans. There is a growing literature that supports an elevation of cortical and hippocampal iron in AD through in vivo MRI.130,131 Such changes may be an early biomarker for amyloid deposition in AD,132 as they precede amyloid deposition in the PS1/APP double transgenic mouse model of AD.133 Iron overload treatment of PS1/APP transgenic mice exaggerates amyloid pathology and worsens cognitive outcomes.134 Both iron,127 zinc108 and copper135 have been reported to be enriched within neurofibrillary tangle-bearing neurons in AD. Fe3+ induces the precipitation of phospho-tau,136–138 which is also interesting because of recent MRI data showing that post-mortem brain tissue from fronto-temporal dementia cases (a severe tauopathy) exhibit particularly increased iron levels,139 as do in vivo MRI readings from affected regions in progressive supranuclear palsy,140 which is a primary tauopathy. Iron-chelation of the PS1/APP model of AD suppresses tau hyperphosphorylation, which is considered pathogenic.141,142 Decreased levels of the iron-exporter ferroportin, and iron export-promoting peptide hepcidin, in the hippocampus of post-mortem AD cases, have been recently reported to contribute to iron accumulation in the affected tissue143 (Fig. 1).
A significant role for tissue iron dyshomeostasis in AD pathogenesis is also supported by genetic observations that transferrin and transferrin receptor alleles affect the risk for AD.144–147 This also may be reflected by abnormal hemoglobin synthesis in AD that increases the risk for anemia in the disorder.148 The cause of impaired hemoglobin production in the bone marrow in AD is not yet clear. Aβ binds heme and hemoglobin,149–153 and hemoglobin accumulates in amyloid pathology in AD.154 Aβ is detected at higher levels on red cells from AD patients compared to healthy controls,155,156 where it can potentially oxidize red cell hemoglobin,157 so potentially contributing to the increased risk of anemia in AD.
There have been a number of studies measuring peripheral metal levels, particularly Cu, with conflicting outcomes. Some groups have reported Cu levels in serum and CSF levels to be significantly higher in patients with AD as compared to age match controls.158,159 Increasing copper levels have the potential to result in increased oxidative stress and the increased exchangeable copper in AD patients has been reported to correlate with higher levels of serum peroxides.160 Conversely, other studies have reported that there is no statistically significant difference in levels of serum copper or ceruloplasmin (a ferroxidase that uses the electrochemistry of 6 copper atoms) between the AD and healthy control groups when adjusting for age, sex and ApoE allelic status.161 This disparity in results with respect to peripheral Cu levels has recently been reviewed in depth162 where a meta-analysis was carried out on all studies from 1996 to 2013. The results of this analysis suggested that Cu not bound to ceruloplasmin is significantly increased in AD subjects when compared to healthy controls. A recent report of a direct measure of this value supports its utility in predicting cognitive deterioration,163 but awaits confirmation in separate population studies.
Aβ interactions with Cu and Zn
Evidence from the early onset genetic forms of AD indicates that Aβ metabolism is associated with the onset of disease. Aβ is an amphipathic peptide of variable length ranging from 38 to 43 residues with the most common forms being 40 and 42 residues long. The 16 N-terminal residues are hydrophilic and are responsible for the interactions with metal ions discussed below. The C-terminal residues are hydrophobic and drive the aggregation processes that the peptide is infamous for. As described above, Cu2+ and Zn2+ are released into the synapse during neurotransmission, and are readily exchangeable. In health these metals are taken back up into cells by chaperones such as metallothionein-3. Aβ is also released into the synapse, potentially to modulate synaptic function,164–166 and so the synapse is a unique location where Aβ comes into contact with relatively high concentrations of “freely” available transition metals. This may explain why Aβ deposition is reported to begin in the synapse.167In AD the deposition of aggregated Aβ is associated with oxidative stress. The interaction of Aβ with Zn and Cu can potentially explain both of these phenomena as Aβ will react with both Zn and Cu to form aggregates, while Aβ will react with Cu to generate reactive oxygen species (ROS).168–172 The finding that age- and female-sex-related plaque formation in Tg2576 transgenic mice was reduced by genetic ablation of the synaptic zinc transporter protein ZnT3173 further highlights the potential role metals play in amyloid plaque formation. Consistent with this, low μM levels of Zn2+ will react with synthetic Aβ to induce protease-resistant aggregation and precipitation of the peptide,174–177 which can be reversed by the use of metal chelators.176 This explains why Zn/Cu-selective chelators dissolve Aβ from the insoluble phase of post-mortem human brain specimens that contain amyloid.124,178,179 The interaction of Zn2+ with Aβ occurs in milliseconds,180 selectively (among other biological metal ions) inducing greater exposure of hydrophobic surfaces and A11 immunoreactivity, which are markers of toxic conformers.181 When Zn2+ is present in stoichiometric or greater concentrations relative to Aβ, it induces non-fibrillar, α-helical aggregates, and suppresses β-sheet formation.177,182,183 When Zn2+ is present at concentrations that are substoichiometric relative to Aβ, it promotes fibrillar, β-sheet enriched aggregates, probably by a seeding mechanism.184 Some oligomeric Aβ species induced by Zn2+ are neurotoxic,185 but this also may be a product of the stoichiometric ratio Zn2+ to Aβ since suprastoichiometric ratios induces a species that is redox-inert, and may be the more abundant species in amyloid plaques in contrast to the histologically-invisible forms in the interstitium, cell bodies and synapse.186 It should be noted that the aggregation and toxicity of Aβ are notoriously subject to variability, and factors such as pH, salinity and the presence of Ca2+ and Mg2+ modulate its physicochemical behaviour in response to metal ions.177,178,187,188 We found, for example, that the presence of NaCl enhances Zn2+ induced precipitation of Aβ1–40, which was greatest at 150 mM NaCl (i.e. isotonic saline).188 Nevertheless, the behaviour of the peptide is frequently reported in non-physiological conditions e.g. absence of NaCl. Furthermore, the type of cells that are tested in toxicity experiments, whether primary neurons, neuronal or non-neuronal cells, as well as the culture conditions, all contribute to the variability of results.
Studies of Aβ–Zn2+ interaction in flies have supported the pathogenic consequences of this reaction.189,190 Endogenous drosophila ZIP1 (dZip1, Zrt-Irt like protein 1, a homologue of the transporters that traffic Zn2+ into the cytoplasm) expression was lower in young adult flies transgenic for the overexpression of human Aβ1–42 as compared to the control.190 In comparison to age-matched flies without Aβ expression, 30 day old Aβ42 flies expressed a 170% elevation of brain Zn level. dZip1 knockdown ameliorated the neurodegeneration associated with Aβ overexpression, while dZip1 overexpression or exposure to elevated zinc, exaggerated neurodegeneration and neuropathology, and chelation or overexpression of MTF-1 (a transcription factor that upregulates metallothioneins) ameliorates the toxic phenotype.189,190
Cu2+ can induce similar aggregation of Aβ under mildly acidic conditions,191,192 but at neutral pH Cu2+ at stoichiometric concentrations or greater stabilizes a dimer of Aβ,187 preventing fibril formation.193–195 At substoichiometric concentrations, Cu2+ promotes Aβ fibril formation.184,194,196 The presence of Cu2+ augments Aβ toxicity in cell cultures,177,179,186,197 even at substoichiometric concentrations.184,196 When Cu2+ coordinates to Aβ the metal is redox active and is easily reduced to Cu+, which must be accompanied by the oxidation of another such as thiols, ascorbate, lipid or Aβ itself.198–203 The side-chains of residues 6, 13, 14 and 35 have all been shown by mass spectrometry to be oxidised by the reduction of Cu by Aβ.200,204,205 One residue that is particularly susceptible to oxidative modification is tyrosine and elevated levels of dityrosine and nitro-tyrosine within the neuronal lesions of AD brains has been reported.206 When Aβ reacts with Cu2+ in the presence of H2O2, the tyrosine residue at position 10 will form covalent cross-links with tyrosine residue on other Aβ molecules within an aggregated assembly.199,207,208 These covalently cross-linked oligomers are resistant to proteolytic degradation and promote further peptide aggregation. Support for the pathogenic relevance of Aβ:Cu assemblies has also come from studies of transgenic drosophila, where copper chelation or suppression of Ctr1C (a copper importer), rescues the toxic phenotype.209 Similar results were achieved by inhibiting another copper importer, Ctr1B, and by overexpressing a copper exporter DmATP7 in the nervous system of the Aβ transgenic flies.209
The ability of Aβ to coordinate and trap Cu in an extracellular environment suggests this may result in a deficiency of Cu in the cytoplasm of neurons resulting in certain cellular functions being inhibited. Indeed some studies have shown that Cu levels are increased in the serum and CSF at the same time they are decreased in the hippocampus and amygdala of AD patients.158,159,210,211 Deficiencies in Cu are likely to result in impairment of Cu dependent enzymes that are essential for normal physiological function. Consistent with this, cytochrome c oxidase (COX) and peptidylglycine α-amidating monooxygenase have significantly decreased activities in AD brain.212 Impaired COX activity will lead to deficits in energy metabolism which is also a feature of the AD brain.213 The activity of Cu/Zn-superoxide dismutase-1 (Cu/Zn-SOD1) has been reported to be decreased in both human AD brain and in transgenic animal models.214 It has been suggested that decreased neuronal Cu levels will result in diminished activation of the PI3K/Akt pathway.215 This pathway plays a role in modulating the signals between extracellular growth factors and induction of cell survival.216 Additionally and perhaps most importantly the pathway also modulates (i.e. inhibits) the activity of GSK3β217 one of the kinases responsible for the phosphorylation of tau, such that lower Cu levels leads to increased GSK3β activity and higher degrees of tau phosphorylation. The role GSK-3β plays in AD pathology has triggered a surge in the development of GSK-3β inhibitors to target disease progression. However as will be discussed in more detail below, therapeutic strategies aimed at overcoming Cu deficiency will target PI3K/AKT pathway resulting in an inhibition of GSK3β which in turn results in a decrease in tau phosphorylation and subsequently improvements in cognitive performance (Fig. 4).
Defining the Aβ coordination site
The first observations that Aβ will interact with metal ions were reported in the early to mid 1990's and almost immediately attempts were made to characterise the Aβ metal coordination site. However despite almost twenty years of research the precise nature of the metal coordination site(s) remains controversial (recently reviewed172). Inspection of the Aβ sequence identifies the N-terminal amine and three histidine residues at sequence numbers 6, 13 and 14 that are likely to facilitate the peptide's interactions with Cu and Zn. A variety of spectroscopic studies have confirmed that these residues are primarily involved in metal coordination, under certain conditions other residues have also been reported to be involved (Fig. 2). However there is a lack of consensus amongst these studies. There are a variety of reasons for this, many attributable to the nature of the Aβ peptide. Various spectroscopic methods require at least micromolar concentrations for accurate measurements to be obtained, at these concentrations Aβ will aggregate. Consequently most of the spectroscopic studies have been carried out on shorter versions of the peptide typically sequences ranging from residues 1–16 or 1–28. While these shorter peptides are more soluble they do to some extent still aggregate. In addition to issues associated with peptide aggregation there are clearly pH dependent effects on metal coordination leading to the observation that coordination mode is pleomorphic.218–221 The lack of consensus in the coordination mode and contradictory nature of the published literature has meant that this topic has been the subject of many in reviews in recent years. These reviews are able to deal with the controversies in more detail than we can here.192,222–224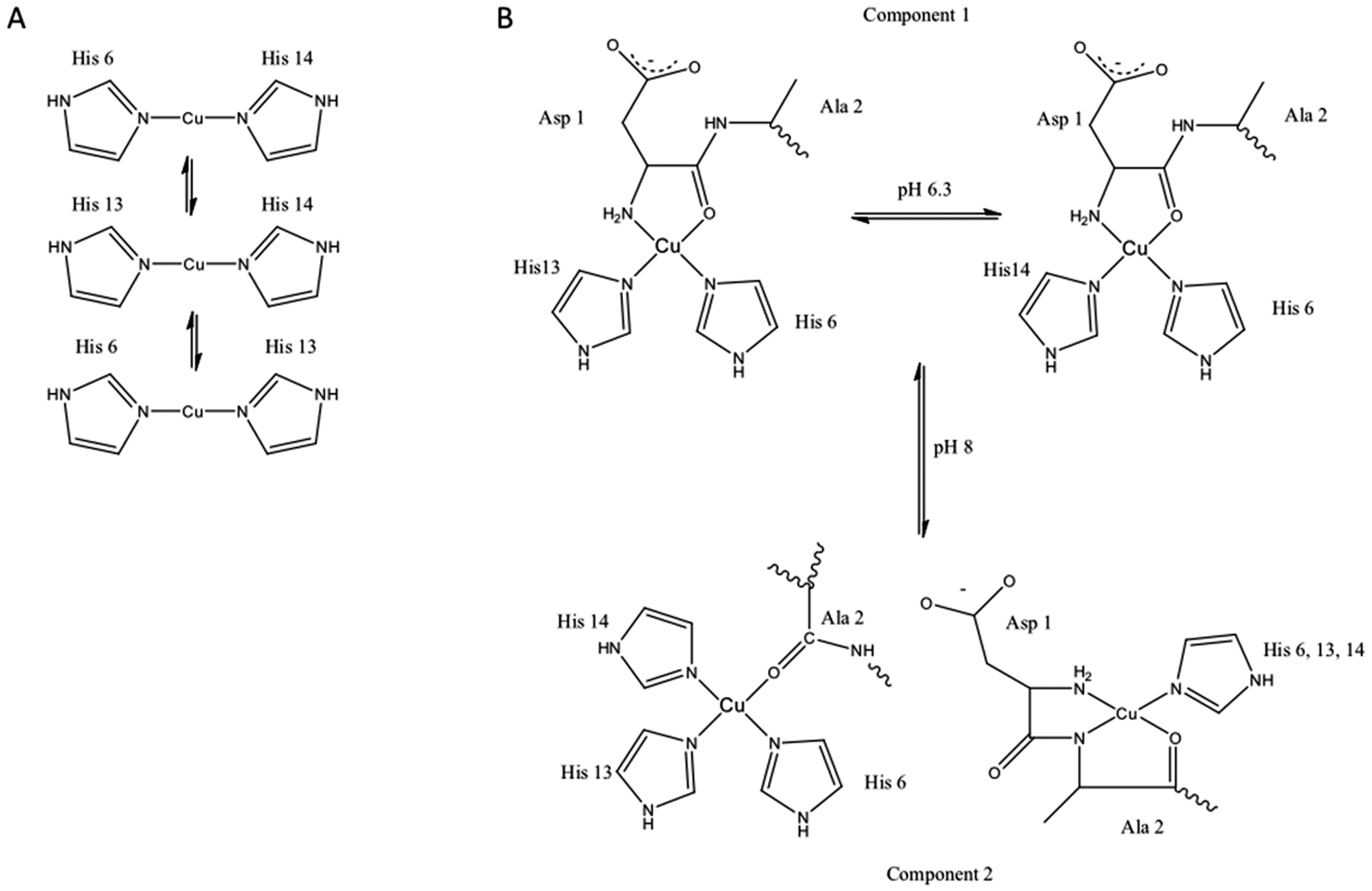 | ||
| Fig. 2 Proposed copper binding sites on Aβ. (A) Cu+ binds Aβ via a linear two-point coordinate mode, where the imidazole sidechains of histidine residues 6, 13, 14 are the ligands. (B) Cu2+ binds Aβ in a square planar 4-point coordination mode, the nature of the ligands bound to the metal are dependent on the solution conditions such as pH. At slightly acidic pH (component I of EPR spectra) the coordination site includes a chelate ring formed by the N-terminal amine and the carbonyl oxygen of residue. The other two coordination sites are occupied by interconverting imidazole sidechains from the histidine residues 6, 13 and 14. At slightly higher pH another coordination mode is characterised in the EPR spectra and has been labelled component 2. Two different structures have been proposed to give rise to this component of the EPR spectrum.192,355,356 | ||
If Aβ–metal interactions drive disease pathology and progression then inhibiting this interaction should be therapeutically beneficial. The simplest way to achieve this would be to use a compound that has a higher metal binding affinity than Aβ (see therapeutics section below). Unfortunately the same difficulties that occur in defining the metal coordination site have also bedevilled attempts to determine Aβ–metal affinities (reviewed in detail in ref. 225) with a wide range of binding constants from high micromolar to attomolar being published. The current best estimates for the binding affinity for Aβ with Zn is in low μM range while for Cu it is in the sub nM range.226,227 Oligomeric species of Aβ bind Cu2+ and Zn2+ with far greater affinity than monomer.228–230 The perturbed equilibrium of the precipitated peptide withdrawing Cu ions from solution may explain the high-affinity apparent constants reported, and explain why the amyloid pathology manages to trap such high concentrations of metal ions. Under conditions where the peptide is kept monomeric or prevented from aggregating the apparent affinity of the binding site is much lower.
The breakdown in metal homeostasis in Parkinson's disease
PD has characteristic proteopathies of Lewy bodies (containing precipitated α-synuclein), as well as neurofibrillary tangles (containing tau, like AD), within the substantia nigra pars compacta, which loses dopaminergic neurons in the disease (reviewed in ref. 97 and 231). As with tangles (vide supra) iron also accumulates within Lewy bodies.232 However, it has not yet been established that α-synuclein binds directly to any metal ion in vivo or in the pathology. Iron may feature in the physiological function of α-synuclein since its 5′UTR contains a functional iron-responsive element that increases α-synuclein expression in the presence of iron.233 Nevertheless, there are several reports of potentially pathogenic interactions of both iron and copper ions with α-synuclein in cell-free systems.234–237 Redox cycling of copper ions has been proposed to be a physiological mechanism by which α-synuclein can be a ferrireductase,238 but can also lead to oxidative damage of the protein,239 and may contribute to toxicity.240,241 While the role of metal interaction with α-synuclein in the pathogenesis of PD remains to be proven, what is more certain is that dysregulation of metal homeostasis in affected tissue in PD that carries the potential of causing abnormal interactions with the protein.Nigral iron elevation in PD is a consistently-reported feature of the disease, and models of the disease. The proposed pathogenic mechanisms for iron-mediated PD degeneration are by increasing oxidative stress,242,243 and by causing α-synuclein aggregation.234,235 A subset of catecholaminergic neurons in the nigra contains a metal-laden pigment (principally bound to iron), neuromelanin, which gives the tissue its black colour.244,245 These neurons are selectively lost in PD, whereas non-pigmented nigral neurons are spared,246 which has drawn suspicion towards the heavy iron (or toxic environmental metal ion) load of this pigment as contributing to neurotoxicity. Its role in pathogenesis is still unclear as there is also evidence for the pigment playing a neuroprotective, antioxidant role, possibly by sequestering redox-active metals.247
Iron accumulation occurs early in PD progression where it is detectable by ultrasonography248 and by MRI.249 Motor phenotype has also been shown to be associated with MRI-detected iron accumulation in specific basal ganglia structures.250 Tissue examination reveals that the iron is elevated in both neurons and glia.251 Genetic lesions that primarily cause substantia nigra (SN) iron retention, such as aceruloplasminemia,252 neuroferritinopathy,253 and neurodegeneration with brain iron accumulation type 1,254 induce SN degeneration and parkinsonism, arguing that iron retention alone is a sufficient cause of Parkinson's disease. Iron accumulation is observed in sporadic PD251 and also with PD familial mutations in genes encoding LRRK2,255 parkin,256 α-synuclein,257 PINK1258 and DJ-1.257 Additionally, iron-chelation with deferiprone or desferrioxamine has also been reported to induce a strong mitophagy response, which is impaired in PD and other neurodegenerative diseases.259 Although the iron accumulation may arise from different mechanisms, the iron accumulation undoubtedly contributes to PD neurodegeneration as the same iron accumulation is a sufficient cause of PD in other, primary brain iron accumulation genetic lesions. The product of iron with dopamine levels in the neurons of the various nuclei in this region explains why the SNpc is most predisposed to combustion, leading to demise in this region with relative sparing of the other iron-containing neurons, which lack the same enrichment of dopamine.260 Iron therefore also represents a common therapeutic target.
A recent Mendelian randomization analysis revealed a significant association between three genetic variants that are known to have major influences on serum iron levels (two alleles of HFE, where mutation causes hemochromatosis; and matriptase, TMPRSS6) and the risk for PD. The findings in this large population suggest a causal association between increased serum iron levels and decreased risk of developing PD. Serum iron explained ≈33% of the risk for PD in people with no other iron abnormality (3% risk reduction per 10 μg dL−1 increase in serum iron, therefore 33%, considering the normal range of serum iron is 65–176 μg dL−1 for men). Since, as explained above, increased brain (nigral) iron may be pathogenic in PD, the apparent inverse relationship between serum and brain iron in the risk for PD could be consistent with the inability for iron to be trafficked out of the brain into the plasma. Most iron in plasma is bound to transferrin and iron is loaded into transferrin principally through the activity of the ferroxidase, ceruloplasmin.261
Ceruloplasmin is expressed in glia in the brain262 as well as being abundant in plasma. The specific ferroxidase activity of ceruloplasmin is decreased ∼80% in the substantia nigra of idiopathic Parkinson's disease cases, possibly related to a marked drop in copper levels in the tissue.263 Copper regulation is systemically perturbed in PD.264 Consistent with ceruloplasmin loss being pathogenic in PD by raising nigral iron, ceruloplasmin knock-out mice develop parkinsonism with elevated nigral iron, and are rescued by iron chelation,263 as is the MPTP model of PD,265 and parkinsonism in tau knockout mice, where iron accumulates in the nigra.97
Several iron chelation approaches have been reported as salutary in PD rodent models.266–269 One of the agents that has successfully reduced iron levels and improved motor function in animal models of PD is M30 [5-(N-methyl-N-propargylaminomethyl)-8-hydroxyquinoline].270 This compound is reported to be multi-functional combining the chelating ability of the 8-hydroxyquinoline moiety with the ability to inhibit monoamine oxidase through the propargyl moiety.
The most advanced compound in the iron-chelator class is deferiprone, since it has recently completed a 12 month double-blind, placebo-controlled randomized clinical trial (30 mg per kg per day) in early-stage PD patients (n = 37 completed in a delayed-start paradigm). The trial revealed that patients responded significantly earlier and sustainably to treatment, as measured by both amelioration of substantia nigra iron accumulation (by MRI) and by Unified Parkinson's Disease Rating Scale motor indicators of disease progression.271 Deferiprone was initially tested in the MPTP mouse mice model of PD using a preventative treatment strategy, i.e. the drug was first administered prior to the creation of the lesion in the SNpc by MPTP. The drug was able to reduce iron levels and increase tyrosine hydroxylase positive cells in the SN, this was accompanied by reductions in oxidative stress markers and improvements in motor function.271 This success encouraged the use of deferiprone in a small clinical trial of PD patients. This trial was an 18 month placebo controlled double blind study that utilized a 6 month delayed start paradigm (i.e. half the cohort started on drug at the beginning of the trial the other half started on placebo, after 6 months the placebo group also began receiving drug). The purpose of such trial design is to differentiate symptomatic effects from true disease modifying ones. The patients were orally administered drug at 30 mg per kg per day, iron levels in the SN were assessed by magnetic resonance imaging and motor function scores were determined using the Unified Parkinson's Disease Rating Scale (UPDRS). For the initial treatment group there were significant reductions in iron after six months that were sustained after 12 and 18 months, for the delayed start group while on placebo there was no change in iron but once treatment with deferiprone commenced this group also saw decreases in iron, at the 18 month time point the decrease in iron was similar for both the initial treatment group and the delayed start. The initial treatment group showed a significant improvement in their UPDRS scores as compared to baseline and placebo, while the scores for this group were always better than baseline there was no further improvement in the scores despite a further 12 months of drug treatment. The delayed start group showed no improvement in their UPDRS at any stage of the trial despite the significantly decreased iron levels. These promising results await phase 3 validation.
Zinc has also recently featured in PD. Elevated nigral zinc has been found in PD patients.272 The PARK9 mutation of ATP13A2, that causes juvenile onset PD, leads to dyshomeostasis of intracellular Zn2+ that contributes to lysosomal dysfunction and the accumulation of α-synuclein.273 Recent evidence in neurospheres derived from patients confirms the Zn2+ dyshomeostasis and shows how this adversely impacts on mitochondrial metabolism.274
Metal dyshomeostasis in Huntington's disease
Huntington's disease (HD) is a rare genetic disorder associated with expanded trinucleotide CAG repeats in the gene encoding the huntingtin protein. When translated into the protein this results in long extended poly glutamine sequences that are particularly prone to aggregation. The disease is associated with both motor impairment and cognitive decline.The pioneering work of Jenner, Marsden and colleagues catalogued tissue metal changes in basal ganglia disorders, demonstrating a robust elevation of iron in the caudate nucleus and copper in the putamen in HD.272,275
Several further studies have confirmed the elevation of striatal iron in HD, as well as co-registration of changes in Magnetic Resonance Imaging (MRI) in post-mortem tissue. MRI studies utilizing field-dependent R2 increase, for a specific index of tissue ferritin content, found an elevation of iron in caudate, putamen, and globus pallidus in HD subjects compared to controls.276,277
Field map (FM) MRI, a novel analytical approach based on Susceptibility Weighted Imaging (SWI), assesses magnetic iron concentrations, and was used recently to analyze various brain regions in HD subjects at various clinical stages of the disease, and compared them to age- and sex-matched healthy controls.278 FM values in the pre-HD and HD groups were significantly higher than those in the control group in the basal ganglia structures, including the caudate, putamen, and globus pallidus, bilaterally starting with preclinical HD. A trend for higher FM values with increasing disease severity was noted, starting with significant increases in preclinical HD. In the cortex, iron levels were significantly elevated in the most advanced stage of disease studied, affecting mainly left superior frontal, left middle frontal, and bilaterally anterior cingulate, paracentral and precuneus. There were no changes in the thalamus, hippocampus, or amygdala. A significant association was found between FM value and CAG triplet repeat length bilaterally in the caudate, accumbens, putamen, pallidum, and anterior cingulate. These investigators confirmed the elevation in iron levels in HD in a separate post-mortem series, which demonstrated elevated iron as well as zinc in the pallidum and putamen.
In vitro studies showed that the amino-terminal 171 residues of huntingtin contain a domain that interacts with and reduces Cu2+ to Cu+, and Fe3+ to Fe2+,279,280 potentially contributing to the oxidative stress that is prevalent in the disorder. In addition, full-length wild-type mouse huntingtin was found to form SDS-resistant aggregates following incubation at 37 °C. This aggregation was promoted by Cu2+, and inhibited by metal-complexing agents EDTA and clioquinol. Copper and iron concentrations in the striata and frontal cortices of 12 month-old knock-in CAG140 and 12 week-old R6/2 transgenic mouse models of HD have also been reported. Despite the absence of neurobehavioural deficits in CAG140 mice cortical iron levels were significantly increased by 15%.279 The R6/2 mice had cognitive deficits as assessed by open-field tests and a 12% reduction in brain weight consistent with advanced disease, and exhibited significantly increased copper in striatum (26%) and cortex (51%). Importantly, the amyloid precursor protein (APP) was significantly decreased in striatum and cortex of the R6/2 HD transgenic mice,279 which may account for the elevation of copper and iron in the brain tissue, since APP promotes the export of copper83 and iron.29,97 A recent in-depth examination of the brains of the R6/2 model revealed evidence of an increased labile iron pool, with accumulation of Fe2+ in secondary lysosomes. Treatment of model with intrathecal desferrioxamine rescued the motor and atrophy phenotypes within 2 weeks of treatment.281 Modulation of copper homeostasis by genetic and dietary interventions strikingly modifies disease progression in a HTT exon 1 Drosophila model of HD.282 Copper interaction with mutant HTT promoted toxicity in this model, which was rescued by substituting key copper-coordinating residues, Met8 and His82.282
The breakdown in metal homeostasis in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a disease in which motor neurons in the spinal cord and brain progressively deteriorate leading to paralysis and death. Currently there is no effective disease modifying therapy for this disease. Approximately 10% of ALS cases have identified genetic causes while the remaining 90% appear to be sporadic in nature. Genes associated with the familial forms of ALS include TARDBP,283FUS,284C9ORF72285,286 and SOD1.287 The proteins (respectively, TDP-43, FUS, C9ORF72 and Cu/Zn superoxide dismutase [SOD1]) encoded by these various genes have been found in aggregated deposits within diseased tissue. How these various genes and/or their translated proteins drive disease progression is still the subject of much debate. One hypothesis is that disease progression is linked with increases in endoplasmic reticulum (ER) stress as a result of the up-regulation in the unfolded protein response (UPR) pathway. One consequence of the elevation of this pathway is the up-regulation of pro-apoptotic cascades and the formation of stress granules, which contain both proteins and RNA. Stress granule formation is also promoted by oxidative stress. The sequestration of both protein and RNA into these intracellular bodies appears to be the mechanism by which cells shut down less essential RNA translation in response to cellular stress. Among the protein products incorporated into stress granules are TDP43 and FUS, which have roles in RNA metabolism.Mutations in SOD1 (of which there are over 100) are the most common cause of familial ALS, currently there is no generally accepted hypothesis as to how these mutations drive disease, but it does appear to be the gain of a toxic function and not the loss of SOD1 activity.288 Pathogenic mutations of SOD1 that cause familial-ALS are also associated with SOD1 aggregation,288 and SOD1 aggregates are also observed in affected spinal neurons in sporadic cases.289 However, as is the case with other proteopathies associated with neurodegenerative disease, it is unclear whether the SOD1 proteopathy is itself the source of toxicity. Since copper is redox-active, and since oxidative damage characterizes ALS, considerable effort has explored the role of copper in ALS pathogenesis caused by mutant ALS. Spinal cord copper levels are elevated in transgenic mice carrying the SOD1G93A mutant.290–292
Both copper chelation, and genetic strategies that lower spinal cord copper, ameliorate the ALS phenotype in mutant SOD1 transgenic mouse models.293–298 Genetic ablation of the copper chaperone of SOD1 (CCS1) did not rescue the ALS phenotype of mutant SOD1 transgenic mice, but this just establishes that if copper plays a role in pathogenesis, then it is not the copper that is normally inserted into the active site.299,300 Supporting this interpretation, copper(II)diacetylbis(N(4)-methylthiosemicarbazonato) (CuII(ATSM)), which loads copper into the active site of SOD1, ameliorates the phenotype.301 There is also evidence that copper ions can adventitially attach to pro-oxidant, pathological surface binding sites on mutant SOD1, which are revealed by oxidation of the disulphide bridge required to maintain tertiary structure and stabilize dimeric SOD1.302,303 Presentation of copper in excess of the ability of the amount that SOD1 can incorporate into its active site may explain why overexpression of CCS1 markedly exacerbated the pathological phenotype of mutant SOD1 transgenic mice.304 Supporting the possibility that copper outside of the active site of SOD1 may be pathogenic, recent findings have shown that transgenic models of ALS expressing SOD1 with mutations that disrupt copper binding at the active site still demonstrate a marked age-dependent rise in spinal cord copper levels in tissue fractions outside of SOD1.305
Recently, it has been demonstrated that mutations in SOD1 associated with familial forms of ALS can drive ER stress, through interactions with Derlin-1, which is a component of the ER-associated degradation machinery.306 Although these mutations are spread throughout the structure of SOD1 in general these mutations lead to a decreased affinity of mutant SOD1 for Zn. Wildtype SOD1 exists predominantly as a homodimer however the loss of Zn from the active site destabilizes the dimer interface shifting the equilibrium between monomer and dimer more towards the monomer.307 It has recently been demonstrated that under conditions of Zn deficiency wildtype SOD1 can also drive ER stress, the loss of Zn from SOD1 causes a change in protein confirmation promoting the interaction with Derlin-1.308 Interestingly, it has previously published that a small cohort of ALS subjects from Guam have decreased cortical Zn levels as compared to controls, unfortunately this study did not look at Zn levels in the spinal cord of these subjects.309
The deposition of aggregated proteins requires the misfolding of the protein. The folding/unfolding pathway of SOD1 has been extensively studied and involves a number of intermediates including the Zn deficient form described above (reviewed310). Cu deficient forms have also been identified although existing at much lower levels than the Zn deficient forms. Interestingly the folding/unfolding pathways of mutant forms of SOD1 have significantly more of the Cu deficient intermediate than does wildtype SOD1 and it has been speculated upon that this form of SOD1 maybe the toxic species that is responsible for the neurodegeneration.311
There is also emerging evidence that Fe metabolism is perturbed in ALS, with serum Ferritin levels shown to be elevated in ALS subjects as compared to controls, it is not clear whether this a contributing factor to the disease or as a response to increased oxidative stress associated with the disease.312 Additionally a H63D mutation in the HFE gene has been shown to be a risk factor for ALS,313 how this mutation increases the risk of developing ALS is not clear at this stage.
Targeting metal homeostasis as a therapeutic strategy
To date there are no drugs that modify the underlying causes of the neurodegenerative diseases. All the approved drugs such as the acetylcholinesterase drugs in AD, the dopamine agonists in PD, and riluzole in ALS, give modest improvements in alleviating disease symptoms rather than affecting the underlying course of the disease.314 These diseases are characterized by cognitive and motor deficits that arise from defective synaptic transmission, a process that involves the synchronized function of neurotransmitters, cell surface receptors, and pre- and postsynaptic cycling of synaptic vesicles. As described earlier, transition metal ions play a major role in this dynamic process, and our overarching hypothesis is that a breakdown of this process is upstream to disease progression. Therefore chemical agents that are able to restore metal homeostasis should be effective disease modifying drugs. One approach is to use metal chelators as have been used clinically for decades to target metal overload diseases such as hemochromatosis (Fe) and Wilson's disease (Cu). However, the chelators used to treat these diseases such as desferrioxamine, penicillamine and trientine are intended to deplete metal from overloaded tissue, and so facilitate excretion of the overloaded metal. To accomplish this, the chelators usually have high metal-binding affinities and are hydrophilic. However, these types of chelators are inappropriate as potential therapeutics for neurodegenerative diseases as they have to cross the blood brain barrier, which generally requires small hydrophobic molecules. Additionally, as we have laboured to explain, metal ions are essential to many essential neurochemical processes and the indiscriminate removal of metals from brain tissue is problematic. In addition, the metal ions that are maldistributed or accumulating in the neurodegenerative diseases that we have reviewed, do not require high-affinity ligands (i.e. classical chelators) in order to be redistributed or sequestered. However, no brain-penetrant moderate-affinity metal ion-ligating molecules are approved for any medical indication. Therefore, those who test the validity of metal-related targets as being of potential value for novel therapeutics have been obliged to invent a new pharmacology, which has not been a palatable prospect for modern big pharmaceutical companies.Metal Protein Attenuating Compounds (MPAC) for Alzheimer's disease
As discussed above, AD could not be described as a traditional metal overload disease. A more accurate description is one where metal homeostasis is perturbed, resulting in mis-compartmentalisation of metal ions. Therefore the goal of the MPAC strategy for AD is to develop compounds that target mis-localized metals and restore them to where they came from, i.e. within neurons, astrocytes or glia. MPAC is a term for an organic small molecule that, without the high affinities of chelators, can ligate adventitial metal ions attached to proteinopathies and redistribute them to a safer compartment. The prototypic MPAC is clioquinol (CQ, 5-chloro-7-iodo-8-hydroxyquinoline) a small hydrophobic molecule with moderate affinity for Cu2+ and Zn2+ (Kd of 8 × 10−11 M and 1.4 × 10−9 M, respectively) and capable of crossing the blood–brain barrier.315In vitro, CQ inhibits metal mediated aggregation and ROS production by Aβ.316 Tg2576 transgenic mice orally treated with CQ for nine weeks had a 49% decrease in brain Aβ burden as compared with non-treated controls. This treatment did not induce a systemic change in metal levels.316These successful animal studies were followed by human studies, with the effect of oral CQ treatment in a randomized, double-blind, placebo-controlled pilot phase II clinical trial of moderately severe AD patients was evaluated.317 CQ treatment was reported to significantly attenuate cognitive deterioration over a 9 month period. Despite these positive outcomes further development of CQ was halted due to manufacturing difficulties as contamination with di-iodo-8-hydroxyquinoline, a known carcinogen, occurred during larger scale chemical synthesis preventing the production of GMP-quality trial stock. As the second generation MPAC PBT2 (a novel chemical entity) was then ready for clinical development, no attempt was made to pursue further work with CQ.
PBT2 is a second generation MPAC,318 like CQ it is an 8-hydroxyquinoline (Fig. 3) but with improved solubility, blood brain barrier penetrability and greater efficacy in preclinical in vivo studies.319 PBT2 rapidly improved cognitive performance in the APP/PS1 and Tg2576 transgenic mouse models of AD, accompanied by changes in a number of disease relevant biomarkers including an increase in synaptophysin levels indicating improved synaptic health, reductions in Aβ load and tau phosphorylation. Perhaps most importantly, there was a significant reduction in soluble interstitial Aβ within hours of drug being administered. PBT2 was subsequently studied in a phase IIa clinical trial in subjects with early AD, which involved a 12 week treatment period, was double blind and placebo controlled. This study demonstrated that the drug was safe and well tolerated at 50 mg and 250 mg daily doses, with reduced CSF levels of Aβ1–42 at the 250 mg dose. Improved cognitive performance in patients taking PBT2 (compared to placebo) was observed when tested on executive function in the Neuropsychological Test Battery.320,321 A small 12 month phase II trial of PBT2 (n ≈ 50 in two randomized arms) has just been completed with positron emission tomography imaging of brain amyloid as the primary readout. The drug was well tolerated, but no significant effect on amyloid burden was found in the primary readout. A post-hoc analysis recently reported that PBT2 slowed the decline of hippocampal atrophy at almost half the rate of the placebos, and there was significant drop in amyloid burden when adjusting for baseline levels of amyloid (http://pranabio.com/wp-content/uploads/2014/07/AAIC-Panel-Presentation_Colin-Masters-1.pdf). While the data remain unpublished and, therefore, unavailable for scrutiny, it is possible that the study was underpowered. However, since the drug was not designed to target amyloid fibrils, it is possible that the result merely confirms that PBT2 does not engage with fibrils.
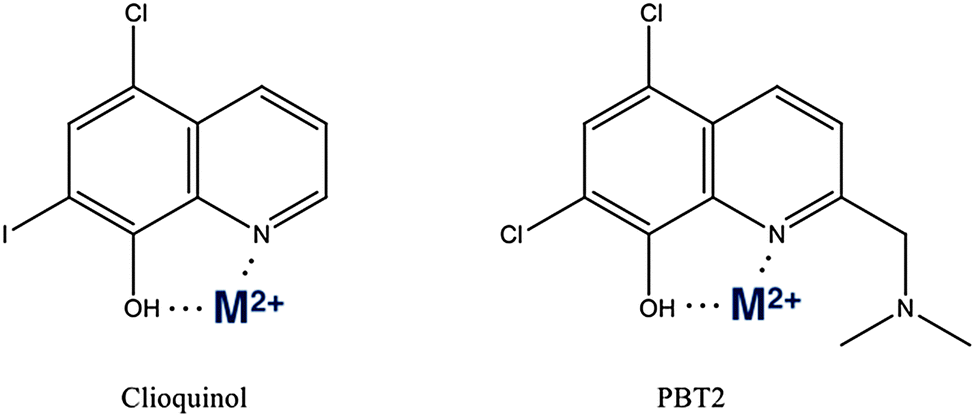 | ||
| Fig. 3 Structures of clioquinol and PBT2.357 The oxygen, nitrogen metal ion coordination sites are shown, which are in common between all molecules utilizing the 8-OH quinolone scaffold. | ||
The original goal in developing the MPAC compounds for the treatment of AD had been to identify compounds that were capable of inhibiting the Aβ–metal interactions that we hypothesized were the underlying cause of AD (see above). However in the development of PBT2 it soon became obvious that simply inhibiting Aβ–metal interactions was insufficient to explain all the observed affects of the drug. Through a variety of investigations it was shown that compounds such as PBT2 and CQ translocate Cu2+ and Zn2+ from the extracellular environment to inside the cell. Once inside the cell, the metals dissociate from the drug to initiate a neuroprotective signalling cascade that includes the activation of PI3K and up regulation of metalloprotease activity, which then degrades Aβ.319,322,323 This signalling cascade also results in an inhibition of tau phosphorylation, via inhibition of GSK3β.324 Other positive consequences of this metal “ionophore” effect include the promotion of neurite extension and increased dendritic spine density.325
These results indicate that the MPACs are not acting as simple chelators, and that their mechanism of action is more sophisticated as they not only prevent the deleterious effects of the breakdown in metal homeostasis, but also rectify the misbalance by restoring metals that are deficient to the cell to carry out salutary actions. As such the MPACs mechanism is similar to that of a metal chaperone.
The positive clinical outcomes for CQ and PBT2 have promoted a growing interest in the ability of metal chelating compounds to have therapeutic potential in AD. This has resulted in an array of chelating compounds with a wide variety properties being synthesized.141,270,326–328 To augment the ability to dissociate metal-induced Aβ oligomers, some medicinal chemistry approaches employ structures that can both attach to the Aβ as well as interact with the pathological metal,177,179,329–335 or have hybrid structures that provide additional salutary biological interactions such as glucose receptor targeting,336 monoamine oxidase B inhibition268,270 or acetylcholinesterase inhibition.337 Most of these compounds have been shown to inhibit Aβ–metal interactions and therefore Aβ aggregation and ROS production, and, apart from clioquinol and PBT2,316,319,338–340 other metal-targeting and hybrid molecules have also shown beneficial effects in transgenic animal studies.141,328,341 These various approaches were reviewed in detail in this journal recently.342
Targeting metals in candidate HD therapeutics
An unbiased screening of >200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds identified the 8-hydroxyquinoline chemical class as the most effective at preventing proteotoxicity caused by misfolded proteins including polyglutamine through a metal-dependent mechanism.343 This was consistent with a prior report that the brain permeable copper and iron chelator clioquinol was neuroprotective, decreased brain aggregate load, and restored fallen insulin levels in R6/2 HD mice.344 Similarly, epigallocatechin-gallate, a green tea flavonoid, and also a chelator, has been reported to inhibit huntingtin misfolding and rescue toxicity in a Drosophila model of HD.345 Confirming a role for copper interaction with mutant huntingtin in promoting disease pathogenesis, lowering central nervous system copper levels with either Ctr1B suppression or DmATP7A overexpression rescued lethality and neuropathology in two polyQ transgenic Drosophila models of HD.209 The Cu+ chelator bathocuproine disulfonate and Cu2+ chelator, clioquinol, significantly rescued the survival rate of the Htt transgenic flies, whereas supplementation of additional CuCl2 worsened the survival defect.209 Ablation of the Cu2+ coordination sites on mutant Htt279 generated a form of the protein that, even when overexpressed, had markedly less toxicity than the parental mutant in a transgenic Drosophila.209
000 compounds identified the 8-hydroxyquinoline chemical class as the most effective at preventing proteotoxicity caused by misfolded proteins including polyglutamine through a metal-dependent mechanism.343 This was consistent with a prior report that the brain permeable copper and iron chelator clioquinol was neuroprotective, decreased brain aggregate load, and restored fallen insulin levels in R6/2 HD mice.344 Similarly, epigallocatechin-gallate, a green tea flavonoid, and also a chelator, has been reported to inhibit huntingtin misfolding and rescue toxicity in a Drosophila model of HD.345 Confirming a role for copper interaction with mutant huntingtin in promoting disease pathogenesis, lowering central nervous system copper levels with either Ctr1B suppression or DmATP7A overexpression rescued lethality and neuropathology in two polyQ transgenic Drosophila models of HD.209 The Cu+ chelator bathocuproine disulfonate and Cu2+ chelator, clioquinol, significantly rescued the survival rate of the Htt transgenic flies, whereas supplementation of additional CuCl2 worsened the survival defect.209 Ablation of the Cu2+ coordination sites on mutant Htt279 generated a form of the protein that, even when overexpressed, had markedly less toxicity than the parental mutant in a transgenic Drosophila.209
Clioquinol is moderate in its affinity for transition metals, redox-silencing, and the prototype for PBT2,319 a novel drug candidate that reported efficacy in a phase 2 clinical trial for Alzheimer's disease in 2008.320,346,347 PBT2 itself significantly delayed the onset of paralysis in a C. elegans model of polyQ over-expression.348 PBT2 oral treatment (from 3 weeks of age) also significantly rescued the motor deficits of R6/2 HD mice and prevented brain atrophy.348 On the basis of these results, PBT2 recently completed a phase 2 clinical trial in a cohort of HD patients over a 6 month period. While the result await peer-reviewed publication, the announcement of the top-line results indicated that the primary readout was safety and tolerability was met satisfactorily, and, in addition, secondary measures indicated some cognitive benefit and possible rescue of brain atrophy.349 These results appear promising, but efficacy waits for phase 3 confirmation.
Utilizing metal chemistry as a therapeutic strategy
Bis(thiosemicarbazone) complexes for AD
The mechanism of action of the MPACs is complex with a number of different activities being potentially salutary. One concept that we have investigated is that the delivery of bioavailable copper and or zinc into cells can activate neuroprotective signalling cascades. To further study whether metal delivery in the absence of any chelating effects has any positive therapeutic benefits we utilised Cu(II) and Zn(II) complexes of bis(thiosemicarbazone) (BTSC). These complexes have been reported to have a broad range of pharmacological activity, with recent interest focused primarily on the ability of selected complexes to deliver radioactive copper isotopes to hypoxic tissue and leukocytes.350,351 Our initial work focused on cell culture experiments where we were able to demonstrate that a range of metal BTSC complexes were able to increase intracellular metal levels in Chinese hamster ovary cells overexpressing APP (APP-CHO).352 The copper complexes had been engineered to either be stable in an intracellular environment, or susceptible to a reduction in the oxidation state of the metal, which, in turn, significantly decreased the stability of the metal ligand complex (i.e. upon reduction in the metal's oxidation state, the copper was now bioavailable). The APP-CHO cells secrete Aβ into the cell culture media, and treatment with metal BTSC complexes where the metal dissociated from the complexes significantly reduced the detectable levels of Aβ. This was consistent with previous reports that found that copper elevation suppressed Aβ production.60,68,86 Complexes that did not release the metal had no effect on levels of extracellular Aβ. This ability to decrease Aβ levels was a consequence of initiating a signaling cascade that included PI3K and c-Jun N-terminal kinase and activation of proteases that degraded Aβ (Fig. 4).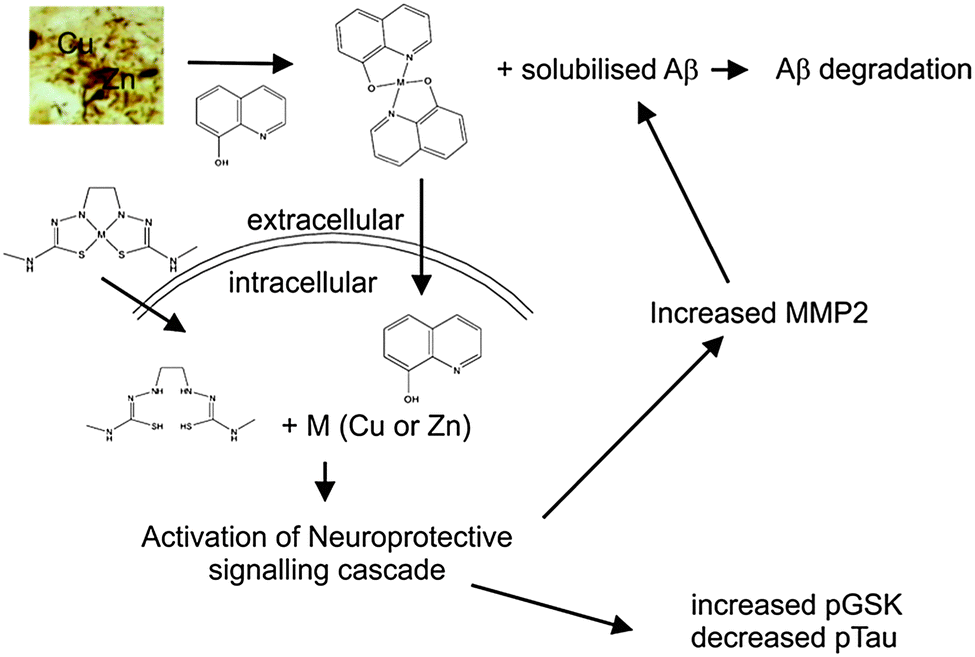 | ||
| Fig. 4 The mechanism of action for metal chaperones in AD. Aβ metal interactions drive Aβ aggregation. Amyloid plaques, the defining pathological feature of AD, are enriched in metals such as Cu and Zn. The MPACs CQ and PBT2 are able to remove metals from plaques in the process solubilising the Aβ, which, in turn facilitates degradation of the peptide. In addition to this activity, the MPACs are able to translocate metal ions across cellular membranes into the cell and in the process activating neuroprotective cell signalling cascades that result in increased phosphorylation of GSK3β and a reduction in the phosphorylation of tau. Bisthiosemicarbazone complexes such as CuII(gtsm), while not able to directly affect Aβ in plaques are able to activate neuroprotective signalling pathways by crossing cell membranes and releasing bioavailable metals. | ||
Two compounds, glyoxalbis(N(4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(gtsm)) and diacetylbis(N(4)-methyl-3-thiosemicarbazonato) copper(II) (CuII(atsm)), known to cross the blood brain barrier,353 were selected to evaluate this class of compound in vivo. In the Cu2+ oxidation state both of these complexes are very stable (logKA ∼ 18)350 but CuII(gtsm) is more susceptible to intracellular reductants and the Cu+ complex is much less stable (logKA ∼ 8). In contrast, CuII(atsm) is resistant to reduction in normal cells, and so the copper remains coordinated to the complex. Both complexes were administered to APP/PS1 by oral gavage for four months. CuII(gtsm) improved cognitive performance in these mice whereas CuII(atsm) had no effect. Consistent with the beneficial effects of CuII(gtsm), mice treated with this drug had reduced levels of Aβ oligomers and phosphorylated tau.354 These data are consistent with the proposed therapeutic mechanism of clioquinol, PBT2 and related molecules, of enhancing the bioavailability of copper and zinc (Fig. 4).
Conclusions
In this review, we have attempted to catalogue the multiple pathological changes that occur in brain metal homeostasis in age-related neurodegenerative disorders, focusing on Alzheimer's disease, Parkinson's disease, Huntington's disease and ALS. This list of diseases is not comprehensive, but where the evidence for metal involvement is most developed. These neurodegenerative diseases are not caused by simple toxicological overload by metals, although the possibility that exposure may alter disease risk remains an important line of investigation. Rather, with aging, and with neurodegenerative disease, there is a breakdown in the homeostasis of essential endogenous biometals. As a result metals both accumulate in microscopic proteopathies, and can be deficient in other cells or cellular compartments. Therefore, bulk measurement of metal content in brain tissue samples reveal only the “tip of the iceberg”, with most of the important changes occurring on a microscopic and biochemical level.Changes in metal distribution, cellular deficiencies, or sequestration in proteopathies all present abnormalities that can be corrected in animal models by small molecules. These biochemical targets are more complex than the simple excess metal levels that are targeted by chelators. A summary of the potential targets for the different types of pharmacological approaches discussed in this review is shown in Fig. 1. It is still relatively early days for this new metal-related pharmacology, and our impression from decades of experience in communicating with colleagues in the mainstream of neurodegenerative disease research is that until metallobiology yields a drug that shows robust benefit in the clinic, most researchers in the field of neurodegenerative disease will remain unfamiliar with the concepts of metalloneurobiology and its pharmacological targets. We nonetheless feel that the targets of metalloneurobiology are rich with pharmacological opportunities. We hope this review may illustrate some of the richness in the science that has developed in the study of metals in neurodegeneration, and encourage further exploration of its novel pharmacology.
Acknowledgements
Supported by funds from the National Health & Medical Research Council, Australian Research Council, and the Cooperative Research Center for Mental Health. The Florey Institute acknowledges the funding support from the Victorian Government's Operational Infrastructure Support program.References
- B. Halliwell and J. M. C. Gutteridge, Biochem. J., 1984, 219, 1–14 CAS.
- D. Harman, J. Gerontol., 1956, 11, 298–300 CrossRef CAS.
- D. Harman, Age, 1993, 16, 23–30 CrossRef.
- C. J. Frederickson, J. Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS PubMed.
- Y. H. Hung, A. I. Bush and R. A. Cherny, JBIC, J. Biol. Inorg. Chem., 2010, 15, 61–76 CrossRef CAS PubMed.
- T. Fukada and T. Kambe, Metallomics, 2011, 3, 662–674 RSC.
- R. D. Palmiter, T. B. Cole, C. J. Quaife and S. D. Findley, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14934–14939 CrossRef CAS.
- G. Danscher and M. Stoltenberg, J. Histochem. Cytochem., 2005, 53, 141–153 CrossRef CAS PubMed.
- J. Qian and J. L. Noebels, J. Physiol., 2005, 566, 747–758 CAS.
- G. A. Howell, M. G. Welch and C. J. Frederickson, Nature, 1984, 308, 736–738 CrossRef CAS.
- S. Y. Assaf and S.-H. Chung, Nature, 1984, 308, 734–736 CrossRef CAS.
- K. Vogt, J. Mellor, G. Tong and R. Nicoll, Neuron, 2000, 26, 187–196 CrossRef CAS.
- C. J. Frederickson, L. J. Giblin, 3rd, R. V. Balaji, R. Masalha, C. J. Frederickson, Y. Zeng, E. V. Lopez, J. Y. Koh, U. Chorin, L. Besser, M. Hershfinkel, Y. Li, R. B. Thompson and A. Krezel, J. Neurosci. Methods, 2006, 154, 19–29 CrossRef CAS PubMed.
- S. L. Sensi, P. Paoletti, A. I. Bush and I. Sekler, Nat. Rev. Neurosci., 2009, 10, 780–791 CrossRef CAS PubMed.
- S. L. Sensi, P. Paoletti, J. Y. Koh, E. Aizenman, A. I. Bush and M. Hershfinkel, J. Neurosci., 2011, 31, 16076–16085 CrossRef CAS PubMed.
- P. Paoletti, P. Ascher and J. Neyton, J. Neurosci., 1997, 17, 5711–5725 CAS.
- L. Mony, L. Krzaczkowski, M. Leonetti, A. Le Goff, K. Alarcon, J. Neyton, H. O. Bertrand, F. Acher and P. Paoletti, Mol. Pharmacol., 2009, 75, 60–74 CrossRef CAS PubMed.
- E. Pan, X. A. Zhang, Z. Huang, A. Krezel, M. Zhao, C. E. Tinberg, S. J. Lippard and J. O. McNamara, Neuron, 2011, 71, 1116–1126 CrossRef CAS PubMed.
- Y. Z. Huang, E. Pan, Z. Q. Xiong and J. O. McNamara, Neuron, 2008, 57, 546–558 CrossRef CAS PubMed.
- L. Besser, E. Chorin, I. Sekler, W. F. Silverman, S. Atkin, J. T. Russell and M. Hershfinkel, J. Neurosci., 2009, 29, 2890–2901 CrossRef CAS PubMed.
- T. Perez-Rosello, C. T. Anderson, F. J. Schopfer, Y. Zhao, D. Gilad, S. R. Salvatore, B. A. Freeman, M. Hershfinkel, E. Aizenman and T. Tzounopoulos, J. Neurosci., 2013, 33, 9259–9272 CrossRef CAS PubMed.
- M. Hershfinkel, K. Kandler, M. E. Knoch, M. Dagan-Rabin, M. A. Aras, C. Abramovitch-Dahan, I. Sekler and E. Aizenman, Nat. Neurosci., 2009, 12, 725–727 CrossRef CAS PubMed.
- A. G. Daniel, E. J. Peterson and N. P. Farrell, Angew. Chem., Int. Ed., 2014, 53, 4098–4101 CrossRef CAS PubMed.
- E. Aizenman, A. K. Stout, K. A. Hartnett, K. E. Dineley, B. McLaughlin and I. J. Reynolds, J. Neurochem., 2000, 75, 1878–1888 CrossRef CAS.
- Y. Zhang, H. Wang, J. Li, D. A. Jimenez, E. S. Levitan, E. Aizenman and P. A. Rosenberg, J. Neurosci., 2004, 24, 10616–10627 CrossRef CAS PubMed.
- M. C. McCord and E. Aizenman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13988–13993 CrossRef CAS PubMed.
- Y. Uchida, K. Takio, K. Titani, Y. Ihara and M. Tomonaga, Neuron, 1991, 7, 337–347 CrossRef CAS.
- Y. Uchida, F. Gomi, T. Masumizu and Y. Miura, J. Biol. Chem., 2002, 277, 32353–32359 CrossRef CAS PubMed.
- J. A. Duce, A. Tsatsanis, M. A. Cater, S. A. James, E. Robb, K. Wikhe, S. L. Leong, K. Perez, T. Johanssen, M. A. Greenough, H. H. Cho, D. Galatis, R. D. Moir, C. L. Masters, C. McLean, R. E. Tanzi, R. Cappai, K. J. Barnham, G. D. Ciccotosto, J. T. Rogers and A. I. Bush, Cell, 2010, 142, 857–867 CrossRef CAS PubMed.
- S. L. Sensi, L. M. Canzoniero, S. P. Yu, H. S. Ying, J. Y. Koh, G. A. Kerchner and D. W. Choi, J. Neurosci., 1997, 17, 9554–9564 CAS.
- M. A. Greenough, I. Volitakis, Q. X. Li, K. Laughton, G. Evin, M. Ho, A. H. Dalziel, J. Camakaris and A. I. Bush, J. Biol. Chem., 2011, 286, 9776–9786 CrossRef CAS PubMed.
- N. T. Watt, D. R. Taylor, T. L. Kerrigan, H. H. Griffiths, J. V. Rushworth, I. J. Whitehouse and N. M. Hooper, Nat. Commun., 2012, 3, 1134 CrossRef PubMed.
- E. D. Gundelfinger, T. M. Boeckers, M. K. Baron and J. U. Bowie, Trends Biochem. Sci., 2006, 31, 366–373 CrossRef CAS PubMed.
- M. K. Baron, T. M. Boeckers, B. Vaida, S. Faham, M. Gingery, M. R. Sawaya, D. Salyer, E. D. Gundelfinger and J. U. Bowie, Science, 2006, 311, 531–535 CrossRef CAS PubMed.
- P. A. Adlard, J. M. Parncutt, D. I. Finkelstein and A. I. Bush, J. Neurosci., 2010, 30, 1631–1636 CrossRef CAS PubMed.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. d. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464–1467 CAS.
- A. M. Grabrucker, M. J. Schmeisser, P. T. Udvardi, M. Arons, M. Schoen, N. S. Woodling, K. I. Andreasson, P. R. Hof, J. D. Buxbaum, C. C. Garner and T. M. Boeckers, Mol. Neurodegener., 2011, 6, 65 CrossRef CAS PubMed.
- J. Telianidis, Y. H. Hung, S. Materia and S. L. Fontaine, Front. Aging Neurosci., 2013, 5, 44 Search PubMed.
- R. El Meskini, K. L. Crabtree, L. B. Cline, R. E. Mains, B. A. Eipper and G. V. Ronnett, Mol. Cell. Neurosci., 2007, 34, 409–421 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- S. C. Dodani, D. W. Domaille, C. I. Nam, E. W. Miller, L. A. Finney, S. Vogt and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5980–5985 CrossRef CAS PubMed.
- D. E. Hartter and A. Barnea, Synapse, 1988, 2, 412–415 CrossRef CAS PubMed.
- J. Kardos, I. Kovacs, F. Hajos, M. Kalman and M. Simonyi, Neurosci. Lett., 1989, 103, 139–144 CrossRef CAS.
- A. Hopt, S. Korte, H. Fink, U. Panne, R. Niessner, R. Jahn, H. Kretzschmar and J. Herms, J. Neurosci. Methods, 2003, 128, 159–172 CrossRef CAS.
- M. L. Schlief, A. M. Craig and J. D. Gitlin, J. Neurosci., 2005, 25, 239–246 CrossRef CAS PubMed.
- M. L. Schlief, T. West, A. M. Craig, D. M. Holtzman and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14919–14924 CrossRef CAS PubMed.
- E. D. Gaier, B. A. Eipper and R. E. Mains, J. Neurosci. Res., 2013, 91, 2–19 CAS.
- V. Vlachova, H. Zemkova and L. Vyklicky, Jr., Eur. J. Neurosci., 1996, 8, 2257–2264 CrossRef CAS.
- P. Q. Trombley and G. M. Shepherd, J. Neurophysiol., 1996, 76, 2536–2546 CAS.
- T. Weiser and M. Wienrich, Brain Res., 1996, 742, 211–218 CrossRef CAS.
- Z. D. Niu, J. T. Chen, S. Wang, M. Wang, X. M. Li and D. Y. Ruan, Toxicol. Lett., 2006, 165, 289–296 CrossRef CAS PubMed.
- Z. D. Niu, K. Yu, Y. Gu, M. Wang, J. Q. She, W. H. Chen and D. Y. Ruan, NeuroReport, 2005, 16, 1585–1589 CrossRef CAS.
- C. Coddou, C. Villalobos, J. Gonzalez, C. Acuna-Castillo, B. Loeb and J. P. Huidobro-Toro, J. Neurochem., 2002, 80, 626–633 CrossRef CAS.
- C. Coddou, B. Morales and J. P. Huidobro-Toro, Eur. J. Pharmacol., 2003, 472, 49–56 CrossRef CAS.
- R. A. Lorca, C. Coddou, M. C. Gazitua, P. Bull, C. Arredondo and J. P. Huidobro-Toro, J. Neurochem., 2005, 95, 499–512 CrossRef CAS PubMed.
- C. Acuna-Castillo, C. Coddou, P. Bull, J. Brito and J. P. Huidobro-Toro, J. Neurochem., 2007, 101, 17–26 CrossRef CAS PubMed.
- N. Doreulee, Y. Yanovsky and H. L. Haas, Hippocampus, 1997, 7, 666–669 CrossRef CAS.
- H. You, S. Tsutsui, S. Hameed, T. J. Kannanayakal, L. Chen, P. Xia, J. D. Engbers, S. A. Lipton, P. K. Stys and G. W. Zamponi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1737–1742 CrossRef CAS PubMed.
- C. Peters, B. Munoz, F. J. Sepulveda, J. Urrutia, M. Quiroz, S. Luza, G. V. De Ferrari, L. G. Aguayo and C. Opazo, J. Neurochem., 2011, 119, 78–88 CrossRef CAS PubMed.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, J. Neurochem., 2011, 119, 220–230 CrossRef CAS PubMed.
- C. J. Maynard, R. Cappai, I. Volitakis, R. A. Cherny, A. R. White, K. Beyreuther, C. L. Masters, A. I. Bush and Q.-X. Li, J. Biol. Chem., 2002, 277, 44670–44676 CrossRef CAS PubMed.
- C. J. Maynard, R. Cappai, I. Volitakis, R. A. Cherny, C. L. Masters, Q. X. Li and A. I. Bush, J. Inorg. Biochem., 2006, 100, 952–962 CrossRef CAS PubMed.
- I. Singh, A. P. Sagare, M. Coma, D. Perlmutter, R. Gelein, R. D. Bell, R. J. Deane, E. Zhong, M. Parisi, J. Ciszewski, R. T. Kasper and R. Deane, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14771–14776 CrossRef CAS PubMed.
- Y. Pushkar, G. Robison, B. Sullivan, S. X. Fu, M. Kohne, W. Jiang, S. Rohr, B. Lai, M. A. Marcus, T. Zakharova and W. Zheng, Aging Cell, 2013, 12, 823–832 CrossRef CAS PubMed.
- D. Religa, D. Strozyk, R. A. Cherny, I. Volitakis, V. Haroutunian, B. Winblad, J. Naslund and A. I. Bush, Neurology, 2006, 67, 69–75 CrossRef CAS PubMed.
- T. A. Bayer, S. Schafer, A. Simons, A. Kemmling, T. Kamer, R. Tepests, A. Eckert, K. Schussel, O. Eikenberg, C. Sturchler-Pierrat, D. Abramowski, M. Staufenbiel and G. Multhaup, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14187–14192 CrossRef CAS PubMed.
- A. L. Phinney, B. Drisaldi, S. D. Schmidt, S. Lugowski, V. Coronado, Y. Liang, P. Horne, J. Yang, J. Sekoulidis, J. Coomaraswamy, M. A. Chishti, D. W. Cox, P. M. Mathews, R. A. Nixon, G. A. Carlson, P. S. George-Hyslop and D. Westaway, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14193–14198 CrossRef CAS PubMed.
- M. A. Cater, K. T. McInnes, Q. X. Li, I. Volitakis, S. La Fontaine, J. F. Mercer and A. I. Bush, Biochem. J., 2008, 412, 141–152 CrossRef CAS PubMed.
- P. Ramos, A. Santos, N. R. Pinto, R. Mendes, T. Magalhaes and A. Almeida, J. Trace Elem. Med. Biol., 2014, 28, 13–17 CAS.
- J. H. Chen, N. Singh, H. Tay and T. Walczyk, Metallomics, 2014 10.1039/c4mt00054d.
- G. Bartzokis, J. Mintz, D. Sultzer, P. Marx, J. S. Herzberg, C. K. Phelan and S. R. Marder, Am. J. Neuroradiol., 1994, 15, 1129–1138 CAS.
- G. Bartzokis, T. A. Tishler, P. H. Lu, P. Villablanca, L. L. Altshuler, M. Carter, D. Huang, N. Edwards and J. Mintz, Neurobiol. Aging, 2007, 28, 414–423 CrossRef CAS PubMed.
- G. Bartzokis, P. H. Lu, K. Tingus, D. G. Peters, C. P. Amar, T. A. Tishler, J. P. Finn, P. Villablanca, L. L. Altshuler, J. Mintz, E. Neely and J. R. Connor, Neuropsychopharmacology, 2011, 36, 1375–1384 CrossRef CAS PubMed.
- E. K. Kastman, A. A. Willette, C. L. Coe, B. B. Bendlin, K. J. Kosmatka, D. G. McLaren, G. Xu, E. Canu, A. S. Field, A. L. Alexander, M. L. Voytko, T. M. Beasley, R. J. Colman, R. H. Weindruch and S. C. Johnson, J. Neurosci., 2010, 30, 7940–7947 CrossRef CAS PubMed.
- A. Takeda and H. Tamano, Front. Aging Neurosci., 2014, 6, 26 Search PubMed.
- S. A. James, I. Volitakis, P. A. Adlard, J. A. Duce, C. L. Masters, R. A. Cherny and A. I. Bush, Free Radical Biol. Med., 2012, 52, 298–302 CrossRef CAS PubMed.
- J. A. Duce and A. I. Bush, Prog. Neurobiol., 2010, 92, 1–18 CrossRef CAS PubMed.
- S. A. Bellingham, D. K. Lahiri, B. Maloney, S. La Fontaine, G. Multhaup and J. Camakaris, J. Biol. Chem., 2004, 279, 20378–20386 CrossRef CAS PubMed.
- J. T. Rogers, J. D. Randall, C. M. Cahill, P. S. Eder, X. Huang, H. Gunshin, L. Leiter, J. McPhee, S. S. Sarang, T. Utsuki, N. H. Greig, D. K. Lahiri, R. E. Tanzi, A. I. Bush, T. Giordano and S. R. Gullans, J. Biol. Chem., 2002, 277, 45518–45528 CrossRef CAS PubMed.
- H. H. Cho, C. M. Cahill, C. R. Vanderburg, C. R. Scherzer, B. Wang, X. Huang and J. T. Rogers, J. Biol. Chem., 2010, 285, 31217–31232 CrossRef CAS PubMed.
- G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406–1409 CAS.
- S. O. Dahms, I. Konnig, D. Roeser, K. H. Guhrs, M. C. Mayer, D. Kaden, G. Multhaup and M. E. Than, J. Mol. Biol., 2012, 416, 438–452 CrossRef CAS PubMed.
- S. A. Bellingham, G. D. Ciccotosto, B. E. Needham, L. R. Fodero, A. R. White, C. L. Masters, R. Cappai and J. Camakaris, J. Neurochem., 2004, 91, 423–428 CrossRef CAS PubMed.
- K. M. Acevedo, Y. H. Hung, A. H. Dalziel, Q. X. Li, K. Laughton, K. Wikhe, A. Rembach, B. Roberts, C. L. Masters, A. I. Bush and J. Camakaris, J. Biol. Chem., 2011, 286, 8252–8262 CrossRef CAS PubMed.
- K. M. Acevedo, C. M. Opazo, D. Norrish, L. M. Challis, Q. X. Li, A. R. White, A. I. Bush and J. Camakaris, J. Biol. Chem., 2014, 289, 11007–11019 CrossRef CAS PubMed.
- T. Borchardt, J. Camakaris, R. Cappai, C. L. Masters, K. Beyreuther and G. Multhaup, Biochem. J., 1999, 344(Pt 2), 461–467 CrossRef CAS.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS.
- Y. H. Hung, E. L. Robb, I. Volitakis, M. Ho, G. Evin, Q. X. Li, J. G. Culvenor, C. L. Masters, R. A. Cherny and A. I. Bush, J. Biol. Chem., 2009, 284, 21899–21907 CrossRef CAS PubMed.
- T. J. Nelson and D. L. Alkon, J. Biol. Chem., 2005, 280, 7377–7387 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS PubMed.
- L. Puglielli, A. L. Friedlich, K. D. R. Setchell, S. Nagano, C. Opazo, R. A. Cherny, K. J. Barnham, J. D. Wade, S. Melov, D. M. Kovacs and A. I. Bush, J. Clin. Invest., 2005, 115, 2556–2563 CrossRef CAS PubMed.
- H. Kessler, T. A. Bayer, D. Bach, T. Schneider-Axmann, T. Supprian, W. Herrmann, M. Haber, G. Multhaup, P. Falkai and F. G. Pajonk, J. Neural Transm., 2008, 115, 1181–1187 CrossRef CAS PubMed.
- D. L. Sparks and B. G. Schreurs, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11065–11069 CrossRef CAS PubMed.
- M. C. Morris, J. Nutr., Health Aging, 2006, 10, 204 Search PubMed.
- R. C. McCarthy, Y. H. Park and D. J. Kosman, EMBO Rep., 2014, 15, 809–815 CrossRef CAS PubMed.
- B. E. Needham, G. D. Ciccotosto and R. Cappai, Metallomics, 2014, 6, 598–603 RSC.
- P. Lei, S. Ayton, D. I. Finkelstein, L. Spoerri, G. D. Ciccotosto, D. K. Wright, B. X. W. Wong, P. A. Adlard, R. A. Cherny, L. Q. Lam, B. R. Roberts, I. Volitakis, G. F. Egan, C. A. McLean, R. Cappai, J. A. Duce and A. I. Bush, Nat. Med., 2012, 18, 291–295 CrossRef CAS PubMed.
- E. H. Gray, K. J. De Vos, C. Dingwall, M. S. Perkinton and C. C. Miller, J. Alzheimer's Dis., 2010, 21, 1101–1105 CAS.
- B. Angeletti, K. J. Waldron, K. B. Freeman, H. Bawagan, I. Hussain, C. C. Miller, K. F. Lau, M. E. Tennant, C. Dennison, N. J. Robinson and C. Dingwall, J. Biol. Chem., 2005, 280, 17930–17937 CrossRef CAS PubMed.
- K. Murakami, N. Murata, Y. Noda, S. Tahara, T. Kaneko, N. Kinoshita, H. Hatsuta, S. Murayama, K. J. Barnham, K. Irie, T. Shirasawa and T. Shimizu, J. Biol. Chem., 2011, 286, 44557–44568 CrossRef CAS PubMed.
- D. R. Taylor and N. M. Hooper, Biochem. J., 2007, 402, 17–23 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- I. Grundke-Iqbal, J. Fleming, Y. C. Tung, H. Lassmann, K. Iqbal and J. G. Joshi, Acta Neuropathol., 1990, 81, 105–110 CrossRef CAS.
- C. R. Jack, Jr., T. M. Wengenack, D. A. Reyes, M. Garwood, G. L. Curran, B. J. Borowski, J. Lin, G. M. Preboske, S. S. Holasek, G. Adriany and J. F. Poduslo, J. Neurosci., 2005, 25, 10041–10048 CrossRef PubMed.
- M. F. Falangola, S. P. Lee, R. A. Nixon, K. Duff and J. A. Helpern, Neurochem. Res., 2005, 30, 201–205 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed.
- J. Dong, C. S. Atwood, V. E. Anderson, S. L. Siedlak, M. A. Smith, G. Perry and P. R. Carey, Biochemistry, 2003, 42, 2768–2773 CrossRef CAS PubMed.
- S. W. Suh, K. B. Jensen, M. S. Jensen, D. S. Silva, P. J. Kesslak, G. Danscher and C. J. Frederickson, Brain Res., 2000, 852, 274–278 CrossRef CAS.
- M. Stoltenberg, M. Bruhn, C. Sondergaard, P. Doering, M. J. West, A. Larsen, J. C. Troncoso and G. Danscher, Histochem. Cell Biol., 2005, 123, 605–611 CrossRef CAS PubMed.
- M. Stoltenberg, A. I. Bush, G. Bach, K. Smidt, A. Larsen, J. Rungby, S. Lund, P. Doering and G. Danscher, Neuroscience, 2007, 150, 357–369 CrossRef CAS PubMed.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS PubMed.
- A. C. Leskovjan, A. Lanzirotti and L. M. Miller, NeuroImage, 2009, 47, 1215–1220 CrossRef PubMed.
- A. L. Friedlich, J. Y. Lee, T. van Groen, R. A. Cherny, I. Volitakis, T. B. Cole, R. D. Palmiter, J. Y. Koh and A. I. Bush, J. Neurosci., 2004, 24, 3453–3459 CrossRef CAS PubMed.
- D. C. Brady, M. S. Crowe, M. L. Turski, G. A. Hobbs, X. Yao, A. Chaikuad, S. Knapp, K. Xiao, S. L. Campbell, D. J. Thiele and C. M. Counter, Nature, 2014, 509, 492–496 CrossRef CAS PubMed.
- J. L. Smith, S. Xiong and M. A. Lovell, Neurotoxicology, 2006, 27, 1–5 CrossRef CAS PubMed.
- G. Danscher, K. B. Jensen, C. J. Frederickson, K. Kemp, A. Andreasen, S. Juhl, M. Stoltenberg and R. Ravid, J. Neurosci. Methods, 1997, 76, 53–59 CrossRef CAS.
- A. Takeda, A. Minami, S. Takefuta, M. Tochigi and N. Oku, J. Neurosci. Res., 2001, 63, 447–452 CrossRef CAS.
- W. Chowanadisai, S. L. Kelleher and B. Lonnerdal, J. Nutr., 2005, 135, 1002–1007 CAS.
- G. Lyubartseva, J. L. Smith, W. R. Markesbery and M. A. Lovell, Brain Pathol., 2010, 20, 343–350 CrossRef CAS PubMed.
- P. A. Adlard, J. M. Parncutt, D. I. Finkelstein and A. I. Bush, J. Neurosci., 2010, 30, 1631–1636 CrossRef CAS PubMed.
- M. A. Lovell, J. L. Smith, S. Xiong and W. R. Markesbery, Neurotoxic. Res., 2005, 7, 265–271 CrossRef CAS.
- J.-Y. Lee, J.-H. Kim, S. H. Hong, J. Y. Lee, R. A. Cherny, A. I. Bush, R. D. Palmiter and J.-Y. Koh, J. Biol. Chem., 2004, 279, 8602–8607 CrossRef CAS PubMed.
- M. J. Callahan, W. J. Lipinski, F. Bian, R. A. Durham, A. Pack and L. C. Walker, Am. J. Pathol., 2001, 158, 1173–1177 CrossRef CAS.
- N. L. Bjorklund, L. C. Reese, V. M. Sadagoparamanujam, V. Ghirardi, R. L. Woltjer and G. Taglialatela, Mol. Neurodegener., 2012, 7, 23 CrossRef CAS PubMed.
- A. Deshpande, H. Kawai, R. Metherate, C. G. Glabe and J. Busciglio, J. Neurosci., 2009, 29, 4004–4015 CrossRef CAS PubMed.
- G. Meloni, V. Sonois, T. Delaine, L. Guilloreau, A. Gillet, J. Teissie, P. Faller and M. Vasak, Nat. Chem. Biol., 2008, 4, 366–372 CrossRef CAS PubMed.
- M. A. Smith, P. L. R. Harris, L. M. Sayre and G. Perry, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9866–9868 CrossRef CAS.
- Q. Pankhurst, D. Hautot, N. Khan and J. Dobson, J. Alzheimer's Dis., 2008, 13, 49–52 CAS.
- C. Bouras, P. Giannakopoulos, P. F. Good, A. Hsu, P. R. Hof and D. P. Perl, Eur. Neurol., 1997, 38, 53–58 CrossRef CAS PubMed.
- G. Bartzokis, D. Sultzer, J. Mintz, L. E. Holt, P. Marx, C. K. Phelan and S. R. Marder, Biol. Psychiatry, 1994, 35, 480–487 CrossRef CAS.
- E. P. Raven, P. H. Lu, T. A. Tishler, P. Heydari and G. Bartzokis, J. Alzheimer's Dis., 2013, 37, 127–136 CAS.
- S. van Rooden, M. J. Versluis, M. K. Liem, J. Milles, A. B. Maier, A. M. Oleksik, A. G. Webb, M. A. van Buchem and J. van der Grond, Alzheimer's Dementia, 2014, 10, e19–e26 CrossRef PubMed.
- A. C. Leskovjan, A. Kretlow, A. Lanzirotti, R. Barrea, S. Vogt and L. M. Miller, NeuroImage, 2011, 55, 32–38 CrossRef CAS PubMed.
- J. Becerril-Ortega, K. Bordji, T. Freret, T. Rush and A. Buisson, Neurobiol. Aging, 2014, 35, 2288–2301 CrossRef CAS PubMed.
- L. M. Sayre, G. Perry, P. L. Harris, Y. Liu, K. A. Schubert and M. A. Smith, J. Neurochem., 2000, 74, 270–279 CrossRef CAS.
- A. Yamamoto, R. W. Shin, K. Hasegawa, H. Naiki, H. Sato, F. Yoshimasu and T. Kitamoto, J. Neurochem., 2002, 82, 1137–1147 CrossRef CAS PubMed.
- B. Bader, G. Nubling, A. Mehle, S. Nobile, H. Kretzschmar and A. Giese, Biochem. Biophys. Res. Commun., 2011, 411, 190–196 CrossRef CAS PubMed.
- G. Nubling, B. Bader, J. Levin, J. Hildebrandt, H. Kretzschmar and A. Giese, Mol. Neurodegener., 2012, 7, 35 CrossRef PubMed.
- J. L. De Reuck, V. Deramecourt, F. Auger, N. Durieux, C. Cordonnier, D. Devos, L. Defebvre, C. Moreau, D. Caparros-Lefebvre, D. Leys, C. A. Maurage, F. Pasquier and R. Bordet, Eur. J. Neurol., 2014, 21, 1026–1031 CrossRef CAS PubMed.
- J. H. Lee, Y. H. Han, B. M. Kang, C. W. Mun, S. J. Lee and S. K. Baik, J. Neurol., 2013, 260, 2094–2101 CrossRef CAS PubMed.
- L. Kupershmidt, T. Amit, O. Bar-Am, M. B. Youdim and O. Weinreb, Antioxid. Redox Signaling, 2012, 17, 860–877 CrossRef CAS PubMed.
- C. Guo, T. Wang, W. Zheng, Z. Y. Shan, W. P. Teng and Z. Y. Wang, Neurobiol. Aging, 2013, 34, 562–575 CrossRef CAS PubMed.
- A. A. Raha, R. A. Vaishnav, R. P. Friedland, A. Bomford and R. Raha-Chowdhury, Acta Neuropathol. Commun., 2013, 1, 55 CrossRef PubMed.
- K. J. Robson, D. J. Lehmann, V. L. Wimhurst, K. J. Livesey, M. Combrinck, A. T. Merryweather-Clarke, D. R. Warden and A. D. Smith, J. Med. Genet., 2004, 41, 261–265 CrossRef CAS.
- D. J. Lehmann, M. Schuur, D. R. Warden, N. Hammond, O. Belbin, H. Kolsch, M. G. Lehmann, G. K. Wilcock, K. Brown, P. G. Kehoe, C. M. Morris, R. Barker, E. Coto, V. Alvarez, P. Deloukas, I. Mateo, R. Gwilliam, O. Combarros, A. Arias-Vasquez, Y. S. Aulchenko, M. A. Ikram, M. M. Breteler, C. M. van Duijn, A. Oulhaj, R. Heun, M. Cortina-Borja, K. Morgan, K. Robson and A. D. Smith, Neurobiol. Aging, 2012, 33(202), e201–e213 Search PubMed.
- F. Giambattistelli, S. Bucossi, C. Salustri, V. Panetta, S. Mariani, M. Siotto, M. Ventriglia, F. Vernieri, M. L. Dell'acqua, E. Cassetta, P. M. Rossini and R. Squitti, Neurobiol. Aging, 2012, 33, 1633–1641 CrossRef CAS PubMed.
- A. C. Crespo, B. Silva, L. Marques, E. Marcelino, C. Maruta, S. Costa, A. Timoteo, A. Vilares, F. S. Couto, P. Faustino, A. P. Correia, A. Verdelho, G. Porto, M. Guerreiro, A. Herrero, C. Costa, A. de Mendonca, L. Costa and M. Martins, Neurobiol. Aging, 2014, 35, 777–785 CrossRef CAS PubMed.
- N. G. Faux, A. Rembach, J. Wiley, K. A. Ellis, D. Ames, C. J. Fowler, R. N. Martins, K. K. Pertile, R. L. Rumble, B. Trounson, C. L. Masters, A. R. G. The and A. I. Bush, Mol. Psychiatry, 2014 DOI:10.1038/mp.2013.178.
- T. Dyrks, E. Dyrks, T. Hartmann, C. Masters and K. Beyreuther, J. Biol. Chem., 1992, 267, 18210–18217 CAS.
- D. Pramanik and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 81–87 CrossRef CAS PubMed.
- M. Seal, S. Mukherjee, D. Pramanik, K. Mittra, A. Dey and S. G. Dey, Chem. Commun., 2013, 49, 1091–1093 RSC.
- D. Pramanik, C. Ghosh, S. Mukherjee and S. G. Dey, Coord. Chem. Rev., 2013, 257, 81–92 CrossRef CAS PubMed.
- S. Chassaing, F. Collin, P. Dorlet, J. Gout, C. Hureau and P. Faller, Curr. Top. Med. Chem., 2012, 12, 2573–2595 CrossRef CAS.
- J. Y. Chuang, C. W. Lee, Y. H. Shih, T. Yang, L. Yu and Y. M. Kuo, PLoS One, 2012, 7, e33120 CAS.
- J. Rogers, R. Li, D. Mastroeni, A. Grover, B. Leonard, G. Ahern, P. Cao, H. Kolody, L. Vedders, W. P. Kolb and M. Sabbagh, Neurobiol. Aging, 2006, 27, 1733–1739 CrossRef CAS PubMed.
- V. L. Villemagne, K. A. Perez, K. E. Pike, W. M. Kok, C. C. Rowe, A. R. White, P. Bourgeat, O. Salvado, J. Bedo, C. A. Hutton, N. G. Faux, C. L. Masters and K. J. Barnham, J. Neurosci., 2010, 30, 6315–6322 CrossRef CAS PubMed.
- R. Jayakumar, J. W. Kusiak, F. J. Chrest, A. A. Demehin, J. Murali, R. P. Wersto, E. Nagababu, L. Ravi and J. M. Rifkind, Biochim. Biophys. Acta, 2003, 1622, 20–28 CrossRef CAS.
- H. Basun, L. G. Forssell, L. Wetterberg and B. Winblad, J. Neural Transm.: Parkinson's Dis. Dementia Sect., 1991, 3, 231–258 CAS.
- R. Squitti, D. Lupoi, P. Pasqualetti, G. Dal Forno, F. Vernieri, P. Chiovenda, L. Rossi, M. Cortesi, E. Cassetta and P. M. Rossini, Neurology, 2002, 59, 1153–1161 CrossRef CAS.
- R. Squitti, P. M. Rossini, E. Cassetta, F. Moffa, P. Pasqualetti, M. Cortesi, A. Colloca, L. Rossi and A. Finazzi-Agro, Eur. J. Clin. Invest., 2002, 32, 51–59 CrossRef CAS.
- A. Rembach, J. D. Doecke, B. R. Roberts, A. D. Watt, N. G. Faux, I. Volitakis, K. K. Pertile, R. L. Rumble, B. O. Trounson, C. J. Fowler, W. Wilson, K. A. Ellis, R. N. Martins, C. C. Rowe, V. L. Villemagne, D. Ames, C. L. Masters, A. r. group and A. I. Bush, J. Alzheimer's Dis., 2013, 34, 171–182 CAS.
- R. Squitti, I. Simonelli, M. Ventriglia, M. Siotto, P. Pasqualetti, A. Rembach, J. Doecke and A. I. Bush, J. Alzheimer's Dis., 2014, 38, 809–822 CAS.
- R. Squitti, R. Ghidoni, M. Siotto, M. Ventriglia, L. Benussi, A. Paterlini, M. Magri, G. Binetti, E. Cassetta, D. Caprara, F. Vernieri, P. M. Rossini and P. Pasqualetti, Ann. Neurol., 2014, 75, 574–580 CrossRef CAS PubMed.
- D. Puzzo, L. Privitera, E. Leznik, M. Fa, A. Staniszewski, A. Palmeri and O. Arancio, J. Neurosci., 2008, 28, 14537–14545 CrossRef CAS PubMed.
- E. Abramov, I. Dolev, H. Fogel, G. D. Ciccotosto, E. Ruff and I. Slutsky, Nat. Neurosci., 2009, 12, 1567–1576 CrossRef CAS PubMed.
- D. Puzzo, L. Privitera, M. Fa, A. Staniszewski, G. Hashimoto, F. Aziz, M. Sakurai, E. M. Ribe, C. M. Troy, M. Mercken, S. S. Jung, A. Palmeri and O. Arancio, Ann. Neurol., 2011, 69, 819–830 CrossRef CAS PubMed.
- R. D. Terry, J. Neuropathol. Exp. Neurol., 1996, 55, 1023–1025 CrossRef CAS PubMed.
- A. I. Bush, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS.
- D. G. Smith, R. Cappai and K. J. Barnham, Biochim. Biophys. Acta, 2007, 1768, 1976–1990 CrossRef CAS PubMed.
- V. Balland, C. Hureau and J. M. Saveant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113–17118 CrossRef CAS PubMed.
- D. Jiang, L. Men, J. Wang, Y. Zhang, S. Chickenyen, Y. Wang and F. Zhou, Biochemistry, 2007, 46, 9270–9282 CrossRef CAS PubMed.
- L. E. Cassagnes, V. Herve, F. Nepveu, C. Hureau, P. Faller and F. Collin, Angew. Chem., Int. Ed., 2013, 52, 11110–11113 CrossRef CAS PubMed.
- J. Y. Lee, T. B. Cole, R. D. Palmiter, S. W. Suh and J. Y. Koh, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7705–7710 CrossRef CAS PubMed.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. d Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464–1467 CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1997, 272, 26464–26470 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- R. A. Cherny, J. T. Legg, C. A. McLean, D. Fairlie, X. Huang, C. S. Atwood, K. Beyreuther, R. E. Tanzi, C. L. Masters and A. I. Bush, J. Biol. Chem., 1999, 274, 23223–23228 CrossRef CAS PubMed.
- J. S. Choi, J. J. Braymer, R. P. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990–21995 CrossRef CAS PubMed.
- D. Noy, I. Solomonov, O. Sinkevich, T. Arad, K. Kjaer and I. Sagi, J. Am. Chem. Soc., 2008, 130, 1376–1383 CrossRef CAS PubMed.
- W. T. Chen, Y. H. Liao, H. M. Yu, I. H. Cheng and Y. R. Chen, J. Biol. Chem., 2011, 286, 9646–9656 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J.-P. Vonsattel, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1997, 272, 26464–26470 CrossRef CAS PubMed.
- Y. Yoshiike, K. Tanemura, O. Murayama, T. Akagi, M. Murayama, S. Sato, X. Sun, N. Tanaka and A. Takashima, J. Biol. Chem., 2001, 276, 32293–32299 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, JBIC, J. Biol. Inorg. Chem., 2004, 9, 954–960 CrossRef CAS PubMed.
- I. Solomonov, E. Korkotian, B. Born, Y. Feldman, A. Bitler, F. Rahimi, H. Li, G. Bitan and I. Sagi, J. Biol. Chem., 2012, 287, 20555–20564 CrossRef CAS PubMed.
- M. P. Cuajungco, L. E. Goldstein, A. Nunomura, M. A. Smith, J. T. Lim, C. S. Atwood, X. Huang, Y. W. Farrag, G. Perry and A. I. Bush, J. Biol. Chem., 2000, 275, 19439–19442 CrossRef CAS PubMed.
- A. I. Bush, W. H. Pettingell Jr., M. D. Paradis and R. E. Tanzi, J. Biol. Chem., 1994, 269, 12152–12158 CAS.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1997, 272, 26464–26470 CrossRef CAS PubMed.
- H. Hua, L. Munter, A. Harmeier, O. Georgiev, G. Multhaup and W. Schaffner, Biol. Chem., 2011, 392, 919–926 CrossRef CAS PubMed.
- M. Lang, L. Wang, Q. Fan, G. Xiao, X. Wang, Y. Zhong and B. Zhou, PLoS Genet., 2012, 8, e1002683 CAS.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817–12826 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- S. Jun, J. R. Gillespie, B. K. Shin and S. Saxena, Biochemistry, 2009, 48, 10724–10732 CrossRef CAS PubMed.
- J. T. Pedersen, J. Ostergaard, N. Rozlosnik, B. Gammelgaard and N. H. Heegaard, J. Biol. Chem., 2011, 286, 26952–26963 CrossRef CAS PubMed.
- M. Mold, L. Ouro-Gnao, B. M. Wieckowski and C. Exley, Sci. Rep., 2013, 3, 1256 Search PubMed.
- C. J. Sarell, S. R. Wilkinson and J. H. Viles, J. Biol. Chem., 2010, 285, 41533–41540 CrossRef CAS PubMed.
- X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- K. J. Barnham, F. Haeffner, G. D. Ciccotosto, C. C. Curtain, D. Tew, C. Mavros, K. Beyreuther, D. Carrington, C. L. Masters, R. A. Cherny, R. Cappai and A. I. Bush, FASEB J., 2004, 18, 1427–1429 CAS.
- K. J. Barnham, G. D. Ciccotosto, A. K. Tickler, F. E. Ali, D. G. Smith, N. A. Williamson, Y. H. Lam, D. Carrington, D. Tew, G. Kocak, I. Volitakis, F. Separovic, C. J. Barrow, J. D. Wade, C. L. Masters, R. A. Cherny, C. C. Curtain, A. I. Bush and R. Cappai, J. Biol. Chem., 2003, 278, 42959–42965 CrossRef CAS PubMed.
- D. A. Butterfield, J. Drake, C. Pocernich and A. Castegna, Trends Mol. Med., 2001, 7, 548–554 CrossRef CAS.
- L. Puglielli, A. L. Friedlich, K. D. Setchell, S. Nagano, C. Opazo, R. A. Cherny, K. J. Barnham, J. D. Wade, S. Melov, D. M. Kovacs and A. I. Bush, J. Clin. Invest., 2005, 115, 2556–2563 CrossRef CAS PubMed.
- S. Turnbull, B. J. Tabner, O. M. El-Agnaf, L. J. Twyman and D. Allsop, Free Radical Biol. Med., 2001, 30, 1154–1162 CrossRef CAS.
- C. Schoneich, Arch. Biochem. Biophys., 2002, 397, 370–376 CrossRef PubMed.
- C. S. Atwood, X. Huang, A. Khatri, R. C. Scarpa, Y. S. Kim, R. D. Moir, R. E. Tanzi, A. E. Roher and A. I. Bush, Cell. Mol. Biol., 2000, 46, 777–783 CAS.
- Y. K. Al-Hilaly, T. L. Williams, M. Stewart-Parker, F. Lenzie, E. Skaria, M. Cole, W. G. Bucher, K. L. Morris, A. A. Sada and L. C. Serpell, Acta Neuropathol. Commun., 2013, 1 Search PubMed.
- C. S. Atwood, G. Perry, H. Zeng, Y. Kato, W. D. Jones, K. Q. Ling, X. Huang, R. D. Moir, D. Wang, L. M. Sayre, M. A. Smith, S. G. Chen and A. I. Bush, Biochemistry, 2004, 43, 560–568 CrossRef CAS PubMed.
- D. P. Smith, G. D. Ciccotosto, D. J. Tew, M. T. Fodero-Tavoletti, T. Johanssen, C. L. Masters, K. J. Barnham and R. Cappai, Biochemistry, 2007, 46, 2881–2891 CrossRef CAS PubMed.
- M. Lang, Q. Fan, L. Wang, Y. Zheng, G. Xiao, X. Wang, W. Wang, Y. Zhong and B. Zhou, Neurobiol. Aging, 2013, 34, 2604–2612 CrossRef CAS PubMed.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS.
- C. J. Maynard, A. I. Bush, C. L. Masters, R. Cappai and Q. X. Li, Int. J. Exp. Pathol., 2005, 86, 147–159 CrossRef CAS PubMed.
- I. Maurer, S. Zierz and H. J. Moller, Neurobiol. Aging, 2000, 21, 455–462 CrossRef CAS.
- P. L. McGeer, E. McGeer, J. Rogers and J. Sibley, Lancet, 1990, 335, 1037 CrossRef CAS.
- T. A. Bayer, S. Schafer, A. Simons, A. Kemmling, T. Kamer, R. Tepest, A. Eckert, K. Schussel, O. Eikenberg, C. Sturchler-Pierrat, D. Abramowski, M. Staufenbiel and G. Multhaup, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14187–14192 CrossRef CAS PubMed.
- T. M. Malm, H. Iivonen, G. Goldsteins, V. Keksa-Goldsteine, T. Ahtoniemi, K. Kanninen, A. Salminen, S. Auriola, T. Van Groen, H. Tanila and J. Koistinaho, J. Neurosci., 2007, 27, 3712–3721 CrossRef CAS PubMed.
- C. A. Grimes and R. S. Jope, Prog. Neurobiol., 2001, 65, 391–426 CrossRef CAS.
- E. A. Ostrakhovitch, M. R. Lordnejad, F. Schliess, H. Sies and L. O. Klotz, Arch. Biochem. Biophys., 2002, 397, 232–239 CrossRef CAS PubMed.
- S. C. Drew, C. L. Masters and K. J. Barnham, J. Am. Chem. Soc., 2009, 131, 8760–8761 CrossRef CAS PubMed.
- S. C. Drew, C. J. Noble, C. L. Masters, G. R. Hanson and K. J. Barnham, J. Am. Chem. Soc., 2009, 131, 1195–1207 CrossRef CAS PubMed.
- C. Hureau, Y. Coppel, P. Dorlet, P. L. Solari, S. Sayen, E. Guillon, L. Sabater and P. Faller, Angew. Chem., 2009, 48, 9522–9525 CAS.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Angew. Chem., 2009, 48, 9273–9276 CAS.
- C. Hureau, V. Balland, Y. Coppel, P. L. Solari, E. Fonda and P. Faller, JBIC, J. Biol. Inorg. Chem., 2009, 14, 995–1000 CrossRef CAS PubMed.
- S. C. Drew and K. J. Barnham, Acc. Chem. Res., 2011, 44, 1146–1155 CrossRef CAS PubMed.
- S. Warmlander, A. Tiiman, A. Abelein, J. Luo, J. Jarvet, K. L. Soderberg, J. Danielsson and A. Graslund, ChemBioChem, 2013, 14, 1692–1704 CrossRef PubMed.
- Z. Xiao and A. G. Wedd, Nat. Prod. Rep., 2010, 27, 768–789 RSC.
- P. Faller, ChemBioChem, 2009, 10, 2837–2845 CrossRef CAS PubMed.
- B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- D. Jiang, L. Zhang, G. P. Grant, C. G. Dudzik, S. Chen, S. Patel, Y. Hao, G. L. Millhauser and F. Zhou, Biochemistry, 2013, 52, 547–556 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- Y. Miller, M. Buyong and R. Nussinov, Coord. Chem. Rev., 2012, 256, 2245–2252 CrossRef CAS PubMed.
- P. Lei, S. Ayton, D. I. Finkelstein, P. A. Adlard, C. L. Masters and A. I. Bush, Int. J. Biochem. Cell Biol., 2010, 42, 1775–1778 CrossRef CAS PubMed.
- R. J. Castellani, S. L. Siedlak, G. Perry and M. A. Smith, Acta Neuropathol., 2000, 100, 111–114 CrossRef CAS.
- J. T. Rogers, S. Mikkilineni, I. Cantuti-Castelvetri, D. H. Smith, X. Huang, S. Bandyopadhyay, C. M. Cahill, M. L. Maccecchini, D. K. Lahiri and N. H. Greig, J. Neural Transm., 2011, 118, 493–507 CrossRef CAS PubMed.
- N. Ostrerova-Golts, L. Petrucelli, J. Hardy, J. M. Lee, M. Farer and B. Wolozin, J. Neurosci., 2000, 20, 6048–6054 CAS.
- Bharathi, S. S. Indi and K. S. Rao, Neurosci. Lett., 2007, 424, 78–82 CrossRef CAS PubMed.
- J. A. Wright and D. R. Brown, J. Neurosci. Res., 2008, 86, 496–503 CrossRef CAS PubMed.
- A. Binolfia, L. Quintanarb, C. W. Bertoncinic, C. Griesingerd and C. O. Fernandezd, Coord. Chem. Rev., 2012, 256, 2188–2201 CrossRef PubMed.
- P. Davies, D. Moualla and D. R. Brown, PLoS One, 2011, 6, e15814 CAS.
- M. C. Miotto, E. E. Rodriguez, A. A. Valiente-Gabioud, V. Torres-Monserrat, A. Binolfi, L. Quintanar, M. Zweckstetter, C. Griesinger and C. O. Fernandez, Inorg. Chem., 2014, 53, 4350–4358 CrossRef CAS PubMed.
- J. A. Wright, X. Wang and D. R. Brown, FASEB J., 2009, 23, 2384–2393 CrossRef CAS PubMed.
- X. Wang, D. Moualla, J. A. Wright and D. R. Brown, J. Neurochem., 2010, 113, 704–714 CrossRef CAS PubMed.
- D. T. Dexter, F. R. Wells, A. J. Lees, F. Agid, Y. Agid, P. Jenner and C. D. Marsden, J. Neurochem., 1989, 52, 1830–1836 CrossRef CAS PubMed.
- J. Sian, D. T. Dexter, A. J. Lees, S. Daniel, Y. Agid, F. Javoy-Agid, P. Jenner and C. D. Marsden, Ann. Neurol., 1994, 36, 348–355 CrossRef CAS PubMed.
- L. Zecca, M. Gallorini, V. Schunemann, A. X. Trautwein, M. Gerlach, P. Riederer, P. Vezzoni and D. Tampellini, J. Neurochem., 2001, 76, 1766–1773 CrossRef CAS.
- K. L. Double, M. Gerlach, V. Schunemann, A. X. Trautwein, L. Zecca, M. Gallorini, M. B. Youdim, P. Riederer and D. Ben-Shachar, Biochem. Pharmacol., 2003, 66, 489–494 CrossRef CAS.
- K. S. Rao, M. L. Hegde, S. Anitha, M. Musicco, F. A. Zucca, N. J. Turro and L. Zecca, Prog. Neurobiol., 2006, 78, 364–373 CrossRef CAS PubMed.
- L. Zecca, C. Bellei, P. Costi, A. Albertini, E. Monzani, L. Casella, M. Gallorini, L. Bergamaschi, A. Moscatelli, N. J. Turro, M. Eisner, P. R. Crippa, S. Ito, K. Wakamatsu, W. D. Bush, W. C. Ward, J. D. Simon and F. A. Zucca, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17567–17572 CrossRef CAS PubMed.
- D. Berg, Mov. Disord., 2009, 24(suppl 2), S677–S683 CrossRef PubMed.
- J. M. Gorell, R. J. Ordidge, G. G. Brown, J. C. Deniau, N. M. Buderer and J. A. Helpern, Neurology, 1995, 45, 1138–1143 CrossRef CAS.
- N. Bunzeck, V. Singh-Curry, C. Eckart, N. Weiskopf, R. J. Perry, P. G. Bain, E. Duzel and M. Husain, Parkinsonism Relat. Disord., 2013, 19, 1136–1142 CrossRef PubMed.
- S. S. P. Oakley, Neurology, 2007, 68, 1820–1825 CrossRef PubMed.
- H. Hochstrasser, J. Tomiuk, U. Walter, S. Behnke, J. Spiegel, R. Kruger, G. Becker, O. Riess and D. Berg, FASEB J., 2005, 19, 1851–1853 CAS.
- J. Burn and P. F. Chinnery, Semin. Pediatr. Neurol., 2006, 13, 176–181 CrossRef PubMed.
- M. Neumann, S. Adler, O. Schluter, E. Kremmer, R. Benecke and H. A. Kretzschmar, Acta Neuropathol., 2000, 100, 568–574 CrossRef CAS.
- N. Bruggemann, J. Hagenah, K. Stanley, C. Klein, C. Wang, D. Raymond, L. Ozelius, S. Bressman and R. Saunders-Pullman, Mov. Disord., 2011, 26, 885–888 CrossRef PubMed.
- J. M. Hagenah, I. R. Konig, B. Becker, R. Hilker, M. Kasten, K. Hedrich, P. P. Pramstaller, C. Klein and G. Seidel, J. Neurol., 2007, 254, 1407–1413 CrossRef CAS PubMed.
- K. J. Schweitzer, T. Brussel, P. Leitner, R. Kruger, P. Bauer, D. Woitalla, J. Tomiuk, T. Gasser and D. Berg, J. Neurol., 2007, 254, 613–616 CrossRef PubMed.
- J. M. Hagenah, B. Becker, N. Bruggemann, A. Djarmati, K. Lohmann, A. Sprenger, C. Klein and G. Seidel, J. Neurol., Neurosurg. Psychiatry, 2008, 79, 1071–1074 CrossRef CAS PubMed.
- G. F. Allen, R. Toth, J. James and I. G. Ganley, EMBO Rep., 2013, 14, 1127–1135 CrossRef CAS PubMed.
- D. J. Hare, P. Lei, S. Ayton, B. R. Roberts, R. Grimm, J. L. George, D. P. Bishop, A. D. Beavis, S. J. Donovan, G. McColl, I. Volitakis, C. L. Masters, P. A. Adlard, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. A. Doble, Chem. Sci., 2014, 5, 2160–2169 RSC.
- H. Yamanishi, S. Iyama, Y. Yamaguchi, Y. Kanakura and Y. Iwatani, Clin. Chem., 2003, 49, 175–178 CAS.
- L. W. Klomp, Z. S. Farhangrazi, L. L. Dugan and J. D. Gitlin, J. Clin. Invest., 1996, 98, 207–215 CrossRef CAS PubMed.
- S. Ayton, P. Lei, J. A. Duce, B. X. Wong, A. Sedjahtera, P. A. Adlard, A. I. Bush and D. I. Finkelstein, Ann. Neurol., 2012, 73, 554–559 CrossRef PubMed.
- F. Larner, B. Sampson, M. Rehkamper, D. J. Weiss, J. R. Dainty, S. O'Riordan, T. Panetta and P. G. Bain, Metallomics, 2013, 5, 125–132 RSC.
- D. Kaur, F. Yantiri, J. Kumar, J. O. Mo, S. Rajagopalan, V. Viswanath, R. Boonplueang, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitakis, L. Ellerby, R. A. Cherny, A. I. Bush and J. K. Andersen, Neuron, 2003, 37, 923–933 CrossRef.
- M. B. Youdim, M. Fridkin and H. Zheng, J. Neural Transm., 2004, 111, 1455–1471 CrossRef CAS PubMed.
- S. Gal, H. Zheng, M. Fridkin and M. B. Youdim, J. Neurochem., 2005, 95, 79–88 CrossRef CAS PubMed.
- H. Zheng, S. Gal, L. M. Weiner, O. Bar-Am, A. Warshawsky, M. Fridkin and M. B. Youdim, J. Neurochem., 2005, 95, 68–78 CrossRef CAS PubMed.
- F. Febbraro, K. J. Andersen, V. Sanchez-Guajardo, N. Tentillier and M. Romero-Ramos, Exp. Neurol., 2013, 247, 45–58 CrossRef CAS PubMed.
- Y. Avramovich-Tirosh, T. Amit, O. Bar-Am, H. Zheng, M. Fridkin and M. B. Youdim, J. Neurochem., 2007, 100, 490–502 CrossRef CAS PubMed.
- D. Devos, C. Moreau, J. C. Devedjian, J. Kluza, M. Petrault, C. Laloux, A. Jonneaux, G. Ryckewaert, G. Garcon, N. Rouaix, A. Duhamel, P. Jissendi, K. Dujardin, F. Auger, L. Ravasi, L. Hopes, G. Grolez, W. Firdaus, B. Sablonniere, I. Strubi-Vuillaume, N. Zahr, A. Destee, J. C. Corvol, D. Poltl, M. Leist, C. Rose, L. Defebvre, P. Marchetti, Z. I. Cabantchik and R. Bordet, Antioxid. Redox Signaling, 2014, 21, 195–210 CrossRef CAS PubMed.
- D. T. Dexter, A. Carayon, F. Javoy-Agid, Y. Agid, F. R. Wells, S. E. Daniel, A. J. Lees, P. Jenner and C. D. Marsden, Brain, 1991, 114(Pt 4), 1953–1975 CrossRef PubMed.
- T. Tsunemi and D. Krainc, Hum. Mol. Genet., 2014, 23, 2791–2801 CrossRef CAS PubMed.
- J. S. Park, B. Koentjoro, D. Veivers, A. Mackay-Sim and C. M. Sue, Hum. Mol. Genet., 2014, 23, 2802–2815 CrossRef CAS PubMed.
- D. T. Dexter, P. Jenner, A. H. Schapira and C. D. Marsden, Ann. Neurol., 1992, 32(suppl), S94–S100 CrossRef CAS PubMed.
- G. Bartzokis, J. Cummings, S. Perlman, D. B. Hance and J. Mintz, Arch. Neurol., 1999, 56, 569–574 CrossRef CAS.
- G. Bartzokis, P. H. Lu, T. A. Tishler, S. M. Fong, B. Oluwadara, J. P. Finn, D. Huang, Y. Bordelon, J. Mintz and S. Perlman, Neurochem. Res., 2007, 32, 1655–1664 CrossRef CAS PubMed.
- H. D. Rosas, Y. I. Chen, G. Doros, D. H. Salat, N. K. Chen, K. K. Kwong, A. Bush, J. Fox and S. M. Hersch, Arch. Neurol., 2012, 69, 887–893 CrossRef PubMed.
- J. H. Fox, J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush and S. Hersch, PLoS One, 2007, 2, e334 Search PubMed.
- J. H. Fox, T. Connor, M. Stiles, J. Kama, Z. Lu, K. Dorsey, G. Lieberman, E. Sapp, R. A. Cherny, M. Banks, I. Volitakis, M. DiFiglia, O. Berezovska, A. I. Bush and S. M. Hersch, J. Biol. Chem., 2011, 286, 18320–18330 CrossRef CAS PubMed.
- J. Chen, E. Marks, B. Lai, Z. Zhang, J. A. Duce, L. Q. Lam, I. Volitakis, A. I. Bush, S. Hersch and J. H. Fox, PLoS One, 2013, 8, e77023 CAS.
- G. Xiao, Q. Fan, X. Wang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14995–15000 CrossRef CAS PubMed.
- J. Sreedharan, I. P. Blair, V. B. Tripathi, X. Hu, C. Vance, B. Rogelj, S. Ackerley, J. C. Durnall, K. L. Williams, E. Buratti, F. Baralle, J. de Belleroche, J. D. Mitchell, P. N. Leigh, A. Al-Chalabi, C. C. Miller, G. Nicholson and C. E. Shaw, Science, 2008, 319, 1668–1672 CrossRef CAS PubMed.
- W. Robberecht and T. Philips, Nat. Rev. Neurosci., 2013, 14, 248–264 CrossRef CAS PubMed.
- M. DeJesus-Hernandez, I. R. Mackenzie, B. F. Boeve, A. L. Boxer, M. Baker, N. J. Rutherford, A. M. Nicholson, N. A. Finch, H. Flynn, J. Adamson, N. Kouri, A. Wojtas, P. Sengdy, G. Y. Hsiung, A. Karydas, W. W. Seeley, K. A. Josephs, G. Coppola, D. H. Geschwind, Z. K. Wszolek, H. Feldman, D. S. Knopman, R. C. Petersen, B. L. Miller, D. W. Dickson, K. B. Boylan, N. R. Graff-Radford and R. Rademakers, Neuron, 2011, 72, 245–256 CrossRef CAS PubMed.
- A. E. Renton, E. Majounie, A. Waite, J. Simon-Sanchez, S. Rollinson, J. R. Gibbs, J. C. Schymick, H. Laaksovirta, J. C. van Swieten, L. Myllykangas, H. Kalimo, A. Paetau, Y. Abramzon, A. M. Remes, A. Kaganovich, S. W. Scholz, J. Duckworth, J. Ding, D. W. Harmer, D. G. Hernandez, J. O. Johnson, K. Mok, M. Ryten, D. Trabzuni, R. J. Guerreiro, R. W. Orrell, J. Neal, A. Murray, J. Pearson, I. E. Jansen, D. Sondervan, H. Seelaar, D. Blake, K. Young, N. Halliwell, J. B. Callister, G. Toulson, A. Richardson, A. Gerhard, J. Snowden, D. Mann, D. Neary, M. A. Nalls, T. Peuralinna, L. Jansson, V. M. Isoviita, A. L. Kaivorinne, M. Holtta-Vuori, E. Ikonen, R. Sulkava, M. Benatar, J. Wuu, A. Chio, G. Restagno, G. Borghero, M. Sabatelli, I. Consortium, D. Heckerman, E. Rogaeva, L. Zinman, J. D. Rothstein, M. Sendtner, C. Drepper, E. E. Eichler, C. Alkan, Z. Abdullaev, S. D. Pack, A. Dutra, E. Pak, J. Hardy, A. Singleton, N. M. Williams, P. Heutink, S. Pickering-Brown, H. R. Morris, P. J. Tienari and B. J. Traynor, Neuron, 2011, 72, 257–268 CrossRef CAS PubMed.
- D. R. Rosen, T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J. P. O'Regan and H. X. Deng, et al. , Nature, 1993, 362, 59–62 CrossRef CAS PubMed.
- B. F. Shaw and J. S. Valentine, Trends Biochem. Sci., 2007, 32, 78–85 CrossRef CAS PubMed.
- D. A. Bosco, G. Morfini, N. M. Karabacak, Y. Song, F. Gros-Louis, P. Pasinelli, H. Goolsby, B. A. Fontaine, N. Lemay, D. McKenna-Yasek, M. P. Frosch, J. N. Agar, J. P. Julien, S. T. Brady and R. H. Brown, Jr., Nat. Neurosci., 2010, 13, 1396–1403 CrossRef CAS PubMed.
- T. Ahtoniemi, G. Goldsteins, V. Keksa-Goldsteine, T. Malm, K. Kanninen, A. Salminen and J. Koistinaho, Mol. Pharmacol., 2007, 71, 30–37 CrossRef CAS PubMed.
- Q. X. Li, S. S. Mok, K. M. Laughton, C. A. McLean, I. Volitakis, R. A. Cherny, N. S. Cheung, A. R. White and C. L. Masters, Aging Cell, 2006, 5, 153–165 CrossRef CAS PubMed.
- E. Tokuda, S. Ono, K. Ishige, A. Naganuma, Y. Ito and T. Suzuki, Toxicology, 2007, 229, 33–41 CrossRef CAS PubMed.
- A. F. Hottinger, E. G. Fine, M. E. Gurney, A. D. Zurn and P. Aebischer, Eur. J. Neurosci., 1997, 9, 1548–1551 CrossRef CAS.
- M. Kiaei, A. I. Bush, B. M. Morrison, J. H. Morrison, R. A. Cherny, I. Volitakis, M. F. Beal and J. W. Gordon, J. Neurosci., 2004, 24, 7945–7950 CrossRef CAS PubMed.
- S. Nagano, Y. Fujii, T. Yamamoto, M. Taniyama, K. Fukada, T. Yanagihara and S. Sakoda, Exp. Neurol., 2003, 179, 176–180 CrossRef CAS.
- S. Nagano, Y. Ogawa, T. Yanagihara and S. Sakoda, Neurosci. Lett., 1999, 265, 159–162 CrossRef CAS.
- E. Tokuda, S. Ono, K. Ishige, S. Watanabe, E. Okawa, Y. Ito and T. Suzuki, Exp. Neurol., 2008, 213, 122–128 CrossRef CAS PubMed.
- S. Petri, N. Y. Calingasan, O. A. Alsaied, E. Wille, M. Kiaei, J. E. Friedman, O. Baranova, J. C. Chavez and M. F. Beal, J. Neurochem., 2007, 102, 991–1000 CrossRef CAS PubMed.
- J. S. Beckman, A. G. Estevez, L. Barbeito and J. P. Crow, Free Radical Biol. Med., 2002, 33, 1433–1435 CrossRef CAS.
- A. I. Bush, Nat. Neurosci., 2002, 5, 919 CrossRef CAS PubMed.
- E. J. McAllum, N. K. Lim, J. L. Hickey, B. M. Paterson, P. S. Donnelly, Q. X. Li, J. R. Liddell, K. J. Barnham, A. R. White and P. J. Crouch, Amyotrophic Lateral Scler. Frontotemporal Degener., 2013, 14, 586–590 CrossRef CAS PubMed.
- H. Kishigami, S. Nagano, A. I. Bush and S. Sakoda, Free Radical Biol. Med., 2010, 48, 945–952 CrossRef CAS PubMed.
- S. Watanabe, S. Nagano, J. Duce, M. Kiaei, Q. X. Li, S. M. Tucker, A. Tiwari, R. H. Brown, Jr., M. F. Beal, L. J. Hayward, V. C. Culotta, S. Yoshihara, S. Sakoda and A. I. Bush, Free Radical Biol. Med., 2007, 42, 1534–1542 CrossRef CAS PubMed.
- M. Son, K. Puttaparthi, H. Kawamata, B. Rajendran, P. J. Boyer, G. Manfredi and J. L. Elliott, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6072–6077 CrossRef CAS PubMed.
- E. Tokuda, E. Okawa, S. Watanabe, S. Ono and S. L. Marklund, Neurobiol. Dis., 2013, 54, 308–319 CrossRef CAS PubMed.
- T. Fujisawa, K. Homma, N. Yamaguchi, H. Kadowaki, N. Tsuburaya, I. Naguro, A. Matsuzawa, K. Takeda, Y. Takahashi, J. Goto, S. Tsuji, H. Nishitoh and H. Ichijo, Ann. Neurol., 2012, 72, 739–749 CrossRef CAS PubMed.
- B. R. Roberts, J. A. Tainer, E. D. Getzoff, D. A. Malencik, S. R. Anderson, V. C. Bomben, K. R. Meyers, P. A. Karplus and J. S. Beckman, J. Mol. Biol., 2007, 373, 877–890 CrossRef CAS PubMed.
- K. Homma, T. Fujisawa, N. Tsuburaya, N. Yamaguchi, H. Kadowaki, K. Takeda, H. Nishitoh, A. Matsuzawa, I. Naguro and H. Ichijo, Mol. Cell, 2013, 52, 75–86 CrossRef CAS PubMed.
- M. Yasui, K. Ota and R. M. Garruto, Neurotoxicology, 1993, 14, 445–450 CAS.
- Y. Sheng, M. Chattopadhyay, J. Whitelegge and J. S. Valentine, Curr. Top. Med. Chem., 2012, 12, 2560–2572 CrossRef CAS.
- P. Ip, V. K. Mulligan and A. Chakrabartty, J. Mol. Biol., 2011, 409, 839–852 CrossRef CAS PubMed.
- Y. Nadjar, P. Gordon, P. Corcia, G. Bensimon, L. Pieroni, V. Meininger and F. Salachas, PLoS One, 2012, 7, e45034 CAS.
- E. F. Goodall, M. J. Greenway, I. van Marion, C. B. Carroll, O. Hardiman and K. E. Morrison, Neurology, 2005, 65, 934–937 CrossRef CAS PubMed.
- A. Takeda, E. Loveman, A. Clegg, J. Kirby, J. Picot, E. Payne and C. Green, Int. J. Geriatr. Psychiatry, 2006, 21, 17–28 CrossRef CAS PubMed.
- E. Ferrada, V. Arancibia, B. Loeb, E. Norambuena, C. Olea-Azar and J. P. Huidobro-Toro, Neurotoxicology, 2007, 28, 445–449 CrossRef CAS PubMed.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS.
- C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi and C. L. Masters, Arch. Neurol., 2003, 60, 1685–1691 CrossRef PubMed.
- K. J. Barnham, E. Gautier, G. Kok and G. Krippner, PCT Publications WO2004007461, 2003.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q. X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS PubMed.
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2008, 7, 779–786 CrossRef CAS.
- N. G. Faux, C. W. Ritchie, A. Gunn, A. Rembach, A. Tsatsanis, J. Bedo, J. Harrison, L. Lannfelt, K. Blennow, H. Zetterberg, M. Ingelsson, C. L. Masters, R. E. Tanzi, J. L. Cummings, C. M. Herd and A. I. Bush, J. Alzheimer's Dis., 2010, 20, 509–516 CAS.
- A. R. White, T. Du, K. M. Laughton, I. Volitakis, R. A. Sharples, D. E. Hoke, R. M. D. Holsinger, G. Evin, R. A. Cherny, A. F. Hill, K. J. Barnham, Q. X. Li, A. I. Bush and C. L. Masters, J. Neurochem., 2006, 98, 90 Search PubMed.
- A. Caragounis, T. Du, G. Filiz, K. M. Laughton, I. Volitakis, R. A. Sharples, R. A. Cherny, C. L. Masters, S. C. Drew, A. F. Hill, Q. X. Li, P. J. Crouch, K. J. Barnham and A. R. White, Biochem. J., 2007, 407, 435–450 CrossRef CAS PubMed.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, J. Neurochem., 2011, 119, 220–230 CrossRef CAS PubMed.
- P. A. Adlard, L. Bica, A. R. White, M. Nurjono, G. Filiz, P. J. Crouch, P. S. Donnelly, R. Cappai, D. I. Finkelstein and A. I. Bush, PLoS One, 2010, 6, e17669 Search PubMed.
- L. Huang, C. Lu, Y. Sun, F. Mao, Z. Luo, T. Su, H. Jiang, W. Shan and X. Li, J. Med. Chem., 2012, 55, 8483–8492 CrossRef CAS PubMed.
- Y. Avramovich-Tirosh, L. Reznichenko, T. Mit, H. Zheng, M. Fridkin, O. Weinreb, S. Mandel and M. B. Youdim, Curr. Alzheimer Res., 2007, 4, 403–411 CrossRef CAS.
- L. Kupershmidt, T. Amit, O. Bar-Am, O. Weinreb and M. B. Youdim, Mol. Neurobiol., 2012, 46, 217–220 CrossRef CAS PubMed.
- W. H. Wu, P. Lei, Q. Liu, J. Hu, A. P. Gunn, M. S. Chen, Y. F. Rui, X. Y. Su, Z. P. Xie, Y. F. Zhao, A. I. Bush and Y. M. Li, J. Biol. Chem., 2008, 283, 31657–31664 CrossRef CAS PubMed.
- J. J. Braymer, A. S. Detoma, J. S. Choi, K. S. Ko and M. H. Lim, Int. J. Alzheimer's Dis., 2010, 2011, 623051 Search PubMed.
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef PubMed.
- M. Jensen, A. Canning, S. Chiha, P. Bouquerel, J. T. Pedersen, J. Ostergaard, O. Cuvillier, I. Sasaki, C. Hureau and P. Faller, Chemistry, 2012, 18, 4836–4839 CrossRef CAS PubMed.
- J. J. Braymer, J. S. Choi, A. S. DeToma, C. Wang, K. Nam, J. W. Kampf, A. Ramamoorthy and M. H. Lim, Inorg. Chem., 2011, 50, 10724–10734 CrossRef CAS PubMed.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS PubMed.
- C. Rodriguez-Rodriguez, N. Sanchez de Groot, A. Rimola, A. Alvarez-Larena, V. Lloveras, J. Vidal-Gancedo, S. Ventura, J. Vendrell, M. Sodupe and P. Gonzalez-Duarte, J. Am. Chem. Soc., 2009, 131, 1436–1451 CrossRef CAS PubMed.
- H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, K. H. Thompson and C. Orvig, Angew. Chem., Int. Ed., 2007, 46, 1716–1718 CrossRef CAS PubMed.
- H. Zheng, M. B. Youdim and M. Fridkin, J. Med. Chem., 2009, 52, 4095–4098 CrossRef CAS PubMed.
- Y. H. Zhang, J. Raymick, S. Sarkar, D. K. Lahiri, B. Ray, D. Holtzman, M. Dumas and L. C. Schmued, Curr. Alzheimer Res., 2013, 10, 494–506 CrossRef CAS.
- T. Wang, C. Y. Wang, Z. Y. Shan, W. P. Teng and Z. Y. Wang, J. Alzheimer's Dis., 2012, 29, 549–559 CAS.
- C. Grossi, S. Francese, A. Casini, M. C. Rosi, I. Luccarini, A. Fiorentini, C. Gabbiani, L. Messori, G. Moneti and F. Casamenti, J. Alzheimer's Dis., 2009, 17, 423–440 CAS.
- J. Y. Lee, J. E. Friedman, I. Angel, A. Kozak and J. Y. Koh, Neurobiol. Aging, 2004, 25, 1315–1321 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- D. F. Tardiff, M. L. Tucci, K. A. Caldwell, G. A. Caldwell and S. Lindquist, J. Biol. Chem., 2012, 287, 4107–4120 CrossRef CAS PubMed.
- T. Nguyen, A. Hamby and S. M. Massa, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11840–11845 CrossRef CAS PubMed.
- D. E. Ehrnhoefer, M. Duennwald, P. Markovic, J. L. Wacker, S. Engemann, M. Roark, J. Legleiter, J. L. Marsh, L. M. Thompson, S. Lindquist, P. J. Muchowski and E. E. Wanker, Hum. Mol. Genet., 2006, 15, 2743–2751 CrossRef CAS PubMed.
- L. Lannfelt, K. Blennow, H. Zetterberg, S. Batsman, D. Ames, J. Harrison, C. L. Masters, S. Targum, A. I. Bush, R. Murdoch, J. Wilson and C. W. Ritchie, Lancet Neurol., 2009, 8, 981 CrossRef CAS.
- N. G. Faux, C. W. Ritchie, A. Gunn, A. Rembach, A. Tsatsanis, J. Bedo, J. Harrison, L. Lannfelt, K. Blennow, H. Zetterberg, M. Ingelsson, C. L. Masters, R. E. Tanzi, J. L. Cummings, C. M. Herd and A. I. Bush, J. Alzheimer's Dis., 2010, 20, 509–516 CAS.
- R. A. Cherny, S. Ayton, D. I. Finkelstein, A. I. Bush, G. McColl and S. Massa, J. Huntington's Dis., 2012, 1, 211–219 Search PubMed.
- http://pranabio.com/news/prana-announces-successful-phase-2-results-huntington-disease-trial#.U9mwxVZGPPM (Accessed July 31, 2014).
- M. A. Green, D. L. Klippenstein and J. R. Tennison, J. Nucl. Med., 1988, 29, 1549–1557 CAS.
- J. Lewis, R. Laforest, T. Buettner, S. Song, Y. Fujibayashi, J. Connett and M. Welch, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1206–1211 CrossRef CAS PubMed.
- P. S. Donnelly, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, R. A. Cherny, R. A. Sharples, A. F. Hill, Q. X. Li, C. L. Masters, K. J. Barnham and A. R. White, J. Biol. Chem., 2008, 283, 4568–4577 CrossRef CAS PubMed.
- M. T. Fodero-Tavoletti, V. L. Villemagne, B. M. Paterson, A. R. White, Q. X. Li, J. Camakaris, G. O'Keefe, R. Cappai, K. J. Barnham and P. S. Donnelly, J. Alzheimer's Dis., 2010, 20, 49–55 CAS.
- P. J. Crouch, L. W. Hung, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, M. Nurjono, A. Caragounis, T. Du, K. Laughton, I. Volitakis, A. I. Bush, Q. X. Li, C. L. Masters, R. Cappai, R. A. Cherny, P. S. Donnelly, A. R. White and K. J. Barnham, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 381–386 CrossRef CAS PubMed.
- S. C. Drew and K. J. Barnham, Acc. Chem. Res., 2011, 44, 1146–1155 CrossRef CAS PubMed.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014 DOI:10.1021/ar400293h.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
| This journal is © The Royal Society of Chemistry 2014 |