Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene
Ekaterina V.
Makshina
*a,
Michiel
Dusselier
a,
Wout
Janssens
a,
Jan
Degrève
b,
Pierre A.
Jacobs
a and
Bert F.
Sels
*a
aCentre for Surface Chemistry and Catalysis, Faculty of Bioscience Engineering, KU Leuven, Kasteelpark Arenberg 23, B-3001, Heverlee, Belgium. E-mail: bert.sels@biw.kuleuven.be; ekaterina.makshina@biw.kuleuven.be; Fax: +32 16 321 998; Tel: +32 16 3 21 59
bDepartment of Chemical Engineering, KU Leuven, Willem de Croylaan 46, 3001 Heverlee, Belgium
First published on 4th July 2014
Abstract
Increasing demand for renewable feedstock-based chemicals is driving the interest of both academic and industrial research to substitute petrochemicals with renewable chemicals from biomass-derived resources. The search towards novel platform chemicals is challenging and rewarding, but the main research activities are concentrated on finding efficient pathways to produce familiar drop-in chemicals and polymer building blocks. A diversity of industrially important monomers like alkenes, conjugated dienes, unsaturated carboxylic acids and aromatic compounds are thus targeted from renewable feedstock. In this context, on-purpose production of 1,3-butadiene from biomass-derived feedstock is an interesting example as its production is under pressure by uncertainty of the conventional fossil feedstock. Ethanol, obtained via fermentation or (biomass-generated) syngas, can be converted to butadiene, although there is no large commercial activity today. Though practised on a large scale in the beginning of the 20th century, there is a growing worldwide renewed interest in the butadiene-from-ethanol route. An alternative route to produce butadiene from biomass is through direct carbohydrate and gas fermentation or indirectly via the dehydration of butanediols. This review starts with a brief discussion on the different feedstock possibilities to produce butadiene, followed by a comprehensive summary of the current state of knowledge regarding advances and achievements in the field of the chemocatalytic conversion of ethanol and butanediols to butadiene, including thermodynamics and kinetic aspects of the reactions with discussions on the reaction pathways and the type of catalysts developed.
 Ekaterina V. Makshina | Dr Ekaterina V. Makshina obtained her PhD degree in 2007 in the frame of a cooperation agreement (Co-tutelle) of Moscow State University (Laboratory of Kinetics and Catalysis) and Littoral University (Laboratory of Catalysis and Environment), Dunkerque. Her PhD thesis dealt with the oxidation of methanol and VOCs using supported oxide catalysts. Since 2010 she has been working as a post-doc fellow in the Center for Surface Chemistry and Catalysis (University of Leuven) in the group of Prof. Sels on biomass transformation into commodity chemicals. She has published 16 papers. |
 Michiel Dusselier | Dr Michiel Dusselier obtained a MSc in Engineering (Catalytic Technology) at KU Leuven in 2009, studying in part at the TU München. He obtained his PhD in 2013 under the guidance of Prof. Sels and Jacobs on the topic of tailoring catalytic routes towards and from lactic acid and examining the synthesis and potential of bio-monomers. He was awarded the ACS Dr J. Breen memorial fellowship in 2013. As a postdoc, he currently focuses on zeolite catalysis and renewable conversion. He has authored 18 papers and 2 patents. Currently, he is staying in the group of Prof. Davis at Caltech. |
 Wout Janssens | Wout Janssens obtained his MSc in Engineering (Catalytic Technology) at KU Leuven in 2010. His master thesis was performed under the guidance of Prof. Sels and dealt with the synthesis and application of metal nanoparticles for catalytic purposes. Currently he is in the process of finishing his PhD under the guidance of Prof. Sels, dealing with the heterogeneously catalysed transformation of ethanol into 1,3-butadiene. |
 Jan Degrève | Jan Degrève obtained his PhD in Chemical Engineering from SUNY Buffalo, USA, in 1989 with a thesis on the numerical simulation of combustion phenomena for the production of ceramic materials. He is presently professor of Chemical Engineering at KU Leuven, Belgium where he teaches courses on the bachelor and master level. His main fields of interest and expertise are design and analysis of chemical reactors, separation processes, chemical process design and flowsheet simulation. He is also the programme director for the advanced Master of Safety Engineering at KU Leuven. |
 Pierre A. Jacobs | Pierre A. Jacobs has been since 2008 professor emeritus with special mandate at KU Leuven. During his active career, he has been professor of Physical-Chemistry and Catalysis at KU Leuven and authored over 650 reviewed papers. The industrial impact of his research is visible in numerous patents and culminated in the recent nomination for the 2013 Alwin Mittasch special prize marking the centennial commissioning of the first ammonia plant. Among the important awards received for his research are the Paul Emmett Award in 1981, for his contribution to fundamental understanding of zeolite catalysts, the Donald Breck Award in 1998 for his work on zeolite-based biomimetic catalysis. In recognition of his contributions to bridge homogeneous, heterogeneous and bio-catalysis, he received the Synetix Award and the Kreitman Award in 2001. He was Karl Ziegler Guest Professor in 2004. |
 Bert F. Sels | Bert F. Sels obtained his PhD in 2000 at KU Leuven in the field of oxidation chemistry with heterogeneous catalysis. He was awarded the DSM Chemistry Award in 2000, and in 2005 the Incentive Award by the Societè Chimique Belge for his research achievements. In 2005 he was appointed assistant professor at KU Leuven, and full professor in 2008. He now teaches courses on spectroscopic tools in organic chemistry, sustainability of chemical processes, spectroscopic tools in surface chemistry and heterogeneous catalysis. He heads a research group in the Center for Surface Chemistry and Catalysis (KU Leuven). His current field of research interest focuses on synthesis, characterization and use of heterogeneous catalysis, mainly in transformation of renewable resources to chemicals. He was recently elected the co-chair of the IZA Catalysis Commission. |
1. General introduction
1.1. Setting the scene
1,3-Butadiene (C4H6 or BD) is the most important conjugated diene, being the basis of a wide variety of synthetic rubbers, elastomers and polymer resins upon polymerisation by itself or in conjunction with other polymerizable monomers. The global BD demand in 2012 was approximately 10 million metric tons with an incremental growth of 1–2% per year.The synthetic BD industry started in 1920–1930; the synthetic rubber was proposed as an alternative for the manufacture of automobile tires. Sodium-butadiene rubber was the first synthetic rubber industrially produced worldwide. In Russia, the S.K. synthetic rubber production (abbreviation from Russian “Sinteticheskii Kauchuk” – synthetic caoutchouc) was made in a one-stage process from grain ethanol, often referred to as the Lebedev process. In addition to Russia, synthetic rubber from ethanol (KER: abbreviation of Polish – “kauczuk erytrenowy” – erythrene caoutchouc) was also produced in Poland in the early 1930s. Germany produced the rubber under the brand name “Buna-S” and two routes starting from coal-derived acetylene were employed. The main route was the condensation of acetaldehyde via 1,3-butanediol with an overall BD yield on acetylene of 60%;1 the second route proceeded via 1,4-butynediol with an overall yield on acetylene of 70–75%.1 The first process involved the hydration of acetylene to acetaldehyde, which is subsequently converted into the aldol product, hydrogenated to 1,3-butanediol and dehydrated to butadiene, while the second (Reppe) process used formaldehyde and acetylene to produce 1,4-butynediol, which was hydrogenated to 1,4-butanediol and dehydrated partly via tetrahydrofuran to BD. These routes are no longer practicable. In the US, BD routes employed were from oil sources and ethanol by a two-step process using a mixture of ethanol and acetaldehyde.2
In the US and most of Europe, the ethanol and acetylene routes have been abandoned after the world war because of the emerging production of BD from petroleum sources. The ethanol route only survived up to now in China and India. In the US, a large percentage of BD production is obtained by dehydrogenation of n-butane and n-butenes.2 Direct extraction from C4 streams arising from naphtha crackers (ethylene plants) is substantially practised in Europe since the early seventies. The separation of BD from C4 streams though requires an expensive extractive distillation with a selectivity of only 4–5% due to its azeotrope formation with butane.
The growth of ethylene production outpaced that of BD demand resulting in an oversupply of BD. Although this situation has led to a shut-down of many on-purpose BD production units, it also stimulated the development of chemical processes using BD as the building block. BD is thus considered as a high value by-product of ethylene production from steam crackers without clear production economics. Since many years, the BD supply and price have been influenced by the demand for ethylene.
Recent trends in steam cracking show a lightened feedstock as a result of lower costs and larger availability of ethane from natural gas like shale gas, this change in feedstock affecting the supply and price of BD. Table 1 illustrates the lower BD yield from lighter feedstock relative to naphtha and light atmospheric gas oil in the steam cracker.2,3 Whereas an ethane-only cracker has the lowest capital cost and the highest selectivity towards ethylene, it shows the lowest BD yield. As a consequence, restrictions on the availability of BD and high BD prices are expected in the near future. Moreover, lightening of the feedstock for the purpose of ethylene production leads to a deficit of C3, C4, C5 and pygas supply. Shortage of these chemicals, partially introduced due to the shale gas revolution, may create new opportunities for the production of biobased chemicals from renewable feedstock like sugars, glycerin etc. One could argue about the price of such chemicals, but their production cost will decrease as the process technologies evolve and the sustainability benefits should also not be overlooked.4,5 The worldwide change in feedstock usage asks for a revisit of on-purpose BD production. Future BD production will probably be diverse, highly depending on the local availability and price of the different feedstocks such as coal, oil, gas and biomass. Most relevant on-purpose BD synthesis strategies are summarized in Scheme 1.
| Feedstock | Butadiene/ethylene |
|---|---|
| Ethane | 0.02 |
| Propane | 0.07 |
| Butane | 0.07–0.11 |
| Naphtha | 0.13 |
| Gas oil | 0.26 |
 | ||
| Scheme 1 Synthesis of 1,3-butadiene (BD) from various carbon-containing feedstocks. The focus of this review is shown in the red dashed line. BDO = butanediols. | ||
Obviously, the success of the ethanol-to-BD route will depend on the cost of the upstream ethanol process. While the exploitation of the Brazilian sugarcane currently delivers the cheapest ethanol, over time cellulosic-based second generation ethanol in Asia, Europe and United States may well surpass the techno-economic performances. Many research efforts are presently undertaken developing more efficient ethanol production technologies from low-cost and non-food/feed biomass feedstocks.10 Ethanol from cellulosic and algal sources, the so-called 2nd and 3rd generation ethanol, is promising in this respect.11–17
While the industrial feasibility of the ethanol-to-BD route has been proven long time ago, a number of improvements both from an engineering and catalytic point of view is still vital to attain high BD yield and high BD productivity. In order to compete with existing oil- and gas-based technologies like C4 dehydrogenation, issues related to carbon efficiency, catalyst cost, toxicity, catalyst performance, feedstock tolerance, back-end optimization, catalyst lifetime and stability require further optimization. So, it seems that extensive research is necessary to improve the viability of the ethanol route to BD vis-à-vis the conventional routes. In 2013, Axens, IFPEN and Michelin have launched their joint research program to develop an economically competitive process to produce bio-sourced synthetic rubber from bioethanol.18,19
Next to ethanol, other bio-derived chemicals like butanols (BuOH), viz. n-butanol and Gevo's isobutanol, 2,3-butanediol (2,3-BDO) and 1,4-butanediol (1,4-BDO) may also be useful to produce BD. Most of the C4 alcohols are now available from renewable feedstocks via fermentation of biomass or syngas (e.g. biomass derived).20,21 For instance, Genomatica – a company designing microorganisms for the conversion of sugars and cellulosic biomass to sustainable chemicals – is currently producing 1,4-BDO at a demonstration plant. The first commercial-scale plant is scheduled, with Novamont as a partner.22,23 Together with Versalis, Eni's chemicals subsidiary leader in the production of elastomers, they recently announced the establishment of a strategic partnership to enable the production of BD from renewable feedstocks. Main incentive of the collaboration is the successful direct production of million pound quantities of bio-based 1,4-BDO from biomass resources in 2011 by Genomatica.24
Acid catalysed dehydration of (bio-based) butanols leads to a mixture of n-butenes, requiring further dehydrogenation to produce BD with existing technology.2 Next to dehydration of monohydric alcohols direct butanediol (BDO) dehydration to BD was also performed. This strategy has the advantage of avoiding the expensive dehydrogenation step. The reaction involves a double dehydration, preferably employing a catalytic process, the catalyst and the reaction conditions employed differing with the position of the alcohol groups. Dehydration of 1,4-BDO is well-known from the acetylene-based Reppe route.2 It is carried out at 280 °C by a rather complex method using an excess of steam and THF in the presence of a sodium phosphate catalyst. The dehydration chemistry of 1,3-BDO is slightly different. High BD yields above 80% were obtained at 400 °C using the same catalyst in the presence of steam. Dehydration of the other diols is less selective. So, 2,3-BDO easily dehydrates to methyl ethyl ketone, while its diacetate can be pyrolysed in high yield to BD upon heating to 475–600 °C, the acetic acid co-product being recycled.25 New catalytic advances in the dehydration of diols and polyols in detail will be described below.
Other routes to BD from natural gas were investigated. They mainly focused on the direct and indirect production of chemical and fermentation ethanol, which is converted to BD. Several papers and reviews have been published on the catalytic conversion of syngas to ethanol and higher alcohols using chemocatalysis.28–31 Three chemocatalytic routes are known in the literature to convert syngas to ethanol: (i) the direct conversion via selective CO hydrogenation on a solid catalyst, (ii) methanol homologation, which involves reductive carbonylation of methanol over a redox catalyst, and (iii) a multistep process, wherein classical methanol production from syngas, is followed by carbonylation to acetic acid, and subsequent hydrogenation to ethanol. None of these routes have been commercially practised. The carbonylation of methanol to ethanol is the most promising route, while the direct formation of ethanol from syngas is probably commercially less interesting due to a low ethanol yield and selectivity.32 A recent example of the methanol–acetic acid route (TCX technology) is reported by Celanese.33
The fermentative production of ethanol from syngas has also gained considerable attention. The syngas could be an industrial waste gas or it may be obtained by natural gas reforming or gasification of biomass. The utilization of the whole biomass of low quality and the elimination of biomass fractionation are major advantages here. The state-of-the art and the most important challenges recently have been critically reviewed.34–38
Dehydrogenation of butane from natural gas is a better economical solution to BD production. This well-established technology has been practiced until the 1990's in Russia and the US. The main disadvantages of the process are the high endothermicity and the high process temperature (600–700 °C) to achieve an economical equilibrium conversion. As the reaction runs at incomplete conversion, separation of feed and products is mandatory. Due to coke formation, catalysts usually run for only 5 to 15 minutes. As heat from catalyst regeneration fuels the endothermic dehydrogenation, the overall lifetime of the catalysts is much longer. The Houdry-Catadiene process using a mixture of Al oxide and Cr oxide typically shows a reactor outlet with 15 vol% butadiene, and about 50% carbon yield.39 Somewhat higher yields (65%) are reported for the two-step Phillips Petroleum technology, involving butane dehydrogenation to n-butenes with Cr/Na/Al oxide, followed by their extractive distillation and conversion to butadiene over Fe/K/Al oxide.39 Application of vacuum or addition of water are used to reduce the partial pressure (Le Chatelier) and to overcome coke formation by stimulating the water gas shift reaction, respectively (see also technologies from Dow and Shell). The conversion and selectivity of the process are also greatly improved by hydrogen removal. In the presence of dioxygen, water is formed, the exothermicity partly being used to compensate the energy required for the dehydrogenation. Whereas the presence of water and O2 increases catalyst lifetime, O2 also helps the difficult abstraction of allylic hydrogen. With Phillips O–X–D at 480–600 °C, 80% conversion and 90% BD selectivity were obtained. The Oxo-D process of PetroTex shows 93% BD selectivity at 65% conversion.2 Both processes run only on n-butenes since the addition of oxygen to butane would result in products oxidation at the temperature required for butane dehydrogenation. Although use of iodine to remove H2 as HI (Idas process, Shell France) has been proposed, serious corrosion has been encountered. The yield of BD on butane was said to be 70%. Butenes could be also resourced from dehydrating biobutanols to produce bio-based BD, though the price of the bioalcohols is currently too high for commercial use.
The dehydrogenation processes are competitive with the cracking process when BD price is high and raw C4 is available. Honeywell UOP in partnership with TPC group and BASF in collaboration with Linde recently announced the development and licensing of such a process for the on-purpose BD production from butane.40–42
1.2. Scope of the review
The introductory section already partly reveals many feedstock possibilities for future on-purpose production of BD. Routes from butane and n-butenes are probably the most developed ones. Although they will not be described in this review, they should be regarded as benchmark to alternative routes. The most promising competitive routes are BD from ethanol and BDOs. They form the main subject of this review.Research on ethanol conversion to BD has been performed intensively in the twenties to sixties of the 20th century. A possible reaction mechanism and promising catalytic systems have been presented in previous reviews.43–48 With the decreasing industrial interest for this reaction at that time, the scientific interest also faded, while only a few publications analysing recent achievements appeared.49–52 Yet, there is now a steadily growing interest of academia and industry in producing bio-ethanol and using it as a platform molecule for various base chemicals,53–61 BD being an important chemical among them. Therefore, a comprehensive review paper of old and recently published reports on the catalytic conversion of ethanol to BD is attempted in the first part. The contribution includes details of the reaction network, mechanistic and thermodynamic considerations and compares catalytic data (in terms of BD yield, ethanol conversion, carbon yield and volume productivity) of various catalyst types. Catalytic properties and structure/activity–selectivity relationships were made as much as possible, if appropriate, in order to guide and inspire the reader.
Routes to BD from 4-carbon diols (BDO) have attracted much recent attention and this evolution is mainly explained by the availability of bio-derived BDOs from renewable and waste feedstocks. Though the chemistry seems less complicated and has already been described in part of the old BD processes from acetylene and ethanol, the selective double dehydration of BDOs to BD under milder conditions remains challenging requiring unique catalysis. The second part of the review is therefore devoted to summarize and describe the most important catalytic achievements to dehydrate such diols including the mechanistic proposals and thermodynamics, the type of catalyst developed and the current status.
2. Ethanol-based butadiene
2.1. The technological options: one vs. two-step processes
In 1903 the catalytic synthesis of BD from ethanol was first performed by Ipatiev in Russia.62 Low BD yield of about 1.5% was obtained by passing ethanol at 550–600 °C vapour over Al powder. Filippov obtained a BD yield of 5% from diethyl ether at 400–500 °C on the same catalyst.63 Ostromyslensky proposed a catalytic route from a mixture of ethanol and acetaldehyde over alumina or clay catalysts at 440–460 °C, yielding 18% BD.64 Maximoff claimed the formation of BD from a mixture of ethanol with acetaldol (or crotonaldehyde) over aluminium hydroxide.65 Later, Lebedev66–68 proposed his first process on a mixture of zinc oxide and alumina at 400 °C. He achieved a BD selectivity of 18%, directly from ethanol according to following reaction (1): | (1) |
Improvements by Lebedev's group with an undisclosed catalyst composition showed higher yields with up to 70% BD selectivity at 350 °C and atmospheric pressure.69 The process was examined both in Germany and in the US, but was not taken to the full scale. I.G. Farbenindustrie tested supported magnesia catalysts promoted by Co or Cr and reported 60% yield at 300 °C and atmospheric pressure.1 In the US a similar catalyst containing 59% magnesia, 2% chromic acid and 39% silica gel was also examined.1 This catalyst gave 38% BD yield per pass with 56% selectivity at 400–425 °C.
In addition to the direct ethanol conversion according to (1), BD from a mixture of ethanol and acetaldehyde (or a so-called two-steps process according to Ostromyslensky's pathway) was studied. Union Carbide and Carbon Chemicals Corporation performed the industrial manufacturing of BD from ethanol in the USA over tantalum oxide on silica using this two-step process (Scheme 2). This process includes the partial dehydrogenation of ethanol to acetaldehyde, followed by the transformation of the mixture of ethanol and acetaldehyde into BD. The yield of ethanol was 18% at a conversion of 30% per pass.70
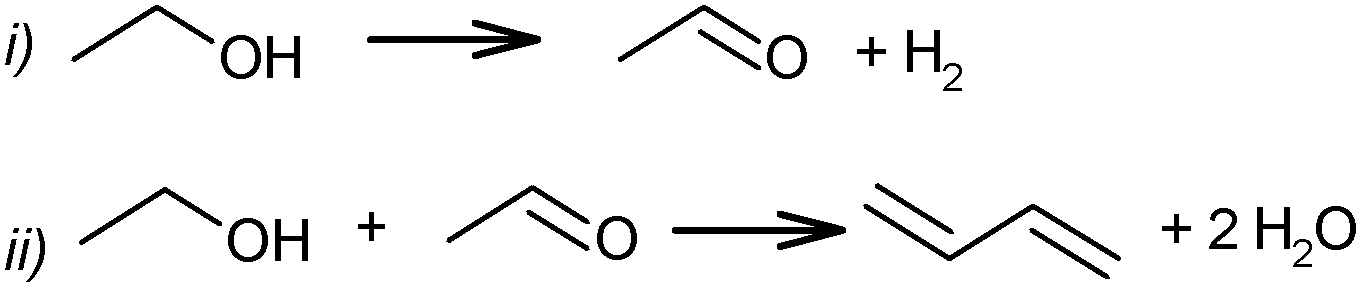 | ||
| Scheme 2 Two-step process of BD from ethanol. | ||
2.2. Routes and chemistry from ethanol to BD
Ethanol as a building block can be converted into BD. The reaction network involves a series of reactions, leading via multiple pathways to a variety of products that are controlled differently by kinetics and thermodynamic restrictions. Before describing the thermodynamic and kinetic aspects, the different reactions involved and the complex reaction network in the synthesis of BD from ethanol will be clarified first.Initially, Lebedev et al. proposed a radical mechanism to describe the direct formation of BD from ethanol, in accordance with the reactions in Scheme 3.68 A few years later, Balandin argued that this radical reaction mechanism was energetically possible.71 All possible combinations of molecules in the reaction mixture were analysed based on the multiplet theory. Whereas the formation of 49 different possible reaction products was predicted, only 21 of them could be detected. Acetaldehyde was identified as a primary product, while BD was assumed to be a third generation product. Baladin concluded that the reaction proceeded via 1,3-BDO according to the following reaction sequence (Scheme 4).
 | ||
| Scheme 3 BD formation mechanism according to Lebedev.68 | ||
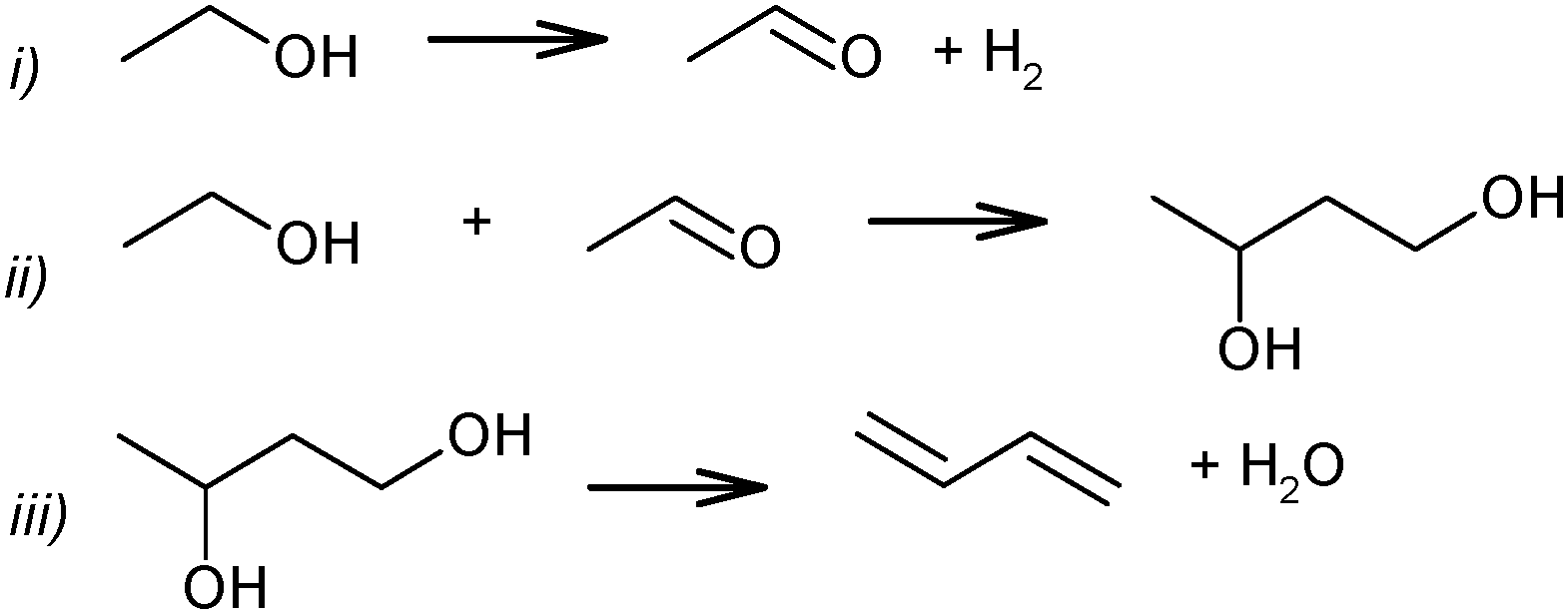 | ||
| Scheme 4 BD formation mechanism according to Balandin.71 | ||
This scheme was essentially similar to the mechanism proposed earlier by Ostromyslensky for his two-step process using a mixture of ethanol and acetaldehyde.64 Egloff and Hulla supported the intermediate formation of 1,3-BDO.44
The above mechanistic proposals were all rejected a few years later by different research groups.69,72–76 By studying the transformation of various reactants and mixtures thereof, the authors considered that the reactions, including the Lebedev and Ostromyslensky synthesis, all proceeded through the intermediate formation of crotonaldehyde.69,72,73,77,78 The mechanism includes the formation of acetaldol via the aldol condensation of two acetaldehyde molecules, followed by dehydration of the aldol to crotonaldehyde, in accordance with reaction (2):
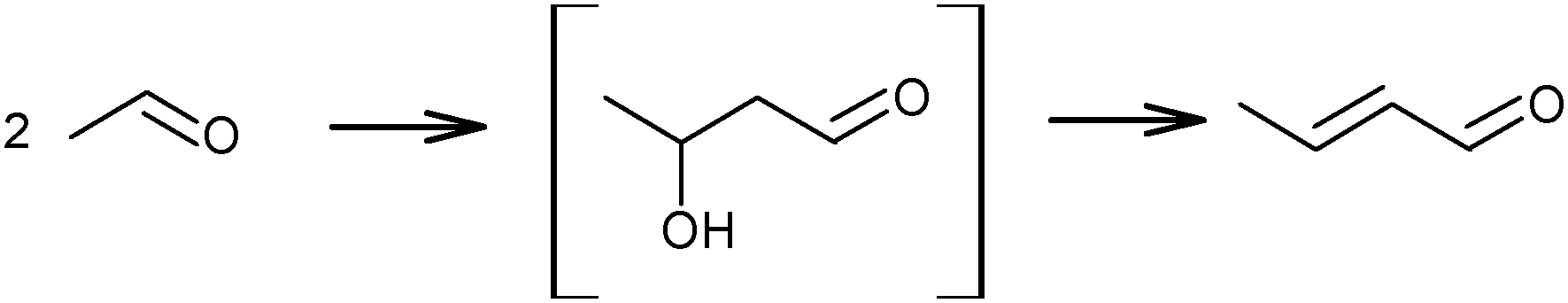 | (2) |
Further transformation of crotonaldehyde to BD has been the subject of many discussions for a long time. According to Quattlebaum et al., crotonaldehyde is deoxygenated with ethanol, as presented in Scheme 5(i).73 Moreover, the authors for the first time attempted a description of the mechanism taking into account the participation of surface atoms of the catalyst. They assumed a synthesis route via hydrogen transfer from ethanol to the enol-form of crotonaldehyde.73 It has been suggested that the reactive bridged siloxane of the silica-based catalyst participated in the deoxygenation process by reacting with ethanol and the enol, thereby supporting an intermolecular hydride shift (Scheme 5ii–iv), while the oxide promoter (e.g., tantalum, zirconium or niobium oxides) performs the aldol condensation. The latter reaction was identified as the rate-controlling step for the Ostromyslensky process.73
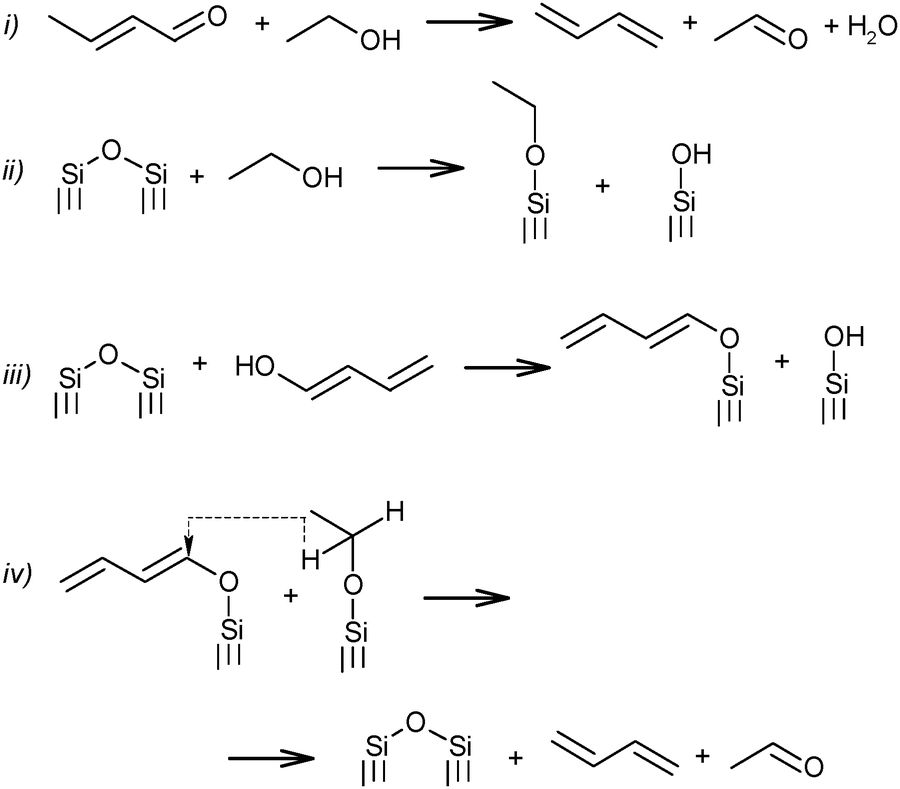 | ||
| Scheme 5 BD formation mechanism according to Quattlebaum et al.73 | ||
Jones et al.76 studied the reaction mechanism of the two-step process over the same catalyst, viz. 2%Ta2O5–SiO2. They suggested an opposite effect from the catalyst's components, the unmodified silica being more effective for acetaldehyde condensation and the 2%Ta2O5–SiO2 material being more active for feed mixtures requiring deoxygenation of the aldehyde. Thus, the proposed mechanism of the process (in Scheme 5i) was supported, while reaction sequence ii–iv was rejected (Scheme 5), because participation of the Ta promoter was not included.
At the same time, a slightly different pathway for crotonaldehyde transformation was proposed (Scheme 6).69,72 Crotonaldehyde was first reduced to crotyl alcohol (Scheme 6i and ii), followed by its dehydration to BD (Scheme 6iii). Ethanol (or any other alcohol present in the system) served as a hydrogen donor.
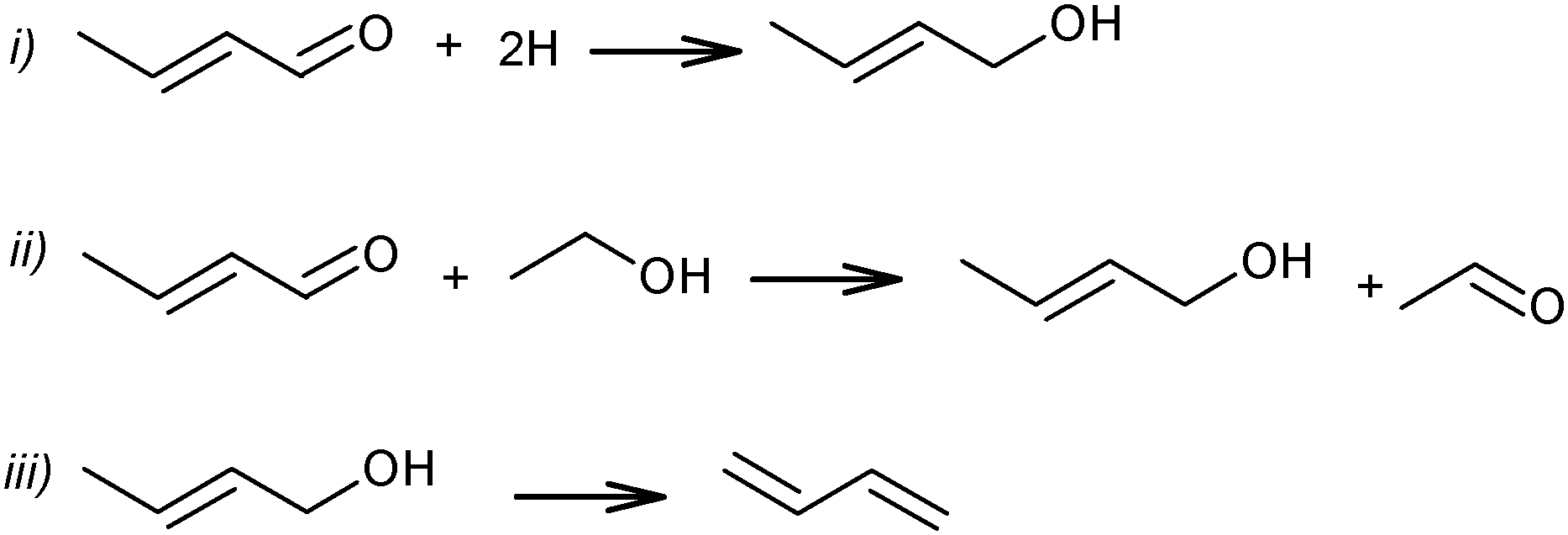 | ||
| Scheme 6 Pathways of crotonaldehyde to BD.69,72 | ||
Kagan et al. over Lebedev type catalysts showed BD production by the direct reduction of crotonaldehyde with hydrogen.72 In view of the overall scheme of the reaction mechanism, it was concluded that this hydrogenation process is less likely than the hydrogen transfer from the alcohol to the aldehyde (Scheme 6ii). Natta and Rigamonti74 and later Bhattacharyya and Sanyal,75 based on thermodynamic calculations of the proposed reaction scheme (Scheme 6), have confirmed that the formation of BD proceeded via the selective reduction of crotonaldehyde by ethanol to crotyl alcohol, the direct reduction of crotonaldehyde by hydrogen being thermodynamically less favourable. In the overall reaction network (Scheme 7), crotonaldehyde formation from acetaldehyde, i.e. the aldol condensation step, was indicated as the rate-controlling step.72,75
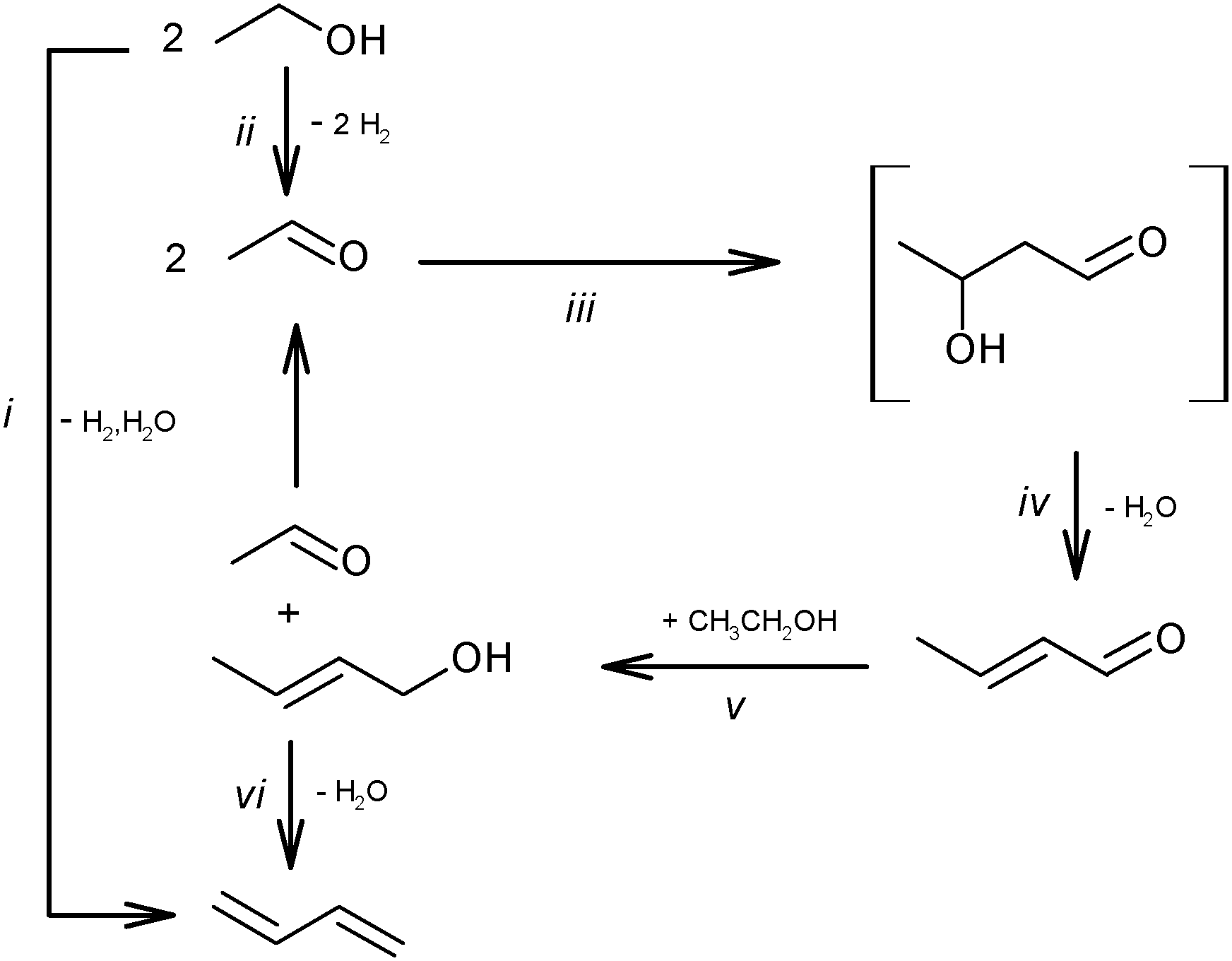 | ||
| Scheme 7 Overall scheme of ethanol conversion to BD. | ||
The reaction network in Scheme 7 was also confirmed by Vinogradova et al. using C14 labelled acetaldehyde.79 They showed the conversion of crotonaldehyde into crotyl alcohol in the presence of an excess of ethanol due to the repartition of hydrogen, which occurs via the formation and decomposition of an intermediate complex between ethanol and the aldehyde.
Later, Niiyama et al. studied the ethanol to BD conversion over MgO–SiO2 catalysts. They assumed the ethanol dehydrogenation as a rate-determining step on the basic catalyst.80 They postulated for the first time that the intermolecular hydrogen transfer between ethanol and crotonaldehyde proceeded via a Meerwein–Pondorf–Verley mechanism (MPV), requiring the participation of acid and basic sites. High BD yields necessitate a balanced ratio between the acidity and basicity.80
Kvisle et al. also using the MgO–SiO2 catalyst81 concluded that the rate-limiting step consists in the condensation of acetaldehyde occurring prior to the hydrogen transfer step, rather than the dehydrogenation of ethanol to acetaldehyde. In the absence of quantitative details, they also emphasized the synergic effect of the catalyst's components for active and selective catalysis.
Separately, the possible formation of 1,3-BDO as an active intermediate was studied in more detail.73,74,82 Based on the transformation of pure butanediol or its mixture with ethanol, it was concluded that the former did not participate in BD formation from ethanol.73,82 The analysis by Natta and Rigamonti supported the thermodynamically impossible formation of 1,3-BDO from ethanol and acetaldehyde.74
Although the route of BD formation (Scheme 7) is generally accepted, alternative schemes were proposed as well. In Scheme 8 it is suggested that BD formation results from an interaction between acetaldehyde and ethylene via the Prins reaction.83,84 Thermodynamically, the Prins mechanism is energetically possible but less favourable than the generally accepted mechanism via aldol condensation.51,74 However, no experimental studies confirming the involvement of the Prins reaction have been performed.
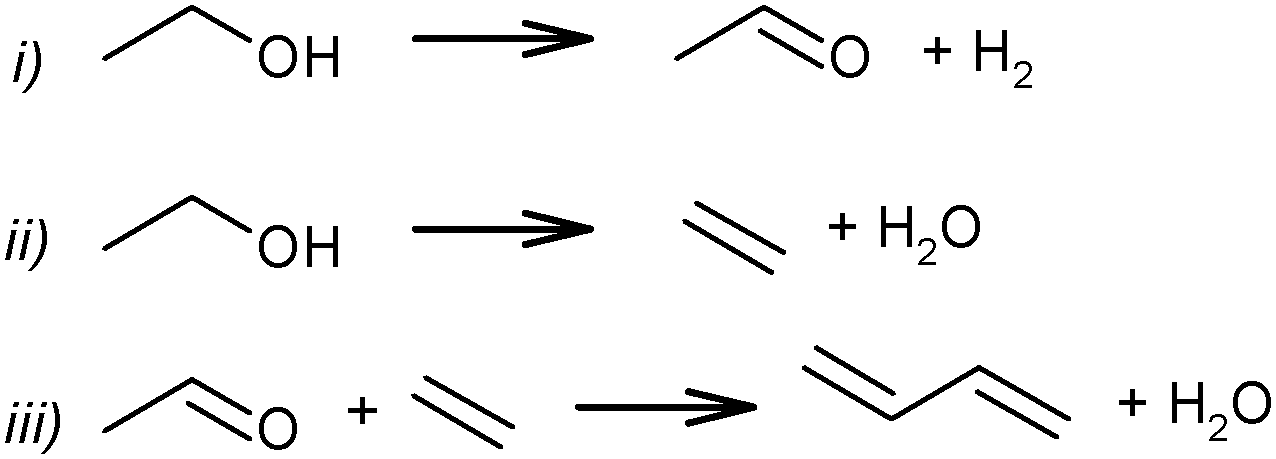 | ||
| Scheme 8 Prins reaction scheme for BD formation from ethanol.83–85 | ||
In the literature, all mechanistic and reaction network studies were based on the experimental results of catalytic tests using various feed combinations, temperatures and contact times. There has been only one attempt studying BD formation from ethanol by IR spectroscopy.86 Ethanol adsorption and its chemical transformation were studied over an alumina–zinc catalyst in the temperature range of 20–400 °C. Above 300 °C a decrease of surface alkoxy-species in the region 1080–1120 cm−1 with increasing reaction temperature was observed at the expense of several new vibration bands. The most intense signal was assigned to the characteristic vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1750 cm−1) and CC–O (1035 and 1680 cm−1), while the origin of other, less intense bands was not specified. However, a conclusive mechanism was not proposed.
O (1750 cm−1) and CC–O (1035 and 1680 cm−1), while the origin of other, less intense bands was not specified. However, a conclusive mechanism was not proposed.
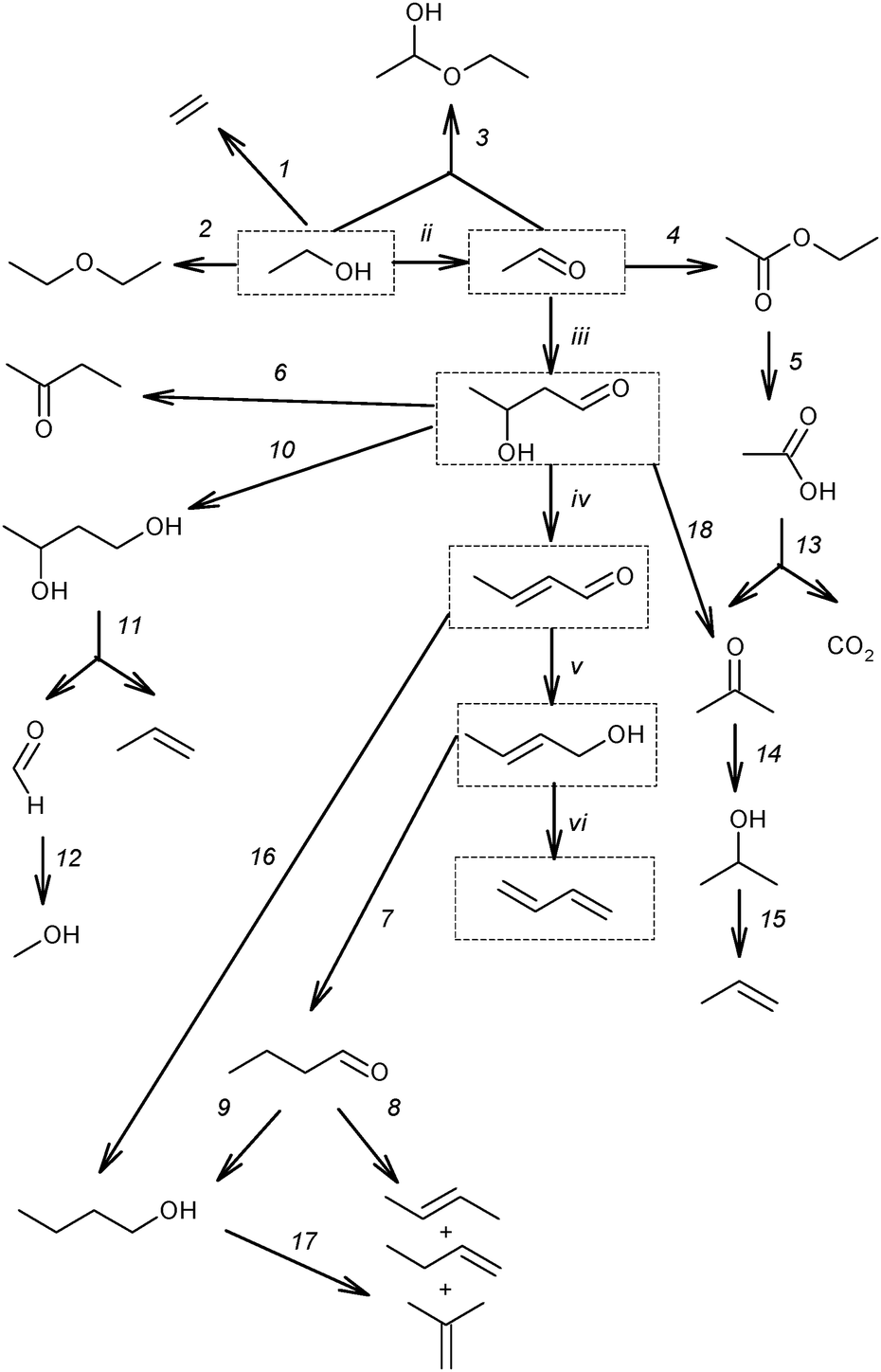 | ||
| Scheme 9 Reaction network of the main by-products.53,59,73,87–89 Reactions (ii)–(vi) and the products (in boxes) represent the generally accepted reaction pathway towards BD. | ||
According to this classification, the first group includes ethylene and diethyl ether, being products of the direct ethanol dehydration (reaction (1) and (2), Scheme 9). Ethyl acetal is a condensation product in the reaction between ethanol and acetaldehyde (reaction (3)). Ethyl acetate is a product of acetaldehyde disproportionation, also known as the Tishchenko reaction (reaction (4), Scheme 9). Finally, acetic acid is a product of ethyl acetate hydrolysis (reaction (5), Scheme 9).73
The second group of by-products consists of crotonaldehyde, a product of acetaldehyde condensation to acetaldol (reactions (iii), Scheme 9) and its subsequent dehydration (reaction (iv)); crotyl alcohol, a product of the reduction of crotonaldehyde (reaction (v)). Methyl ethyl ketone (butan-2-one) could be a product of deoxygenated acetaldol with subsequent rearrangement (reaction (6))73 or it may result from dehydration of 1,3-BDO (see below, Section 3.2), or it could result from crotyl alcohol isomerization.90 Butanal can be an isomerization product of crotyl alcohol (reaction (7)), while butenes and butanol may be produced from butanal via deoxygenation (reaction (8)) and reduction (reaction (9)), respectively.73 1,3-BDO was considered as a product of acetaldol reduction (reaction (10)). Furthermore, C6-hydrocarbons and C6-oxygenated products were formed by the condensation of C4-aldehydes such as butanal and crotonaldehyde with acetaldehyde via similar mechanisms (reaction sequence, ii–vi).73
The presence of molecules containing three and five carbon atoms was rationalized by the cleavage of 1,3-BDO into propylene and formaldehyde (reaction (11), Scheme 9). The occurrence of the last reaction explains the formation of C3-hydrocarbons and C3-oxygenated compounds as a result of an interaction of formaldehyde with ethanol and acetaldehyde, as well as C5-hydrocarbons and C5-oxygenated products as a result of an interaction of formaldehyde with C4-oxygenated compounds.73 Also the reduction of formaldehyde could be a source of methanol (reaction (12)), which further reacts with other alcohols resulting in ether formation and ultimately in aromatics according to the Methanol-to-Gasoline concept.91
An almost similar scheme of by-product formation was proposed by Gorin et al.87,88 Despite the experimental evidence of reaction (11) (Scheme 9),82 an alternative mechanism was proposed to explain the formation of by-products with an odd number of carbon atoms.88 The steps proposed are the ketonization of acetic acid, formed via reaction (5), resulting in acetone with release of CO2 (reaction (13)). Acetone is further reduced to isopropyl alcohol (reaction (14)), which is likely the origin of propylene via dehydration (reaction (15)). Acetone and isopropanol could participate in the formation of the C5-hydrocarbon pool92,93via mechanisms similar to those proposed for BD formation (Scheme 9, reactions (ii)–(vi)).
Alternatively, it has been suggested that n-butanol is formed directly via hydrogenation of crotonaldehyde (reaction (16), Scheme 9)53,59,89 by surface hydrogen atoms resulting from ethanol dehydrogenation (reaction (ii)), whereas the butenes can be obtained by dehydration of n-butanol (reaction (17)).59 Furthermore, it has been also proposed that acetone could be produced from acetaldol (reaction (18)).94–96 Reaction proceeds via formation of surface carboxylate intermediate CH3CH(OH)CH2COO(s) followed by dehydrogenation and decarboxylation.
All compounds identified as by-products in both Lebedev's and Ostromyslensky's processes are summarized in Table 2.
| Number of carbon atoms | Hydrocarbons | Oxygenated compounds |
|---|---|---|
| 1 | Methane |
Carbon monoxide
Carbon dioxide Formaldehyde Methanol |
| 2 |
Ethylene
Ethane |
Acetaldehyde
Acetic acid |
| 3 |
Propylene
Propane |
Methyl ethyl ether
Acetone Propanol and isopropanol |
| 4 |
Butadiene
Butenes: 1-butenes, trans-2-butene, cis-2-butene; iso-butene |
Ethers: ethyl ether, ethyl vinyl ether
Esters: ethyl acetate Aldehydes: butanal, crotonaldehyde Ketones: butanon Alcohols: butanol, crotyl alcohol Ethyl acetal |
| 5 |
Pentenes: 2-pentene, 1-pentene, 2-methyl-2-butene, 2-methyl-1-butene and 3-methyl-1-butene
Pentadienes: 1,3-pentadiene; 2-methyl-1,3-butadiene |
Amyl alcohol |
| 6 |
Hexanes: both branched and strait-chain isomers
Hexadienes: 1,3-cyclohexadiene, 2,4-hexadiene, 3-methyl-1,3-pentadiene and 1,3-hexadiene benzene |
Ethers: butyl acetate
Aldehydes: hexaldehyde and hexenal Alcohols: hexanol; 2-hexen-1-ol |
| 7 | Toluene | |
| 8 | 4-Vinylcyclohexene; p-xylene | n-Octyl alcohol |
2.3. Thermodynamic considerations
The thermodynamics for the conversion of ethanol to BD have been described by several authors.43,51,74,75 The first attempt was performed by Natta and Rigamonti74 in 1947, aiming at unravelling the reaction mechanism. Seven different mechanisms were discussed in this study. Based on the experimental data and on the data for the Gibbs free energy of the different reaction steps in the temperature range 400–430 °C, the authors concluded that the reaction sequence of reactions (ii) to (vi) in Scheme 7 is the most probable pathway to form BD from ethanol. Later, several groups carried out a more comprehensive thermodynamic analysis as shown in Scheme 7.43,51,75An Ellingham-type plot (Fig. 1) displays the change in Gibbs free energy (ΔG, kJ mol−1) between 25 and 530 °C for the different reactions shown in equation (i) to (vi). Calculations were done in Aspen Plus® software for pure components in their real, and not standard states at the given reaction temperature and pressure. The ethanol to BD reaction is endothermic by 102–109 kJ mol−1 from 200 to 500 °C (not shown). The Gibbs free energy change for the direct conversion of ethanol to BD shows that the reaction (i) is favourable above 150 °C. Higher temperature shifts the equilibrium of the endothermic reaction to higher conversions. Since the number of moles of products is higher than that of the reagent, the reaction is pressure dependent, low pressures being favourable for high equilibrium conversion.
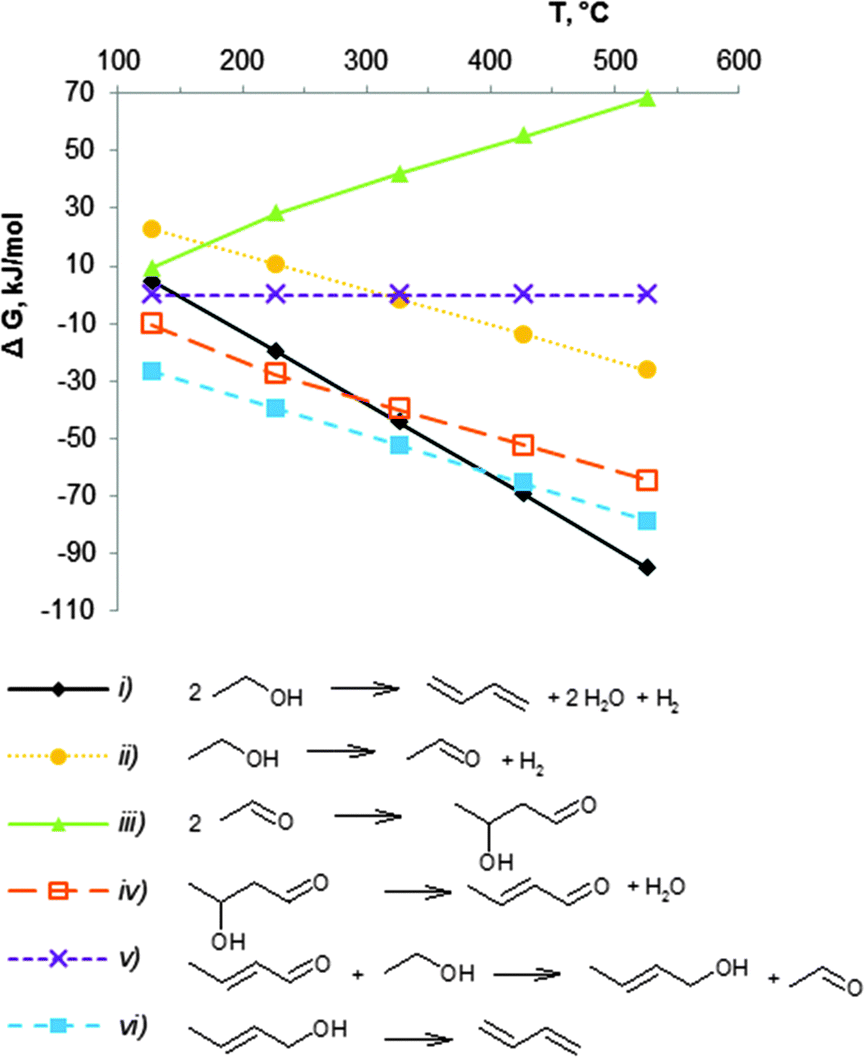 | ||
| Fig. 1 Calculated thermodynamic data for ethanol transformation into BD, ΔG values of the overall reaction (i) and the intermediate steps (ii–vi). The calculations were performed with Aspen Plus® software for mixtures of pure components at given temperature, p = 1 atm (ΔG is calculated in the real state and thus different from ΔG0). | ||
Inspection of the individual reactions shows different thermodynamic behaviour. The condensation of acetaldehyde to crotonaldehyde ((iii) + (iv) in Fig. 1 and Schemes 7, 9) is characterized by very low values of the Gibbs energy (ΔG). The reaction is unfavourable above 200 °C, whereas the formation of acetaldehyde from ethanol (ii) and the dehydration of crotyl alcohol to BD (vi) are favourable above 250–300 °C. Thus, in the usual temperature range applied for BD synthesis, viz. 350 to 430 °C, the equilibrium of reaction (vi) (Fig. 1) is clearly shifted to BD formation. One reaction, viz. the bimolecular MPV ((v) in Fig. 1 and Schemes 7, 9), forming crotyl alcohol (CrOH), shows very low ΔG values over the entire temperature range. However, as long as CrOH is consumed by dehydration in the consecutive reaction to BD (vi), the MPV reaction (v) will be consuming crotonaldehyde (CrH) to form new CrOH. Furthermore, as the equilibrium of reaction (ii) is in favour of acetaldehyde formation, an excess of acetaldehyde in the system also stimulates reaction (iii) + (iv) to continuously produce crotonaldehyde.
It may thus be concluded that the choice of the temperature range for BD formation from ethanol will be defined by kinetic factors, and thus by the ability of the catalyst components to accelerate the individual reactions.43
Fig. 2 shows the equilibrium compositions (mole fractions) in the direct conversion of ethanol to BD. These data were calculated at one atmosphere pressure using the Equilibrium based reactor (REquil) in Aspen Plus® software. The results indicate that an almost complete EtOH conversion could be obtained above 227 °C (Fig. 2a). A very similar result is obtained when the thermodynamic analysis was approached via the reaction sequence (ii) to (vi) in Schemes 7 and 9 (Fig. 2b). Only a slight decrease in BD concentration was found for temperatures higher than 340 °C due to unconverted acetaldehyde and crotonaldehyde (Fig. 2b).
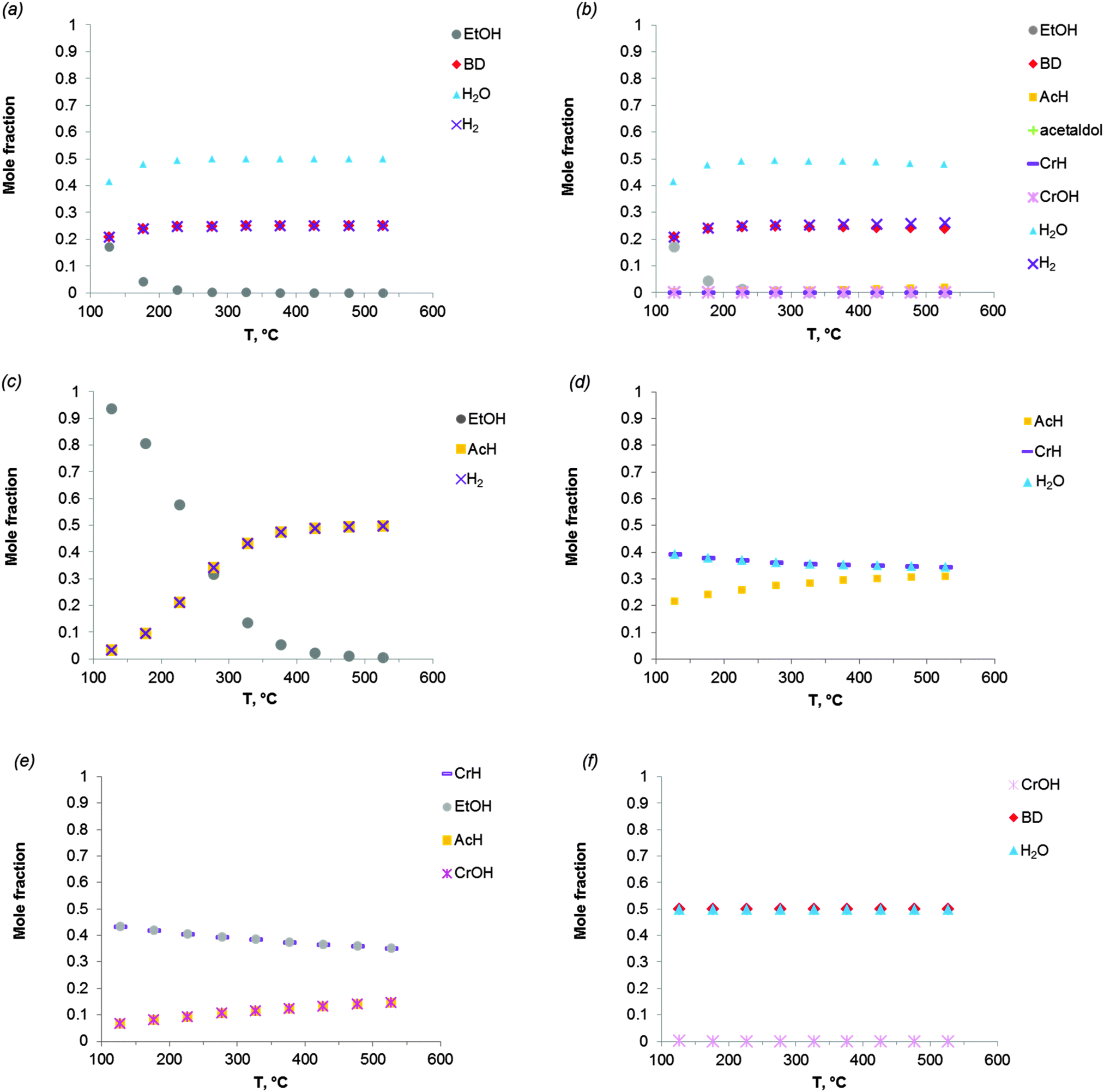 | ||
Fig. 2 Equilibrium compositions calculated for: (a) the overall reaction of ethanol to BD, reaction (i); (b) the reaction of ethanol to BD taking into account the reaction sequence, reactions (ii) to (vi); (c) ethanol dehydrogenation to acetaldehyde, reaction (ii); (d) aldol condensation of acetaldehyde to crotonaldehyde, reaction (iii) + (iv); (e) MPV reaction of ethanol with crotonaldehyde to synthesize crotyl alcohol, reaction (v), with a mixture of ethanol and crotonaldehyde (molar ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); (f) dehydration of crotyl alcohol to BD, reaction (vi). The pressure in the calculations is assumed 1 atmosphere. The simulation is performed using Equilibrium based reactor (REquil) in Aspen Plus® software. The reactions (i) to (vi) are in line with the reactions in Fig. 1 and in Scheme 7. (CrOH = crotyl alcohol; CrH = crotonaldehyde; AcH = acetaldehyde; BD = 1,3-butadiene). :1); (f) dehydration of crotyl alcohol to BD, reaction (vi). The pressure in the calculations is assumed 1 atmosphere. The simulation is performed using Equilibrium based reactor (REquil) in Aspen Plus® software. The reactions (i) to (vi) are in line with the reactions in Fig. 1 and in Scheme 7. (CrOH = crotyl alcohol; CrH = crotonaldehyde; AcH = acetaldehyde; BD = 1,3-butadiene). | ||
The different steps of the reaction network (Schemes 7 and 9) were also individually analysed with respect to the equilibrium compositions and conversion. Ethanol dehydrogenation is thermodynamically favored by an increase in temperature,43,51,74,75 a temperature higher than 430 °C being required to obtain full ethanol conversion (Fig. 2c). In the commercially applied temperature range of 325 to 430 °C, a 76 to 96% (on carbon basis) thermodynamic yield of acetaldehyde (AcH) can be reached.
Further transformation of acetaldehyde into crotonaldehyde proceeds via acetaldol (Scheme 7(iii)). The aldol reaction is thermodynamically unfavourable (Fig. 1),51,75 but the subsequent aldol dehydration into crotonaldehyde (CrH) (iv) is highly favorable (Fig. 1). The thermodynamics show full conversion of the aldol over the entire temperature domain. The overall transformation of acetaldehyde into CrH (reactions (iii) + (iv)) inhibited by an increase in temperature shows low ΔG values over the entire temperature domain. Consequently, the equilibrium conversion of acetaldehyde is 72% at 330 °C (on carbon basis) and decreases with increasing temperature (Fig. 2d). Subtle acid–base catalysis is required to assist this condensation reaction.
The subsequent step with crotonaldehyde is the MPV reaction (v), which is also characterized by very low ΔG values.43,51,74 The conversion of crotonaldehyde (CrH) into crotyl alcohol (CrOH) is 23–27% (from 325 to 430 °C, calculated on CrH basis) in the case of an equimolar substrate feed (Fig. 2e). The equilibrium conversion could be increased by increasing the molar ratio of EtOH to CrH, with a value of 77%, when the molar ratio of EtOH to CrH is 20. CrOH formed as a product of the MPV reaction is then dehydrated to BD (Fig. 2f). This reaction is thermodynamically favorable irrespective of the temperature applied.
The thermodynamics were also analysed taking into account the presence of side-products (Table 2). The effect of ethylene and diethyl ether (DEE) on the thermodynamics was specified first. Fig. 3a shows the equilibrium composition of the reaction mixture in the case where ethanol dehydration occurs together with ethanol-to-BD. Ethylene is clearly the most thermodynamically stable product, whereas diethyl ether is formed at temperatures below 150 °C. The BD yield is considerably lower when ethanol dehydration is included in the thermodynamic analysis (compare Fig. 3a with Fig. 2a and b). When the temperature increases from 325 to 430 °C, in the presence of ethylene in the product mixture, the thermodynamic yield of BD is reduced to 24–27% (on carbon basis). When the reaction temperature is increased to 530 °C, the BD yield only slightly increases (up to 30%). Thus dehydration of ethanol to ethylene should be avoided kinetically to attain high BD yields. As the reaction is catalysed by the acidity of the catalyst or the support, the choice of the appropriate catalyst plays a significant role in the BD selectivity of the overall ethanol-to-BD process.
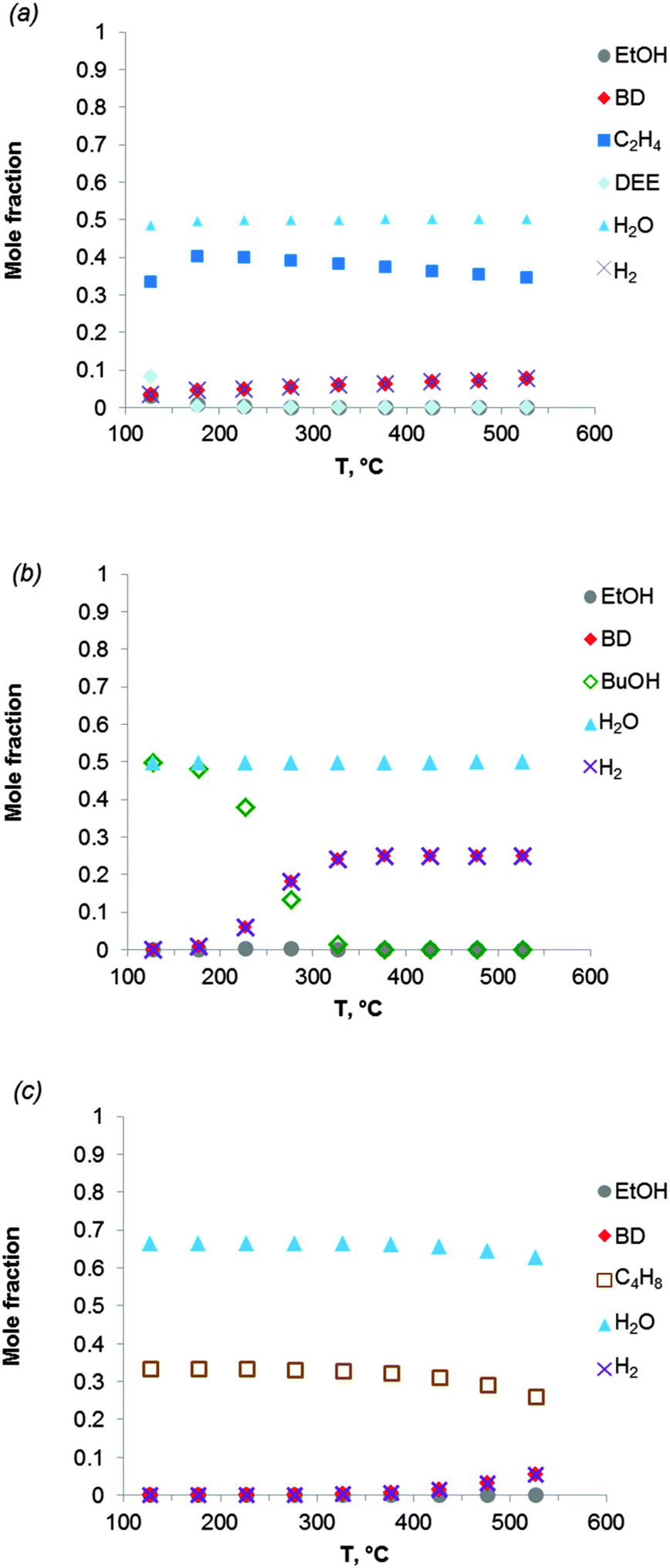 | ||
| Fig. 3 Equilibrium composition calculated for the conversion of ethanol to BD in case following by-products are allowed: (a) diethyl ether and ethylene; (b) 1-butanol; (c) butenes (1-butene, cis-2-butene, trans-2-butene). The pressure in the system is 1 atmosphere. Simulation is performed using the Equilibrium based reactor (REquil) in Aspen Plus® software. (BuOH = 1-butanol; DEE = diethyl ether). | ||
For reaction temperatures ranging from 350 to 430 °C, co-production of 1-BuOH in the ethanol-to-BD reaction is of little importance, BD being formed preferentially according to the thermodynamics (Fig. 3b). Conversely, all isomers of butenes thermodynamically more favorable than BD (Fig. 3c) may be formed by acid catalysed dehydration of butanols. Thus, optimization of catalyst properties is of key importance to obtain high BD yields.
Thermodynamic simulation was also carried out for the two-step Ostromyslensky process, co-feeding a mixture of ethanol and acetaldehyde to the catalyst (Fig. 4). Butadiene is the most favorable product in the reaction mixture. It seems that there is no thermodynamic advantage when performing ethanol-to-BD in two steps. The increase in BD yield and selectivity of the two-stage process can thus only be explained by kinetic factors, related to the properties of the catalyst.
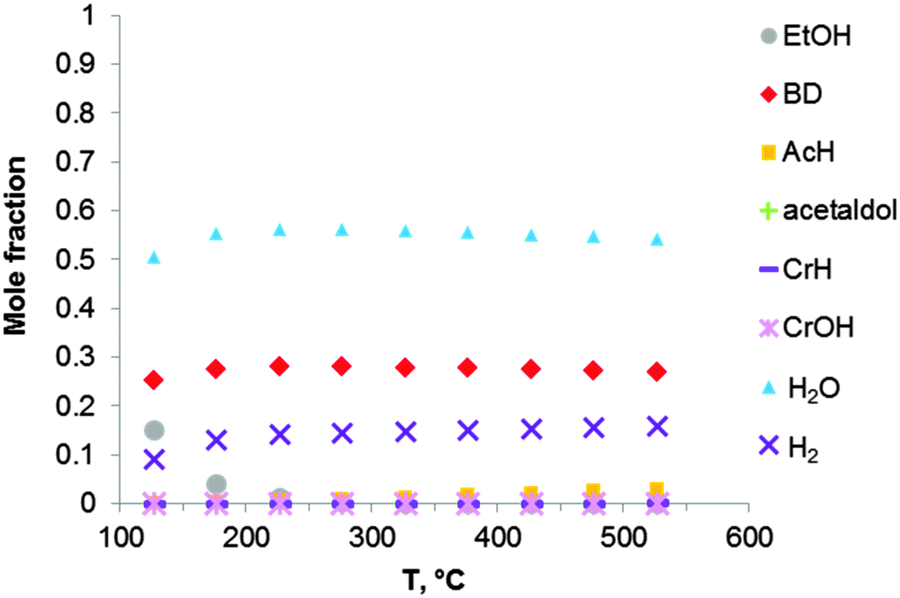 | ||
| Fig. 4 Equilibrium compositions calculated for the synthesis of BD from an ethanol–acetaldehyde mixture with a molar ratio of EtOH to AcH of 3, via the reaction sequence (ii) to (vi) of Fig. 1 and Scheme 7. The pressure in the system is 1 atmosphere. The simulation is performed using the Equilibrium based reactor (REquil) in Aspen Plus® software. | ||
Summarizing the thermodynamic aspects, reaction temperatures between 325 and 430 °C are preferred, since lower temperatures lead to restricted acetaldehyde formation, whereas higher temperatures disfavor the aldol condensation. In the preferred temperature range, BD yield and selectivity are kinetically determined, the choice of the active catalyst being of utmost importance.
2.4 Catalyst development and reaction conditions
The preceding analysis does not include the choice of the catalyst. The sequence of reactions (ii) to (vi) (Schemes 7 and 9) converting ethanol to BD is favoured at high temperature, most side-reactions (hydrogenation and dehydration) being more affected. Therefore the catalyst must be particularly active in reactions like dehydrogenation, aldol condensation and MPV, while avoiding side-reactions such as ethylene and butene formation.The data in the literature on agents capable of activating the ethanol-to-BD conversion are abundant. They will be summarized for both the one-step and two-step processes in the next section. Further classification is based on the support type and the increasing number of modifiers in the catalyst composition. Where possible, focus will be on structure–activity/selectivity relationships.
In the first patents, Lebedev et al.66,67 have been referring to a mixture of zinc oxide and alumina, yielding BD with a selectivity of 18 wt%. Other compositions containing uranium oxide98 and mixtures of aluminium hydrosilicate98,99 or floridin (Fuller's earth)98,100 with zinc or manganese oxide were suggested as well. The dehydrogenation and dehydration components should be present in an optimal ratio, viz. 25% dehydration and 75% dehydrogenation component, in order to attain a high BD yield.68 However, this optimal ratio strongly depending on the catalyst nature has been the subject of many discussions in the more recent literature. Unfortunately, the elemental composition of the preferred catalyst, showing up to 42 wt% BD selectivity (or close to 70% on carbon basis), remained undisclosed.68,69 The composition of a Lebedev industrial catalyst became available in 1941 after occupation of a factory for synthetic rubber production in the Russian city of Yefremov by the German army.43,74 Chemical analysis revealed the presence of 44.6 wt% of magnesia and 10 wt% of silica, next to a large number of small amounts of different elements.43,74 XRD analysis has shown the presence of magnesium oxide, silica and kaolin. Later on, in the literature an abundant number of catalyst combinations has been described and tested. Note that the definition of a “Lebedev catalyst” is ambiguous as in the literature two compositions, viz. ZnO–Al2O3 and MgO–SiO2, have often been used by different authors as a reference to Lebedev's catalyst. The present review categorises the catalysts into three groups, encompassing (i) doped alumina catalysts (Table 3 and Fig. 5), (ii) pure and promoted magnesia–silica containing catalysts (see Table 4 and 5; Fig. 6–11), and (iii) other catalysts (Table 6). Comparison between the catalysts is made based on BD selectivity and yield, total carbon conversion in the product stream (TC, %), activity (gBD gcat−1 h−1) and volume productivity (gBD lcat−1 h−1).
| Entry | Catalyst | T/°C | WHSV/h−1 | LHSV/h−1 | TCa/% | Yields/C% | Selectivity/C% | Activity/gBD gcat−1 h−1 | Volume productivity/gBD lcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a TC = Total conversion of ethanol. | ||||||||||
| 1 | ZnO–Al2O3 | 400 | — | — | — | — | 31 | — | — | 67 |
| 2 | 40%ZnO–60%Al2O3 | 360–380 | — | 0.3 | — | 5.6 | — | — | 8 | 74 |
| 3 | 25%ZnO–75%Al2O3 | 425 | 0.25 | 0.4 | 67 | 10 | 15 | 0.01 | 17 | 101 |
| 4 | 75%ZnO–25%Al2O3 | 445 | — | 0.3 | 75 | 15 | 20 | — | 23 | 102 |
| 5 | 80%MgO–20%Al2O3 | 430 | — | 0.3 | 95 | 10 | 11 | — | 12 | 74 |
| 6 | 2%PbO–98%Al2O3 | 425 | 0.6 | 0.6 | — | 18 | — | 0.06 | 46 | 101 |
| 7 | 1%TiO2–9% ZrO2–90%Al2O3 | 425 | 0.5 | 0.6 | — | 17 | — | 0.05 | 43 | 101 |
| 8 | 30%Sb2O3–70%Al2O3 | 425 | 0.9 | 0.6 | — | 14 | — | 0.07 | 36 | 101 |
| 9 | 10%Cr2O3–90%Al2O3 | 425 | 0.5 | 0.6 | — | 8 | — | 0.02 | 20 | 101 |
| 10 | 70% MgO–30%Al2O3 | 450 | 2.0 | — | 86 | 18 | 21 | 0.20 | — | 59 |
| 11 | 40%ZnO–60%Al2O3 | 425 | 1.5 | 1.6 | 94 | 56 | 59 | 0.50 | 421 | 104 |
| 12 | 20%MgO–80%Al2O3 | 425 | 1.5 | 1.5 | — | 48 | — | 0.40 | 322 | 104 |
| 13 | 40%Cr2O3–60%Al2O3 | 450 | 1.5 | 1.6 | — | 47 | — | 0.40 | 365 | 104 |
| 14 | 30%ZnO–1%K2O–4%SiO2–4%MgO–Al2O3 | 420 | — | 2.5 | 43 | 34 | 80 | — | 395 | 105 |
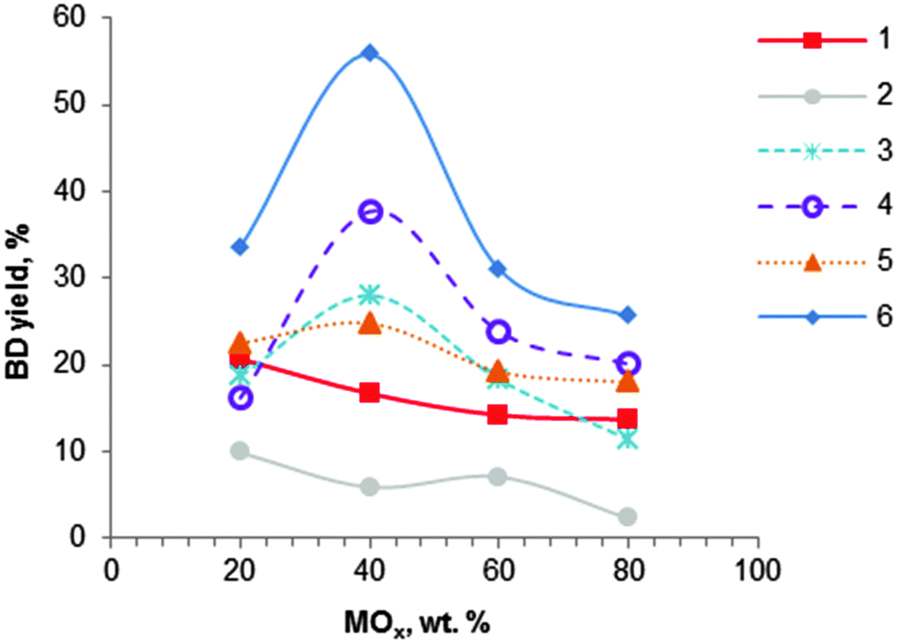 | ||
| Fig. 5 Effect of MOx–Al2O3 catalyst composition on butadiene yield: (1) MgO–Al2O3 (400 °C); (2) SiO2–Al2O3 (425 °C); (3) ZrO2–Al2O3 (425 °C); (4) Fe2O3–Al2O3 (425 °C); (5) Cr2O3–Al2O3 (425 °C); (6) ZnO–Al2O3 (425 °C), WHSV ∼ 1.5 h−1.104 | ||
| Entry | Mg/Si | Preparation | T/°C | WHSV/h−1 | LHSV/h−1 | TCa/% | Yields/C% | Selectivity/C% | Activity/gBD gcat−1 h−1 | Volume productivity/gBD lcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a TC = Total conversion of ethanol. | |||||||||||
| 1 | 2.2 | Wet-kneading, MgO + SiO2 | 350–400 | — | 0.3 | — | — | 30–40 | — | — | 109–111 |
| 2 | 2.3 | Impregnation of SiO2 hydrogel by Mg-salt | 410 | 0.5 | 0.4 | 25 | 9 | 36 | 0.02 | 15 | 101 |
| 3 | 2.2 | Wet-kneading, MgO + SiO2 | 430 | — | 0.3 | 84 | 35 | 41 | — | 48 | 112 |
| 4 | 2.2 | Wet-kneading, MgO + SiO2 with NH4OH | 415 | — | 0.3 | 55 | 27 | 50 | — | 37 | 112 |
| 5 | 2.2 | Wet-kneading, MgO + SiO2 with H3COOH | 415 | — | 0.3 | 34 | 18 | 54 | — | 25 | 112 |
| 6 | 5 | Mg(OH)2 + SiO2 | 400 | — | 0.3 | 52 | 28 | 54 | — | 33 | 113 |
| 7 | 5 | Same as entry 6, after hydrothermal treatment | 400 | — | 0.3 | 64 | 37 | 58 | — | 44 | 113 |
| 8 | 2 | Commercial | 440 | 0.30 | 0.2 | 70 | 37 | 48 | 0.06 | 31 | 114 |
| 9 | 3 | Mg(OH)2 + colloidal SiO2 | 380 | 0.40 | — | 46 | 28 | 62 | 0.06 | — | 80 |
| 10 | 1 | Wet-kneading, Mg(OH)2 + SiO2 | 350 | 0.15 | — | 50 | 42 | 84 | 0.04 | — | 115 |
| 11 | 0.83 | Wet-kneading, Mg(OH)2 + SiO2 | 350 | 0.03 | — | 53 | 16 | 30 | 0.003 | — | 81 |
| 12 | 1 | Wet-kneading, Mg(OH)2 + SiO2 | 350 | 0.20 | — | 76 | 15 | 20 | 0.02 | — | 116 |
| 13 | 6 | Wet mixing MgO + SiO2 | 400 | — | — | 66 | 32 | 48 | — | — | 117 |
| Entry | Catalyst composition | T/°C | WHSV/h−1 | LHSV/h−1 | TCa/% | Yield/C% | Selectivity/C% | Activity/g gcat−1 h−1 | Volume productivity/g lcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a TC = Total conversion of ethanol. | ||||||||||
| 1 | 2%Cr2O3–59%MgO–39%SiO2 | 415 | — | 0.2 | 82 | 43 | 52 | — | 42 | 112 |
| 2 | 2%Cr2O3–59%MgO–39%SiO2 | 400 | 0.4 | 0.4 | 68 | 38 | 56 | 0.08 | 65 | 101 |
| 3 | 2%Cr2O3–79%MgO–19%SiO2 | 435 | — | 0.3 | 75 | 40 | 52 | — | 58 | 102 |
| 4 | 2%Cr2O3–79%MgO–19%SiO2 | 400 | — | 0.3 | 60 | 37 | 62 | — | 44 | 118 |
| 5 | 3%Cr2O3–54%MgO–43%SiO2 | 350 | 0.2 | — | 65 | 34 | 53 | 0.04 | 23 | 116 |
| 6 | 2%Cr2O3–76%MgO–11%SiO2–11% kaolin | 435 | — | 0.3 | 76 | 48 | 63 | — | 82 | 102 |
| 7 | 2%Cr2O3–76%MgO–11%SiO2–11% kaolin | 400 | — | 0.3 | 75 | 47 | 63 | — | 56 | 118 |
| 8 | 30%ZnO–30%MgO–40%SiO2 | 365 | — | 0.2 | 95 | 26 | 27 | — | 25 | 74 |
| 9 | ZnO–MgO–SiO2–kaolin | <410 | — | 0.3 | 67 | 46 | 69 | — | 64 | 121 |
| 10 | 3%ZnO–56%MgO–42%SiO2 | 400 | 0.7 | — | 98 | 45 | 46 | 0.19 | 117 | 116 |
| 11 | 2%Ta2O5–60%MgO–38%SiO2 | 420 | 0.4 | 0.4 | 68 | 34 | 50 | 0.08 | 58 | 101 |
| 12 | 10%NiO–28%MgO–62%SiO2 | 280 | — | — | 59 | 53 | 90 | — | — | 122 |
| 13 | 3%CuO–56%MgO–42%SiO2 | 400 | 0.7 | — | 86 | 44 | 53 | 0.20 | 123 | 116 |
| 14 | 4%Ag–55%MgO–41%SiO2 | 400 | 0.7 | — | 92 | 49 | 54 | 0.22 | 130 | 116 |
| 15 | MgO/bentonite | 440 | — | 0.4 | — | — | 31 | — | — | 123 |
| 16 | Li+-containing fluorohectorite | 375 | 0.3 | — | 32 | 16 | 48 | 0.03 | — | 124 |
| 17 | 6%Ag/Al–sepiolite | 280 | 0.8 | — | 63 | 5.5 | 9 | 0.03 | — | 83 |
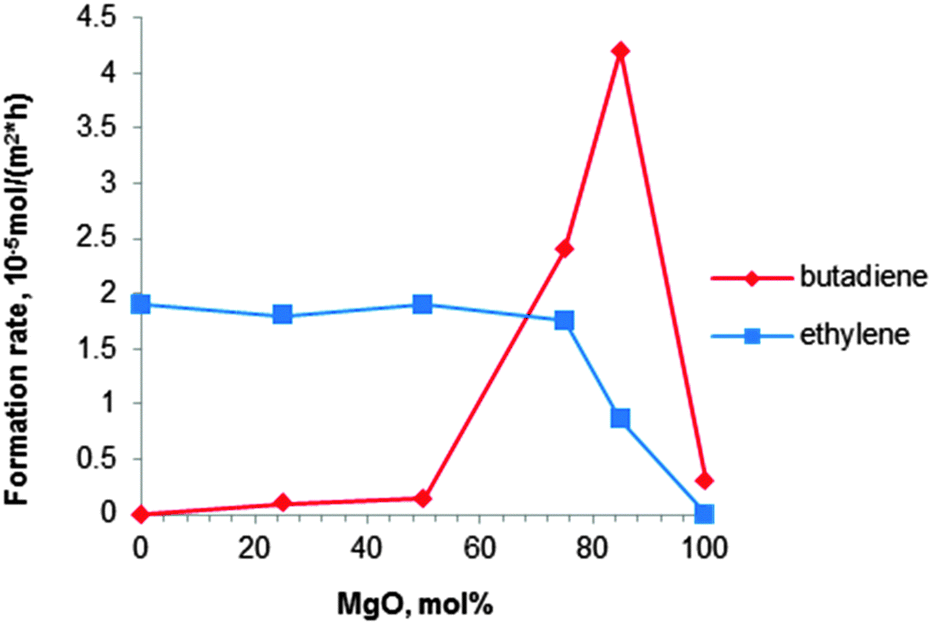 | ||
| Fig. 6 Formation rates of BD and ethylene at 380 °C with a MgO content of MgO–SiO2 catalysts (ethanol concentration of 10 vol%).80 | ||
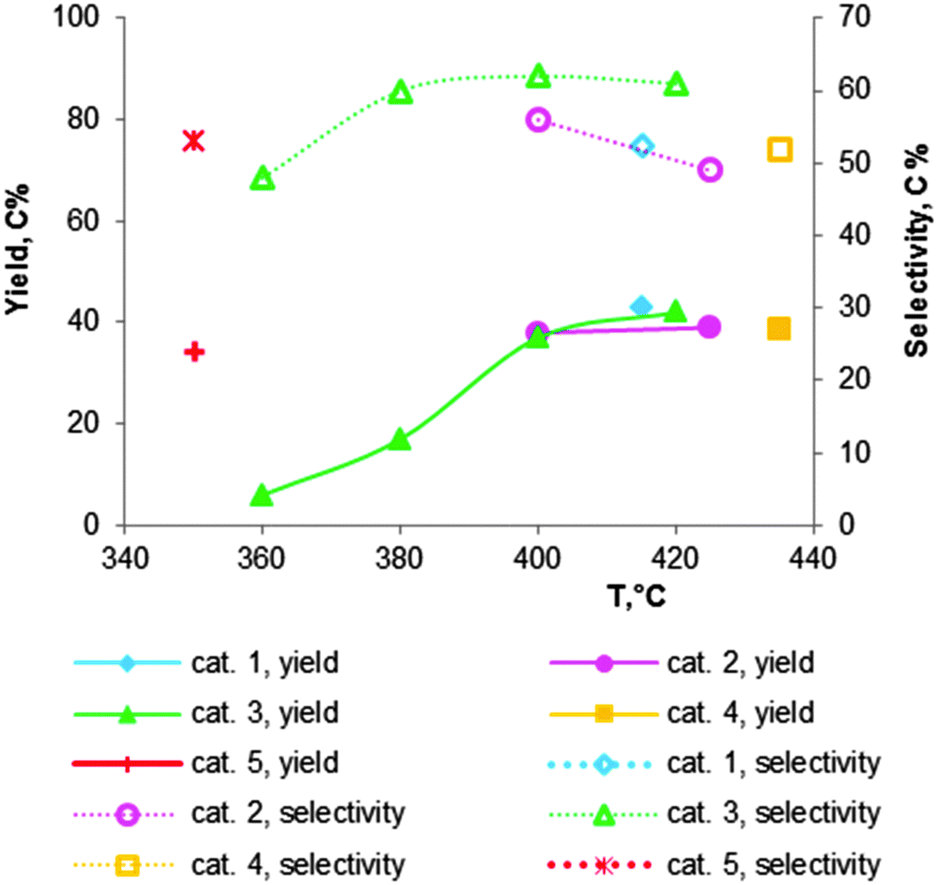 | ||
| Fig. 7 Catalytic performance of chromium oxide doped MgO–SiO2 materials, containing 2 wt% of Cr2O3: cat. 1 – Mg:Si = 2.2;112 cat. 2 – Mg:Si = 2.3;101 cat. 3 – Mg:Si = 6;118 cat. 4 – Mg:Si = 6;102 cat. 5 – Mg:Si = 2116 (see Table 5 for the reaction conditions). | ||
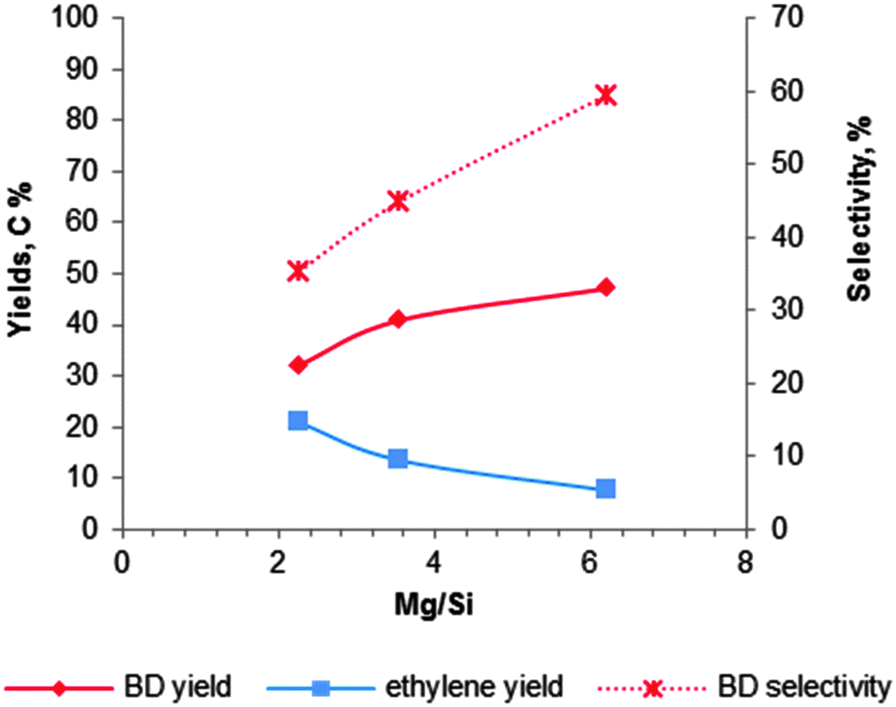 | ||
| Fig. 8 Influence of the magnesium content on the activity and BD selectivity of 2%Cr2O3–MgO–SiO2 at 435 °C and a WHSV of 0.3 h−1.102 | ||
 | ||
| Fig. 9 Influence of the chromium loading on the BD selectivity for Cr2O3–MgO–SiO2 materials with an atomic Mg:Si ratio of 3, carried out at 400 °C.119 | ||
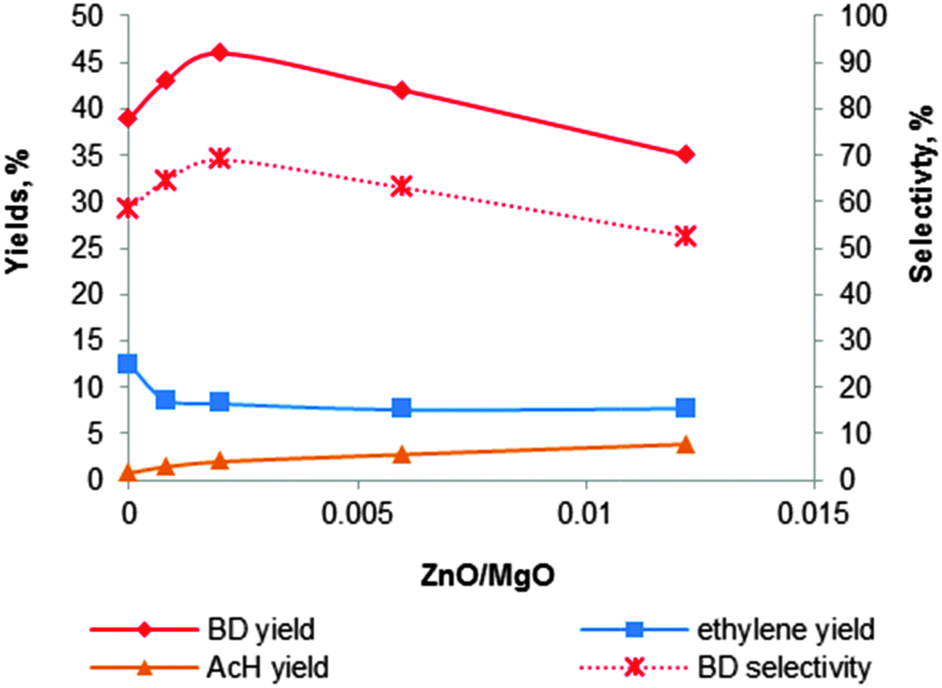 | ||
| Fig. 10 Effect of the content of zinc promoter on the activity and selectivity of the MgO–SiO2 catalyst at a total ethanol conversion of 67% with a WHSV of 0.3 h−1.121 | ||
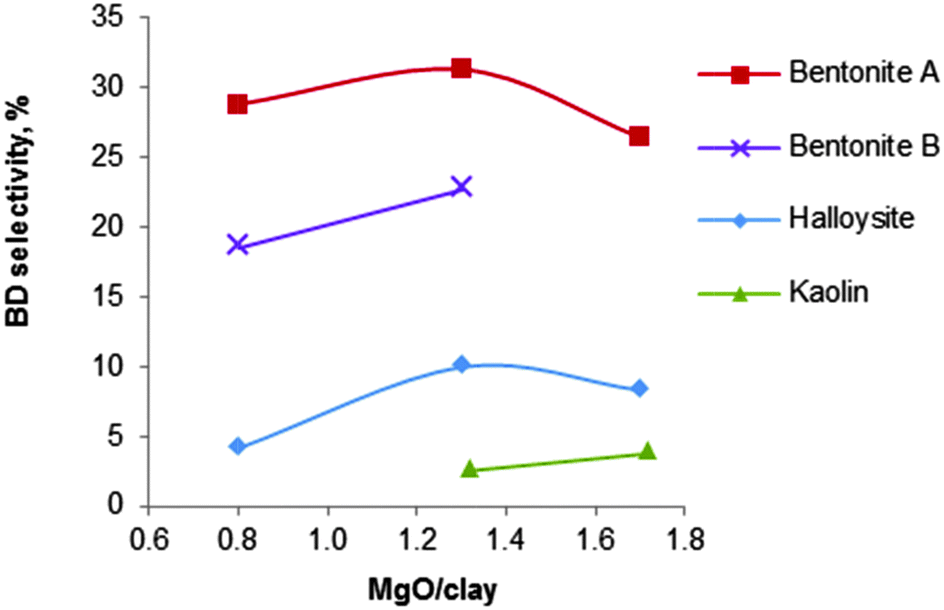 | ||
| Fig. 11 Influence of the nature of various clay minerals on the selectivity of the MgO/silicate catalysts (T = 440 °C, LHSV = 0.4 h−1).123 | ||
| Entry | Catalyst composition | T/°C | WHSV/h−1 | LHSV/h−1 | TCa/% | Yield/C% | Selectivity/C% | Activity/g gcat−1 h−1 | Volume productivity/g lcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a TC = Total conversion of ethanol. b Yield and selectivity are calculated in mol%. | ||||||||||
| 1 | 20%ZnO/diatomaseous earth | 450 | — | 1 | 50 | 29 | 58 | — | 127 | 138 |
| 2 | ZnO + Zn–phosphate/diatomaseous earth | 415 | — | 1.3 | 35 | 22 | 63 | — | 123 | 138 |
| 3 | 10%ZnO–90%SiO2 | 425 | — | 0.6 | — | 20 | — | 0.13 | 51 | 101 |
| 4 | 21% ZrO2/diatomaseous earth | 445 | — | — | — | — | 44 | — | — | 127,139 |
| 5 | 21% ThO2/diatomaseous earth | 445 | — | — | — | — | 38 | — | — | 127,139 |
| 6 | 9,5%ZrO2–90,5%SiO2 | 425 | 1.0 | 0.6 | — | 23 | — | 0.13 | 59 | 101 |
| 7 | ZrO2/ZnO/SiO2–60Å | 375 | — | 1.5 | 46 | 18b | 39b | — | — | 140 |
| 8 | ZrO2/ZnO/SiO2–150Å | 375 | — | 1.5 | 48 | 23b | 48b | — | — | 140 |
| 9 | CuO/ZrO2/ZnO/SiO2–60Å | 375 | — | 1.5 | 39 | 19 | 50b | — | — | 140 |
| 10 | Ag/ZrO2/SiO2 | 325 | 0.3 | — | 34 | 24 | 72 | 0.04 | — | 141 |
| 11 | 2%Ta2O5–98%SiO2 | 425 | — | 0.6 | — | 16 | — | — | 41 | 101 |
| 12 | 1.1% CuO on 2%Ta2O5–98%SiO2 | 425 | 0.5 | 0.4 | 83 | 25 | 30 | 0.06 | 43 | 101 |
| 13 | ZrO2–Fe2O3 (40:60) |
425 | 1.5 | — | — | 40 | — | 0.34 | — | 104 |
Doped alumina catalysts. Early publications by Lebedev have claimed a BD selectivity of 31% with a ZnO–Al2O3 catalyst (Table 3, entry 1).67 Several authors failed to reproduce this BD selectivity. Natta and Rigamonti reported a BD yield of 5.6% with 40%ZnO–60%Al2O3, although a high selectivity to ethylene was observed (Table 3, entry 2).74 Reduction of the dehydration activity of the alumina by treatment with sodium carbonate increased the BD yield to 9%. Variations of Zn and Al ratios were investigated. The catalyst 25%ZnO–75%Al2O3 showed a BD yield of 10% (Table 3, entry 3),101 while with a 75%ZnO–25%Al2O3 catalyst a BD yield of 15% with a selectivity of 20% has been reported (Table 3, entry 4).102 Despite the improvements, the selectivity was lower than commercially acceptable levels. Many authors therefore considered the unpromoted ZnO–Al2O3 composition as “not very promising”.
Natta and Rigamonti reported enhanced catalytic results with MgO–Al2O3 catalysts rich in magnesia, yielding 10% BD with a selectivity of 11% (Table 3, entry 5).74 This result was explained by the higher condensation activity of magnesia, compared to zinc oxide. Corson et al.101 have varied the MgO:Al2O3 ratios between 0.3 and 2.2 by co-precipitation of magnesium and aluminium salts. However, such mixed oxides showed poor activity, yielding less than 7% BD. Among other alumina containing catalysts, the following oxide systems were found to be more active producing BD in yields higher than 8%: PbO–Al2O3, TiO2–ZrO2–Al2O3, Sb2O3–Al2O3 and also Cr2O3–Al2O3 (Table 3, entries 6–9). Conversely, SiO2–Al2O3, ZrO2–Al2O3, Fe2O3–Al2O3 and NiO–Al2O3 yielded less than 7% BD. Thus, none of the alumina-based materials were considered as a promising catalyst.101
Hydrotalcite-derived mixed oxides are often used as basic catalysts, especially when a high dispersion of the elements in the composition is needed.103 León et al.59 using a MgAl hydrotalcite precursor with a Mg:Al atomic ratio of 3:1 obtained at 450 °C using a WHSV of 2 h−1 a moderately high BD selectivity of about 21% at an ethanol conversion of 86%. Tuning the acid–base properties of the catalyst by changing the synthesis procedure by precipitation of hydrotalcites under low or high supersaturation conditions or under sonication, over catalysts with low acidity and basicity an accumulation of acetaldehyde was found, besides a low activity to ethylene and C4 side-products. Conversely, catalysts with moderate basicity and high acidity showed next to superior BD yields also high amounts of other thermodynamically stable dehydration products like ethylene and butenes (Section 2.3). Substitution of Fe for Al in the hydrotalcite precursor, adjusting the acidity without affecting the MgO-related basicity, yielded less acidic catalysts showing at the same conversion high 1-butanol yields amounting to 3 and 11% with Mg–Fe and Mg–Al catalysts, respectively, as well as decreasing BD yield. Clearly, tuning of the acid–base properties of the catalysts both in strength and number seems to be the preferred way to attain high BD yields.
Extensive work on alumina-based catalysts containing mono-, binary and ternary oxides has been performed by Bhattacharyya et al.75,104,106–108 None of the studied ternary oxide-based catalysts gave better results than the binary oxides. Among the binary oxides, mixtures of alumina with magnesia, zirconia, chromium oxide, calcium oxide, manganese oxide, silica, iron oxide, zinc oxide and nickel oxide were investigated (Fig. 5).104 Most binary systems showed an optimum composition corresponding to a 40:60 weight ratio for MOx:Al2O3: for MgO–Al2O3 and SiO2–Al2O3 materials, catalysts containing 80% of alumina showed enhanced catalytic performance (Fig. 5 and Table 3, entries 11–13). The best catalytic results were found with ZnO-containing alumina, yielding at 425 °C and atmospheric pressure 56% BD with a selectivity of 59% to BD, corresponding to a volume productivity of 421 gBD L h−1 (entry 11). The catalyst showed a very high activity of 500 gBD kgcat h−1. Iron oxide on alumina was the second best catalyst (Fig. 5). In addition, the materials containing nickel oxide and manganese oxide (not shown) were found to be active only in the dehydrogenation of ethanol, irrespective of the catalyst preparation method and the composition.
Binary oxide systems prepared by co-precipitation of the corresponding salts with aqueous ammonia showed higher activity relative to mechanically blended samples and/or individual oxides, likely due to synergetic effects of the various active sites.104 Unfortunately, no physicochemical information of the different catalytic systems was available proving this statement. Furthermore, metal nitrate precursors are preferred over chlorides and sulphates, because the anion could be completely removed by washing and calcination. In addition, substitution of aqueous ammonia as a precipitating agent by sodium or potassium hydroxides resulted in adverse effects.107 Optimal contact times attaining maximum BD yields were reported for the binary systems. A WHSV of 1.5 h−1 showed the highest BD yields for most catalysts, except for the SiO2–Al2O3 catalyst which showed the highest BD yields at a WHSV of 1 h−1. Note that many authors were using much higher contact times.74,101,102 Bhattacharyya et al. have clearly demonstrated the remarkable importance of an optimal WHSV. In the case of ZnO–Al2O3, BD yield dropped from 56 to 34% with a WHSV decreasing from 1.5 to 1 h−1, or to 12% with an increase of WHSV from 1.5 h−1 to 2 h−1.104 It is worth mentioning that high BD selectivity and yields at a high EtOH feed rate is beneficial because high space-time yields can be reached (Table 3, compare entries 11–13 with entries 2–9). With regard to the temperature, the highest BD yield was observed at 425 °C for most binary systems. Over the ZnO–Al2O3 catalyst a decrease of the temperature from 425 to 375 °C showed a BD yield decrease from 56 to 14%.104 Furthermore, it has been shown that use of a fluidized bed reactor allowed us to significantly increase BD yield up to 73% compared to 56% obtained with a fixed bed reactor.108
Tret'yakov et al. reported a similar zinc–alumina catalyst promoted with a small percentage of potassium oxide, silica and magnesia, viz. 30%ZnO–1%K2O–4%SiO2–4%MgO–Al2O3.105 They reported 34% BD yield with 80% selectivity at 420 °C and low contact time, e.g. LHSV of 2.5 h−1 (Table 3, entry 14). Catalysts were synthesised by mixing of zinc, aluminium and potassium oxides with suspension of magnesia and silica. The catalyst composition contained all the necessary active elements to catalyse the sequence of transformations such as dehydrogenation (reaction (ii), Scheme 7) with Zn, the base-catalysed condensation (iii and iv) with MgO, the MPV reaction (v) with Zn or MgO, and the dehydration (vi) with SiO2. The dehydration of ethanol to ethylene is avoided possibly due to poisoning of the most acidic sites by K2O. Addition of hydrogen peroxide (0.8–1.5 wt%) in the feed significantly improved the catalytic results. A BD yield of 48% was found over the same catalyst under similar conditions showing 90% selectivity for BD, corresponding to a productivity of 550 gBD lcat−1 h−1. The promoting effect of peroxide was explained, on one hand, by hydroxylation of the catalyst surface during the reaction with peroxide and, on the other hand, by cleaning of the surface from coke by radicals resulting from peroxide decomposition.
In conclusion, numerous catalysts of the Zn–alumina type were not retained because of low activity or selectivity. High activity was reached after modification with promoters, facilitating simultaneously the five elemental reactions for fast BD formation, avoiding formation of the thermodynamically more favourable side-products such as ethylene and butenes. High temperature and low contact times were beneficial for the BD yield. Synthesis procedures and choice of precursors were essential to reproduce the best performing catalysts.
Magnesia–silica catalysts. Magnesia on silica has long been studied for converting ethanol in BD. Many catalytic studies have been reported to reproduce the catalytic experiments and to better understand the active sites on the magnesia–silica catalyst type. Table 4 compares the most important catalytic data for un-promoted systems.
Szukiewicz109–111 has reported between 350 and 450 °C the production of BD from ethanol over a MgO–SiO2 catalyst with a BD selectivity of 30–40%, using a LHSV of 0.3 h−1 (Table 4, entry 1). Unfortunately, ethanol conversion data were absent. The catalyst had a MgO:SiO2 weight ratio of 60:40 and was prepared by wet-kneading of a physical mixture of the dry oxide powders in water, the amount of water added only specified as 1.5–4 parts of water to one part of powder mixture by weight. The aging time (2 to 12 h) was not essential. At 410 °C and with a LHSV of 0.4 h−1, Corson et al.,101 with a similar MgO–SiO2 catalyst, having a molar Mg/Si ratio of 2.2, were able to reproduce the data, viz. a BD yield of 9% and a BD selectivity of 36% (Table 4, entry 2).
A variety of procedures is known to synthesize magnesia–silica catalysts. A comprehensive study of the preparation of MgO–SiO2 catalysts (with constant Mg to Si molar ratio of 2.2 according to the method proposed by Szukiewicz109–111) was carried out by Natta and Rigamonti.112 It was postulated that the activity and selectivity of the magnesia–silica catalysts strongly should be dependent on the synthesis procedure, including the amount of water added, the time and temperature of aging, the drying and calcination procedure. XRD-analysis demonstrated the requirement of a high dispersion of magnesia on silica and the presence of a limited amount of amorphous magnesia hydrosilicate phase (less than 10 wt% based on Mg), resulting from the interaction of dissolved Mg2+ with the silanols of the silica surface. Wet kneading, followed by calcination at 450 °C, thus provided superior magnesia silica catalysts, at 430 °C showing an ethanol to BD yield of 35% and a selectivity of 41% or volume productivity of 48 gBD l−1 h−1 (Table 4, entry 3). Mechanical blending or mixing of the oxide powders in alcohol preventing silicate formation showed poor catalytic performance.
Stability is one of the key problems of magnesia–silica catalysts. The formation of crystalline silicate from the amorphous magnesium hydrosilicate during the catalytic process was found to be responsible for the decrease in BD yield after regeneration.112 During catalyst synthesis, prevention of extensive formation of magnesium hydrosilicate is possible by reduction of the Mg2+ solubility when cold water is used or acetic acid or aqueous ammonia is added. Such catalysts showed a high stability upon regeneration. Though slightly less active, a high BD selectivity between 41 and 54% is reached (Table 4, entries 3–5).
Hydrothermal treatment between 100 and 200 °C significantly improved activity and selectivity of magnesia–silica catalysts.113 Such treatment increased the specific surface area and pore volume of the catalyst. Hydrothermal treatment of a magnesia–silica catalyst, at 400 °C yielded 37% BD with a selectivity of 58% for a LHSV of 0.3 h−1 (Table 4, compare entries 6 and 7). This mild hydrothermal treatment was also proposed for regeneration of deactivated catalysts. The catalytic result is close to the commercial MgO–SiO2 catalyst (with a molar ratio of Mg to Si of 3) (Table 4, entry 8). A 2 h pretreatment between 400 and 500 °C was defined as preferable. Higher temperatures resulted in a decreased BD yield.114
Niiyama et al.80 studied the effect of the Mg/Si molar ratio in the catalyst. Catalysts were prepared by kneading magnesium hydroxide with colloidal silica, followed by drying and calcination at 600 °C for 3 h. Note that this pre-treatment temperature is higher than the previously recommended one.112,114 At 380 °C a butadiene yield of 28% with a selectivity of 62% was reported over the catalyst with a Mg to Si molar ratio of 3 (Table 4, entry 9). The acid–base properties of the magnesia–silica catalysts in terms of the Mg to Si ratio have been investigated as well.80 Whereas the formation rate of ethylene was too high for samples with MgO content below 75 mol% (67 wt%), pure MgO was inactive for both dehydration and BD formation, the latter requiring a balance between acidic and basic sites. The catalyst with 85 mol% (79 wt%) of MgO (Mg to Si molar ratio of 5.7) possessed an optimal acid–base balance reaching a high BD formation rate of 42 μmolBD m−2 h−1 (Fig. 6). Unfortunately, BD yield and selectivity were not reported.80
Ohnishi et al. probably reported the highest BD yields ever over magnesia–silica.115 At 350 °C after 10 min-on-stream the butadiene yield and selectivity amounted to 42 and 84%, respectively (Table 4, entry 10). The catalysts were prepared by wet-kneading of precipitated magnesium hydroxide and silica gel derived from tetraethyl orthosilicate (TEOS). The nature of the reagents was shown to be important, as substitution of magnesium nitrate and nitric acid for magnesium chloride and hydrochloric acid, respectively, showed reduced BD formation. This was rationalised by incomplete removal of chloride ions during washing of the precipitates, in agreement with earlier results reported by Bhattacharyya et al.107 for alumina catalysts. Maximum activity and BD selectivity were obtained for a Mg to Si molar ratio of 1.
Many authors attempted reproducing such excellent data. In the hands of Kvisle et al.81 samples prepared according to the Ohnishi method failed to show excellent performance. The better results at 350 °C only amounted to a BD yield of 16% with a selectivity of 30% (Table 4, entry 11).
From time-on-stream studies only showing steady-state behaviour and acceptable carbon mass balance after 20 to 50 minutes on-stream, it was concluded that any catalytic result corresponding to shorter times-on-stream, viz. 10 min as used by Ohnishi et al., should be interpreted with caution. Recently, with a similar unmodified magnesia–silica catalyst, we only could reach at 350 °C and a WHSV of 0.2 h−1 a BD yield of 15% with a BD selectivity of 20% (Table 4, entry 12).116 This catalyst performance is similar to that reported by Kvisle et al.81 Analysis with UV-vis and XRD showed that wet-kneading of the sample induces structural perturbations in the highly dispersed MgO, forming Mg–O–Si interactions like in amorphous magnesia hydrosilicate.81 Both criteria were already proposed by Natta112 and Kovařík113 as a fingerprint of active and selective catalysts.
Recently, Tong et al. reported the formation of BD from ethanol with 48% selectivity over magnesia–silica catalysts obtained by wet-mixing (Table 4, entry 13).117 An optimal calcination temperature of 500 °C was suggested, which is in good agreement with data reported earlier. The best performance was obtained with a MgO content of 80 wt% (or a molar Mg to Si ratio of 6).
In addition to the catalyst composition and synthesis procedure, reaction conditions also affect catalytic activity and BD selectivity. With the commercial catalyst (Table 4, entry 8) increasing BD yields are obtained at reaction temperatures up to 460 °C in agreement with thermodynamics, while the BD selectivity reaches a maximum of 48% at 430–440 °C and then decreases.114
Kvisle et al. have investigated the effect of the ethanol concentration on its conversion to BD at 350 °C with their magnesia–silica catalyst (entry 11) at a constant carrier gas flow rate of 18 ml min−1. The authors observed higher BD yields at lower ethanol partial pressures (3000 ppm), in agreement with the increase in the number of product molecules in the chemical reaction of ethanol to BD (eqn (1)).81 Lowering the partial pressure obviously leads to lower volume productivity, while an increase of the carrier gas flow rate (or decrease of the contact time) at constant ethanol concentration in the feed leads to a lower butadiene yield.80
Summarizing the results mentioned, it follows that high BD yields (up to 30–40%) and selectivity (40 to 60%) are possible over the un-promoted binary magnesia–silica materials of Table 4 at temperatures ranging from 350 to 400 °C, using space velocities from 0.3 to 0.5 h−1. For low partial pressures of ethanol, a value of 10 vol% is a compromise between BD yield (selectivity) and volume productivity. Higher partial pressures require higher reaction temperatures resulting in the formation of more side-products. Although contradictory data are reported on the optimal Mg to Si molar ratio and the preferred synthesis recipe, it appears that the catalytic performance of the MgO–SiO2 catalysts is very sensitive to the acid–base properties of the synthesized material. There is agreement that the synthesis procedure should promote formation of Mg–O–Si species, at the same time keeping the remaining MgO highly dispersed. However, for the design of improved catalysts, more systematic characterization of the surface properties of the active catalysts is a prerequisite.
Magnesia–silica materials doped with a dehydrogenation promoter. The commercial magnesia–silica catalysts industrially used in USSR contained various dopants affording high and stable BD yields. These dopants were initially thought to improve the dehydrogenation capacity of the commercial catalyst, resulting in faster conversion of ethanol to acetaldehyde. According to the patents of Szukiewicz,109–111 modification with chromium oxide up to 10 wt% led to a significant increase in BD selectivity of about 5%. The Cr modification reduced the formation of some by-products like ethylene, while others, like butenes, were increasing. The effect of different dopants on the catalytic performance has been studied in detail.
Natta and Rigamonti74,112 have employed the addition of chromic acid as a promoter of a MgO–SiO2 catalyst with an atomic Mg:Si ratio of 2.2. At 415 °C and with a LHSV of 0.2 h−1 (entry 1, Table 5) ethanol was converted to a high yield and selectivity of BD of 43 and 52%, respectively. With the Cr-free catalyst only the respective values of 35 and 41% were obtained. The positive effect of the Cr-promotion was connected to the formation of magnesium chromate preventing excessive formation of magnesium silicate. It appears that modification with chromium oxide has an effect similar to that of ammonia or acetic acid (see before).
Corson et al.101 showed that among the more than 500 tested materials, the 2%Cr2O3–59%MgO–39%SiO2 catalyst was superior. In a fixed-bed reactor at 400 °C with a WHSV of 0.4 h−1, it exhibited a BD yield of 38% with 56% selectivity (Table 5, entry 2), corresponding to a volume productivity of 65 gBD lcat−1 h−1.
Fig. 7 shows an overview of the promotion with 2 wt% Cr of MgO–SiO2 catalysts with varying atomic ratios of Mg:Si, as prepared and tested by different authors.101,102,112,116,118 Modification of magnesia–silica materials with a small amount of chromium oxide clearly results in higher BD yield and selectivity in comparison with the unmodified MgO–SiO2 samples (compare Table 5, entries 1–5 and the corresponding data in Table 4). Some authors reported a decrease in selectivity at temperatures higher than 400 °C, despite the higher BD yield.101,118 Comparison of the catalysts is difficult due to the different reaction conditions.
László and co-workers102 have studied the Mg content for 2 wt% Cr2O3 doped MgO–SiO2 materials at 435 °C with a LHSV of 0.3 h−1. The catalytic data are summarized in Fig. 8. They observed that a lower silica content leads to a higher BD yield and selectivity, while ethylene formation decreased. The observation again proofs the requirement of an optimal balance of the surface acid–base properties. They obtained the highest BD yield of 40% with a selectivity of 52% over the magnesia–silica catalyst with an atomic Mg:Si of 6 (Fig. 8; Table 5, entry 3).
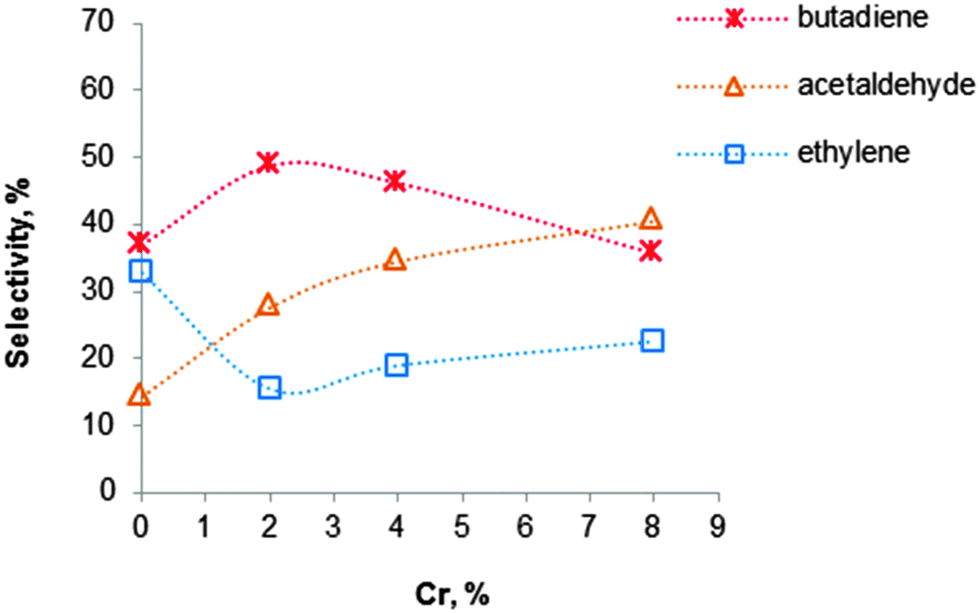
An increase of the chromium content of these catalysts resulted in an increase of the BD yield.102 Silva et al.119 have been studying the influence of the chromium loading on the catalytic performance of MgO–SiO2 materials in the conversion of ethanol to BD at 400 °C. The Cr2O3–MgO–SiO2 materials were prepared by conventional impregnation on magnesia–silica catalysts. A steadily increasing activity was observed with increasing Cr content, the effect on the BD selectivity being displayed in Fig. 9. Clearly, an increase of Cr enhances acetaldehyde selectivity. Whereas BD selectivity first increases until 50% upon addition of Cr up to 2 wt%, it drops with further enhanced Cr content.
The opposite effect is seen for the ethylene selectivity. Cr was noted to promote ethanol dehydrogenation and to adjust the acid–base properties of the magnesia–silica material, by eliminating strong acid sites with H0 ≤ 3.3, thus avoiding the formation of thermodynamically more favourable ethylene. Too high Cr contents should be avoided as well, since they introduce new acid sites related to the presence of Cr(III) species with distorted octahedral symmetry or to Cr(VI) species, as evidenced by UV-reflectance bands at 27000 and 37000 cm−1, respectively.119
Zinc oxide has also been used as a promoter for Lebedev's industrial magnesia–silica catalyst.120 Natta et al.,74 partially substituting zinc oxide for magnesium oxide, reported lower BD yield and selectivity in comparison to unmodified MgO–SiO2. At 365 °C and a LHSV of 0.2 h−1, with a 30%ZnO–30%MgO–40%SiO2 catalyst, a BD yield of about 26% at almost complete ethanol conversion was obtained (compare entry 3 of Table 4 with entry 8 in Table 5). The ZnO modified materials were prepared either by co-precipitation of the carbonates of magnesium and zinc or by mechanical mixing of the oxides, followed by wet-kneading with silica. The lower BD yield and selectivity were due to a reduced dehydrogenation and condensation capacity as a result of the formation of a solid solution between MgO and ZnO.
Berak et al., studying the influence of the zinc oxide promoter loading on the performance of the magnesia–silica catalysts,121 reported an increase of the activity with increasing Zn content.
The reaction temperature to reach 66% ethanol conversion decreased with an increase of the zinc oxide content. Fig. 10 shows the effect of the Zn promoter loading on the BD yield and product selectivity. With increasing Zn content, the BD yield first increased to 46% with a selectivity of 69% at a molar ZnO:MgO ratio of 0.002 (entry 9, Table 5). Further increase of the Zn loading resulted in a decreased BD yield at the expense of acetaldehyde, ethylene and other by-products formation.
Makshina et al.116 recently confirmed that the modification of silica by co-impregnation with both zinc and magnesium salts results in a poor activity, yielding 17% of BD with a selectivity of 31%, in comparison to similar materials obtained by consecutive modification of silica with MgO and ZnO.116 At 400 °C and with a WHSV of 0.7 h−1, the latter catalyst showed a BD yield of 52% at almost full ethanol conversion, corresponding to a record volume productivity of more than 100 gBD lcat−1 h−1 (entry 10, Table 5). Inspection of the side products pointed to high levels of unconverted acetaldehyde with the co-impregnated catalyst, indicating that impregnation leads to an un-balance between the dehydrogenation and aldol condensation capacity. Possibly, the formation of new phases in the co-impregnated sample blocks or prevents the creation of the active basic sites. XRD analysis showed the presence of mixed Zn–Mg silicates in the co-impregnated sample. Further spectroscopic work is needed to confirm the rationalisation.
Furthermore, Ta, Ag, Ni, Mn, Cu, Mo, Fe, Co, and Zr oxides were also used as promoters for MgO–SiO2 catalysts in the ethanol to BD reaction.101,113,116,122,125–128
Among these redox dopants, tantalum oxide was found to be an efficient promoter for magnesia–silica catalysts.101 These catalysts showed a butadiene yield of 34% with a selectivity of 50% at 420 °C with a WHSV of 0.4 h−1 (Table 5, entry 11), corresponding to a volume productivity of 58 gBD L−1 h−1. Similar to chromium oxide, an amount as low as 2 wt% of tantalum oxide was sufficient to boost the butadiene yield to higher values relative to the unmodified catalyst. Variation of the magnesium content was also studied for the Ta promoted samples. By changing the atomic Mg:Si ratio from 0.6 to 2, an increase of the BD yield from 7 to 34% has been reported.
Kitayama et al.122 have shown that NiO promoted MgO–SiO2 is highly selective to BD. At 280 °C about 90% BD selectivity at 59% ethanol conversion (entry 12, Table 5) was found with a 10 wt% NiO supported on MgO–SiO2 with an atomic Mg:Si ratio of 0.7. Catalysts were synthesized via impregnation of Mg(OH)2–aerosil mixtures with nickel nitrate followed by calcination at 400 °C for 2 h. They were shown to possess optimal acid–base properties, although X-ray analysis still revealed the presence of magnesium silicate. Whereas higher levels of Mg resulted in the appearance of pure MgO phases, a decreased surface area was found, leading to lower activity. It was claimed that substituted Ni is present as Ni2+ cations, dispersed into the silicate and MgO lattice. However, no characterisation of acid–base properties has been provided.
Makshina et al.116 have performed a comparative study of magnesia–silica materials modified with various transition metal oxides and metallic silver. Among the oxides, Ni, Cu and Zn were promising with regard to the BD yield and selectivity, while Fe and Mn showed insufficient performance. At 400 °C and a WHSV of 0.7 h−1, Cu-loaded magnesia silica with an atomic Mg:Si ratio of 2 showed a BD yield of 47% at almost complete ethanol conversion, corresponding to a remarkable volume productivity of 123 gBD lcat−1 h−1 (entry 13, Table 5). Sequential impregnation is essential for keeping the specific activity of the individual elements as high as possible. The atomic Mg:Si ratio influenced selectivity and activity, a value of 2 being optimal. The presence of metallic Ag had a similar beneficial effect on the catalytic performance. under similar experimental conditions, the Ag promoted catalyst exhibited a high selectivity of 54% for BD at full conversion, corresponding to a formation rate of 220 g BD per kilogram catalyst per hour (entry 14, Table 5).
Recently it has been reported that modification of the magnesia–silica catalyst with Zr and Zn oxides (1.5 and 0.5 wt% respectively) leads to a significant increase in selectivity, e.g. at 325 °C and WHSV = 0.3 h−1 doped catalyst with a Mg:Si ratio of 85:15 showed 63 mol% selectivity towards BD at 40% of ethanol conversion while unpromoted material yielded butadiene with only 26 mol% selectivity at the same level of conversion.128
Concluding this part, promotion of the magnesia silica catalyst with elements showing dehydrogenation activity, generally improves both the catalyst activity and selectivity to BD. The higher activity may be rationalised by faster acetaldehyde formation. However, the promoter loading is crucial since a balance with the basic sites, related to the highly dispersed MgO, is important as unconverted acetaldehyde otherwise causes a decrease in BD yield. Some of the dopants may introduce strong acidity or may block the mild acidity of the silica, e.g., by silicate formation. These effects need to be avoided to prevent diethyl ether but mainly ethylene formation from ethanol or to facilitate the dehydration of the aldol and crotyl alcohol to BD. Once used in the optimal content, the nature of the element seems somewhat less crucial, since Cr, Zn, Ta, Cu, Ni, Zr oxides and Ag show beneficial effects. However, a sequential loading of the silica with the base, and the dopant using intermediate calcination or not, is crucial to attain high yield of and selectivity to BD. Co-impregnation often forms additional silicate phases or causes blockage or creation of acid sites. There is no consensus on the optimal atomic ratio of Mg:Si in the doped catalysts, probably resulting from differences in the preparation methods and in the nature of the dopant. It is therefore recommended to compare the different catalysts based on their surface properties like acid–base relationship (in terms of amounts, strength and location) rather than on the basis of the elemental composition only. Today, systematic studies with advanced surface characterization tools in combination with catalytic measurements are missing for the ethanol-to-BD reaction.
Magnesia–silica catalysts promoted by alkaline metal cations and clay minerals. As ethylene is thermodynamically favoured in the reaction of ethanol to BD, promotion of magnesia-silica with alkaline metal oxides has been studied. Accordingly, Butterbaugh and Spence125,126 have reported active MgO–SiO2 catalysts, yielding BD from ethanol with a selectivity up to 48% at 435 °C, by contacting the precursor mixture of magnesium hydroxide and silica with an alkaline solution at 90–100 °C for 1–2 h. The alkaline treatment improved the BD selectivity at the expense of the ethylene selectivity. The most promising results were obtained with alkali-digested MgO–SiO2 materials containing 20 to 50 wt% magnesia, corresponding to an atomic Mg
:Si ratio of 0.4 to 1.5.
Later, Ohnishi et al. used post-impregnation with sodium or potassium hydroxide followed by calcination at 500 °C, resulting in the formation of highly active MgO–SiO2 catalysts. At 350 °C and with a WHSV of 0.2 h−1, Na2O/MgO–SiO2 and K2O/MgO–SiO2 yielded 87 and 70% BD, respectively.115 Though the post-synthetic selective poisoning of acid sites by addition of alkali is similar to that proposed by Butterbaugh and Spence,125,126 the catalytic performance reported by Ohnishi et al. was almost twice as high. The different time-on-stream behaviour of the catalyst possibly is at the origin of the different catalytic data. In our experience, early sampling under non-steady state conditions often leads to higher conversions. Within one to two hours BD yield and selectivity converge to lower steady state values, followed by slow deactivation at longer time-on-stream.
László et al.102 have studied ethanol to BD conversion with magnesia–silica catalysts mixed with clays or clay-related minerals. Replacement in a 2 wt% chromium oxide promoted catalyst of half of the silica by kaolin, at 435 °C and with a LHSV of 0.3 h−1 showed a BD yield and selectivity increase from 40 to 48% and from 54 to 63%, respectively, while the ethylene selectivity decreased (compare entries 3 and 6, Table 5). Kovařík et al.118 using a similar catalyst reported a similar increase of the BD yield with partial substitution of silica for kaolin. At 400 °C and with a LHSV of 0.3 h−1, 37 and 47% BD yields were obtained with 2%Cr2O3–MgO–SiO2 and 2%Cr2O3–MgO–SiO2–kaolin, respectively (compare entries 4 and 8, Table 5). A complete substitution of silica for kaolin resulted in a reduced BD yield and enhanced ethylene yield. Under the same conditions, a MgO–kaolin catalyst yielded 22.5% BD, while the MgO–SiO2 catalyst produced 35% BD yield.74 Complete substitution of silica by other layered silicates such as talc and kaolinite showed similarly reduced BD yields.129 Generally, both MgO and SiO2 components being essential in the catalyst composition should not be replaced for more than 20 wt% by the layered silica to attain BD yields of 35 to 40%.129
The situation is somewhat different when MgO·3SiO2·3H2O, an amorphous magnesium hydrosilicate, is used as a catalyst precursor.129 At 400 °C and with a LHSV of 0.3 h−1 it gives up to 80% yield of ethylene from ethanol. After precipitation of magnesium hydroxide with aqueous ammonia or sodium hydroxide (MgO > 60 wt%) and subsequent calcination at 550 °C, BD yields of over 50% were noticed. Based on X-ray diffraction, a partially crystalline phase, viz. composition, 3MgO·4SiO2·2H2O, could be identified next to MgO. Whereas the presence of free MgO has been suggested to be essential to ensure fast dehydrogenation of ethanol to acetaldehyde, the precise catalytic role of the Mg silicate phase has not been clarified.
Aleixandre et al.123 have reported the effect of clay-modified MgO on the BD selectivity. At 440 °C and using a LHSV of 0.4 h−1, four different clay minerals, viz. bentonite A and B, halloysite, and kaolin, were physically mixed with a variable amount of MgO (Fig. 11). Bentonite B was obtained from bentonite A by extracting free silica. The MgO–clay catalyst containing 57 wt% of bentonite A exhibited the highest BD selectivity (31%). Unfortunately, no ethanol conversions were reported (entry 15, Table 5). Catalytic differences were rationalised by taking into account the number of accessible active acid–base sites on the clay mineral. Compared to halloysite and kaolin, montmorillonite – the main component of bentonites – is known for its large interlamellar voids and high cation-exchange capacity. However, the presence of different impurities in bentonites like iron oxides which likely could promote ethanol dehydrogenation was not taken into consideration.
In conclusion, the catalytic performance of magnesia–silica catalysts can be promoted with alkaline metal cations, by suppressing (too) high acidity, thus reducing/blocking the ethanol to ethylene dehydration. Whereas modification of the magnesia–silica catalyst with layered silicates or clay minerals also results in an increase of BD selectivity, the catalytic role of the silicates is less clearly defined. Despite these improvements, addition of dehydrogenation modifiers is more effective to reach high BD yields.
Magnesium silicate clay minerals. A series of unmodified Mg and Si containing clay minerals have also been used in the ethanol to BD reaction. Dandy et al. demonstrated BD formation over sepiolite for temperatures ranging from 200 to 300 °C.130
Higher BD yields were generally obtained after modification of the clay with dopants, affecting the acid–base and dehydrogenation properties of the catalyst. Suzuki et al.124 have employed synthetic Li modified fluorohectorite as a catalyst in the ethanol to BD reaction at 375 °C using a LHSV of 0.3 h−1. The catalyst exhibited an excellent BD selectivity of 48% at 33% ethanol conversion (Table 5, entry 16). An IR study with adsorbed pyridine and CO2 showed that the presence of Li+ and F− in the catalyst modified the Lewis and Brønsted acid sites, thus optimising the dehydration properties of the catalyst. Oxygen atoms bound to Mg2+ at the edge of the layers were responsible for the dehydrogenation activity.
To further improve the dehydrogenation ability of the clay catalysts, various metal oxide dopants have been explored. Cd, Zn and Cu oxide promoters have been tested to improve the BD yields with smectite and bentonite clays.131 Smectite clay modified with Cd showed better results than other catalysts yielding BD with 17% selectivity. Kitayama et al.132–137 used sepiolites modified by impregnation with various transition metal oxides as ethanol conversion catalysts in a recirculation reactor at 320 °C. For the unmodified sepiolite ethylene selectivity was very high, while after doping BD was one of the major products, the nature and amount of the precursor salt of the promoter determining the BD yield. With manganese content increasing from 39 to 79 mol% in the MnO2–sepiolite, the BD yield increased spectacularly from 8.4 to 33.4%. The catalyst prepared from manganese acetate showed better activity than that from manganese chloride.135 Superior results were obtained with ZnO modified sepiolite containing 4.4 wt% Zn, yielding 63.5% of BD at full conversion after 2 h of reaction at 260 °C.133 High BD yields ranging from 33 to 58% were obtained over sepiolites modified with copper, nickel, cobalt and vanadium oxides132,134,136 or with mixtures of vanadium oxide with tungsten or molybdenum oxides.137
Gruver et al.83 have reported that a 6 wt% Ag promoted sepiolite is able to produce BD from ethanol. At 280 °C and a WHSV of 0.8 h−1, they obtained about 9% BD yield at 63% ethanol conversion. As the major product was acetaldehyde, obtained at about 20% yield, the catalyst probably lacks appropriate basicity needed to compensate for the fast dehydrogenation caused by the presence of metallic silver (entry 17, Table 5).
In summary, clay minerals like magnesia–silica catalysts require careful promotion of the dehydrogenation activity and fine-tuning of the acid–base properties in order to achieve high BD yields from ethanol. While the aforementioned modified magnesia–silica catalysts generally show better BD yields and selectivity than the promoted clays, individuals like ZnO promoted sepiolite also exhibited promising catalytic properties.
Other catalysts. Catalysts free of Mg2+ and Al3+ have been reported as well. Spence and Butterbaugh138 have reported the use of 20 wt% zinc oxide supported on silica (like diatomaceous earth) for butadiene formation from ethanol, showing a selectivity of 58% and a yield of 29% at 450 °C using a LHSV of 1 h−1 (Table 6, entry 1). The reaction temperature for BD formation was lowered by modifying the Zn catalyst with phosphate or tungstate. ZnO on silica and ZnO/Zn(II)phosphate on silica showed BD yields of 29 and 22% and a BD selectivity of 58 and 63%, at 450 and 415 °C, respectively. Corson et al.101 have found a BD yield of 20% at 425 °C using 0.6 h−1 LHSV with a 10 wt% ZnO on a silica catalyst (Table 6, entry 3).
Thorium and zirconium oxides on silica exhibit catalytic activity for BD formation from ethanol as well, at 445 °C amounting to BD selectivities of 35 and 44%, respectively (Table 6, entries 4 and 5). Unfortunately, no details about ethanol conversion were reported.127,139 Corson et al. reported 23% of BD yield using a ZrO2–SiO2 catalyst (Table 6, entry 6).101
Spence et al.127,139 have claimed copper and molybdenum oxides as potential dopants for the ZrO2–SiO2 catalyst. Unfortunately, most of the catalytic data refer to catalytic compositions for which detailed information on the synthesis procedure and the physicochemical properties of the catalyst surface is missing. Conversion of ethanol to BD has recently been reported by Jones et al.140 over silica doubly promoted with metal oxides of zinc, zirconium, copper, cobalt, manganese, cerium and hafnium. Emphasis was on the effect of the nature of the promoters and the texture of the silica on the catalytic activity and BD selectivity. The best results were obtained over a ZnO–ZrO2/SiO2 catalyst containing 1.5 and 0.5 wt% ZrO2 and ZnO, respectively. Higher ethylene selectivity occurred in parallel with higher promoter contents. At 375 °C and a space velocity of 1.5 h−1, the catalyst showed 18 mol% BD yield with a BD selectivity of 39 mol%, corresponding to a high volume productivity of 124 gBD lcat−1 h−1 (entry 7, Table 6). Using this catalyst as reference, it could be shown that impregnation of the promoters rather than sol–gel synthesis is the preferred synthesis method to achieve high BD yields. High ethylene yields were observed as well.
Selective poisoning of acid sites with alkali metal cations, as proposed by Ohnishi115 for the MgO–SiO2 system, was not very successful for the Zn/Zr oxide silica system. In contrast, adding a third element like Cu oxide to the Zn–Zr doped silica significantly improved the BD selectivity, yielding 19 mol% BD with a BD selectivity of 50 mol% (compare entries 7 and 9 in Table 6). Finally, an increase in BD yield and selectivity with pore size of the silica support increasing from 40 to 150 Å was noticed for the Zn–Zr modified silica. Doped large pore silica yielded 23% BD with 48% BD selectivity under the same reaction conditions (compare entries 7 and 8, Table 6). According to authors140 the large pore silica catalyst contained lower proportion of acidic silanols, as measured by the fractions of Q4vs. Q2 and Q3 in 29Si MAS NMR that results in the decrease of ethylene yield. However diffusional effects and dispersion of supported metal oxides particles were not taken into account to explain the changes in selectivity. Catalytic tests at different space velocities showed that a decrease of the space velocity from 1.5 to 0.75 h−1 results in a slight increase in BD yield, albeit with lower BD productivity. No explanation of the contact time effect was advanced. It should be stressed that the preferred LHSV of 1.5 h−1 used is similar to that used by Bhattacharyya et al.104,107,108 for the superior modified alumina catalysts. This contact time is several times higher than that usually applied for the magnesia–silica and other related catalytic systems (compare Table 6, entries 7–9 with the entries in Tables 4 and 5).
Recently, Ordomskiy et al.141 have claimed that silver, cerium, tin and antimony oxide promoted ZrO2–SiO2 are highly selective for BD. At 325 °C and with a WHSV of 0.3 h−1, 72% BD selectivity at an ethanol conversion of 34% was obtained over a 0.4 wt% Ag modified catalyst, corresponding to a productivity of 40 g kgcat−1 h−1 (entry 10, Table 6). A remarkably low temperature was used compared to the usual temperature window for ethanol conversion to BD.
Furthermore, a high BD yield has been observed over a tantalum oxide doped silica – the commercial catalyst developed for the two-step process.101 Good yields of BD up to 16% have been observed over the catalysts with 2 wt% Ta2O5 on silica at 425 °C and 0.6 h−1 (Table 6, entry 11).101 Under similar reaction conditions, an increase in BD yield up to 25% with 30% BD selectivity was found upon doping the Ta oxide catalyst with Cu oxide, likely due to an improved dehydrogenation capacity of the catalyst (Table 6, entry 12). Although lead and cadmium oxide silica promoters have been proposed, no systematic approach to variations in catalyst composition was present nor was detailed information concerning the preparation methods available. Finally, although it has been postulated that modification of silica gel with 0.5–10 wt% titanium oxide should give suitable catalysts for ethanol transformation into BD, no catalytic data were presented to support this.142
Next to the reported silica supported oxide materials, silica-free catalysts have been found to be active in the direct conversion of ethanol to BD. Bhattacharyya et al. studied binary systems containing zirconium or thorium oxides.104 Among the tested materials, ZrO2–Fe2O3, having a weight ratio of 40 to 60, showed a high BD yield of 38% with 45% selectivity at 450 °C and a space velocity of 1.5 h−1 (Table 6, entry 13). Arata et al.143 have shown that promotion of zirconia with weakly basic TiO2 catalysed the ethanol to BD reaction, yielding 12.4% BD at 330 °C.
Tsuchida et al.53 have employed hydroxyapatite to produce BD from ethanol at 350 °C and 1 h−1. The acid–base properties of the catalyst surface were controlled by changing the Ca to P ratio in the hydroxyapatite composition. Catalysts with a molar Ca:P ratio larger than 1.62 possessed the appropriate acid–base balance, yielding 24% BD at an ethanol conversion of 20%.
Commercialized by Union Carbide, the process has been supplying the major amount of synthetic BD in the US during 1943 to 1944.1 Although many studies on the two-step process were not showing much pertinent information, most of the available bibliographic data refer to reactions of co-fed ethanol and acetaldehyde in the presence of catalysts, previously developed for the one-step process. It was attempted mostly to elucidate the reaction mechanism or to evaluate the effect of the presence of the different components on the catalyst composition. In this respect, various authors noticed that addition of acetaldehyde results in an increase of BD yield. However, it remains undecided whether the two-step process should be preferred over the one-step process.
Corson et al. have reported that BD formation from ethanol via the two-step process is preferable.101 This preference is based on the enhanced flexibility for selection of appropriate conditions for each reaction separately and on the decreased catalyst complexity with respect to composition and properties of the two catalyst types used in each stage. The two-step process allowed a reduction of the reaction temperature from 400–425 °C to 350 °C. The purity of BD extracted from the 4-carbon cut amounting to 98% BD in the two-step process was only 80% in the one-step process.
Conversely, Bhattacharyya et al. have claimed that the one-step process is preferable.108 Under appropriate conditions, the amount of formed BD is high enough so that it is easily separated from other gases.108 Jones et al. showed that the addition of acetaldehyde next to ethanol does not result in any improvement of BD yield for catalysts that are able to produce significant amounts of acetaldehyde (AcH) from ethanol.140
Ostromyslensky has reported that a higher yield of BD was obtained over aluminium oxide at 440–460 °C using an equimolar mixture of EtOH and AcH.64,144 Later, Maximoff and Canonici145,146 have shown that aluminium sulphate, and basic aluminium sulphate alone or deposited on pumice-stone or Kieselguhr, were capable of catalysing the formation of BD from EtOH–AcH mixtures at a reaction temperature ranging from 280 to 450 °C yielding a BD selectivity of 33% with a molar EtOH to AcH ratio of 2.
The two-step process of BD formation has been studied intensively at Union Carbide and Carbon Chemical Corp. as early as 1947 with tantalum, zirconium or niobium oxide promoted silica.70,78 The first step proceeded over a standard copper dehydrogenation catalyst. The best results of the second step were obtained over a catalyst containing 2 wt% tantalum oxide on silica. At 325–350 °C and a space velocity of 0.4–0.6 h−1, the catalyst produced 35% BD yield with a selectivity of 67% using a mixture of 69% by weight ethanol, 24% acetaldehyde and 7% water. Higher Ta2O5 contents in the catalyst resulted in a further increase of the BD yield but at the expense of BD selectivity. It has been stated that the activity of catalysts containing less than 1 wt% of tantalum oxide was not reproducible.101
The catalytic activity of zirconium and niobium oxides on silica was lower than the Ta2O5–SiO2 material. 1.6 wt% ZrO2 on SiO2 showed a BD selectivity of 59% under comparable reaction conditions.78 Corson et al. have reported that hafnia promoted silica gel was selective and active in the two-step production of BD. At 300 °C a BD yield of 30–40% and a BD selectivity of 50–60% could made from an ethanol–acetaldehyde mixture.101
Ordomskiy et al. have claimed that zirconium, titanium, tantalum, niobium or magnesium oxides supported on silica promoted by silver, gold, copper, cerium, tin or antimony oxides are highly active and selective catalysts for the production of BD from a mixed acetaldehyde–ethanol feed at 325 °C.141 Among the Ag promoted MOx–SiO2 catalysts, mildly basic titanium oxide was found to be the most selective catalyst showing a BD yield of 25% and a BD selectivity of 72%. Ag promoted MgO-containing catalyst produced more BD, yielding 29%, but with a BD selectivity of only of 64%. Among the zirconium containing materials, M–ZrO2–SiO2 doped with different metals, gold was claimed to be the best choice producing BD with a yield of 25% and a selectivity of 82%. Promotion of a Ag modified ZrO2–SiO2 with cerium oxide further increased the BD yield when compared to tin, antimony and sodium dopants, yielding 33% BD with 81% selectivity. Silica was the best support, and use of promoted alumina and aluminosilicate resulted in lower BD yields, 18 and 25%, respectively, and selectivity was 57 and 74%, respectively.
Chae et al.147 have studied the effect of the pore size, pore volume and pore organisation on the catalytic conversion of an ethanol–AcH mixture at 350 °C and a LHSV of 1 h−1 by using 2 wt% Ta2O5 on several mesoporous silica like SBA-15, KIT-6 and MCM-41 type materials. The BD yield increased from 22–24 till 37% when a SBA-15 silica support was used instead of a commercial silica gel, while the BD selectivity increased from 72–75 to 80%. Moreover, Ta2O5 supported on ordered silica exhibited a better coke tolerance and stability with time-on-stream. The best catalytic results in terms of activity and selectivity were obtained using silica support material with nano-sized morphology and large pore sizes, indicating the importance of efficient pore diffusion. 2D or 3D organization of the mesopores like in SBA-15 or KIT-6 was found to be less important. The advantage of hierarchical mesoporous silicas is related to the ability of reaching higher dispersion of tantalum making more active sites available and preventing agglomeration of Ta oxide during synthesis and catalytic reaction.
Romanovsky and Jordan148–150 have reported that the addition of phosphate modifiers to magnesia–rich MgO–silica catalysts with an atomic Mg to Si ratio of 5 resulted in a higher conversion of the ethanol–acetaldehyde mixture and a higher BD selectivity. Key promoters for silica–magnesia were 5 to 25 wt% calcium or calcium–nickel phosphate complexes having molar Ca to Ni ratio of 7.5–9.2. The best results were obtained at 400 °C using a LHSV of 0.4–1.0 h−1, yielding 21 and 23% BD with 56 and 58% selectivity, respectively.
In addition to the catalyst composition, the reaction operating conditions like temperature, contact time and feed molar ratio of ethanol to AcH also played an important role in the catalytic activity and BD selectivity. The old commercial process operating between 325 and 350 °C used a mixture of 7 wt% water and 93 wt% feed with a molar ethanol to AcH ratio of 2.7.
Toussaint et al.78 investigated the influence of the reaction temperature, the molar ratio of EtOH to AcH and the space velocity on the catalyst activity and BD selectivity of a commercial 2 wt% Ta2O5 on silica. High BD selectivity was obtained at 325 °C for a feed mixture with a EtOH to AcH ratio of 3, whereas better results were found at 350 °C with a feed mixture ratio of EtOH to AcH of 2–2.5. Catalyst deactivation was less pronounced at lower temperature. Though an increase in BD yield was observed at 350 °C with a EtOH to AcH feed molar ratio of 2, a considerable loss in activity was detected after 160 h on stream, when compared to a reaction at 325 °C using a feed molar ratio of 3, the latter conditions being considered as optimal. Corson et al. have reported that for a 2%Ta2O5–SiO2 the optimal conditions for BD production were achieved at 350 °C, using a LHSV of 0.6 h−1 and a molar EtOH to AcH molar feed ratio of 2.75.151 The amount of coke deposited on the catalyst was significantly higher when the EtOH to AcH molar ratio was decreased below 2.75, resulting in faster catalyst deactivation.
The old commercial process with 2 wt% Ta2O5 on silica used 7 wt% water in the feed. Romanovsky and Jordan employed 40 to 60 wt% water with the phosphate supported magnesia–silica catalyst to reach the highest BD yield of 23% at 410 °C using a LHSV of 0.4 h−1. However, the precise role of water has not been investigated.148–150
In conclusion, 2 wt% Ta2O5 on silica seems to be the preferred catalyst for converting a mixture of ethanol and acetaldehyde to BD contains. High ethanol to acetaldehyde molar ratios are required to obtain high BD selectivity and to avoid coke formation. Likely, water in the feed mixture has a beneficial though not further determined role in the catalysis. High catalyst stability and high BD yield are obtained for reaction temperatures between 325 and 350 °C, the preferred contact times corresponding to a LHSV ranging from 0.3 to 1 h−1. Use of large pore size mesoporous silica is beneficial for the BD yield and selectivity due to high Ta oxide dispersion and efficient pore diffusion. Best BD yield and selectivity were reported to be about 25 and 80% respectively, corresponding to a volume productivity of 350 gBD l−1 h−1.
3. Dehydration of C4 alcohols to BD
3.1 Introduction and the feedstock origin
Next to the ethanol-based route, alternative paths to on-purpose production of BD have been demonstrated. These routes are based on the (deoxy)dehydration of mono- and polyols with a four-carbon (C4) backbone. In the literature the catalytic processing of 1-butanol, several butanediols (BDOs) and even tetritols were reported for BD production via a direct or multistep process. Scheme 10 provides an overview of the C4-based alcohol routes and summarizes the major chemical transformations involved.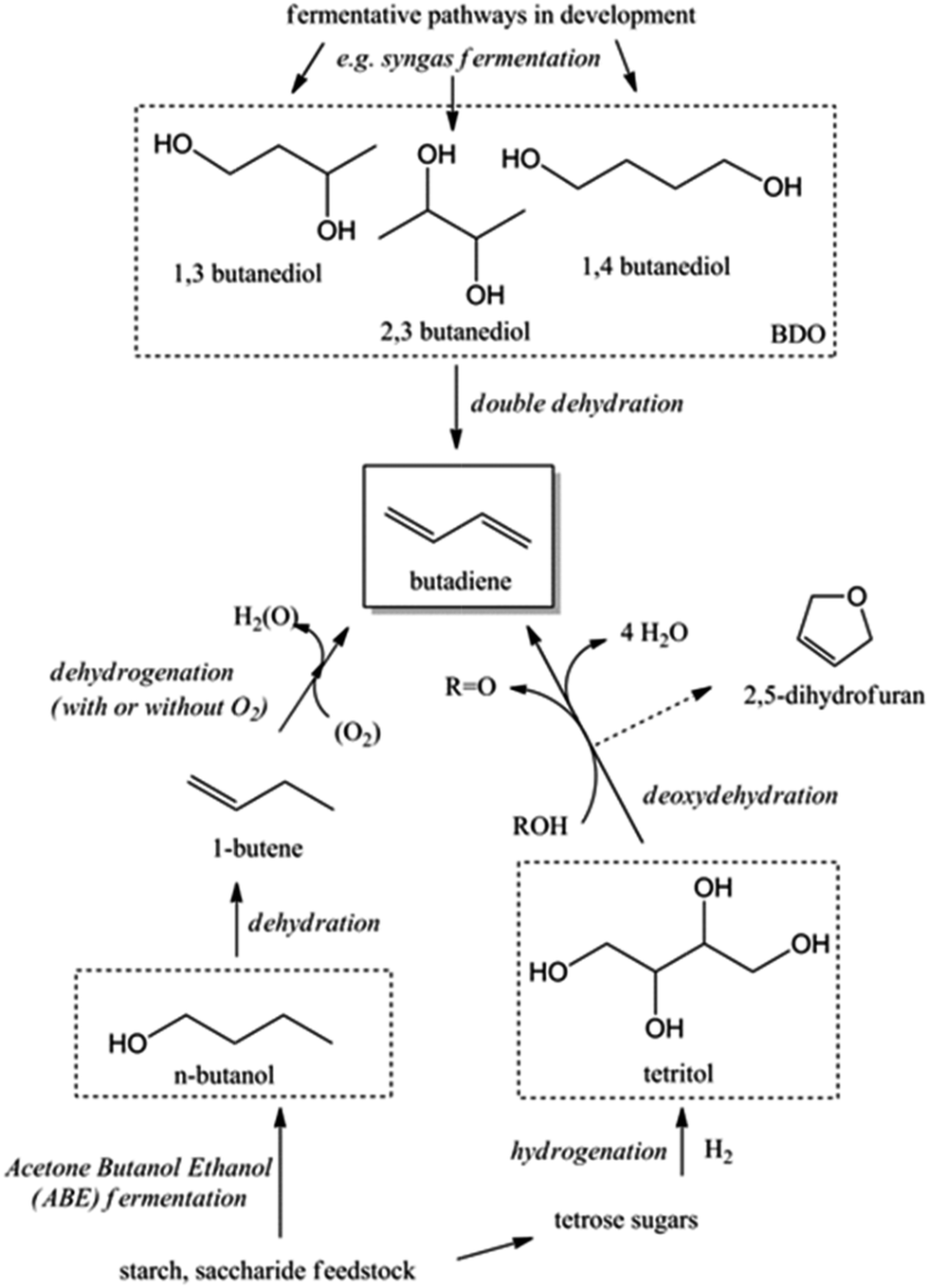 | ||
| Scheme 10 Overview of 4-sugar alcohol based BD production routes. | ||
Double dehydration of several BDOs, viz. 1,3-, 2,3- and 1,4-BDO, leads to the formation of BD. In the search for novel on-purpose BD routes independent of the fossil oil feedstock, the origin of the different BDOs is addressed first. 1,4-BDO is a versatile intermediate in the chemical industry with an annual production in the range of 1 million metric tons.2 The main synthesis route is based on the Reppe process starting with the reaction of acetylene with formaldehyde to form 2-butyne-1,4-diol, followed by hydrogenation to 1,4-BDO. The latter is not only a feedstock for tetrahydrofuran (THF), but it is also used for polyurethanes and poly(butylene) terephthalate. Very recently, Genomatica unveiled an inventive bio-based route towards 1,4-BDO, using a metabolically engineered Escherichia coli, which is capable of directly producing 1,4-BDO by fermenting different biomass-derived sugars.152–154 Although this technology is immature, it has been proven on a 2000 ton scale. Major players have already licensed the technology for scale-up, paving the path for replacing the non-renewable 1,4-BDO production.24
Next to the 1,4-isomer, 2,3-BDO (Scheme 10) is also a potential feedstock for BD. Though 2,3-BDO production by bacterial sugar fermentation has been reported,155–157 its production was replaced by petrochemical routes based on sequential butene chlorohydrination and chlorohydrin cyclization, followed by hydrolysis of the resulting epoxides and their subsequent fractionation.155 A recently discovered approach to 2,3-BDO is gaining more importance. The route is based on the fermentation of syngas by acetogenic organisms.158 Feedstock syngas not only can be derived from biomass gasification, but also from industrial gas waste, e.g. from a steel mill. Several companies are actively exploring gas fermentation to liquid fuels, mainly to ethanol, Lanzatech being a pioneer in gas fermentation to 2,3-BDO.26,159
1,3-BDO is the third BDO isomer in Scheme 10. This diol is the intermediate in the old synthesis of BD based on acetaldehyde from acetylene, followed by aldol condensation and reduction of the intermediately formed 3-hydroxybutanal, a route also included in Scheme 1.155 For this diol, no sustainable production technology has been demonstrated at the commercial level. However, numerous patents were filed in this domain, using engineering of fermentative pathways.21,160–162
On the left-hand side of the lower part of Scheme 10, another route to BD is shown, based on butanol dehydration. n-Butanol and its isomers are dehydrated via a straightforward acid-catalysed gas phase approach, leading to 1-butene (and isomers).163–166 This could be an alternative way to biomass-derived BD, as it plugs directly into the state-of-the-art industrial dehydrogenation process of 1-butene.2,39 Though the latter process has been practiced by Shell, Phillips and Petro-Tex, most plants have been closed due to the high operational costs originating from the elevated temperatures (up to 700 °C) needed to compensate for the endothermicity. The catalyst is composed either of oxides of Cr, Fe and/or Al (e.g., Fe2O3/bauxite) or of (Ca–Ni) phosphates.2,39 Often, besides steam, oxygen is added to reduce coking, oxidize hydrogen gas to water and generally to shift the equilibrium away from the hydrocarbons. The oxidative variant usually runs with mixed oxides based on Bi/Mo or Sn/Sb.2
The production of renewable butanol is experiencing a true revival. The production of butanols relies on the fermentation of carbohydrates in the so-called ABE fermentation,165,167 named after its major products, viz. acetone, butanol and ethanol. The process was operated commercially in the first half of the 20th century, but abandoned again in favour of fossil alternatives.168 The focus of recent developments is aimed at the synthesis of butanol, due to its high added-value and its proven efficiency as a liquid transportation fuel with a high substitution potential for both ethanol and biodiesel.168–170 Solventogenic clostridia organisms are used to produce the acetone:butanol:ethanol mixtures in a fixed 3:6:1 molar ratio. Large scale economic breakthrough of this technology is hampered by the high cost of the carbohydrate feedstock. Use of cheaper lignocellulosic biomass may present a great opportunity, though considerable efforts are required to improve the performance of the fermentation and to make the butanol recovery more sustainable.168–170 As both ABE fermentation and the oxidative dehydrogenation of butenes to BD have been applied commercially in the past, a combined revival of these technologies could potentially lead to bio-based BD, given the current drivers in this market.171 One should however include the additional catalytic dehydration to transform butanol to butenes. Although this reaction is considered rather easy, butanol is produced in water and its preceding recovery is a major issue.21,165,166 Due to its biofuel potential and incentives therein, bio-butanol production could likely increase dramatically on a short timescale,165 rapidly followed by BD production from the butanols.
Scheme 10 also shows a one-step route based on the deoxydehydration (DODH) reaction of 4-carbon sugar alcohols. These tetritols are produced via hydrogenation of natural tetrose sugars. As recently demonstrated in an efficient way by Shiramizu and Toste, the liquid phase one-pot double DODH approach towards BD is based on homogeneous rhenium catalysts and involves the (transfer) hydrogenation with a sacrificial reductant. Unfortunately, tetrose sugars are not produced on a large scale today and remain an expensive and elusive feedstock.154,172–175
In conclusion, the introduction clearly points to the availability of on-purpose routes towards BD via 4-carbon mono-, di- and tetrols, which are derived from a renewable feedstock or syngas. The main driver for the breakthrough is the success of engineering organisms to ferment carbohydrates or syngas to available bio-butanols and BDOs. Selective dehydration is thus an essential step. The purpose of the next part is therefore to review and assess various catalytic dehydration processes for the conversions of BDOs reported in the recent past with an emphasis on BD production, including the chemistry and thermodynamics of the reactions, the reaction network and the type of catalysts developed. In what follows, the DODH approach applied to tetritols will be assessed as well.
3.2. Mechanistic, catalytic and thermodynamic aspects of BDO to BD dehydration chemistry
Double catalytic dehydration of 1,3-BDO, 1,4-BDO and 2,3-BDO leads to BD. The dehydration reaction with the different BDOs leads to a mix of overlapping intermediates – originating from the first dehydration – and side-products, resulting from competing reactions. The routes and chemistry of BDO double dehydration are schematically represented in Scheme 11, and will be discussed for each type of BDO.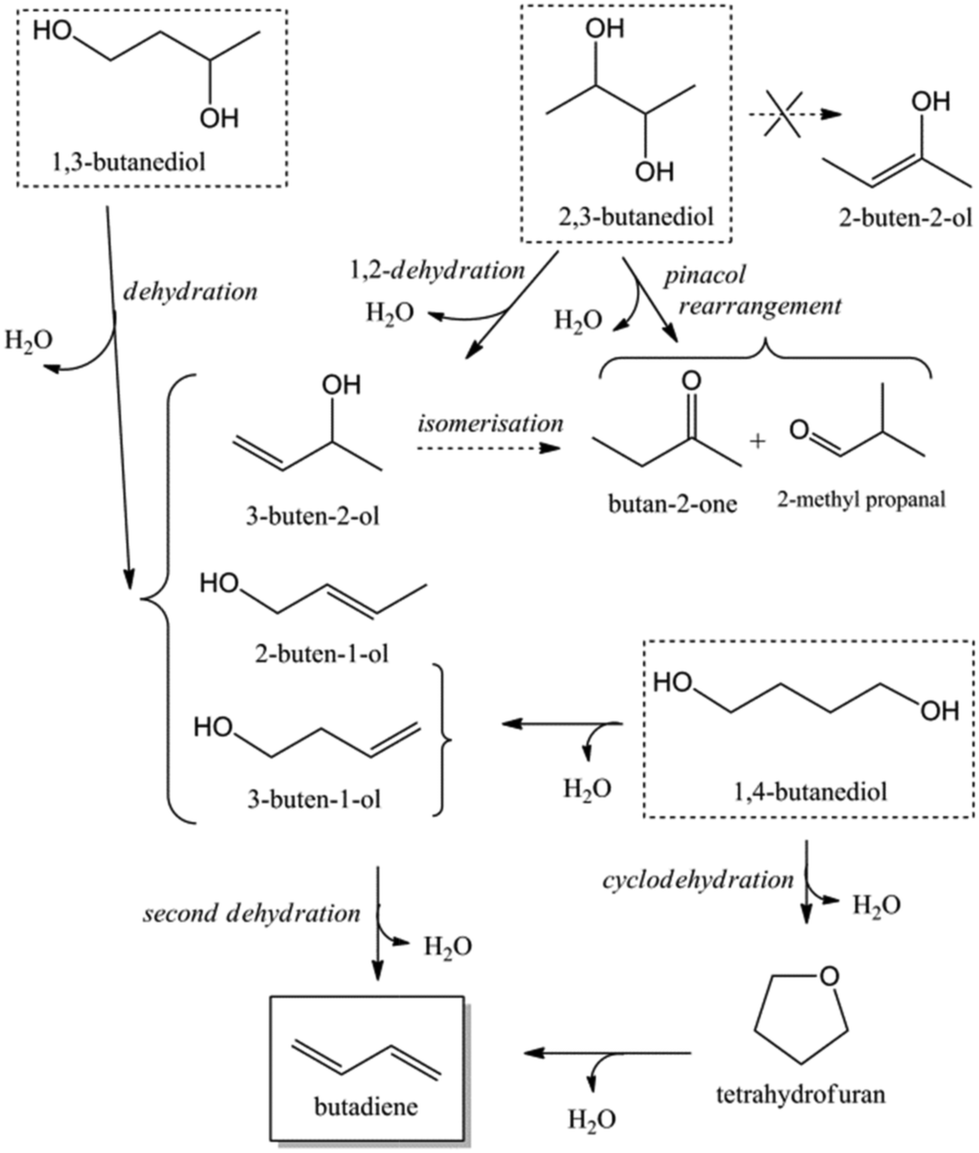 | ||
| Scheme 11 BDO to BD reaction scheme with the major intermediate and side products. | ||
Next to butan-2-one formation via the pinacol route, dehydration of 2,3-BDO also follows another reaction mode, often denoted as 1,2-elimination, forming an alkene upon elimination of water. If one water molecule is removed, the unsaturated 3-buten-2-ol will be formed, which may further dehydrate to BD. The ratio of the two dehydration types, presented in parallel in Scheme 11, depends on the chemical structure of the diol, the acidic property of the catalysts and the experimental conditions.178,179 Most reports on the conversion of 2,3-BDO in the gas or liquid phase show a predominant pinacol type dehydration. The pinacol reaction is accompanied by BD formation in the liquid phase only under non-classical conditions. For instance, Waldmann et al. have studied the dehydration of 2,3-BDO to BD in excess of pthalic anhydride with benzenesulfonic acid.178,180 The pyrolysis of 2,3-acetoxy derivatives of 2,3-BDO into BD is also known.181 The use of amines also helps in guiding the dehydration to BD, while avoiding butan-2-one formation from 2,3-BDO.182 Hereto, a vapour phase reaction over a catalyst, containing silica and tungsten oxide on alumina, succeeded yielding 48% of BD from 2,3-BDO in a single pass at 330 °C. However, this high yield was only possible by co-feeding butan-2-one, water and triethylamine. Later, a patent was filed on a similar catalyst composition regarding the formation of 2-methyl-butadiene (isoprene) in high yields (>90%) from 2-methyl-2,3-butanediol in the vapour phase without co-feeding additional reagents. Unfortunately, the patent did not demonstrate the conversion of 2,3-BDO to BD.178,183
The first real attempts on the single-step 2,3-BDO-to-BD conversion without sacrificial or stoichiometric reagents date back to the 1940s. Bourns et al. have assessed a Morden bentonite clay as a catalyst and they noticed full conversion of 2,3-BDO into butan-2-one below 350 °C. Between 450 and 700 °C, more gaseous products were formed containing small quantities of BD. Dilution of the feed stream with steam increased the BD yield to 25%.184 Winfield was the first who truly aimed at making BD form 2,3-BDO in as single step and thoroughly screened a range of catalysts including several metal oxides and salts such as SiO2, B2O3, ThO2, BeO, CaHPO4, CaCO3 and CaSO4 in a vapour phase reactor setup in 1945.185 The author aimed at a reaction step, proceeding via 1,2-elimination towards 3-buten-2-ol, and further dehydration to BD (Scheme 11). While most catalysts promoted pinacol dehydration with almost exclusive formation of butan-2-one, ThO2, prepared according to a specific synthesis protocol, produced selectively BD. The best result was obtained over the ThO2 catalyst at 350 °C in a fixed-bed reactor under reduced pressure, yielding 60% BD and 20% 3-buten-2-ol. According to the authors, the rate of the second dehydration step, converting 3-buten-2-ol to BD, appears inversely proportional to the applied pressure. Winfield then thoroughly investigated the adsorption equilibria of his catalytic system to elucidate the profound effect of water adsorption and the concomitant retardation of the second dehydration.186
The work of Winfield on ThO2 may be considered as the breakthrough in converting 2,3-BDO to BD, since most other catalytic materials produced butan-2-one.178,187 Bartok et al. have reported that the pinacol route is the preferred dehydration pathway in the presence of zeolites like Na+ and Na+/H+ exchanged X and Y type faujasites at 250 °C.188,189 The authors noticed that a lower acidity within X-type zeolites not only led to more pronounced 1,2-elimination and thus BD, but also to a decreased conversion rate.188 For instance, at 250 °C, use of the NaH–X zeolite showed 59% conversion, distributed over 78% butan-2-one, 4% 2-methyl-propanal and 16% BD.179,188 Zeolites generally require high reaction temperatures for dehydration and therefore they also tend to catalyse fragmentation of the carbon backbone.189 Other zeolite topologies have also been investigated. Lee et al. have investigated a series of zeolites with different Si/Al ratios within MFI, MOR, BEA and FAU topologies. They noted that the small pore zeolites favoured butan-2-one formation,190 whereas the large pore zeolites mostly yielded acetals and ketals. However, no significant formation of BD was reported. Very recently, Zhang et al. have studied the influence of boric acid modification of H-ZSM-5 zeolites191 in a study concerned with the effect of acid site density and strength. In all cases however, BD yields were in the range of 1%.190–192
Olah et al. in a study on the use of H-Nafion, a perfluorinated resin with sulfonic acid groups, reported that this superacid mainly catalysed the pinacol route.179 In a reactive distillation setup, 1,3-dioxolane derivatives were predominantly formed via acetalisation of butan-2-one with 2,3-BDO, while butan-2-one was the main product in a continuous flow reactor setup. At 150 °C, the conversion of 2,3-BDO was about 78%, while the selectivity to BD only reached 4%. Butan-2-one and its acetals accounted thus for about 94% of the recovered products. At 175 °C, the yield of BD increased to 8% at full BDO conversion.
Other strong acids like heteropolyacids examined by Török et al. showed a dominant pinacol pathway.193 The preference for pinacol formation with acid catalysts is attributed to the strength of the acid sites. Strong acidity better stabilises the oxonium ion transition state (thus the carbocation formed from 2,3-BDO), hence favouring the 1,2-hydride (and also some 1,2-methyl) shift. Ironically, when 2,3-dimethyl-2,3-butanediol (actual pinacol) is the substrate instead of 2,3-BDO, pinacol rearrangement is less dominant and diene formation is more pronounced. Ironically, when 2,3-dimethyl-2,3-butanediol (actual pinacol) is the substrate instead of 2,3-BDO, pinacol rearrangement is less dominant and diene formation is more pronounced. This change in the reaction pathway is likely related to the relative ease of pinacol rearrangement with 2,3-BDO, since the migration of the hydride as in 2,3-BDO is easier than the shift of a methyl group as in 2,3-dimethyl-2,3-butanediol.188,189
Sato et al. have reported the use of basic CeO2 to dehydrate 2,3-BDO at 425 °C, noting the formation of butan-2-one as the major product, while no BD was formed (Table 7, entry 7B).
| Entry | Feedstock | Catalyst | T/°C | WHSV/h−1 | TC/% | S BD/mol% | S THF/mol% | S 3-buten-1-ol/mol% | S 3-buten-2-ol/mol% | S 2-buten-1-ol/mol% | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a 2,3-BDO as feed: major product is 64 mol% butan-4-one (thus pinacol rearrangement). b Other major product was 3-buten-2-one: 19 mol%/11 mol%. c Other major product was 3-buten-2-one: 19 mol%/11 mol%. d Major product is 58% propene instead of formaldehyde. TC presents the total conversion of substrate (in %) | |||||||||||
| 1 | 1,4-BDO | Al2O3 | 200 | 9.0 | 17 | 70 | 30 | 0 | — | — | 202 |
| 2 | 1,4-BDO | Al2O3 | 275 | 9.0 | 100 | 0 | 99 | 0 | — | — | 202 |
| 3 | 1,4-BDO | SiO2–Al2O3 | 200 | 9.0 | 100 | 5 | 92 | 0 | — | — | 202 |
| 4 | 1,4-BDO | ZrO2 | 275 | 9.0 | 15 | 49 | 37 | 0 | — | — | 202 |
| 5 | 1,4-BDO | CeO2 | 275 | 6.0 | 6 | 87 | 0 | 0 | — | — | 202 |
| 6 | 1,4-BDO | CeO2 | 425 | 6.0 | 73 | 5 | 7 | 56 | — | — | 202 |
| 7A | 1,4-BDO | CeO2 | 425 | 1.8 | 91 | 24 | — | 36 | — | — | 202 |
| 7Ba | 2,3-BDO | CeO2 | 425 | 13.2 | 51 | 0 | — | — | 3 | — | 203 |
| 8 | 1,4-BDO | Yb2O3 | 375 | 20.3 | 40 | — | 3 | 82 | — | — | 208 |
| Entry | Feedstock | Catalyst | T/°C | WHSV/h−1 | TC/% | Sel.BD/mol% | S formaldehyde/mol% | S 3-buten-1-ol/mol% | S 3-buten-2-ol/mol% | S 2-buten-1-ol/mol% | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 1,3-BDO | SiO2–Al2O3 | 200 | 11.4 | 19 | 20 | 0 | 44 | 2 | 14 | 214 |
| 10 | 1,3-BDO | SiO2–Al2O3 | 250 | 11.4 | 74 | 36 | 8 | 28 | 3 | 3 | 214 |
| 11 | 1,3-BDO | Al2O3 | 250 | 11.4 | 49 | 0 | 28 | 15 | 0 | 4 | 214 |
| 12b | 1,3-BDO | TiO2 | 350 | 11.4 | 56 | 0 | 5 | 21 | 3 | 11 | 214 |
| 13c | 1,3-BDO | ZrO2 | 350 | 11.4 | 57 | 3 | 3 | 18 | 13 | 15 | 214 |
| 14 | 1,3-BDO | CeO2 | 325 | 6.7 | 73 | 0 | 0 | 0 | 58 | 36 | 216 |
| 15 | 1,3-BDO | Yb2O3 | 325 | 6.7 | 33 | 0 | 0 | 1 | 51 | 35 | 216 |
| 16 | 3-Buten-1-ol | SiO2–Al2O3 | 250 | 5.7 | 42 | 13 | 58d | — | — | — | 214 |
| 17 | 2-Buten-1-ol | SiO2–Al2O3 | 250 | 5.7 | 77 | 93 | — | — | — | — | 214 |
| 18 | 3-Buten-2-ol | SiO2–Al2O3 | 250 | 5.7 | 71 | 93 | — | — | — | 3 | 214 |
Next to the unique ThO2 catalyst of Winfield, a recent patent showed promising dehydration of 2,3-BDO to BD in the presence of an hydroxyapatite–alumina catalyst at 320 °C, yielding BD yields above 45%.194
Finally, an alternative and original strategy was recently proposed, which tries to bridge the fermentative production of 2,3-BDO with its conversion to BD. The strategy is composed of (i) the esterification of the diol with formic or acetic acid – which are naturally present in glucose-to-BDO fermentation liquors-, followed by (ii) the pyrolysis of the di-esters formed with production of BD at 500 °C. The yields of step 1 are in the range of 70–85% depending on the acid, whereas the pyrolysis of the pure di-esters yields BD in the range of 82–94%. This appears to be an effective way of avoiding pinacol type reactions, but requires pyrolysis at elevated temperatures.195
In conclusion, direct dehydration of 2,3-BDO into BD in high yields is very challenging due to the competitive butan-2-one formation according to the pinacol rearrangement. While the latter is catalysed preferably by most acid catalysts, Winfield's work of 1945 using ThO2 and recent work with hydroxyapatite prove that the competitive dehydration of 2,3-BDO to BD is possible, provided that the appropriate catalyst is used. Therefore, research urgently needs further and deeper exploration to clarify the required catalytic properties and surface reactions. Some companies159 very recently have announced development of such new catalytic technology, in light of the recent breakthroughs in the production of 2,3-BDO from syngas and biomass fermentation.158
Production of BD from 1,4-BDO demands a higher reaction temperature. The formation of THF over Ca3(PO4)2 is selective in the temperature window of 250 to 320 °C. At higher temperatures, the formation of some BD was observed along with high concentrations of unsaturated alcohols like 3-buten-1-ol and 2-buten-1-ol. Further increase of the temperature above 400 °C led to the predominant formation of BD.178 According to Freidlin et al., with phosphate type catalysts THF was said not to isomerise into the butenols, but to convert directly into BD at temperatures above 360 °C.197
BD is thus formed both from THF and 3-buten-1-ol, as visually presented in Scheme 11. The primary pathway is said to occur via 3-buten-1-ol, which is the product of dehydration via the catalysed 1,2-elimination mechanism.178 The work by Reppe et al. has confirmed that the 1,4-cyclodehydration is mildly exothermic and rather easy, while the conversion of THF to BD reaction is highly exothermic and is much more difficult.198 Nevertheless, they have described a process converting 1,4-BDO vapours mixed with steam, over a supported H3PO4 catalyst bed, containing sodium phosphates. The residence time could be optimized to form almost quantitative yields of THF, including small percentages of BD. By separating BD and recycling THF over the catalyst bed, BD yields of over 90% were shown.198,199 The reverse process, viz. 1,4-BDO production from BD, is currently commercially practiced. This process proceeds via 1,4-diacetoxybutene formation from BD and acetic acid and subsequent hydrogenation followed by a final acid hydrolysis. Acetic acid can be recycled and, next to 1,4-BDO, THF is formed.2,200,201
Sato et al. have studied the dehydration of 1,4-BDO to unsaturated alcohols. A wide range of oxides such as Al2O3, SiO2–Al2O3, ZrO2, MgO were screened as well as plethora of lanthanide oxides, with a special focus on CeO2.202,203 Logically, the different catalytic behaviour of SiO2–Al2O3 and Al2O3 showed that acidity influences the dehydration activity. The dehydration reaction over alumina at 200 °C (entry 1 in Table 7) shows a conversion of 1,4-BDO of 17%, whereas full conversion is obtained with SiO2–Al2O3 at the same temperature (entry 3, Table 7). While at low conversions the selectivity to BD was high, viz. 70% for alumina, THF is the dominant product at full BDO conversion (entries 2 and 3), while no unsaturated alcohols were formed.
Yamamoto et al.204,205 have investigated the use of ZrO2 for the selective dehydration of 1,4-BDO to produce THF and 3-buten-1-ol at 275 °C and a WHSV of 9 h−1 (entry 4, Table 7). The competitive formation of both products is likely due to the presence of both acid and basic sites on ZrO2.204 This surface composition is able to compete for 1,4-cyclodehydration to THF and 1,2-elimination to unsaturated alcohols. In order to avoid the formation of THF, the authors decided to investigate purely basic cerium oxides. Usage of CeO2 completely avoided the formation of THF. At 275 °C and a WHSV of 6.0 h−1, only 6% 1,4-BDO conversion was noticed with a high BD selectivity close to 90%, while a 73% conversion was obtained at 425 °C albeit with a selectivity shift from BD to 3-buten-1-ol (compare entries 5 and 6 of Table 7). Upon increasing the contact time with a factor of three, BD selectivity increased from 5 to 24% at the expense of 3-buten-1-ol (entry 7A, Table 7), while a 91% conversion of 1,4-BDO was obtained. These results highlight the importance of tuning the contact time to promote single or double dehydration. The excellent redox properties of CeO2 is responsible for a selective stimulation of dehydration to the unsaturated alcohol.202,206 A similar behaviour was noticed with In2O3207 and weakly basic, heavy rare earth oxides202,203,206,208–210 such as Yb2O3 (entry 8, Table 7). The latter catalyst was very active and selective for dehydrating 1,4-BDO selectively to 3-buten-1-ol at 375 °C and a WHSV of 20.3 h−1 with a selectivity of 82% at a conversion of 40%. The very high WHSV (and thus low contact time) reveals a high activity of Yb2O3. Although rather basic in nature, the catalytic action of the rare-earth oxides likely proceeded via a concerted acid–base mechanism, as hinted from gas poisoning experiments.211 Sato et al. recently reviewed their efforts in the dehydration of diols with rare-earth oxides.211
In conclusion, the most favourable dehydration route of 1,4-BDO is cyclodehydration, rendering the formation of THF. Although THF may be further dehydrated to BD, this reaction requires severe thermal conditions. The more straightforward route from 1,4-BDO follows the double 1,2-elimination with intermediate formation of 3-buten-1-ol. This pathway is less dominant over acidic catalysts. Neutral to basic catalysts like phosphates, ZrO2, CeO2 and Yb2O3 favour the formation of the unsaturated alcohol, BD being formed at high enough contact time and increased reaction temperature. However, since the formation of BD was never the purpose of the aforementioned catalytic studies, we anticipate that much better catalytic results are expected soon once the contact time, catalyst composition and other engineering parameters are fine-tuned with focus on 1,4-BDO dehydration to BD.
Different products such as BD, intermediately dehydrated enol products, and other side-products have been encountered in the dehydration of 1,3-BDO. Their pathways are depicted in Scheme 11. The intermediate products are the result of a different regioselectivity in the first dehydration step. According to the classic acid-catalysed 1,2-dehydration mechanism, the diol should transform to 2-buten-1-ol, the most stable alkene in agreement with Zaitsev's rules. The anti-Zaitsev product, 3-buten-1-ol, is formed abundantly with 1,3-BDO. In addition, while the elimination of water from a secondary alcohol is usually easier than from a primary one due to the higher stability of the more substituted carbocation, the terminal hydroxyl in the case of 1,3-BDO also undergoes serious dehydration, rendering the formation of 3-buten-2-ol, as a third enol product in the mixture. BD formation via dehydration of the three enols occurs with different reactivity for each enol type. Competitive isomerization with the formation of butan-2-one is an unwanted side-reaction in view of BD production. Another mode of dehydration, namely 1,3-cyclodehydration resulting in the formation of the cyclic 2-methyl oxirane, is usually not encountered for 1,3-BDO. Next to dehydration, fragmentations may occur via retro-aldolisation if one of the hydroxyls is able to undergo dehydrogenation to yield a carbonyl group.
Patent literature on the synthesis of BD from 1,3-BDO is ample. The patents describe the conditions and catalyst compositions, which favour BD formation such as supported and unsupported acidic and neutral phosphates like Na-(poly)phosphates (at 270 °C)2 and CePO4 (at 320 °C).212 Supported H3PO4 and more complex compositions have also been used.213 BD yields over 90% are not uncommon in these patents.
Sato et al.211 have thoroughly investigated the conversion of 1,3-BDO in the vapour phase with focus on the production of the unsaturated alcohol, 3-buten-2-ol, rather than on BD (Scheme 11). Such enols are important raw materials for the synthesis of custom and added-value specialty chemicals. Although not aimed at producing BD, these studies are relevant for gaining mechanistic insight into the dehydration of 1,3-BDO over solid acid and base catalysts. A selection of the catalytic data is displayed in Table 7 (entries 9 to 18). Strongly acidic catalysts such as SiO2–Al2O3 were more active and yielded BD next to the enols (entries 9 to 13, Table 7). 19% of 1,3-BDO could be converted into 20% BD, 44% 3-buten-1-ol and 14% 2-buten-1-ol over SiO2–Al2O3 at 200 °C and WHSV of 11.4 h−1. Increasing the reaction temperature to 250 °C, both the conversion and the BD selectivity increased to 74% and 36%, respectively, (compare entries 9 and 10, Table 7). Interestingly, and in contrast to dehydration of 1,4-BDO, dehydration of 1,3-BDO is more selective to BD in the presence of acid catalysts.
Less acidic oxides like alumina tend to catalyze other reaction channels. In the presence of alumina, formation of formaldehyde and 4-methyl-1,3-dioxane with 28 and 18% selectivity was noticed next to some 3-buten-1-ol (entry 11). Formaldehyde likely originated from 1,3-BDO dehydrogenation towards 4-hydroxy-butan-2-one, followed by retro-aldol fragmentation. Formaldehyde further reacts with a new 1,3-diol molecule to form 1,3-dioxane.214
In the case of Zr and Ti oxides (entries 12 and 13), mainly unsaturated alcohols were found, next to 3-buten-2-one, indicative of the formation of 4-hydroxy-butan-2-one and its dehydration, reaction temperatures as high as 350 °C being required to achieve comparable 1,3-BDO conversions. CeO2 and Yb2O3 were very selective for the formation 3-buten-2-ol (from primary alcohol dehydration) and 2-buten-1-ol (Zaitsev's product from a secondary alcohol dehydration) (entries 14 and 15). Unfortunately no BD was formed over the mildly basic oxides.206,211,214,215 At 325 °C and a WHSV of 6.7 h−1, 73% 1,3-BDO is converted into 3-buten-2-ol and 2-buten-1-ol with a selectivity of 58 and 36%, respectively.
The same authors have also investigated the dehydration of the intermediate enols to BD using acidic SiO2–Al2O3. Interestingly, 3-buten-2-ol and 2-buten-1-ol are selectively converted to BD at 250 °C and a WHSV of 5.7 h−1, 2-buten-1-ol being slightly more reactive, while the anti-Zaitsev's product 3-buten-1-ol is less reactive, primarily forming propylene under the same conditions (entries 16 to 18, Table 7). Clearly, the intermediate formation of the latter enol should be limited as much as possible, as almost no BD is formed from it.
In conclusion, dehydration of 1,3-BDO has been commercially practiced at 400 °C in the presence of steam using a mixture of NaPO3 and Na2P2O7, the pyrophosphate being the dehydrating agent, yielding 80% of BD. Conversion of 1,3-BDO in the presence of acid catalysts mainly follows three parallel pathways in the first dehydration step, leading to a peculiar product regioselectivity with three enol intermediates. While 3-buten-2-ol and 2-buten-1-ol are selectively converted to BD, intermediate formation of 3-buten-1-ol should be avoided since its transformation into BD is unselective. Here, the use of mildly basic rare earth metal oxides like CeO2 may be interesting to apply in a two-stage approach with Brønsted acids like silica–alumina, since it converts 1,3-BDO exclusively into the two preferred enols, which are further dehydrated over the silica–alumina to attain high yields of BD. Despite the promising dehydration of 1,3-BDO to BD, production of 1,3-BDO from sustainable resources is less developed when compared to the other BDOs. Its limited availability via renewable routes presents a major bottleneck.
| C4H10O2 → C4H6 + 2H2O | (3) |
The equilibrium composition shown in Fig. 12 for the dehydration of 1,3-BDO to its three enol products shows that 2-buten-1-ol is favoured thermodynamically at low temperature, whereas the formation of 3-buten-1-ol gains importance at higher temperature. 3-Buten-2-ol will be highly unfavourable over the whole temperature range, if formation of the other two enols is possible. Clearly, the selective formation of BD from 1,3-BDO is a kinetic issue only.
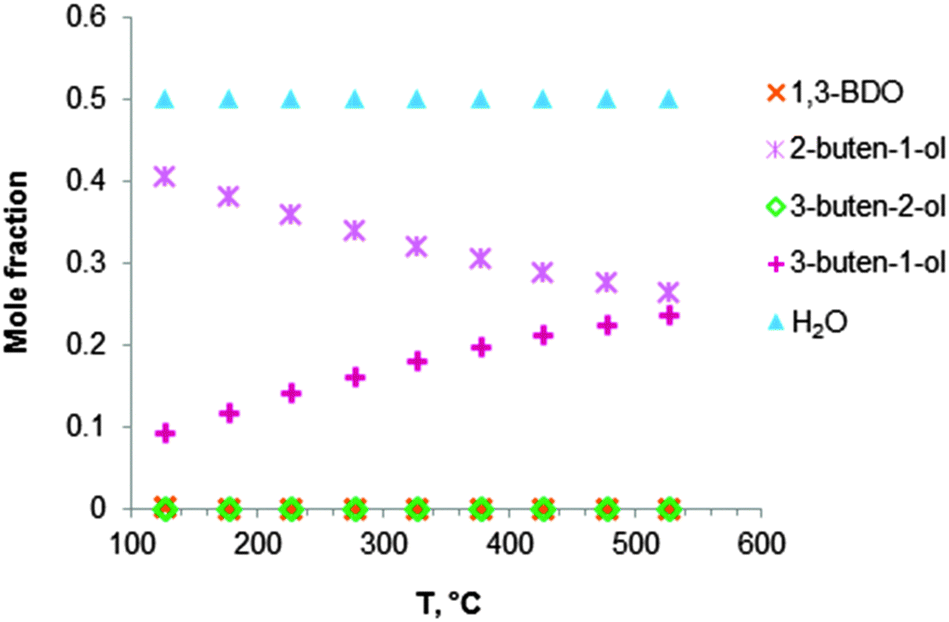 | ||
| Fig. 12 Equilibrium composition calculated for 1,3-BDO to x-buten-y-ol, performed using the Equilibrium based reactor (REquil) in Aspen Plus® software. | ||
As explained before, dehydration of 2,3-BDO shows the dominant formation of butan-2-one rather than BD, due to the presence of an easy pinacol reaction channel. Fig. 13 shows the variation of the equilibrium composition with respect to temperature. It should be noted that butan-2-one is the most favourable product up to 500 °C. At higher temperatures, further dehydration according to the 1,2-elimination mechanism is favoured, resulting in the formation of BD. This means that tuning the catalytic properties towards 1,2-elimination is of utmost importance to attain BD selectively. If not, butan-2-one will be predominantly formed.
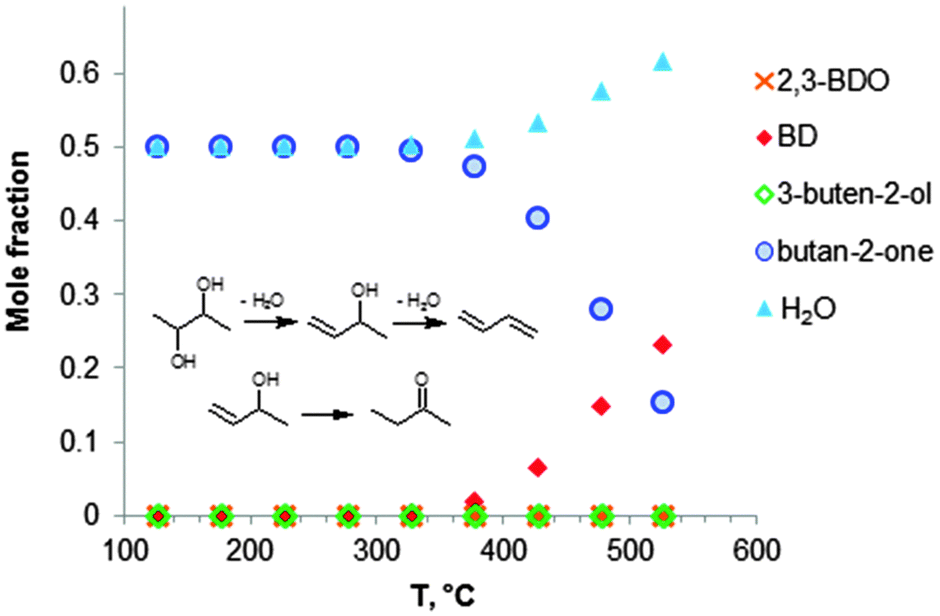 | ||
| Fig. 13 Equilibrium composition calculated for 2,3-BDO to 3-buten-2-ol, BD and butan-2-one, using the Equilibrium reactor (REquil) in Aspen Plus® software. | ||
In the thermodynamic simulations of 1,4-BDO dehydration to BD, the presence of THF in the equilibrium composition was unimportant (data not shown). Thus THF is not thermodynamically favoured, while its abundant formation in the catalytic experiments demonstrates the fast kinetics of the 1,4-cyclodehydration. In the long run, BD formation is favoured, albeit with a high energy barrier. Lowering the energy barrier will be a great challenge for catalytic scientists if production of BD from 1,4-BDO is aimed at.
3.3. Mechanistic and catalytic chemistry of deoxydehydration
Besides the dehydration route for converting BDOs into BD, there also exists a catalytic deoxydehydration (DODH) methodology, involving dehydration with concomitant oxygen transfer to a reductant (Red.). This approach is generalized for vicinal diols in eqn (4), L and M being ligand and (transition) metal, respectively. | (4) |
The metal-catalysed single stage DODH has first been reported by Cook et al. based on a 1,2,3,4,5-pentamethylcyclopentadienyl–ReO3 catalyst (Cp*ReO3) and triphenyl phosphine (PPh3) as the sacrificial reductant.217 Although some homogeneous catalysts based on Ru,218 Mo219 or V220 have since then been examined for DODH potential, most reports focus on the use of Re221–231 as the catalytic centre. Most DODH work was published in view of deoxygenation of biomass derived oxygen-rich substrates to produce new biofuels.232,233 Although erythritol was included as a substrate in the original report by Cook and Andrews,217 the application of DODH to sugar alcohols and real biomass substrates such as glycerol and tetritols and not merely to aliphatic or aromatic 1,2-diols, recently received much interest.221,222,227,229,234,235 Cook et al. have worked under biphasic conditions for DODH of erythritol at 135 °C, using chlorobenzene as a solvent, yielding after 28 h about 80% of BD (entry 1, Table 8). Next to BD as the major product, 3-butene-1,2-diol and cis-2-butene-1,4-diol were formed as well in a molar ratio of 85:15. The latter enolic products are the result of a single DODH reaction on either one of the 1,2 or 2,3-diol couple. Scheme 12 i summarizes all possible enol products that can be derived from erythritol via a single DODH reaction, viz. 3-butene-1,2-diol (1), cis-2-butene-1,4-diol (2) and trans-2-butene-1,4-diol (3). Since 3-butene-1,2-diol is a vicinal diol, it may undergo a second DODH resulting in BD formation. Cook and Andrews have verified the BD formation route by feeding 3-butene-1,2-diol to the catalytic system.217 They observed fast BD formation, whereas no direct DODH was observed when feeding cis-2-butene-1,4-diol. They have noticed some isomerisation of cis-2-butene-1,4-diol into 3-butene-1,2-diol under the catalytic conditions indicating indirect formation of BD, albeit very slow. In the end, because the solubility of the three butenediols is much higher in chlorobenzene than the erythritol reagent, sequential DODH is favoured promoting BD formation. Cook et al. also applied the DODH protocol to xylitol.217
| E | Catalyst | Solvent/red. | Feed | BDa | 1 | 2 | 3 | 4 | 5 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Yields of BD and compounds 1–5, as seen in Scheme 12i, are expressed in mol%. b 0.044 M erythritol (ery), 1.5 mol% Re, 0.044 M PPh3, 135 °C, 28 h obs = observed. c 1 M ery, 1.25 mol% Re, 1.7 mol% TsOH, 160 °C, 12 h, air. d 0.2 M ery, 10 mol% Re, 0.3 M Na2SO3, 155 °C, 100 h. e 0.3 M ery, 2.5 mol% Re, 170 °C, 1.5 h, N2. f 3-Octanol plays the role of solvent and reductant. g Reaction with DL-threitol instead of erythritol, main side product is a diastereoisomer of compound 4. h Conditions as e but at 155 °C for 5.5 h. i Resp. intermediates from Scheme 12i as substrate, 0.5 h of reaction. j 0.1 M erythritol, 2 mol% Re, 0.22 M PPh3, 180 °C, 24 h, N2. k X = 1,2,4-tri(tert-butyl)cyclopentadienyl. l Conditions as j but 15 h. E = Entry. | ||||||||||
| 1b | Cp*ReO3 | C6H5Cl/PPh3 | ery | 80 | obs.b | obs.b | — | — | — | 217 |
| 2c | Re2(CO)10 | 3-Octanolf | ery | — | — | — | — | — | 62 | 221 |
| 3d | Bu4NReO4 | benzene/SO3 | ery | 27 | 0 | 3 | 0 | 0 | 6 | 222 |
| 4e | MeReO3 | 3-Octanolf | ery | 89 | 0 | 0 | 0 | 0 | 11 | 234 |
| 5e | MeReO3 | 3-Octanolf | threig | 81 | 0 | 0 | 0 | 13 | 0 | 234 |
| 6h | HReO3 | 3-Octanolf | ery | 73 | 0 | 0 | 0 | 0 | 7 | 234 |
| 7e | MeReO3 | 3-Octanolf | 2i | 70 | 0 | 0 | 0 | 6 | 0 | 235 |
| 8e | MeReO3 | 3-Octanolf | 3i | 70 | 0 | 0 | 0 | 0 | 0 | 235 |
| 9j | XReO3k | PhCl/PPh3 | ery | 18 | 0 | 6 | 2 | — | — | 229 |
| 10l | XReO3k | Pyridine/PPh3 | ery | 30 | 4 | 3 | 5 | — | — | 229 |
| 11e | XReO3k | 3-Octanolf | ery | 67 | 0 | 0 | 0 | — | 7 | 229 |
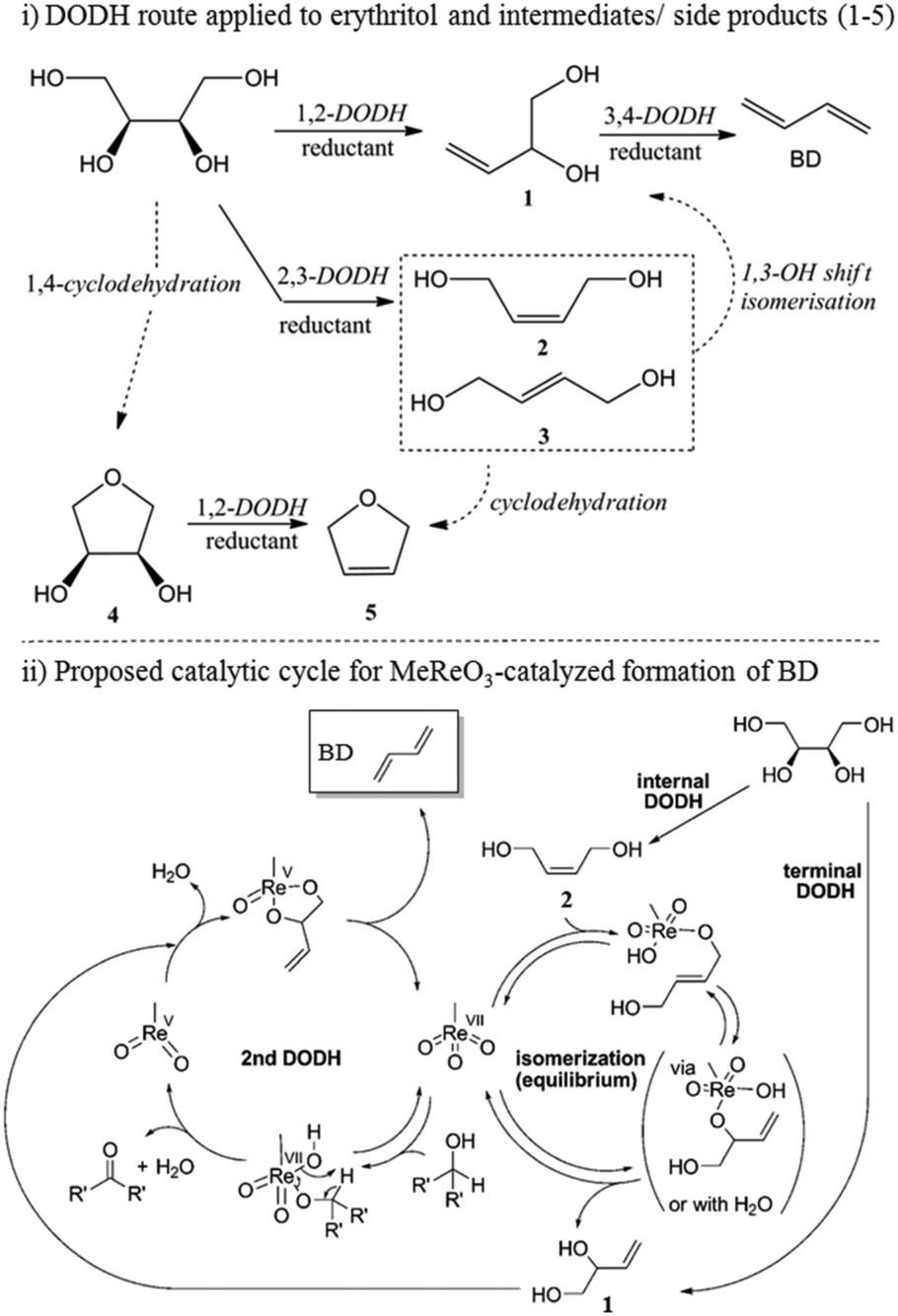 | ||
| Scheme 12 Tetritol to BD route: (i) main products: 3-butene-1,2-diol (1); cis-2-butene-1,4-diol (2), 3: trans-2-butene-1,4-diol (3); 1,4-anhydroerythritol (4); 2,5-dihydrofuran (5). (ii) Proposed catalytic cycle, adapted from Shiramizu et al.235 | ||
Arceo et al.208 have studied the use of Re2(CO)10 in 3-octanol, acting as solvent and the reductant for the conversion of erythritol in the presence of p-toluenesulfonic acid. The reaction (entry 2, Table 8) mainly showed 2,5-dihydrofuran formation. Erythritol underwent a sequential 1,4-cyclodehydration yielding 1,4-anhydroerythritol (4 in Scheme 12), followed by a vicinal DODH reaction leading to the formation of 2,5-dihydrofuran (5 in Scheme 12). The presence of the acid additive was suggested to be the driver for cyclisation to 1,4-anhydroerythritol. Such a mechanism corroborates with the well-established substitution-independent acidic cyclisation of γ-diols (see the 1,4-BDO section).178
In 2011, Ahmad et al. have investigated a sulphite driven oxorhenium catalysed approach.222 Bu4NReO4 (perrhenate salts) was tested with a Na2SO3 reductant in benzene for the DODH of erythritol (entry 3, Table 8) at 155 °C. BD, cis-2-butene-1,4-diol and 2,5-dihydrofuran were encountered, with BD being the major product (24%). MeReO3 was also used in combination with sulphites, but this Re catalyst performed less well. The reaction products of these DODH reactions are fully in line with the proposed Re reaction system of Cook et al. with PPh3 (entry 1), and helped corroborating the reaction path in Scheme 12i.
However, Nicholas et al.209 attributed the formation of 2,5-dihydrofuran via fast 2,3-DODH, followed by a 1,4-cyclodehydration. Given the long reaction times, the sulphite225 system seemingly presents a less active DODH system for tetritols. Mechanistically, the first 1,2-diol DODH transforming erythritol into 3-butene-1,2-diol is generally considered faster than a 2,3-DODH (Scheme 12i). The second DODH, of 3-butene-1,2-diol into BD, is considered to be even faster, because this diol was never noticed in the initial reaction stage. Although considerably slower, and thus not a major pathway, the initial 2,3-diol DODH leading to the cis rather than to the trans form of 2-butene-1,4-diol, further confirms the preference of oxorhenium for a syn-oriented elimination.
Both Shiramizu and Toste234 and Yi et al.227 independently have further explored the DODH reaction on erythritol, both with a less complex and commercially available MeReO3 catalyst. The former authors have provided a breakthrough in the frame of BD production, by adopting a closed system in excess of a sacrificial alcohol reductant – as in the work of Bergman221 – with the significantly simpler MeReO3 catalyst to assist the DODH. This transfer hydrogenation, here demonstrated with 3-octanol to 3-octanone, is useful in view of the production of bio-alcoholic solvents like isobutanol. Moreover, the formed ketone is easily reduced and recycled, if necessary, or may be used in other reactions like aldol condensation. The authors have reported very efficient double DODH on erythritol and DL-threitol at 170 °C, yielding BD in 89% yield at high conversion (entries 4 and 5, Table 8). The major side-product from erythritol was 2,5-dihydrofuran in line with the Bergman's report, suggesting a dominant 1,4-cyclodehydration activity (see Scheme 12i). In the case of DL-threitol, the major side-product was a trans isomer of 1,4-anhydroerythritol, a cyclic internal trans-diol. Its formation demonstrates the need for cis-diol substrates to get an efficient vicinal-directed DODH. To check whether the introduction of Brønsted acidity would result in an increase of the 2,5-dihydrofuran yield via the acidic 1,4-cyclodehydration path (Scheme 12i), a reaction with HReO4 as a catalyst was performed under similar conditions (entry 6). However, no changes in the product distribution were noted, while BD remained the main product. This points to the high preference of catalytic DODH rather than cyclodehydration under the conditions used.
Yi et al. also used MeReO3, but mainly reported on a solventless DODH approach, practiced with glycerol.227 To remove and collect the gaseous products, a continuous distillation setup was used, mainly producing allyl alcohol from glycerol. The latter is not only the reactant, it also participates in transfer hydrogenation with co-production of dihydroxyacetone. With 2 mol% of MeReO3 and 1.5 molar equivalents of 1-heptanol as a transfer reagent, erythritol and threitol were converted mainly into gaseous products.214 Using the continuous distillation procedure, no BD was condensed in the collector. Erythritol mainly yielded 2,5-dihydrofuran and cis-but-2-enal, while meso-threitol was converted into (3S,4S)-tetra-hydrofuran-3,4-diol, an isomer of compound 4 in Scheme 12i.227 Differences with the results of Shiramizu and Toste234 (entries 4 and 5, Table 2) may originate either from the excess of sacrificial alcohol used or, more likely, from the differences in setup; viz. an open distillative system versus a closed approach. It appears that the open reactor promotes 1,4-cyclodehydration, next to DODH chemistry, preferably leading to the cyclic species of Scheme 12 rather than to BD. The resemblance with the product spectrum of the Bergman system is obvious.221 However, rather than being the effect of Brønsted acidity (Bergman), here a plausible explanation could be a shift in reaction equilibria, caused by the open distillation, hereby promoting dehydration.
To expand the scope of the oxorhenium catalyzed DODH, Shiramizu and Toste have investigated different substrates.235 Oxorhenium compounds not only catalyse DODH chemistry, but in parallel accelerate 1,3-OH shifts in allylic alcohols. The latter reaction thus formally presents 1,4-DODH and 1,6-DODH (eqn (5) and (6), respectively), rather than classic cis-vicinal or 1,2-DODH (eqn (4)).
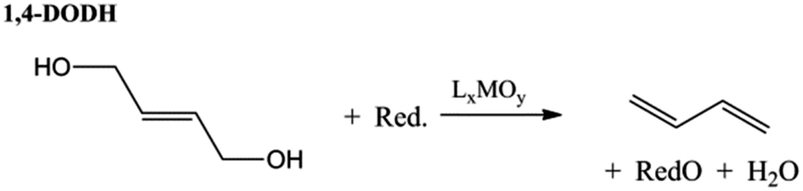 | (5) |
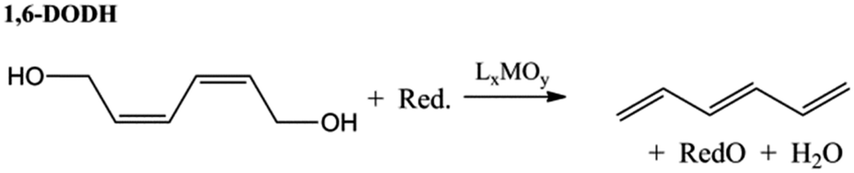 | (6) |
For intermediates like cis- and trans-2-butene-1,4-diol (2 and 3, Scheme 12i) tested under conditions similar to those of the original tetritol, very smooth progression of 1,4-DODH chemistry has been noticed for both isomers.222 The catalytic results (entries 7 and 8, Table 8) indicate that the formation of a 7-membered rhenium-diolate from the catalyst and the 1,4-diol is excluded as both isomers showed identical reactivity. On this basis, the 1,3-OH shift causing isomerisation of allylic alcohols may be included in Scheme 12i (dotted line). Note that Cook et al.217 already noticed the formation of 3-butene-1,2-diol from cis-2-butene-1,4-diol, though at a much slower rate than the DODH reaction. Shiramizu and Toste have proposed a catalytic cycle for their tandem isomerisation/DODH protocol, which is in fact the combination of a 1,4 and 1,2-DODH (Scheme 12ii). In the tentative catalytic cycle it is proposed that the 1,4-DODH reaction proceeds through a 5-ring rheniumV-diolate before expelling the isomerised olefin, i.e. 3-butene-1,2-diol (1) in the case of erythritol. This intermediate is identical to the one in the last step of the final 1,2-DODH reaction, just before BD expulsion (Scheme 12ii). This merged isomerisation-DODH mechanism might explain the high overall DODH efficiency of the MeReO3–3-octanol system, in conjunction with the high substrate solubility. Theoretical density functional studies by Qu et al. have demonstrated the possibility of a pathway involving a MeReO(OH)2 species formed from MeReO3 and the reducing alcohol,231 coordinating diols through double dehydration. Thus, the alcohol may act as a shuttle facilitating various hydrogen-transfer steps. Other mechanistic proposals, elaborated for the synthesis of trans-stilbene, involve a rheniumIII-diolate, formed from 3-octanol reduction of methyldioxorheniumV.228 Finally, Raju et al. reported the conversion of erythritol via DODH with a bulky 1,2,4-tri(tert-butyl)cyclopentadienyl-ReO3 catalyst.229 Entries 9 and 10 in Table 8 show these results, using PPh3 as a reductant in chlorobenzene or pyridine. Low BD yields were noticed and several intermediates, including trans-2-butene-1,4-diol, were obtained, pointing to an inefficient DODH protocol. Under the conditions of Shiramizu and Toste, 67% BD yield was noticed with the aforementioned bulky catalyst (entry 11), underlining the importance of solvent and the reductant in DODH reactions, as well as the appropriateness of using a sacrificial alcohol such as 3-octanol.221,223,234
As mentioned before, the four-carbon sugar-based feedstock is currently not produced on a large scale and certainly not on the scale requested for BD production. Therefore, it seems unlikely that it will become shortly a viable feedstock for BD via DODH chemistry. Alternatively, the development has been reported for new routes to tetritols via decarbonylation,236 or to tetroses via decarboxylation237 of larger carbohydrates such as pentoses and hexoses and via aldol condensation of glycolaldehyde.173 Pentose and hexoses are abundantly present in (hemi-)cellulosic biomass,238–240 while glycol aldehyde is a dominant compound in bio-oils from biomass pyrolysis.173,241,242 Interestingly, the elegant DODH approach was also performed with glucose-derived sugar alcohols such as mannitol and sorbitol, leading to a hexatriene as the major product.234 Such hexitol feedstocks can be directly derived from cellulosics.175,243,244
A second issue of current DODH next to feedstock availability and cost lies in the use of the expensive complex Re catalysts. Taking into account their turnover rate, their use on a large scale appears uneconomical for the production of BD. Note however that NH4ReO4 has a significantly lower cost.223 For an industrial breakthrough of the DODH approach, research should be directed to the development of stable and practical heterogeneous catalysts. Denning et al. have recently ventured into this area, with the synthesis of a carbon supported perrhenate DODH catalyst.224 In addition, the solventless distillation of Ji et al.,214 which was mainly applied to convert glycerol, could be envisioned occurring in a full continuous mode. It seems that such open and continuous DODH systems are worthy of exploring for BD production from tetritols, provided 2,5-dihydrofuran formation can be suppressed.
4. Conclusions and perspectives
On-purpose synthesis of BD has received a recent revival of interest because its availability from naphtha and gas crackers is diminishing. Worldwide investment in BD extraction capacity at the crackers and dehydrogenation of the butane fraction, either as a component of natural gas and shale gas or as a waste fraction from petrorefinery, are two obvious ways to satisfy the needs of a firm BD market. Although the butane dehydrogenation route to BD is an energy consuming process because of its endothermicity, state-of-the-art technology has proven its reliability in the past and new initiatives were recently announced. However, driven by the shortage of light alkanes due to lightening of cracker feeds and the increasing global demand for bio-derived chemicals, biomass-based conversion might offer new avenues for on-purpose BD production from renewable feedstock. The major technical challenge here is to produce BD from biomass and/or biomass-derived syngas that will be cost competitive with fossil oil-based BD. Several research and development needs are required to answer critical questions regarding the technical, viz. process (operational) costs, and economic, viz. feedstock price and BD price, feasibility to integrate a bio-derived BD in the chemical industry.Ethanol can be converted to BD. This route has been practised on the industrial scale long before fossil oils and gases found general use. The industrial production of bio-ethanol is under extensive development, enabling production of large amounts of ethanol in the near future. Other encouraging developments show ethanol production from syngas, either derived from natural gas, waste gas or biomass feedstock. Also methanol carbonylation, followed by acetic acid hydrogenation might turn out promising for ethanol production. Meanwhile, fermentation became a very competitive process to ethanol.
The conversion of ethanol to BD is thermodynamically feasible and proceeds via a sequence of reactions, receiving general acceptance, viz. ethanol dehydrogenation, aldol condensation, MPV reduction, and dehydration. Clearly, a subtle balance of the four reactions is required to achieve high BD yield.
The literature overview indicates that a strong base like MgO or ZnO is essential to catalyse the aldol coupling, while Ag and CuO are good candidates to assist the dehydrogenation step. The catalytic system tolerates the presence of weak Brønsted acidity, e.g. surface hydroxyls of silica, alumina or clays, catalysing dehydration, though its amount and strength should be limited, to avoid the thermodynamically favourable ethylene as the side-product. The activity of the MPV hydrogen transfer reaction is likely most difficult to understand at the molecular level, but it is related to the presence of Lewis acid sites, on incompletely coordinated Mg2+ in MgO and some clays or on Al3+ in alumina and clays. The addition of ZrO2 is an interesting option to assist aldol condensation and MPV reduction.
Current BD selectivity with such catalysts ranges from 50 to 80%, the latter being obtained at modest conversion, thus showing room for improvement and development. Hydrocarbons like ethylene and butenes and oxygenates like acetaldehyde are produced, thereby reducing total BD selectivity. While ethylene may be valorised, the formation of butenes is unfortunate because of separation issues with BD. Another challenge of the ethanol-to-BD reaction consists in its low volume productivity and often low BD concentration in the product stream. Current values range from 50 to 400 gBD h−1 per litre catalyst volume, with values below 100 being reported most frequently. Thus one faces the challenging task to develop more active and selective catalysts at higher ethanol concentration. A low ethanol conversion should not always be an inconvenience per se as efficient recycling of unconverted ethanol is optional, as well as a product recycle since many by-products can be formed from the various intermediates in the reaction sequence. Besides recycling reactors, reactor types like a fluidized bed could favour the cascade of consecutive reactions to BD due to better heat supply and back-mixing;108 there has been no systematic study of the influence of the reactor design and reactor type on the production of BD from ethanol. Most of the research and catalyst developments were devoted to experiments in plug flow reactors.
There is also little information available about the importance of the purity of the ethanol for BD production. Sporadically, a beneficial effect of water is reported, possibly as a dilute of ethanol or as a reactant that poisons strong acid–base sites. Others reported on the usage of diethyl ether in the feed stream as a better reagent for BD production. The presence of impurities of larger alcohols like isoamyl alcohol in the bio-ethanol feed has not been investigated in the context of on-purpose BD production.
Catalysts of particular interest for further improvement would include a combination of the abovementioned elements. Catalyst preparation and pretreatment techniques are very important in order to maintain high dispersion and ideal mixtures/phases of the different functions. Structural modifiers are likely required to stabilize the high dispersion against sintering. Scalability and cost of catalyst preparation/modification are important factors to consider as well with these multi-component catalysts.
Whereas whole series of catalyst compositions have been tested, many reports unfortunately lack thorough physicochemical characterization and kinetic approaches defining the ideal catalyst in terms of acid–base and redox properties. Studies of individual reaction types in the conditions of BD formation are highly welcome,90,245,246 but this is not always possible due to the thermodynamic constraints. They will form a first step in the deeper understanding of the correlation between the active sites. A better understanding of the adsorption phenomena and surface chemistry is crucial, especially of the critical reaction steps determining selectivity and rate limiting events. Advanced (in situ and operando) spectroscopic tools are available today to better monitor the reaction processes at the microscopic and molecular level and the catalyst behaviour with nanoscale resolution under reaction conditions. Crucial information on the overall catalyst behaviour, in terms of textural and chemical (redox, acid–base) properties, is needed, ultimately helping improving overall catalytic performance.
Next to ethanol, there have been recent developments in producing bio-derived 4-carbon alcohols, like n-butanol, isobutanol and various butanediols such as 1,3-BDO, 1,4-BDO and 2,3-BDO using fermentation of sugars. Also fermentation of syngas from natural gas or biomass has been demonstrated to provide some of the diols. Dehydration of the monohydric alcohols provides bio-derived butenes, which could be dehydrogenated to BD, preferably in the presence of oxidants to compensate for the endothermicity. Availability of cheap alcohols will allow commercialization of the ODH technology.
Dehydration of butanediols to BD has received increasing interest given the availability of bio- and gas-derived BDO's. Each diol has its own unique features and challenges. Dehydration of 2,3-BDO faces a facile pathway to butan-2-one, following a pinacol-type rearrangement. Whereas higher temperatures will stimulate BD formation thermodynamically, kinetic control is key to achieve high BD yields. This remains the key challenge for the catalytic chemist, especially with the renewable production of 2,3-BDO just around the corner. For as such unknown reasons, a ThO2 catalyst currently delivers the best catalytic performances.
Dehydration of 1,4-BDO suffers from the production of THF. Again, use of higher temperatures is part of the thermodynamic solution, although kinetics to a major extent still determine the selectivity. THF can be further processed into BD, albeit at high temperatures. There is another challenging route proceeding according to a double dehydration with intermediate formation of 3-buten-1-ol. Suitable catalysts are neutral to basic like CeO2 and Yb2O3. Combination of effective catalysis with the recent breakthrough in fermentative 1,4-BDO production is a route to BD that probably will be picked up in the very near future.
Dehydration of 1,3-BDO is a classical route, as this diol was the key intermediate in the acetylene-based industrial BD production long time ago. Therein, cheap Na–polyphosphate catalysis allowed good catalytic dehydration results.2 Alternatively, a two-step reaction with CeO2 may be envisioned producing selectively 2-buten-1-ol and 3-buten-2-ol, followed by their dehydration to BD using common Brønsted acid catalysts like silica–alumina. However, the bottleneck in this approach resides in the development of a renewable production of 1,3-BDO.
Finally, the pioneering work of deoxydehydration of butanediols and tetritols is an elegant chemistry to produce BD. Although this route is probably far from commercialisation, it shows that creative thinking could lead to original catalytic pathways towards useful drop-in chemicals from bio-derived chemicals. Development of cheap catalysts is essential though, and perhaps heterogeneous catalysis may offer the solution.
Acknowledgements
E.V.M. acknowledges the KU Leuven (Spiro fellowship) for financial support. M.D. thanks the Research Foundation Flanders (FWO Vlaanderen) for a postdoctoral fellowship. W.J. thanks KU Leuven for PhD financing (FLOF). IAP (Belspo) and the Belgium government are acknowledged for general financial support of the research of B.F.S.Notes and references
- R. F. Goldstein and A. L. Waddams, The petroleum chemicals industry, E.&F.N. Spon LTD, London, 1967 Search PubMed.
- H.-J. Arpe, Industrial Organic Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- H. N. Sun and J. P. Wristers, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000, pp. 365–392 Search PubMed.
- K. Laird, Bio-based chemicals receive boost from shale gas boom in North America, 2014, http://www.plasticstoday.com/articles/bio-based-chemicals-receive-boost-from-shale-gas-boom-in-north-america-140523?cid=nl.plas08 Search PubMed.
- M. Hackett, Chemical Building Blocks from Renewables, 2014, IHS Chemical Special Report, http://www.ihs.com/products/chemical/planning/special-reports/chemical-renewables.aspx.
- G. Centi and R. A. van Santen, Catalysis for Renewables, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 387–411 Search PubMed.
- RFA's Ethanol Industry Outlook, 2013, http://www.ethanolrfa.org/pages/annual-industry-outlook.
- C. A. Cardona and Ó. J. Sánchez, Bioresour. Technol., 2007, 98, 2415–2457 CrossRef CAS PubMed.
- L. Panella, Sugar Tech, 2010, 12, 288–293 CrossRef CAS PubMed.
- B. Kamm, P. R. Gruber and M. Kamm, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- C. S. Goh and K. T. Lee, Renew. Sustainable Energy Rev., 2010, 14, 842–848 CrossRef CAS PubMed.
- R. P. John, G. S. Anisha, K. M. Nampoothiri and A. Pandey, Bioresour. Technol., 2011, 102, 186–193 CrossRef CAS PubMed.
- M. Y. Menetrez, Environ. Sci. Technol., 2012, 46, 7073–7085 CrossRef CAS PubMed.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS PubMed.
- R. C. Saxena, D. K. Adhikari and H. B. Goyal, Renew. Sustainable Energy Rev., 2009, 13, 167–178 CrossRef PubMed.
- A. Singh, P. S. Nigam and J. D. Murphy, Bioresour. Technol., 2011, 102, 10–16 CrossRef CAS PubMed.
- J. O. B. Carioca, Biotechnol. J., 2010, 5, 260–273 CrossRef CAS PubMed.
- Axens, IFPEN and Michelin Join Forces to Create a Synthetic Rubber Production Channel Using Biomass, 2013, http://www.axens.net/news-and-events/news/297/axens-ifpen-and-michelin-join-forces-to-create-a-synthetic-rubber-production-channel-using-biomass.html.
- Axens partners with Michelin, IFP in French research initiative for biobased butadiene, 2013, IHS Chemical Week, http://www.chemweek.com/markets/basic_chemicals/petrochemicals/butadiene/Axens-partners-with-Michelin-IFP-in-French-research-initiative-for-biobased-butadiene_56616.html.
- E. Celinska and W. Grajek, Biotechnol. Adv., 2009, 27, 715–725 CrossRef CAS PubMed.
- Z.-L. Xiu and A.-P. Zeng, Appl. Microbiol. Biotechnol., 2008, 78, 917–926 CrossRef CAS PubMed.
- M. J. Burk, A. P. Burgard, R. E. Osterhout, J. Sun and P. Pharkya, WO 2012/177710, 2012.
- R. E. Osterhout, A. P. Burgard and M. J. Burk, WO 2013/028519, 2013.
- Genomatica, Successful commercial-scale production of BDO, 2013, http://www.genomatica.com/products/bdo/.
- R. Hill and E. Isaacs, GB 483989, 1938.
- M. Köpke, C. Mihalcea, F. Liew, J. H. Tizard, M. S. Ali, J. J. Conolly, B. Al-Sinawi and S. D. Simpson, Appl. Environ. Microbiol., 2011, 77, 5467–5475 CrossRef PubMed.
- M. Mohammadi, G. D. Najafpour, H. Younesi, P. Lahijani, M. H. Uzir and A. R. Mohamed, Renew. Sustainable Energy Rev., 2011, 15, 4255–4273 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- Y. Liu, K. Murata, M. Inaba, I. Takahara and K. Okabe, Catal. Today, 2011, 164, 308–314 CrossRef CAS PubMed.
- J. R. Rostrup-Nielsen, Catal. Rev. Sci. Eng., 2004, 46, 247–270 CAS.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- M. Scates, R. D. Shaver, J. Zink, R. Zinobile and O. Oyerinde, WO 2013/070216, 2013.
- F. R. Bengelsdorf, M. Straub and P. Duerre, Environ. Technol., 2013, 34, 1639–1651 CrossRef CAS.
- D. W. Griffin and M. A. Schultz, Environ. Prog. Sustainable Energy, 2012, 31, 219–224 CrossRef CAS.
- A. M. Henstra, J. Sipma, A. Rinzema and A. J. M. Stams, Curr. Opin. Biotechnol., 2007, 18, 200–206 CrossRef CAS PubMed.
- M. Köpke, C. Mihalcea, J. C. Bromley and S. D. Simpson, Curr. Opin. Biotechnol., 2011, 22, 320–325 CrossRef PubMed.
- P. C. Munasinghe and S. K. Khanal, Bioresour. Technol., 2010, 101, 5013–5022 CrossRef CAS PubMed.
- J. Grub and E. Löser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- Linde and BASF intend to jointly develop on-purpose butadiene technology, 2014, http://www.basf.com/group/pressrelease/P-14-249.
- Honeywell UOP Licensing TPC Group's Advanced Process Technology to Produce Key Ingredient for Synthetic Rubber, 2014, http://www.uop.com/?press_release=honeywell-uop-licensing-tpc-groups-advanced-process-technology-to-produce-key-ingredient-for-synthetic-rubber.
- T. Black and J. Kaskey, Honeywell, BASF tout rival solutions for global butadiene shortage, 2014, http://www.hydrocarbonprocessing.com/Article/3351368/Channel/194955/Honeywell-BASF-tout-rival-solutions-for-global-butadiene-shortage.html.
- J. M. Berak, R. Guczalski, J. Wójcik and S. Zalwert, Przem. Chem., 1962, 41, 80–84 CAS.
- G. Egloff and G. Hulla, Chem. Rev., 1945, 36, 63–141 CrossRef CAS.
- J. A. Gamma and T. Inouye, Chem. Metall. Eng., 1942, 49, 97–100 CAS.
- J. A. Gamma and T. Inouye, Chem. Metall. Eng., 1943, 50, 94–97 CAS.
- A. Talalay and M. Magal, Synthetic Rubber from Alcohol. A Survey Based on the Russian Literature, Interscience Publishers, Inc., New York, 1945 Search PubMed.
- A. Talalay and L. Talalay, Rubber Chem. Technol., 1942, 15, 403–429 CrossRef CAS.
- L. Tong, Z.-Z. Liu and M.-h. Zhang, Chem. Ind. Eng., 2012, 29, 38–44 CAS.
- Y. Wang and S. Liu, J. Bioprocess Eng. Biorefinery, 2012, 1, 33–43 CrossRef PubMed.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- J. Sun and Y. Wang, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008, 259, 183–189 CrossRef CAS PubMed.
- T. Tsuchida, S. Sakuma, T. Takeguchi and W. Ueda, Stud. Surf. Sci. Catal., 2007, 172, 543–544 CAS.
- T. Tsuchida, S. Sakuma, T. Takeguchi and W. Ueda, Ind. Eng. Chem. Res., 2006, 45, 8634–8642 CrossRef CAS.
- V. F. Tretyakov, Y. I. Makarfi, R. M. Talyshinsky, N. A. Frantsuzova, V. N. Torhovsky, S. N. Antonuk and K. V. Tretyakov, Fine Chem. Technol., 2010, 5, 77–86 Search PubMed.
- L. D. Feng, X. S. Chen, B. Sun, X. C. Bian and Z. M. Chen, Polym. Degrad. Stab., 2011, 96, 1745–1750 CrossRef CAS PubMed.
- S. Ogo, A. Onda, Y. Iwasa, K. Hara, A. Fukuoka and K. Yanagisawa, J. Catal., 2012, 296, 24–30 CrossRef CAS PubMed.
- M. León, E. Díaz and S. Ordóñez, Catal. Today, 2011, 164, 436–442 CrossRef PubMed.
- T. Haishi, K. Kasai and M. Iwamoto, Chem. Lett., 2011, 40, 614–616 CrossRef CAS.
- M. León, E. Díaz, A. Vega, S. Ordóñez and A. Auroux, Appl. Catal., B, 2011, 102, 590–599 CrossRef PubMed.
- V. N. Ipatieff, J. Russ. Phys.-Chem. Soc., 1903, 35, 449–452 Search PubMed.
- O. G. Filippov, J. Russ. Phys.-Chem. Soc., 1910, 42, 364–365 Search PubMed.
- I. I. Ostromyslensky, J. Russ. Phys.-Chem. Soc., 1915, 47, 1472–1506 Search PubMed.
- A. T. Maximoff, US 1,682,919, 1928.
- S. V. Lebedev, FR 665917, 1929.
- S. V. Lebedev, GB 331482, 1930.
- S. V. Lebedev, Zh. Obshch. Khim., 1933, 3, 698–717 CAS.
- Y. A. Gorin, Zh. Obshch. Khim., 1946, 16, 283–294 CAS.
- W. J. Toussaint and J. T. Dunn, US 2,421,361, 1947.
- A. A. Balandin, Zh. Fiz. Khim., 1935, 6, 357 Search PubMed.
- M. Y. Kagan, G. D. Lyubarskii and O. M. Podurovskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1947, 173–181 CAS.
- W. M. Quattlebaum, W. J. Toussaint and J. T. Dunn, J. Am. Chem. Soc., 1947, 69, 593–599 CrossRef CAS.
- G. Natta and R. Rigamonti, Chim. Ind., 1947, 29, 195–200 CAS.
- S. K. Bhattacharyya and S. K. Sanyal, J. Catal., 1967, 7, 152–158 CrossRef CAS.
- H. E. Jones, E. E. Stahly and B. B. Corson, J. Am. Chem. Soc., 1949, 71, 1822–1828 CrossRef CAS.
- Y. A. Gorin, Zh. Obshch. Khim., 1946, 16, 1089–1100 CAS.
- W. J. Toussaint, J. T. Dunn and D. R. Jackson, Ind. Eng. Chem., 1947, 39, 120–125 CrossRef CAS.
- O. M. Vinogradova, N. P. Keier and S. Z. Roginskii, Dokl. Akad. Nauk SSSR, 1957, 112, 1075–1078 CAS.
- H. Niiyama, S. Morii and E. Echigoya, Bull. Chem. Soc. Jpn., 1972, 45, 655–659 CrossRef CAS.
- S. Kvisle, A. Aguero and R. P. A. Sneeden, Appl. Catal., 1988, 43, 117–131 CrossRef CAS.
- Y. A. Gorin, K. N. Charskaya and A. V. Bochkareva, Sb. Statei Obshch. Khim., 1953, 2, 818–822 CAS.
- V. Gruver, A. Sun and J. J. Fripiat, Catal. Lett., 1995, 34, 359–364 CrossRef CAS.
- J.-B. d'Espinose de la Caillerie, V. Gruver and J. J. Fripiat, J. Catal., 1995, 151, 420–430 CrossRef.
- E. Arundale and L. A. Mikeska, Chem. Rev., 1952, 51, 505–555 CrossRef CAS.
- A. A. Davydov and Y. M. Shchekochikhin, Kinet. Katal., 1971, 12, 383–389 CAS.
- Y. A. Gorin, Zh. Obshch. Khim., 1950, 20, 1596–1614 CAS.
- Y. A. Gorin and N. A. Kalinicheva, Zh. Obshch. Khim., 1952, 22, 1256–1266 CAS.
- J. I. Di Cosimo, V. K. Diez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova, S. Tolborg and E. Taarning, J. Catal., 2014, 316, 121–129 CrossRef PubMed.
- M. Stöcker, Zeolites and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 687–711 Search PubMed.
- Y. A. Gorin and I. K. Gorn, Zh. Obshch. Khim., 1948, 18, 646–655 CAS.
- Y. A. Gorin, Zh. Obshch. Khim., 1948, 18, 1065–1068 CAS.
- D. J. Elliott and F. Pennella, J. Catal., 1989, 119, 359–367 CrossRef CAS.
- K. Inui, T. Kurabayashi and S. Sato, J. Catal., 2002, 212, 207–215 CrossRef CAS.
- T. Nishiguchi, T. Matsumoto, H. Kanai, K. Utani, Y. Matsumura, W.-J. Shen and S. Imamura, Appl. Catal., A, 2005, 279, 273–277 CrossRef CAS PubMed.
- G. M. Sinaiskii, T. V. Ratner, V. P. Makarova, Y. A. Gorin, V. S. Ivanov and L. V. Alferova, Zh. Obshch. Khim., 1957, 27, 2927–2931 CAS.
- S. V. Lebedev, SU 24,393, 1931.
- S. V. Lebedev, DE 577,630, 1933.
- S. V. Lebedev, SU 35,182, 1934.
- B. B. Corson, H. E. Jones, C. E. Welling, J. A. Hinckley and E. E. Stahly, Ind. Eng. Chem., 1950, 42, 359–373 CrossRef CAS.
- I. R. László, B. Falkay and L. Hegyessy, Magy. Kem. Foly., 1954, 60, 54–74 Search PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev., 2001, 43, 443–488 CAS.
- S. K. Bhattacharyya and N. D. Ganguly, J. Appl. Chem., 1962, 12, 105–110 CrossRef CAS.
- V. F. Tret'yakov, S. N. Khadzhiev, R. M. Talyshinskii, A. L. Maksimov and A. M. Ilolov, RU 2459788, 2012.
- S. K. Bhattacharyya and N. D. Ganguly, J. Sci. Ind. Res., 1960, 19, 33–34 Search PubMed.
- S. K. Bhattacharyya and N. D. Ganguly, J. Appl. Chem., 1962, 12, 97–104 CrossRef CAS.
- S. K. Bhattacharyya, N. D. Ganguly and B. N. Avasthi, Kinet. Katal., 1965, 6, 144–148 CAS.
- W. Szukiewicz, US 2,357,855, 1944.
- W. Szukiewicz, GB 573,631, 1945.
- W. Szukiewicz, CA 448,963, 1948.
- G. Natta and R. Rigamonti, Chim. Ind., 1947, 29, 239–243 CAS.
- B. Kovařík, Collect. Czech. Chem. Commun., 1959, 24, 1260–1267 CrossRef.
- H. Leszczyńska, Z. Leszczyński and E. Treszczanowicz, Przem. Chem., 1955, 34, 47–54 Search PubMed.
- R. Ohnishi, T. Akimoto and K. Tanabe, J. Chem. Soc., Chem. Commun., 1985, 1613–1614 RSC.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344 CrossRef CAS PubMed.
- L. Tong and Z.-Z. Liu, Xiandai Huagong, 2012, 32, 39–44 CAS.
- B. Kovařík, J. Beníšek and J. Zavřel, Chem. Prum., 1960, 10, 81–83 Search PubMed.
- R. I. Silva, R. Frety and Y. L. Lam, Actas do 9.o simpósio iberoamericano de catálise, Lisbon, Portugal, 1984, pp. 604–613 Search PubMed.
- O. B. Krylov, Heterogeneous Catalysis, Akademkniga, Moscow, 2004 Search PubMed.
- J. M. Berak, R. Guczalski and J. Wójcik, Acta Chir. Acad. Sci. Hung., 1966, 50, 163–166 CAS.
- Y. Kitayama, M. Satoh and T. Kodama, Catal. Lett., 1996, 36, 95–97 CrossRef CAS.
- V. Aleixandre and T. Fernandez, An. Edafol. Fisiol. Veg., 1958, 17, 805–832 Search PubMed.
- E. Suzuki, S. Idemura and Y. Ono, Appl. Clay Sci., 1988, 3, 123–134 CrossRef CAS.
- D. J. Butterbaugh and L. R. U. Spence, US 2,447,181, 1948.
- D. J. Butterbaugh and L. R. U. Spence, US 2,423,681, 1948.
- L. R. U. Spence, D. J. Butterbaugh and D. G. Kundiger, US 2,436,125, 1948.
- M. Lewandowski, G. S. Babu, M. Vezzoli, M. D. Jones, R. E. Owen, D. Mattia, P. Plucinski, E. Mikolajska, A. Ochenduszko and D. C. Apperley, Catal. Commun., 2014, 49, 25–28, DOI:10.1016/j.catcom.2014.02.003.
- B. Kovařík, Collect. Czech. Chem. Commun., 1961, 26, 1918–1924 CrossRef.
- A. J. Dandy and M. S. Nadiye-Tabbiruka, Clays Clay Miner., 1982, 30, 347–352 CAS.
- V. Aleixandre and T. Fernandez, An. Edafol. Fisiol. Veg., 1958, 17, 305–319 Search PubMed.
- T. Kitayama and T. Wada, JP 57 102822, 1982.
- Y. Kitayama and A. Abe, J. Chem. Soc. Jpn., Chem. Ind. Chem., 1989, 11, 1824–1829 CrossRef.
- Y. Kitayama, T. Hoshina, T. Kimura and T. Asakawa, Proceedings of the 8th International congress on catalysis, Verlag Chemie, Weinheim, FRG, Berlin (West), 1984, pp. V647–V657 Search PubMed.
- Y. Kitayama and A. Michishita, J. Chem. Soc., Chem. Commun., 1981, 401–402 RSC.
- Y. Kitayama, K. Shimizu, T. Kodama, S. Murai, T. Mizusima, M. Hayakawa and M. Muraoka, Stud. Surf. Sci. Catal., 2002, 142, 675–682 CrossRef.
- Y. Kitayama and T. Wada, JP 58 059928, 1983.
- L. R. U. Spence and D. J. Butterbaugh, US 2,423,951, 1947.
- L. R. U. Spence, D. J. Butterbaugh and D. G. Kundiger, US 2,438,464, 1948.
- M. D. Jones, C. G. Keir, C. D. Iulio, R. A. M. Robertson, C. V. Williams and D. C. Apperley, Catal. Sci. Technol., 2011, 1, 267–272 CAS.
- V. V. Ordomskiy, V. L. Sushkevich and I. I. Ivanova, WO 2012/015340, 2012.
- W. Knepper and G. Maaß, DE 946886, 1956.
- K. Arata and H. Sawamura, Bull. Chem. Soc. Jpn., 1975, 48, 3377–3378 CrossRef CAS.
- I. I. Ostromyslensky, J. Russ. Phys.-Chem. Soc., 1915, 47, 1509–1529 Search PubMed.
- A. Maximoff and O. Canonici, GB 535,678, 1941.
- A. Maximoff and O. Canonici, US 2,297,424, 1941.
- H.-J. Chae, T.-W. Kim, Y.-K. Moon, H.-K. Kim, K.-E. Jeong, C.-U. Kim and S.-Y. Jeong, Appl. Catal., B, 2014, 150–151, 596–604 CrossRef CAS PubMed.
- C. Romanovsky and T. E. Jordan, US 2,800,517, 1957.
- C. Romanovsky and T. E. Jordan, US 2,819,327, 1958.
- C. Romanovsky and T. E. Jordan, US 2,819,327, 1958.
- B. B. Corson, E. E. Stahly, H. E. Jones and H. D. Bishop, Ind. Eng. Chem., 1949, 41, 1012–1017 CrossRef CAS.
- M. J. Burk, D. S. J. Van, A. Burgard and W. Niu, WO 2008/115840, 2008.
- W. Clark, M. Japs and M. J. Burk, WO 2010/141780, 2010.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. B. Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. Van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef CAS PubMed.
- H. Gräfje, W. Körnig, H.-M. Weitz, W. Reiß, G. Steffan, H. Diehl, H. Bosche, K. Schneider and H. Kieczka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- X.-J. Ji, H. Huang and P.-K. Ouyang, Biotechnol. Adv., 2011, 29, 351–364 CrossRef CAS PubMed.
- M. J. Syu, Appl. Microbiol. Biotechnol., 2001, 55, 10–18 CrossRef CAS.
- J. Daniell, M. Köpke and S. Simpson, Energies, 2012, 5, 5372–5417 CrossRef CAS PubMed.
- LANZATECH, INVISTA and LanzaTech Sign Joint Development Agreement for Bio-Based Butadiene http://www.lanzatech.com/sites/default/files/imce_uploads/news_release_-_invista_announces_lanzatech_partnership_-_embargoed_final_0.pdf.
- A. P. Burgard, M. J. Burk, R. E. Osterhout, P. Pharkya and J. Li, WO 2013/036764, 2013.
- R. E. Osterhout, A. P. Burgard and M. J. Burk, WO 2013/028519, 2013.
- A. P. Burgard, M. J. Burk, R. E. Osterhout and P. Pharkya, WO 2010/127319, 2010.
- P. Brandão, A. Philippou, J. Rocha and M. Anderson, Catal. Lett., 2002, 80, 99–102 CrossRef.
- M. A. Makarova, E. A. Paukshtis, J. M. Thomas, C. Williams and K. I. Zamaraev, J. Catal., 1994, 149, 36–51 CrossRef CAS.
- M. Mascal, Biofuels, Bioprod. Biorefin., 2012, 6, 483–493 CrossRef CAS.
- R. M. West, D. J. Braden and J. A. Dumesic, J. Catal., 2009, 262, 134–143 CrossRef CAS PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef CAS PubMed.
- V. García, J. Päkkilä, H. Ojamo, E. Muurinen and R. L. Keiski, Renew. Sustainable Energy Rev., 2011, 15, 964–980 CrossRef PubMed.
- P. Dürre, Curr. Opin. Biotechnol., 2011, 22, 331–336 CrossRef PubMed.
- E. M. Green, Curr. Opin. Biotechnol., 2011, 22, 337–343 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, F. de Clippel, J. Dijkmans, D. W. Gammon and B. F. Sels, ChemCatChem, 2013, 5, 569–575 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, S. De Smet, R. De Clercq, L. Verbelen, P. Van Puyvelde, F. E. Du Prez and B. F. Sels, ACS Catal., 2013, 3, 1786–1800 CrossRef CAS.
- B. M. Kabyemela, T. Adschiri, R. M. Malaluan, K. Arai and H. Ohzeki, Ind. Eng. Chem. Res., 1997, 36, 5063–5067 CrossRef CAS.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed.
- R. Fittig, Justus Liebigs Ann. Chem., 1860, 114, 54–63 CrossRef.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, 2007 Search PubMed.
- M. Bartók and Á. Molnár, Ethers, Crown Ethers, Hydroxyl Groups and their Sulphur Analogues (1981), John Wiley & Sons, Ltd., 2010, pp. 721–760 Search PubMed.
- I. Bucsi, Á. Molnár, M. Bartók and G. A. Olah, Tetrahedron, 1994, 50, 8195–8202 CrossRef CAS.
- H. Waldmann and F. Petr
![[u with combining dot above]](https://www.rsc.org/images/entities/char_0075_0307.gif) , Chem. Ber., 1950, 83, 287–291 CrossRef CAS.
, Chem. Ber., 1950, 83, 287–291 CrossRef CAS. - J. van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 CrossRef CAS.
- W. J. Hale and H. Miller, US 2400409, 1946.
- W. J. Hale, US 2441966, 1948.
- A. N. Bourns and R. V. V. Nicholls, Can. J. Res., Sect. B, 1947, 25B, 80–89 CrossRef CAS PubMed.
- M. E. Winfield, J. C. S. I. R. (Aust.), 1945, 18, 412–423 CAS.
- M. Winfield, Aust. J. Chem., 1950, 3, 290–305 CrossRef.
- S. V. Kannan and C. N. Pillai, Indian J. Chem., 1969, 7, 1164–1166 CAS.
- Á. Molnár, I. Bucsi and M. Bartók, Stud. Surf. Sci. Catal., 1988, 41, 203–210 CrossRef.
- A. Molnar, I. Bucsi and M. Bartok, Acta Phys. Chem., 1985, 31, 571–579 CAS.
- J. Lee, J. B. Grutzner, W. E. Walters and W. N. Delgass, Stud. Surf. Sci. Catal., 2000, 130C, 2603–2608 CrossRef CAS.
- W. Zhang, D. Yu, X. Ji and H. Huang, Green Chem., 2012, 14, 3441–3450 RSC.
- A. Multer, N. McGraw, K. Hohn and P. Vadlani, Ind. Eng. Chem. Res., 2013, 52, 56–60 CAS.
- B. Török, I. Bucsi, T. Beregszászi, I. Kapocsi and Á. Molnár, J. Mol. Catal. A: Chem., 1996, 107, 305–311 CrossRef.
- Y. H. Han and H. R. Kim, KR 2012107353, 2012.
- J. Baek, T. Y. Kim, W. Kim, H. J. Lee and J. Yi, Green Chem., 2014 10.1039/C4GC00485J.
- I. Bucsi, Á. Molnár, M. Bartók and G. A. Olah, Tetrahedron, 1995, 51, 3319–3326 CrossRef CAS.
- L. K. Freidlin and V. Z. Sharf, Izv. Akad. Nauk SSSR, Ser. Khim., 1960, 1700–1702 CAS.
- W. Reppe, Justus Liebigs Ann. Chem., 1955, 596, 80–158 CrossRef CAS.
- W. Reppe, A. Steinhofer and G. Daumiller, DE 899350, 1953.
- H. Katori and N. Murai, JP 07082191, 1995.
- T. Haas, B. Jaeger, R. Weber, S. F. Mitchell and C. F. King, Appl. Catal., A, 2005, 280, 83–88 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, T. Sodesawa and N. Honda, J. Mol. Catal. A: Chem., 2004, 221, 177–183 CrossRef CAS PubMed.
- N. Yamamoto, S. Sato, R. Takahashi and K. Inui, Catal. Commun., 2005, 6, 480–484 CrossRef CAS PubMed.
- N. Yamamoto, S. Sato, R. Takahashi and K. Inui, J. Mol. Catal. A: Chem., 2006, 243, 52–59 CrossRef CAS PubMed.
- A. Igarashi, N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, Appl. Catal., A, 2006, 300, 50–57 CrossRef CAS PubMed.
- M. Segawa, S. Sato, M. Kobune, T. Sodesawa, T. Kojima, S. Nishiyama and N. Ishizawa, J. Mol. Catal. A: Chem., 2009, 310, 166–173 CrossRef CAS PubMed.
- A. Igarashi, S. Sato, R. Takahashi, T. Sodesawa and M. Kobune, Catal. Commun., 2007, 8, 807–810 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, M. Kobune, H. Inoue, Y. Izawa, H. Ohno and K. Takahashi, Appl. Catal., A, 2009, 356, 64–71 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, T. Sodesawa, N. Honda and H. Shimizu, Catal. Commun., 2003, 4, 77–81 CrossRef CAS.
- S. Sato, F. Sato, H. Gotoh and Y. Yamada, ACS Catal., 2013, 3, 721–734 CrossRef CAS.
- W. Reppe and U. Hoffmann, DE 578994, 1933.
- L. P. Kyriakides, J. Am. Chem. Soc., 1914, 36, 987–1005 CrossRef.
- N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2006, 256, 106–112 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, T. Sodesawa, A. Igarashi and H. Inoue, Appl. Catal., A, 2007, 328, 109–116 CrossRef CAS PubMed.
- H. Gotoh, Y. Yamada and S. Sato, Appl. Catal., A, 2010, 377, 92–98 CrossRef CAS PubMed.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- S. Stanowski, K. M. Nicholas and R. S. Srivastava, Organometallics, 2012, 31, 515–518 CrossRef CAS.
- L. Hills, R. Moyano, F. Montilla, A. Pastor, A. Galindo, E. Álvarez, F. Marchetti and C. Pettinari, Eur. J. Inorg. Chem., 2013, 3352–3361 CrossRef CAS.
- G. Chapman and K. M. Nicholas, Chem. Commun., 2013, 49, 8199–8201 RSC.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed.
- I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef CAS.
- C. Boucher-Jacobs and K. M. Nicholas, ChemSusChem, 2013, 6, 597–599 CrossRef CAS PubMed.
- A. L. Denning, H. Dang, Z. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef CAS.
- P. Liu and K. M. Nicholas, Organometallics, 2013, 32, 1821–1831 CrossRef CAS.
- J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M. Abu-Omar, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed.
- J. Yi, S. Liu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 1401–1404 CrossRef CAS PubMed.
- S. Liu, A. Senocak, J. L. Smeltz, L. Yang, B. Wegenhart, J. Yi, H. I. Kenttämaa, E. A. Ison and M. M. Abu-Omar, Organometallics, 2013, 32, 3210–3219 CrossRef CAS.
- S. Raju, J. T. B. H. Jastrzebski, M. Lutz and R. J. M. Klein Gebbink, ChemSusChem, 2013, 6, 1673–1680 CrossRef CAS PubMed.
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Chem. – Eur. J., 2013, 19, 13224–13234 CrossRef CAS PubMed.
- S. Qu, Y. Dang, M. Wen and Z.-X. Wang, Chem. – Eur. J., 2013, 19, 3827–3832 CrossRef CAS PubMed.
- S. Dutta, ChemSusChem, 2012, 5, 2125–2127 CrossRef CAS PubMed.
- J. O. Metzger, ChemCatChem, 2013, 5, 680–682 CrossRef CAS.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed.
- R. N. Monrad and R. Madsen, J. Org. Chem., 2007, 72, 9782–9785 CrossRef PubMed.
- G. Hourdin, A. Germain, C. Moreau and F. Fajula, J. Catal., 2002, 209, 217–224 CrossRef CAS.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- M. S. Mettler, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7797–7809 CAS.
- C. R. Vitasari, G. W. Meindersma and A. B. de Haan, Green Chem., 2012, 14, 321–325 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93 CrossRef CAS PubMed.
- V. L. Sushkevich, I. I. Ivanova and E. Taarning, ChemCatChem, 2013, 5, 2367–2373 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |