 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical methods for sensing and imaging oxygen: materials, spectroscopies and applications†
Xu-dong
Wang
and
Otto S.
Wolfbeis
*
Institute of Analytical Chemistry, Chemo- and Biosensors, University of Regensburg, D-93040 Regensburg, Germany. E-mail: otto.wolfbeis@ur.de; Fax: +49 (0)941 943 4064; Tel: +49 (0)941 943 4065
First published on 18th March 2014
Abstract
We review the current state of optical methods for sensing oxygen. These have become powerful alternatives to electrochemical detection and in the process of replacing the Clark electrode in many fields. The article (with 694 references) is divided into main sections on direct spectroscopic sensing of oxygen, on absorptiometric and luminescent probes, on polymeric matrices and supports, on additives and related materials, on spectroscopic schemes for read-out and imaging, and on sensing formats (such as waveguide sensing, sensor arrays, multiple sensors and nanosensors). We finally discuss future trends and applications and summarize the properties of the most often used indicator probes and polymers. The ESI† (with 385 references) gives a selection of specific applications of such sensors in medicine, biology, marine and geosciences, intracellular sensing, aerodynamics, industry and biotechnology, among others.
 Xu-dong Wang (left) and Otto S. Wolfbeis (right) | Xu-dong Wang, born in 1985, received his PhD degree in Chemistry from Xiamen University (Xiamen, China) in 2011. He was an Alexander von Humboldt fellow in the group of Prof. Wolfbeis and is currently working at the Karlsruhe Institute of Technology. His research interests include the design of optical chemical and biosensors, nanosensors and molecular probes, developing novel (bio)sensing schemes, novel (nano)-materials and methods, and their applications to biological and chemical analysis. He has authored or coauthored more than 20 articles in the past five years, and his current h-index is 13 (as of Jan 2014). |
Otto S. Wolfbeis was a Full Professor of Analytical and Interface Chemistry at the University of Regensburg from 1995 to 2012. He has authored numerous papers on optical (fiber) chemical sensors, fluorescent probes, labels and assays, on nanomaterials for use in sensing schemes and in spectroscopic methods including fluorescence (lifetime) imaging. He has acted as the (co)organizer of several conferences related to fluorescence spectroscopy (MAF) and to chemical sensors and biosensors (Europtrode). His current h-index is 75. He is one of the 10 curators of Angewandte Chemie and the editor of two other journals. Also see: http://www.wolfbeis.de. |
1. Introduction
Almost all living organisms utilize oxygen‡ for energy generation and respiration. Discovered by Joseph Priestley in the 1770s, this event was elected as one of the top 100 greatest science achievements because of the significance of oxygen in science and technology.1 Oxygen is involved in practically all forms of living organisms and played a major role in evolution.2 Adult humans metabolize ∼200 g of oxygen per day. Human beings can live up to 1 month without food, up to 2 weeks without water, but not longer than maximally 10 min without supply of oxygen. It therefore does not come as a surprise that the (analytical) chemistry of oxygen plays a major role in biology and medicine. Sensing (i.e., continuous monitoring) of oxygen has attracted particular attention as can be seen from the numbers of respective references. A search for an “oxygen sensor” in SciFinder yields ∼8670 references, and ∼27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 references are found that contain the concept “oxygen sensor” (search performed on August 1st, 2013).
500 references are found that contain the concept “oxygen sensor” (search performed on August 1st, 2013).
There are four major methods known for determination of oxygen. These are (a) the classical Winkler titration,3 (b) electroanalytical,4 (c) pressure-based, and (d) optical methods. The latter can be subdivided into numerous single methods that range from direct spectroscopy to indicator based methods. Each method has its specific merits and applications.5–7 The Winkler method is precise but does not enable continuous sensing. The Clark electrode8 is the state of the art for the determination of oxygen at room temperature and in small volume. It works both in gaseous and fluid samples (whole blood included). Clark electrodes can be applied at temperatures up to ∼200 °C. They perform excellently but consume the analyte and are interfered by gases such as chlorine, ozone or nitrogen oxides. The quantitation of oxygen in (car) exhaust gases (which is challenging in view of the high temperatures of such samples) is performed using solid-state electrically conducting sensor materials to provide a feedback signal in catalytic converters. This so-called lambda probe (for a review, see ref. 9) was developed by the Bosch company during the late 1960s and is based on a zirconia ceramic coated on both the exhaust and reference sides with a thin layer of platinum to form a solid-state electrochemical fuel cell, where CO (if present) is oxidized by oxygen to form CO2. Both heated (>300 °C) and (less often) nonheated forms are known. The sensor does not actually measure oxygen concentration, but rather the difference between the amount of oxygen in the exhaust gas and the amount of oxygen in the supplied air.
Optical oxygen sensors have become attractive in the past four decades because of features such as (a) the lack of oxygen consumption during measurements; (b) full reversibility; (c) good precision and accuracy; (d) the possibility of remote sensing using optical fibers; (e) the ease of miniaturization (down to the size of nanosensors); (f) the option of performing non-invasive measurements; and (g) the highly attractive feature of enabling imaging of oxygen both over large areas and on a micrometer scale. The success of optical sensors for oxygen is corroborated by the number of companies that are manufacturing respective instrumentation, examples being Presens (probably the largest; www.presens.de); Centec (www.centec.de); Ocean Optics, Inc. (www.oceanoptics.com); Oxysens, Inc. (www.oxysense.com); Finesse, Inc. (www.finesse.com); PyroScience (www.pyro-science.com); and Hach-Lange (www.hach-lange.de), to mention the larger ones. In the medical field, OptiMedical Systems, Inc. (www.optimedical.com) and Terumo (www.terumo-cvs.com/products/) probably are the largest.
There is some confusion in terms of definitions and terminology. We refer to a sensing element as a material composed of both an oxygen-sensitive probe (OSP) and an appropriate (polymer) matrix that acts as a host or support (see Table 1). A complete sensor device will also incorporate a readout unit to give an electrical (or digital) signal. Ideally, the following (“Cambridge”) sensor definition applies: a sensor is a (small) instrumental system capable of (optically) detecting and quantifying a physical or chemical parameter over time and with high specificity (under the given circumstances). The vast majority of sensors for oxygen consists of an oxygen sensitive probe, a polymer or matrix for hosting the oxygen sensitive probe, a read-out (electronic) system, and a device to process data. A discussion on the definitions of sensors, probes and labels was presented.10
| Term | Definition |
|---|---|
| (Optical) probe, indicator, molecular probe | A molecule that displays an optical effect with a specific analyte such as oxygen (or pH, or a metal ion). Whatever their name: they are not sensors. OSP is used here as an acronym for oxygen-sensitive probe. |
| Polymer, host, binder, matrix, paint | These terms relate to a polymer that hosts a probe (an indicator), for example an OSP. The choice of the polymer (and any additives) is a very critical step in sensor development. |
| Sensor chemistry, “stimulus-responsive polymer” | A combination of materials (typically a polymer containing a probe and, possibly, additives) that enables continuous chemical sensing of oxygen; can be manufactured in various formats such as in the form of a sensor film, of sensor nanoparticles, or as a coating on (or at the distal end of) an optical waveguide. |
| Sensor cocktail, ink, paint | A solution of a sensor “chemistry” in an appropriate solvent. |
| Sensor | The definition of a sensor as a “miniaturized device that can deliver real-time and on-line information on specific parameters” was used for decades until organic chemists spoiled it by referring to molecules, probes, indicators and the like as “sensors”. Sensors are devices according to this definition, and they are produced by the millions. One may differentiate between physical sensors (such as those for temperature, pressure, acceleration) and chemical sensors (such as for pH, oxygen, methane, NOx, glucose). Sensors incorporate a readout system and are expected to provide a digital signal. |
In addition to direct spectroscopic methods (see Section 3), oxygen can be sensed via absorptiometric probes, i.e. those that undergo a color change upon exposure to oxygen (see Section 4), but the vast majority of optical sensors for oxygen are based on luminescent probes (Section 6) that are contained in an oxygen-permeable polymer (Section 7.1). Fig. 1 shows a cross-section of a typical planar luminescent sensor for oxygen. It consists of a solid but optically transparent support and a polymer matrix (referred to as the “sensing layer”) permeable to oxygen that contains a quenchable probe. Optionally, a black cover (the so-called “optical isolation”, see Section 8.4) is placed on top to prevent sample luminescence originating from blood or (plant) tissue, for example, from interfering. The sensor layer is illuminated on one side and luminescence is collected on the same side. The sample to be analyzed is in contact with the sensing element on the other side, in Fig. 1 placed on top.
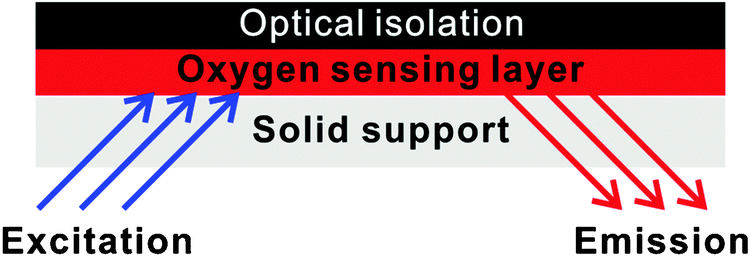 | ||
| Fig. 1 Schematic cross-section of a typical planar luminescent sensor layer (not to scale). | ||
Most oxygen-sensitive probes (OSPs) are used either in surface-absorbed or in polymer-dissolved form. In their pioneering work back in 1931, Kautsky and Hirsch11 used probes such as trypaflavin adsorbed on a silica gel solid support. The use of solid (polymeric) supports not only separates the OSP from the sample, but also can increase the QY of an OSP (due to rigidization) and may also result in the formation of long-lived phosphorescent emissions resulting from long-lived triplet states.12–15 This is quite beneficial because long decay times increase quenching efficiency.16,17 The immobilization of OSPs in matrices offers additional advantages such as improved diffusion and permeability17 so that sensitivity and detection range can be adjusted. In addition, the sensing element can be used for a long time, and the polymer matrix can shield the OSPs from interferences by other quenchers. In fact, some polymers have excellent permeation selectivity. On the other hand, organic polymers and (metal) organic probes tend to decompose at temperatures <200 °C. There is a vast variety of luminescent and other methods to readout the effects caused by oxygen (see Sections 4, 5 and 9).
Once an appropriate material has been identified for use in a sensing element, it can be applied in various formats as outlined in Section 11. The materials are often applied in the form of a (viscous) solution in a (usually organic) solvent, the so-called “sensor cocktail”, which then may be deposited on a mechanical support such as a thin film of an inert and transparent polymer to form a sensor film (or sensor layer, or “paint”). Following solvent evaporation, the resulting (solid) sensor element can then be investigated by various kinds of optical spectroscopies. The cocktail may also be deposited at the tip of an optical fiber, on another kind of waveguide, or even be incorporated into its core or cladding. This will result in so-called fiber optics or waveguide sensors. If placed on the clad of an optical fiber at intervals over a certain distance, so-called distributed sensors are obtained that can be investigated by time-resolved methods of spectroscopy. They allow for continuous and spatially resolved sensing of oxygen along an optical fiber. Sensor chemistries may also be shaped in the form of (nano)particles as will be outlined in Section 11.6. If the sensor material is deposited on the whole object or area of interest, 2-dimensional imaging of oxygen becomes feasible, for example in so-called pressure-sensitive paints that are used to measure the air pressure on cars and aircrafts, to sense oxygen in (cancerous) skin, to monitor photolytic or photosynthetic processes, or the consumption of oxygen in fuel cells.
In this review, we summarize the state of the art in optical sensing and imaging of oxygen. More specifically, we review sensors based on the measurement of absorbance, reflectance and luminescence (including fluorescence, phosphorescence, bioluminescence and chemiluminescence). The spectral range spans the ultraviolet, visible, and near infrared regions. On the other hand, X-ray fluorescence, X-ray photoelectron spectroscopy, and electron paramagnetic resonance are not covered. Several reviews5–7,18–25 and book chapters26–28 related to aspects of optical oxygen sensing have appeared, but they usually cover a narrow field or a limited time frame. Some earlier but useful work has resided hidden for a long time but is cited here.
Papkovsky et al.29 have summarized applications of optical oxygen sensors in biosciences, with a focus on enzymatic assays, respiration, food and microbial safety, bioreactors and fluidic chips. We summarize the subject as a whole and are presenting a comprehensive and hopefully clear blueprint for optical sensing of oxygen. It covers the state of the art from the first reported optical sensing scheme for oxygen11 to the present. Quaranta et al.30 have summarized the wealth of metal ligand complexes that is available for use in optical sensing of oxygen.
2. A look back
Quenching of luminescence by oxygen and other quenchers was long known but not understood until Stern and Volmer derived their famous equation.31 Optical continuous sensing of oxygen probably started back in the 1930's when Kautsky and Hirsch reported11,32 that the room-temperature phosphorescence (RTP) of certain dyes adsorbed on silica particles is quenched by even trace quantities of oxygen. Sensing beads were prepared by soaking silica gel particles with a solution of dyes such as trypaflavine or fluorescein. They were then dried and placed in a flow-through cell schematically shown in Fig. 2. RTP was monitored using a fluorometer and it was found that even ppm quantities of oxygen gas (equivalent to a pO2 of as little as 0.5 × 10−3 Torr) were detectable. The effect was found to be fully reversible within 1–2 s. The device led to the discovery of the Kautsky effect, i.e. the delay in the production of oxygen following the illumination of a leaf. This is a good example on how chemical sensor technology can impact other kinds of science.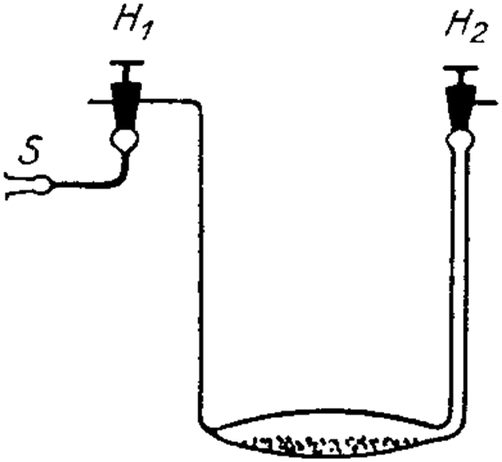 | ||
| Fig. 2 Schematic of the first continuous sensor for oxygen. A sample gas containing oxygen (S) is passed via valve H1 over silica beads dyed with a quenchable probe for oxygen and placed in a small flow-through cell. The room temperature phosphorescence of the beads on the bottom is quenched by even traces of oxygen and was monitored with a simple fluorometer. | ||
This RTP method was later applied by others33,34 to monitor oxygen in seawater. The first fluorescent sensor system was described by Bergman35 in 1968 and comprised a UV light source, an oxygen-sensitive fluorescent layer composed of porous glass (or a thin film of polyethylene) soaked with the oxygen-sensitive probe fluoranthene, and a photodetector. Thus, it contained all the elements of a modern optical sensor. Oxygen quenches the fluorescence of fluoranthene, and intensity served as the analytical information. The system responds to oxygen at levels above 1 Torr.
The milestone paper by Lakowicz and Weber36 in 1973 on the potential of probing structural fluctuations in proteins with oxygen-quenchable probes also revealed, for many, the potential of such probes for sensing oxygen in unknown (gaseous or fluid) samples. A device similar to the one of Bergman was described, a few years later, in a patent.37 It also mentions the possibility of using a radioactive source (rather than a UV light source) to excite fluorescence. In 1974, Hesse38 described a device that appears to have been the first fiber optic chemical sensor. An oxygen-sensitive chemistry was placed in front of a fiber optic light guide through which exciting light was guided. The fluorescence emitted was guided back through either the same fiber, or through other bundle of fibers. The system is based on the measurement of either fluorescence intensity or fluorescence decay time, both of which are affected by oxygen. In 1975 and 1976, Lübbers and Opitz39,40 described instruments capable of monitoring oxygen or carbon dioxide. They first referred to it as an “optrode” (by lingual analogy to the electrode), and later as an “optode” (oπτιχoσ oδoσ; Greek for “optical way”). They were also the first to apply an oxygen sensor as a transducer in an enzyme-based biosensor.41
The fiber optic oxygen sensor described by Peterson et al.42 in 1984 was a milestone in optical fiber sensor technology for use in medicine. The sensor was used to monitor oxygen in the blood of an ewe in vivo. A planar sensor for oxygen based on the covalent immobilization of the pyrene probe on a porous glass support and with milliseconds response time was reported in the same year.43 A chemiluminescence (CL) based sensor for oxygen was reported by the Seitz group44 that exploits the CL produced in the reaction of oxygen with an electron-rich ethylene. The AVL company has filed a patent45 that describes oxygen sensitive materials based on polycyclic aromatic hydrocarbons dissolved in a film of poly(vinyl chloride) containing a plasticizer. Deposited on an acrylate glass and covered with microparticles of ferric oxide acting as optical isolations to prevent interferences caused by the intrinsic fluorescence of biological matter, they can be used to monitor oxygen in bioreactors, in blood samples and in breath gas.
The first dual sensor (for simultaneous determination of oxygen and the inhalation narcotic halothane) was reported46 in 1985. It is based on dynamic quenching of the fluorescence of the probe decacyclene in silicone rubber by both oxygen and halothane. Interferences by oxygen are taken into account by a second sensor layer covered with polytetrafluoroethylene (which is impermeable to halothane) and responds to oxygen only. Halothane concentrations can be calculated with the help of an extended Stern–Volmer relation (Section 9.8.1). The probe is practically specific for the two analytes, since other gases present in inhalation gases or blood do not interfere.
Quenchable ruthenium ligand complexes for use in optical oxygen sensing were reported47 in 1986. Ruthenium tris(bipyridyl) was adsorbed onto silica gel and placed in a silicone membrane. Ruthenium probes with better QY and known from the work of Alford et al.48 were applied as OSPs by Demas and coworkers49 in 1987. Such complexes have decay times of the order of a few microseconds. The first article50 on luminescence decay time-based sensing of oxygen appeared in 1988. The Wilson group51 introduced a quenchometric scheme for oxygen based on the phosphorescence of metalloporphyrins in aqueous solution and bound to albumin, later to dendritic molecules. Both intensity and decay time were measured as a function of oxygen partial pressure. (Metallo)porphyrin-based sensor membranes were developed in the 1980s by the Gouterman group52 that are suitable for phosphorescent sensing of oxygen, and a strong patent was published53 that covers pressure-sensitive paints based on the same effect. A 2-volume book that appeared in 1991 gives an account of the work on fiber optic chemical sensors and biosensors.27 The first reliable fiber optic oxygen microsensors were introduced54 in 1995. A look back on the history of optical chemical sensor technology up to the year 2000 has been published.55
3. Methods for direct spectroscopic sensing of oxygen
Molecular oxygen has two main absorption bands in its UV-vis spectrum, one deep in the UV, the other at 760 nm which is very weak. It also has an intrinsic emission peaking at 1270 nm. Geddes et al.56 in 1992 monitored breath gas oxygen via its UV absorption at 145 nm. Not unexpectedly, water vapor and carbon dioxide (and probably many other species) interfere. The weak absorption lines at ∼760 nm can be exploited to sense oxygen in high concentration or if large penetration lengths can be accomplished such as in astronomy.57,58 This direct spectroscopic method also enables monitoring of 16O18O and 16O17O isotopes, which provides valuable data with respect to the composition of the protoatmosphere via the trapped gas in the polar ice. The oxygen line absorption was also used to monitor oxygen in harsh environments at high temperature, such as exhaust gas in combustion engines or burners.59 Respective instrumentation requires high-power lasers, long optical pathways (∼100 m), and highly sensitive detectors.Oxygen was also sensed directly by employing gas correlation absorption spectroscopy using multimode diode lasers.60 A diode laser was applied that has an emission spectrum that overlaps the oxygen absorption lines of the absorption band at 760 nm. A detection limit of 700 ppm was achieved with good accuracy (2%) and linearity (R2 = 0.999). For comparison, measurements of ambient oxygen were also performed by tunable diode laser absorption spectroscopy (TDLAS) employing a vertical cavity surface emitting laser. The sensor is based on correlation spectroscopy and displays good stability, is easy to use, and instrumentation is more simple than the TDLAS-based instrumentation. On the other hand, it can only be applied to gaseous samples.
In rather related work, wavelength modulation absorption spectroscopy of oxygen at 760.241 nm was applied61 to determine its concentration in the 0–100% range at ambient pressure. TDLAS was applied, and the oxygen absorption was scanned with a tunable laser, while wavelength modulation spectroscopy was used to obtain the harmonics (1f, 2f, 3f and 4f) of the oxygen absorption signal. The modulation parameters such as the modulation voltage, modulation frequency, reference phase, time constant of the lock-in amplifier, the tuning voltage, and the tuning frequency were optimized to obtain the harmonics of high amplitude and narrow half width. Oxygen concentrations were measured by the following three methods: (i) using only the 2nd harmonic; (ii) using the 2nd and 4th harmonics; and, (iii), using the 1st and 2nd harmonics.
Direct sensing of oxygen via the intrinsic luminescence of singlet oxygen (1Δ) with its peak at 1270 nm has also been reported,62,63 but emission is weak (QY in water is 9.3 × 10−7).64 Sensing of liquid oxygen at high pressure and at high flow rates is challenging. Dynamic quenching of the luminescence of OSPs is very strong at high oxygen concentrations as they occur in fluids, and therefore is not the method of choice to sense liquid oxygen. Here, Raman spectroscopy is superior, as shown by Tiwari et al.65 who designed an integrated fiber optic Raman sensor that employs a frequency-doubled 532 nm cw Nd:YAG laser as the light source. A standard spectrometer can be used to collect the Raman spectrum of liquid oxygen or its mixtures with liquid nitrogen (such as cryogenic fluids used in the supercritical environment of rocket engines). The method is safe, but background luminescence can interfere.
While direct spectroscopic sensing of oxygen appears to be much easier in terms of handling and effort, one has to face the fact that bulky and expensive instrumentation is needed to obtain a practical sensor because the UV absorbance and the emission of singlet oxygen are weak. This makes such approaches less economical and much less convenient. In addition, omnipresent substances such as water and carbon dioxide may interfere. Thus, these approaches cannot be recommended for practical uses except for very special situations.
4. Sensing oxygen using absorptiometric probes
Such sensors are based on the use of optical probes that undergo a chromogenic reaction with oxygen (mostly certain leuco dyes) or where oxygen causes a color shift (such as in the hemoglobin–oxyhemoglobin system). Many of these sensors respond irreversibly. Readout usually is performed by reflectometry, not by absorptiometry. The law of Kubelka and Munk relates the intensity of reflectance at a specific wavelength to the concentration of the absorber dye formed with oxygen in a chromogenic reaction: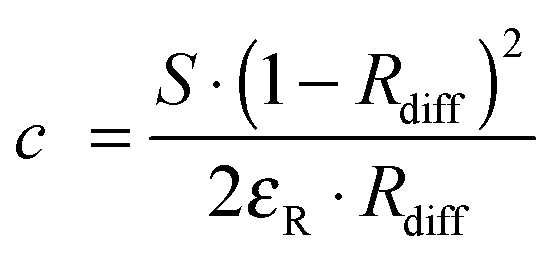 | (1) |
Chromogenic (irreversible) test stripes are widely used in semiquantitative analysis, for example to determine pH values, blood glucose, or nitrate in water samples. The reflectivity of a sensing area typically is read out via small (mostly hand-held) reflectometers comprising (i) a first LED light source operated at the analytical wavelength, (ii) a second LED operated at a wavelength where reflectivity does not change with analyte concentration; (iii) a photodiode detector that alternatively (in ms intervals) reads the reflectivity of the colored and non-colored area; (iv) a power source (usually a battery); (v) an electronic circuit that amplifies the signal of the photodiode; and (vi) a microprocessor that converts the amplified signal into comprehensive information such as a concentration unit.
Absorbance/reflectance-based oxygen sensors can be divided into three sub-groups, viz. (a) those using biological OSPs (such as hemoglobin), (b) those using synthetic oxygen binders (such as certain cobalt complexes), and (c) those using redox chemistry (using the oxidative power of oxygen). The probes can undergo a change in absorbance (intensity) or peak wavelength, or both. All suffer from the fact that they can readily detect the presence of oxygen, sometimes even quantify it, but that they are hardly capable of detecting an increase in the concentration of oxygen over time, and – even less easily – a decrease. The various absorptiometric (reflectometric) methods will be discussed in the following.
4.1. Hemoglobin and myoglobin as optical probes for oxygen
Hemoglobin and myoglobin, when binding oxygen, undergo a large spectral change. Seitz et al.66 constructed an optical oxygen sensor by immobilizing deoxyhemoglobin on a cation exchange resin. The Soret absorption band of deoxyhemoglobin shifts to longer wavelengths when exposed to oxygen to form oxyhemoglobin. The ratio of reflectances at 405 and 435 nm serves as the analytical information. This ratiometric method is suitable to measure oxygen partial pressure from 20 to 100 Torr. However, the response time is long (around 3 min). This kind of oxygen sensor has several limitations: (1) the useful lifetime is short because of irreversible degradation of hemoglobin; (2) response to oxygen partial pressure is nonlinear and not readily described mathematically; (3) the sensor is interfered by gases that can also bind to hemoglobin (such as carbon monoxide), and by pH, because the affinity of hemoglobin for oxygen is a function of pH and ionic strength.Myoglobin was used in another absorbance based oxygen sensor. It reversibly binds dissolved oxygen to form oxy-myoglobin whose absorption spectrum is quite different. Valentine et al.67 used the effect by encapsulating myoglobin in a sol–gel glass matrix to prepare a reversible sensor for dissolved oxygen (DO). The absorbances at 418, 432 and 436 nm change linearly on exposure to dissolved oxygen, but the signal change is small. McCurley et al.68 also encapsulated myoglobin in a sol–gel, reduced it to deoxy-myoglobin by bathing the gel in a dithionite solution, and exposed the sensor gel to DO upon which deoxy-myoglobin is oxidized to oxy-myoglobin. A fluorescent dye, brilliant sulfaflavine, was added to the system. It absorbs light at 430 nm and emits radiation at 520 nm. The excitation light for the dye is passed through the myoglobin-containing gel. The emission of the fluorescent dye changes as the absorbance of the myoglobin at 430 nm changes in response to DO. Carbon monoxide, HCN and SO2 are likely to interfere in both methods.
4.2. Molecular absorptiometric probes for oxygen
Several cobalt–organic compounds are capable of reversibly binding molecular oxygen. The respective absorption spectra change with oxygenation. Baldini et al.69 screened a large number of oxygen carriers and found some of them to be viable optical probes, while others do not give large signal changes or have slow response. The bis(histidinato)cobalt(II) complex [Co(His)2] is well suited. Its absorbance at 408 nm increases strongly with oxygen concentration. An optical fiber sensor was designed that consisted of a polymeric hollow-fiber membrane filled with a solution of Co(His)2. However, its response time is quite long (up to 30 min) and the color of Co(His)2 depends on pH. In order to improve performance, the probe was absorbed on thin layer chromatography plates and then coated with silicone rubber.70 If exposed to an oxygen free environment, the color of the plate changes markedly and can be detected with bare eyes. The effect is reversible and the response time is shorter.The oxygen carrier cobalt(II)-tetrakis(o-pivalamidophenyl)porphinato (CoP) has a unique structure in that it possesses a cavity for reversibly binding oxygen and a coordination site with a nitrogenous ligand to further increase the affinity for oxygen. It was used71 along with two methacrylate-co-vinylimidazole copolymers (one partially fluorinated) to reversibly bind oxygen. The oxygenated complex exhibits a maximum absorbance at 547 nm and an isosbestic point at 536 nm. The sensor was used to determine oxygen in the 1–1000 hPa range, has a short response time (5–15 s) and good long-term stability, but is interfered by humidity. Tsuchida et al.72 synthesized polymers with nitrogenous ligands capable of coordinating CoP. They found that the binding of oxygen by CoP is strongly affected by the kind of polymer depending on their nitrogen ligands. The CoP coordinated to fluorinated polymers is well suited to sense oxygen in water.
The iridium complex Ir(CO)Cl(PPh3)2 undergoes a reversible(!) reaction with oxygen to form Ir(CO)Cl(O2)(PPh3)2 (which however is photosensitive), and this is accompanied by substantial changes in the spectral properties of the complex.73 Various other oxygen-carriers (mainly complexes of cobalt, iron, manganese, platinum and iridium for potential use in oxygen transport) have been described.74,75 The Vaska iridium complex has also been studied76 but was found to react too slowly and not to be sensitive enough.
An optical fiber sensor77,78 utilizes the change in the contact charge-transfer absorption (CCTA) of N,N-dimethyl-p-toluidine in the presence of oxygen. The probe has a broad CCTA band in the UV/Vis region whose absorbance increases with increasing oxygen concentration. The band disappears if oxygen is removed. There is a linear relationship between absorbance and oxygen concentration in accordance with the Beer–Lambert law. Response is reversible, and the sensitivity is higher at shorter wavelength. However, the slope decreases at high oxygen levels, and elemental chlorine and SO2 interfere like in many other sensors for oxygen.
Absorption is not limited to transitions from the ground state to an excited state. The group of Amao79–81 used the transient triplet–triplet absorption of the excited states of the fullerenes C60 and C70 to sense oxygen. The efficiency of this absorption depends on pO2. The method of triplet–triplet (excited state) absorption was also applied to platinum(II) complexes such as PtOEP,82 PtTFPP,82 and certain zinc porphyrins.83 The photoacoustic response84 may also be utilized to measure transient absorptions and thus to optically sense oxygen. This technique is based on photoacoustic probing of the excited state lifetime of Methylene Blue (MB). MB has an absorption peak at 660 nm. A double pulse laser system is used to excite the dye and probe its transient absorption by detecting photoacoustic emission. The relaxation rate of MB depends linearly on oxygen concentration. The measurements show high photoacoustic signal contrast at a wavelength of 810 nm, where the excited state absorption is more than four times higher than the ground state absorption.85
4.3. Methods based on the oxidative power of oxygen
Oxygen is a powerful oxidant that can convert certain colorless species into colored products, or can cause a color change of a dye due to oxidation.86 The reduced (“leuco”) form of Methylene Blue (MB) is colorless and quickly converted back to blue MB by oxygen, a reaction that occurs at room temperature.20 In some cases it has been reported that the chromogenic reaction can be reversed by applying reducing agents or by electroreduction. Early work87 involved the use of leuco MB to determine low levels (<100 μg L−1) of oxygen in power station water. Iron(II) and copper(II) ions interfere and must be removed before analysis by passing the water sample through a cation-exchange column. Others88 have determined dissolved oxygen using photoreduced leuco phenothiazine dyes. Perlman and Linschitz89 have tested numerous oxygen indicators (by placing them in or on irreversibly responding test stripes) for use in packaging, including the leuco forms of MB. The leuco forms were generated by reaction with (strongly smelling) mercaptoethanol and deposited on filter paper, silica particles, nitrocellulose and other solid supports. Other leuco forms include those of thionine and methyl violet. Blue MB can be chemically reduced back to the leuco form with reducing agents such as dithionite, ferrous compounds, sulfite, ascorbic acid, or glucose in alkaline medium.The Mitsubishi Gas Company has patented90,91 and commercialized respective chromogenic oxygen indicators for use in food packaging (called the “Ageless Eye”, Fig. 3). It is also based on leuco MB which was chemically reduced using glucose in alkaline medium. The exposure of the Ageless Eye to oxygen can oxidize the leuco form into the blue form.
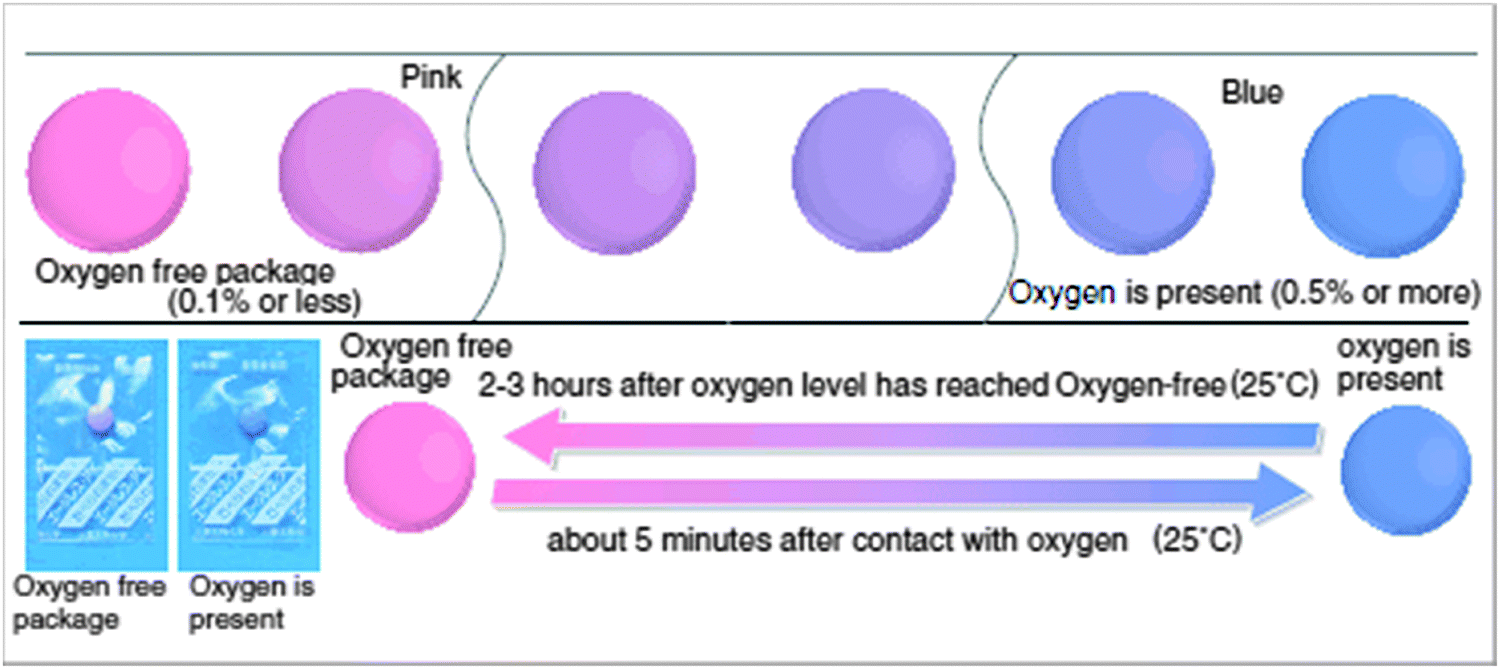 | ||
| Fig. 3 The color of Ageless Eye oxygen indicator changes from pink to blue in the presence of oxygen. | ||
MB can also be reduced by irradiation with UV light.92,93 Re-usable oxygen-sensitive inks were obtained by immobilizing TiO2, the sacrificial electron donor triethanolamine (TEOA), and MB in a hydroxyethyl cellulose (HEC) matrix. The resulting blue TiO2-TEOA-MB-HEC film bleaches under UV irradiation, but not under ambient room light or visible light. If exposed to oxygen, the colorless film is irreversibly oxidized to give a blue film which again can be converted into its colorless form by UV irradiation for about 2.5 min. This cycle can be repeated at least 5 times. The working principle of this scheme is based on the formation of electron–hole pairs in the TiO2 semiconductor particles by UV light. The holes oxidize the TEOA, and the photogenerated electrons reduce the MB dye into its leuco form. Its irreversibility and reusability make this intelligent ink useful for applications in packed food technology. The feature of generating the leuco form by UV irradiation after packing simplifies the process, because handling of leuco dyes is complicated as it requires the complete absence of oxygen. Semiconductors such as ZnO or SnO2 can also be applied for preparing the UV activated oxygen indicator film, but TiO2 works best. The response time can be adjusted by varying the thickness of the sensor film or by using polymers with different oxygen permeability.94
The same group95 later modified the hydrophilic, water-soluble and cationic indicator MB into a hydrophobic MB by exchanging the anion. The hydrophobic indicator is soluble in organic solvents and the blue ink can be printed directly on food package. A film made of TiO2, ion-paired MB, glycerol and a polymer named zein (a prolamine-type of protein found in maize) loses its color rapidly (<30 s) upon exposure to UVA light and remains colourless in an oxygen-free atmosphere, returning to its original blue color upon exposure to air. In the latter step the rate of color recovery is proportional to the level of ambient oxygen and the same film can be UV-activated repeatedly (see Fig. 4). This makes the ink useful for direct printing on food package.
 | ||
| Fig. 4 Sensor film printed with an ink made of titanium dioxide, Methylene Blue, glycerol and a polymer. The film loses its color upon exposure to UV light but becomes blue again on exposure to air. (Reprinted with permission from ref. 95 Copyright Royal Society of Chemistry, 2008). | ||
A photoinitiator was employed96,97 to photoreduce MB via UV irradiation in an acrylate matrix. Irradiation generates radicals that reduce blue MB to its leuco form. Simultaneously, the acrylate monomer is polymerized to form a solid film that contains the indicator. This kind of photoreduction is reversible due to the cyclic processes of (1) oxidation on air, and (2) reduction in an oxygen-free environment under UV light. However, the cycles come to an end once the photoinitiator is consumed. The absorption of MB decreases with the number of cycles on air because of partial photo-decomposition of the indicator.
The polyviologens form a class of less sensitive irreversible oxygen indicators.98 They can also be reduced to colorless forms by exposure to UV light via mediated reduction in the presence of EDTA and TiO2 as described above for MB. However, the rate for reduction is much faster than in the case of MB. The reduced (leuco) forms of thionine and 2,2′-dicyano-1,1′-dimethylviologen were seen to persist until the oxygen concentration exceeded 2.3% and 4.0%, respectively. It was also reported99 that beige-colored anthraquinone β-sulfonate can be reduced to its red phenolate dianion by sodium thiosulfate in alkaline solution. When contacted with oxygen, the red color turns back to beige. This was suggested to serve as a time label to monitor the freshness of food. The rate of the color change can be adjusted by varying the chemical composition of the polyacrylate matrix.
In a fiber optic oxygen sensor for medical use, a viologen indicator is employed that becomes a strong absorber after brief stimulation with UV light. Its color thereafter disappears over time, and the rate of indicator return to transparency is proportional to the local concentration of oxygen.79 Absorbance is monitored with a red LED and a photodiode, and data are processed by a dedicated processor. The solid-state sensor system has a performance that compares to existing oxygen techniques and may be applied to both in vitro and in vivo oxygen assays.
Colorimetric oxygen-sensitive films can also be based on the redox chemistry of 2,6-dichlorophenolindophenol (2,6-DCPIP) in the presence of fructose and an organic base in a thin film of ethyl cellulose.100 The respective sensor film is colorless in the absence of oxygen, but turns to blue in its presence at levels of 30 Torr and above. This represents a simple means for colorimetric detection of oxygen. The oxidized form of 2,6-DCPIP can be reduced by fructose in the presence of a base contained in the polymer film. This “sensor” reversibly responds to oxygen over 10 nitrogen–oxygen switching cycles within a period of ∼5 h. At oxygen pressures between 0 and 50 Torr, there is a linear relationship between the absorbance of the film and oxygen partial pressure, and response occurs within 20 s.
The leuco forms of indigo and thioindigo were immobilized101 in poly(ethylene glycol), polyurethane hydrogel and poly(styrene-co-acrylonitrile) respectively, in order to adjust the permeability for oxygen and, thus, the response time. The reduced (leuco) forms are better soluble and almost colorless. On interaction with oxygen, a vivid blue or red color develops as can be seen in Fig. 5. The color change can be used to irreversibly detect oxygen. The sensor can be reversed (converted into the faintly colored form) by treatment with dithionite. In one further approach,102 an indicator system was described that gives color changes that result from the fact that the interaction of oxygen with the indicator layer causes a change in the local pH value. Numerous indicators were presented.
 | ||
| Fig. 5 Left: real color images of an irreversibly responding test stripe (consisting of a solution of leuco indigo in a polyurethane hydrogel; LI–D4) for oxygen over a time interval of 20 min. Right: real color images of an irreversibly responding test stripe (consisting of a solution of leuco thioindigo in poly(styrene-co-acrylonitrile; LTI-PSAN)) for oxygen over a time interval of 36 h. (Reprinted from ref. 101 with permission from Elsevier). | ||
The same sensor material was also filled in thin capillaries that can serve as opto-chemical timers. The “clock” is started by opening one end of the capillary filled with leuco (thio)indigo.103 The length of the colored section increases over time as oxygen diffuses in. By using different molecular weight poly(ethylene glycols), one can control the permeability for oxygen and, thus, the time frame of the clock. Significant properties of several absorption-based irreversible probes for oxygen are summarized in Table 2. The above technologies are also covered by various patents.
| Dye/matrix | Color transition | Comments | Ref. |
|---|---|---|---|
| Phenothiazine | Colorless → blue | Photoreduced at pH above 9.5; Methylene blue is best because of its stability; the Methylene blue-EDTA solution undergoes more than 150 cycles of photoreduction-air oxidation without apparent degradation of the dye. | 88 |
| Malachite green (leuco form) on silica gel | Colorless → greenish blue | Oxidized by reactive oxygen species only. | 32 |
| 2,6-Dichloroindophenol (leuco form) in ethyl cellulose | Colorless → blue | Indicator is co-immobilized with fructose and base; reversible response with short response time (∼20 s); applicable to oxygen partial pressure between 0–50 Torr, linear relationship between absorbance and oxygen partial pressure. | 100 |
| Methylene blue (leuco form), TiO2 and triethanolamine in hydroxyethyl cellulose | Colorless → blue | UV-activated reusable ink; irreversible oxygen response but regenerable via UV irradiation; regeneration time 2.5 min; stable in an oxygen-free environment; colorimetric determination; response time adjusted via the thickness of the matrix or using polymers with different permeabilities. | 92, 94, 104 |
| Anthraquinone β-sulfonate in pHEMA | Red → beige | Reduced form (dianion) is red; color fades into beige with oxygen; useful for fadable printed signs for food packaging | 99 |
| Indigo (leuco form) and PEG | Colorless → blue | Reduced in basic solution with Na2S2O4; time frame adjusted by using different molecular weight PEG; fairly stable but undergoes some photobleaching upon exposure to visible light; also used as a timer material; periods can be adjusted to up to 40 years. | 101, 103 |
| Thioindigo (leuco form) and PEG | Colorless → bed | Reduced in basic solution with Na2S2O4; the timer covered time frame could be adjusted by using different molecular weight PEG; fairly stable but undergoes some photobleaching on exposure to visible light. | 101, 103 |
| Polyviologens (leuco form), TiO2, EDTA | Pale yellow → purple | More quickly reduced than Methylene Blue upon UV exposure; high anodic redox potential; suitable for oxygen at above 0.5%, even above 4% (the upper LOD for MB is 0.1% only). | 98 |
4.4. Inorganic chromogenic materials
Butler and Ricco105 coated optical fibers with nm-thin films of metals (such as nickel) and found that their reflectivity at 860 nm changes on exposure to oxygen as a result of the formation of a thin layer of an oxide. The film thickness also changes and so affects reflectivity. Instrumentation and techniques are simple and applicable to monitor the corrosion rates of metals. An oxygen sensor for car exhaust gases106 comprises a supported film of a heat-responsive inorganic sensing material having a light reflective surface which reversibly reacts with oxygen to form an oxide which causes a change in the reflectivity of the surface. The sensor also includes a temperature sensing means and a 3-way optical system.5. Oxygen as a quencher of luminescent probes
Most sensors for oxygen are based on the effect of the quenching of the luminescence (fluorescence or phosphorescence) of oxygen-sensitive probes (OSPs). The process involves dynamic collision between molecular (triplet) oxygen and the excited electronic state of the OSP and leads to a reduction of its intensity and decay time. Quenching often is accompanied by the formation of singlet oxygen. It is noted in this context that not a single sensor is known for singlet oxygen. However, several irreversibly responding molecular probes have been reported that can be used to quantify singlet oxygen.107The kinds of methods used in luminescence spectrometry vary to a large extent as will be briefly discussed in this section. The analytically responsive (“dynamic”) range of a sensor material can be easily adjusted by (a) proper choice of the OSP (discussed in Section 6), (b) the matrix materials (discussed in Section 7), and (c) by various additives (Section 8). A discussion of the various read-out schemes and geometries of sensors will then be presented in Section 9.
Dynamic (collisional) quenching by oxygen is a photophysical (rather than a photochemical) process. It is fully reversible, does not alter the optical probe, and thus has no effect on its absorption spectrum. Rather, it leads to a drop in luminescence intensity and decay time.108,109 The relationship between intensity (or decay time) and the concentration of oxygen ([O2]) is reflected by the Stern–Volmer equation which, in its most simple form, reads as
| F0/F = τ/τ0 = 1 + KSV[O2] | (2) |
In an ideal quencher system, there is a linear relationship between F0/F (or τ0/τ) and oxygen concentration as shown in Fig. 6. Unfortunately, data for KSV are given by authors in various units including inverse (partial) pressure, (ppm)−1, %−1, inverse molarity, and others. If data are given in %−1 or (ppm)−1 unit, the barometric pressure most be indicated.
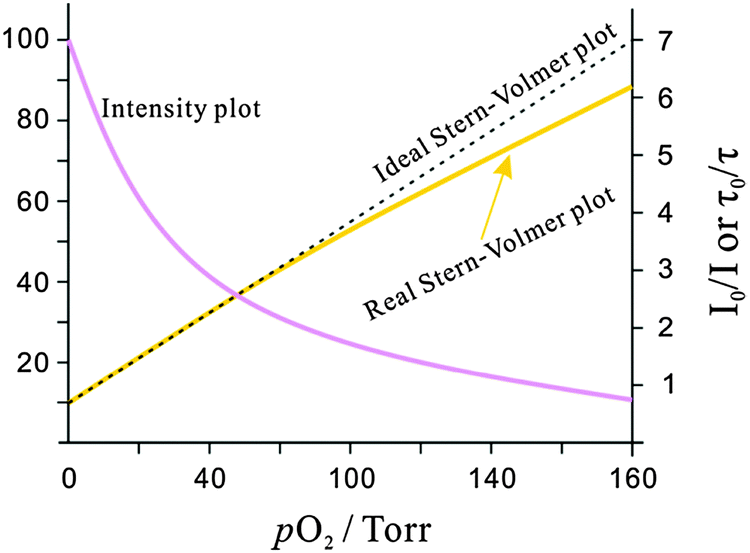 | ||
| Fig. 6 Intensity plot of the quenching of luminescence intensity by oxygen, and respective Stern–Volmer plot. | ||
Stern–Volmer plots (SVPs) can be established by measurement of either luminescence intensity or decay time. However, luminescence intensity data can be adversely affected by poor stability of the light source, variations in the efficiency of the transmission optics, drifts in detector sensitivity, leaching and photodecomposition of probes, inhomogeneous probe distribution, background luminescence and stray light. In order to correct for these effects, an inert reference fluorophore emitting at a different wavelength is often used. See Section 9.9 on referenced sensing.
The Stern–Volmer constant determines the limits of detection (LODs) of a sensor. The LOD is governed by both the initial slope (KSV) of the quenching plot and by the resolution of the instrument. Assuming a ±0.1% uncertainty in light intensity measurement (which is the lower limit and requires a well-thermostatted device), the detection limit is 0.003/KSV (KSV expressed in Torr−1 units and at a signal-to-noise ratio of 3). Phosphorescence based sensors, in contrast to fluorescent sensors, often have much larger KSV values and therefore have much lower LODs.
Many SVPs of sensor films exhibit downward curvature as shown in Fig. 6. This usually is indicating contributions by less efficient mechanisms of quenching. A general discussion of situations where both static and dynamic quenching occur can be found in respective textbooks.108,109 Specifically for the situation of sensing oxygen, Demas et al.110,111 have investigated the photophysics and photochemistry of the quenching by oxygen of several Ru(II) polypyridyl complexes in various polymers. Their results showed that downward curved SVPs originate from the heterogeneity of the microenvironment of the OSPs. They assumed that the Ru(II) polypyridyl complex exists in (at least) two distinctly different environments, one being quenchable, the other either not being quenched at all, or being quenched at a very different rate. A two-site model111,112 was introduced to fit the curved SVPs, which has been widely used ever since. In the conventional form, it reads as follows (eqn (3)),
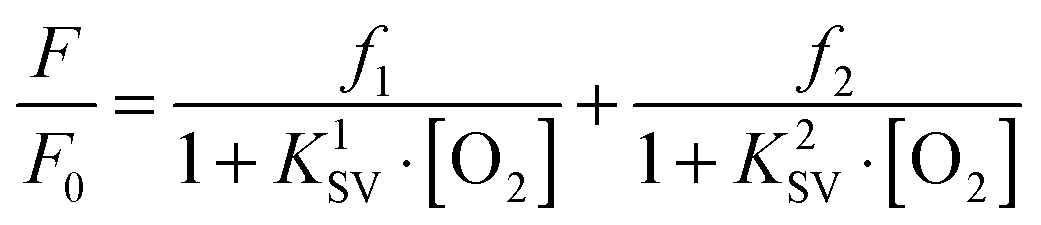 | (3) |
It shall be reminded here that this model is sensitive to signals that contain contributions by stray light, ambient light and background luminescence. There are several reports on the literature where rather low values for K2SV have been calculated, and where contributions from these two sources of error to the overall second quenching constant cannot be excluded. Stray light and background luminescence, but not ambient light, can be reduced – if not eliminated – if gating (time-resolved detection) is applied. Gating must not be confused with lifetime detection and is possible only if long-lived (>1 μs) OSPs are used.
Another model was presented113 that relies on the analysis of the multi-exponential luminescence decay of a luminophore, specifically of Ru(dpp), in various polymers. A fit with a sum of exponentials gives physically unreasonable dependences of the pre-exponential factors of the decay components on the oxygen pressure. This is clear evidence that the decay profile is the total of many single but different relaxation rates. A spatial disorder model can relate the distribution of relaxation rates to a distribution of different distances between the OSP and the sites of interaction in the polymer. The two models for multiple-site quenching have been applied to different situations. Generally, the two-site model is well-suited to describe quenching in heterogeneous systems, while the spatial disorder model better fits homogeneous systems. On the other side, the two-site model can also describe nonlinear quenching in homogeneous systems.114,115
Phosphorescence lifetime analysis was also accomplished using a quadratic programming algorithm that can provide information on the distributions of quenchers in heterogeneous systems.116 The method is based on decomposition of the data vector to a linearly independent set of exponentials and uses quadratic programming principles. Solution of the resulting algorithm requires a finite number of calculations (it is not iterative) and is computationally fast and robust. The algorithm has been tested on various simulated decays and for analysis of phosphorescence data of palladium(II) porphyrins with discrete distributions of lifetimes. The technique is recommended for resolution of the distributions of quencher concentration in heterogeneous samples, of which oxygen distributions in tissue is an important example. Improved calibration of phase-fluorometric oxygen sensors has been demonstrated on the basis of physical models,117 and the response of phase-fluorometric oxygen sensors can be modeled with respect to effects of temperature and operational requirements.118
6. Luminescent probes for oxygen
These form by far the largest group of oxygen-sensitive probes (OSPs). Kautsky and coworkers11,16,32 in the 1930s observed that both the phosphorescence and fluorescence of surface-adsorbed OSPs such as acriflavin, trypaflavin, benzoflavin, safranin, chlorophyll, porphyrins and others are quenched by molecular oxygen. This effect was used to detect the formation of oxygen as a result of photosynthesis, and in turn led to the discovery of the Kautsky effect. After more than 80 years of development, numerous OSPs are available now,30 with excitation and emission maxima ranging from the UV to the NIR, and with excited-state lifetimes ranging from a few nanoseconds to milliseconds if not seconds in the case of phosphorescent probes.Luminescent OSPs can be classified into four subtypes: (1) organic OSPs (mainly polycyclic aromatic hydrocarbons and fullerenes); (2) metal–ligand complexes (mainly transition metal–ligand complexes and metalloporphyrins); (3) luminescent nanomaterials; and (4) multiple emitters and related species. Absorption-based (non-luminescent) OSPs are not included in this part but were discussed in Section 4. Luminescence-based oxygen sensor technologies are covered by numerous patents which cannot be included here.
6.1. Organic probes
| Dye/matrix | λ exc/λem | Sensitivitya | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a The “equal to” symbol (=) indicates a value given by the authors. The “≈” symbol indicates a value that was estimated by the authors of this review from data presented in the respective article. A blank indicates that sensitivity could not be easily calculated for various reasons. | |||||
| Fluoranthene in polyethylene | 330/390 | I 0/I100 ∼ 1.11 | First PAH-based optical sensors for oxygen. | I | 35 |
| Pyrene-1-butyric acid in gel | 345/460 | Short wave excitation; pyrene-1-butyric acid dissolved in organic solvent and then encapsulated in an oxygen-permeable gel to form nanoparticles | I | 39, 158 | |
| Pyrene in silicone rubber (PDMS) | 335/372,384 | I 0/I100 ∼ 6 | Acrylate-PDMS composite; non-linear SVPs; downward curvature is explained by the domain model; other PDMSs also investigated. | I | 159 |
| Pyrene-1-butyric acid on oxidized alumina | 355/474 | I 0/I100 = 6.14 | Anodic oxidized alumina plate; photostable; non-linear SVPs; quenching of the excimer. | I | 129 |
| Pyrenedecanoic acid on alumina plate | 340/376 | I 0/I100 = 18.6 | Anodic oxidized alumina plate; perfluorohydrocarbon added to improve sensitivity and response time; suffers from UV absorption and emission. | I | 134 |
| Pyrendecanoic acid and perfluorodecanoic acid on an alumina plate | 340/396 | I 0/I100 = 20.2 | |||
| Perylenedibutyrate on silica | 468/514 |
I
0/I100 = 1.67
I 0/I21 = 1.19 |
Good stability, low toxicity, loss of sensitivity of about 3% per day during dry storage; loss is 0.1% per day when stored in water. | I | 42 |
| (a) Pyrene in silicone rubber | 320/400 | I 0/I21 = 1.90 | Fluorescence of the energy transfer system (pyrene–perylene) is more efficiently quenched by oxygen than the fluorescence of pyrene itself. | I | 140 |
| (b) Perylene in silicone rubber | 400/474 | I 0/I21 = 1.14 | |||
| (c) Pyrene/perylene in silicone rubber | 320/474 | I 0/I21 = 4.36 | |||
| Decacyclene in PDMS | 385/510 | I 0/I100 = 7.8 | Photostable; excitable in the blue range, large Stokes shift; lifetime is ∼21 ns; QY 0.29 in toluene; decacyclene used in the form of its silicone-soluble tert-butyl derivative; black Teflon used as an optical isolation; also used to sense halothane. | I | 6, 46, 142 |
| Camphorquinone in PS | 470/560 | I 0/I6% = τ0/τ6% ∼ 19 | Monoexponential decay; time and intensity plots coincide for all the polymer matrices; not photostable. | I-RTP; L-RTP | 153 |
| Camphorquinone in PVC | 470/560 | I 0/I21 ∼ 2.5 | Photostable, but intensity drifts. | ||
| Camphorquinone in PMMA | 470/560 | I 0/I21 ∼ 7.7 | |||
| Erythrosine B on amino-modified silica | 547/695 | I 0/I0.05% ∼ 4.2 | Works well in the dry state only; good photostability, fast response (<2 s); suitable for sensing extremely low oxygen concentration; non-linear SVP; decay time >0.2 ms. | I-RTP | 154 |
| Erythrosine B immobilized in/on ormosil | 520/660 | I 0/I0.015% = 2.77 | Probe covalently immobilized; oxygen sensitivity influenced by pH, TMOS/MTMOS ratio, and alcohol media; good photostability. | I-RTP | 160 |
| Erythrosine B in sol–gel | 532/570,691 | I 0/I100 ∼ 123 | Unusual (and non-linear) SVPs; intensity of delayed fluorescence (at 570 nm) increases with temperature, but phosphorescence (at 691 nm) decreases with increasing temperature, response time ∼100 ms; works from −50 to 200 °C; τphos 280 μs, QYphos 2%. | I-RTP | 157, 161 |
| Fluorescent Yellow on silica gel | 466/519 | I 0/I21 ∼ 3 | Used to visualize surface gas flows. | I | 162 |
| C60 fullerene in PS | 532/750 | τ 0/τ100 ∼ 51 | Quenching of the excited triplet state; τ0 ∼ 100 μs; high sensitivity; linear SVPs. | L, TA | 79, 81 |
| C70 fullerene in PS | 532/860 | τ 0/τ100 > 400 | Quenching of the excited triplet state; τ0 = 414 μs; very high sensitivity; linear SVPs. | L, TA | 80 |
| C70 fullerene in ormosil | 470/650–710 | τ 0/τ300ppm ∼ 8.5 | E-type delayed fluorescence; lifetime >25 ms; response (<0.1 s); fully reversible, also used at elevated temperatures. | I; LIM | 147 |
| C70 fullerene in ethyl cellulose | τ 0/τ300ppm ∼ 12.5 | ||||
| 13C70 fullerene in polystyrene | 470/600–750 | τ 0/τ150ppm ∼ 3.0 | Suitable for trace oxygen sensing; fully reversible; fluorescence lifetime imaging; temperature influences. | I; LIM | 149 |
| 13C70 fullerene in ormosil | τ 0/τ150ppm ∼ 4.5 | ||||
| 13C70 fullerene in ethyl cellulose | τ 0/τ150ppm ∼ 5.5 | ||||
Decacyclene and benzo(g,h,i)perylene121 have much better photostability. Decacyclene was converted into a silicone-soluble derivative by alkylation with t-butyl groups.121 The resulting sensor film was used in a steam-sterilizable fiber waveguide sensor to monitor oxygen concentration in a bioreactor. The sensor layer was covered with a black layer that acts as an optical isolation to avoid interferences by the fluorescence of cellular matter. When immersed into a bioreactor, response times were 9–65 s, and the drift was −0.01 to −0.09% (signal loss per hour). No effects of stirring were observed. Okazaki et al.122 have frequency-doubled the 780 nm emission of a semiconductor laser to obtain a 390 nm line with 50 nW intensity which is suitable to excite benzo(g,h,i)perylene. The beam was launched into a fiber which guided light to the sensing material (the indicator dissolved in silicone grease) placed at its end. A second fiber was used to collect fluorescence at 430 nm which depends on pO2 in the 0–30% range at atmospheric pressure.
Perylenedibutyrate42 on polystyrene beads was used in the first fiber optic sensor for monitoring oxygen in blood. It is quite photostable and has excitation and emission maxima of 468 and 514 nm, respectively. Its fluorescence is strongly quenched by oxygen, thereby allowing a resolution of ±1 Torr up to 150 Torr (20 kPa) of oxygen. A sensor was constructed that consisted of two thin fibers, one guiding blue excitation light to the dyed particles in a tubing at the common end of the two fibers, the other guiding scattered blue light and green fluorescence to a photodetector. The dyed polystyrene beads are contained in a 25 μm polypropylene tubing whose end was sealed with epoxy. The sensor measures the ratio of scattered blue light (F0) and green fluorescence (F). An electronic circuit processes the blue and green signal intensities according to the following relationship:
| pO2 = (gain)(F0/F − 1)m | (4) |
Note that this is the Stern–Volmer equation re-arranged with an exponent m added for curvature because plots are not linear. This relationship is an alternative to the mathematical models for non-linear SVPs as described in Section 5. This ratiometric method can also eliminate temperature effects and drifts resulting from photobleaching. Coronene on polystyrene,42 and coronene and diphenylanthracene in polydimethylsiloxane were also used123 but require more shortwave excitation wavelengths.
Pyrene and its derivatives, in contrast, have fairly good photostabilities, quantum yields of >0.3, and surprisingly long excited state lifetimes of up to 200 ns. They also display good sensitivity to oxygen and a low temperature coefficient at ambient temperatures. However, pyrene – like other PAHs – lacks good solubility in polymers and tends to aggregate.124 Thus, pyrene derivatives bearing lipophilic side chains have been prepared to overcome these drawbacks.125 The better water-soluble derivative pyrenebutyric acid has an absorption maximum at 355 nm, a monomer emission maximum at 396 nm, and another band peaking at ∼450–470 nm which is attributed to the excimer. Its fluorescence lifetime is reported6 to be ∼200 ns. The probe was used, in dimethylformamide (DMF) solution,39 in a device called “optode” or “optrode”, and later was immobilized on glass.38 In closely related work,126 pyrenebutyric acid was chemically bound to controlled pore glasses, and the quenching by oxygen of its regular emission and its excimer emission was studied, also at −196 °C. Pyrenes later have been immobilized on various kinds of other supports.43,127–134 Pyrene was also covalently immobilized, along with the enzyme glucose oxidase (GOx), on poly(vinyl alcohol) (PVA) via “click” chemistry.135 The resulting composite (PVA–pyrene–GOx) is a water-soluble polymer to which both the OSP and the enzyme are firmly attached and not leach at all. It was used as a biosensor for glucose where the consumption of oxygen is measured via the increase in the fluorescence of pyrene. For other biosensors of that kind see the ESI.† The major drawback of pyrene and its derivatives is the need for photoexcitation at ∼330 nm where many other synthetic and natural materials also give strong fluorescence.
Numerous other probes out of the group of the PAHs were found to be quenched by oxygen.27 Strongly quenching makes an OSP better suited for ambient levels of oxygen, while weak quenching makes them suitable for sensing rather high barometric pressures as they occur, for example, in wind tunnels. The following PAHs (with excitation/emission wavelengths and the % reduction in fluorescence on going from nitrogen gas to air at atmospheric pressure) have been reported: decacyclene in silicone (390–420/510 nm; −55%); diphenylanthracene in xylene (394/435 nm; −25%); benzo(ghi)perylene in silicone (380–410/430 nm; −60%); anthracene in silicone (385/440 nm; −10%); coronene in silicone (340/446 nm; −70%); carbazole in silicone (345/360 nm; −28%); indenopyrene in xylene (410–430/480, 510 nm; −15%); pyrenebutyric acid in silicone (345/400 nm; −68%); dibenzoanthracene in xylene (350/420 nm; −58%); fluoranthene in xylene (360/425–525 nm; −30%); chrysene in xylene (320/430 nm; −68%); and benzo(a)anthracene in xylene (360/436 nm; −62%). Decacyclene and benzo(g,h,i)perylene are most suitable given their relatively long excitation wavelength, good quenchability, low toxicity, and fair photostability.
Similarly, various blended films prepared by combination of eight polycyclic aromatic hydrocarbons and thirteen kinds of polymers were tested136 for use in quenching-based oxygen sensors. The sensor based on a modified polysiloxane and 1-pyrenebutyric acid was reported to perform best in terms of sensitivity, response time, and reproducibility. Chemical structures of common probes are given in Fig. 7.
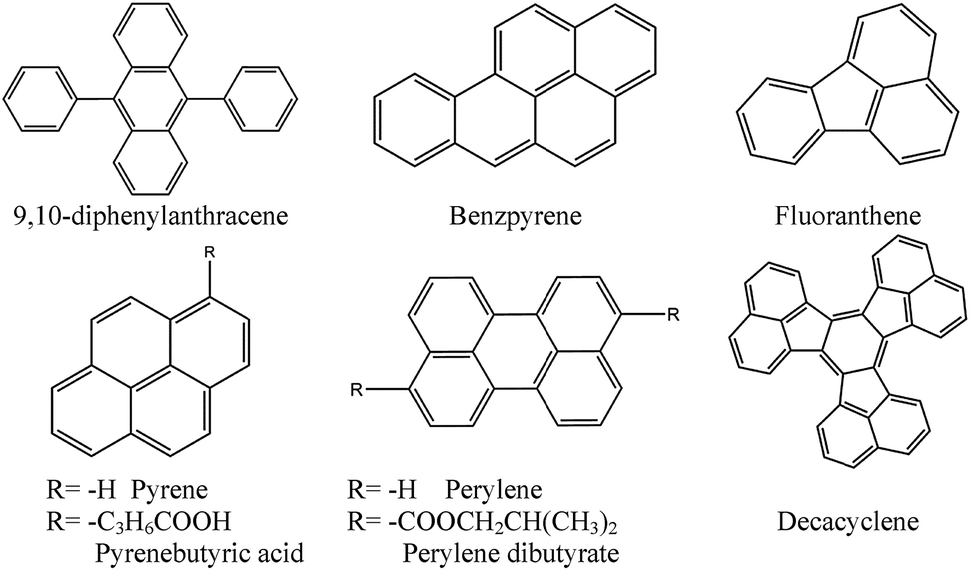 | ||
| Fig. 7 Chemical structures of common PAH-based oxygen-sensitive probes (OSPs). | ||
An unusual effect was found in that the fluorescence of certain aromatic fluorophores is enhanced (rather than quenched) by oxygen in exceptional cases. The fluorescence of dibenzanthracene in a poly(vinyl acetate) matrix is reported to undergo a 10% fluorescence enhancement on exposure to air compared to evacuated samples.137 Similar results were obtained for other PAHs in PVA and other polymers.138 Arguable explanations for the effect were given.139 Another unusual finding is the observation that the fluorescence of the Förster resonance energy transfer (FRET) system formed between pyrene and perylene is much more strongly quenched than the fluorescence of each single component.140 The ratio of the Stern–Volmer quenching constants between pyrene, perylene, and the FRET system is 165:12:520.
Photostable heterocyclic OSPs have been reported141 with longwave excitation wavelengths between 465 and 566 nm, and emissions in the 511–652 nm range. Such probes can be excited with light emitting diodes (LEDs), and yield intense fluorescence, but they are suitable for higher pressures of oxygen only. Decacyclene (Fig. 7) is an attractive visible-light excitable organic probe for oxygen sensing because it works well for ambient levels of oxygen. It can be photoexcited with (purple) LEDs, has a large Stokes shift, and displays good photostability. Its absorption maximum is at 380 nm, but extends far into the visible and ends at around 480 nm. It was also used as a probe for simultaneous sensing of oxygen and halothane,46 and in an oxygen transducer to sense glucose.142 Its poor solubility in apolar polymers (such as silicone) was overcome by alkylating it with tertiary butyl groups which renders decacyclene well soluble even in silicone rubber.143 This sensor was used for many years in clinical oxygen analyzers until it was replaced by certain ruthenium-type probes.
Strongly fluorescent derivatives of coumarin were also used as OSPs in oxygen sensors,144,145 but they are not strongly quenched by oxygen. Thin-films of a substituted terfluorene146 have a fluorescence that was found to be quickly quenched by oxygen due to its specific morphology. Fast response (≪100 ms), full reversibility and high efficiency are typical features. Sensitivity can be further enhanced up to 10-fold and 20-fold in amplified spontaneous emission and lasing action, respectively. A variety of other dyes was tested for use as OSPs. Excellent quenching by oxygen was observed27 with the following fluorophores (the excitation/emission maxima given in brackets) when adsorbed on silica-based chromatographic plates: acridine yellow (458/530 nm), perylene-tetracarboxylic acid N-alkylimide (PTAI) (495/555 nm) and Greengold (another perylene dye; 460/520 nm), and anthranilic acid (330/410 nm). In a typical experiment for quenching studies, PTAI is immobilized via established silyl reagent chemistry to give a sensing material that can be excited at 480–500 nm with blue LEDs. Response times are in the order of 1 s. The probe PTAI displays two emissions (with peaks at 540 and 575 nm, respectively) whose respective quenching constants are different (0.022 and 0.028%−1 O2 at 760 Torr). Similar effects were found with the other dyes. However, a marked influence of humidity was observed in all cases which makes the material better suitable for sensing oxygen in nonaqueous samples. Possibly, the use of hydrophobic supports may eliminate the cross-sensitivity to moisture.
Sensors with even lower limit of detections (LODs) were obtained149 by employing the quenching of the thermally activated delayed fluorescence of isotopically enriched carbon-13 fullerene C70 (13C70). This fullerene was dissolved in either polystyrene (PS), ethyl cellulose (EC) and an organically modified silica gel (“ormosil”). The respective sensor films (5–10 μm thick), on photoexcitation at 470 nm, display a strong delayed photoluminescence with peaks between 670 and 700 nm. Its quenching by molecular oxygen was studied at 25 °C and 60 °C, and in concentrations from zero up to 150 ppmv of oxygen in nitrogen. The rapid lifetime determination method was applied to determine oxygen-dependent lifetimes (which are in the order of 15–45 ms) and for lifetime imaging of oxygen. The lower limits of detection (at 1% quenching) vary with the polymer used (ethyl cellulose ∼250 ppbv, ormosil ∼320 ppbv, polystyrene ∼530 ppbv at 25 °C) and with temperature. These oxygen sensors are the most sensitive ones (in terms of limits of detection) described so far.
Fullerenes such as C60 and C70 also display triplet–triplet absorption in the IR region that is sensitive to oxygen. The QY of intercrossing system from the photoexcited singlet state to the triplet state of fullerene is estimated to be 1.0, and the triplet state is efficiently quenched by oxygen. Thus, fullerene is an attractive probe for optical oxygen sensing using the T1–Tn absorption based on laser flash photolysis. Amao and Okura et al.79 studied the decay of T1–Tn absorption of C60 in films of polystyrene and found the photoexcited triplet state of C60 to be strongly quenched by oxygen. This resulted in an oxygen sensor with very low limits of detection. The lifetime of the excited triplet state of C60 is ∼100 μs, and the oxygen sensitivity (expressed as τ0/τ100) is ∼51. In the same year, this group80 also studied the use of fullerene C70 for oxygen sensing and found that the lifetime of the photoexcited triplet state of C70 (∼410 μs) is about four times longer than that of C60, which results in much higher sensitivity than with C60 in view of a τ0/τ100 ratio of >400.
An even more sensitive oxygen sensor is based on the finding150 that the slow-decaying phosphorescence observed at temperatures below −70 °C is converted into a rapidly decaying fluorescence when oxygen is admitted. In a typical experiment, trypaflavine was adsorbed on silica and placed in an evacuated flask in liquid air or solid CO2. When excited with visible light, the material has orange phosphorescence with a decay time as long as 40–50 s(!). If, after switching off the excitation light source, traces of oxygen are allowed to enter the flask, phosphorescence is instantaneously quenched, but a flash of green fluorescence is observed. The effect is observed with traces of oxygen and has been exploited to detect it at pressures around 5 μTorr (i.e., 10 pmol of oxygen in a volume of 50 mL). The effect is the same under vacuum, hydrogen, or nitrogen, and the working range is 10−6 to 10−4 Torr under visual absorption. Rhodamines B and 6G, uranine, phosphine, benzoflavine, 1-hydroxypyrene-3,6,8-trisulfonate (HPTS) and 7-hydroxycoumarins also showed this effect at −180 °C. Pollack et al.33 have modified the Kautsky method and were able to detect the production of 4 × 1012 oxygen molecules when a green leaf was exposed to a flash of light. Another field of application was found in the detection of the onset of oxygen production in water electrolysis when the voltage is raised from 0 to 2.6 volts.
Zakharov and Grishaeva34 have utilized the phosphorescence quenching effect of oxygen to devise an optosensor for low levels of oxygen in water. It was found that with their material, the phosphorescence was retained even if immersed in water or various organic solvents, with decay times ranging from 50 to 100 ms. Dye-loaded silica gels of various structures and celluloses of varying viscosity phosphoresce in water, ethanol, isoamyl alcohol, heptane and chloroform. Deoxygenation led, in most cases, to a significant increase in intensity. In addition, the emission intensity depends on the structure of silica gels in that those having fine pores display the strongest intensity. The findings were applied to sense oxygen in water. The afterglow of acridine orange adsorbed on Silochrome S-120 was most sensitive to quenching by water-dissolved oxygen and used for its continuous determination in the 0.06 to 1.0 μg L−1 concentration range.34 With acriflavine, the detection limit is 0.35 μg L−1 when adsorbed on silica gel, and from 10 to 100 μg L−1 when adsorbed on cellulose. Other kinds of supports have also been studied and dynamic ranges from 0.4 to 400 μg L−1 were reported.151 Sensitivity can considerably be improved by using hydrophobic supports such as silanized silica gel.15 Detection limits then are 5 × 10−4 μg L−1, but it was observed152 that the dye undergoes “photoabsorption of oxygen” which resulted in a distortion of the SVPs at high illumination intensity. This effect is negligible if the exciting light is sufficiently attenuated.
Camphorquinone (CQ) (see Fig. 8) displays intense room temperature phosphorescence (RTP) when immobilized in a polymer matrix. The excitation and emission bands lie in the visible and the RTP is quenched by oxygen. This effect was exploited153 to sense oxygen via the RTP of CQ immobilized in several polymer matrices. The sensors can be operated in both the intensity and the lifetime mode, and oxygen can be determined in the gas phase in the 0.1–25% concentration range. However, CQ suffers from photodecomposition, which causes signal loss over time and, thus, compromises the accuracy in intensity-based measurements. Erythrosine B is another commonly used phosphorescent OSP. It has a decay time of almost a microsecond in the dry state. If immobilized154 on a silica-based ion exchanger, a material is obtained which under excitation at 547 nm displays an intense red phosphorescence (peaking at 695 nm) that is strongly quenched by oxygen. The response time is <2 s on both the forward and reverse direction which is unusual because the response time of most sensors on going from oxygen-free environment to air or pure oxygen is longer than in the reverse direction.155 This sensor has good photochemical stability and does not photodegrade if continuously irradiated with a xenon discharge lamp for more than 24 h. The detection limit is 0.6 ppm of oxygen in dry argon. Humidity may interfere. The related dye pyronine in various polymers may also be used.156
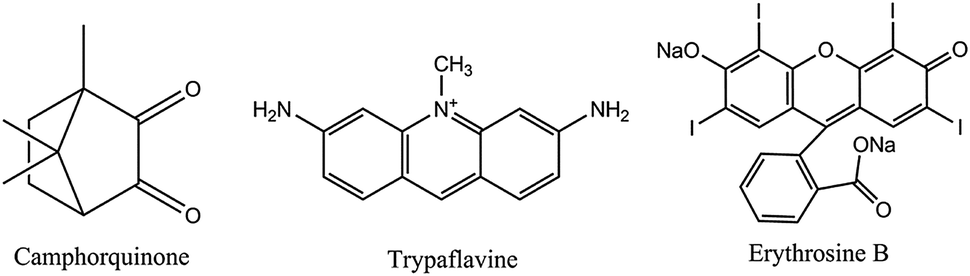 | ||
| Fig. 8 The chemical structures of the RTP oxygen-sensitive probes. | ||
Erythrosine B was doped into a sol–gel film where its RTP is strongly quenched (I0/I100 ∼ 123) by oxygen.157 It has two emission peaks, a weak one at 570 nm (attributed to delayed fluorescence), and a phosphorescent peak at 691 nm. The delayed fluorescence and the phosphorescence are oppositely dependent on temperature in that the phosphorescence intensity decreases as temperature increases. By contrast, the intensity of the delayed fluorescence is enhanced with an increase of temperature. This unique behavior may be applied for temperature self-compensation, and is observed within the temperature range from −50 to 200 °C. Porphyrins may also be used as OSPs but will be treated in Section 6.2.5.
6.2. Metal–ligand complexes
Probes with long decay times are well suited for oxygen sensing because their long-lived excited state makes collisional quenching by oxygen more likely. It can be stated that the detection limits of an OSP mainly depend on the lifetime of its emitting state in that longer lifetimes favor quenchability. However, there is no linear relationship between quenchability and decay time. Luminescent transition-metal complexes, unlike PAHs, have relatively long lifetimes, absorption in the visible, large Stokes shifts, and often are photostable. Their spectroscopic and chemical properties can be tuned to some extent via modification of the structure of the complexes. Molar absorption coefficients (ε; these are important with respect to brightness which is defined as the product of QY·ε) are moderate except for the Soret bands of porphyrins.On photoexcitation, transition metal complexes usually undergo metal–ligand charge transfer to form an excited triplet (or higher multiplicity) state. Triplet–singlet back transitions are spin-forbidden, and the triplet-state lifetimes therefore are much longer than those of a singlet state. This provides enough time for oxygen to collide with the excited-state molecule and results in high sensitivity to oxygen. The triplet states have lower energy than singlet states so that the wavelength of phosphorescence is longer than that of fluorescence. The resulting large Stokes shifts facilitate spectral separation. Early quenching studies in fluid solution163 revealed that the luminescence of complexes of ruthenium(II), osmium(II) and iridium(III) is strongly quenched by oxygen. Numerous other OSPs based on transition-metal complexes have been synthesized meanwhile. Depending on the central metal ion, we have categorized them into six groups, viz. the complexes of ruthenium(II), iridium(III), osmium(II), rhenium(II), the trivalent lanthanides, and the large group of metalloporphyrins.
 | ||
| Fig. 9 Chemical structures of representative Ru(II) complexes for use in oxygen sensing. | ||
The water-soluble probe Ru(bpy) in the form of its dichloride is the earliest ruthenium-based OSP ever used in a sensor for oxygen.47 It was immobilized by adsorbing it on silica microparticles which then were incorporated into silicone rubber. Silicone was chosen as a matrix because of its excellent permeability to oxygen and its flexibility in terms of designing sensor geometries and formats (fiber optics included). The results obtained with this sensor already revealed typical issues such as downward curved Stern–Volmer plots (SVPs), differences in the forward and backward response times, and sensitivity to relative humidity in the case of gaseous samples (which of course play no role in the case of aqueous or blood samples). The probe has an absorption maximum at 460 nm and can be excited with blue LEDs. Its molar absorption coefficient is moderate (<20000 M−1 cm−1), and its emission maximum peaks at 610–630 nm depending on the (polymer) solvent. The large Stokes shift of ∼160 nm facilitates the separation of the analytical signal (the luminescence) from scattered excitation light and reduces self-absorption of luminescence. However, this probe has a fairly low QY (∼0.02), and quenching by oxygen is moderately efficient.
Enhanced stability and oxygen sensitivity of the Ru(bpy) complex is found if the probe is placed in an ionic liquid doped into electrospun fibers made from poly(methyl methacrylate) and ethyl cellulose.164 The use of electrospun polymer microfibers results in increased surface area and sensitivity. However, ionic liquids tend to leach if the sensor is immersed into aqueous solutions or organic solvent. Steady state and lifetime data were used to sense oxygen in the gas phase. The luminescence intensity of the sensor is said not to drift over 44 months.
Similar to Ru(bpy), the ionic probe Ru(phen)3Cl2 is fairly water-soluble and has an unquenched lifetime of 1 μs. Its spectra are quite similar to those of Ru(bpy), but the molar absorption coefficient and QY (∼0.05) are somewhat better.165–170 It has the highest temperature sensitivity171 among the Ru(II) polypyridyl complexes. In fact, it was incorporated in the gas-blocking polymer polyacrylonitrile and used to sense temperature.171–173
The probe Ru(dpp) has a fairly strong absorption in the visible region (λmax = 463 nm, ε = 28400 M−1 cm−1) which makes it compatible with blue LEDs and with the argon ion laser (488 nm). Its luminescence QY is much higher (up to 0.5), and the unquenched excited-state lifetime is ∼6 μs (under nitrogen). This results in high brightness (ε·QY; here 10200 M−1 cm−1). It also exhibits good photostability.174,175 These features make Ru(dpp) the most commonly used ruthenium-based OSP. The better soluble perchlorate of Ru(dpp) was incorporated into silicone rubber to yield a widely used sensor material.49
The probe later has been intensely studied in various matrices including organic polymers, inorganic sol–gel films, and organically modified silicate (ormosil) matrix. Typical data are given in Table 4. Oxygen sensing can, in fact, be well adjusted via the choice of the polymer matrix. Ru(dpp) with counter anions such as chloride, perchlorate, or sulfate has poor solubility in hydrophobic polymers such as silicone rubbers. If the inorganic anion is replaced by lipophilic organic counter anions such as dodecyl sulfate or trimethylsilylpropansulfonate,176 distinctly improved solubility in silicones and other apolar polymers is warranted. The introduction of the new counterion does not cause measurable changes in the absorption and emission spectra, but the luminescence intensity of sensing films is increased. The ligand dpp may also be modified with a lipophilic chain (such as 5-octadecanoylamido)177 or 4-octylphenyl groups178 to obtain hydrophobic luminescent probes that are fairly well soluble in silicone.
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a dpp = 4,7-diphenyl-1,10-phenanthroline. b TMS = trimethylsilylpropane sulfonate; DS = dodecyl sulfate. c 5-odap = 5-octadecanamide-1,10-phenanthroline. d Ph4B = tetraphenyl borate, TBP = tri-n-butylphosphate. e GP-163: an acrylate containing polydimethylsiloxane rubber (PDMS); 7FBMA = 1H, 1H-heptafluorobutyl methacrylate. f Octyl-triEOS = n-octyltriethoxysilane, TEOS = tetraethoxysilane. g PD = photodiode. h PTBS = poly(4-tert-butylstyrene); PTFE-co-VP = poly(tetrafluoroethylene-co-vinylidenefluoride-co-propylene); PSAN = poly(styrene-co-acrylonitrile); PVMK = poly(vinyl methyl ketone). i 8-dpp = 4,7-bis(4-octylphenyl)-1,10-phenanthroline. j DMCH = 6,7-dihydro-5,8-dimethyl-dibenzo(i,j)(1,10-phenanthroline). k s2d = 1,10-phenanthroline-4,7-bis(phenylsulfonate); acap = 5-acetamide-1,10-phenanthroline. l Ru dye = Ru(II)dichloro-(2,6-bis[1-(4-dimethylaminophenylimino)ethyl]pyridine). m DO = Dissolved Oxygen. n AOT = bis(2-ethylhexyl)sulfosuccinate. o Ox satu = oxygen saturated solution. p tfpb = tetrakisbis-3,5-(trifluoromethyl)phenylborate. | |||||
| Ru(dppa)3(ClO4)2 in plasticized PVC | 465/610 | I 0/I100 ∼ 3.5 | Decay is not single exponential; double-exponential fit possible for PVC membranes. | I | 113 |
| Ru(dpp)3(laurylsulfate)2 in silicone | I 0/I100 ∼ 5.3 | ||||
| Ru(dpp)3(ClO4)2 in ethyl cellulose (46%) | I 0/I100 ∼ 3.2 | ||||
| Ru(dpp)3(ClO4)2 in PS | I 0/I100 ∼ 2.0 | ||||
| Ru(dpp)3(ClO4)2 in PS | 470/610 | I 0/I100 ∼ 2.3 | Dye aggregates at higher concentrations, and QY decreases; QYphos = 0.6 for low concentrations. | I | 114 |
| Ru(dpp)3(ClO4)2 in PS, TiO2 particles added | 455/610 | I 0/I100 ∼ 1.25 | TiO2 is used as a scatterer to increase the brightness of the microsensor; excellent long-term stability and storage stability. | I | 54 |
| Ru(dpp)3(ClO4)2 in PVC + plasticizer (2-nitrophenyl octyl ether) | 480/610 | I 0/I21 = 2.0 | Fast response times, but severe leaching of the plasticizer in water containing detergents; used for determination of biochemical oxygen demand. | I | 193 |
| Ru(dpp)3Cl2 in polyacrylamide | 488/610 | I 0/I100 = 3.2 | Dye encapsulated in polyacrylamide during the polymerization process on optical fiber; used to sense oxygen in solution. | I | 194 |
|
Ru(dpp)3(TMSb)2 in silicone, TiO2 added
Ru(dpp)3(DS)2 in silicone, TiO2 added |
465/610 | I 0/I21 = 2.5 | Self-quenching not observed at high dye concentration; other silicones also investigated; TiO2 added as a light scatterer and as an optical isolation. | I | 176 |
| Ru(5-odapc)3 on controlled pore glass | 449/600 | I 0/I140Torr ∼ 3.5 | QY 0.027, τ0 0.5 μs; highly soluble in silicone; linear SVPs in pure silicone. | I | 177 |
| Ru(5-odap)3 on silica gel | 449/600 | I 0/I140Torr ∼ 2.8 | |||
| Ru(5-odap)3 on XAD4 | 449/600 | I 0/I140Torr ∼ 1.5 | |||
| Ru(5-odap)3 on C18–silica | 449/600 | I 0/I140Torr ∼ 1.3 | |||
| Ru(5-odap)3 in silicone | 449/600 | I 0/I140Torr ∼ 1.1 | |||
| Ru(dpp)3 on controlled pore glass | 462/618 | I 0/I140Torr ∼ 6.0 | More sensitive than Ru(5-odap)3 in all matrices downward curved SVPs; QY 0.366; τ0 5.9 μs. | I | 177 |
| Ru(dpp)3 on silica gel | 462/618 | I 0/I140Torr ∼ 6.0 | |||
| Ru(dpp)3 on C18 silica | 462/618 | I 0/I140Torr ∼ 2.7 | |||
| Ru(dpp)3 in silicone | 462/618 | I 0/I140Torr ∼ 1.2 | |||
| 462/618 | |||||
| Ru(dpp)3(Ph4B)2 in CAB + TBPd | 430/596 | I 0/I100 ∼ 6.8 | Effect of a plasticizer also investigated; the fraction of the plasticizer increases sensitivity and decreases response and recovery times; films are very photostable; tributylphosphate is the most effective plasticizer. | I | 195 |
| Ru(dpp)3(Ph4B)2 in PVA + TBP | 430/596 | I 0/I100 ∼ 3.8 | |||
| Ru(dpp)3(Ph4B)2 in PS + TBP | 430/596 | I 0/I100 ∼ 3.3 | |||
| Ru(dpp)3(Ph4B)2 in PMMA + TBP | 430/596 | I 0/I100 ∼ 3.1 | |||
| Ru(dpp)3(Ph4B)2 in PVC + TBP | 430/596 | I 0/I100 ∼ 3.0 | |||
| Ru(dpp)3 in polyacrylic acid on alumina plate | 443,470/610 | I 0/I100 = 2.3 | Electrostatically attached to anionic polymers, then chemisorbed on an alumina plate; non-linear SVPs; Ru(phen) and Ru(bpy) also studied, but display low sensitivity; long-term stability not tested. | I | 168 |
| Ru(dpp)3 on poly(sodium 4-styrene) sulfonate | 447,475/610 | I 0/I100 = 3.73 | |||
| Ru(dpp)3 in poly(acrylic acid) on alumina plate | 470/610 | I 0/I100 = 3.7 | Ru(dpp)3 immobilized in poly(acrylic acid) by the electrostatic effect and chemisorbed on an aluminum plate via the carboxy group; non-linear SVPs; response time ∼4.6 s; good photostability. | I | 196 |
| Ru(dpp)3 in silicone rubber | I 0/I100 = 2.7 | ||||
| Ru(dpp)3Cl2 in PDMS-co-7FBMAe | 450/610 | I 0/I100 ∼ 17 | PDMS copolymerized with methacrylate based monomers (including fluorinated and nonfluorinated); close to linear SVPs; good mechanical properties; the effect of the hydrocarbon chain and fluorination also studied. | I | 197 |
| Ru(dpp)3Cl2 on SiO2 in PDMS (silanol–silanol condensed) | 470/>550 | I 0/I21 = 1.4 | PDMS is silanol–silanol condensed; dye was adsorbed on SiO2 and dispersed in PDMS; quenching is sensitive to humidity and temperature. | I | 198 |
| Ru(dpp)3Cl2 on SiO2 in PDMS (cross-linked) | 470/>550 | I 0/I21 = 1.68 | PDMS is vinyl-hydride cross-linked; more stable but quenching properties change with time. | I | 198 |
| Ru(dpp)3(ClO4) in RTV silicone | 450/610 | I 0/I100 = τ0/τ100 = 7.5 | SVPs close to linear, lifetime and intensity plots coincide; sensitivity is higher in RTV-118 silicone than in any other silicone. | I, L | 49 |
| Ru(dpp)3(ClO4)2 on SiO2 in silicone | 467/598 | I 0/I100 = 14.6 | Dye first adsorbed on SiO2, then mixed into silicone; more oxygen sensitive and more linear SVPs than for Ru(dpp)3(ClO4)2 in silicone due to better solubility; recovery time longer; SiO2 also acts as a light scatterer. | I | 199 |
| Ru(dpp)3(ClO4)2 on SiO2 in gelatine | 469/612 | I 0/IDO,10mM ∼ 7.5 | Dye first adsorbed on SiO2, then mixed into gelatine. Used for determination of oxygen in organic solvents. | I | 200 |
| Ru(dpp)3 in ormosil | 450/620 | I 0/I100 ∼ 8.8 | Sensitivity to dissolved oxygen increased due to increased hydrophobicity. | I | 201 |
| Ru(dpp)3 in sol–gel | I 0/I100 = 3.7 | Sensitivity adjusted by changing the pH of sol–gel precursor solution; good stability; sol–gel films are not stable in aqueous solution on long-term storage. | I | 202 | |
| Ru(bpy)3 in sol–gel | I 0/I100 = 2.0 | ||||
| Ru(dpp)3 in ormosil | 470/610 | τ 0/τair = 1.4 | Phase modulation; sensor properties slowly change over time. | L | 203 |
| Ru(dpp)3Cl2 in TEOS and octyl-triEOS ormosilf | 475/>570 | I 0/I100 ∼ 15 | Sensitivity and long-time stability are higher than for pure TEOS-based films; single exponential decay; uniform and crack-free sensing films. | I, L | 204 |
| Ru(dpp)3 in ormosil | 505/red PDg | τ 0/τair = 1.53 | Ormosil dissolves in organic solvents, but is insoluble in water, methanol, ethanol; no aging; high photostability; linear SVPs; response time 250 ms; τ0 5.1 μs. | I, L | 205 |
| Ru(dpp)3 in DMOS ormosil | 467/592 | I 0/I100 ∼ 14 | Good photostability; fast response (10 s); linear SVPs if cured at high temperature. | I | 206 |
| Ru(dpp)3Cl2 + Oregon Green in sol–gel nanoparticles | 488/610 | I 0/Iair ∼ 6 | Particles size 50–300 nm; PEG added as a sterical stabilizer; also good for sensing in solution; stable to leaching and decomposition; reference dye Oregon Green is pH sensitive. | I | 174 |
| Ru(dpp)3(TMS)2 in ethyl cellulose (46%) | 470/>570 | τ 0/τair ∼ 1.95 | Phase modulation techniques used to measure lifetime; combinatorial approach for rapid screening of probes and polymers. | I, L | 207 |
| Ru(dpp)3(TMS)2 in ethyl cellulose (49%) | 470/>570 | τ 0/τair ∼ 1.67 | |||
| Ru(dpp)3(TMS)2 in PTBSh | 470/>570 | τ 0/τair ∼ 1.8 | |||
| Ru(dpp)3(TMS)2 in cellulose acetate | 470/>570 | τ 0/τair ∼ 1.65 | |||
| Ru(dpp)3(TMS)2 in PTFE-co-VP | 470/>570 | τ 0/τair ∼ 1.2 | |||
| Ru(dpp)3(TMS)2 in PSAN | 470/>570 | τ 0/τair ∼ 1.15 | |||
| Ru(dpp)3(TMS)2 in PVMK | 470/>570 | τ 0/τair ∼ 1.07 | |||
| Ru(8-dppi)3(ClO4)3 in PS | 460/620 | I 0/I100 = 3.5 | Good solubility in polar polymers; QY 0.19; green emitting Ir(III) complexes also reported. | I | 178 |
| Ru(bpy)(DMCHj)2(PF6)2 on SiO2 silicone | 528/736 | I 0/Iair = 1.45 | Low QY (0.005); longer wavelength than other Ru probes; almost linear SVP; QYphos ∼ 0.01; silicone soluble ion pairs; QYphos ∼ 0.01. | I | 179 |
| Ru(dpp)(DMCH)2(PF6)2 in PVC | 563/738 | I 0/Iair = 1.39 | |||
| Ru(dpp)2(DMCH)(DS)2 in silicone | 532/732 | I 0/Iair = 1.37 | |||
| Ru(5-acrylamido-phen)3Cl2 in polyacrylamide | 488/600 | I 0/I100 ∼ 6 | Compound co-polymerized with acrylamide; no leaching; high stability in aqueous and organic solutions. | I | 180 |
| Ru(phen)3 in poly(n-butylaminothionylphosphazene) | 450/610 |
I
0/Iair ∼ 1.75
τ 0/τair ∼ 1.75 |
Indicator covalently attached to the polymer; intensity-based and lifetime-based SVPs are linear and coincide; sensitivity independent of dye concentration; τ0 1.23 μs (multi-exponential). | I, L | 184 |
| Ru(4,7-Me2phen)3(ClO4)2 in RTV silicone | 450/— | I 0/Iair = 5.6 | SVPs are non-linear; lifetime plots deviate from intensity-based plots. | I, L | 111 |
| Ru(4,7-Me2phen)3(Ph4B)2 in RTV silicone | 450/— | I 0/Iair = 4.3 | |||
| Ru(phen)2(CN)2 in RTV silicone | 450/— | I 0/Iair = 4.0 | |||
| Ru(dpp)3(ClO4)2 in RTV silicone | 450/620 | I 0/Iair = 7.4 | |||
| Ru(bpy)3 in RTV silicone | 450/620 | I 0/I100 = 2.6 | |||
| [Ru(s2d)3]4− on controlled pore glass | 475/620 | For determination of oxygen in organic solvents; QY 0.17 for aqueous solution; τ ∼0.3 μs in aerated methanol; long response time. | I, L | 181 | |
| [Ru(s2d)2(acap)]2− on controlled pore glassk | 475/615 | I 0/I100 ∼ 5 | |||
| Ru dyel in silicone and C10F21CH2COOH | 340/597 | I 0/I100 = 12.6 | Silicone not specified; pH sensitive; sensitivity increases with addition of perfluorochemicals; poor article. | I | 208 |
| Ru(bpy)3 in silicone rubber | 460/610 | I 0/I100 = 1.49 | Weak signal and quenching effect in silicone only matrix; OSP tends to aggregate. | I | 47 |
| Ru(bpy)3 on SiO2 in silicone | I 0/I100 = 3.57 | Intense signal and good quenching effect in silica gels, in silicone matrix; QY ∼10%; non-linear SVPs. | I | 47 | |
| Ru(bpy)3Cl2 in silicone + TiO2 | 460/610 |
I
0/I100 ∼ 4.5
τ 0/τ100 ∼ 3.6 |
First lifetime sensor; LED excitation; phase modulation; TiO2 acts as light scatterer and optical isolation. | I, L | 50 |
| Ru(bpy)3 in poly(hydroxyethyl methacrylate) | 450/593 | τ 0/τ100 = 1.01 | Virtually no quenching. | I | 209 |
| Ru(bpy)3 in poly(hydroxyethyl methacrylate) and hydroxyethyl methacrylate | 450/593 | τ 0/τ100 = 1.98 | Contains monomer; sensitivity increases with monomer concentration; τ vs. O2 concentration plots are close to linearity up to 100% O2 (non-monoexponential behavior probably the reason); sensitivity decreases with aging. | ||
| Ru(bpy)3 inside zeolite Y in silicone rubber | 480/610 | I 0/I100 = 3.44 | Sensitivity dramatically increased compared to non-zeolitic matrix; no self-quenching and leaching; excellent storage stability. | I | 210 |
| Ru(bpy)3 on SiO2 in silicone | 480/610 | I 0/I100 = 1.69 | Change in sensitivity on storage. | ||
| Ru(bpy)3 on SiO2 spheres | 360/600 | I 0/I100 ∼ 2.1 | Sphere diameter 0.1–0.2 mm; properties only for the spheres without matrix; non-linear SVPs; long response time (2–4 min); sensitive to humidity. | I | 211 |
| Ru(bpy)3 in sol–gel | 500/600 | I 0/I100 ∼ 1.28 | Poor sensitivity and reproducibility. | I | 212 |
| Ru(bpy)3 on Nafion | 444/595 | I 0/IDOm,8mM ∼ 1.5 | Many parameters change properties. τ0 has 3 components; spectral properties depend on the solvent; quenching increases dramatically in swelling solvents (methanol, water). | I | 213 |
| Ru(phen)3 on Nafion | 442/583 | I 0/IDO,8mM ∼ 1.4 | |||
| Ru(dpp)3 on Nafion | 460/598 | I 0/IDO,8mM ∼ 6.0 | |||
| Ru(odap)3 on Nafion | 440/583 | I 0/IDO,8mM ∼ 5.1 | |||
| Ru(bpy)3 in MTEOS ormosil | 450/610 | I 0/I100 = 1.5 | Probe functionalized with the triethoxysilane group, and covalently incorporated in sol–gel film; short response times; linear SVP. | I | 182 |
| Ru(bpy)3 in AOTn in a water–gelatine matrix | 416/580 | I 0/IDO,15ppm ∼ 1.4 | For use in organic solvents; reversed micelle structure; properties depend on the concentration of all components. | I | 214 |
| Ru(bpy-pyr)(bpy)2 in aqueous solution | 456/632 | I 0/IOx satuo = 13 | Water soluble probe, used for intracellular oxygen sensing; the same photostability as Ru(phen), τ0 1.3 μs, QY 0.5. | I, L | 215 |
| Ru(bpy-pyr)(bpy)2 in lipobeads | 456/632 | I 0/IOx satu = 2.5 | 2.1 μm lipobeads; for intracellular oxygen; high photo- and chemical stability; QY 0.1. | I | 216 |
| [Ru(phen)3](tfpb)2 as a crystalp | 400/518 | τ 0/τ100 = 3.43 | Crystal based solid state oxygen sensors; good sensitivity; fast response; strictly linear SVPs except for [Ru(phen)3](PF6)2 crystal; intense luminescence with high QYs and long lifetimes; sensitivity and photostability do not change after more than a year of storage. | I, L | 187–189 |
| [Ru(5,6-Me2Phen)3](tfpb)2 as a crystal | —/572 | τ 0/τ100 = 1.83 | |||
| [Ru(phen)3](PF6)2 as a crystal | 465/600 | τ 0/τ100 ∼ 2.9 | |||
Most Ru(II) polypyridyl complexes can be excited with blue light to emit in the red region, but more long-wavelength absorbing and emitting OSPs are desired for biological applications. Klimant et al.179 synthesized several long-wavelength Ru(II) complexes by replacing one or two ligands of Ru(bpy) and Ru(dpp) with 6,7-dihydro-5,8-dimethyl-dibenzo[i,j][1,10]phenanthroline. These long-wavelength Ru(II) complexes can be photoexcited between 450 and 580 nm and have large Stokes shifts. The emission bands sometimes even extend into the NIR region. These OSPs are photostable and compatible with green LEDs but have not been investigated ever since, possibly because their QYs are at <0.01.
In order to ultimately overcome leaching from the polymer matrices, a Ru(II) complex bearing a polymerizable acrylate group can be covalently linked to a matrix.180 The introduction of the acrylate group into the phenanthroline ring does not affect the metal–ligand charge transfer (MLCT) and the spectral properties. In fact, the probe fully maintains its oxygen sensing capability. The co-polymerized sensing films are stable in aqueous solutions and even in hydrophobic organic solvents. Moreno-Bondi et al.181 prepared three sulfonated Ru(II) complexes and linked them to amino-functionalized matrices such as amino-modified porous glass. The material displayed strong emission with a peak at 610 nm if excited at 475 nm. Again, the material was completely resistant to leakage even on long-term usage. It is suitable for sensing oxygen in aqueous solution and in organic solvents. Similarly, a Ru(II) polypyridyl complex was functionalized182 with a triethoxysilane group via reaction with (3-aminopropyl)trimethoxysilane in acidic solution. The probe was condensed into sol–gel glass films and the resulting sensing films revealed enhanced stability in water and organic solvents compared to similar films where probes were incorporated physically. Ru(bpy) modified with a triethoxysilane group was also condensed183 into ormosil films. The Winnik group184 have synthesized alkylamino functionalized Ru(phen) and linked it to a sulfur–nitrogen–phosphorus polymer. The respective sensing film exhibited almost linear Stern–Volmer behavior and good quenchability.
In a study185 on the performance of the probe Ru(dpp) in polystyrene films and of Ru(bpy) in fumed silica gel it was found that the response of the Ru(dpp)–polystyrene system to oxygen can be well fit with the model of a spatially disordered environment (see Section 5). In contrast, the SVPs of the Ru(bpy)–silica-gel system are nonlinear, and this was explained by microheterogeneity. Additionally, in fumed silica gels, oxygen reaches the probe via surface diffusion and absorption isotherms are expect to provide an additional effect on the curvature of such plots. It was attempted186 to eliminate effects of microheterogeneity by synthesizing a Ru(bpy)-centered polystyrene-derived star polymer and expecting that the resulting probes may be evenly surrounded inside the star polymer so that effects of microcrystallization and leaching may be avoided. However, microheterogeneity could not be completely eliminated in that the decays profiles were still multi-exponential. Heterogeneity possibly is caused by the wide range of molecular weights of the polymer prepared during the polymerization.
Solid-state OSPs usually cannot be used directly for sensing oxygen because of strong inner filter effects, self-quenching, and poor accessibility for oxygen. It was shown,187–189 however, that crystals of Ru(II) polypyridyl complexes of the type ([Ru(phen)3]tfpb2, [Ru(5,6-Me2Phen)3]tfpb2 and [Ru(phen)3](PF6)2) can be used directly as solid-state probes for oxygen. Obviously, the crystal lattices contain enough void space to allow oxygen to diffuse in and to quench luminescence. The crystals give strictly linear SVPs, and sensitivity does not change if crystals are stored for more than a year. A single crystal can be used directly as an oxygen sensor rather than using hosting materials to immobilize any OSPs.
The polystyrene-soluble probe [Ru(dpp)](TMS)2 (where TMS stands for the trimethylsilylpropylsulfonate anion) was shown190 to enable sensing of oxygen via two-photon excitation (2-PE). Amino-modified polystyrene nanoparticles (85 ± 19 nm i.d.) were soaked with a solution of the probe in a water–tetrahydrofuran mixture to obtain monodisperse nanoparticles with an average diameter of 121 ± 3 nm and a polydispersity index of 0.04. The particles can be conventionally photoexcited with the 488 nm line of an argon laser. If, however, a mode-locked Ti:sapphire laser with an output wavelength of 830 nm and an average pulse duration of 75 fs is applied, strong 2-PE luminescence is observed. As expected, luminescence intensity increases nonlinearly with the power of the laser. Double-logarithmic plots of laser power (between 25 and 125 mW) versus the emission intensity of the oxygen show the slope for 1-PE to be ∼1 and that for 2-PE to be ∼2. The emission spectra at 2-PE are the same as obtained by 1-PE. The SVPs obtained after excitation in the visible and the NIR (i.e., 2-PE) are identical and both are strictly linear (R2 = 1.000).
Ruthenium probes such as Ru(dpp) are well compatible with present-day low-cost electronics (such as LEDs and silicon photodiodes). These can be well packed into small-sized instrumentation as shown191 for a hand-held optical sensor for dissolved oxygen. A blue LED served as a light source to photoexcite the luminescence of Ru(dpp) absorbed on silica gel in a silicone rubber membrane. The respective instrument has a size of 48 × 148 × 20 mm and weighs only 96 g. It is powered by batteries and gives luminescence intensity data on dissolved oxygen. A miniaturized sensor with a size smaller than a fingernail was obtained192 by integrating an oxygen-sensitive ormosil film with a low-power complementary metal oxide semiconductor (CMOS) chip.
 | ||
| Fig. 10 Basic chemical structures of cyclometalated Ir(III) complexes and of commonly used ligands. | ||
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a ppy = 2-phenylpyridine anion; TFEM = 2,2,2-trifluoroethyl methacrylate. b nBuPTP = poly[(n-butylamino)thionylphosphazene]. c dpt-NH2 = 4-amino-3,5-di-2-pyridyl-4H 1,2,3,4-triazole; pPEGMA = poly(polyethylene glycol ethyl ether methacrylate). d L = 2,6-bis(7′-methyl-4′-phenyl-2′-quinolyl)pyridine; L1 = monoanion of L. e vpy = vinylpyridine. f CS = 3-(benzothiazol-2-yl)-7-(diethylamino)-coumarin; CN = 3-(1-methylbenzoimidazol-2-yl)-7-(diethylamino)-coumarin; CO = 3-(5-chlorobenzoxazol-2-yl)-7-(diethylamino)-coumarin; CN–Me = 3-(benzothiazol-2-yl)-7-dimethylaminocoumarin, acac = acetylacetone. g CS-Jul = 10-(2-benzothiazolyl)-2,3,6,7-tetrahydro-1,1,7,7-tetramethyl-1H, 5H, 11H-(1)benzopyrano(6,7–8i,j)quinolizin-11-one; PS-PVP = Poly(styrene-block-vinylpyrrolidone). h fppy = 4-(2-pyridyl)benzaldehyde, t-Bu-NC = tert-butyl isocyanide, tpy = 2-p-tolylpyridine. i btpy = 2-(benzo[b]thiophene-2-yl)pyridinato-C3,N. j C6 = coumarin 6; vacac = vinylacetoacetate, ppy = 2-phenylpyridine, and vppy = 2-(4-vinyl)-phenylpyridine. k Ir-N-948 = Ir(2-phenylpyridine)2(4,4′-bis(2-(4-N,N-methylhexylaminophenyl)ethyl)-2-2′-bipyridine)Cl. | |||||
| Ir(ppy)3 in poly(styrene-co-TFEM)a | 376/512 | I 0/I100 = 15.3 | SVPs; almost linear; short response times (t95 = 7 s); highly photostable. | I | 221 |
| Ir(ppy)3 in PS | 376/512 | I 0/I100 = 1.1 | QY ∼ 90%. | ||
| Ir(ppy)3 in nBuPTPb polymer | 370/510 | I 0/I159Torr = 3.36 | High sensitivity; τ0 = 1.4 μs. | I | 222 |
| Bu4N[Ir(ppy)2(CN)2] in nBuPTP polymer | 340/470 | I 0/I159Torr = 6.04 | High sensitivity; τ0 = 4.78 μs. | ||
| Ir(ppy)2(dpt-NH2)PF6 in pPEGMAc | 380/490(530) | I 0/I100 = 2.5 | QYphos = 0.25 in acetonitrile; τ0 = 1.3 μs | I | 217 |
| Ir(L)(L1)(PF6)2d in pPEGMA | —/∼605 | τ 0/τ100 = 2.76 | τ 0 = 2.1 μs. | I, L | 220 |
| Ir(L1)2PF6 in pPEGMA | —/∼615 | τ 0/τ100 = 1.61 | τ 0 = 1.4 μs. | ||
| [(ppy)Ir(dpt-cy-dpt)Ir(ppy)](PF6)2 in pPEGMA | —/∼520 | τ 0/τ100 = 2.90 | τ 0 = 1.13 μs. | ||
| Size and charge of the chromophore affect sensitivity (slope). | |||||
| Ir(ppy)2(vpye)Cl in PDMS silicone | 265,396/509 | I 0/I100 ∼ 1.65 | Covalently attached to H-terminated PDMS; QYphos = 0.32, τ0 = 190 ns; polymerizable probe; blending with PS improves sensitivity. | I | 235 |
| Ir(CS)2(acac)f in PS | 448,477/566 | τ 0/τ175hPa ∼ 1.51 | Brightness is 5 times that of Ru(dpp)3 or PtTFPP; strong visible absorption; less temperature-sensitive than Ru(dpp)3; spectral properties fine-tuned by varying the ligand; non-linear SVPs; poor photostabilities. QYs and lifetimes also reported in detail. | L | 225 |
| Ir(CN)2(acac) in PS | 421,455/544 | τ 0/τ175hPa ∼ 1.5 | |||
| Ir(CO)2(acac) in PS | 444,472/554 | τ 0/τ175hPa ∼ 1.59 | |||
| Ir(CS-Me)2(acac) in PS | 446,475/566 | τ 0/τ175hPa ∼ 1.7 | |||
| (CS)2Ir(μ-Cl)2Ir(CS)2 in PS | 457,484/588 | τ 0/τ175hPa ∼ 1.96 | |||
| (CN)2Ir(μ-Cl)2Ir(CN)2 in PS | 432,463/567 | τ 0/τ175hPa ∼ 1.85 | |||
| Ir(CS-Jul)2(acac) in PS-PVP nanoparticlesg | 452,481/572 | τ 0/τ20kPa ∼ 3.5 | QY = 0.52; τ0 = 20.8 μs, (longer than other Ir(III) coumarin complexes); high brightness; poor photostability. | L | 228 |
| Ir(ppy-NPh2)3 in 2-methyl-tetrahydrofuran | 405/525,565 | τ 0/τair sat ∼ 170 | QY = 0.70; τ0 = 4.3 μs; good solubility in organic solvents and polymers; compatible with 405 nm LEDs; low temperature dependence. | L | 224 |
| Ir(ppy-NPh2)3 in ethyl cellulose | τ0/τair ∼ 4.5 | ||||
| [Ir(fppy)2(t-Bu-iCN)2]CF3SO3 on silamineh | 258,367/496 | τ 0/τair ∼ 3 | QY = 0.35; τ0 = 17.3 μs. | I, L | 233 |
| [Ir(tpy)2(t-Bu-iCN)2]CF3SO3 on silamine | 260,348/458 | τ 0/τair ∼ 3 | QY = 0.28; τ0 = 35.6 μs. | ||
| Can be covalently bound to an amino-modified polymer; non-linear SVPs; temperature dependent. | |||||
| Ir(btpyi)3 in cellulose acetate butyrate | 366,408/596,645 | τ 0/τair ∼ 1.9 | τ 0 ∼ 8.6 μs; temperature dependent; excitable with purple LED. | L | 238 |
| Ir(C6)2(vacac)j | 445,474/568 | τ 0 = 6.0 μs; QY = 0.22. | I, L | 232 | |
| Ir(ppy)2(vacac) | 257,400/520 | τ 0 = 0.1 μs; QY = 0.02. | |||
| fac-Ir(ppy)2(vppy) | 287,384/542 | τ 0 = 0.4 μs; QY = 0.2. | |||
| mer-Ir(ppy)2(vppy) | 274,390/535 | τ 0 = 0.2 μs; QY = 0.03. | |||
| All these probes carry vinyl groups and can be polymerized into hydride-containing silicones. | |||||
| Ir-N-948k in PS | 494/665 | τ 0/τair ∼ 8.0 | First Ir(III) complex emitting at >650 nm; high QY and long lifetime (QY = 0.58, τ0 = 102 μs); good long-term storage stability; non-linear SVPs. | I, L | 229 |
| Caged iridium(III) complex in DMF | 462/570 | τ 0/τair = 7.7 | τ 0 = 1.27 μs. Suitable for high levels of oxygen. | I, L | 231 |
| Hemi-caged iridium(III) complex in DMF | 462/580 | τ 0/τair = 9.6 | τ 0 = 1.20 μs. Suitable for high levels of oxygen. | ||
| Ir(ppy)3 in DMF | 462/523 | τ 0/τair = 23.7 | τ 0 = 1.87 μs. | ||
The probe [Ir(ppy)2(dpt-NH2)](PF6) (see Fig. 10) has a QY of ∼0.25 in deoxygenated acetonitrile solutions, exhibits two emission peaks (at 490 and 530 nm), and displays good photostability, fully reversible response, and strictly linear Stern–Volmer plots (SVPs).217 Mononuclear and dinuclear Ir(III) cyclometalated complexes were synthesized and immobilized in polymer matrices.220 The resulting sensor materials emit intense luminescence with maxima in the 500–650 nm range and linear SVPs. The authors came to the conclusion that quenching by oxygen is affected by (a) the lifetime of the chromophore, (b) the permeability of the polymer, (c) the size, and (d) the charge of the chromophore. With increased molecular sizes and lower charges of the chromophores, sensitivity is enhanced.
The easily accessible probe Ir(ppy)3 was immobilized221 in the fluoropolymer poly(styrene-co-TFEM) where it displays a strong green luminescence, with a QY as high as 0.90 and a relatively long lifetime (1.5 μs). The sensor is photostable, and its fluorescence is strongly quenched by oxygen (I0/I100 = 15.3). The sensitivity of the probe depends on the matrix. If the fluoropolymer is replaced by polystyrene, its sensitivity (I0/I100) drops to 1.1 and serious photodecomposition does occur. A very high sensitivity is also found if the probe is incorporated into poly((n-butylamino)thionylphosphazene).222 The same Ir(III) OSP was functionalized with amino acids to enable phosphorescence-based imaging of oxygen in living cells,223 but this modification causes the QYs to drop to 0.2. If the ppy ligands of the Ir(III) complexes are substituted by ppy-NPh2, an OSP is obtained224 that has better solubility in organic solvents and polymers, and a QY of 0.70. The broad excitation range makes it compatible with 405 nm lasers or LEDs, and the large dynamic range makes the probe well suitable for sensing oxygen. The decay time in the absence of oxygen is almost independent of temperature in the 1–24 °C range.
Borisov et al.225 have synthesized other cyclometalated Ir(III) coumarin complexes. They possess large molar absorption coefficients (in the order of 30000 to 60000 M−1 cm−1 which are even higher than those of Ru(dpp) and of the longwave bands of porphyrins), lifetimes on the order of 8–13 μs in the unquenched state, QYs between 0.3 and 1.0, and absorption bands that are compatible with purple and blue LEDs. Their brightness (ε × QY) is nearly 5 times of that of Ru(dpp) or PtTFPP so that the signal-to-noise ratio is much better. Spectral properties and oxygen sensitivity can be fine-tuned by varying the coumarin ligand, or by using respective monomeric or dimeric complexes. In addition, these OSPs show lower sensitivity to temperature than the widely used probe Ru(dpp). On the other side, they are less photostable, and this restricts their application to short-term sensing. The photophysical properties of cyclometalated Ir(III) complexes were described,226 and the utility of these [Ir(ppy)2(N–N)]Cl complexes (where ppy stands for 2-phenylpyridine and N–N is a substituted bipyridine, biquinoline, or phenanthroline) were assessed, but the probes have not been converted into sensor materials by incorporating them into an appropriate solid support. A cyclometalated Ir(III) complex with red luminescence was also reported.227
Dimeric Ir(III) coumarin complexes were also investigated. Both their absorption and emission peaks are red-shifted, but their QYs are lower than the respective monomeric complexes. The same group228 has also synthesized Ir(CS-Jul)2(acac), which can be even excited by the 488 nm argon laser. It has a high QY (0.52), and intense absorption peaks are found at 481 nm and 452 nm. Emission peaks are found at 572 nm. The photophysical properties of this probe are quite similar to those of the Ir(III) coumarin complexes except for the long luminescence lifetime (20.8 μs). Hence, the probe is highly sensitive to oxygen, but its photostability is moderate.
A luminescent Ir(III) complex was reported229 that has an unusually longwave emission maximum at 650 nm. The phosphorescence of the probe (referred to as Ir-N-948) also has a useful QY (0.58), a long decay time (102 μs), strong absorption bands between 400 and 500 nm, and a good sensitivity to oxygen. It is soluble in apolar polymers such as polystyrene, and the respective sensor films are quite stable on storage. Its outstanding photophysical properties make the material most suitable for sensing oxygen. Various other Ir(III) complexes are presented in the supplementary information of the article cited.229 In the same year, this group178 also reported on three green-emitting Ir(III) OSPs with similarly high QYs. Organometallic osmium and iridium complexes were reported for use in barometric coatings (pressure-sensitive paints).230 These charge-neutral phosphorescent dyes dissolve well into the host polymer FIB, a highly oxygen-permeable fluoropolymer (see Section 7). The photo-degradation, temperature sensitivity, and SVPs of pressure sensitive paints based on these complexes were studied in detail.
The luminescence intensity of an OSP exponentially decreases with linearly increasing concentration (or partial pressure) of oxygen (see Fig. 6). Thus, the sensitivity of OSPs is best at low levels of oxygen but decreases as the oxygen tension increases. This is disadvantageous when it comes to sense oxygen at levels such as at air saturation. A smart approach was made231 by synthesizing an Ir(III) complex with a caged ligand structure. Quenching of the caged Ir(III) complex by oxygen was remarkably retarded initially due to the caged ligand structure, and this makes the probe better suitable for high-level oxygen sensing. This OSP has a fairly good QY (∼0.5 in dimethylformamide) and a longwave emission (peaking at 570 nm), while the quenching efficiency – compared to Ir(ppy)3 – is decreased by 80%.
Similar to the situation with Ru(II) polypyridyl complexes, Ir(III) complexes have been synthesized that can be covalently linked to a polymer matrix to overcome leaching. For example, Ir(III) complex carrying vinyl groups have been reported232 that are non-ionic, have long luminescence lifetimes, and good QY. They can be covalently attached to hydride-containing silicones via transition-metal-catalyzed hydrosilylation, or chemically bound to polymer backbones during polymerization of vinylic monomers. The same group233 has synthesized amino-reactive cyclometalated Ir(III) complexes which then were covalently bound to a water-soluble amino-functionalized polymeric silamine. The resulting materials were blended with different ratios of colloidal microcrystalline cellulose to prepare a water-soluble PSP. Both probes have long lifetimes (17.3 and 35.6 μs) and QYs of typically 0.3. Their sensitivities (I0/Iair ∼ 3) make them fairly well suitable for sensing purposes. However, luminescence intensities are strongly affected by temperature. Similar Ir(III) complexes were functionalized with silane groups through which they can be covalently bound to sol–gel or ormosil matrices.234 Such materials can be used to generate singlet (1Δ) oxygen during photodynamic therapy but also are likely to act as sensor materials. The non-ionic cyclometalated iridium(III) complex Ir(ppy)2(vpy)Cl contains a vinyl group and therefore can be covalently linked to a silicone backbone during polymerization.235 This results in a sensor material that displays high sensitivity to oxygen, physical stability, and a QY of 0.32. However, it has to be excited in the UV.
Four oxygen-sensitive iridium(III)-containing coordination crystals were synthesized236 using Ir(ppy)2(H2dcbpy)PF6 as the bridging ligand (H2dcbpy = 4,4′-dicarboxy-2,2′-bipyridine). The bridging ligand was crystallized with M(ClO4)2 (M = Zn, Cd, Co and Ni, respectively) to form Ir(III)-containing single crystals. These have emission QYs in air of 0.27, 0.19, 0.001 and 0.002, respectively, and give linear SVPs. The corresponding KSV values were found to be 0.83, 2.82, 1.33, 1.11, and 2.48%−1, respectively. The lowest detection limit was 0.5% for oxygen in nitrogen gas. However, the response time of these crystals is rather long (>70 s), and – like in other Ir(III) complexes – the photostability of the crystals is moderate.
Nanocomposites containing cyclometalated Ir(III) emitters have also been used to sense oxygen.237 They consist of an aluminum oxide–hydroxide nanostructured solid support and a cyclometalated Ir(III) complex as a cover layer. The results were compared to data on the same dyes immobilized in polystyrene films. Since the photoluminescence of the complexes is totally quenched for oxygen concentrations just over 10%, these systems are promising for oxygen detection at low concentration. In particular, dyes on the aluminum support provide sensitivity to oxygen down to 1% of oxygen in a sample gas.
Water-based oxygen sensor films were reported,233 where a luminescent cyclometalated Ir(III) complex was bound to the water-soluble amino-functionalized polymer silamine. Oxygen-sensor films were prepared using aqueous blends of the conjugate with silamine and colloidal microcrystalline cellulose. Like in other cases, the diffusion of oxygen decreases with increasing fraction of filler and thereby decreases sensitivity. These novel materials offer an environmentally friendly alternative to the preparation of oxygen-sensor films.
Metal–ligand complexes of Re(I) typically have excited-state lifetimes between 0.5 and 5 μs, large Stokes shifts, and good photostability. Quenching of the luminescence by oxygen shows a high degree of heterogeneity in silicone polymers.242,243 Such a microheterogeneity induces nonlinear Stern–Volmer plots that can be described by the two-site model (see Section 5). The Re(I) complexes display exceptionally strong luminescence (with QYs of up to 0.7) and strong quenching by oxygen. However, they easily photodegrade on exposure to oxygen. The same group244 also synthesized and tested the complexes ReL(CO)3Cl and ReL(CO)3CN (where L represents the ligand, these including bpy, terpy, dpp, Me2Ph2phen, and Me4phen). The cyano complexes are poorly photostable in polymers,222 but the chloro complexes are fairly photostable and have both long excited-state lifetimes and good QYs.
The excited-state properties of such Re(I) complexes are strongly influenced by ligand variation. The introduction of a phenanthroline ligand increases sensitivity to oxygen and lengthens the decay time. The introduction of t-butyl groups into the bpy ligand also results in greater sensitivity and a longer decay. However, the addition of two methyl groups to the Ph2phen group has an adverse effect. The Re(I) complexes with terpy as the ligand have the shortest lifetimes and lowest sensitivities to oxygen. The ligand terpy is also a poor ligand for luminescent Ru(II) complexes, and the Ru(terpy)22+ complex even is non-luminescent at room temperature.
All these OSPs display nonlinear SVPs and relatively low sensitivity to oxygen. This makes them suitable, however, for sensing high-level barometric (in fact oxygen) pressure such as in wind tunnels. The Lakowicz group has described245 hydrophobic probes such as cis-Re(CO)2(c-dppene)(dpp) and the water-soluble equivalent cis-Re(CO)2(c-dppene)(SO3-dpp), where c-dppene stands for cis-[bis(diphenylphosphino)ethylene]. Both OSPs are highly emissive, have excited-state lifetimes of around 10 μs, and are strongly quenched by oxygen, but photostability is moderate. A recent article246 describes the first Re(I)-dipyrrinato complexes for sensing dissolved oxygen. They have fairly large molar absorption coefficients (2.5–4.2 × 104 M−1 cm−1) and Stokes shifts, but their QYs are low (<0.01) so that brightness is inadequate. An oxygen-quenchable luminescent Re(I) complex containing a carbazole “shield” in its diamine ligand was applied247 in a poly(vinylpyrrolidone) host to sense oxygen with high sensitivity (I0/I100 = 20.5) and short response/recovery times (6 s and 28 s), and a Re(I) complex was used to dope nanofibers so to obtain optical nanosensors for oxygen.248 The fabrication, photophysical parameters and molecular oxygen sensing capability of a rhenium(I) complex with enlarged active area was reported very recently.249
All Re(I) complexes show high emission anisotropies which makes them attractive probes for polarization. On the other hand, they lack significant absorption beyond 400 nm and their molar absorptions often are 3 times smaller (at 370–380 nm) than those of Ru(II) complexes at 450 nm. Figures of merit for oxygen sensors based on osmium and rhenium complexes are summarized in Table 6.
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a trpy = 2,2′,2′′-terpyridine, dppene = cis-bis(1,2-diphenylphosphino)-ethylene, Me2PPh = dimethylphenylphosphine, dppm = bis(diphenylphosphino)methane, das = o-phenylenebis(dimethylarsine), Me2SO = dimethyl sulfoxide. b GP-163 = acrylate containing PDMS; T3642 = trimethylsilylmethyl methacrylate. c nBuPTP = poly((n-butylamino)thionylphosphazene). | |||||
| Os(dpp)3(PF6)2 in PDMS rubber | 480(502)/729 | I 0/Iair = 1.1 | PDMS contains (methacroyloxy)propyl side chains; τ0 = 340 ns. | I, L | 241 |
| Os(dpp)3(PF6)2 in PDMS rubber | 480(502)/729 | I 0/Iair = 1.7 | Methyldiacetoxy terminated PDMS; τ0 = 200 ns. | ||
| Os(trpya)22+ | 698/729 | τ 0/τair > 6.73 | — | I, L | 239 |
| (trpy)(dppene)OsCl+ | 624/718 | τ 0/τair >2.53 | — | ||
| Os(phen)3 | 650/720 | τ 0/τair = 1.39 | QY = 0.013 | ||
| (phen)2Os(MeCN)22+ | 600/688 | τ 0/τair = 4.28 | QY = 0.035 | ||
| (phen)2Os(Me2PPh)22+ | 590/672 | τ 0/τair = 1.84 | QY = 0.072 | ||
| (bpy)2Os(dppm)2+ | 480/652 | τ 0/τair = 1.64 | QY = 0.046 | ||
| (phen)2Os(dppm)2+ | 500/624 | τ 0/τair = 4.85 | QY = 0.12 | ||
| (phen)2Os(dppene)2+ | 455/609 | τ 0/τair = 5.00 | QY = 0.20 | ||
| (bpy)2Os(das)2+ | 450/592 | τ 0/τair = 2.95 | QY = 0.22 | ||
| (bpy)2Os(Me2SO)22+ | 443/575 | τ 0/τair = 2.23 | QY = 0.31 | ||
| (bpy)Os(dppene)22+ | 400/537 | τ 0/τair = 1.22 | QY = 0.47 | ||
| (3,4,7,8-Me4phen)2Os(dppene)2+ | 450/594 | τ 0/τair = 5.59 | QY = 0.17 | ||
| All data for acetonitrile solutions. Excellent photostability; some emitting in the NIR region. | |||||
| Re(phen)(CO)3CN in silicone RTV rubber | 385/520 | I 0/Iair = 2.5 | Other polypyridyl ligands also tested; photolabile; multi-exponential decay; non-linear SV plots | I, L | 242 |
| Re(bpy)(CO)3Cl in GP-163-co-T3642b | 388/540 | I 0/I100 = 1.5 | CN complexes are not photostable; three component emission decays for all the dyes; best suited for high pressure oxygen sensing. | I, L | 244 |
| Re(dpp)(CO)3Cl in GP-163-co-T3642 | 388/540 | I 0/I100 = 3.9 | |||
| Re(Me4phen)(CO)3Cl in GP-163-co-T3642 | 388/540 | I 0/I100 = 3.7 | |||
| Re(t-Bu)2bpy(CO)3Cl in GP-163-co-T3642 | 388/540 | I 0/I100 = 1.7 | |||
| Re(terpy)(CO)2Cl in GP-163-co-T3642 | 388/540 | I 0/I100 = 1.2 | |||
| Re(Me2Ph2phen)(CO)3Cl in GP-163-co-T3642 | 388/540 | I 0/I100 = 3.4 | |||
| [Re(CO)3(bpy)(CN-t-Bu)]Cl in nBuPTPc polymer | 340/507 | — | Poor sensitivity and serious photodecomposition. | I | 222 |
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a tta = thenoyltrifluoroacetonate; fod = 1,1,1,2,2,3,3-heptafluoro-7,7-dimethyl-4,6-octadionate; pta = 1,1,1-trifluoro-5,5-dimethyl-2,4-hexandionate. b phencarz = 2-(N-ethylcarbazolyl-4)imidazo[4,5-f]1,10-phenanthroline. c acac = acetylacetone. | |||||
| Eu(ttaa)3phen in poly(styrene-co-TFEM) | 342/612 | I 0/I100 = 2.4 | Moderate sensitivity to oxygen; potentially useful at high air pressures such as in wind tunnels; linear SVPs; response time 6–7 s. | I | 250, 251 |
| Eu(tta)3 in poly(styrene-co-TFEM) | 342/612 | I 0/I100 = 1.5 | |||
| Eu(fod)3 in poly(styrene-co-TFEM) | 293/612 | I 0/I100 = 1.27 | |||
| Eu(pta)3 in poly(styrene-co-TFEM) | 295/612 | I 0/I100 = 1.19 | |||
| Eu(tta)3(phencarz)b in polystyrene nanofiber | 326/612 | I 0/I100 = 3.38 | Nanofiber was prepared via electrospinning; response time 5.0 s, recovery time 8.0 s; downward curved SVPs. | I | 260 |
| Tb(acacc)3(phen) on silica alumina plate (for TLC) | 268/546 | I 0/I100 = 2.68 | Static quenching dynamic competes with quenching; non-linear SVP; highly photostable; UV excitation; QYphos = 0.11. | I | 256, 257 |
| Tb(III) azaxanthone complex | 355/480,540 | I 0/I100 ∼ 1.5 | Corresponding Eu(III) complex is insensitive to oxygen; mixture of Eu(III) and Tb(III) complex used for ratiometric oxygen measurement; efficient generation of singlet oxygen; cell permeable. | I | 258 |
| Ga(TTA)3 in poly(styrene-co-4-N-acridono-methylstyrene) | 400/514 | τ 0/τ100kPa ∼ 1.13 | Both the Ga(TTA)3 and Eu(TTA)3 were encapsulated into the poly(styrene-co-4-N-acridonomethylstyrene) film, the acridone group in the polymer acts as an antenna to absorb blue light and the energy transferred to both probes; high brightness, compatible with a 405 nm LED. | I, L | 259 |
| Eu(TTA)3 in poly(styrene-co-4-N-acridono-methylstyrene) | 400/617 | I 0/I8.5kPa ∼ 8.0 | |||
Terbium(III) complexes emit green luminescence peaking at 546 nm. The Tb(acac)3(phen) complex was synthesized and adsorbed on silica (an alumina plate as used for TLC) to test it for the quenching of its luminescence by oxygen.256,257 Its QY is 0.5, and lifetimes can be as long as several ms. Its luminescence is more efficiently quenched by oxygen than that of the europium complex. Terbium(III) complexes with an azaxanthone sensitizer and a naphthyl group are also sensitive to oxygen, while the corresponding europium complexes are not.258 This finding may be used for ratiometric (2-wavelength) sensing of oxygen. However, most lanthanide OSPs have to be excited in the UV region. OSPs that can be excited with blue light were first reported by Borisov et al.259 The complexes of Eu(III) and Gd(III) were incorporated into a film of poly(styrene-co-4-N-acridonomethylstyrene) film, where the acridone group act as light-harvesting antenna and the absorbed energy is transferred to the Eu(tta)3 and Gd(tta)3 complexes. These Eu(III) complexes possess bright red room temperature luminescence in polymers (with a QY of ∼20% and a lifetime ∼100 μs), while the Gd(III) complexes emit orange light (QY ∼50%, lifetime ∼2 ms). Both emissions are efficiently quenched by molecular oxygen.
000 M−1 cm−1) between 390 and 420 nm (the so-called Soret bands), and weaker (>10000 M−1 cm−1; the so-called Q-bands) at >500 nm. Obviously, the brightness of the indicators is much higher if they are photoexcitated via the Soret band.
Tetraphenylporphyrin (TPP) without a central metal ion can be photoexcited at wavelengths between 520 and 550 nm, for example with LEDs. Adsorbed on porous polystyrene,261 two fluorescence bands with peaks at 650 and 720 nm can be seen that probably originate from the monomer and dimer species. Fluorescence is quenched by ca. −60% and −55% at 650 and 720 nm, respectively, in going from an oxygen-free environment to pure oxygen. TPP on polystyrene is moisture-insensitive, but the emission strongly depends on temperature. TPPs are not often used for purposes of sensing oxygen because of their poor photostability and for other reasons.
6.2.5.1. Conventional metalloporphyrins. Metalloporphyrins (for chemical structure see Fig. 11), while completely insoluble in water unless chemically modified, display fairly good solubility in many organic polymers but tend to aggregate at higher concentrations. Other features include (1) intense and sharp red luminescence with Stokes shifts of up to 250 nm; (2) luminescence lifetimes in the order of hundreds of microseconds in the absence of oxygen; (3) good chemical stability, in particular if fluorinated; and (4) tunable excitation and emission spectra by peripheral substitution.262 Eastwood and Gouterman263 screened a large number of cobalt(II), nickel(II), platinum(II) and palladium(II) porphyrin complexes. The Pt(II) and Pd(II) metalloporphyrins display strong phosphorescence and long lifetimes, both of which are strongly reduced in the presence of oxygen. They suggested the use of PtTPP and PdTPP as OSPs. Ever since, such kinds of metalloporphyrins have been widely used.
 | ||
| Fig. 11 Chemical structures of major OSPs based on metalloporphyrins. M stands for a metal ion such as Pt(II) or Pd(II). Complexes with trivalent ions such as Ir(III) have also been reported but require an additional (axial) ligand. | ||
In general, the Pd(II) metalloporphyrins have much longer lifetimes and higher sensitivity to oxygen than their Pt(II) analogs. This can be attributed to increased spin–orbit coupling of the heavier metal ion.263 On the other side, the Pt(II) complexes have 2–3 times higher emission QYs than those of the respective Pd(II) metalloporphyrins. Initially, the Pt(II) and Pd(II) complexes of coproporphyrin (CP), octaethylporphyrin (OEP), and TPP have been used. Wilson et al.51,264 studied the quenching of the phosphorescence of PdCP to image oxygen in cells and tissue, and PdTPP was deposited in organized monolayer assemblies using the Langmuir–Blodgett technique.265 The monolayers were coated onto an optical fiber to give the respective sensor. PtOEP, dissolved in silicone rubber, was also used in the first paint for sensing air pressure in wind tunnel tests.266 PtOEP has a QY of 0.42, and its red luminescence (peaking at 650 nm) is strongly quenched by oxygen.267 However, PtOEP easily photodegrades on exposure to shortwave light due to photooxidation of the porphyrin ring.266
In order to improve photostability, several halogen-substituted platinum porphyrins (PtTDCPP, PtTFMPP, PtBr8TMP; see Table 8) were synthesized and studied, in silicone rubber films,268 with respect to photostability and suitability to sense oxygen. The incorporation of halogenated substituents into the porphyrin ligand can substantially increase photostability, but lifetimes and QYs are decreased. Not unexpectedly, the introduction of a heavy halogen atom such as bromine causes a further decrease in QY and lifetime. Fluorine, in contrast, is an excellent choice as a substituent to improve photostability. It generally renders porphyrins and other dyes more photostable. The fluorine-substituted porphyrin PtTFPP has become the most often used OSP among the metalloporphyrins.53 It has three main absorption bands, viz. an intense Soret band peaking at 395 nm, and two Q absorption bands at 508 and 540 nm, respectively, which are much weaker. The four electron-withdrawing pentafluorophenyl substituents raise the redox potentials and the electron density of the probe. This renders PtTFPP even more photostable than Ru(dpp). In addition, PtTFPP is less sensitive to temperature than Ru(dpp).
| Dye/Matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a TPP = meso-tetraphenylporphyrin, OEP = octaethylporphyrin. b IBM = isobutyl methacrylate, TFPM = tetrafluoropropyl methacrylate. c PFS = pentafluorostyrene. d PFE-VFP = poly(tetrafluorethylene)-co-(vinylidene fluoride)-co-(propylene). e CAB = cellulose acetate butyrate. f C4PATP = poly[(n-butylamino)thionylphosphazene]. g DiO = 3,3′-dioctadecyloxacarbocyanine perchlorate, PTMS = phenyl trimethoxysilane, MTMS = methyl trimethoxysilane. h 8F-PEKEK(Ar) = poly(aryl ether ketone) where Ar is 2-2-bis(4-hydroxyphenyl)-1,1,1,3,3,3-hexafluoropropane (6FBA), or 9,9′-bis(4-hydroxyphenyl)propane (HF). i TBP = tributyl phosphate. j PTP = poly(aminothionylphosphazene). k OEPK = octaethylporphyrin ketone. l TFPP = 5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorophenyl)porphyrin. m tBS = 4-tert-butylstyrene, TFEM = trifluoroethyl methacrylate. n PtTFPL = platinum tetra(pentafluorophenyl)porpholactone; FIB = copolymer of heptafluoro-n-butyl methacrylate and hexafluoroisopropyl methacrylate. o PdPC10COOH = [5-(1-carboxydecyl-4-pyridyl)-10,15,20-tritolylporphyrinato]palladium. p TCPP = tetrakis(4-carboxyphenyl)porphyrin. q TMPyP = meso-tetrakis (4-N-methylpyridyl) porphyrin. r CPTEE = coproporphyrin tetraethyl ester, EP = etioporphyrin. s TDCPP = meso-tetra(2,6-dichlorophenyl)porphyrin, TFMPP = meso-tetra(3,5-bis(trifluoromethy1)phenyl)porphyrin, Br8TMP = meso-tetramesityl-β-octabromoporphyrin. t TPTBP = meso-tetraphenyltetrabenzoporphyrin; TPTBPF = meso-tetra(4-fluorophenyl)tetrabenzoporphyrin; TPTBPF2 = meso-tetra(3,5-difluorophenyl)tetrabenzoporphyrin. u NTBP = 6-aza-13,20,27-triphenyltetrabenzoporphyrin, N2-cisTBP = 6,13-diaza-20,27-diphenyltetrabenzoporphyrin. v 1NF = meso-tetra-(4-fluorophenyl)mononaphthotribenzoporphyrin, 2NF = meso-tetra-(4-fluorophenyl)dibenzodinaphthoporphyrin, 3NF = meso-tetra-(4-fluorophenyl)monobenzotrinaphthoporphyrin. w n-ButIm = N-(n-butyl)imidazolo, CarbIm = 1-imidazoleacetic acid. x DDA = N-dodecyl acrylamide; LB = Langmuir—Blodgett. y Pc(OH) = 2,9,16,23-tetraphenoxy-29H,31H-phthalocyanine hydroxide. | |||||
| H2TPPa in plastic-clad silica particles | 590/650 | I 0/I100 ∼ 1.54 | Dye adsorbed on particles from solution; some photobleaching; intensity decreases on storage due to diffusion of the dye in the polymer. | I | 316 |
| PtOEP in TEOS sol–gel | 586/645 | I 0/I100 = 41 | Preparation time ∼1 month; response time up to 9 min. | I | 317 |
| PtOEP in TEOS sol–gel | 535/646 | I 0/I100 ∼ 8.5 | Addition of a surfactant improves film homogeneity and results in crack-free monoliths. | I | 318 |
| PtOEP in polystyrene (PS) | 535/640 | τ 0/τair = 5 | Phase modulation technique employed for lifetime determination. | L | 319 |
| PtOEP in PMMA | 535/640 | τ 0/τair = 1.54 | |||
| PtOEPK in PS | 592/760 | τ 0/τair = 4 | Poor photostability. | ||
| PdOEP in PS | 545/670 | τ 0/τair = 50 | Excellent photostability. | ||
| Ru(dpp)3 in PS | 450/600 | τ 0/τair = 1.28 | |||
| PtOEP in poly-IBM | 535/644 | I 0/I100 = 69.3 | Linear SVPs. | I | 320 |
| PtOEP in poly(IBM-co-TFPM)b | 535/644 | I 0/I100 = 86.4 | Linear SVPs. | ||
| PtOEP in poly-IBM | 535/646 | I 0/I100 = 69 | Extremely high sensitivities and linear SVPs in fluoropolymers; good photostability. | I | 321 |
| PtOEP in poly(IBM-co-TFEM) | 535/646 | I 0/I100 = 288 | |||
| PtOEP in PS | 535/646 | I 0/I100 = 4.5 | |||
| PtOEP in poly(styrene-co-TFEM) | 535/646 | I 0/I100 = 296 | |||
| PtOEP in PS | 535/645 | I 0/I100 = 4.5 | Sensitivity in poly(styrene-co-PFS) is much higher than in PS; linear SVPs for PdOEP in the 0–20% oxygen range. | I | 322 |
| PtOEP in poly(styrene-co-PFS)c | 535/645 | I 0/I100 = 25 | |||
| PdOEP in PS | 546/664 | I 0/I100 = 46.0 | |||
| PdOEP in poly(styrene-co-PFS) | 546/664 | I 0/I100 = 93.6 | |||
| PtOEP in poly(1-trimethylsilyl-1-propyne) | 535/646 | I 0/I100 = 225, I0/Iair ∼ 80 | High photostability; suitable for trace oxygen determination; extremely gas permeable polymer; non-linear SVPs. | I | 323 |
| PtOEP in PS | I 0/I100 = 4.5 | ||||
| PtOEP in PDMS rubber | I 0/I100 = 5.5 | ||||
| PtOEP in PFE-VFPd | 510/630 | I 0/IO2satur = 1.8 | For dissolved oxygen; poor sensitivity; dye aggregates during film preparation; non-linear SVPs. | I | 324 |
| PtOEP in pPEGMA | 535/644 | I 0/I100 ∼ 56 | τ 0 100 μs; more photostable than other complexes; linear SVP. | I, L | 325 |
| PdOEP in pPEGMA | 545/663 | I 0/I100 ∼ 300 | τ 0 770 μs; linear SVPs. | ||
| RuOEP in pPEGMA | 532/656 | I 0/I100 ∼ 4.5 | τ 0 140 μs; low phosphorescence QY; poor photostability | ||
| PdOEP in ethyl cellulose/CABe/PVC | 546/670 | I 0/Iair ∼ 42/18/2.4 | τ 0 1.41/1.45/1.34 ms. | I, L | 326 |
| PtOEP in ethyl cellulose/CAB/PVC | 535/644 | I 0/Iair ∼ 20/12/2 | τ 0 81/100/90 μs. | ||
| PtOEP in PDMS; SiO2 added as scatterer | 534/641 | I 0/I100 = 175–8 | Larger fractions of SiO2 result in less sensitivity and larger τ0 (65–91 μs); dye adsorbed on SiO2 and aggregated. | I, L | 327 |
| PtOEP in C4PATPf; SiO2 added | 535/645 | I 0/I100 = 60–50 | Small dependence on SiO2 content; τ0 104 μs; the dye is located in the matrix polymer. | ||
| PtOEP + DiO in PTMOS MTMOS ormosil nanoparticlesg | 570/640 | I 0/IO2 satur ∼ 33.3 | Ratiometric nanosensors (120 nm in diameter); DiO and OEP are reference dyes; linear SVPs; good storage stability, no leakage; reference dyes are less photostable. | Ir | 328 |
| PtOEPK + OEP in PTMOS MTMOS ormosil nanoparticles | 568/750 | I 0/IO2 satur ∼ 33.3 | |||
| PtOEP/PTMOS + MTMOS ormosil | 505/red PD | τ 0/τair = 10.6 | τ 0 88 μs; linear SVPs; short response times (t90 250 ms). | I, L | 205 |
| PdOEP in 8F-PEKEK(6FBA/HF)h | 546/664 |
I
0/I100 = 240/227
I 0/Iair ∼ 80/75 |
SVPs close to linear up to air saturation; suitable for trace oxygen. | I | 329 |
| PtOEP in 8F-PEKEK(6FBA/HF) | 535/646 | I 0/I100 = 32.4/23.6 | SVPs close to linear up to 100% of O2. | I | 329 |
| Poly-PtOEP | 540/644 | τ 0/τair satur = 41 | τ 0 110 μs; response times <100 ms; film thickness 0.2 μm; highly photostable. | L | 330 |
| Poly-PtTPP | 514/648 | τ 0/τair satur = 57 | τ 0 69 μs; linear SVPs; response times <20 ms; film thickness 0.2 μm; highly photostable. | ||
| Poly(PtTPP-co-IBM-co-TFEM) | 402,510/665 | I 0/I100kpa > 10 | Polymerizable PtTPP probe; highly sensitive to oxygen; linear SVPs. | I | 301 |
| PtOEP in CAB (+TPB)i | I 0/Iair = 13.4 (170.2) | Addition of the plasticizer increases sensitivity and accelerates response and recovery. | I | 331 | |
| PdOEP in CAB (+TPB) | I 0/Iair = 106.5 (631) | ||||
| PtOEP in PMMA (+TPB) | I 0/Iair = 1.78 (57.5) | ||||
| PdOEP in PMMA (+TPB) | I 0/Iair = 21 (671) | ||||
| PtOEP in PTPj | 380/645 | I 0.01atm/I1.00atm = 58 | τ 0 79 μs; linear SVPs. | I, L | 332 |
| Ru(dpp)3 in PTP | 450/610 | I 0.01atm/I1.00atm = 2.55 | τ 0 6.9 μs; slight downward curvature of SVPs. | ||
| PtOEP Schiff base in PVC | 398,443/650 | I 0/I21KPa ∼ 3.0 | Oxygen and pH dual sensor based on a single probe; absorption changes with pH; luminescence and lifetime change with oxygen; non-linear SVPs; PdCP Schiff-base suitable for low oxygen. | L | 333 |
| PdCP Schiff base in PVC | 398,443/650 | I 0/I21kPa ∼ 8.5 | |||
| PtOEPKk in PS | 592/759 | τ 0/τair = 3.6 | QYphos 0.12; τ0 61 μs; photostability 9 times better than for PtOEP. | L | 280 |
| PdOEPK in PS | 602/790 | QYphos 0.04; τ0 480 μs. | |||
| PtOEPK in PS | 591/760 | τ 0/τair = 3.76 | QYphos ∼ 0.1; τ0 64 μs; linear SVP; sterilizable in ethanol or by heat. | L | 281 |
| PdOEP in PS | 546/670 | τ 0/τair ∼ 198 | QYphos 0.2; τ0 990 μs; linear SVP. | ||
| PtOEPK in PVC | 592/758 | τ 0/τ728Torr ∼ 2.05 | τ 0 ∼ 60 μs; linear SVP in PVC, slight downward curvature in PS. | I, L | 334 |
| PtOEPK in PS | 592/758 | τ 0/τ728Torr ∼ 20 | |||
| PdOEPK in PS | 602/790 | τ 0/τ728Torr ∼ 9.5 | τ 0 ∼ 450 μs; linear SVP in PVC, slight downward curvature in PS. | ||
| PdOEPK in PVC | 602/790 | τ 0/τ728Torr ∼ 29 | |||
| PtOEPK in PVC + plasticizer | 592/758 | I 0/I100 ∼ 13.6 | Response time <100 ms; addition of plasticizer reduces response time to 66 ms. | I | 335 |
| PdOEPK in Teflon AF | 590/760 | I 0/I0.01% ∼ 1.17 | Phase modulation technique; linear SVP; for trace oxygen; polymer is 130-times more permeable for oxygen than PS; highly oxygen permeable (130 times better than PS); highly photostable. | L | 336 |
| PtTFPPl in PS | 541/650 |
I
0/I100 = 3.0
I 0/Iair = 1.9 |
SVPs linear up to 20% oxygen; response time 18 s; much more photostable than PtOEP. | I | 269 |
| PtTFPP in poly(tBS-co-TFEM)m | 465/650 | I0/I14.7PSI = 11 | Polymer has good oxygen permeability even better than poly(styrene-co-TFEM); excellent mechanical properties, adheres to glass and metal substrates. | I | 337 |
| PtTFPP in PTFEM | 544/648 | I 0/I100 = 15.4 | Excellent photostability; linear SVPs; response time 5.6 s. | I | 270 |
| PtTFPP in PS | 544/648 | I 0/I100 = 3.0 | Good photostability; non-linear SVP; moderate sensitivity. | ||
| PtTFPP in octyl-triEOS TEOS ormosil | 380/650 | I 0/I100 = 22 | Response time 0.6 s; good stability for long-term usage. | I | 338 |
| PtOEP in octyl-triEOS TEOS ormosil | 380/645 | I 0/I100 = 47 | |||
| PtTFPP in octyl-triEOS TEOS ormosil and silica nanoparticles | 405/650 | I 0/I100 = 106 | Addition of silica nanoparticles improves oxygen quenching; response time 1.3 s; non-linear SVPs. | I | 339 |
| PtTFPP in n-propyl-TriMOS and TFP-TriMOS ormosil | 400/650 | I 0/I100 = 68.7 | Fluorinated ormosil; response time ∼3 s; linear SVPs. | I | 340 |
| PtOEP in n-propyl-TriMOS and TFP-TriMOS ormosil | 400/646 | I 0/I100 = 82.5 | |||
| PtTFPP in ormosil made from 3 precursors | 405/650 | I 0/I100 from 101 to 155 | Three precursors used to make ormosil films; extremely high sensitivities; response time ∼3 s; good oxygen mechanical flexibility; linear SVPs. | I | 341 |
| Poly(PtTFPP-co-HEMA) | 405/650 | I 0/I100 = 1.1 | PtTFPP modified with polymerizable group or silane group; covalently bound to polymer or sol–gel; excellent stability; no leaching; photostable; fast response. | I | 305 |
| Poly(PtTFPP-co-styrene) | 405/650 | I 0/I100 = 5.8 | |||
| PtTFPP in sol–gel film | 405/650 | I 0/I100 = 70 | |||
| PtTFPP in silica gel beads in silicone | 405/650 | I 0/I100Pa ∼ 1.4 | Covalently immobilization; highly photostable; low temperature dependence. PdTFPP has an exceptionally long lifetime (∼1 ms). | I, L | 272 |
| PdTFPP in silica gel beads in silicone | 405/680 | I 0/I1000Pa ∼ 5 | |||
| PtOEP in poly(styrene-co-TFEM) | 535/654 | I 0/I100 = 87.6 | Sensitivity decreases with increasing PS/TFEM ratio; for trace O2 | I | 342 |
| PdOEP in poly(styrene-co-TFEM) | 546/664 | I 0/I100 = 661.7 | |||
| PtTFPL in FIBn | 400(574)/738 | I 0/Iair ∼ 5 | Sensitivity comparable to PtTFPP; lower dependence on temperature than PtTFPP; τ0 72 μs, QYphos is ∼60% of PtTFPP (τ0 120 μs). | I, L | 273 |
| PdPC10COOHo on Al2O3 | 418/708 | I 0/Iair ∼ 15 | Probe has long alkyl chain and self-assembles on aluminum plate; non-linear SVPs; seems to be photostable; strong quenching | I | 343, 344 |
| PtTCPPp on a alumina plate | 538/665 | I 0/Iair ∼ 10 | Non-linear SVPs for both sensors | I | 296–298 |
| PdTCPP on a alumina plate | 523/701 | I 0/Iair ∼ 14 | |||
| PdTMPyP4+ in Nafionq | 442/668 | I 0/I100 = 70 | Other metalloporphyrins are also tested; linear SVPs; single exponential phosphorescence decays for all metalloporphyrins; phosphorescence lifetimes increased in Nafion; low QYs. | I-RTP, L | 288 |
| PtTMPyP4+ in Nafion | 442/645 | I 0/I100 = 6 | |||
|
ZnTPP on silica gel
ZnTPP in NaCl crystal |
551/697 | – | In silica gel, phosphorescence quenching is temperature dependent. In NaCl crystal, quenching obeys Stern–Volmer kinetics; low temperature dependence. | I-RTP | 345 |
| PtCPTEEr in PS | 382(535)/647 | τ 0/τair satur = 3.69 | Hydrophobic probes; QYs between 0.6 and 0.9; high concentration of probes used in polymers. | I-RTP, L | 346 |
| PtCPTEE in silicone rubber | 376(535)/647 | τ 0/τair satur = 13.0 | |||
| PtTPP in PS | 404(508)/662 | τ 0/τair satur = 4.56 | |||
| PtOEP in PS | 383(535)/647 | τ 0/τair satur = 3.60 | |||
| PtEP | 380(535)/647 | ||||
| Pt coproporphyrin in PMMA | 378(512,544)/667 | τ 0/τair ∼ 6 | Low toxicity; soluble in basic aqueous solution, DMF, pyridine; response not only depends on polymer, but also on the solvent used for dissolving the polymer. | L | 289 |
| Pt coproporphyrin in PVC | τ 0/τair ∼ 4.3 | ||||
| Pt coproporphyrin in PS | τ 0/τair ∼ 11.7 | ||||
| Pt coproporphyrin in silicone rubber | τ 0/τair ∼ 46 | ||||
| PtTDCPPs in silicone rubber | 394/650 | I 0/I100 = 51 | Good photostability; downward curved SVP; response time <10 s; self-quenching may occur at high concentration. | I | 268 |
| PtTFMPP in silicone rubber | 395/646 | I 0/I100 = 42 | |||
| PtBr8TMP in silicone rubber | 425/721 | I 0/I100 = 45 | |||
| PtTPTBP in PSt | 432,615/772 | τ 0/τair = 3.08 | Strong red Q band enables the probes to be excited using red LEDs or lasers; high QYs and photostability (especially Pt complexes), low tendency to aggregation in nonpolar polymers; non-linear SVPs; palladium complexes are more prone to thermal quenching; all probes also can be incorporated in PS-co-PVP or polysulfone nanoparticles. | L | 277, 278 |
| PtTPTBPF in PS | 431,617/777 | τ 0/τair = 3.31 | |||
| PtTPTBPF2 in PS | 425,619/785 | τ 0/τair = 2.96 | |||
| PdTPTBP in PS | 445,630/800 | τ 0/τair = 5.78 | |||
| PdTPTBPF in PS | 444,631/801 | τ 0/τair = 5.34 | |||
| PdTPTBPF2 in PS | 439,630/812 | τ 0/τair = 6.48 | |||
| PtNTBPu in PS | 406,630/844 | τ 0/τair = 2.34 | Intense Q-band absorption; compatible with red laser diode; less bright and less sensitive than the TPTBP complex; good photostabilities; negligible thermal quenching at RT; QYs of palladium complexes <10%. | I | 282 |
| PtN2-cisTBP in PS | 388,621/841 | τ 0/τair = 1.72 | |||
| PdNTBP in PS | 421,642/875 | τ 0/τair = 7.31 | |||
| PdN2-cisTBP in PS | 380,631/873 | τ 0/τair = 3.92 | |||
| Pt1NF in PSv | 434,628/815 | τ 0/τ20kPa = 3.6 | τ 0 44 μs; QY 0.53. | L | 284 |
| Pt2NF in PS | 438,652/835 | τ 0/τ20kPa = 3.2 | τ 0 28 μs; QY 0.27. | ||
| Pt3NF in PS | 441,667/870 | τ 0/τ20kPa = 2.6 | τ 0 21 μs; QY 0.25. | ||
| Pd1NF in PS | 450,641/849 | τ 0/τ20kPa = 13 | τ 0 203 μs; QY 0.18. | ||
| Pd2NF in PS | 452,666/868 | τ 0/τ20kPa = 11 | τ 0 138 μs; QY 0.12. | ||
| Pd3NF in PS | 456,681/882 | τ 0/τ20kPa = 10.4 | τ 0 106 μs; QY 0.07. | ||
| Ir-OEP-CO-Cl in PS | 404,550/672 | τ 0/τ20kPa ∼ 9 | τ 0 97 μs; QY 0.14. | L | 286, 287 |
| Ir-OEP-Py2 | 389,539/655 | — | τ 0 40 μs; QY 0.195 | ||
| Ir-OEP-n-butIm2 in bufferw | 390,541/655 | τ 0/τ0.5kPa ∼ 8 | τ 0 27 μs; QY 0.20 | ||
| Ir-OEP-n-barbIm2 in silica gel | 388,538/652 | τ 0/τ0.4kPa ∼ 1.7 | τ 0 37 μs; QY 0.21. | ||
| Poly(DDA-co-PtTPP) in LB filmx | 400/660 | I 0/I100 = 74 | PtTPP covalently bound to a polymer to form LB films; addition of silver nanoparticles enhances luminescence, but decreases sensitivity; nearly linear SVPs. | I | 347 |
| Poly(DDA-co-PtTPP) in LB film and silver nanoparticles | I 0/I100 = 25 | ||||
| AlPc(OH)y in PS | 606/706 | I 0/I10 = 1.04 | Poor sensitivity; non-linear SVP; τ0 11.7 ns. | I | 348 |
PtTFPP is also highly photostable in polymers such as polystyrene and therefore well suited for long-term continuous monitoring of oxygen.269 If continuously photoexcited at the Q bands at 508 or 540 nm for 50 h, the decreases in emission intensity (at 650 nm) are only 9% and 15%, respectively. PtTFPP was also immobilized in the fluoropolymer PTFEM (see Section 7) where it was found270 to be quite photostable. Upon continuous irradiation for 24 h, only minimal deterioration (∼0.5%) was observed. The resulting oxygen sensor material has better sensitivity (the I0/I100 ratio is 15.4) compared to that of the same probe in polystyrene (I0/I100 = 3.0), and the response time was reduced to 5.6 s.
The brightness of PtTFPP can be further enhanced using light-harvesting antennas. This approach is particularly beneficial because luminescent sensor films as thin as <0.5 μm can be produced that still possess an adequate signal-to-noise ratio.271 The palladium analogue (PdTFPP) has an exceptionally long lifetime (∼1 ms at room temperature), and has such a high sensitivity that it can only be used to sense oxygen at trace levels (from 0.02 to 100 Pa).272 Also see the section on low-level oxygen sensors.
6.2.5.2. Metalloporphyrins with large Q-band absorption and NIR emission. As was stated before, the probe PtTFPP has outstanding photophysical properties in terms of oxygen sensing, the weak Q bands being the only shortcoming. This situation was (partially) improved by synthesizing metalloporphyrins possessing much stronger Q-band absorptions, for examples the various porphyrin lactones, porphyrin ketones, benzoporphyrins, and azatetrabenzoporphyrins.273–275 Simultaneously, the emission bands were shifted to the NIR. For example, the probe PtTFPL has a fairly strong absorption of its Q-band centered at 550 nm, while the emission peak is red-shifted to 730 nm (compared to that of PtTFPP at 650 nm). The probe is more sensitive to oxygen than PtTFPP. Its unquenched lifetime is 72 μs, and luminescence is less sensitive to temperature than PtTFPP. The Pd(II) analog (PdTFPL) has an even longer lifetime of ∼1 ms. Both probes are quite resistant to photo-oxidation because of the presence of a lactone functionality.276
Fluorinated complexes of Pt(II) and Pd(II) with meso-substituted benzoporphyrins represent another group of longwave absorbing and NIR-emitting probes.277–279 Such fluorinated probes are more photostable, and their emission bands extend to above 800 nm. This is an analytical wavelength that is hardly interfered by background luminescence. They possess intense Soret bands and fairly strong Q-band absorptions, and thus can be effectively excited with blue or red LEDs or lasers. The meso-substituted benzoporphyrins do not tend to aggregate in nonpolar polymers. Compared to classical metalloporphyrins such as PtTFPP and PtOEP, the benzoporphyrins display a more intense second Q-band absorption and higher QYs, some having QYs exceeding those of PtOEP (0.415).
As was to be expected in view of their longer lifetimes, the respective Pd(II) complexes are more sensitive to oxygen than the Pt(II) probes. Dissolved in polystyrene, the latter are ideally suited for sensing oxygen partial pressures up to air pressure, and both of gaseous and fluid samples. The Pd(II) complexes, on the other hand, are more adequate for sensing low levels of oxygen because their luminescence is almost completely quenched at air saturation. The Pd(II)benzoporphyrins are more prone to thermal quenching than the Pt(II) complexes. All these OSPs have good photostability, the Pt(II) complexes in particular. Fluorination of the benzo ring resulted in a further red shift of the Q-band and the emission band, and also improves photostability, however at the expense of phosphorescence QY. Among the fluorinated metal benzoporphyrin complexes, Pt(II) meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (PtTPTBPF) has the highest brightness and photostability. Its synthesis is difficult, however.
Papkovsky280,281et al. have synthesized the blue Pt(II) and Pd(II) complexes of certain ketoporphyrins (PtOEPK). Their absorptions peak at around 590 nm, and this makes them compatible with yellow LEDs. In addition, they display large Stokes shifts, almost linear Stern–Volmer plots in polystyrene, and excellent sensitivity to oxygen. All Pt(II) and Pd(II) complexes of porphyrin lactones and ketoporphyrins have relatively intense absorptions at 570–600 nm, but QYs are moderate. Borisov et al.282 reported on Pt(II) and Pd(II) complexes of azatetrabenzoporphyrins. The strong Q-band absorptions were found to be excellently compatible with the 635 nm laser diode (Fig. 12), and the strong NIR phosphorescence is efficiently quenched by oxygen. The luminescence decay times are shorter than those of the respective TPTBP complexes, and this enabled the design of less sensitive probes with wider analytical range. The QYs are lower (∼0.1) than those of the TPTBP complexes, especially for the case of Pd(II) azatetrabenzoporphyrins, their synthesis is more complicated, and the solubility in organic polymers is limited.
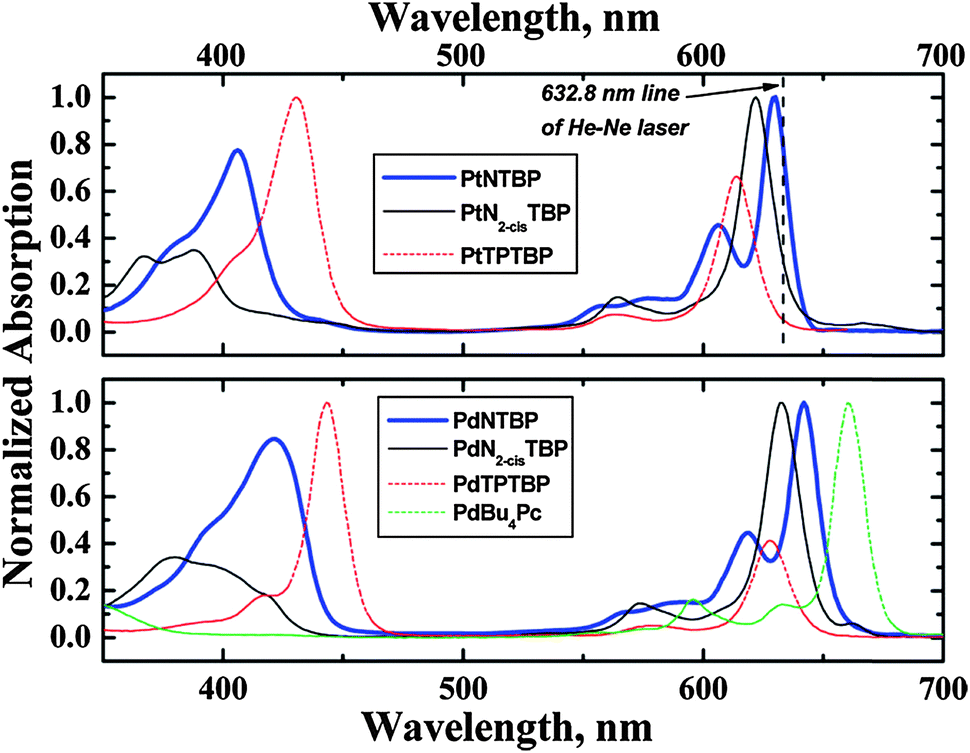 | ||
| Fig. 12 Absorption spectra of the Pt(II) and Pd(II) complexes with azatetrabenzoporphyrins NTBP and N2-cisTBP. The Q-band absorption is very intensive and compatible with laser diodes. (Reprinted from ref. 282 with permission from the American Chemical Society). | ||
The absorption and emission peaks of porphyrins were further longwave shifted by extending the π-system. The Vinogradov group283 described metal complexes of tetranaphthoporphyrin (TNP) which, compared to the respective tetrabenzoporphyrins, have red-shifted Q-band absorptions and longwave emissions, but their QYs are smaller (0.22 for PtTPTNP; 0.08 for PdTPTNP), as are their photostabilities. Other π-extended hybrid benzo- and naphthoporphyrin complexes of Pt(II) and Pd(II) were reported284 with tuned spectral properties, intense absorption in the NIR region (628–691 nm), and emissions peaking between 815 and 882 nm. It was observed that both the QY and luminescence decay time decrease with increasing number of naphtho groups in the system.
Many tetraarylporphyrins and their Pt(II) complexes were investigated with respect to uses in sensors for oxygen.285 The Pt(II) complexes of tetraphenylporphyrin, tetranaphthoporphyrin, and tetrapyrenyl-porphyrins possess rather similar photophysical properties. Placed in polymer films, both the emission intensity and lifetime are quenched by oxygen. The naphthyl derivatives display better sensing capability, with a quenching constant (KSV) of 0.068 Torr−1 (versus 0.040 Torr−1 for the parent tetraphenyl derivative).
The Ir(III) porphyrin complexes form a rather recent class of OSPs.286 The photophysical properties of these hexa-coordinated complexes can be tuned by changing the axial ligands. The neutral Ir–OEP–CO–Cl complex has a good QY (0.21) and its emission is quite sensitive to oxygen, and the lifetime is 97 μs. By proper choice of the axial ligand, the solubility of the complexes in polymers can be improved. Respective structures are shown in Fig. 13. The axial ligand may also be modified with functional groups for covalent coupling, for example to a peptide.287
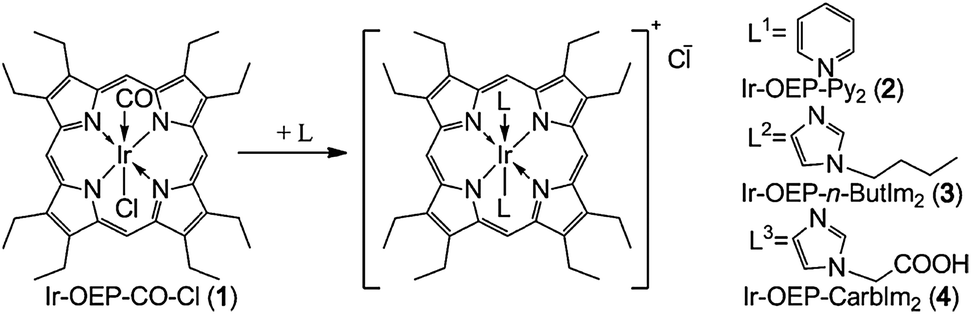 | ||
| Fig. 13 Chemical structures of the four Ir–octaethylporphyrin complexes. The change of axial ligand with functional group is beneficial for post-modification and coupling. (Reprinted from ref. 286 with permission from Wiley-VCH). | ||
6.2.5.3. Water-soluble metalloporphyrins. Water-soluble porphyrins, in contrast to the former ones, are useful for sensing dissolved oxygen in aqueous solutions. Vasil'ev and Borisov288 synthesized water-soluble cationic complexes of Pt(II), Pd(II) and Rh(III) with the water-soluble porphyrins meso-tetrakis (4-N-methylpyridyl)porphyrin and meso-tetrakis (4-N,N,N-trimethylaminophenyl)porphyrin. The resulting cationic metalloporphyrins have been immobilized on (anionic) ion-exchange films of the Nafion type to result in sensitive, stable and fast-responding sensor films. Linear Stern–Volmer plots were obtained over the entire range of oxygen partial pressure.
The Pt(II) and Pd(II) complexes of coproporphyrins represent another kind of water-soluble OSPs.289,290 They are non-toxic, have high phosphorescence QY and relatively long decay times at room temperature, but photostabilities are not excitingly good. Papkovsky et al.291 synthesized the hydrophilic coproporphyrin ketones PtCPK and PdCPK. They are water-soluble, display acceptable photostability and red-shifted emission in the NIR (PtCPK: λem 767 nm; PdCPK: λem 796 nm). Moreover, they can be photo-excited at 370–410 nm (Soret bands) or, less efficiently, at 580–600 nm (Q bands). Compared to the Pt(II) and Pd(II) coproporphyrins, the PtCPK and PdCPK probes have enhanced Q-band absorbance, and the emissions are red-shifted by ∼60 nm. The relatively strong Q-band absorbance and intense NIR emission make these probes viable intracellular OSPs.292 Unlike PtOEPK or PdOEPK (which have very high quenchability), the two probes exhibit moderate sensitivities to oxygen (τ0/τ100 is 1.7 and 4.44 for PtCPK and PdCPK, respectively), which is kind of unexpected.
The water-soluble Pt(II) and Pd(II) meso-tetra[4-carboxyphenyl]porphine (TCPP) were also shown to be useful OSPs. Both the dissolved form (for intracellular studies293–295) and the adsorbed form (on silica296–298) for sensing gaseous oxygen were studied. They do not exhibit high QYs or long lifetimes. However, the carboxy groups enable further modifications in that they can be functionalized with positively charged groups for promoting the loading efficiency into cells.299 Alternatively, they may be covalently linked to peptides and loaded into cells without any transfer reagent.300
6.2.5.4. Functionalized metalloporphyrins. In order to render metalloporphyrins conjugatable to solid matrices they have to be functionalized. For example, a PtTPP derivative with a polymerizable methacrylate group was prepared and copolymerized with the host polymer IBM-co-TFEM (see Section 7).301 The introduction of the methacrylate group and copolymerization did not influence its oxygen sensing properties. The resulting copolymer has good oxygen sensitivity, and the SVPs are linear, which indicates that chemical binding to the polymer backbone does not result in large microheterogeneity. In contrast, a sensing film prepared by dissolving PtTPP in poly-IBM-co-TFEM has a downward curved Stern–Volmer plot (SVP). Similar findings were reported by Tian et al.302 who modified PtTPP with a polymerizable group to obtain an oxygen sensitive monomer. The monomer was copolymerized with hydroxyethyl methacrylate and acrylamide to give a hydrophilic sensor film. A polymerizable pH probe was also incorporated so that a dually (pH- and oxygen-)responsive material was obtained that has green and red emission spectra. No toxicity to HeLa cells was detectable after 40 h of incubation. The groups of Tian303 and Lu et al.304 have chemically modified PtTFPP with hydroxyethyl methacrylate (HEMA) moieties in order to cross-link it with acrylamide and styrene. The poly-HEMA matrix is more suitable than polystyrene (PS) for sensing dissolved oxygen, because sensitivity in poly-HEMA is higher than that in PS, and response is faster. Compared to sensing films prepared by physical entrapment of PtTFPP in polymers, chemical cross-linking alleviated the leaching problem and improved photostability. PtTFPP was also modified305 with a silane group and covalently attached to the network of a sol–gel and an organically modified sol–gel. The modification of PtTFPP or PdTFPP with thiol group enables them to be covalently attached to polymer or a silica matrix.306 No leakage of the OSPs does occur as a result.
6.2.5.5. Metalloporphyrins that undergo two-photon excitation. The Soret band of Pt(II) and Pd(II) porphyrin-based phosphorescent OSPs is in the near UV region, and this causes substantial autofluorescence in the case of biological samples. The Q band may also be used for excitation, but at the expense of brightness because the molar absorption coefficients of the Q bands are much weaker. Two-photon excitation (2-PE; typically performed at >700 nm) provides an alternative, but commonly used metalloporphyrins exhibit extremely low two-photon absorption cross-sections. Vinogradov et al.307 designed several water-soluble Pt(II) porphyrin-coumarin based dendrimers, in which 2-photon absorbing antennas (the coumarin moieties) were coupled to the metalloporphyrin core (see Section 9.5). The introduction of an antenna largely improves 2-PE via intermolecular energy transfer. The resulting metalloporphyrin-antenna construct was further incorporated in a protecting dendritic jacket to isolate the core from interactions with biological macromolecules, to control diffusion of oxygen, and to make the entire sensor water-soluble. Combined with multi-photon laser scanning microscopy, 3D imaging of oxygen with submicron spatial resolution was accomplished. The same group308 synthesized the highly phosphorescent Pt(II) porphyrin poly(arylglycine) dendrimer and used it for mapping oxygen in brains of mice and rats. Again, several coumarin units were attached at the periphery of the dendrimer to efficiently capture 2-photons and to transfer their energy nonradiatively to the Pt(II) porphyrin. Upon excitation with light of wavelength 780 nm, Pt(II) porphyrin undergoes fast intersystem crossing to its triplet state and emits phosphorescence which is quenched by molecular oxygen. Another phosphorescent probe, referred to as PtP-C343 (Fig. 14), is based on the photonic energy transfer from a coumarin antenna unit to a Pt(II)tetrabenzoporphyrin, and this also enables 2-PE in the NIR region.309,310 Lecoq et al.311 employed this in combination with phosphorescence lifetime microscopy to image oxygen partial pressure in the brain at depths up to 300 mm with μm-resolution. In addition, the method allowed simultaneous measurements of blood flow and of oxygen in capillaries. Sensory stimulation evokes functional hyperemia, accompanied by an increase in pO2 in capillaries. Two-photon laser scanning microscopic imaging of pO2 opens new avenues for studies of brain metabolism and blood flow regulation.
 | ||
| Fig. 14 Two-photon-enhanced oxygen probe PtP–C343 consisting of phosphorescent Pt meso-tetraarylporphyrin (PtP, red), several coumarin-343 units (C343, blue), polyarylglycine dendrimer (black) and peripheral oligoethyleneglycol residues (green). Arrows in the cartoon depict excitation of the C343 antenna via 2PA (brown), FRET (yellow) and phosphorescence of PtP-core (red). (Reprinted from ref. 309 with permission from Wiley-VCH). | ||
6.2.5.6. Metalloporphyrins for sensors with visual (bare-eye) read-out. Sensing oxygen with bare eyes via luminescent probes becomes possible if luminescence occurs in the visible and has strong intensity which is the case when using porphyrin-based OSPs. The human eye can identify ∼10 million color types. This was exploited312 in a two-color sensor where the green (and constant) luminescence of an inert dye serves as the color background, while the red emission of a porphyrin OSP varies in color according to the actual oxygen pressure. This sensing scheme has a kind of “traffic light response”. The contrast can be further improved313 by using the emission of a green LED as the background and the red emission of PtOEP as the signal. The green LED also photoexcites the luminescence of the OSP. In yet another kind of visual-readout oxygen sensor,314 quantum dots with green background luminescence were placed in a sol–gel film, while the red emission of the oxygen probe PtTFPP yields the analytical information. The colors of the sensors at various oxygen concentrations are shown in Fig. 15. A resolution of up to 1.0% is accomplished even with visual inspection. All these approaches are making the quantitation of oxygen simple and convenient.
 | ||
| Fig. 15 The apparent colors of luminescence colorimetric oxygen sensor without (upper) and with (lower) green-color background at different oxygen concentrations. (Reprinted from ref. 314 with permission from Wiley-VCH). | ||
Quantitative oxygen sensing and imaging was also accomplished using a photographic technique315 in which a colorimetric oxygen sensor is used to quantitatively image (“photographing”) oxygen. The technique is convenient in that it can be performed by unskilled persons, and simple in that a commercial camera (along with software that processes the data stored in the memory of the camera) can be applied (see Section 9.10.1). A similar system is commercially available (http://www.presens.de/).
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a dbp = 2,9-di-tert-butyl-1,10-phenanthroline; dmp = 2,9-dimethyl-1,10-phenanthroline. b dipp = 2,9-diisopropyl-1,10-phenanthroline. c POP = bis[2-(diphenylphosphino)phenyl]ether; xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene. d CN-xylyl = 2,6-dimethylphenylisocyanide. e PTO = 2-(pyridin-2-yl)-5-p-tolyl-1,3,4-oxadiazole. f Pt(thpy)2 = cis-bis(2-(2′-thienyl)pyridine)platinum(II). g pop = P2O5H22−. h Au complex = bis(μ-(bis(diphenylphosphino)octadecylamine-P,P′))diiodo gold(I). i Pb(7-SO3-Ox) = the 8-hydroxy-7-quinolinesulfonate chelate complex of lead(II). j ferron = 8-hydroxy-7-iodo-5-quinolinesulfonate. | |||||
| [Cu(dbp)(dmp)]PF6 in PSa | 450/610 | I 0/I100 = 1.60 | Low QY and lifetime (QYphos 0.01); τ0 0.73 μs. | I | 365 |
| [Cu(dipp)2]BF4 as a crystalb | 400/670 | I 0/I100 = 1.30 | Solid-state oxygen sensors, other Cu(I) crystals also tested; strictly linear SVPs; high solid-state QYs and lifetime (10 times larger than those in solution); response time 280 ms. | I | 357 |
| [Cu(dipp)2]tfpb as a crystal | 400/679 | I 0/I100 = 1.15 | |||
| [Cu(dmp)(dbp)]tfpb as a crystal | 400/608 | I 0/I100 = 1.10 | |||
| [Cu(POP)(dmp)]tfpb as a crystalc | 400/517 | I 0/I100 = 4.60 | Solid state oxygen sensors, other Cu(I) crystals also tested; strictly linear SVPs; QYs >0.66; lifetimes ∼30 μs; fast response (51 ms). | I, L | 358 |
| [Cu(xantphos)(dmp)]tfpb as a crystal | 375/540 | I 0/I100 = 6.65 | |||
| [Cu(xantphos)(dipp)]tfpb as a crystal | 400/513 | I 0/I100 = 4.41 | |||
| ([Cu(CNxylyl)2(dmp)]tfpb) as a crystald | 375/520 | I 0/I100 = 91.9 | Solid state oxygen sensors, other Cu(I) crystals also tested (lifetime of other crystal is from 100 μs to 1.2 ms); strictly linear SVPs; QYs around 0.23; fast response (67 ms). | I, L | 360 |
| [Cu(PTO)(PPh3)2]BF4 crystal in MCM-41 silicae | 355/515 | I 0/I100 = 5.95 | Crystals doped into MCM-41 silica to obtain an oxygen sensor; downward curved SVPs; good photostability; lifetime ∼64.4 μs; sensitivity depends on the doped concentration of Cu(I) crystal in silica. | I | 356 |
| Pt(dpp)(CN)2 in silicone RTV rubber | 290/520 | I 0/I100 ∼ 14 | Highly photostable; absorption in UV; forms an excimer at high concentration; response time <3 s. | I | 362 |
| Pt(dtbpy)(CN)2 in silicone RTV rubber | 302/486 | I 0/I100 ∼ 5 | |||
| Pt(thpy)2f in PDMS rubber | 470/>530 | I 0/I100 ∼ 10 | QYphos 0.36, τ0 4.8 μs; low absorption (2000 M−1 cm−1 at 470 nm); PDMS not specified, used with the curing agent; dye crystallizes at high concentrations; non-linear SVPs. | I, L | 366 |
| Pt(thpy)2 in PS | 470/>530 | I 0/I100 ∼ 3 | Intensity is 25 times higher than in PDMS; SVP almost linear. | ||
| [Pt2(pop)4](Bu4N)4 in silicone RTV rubberg | 368/509 | I 0/I100 ∼ 2.2 | Water-soluble probe; intense luminescence; high QY, excellent photostability in silicone rubber film. | I | 367 |
| [Pt2(pop)4](Bu4N)4 in PVA | I 0/I100 ∼ 1.1 | ||||
| Au complexh in PS (high molecular weight) | 418/530 | I 0/Iair = 1.85 | Linear SVPs for all the polymers; sensitivity depends on polymer molecular weight; dye photodegrades in sunlight within several hours. | I | 354, 355 |
| Au complex in PS (low molecular weight) | I 0/Iair = 1.30 | ||||
| Au complex in silicone RTV rubber | I 0/Iair = 1.38 | ||||
| Au complex in CAB | I 0/Iair = 1.46 | ||||
| Au complex in PMMA | I 0/Iair = 1.17 | ||||
| Pb(7-SO3-Ox)i in Dowex 1-X2-200 basic anion-exchange resin beads | 385/625 | I 0/I3% = 1.28, I0/Iair sat ∼ 2.85 | RTP emission; good photostability; no leaching; no quenching if dry gases are used; τ0 154 μs under moist argon. | I | 350 |
| Nb–ferron on Dowex resin | 365/570 | I 0/Iair = 6.3 | RTP emission only in anion-exchange resin; high photostability; do not easily leach; for aqueous and organic solutions; linear SVPs. | I, L | 349 |
| Zr–ferron on Dowex resin | 380/585 | I 0/Iair = 5.8 | |||
| Al–ferron on Dowex resin | 390/600 | I 0/Iair = 5.5 | |||
| Ga–ferron on Dowex resin | 390/600 | I 0/Iair = 5.2 | |||
| Al–ferron complexj in TMOS MTMOS ormosil | 380/580 | I 0/Iair ∼ 6 | Sensitivity depends on the concentration of indicator; lifetime SVPs are downward curved; τ0 460 μs. | I | 351 |
| τ 0/τair ∼ 3.7 | 352 | ||||
The copper(I) complex of a pyridinyl-5-tolyl-1,3,4-oxadiazole has a yellow luminescence with a lifetime of 64 μs under pure nitrogen atmosphere.356 The complex was doped into a silica supporting matrix (MCM-41) to construct an oxygen-sensitive material that has a sensitivity (I0/I100) of 6.0, a response time of 10 s, and good photostability. Several crystalline highly emissive copper(I) complexes with high QY (0.66), long lifetime (∼30 μs), fast response (51 ms) and strictly linear SVPs can also act as OSPs.357,358 The crystals can be excited in the near-UV at ∼375 nm to emit yellow-green luminescence with a maximum at 540 nm. Another phosphorescent copper(I) complex359 with an RTP phosphorescence peaking at 545 nm and a long excited state lifetime (4.7 μs) was incorporated into electrospun composite nanofibers of polystyrene to obtain an oxygen sensing material with a sensitivity (I0/I100) of 7.2 and a response time of 7 s. No photobleaching was detectable. The exceptionally long-lived luminescence of certain [Cu(I)(isocyanido)2(phen)]+ complexes in the form of nanoporous crystals may also be exploited for sensing oxygen.360 They display strong emissions with peaks at ∼530 nm if excited at 375 nm, and lifetimes between 100–120 μs. Both intensity and decay times are quenched by oxygen. The respective SVPs are strictly linear, and sensitivity is almost 38 times better than that of Ru(phen) complexes.
A phosphorescent Cu(I) complex with pyridine and triphenylphosphine ligands was reported361 that displays a yellow phosphorescence peaking at 552 nm and a long excited state lifetime (83.5 μs) under nitrogen. It was doped into a mesoporous silica matrix to obtain an oxygen-sensing material with a sensitivity (I0/I100 = 4.48) for oxygen, a short response time (due to a large surface-to-volume ratio), and no photobleaching. While Cu(I) complexes are affordable, they suffer from the drawbacks of UV excitation and – sometimes – low brightness.
While Pt(II)porphyrins are widely used as OSPs, other complexes of Pt(II) have been rarely used. Wong et al.362 demonstrated that the Pt(dtbpy)(CN)2 and Pt(dpp)(CN)2 complexes dissolved in a silicone rubber matrix are viable materials for optical sensing of oxygen. The probes do not carry a charge, can be mixed with silicone matrix more easily than Ru(bpy), and do not leach into aqueous solutions. The resulting sensors have fast response (<3 s), and alkyl halides do not interfere. Triphenylamine-based cyclometalated Pt(II) complexes were also shown to be efficient OSPs.363 They contain a pyridyl moiety and a cyano group and were dissolved in a polymer film. The luminescence of these new OSPs is quenched by oxygen with highest efficiency and an apparent Stern–Volmer constant of 0.102 Torr−1.
Numerous other luminescent metal complexes are known364 but have not been studied so far. Many of them are likely to be viable probes for oxygen that may cover various ranges of oxygen levels. However, it needs to be seen whether they are better than some of the existing probes that perform quite well.
6.3. Intrinsically luminescent nanomaterials as probes for oxygen
Numerous kinds of intrinsically (non-doped) luminescent nanomaterials including nanocrystals have been discovered in the past three decades, including quantum dots (QDs), carbon dots, silicon dots, luminescent (conjugated) polymer dots, luminescent noble-metal nanoparticles, luminescent graphenes and photon upconversion nanoparticles. However, quenching by oxygen has hardly been studied. Zhu et al.368 reported that the luminescence cadmium telluride QDs capped with cysteine is reversible but weakly quenched by oxygen. The particle size, capping molecules and pH value govern the quenching process and thereby strongly affect the sensing capability. In addition, effects such as surface oxidation, oxidation of capping molecules, blinking (up to seconds), and cytotoxicity are not well under control. The use of these materials for oxygen sensing is still challenging. Nanoparticles doped with OSPs, on the other hand, play an important role and will be treated in Section 7.3.6.4. Multiple emitter probes
The need for OSPs that enable ratiometric sensing has resulted in the design of luminescent dyes that have at least two distinct emission bands, one being sensitive to oxygen, the other not. Such dyes are useful for referenced sensing of oxygen. See the respective section. Franck and Pringsheim17 studied the effect of oxygen on the fluorescence and room-temperature phosphorescence (RTP) of trypaflavine absorbed on (dry) silica gel. Trypaflavine exhibits fluorescence in aqueous solution but becomes phosphorescent if absorbed on silica gel, paper, or gelatine. The RTP is very sensitive to oxygen, while fluorescence is not. Albeit weak, it may serve as a reference signal. Similarly, certain bromonaphthyl ketones immobilized on cellulose display oxygen-dependent RTP along with oxygen-independent fluorescence in the dry state.369 This finding resulted in a ratiometric sensing system with a large change in intensity ratios. However, the sensor can only be used in dry state. Air of 100% relative humidity completely quenches RTP.A probe referred to as dppe-Pt2P has two distinct emission peaks of differing sensitivity to oxygen.357,358 The long-lived triplet emission (15 μs) with a peak at 680 nm is sensitive to oxygen while the short-lived singlet excited state (with an emission maximum at 570 nm and a decay time of 0.5 ns) is not quenched. It was suggested that this dual-emission probe can be used for ratiometric intensity based sensing, for frequency-modulation lifetime discrimination,370,371 and for fluorescence polarization based372 sensing. Similar dual emitters were reported by Zhao et al.373,374 who synthesized several cyclometalated Pt(II) complexes of general formula C∧NPt(acac). These exhibit both fluorescence (500–650 nm; QY ∼0.95; lifetimes ∼60 ps) and phosphorescence (650–850 nm; QY ∼0.22; decay time ∼1.53 μs), and can be applied to ratiometric sensing of oxygen. In addition, several Cu(I) complexes with indole derivatives were also reported to possess dual-emission and to be useful for ratiometric oxygen sensing.375
Following the observation Parker and Barnes376 that the UV-excitable fluorescence of the borate–benzoin complex is quenched by oxygen, a dually emissive iodine-substituted difluoroboron dibenzoylmethane was conjugated to poly(lactic acid) to form a solid-state oxygen sensing material (Fig. 16).364,365 It displays intense blue fluorescence and an unusual, long-lived green RTP. While its fluorescence is insensitive to oxygen, its RTP is quenched by oxygen. The ratio of fluorescence to RTP can be adjusted by systematic variation of the chain length of the poly(lactic acid). Films made from low-molecular weight poly(lactic acid) have weak fluorescence and strong RTP and therefore are single-wavelength probes, while higher molecular-weight polymers give balanced fluorescence and phosphorescence intensities so to enable dual-wavelength ratiometric sensing. The RTP has a decay time of around 4 ms which is easy to determine. This material was also applied to fabricate nanoparticles for intracellular imaging of hypoxia. The only limitation of these attractive OSPs is their excitation wavelength in the UV where biomatter has poor optical transparency and background luminescence can be strong. A related dual emissive probe was used in polylactide nanoparticles of <100 nm in size. The material displays intense blue fluorescence, two-photon absorption, and long-lived room temperature phosphorescence (RTP) which strongly quenched by oxygen.377 The nanoparticles are internalized by cells and can be visualized by fluorescence microscopy. Multiple emitter probes can also be synthesized by combination of oxygen-insensitive fluorescent moieties and functionalized OSPs. A copolymer was synthesized by polymerization of functionalized PtTFPP monomer and fluorine monomer.378 The luminescence intensity from polyfluorine is not oxygen dependent but that of the PtTFPP moiety is sensitive to oxygen concentration. The ratio of the two emissions (can be adjusted by changing the initial feed ratio of two monomers) serves as a ratiometric dual emissive oxygen sensing system.
 | ||
| Fig. 16 Synthetic route and chemical structure of a luminescent polymer (a), its spectra (showing dual-emission), and its quenching by oxygen. The emission spectra of polymers in air (b) and in nitrogen (c). (Reprinted from ref. 379 with permission from Nature Publication group). | ||
6.5. Various other probes for oxygen
Hendricks380 observed the thermally stimulated luminescence of poly(ethylene 2,6-naphthalenedicarboxylate) (PEN). If irradiated with 330 or 390 nm UV light and exposed to oxygen (or air), the luminescence of the polymer is activated. After switching off the light source and heating to above 71 °C, PEN begins to emit luminescence that increases with temperature up to a maximum output at 130 °C. Its intensity depends on pO2, but even more on temperature. The samples would not luminesce if kept in darkness or in a vacuum. The mechanism of this thermally stimulated luminescence is still unknown. It was tentatively attributed to a photochemical reaction involving UV light, oxygen, and PEN. From a present day view it cannot be excluded that the actual species that causes luminescence to occur is ozone rather than oxygen, since ozone is known to react with various polymers. Subsequent thermal decomposition of the endoperoxides formed may result in chemiluminescence. Ozone may be present in ambient air but also may be produced in situ by the UV light of the lamp.The fluorescence of a thin film of photo-oxidized poly(9,9-dioctylfluorene) reversibly responds to oxygen if excited with a 325 nm He–Cd laser.381 However, sensitivity is moderate, and the sensor degrades irreversibly after 12 h of continuous illumination. Monolithic silica aerogel absorb light in the UV region (300–360 nm) and emit visible light at 400–600 nm if treated with ammonia and 2.45 GHz microwave radiation in a reducing atmosphere.382 The aerogel has a high surface area and porosity that allows rapid diffusion of oxygen, which quenches its luminescence. The material has a high sensitivity (with an I0/I100 of >12) and can be used to sense gaseous and dissolved oxygen. An aqueous solution containing 1-bromonaphthalene and a cyclodextrin was found383 to display intense RTP while adding various alcohols. Phosphorescence decreases with increasing concentrations of oxygen. The sensitivities and analytical ranges strongly depend on the sort of alcohol. Under optimized conditions, the system responded to dissolved oxygen in the 0 to 40.2 mM concentration range.
Conventional organic OSPs and metal–ligand OSPs cannot be used for high temperature sensing due to their thermal lability. Ghosh et al.384–386 found on the Mo6Cl12 cluster to possess a wide absorption band that extends from 300 to 400 nm, an emission extending from 600 to 900 nm, a lifetime of >100 μs, and a large Stokes shift (>300 nm). The red luminescence is reversibly quenched by oxygen. The cluster can withstand repeated cycling between air and nitrogen and showed no signs of decomposition at temperatures even higher than 600 °C. This makes it suitable for monitoring oxygen in power plants and exhaust gas in cars. It can sense oxygen with high resolution (0.1% absolute oxygen concentration change) both in the gas phase and in aqueous solution. Remillard et al.387 noticed that the fluorescence of Cu-ZSM-5 zeolites reversibly changes on cycling between oxidative and reduced atmospheres, and this was exploited to sense oxygen at temperatures as high as 500 °C. However, the material has to be regenerated by exposing it to reducing gases, and this limits practical usage. Table 10 summarizes the kinds and properties of the various other luminescent OSPs.
| Dye/matrix | λ exc/λem | Sensitivity | Comments | RO | Ref. |
|---|---|---|---|---|---|
| a dppe-Pt2P = [1,2-bis(diphenyl phosphino)ethane-Pt[S2C2(CH2–CH2–N)-2-pyrimidine](BPh4)], TEC = triethyl citrate. b For the detailed chemical structure see the corresponding reference. | |||||
| dppe-Pt2Pa in cellulose acetate and plasticizer | 470/570,680 | I 0/I100 ∼ 3.5 | RTP at 680 nm with lifetime of 15 μs is sensitive to oxygen; singlet emission with lifetime of 0.5 ns is insensitive to oxygen; both ratiometric and lifetime discrimination are used; QYfl 0.002, QYphos 0.01; τ0 28.8 μs. | I | 370, 371 |
|
L1-Pt(acac)b
L2-Pt(acac) |
459/502,683
474/521,692 |
I 0/I0.2bar ∼ 2 I0/I0.2bar ∼ 6 | Dual emitter; fluorescence (500–650 nm, QY ∼0.95, lifetime ∼0.06 ns) is not sensitive to oxygen, but phosphorescence (650–850 nm, QY ∼0.22, lifetime ∼1.53 μs) is quenched by oxygen. Data for probes in organic solvent. | I | 373, 374 |
| BF2dbm(I) in PLA polymer | 405/460,525 | I 0/I100 ∼ 1.8 | Fluorescence peaks at 460 nm, RTP at 525 nm; luminescent polymer; fluorescence and RTP emission ratio can be varied by variation of polymer molecular weight; RTP lifetime ∼4 ms, suitable for lifetime based imaging; also fabricated in the form of nanoparticles. | I, L | 379 |
| Mo6Cl12 in poly(1-trimethylsilyl-1-propyne) | 325/>590 | I 0/Iair = 5.5 | τ 0 50–180 μs; slow photodegradation even at high temperature (up to 600 °C); also be used for dissolved oxygen sensing. | I, L | 384, 386 |
| Photoluminescent silica aerogel | 330/>450 | I 0/Iair ∼ 4.1 | Prepared by irradiation of aerogel with 2.45 GHz in the presence of ammonia. | I | 382 |
| Cu-ZSM-5 (zeolite) | UV/470,550 | I 0/I2.0% ∼ 1.22 | Copper-containing zeolite; operated at 300–575 °C; response depends on the redox nature of the gases. | I | 387 |
7. Polymeric matrices (hosts) and supports
7.1. General considerations
Polymer chemistry forms an integral part of chemical sensor technology. The function of polymers most often is that of (i), a solid support in which, or onto which, OSPs can be immobilized, and (ii), to warrant some permeation selectivity for oxygen, while rejecting other species. The use of polymers in various sensor applications has been reviewed.388 Korotcenkov389 has summarized the wealth of composite materials that is available to manufacture oxygen sensitive materials.Online monitoring of oxygen in air or (flowing) liquid samples requires an OSP to be firmly immobilized in a host matrix while its luminescence is measured over time. This is important in the case of medical sensing where OSPs must not leach out to enter blood or skin. The incorporation of OSPs into an oxygen-permeable host polymer has severe additional beneficial effects: (a) the host provides a constant microenvironment and protects the OSPs from potential interferents. (b) Quenching constants can be adjusted by proper choice of materials. (c) The interaction of the OSP with sample materials (such as protein) is prevented. Combinatorial methods207 were applied to speed up optical sensor material (OSM) development by studying numerous combinations of polymers, solvents, indicators, and additive plasticizers in different ratios. The combinatorial approach creates a large database that facilitates the selection of appropriate OSMs. It offers an alternative to the cumbersome search for new materials on the basis of theoretical predictions which so often is hampered by practical limitations such as poor compatibility of solvents, polymers, indicator dyes, and plasticizers, but also by poor solubility.
There are several requirements for the matrix to be a viable host for the OSPs: (1) the OSPs and the matrices are to be compatible in terms of solubility so that the OSPs will enter and remain inside the matrix without leaching or aggregation. (2) The matrix has to be optically transparent, possess good adhesion to a conceivable mechanical support, and should be easy to handle. (3) Its permeability and diffusion rate for oxygen should match the specific needs of the sensor with respect to quenchability and response time. (4) The polymer is expected to possess good long-term stability and not to degrade. Certain sol–gels, for example, change their microstructure over even months. (5) The host should not significantly alter the photophysical properties (such as spectra or decay times) of the OSPs. (6) If used in vivo, biocompatibility should be warranted.
Methods for immobilization of OSPs in an oxygen-permeable polymer can be divided into three main subgroups. In method A, the OSP is homogeneously dissolved in a polymer. In method B, the OSP is first adsorbed on the surface of a (particle) support and then incorporated into a polymer. In method (C), the OSP is first homogeneously incorporated into micro/nanoparticles which then are incorporated into a – usually other kind of – host polymer. This is schematically shown in Fig. 17.
 | ||
| Fig. 17 Schematic of the three most common methods for immobilizing oxygen-sensitive probes (OSPs) in a polymer host. In (A), the OSP molecules (symbolized by blue dots) are homogeneously distributed in the (yellow) host polymer. It can be present in dissolved form or covalently bound to the polymer. In (B), the OSP molecules are adsorbed on the surface of the (green) micro- or nanoparticles (silica or polystyrene, for example) which, in turn, are incorporated into the host polymer. In (C), the OSP molecules are first homogeneously distributed in micro- or nanoparticles (pink) which then are incorporated into the host polymer. | ||
Method A, in its most simple form, involves the dissolution of an OSP in a polymer which obviously has to be a solvent for the OSP. This is easily performed and therefore the preferred method. Alternatively, the OSP may be covalently linked to the host polymer as described further below. In a typical process, the OSP is dissolved in a suitable solvent, mixed with a solution of a host polymer to for a sensor cocktail, spread on a support or waveguide, and dried so to form an optical oxygen sensor layer. The leakage of dyes from such sensor layers only depends on the strength of the interaction of the OSPs with the hosting matrix material. Most often, this interaction is of hydrophobic nature. This method is convenient and time- and labor-saving. It works well if both the probe and the host polymer have similar polarity. It does not work if, for example, the polymer is highly hydrophobic (such as most silicones) and the OSP is rather polar if not ionic.
OSPs have also been covalently linked to the host polymer to yield homogeneous sensing layers or micro/nano-particles with firmly bound (and thus non-leaching) probes. OSPs bearing alkene groups186,235,301–303 or amino, carboxy, or siloxane functional groups43,181,184,234 have been covalently immobilized in this way. Respective OSMs display excellent stability in terms of leakage, and the OSPs are evenly distributed in the hosting matrix. If properly executed, the OSP do not aggregate so that there is no self-quenching.
In method B, viz. the adsorption of a (charged) OSP on the surface of a (particle) support, the probe is deposited on particles with polar surface, examples being alumina, silica, or porous glass beads (all possessing a negative charge at pH 7), or amino-modified polystyrene particles (possessing a positive charge at pH 7). The OSP is firmly bound, usually due to electrostatic interaction. Examples include the immobilization of phosphorescent dyes on silica as described by Kautsky and others (see Table 3), or of ionic ruthenium probes on silica particles (see Table 4). The particles can be used as such (mainly for gaseous samples), or incorporated into a host matrix such as silicone rubber or polystyrene. Unlike in method A, the bulk polymers need not be a solvent for the OSP. In fact, they should not at all. In a typical process, the OSP is dissolved in water or ethanol, and the silica particles are suspended in this solution for up to one hour. The particles are filtered off, washed, dried, and dispersed into an organic solution of the host polymer of choice. The resulting cocktail can then used to form an optical oxygen sensor layer of almost any desired shape. Leakage of ionic dyes from such composite sensor layers is negligible. On the other hand, surface-adsorbed dyes without a host polymer easily leach if exposed to liquid solutions but are adequate for sensing oxygen in the gas phase. Surface-absorbed OSPs also tend to be sensitive to the relative humidity of gaseous samples.
In method C, the OSP is first dissolved in micro/nanoparticles (rather than adsorbed on a particle's surface) which acts as a “solvent” for the OSP. The particles then are incorporated in a (different) host polymer that needs not be a solvent for the OSP. It has become quite common in recent years. This is due to several advantages in that (a) fluorescence resonance energy transfer between OSPs and any other dyes (such as reference dyes or other probes) is prevented; (b) the quantities of particles (sometimes more than one kind) can be precisely controlled; (c) the polymers can be optimized in that a highly oxygen permeable polymer is used for the beads, while a biocompatible host polymer or well-adhering polymer may be chosen as the host; (d) the host can be chosen such that leaching of the probes out of the beads is prevented; and (e), the method is also applicable to simultaneously immobilize enzyme-loaded beads inside a sensor layer. This bead-based approach is also quite suitable in the case of multiple-analyte sensing.
Methods for preparing dyed sensor microparticles390 include silane reagent-based techniques (including the Stöber process for making sol–gel particles), swelling of polymer nanoparticles in the presence of OSPs, nanoprecipitations of polymers in the presence of OSPs, and direct (emulsion) polymerization. The resulting oxygen-sensitive micro/nano-particles may also be used as such, i.e. without incorporating them into sensor layers.
It is obvious from these considerations that the selection of a favorable matrix is a key to successful sensor design as will be further outlined in the following Subsections 7.3. to 7.5. According to the chemical properties of host matrices, the polymers were subdivided into three main classes, viz. organic polymers, inorganic materials (including organic modified silicates and organic polymers with inorganic fillers), and nanomaterials.
7.2. Permeability, solubility, and diffusion of gases in polymers
There are three important parameters that characterize a polymer and govern the response of sensors for oxygen: permeability (P), solubility (S), and diffusion (D). These are related by the equation P = S·D and shall be briefly discussed here. When oxygen permeates through a polymer membrane, the rate of permeation is given by the permeability coefficient P. Typical data for P and S for oxygen in various polymers are compiled in Table 11. The unique properties of silicone are obvious.| Polymer | P | S | D |
|---|---|---|---|
| a With 10% filler. b At 30 °C. c At 20 °C. | |||
| Polydimethylsiloxanea | 200–400 | 138 | 1500–2500 |
| Polyethylene (low density) | 0.7–0.9 | 21 | 46–60 |
| Polyethylene (high density) | 0.13–2.3 | 8.0 | 17 |
| Polypropylene | 0.77 | — | — |
| Polyisoprene | 7.7 | — | — |
| Polystyrene | 0.88 | 25 | 11 |
| Polycarbonate | 0.5 | 2.3 | 2.1 |
| Poly(vinyl acetate) | 0.4 | 6.2 | 5.5b |
| Poly(vinyl alcohol) | 0.007b | — | — |
| Poly(vinyl chloride); not plasticized | 0.015–0.03 | 13 | 1.2 |
| Poly(vinylidene dichloride) | 0.0018 | — | — |
| Teflon | 1.65 | 1.3–2.5 | 15 |
| Polyacrylonitrile | 0.00007 | — | — |
| Natural rubber | 7.8 | 50 | 173 |
| Nylon 6 | 0.013–0.03 | — | — |
| Cellulose acetate (not plasticized) | 0.2–0.6 | — | — |
| Poly(ethyl methacrylate) | 0.4–0.8 | 38 | 10 |
| Poly(hydroxyethyl methacrylate) | — | — | 0.03–0.06c |
| Water | — | — | 0.025 |
The concentration c of oxygen in a polymer can be described by the equation c = S·p, where p is its partial pressure and S is the solubility coefficient of oxygen in a polymer. It decreases with increasing temperature above the glass temperature. The temperature dependence of both P and S again can be described by an Arrhenius type of equation. The concentration of oxygen in air-saturated water is 8 ppm (8 mg L−1; equal to 0.25 mM at 22 °C, and 1.24 mM if saturated with 100% oxygen at atmospheric pressure.
The oxygen partial pressure of the arterial blood of healthy and normally breathing persons is around 90 Torr, this corresponding to a 143 μM oxygen concentration only. Blood solubility may also be expressed in terms of gas volumes: 100 mL blood dissolve 3.1 μL of oxygen at 37 °C per Torr of oxygen pressure applied. The largest fraction of oxygen in blood, however, is bound to hemoglobin. A typical value for bound oxygen is 20–21 mL of oxygen per 100 mL blood.
The solubility coefficient (S) of oxygen in all polymers decreases with increasing temperature above the glass temperature and obeys an Arrhenius relationship
| S(T) = S0e(−δH/RT) | (5) |
The diffusion coefficient (or diffusion constant) of oxygen in silicone also obeys an Arrhenius relationship with
| D = D0exp(−ED/RT) | (6) |
Experiments on the diffusion of oxygen in poly(methyl methacrylate) (PMMA) have been performed393 using the room temperature phosphorescence technique. The diffusion of oxygen through PMMA as studied by phosphorimetry is very slow, with coefficients varying from 2.7 to 5.5 × 10−9 cm2 s−1 at 20 °C. It is constant within the used dye concentration and independent of the nature of the OSP. The activation energy (D0 in eqn (6)) is 250 cal mol−1.
Oxygen diffusion in the much more hydrophilic polymer poly(hydroxyethyl methacrylate) (polyHEMA) has been studied by fluorescence quenching techniques.394 The diffusion constant is 1.36 × 10−7 cm2 s−1 at 20 °C. It was calculated that the maximum possible oxygen concentration in an aqueous polyHEMA matrix is 0.4 mM under pure oxygen at atmospheric pressure. The value of this study lies in the fact that a material was studied that closely resembles the composition of a sensing membrane composed mainly of hydrogel and water. This type of material was used to incorporate hydrophilic sensor micro- and nanoparticles, and in biosensors using immobilized enzymes. Quenching of fluorescence obeys Stern–Volmer kinetics with a KSV of 0.016 Torr−1. Diffusion data for other hydrogels have also been determined.395 Draxler et al.113 have studied numerous polymers with respect to their permeability for oxygen via its effect on the quenching of polymer-dissolved ruthenium-based OSPs.
Badocco et al.396 have studied the experimental parameters that characterize the sensitivity and precision of sensor materials composed of polysulfone and Ru(dpp). The asymmetric shape of the emission band was used to define two parameters (the asymmetry factor and the percent variation of emission intensity) to characterize the sensitivity (=slope) of the sensor. Correlations between asymmetry factor and variation in intensity were established, and a double working curve was introduced to optimize precision. Mathematical modeling enabled the calculation of oxygen diffusion coefficients inside the sensor layer (2 × 10−8 cm2 s−1), and of oxygen solubility (2.2 × 10−3 mol atm−1 cm−3) in the polymer matrix by using experimental data for membrane thickness, response time (t90), and luminescence lifetime. In subsequent work,397 the group has determined the activation energies of the non-radiative decay and the gas solubility enthalpy inside a PtTFFP/polysulfone-based oxygen sensor layer using experimental data such as KSV, decay time, diffusion coefficients and solubility. The dependence of the three parameters on temperature ranging between 40 and 90 °C was taken into account.
7.3. Organic host polymers
The permeabilities of many organic polymers for various gases vary to a very wide extent. Respective data are compiled in the Polymer Handbook, for example.398 Data (as far as available) on polymers often used for sensing oxygen are summarized in Table 11. Organic polymers usually are preferred because they often are commercially available or can be fabricated fairly easily and at affordable costs, come in a large variety, and can be easily further modified. Fig. 18 gives chemical structures (on monomeric building blocks) of commonly used polymers. | ||
| Fig. 18 Chemical structures (of building blocks) and respective acronyms of polymers frequently used as hosts in optical sensors. Many polymers contain fluorine or silica and are hydrophobic (except for Nafion™). EC: ethyl cellulose; PDMS: poly(dimethylsiloxane); PMMA: poly(methyl methacrylate); PS: polystyrene; PTMSP: poly(1-trimethylsilyl-1-propyne); poly-styrene-co-PFS: poly(styrene)-co-(pentafluorostyrene); polystyrene-co-TFEM: poly(styrene)-co-(trifluoroethyl methacrylate); poly-IBM-co-TFEM: poly(isobutyl methacrylate)-co-(trifluoroethyl methacrylate); poly-tBS-co-TFEM: poly(4-tert-butylstyrene)-co-(trifluoroethyl methacrylate); FIB: poly(hexafluoroisopropyl methacrylate)-co-(heptafluoro-n-butyl methacrylate); PSAN: poly(styrene-co-acrylonitrile). | ||
Many organic polymers are good solvents for OSPs but also act as barriers against notorious quenchers such as heavy metal ions or anions such as iodide or salicylate. Most polymers for use in oxygen sensors are hydrophobic in nature because oxygen dissolves and diffuses quite well through such polymers. If hydrophilic matrices are used, they usually act as a host matrix for oxygen-sensitive particles made from hydrophilic materials. An overview on representative host polymers is given in Table 12.
7.3.1.1. Silicone rubber. Silicones are very hydrophobic and have unique properties in possessing a higher permeability for oxygen than most other polymers, but being impermeable to ions including the proton. Silicones also have excellent optical and mechanical properties, and a unique solubility for oxygen. This is particularly true for fluorosilicones. This need to be considered in the case of very small sample volumes because such polymers can deplete the aqueous sample of oxygen, and this may result in substantial analytical bias.
Silicone rubbers were among the earliest used homopolymers for constructing optical oxygen sensors. Silicone rubbers (polysiloxanes) such as poly(dimethyl siloxane) (PDMS) are cross-linked polymers with excellent gas permeability, high thermal stability, excellent chemical and mechanical stability, ease of handling, good adhesion to glass, inertness to biological samples, and good optical transparency. Many silicones are of the room-temperature vulcanizing (RTV) type that cure upon exposure to moist air to release products such as acetic acid or amines. Other silicones cure as a result of additions reactions, for example by hydride addition under the catalytic effect of certain metal organic compounds. These silicones are of the two-component type. No acid or base is released in this case. The respective (commercial) prepolymers can be dissolved in aprotic solvents such as toluene or chloroform which facilitates handling.
Silicones cannot be easily plasticized by conventional plasticizers, but form copolymers which may be used instead. Their hydrophobic nature makes silicone rubbers rather poor solvents for ionic or highly polar OSPs.102,103 Methods for solubilizing ionic species in silicone matrices have been developed that rely on the exchange of the inorganic counter ion (anionic or cationic) by an organic counter ion.176,399 Another way to overcome poor solubility of probes in general is to adsorb the (charged) OSP on (silica) beads, which then are dispersed into the silicon prepolymer before curing.47 As can also be seen in Tables 3–8, silicones have been used for immobilizing OSPs quite often.49,123,128,400,401 Respective sensors may be sterilized by standard methods, for example by steam sterilization at 127 °C, or with hydrogen peroxide.
Silicone rubbers are elastomers and many of them have rather low glass transition temperatures (e.g., PDMS; −127 °C). Silicone films often are mixed with hardening additives (“binder”, “filler”) such as silica or alumina. Curing time, temperature, and humidity affect the properties of the final polymer and therefore the oxygen sensing capability. Commercially available prepolymers may contain solvents, fillers, low molecular weight cross-linkers, catalysts and other additives. These ‘unknown’ components may cause unexpected results.176,177 In some cases, silicones were blended with other polymers. An Ir(ppy)-labeled PDMS was blended with polystyrene to form a sensing film for oxygen.402 The morphology and oxygen sensing properties of blend film were studied and results revealed that the oxygen permeability in the blend film is spatially heterogeneous. There are circular, 2–5 μm diameter regions of high oxygen permeability surrounded by a majority phase that is luminescent, but relatively less sensitive to oxygen quenching. This morphology is due to micro scale phase segregation between the Ir-labeled PDMS and the polystyrene.
7.3.1.2. Ethyl cellulose. Ethyl cellulose (EC) is a derivative of cellulose in which most of the hydroxy groups on the repeating glucose units are ethylated. EC is very hydrophobic and has been widely used as matrix to host OSPs.148,369,371,403,404 EC is also used because of its oxygen permeability, optical transparency, mechanical strength, and photo- and thermal stability. It is a better solvent for OSPs than silicone rubber, but its permeation selectivity is less expressed. There is one report that the sensing performance of these matrixes was influenced by humidity.403 This effect can be reduced by plasticizing the EC matrix. Oxygen-quenching often does not follow a linear Stern–Volmer equation, but rather is downward curved. In order to improve the sensing performance, a plasticizer is often added.405 Like poly(vinyl chloride), polyethylene, polystyrene and poly(tetrafluoroethylene), EC efficiently reject ionic species. Except for polystyrene, they all are difficult to chemically modify so that their function is confined to that of a “solvent” for oxygen and a permeation-selective matrix, while electrostatic or covalent immobilization is tedious.
7.3.1.3. Poly(vinyl chloride). Plasticized PVC has been used to incorporate OSPs, mainly by physical entrapment, rarely by covalent immobilization. Sensor films are readily produced by casting solutions of PVC in tetrahydrofuran and possess good optical and mechanical properties. Pure (non-plasticized) PVC suffers from oxygen permeabilities that are 200 to 10
000-fold lower than those of polysiloxanes.406 In addition, hydrolysis of plasticizers of the ester type can occur in basic solution when applying common sterilization techniques. Useful plasticizers include dioctyl sebacate, tributyl- and trioctyl phosphate, dioctyl- and dinonyl phthalate, 2-cyano- and 2-nitrophenyloctyl ether, and related long-chain (and often branched) esters and ethers. Nitrophenyloctyl ether unfortunately can act as a quencher of luminescence, and 2-cyanophenyloctyl ether was recommended instead.407 Plasticizers are added to PVC in fractions up to 66% and can be used to govern quenching constants. Plasticized PVC has been used as a host in oxygen sensors in few cases, not the least because the plasticizers tend to leach into the water phase and, in particular, into blood. This is inacceptable and also causes the quenching constants to shift over time.
7.3.1.4. Polymethacrylates. Thermoplastic polymers such as poly(methyl methacrylate) (PMMA; plexiglass)195 need not be cured, their composition can be easily varied, and oxygen sensitivity can be adjusted by addition of a plasticizer, however with the same compromises as in the case of PVC. The glass transition temperature of PMMA is rather high (110 °C), but despite its excellent optical properties, it has not often been used in pure form as a sensor matrix, mainly because of low oxygen permeabilities which are 200–104 fold lower than those of polysiloxanes.406 Permeability increases with the length of the alkyl ester, and in particular in the case of silyl esters. The quenching of the RTP of the aromatic hydrocarbons triphenylene and phenanthrene in PMMA has been studied393 in order to determine the diffusion coefficients of oxygen. It was found that reduction of the phosphorescence lifetime of the probes in the presence of oxygen is much smaller (by −10% only) than the reduction of phosphorescence intensity (−80%). This was explained in terms of both static and dynamic quenching.
The quenching of the luminescence of a bis(phenylethynyl)anthracene by oxygen at pressures of up to 25 atm in thin films of PMMA was reported408 to be due to the structural inhomogeneity of the polymer and the migration of electronic excitation. The local and volume-averaged diffusion coefficients for oxygen were determined. Demas et al.244,409 report that the addition of small amounts of trimethylsilylmethyl methacrylate to a methacryloxy containing PDMS polymer can dramatically improve the quenching of the luminescence of Re(I) complexes by oxygen. In contrast to polymeric esters of (meth)acrylic acid, the respective nitriles, polyacrylonitrile included, are virtually impermeable to oxygen. In fact, they are well suited to shield any kind of probe (for temperature, for example) from oxygen (see Table 12).
7.3.1.5. Polystyrene. Polystyrene (PS) has good optical properties and acceptable permeability and solubility coefficients for oxygen. Sensor films typically are cast from solutions of PS and an OSP in toluene, ethyl acetate, or acetone. The resulting sensing films show excellent shelf-life and operational stability also in aqueous solutions. Given its moderate solubility for oxygen, it requires probes that are efficiently quenched. PS is not a good solvent for ionic OSPs such as the ruthenium diimine complexes unless the anion is made hydrophilic as discussed in the section on silicones. PS-based sensors may be sterilized by standard methods. Quenching constants are smaller in PS than in silicone by a factor of typically 10–100. Its glass transition temperature is ∼90 °C.
The dynamic range and sensitivity of optical oxygen-sensors based on the probe PtTFFP can be fine-tuned by employing differently substituted PSs.410 Poly(2,6-dichlorostyrene) gives linear SVPs, while poly(4-tert-butylstyrene) and poly(2,6-fluorostyrene) yield sensors with increased sensitivity (quenchability) but non-linear SVPs. It was noticed411 that PS can be emulsified with silicone to yield films with improved mechanical properties and good adhesion to an optical isolation layer (black silicone).
7.3.1.6. Other homopolymers. Winnik et al.184,332 reported on a new class of sulfur–nitrogen–phosphorus polymers called poly(thionylphosphazenes) (PTPs) for use in sensor films for oxygen that were referred to as pressure-sensitive paints. The PTPs possess a large free volume, and the polymer structure and the sensitivity can be adjusted over a wide range. High quality films can be obtained without the need of cross-linking. The probe PtOEP was encapsulated in this polymer where it displays extremely high sensitivity to oxygen, with an I0.01atm/I1atm value of 58. The material was also used to synthesize gas permeable block copolymers.412 One of the first articles ever on fluorescent sensing of oxygen describes a sensor film made from polyethylene and doped with various polycyclic aromatic hydrocarbons.35 Polypropylene in the form of non-woven membranes and doped with various porphyrin type OSPs was recently used to sense oxygen.413 The resulting sensors exhibited high brightness, optimal lifetime signals (22–30 μs at 21 kPa and 50–60 μs at 0 kPa O2), linear Stern–Volmer plots, temperature dependence and low cross-sensitivity to humidity.
Polyacetylene and its derivatives (often containing trimethylsilyl groups) form another class of highly gas-permeable polymers).414 Poly(1-trimethylsilyl-1-propyne) probably has the highest oxygen permeability of all known polymers. The probe PtOEP in this polymer has a quenching factor (I0/I100) of 225, which makes this combination most useful for trace oxygen sensing.323 However, the long-term stability of this polymer is rather poor and the sensor degrades fast. This is accompanied by pronounced changes in response function. The polysulfones form a group of thermoplastic polymers known for their toughness and stability at high temperatures. They have been used to encapsulate Ru(dpp) with octylsulfonate counterion for oxygen sensing.415 The effects of probe concentration and thickness of film on the sensitivity and response time of the obtained sensors have been systematically studied. Highest sensitivity was obtained with a film thickness of 1.6 μm, and the sensor exhibited linear SVP in the whole range of oxygen concentration. The spectral asymmetry of Ru(dpp) in polysulfone was studied in some detail and used to improve the precision of respective sensors.396 Polysulfone was also found to be a good host for the probe PtTFPP.397
7.3.1.7. Hydrophilic polymers. Hydrophilic polymers are less permeable to oxygen than most hydrophobic polymers. They are less often used but offer advantages in the case of biosensing, in particular in combination with enzymes. They often serve as a host for hydrophobic sensor microparticles.315,416–418 Many polymers of that kind (polyurethanes, for example) are soluble in ethanol or dimethylsulfoxide. The solvent can be easily evaporated at room temperature, so that the preparation of sensing film is convenient and environmentally friendly. Díaz-García et al.214 used gelatin organogels originating from reverse micelle solution to immobilize Ru(bpy). Such organogels are optically transparent and stable in contact with various organic solvents, and have good long-term stability without swelling and/or solubility problems in contact with organic solutions. Grafted hydrophilic polymers were also studied with respect to applications in optical sensor membranes.419 Specifically, poly(hydroxyethyl methacrylate) (poly-HEMA) containing 9,10-diphenylanthracene as the oxygen-sensitive material was covalently grafted via silane coupling techniques onto fused silica optical fibers. Fluorescence was stimulated by the evanescent wave of a UV laser or a xenon lamp and quenched by −20% when the sensor went from an oxygen-free to an oxygen-saturated (40 ppm) aqueous environment. In additional experiments, glucose oxidase was co-polymerized with HEMA, and a hydrophilic material was obtained that eventually was used as a sensing material for glucose by coupling it to an oxygen probe.
Chitosan is a highly biocompatible polymer composed of aminoglucose monomers that was widely used as a hydrophilic matrix because it is easily accessible. In a typical example, dendrimeric and free forms of the OSP Pd(II) meso-tetra(4-carboxyphenyl)porphyrin were incorporated into a matrix of chitosan-based colloidal particles.420 A UV-cured inorganic–organic polymer composite was used as a host for chitosan-coated magnetic microparticles or on commercial microbeads modified with a ferrofluid and for a ruthenium-based probes.421 Chitosan is also an excellent host for enzymes. In the given case, the enzyme diamine oxidase was immobilized in the matrix where it catalyzes the oxidation of the amines putrescine and cadaverine under consumption of oxygen. See the ESI† for uses of oxygen sensors in enzyme-based biosensors.
Polyurethanes (PUs) form another large class of hydrogels. Hydrophilic supports such as PU are characterized by a large number of hydrogen-bridging functions such as hydroxy, amino, or carboxamide groups, or by anionic groups (mainly carboxy and sulfo) linked to the polymer backbone. Typical other examples include polyacrylamides and polyglycols. Depending on the degree of polymerization and cross-linking, they are water-soluble or water-insoluble. All swell in water. Throughout, they are easily penetrated by aqueous solutions but have been rarely used in sensors for oxygen due to the lack of permeation selectivity and slow diffusion of oxygen. These materials are also well biocompatible, and various enzymatic biosensors have been reported (see the ESI†) with oxygen transduction, where both the enzyme and the OSP are immobilized in the polyurethane. A sensor for uric acid is a typical example.422 Polyurethanes were also used as paints into which hydrophilic oxygen sensor particles were incorporated,418,423,424 and in irreversibly responding sensors for oxygen using leuco dyes.103
Poly(ethylene glycol) (PEG) hydrogel films were applied425 as a matrix to incorporate oxygen-responsive ruthenium OSPs which then was grafted and patterned by photopolymerization. An optofluidic sensing platform was obtained that contained embedded oxygen sensing elements that exhibited excellent performance when sensing dissolved oxygen. In another kind of oxygen sensor,426 the probe dichloro(tris-1,10-phenanthroline) ruthenium(II) was immobilized in a photo-polymerized hydrogel made from PEG diacrylate, a polymer known to minimize protein and cell adhesion. A low-average molecular weight polymer was employed to prevent fluorophore leaching. The PEG sensors were stored in water for several months and retained their physical shape and sensitivity to oxygen (0–9 ppm). Porphyrins can be used as efficient cross-linkers to generate a new class of hydrogels for use in sensing and imaging of oxygen.427 A porphyrin carrying four carboxy groups was reacted with a poly(ethylene glycol) possessing terminal amino groups to form a polyamide in a range of appropriate conditions. The network structure of the hydrogel maintained a porphyrin spacing that prevented excessive fluorescence self-quenching despite high (5 mM) porphyrin density. The near-infrared properties readily enabled low background, noninvasive fluorescence monitoring of the implanted hydrogel in vivo. An oxygen-insensitive copper(II)porphyrin hydrogel was used as a reference dye.
Poly(vinyl alcohol) (PVA) is also easily available and tunable in terms of hydrophilicity but was rarely used in unmodified form because it does not well retain probes and other dyes. Rather, composites were employed, for example428 in a glucose sensor based on oxygen transduction where an organically modified silicate film was doped with Ru(dpp) and covered with a poly(vinyl alcohol) composite with a silica sol–gel containing immobilized glucose oxidase. PVA also served as a support in an optical fiber sensor for biochemical oxygen demand (BOD) because the bacteria needed in such a sensor can be physically immobilized in a PVA matrix.193 Covalent immobilization of dyes is more promising, and the OSP pyrene was covalently immobilized on PVA via “click” chemistry, a copper(I)-catalyzed azide/alkyne cycloaddition reaction proceeding at room temperature and in water solution at neutral pH values.135
Silamines, a group of water-soluble (di)alkylpolysiloxanes with terminal amino groups (described earlier in Section 6 on iridium-based OSPs) were used in hydrophilic oxygen sensor films along with microcrystalline cellulose.233 These materials are environmentally friendly and particularly well suited if combined with biomolecules to form biosensors. In subsequent work,429 charged and uncharged iridium luminophores were covalently bound to the silamine by either reductive amination or coupling reactions. Paints sensitive to barometric pressure were obtained by blending the materials with microcrystalline cellulose, and luminophore charge effects were studied by (lifetime) fluorometry. Rather than covalently immobilizing the OSP (and more often in recent years), the oxygen transducer may be first incorporated into hydrophobic micro- or nano-particles which then can be placed in the (silamine) hydrogel matrix (see Fig. 17; method C).
Fluorinated copolymers (see Table 12) are useful for the construction of optical oxygen sensors of extremely high oxygen permeability. The solubility of oxygen in fluorocarbons is 3–10 times better than in the parent hydrocarbons. The C–F chemical bond is short and strong (with a binding energy of 116 kcal mol−1), and the fluoropolymers therefore are quite resistant toward photooxidation. The group of Amao431 has developed several fluoropolymers for oxygen sensing with extremely high sensitivity, these include poly(styrene-co-pentafluorostyrene),322 poly(styrene)-co-(trifluoroethyl methacrylate) (“poly-styrene-co-TFEM”),321,342 poly(isobutyl methacrylate)-co-(trifluoroethyl methacrylate) (“poly-IBM-co-TFEM”),432 and fluorinated poly(aryl ether ketone).329 They observed that the sensitivity of the polymer increased with the degree of fluorination and that the use of fluoropolymers generally leads to low limits of detection and more rapid response.432 The probe PtOEP displays extremely high sensitivities to oxygen, with an I0/I100 of 288 and 296 for poly-IBM-co-TFEM and poly-styrene-co-TFEM, respectively.321,342 The sensing films are fairly photostable and have response times of ∼5 s. Schanze et al.172 replaced the styrene monomer by 4-tert-butylstyrene (tBS) and copolymerized it with TFEM to obtain the copolymer poly-tBS-co-TFEM with even higher gas permeability than poly-styrene-co-TFEM, and the immobilized probe PtTFPP also displays very high sensitivity.
Poly[(hexafluoroisopropyl methacrylate)-co-(heptafluoro-n-butyl methacrylate)] (“FIB”) is a unique fluoropolymer for producing sensors and pressure-sensitive paints.230,273,274,433 It has excellent permeability for oxygen and a temperature dependence that is much smaller than that of other polymers. Ertekin et al.208 have doped silicone host matrices with four kinds of perfluorinated hydrocarbons having different degrees of fluorination. It was found that the sensitivity of the ruthenium probe was greatly enhanced when increasing the “fluorine number”. Even the medically used oxygen carrier F19-decanoic acid can serve as a sensor matrix,434 and the luminescence of ruthenium-based oxygen probes was found to be strongly quenched therein. In subsequent work, the sensitivity and stability of TPP-based optical oxygen sensing films made from polystyrene, ethyl cellulose, poly(1-trimethylsilyl-1-propyne) and poly(isobutylmethacrylate) was also improved by adding perfluorochemicals.435
Nafion is a fluorinated copolymer based on sulfonated tetrafluoroethylene. It is not soluble in, but highly permeable to water. Nafion is a chemically inert, gas-permeable ionomer material that can be easily cast (from commercially available solutions) in the form of thin films or other coatings. Ionic and polar probes can be easily immobilized on the surface of cast films of commercially available membranes in order to construct oxygen sensors, for example via adsorption436,437 or cation exchange.213 However, the spectral properties of the obtained sensing films are sensitive to the degree of Nafion swelling by organic solvent, and the leakage of ionic probes from the film cannot be excluded. Cationic metalloporphyrins were irreversibly immobilized on Nafion by a simple method.288
While fluoropolymers increase quenchability, the addition of gas-blocking polymers can reduce it. This enables quenching to be adjusted to a desired efficiency. The copolymer poly(styrene-co-acrylonitrile) (PSAN), for example, enables the sensitivity of oxygen sensors to be tuned by varying the fraction of poly(acrylonitrile).416–418 Compared to PS, the oxygen sensitivity in PSAN is significantly decreased. Tian et al.303,305 co-polymerized 2-hydroxyethyl methacrylate and styrene with PtTFPP bearing four methacrylate groups. The dye is covalently attached and sensitivity can be adjusted via the fraction of the methacrylate.
Stubenrauch et al.438 have immobilized PtTFPP in various poly(norbornenes) that were obtained by ring opening metathesis copolymerization. These materials have high glass transition temperatures and high optical clarity. The oxygen permeability was increased by introducing bulky side groups. Sensing layers based on polymers with bulky groups exhibited high sensitivity to oxygen (I0/I35hPa ∼ 2.75) and were used for trace oxygen sensing. Di Marco et al.217 co-polymerized the macromonomer poly(ethylene glycol) with an ethyl ether methacrylate to obtain the co-polymer pPEGMA. This polymer is affordable, chemically stable, fully amorphous, and has a low glass transition temperature. This results in a good permeability and adhesion. The quenching of the luminescence of the probe [Ir(ppy)2(dpt-NH2)](PF6) incorporated into this polymer follows a strictly linear Stern–Volmer plot. The same group325 also studied the effect of molecular weight of pPEGMA on the oxygen sensing performance. The Winnik group439 has prepared nonaqueous dispersions of poly(vinyl acetate) particles that were sterically stabilized with poly(ethylhexyl methacrylate). Trace quantities of phenanthrene were covalently bound to either polymer. Their fluorescence is very similar in degassed samples, but strong differences were noted in the presence of oxygen. The effect was used to study effects of morphology and swelling. In a very different and new approach, very small amounts of Ru(II) were doped into a flexible, ultramicroporous, fluorescent Zn(II) azolate coordination polymer to produce phosphorescent materials with very high and tunable oxygen quenching efficiency. This approach resulted in a simple color-changing ratiometric oxygen sensor.440 However, this sensor is more complex (both in terms of materials synthesis and sensor fabrication) than known sensors with either instrumental or visual read-out.
| Polymer [acronym] | P | Comments | Ref. |
|---|---|---|---|
| a Calculated from S. Asakawa, Y. Saitoh, K. Waragai and T. Nakagawa, Gas Sep. Purif., 1989, 3, 117–122. b Oxygen permeability of dry Nafion 117 at 35 °C and 1 atm. Data from J. S. Chiou and D. R. Paul, Industr. Eng. Chem. Res., 1988, 27, 2161. | |||
| Ethyl cellulose [EC] | 11.0 | Biocompatible; good optical transparency; good mechanical strength. | 441 |
| Poly(dimethyl siloxane) [PDMS] | 695 | Excellent gas permeability, high thermal stability, excellent chemical and mechanical stability, ease of handling, good adhesion to glass fibers, inertness to biological samples, optically transparent; hydrophobic; low glass transition temperature; many commercially available prepolymers contain solvents, fillers, low molecular weight cross-linkers, catalysts and other additives; need for curing. | 49, 123, 128, 400, 401 |
| Poly(methyl methacrylate) [PMMA] | 0.116 | Thermoplastic; easy to manufacture and reproducible; low cost; poor oxygen permeability; only useful for sensing oxygen at high partial pressure; photostable; good optical transparency; no need for curing. | 195, 289, 331, 337 |
| Polystyrene [PS] | 1.9 | Easy to manufacture; low cost; moderate oxygen permeability; allows oxygen sensing at high concentration; may be sterilized; excellent shelf time and stability in aqueous solutions; good optical transparency; no need for curing; much lower sensitivity than PDMS. | 269, 442, 443 |
| Poly(1-trimethylsilyl-1-propyne) [PTMSP] | 11251a |
High gas permeability; no need for cross-linking; good photostability; useful for trace oxygen sensing. | 323 |
| Poly(styrene)-co-(pentafluorostyrene) [poly-styrene-co-PFS] | n.d. | Extremely high gas permeability; excellent photostability; short response time; useful for trace oxygen sensing. | 322, 431 |
| Poly(styrene)-co-(trifluoroethyl methacrylate) [poly-styrene-co-TFEM] | n.d. | Extremely high gas permeability; excellent photostability; short response time; useful for trace oxygen sensing. | 221, 342 |
| Poly(isobutyl methacrylate)-co-(trifluoroethyl methacrylate) [poly-IBM-co-TFEM] | n.d. | Extremely high gas permeability; excellent photostability; short response time; useful for trace oxygen sensing. | 321, 432 |
| Poly(4-tert-butylstyrene)-co-(trifluoroethyl methacrylate) [poly-tBS-co-TFEM] | n.d. | Extremely high gas permeability (even higher than poly-styrene-co-TFEM); excellent photostability; short response time; useful for trace oxygen sensing. | 172 |
| Poly(hexafluoroisopropyl methacrylate)-co-(heptafluoro-n-butyl methacrylate) [FIB] | n.d. | Extremely high gas permeability; excellent photostability; short response time; small temperature dependence; suitable for application in wind tunnels with temperature compensation. | 273, 274, 433 |
| Poly(styrene-co-acrylonitrile) [PSAN] | 0.0032 | Moderate gas permeability that can be tuned by changing the ratio of monomers; good stability; easy to handle. | 273, 274, 433 |
| Nafion | 0.81b | Ionic polymer; chemically inert; good gas permeability; probes can be absorbed on the polymer; sensitivity depends on the swelling in different solvent. | 213, 288, 436, 437 |
Multilayer fluorescent thin film chemical sensors were also obtained by electrostatic layer-by-layer self-assembly.445 Ruthenium-based oxygen indicators were used as model fluorophores. Three techniques for building fluorescent sensing films were considered, viz. direct electrostatic assembly of charged fluorescent indicators, fluorophore/polyion premixing, and conjugation of indicator to the polyelectrolyte. In all cases, the films retained oxygen sensitivity and did not exhibit significant self quenching. Another polyion with entrapped OSP was applied to manufacture spin-assembled oxygen sensitive nanofilms.446 The sensor demonstrated good correlation to the Stern–Volmer relationship, with linear decay in sensitivity of approximately 1.6% per week. Response to gaseous oxygen was linear over a 0–100% concentration range, with a resolution of ±0.2%.
Self-assembled ionic nanofilms on top of fluorescent nanoparticles represent another type of nanoscale oxygen sensors.447 The probe Ru(dpp) was immobilized within polyelectrolyte multilayers deposited on the surface of nanoparticle templates by the layer-by-layer (LbL) technique. The ionically bound nanofilms create a porous scaffold into which controlled precipitation of the OSP is achieved using a combination of electrostatic attraction and dye insolubility in water. The fluorescent nanoparticles act as physical scaffolds and also provide a complementary spectral signature for use as an internal reference. Oxygen sensors created on 100 nm yellow-green fluorescent particles exhibit a linear Stern–Volmer response. The nanosensors were chemically delivered into human dermal fibroblasts with no apparent loss in cell viability.
Papkovsky et al.448 described oxygen sensitive materials based on nanostructured films of high-density polyethylene and polypropylene. The polymer substrates were subjected to solvent crazing to produce well-defined 3D networks of nanopores (size <15 nm) and with a volume porosity of up to 60%. OSPs such as PtTFPP and PtOEPK were added during or after the solvent crazing process, and found to be firmly trapped in the nanopores. Such sensing materials are chemically and mechanically stable, and their preparation is simple.
7.4. Inorganic supports and matrix materials
Inorganic materials are attractive materials for fabricating optical oxygen sensors because they have good mechanical strength, high thermal and oxidative stability, and often can be produced at room temperature. The following sections cover the most important groups of solid materials, viz. (porous) glass, silica, sol–gels, organically modified silicates, but also metals and metal oxides.Porous glass has good optical transparency and excellent mechanical strength. Because of the inner porous structure, it has a high specific surface. Bergman35 absorbed the OSP fluoranthene in porous glass to sense the atmospheric oxygen. Pyrenebutyric acid was covalently coupled to amino-functionalized controlled pore glass (CPG),43 and a sulfonated Ru(II) complex was covalently linked onto CPG particles which allowed direct monitoring of oxygen in many solvents and without leaching.181
Zeolites and mesoporous silica can be used for absorbing OSPs into their inner pores so to develop another kind of oxygen sensor. Wolfbeis et al.210 encapsulated Ru(bpy) in zeolite Y cages and mixed the particles with silicone rubber prepolymers to form oxygen sensing films after vulcanization. The sensing films display high quenching efficiency (with an I0/I100 value of ∼4 which, surprisingly, is higher than without the zeolite cage) and excellent stability. Even if stored in water at ambient temperature for several months, there is no obvious change in the sensing properties. Such sensors can be operated at temperatures up to 200 °C. The positively charged probe Ru(bpy) was also absorbed in mesoporous silica materials such as C18-FSM,453 MCM-41,454 MCM-48455 and silica spheres.211 However, the results of Pang et al.211 who absorbed Ru(bpy) in 100–200 μm mesoporous silica spheres revealed that it displays rather poor sensitivity and slow response. Very efficient quenching of luminescence by oxygen was observed456 with functionalized mesoporous silica of types SBA-15 and MCM-41 and doped with a novel Pt(II)–porphyrin complex. The ratios of luminescence intensities under 0% and 100% oxygen are >8700 for the doped SBA-15 material, and >3800 for MCM-41 (at a doping of 20 mg g−1).
On the other hand, Wang et al.457 reported on a multifunctional mesoporous nanocomposite with magnetic, optical, and excellent oxygen-sensing performance. This multifunctional nanomaterial was prepared by covalently grafting Ru(II) polypyridyl complexes into the channels of magnetic mesoporous silica nanocomposites. The super-paramagnetic nanocomposites allow oxygen to be sensed with good sensitivity (I0/I100 = 5.2) and short response and recovery times. Similar findings on flexible ultramicroporous frameworks were reported by others.440 A synthetic layered porous clay mineral (Sumecton) was doped with Pt(II) and Pd(II) porphyrins.458 These are known to be very efficient agents for sensing applications because of a “heavy atom effect”. The combination of metalloporphyrin and layered materials results in good sensitivity at aerobic conditions, but the sensors are very slow.
Sol–gel glasses have favorable mechanical properties, chemical stability, and superior optical clarity. They are prepared by room-temperature reaction of organic precursors, and their chemical properties can be flexibly tuned by using different precursors. The properties of sol–gels made from tetraalkoxy silanes resemble those of controlled porous glass (CPG). The bulk phases (unlike the pores) of conventional sol–gels and CPGs are impermeable to oxygen, and OSPs therefore have to be deposited on their surface which, however, can be quite large in view of the micro/nano-porous structure of sol–gels and CPGs. Moreover, with alkyl(aryl)triethoxy silanes or dialkyl (or aryl alkyl)diethoxy silanes can be applied to yield organically modified silicates (“ormosils”, see Section 7.2.2). These are permeable to oxygen (higher fractions of alkyl and – in particular – aryl siloxanes favoring oxygen permeability and probe solubility), so that the OSPs can be directly incorporated into the matrix. Drying and heat treatment have been applied to progressively densify the gel by elimination of solvents and water. By adding the OSPs to the sol and by appropriate choice of initial pH value and reaction temperature, the OSPs can be trapped in the nanocages formed during the gelation. At the sol stage, films can be coated onto a variety of substrates including optical fibers, capillaries and (waveguide) planar glass.
MacCraith et al.202 used the sol–gel technique to prepare microporous glass films with immobilized ruthenium probes for use in oxygen sensing. The sensor preparation (hydrolysis of tetraethyl orthosilicate in acidic solution at room temperature) is simple and reliable. The films are tough, inert, and often chemically bound to the supporting substrate which further improves physical stability. Sensitivity can be adjusted by varying the pH of the precursor solution to increase the overall pore volume. Lee and Okura317,318 doped PtOEP into sol–gel glasses prepared by hydrolysis of tetraethoxysilane. The addition of the surfactant Triton X-100 improved the homogeneity of the silica sol and results in crack-free sensing films. The probe PtOEP was immobilized in this material where it is efficiently quenched (with an I0/I100 of >40). Again, the preparation of the sensing films takes >2 weeks. Fluorinated xerogels doped with quenchable Pt(II) complexes were reported340 to have fast response (4–7 s), good sensitivity, and linear SVPs. Rather similar materials resulted in even faster responses as reported in a second paper by this group.338 However, most other oxygen sensors based on mixed polymer materials display nonlinear Stern–Volmer relationship, and this can be described by the so-called two-site model (see eqn (3)).
It was also noticed201 that the structure and oxygen quenching capability of sol–gel sensor films is strongly related to parameters such as the speed of dip-coating, the water-to-precursor ratio, pH value, and aging time. The porosity and average pore size of the films strongly depend on the water-to-precursor ratio, and this affects the diffusion of oxygen. On the positive side, this enables the quenching efficiency of sol–gel films to be fine-tuned and optimized. On the less positive side, this implies compromised reproducibility. Kuncová and coworkers166 immobilized Ru(phen) in xerogels. The preparation of the film is simple, but takes 1–2 weeks. However, the xerogel – like many sol–gels and ormosils – is not stable over time in aqueous solution, probably the result of ongoing condensations in the network.
The response to oxygen of Ru(dipy) in two room-temperature ionic liquids (RTILs) has been evaluated in terms of sensitivity, stability, regeneration ability and response time of a sensor.459 The response times range from 2 to 5 s. The RTILs warrant good oxygen solubility, enhanced brightness and photostability. The stability the Ru(II) complex in RTILs was tested over 12 months when stored on laboratory air. The sensing performance of a sensor composed of Ru(bpy) in a sol–gel matrix modified with an RTIL is reported to be much better than that of a plain sol–gel.460 An ionic liquid was added during acid-catalyzed formation of the sol–gel from TEOS to give sol–gel sensor films that exhibit larger quenching constants, linear Stern–Volmer plots, and crack-free surfaces. The addition of the ionic liquid also caused a 23 nm red-shift in the emission peak wavelength, but the composite is only applicable to gas phase measurements.
MacCraith et al.462 systematically modified the water-to-precursor ratio in order to tailor the microstructure and sensitivity of ormosil sensing films for gaseous and dissolved oxygen. Hydrophobicity was increased by using organically modified precursors such as methyltrimethoxysilane (MeTriMOS) or ethyltrimethoxysilane. The addition of MeTriMOS was found to strongly increase the sensitivity for dissolved oxygen. Murtagh et al.463 made similar observations in a study on the oxygen sensing properties of Ru(dpp) immobilized in MeTriMOS-based ormosil gels. The incorporation of platinum porphyrins into the ormosil coating has also been reported464 to give luminescent sensor materials for oxygen.
Sensors for oxygen based on an ormosil matrix were presented465 along with a large set of luminescence decay time based chemical sensors for clinical analytes including oxygen, pH, CO2, K+ and glucose. Sanz-Medel et al.351 prepared sol–gel films from TMOS and MeTriMOS to immobilize the Al(III)–ferron complex. The resulting RTP-based oxygen sensor was used for monitoring oxygen in gas phase, in water and in organic solvents. The sensing films are quite rigid (which favors RTP and prolongs the triplet lifetime of the probe). The sensors have fast response, good reproducibility and detection limits as low as 5 ppm (v/v) in the gas phase. The use of MeTriMOS increases film stability in both aqueous and organic solutions. García and coworker160 applied the same ormosil to entrap erythrosine B (via its isothiocyanate) to obtain another kind of RTP oxygen sensor. The physical properties and oxygen sensing performance were governed by parameters such as the ratio of TMOS-to-MeTriMOS and even the fraction of alcohol when dissolving the precursors. If prepared at high pH, highly porous silica glass is obtained, while low pH values lead to rather dense sol–gel films.
Phenyl-substituted ormosils have particularly attractive properties with respect to sensing oxygen. An ormosil that is soluble in organic solvents such as chloroform was prepared205 from phenyltrimethoxysilane (Ph-TriMOS) and the end-capping agent trimethylmethoxysilane (TMMOS). The mechanical and photophysical properties of the sensing films were tuned by adjusting the ratio of Ph-TriMOS and TMMOS. Sensing films with a low content of TMMOS display high oxygen sensitivity and fast response time (250 ms), most likely because the presence of phenyl groups improves oxygen permeability. The solubility in polar organic solvents is a unique feature that facilitates the preparation of various kinds of sensor formats including fiber optic, planar, or evanescent wave sensors. The material is used in a commercial sensor for oxygen. On the other side, it is insoluble in water, ethanol and methanol, and thus well suitable for sensing bioliquids. Similar ormosil films were prepared later206 from dimethoxydimethylsilane as the organic modifier. This kind of sensor is also highly sensitive and has a fast response and long-term stability.
The polycondensation of n-octyltriethoxysilane (octyl-TriEOS) and TEOS in the presence of the probe Ru(dpp)leads to ormosils that display purely single-exponential luminescence decay times and linear Stern–Volmer plots (SVPs).204 Unlike in sensing films derived from TEOS, where sensitivity decreases by a factor of almost 5 after 11 months, response remained virtually constant. The authors also studied466 the effect of alkyl chain length of the organically modified precursors on the sensors' performance. The same ormosils served as a host matrix for the probes PtTFPP and PtOEP, and the resulting sensors were found338 to possess high sensitivity (I0/I100 = 22.3 for PtTFPP, and I0/I100 = 47.8 for PtOEP) and short response time (0.6 s). It was also observed339 that the addition of silica nanoparticles (prepared by the Stöber method) to the ormosil material enhances quenching by oxygen. Sensing films based on PtTFPP, silica nanoparticles and ormosil display, in fact, extremely high sensitivities (I0/I100 = 166), and very short response time (1.3 s). The probe PdTFPP was doped467 later into an ormosil prepared from n-propyltrimethoxysilane (n-propyl-TriMOS), tetraethylorthosilane, and n-octyltriethoxysilane ormosil, and the material shows very high sensitivity (I0/I100 = 263).
Sensitivity (in terms of detection limits) can be substantially improved by employing fluorinated precursors.468 The precursors n-propyl-TriMOS and 3,3,3-trifluoropropyltrimethoxysilane were co-condensed under acidic conditions and doped with Ru(dpp) to give a sensor material of high sensitivity (I0/I100 = 35) and very small signal drift (<2% over 6 months). The same material was doped with probes PtTFPP and PtOEP,340 this again leading to improved sensitivities (I0/I100 = 68.7 for PtTFPP and I0/I100 = 82.5 for PtOEP) and response times ∼3 s. The same group341 used three other precursors (partially fluorinated). The introduction of both fluoro substituents and octyl groups made the sensing film even more sensitive (I0/I100 up to 155). An overview of fluorinated ormosil xerogels as optical chemical sensors has been provided469 and showed how, together with the dye quenching rate, the subtle structural features of an organofluorosilica matrix are of fundamental importance in determining the overall sensor performance.
Fluorinated xerogel films containing Ru(bpy) were employed470 in fiber optic sensors for dissolved oxygen. The response time is 4 s, and 0.04 ppm of oxygen can be detected in water. The SVP is linear so that two-point calibration is possible. Another fiber optic (and evanescent wave) sensor is making use of a fluorinated ormosil doped with a ruthenium probe, and a fluorinated ormosil was obtained471 by co-condensation of n-propyltrimethoxysilane and 3,3,3-trifluoropropyl-trimethoxysilane. The OSP in the fluoro-ormosil was photoexcited by the evanescent wave produced on the surface of the fiber core using a blue LED as the light source, and luminescence detected with a miniaturized photomultiplier tube. The LOD of this sensor material is 0.01% of oxygen, and the response time is about 1 s. The same xerogel was used in an integrated micro-volume fiber optic sensor based on Ir(III) complexes.472
Xerogel ormosils doped with platinum(II) porphyrin dyes represent other sensing materials with a widely adjustable sensitivity, reversible and fast response (1 s), linear calibration plots, and long-term stability.473 They have been prepared from pentafluorophenylpropyl-trimethoxysilane, n-octyltrimethoxysilane, tetramethoxysilane and 3,3,3-trifluoropropyl-trimethoxysilane. The sensitivity of the materials to oxygen was tuned by adjusting the xerogel composition and the luminophore.
In one further kind of highly sensitive fluorinated sol–gel sensor film,474 the matrix was prepared from 3,3,3-trifluoropropyltrimethoxysilane and TMOS, and the well-known OSP Ru(dpp) was entrapped in a wide range of ormosils in different precursor ratios. The influence of matrix composition on sensitivity, interference by humidity, and on long-term stability was investigated, and performance was compared to that of similar but non-fluorinated films. The lowest limit of detection is 0.002% of oxygen. It is obvious from the compilation in this section that the number of ormosils for use in sensors for oxygen (and other gases) is virtually unlimited. In fact, the number seems to exceed the current need.
Ormosils with covalently linked (rather than physically embedded) OSPs were obtained by co-condensation of OSPs carrying trimethoxysilane groups with various siloxanes. A triethoxysilyl-functionalized Ru(bpy) was chemically linked to a conventional ormosil matrix,183 and the probe PtTFPP was modified in the same way.305 The sensing films obtained have excellent stability and high sensitivity (I0/I100 = 70). Mesostructured silica covalently grafted with Ru(II)(bpy)2(phen) was reported475 to represent an excellent sensor material. The network was prepared via a sol–gel approach with the help of cetyltrimethylammoniumbromide (CTAB) surfactant. 1,10-Phenanthroline was covalently grafted to 3-(triethoxysilyl)propyl isocyanate which not only is the sol–gel precursor but also acts as the second ligand of the Ru(II) complex. The bulk xerogels were spin-coated to form thin films that show good homogeneity and sensitivity to oxygen. The covalently grafted OSPs are said to be superior to the physically incorporated ones.
The effects of mineral fillers of nanometer dimensions on the performances of optical oxygen sensor based on organic polymers were studied by the groups of Cox391,392 and Winnik.476 It was found391 for a series of poly(dimethyl siloxanes) of various viscosity that oxygen has a large diffusion constant D (0.115 cm2 s−1) and a low activation energy ED (4.8 kcal mol−1) which is not temperature-dependent between 5 and 45 °C. For comparison, the diffusion coefficient for oxygen in water is 2.5 × 10−5 cm2 s−1 at 25 °C. The diffusion coefficient is independent of oxygen concentration and fluorophore concentration in silicone over the usual pressure and temperature range. In the presence of small weight fractions of fumed silica fillers, the diffusion of oxygen is reduced, but the activation energy is not affected at all.392
If OSPs are poorly soluble in polymers, inorganic particles can serve as carriers to transfer the OSP molecules into polymer matrix. It is thought that the probes are bound to the particle surface or trapped inside the pores of silica or porous glass. In its most simple form,47 a probe such as Ru(bpy) is placed in and on silica gel, and the dye-doped particles are then dispersed into a silicone rubber matrix to obtain an oxygen-responsive material. In this particular case, silica acts as (a) a filler, (b) a mechanical support of the probe, and (c) as a scattering material. It is mandatory, however, to warrant an even distribution of the probe on the particle, but even if so, the SVPs are mostly nonlinear. This method has been widely used in subsequent years and has experienced many modifications.210 Wong et al.199 have compared the oxygen sensing properties of silica-containing silicone rubber sensing films with silicone films without silica fillers. The probe Ru(dpp) was first absorbed onto fumed silica and then dispersed in silicone rubber to form sensing films. Compared to films without silica, the response time of the silica-containing silicone rubber films both in the backward direction is much longer than in films without silica. This was interpreted in terms of strong adsorption of oxygen so that the desorption kinetics is slow.
Similarly,200 Ru(dpp) was absorbed on silica gel and dispersed in a film of gelatin to obtain an oxygen sensor that works in organic solvents. The sensing material is photochemically inert and can be easily molded into different shape with good mechanical and chemical stability. It also has good sensitivity and low detection limits. The same probe was also encapsulated173,477 in ormosil microbeads, which then were immobilized in a hydrogel to construct planar oxygen sensors.
Winnik et al.327 have systematically examined the influence on oxygen diffusion and permeation in the linear hybrid polymers obtained from PDMS and poly(n-butylaminothionylphosphazene) (C4PATP) in the presence of 10 nm silica particles. The results revealed improved mechanical properties of both polymers, but perturbed oxygen diffusion and permeability. The quenching constants depended on the fraction of silica filler. The luminescence both in silica-free and in silica-filled C4PATP films decays mono-exponentially in the absence of oxygen, which proves that the PtOEP remain dispersed molecularly in the polymer rather than being absorbed on silica nanoparticles.
7.5. Nanoparticles
Oxygen sensitive nanoparticles (NPs) have experienced a large success because they enable micro- and nanostructured systems such as cells, tissue, or nanoscale devices to be studied. In terms of sensing oxygen, there is no alternative to such NPs at present. Due to their small size, there is virtually no diffusion barrier for oxygen, and response therefore is fast. Nanobead sensors are also widely used now for imaging oxygen. Other nanostructured materials (including nanofilms) have been discussed in Section 6.3. Applications are summarized in the ESI.†Many conventional bulk polymers can be converted into nanosized particles using the so-called precipitation method.480 This method is simple in that it only requires the OSPs and polymers to be dissolved in a – usually water-miscible – solvent and to precipitate the nanoparticles by slow addition of water. Polyacrylonitrile (PAN) NPs dyed with Ru(phen) were obtained481 by precipitating them from a solution in dimethylformamide. Such particles are impermeable to oxygen so that the indicator in the particles is not quenched. Rather, such particles may serve as labels, as reference fluorophores, or as oxygen-insensitive probes for temperature. Borisov et al.277 incorporated Pt(II) and Pd(II) benzoporphyrins into polysulfone nanobeads prepared by the precipitation method. The group also applied used this method480 to transform the following bulk polymers into the respective NPs for use in nanosensors: Poly(styrene-co-maleic anhydride), polysulfone, cellulose acetate, poly(vinylidene chloride)-co-(acrylonitrile), poly(methyl methacrylate)-co-(methacrylic acid), poly(hydroxyethyl methacrylate), polyurethane, hydrothane, ethyl cellulose, Nafion and Eudragit. The resulting uncharged beads (with an average diameter of ∼380 nm) are stable in water and do not aggregate. The biocompatible cationic Eudragit was converted482 into nanoparticles by the precipitation method in the presence of the probe PtTFPP to give sensor NPs with an average particle size of ∼30 nm. Such particles are easily internalized by cells.
Commercially available polymer NPs are another source for fabricating oxygen nanosensors. They can be swollen in an organic solvent, and the OSPs also contained in the solvent are taken up by the NPs to give oxygen-sensitive nanobeads. Schmälzlin et al.442,483 have encapsulated PtTFPP in carboxylated microbeads of polystyrene (PS) and used them to determine dissolved oxygen in plant cells. The hydrophobic and inert nature of PS prevents dye leakage, and the beads are well compatible with living cells. Wang et al.484 stained amino-functionalized polystyrene (PS) nanoparticles with PtTFPP along with a reference dye via the swelling method to obtain a NPs for imaging intracellular oxygen. Both dyes are retained by the nanoparticles, and PtTFPP has a good sensitivity to oxygen. Commercially available poly(styrene-co-vinylpyrrolidone) NPs are excellent matrices for simple preparation of optical nanosensors.277 They are uncharged, have an average diameter of ∼245 nm, are stable even in complex media such as those used in biotechnology, and do not aggregate at high ionic strength. These NPs possess two sites for probe immobilization, viz. a hydrophobic polystyrene core and a hydrophilic shell consisting of poly(vinyl pyrrolidone).485 Hydrophobic probes prefer the hydrophobic polystyrene core, while hydrophilic probes prefer the shell. Borisov et al.228 incorporated the OSPs PtTFPP, PdTFPP, Ru(dpp) and Ir(III) coumarin complexes in this host. The resulting nanosensors displayed high sensitivity to oxygen and very fast response, especially for nanosensors based on PtTFPP and the Ir(III) complex which is particularly bright.
Polymer NPs can also be prepared via polymerization of monomers. The technique for polymerizing styrene is quite established, and the size of resulting PS NPs can be adjusted via the experimental conditions during polymerization. In addition, the charge of the surface can be governed and chemical functions can easily be introduced. Cywinski et al.486 synthesized 20 nm sized NPs for encapsulating PtTFPP (possessing red luminescence) and a perylene reference dye (with green fluorescence) via microemulsion polymerization of styrene. The resulting NPs respond quickly and sensitively and can be used for ratiometric sensing of oxygen in cell culture media. Monodisperse PS beads loaded with the probe PtOEP and a reference dye were synthesized using dispersion polymerization.487,488 The PS beads were monodisperse with very uniform size, and the size was controlled by adjusting the concentration of initiator or steric stabilizer. Loaded with PtOEP, the particles exhibit high sensitivity to oxygen, while the fluorescence of the reference dye is not quenched by oxygen. Because of the hydrophobic nature of dyes and polymer, both dyes are firmly encapsulated in the beads, which enable both ratiometric and colorimetric readout. However, such beads cannot be directly used in aqueous media.
Kopelman et al.489 introduced on spherical optical nanosensors (called PEBBLEs) of 20–200 nm in diameter. Polyacrylamide NPs were stained with hydrophilic fluorescent probes. Such PEBBLEs are biocompatible, and the probes are well protected from potential interferents as present in the cytosol of cells. However, some probe leakage in aqueous samples was observed. The hydrophobic probe PtOEPK and the reference dye OEP were also doped into emulsion polymerized poly(decyl methacrylate) NPs with a diameter of about 150–250 nm.490 The nanosensors thus obtained are more hydrophobic, have good stability and probes do not leach. The same group491 later prepared polyacrylamide hydrogel NPs by a microemulsion polymerization to encapsulate the NIR-probe PdTPTBP (a dendrimer) along with a reference dye. Hydrophilicity, surface charge and oxygen permeability were controlled by using monomers with different functional groups to form copolymers. The dyes were covalently bound to the polymer so that enhanced loading was possible, whilst leaching was eliminated. The NPs have a size of 30 nm and functional groups on their surface. This may be used for conjugation to peptides for targeted oxygen level monitoring in live cancer cells. Polyacrylamide NPs were also loaded with Ru(dpp) with a hydrodynamic diameter of 45 nm. The nanoparticles were prepared by radical polymerization of an inverse microemulsion, and the OSP was encapsulated during polymerization.492 The NPs were used to image intracellular oxygen. In a related approach, a PtTFPP-bearing a styrene group was copolymerized with 2,2′-N-isopropylacrylamide (NIPAM) and acrylic acid to form core–shell nanoparticles with an average size of 45 nm.493 These nanosensors have biocompatible shells which improve storage stability and make them suitable for intracellular application. In addition, the oxygen probe PtTFPP is chemically attached inside the nanoparticle and evenly distributed. Its response to dissolved oxygen was investigated and showed that dynamic range of phosphorescence lifetime to be improved (up to 44 μs). The extended dynamic range also warrants better sensitivity.
OSPs have also been linked to biological macromolecules to obtain oxygen nanosensors. Johnson et al.293 and Papkovsky et al.290,291 physically attached several water-soluble metalloporphyrins (referred to as PtCP, PdCP, PtCPK and PdCPK) to albumin so to form a probe-albumin complex for monitoring oxygen in microvascular vessels in a rat muscle. The results proved that such “sensor probes” enable rapid collection of oxygen concentration data in the microcirculatory system and can assist in the understanding of relationships between oxygen tension, metabolism and blood flow regulation. However, the probe-albumin complex is not very stable in that the probe can dissociate from the albumin. To overcome this limitation, the probes PtCP and PdCP were modified with isothiocyanate (NCS) groups and then covalently linked to the amino groups of albumin proteins.494,495 The Rosenzweig group introduced216,496 nanosized lipobeads with encapsulated ruthenium probes as another kind of nanosensors for oxygen. The lipobeads consisted of a polymer particle coated with a phospholipid membrane. Lipophilic OSPs were then immobilized in the hydrophobic regions of the membrane. Such lipobead nanosensors are capable of real-time monitoring of oxygen in tissue and single cells with an excellent signal-to-noise ratio and high sensitivity. Polymeric lipid vesicles (150 nm in diameter) may also serve as a host particle for the probe Ru(phen).497
Present-day methods of surface chemistry enable the net charge, particle size (from a few nm to several μm), pore size and hydrophobicity of silica NPs to be adjusted. OSPs may be covalently immobilized inside pores499 or on the outer surface of particles.500 The OSP pyrenebutyric acid was covalently immobilized in the pores of porous glass beads43 to obtain a sensor with fast response. The probe PtTCPP was covalently conjugated499 into the nanopores of mesoporous silica NPs with diameters of 70–100 nm. McShane et al.500 chemically linked the probe PtTCPP and NIR quantum dots on the surface of ∼10 μm silica microparticles and used these for ratiometric oxygen sensing. In a related approach,501 a sensor for oxygen was obtained using a quantum dot scaffold. It was applied to generate concentration profiles of oxygen in tumors (along with those for glucose and pH).
Micro-sized silica gels have a porous structure comparable to that of controlled porous glass. They have been exploited in nanomaterials to immobilize OSPs. Acosta et al.502 absorbed Ru(dpp) and the reference dye Nile red in ∼10 nm silica gel NPs, and then coated them with PDMS to prevent dye leaching. The resulting micro-sized oxygen sensors can be suspended in optically transparent biomaterial and enabled ratiometric determination of oxygen in culture media. However, microscope images revealed that the OSP and reference dye were not uniformly distributed inside the microparticles. Thus, the spatial resolution of the sensor is moderate. It shall be remembered here that most sensors based on silica-type of materials are cross-sensitive to humidity. In fact, this was exploited to design a sensor for humidity.503
8. Additives, coatings, and other components
In order to improve and enhance the performance of oxygen sensitive materials (OSMs), various kinds of additives have been developed and applied. These include (a) plasticizers, (b) photostabilizers that reduce the rate of photodegradation of the OSPs, (c) optical isolations that reduce or eliminate interference by ambient light, (d) scattering particles that give rise to more homogeneous excitation and emission light fields, and (e) various other additives to enhance signals, sensing performance, or mechanical integrity. Antifouling polymers containing phosphorylcholine (PC)-substituted methacrylate units have been reported504 were obtained by copolymerization of the PC-methacrylates with dodecyl methacrylate and used to coat luminescent oxygen sensors. Nanometer-sized coatings of such materials can reduce the adhesion of marine bacteria by more than 70%, and of thrombocytes by more than 90% to the surface silicone sensor layers doped with Ru(dpp). The PC-coated and the uncoated sensor films perform very well in aqueous media and are mechanically stable for more than one year of continuous immersion. If blood clotting is to be prevented, sensors typically are coated with a thin layer containing covalently bound heparin.8.1. Plasticizers
Plasticizers are added to polymers in order to improve the efficiency of quenching by oxygen and to facilitate the handling of brittle polymers. Polystyrene, ethyl cellulose, polysiloxanes, poly(meth)acrylamides and polyurethanes are rarely softened by addition of plasticizers. Rather, they are intrinsically plasticized by using monomers that possess long-chain alkyl and (ideally) bulky substituents that prevent pseudo-crystalline or liquid crystal kind of domains to be formed. that would retard diffusion of oxygen. Poly(vinyl chloride) and several other brittle polymers are almost always plasticized by addition of viscous and hydrophobic materials such as long-chain alkyl phthalates, phosphates, sebacates, or phosphine oxides. Nitrophenyloctyl ether (which can act as a quencher unfortunately) and o-cyanophenyloctyl ether have also been used. If plasticizers can be avoided, this is recommended because they tend to leach which has two major consequences: The first relates to the response function which will change as a result of leaching. The second is related to the approval of oxygen sensor instrumentation for in vivo use which is unlikely to be granted if (potentially toxic) plasticizers cannot be prevented from leaching into a sample matrix such as blood. Mills405 has performed a study on the effect of the viscosity of plasticizers (mainly triethyl phosphate) on the sensitivity of Ru(bpy)-dyed oxygen sensor films made from either cellulose acetate butyrate or poly(methyl methacrylate).The Stern–Volmer constant is found to be inversely dependent upon the viscosity of the quenching medium, although the natural lifetime of the electronically excited state of the OSP is largely independent of medium.8.2. Scattering particles
The addition of scattering particles to sensing films can much improve the uniformity of excitation and emission light fields. The addition of SiO2, TiO2 or BaSO4 particles, for example, enables a more efficient excitation of the OSP due to multiple scattering of the excitation light in the sensor material layer and can improve the mechanical stability of the respective sensor films. Klimant et al.498 reported on the use of TiO2 nanoparticles as scattering centers in planar sensors based on magnetic oxygen-sensitive microbeads. The addition of the black magnetic particles causes an inner filter effect and absorption of luminescence. The addition of scattering TiO2 particles results, however, in better excitation of the OSP and in enhanced signals. The use of white cellulose powder may also improve light scattering and signal collection.505 Ricketts and Douglas313 constructed a colorimetric oxygen sensor using a green LED that gives a background signal. The LED was covered with an ethyl cellulose sensor film whose red emission (originating from PtOEP) serves as the oxygen-sensitive signal. Scattering agents such as TiO2 or ZnO were added to the sensing matrix to produce spatially uniform luminescence. Microporous light-scattering supports have also been used.506 Noble-metal nanoparticles have strong surface plasmon resonance absorption, and silver nanoparticles have been suggested347 as additives to luminescent Langmuir–Blodgett films containing the probe PtTPP where they enhanced luminescence intensity by a factor of 10, however at the expense of quenchability by oxygen. Practically all sensors containing fillers display non-linear SVPs because the probes located near the particles are less amenable to quenching than probes located in the bulk polymer.8.3. Photostabilizers
The photo-degradation of OSPs in most cases is due to the formation of singlet oxygen (1Δ) that is produced from ground state (triplet; 3Σ) oxygen in the quenching process.175 The production of singlet oxygen (1Δg) by ruthenium(II) complexes in microheterogeneous systems was studied in some detail.507 Similarly, the formation of singlet oxygen by three kinds of poly(ethylene glycol)-coated palladium(II) tetrabenzoporphyrins (“Oxyphors”; a widely used group of dendritic oxygen nanosensors) was investigated.508 It was found that an increasing size of the dendrimer has practically no effect on singlet oxygen sensitization efficiency in spite of the strong attenuation of the triplet quenching rate with an increase in size. While generating singlet oxygen, the dendritic probes are non-phototoxic which is in contrast to the commonly used photodynamic drug Photofrin™. The lack of phototoxicity is presumably due to the inability of PEGylated probes to associate with cell surfaces and/or penetrate cellular membranes. In contrast, conventional photosensitizers bind to cell components and act by generating singlet oxygen inside, or in the immediate vicinity of cellular organelles.Singlet oxygen can react with OSPs to give non-luminescent products. It can also attack polymer backbones. Photodecomposition is particularly efficient under strong UV radiation and when powerful lasers are used as light sources. It can be reduced by applying pulsed light excitation or low radiant powers. Davidson et al.509 found that olefinic systems having tertiary amino-substituent groups are very stable towards oxidation by 1O2. They have reported that the tertiary amino group effectively quenches 1O2 and protects olefinic systems from oxidation. The addition of compounds bearing tertiary amino-group to the hosting materials was suggested to prevent OSPs from being oxidized by 1O2. Other donors that can quench 1O2 include 1,4-diazabicyclo-[2.2.2]octane (DABCO),313,510 other tertiary amines,509,511 certain sulfur compounds,512 carotene,513 allylurea and azides.514,515 In a systematic study on the stability of OSP towards oxidation by singlet oxygen,516 singlet oxygen was produced in situ via a red light-excitable metalloporphyrin sensitizer, and the degradation of the OSPs was studied via UV-Vis spectroscopy. Electron paramagnetic resonance was also applied to monitor singlet oxygen. Practically all dyes faded away in the presence of singlet oxygen.
8.4. Optical isolations
In order to eliminate interferences caused by (a) ambient light, (b) straylight from light sources, (c) sample fluorescence, and (d) changing sample refractive index (which affects the numerical aperture of an optical waveguide), a non-transparent optical isolation layer can be applied when aiming for high-performance optical sensors (see Fig. 1). Such an optical isolation is expected to be impermeable to exciting light, but permeable to the analyte and to possess good adhesion to the sensing layer (unless the sensor layer is directly blackened). Optical isolations can cause an increase in response time by increasing the diffusion path length of oxygen from the sample into the sensing layer. Most commercial optical sensors for oxygen contain optical isolations.Kroneis and Marsoner121 used silicone rubber pigmented with (black) Fe3O4 for optical isolation and constructed a long-term stable optical oxygen sensor for monitoring oxygen in bioreactors. A thin cover layer (ca. 20 μm thick) composed of red (ferric oxide-pigmented) silicone may also be used.50,517 Klimant and Wolfbeis176 tested various types of other potentially useful optical isolations. The first was a layer of silicone rubber blackened with carbon black and deposited in 10 μm thickness on the sensor. It has the advantages of excellent adhesion and oxygen permeability, does not affect the sensitivity, but the signal intensity dropped by some 15%. The second was a black 12 μm Teflon membrane, uniformly spread over the sensing layer. Compared to pure silicone matrix, it shows distinctly lower oxygen permeability. The third was white titanium dioxide (TiO2) powder dispersed into the silicone matrix. It turned out that the white particles not only act as an optical isolation but also as scattering centers, thereby improving the excitation efficiency and the strength of the signal (fluorescence). The TiO2 particle may be replaced by barium sulfate.
Klimant et al.54 coated the tip of a fiber optic sensor for oxygen with a layer of black silicone to avoid interferences from ambient light, and to make the signal independent of background fluorescence from marine sediments and biofilms (e.g. from chlorophyll). Especially in natural systems with a high density of biomass, the optical signal can decrease a lot if an uncoated sensor tip enters a marine mat. The black silicone coating also acts as a barrier to potential quenchers such as heavy metal ions. There are also reports on the use of black layers of commercial charcoal193 or carbon black518 as an optical isolation. The features of various (including white) materials have been discussed.176
Contributions by straylight to the overall signal can be eliminated mathematically. The apparent (total) signal I′ can be considered as being composed of I (the true fluorescence at a given pO2) and straylight (Ix). A fairly simple method has been worked out to precisely determine KSV and I from three sets of intensity data.142
8.5. Materials acting as inert solid supports
Solid supports (see Fig. 1) are used to facilitate the handling of the rather thin (0.5 to 10 μm) sensing film by depositing them on their surface. Supports are expected to be impermeable to oxygen, and also to be optically transparent at the investigated wavelength unless the sensor material is illuminated from top. In the case of imaging the oxygenation of skin, the oxygen-impermeable support forms the upper layer that also prevents air from entering the sensor layer placed on skin.Most initial optical sensors for oxygen were of the planar sensor type and prepared by depositing the sensor chemistry on quartz, on other high-quality glass, or on poly(methyl methacrylate) (known as “plexiglass”). Films of poly(ethylene terephthalate) (Mylar™), typically of a thickness of 100–200 μm, were later found to be better suited517 in view of the ease of mass fabrication of sensor spots by simply punching them out. This material is also optically well transparent, flexible, cheap, inert, and oxygen-blocking, and thus has been widely used thereafter.101,176,193,210,518,519 Care has to be taken not to use materials that exhibit intrinsic and usually shortwave fluorescence that may interfere with the luminescence of the sensor chemistry. Care also has to be taken not to use solvents (for sensor cocktails) that dissolve the solid support. Other, and less common supports, include anodized alumina and the various materials used in objects and models investigated in wind tunnels to measure oxygen partial pressure. This is described in more detail in the ESI.†
9. Spectroscopies and readout schemes in luminescent sensors
Luminescence spectroscopy knows numerous methods that can be exploited for purposes of sensing. All have found applications, and this has resulted in quite a number of read-out schemes that shall be discussed in the following.9.1. Sensing based on the measurement of luminescence intensity
The ratio between luminescence intensity (F) and the concentration of a fluorophore is well described by Parker's law. In its most simple version – that is valid for absorbances of >0.05 M−1 cm−1 only – it is stating that F is directly related to the intensity of the light source (I), the molar absorbance of the probe (ε), its concentration (c), the penetration length (l), and the quantum yield (QY) of the probe (Φ) in the following way:| F = I·ε·c·l·QY·k |
The geometrical factor k accounts for effects caused by the instrumental arrangement. The product of ε and QY is sometimes termed brightness and represents a practical parameter that indicates how much of the incident light is absorbed and then converted into luminescence. The relationship between quencher concentration and fluorescence intensity is given by the Stern–Volmer law and its various modifications as outlined in Section 5.
Measurement of luminescence intensity may be combined with time-resolution if the OSPs possess a comparably long decay times, typically >1 μs. Time-resolved (“gated”) fluorometry enables background fluorescence (with decay times less than 1/10 of the decay time of the OSP) to be suppressed. In this scheme, luminescence is excited with a short pulse of light, but rather than detecting luminescence immediately after the light source has been turned off, it is detected only after certain time interval during which the short-lived intrinsic fluorescence has decayed. Fig. 19 shows a schematic of sensing based on time resolved fluorometry.
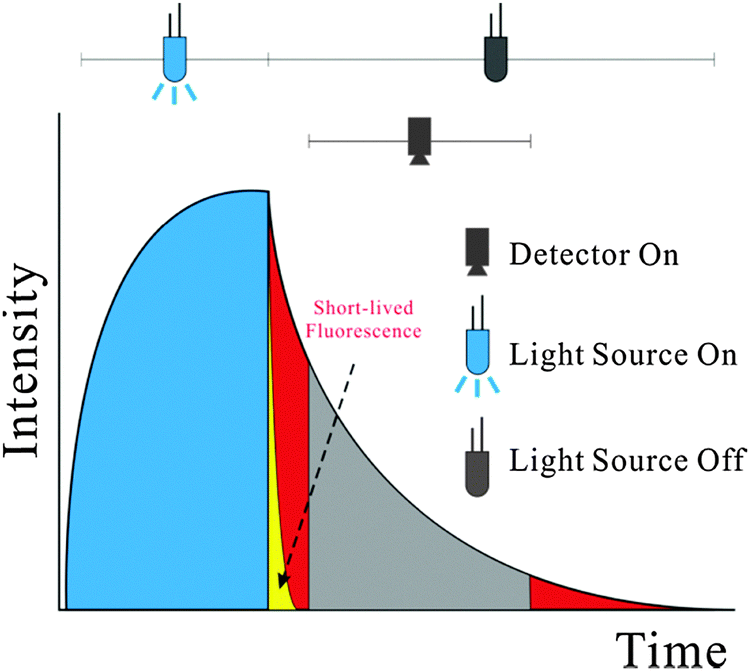 | ||
| Fig. 19 Time resolved fluorometry. Following an excitation pulse (during which luminescence rises to a maximum), the photodetector is opened only after a certain delay time during which background luminescence (yellow) is allowed to decay. The luminescence of the oxygen probe, in contrast, decays much slower (red) and can be detected after the delay time at virtually zero background. The method does not compensate, however, for constant levels of ambient light or long-lived background luminescence. | ||
9.2. Sensing based on the measurement of luminescence decay time
Luminescence lifetime is an intrinsically referenced parameter of a luminophore. It is independent of probe concentration, photobleaching (to a wide extent), drifts in source intensity, detector sensitivity, and inner filter effects, all of which are major limitations of intensity-based sensors. Finally, the effects of fiber bending remain small. It is obvious that this kind of sensor has considerable stability and advantages over sensors based on measurement of intensity.Fluorescence lifetime can be measured in either the time-domain or the frequency-domain mode (Fig. 20). In frequency-domain fluorometry, the lifetime is determined via the phase shift between the phase angle of the luminescence and that of the sinusoidally modulated excitation light. If long-lived (metal–organic) excited-state luminophores are used, modulation frequencies are comfortably low (1–10 kHz) and instrumentation is small. Time-domain fluorometry often uses time-correlated single-photon counting techniques to record the luminescence decay profile, and the decay curve is analyzed in terms of one or more decay times.
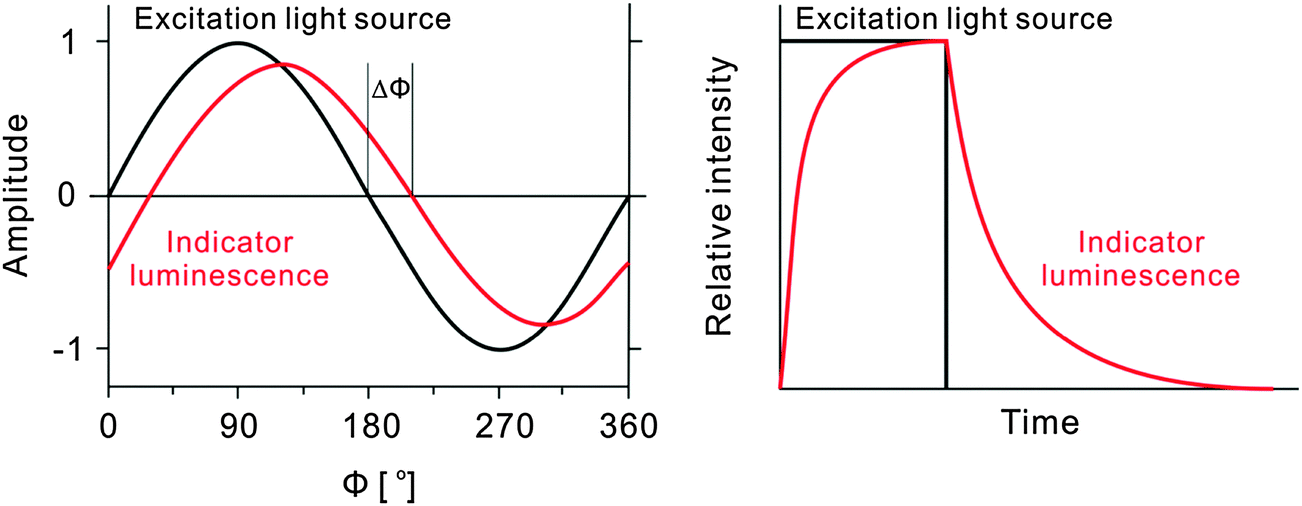 | ||
| Fig. 20 Left: the principle of phase fluorometry. Luminescence is photoexcited with sinusoidally modulated light (black trace), and the luminescence of the indicator probe (red trace) is phase shifted as a function of oxygen by an angle of ΔΦ. The phase shift can be related to pO2. Right: photoluminescence is excited with a short pulse of light (black trace) upon which it rises to a constant (steady-state) value (red trace). Once the light source is turned off, the indicator luminescence decays as a function of oxygen particle pressure. | ||
| tanϕ = 2πfτ | (7) |
The modulation frequency (f) is kept constant so that the tangent of the phase shift is directly related to the decay time, which in turn is related to the oxygen concentration via the Stern–Volmer equation. The selection of the appropriate modulation frequency is critical with respect to precision and sensitivity. The optimal modulation frequency in some cases is different from the theoretically calculated frequencies.520,521
Long lifetime OSPs require less complicated instruments for the determination lifetime. The first lifetime-based oxygen sensor50 was based on phase fluorometry. The probe Ru(bpy) was absorbed on a silica gel and then immobilized in silicone rubber to construct a sensor film. A frequency-modulated blue LED served as the excitation light source. The system was later miniaturized and eventually led to a portable device. Quite similar systems were reported recently.522 The group of MacCraith520,523 developed similar systems using sol–gel based sensor chemistries. Two lifetime-based sensor arrays for multi-site fiber optic measurements were reported by the groups of Holst524 and Klimant319 and used to determine oxygen in biofilms and aquatic sediments. A solid state electroluminescent lamp (ELL) with a peak emission at 454 nm represents an inexpensive intensity-modulated excitation light sources for phase fluorometric sensing of oxygen.525 Planar surface ELLs can be produced in various shapes and in large sizes. Accordingly, the overall optical output power emitted by ELLs is much higher than that of blue LEDs. For a sinusoidal driving voltage at a frequency f, the ELL output light is modulated at 2f and at harmonics of 2f. Because of this nonlinear modulation characteristics, a square wave driving signal can be applied and results in a pulsed light output at a repetition rate twice the square wave frequency. The shortest light pulses have a typical half width of 1 μs and this cover practically all OSP except for the PAHs. However, the high driving voltage requires effective shielding to avoid electromagnetic crosstalk between the light source and the photodetector. Also, the optical output power of the ELLs decreases rapidly during the first 100 h of operation.
In addition to sinusoidally modulated excitation, light sources may also be modulated in the shape of a square-wave function. Trettnak et al.411 constructed a small sized (120 × 60 × 30 mm) sensor module based on the use of LEDs and photodiodes. The oxygen-dependent lifetime was calculated from the phase shift between the square-wave excitation and the quasi-square-wave of the emission of a metalloporphyrin probe.
Phase modulation fluorometry is more prone to interference caused by autofluorescence than pulse fluorometry. Schmälzlin et al.442 noted, for example, that the green and red autofluorescence of green plant cells strongly interferes with the luminescence of the platinum(II) porphyrin OSP. This was overcome by using a multi-frequency phase-modulation technique which enabled the separation of the analytical signal from autofluorescence. The method was applied in later work483 to sense oxygen in individual gland tubules during hormone-induced secretory activity of a blowfly. Multi-frequency phase modulation was also shown to work in ratiometric oxygen sensing370,371 and imaging.526
A frequency domain instrument was described527 for real-time measurements of phosphorescence lifetime distributions in microheterogeneous samples. An array of harmonics (typically 100–200 frequencies) was used to modulate the excitation source, a light emitting diode. The dependence of the phase/amplitude factor on the modulation frequency was determined by linear least-squares analysis of the multiple decay profiles of emission. The instrument may also be applied to measure phosphorescence in a single-frequency mode. This is useful for rapid evaluation of apparent luminescence lifetimes. The instruments were tested in Stern–Volmer calibrations of Pd–porphyrin based phosphorescent OSPs and can determine lifetimes in the range of 10–3000 μs. In the single-frequency mode, the measurement time was reduced to about 0.2–0.5 s. The instruments provide complete correction for the in-phase signal of up to 40% of the total emission intensity. Time resolved fluorometry is the preferred method in high-precision oxygen sensors. Fig. 21 shows a commercial lifetime spectrometer that is used as a detector in fiber optic oxygen microsensors.
 | ||
| Fig. 21 A commercial lifetime spectrometer as used in a fiber optic oxygen microsensor (Fibox 4). Each fiber sensor linked to this reader has a unique ID and can measure oxygen at levels between 1 ppb and 100% (in gas or in gas-saturated water). Calibration is performed via a barcode scan, and effects of temperature, pressure and salinity are electronically compensated for. From Presens GmbH (http://www.presens.de/) with permission. | ||
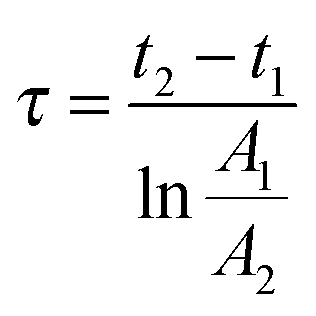 | (8) |
Demas et al.529 evaluated the precision of the RLD method for a single exponential decay as a function of integration time relative to the lifetime and the total photon counts. Their results showed the RLD method to exhibit good precision and accuracy. Its speedy and its simple instrumentation make RLD an excellent tool for data acquisition even under conditions of real-time analysis. On the other hand, it can produce large errors under the certain conditions.530 Unexpected and uncharacteristic Stern–Volmer quenching plots may be obtained for two-component systems. Artifacts include bimodal quenching curves as well as “anti-quenching” curves. These phenomena are further exacerbated in the presence of fractions of unquenched long-lived components. As the RLD method is always combined with gated (time-resolved) detection, it enables short-lived intrinsic fluorescence of samples to be suppressed. This method is particularly useful for purposes of imaging.
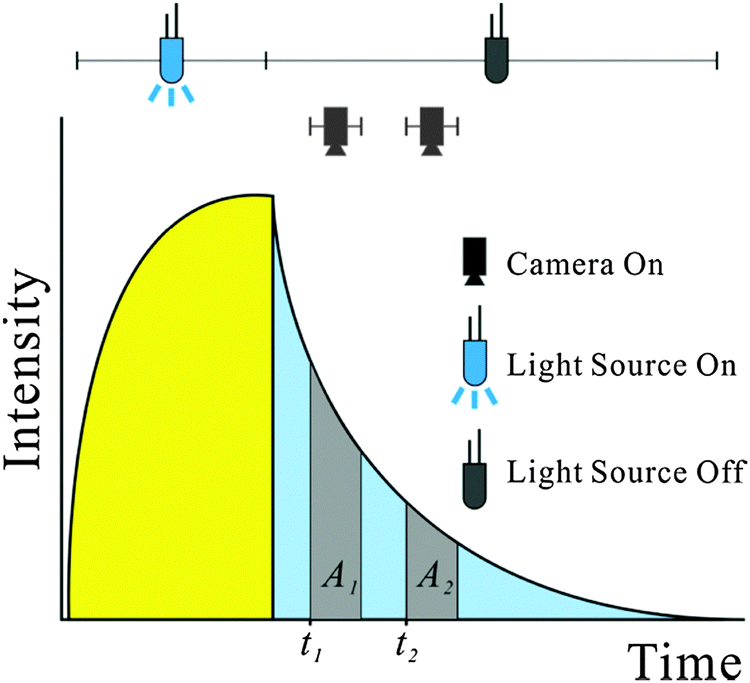 | ||
| Fig. 22 Schematic presentation of the RLD method. The integrated areas A1 and A2 are used to calculate decay time rapidly. Note that short-lived background luminescence (until time t1) is suppressed. | ||
 | ||
| Fig. 23 The principle of DLD determination. Following an excitation phase (yellow), the detection gate is opened 4 times to give 4 integrated areas (A1 to A4) that can be used to calculate two lifetimes. | ||
9.3. Sensing based on measurement of luminescence resonance energy transfer
Fluorescence (luminescence) resonance energy transfer (FRET) occurs if two fluorophores with spectral match between the emission of the first (the donor) and the absorption of the second (the acceptor) come in close proximity, typically <10 nm. In this case, photonic energy pumped into the donor is transferred to the acceptor whose luminescence is then observed. FRET systems are widely used in biophysical studies and can serve as molecular bars on a nanoscale. FRET is also one way to enlarge the Stokes shift and amenable to ratiometric (mostly 2-wavelength) sensing.Wolfbeis & Sharma140,531 reported on a FRET system where the oxygen quenchable fluorophore pyrene acts as the donor, and the oxygen insensitive fluorophore perylene as the acceptor. If photoexcited at 320 nm, the FRET system gives a strong luminescence at 476 nm. Unlike in many FRET sensors (for pH etc.), in which the acceptor is analyte-sensitive, the donor is analyte-sensitive here. The system strongly responds to oxygen, and the quenching efficiency even exceeds the quenching efficiency of pyrene alone. In fact, a 4-fold increase in the Stern–Volmer quenching constant was observed in the FRET system compared to the quenching of pyrene alone. This led to a strong increase in signal resolution and sensitivity.140
Like in other sensors, FRET sensors are preferred that emit longwave light. Bawendi and Nocera et al.532 conjugated oxygen probes of the Os(II) polypyridyl type to quantum dots. The Os(II) complexes have broad absorptions that extend up to 600 nm, and this feature makes them viable acceptors of resonance energy from QDs donors. The QDs display strong two-photon absorption, so that the conjugates can be excited with NIR light. The emission of the QDs is insensitive to oxygen, but that of the Os(II) polypyridyl complexes is quenched. The QDs emission can be used as internal reference to correct for any fluctuations in the photoluminescence intensity. These features make the two-photon excitable oxygen sensors quite suitable for application in colored or turbid media. However, the quenching efficiency of this conjugate is rather low (τ0/τ100 = 1.45), and both the Os(II) complexes and the QDs are highly toxic. 2-PE-based sensing was also accomplished533 by using CdSe quantum dots modified with Pd(II)porphyrins with meso-pyridyl substituents. Spectral overlap of the emission of the QDs and of porphyrin absorption results in high FRET efficiency which serves as the mechanism for signal transduction in these constructs. The nanosensors respond to oxygen in the pressure range from 0–160 Torr under both one- and two-photon excitation.
Photon upconversion nanoparticles (UCNPs) were used534 to photoexcite the probe Ir(CS)2(acac) (see Section 6.2.2) to obtain an NIR-excitable oxygen sensor. The use of UCNPs has outstanding features such as (a) excitation at 980 nm which results in zero luminescence background in the visible, (b) deep penetration of excitation light into biomaterials, and (c) no photodamage. The UCNPs are nontoxic and do not suffer from size-dependent emission and blinking. Their emission is not at all quenched by oxygen. Rather, they act as nanolamps for exciting the iridium OSP because its absorption band perfectly matches the blue emission of the NaYF4:Yb, Tm UCNPs. The green emission of the Ir(II) probe is quenched by oxygen. Thus, an oxygen sensing system is obtained that can be excited using an NIR laser, even though the OSP is not directly excitable in the NIR. This scheme represents one further approach to overcome the lack of NIR-excitable oxygen probes. However, the oxygen quenching sensitivity of the sensor film is moderate. It was also reported that the Stern–Volmer quenching constants are different depending on whether the luminescence of the probe Ir(CS)(acac) was directly excited with blue light or via the up-converting particles. In a related approach,169 UCNPs of the type NaYF4:Yb,Tm were coated with Ru(phen)-doped silica. The resulting particles have an average size of around 72 nm, and sharp blue emission peaks at 450 and 474 nm. They were used to photoexcite the Ru(phen) probe in the shell. These nanoparticles have high sensitivity (I0/I100 = 3.9) and their hydrophilic surface can be functionalized with reactive groups for further conjugations.
9.4. Sensing based on measurement of fluorescence anisotropy
Fluorescence anisotropy r is defined as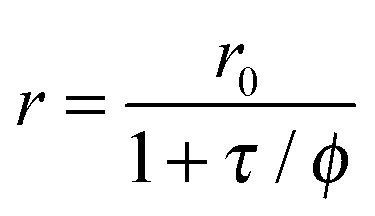 | (9) |
 | (10) |
Sensing of oxygen via anisotropy is based on the fact that polarization depends on the decay time of the OSP108,109 as can be seen from eqn (9). The decay time of the OSPs is reduced by oxygen so that anisotropy can be related to the concentration (or partial pressure) of oxygen. Rao and Lakowicz535 showed that the anisotropy of immobilized Ru(dpp) and an inert reference dye, both contained in a stretched film of poly(vinyl alcohol), depend on the local concentration of oxygen. The reference dye has a linear structure, and its emission is highly polarized. The global anisotropy is the weighed sum of the anisotropy contributions of the two emissions. An increase of the fraction resulting from the oxygen sensing luminophore results in decreased anisotropy, and vice versa. As expected, the polarization values displayed large changes, from −0.33 to +0.45 on changing from 0 to 100% oxygen. Since polarization values can be routinely determined to better than ±0.01, the accuracy of sensing oxygen is around 1% or better. The group later reported372 on a sensor based on the changes in the emission polarization of a dual-emitter dye (dppe-Pt2P, see Table 10) that exhibits both a short and a long lifetime emission.
Fluorescence polarization was also used to separate the analytical luminescence signal from scattered excitation light.536 Optical signals were separated by cross-polarization, which is simple and wavelength-independent. A stable and sensitive optical oxygen sensor was constructed based on a consumer CMOS image sensor array and polarization signal isolation. The image sensor is inherently color discriminating, while the polarization is a wavelength-independent scheme for filtering excitation light. The combination of the two components generates a compact, multi-color detection system that was applied to point-of-use oxygen sensing based on the quenching of the PtOEP luminophore by oxygen. The method has a high extinction ratio and can separate even weak luminescence from a strong background signal.
9.5. Sensing based on two-photon excitation
Two-photon excitation (2-PE)307,308,532 enables OSPs to be excited with long-wavelength (even NIR) light. This is beneficial in terms of biological research because most biomatter has strong absorption and fluorescence at <500 nm. However, if cross-sections for 2-PE are low, intense lasers are needed that may burn samples. A major benefit of 2-PE in the case of phosphorescent probes is the confinement of the triplet state to the immediate vicinity of the laser focus, thereby eliminating oxygen consumption and formation of reactive oxygen species along the excitation path.The two-photon absorption (2-PA) cross-sections of porphyrins are low, typically in the order of a few Goeppert-Meyer (GM) units (where 1 GM equals 10−50 cm4 s per photon). In order to increase cross-sections, light-harvesting antennas were used to capture two-photon energy, and the captured energy was transferred to the OSPs via resonance energy transfer. Commonly used metalloporphyrins exhibit extremely low 2-PA cross-sections. Vinogradov et al.307 designed several water-soluble Pt(II) porphyrin–coumarin based dendrimers, in which the antenna (a coumarin) was coupled to the metalloporphyrin core (Fig. 24). The resulting metalloporphyrin-antenna construct was further incorporated in a protecting dendritic jacket to isolate the core from interactions with biological macromolecules, to control diffusion of oxygen, and to make the entire sensor water-soluble. The same group537 also studied photophysical processes of rhodamine antennas in combination with various metalloporphyrins. The probe PtP–C343 was found to possess a high 2-PA coefficient because of the use of C343 coumarin as the antenna. It enabled 2-PE-based imaging of oxygen and blood flow in deep cerebral vessels.311
 | ||
| Fig. 24 Mechanisms of the antenna-based two-photon excitation of metalloporphyrins for intracellular sensing of oxygen. (Reprinted from ref. 307 with permission from American Chemical Society). | ||
In yet another kind of 2-PE system, Kondrashina et al.538 have used a highly photostable platinum(II)porphyrin along with poly(9,9-dioctylfluorene). The latter acts as a two-photon antenna and as a FRET donor. The Ru(dpp) complex has a 2-PA cross-section (210 GM units) that is much higher than that of porphyrins. Incorporated into polystyrene microparticles, Ru(dpp) was shown190 to enable 2-PE-based sensing of oxygen (also see Section 6.2.1.). Quantum dots also possess high brightness and large 2-PA cross-section. They were coupled to Os(II) polypyridyl type oxygen probe to give nanosensors for oxygen.358 The QDs can capture two-photon energy from excitation laser and transfer it to the Os(II) complex. The method enables oxygen being ratiometrically detected via NIR excitation. However, the quenching effect is not satisfied, and both QDs and Os(II) complex are highly toxic. 2-PE-based sensing was also accomplished539 by using CdSe quantum dots modified with Pd(II)porphyrins with meso-pyridyl substituents. Spectral overlap of the emission of the QDs and of porphyrin absorption results in high FRET efficiency which serves as the mechanism for signal transduction in these constructs. The nanosensors respond to oxygen in the pressure range from 0–160 Torr under both one- and two-photon excitation.
9.6. Sensing based on chemi- and bioluminescence
Sensors based on measurement of chemiluminescence (CL) or bioluminescence do not require external light sources and optical filters, but chemical reagents that are consumed over time. In addition, oxygen is consumed which is in contrast to quenchometric sensors. CL methods in sensor technology (oxygen included) have been reviewed.540 Seitz et al.44 reported that the cyclic aminoethylene EIA (which can be easily prepared) reacts with oxygen under emission of chemiluminescence (CL). Tetraaminoethylenes without aromatic functions also react with oxygen to produce CL. Its intensity is directly proportional to the concentration of oxygen. Low levels of CL can be easily measured, and a device has been presented in which the oxygen diffuses through a Teflon membrane before reacting with the CL reagent (a 10% hexane solution of 1,1′,3,3′-tetraethyl bisimidazoline). Steady state CL intensity is proportional to pO2, but response to oxygen decays gradually over a 12 h period because of reagent consumption. The detection limit of oxygen in the gas phase is estimated to be as low as 1 ppm (v/v). An increase in the reaction temperature causes an increase in signal. Response times are in the order of 10–20 s.Another CL-based detection scheme is based on the luminol reaction541 but is not practical. All CL-based methods suffer from the disadvantages of (a) storing the reagent under exclusion of oxygen; (b) being interfered by ambient light; (c) poor reversibility, and (d) consumption of oxygen. This makes them applicable to a limited number of applications only.
For bioluminescence (BL) to occur in luminous biospecies, the presence of oxygen is mandatory.542–545 The intensity of the BL therefore can be unambiguously related to the oxygen concentration. Beijerinck546 back in 1902 utilized bacterial BL to detect oxygen during photosynthesis with low detection limits. Other studies542 revealed the relationship between the BL intensity of luminous bacteria and fungi and the concentration of oxygen, and between the BL of P. phosphoreum and oxygen.543 They resulted in convenient and sensitive methods for quantifying dissolved oxygen with high accuracy at 10−6 to 10−8 mol L−1 levels. Williams et al.545 used Photobacterium fischeri to quantify dissolved oxygen. A suspension of the bacterium was covered with oxygen-permeable membranes (polypropylene or silicone rubber), and BL was detected as a function of pO2. The sensor responds quite rapidly (<8 s), and there is a linear relationship between BL intensity and dissolved oxygen concentration in the range from 35 nmol L−1 to 8.4 μmol L−1. Response is fully reversible both in the gas and the liquid phase. On the less favorable side, luminous bacteria often require the presence of high salinity, and their own respiration may compromise accuracy. The BL of cells is also sensitive to metabolic inhibitors, and depends on the age of bacteria.
A solid phase reagent (composed of a fluorinated polyalcohol, luminol and ferric sulfate) was reported547 to display CL if exposed to a stream of oxygen. It can sense oxygen in moderately low concentration (2.4 ppm in water). In an electrochemiluminescence (ECL) based sensor for oxygen,548 ECL was generated at a disposable CdS-modified screen printed carbon electrodes during cathodic pulse polarization. Two emissions, with peaks located at 520 nm and 580 nm, are found. The intensity of ECL is linearly related to the concentration of dissolved oxygen in the range of 1.7–33 mg L−1, with a detection limit of 0.02 mg L−1. The method was applied to detect the dissolved oxygen concentration and biochemical oxygen demand.
9.7. Other luminescent sensing schemes for oxygen
Borisov et al.549 have introduced a sensing scheme for oxygen that exploits the oxygen-sensitive anti-Stokes emission of OSPs. This emission is generated via triplet–triplet annihilation-based upconversion using the probe pair of PtTPTBP and diisobutyl 3,9-perylenedicarboxylate. Porous glass beads carry the “sensing chemistry” that includes a sensitizer, an annihilator, both dissolved in a high boiling solvent. The beads are dispersed in silicone rubber or Teflon and deposited in the form of sensor films. LED light sources are used for excitation, and the upconverted emission is very sensitive to oxygen both in terms of decay time and intensity. The latter follows a quadratic Stern–Volmer relationship which is quite a novel finding. The NIR luminescence of the sensitizer is also quenched but to a much lesser extent. Oxygen can be sensed over a wide dynamic range and at very low levels.A fluorescence turn-on sensing scheme for dissolved oxygen was introduced550 that is based on the use of a perylene diimide dye (PDD) which is electro-chemically reduced to its non-fluorescent dianion form (PDD2−). In the presence of oxygen, the dianion is oxidized to its initial form via an electron transfer reaction with oxygen upon which fluorescence is recovered. As a result, the fluorescence intensity of the dianion in solution increases upon the addition of oxygen gas. High sensitivity is obtained when the emission intensity reaches its maximum by the addition of 2.4% (v/v) oxygen gas. Electrochemical reduction can regenerate this “sensor”. The limit of detection is 0.14% of oxygen in argon gas of atmospheric pressure.
A surface plasmon-coupled emission (SPCE) based oxygen sensor was presented.551 It is making use of a ruthenium OSP electrostatically attached to a protective layer of SiO2 above a silver film deposited on a glass slide. SPCE is a highly localized phenomenon occurring when excited fluorophores are within 200 nm of thin, continuous metallic films. The observed SPCE of the probe is highly directional (at an angle of 45.5°) and results in a 27-fold enhancement in intensity over the free space fluorescence. Like in certain evanescent types of arrangements, this so-called Kretschmann configuration allows for an optical filter-free detection of emission. Quenching efficiency is somewhat reduced due to the metallic effects on fluorescence lifetimes but response is very fast. Baluschev et al.552 have presented first results of ultralow excitation intensity continuous-wave excited up-conversion fluorescence in a Kretschmann SPR geometry. The active system is a blue-emitting polymer matrix of polyfluorene blended with metalated porphyrines such as PtOEP. The up-conversion fluorescence is a consequence of a two-particle triplet–triplet annihilation process near a silver surface and at very low (a few Watt cm−2) laser intensity. The cw luminescence spectra reveal the fluorescence of the polyfluorene, the fluorescence of PtOEP, and the phosphorescence of PtOEP. A working prototype fiber-optic intensity-based oxygen sensor was also briefly described.
9.8. Multiple sensing
Optical sensing has the unique feature of enabling multiple (= multi-analyte) sensing and imaging, examples being the simultaneous sensing (or imaging) of temperature and oxygen, or even of three parameters (pH, temperature and oxygen). Multiple sensors have the advantage of gathering substantially more information at a single sensing site. Space is limited in the case of fiber optic microsensors, inside cells, and in the case of nanoparticle-based sensors. The state of the art in luminescence based multiple chemical sensing and imaging has been reviewed.390 Two main methods have to be differentiated, viz. (a) multiple sensing using single probes, and (b), multiple sensing using more than one probe. They shall be discussed in the following.Sodium fluorescein (uranine) is an example for a probe that is affected by both pH and very low levels of oxygen. When adsorbed on silica gel, it has a pH-dependent absorption and displays a phosphorescence intensity and decay time that are strongly quenched by even traces of oxygen. Typical couples of other parameters that may be simultaneously determined by measurement of intensity (I) and lifetime (τ) of a single (!) indicator are the following: NH3/oxygen, CO2/oxygen, or oxygen/temperature.553 Single-probe dual sensing also includes methods where probes are used that have dual emission bands that are differently modulated by two analytes.369
In yet another version, a single probe is placed in a polymer host that has varying permeability for quencher analytes. The simultaneous determination of oxygen and the inhalation narcotic halothane, both of which act as dynamic quenchers of the fluorescence of the OSP decacyclene is a typical example.46 In this case, the Stern–Volmer equation (eqn (2)) has to be extended554 to give eqn (11) which accounts for the contribution of an additional quencher (Q)
| F0/F = 1 + QKSV[Q] + OKSV[O2] | (11) |
Here, [Q] and [O2], respectively, are the concentrations of the second quencher and of oxygen, respectively. QKSV and OKSV are the respective Stern–Volmer constants. The equation can be solved if use is made of two sensors (referred to as A and B here) with different sensitivity and different quenching constants (i.e. made from different materials) for the two quenchers. Let sensor A display a total response function (F0/F − l) termed α, and sensor B display a response function (F0/F − l) termed β. The Stern–Volmer quenching constants of sensors A and B for oxygen (OKA and OKB) and the second quencher (QKA and QKB), respectively, have to be obtained first by calibration.
The concentration of the oxygen can be calculated then according to
| [O2] = (αQKB − βQKA)/(OKA·QKB − QKA·OKB) | (12) |
To obtain [Q], a similar expression can be given (eqn (13)):
| [Q] = (αOKB − βOKA)/(QKA·OKB − OKA·QKB) | (13) |
As a consequence, measurement of fluorescence quenching with two different sensors allows the determination of oxygen and/or the other quencher simply by measuring α and β.
Sometimes, one of the two sensors can be made specific for oxygen (for example by covering it with a thin membrane consisting of polytetrafluoroethylene (PTFE)). As a result, the quenching constant for the second quencher (QKB) becomes zero, and eqn (12) is considerably simplified
| [Q] = (αOKB − βOKA)/(QKA·OKB) | (14) |
Simultaneously, eqn (13) can be transformed to give
| [O2] = β/OKB | (15) |
The most accurate sensor type consists of two identical sensing membranes, one of which is covered with a membrane that is impermeable to the second quencher (PTFE in the case of halothane). In this case the two quenching constants for oxygen (OKA, and OKB in eqn (14)) become identical, which leads to the final and very simple equation that describes the relationship between the two sensor signals and the concentration of the second quencher:
| [Q] = (α − β)/QKA | (16) |
 | ||
| Fig. 25 Cross section of a dual optical chemical sensor. The particles (P1, P2) are responsive to the respective analytes and yield the optical information provided their optical emissions can be discriminated (by color, decay time, or the like). The particles are incorporated into a matrix polymer (the binder) whose choice is critical. (Reprinted from ref. 390 with permission from The Royal Society of Chemistry). | ||
Numerous kinds of methods for dual sensing of oxygen and a second species have been reported. Examples include oxygen/temperature,148,173,238,417,555–557 oxygen/pH,173,424 oxygen/CO2 in blood,517 in fluidic circuits,558 and in aquatic systems,559 and oxygen/glucose.165 A triple sensor has been reported45 for pH, oxygen and temperature, and a quadruple sensor for simultaneous determination of oxygen, CO2, pH, and temperature.390,560 Temperature always exerts an effect (see Section 10.3.) and often has to be measured in parallel in order to correct the oxygen signal for the effect of temperature. Table 13 summarizes the state of the art in multiple sensing.
| Analytes | Sensor materials | Sensor format | Spectroscopy | Remarks | Ref. |
|---|---|---|---|---|---|
| O2 | Decacyclene in silicone rubber/ polytetrafluoroethylene | Fiber optics | Intensity | Two sensing films. Film A: decacyclene in silicone rubber for sensing oxygen and halothane: film B: decacyclene in silicone rubber covered with Teflon; senses oxygen only. Both parameters calculated via equ. 9.8 and 9.9. | 46 |
| Halothane | Decacyclene in silicone rubber | ||||
| O2 | Ru(dpp) in siloxane coated with polyHEMA hydrogel | Fiber optics | Intensity | Distal end of an imaging fiber (350 μm) deposited with individual sensor materials for oxygen and glucose; sensor maintains the sensitivity for 2 days; detection limit for glucose: 0.6 mM. | 165 |
| Glucose | Ru(dpp) in siloxane coated with polyHEMA hydrogel containing glucose oxidase | ||||
| O2 | PtTFPP in tBS/p-tBS-co-TFEM | Film | Ratiometric intensity | Spectrally well separated; used in a temperature-compensated pressure sensitive paints. | 172 |
| T | Ru(phen) in PAN particles | ||||
| O2 | PdTFPP in PSAN particles | Film | Lifetime | Particles immobilized in hydrogel; signal separated via different lifetimes. | 417 |
| T | Ru(phen) in PAN particles | ||||
| O2 | Ru(dpp) in sol–gel | Film | Lifetime | PSP application, temperature compensation; excited with a single LED at 460 nm; time gated technique to temporally separate signals. | 563 |
| T | Magnesium fluoro-germanate in sol–gel | ||||
| O2 | PtTFPP in FIB | Film | Lifetime | PSP application, sprayable sensor; temperature compensation. | 564 |
| T | La2O2S:Eu3+phosphor in FIB | ||||
| O2 | PtTFPL in FIB | Film | Lifetime | Sprayable sensor for PSP applications; signals separated via their colors or via time-gating; both probes excited at 400 nm. | 273 |
| T | EuD2 in FIB | ||||
| O2 | PdTFPP in PSAN microbeads | Film | Lifetime | Sensor particles immobilized in hydrogel; excited at 405 nm; signal separated using optical filters or decay time with certain delay; not influenced by humidity, suitable for PSP, biological and medical applications. | 416 |
| T | Eu complex in poly(4-tert-butyl styrene) microbeads | ||||
| O2 | C70 in ethyl cellulose or ormosils | Film | Lifetime | Excited using a single LED at 470 nm; extremely sensitive to oxygen (ppbv range); signal separated either by optical filters or lifetime. | 148 |
| T | Ru(phen) in PAN | ||||
| O2 | PtTFPP in PSAN microparticles | Film | Lifetime | Both particles immobilized in hydrogel to form composite material; excited using a single LED at 405 nm; dual lifetime determination (DLD) used for signal separation and analysis; high spatial resolution. | 555 |
| T | Eu(tta)3(dpbt) in PVC particles | ||||
| O2 | Ir(btpy)3 in cellulose acetobutyrate | Film | Lifetime | Signal separated using optical filters; both probes were excited using a 405 nm LED. | 238 |
| T | Ir(ppy)2(carbac) in PAN microparticles | ||||
| O2 | PtTFPP in polystyrene | Film | Lifetime | Two layer approach; oxygen sensitive layer on top of the temperature sensitive layer; excited with a single 405 nm LED; DLD used for signal separation and analysis; used as a transducer for monitoring glucose. | 557 |
| T | Eu(tta)3(dpbt) in poly(vinyl methylketone) | ||||
| O2 | PtTFPP in PS–PVP core–shell nanoparticles | Particles | Lifetime | Particles dispersed in water, and use as water-sprayable paint; can be washed away easily; both probes excited at 405 nm; signals separated using optical filters. | 556 |
| T | Ir(ppy)2(carbac) in PAN microparticles | ||||
| O2 | PtTFPP in PS–PVP core–shell nanoparticles | Film | Ratiometric intensity | Particles immobilized in hydrogel; signal separated and recorded using a color CCD digital camera; image data processed using photographing technique. | 423 |
| T | Pt(Br-thq)(acac) in poly(VDC-co-AN) particles | ||||
| O2 | PtTFPP in silica nanoparticles | Film | Intensity | Both dyes encapsulated in core–shell silica nanoparticles and excited at 409 nm; the nanoparticles immobilized in ormosil film; signals spectrally separated. | 565 |
| T | CdSe QDs in silica nanoparticles | ||||
| O2 | Ru(dpp) in sensing film (commercial product) | Film | Lifetime | Two layer structure; frequency domain technique used to measure oxygen, time domain RLD used for temperature; single photodetector; requires fast electronics. | 566 |
| T | Tris(dibenzoylmethane)mono(5-amino-1,10-phenanthroline)europium(III) in PMMA | ||||
| O2 | Ru(dpp) in ormosil microparticles | Film | Lifetime intensity | Both microparticles immobilized in hydrogel; signal spectrally separated using optical filters; selectivity and sensitivity can be adjusted; few cross-sensitivities. | 477 |
| pH | Carboxyfluorescein covalently attached to polyHEMA microbeads | ||||
| O2 | PtTFPP in hydrogel | Film | Lifetime | pH measured via DLR; signals separated using optical filters; cross-sensitivity to ionic strength is small; used for image pH and oxygen distribution in a marine sediment. | 559 |
| pH | 2′-Chloro-7′-hexylfluorescein octadecyl ester in hydrogel | ||||
| O2 | Pt-octaethylporphyrin (PtOEP)-Schiff-base group (SB) and Pd-coproporphyrin-I tetraester (PdCP)-SB in PVC | Film | Emission intensity | Dual sensing of oxygen and pH based on one probe; absorption intensity for pH sensing; emission intensity or lifetime used for ratiometric oxygen sensing. | 333 |
| pH | Absorption | ||||
| O2 | PtTPP copolymerized with HEMA | Film | Ratiometric intensity | Probes copolymerized into planar sensing film to prevent dye leakage; pH-sensitive emission and oxygen-sensitive emission spectrally separated; not cytotoxic. | 302, 304 |
| pH | 4-Amino-1,8-naphthalimide based monomer polymerized with HEMA | ||||
| O2 | PtTFPP in polystyrene particles | Film | Ratiometric intensity | Particles immobilized in hydrogel; signal separated and recorded using a color CCD digital camera; image data processed using photographing technique. | 567 |
| pH | FITC on amino-cellulose particles | ||||
| O2 | PtTPTBP in pluronic–silica nanoparticles | Nano-particles | Ratiometric | Ultra-small size (12 nm) nanosensors; PtTPTBP in hydrophobic core, and FITC covalently attached on the surface of the nanoparticles; signals spectrally separated; used for sensing oxygen and pH in cytosol. | 568 |
| pH | FITC in pluronic–silica nanoparticles | ||||
| O2 | PtOEP in a polystyrene/poly(ethylene glycol) | Film | Ratiometric | Uses multicolor microcavity OLEDs as a light source, sensor film composed of a polystyrene/poly(ethylene glycol) blend. | 561 |
| pH | Blend | ||||
| O2 | PtTFPP in polystyrene | Film | Lifetime (frequency domain) | Two-layer structure; CO2-sensitive layer spread on the oxygen sensitive layer; signal separated using optical filters; excited at two wavelengths; CO2 measured using dual lifetime referencing method. | 558 |
| CO2 | HPTS(TOA)3 in ethyl cellulose particle | ||||
| O2 | Ru(dpp) in ethyl cellulose | Film | Lifetime | Signal separated using optical filters or gated lifetime; sensor used for intrinsically referenced mapping of CO2 and O2 in aquatic biofilms. | 569 |
| CO2 | HPTS(TOA)3 in Ethyl Cellulose | Intensity | |||
| O2 | PtTFPP or PtTFPL in PSAN microparticles | Film | Lifetime | First triple sensor; particles immobilized in hydrogel; excited with a 405 nm LED; signal separated using optical filters or lifetime with certain delay. | 418 |
| T | Eu(tta)3(dpbt) in PVC particles | DLD | |||
| pH | HPTS covalently attached to amino-modified polyHEMA microparticles | Intensity | |||
| O2 | Ir(CS)2(acac) in PSAN particles | Film | Lifetime | First quadruple sensor; three layer structure, oxygen-sensitive particles and CO2-sensitive particles immobilized at the bottom, temperature-sensitive particle and pH indicator were immobilized on top; the two layers separated by a layer of silicon contain SiO2 beads as a scatterer; signals separated via optical filters. | 560 |
| CO2 | HPTS(TOA)3 in EC49 particles | Intensity | |||
| T | Cr-YAB particles | Lifetime | |||
| pH | SNARF-derivative in hydrogel | Intensity | |||
Dual fiber optic microsensors for oxygen and pH, and for oxygen and temperature based on optical fibers were reported173 that have sensor tips with a diameter of ∼140 μm. The sensor chemistries consist of luminescent microbeads that respond to the respective parameters by a change in the decay time, or the intensity of luminescence, or both. The use of microbeads enables the ratio of the signals to be easily varied, reduces the risk of fluorescence resonance energy transfer between indicator dyes, and reduces the adverse (oxidative) effect of singlet oxygen that is generated in the oxygen-sensitive beads. Tian et al.302 have used a PtTPP probe (for oxygen) and a green emitting polymerizable pH probe to obtain a (hydroxyethyl methacrylate)-co-acrylamide copolymer with a dual response. Simultaneous monitoring of oxygen and pH was also accomplished561 using a multicolor microcavity organic LEDs as a light source along with small-molecule- or polymer-based detectors with selective spectral response, and a sensor film composed of a structured high molecular weight polystyrene/poly(ethylene glycol) blend.
A dual sensor for oxygen and T is based on a referenced oxygen-sensitive and temperature sensitive paint with color camera read-out and full compensation for effects of T, in one version based on a lipophilic polymer,423 in another on a water-based and sprayable paint.556 Two iridium(III) complexes were shown to enable dual sensing in that the red emission of the first and the green emission of the second can be directly related to pO2 and T, respectively.238 Another method for simultaneous luminescent lifetime-based sensing of T and pO2 was applied to monitor an enzymatic reaction at varying T.557 In this sensing scheme – which was referred to as dual lifetime determination (DLD; see Section 9.2.3) – two lifetimes can be extracted from a complex decay profile. DLD was applied to sense oxygen and temperature independently and to monitor the consumption of oxygen in the glucose oxidase-catalyzed oxidation of glucose at varying temperatures. Other sensor materials for simultaneous imaging of oxygen and T on surfaces are known,417,555 and a T-sensitive europium(III) probe along with PdTFPP was used for simultaneous sensing of T and oxygen.416
Dual luminescent arrays sensors were also fabricated by inkjet-printing of pressure-sensitive and temperature-sensitive paints.562 This dual sensor consists of discrete dot arrays of a PSP and a TSP and has been developed for a precise measurement of barometric pressure on the solid surface of aircraft models. The PSP and TSP luminophores are spatially isolated from each other so that there is no interaction between the two luminophores. This technology enables an optimal solvent and binder to be found for each luminophore. In this study, a solution of PtTFPP in 2-propanol and a solution of ZnS–AgInS2 (ZAIS) nano-particles in toluene were employed as PSP and TSP solutions, respectively.
In yet another version of a dual sensor for oxygen and T, the probes Eu(tta)3 and Gd(tta)3 were placed in an acridone–polystyrene matrix to form ultra-thin (250 nm) sensing layers.259 The acridone in the polymer acts as antenna to capture excitation energy which is then transfered to the luminescent lanthanide complex. The luminescence of an Eu(tta)3-doped sensing layer is only slightly affected by oxygen but highly sensitive to T in the physiological range (293–313 K). The Gd(tta)3 has long phosphorescence lifetimes (up to 1 ms) that are very sensitive to oxygen.
The rates of enzymatic reactions strongly depend on temperature and pH value, and a triple sensor therefore was reported390 that can sense glucose (via O2) but also any (undesired) deviations in temperature and local pH value. A cross section of such a sensor is shown in Fig. 26. In another kind of dual sensor for real-time monitoring of glucose and oxygen,570 a blue emitter is used as the glucose probe, a red emitter as the oxygen probe, and a yellow emitter as a built-in reference probe.
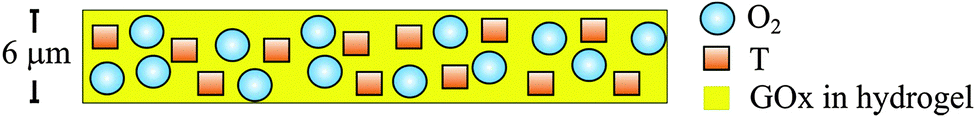 | ||
| Fig. 26 Cross-section of a triple sensor for glucose (via oxygen), temperature and pH. The sensor layer consists of polyurethane hydrogel into which luminescent sensor beads (typically 3 μm in diameter) for temperature (T) and oxygen (O2) were incorporated along with cross-linked glucose oxidase (GOx). The size of the beads is 3 ± 1 μm. The yellow coloration of the hydrogel matrix symbolizes the presence of GOx that converts glucose into gluconolactone and simultaneously consumes oxygen which is measured. (Reprinted from ref. 390 with permission from The Royal Society of Chemistry). | ||
9.9. Referenced sensing and imaging
Single-wavelength intensity-based luminometry suffer from a number of drawbacks such as drifts of the opto-electronic system (source and detectors), variation in the optical properties of the sample (including probe concentration, leaching, photobleaching, turbidity, coloration and varying refractive index), inner filter effects, interfering ambient light, and background luminescence. In order to (partially) overcome these, referenced analytical techniques have been developed. The following referenced sensing schemes are most common: Two-wavelength ratiometry, luminescence resonance energy transfer, measurement of oxygen-dependent decay time (“lifetime”), dual lifetime referencing, and anisotropy. Some will be discussed in the following, some have been treated in earlier sections.Two-wavelength referencing is quite common. One method is based on the use of a single OSP possessing two emission bands that are differently quenched by oxygen. In another scheme, the oxygen-dependent luminescence is referenced to a Raman signal that obviously is independent of oxygen. In a third, an OSP is used along with an inert fluorophore. An inert reference fluorophore is not needed in methods based on the measurement of decay time, Raman scattering, or anisotropy. Their merits have been discussed in the respective Sections 9.2 and 9.4 earlier in this review.
Schaeferling and Duerkop571 have surveyed aspects of referenced fluorometric detection schemes. Among those, two-wavelength referencing is about as old as luminescent sensing of oxygen. The signal of the OSP is divided by that of a reference dye, and this simple method can suppress the adverse effects of fluctuations of opto-electronic components (source, detector), effects of varying distance, of bleaching and leaching (provided both dyes do so and at the same rate), and – in the case of fiber sensors – effects of fiber bending. However, working at two wavelengths also makes the optical configurations more complicated, and at least two optical filters are required. If the OSP and the reference dye photobleach (or even leach) at a different rate, the method is hardly applicable.
Two-wavelength ratiometric oxygen sensing572 is rather simple. Ideally, the two luminophores have the same excitation wavelength but different emission bands. Lübbers et al.573 used pyrenebutyric acid as the OSP and an inert fluorescent that gives a constant reference signal. Another way42 is to relate more longwave fluorescence to the intensity of blue scattered light originating from the excitation light source. This can compensate for lamp fluctuations and detector sensitivity drifts. Zhang and Seitz66 determined the ratio of reflected light (at 405 and 435 nm) of an oxygen-sensitive layer composed of immobilized hemoglobin plus deoxyhemoglobin. One may also use indicators with two luminescence bands, one of which is quenched by oxygen while the other remains unaffected.369 The probe 4-bromo-1-naphthoic acid in a γ-cyclodextrin cavity was linked to cellulose where it shows oxygen-dependent phosphorescence and oxygen-independent fluorescence. The ratio of luminescence intensities can be measured at two wavelengths and yields a parameter for oxygen pressure. It was also suggested to use the ratio of the intensities of monomer or excimer bands of pyrenes (which are differently affected by oxygen).
Referencing is almost mandatory in the case of intensity-based oxygen nanosensors. Kopelman et al.174 immobilized Ru(dpp) and the reference dye Oregon Green 488 in PEG-stabilized silica nanoparticles. The fluorescence of Oregon Green is not affected by oxygen. This makes it an ideal reference dye with good spectral match. The resulting ratiometric nanosensors have excellent reversibility and enable real-time monitoring of intracellular oxygen at pH values above 6. The group also reported490 on the use of the oxygen-insensitive dye octaethylporphyrin (OEP) and the probe PtOEPK in poly(decyl methacrylate) nanoparticles to obtain plastic oxygen nanosensors. Because of the hydrophobic nature of both dyes, they do not leach and are well protected from non-specific binding to proteins inside the nanoparticles.
OEP is not a good reference dye because it suffers from photobleaching. A combination of PtOEPK and a bodipy dye is better and was used in a ratiometric sub-μm fiber optic oxygen sensor.574 Ormosil-based nanoparticles have also been reported328 containing dye pairs (PtOEP and 3,3′-dioctadecyloxacarbocyanine, or PtOEPK and OEP). Both exhibit high sensitivity to oxygen (I0/IO2 satur > 30) and linear SVPs over the entire range of water-dissolved oxygen (0–43 ppm). However, the reference dyes suffer from photobleaching. The same group later491 encapsulated the NIR emitting probe PtTCPTBP and the reference dyes Alexa 647 or Hilyte 680 in hydrophilic nontoxic polyacrylamide nanoparticles. The nanoparticles were further functionalized with peptides for targeted monitoring of oxygen in cancer cells. The use of a NIR probe has the advantage of deeper penetration of light into tissue and generating less autofluorescence. McNeill et al.575 doped PtOEP into a luminescent conjugated polymer and prepared luminescent polymer nanoparticles with a diameter of around 25 nm. The blue emission of the conjugated polymers is not quenched by oxygen and serves as a reference, whilst the red phosphorescence of PtOEP is strongly quenched by oxygen.
Gouterman and Khalil et al.274,275 reported on a referenced pressure-sensitive paint which, in fact, is an oxygen-sensitive paint. It is making use of the longwave emitting oxygen probe PtTFPL and a reference dye that has a short lifetime and a fluorescence peaking at 650 nm. The paint was used for ratiometric measurement of oxygen partial pressure in wind tunnels. Tian et al.304 copolymerized the probe PtTFPP containing a polymerizable group with a luminescent monomer to obtain oxygen sensing polymer films possessing two emission bands. The blue luminescence of the conjugated polymer is insensitive to oxygen and can be used as reference signal. Jorge et al.576 employed luminescent core–shell quantum dots (QDs) as reference luminophores and Ru(bpy) as the OSP. The QDs have a wider excitation range and narrower emission spectra. This enables the reference dye and the OSP to be excited with a single excitation source. The narrow emission favors spectral separation, and the color of the emission of the QDs can be tuned via their size.
Other reference dyes include the silica complex of OEP which was used in PS beads,487 certain stilbenes,488 and a yellow perylene dye in 20 nm PS nanoparticles.486 The perylene dyes are probably the most stable reference dyes. Even smaller particles (only ∼8 nm in diameter) were obtained577 by partially exchanging the capping ligand of QDs by a pyrenylimidazole. The resulting nanosensors display emissions from both the QDs and the pyrene derivative. The red emission of the former is insensitive to oxygen and can act as a reference signal, while the blue emission of the pyrene derivative is sensitive to oxygen. However, the hydrophobic nature of capping agents renders these NPs poorly soluble in water, and this may compromise applications in biosystems. McShane et al.500 covalently linked PtTCPP and carboxy-modified QDs onto amino-modified silica particles with a diameter of around 10 μm. Again, the NIR emission of the QDs is not quenched by oxygen, but the luminescence of PtTCPP is, and this was used for ratiometric sensing. Unfortunately, almost all authors working with QD-based sensors for oxygen do report on conceivable interferences by other species also known to quench the fluorescence of QDs.
A ratiometric fiber optic oxygen sensor578 reported on that is based on a sol–gel matrix doped with two fluorophores. The Pt(II) or Pd(II) complexes of a pentafluorophenylporphyrine acted as the red-emitting OSPs, while the blue-emitting laser dye 7-amino-4-trifluoromethylcoumarin acted as the reference dye. This ratiometric sensor has good sensitivity, a response time of ∼3 s, and is fully reversible. Effects of spurious fluctuations in the intensity of the excitation source and optical transmission properties of the optic fiber are eliminated. Table 14 summarizes and assesses the common spectroscopic methods for readout in sensors for oxygen based on the use of optical probes.
| Method | Advantages | Disadvantages | Compensation of interferences bya | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Optical components (e.g. filters, optical gratings) | Instrumental drift (e.g. light source, detector) | Optical misalignment | Background fluorescence from samples | Light scatted by sensor materials | Intrinsic color of samples | Dye leaching and bleaching | Inhomogeneous dye loading | Temperature | |||
| a (++) Efficient compensation; (+) partial compensation; (−) no compensation; (0) no effect. | |||||||||||
| Absorption; reflectance | Simple instrumentation; low-cost; portable | Moderate sensitivity; many interferents (such as ambient and background light) | – | – | – | – | – | – | – | – | – |
| Luminescence intensity | Sensitive; low-cost; portable devices; enables imaging | Interfered by many parameters | – | – | – | – | – | – | – | – | – |
| Luminescence decay time | Sensitive; precise; self-referenced; enables imaging; 1-point calibration | Relatively expensive; works best in the case of OSPs with long lifetime | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | – |
| Anisotropy | Applicable to solution with high viscosity; 1-point calibration | Relatively poor sensitivity and precision; requires OSPs with high anisotropy | + | ++ | + | – | – | ++ | ++ | ++ | – |
| Two-wavelength referencing | Good precision | Two dyes needed; photodecomposition and leaching of the dyes may be different | – | + | ++ | – | + | – | – | – | + |
| Dual lifetime referencing (DLR) | Sensitive; precise; enables imaging; simple calibration | Two dyes needed with different excited-state lifetimes; more complex | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | + |
| Fluorescence resonance energy transfer | Low-cost instrumentation | Two dyes (donor and acceptor dye) needed that have to be in close proximity; and to spectrally overlap | – | + | – | – | + | – | – | 0 | – |
| Luminescent colorimetry | Low cost; visual inspection also possible; good sensitivity and selectivity; | Two dyes needed; semiquantitative in the case of visual read-out; influenced by ambient light | 0 | 0 | 0 | – | – | – | – | – | – |
| Photographing (RGB) | Simple; low-cost; easy of imaging; works best of two dyes are employed | Probes emissions have to match the RGB channels of digital cameras | 0 | + | ++ | – | + | – | – | – | – |
Multiple emitters (see Section 6.4) can also be used to construct self-referenced ratiometric oxygen sensors.370,579 These intrinsic ratiometric OSPs normally possess multiple emission bands, and each band has different oxygen response. Sensors based on such OSPs not only compensate for effects of common interferences in two-wavelength referencing, but also for photobleaching. Another ratiometric probe was reported580 that may also be used in a sensor, but this has not been demonstrated so far. Schemes for the design of fluorescent ratiometric nanosensors for oxygen (and other species) have been reviewed.581
A phosphoroscope with no moving parts was described that enables detection of both fluorescence and phosphorescence which is useful in ratiometric sensing.582 The total luminescence, the long-lived phosphorescence, and the short-lived fluorescence can be determined in a single scan. A 50% duty cycle excitation from a diode laser was used to excite the sample. Phosphorescence is extracted from the off period of the digitized waveform, the total emission from the full cycle, and fluorescence from the on-period corrected for the phosphorescence contribution. The performance of the system was demonstrated by acquiring the RTP spectra of (a) organic dyes in boric acid glasses, (b) a multi-emissive and oxygen-quenchable boron–polymer dye, and (c) a europium chelate.
A patent583 describes a smart referenced sensor element that is schematically shown in Fig. 27. In essence it consists of an LED light source (14) whose emission is reflected (in elements 29 and 31) into two minute prism-shaped waveguides (22 and 27), one being covered with analyte-sensitive chemistry (30), the other not. Attenuated light and fluorescence are guided to another pair of reflectors (28 and 32) and directed to photodiode detectors (16 and 18). This system is simple, widely self-referenced, and can be mass manufactured at low costs from semiconductor and plastic (plexiglass) components. Several other versions are also described.
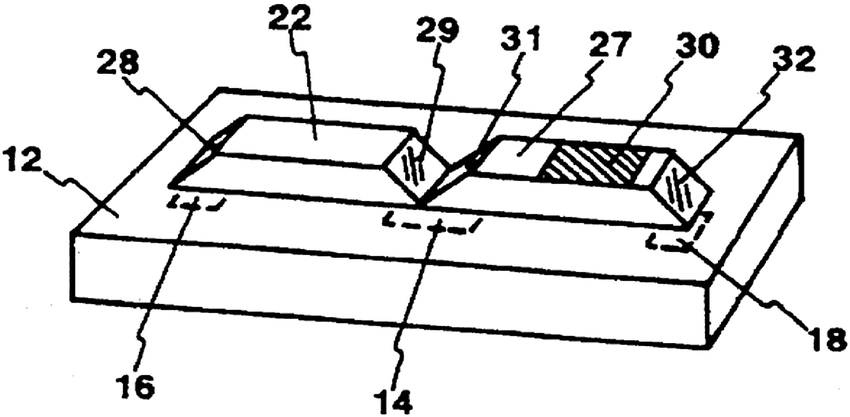 | ||
| Fig. 27 A smart waveguide sensor element for referenced indicator-based chemical sensing. Reprinted from ref. 583 light from an LED (14) is directed by reflective end faces 29 and 31 into the two arms (22, 27; one acting as the sensor arm, the other as a reference) and then is reflected again (28, 32) to hit the detector (18) and reference detector (16), respectively. The sensing arm also contains a sensor layer (30) that affects the intensity of light passing it (27). | ||
9.10. Specific aspects of luminescent imaging of oxygen
This section covers methods for imaging oxygen using either planar sensor films or nanoparticles. The art of fluorescence imaging using chemical sensors (including oxygen sensors) has been reviewed.584 The use of molecular probes, though often used, has limitations, the most serious one being the complex interaction of such probes with membranes, cell compartments and proteins. This results in complex quenching constants and uneven distribution if not aggregation of molecular probes. Nanosized sensor particles do not suffer from such limitations in a first approximation.Peterson and Fitzgerald162 probably were the first to show that quenching by oxygen can be used to visualize the flow of gas blown onto a surface. The OSP Fluorescent Yellow was absorbed on silica gel to form a planar oxygen sensor layer. Streams of oxygen and nitrogen were directed onto the surface of the sensor to result in patterns of different brightness. This approach paved the way to quantitatively imaging oxygen and – indirectly – air pressure, but is sensitive to humidity. Considerably improved results were obtained when porous polystyrene particles were applied as a support.261 Water vapor and N2O do not interfere. The response time is <5 ms for 90% of the total signal change, which is distinctly quicker than many other oxygen sensors. Following this initial work, numerous other methods and materials have been reported thereafter. This will be discussed in the following.
Imaging of phosphorescence was introduced264 in 1988 as a novel method for imaging its distribution in perfused tissue. Isolated rat liver was perfused through the portal vein with media containing palladium coproporphyrin whose phosphorescence was used to image the oxygenation of liver at various perfusion rates. Because oxygen is a powerful quenching agent for phosphors, the transition from well-perfused liver to anoxia (no flow of oxygen) resulted in a large increase of phosphorescence intensity. During stepwise restoration of the flow of oxygen, the images showed marked heterogeneous patterns of tissue reoxygenation, which indicated that there were regional inequalities in oxygen delivery. While not using sensors along with lifetime imaging (that would have warranted more precise data that are not affected by probe binding by proteins and by uneven spatial distribution), this method paved the way for sensing oxygen in medicine. Hartmann and Ziegler585 reported on an all solid-state method (LED light sources; photodiode detectors) for imaging of oxygen using a sensor foil containing silica particles dyed with the Ru(phen) complex and dispersed in a layer of silicone. The spatial resolution was 1 mm, and the oxygen resolution was between 0.4 and 2.5 Torr depending on the actual pO2.
Several instrumental approached are common at present. The first is based on the use of CCD cameras and works in the intensity mode. The second is in the lifetime mode if the CCD device has an adequately fast response. Both methods also work in combination with microscopy. Advanced instrumentation is commercially available. A substantial collection of protocols on the use of hand-held optical imagers is available on the internet.586
A pixel-by-pixel calibration method was developed to overcome these problems and achieve quantitative imaging.587 Each pixel on the CCD or CMOS chip was treated as an individual oxygen sensor and calibrated in situ. The method enables oxygen imaging with a spatial resolution of 3.0 μm and eliminates the effects of uneven probe distribution and light source distribution. It is mandatory, though, that the sensing film is firmly fixed to prevent major errors caused by pixel movement. The influence of excitation light distribution can be prevented by using cross-aligned polarizers, which led to the design of a compact oxygen sensor without using any optical filters.536 A more simple approach consists of the use of a reference dye to correct for such interferences.275
Color CCD and CMOS cameras even better serve the purpose of sensing and imaging oxygen via luminescent sensor layers588 A read-out scheme by means of digital color camera and the application of a Bayer filter (pixels or layers that are sensitive to different ranges of wavelengths, typically red, green and blue (RGB) is schematically shown in Fig. 28. Such detectors may be considered as very simple (3-color) spectrophotometers that can be used to separate multi-color sensor signals or referenced signals, the only stipulation being the availability of indicators that emit at different wavelengths.
 | ||
| Fig. 28 Scheme of color CCD equipped with red-green-blue optical filters. | ||
RGB-based detectors have been often explored for imaging of oxygen.315,488,589–591 If the chosen OSP and the reference dye match the spectral characteristics of RGB filters, the luminescence intensities of the OSP and the reference dye can be recorded in the RGB channels of color pictures and easily referenced using routine software. In a typical example, a ratiometric intensity-based imaging scheme was reported315 where a red-emitting probe and a green fluorescent reference dye (whose luminescences match the red and green optical filters on the color CCD in a digital camera, respectively) were incorporated into a hydrogel matrix. The method enables photographing of the oxygen distribution by taking a picture using a digital camera (Fig. 29). It was also used to image oxygen-deficiency by taking pictures of a sensory cloth.488 By introducing a blue-emitting reference dye, adverse effects of inhomogeneous illumination and non-uniform film thickness can be referenced out, but it is mandatory that the concentrations of probe and reference dye are the same at each data point/pixel.
 | ||
| Fig. 29 Imaging of oxygen distribution using planar sensing films and photographic (RGB) readout. The sensing film was first put in nitrogen environment. Exposure to a stream of oxygen causes the luminescence intensity change of the red dye and imaged using the photographic technique. (A) Regular photo (14-bit color depth per channel) obtained for colorimetric readout; (B) pseudo color image of the G/R ratio calculated with data of the green channel and the red channel. (Reprinted from ref. 315 with permission from Wiley-VCH). | ||
A similar approach has been reported by Park et al. In the first,591 a light panel screen with light-emitting diode backlight was used as a light source to excite a sensor film containing PtOEP as the OSP in an ethyl cellulose host polymer. A green LED light uniformly displayed from the light panel excited the sensor film to exhibit two-dimensional luminescence distribution corresponding to the pressure of gaseous oxygen. A color camera was used as a photodetector for registering oxygen images and analyzing the oxygen contents quantitatively. In the second,592 commercial RedEye oxygen sensor patches were applied. Two methods of color intensity analysis are investigated and compared. The first is to analyze the total Red-Green-Blue (RGB) color intensity of the original color image. The second involves extracting the red color element to enhance the sensitivity of oxygen measurement. Results showed the linearity and sensitivity of the red intensity analysis to be improved over those of spectrometric measurement and total color intensity analysis. However, the method do not use a reference dye for ratiometric readout, and the distribution of excitation light and dye, ambient light and background luminescence may compromise the precision of imaging.
Intensity-based imaging of oxygen with a display screen and a color camera was demonstrated590 and can give both quantitative and qualitative results. A liquid crystal display (LCD) screen was employed as a light source for photoexcitation, and a color CCD camera as a photodetector for measurement of emission. Small fluidic channels and oxygen sensor films were integrated. A ruthenium-based OSP was used in the sensor films that can be photoexcited with blue light emitted by the LCD screen. The color camera can map the distribution of red fluorescence emission. This combination of ubiquitous LCD and color camera enables a potential capability of uniform illumination and distribution recording over a large area with variable wavelength ranges. Possible niche application areas include multiple-analyte and high throughput analysis over a large-area fluidic network.
The photographic approach was coupled to microscopy to image oxygen distribution in microfluidic devices with integrated optical sensors.589 The same principle led to the development of hand-held compact imaging devices, which are beneficial to fast oxygen imaging in vivo (Fig. 30).593–595 Imaging was shown to possess the same precision and accuracy as the ratiometric intensity-based measurement using a fluorometer.596 It was later applied to ratiometric imaging of the oxygenation of mouse skin.597
 | ||
| Fig. 30 A commercial hand-held and portable oxygen imager based on the RGB technique. The imager is linked to a computer (via a USB interface) where calibration functions are used to convert the data into a pseudo-color picture. From Presens GmbH (http://www.presens.de/). It is particularly useful for ratiometric (2-dye) imaging. | ||
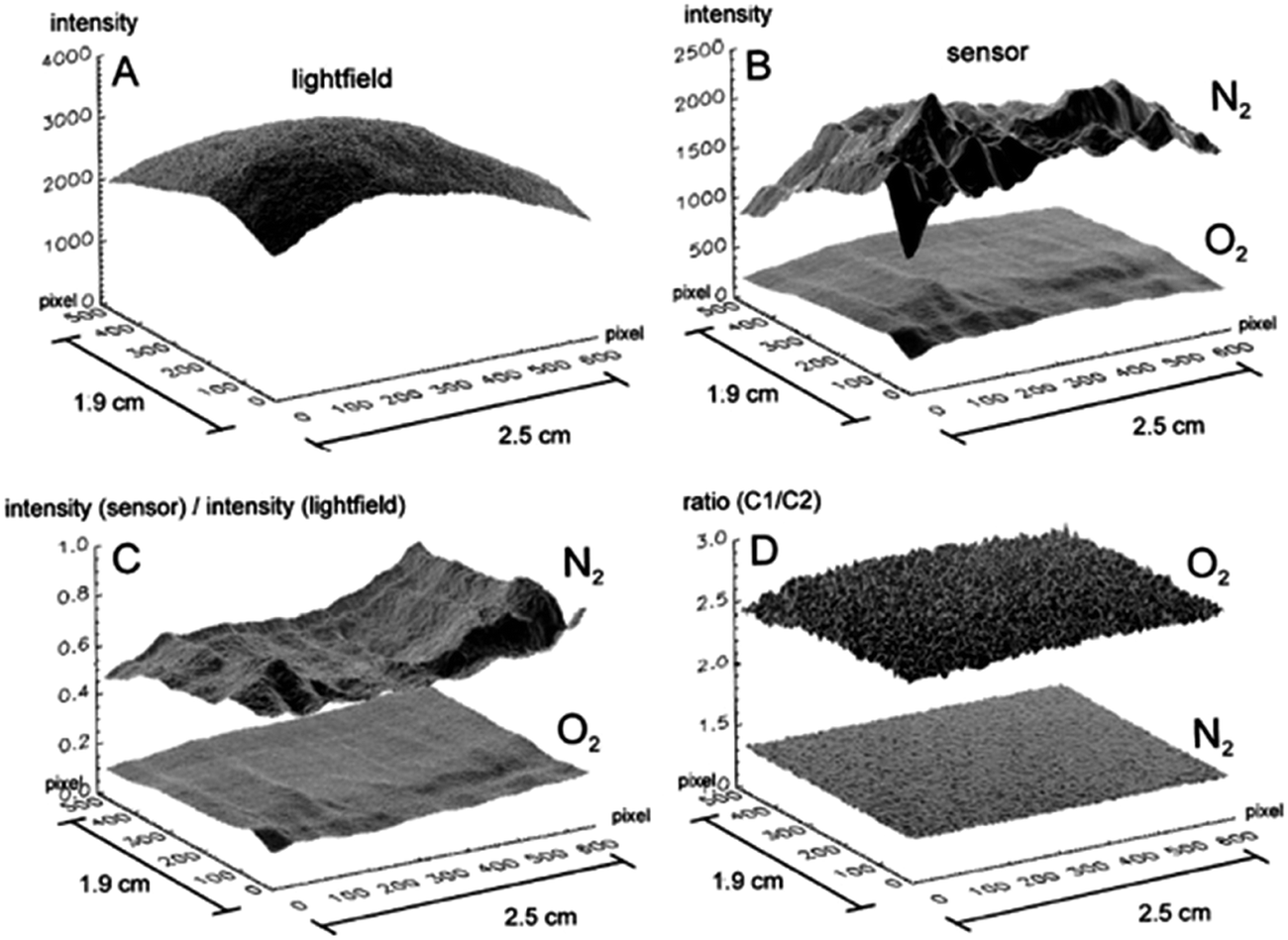 | ||
| Fig. 31 In rapid lifetime-based imaging, effects caused by inhomogeneous distribution of excitation light and of probe concentration are suppressed. (A) Luminescence intensity of an oxygen sensor foil which more or less records the inhomogeneity of the light field on illumination with a point source. (B) Uncorrected intensity response of an oxygen sensor in the presence (bottom) and absence (top) of oxygen. (C) Intensity response of the oxygen sensor corrected for the light field using the data of graph A. (D) Time-resolved (“gated”) images of the same sensor film showing even signal distribution both under nitrogen and oxygen. (Reprinted from ref. 441 with permission from the Society for Applied Spectroscopy). | ||
Oxygen imaging based on the RLD method can also be performed with time-gated CCD devices. A simple fluorescence lifetime imaging system using a gated microchannel plate image intensifier coupled to a CCD camera has been developed as early as 1991.599 Nanosecond-level time-resolved fluorescence images of a sample under pulsed light excitation can be detected directly. Coupled to the RLD method for multi-gate detection, fluorescence lifetime imaging can be easily performed. Holst et al.598 applied the RLD method to luminescence lifetime imaging using a fast gateable CCD camera. Probes with lifetimes from 1 μs to 1 s may be applied in this method. Other features include good contrast enhancement and suppression of background. A hand-held time-gated imaging device for oxygen imaging based on the RLD method (Fig. 32) was reported later.441 The method takes less than 1 min to acquire oxygen images. A modular luminescence lifetime imager (MOLLI) was presented600 that enables both luminescence intensity images and luminescence lifetime images to be acquired. By making use of transparent sensor layers (i.e., films not containing scattering particles or binders), the method was applied to study (a) coral sand sediment samples (a macrolens application with an resolution of approximately 50 μm per pixel); (b) lichen with cyanobacteria as symbionts (an endoscope application with a resolution of approximately 15–62 μm per pixel); and (c) foraminifer with diatoms as symbionts (a microscope application with a resolution of approximately 4 μm per pixel).
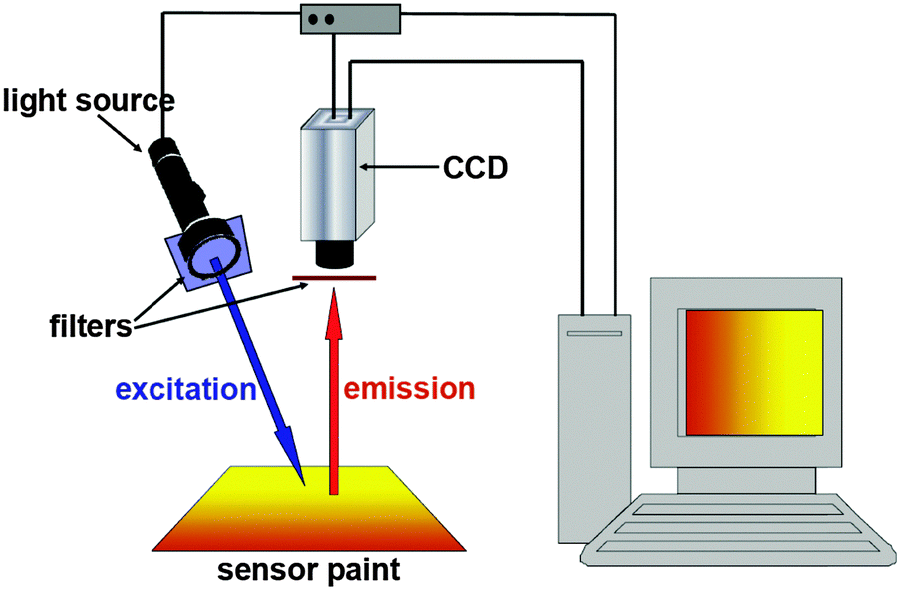 | ||
| Fig. 32 Schematic of a CCD array for imaging sensor films (“paints”) deposited on the subject of interest. | ||
Frequency domain based lifetime imaging can be performed using phase-sensitive CCD cameras. Lakowicz et al.601 reported the creation of two-dimensional fluorescence lifetime images based on a sinusoidally modulated image intensifier that is operated as a radio-frequency phase-sensitive CCD camera. By combining the image intensifier with a CCD camera and applying digital image processing, lifetime-selective signal suppression was realized even for fluorophores with comparable lifetimes, such as rhodamine 6G (4 ns) and rhodamine B (1.5 ns). Because the lifetimes of many dyes are sensitive to the chemical environments of the fluorophore, FLIM can reveal the local chemical composition and properties of the molecular environment that surrounds the fluorophore. Thus, the system has the potential use for oxygen imaging. Lo and coworkers602 later built a modulation system for the phase-resolved two-dimensional fluorescence phase imaging of oxygen using a sensing film composed of PtTFPP-doped sol–gel. Two-dimensional oxygen distribution imaging was obtained using the modulation system which is suitable to sense gaseous oxygen at levels between 0 and 20%.
10. Selectivity, sensitivity, and effects of temperature
10.1. Selectivity and interferences
Molecular triplet oxygen (3Σ) is not the only powerful quencher of the luminescence of luminophores. Depending on the kind of emitter, numerous other species can also act as quenchers, albeit to a largely different extent. Examples include elemental chlorine, bromine and iodine, SO2 and – less so – SO3, all NOx species, brominated and iodinated organic species (including the inhalation narcotic halothane), all nitro compounds, and many olefines and quinones. These species are electrically neutral and can hardly be excluded from exerting a quenching effect because they (slowly) penetrate polymeric matrices hosting OSPs.On the other hand, heavy metal ions, bromide and iodide are notorious ionic quenchers, whilst sulfite and thiocyanate have not been well studied but are likely, though weak, interferents. Sulfide can “quench” by undergoing a chemical addition reaction with the fluorophore.603 The effect of ionic species can, however, be eliminated by incorporating the OSPs in an ion-impermeable matrix material (such as silicone, polystyrene or ethyl cellulose) as described in Section 7.1. If hydrophilic matrix materials have to be employed for any reason, ionic quenchers will interfere. Many of the above quenchers do not occur in biological matrices, however.
No (or negligible) interferences are to be expected from gases such noble gases, hydrogen, nitrogen, CO2 (with limitations as it is a weakly acidic gas), alkanes (gaseous and fluidic), vapors of alcohols (with limitations), ethers, and of chlorinated solvents. Some organic solvents can destroy sensor films, however. All alkali and earth alkaline ions remain inert as do the anions sulfate, nitrate, nitrite, bicarbonate, carbonate, chlorate, acetate and the like. All these species do not enter hydrophobic polymer membranes anyway. Similarly, solutions of main group element ions and all ionic surfactants remain fairly inert. Bioorganic species such as saccharides, lipids and proteins do not act as efficient quenchers. Their quenching constants (KSV) are below 1 M−1. However, tryptophan in proteins can act as an energy acceptor in FRET system if very shortwave absorbing probes are used which is not recommended anyway in optical sensor technology.
Humidity, often referred to as “relative humidity” (RH), interferes in sensors based on surface-adsorbed probes, in particular conventional silica which strongly absorbs water, and with some sol–gels as outlined in Section 7.3. Practically all suffer from the presence of water vapor in gaseous samples because RH is a notorious quencher of luminescence. This is, of course, not the case if (water-)dissolved oxygen is to be sensed, but in such cases the luminescence intensity of the sensors can be weak as a result of quenching by adsorbed water. The effect of RH on the response characteristics of luminescent PtOEP-based oxygen sensors has been studied in some detail.403 If hydrophilic host polymers are replaced by more hydrophobic hosts such as ethyl cellulose, the effects is strongly reduced but not eliminated. Sensors based on host materials such as silicones, polystyrene, or poly(vinyl chloride) are not severely affected. Plasticizers also exert a beneficial effect in reducing the effects of RH. Lifetime data suggested that the effect of RH is due to its effect on oxygen permeability. The use of a silver-octadecanethiol coating along with a reference gas correction algorithm can minimize the effect of RH in fluorescent sensors for oxygen.604
10.2. Sensitivity, precision, and limits of detection
Sensitivity is defined as the slope of a calibration plot, in the simplest form as the signal change (ΔS) divided by the change in concentration (ΔC) of oxygen. However, many authors refer to sensitivity as the lowest detectable concentration of oxygen (or any other analyte). This parameter is defined by IUPAC, however, by the limits of detection (LOD) and of quantification (LOQ). The LOD usually is expressed as the concentration where the analytical signal is 3 times that of the noise, in other words at a signal-to-noise ratio (SNR) of 3.The analytical range of a luminescence based sensor for oxygen is governed by the shape of the respective quenching plot and the Stern–Volmer constant (which, of course, is the slope of the plot). Most sensors have been adjusted (by proper choice of OSP and polymer) to perform best in the range between 0 to 300 Torr. Phosphorescence-based sensors are much more sensitive than fluorescent sensors by principle (not the least due to much longer decay times), with typical limits of detection as low as 0.004 Torr. Higher quenching constants result in (a) high accuracy at low levels (because of a larger relative signal change per Torr oxygen), and (b) in dynamic ranges not exceeding ∼200 Torr after which signal changes become vanishingly small. The proper adjustment of a quenching constant always is a compromise between analytical range and accuracy. A bi-label oxygen sensor was reported605 where two luminophores were incorporated into a polymeric matrix with the aim to enlarge the analytical range of sensors. The luminophores Pt(II)-TPP, Pd(II)-TFPP and Ru(dpp) octylsulfate in plasticized poly(vinyl chloride) or polysulfone behave as if they were independent. This lifetime-based approach also resulted in improved precision.
The sensitivity (“slope”) of all such sensors is highest at the initial part of the curve, i.e., at low oxygen tensions where ΔS/Δ[O2] is maximal and almost linear. At high pO2, on the other hand, the slope is small and sensitivity poor. One might therefore assume that luminescent based sensors are most precise in this range. This is largely true. However, even minute experimental errors in the pO2 at low pO2 result in large errors in the calibration step.
The precision and accuracy of any optical sensor for oxygen are governed by the uncertainties in the determination of F0 (or τ0) at zero oxygen, of KSV, and of F or τ at a given pO2. As a rule of thumb, the first two govern the precision, and F(τ) values govern accuracy. Precise determination of F or τ obviously is essential for obtaining precise calibration plots and KSV values as needed to establish the Stern–Volmer relationship. F0(τ0) are typically obtained with oxygen-free gases, or with solutions bubbled (“tonometered”) with nitrogen or argon, or containing sodium dithionite which completely removes oxygen by reduction. If Stern–Volmer plots are linear, the quenching plot may be established with data obtained with two solutions (or gases) containing lower and higher concentration of oxygen. A final source of error results from contributions of straylight to the fluorescence signal which gives an apparent signal F′ composed of F (the true intensity at given pO2) and the contribution by straylight (Ix). A fairly simple method has been worked out to precisely determine KSV and F0 from three sets of intensity data.142 Frequency-domain lifetime fluorometry is also affected by straylight, but time-domain lifetime fluorometry is not.
The detection limits of a sensor are governed by both the initial slope of the quenching curve and instrumental resolution. Assuming a ±0.1% uncertainty in light intensity measurement (which is the optimum in a well-thermostatted device), the detection limit is 0.003/KSV, where KSV is expressed as Torr−1 and the signal-to-noise ratio is 3. KSV values ranging from 0.001 to 0.1 Torr−1 have been reported for fluorosensors, which results in detection limits from 3 to 0.03 Torr. Phosphorescence based sensors, in contrast, are much more sensitive for the reasons outlined before.
The imprecision of sensors resulting from the signal drift in intensity-based detection was studied more recently606 for the case of a polysulfone-based sensor membrane incorporating PtTFPP as the luminescent probe. Photochemical, thermal and oxidative degradation of luminophore and host polymer were examined. The respective contributions were quantified. Oxidative degradation played no role, but thermal and photochemical degradation of the luminophore cause a drift that affects precision independent of whether intensity is measured, or lifetime in the frequency domain or time domain. The design of an intensity-based sensor with high accuracy and precision is suggested. In an extension of this work,607 an iterative algorithm for drift correction and its experimental verification in intensity-based detection was presented. The signal of a reference oxygen mixture (on air) is needed. The “classical” Stern–Volmer approach is not useful here because it produced a large positive systematic error that increases over time and with increasing fractions of oxygen. The algorithm resulted in accuracy as good as that of phase shift-based commercial sensors.
10.3. Effects of temperature and other parameters
It may be stated that any sensor in this world measures temperature (T) and – ideally – another species too. Unless T itself is to be sensed, its effects are not truly welcome. Remarkably, there are relatively few reports on the T dependence of sensors for oxygen. This may be due to the fact that T exerts rather complex effects on solubility, diffusion and permeability of gases in polymers, on fluorescence QYs and decay times and quenching constants. As a result, mathematical modeling of its effects is complicated. Temperature is probably the single biggest source of error in optical sensors for oxygen. It was stated in one case576 that ratiometric (2-wavelength) sensing can widely reduce effects of T, but this probably was an exception. A review on optical probes and sensors for T is available.608The determination of T coefficients (“tempcos”) of sensors is a complex task. T is known to affect the luminescence QY of the dye (usually with a negative tempco), (b) the quenching constant(s) (with a positive tempco), (c) the solubility of oxygen in the membrane (negative tempco), (d) the diffusion of oxygen into the membrane (positive tempco), and (e), in the case of blood measurement, the oxygen/hemoglobin binding curve (negative tempco). Furthermore, increasing temperatures usually shorten fluorescence lifetimes (τ) and facilitates S1 → T1 and T1 → S1 transitions. All these factors contribute to the “apparent tempco” of a sensor. Most of the reports on the tempco of sensors refer to one of these effects only. Throughout, increased temperature shortens the response time of sensors. Typically, the 37 °C response time is around 2/3 of that of the 22 °C value.
In a study609 on the tempco of the quenching constant of the OSP benzo(g,h,i)perylene in silicone rubber over the 25 to 60 °C range, it was found that KSV increases from 0.0112 to 0.0140 Torr−1, and the slope of a plot of temperature vs. KSV is almost the same as the slope of a plot of oxygen permeability vs. temperature. Peterson et al.42 on the other hand, observed a 0.6% decrease in pO2 indication per 1 °C increase with a dye adsorbed on resin beads. Under nitrogen, the signal was found to be temperature-independent. Bacon and Demas49 reported the lifetime of a ruthenium probe to decrease from 5.8 μs at 0 °C to 3.3 μs at 60 °C. The fluorescence of surface-adsorbed Ru(bpy) drops by almost −30% on going from 25 to 37 °C, but the quenching constant remains practically unchanged.
For 9,10-diphenylanthracene in poly(dimethyl siloxane), a decrease of ca. −37% in the quenching constant was reported123,392 on increasing temperature from 2 to 48 °C. This is a very large effect that can severely compromise the precision of such optical sensors. At constant oxygen level, a temperature change of 1 °C, therefore introduces an error of 4% in oxygen determination when assuming a T-independent fluorescence QY. A study on the effects of viscosity on the quenching of the fluorescence of diphenylanthracene by oxygen in poly(dimethylsiloxane) solvent revealed123 a distinct viscosity dependence of fluorescence intensity, but the slope of the SVP is not a function of viscosity, as it was constant for fluids and fully polymerized silicone. It shall also be kept in mind that bimolecular quenching is a complex process that is also affected by parameters such as solvent cage effects in addition to viscosity and temperature.610
The fluorescence of silicone membranes doped with pyrenebutyric acid displays an unexpectedly small effect of T (a 20% signal loss only on changing from 20 to 200 °C in air).127 The overall quenching constant, in contrast, varies strongly with T. The fiber optic oxygen probe described by Miller et al.611 using decacyclene as an OSP displays an error of ±7 Torr at 100 Torr oxygen, when the temperature used in calculations is 36 rather than 37 °C.
Erythrosine B contained in a sol–gel matrix displays both delayed fluorescence and strong phosphorescence.157 The two emissions are oppositely dependent on temperature, and this unique behavior was applied for temperature self-compensation within the temperature range from −50 to 200 °C. The effect of T on the luminescence of the oxygen probe tetraphenylporphyrin (without a central metal ion) is also rather high.261
Most metal–ligand and metal organic complexes have a luminescence that strongly depends on T. The sensitivity of a cyclometalated iridium complex to T is −1.1 and −1.4% per °C at vacuum and 1 bar atmospheric pressure, respectively.233 Such metal complexes are well suited for use in T-sensitive paints (TSPs). For other data see the comments in the tables on oxygen-sensitive probes (Section 6.2.4) and the review by Wang et al.608 Given its adverse effect, attempts have been made to measure T simultaneously with oxygen, in particular in the context of imaging oxygen over large areas where T can vary locally. Multiple sensing was discussed in Section 9.8.
Optical probes stable enough to enable sensing of oxygen at high-temperatures are rare. Ghosh et al.281,282 report on a Mo6Cl12 cluster that has a broad absorption at 300–400 nm, an emission between 600 and 900 nm, a long cluster lifetime (>100 μs), and a large Stokes shift (>300 nm). Its red luminescence is reversible quenched by oxygen. The cluster can withstand repeated cycling and showed no signs of decomposition to temperatures higher than 600 °C. This makes it suitable for monitoring oxygen in power plants and exhaust gases. It can sense oxygen with high resolution (0.1% absolute oxygen concentration change) both in the gas phase and in aqueous solution. Remillard et al.387 noticed that the fluorescence of Cu-ZSM-5 zeolites reversibly changes on cycling between oxidative and reduced atmospheres, and this was exploited to sense oxygen at temperatures as high as 500 °C. However, the material has to be regenerated by exposing it to reducing gases, and this limits practical usage. For the effects of T on the diffusion, permeability and solubility of oxygen in polymers, see Section 7.1 on “Organic Polymers”.
The feasibility of sensing oxygen between 25 and 220 °C has been investigated127 using a silicone membrane doped with pyrenebutyric acid and attached to the end of a quartz light guide that was exposed to gases of varying temperatures. The fluorescent signal decreases almost linearly on going from 20 to 200 °C under either nitrogen, 10% oxygen, or 100% oxygen, with a total signal change of −18%). However, after being corrected for straylight (which contributes to more than 50% of the signal), the behavior is more complex. Chu et al.612 have introduced a method for temperature compensation in the range of 25 and 70 °C by making use of two PtTFPP-based oxygen sensors, and an algorithm was developed that provides an effective means for improving the accuracy of (fiber optic) sensor without an additional temperature sensor.
10.4. Low-level oxygen sensors
The interest in sensors for detecting extremely low levels of oxygen is so high that it deserves an extra section. Low-oxygen situations are encountered in the chemical industry, in the production of gases, in space research, and numerous other and often unexpected fields. Extremely low levels of oxygen in a nitrogen carrier gas were determined by Kautsky back in 1931 by exploiting the quenching of the phosphorescence of surface-adsorbed trypaflavin by traces of oxygen.11,32 Eventually, this led to the discovery of the Kautsky effect, i.e. the delay in the production of oxygen by plants following illumination with light. Low levels of oxygen in water (0.06 to 1 μg L−1) typically are determined via quenching of phosphorescence of organic dyes adsorbed on silica type of supports. Analytical ranges between 0.4 and 400 μg L−1 can be accomplished with other kinds of supports as shown by the Zakharov group.613 The lowest detection limits reported by this group for oxygen in water is 0.5 ng L−1 of dissolved oxygen.The use of long lifetime metalloporphyrins (such as palladium porphyrin with a lifetime of ∼1 ms) and high gas-permeable matrices (such as fluorinated polymers) will substantially improve the detection limits for gaseous and dissolved oxygen. Many sensors also work in harsh organic solvents where the survival of sensor material becomes critical because polymers may become dissolved or deteriorated by strong acids or bases. Again, the selection of a proper sensing material is critical for sensor design and still represents a challenge. Table 15 summarizes the current state.
| Probe | Host polymera | LOD | Ref. |
|---|---|---|---|
| a TFEM: poly(trifluoroethyl methacrylate); PFS: polypentafluorostyrene; 8F-PEKEK: poly(aryl ether ketone); 6FBA: 2-2-bis(4-hydroxyphenyl)-1,1,1,3,3,3-hexafluoropropane; HF: 9,9′-bis(4-hydroxyphenyl)fluorine; octyl-triEOS: n-octyltriethoxysilane-based sol–gel; TEOS: tetraethoxylsilane-based sol–gel; TFP-triMOS: 3,3,3-trifluoropropyltrimethoxysliane-based sol–gel. | |||
| Acridine orange | Adsorbed on silochrome S-120, afterglow | 0.04 | 613 |
| Acriflavine | Adsorbed on silica gel | 0.25 | 151 |
| Acriflavine | Silanized silica gel | 0.35 ppbv | 15 |
| Erythrosine B | Sol–gel silica | 14 | 161 |
| Organic dyes | Trypaflavin, chlorophyll, porphyrins etc. absorbed on silica gel or aluminium oxide gel; oxygen detected in ppm concentrations in a flow of nitrogen | — | 11, 32 |
| Organic dyes | Various organic phosphorescent dyes absorbed on silica gel; can detect the formation of 50 pL (!) of oxygen per minute | — | 33 |
| 12C70 | Polystyrene | 23 | 147 |
| 13C70 | Polystyrene | 0.53 | 149 |
| 13C70 | Ethyl cellulose | 0.25 | 149 |
| PtOEP | Poly(styrene-co-TFEM) | 33 | 321 |
| PtOEP | Cellulose acetate butyrate + poly(tetraphenyl butadiene) | 12 | 331 |
| PdOEP | Poly(methyl methacrylate) + poly(tetraphenyl butadiene) | 3 | 331 |
| PdOEP | Polystyrene | 40 | 281 |
| PtTFPP | Octyl-triEOS/TEOS, doped with dye entrapped core–shell silica particle | 28 | 339 |
| PtTFPP | n-Propyl-triMOS/TEOS/octyl-triEOS | 67 | 341 |
| PtTFPP | Polydimethylsiloxane (PDMS) | 27 | 614 |
| PtOEP | Poly(1-trimethylsilyl-1-propyne) | 18 | 323 |
| PdOEP | 8F-PEKEK(6FBA/HF) | 26 | 329 |
| PdOEP | Poly(styrene-co-TFEM) | 14 | 342 |
| PdOEPK | Teflon | 6 | 336 |
| PtTFPP | Covalently labeled on the surface of silica gel | 1.5 | 272 |
| Al(III)–ferron | Sol–gel silica from TMOS and MeTriMOS | 5 | 351 |
11. Sensing formats
Once an appropriate sensor material and a method for read-out have been identified, they may be applied in numerous formats. These are divided here into the following four main categories: (a) Planar films for sensing and imaging (also over large areas); (b) fiber optic sensors (including point sensors and distributed sensors); (c) microparticle based sensors, and (d) nanosensors, mainly for biological (intracellular) research and in microscopy.11.1. Planar sensors
The planar film sensing format is most widely used. Sensing films are prepared by dissolving the hosting material and the OSPs in certain solvents, spreading the resulting “cocktail” on a mechanical support, and allowing it to dry (or polymerize or cure) to form a thin film (see Fig. 1) that may be applied as a coating or from which sensor spots may be punched. They can be prepared at low cost using standard film technology. The sensor spots often are used in disposables. Fig. 33 shows the portable blood gas analyzer and a disposable kit with integrated sensor spots for several species (O2 included) in a microfluidic channel. | ||
| Fig. 33 Portable blood gas analyzer and a disposable kit with integrated sensor spots for several species. Left: portable analyzer for blood gases (incl. oxygen), blood electrolytes, and glucose. Blood is inserted via the syringe on the right. The disposable kit is placed under the lid. Right: the disposable contains an integrated microfluidic system and 6 (dark) sensor spots for sensing oxygen (along with pH, CO2, Na+, K+), glucose and chloride in whole blood. The pH sensor is of yellow orange color, the others are black because they are covered with optical isolations (see Section 8.4) to prevent blood fluorescence to interfere. The instrument is widely used in ambulance cars and peripheral stations. © OptiMedical Inc. | ||
11.2. Fiber optic and other waveguide sensors
Optical fiber sensors (OFSs) enable oxygen to be determined remotely, at poorly accessible sites, and in harsh or hazardous environment. There is a substantial literature on fiber optic chemical sensors and biosensors.24,615–620 Waveguide sensors for oxygen are obtained by depositing the sensor materials (or “cocktails”) at the tip (the “distal” end) of an optical fiber, and integrated optical system, or a capillary waveguide, for example. Again, the choice of the sensor materials governs selectivity, sensitivity and analytical ranges. Fig. 34 shows a typical tip of a fiber optic sensor. | ||
| Fig. 34 Typical terminal end of a fiber optic sensor for oxygen. The tip is coated with sensor chemistry and covered with black silicone which acts as an optical isolation (see Section 8.4.) that prevents ambient light and sample fluorescence to interfere. | ||
Plastic, glass, and quartz fibers may be used, depending on the analytical wavelength applied. Plastic fibers have a larger aperture, are flexible and easy to work with, but have poor transmission below 420 nm and do not tolerate multiple heat sterilization. Glass fibers are available in small size and have low attenuation, but have a small aperture. They are suitable only down to ∼380 nm. Quartz fibers transmit in the UV, but their aperture is not better than that of glass. Both glass and quartz fibers are fragile and present a risk when breaking in vivo.
Needle-type OFSs for oxygen are used to measure oxygen partial pressure in the vascular system, tissue, bacterial mats, in plants and in numerous other situations. Typical examples are summarized in the ESI† on this review. If inserted into blood vessels or tissue, such sensors must not cause local disturbance, be safe and non-toxic. Peterson et al.42 were the first to demonstrate the feasibility of fiber optic sensing of oxygen in vivo. The probe perylene dibutyrate was incorporated into porous polypropylene that was placed at the tip of an optical fiber, which then was inserted into the bloodstream of a ewe. The results obtained with the OFS agreed well with those of a blood oxygen analyzer. Miller et al.611 developed a disposable triple sensor for simultaneous monitoring of pH, pCO2 and pO2 directly in the artery of the forearm. The sensor was biocompatible and nontoxic, sterilizable and usable for 72 h of operation. The triple sensor consists of a bundle of three 140 μm optical fibers with respective sensor chemistries. Its performance was carefully investigated,621 and precision, stability, consistency and accuracy were found to be excellent and more than adequate. However, and unlike the extracorporeal (non-invasive) triple sensor (GasStat™) from the same company (later to become part of 3M Inc., and even later of Terumo), this fiber optic triple sensor has not been commercialized. This was due to difficulties in the proper placement of the sensor tip in the artery where an oxygen gradient exists in that oxygen levels are higher in blood flowing in the center of the artery but lower near its wall. The performance of continuous systems of ex vivo and in vivo monitoring of blood gases was made by Fogt.622 Lübbers623 has highlighted the specific features of optical sensors for clinical monitoring, and Leiner has reviewed the performance of optical sensors for blood gas analysis.624 A recent review covers fluorescent optical fiber sensors for oxygen and carbon dioxide.612
An OFS for oxygen in respiration gases of guinea pigs was also reported.625 The fiber optic sensor has a response time of less than 20 ms, which is short enough to continuously monitor oxygen in breath air. A fiber optic oxygen microsensor using Ru(dpp) in polystyrene was applied54 to measure oxygen gradients in marine sediments and microbial mats. Kopelman and Rosenzweig194 reported on a similar OFS, but using polyacrylamide as the host material which has poor permeation selectivity. The microsensor has a diameter of 0.8 μm, requires 100 fL only of a sample for analysis, and is fairly sensitive to oxygen (I0/Iair = 3.2). It enables a spatial resolution of 1 μm. Subsequent work180 describes the immobilization of the oxygen probe, and a referenced readout scheme was applied574 in a ratiometric sub-μm fiber optic sensor. A cylindrical-core FOS was developed626 where the probe PtOEP was immobilized in a poly(ethyl methacrylate) film that was deposited on the cylindrical core of a silica optical fiber. The response time of this sensor is <50 ms, but the choice of materials is not perfect in that PtOEP is photolabile and polymers with better permeability are known.
In an FOS for oxygen based on measurement of decay time,45,452,627 a ruthenium probe was employed having an unquenched decay time in the order of 2 μs. Phase fluorometry was performed in the MHz domain. Numerous other versions for lifetime based OFS for oxygen have been described thereafter.628 Commercial systems also use this approach. Needle-type fiber optic sensors using metal–ligand probes have had the largest success. Fig. 35 shows the tip of a decay time-based OFS for oxygen. The red luminescence of the ruthenium-based sensor material is generated by photoexcitation with a blue LED, and decay time is measured as a function of the local pO2 at the tip of the fiber.
 | ||
| Fig. 35 Tip of an optical fiber sensor viewed through a 600 nm longpass filter. Blue light from the light source (not seen here) photoexcites the luminescence of the sensor chemistry (a ruthenium OSP in a sol–gel) at the tip. Both intensity and decay time are a function of the concentration (partial pressure) of oxygen in the sol–gel. | ||
A configuration with crossed optical fibers was reported629 that comprises silver nanoparticles covalently attached to the core of a fiber and labeled with luminescent ruthenium molecules. A second optical fiber (containing the ruthenium probe incorporated into nanoparticle) was placed at right angle of the first fiber to form a fiber–fiber junction. The system was used to detect the luminescence from the ruthenium molecules bound to the first fiber. To employ the effect of metal-enhanced luminescence, the ruthenium complex was kept at an appropriate distance from the silver nanoparticles by polyelectrolyte spacer layers. Luminescence enhancement factors were determined for silver nanospheres, nanotriangles and nanorods and for spacer-layer thicknesses from 214 nm. A 27-fold enhancement was found when the ruthenium complex was placed 4 nm away from silver nanotriangles. The same group630 used the crossed optical fiber with a covalently immobilized ruthenium complex to sense dissolved oxygen via either luminescence intensity ratios or lifetime, the latter performing much better. The sensing properties of an Pt(II) complex were much improved by using metal-coated silica nanoparticles.631 An optical fiber was coated at one end with the probe PtTFPP and silver metal-coated nanoparticles embedded in an n-octyltriethoxysilane-triethoxysilane xerogel. Its sensitivity I0/I100 is as high as 167 which is much better than that of a sensor without silver coating.
Unlike most of the OFSs treated so far, one may also prepare sensors where the core (rather than the clad/coating or the tip) is the sensing material.632 When placed on a suitable substrate, light can propagate directly through the reagent film which is effectively serving as the new “core” of a waveguide. This requires the index of refraction of the sample to be lower than that of the “old” core. The advantage of such a system is the high efficiency with which exciting light is used, the large sampling areas that can be achieved, and the possibility of working with sensing materials containing very low amounts of indicators (some of which are poorly soluble). Side-illuminated polymer optical fiber sensors were described633 that consist of a tapered optical fiber with a ruthenium complex directly embedded in the fiber. The complete setup is based on a side-illumination scheme, and both intensity- and lifetime-based measurements were demonstrated. Sub-μm silica gel fiber made from an organic sol–gel were reported by Yang et al.634Table 16 summarizes the various kinds of OFS for oxygen reported in the literature.
| Waveguide type | Sensor material | Core diameter (μm) | Sensing scheme | Ref. |
|---|---|---|---|---|
| Multi-mode | Perylene dibutyrate in polypropylene | 250 | Intensity | 42 |
| Single-mode fiber optic | Oxygen sensitive ruthenium dye in silicone | 140 | Intensity | 611 |
| Multi-mode | TPP or pyrene absorbed on 0.1 mm Porapak Q and fixed on a film | 3000 | Intensity | 625 |
| Multi-mode | Ru(dpp) in polystyrene | 100 | Intensity | 54 |
| Single-mode | Ru(phen) in polyacrylamide | 100, 3–5, 0.8 | Intensity | 194 |
| Multi-mode | PtOEPK and OEP in PVC | 0.1–0.5 | Ratiometric intensity | 180 |
| Cylindrical core silica | PtOEP in poly(ethyl methacrylate) | 200 | Intensity | 626 |
| Bifurcated fiber | Ru(phen) absorbed in silica gel and immobilized in silicone | 50 | Lifetime (frequency domain) | 452 |
| Multi-mode | Ru(phen) in poly(ethylene glycol)-diacrylate | 200 | Lifetime | 629 |
| Multi-mode | Ru(phen) and rhodamine 110 in poly(ethylene glycol)-diacrylate | 200 | Ratiometric intensity | 630 |
| Multimode fiber optic dual sensor | Ru(dpp) in ormosil microbeads (oxygen and pH), or Pt-TFPP in polystyrene (oxygen and temperature) | 80 | Lifetime and DLR | 173 |
| Multimode sensor for biochemical oxygen demand | Ru(dpp) in organic polymer; bacteria in hydrogel layer | 1 mm | Intensity | 193 |
| Bifurcated fiber biosensor for glutamate using oxygen transducer | Decacyclene in silicone | 3 mm | Intensity | 636 |
| Multimode fiber glucose biosensor using oxygen transducer | Ru(phen) on silica particles in silicone matrix at the tip of the fiber | 3 mm | Intensity | 518 |
| Bifurcated fiber optic cholesterol biosensor using an oxygen transducer | Decacyclene in silicone matrix, enzyme immobilized on Nylon | 1 mm | Intensity | 637 |
| Dual sensor (oxygen and halothane) | Decacyclene in silicone; covered with black Teflon | 2 mm | Intensity | 46 |
| Multimode | Ru(bpy) in silicone | 1 mm | Lifetime | 50 |
In addition to the nanofiber sensor reported in earlier sections, there is a recent report on such a sensor consisting of a polydimethylsiloxane core and a polycaprolactone shell.635 The nanofibers containing oxygen-sensitive probes were prepared by electrospinning and can be applied as optical oxygen sensors for both gaseous and dissolved oxygen. The protective shell layer maintains the fiber's morphology during the slow curing process of the silicone and renders the surface more biocompatible. The response time is very short (0.5 s). The core–shell fibers were integrated into standard cell culture plates of glioma cell lines and glioma-derived primary cells and did not display strong cytotoxic effects.
Fiber optics represent but one kind of optical waveguides. Other formats include planar waveguides such as the versatile chip level waveguide sensor described in a patent583 and consisting of on a chip package which contains an LED light source and a photodiode detector. Simple waveguide elements are mounted on the chip. The waveguide can be coated with various sensor chemistries to form a chemically sensitive element as schematically shown in Fig. 27. Only very recently, a capillary oxygen sensor was described that is based on luminescence lifetime measurements and utilizing monolithically integrated organic photodiodes.638 Such sensors also may be considered as a kind of evanescent sensor as described in the following section.
11.3. Evanescent wave and plasmonic sensing
In an optical waveguide (such as an optical fiber, a planar waveguide, or inside capillaries), light is totally reflected at the interface between the optically more dense medium and the optically rare medium if the angle of refraction is larger than a critical angle α. However, light is not instantaneously reflected at the interface. Rather, it penetrates to some extent into the optically rare phase (Fig. 36). The amplitude of the electric field in the rare phase does not drop abruptly to zero somewhere near the boundary, but rather has a tail that decreases exponentially in the direction of an outward normal to the boundary. This penetration phenomenon is referred to as an evanescent wave.639 In absorptiometry, the attenuation of the propagating beam is measured. In emission spectroscopy, the evanescent wave can be used to generate luminescence (of an OSP, for example) or Raman scatter. The wavelength range extends from the UV to the far infrared depending on the quality of the waveguide. Evanescent wave spectroscopy and evanescent wave sensing (EWS) is closely related to plasmonic resonance spectroscopy and cavity ring-down spectroscopy which is outside the scope of this article, however. | ||
| Fig. 36 Schematic presentation of an evanescent wave in a waveguide in contact with the sample (such as an oxygen sensor coating). Redrawn from www.tau.ac.il/∼applphys/research_fews.htm (with permission from the Tel Aviv University School of Physics and Astronomy). | ||
The depth of penetration (dp) of an evanescent wave is defined as the distance within which the electric field of the wave drops to 1/e. In a first approximation, dp depends on the wavelength of the light and the refraction indices of the two media. Typically, dp ranges from 300 to 1200 nm for visible light, which often is in the same order as the thickness of a sensor layer immobilized on the surface of the waveguide. Light protruding into the reagent phase will be absorbed and – hence – induce the fluorescence of the fluorophore. EWS is particularly attractive when sensing colored species such as blood because the evanescent wave is penetrating the sensor layer on top of the waveguide only, but not the (blood) sample.
EWS along with time resolution techniques640,641 can considerably improve the selectivity of the method (via gated measurements) and has specific features in lifetime-based sensing of oxygen. On the other hand, EWS is adversely affected by changes in the refractive index of the outer medium (usually the sensor layer). Such changes may be caused if organic solvents (alcohol included) enter the sensor layer. The potential of EWS is based on the fact that all the methods described so far for sensing oxygen can be used also in combination with the evanescent wave mode.
Given such features, it does not come as a surprise that EWS has been applied rather early in a fiber optic sensor for oxygen.642 Fiber optic EWS is best demonstrated643 by work on sensing dissolved oxygen (DO) where the declad portion of an optical fiber was coated with a microporous sensor cocktail prepared from trifluoropropyl trimethoxysilane, n-propyl trimethoxysilane, and a ruthenium complex. Excitation by the evanescent wave that is generated at the surface of the core (see Fig. 37) induces luminescence whose intensity depends on the level of DO. The sensor has a detection limit as small as 0.05 ppm of DO and a short response time (15 s).
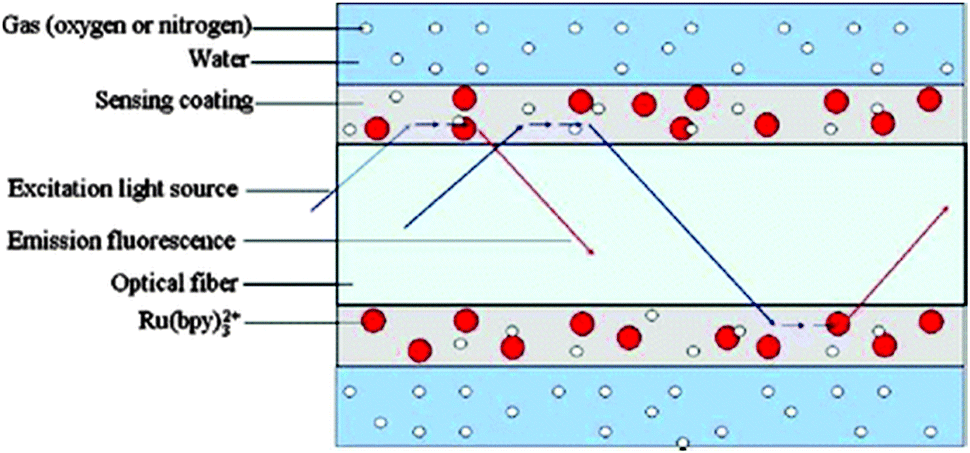 | ||
| Fig. 37 Schematic of evanescent wave excitation using a fiber optic waveguide that carries both the exciting light and back-reflected luminescence. (Reprinted from ref. 643 with permission from Springer.) | ||
Earlier examples include oxygen sensors for breath gas analysis (Section 12.1 in the ESI†),609 and a fiber optic sensor419 that uses the probe 9,10-diphenylanthracene in a poly(hydroxyethyl methacrylate) host. This coating was covalently grafted via silane coupling techniques onto fused silica fibers. Fluorescence was stimulated by the evanescent wave of a UV dye laser or a xenon lamp and quenched by −20% on going from an oxygen-free to an oxygen-saturated (40 ppm; 100% oxygen saturation) aqueous environment. The group of MacCraith202 reported on a fiber optic oxygen sensor based on the quenching of the luminescence of evanescent-wave excited ruthenium complexes in a sol–gel coating. The coating was placed on a declad portion of an optical fiber via a microporous sol–gel film. The OSP was photoexcited by the evanescent field of the 488 nm radiation guided by the optical waveguide. The method later was extended to phase fluorometric sensing of oxygen using an LED-compatible sensor material and evanescent wave excitation.520 Singer at el.644 have deposited a ruthenium complex on an optical fiber by adsorption and then coated with a membrane. It was photoexcited both by the evanescent field associated with light guided in the optical fiber and by direct irradiation, dependent on the refractive index profile of the fiber-membrane-medium system. The emitted light is collected by the same fiber. The thermostatted sensor was exposed to a liquid containing defined concentrations of oxygen which was detectable in the range from 0 to 800 Torr, with a resolution of 2 Torr in the 0–100 Torr range, and of 2% for oxygen partial pressures above 100 Torr. Response times are of the order of 30 s.
In an optical fiber evanescent wave absorptiometric sensor for (irreversible) detection of oxygen deficiency detection,645 the leuco form of the dye Methylene blue (MB) was immobilized in the cladding of the fiber using a sol–gel process. On exposure to oxygen, the dye is oxidized to form MB. The sensor is said to (irreversibly) respond to oxygen at levels between 0.6 and 20.9% in nitrogen. The same sensor chemistry was later applied in an integrated planar Bragg grating probe for (irreversible) detection of oxygen (Fig. 38).646 An optical waveguide containing a Bragg grating is an effective method of detecting minute changes in the refractive index (RI). Removal of a part of the waveguide's clad allows the evanescent field of the optical mode to interact with a sensor layer. The device works by detecting the change in the RI when the sensor coating is exposed to an analyte. This change across the Bragg grating causes a shift in the Bragg wavelength of the grating. The sensory coating of this particular sensor was obtained by modifying the surface of the waveguide with a silica sol–gel containing immobilized leuco MB. On exposure to molecular oxygen, the leuco dye turns blue, and this is “seen” via the absorption of the evanescent wave by the coating. Such Bragg grating based sensors can be small and yet are very sensitive. Fig. 38 shows a photograph of the sensor and how the interaction of the sensitive coating with the analyte leads to a change in the RI and, thus, the Bragg wavelength.
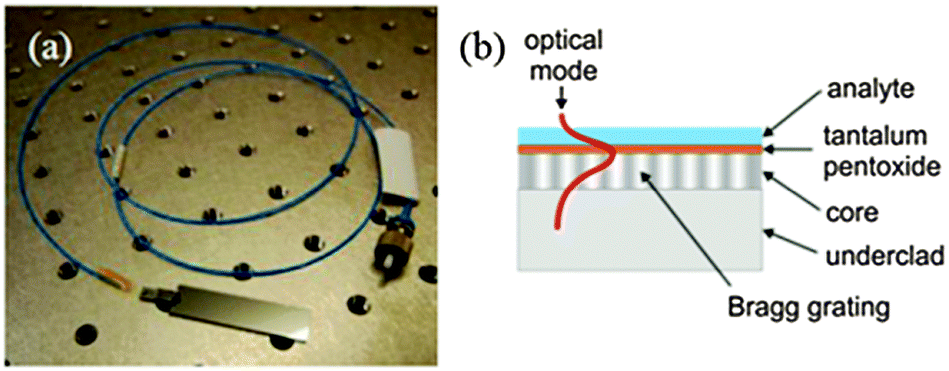 | ||
| Fig. 38 Schematic of an oxygen-sensitive integrated planar Bragg grating waveguide. (a) Photograph of a pigtailed etched Bragg grating device. (b) Schematic representation of the structure and function of a chemically sensitive waveguide integrated Bragg grating with an analyte-sensitive coating. (Reprinted from ref. 646 with permission.) | ||
The scope of EWS fiber optic sensors can be enlarged by nanoparticle scattering deposited in the evanescent field of the sensing fiber.647 The extent of scattering is dominated by the dimensions of the particles. Increasing the radius enhances the scope of the evanescent field and causes a red shift of scattered light. The scattering efficiency accounts for about 74% of the extinction efficiency when the radius is 50 nm at an excitation wavelength of 420 nm. Moreover, the scope of the evanescent field can be extended to approximately 3 μm by silver nanoparticle scattering, which – in fact – enhances the evanescent field to a relatively far-field scope.
So far, mainly (multimode) optical fibers have been discussed, but these represent only one kind of optical waveguides. Other formats include planar waveguides,648 capillary waveguides,649 or integrated optics.650 All these can be operated in the evanescent wave mode, in principle, and used for purposes of sensing. Application of optical waveguide sensors in analytical chemistry, with aspects on instrumentation and applications, has been reviewed.651 A versatile chip level waveguide sensor was described in a patent of Texas Instruments652 as shown in Fig. 27. It can be applied to both absorptiometry and fluorescence.
The number of EWS sensors for oxygen other than fiber optic sensors is small. Kroneis reports on a breath gas analyzer consisting of a planar waveguide coated with a silicone/decacyclene-based sensor chemistry.609 Another article653 reports on the use of lipophilic palladium porphyrins (in the form of self-assembled monolayers) in such sensors. The films were deposited on gold-coated glass slides and photoexcited via the evanescent field of the laser beam passing the glass slab. An integrated optic oxygen sensor was presented654 that serves as an example for a novel and generic evanescent wave sensing platform based on fluorometry. The sensor element comprises a multimode ridge waveguide patterned with an analyte-sensitive fluorescent spot (a ruthenium OSP in a sol–gel) which is photoexcited by an LED. Intensity-based calibration data were generated from the oxygen-dependent waveguide output. This sensor has a limit of detection (LOD) of 0.62% and a resolution of less than 1% of gaseous oxygen. A further kind of integrated optical oxygen sensor655 exploits the evanescent wave interaction of light confined in the waveguide that forms part of a microfluidic system. A ruthenium OSP was immobilized on the waveguide by a combined spin-coating and electrostatic LbL technique. Exposure of the OSP to a fluid containing dissolved oxygen in the microfluidic system enables pO2 to be sensed across a wide range of interest and with an LOD of 0.6 ppm.
The filtering effect of a gold film was exploited in a method for enhancing the luminescence of probes and of sensor materials in general.656 The oxygen probe Ru(dpp) was incorporated into a trimethoxysilane xerogel and spin-coated in a thickness of ∼100 nm. Off-axis illumination of the sensor film results in a luminescence that can be detected with a photodiode placed below the plane of the gold film and the doped sensor film. The use of a gold nanofilm improves the performance of the sensor via two mechanisms: firstly, the transmission of the gold film is high at 520 nm but weak at 450 nm (the wavelength of excitation light), thereby reducing straylight. Secondly, even relatively insensitive luminophores within a distance of ∼10 nm of the gold film are well quenched, and this results in noise reduction by removal of unquenched luminescence of the metal ion–ligand complex. It was questioned whether an element of radiative-decay-engineering of luminescence is occurring with luminophore emission coupling to the gold film and initiating plasmonic emission from the underside of the film, but no evidence either in terms of solely p-polarization or increased intensity of luminescence was evidenced. This confirmed that the sensor enhancement mechanism is primarily the function of metal quenching as revealed by reduced blank intensity and improved response time. A related plasmon-enhanced sensor was reported347 where ultrathin hybrid polymers doped with a Pt(II) porphyrin were used to image oxygen. Langmuir–Blodgett films containing a Pt(II) porphyrin probe were assembled plane-to-plane with a silver nanoparticle array. The use of nanoassemblies resulted in a >10-fold luminescence enhancement. Luminescence intensity and lifetime were studied as a function of the number of layers, and the strong enhancement enables imaging of oxygen on the micrometer scale and with little noise.
11.4. Distributed sensing of oxygen
The possibility of performing spatially resolved measurements is a specific feature of fiber optic based sensing.657 In this scheme, the sensor chemistry is placed as a clad on an optical waveguide, and this will enable distributed sensing of oxygen over large distances, for example via time-domain reflectometry.170,657 This method (commonly used to characterize the loss and length of optical fibers) enables (a) the localization of zones along the fiber where attenuation or fluorescence occurs, and (b) the quantification of the local concentration of chemical species (such as oxygen) by measuring the magnitude of the variation of the signal.658 The luminescence emitted by the probe for oxygen usually returns through the same (or another) waveguide and is received as a signal very much like an acoustical echo. The time after which the echo is registered is a measure of the distance between light source and sensor spot, while its intensity depends on (a) the distance where the echo is generated (due to the usual and linear attenuation along an optical waveguide), and (b) on the extent to which the signal is affected by the local concentration of oxygen. Optical time domain reflectometry (OTDR) instrumentation is fairly small and affordable. Fig. 39 gives a typical signal as obtained by OTDR.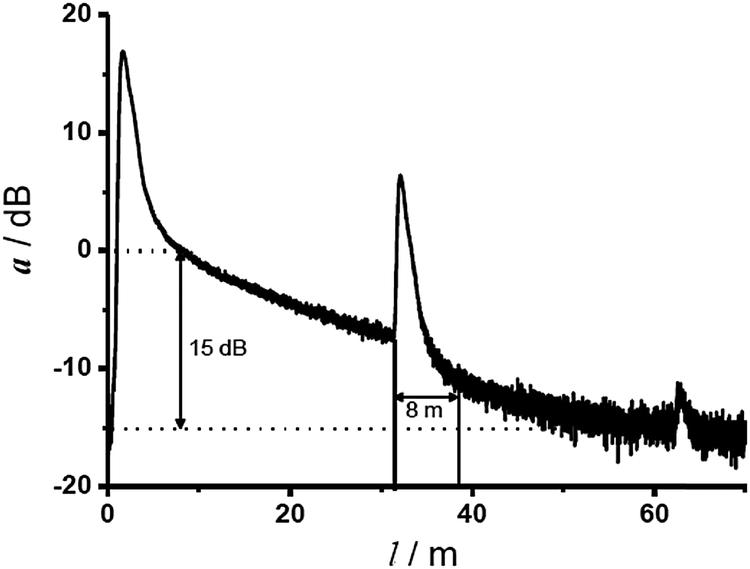 | ||
| Fig. 39 A typical signal trace as obtained by optical time domain reflectometry which is widely used in distributed sensing of gases. From ref. 659 (open access). | ||
The decay time of a probe is a critical parameter in that it compromises the spatial resolution of distributed sensors. Spatial resolution obviously is limited by lifetime since a minimum separation of the fluorophores is required to resolve returning light pulses. Long lifetime probes will not allow for good spatial resolution. Since the decay time of most OSPs based on polycyclic aromatic hydrocarbons is only 20 ns or lower, they are preferred when aiming for high spatial resolution.316 Extended-length fibers have been prepared, for example, where spatially resolved sensing was accomplished by time-of-flight fluorescence detection.660 Sensing was also performed using a fluorescent triangular phenylene dye (whose fluorescence decay times is around 80 ns in the absence of oxygen and around 20 ns in the presence of air in a polymer film), and good spatial resolution was reported using time-correlated single photon counting.661 Distributed sensing of oxygen by OTDR was also accomplished659 by using a fluorescent triangular phenylene dye as the OSP. Immobilized in a polymer matrix, it has a decay time of 86 ns only. This makes it a viable probe to achieve a spatial resolution of a few meters.
If probes with long decay times are used, spatial resolution is more difficult. However, a closer spacing of sensor regions is desirable for many applications. This can be accomplished662 by using a second fiber as an optical delay line, and the minimum spacing between adjacent sensor regions then can be well below the fluorescence lifetime limit. Since the coupling between the two fibers is evanescent, the attenuation of the excitation pulse is low, making long arrays of sensor regions feasible.170,657 Following pulsed, evanescent excitation of the sensor dyes through the first fiber, the second fiber captures their fluorescence and delivers it to the detector. Crossed optical fibers were also shown to enable high-spatial-resolution sensing of dissolved oxygen.630 The luminescent probe (a ruthenium dye) was located in certain spots along the optical fiber in the cladding, and a short laser pulse propagating through the fiber core allows for the determination of the location of the luminophores. The spatial resolution in such quasi-distributed fiber optic sensor arrays is increased beyond the fluorescence-lifetime limit by such a sensing platform. Both luminescence-intensity and luminescence-lifetime changes of the OSP (in response to changes in the concentration of dissolved oxygen) can be used as the analytical information. In the case of intensity measurements, a second adjacent sensor region can be employed as a reference to account for laser pulse energy fluctuations.
Distributed sensing was also performed in another kind of referenced mode660 where an analyte-insensitive fluorophore was added to the sensor material, and intensity was related (referenced) to that of the oxygen probe. In yet another scheme,663 time-correlated single photon counting and stroboscopic detection were combined with an evanescent-wave fiber optic sensing. While demonstrated in this case for sensing pH values, the scheme awaits its extension to distributed lifetime-based sensing of oxygen. Methods have also been developed for optical time-of-flight discrimination of multiple signals along a fiber waveguide.664
11.5. Microparticle-based sensing
Microparticle (μP)-based sensors are mainly used as a bulk material or incorporated into an oxygen-permeable host matrix. μP-sensors are not often used for intracellular sensing because of their size, which is often larger than that of a cell (see Table 17). Sensors beads made from silica μPs were reported16 as early as in 1935. In this case, the dyes were adsorbed on the surface. Other kinds of μP sensors were prepared by polymerization, precipitation, or by doping polymer microparticles with OSPs as outlined in more detail in Section 7.3. Such microsensors were also dispersed in hybrid sensing films, in hydrogels and in silicone matrices. Many of them can be sterilized. Klimant et al.498 prepared magnetically separable sensor μPs from phenyltriethoxysilane, Ru(dpp), Fe3O4, and TiO2 nanoparticles. Because of their magnetic nature, handling is simple and flexible. If placed inside a glass or plastic bioreactor, the sensor film can be magnetically drawn to almost any desired position inside the vessel and the optical signal can be read from outside (Fig. 40, left). Specifically, contactless monitoring of oxygen in cultures of Escherichia coli was demonstrated. These features also make such sensor beads attractive for use in flow-through systems, shaking flasks, and microtiter plates. The same group665 reported on spray-coated magnetic stainless steel macrospheres with polymeric sensor matrices containing the iridium–coumarin complex Ir(CS)2(acac) or PtTPTBP to obtain spheres that serve the same purposes (see Fig. 40, right).| Matrix | Probe | Size (μm) | Remarks | Ref. |
|---|---|---|---|---|
| Silica gel | Trypaflavin | 10 | Room temperature phosphorescence | 16, 17 |
| Silica gel | Ru(bpy) | 2–5 | Room temperature phosphorescence | 47 |
| Silica gel | Ru(dpp) | 9.5–11 | Luminescence quenched by oxygen | 502 |
| Fumed silica | Ru(dpp) | 0.2–0.3 | Dye absorbed on the surface of hydrophilic fumed silica | 199 |
| Silica | PtTCPP | 10.3 | Amino-modified silica microsphere, co-encapsulated with QDs for ratiometric oxygen sensing | 500 |
| Phospholipid coated polystyrene | Ru(bpy-pyr)(bpy)2 | 2.1 | Dye dissolved lipid and coated on polystyrene microparticle | 216 |
| Porous glass beads | Fluoranthene | ∼10 | Fluorescence quenched by oxygen | 35 |
| Ormosil | Ru(dpp) | ∼10 | Magnetic microparticles, luminescence quenched by oxygen, | 498 |
| Ormosil | Ru(dpp) | 1–2 | Luminescence quenched by oxygen | 477 |
| Polystyrene | PtOEP | 1.0 | PtOEP encapsulated during polymerization, reference dye was co-encapsulated for ratiometric readout. | 437, 487 |
| Polystyrene | PtTFPP | 0.3–1.0 | Fluorescence lifetime (frequency domain) based measurement | 442, 483 |
| Poly(styrene-co-acrylonitrile) | Metalloporphyrin | ∼1.0 | Prepared using precipitation method, sensitivity can be tuned by changing the ratio of acrylonitrile in the polymer. | 416–418, 556 |
| Polystyrene or polysulfone | Ir(CS)2(acac) PtTPTBP | 3.2 mm | Magnetic stainless steel macrospheres, luminescence quenched by oxygen | 665 |
| FIB | PtTFPL | 5 cm | Glass spheres | 390 |
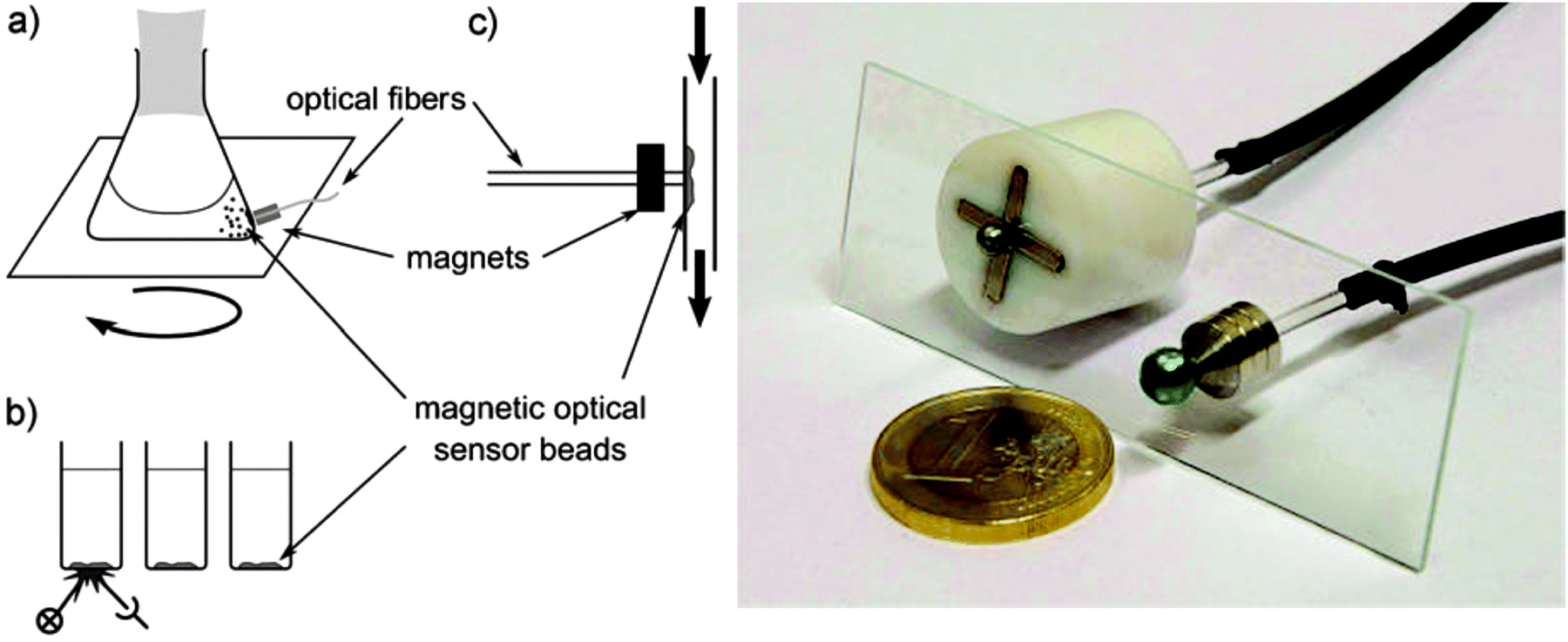 | ||
| Fig. 40 Magnetic oxygen sensitive macrospheres. Left: schematic representation of the potential range of application of magnetic sensor beads in: (a) a shaking flask, (b) microtiter plate measurements, and (c) flow-through systems. Right: magnetic sensor macrospheres (brown and green spheres) captured in front of an optical fiber with a radial or axial separator. (Reprinted from ref. 498 (left) and ref. 665 (right) with permission from Wiley-VCH and American Chemical Society, respectively.) | ||
“Smart tattoo” μP sensors (comprising fluorescent microspheres that can be implanted intradermally and investigated noninvasively using light) were developed666 as potential tools for in vivo biochemical monitoring. Particles were prepared by immobilizing PtOEP in hybrid silicate microspheres along with glucose oxidase. Rhodamine B-doped multilayer nanofilms were subsequently assembled on the surfaces of the particles to provide a reference signal and provide critical control of glucose transport into the particle. The enzymatic oxidation of glucose within the sensor results in the glucose concentration-dependent depletion of local oxygen levels, enabling indirect monitoring of glucose by measuring relative changes in the emission of PtOEP. The sensors respond fairly rapidly (t95 ∼ 1.5 min) and fully reversibly to changes in bulk glucose levels, while demonstrating high baseline stability.667 The role of the porosity in tuning the response range of the microsphere sensors was investigated.668
Mesoporous alginate–silica μPs represent another kind of hybrid matrix.669 The probe PtOEP was loaded into microspheres using a solvent-mediated precipitation method. The beads were then loaded with the enzyme cholesterol oxidase by covalent conjugation. The surface was then “closed” by layer-by-layer self-assembly of alternatively charged polyelectrolytes. Similarly, glucose oxidase was entrapped into calcium alginate along with a ruthenium probe to create a sensor for glucose.670 The μPs were again coated with polyelectrolyte multilayers and used as implantable biosensors — so-called “smart tattoos.” The multilayer nanofilms on the surface of the microspheres can stabilize enzyme entrapment and control substrate diffusion.
Oxygen-responsive fluorescent microbeads embedded into a biocompatible polyacrylamide gel sheet have been attached to target cells for fluorescent imaging of metabolic activity.671 This resulted in a patch-type oxygen imaging sheet useful for in vitro cellular metabolic assays. The sensor beads were deposited in a microfluidic device using electrical manipulation techniques, this followed by coverage with a hydrogel. Fluorescent imaging of oxygen-consuming activity was demonstrated for glucose oxidase-modified μPs as cellular models to show the applicability of the imaging sheet to bioassays.
11.6. Microsensors (not fiber optic, not microparticle-based)
This section covers microsensors that are not of the fiber optic and not of the microparticle type. Such sensors typically consist of a micro-sized semiconductor covered with an oxygen-sensitive coating. In a typical example, such a microsensor was obtained672 by covalently functionalizing a GaN semiconductor with a ruthenium(II) OSP. This resulted in an innovative sensing scheme in which the semiconductor plays the role of both the support and the light source for photoexcitation. Luminescence decay time studies and time-resolved emission spectra confirmed the presence of the dye on the semiconductor surfaces. The sensitivity of the device to oxygen is comparable to that of other ruthenium-based sensor systems. Such microsensors are very compact and intriguing because of the full integration of semiconductor components. In subsequent work, the indicator dyes were grafted onto a blue LED673 following plasma activation of its surface. Confocal fluorescence microscopy with single-photon-timing detection revealed emission lifetimes of ∼2 μs under nitrogen, which however dropped, in the case of n-GaN-functionalized surfaces to 0.6 μs. This was interpreted in terms of photoinduced electron injection from the dye to the semiconductor conduction band, followed by a fast back electron transfer. Hence, the n-GaN–dye system is less suitable. The decay time of the p-GaN/dye material, in contrast, decreases in the presence of oxygen.A completely embedded microsensor array was introduced674 that can be read out with an imaging system to sense oxygen (with a ruthenium OSP) and pH values (with a carboxyfluorescein pH probe; both in a hydrogel) in parallel. The system consists of an ultra-bright blue LED source, coupling optics, interference filters and a compact moisture-resistant CCD camera. The microarray was created by photoreaction injection molding and contains two separate hydrogel based sensing elements (see Fig. 41). The standard error when sensing both dissolved oxygen and pH values during a bioprocess was 0.75% and 0.092, respectively. This microsensor approach may also be considered as a lab-on-a-chip type sensor. High-throughput micropatterning of optical oxygen sensors was also reported and applied to single cell analysis.675 Other work related to microsensors and arrays relates to photolithographic patterning of polymer-encapsulated optical oxygen sensors,676 to micro-patterned sensors and cell self-assembly for measuring the oxygen consumption rate of single cells,677 and to photo-patternable polymeric membranes for optical oxygen sensors.678
 | ||
| Fig. 41 Image of the sensor as captured by the camera. The top row contains a fluorescent pH microsensor, and the bottom row an oxygen sensor layer. It can be seen that signal loss on the edges of the oxygen sensors is evident due to swelling and leaching and therefore box A is used as the average fluorescent signal. (Reprinted from ref. 674 with permission from Elsevier.) | ||
Oxygen sensors have also been designed in the format of a screen-printed flexible radiofrequency identification tag.679 It consists of an oxygen-sensitive membrane containing the oxygen-sensitive probe PtOEP (which is not the best choice) along with an electronic system for RFID communication, all printed on a flexible plastic substrate. Luminescence is excited by an LED operated at 385 nm wavelength, and its intensity is registered by means of a digital color detector. The output data corresponding to red coordinate of the RGB colour space is directly related to the concentration of gaseous oxygen, and it is sent to a microcontroller. The RFID tag is designed for the wireless transmission of the data to a remote reader.
Optical sensing films adaptable to microdevices were also obtained by electrophoretic deposition (EPhD).680 Nanoparticles made from poly(styrene-co-maleic anhydride) and containing the OSP Pt(II) meso-tetra(pentafluorophenyl)porphine were deposited by EPhD which is possible because they carry a charge at near-neutral pH values. Compared to other deposition methods, EPhD is simple and allows control over the rate of deposition. This is crucial for the implementation of optical sensing films in microdevices.
11.7. Dendrites and nanosensors
Optical oxygen sensors can be miniaturized to the nanodimension which is in striking contrast to the size of even the smallest electrochemical oxygen sensors. Such nanosensors can have radii as small as 10 nm and therefore occupy only ∼1 ppb of the volume of an average mammalian cell, thereby causing rather negligible mechanical and functional perturbation. | ||
| Fig. 42 Chemical structure of Oxyphor G2. (Reprinted from ref. 682 with permission from Elsevier.) | ||
In a phosphorescence imaging system for monitoring oxygen distribution in rat liver under ischemia and oxygen reperfusion686 using the probes Oxyphor R2 and Oxyphor G2, the changes of hepatic oxygen pressure under ischemia and subsequent reperfusion were monitored in real-time. The images indicated a transient recovery of hepatic oxygen level during the reperfusion. The lack of complete restoration of oxygen level resembles a similar pattern of hepatic blood flow observed during reperfusion in previous studies.
The third generation of Oxyphor (referred to as Oxyphor G3)684 is based on Pd-tetrabenzoporphyrin dendrimers having both a polyarylglycine scaffold and a poly(ethylene glycol) surface coating as shown in Fig. 43. Oxyphor G3 has a molecular mass of 16.1 kDa and is designed not to interact with albumin and other biomolecules. It folds tightly around the porphyrin core in aqueous media and controls accessibility of oxygen to the phosphor. Its QY is around 10%, and the lifetime is 270 μs in deoxygenated aqueous solution.
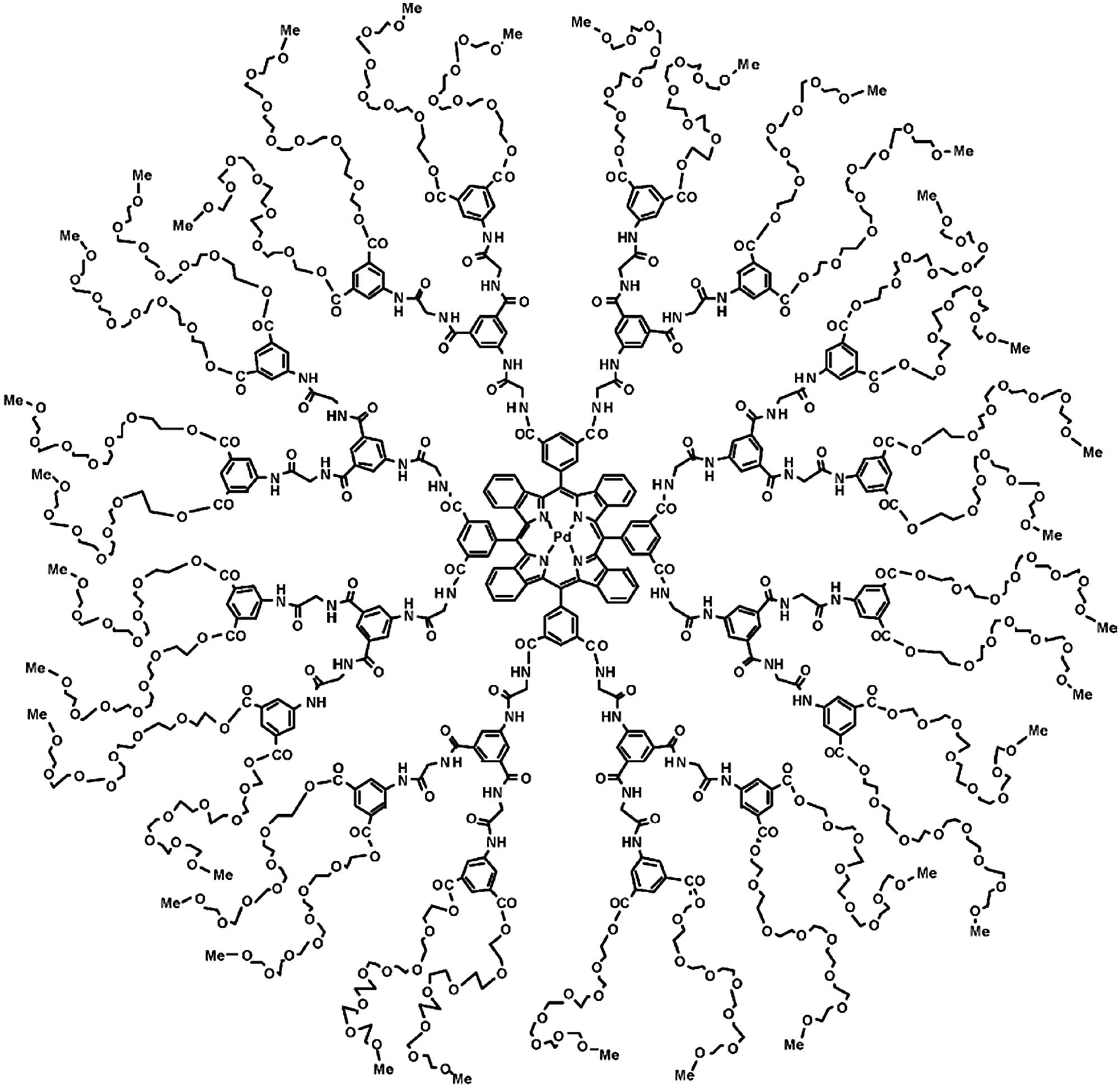 | ||
| Fig. 43 The chemical structure of Oxyphor G3. (Reprinted from ref. 684 with permission from the American Chemical Society.) | ||
The Oxyphor generation four (referred as Oxyphor R4 and Oxyphor G4)687 is the latest and was applied to tumor imaging. Like the Oxyphors of generation 3, they are derived from phosphorescent palladium(II) complexes of tetraphenylporphyrin or tetraphenyl-tetrabenzoporphyrins, respectively, but their phenyl groups have been replaced by 3,5-dicarboxyphenyl groups. These are negatively charged at pH values around 7, and this renders the probes better soluble and, at the same time, cell-impermeable. They possess all the other features that are common for protected dendritic probes, i.e., hydrophobic encapsulation of the luminescent probe and a hydrophilic PEGylated periphery.
Metalloporphyrin-containing dendrimers were also designed for imaging oxygen in vivo based on two-photon excitation.688 Antenna dyes which capturing the two-photon energy were covalently attached to the dendrimers to form nanosized architectures. In these constructs, the generation of porphyrin triplet states following two-photon excitation is induced by an intramolecular Förster-type resonance energy transfer from a covalently attached antenna dye (a coumarin) to a phosphorescent Pt(II) porphyrin. The dendrimer forms an insulating layer between the porphyrin and the antenna dye and simultaneously controls the rate of quenching by oxygen. Modification of the dendrimer periphery with oligo(ethylene glycol) made the signal insensitive to the presence of albumin and other macromolecular solutes. Combined with multi-photon laser scanning microscopy, 3D imaging of oxygen with submicron spatial resolution was accomplished.
The same group308 later synthesized the highly phosphorescent Pt(II) porphyrin poly(arylglycine) dendrimer with several coumarin units attached to the periphery to capture two-photon excitation energy. The dendrimers were used to measure pO2 in the cortical microvasculature and tissue at depths of up to 250 μm and with sub-second temporal resolution. High-resolution images of oxygen distribution were also obtained309 by two-photon laser scanning microscopy using such dendritic nanoprobes in various kinds of cells. Other applications include diffuse tomographic imaging689 and monitoring of oxygen in cortical microvessels by confocal microscopy690 and are summarized in the ESI† in Section S1.
In addition to more conventional host materials of the silica gel or hydrogel type, conjugated fluorescent polymers were also doped575 with a Pt(II) porphyrin probe, and the resulting NPs exhibited bright phosphorescence that is highly sensitive to oxygen. The small size, extraordinary brightness, excellent sensitivity, and ratiometric emission, together with the demonstration of single-particle sensing and cellular uptake, indicate the potential of the NP sensors for quantitative mapping of local molecular oxygen concentration. In another (and cell-permeable) phosphorescent nanosensor, called MM2 and being commercially available, the highly photostable probe PtTFPP and the conjugated polymer poly(9,9-dioctylfluorene) act as a Förster resonance energy transfer donor and as a two-photon antenna, respectively.538 Both fluorophores were placed in NPs with an average size of 70 nm and made from a cationic hydrogel. These nanosensors can be internalized into cells, and this paves the way for high-resolution imaging of cellular oxygen. Both ratiometric (2-wavelength) intensity sensing and phosphorescence lifetime-based imaging under single-photon and two-photon excitation were demonstrated. The nanosensors were placed inside adherent cells and neurospheres to image the oxygen distribution and fluctuation during treatment with drugs.
Ultra-small nanosensors were described for dual sensing of oxygen and pH directly in the cytosol.568 These dually-responsive nanosensors have a unique nanostructure in that a soft core structure is rigidized with a silane reagent to form a mesoporous silica shell, while poly(ethylene glycol) chains form an hydrophilic outer surface (Fig. 44). Lipophilic oxygen-sensitive probes and reference dyes were placed inside the hydrophobic core, while a pH-sensitive probe is covalently attached to the terminal of the poly(ethylene glycol) chains on the surface. The core–shell structure renders the nanosensors well dispersed and highly stable in various kinds of aqueous media. Their average size is 12 nm, and they respond to both pH and oxygen in the physiological range. They do not pass cell membranes, but can be internalized into the cellular cytosol by electroporation, upon which they enable sensing and imaging of pH values and oxygen directly in the cytosol with high spatial resolution. Table 18 gives selected examples of nanosized optical sensors for oxygen.
 | ||
| Fig. 44 Ultra-small dual nanosensors for sensing pH and oxygen values in cytosol. From left to right: the structure of dual nanosensors, oxygen indicator and reference dye in the hydrophobic core, and the pH-sensitive FITC covalently attached at the PEG end; nanosensors delivery in cytosol using electroporation, the green luminescence from FITC, red luminescence from reference dye, and the NIR luminescence (in grey) resulting from the OSP. (Reprinted from ref. 568 with permission of the American Chemical Society.) | ||
| Probe | Matrix | Size (nm) | Readout | Sensitivity | Measurement range | Ref. |
|---|---|---|---|---|---|---|
| Ru(phen) | Liposome | 36 | I | I 0/I100 = 7 | 0–100% | 496 |
| Ru(phen) | NaYF4:Yb3+, Tm3+–SiO2 core–shell nanoparticle | 40–100 | I | I 0/I100 = 3.9 | 0–100% | 169 |
| Ru(dpp) | Silica | 50–300 | RI | I 0/I100 = 6 | 0–100% | 174 |
| Ru(phen) | Phospholipid vesicles | 150 | RI | I 0/I40mgL−1 ∼ 3.8 | 0–40 mg L−1 | 497 |
| Ru(dpp) | Polystyrene (amine-functioned) | 100 | I | n.m. | n.m. | 692 |
| Ru(dpp(SO3Na)2)3 | Polyacrylamide | 45 | L | τ 0/τ100 = 2.14 | 0–100% | 492 |
| PtOEP | Ormosil | 120 | RI | I 0/I42.5ppm = 33.3 | 0–42.5 ppm | 328 |
| PtTFPP | Ormosil | 170 | I | I 0/I100 = 166 | 0–100% | 339 |
| PtTFPP | Poly(styrene-block-vinylpyrrolidone) | 245 | L | I 0/I20kPa ∼ 4.0 | 0–20 kPa | 228, 485 |
| PtTFPP | Eudragit RL-100 | 35 | L | τ 0/τ209μM = 2.33 | 0–209 μM | 482, 538 |
| PtTFPP | Polystyrene (amine-functioned) | 100 or 500 | RI | I 0/I100 ∼ 4.5 | 0–100% | 484 |
| PtTFPP with styrene group | 2,2′-N-Isopropylacrylamide and acrylic acid based copolymer | 45 | L | τ 0/τ20 ∼ 2.5 | 0–20% | 493 |
| PdTCPP | Mesoporous silica | 70–100 | n.m. | n.m. | n.m. | 499 |
| PtOEP | Conjugated fluorescent polymer PFO and PDHF | 50 | RI | I 0/I100 = 20 | 0–100% | 575 |
| PtOEP | Poly(methyl methacrylate) | 150–170 | RI | I 0/I100 = 4.35 | 0–100% | 693 |
| PtOEP | Polystyrene–poly-L-lysine core–shell nanoparticle | 130 | RI | I 0/I42.5ppm = 16.67 | 0–42.5 ppm | 694 |
| PtOEPK | Poly(decyl methacrylate) | 150–250 | RI | I 0/I42.5ppm = 40 | 0–42.5 ppm | 490 |
| PtTPTBP | Pluronic F127–silica core–shell nanoparticle | 12 | RI | I 0/I42.5ppm = 10.05 | 0–42.5 ppm | 568 |
| Oxphor G2 | Polyacrylamide | 30 | RI | I 0/I100 = 28 | 0–100% | 491 |
| No need | Boron polylactide polymer nanoparticle, BF2dbmPLA | 96 | RI | I 0/I1% ∼ 1.41 | 0–1% | 377, 379 |
| Ir(CS)2(acac) |
Eudragit RL-100 polymer
Eudragit RS-100 polymer |
∼360 | L |
τ
0/τair = 2.36
τ 0/τair = 2.03 |
0–21% | 480 |
Other nanoparticles (NPs) of the core–shell type were described493 that consist of a biocompatible shell and a hydrophobic core (containing the covalently immobilized oxygen probe PtTFPP) that was obtained via radical-initiated microemulsion co-polymerization. The NPs can be readily loaded into cells and then are capable of recording intracellular oxygen levels. The NPs have a diameter of <50 nm and luminescence decay times of up to 44 μs. Most importantly, the dye is not leached and the outer shell is biocompatible so that cytotoxicity is strongly reduced.
12. Applications
Numerous applications of optical sensors for oxygen have been described, and some are presented in the ESI† which contains the following sections:(S1) Sensing of oxygen in blood and in the vascular system
(S2) Sensing oxygen in breath gas: respiratory diagnosis
(S3) Sensing and imaging of oxygen in skin (tumor) research, diagnosis and therapy
(S4) Sensing oxygen in medicine and lab (and test) animals (except skin)
(S5) Intracellular sensing of oxygen
(S6) Oxygen sensors in tissue engineering and stem cell research
(S7) Oxygen sensors in animal biology
(S8) Oxygen sensors in plant biology
(S9) Oxygen sensors in drug screening, toxicity testing, mitochondrial activity studies, and related areas
(S10) Oxygen sensors in biotechnology, fermentation and microbiology
(S11) Oxygen sensors in marine and freshwater research
(S12) Oxygen sensors in environmental and geosciences
(S13) Oxygen sensors in food technology and packaging
(S14) Oxygen sensors in gas pressure-sensitive paints
(S15) Oxygen sensors as transducers in biosensors
(S16) Oxygen sensors integrated into microfluidic devices and lab-on-a-chip systems
(S17) Oxygen sensors in industry, material research, production and surveillance
13. Conclusions and outlook
Optical sensing of oxygen is at a rather advanced state. At present, technology is driven by new materials, new nanotechnologies and spectroscopies. Exciting current research activities cover probe design, nanosensors and nanofiber sensors, new sensing schemes such as (localized) surface plasmon resonance, or effects that occur on surfaces of noble metals. These activities will not only further enlarge the range of sensors and the fields of applications, but also improve the performance of current sensors. It can also be stated that the design of sensor materials still is a kind of trial-and-error game because their performance cannot be modeled mathematically so far.By now, optical sensors for oxygen have been applied to numerous impressive applications, but one may state that existing sensors not so much are competing with the classical electrochemical sensor (the Clark electrode). Rather, they have been applied (and are being applied) to fields and research challenges for which appropriate electrochemical or other methods did not exist so far. Typical examples include (a) fiber optic sensors for sensing over large distances, under harsh conditions or in strong electromagnetic fields; (b) imaging of oxygen over large areas, mainly in (skin) medicine, aerodynamics, marine sciences, and biotechnology; (c) nanosensors for use inside cells, in interstitial or vascular fluids, or directly in the flow of a microfluidic system; and (d) contactless sensing of oxygen inside a bioreactor by placing the sensor spot on the inner side of a transparent window and monitoring its optical properties through this window. It is another matter of fact that optical sensors for oxygen (“optodes” as they sometimes are referred to) are in the process of replacing the classical Clark electrode which is the gold standard at present. We predict the oxygen optode to displace the Clark electrode within 10 years from around 50% of its current market.
Any question for the “best” sensor for oxygen cannot be answered unless the problem is specified. Unlike the Clark electrode which comes in a few variations only, hundreds of kinds of optical sensors for oxygen are known or may be envisioned. While sensing of oxygen in engine exhaust gases at temperatures of >400 °C is best performed with a sensor material such as Mo6Cl12 clusters, the detection of oxygen at ppb levels at temperatures between −100 °C and +100 °C is better performed by exploiting the strongly quenched delayed fluorescence of certain fullerenes. It is a fortunate situation that most sensors for oxygen based on the use of luminescent probes contained in a polymer host do survive chemical and/or thermal sterilization. Other situations (such as barometric sensing) require sensors with very fast response times so as to acquire images at a kHz rate. This requires sensor materials that are very different from those where response time plays no role. As for in vivo studies, oxygen sensors whose excitation and emission bands located in the NIR are preferred to prevent interferences from biological luminescence background. NIR oxygen sensors are developing very rapidly in recent years and this trend will continue and further enlarge the database in this field. Sensors with long decay times offer an alternative because they are amenable to time-resolved fluorometry which can suppress background luminescence, but this has limitations if data or images are to be required at fast rates or with high spatial resolution such as in distributed sensing of oxygen along a large distance fiber. We have summarized the most frequently used molecular probes (or indicators; OSPs) and the most frequently used polymer hosts in Tables 19 and 20, respectively. These are widely used but it shall be kept in mind that special situations may require other combinations of materials and, of course, spectroscopies.
| Probe | Exc./Em. maxima | Comments |
|---|---|---|
| Ru(dpp)3 2X− | 460/597 | Fairly photostable; well established; cationic; counter anions (X−) include those of long chain fatty acids, trimethylsilyl propanesulfonate, or tetrafluoroborates; moderate brightness; well soluble in hydrophilic polymers but a lipophilic counterion is needed if to be used in non-polar polymer hosts such as silicones; fairly long decay time (0.5–6 μs). |
|
PtTFPP
PdTFPP |
392(540)/650
407(552)/670 |
Very photostable; well established; uncharged; well soluble in apolar polymers except silicones; lifetimes in the upper μs to ms range; excellent brightness under UV excitation, moderate brightness under excitation with visible light; these probes have widely replaced the formerly used probes PtOEP and PdOEP which suffer from poor photostability. |
|
PtTPTBP
PdTPTBP |
430(614)/770
444(629)/797 |
Very photostable; uncharged; well soluble in apolar polymers except silicones; long lifetime (the Pd complex in particular); emission in NIR, excellent brightness; the Q absorption band of the Pd(II) porphyrin is compatible with red He–Ne laser and laser diode. |
|
PtOEPK
PdOEPK |
398(598)/760
408(601)/791 |
Fairly photostable; uncharged; well soluble in apolar polymers except silicones; long lifetime (the Pd complex in particular); emission in the NIR, moderate brightness. |
| PtTFPL | 400(574)/738 | More photostable than the PtOEPK complex, but otherwise rather similar. |
| Dendrimeric (benzo)porphyrins | 415(524,632,678)/700–800 | Can be well dispersed in watery systems; very fast response; known as Oxyphors; widely used in physiological studies (see Section 12); dendritic structure with hydrophilic outer shell and a core consisting of a Pd(II) (benzo)porphyrin; molecular mass of nanoparticles typically is 15–20 MDa; can be negatively charged to prevent cell permeation, but charge can be varied to prevent interaction with albumins. |
| Ir(III)–coumarin complexes | 421–484/544–588 | High brightness (high quantum yield and absorption coefficient); uncharged; moderately long lifetime; moderate photostability. |
| Polycyclic hydrocarbons: (a) pyrene; (b) decacylene |
(a) 335/384,450
(b) 385/510 |
(a) Fair stability; requires UV excitation; used in fast-responding pressure sensitive paints; less suitable for biological application due to UV excitation; moderate sensitivity and brightness.
(b) Good photostability; compatible with blue LEDs; good oxygen quenching efficiency; large Stokes shift. |
| Fullerenes |
C60: 532/750
C70: 470,532/860 |
Very good stability; extremely high sensitivity; very long lifetime (ms); E-type delayed fluorescence; works at temperatures up to 240 °C, moderate brightness. |
| Polymer | Comments |
|---|---|
| Ethyl cellulose | Biocompatible; good optical transparency; good mechanical strength; ease of handling. |
| Silicone rubbers (incl. fluorosilicones) | Excellent gas permeability, high thermal stability, excellent chemical and mechanical stability, ease of handling, good adhesion to glass fibers, inertness to biological samples, optically transparent; hydrophobic; many commercially available prepolymers contain solvents, fillers, low molecular weight cross-linkers, catalysts and other additives; need for curing. Poor compatibility with many indicators due to string tendency for aggregation; fluorosilicones have very high chemical resistances and ebven better oxygen permeability. |
| Fluorinated copolymers: (a) poly-styrene-co-PFS and poly-styrene-co-TFEM; (b) poly-IBM-co-TFEM and poly-tBS-co-TFEM; (c) FIB | (a)–(c): High gas permeability; excellent photostability; useful for trace oxygen sensing; (a)–(b): gas permeability can be tuned by varying different monomers and the ratio of monomers; (c) poly(hexafluoroisopropyl methacrylate)-co-(heptafluoro-n-butyl methacrylate; high gas permeability; excellent photostability); small temperature dependence; suitable for application in wind tunnels (barometry) with temperature compensation. |
| Polyurethane hydrogels | Highly biocompatible; ease of handling; good optical transparency; more often used as a matrix to host other kinds of sensor particles; swellable. |
| Polystyrene | Easy to manufacture; low cost; moderate oxygen permeability; can be sterilized; excellent shelf time and stability in aqueous solutions; good optical transparency; no need for curing. Copolymers can be used to prepare water-dispersible oxygen-sensitive nanoparticles. |
| PMMA (plexiglass) | Ease of manufacture; low cost material; moderate oxygen permeability; suitable for sensing oxygen above air saturation; copolymers known for preparing water-dispersible sensor nanoparticles; moderate hydrolytic stability under harsh conditions; background fluorescence under UV excitation. |
| Poly(1-trimethylsilyl-1-propyne) (PTMSP) | Extremely high gas permeability; no need for cross-linking; useful for trace oxygen sensing; prone to aging affects (decrease of oxygen permeability with time). |
| Polystyrene-co-acrylonitrile (PSAN) | Gas permeability can be tuned by changing the ratio of monomers; good stability; easy to handle; the monomer acrylonitrile is toxic. |
| Silica gels | Commercially available in different size and porosity; can physically absorb oxygen sensitive probes; easy handling; good optical transparency and low fluorescence background; biomolecules can be incorporated. |
| Ormosils (organically-modified silica gels) | Easily prepared at room temperature; hydrophobicity is tunable by changing precursors; fluorinated ormosils have very high gas permeability that may change over time; good mechanical and chemical stability. |
Despite these attractive features, one cannot ignore the fact that practically all reversible sensors for oxygen are based on the quenching of luminescence by oxygen. This can be considered as being a disadvantage because the human eye can much better differentiate the color and intensity of reflected light than the color and intensity of luminescent light. Practically all commercially available optical tests (like the stripes used for measurement of pH values, or the widely used pregnancy tests) are color-, i.e. reflectometry-based for this reason. Unfortunately, reflectometric test stripes for reversible(!) sensing of oxygen do not exist. This would be great to have but has not been realized so far. There are, however, sensor known that change color (such as from colorless to blue) on exposure to oxygen (see Section 4), but the respective color changes are irreversible. Such “sensors” (better “probes”) cannot monitor the decrease of oxygen concentration over time, for example. Some can be chemically regenerated, though. Single-shot colorimetric probes for oxygen predictably will find more applications in the future, typically in areas where millions of products need to be quickly monitored, for example in the food and pharmaceutical industry. Another drawback of oxygen sensors based on luminescence quenching is their inadequate performance when measuring high concentrations of oxygen. This “market” is still dominated by electrochemical sensors, but there is an urgent demand for such sensors, not the least for safety reasons.
It is noted that optical imaging of oxygen has experienced particularly exciting applications. Field of applications include (a) the use of nanosensors for microscopic imaging of the distribution of oxygen inside cells, (b) the use of sensor paints to image the distribution of barometric pressure on vehicles including aircrafts, (c) the detection of hypoxia in skin tumors, (d) the distribution of oxygen in benthic systems, (e) the read-out of microtiterplates or arrays of micro-vials with oxygen sensors placed on their bottom, to give a few representative examples only.
Fluorescent sensors usually are read out by instrumental means, and it is a fortunate situation that respective readers, due to advances in optoelectronics, are truly small, typically of the size smaller than a cigarette pack. This also holds for imagers. Predictably, sensors will become much smaller (<1 cubic mm) in the future, and this will pave the way to mass production. Such sensors will find large scale applications such as in fuel cells, in mobile phones and computers, or as microbarometers in aircrafts to measure cabin pressure. All this is indication that research in this field will continue to go on strongly and in many directions.
Acronyms
| 2-D | 2-Dimensional |
| APTES | (3-Aminopropyl)triethoxysilane |
| CAB | Cellulose acetate butyrate |
| CCD | Charge coupled device |
| CCTA | Contact charge-transfer absorption |
| CL | Chemiluminescence |
| CMOS | Complementary metal oxide semiconductor |
| CPG | Controlled porous glass |
| DiMe-DMOS | Dimethoxydimethylsilane |
| DLD | Dual lifetime determination |
| EC | Ethyl cellulose |
| Et-TriMOS | Ethyltrimethoxysilane |
| OFS | Optical fiber sensor |
| FRET | Fluorescence resonance energy transfer |
| LB | Langmuir–Blodgett |
| LED | Light-emitting diode |
| MAP | Modified atmosphere packaging |
| MLCT | Metal–ligand charge transfer |
| μP | Microparticle |
| Me-TriMOS | Methyltrimethoxysilane |
| NIR | Near infrared |
| NP | Nanoparticle |
| n-propyl-TriMOS | n-Propyl-trimethoxysilane |
| Octyl-triEOS | n-Octyltriethoxysilane |
| ormosil | Organically modified silicate |
| OSM | Optical sensor material |
| OSP | Oxygen sensitive probe |
| PAHs | Polycyclic aromatic hydrocarbons |
| PDMS | Poly(dimethyl siloxane) |
| PFCs | Perfluorochemicals |
| pHEMA | Poly(2-hydroxyethyl methacrylate) |
| PMMA | Poly(methyl methacrylate) |
| pPEGMA | Poly(ethylene glycol) ethyl ether methacrylate |
| PS | Polystyrene |
| PSAN | Poly(styrene-co-acrylonitrile) |
| PSP | Pressure sensitive paints |
| Ph-TriMOS | Phenyltrimethoxysilane |
| pTMSP | Poly(1-trimethylsilyl-1-propyne) |
| PTPs | Poly(thionylphosphazenes) |
| PVA | Polyvinyl alcohol |
| PVC | Polyvinyl chloride |
| QDs | Quantum dots |
| RLD | Rapid lifetime determination |
| ROMPs | Ring opening metathesis polymers |
| RSD | Relative standard deviation |
| RTP | Room-temperature phosphorescence |
| RTV | Room-temperature vulcanizing |
| SVP | Stern–Volmer plot |
| TEOS | Tetraethyl orthosilicate |
| TFP-TriMOS | 3,3,3-Trifluoropropyltrimethoxysilane |
| TLC | Thin layer chromatography |
| Tri-MOS | Trimethylmethoxysilane |
| UCNPs | (Photon)up-conversion nanoparticles |
| UV | Ultraviolet |
Acknowledgements
This work was financially supported by the Alexander von Humboldt Foundation (Bonn) through a fellowship for Dr Xu-dong Wang which is gratefully acknowledged.References
- G. L. Semenza, Science, 2007, 318, 62–64 CrossRef CAS PubMed.
- H. Decker and K. E. Van Holde, Oxygen and the Evolution of Life, Springer Verlag, Berlin Heidelberg, 2011 Search PubMed.
- L. W. Winkler, Ber. Dtsch. Chem. Ges. B, 1888, 21, 2843–2854 CrossRef.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley-Interscience, 1992 Search PubMed.
- R. Ramamoorthy, P. K. Dutta and S. A. Akbar, J. Mater. Sci., 2003, 38, 4271–4282 CrossRef CAS.
- Y. Amao, Microchim. Acta, 2003, 143, 1–12 CrossRef CAS.
- X. D. Wang, H. X. Chen, Y. Zhao, X. Chen and X. R. Wang, TrAC, Trends Anal. Chem., 2010, 29, 319–338 CrossRef CAS PubMed.
- J. R. Stetter and J. Li, Chem. Rev., 2008, 108, 352–366 CrossRef CAS PubMed.
- R. Moos, Springer Series on Chemical Sensors and Biosensors, 2012, vol. 11, pp. 173–188 Search PubMed.
- O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 9864–9865 CrossRef CAS PubMed.
- H. Kautsky and A. Hirsch, Ber. Dtsch. Chem. Ges. B, 1931, 64, 2677–2686 CrossRef.
- G. N. Lewis and M. Calvin, Chem. Rev., 1939, 25, 273–328 CrossRef CAS.
- G. N. Lewis, D. Lipkin and T. T. Magel, J. Am. Chem. Soc., 1941, 63, 3005–3018 CrossRef CAS.
- I. A. Zakharov and T. I. Grishaeva, J. Appl. Spectrosc., 1979, 31, 1295–1298 CrossRef.
- I. A. Zakharov and T. I. Grishaeva, Zh. Prikl. Spektrosk., 1982, 36, 697–701 Search PubMed.
- H. Kautsky and A. Hirsch, Z. Anorg. Allg. Chem., 1935, 222, 126–134 CrossRef CAS.
- J. Franck and P. Pringsheim, J. Chem. Phys., 1943, 11, 21–27 CrossRef CAS PubMed.
- A. Mills, Platinum Met. Rev., 1997, 41, 115–126 CAS.
- J. N. Demas, B. A. DeGraff and P. B. Coleman, Anal. Chem., 1999, 71, 793A–800A CrossRef CAS.
- A. Mills, Chem. Soc. Rev., 2005, 34, 1003–1011 RSC.
- O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657–2669 RSC.
- C. O'Donovan, J. Hynes, D. Yashunski and D. B. Papkovsky, J. Mater. Chem., 2005, 15, 2946–2951 RSC.
- O. S. Wolfbeis, Adv. Mater., 2008, 20, 3759–3763 CrossRef CAS.
- Y. Amao and I. Okura, J. Porphyrins Phthalocyanines, 2009, 13, 1111–1122 CrossRef CAS.
- A. P. Demchenko, Methods Appl. Fluoresc., 2013, 1, 022001 CrossRef.
- N. Optiz and D. W. Lübbers, Int. Anesthesiol. Clin., 1987, 25, 177–197 CrossRef PubMed.
- O. S. Wolfbeis, Fiber optic chemical sensors and biosensors, CRC Press, Boca Raton, Florida, 1991 Search PubMed.
- X. Lu and M. A. Winnik, in Mol. Supramol. Photochem., ed. V. Ramamurthy and K. S. Schanze, CRC, 2000, vol. 6, pp. 311–352 Search PubMed.
- D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700–8732 RSC.
- M. Quaranta, S. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed.
- O. Stern and M. Volmer, Phys. Z., 1919, 20, 183–188 CAS.
- H. Kautsky, Trans. Faraday Soc., 1939, 35, 216–219 RSC.
- M. Pollack, P. Pringsheim and D. Terwoord, J. Chem. Phys., 1944, 12, 295–299 CrossRef CAS PubMed.
- I. Zakharov and T. Grishaeva, Zh. Anal. Khim., 1980, 35, 481 CAS.
- I. Bergman, Nature, 1968, 218, 396 CrossRef CAS.
- J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS.
- P. R. Stevens, US Pat., 3.612.866, 1982 Search PubMed.
- H. H. Hesse, East Ger. Pat., 106086, 1974 Search PubMed.
- D. W. Lübbers and N. Opitz, Z. Naturforsch., C: J. Biosci., 1975, 30, 532 Search PubMed.
- D. W. Lübbers and N. Opitz, Adv. Exp. Med. Biol., 1976, 75, 65–68 CrossRef.
- K. P. Völkl, N. Opitz and D. W. Lübbers, Fresenius' J. Anal. Chem., 1980, 301, 162–163 CrossRef.
- J. I. Peterson, R. V. Fitzgerald and D. K. Buckhold, Anal. Chem., 1984, 56, 62–67 CrossRef CAS.
- O. S. Wolfbeis, H. Offenbacher, H. Kroneis and H. Marsoner, Mikrochim. Acta, 1984, 82, 153–158 CrossRef.
- T. M. Freeman and W. R. Seitz, Anal. Chem., 1981, 53, 98–102 CrossRef CAS.
- H. Marsoner and H. Kroneis, Eur. Pat. Appl.,, 109958 & 109959, 1984 Search PubMed; H. Marsoner and H. Kroneis, US Pat., 4.587.101, 1984 Search PubMed.
- O. S. Wolfbeis, H. E. Posch and H. W. Kroneis, Anal. Chem., 1985, 57, 2556–2561 CrossRef CAS.
- O. S. Wolfbeis, M. J. P. Leiner and H. E. Posch, Mikrochim. Acta, 1986, 90, 359–366 CrossRef.
- P. C. Alford, M. J. Cook, A. P. Lewis, G. S. G. McAuliffe, V. Skarda, A. J. Thomson, J. L. Glasper and D. J. Robbins, J. Chem. Soc., Perk. Trans. 2, 1985, 705–709 RSC.
- J. R. Bacon and J. N. Demas, Anal. Chem., 1987, 59, 2780–2785 CrossRef CAS.
- M. E. Lippitsch, J. Pusterhofer, M. J. P. Leiner and O. S. Wolfbeis, Anal. Chim. Acta, 1988, 205, 1–6 CrossRef CAS.
- J. M. Vanderkooi, G. Maniara, T. J. Green and D. F. Wilson, J. Biol. Chem., 1987, 262, 5476–5482 CAS.
- H. E. Kahil, M. P. Gouterman and E. Green, PCT Appl., WO 87.00023, 1987 Search PubMed.
- G. E. Khalil, M. P. Gouterman and E. Green, US Pat., 4,810,655, 1989 Search PubMed.
- I. Klimant, V. Meyer and M. Kuhl, Limnol. Oceanogr., 1995, 1159–1165 CrossRef CAS.
- R. Narayanaswamy and O. Wolfbeis, Optical Sensors for Industrial, Environmental and Clinical Applications, Springer, 2003, pp. 1–34, ISBN 33-540-40888-X Search PubMed.
- P. B. Arnoudse, H. L. Pardue, J. D. Bourland, R. Miller and L. A. Geddes, Anal. Chem., 1992, 64, 200–204 CrossRef CAS.
- L. Gianfrani, A. Sasso and G. M. Tino, Sens. Actuators, B, 1997, 39, 283–285 CrossRef CAS.
- V. Weldon, J. O'Gorman, J. J. Pérez-Camacho, J. Hegarty, J. C. Connolly and N. A. Morris, Sens. Actuators, B, 1997, 42, 163–168 CrossRef CAS.
- C. Jia, H. Andreas, S. Rainer, F. Maximilian and A. Markus-Christian, Conference on Lasers and Electro-Optics/International Quantum Electronics Conference, 2009.
- X. Lou, G. Somesfalean, B. Chen and Z. Zhang, Appl. Opt., 2009, 48, 990–997 CrossRef CAS.
- S. Neethu, R. Verma, S. S. Kamble, J. K. Radhakrishnan, P. P. Krishnapur and V. C. Padaki, Sens. Actuators, B, 2014, 192, 70–76 CrossRef CAS PubMed.
- G. E. Khalil, A. Chang, M. Gouterman, J. B. Callis, L. R. Dalton, N. J. Turro and S. Jockusch, Rev. Sci. Instrum., 2005, 76, 1–8 CrossRef PubMed.
- S. Jockusch, N. J. Turro, E. K. Thompson, M. Gouterman, J. B. Callis and G. E. Khalil, Photochem. Photobiol. Sci., 2008, 7, 235–239 CAS.
- A. P. Losev, I. M. Byteva and G. P. Gurinovich, Chem. Phys. Lett., 1988, 143, 127–129 CrossRef CAS.
- V. S. Tiwari, A. T. Luanje, R. R. Kalluru, F. Y. Yueh and J. P. Singh, Rev. Sci. Instrum., 2011, 82, 043108 CrossRef PubMed.
- Z. Zhang and R. W. Seitz, Anal. Chem., 1986, 58, 220–222 CrossRef.
- K. E. Chung, E. H. Lan, M. S. Davidson, B. S. Dunn, J. S. Valentine and J. I. Zink, Anal. Chem., 1995, 67, 1505–1509 CrossRef CAS.
- M. F. McCurley, G. J. Bayer and S. A. Glazier, Sens. Actuators, B, 1996, 36, 491–496 CrossRef CAS.
- F. Baldini, M. Bacci, F. Cosi and A. Del Bianco, Sens. Actuators, B, 1992, 7, 752–757 CrossRef CAS.
- A. Del Bianco, F. Baldini, M. Bacci, I. Klimant and O. S. Wolfbeis, Sens. Actuators, B, 1993, 11, 347–350 CrossRef CAS.
- S. Röösli, E. Pretsch, W. E. Morf, E. Tsuchida and H. Nishide, Anal. Chim. Acta, 1997, 338, 119–125 CrossRef.
- Y. Suzuki, H. Nishide and E. Tsuchida, Macromolecules, 2000, 33, 2530–2534 CrossRef CAS.
- L. Vaska, Science, 1963, 140, 809–810 CAS.
- L. Vaska, Acc. Chem. Res., 1976, 9, 175–183 CrossRef CAS.
- R. D. Jones, D. A. Summerville and F. Basolo, Chem. Rev., 1979, 79, 139–179 CrossRef CAS.
- F. Baldini and A. D. Bianco, Fiber Integr. Opt., 1992, 11, 123–133 CrossRef CAS.
- M. F. Choi and P. Hawkins, Sens. Actuators, B, 1996, 30, 167–171 CrossRef CAS.
- M. F. Choi and P. Hawkins, Talanta, 1995, 42, 987–997 CrossRef CAS.
- Y. Amao, K. Asai and I. G. Okura, Chem. Lett., 1999, 1, 95–96 CrossRef.
- Y. Amao, K. Asai and I. Okura, Bull. Chem. Soc. Jpn., 1999, 72, 2223–2227 CrossRef CAS.
- Y. Amao, K. Asai and I. Okura, Analyst, 2000, 125, 523–526 RSC.
- Y. Amao, K. Asai and I. Okura, J. Porphyrins Phthalocyanines, 2000, 4, 292–299 CrossRef CAS.
- T. Furuto, S.-K. Lee, Y. Amao, K. Asai and I. Okura, J. Photochem. Photobiol., A, 2000, 132, 81–86 CrossRef CAS.
- S. Ashkenazi, S. W. Huang, T. Horvath, Y. E. L. Koo and R. Kopelman, J. Biomed. Opt., 2008, 13, 034023 CrossRef PubMed.
- S. Ashkenazi, J. Biomed. Opt., 2010, 15, 040501 CrossRef PubMed.
- H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments, Wiley-VCH, 2004 Search PubMed.
- G. I. Goodfellow and H. M. Webber, Analyst, 1979, 104, 1105–1118 RSC.
- P. A. Hamlin and J. L. Lambert, Anal. Chem., 1971, 43, 618–620 CrossRef CAS.
- D. Perlman and H. Linschitz, US Pat., 4,526,752, 1985 Search PubMed.
- Y. Yoshikawa, T. Nawata, M. Goto and Y. Fujii, US Pat., 4,169,811, 1979 Search PubMed.
- Y. Yoshikawa, T. Nawata, M. Goto and Y. Kondo, US Pat., 4,349,509, 1982 Search PubMed.
- S.-K. Lee, A. Mills and A. Lepre, Chem. Commun., 2004, 1912–1913 RSC.
- D. Gutiérrez-Tauste, X. Domènech, N. Casañ-Pastor and J. A. Ayllón, J. Photochem. Photobiol., A, 2007, 187, 45–52 CrossRef PubMed.
- S.-K. Lee, M. Sheridan and A. Mills, Chem. Mater., 2005, 17, 2744–2751 CrossRef CAS.
- A. Mills and D. Hazafy, Analyst, 2008, 133, 213–218 RSC.
- Y. Galagan and W. F. Su, J. Photochem. Photobiol., A, 2008, 195, 378–383 CrossRef CAS PubMed.
- Y. Galagan, S.-H. Hsu and W.-F. Su, Sens. Actuators, B, 2010, 144, 49–55 CrossRef CAS PubMed.
- L. Roberts, R. Lines, S. Reddy and J. Hay, Sens. Actuators, B, 2011, 152, 63–67 CrossRef CAS PubMed.
- Y. Galagan and W. F. Su, Food Res. Int., 2008, 41, 653–657 CrossRef CAS PubMed.
- K. Eaton, Sens. Actuators, B, 2002, 85, 42–51 CrossRef CAS.
- S. Wilhelm and O. S. Wolfbeis, Sens. Actuators, B, 2011, 153, 199–204 CrossRef CAS PubMed.
- K. C. Krumhar and M. Karel, US Pat., 5,096,813, 1992 Search PubMed.
- S. Wilhelm and O. Wolfbeis, Microchim. Acta, 2010, 171, 211–216 CrossRef CAS.
- A. Mills, K. Lawrie, J. Bardin, A. Apedaile, G. A. Skinner and C. O'Rourke, Analyst, 2012, 137, 106–112 RSC.
- M. A. Butler and A. J. Ricco, Sens. Actuators, 1989, 19, 249–257 CrossRef CAS.
- G. A. Nyberg, US Pat., 4,764,343, 1988 Search PubMed.
- H. Lin, Y. Shen, D. Chen, L. Lin, B. Wilson, B. Li and S. Xie, J. Fluoresc., 2013, 23, 41–47 CrossRef CAS PubMed.
- J. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3 edn, 2006 Search PubMed.
- B. Valeur, Molecular fluorescence: principles and applications, Wiley-VCH, Weinheim, 2002 Search PubMed.
- E. R. Carraway, J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63, 332–336 CrossRef CAS.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 337–342 CrossRef CAS.
- J. N. Demas, B. A. DeGraff and W. Xu, Anal. Chem., 1995, 67, 1377–1380 CrossRef CAS.
- S. Draxler, M. E. Lippitsch, I. Klimant, H. Kraus and O. S. Wolfbeis, J. Phys. Chem., 1995, 99, 3162–3167 CrossRef CAS.
- P. Hartmann, M. J. P. Leiner and M. E. Lippitsch, Anal. Chem., 1995, 67, 88–93 CrossRef CAS.
- W. Xu, R. C. McDonough, B. Langsdorf, J. N. Demas and B. A. DeGraff, Anal. Chem., 1994, 66, 4133–4141 CrossRef CAS.
- S. A. Vinogradov and D. F. Wilson, Biophys. J., 1994, 67, 2048–2059 CrossRef CAS.
- V. I. Ogurtsov, D. B. Papkovsky and N. Y. Papkovskaia, Sens. Actuators, B, 2001, 81, 17–24 CrossRef CAS.
- V. I. Ogurtsov and D. B. Papkovsky, Sens. Actuators, B, 2006, 113, 917–929 CrossRef CAS PubMed.
- E. J. Bowen and A. Norton, Trans. Faraday Soc., 1939, 35, 44–48 RSC.
- H. Weil-Malherbe and J. Weiss, Nature, 1942, 149, 471–472 CrossRef CAS.
- H. W. Kroneis and H. J. Marsoner, Sens. Actuators, 1983, 4, 587–592 CrossRef CAS.
- T. Okazaki, T. Imasaka and N. Ishibashi, Anal. Chim. Acta, 1988, 209, 327–331 CrossRef CAS.
- M. E. Cox and B. Dunn, Appl. Opt., 1985, 24, 2114–2120 CrossRef CAS.
- B. J. Basu, C. Anandan and K. S. Rajam, Sens. Actuators, B, 2003, 94, 257–266 CrossRef CAS.
- B. J. Basu, A. Thirumurugan, A. R. Dinesh, C. Anandan and K. S. Rajam, Sens. Actuators, B, 2005, 104, 15–22 CrossRef CAS PubMed.
- J. Zilberstein, A. Bromberg and G. Berkovic, J. Photochem. Photobiol., A, 1994, 77, 69–81 CrossRef CAS.
- N. Opitz, H. J. Graf and D. W. Lübbers, Sens. Actuators, 1988, 13, 159–163 CrossRef CAS.
- T. Ishiji and M. Kaneko, Analyst, 1995, 120, 1633–1638 RSC.
- Y. Amao, I. Okura and T. Miyashita, Bull. Chem. Soc. Jpn., 2001, 74, 1159–1160 CrossRef CAS.
- Y. Fujiwara and Y. Amao, Sens. Actuators, B, 2002, 85, 175–178 CrossRef CAS.
- Y. Fujiwara, I. Okura, T. Miyashita and Y. Amao, Anal. Chim. Acta, 2002, 471, 25–32 CrossRef CAS.
- Y. Fujiwara and Y. Amao, Sens. Actuators, B, 2003, 89, 58–61 CrossRef CAS.
- Y. Fujiwara and Y. Amao, Sens. Actuators, B, 2003, 89, 187–191 CrossRef CAS.
- Y. Fujiwara and Y. Amao, Sens. Actuators, B, 2004, 99, 130–133 CrossRef CAS PubMed.
- D. Odaci, B. N. Gacal, B. Gacal, S. Timur and Y. Yagci, Biomacromolecules, 2009, 10, 2928–2934 CrossRef CAS PubMed.
- T. Abe, M. Matsuguchi and Y. Sakai, Chem. Senses, 2000, 16(suppl. B), 109–111 CAS.
- N. Geacintov, G. Oster and T. Cassen, J. Opt. Soc. Am., 1968, 58, 1217–1228 CrossRef CAS and references cited therein.
- P. H. Bolton, R. D. Kenner and A. U. Khan, J. Chem. Phys., 1972, 57, 5604–5605 CrossRef CAS PubMed.
- P. F. Jones and R. S. Nesbitt, J. Chem. Phys., 1973, 59, 6185–6186 CrossRef CAS PubMed.
- A. Sharma and O. S. Wolfbeis, Anal. Chim. Acta, 1988, 212, 261–265 CrossRef CAS.
- O. S. Wolfbeis and F. M. Carlini, Anal. Chim. Acta, 1984, 160, 301–304 CrossRef CAS.
- W. Trettnak, M. J. P. Leiner and O. S. Wolfbeis, Analyst, 1988, 113, 1519–1523 RSC.
- H. Marsoner, H. Kroneis and O. S. Wolfbeis, US Pat., 4,657736, 1987 Search PubMed.
- P. Y. F. Li and R. Narayanaswamy, Analyst, 1989, 114, 663–666 RSC.
- P. Y. F. Li and R. Narayanaswamy, Analyst, 1989, 114, 1191–1195 RSC.
- H.-W. Lin, M.-H. Huang, Y.-H. Chen, W.-C. Lin, H.-C. Cheng, C.-C. Wu, T.-C. Chao, T.-C. Wang, K.-T. Wong, K.-C. Tang and P.-T. Chou, J. Mater. Chem., 2012, 22, 13446–13450 RSC.
- S. Nagl, C. Baleizao, S. M. Borisov, M. Schaferling, M. N. Berberan-Santos and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2007, 46, 2317–2319 CrossRef CAS PubMed.
- C. Baleizao, S. Nagl, M. Schaferling, M. N. Berberan-Santos and O. S. Wolfbeis, Anal. Chem., 2008, 80, 6449–6457 CrossRef CAS PubMed.
- S. Kochmann, C. Baleizão, M. N. Berberan-Santos and O. S. Wolfbeis, Anal. Chem., 2013, 85, 1300–1304 CrossRef CAS PubMed.
- H. Kautsky and G. O. Müller, Z. Naturforsch., A: Astrophys., Phys. Phys. Chem., 1947, 2, 167–172 Search PubMed.
- I. A. Zakharov and T. I. Grishaeva, Zh. Prikl. Spektrosk., 1984, 57, 1240 CAS.
- I. A. Zakharov and T. I. Grishaeva, Zh. Anal. Khim., 1982, 37, 1753 CAS.
- J. M. Charlesworth, Sens. Actuators, B, 1994, 22, 1–5 CrossRef CAS.
- R. Badía, M. E. Díaz-García and A. Sanz-Medel, Microchim. Acta, 1995, 121, 51–61 CrossRef.
- A. Yekta, Z. Masoumi and M. A. Winnik, Can. J. Chem., 1995, 73, 2021–2029 CrossRef CAS PubMed.
- N. Kraus, G. Israel, F. W. Müller, K. Schiller and M. Rätzsch, Angew. Makromol. Chem., 1991, 192, 93–101 CrossRef CAS.
- S. K. Lam, M. A. Chan and D. Lo, Sens. Actuators, B, 2001, 73, 135–141 CrossRef CAS.
- D. W. Lübbers, N. Opitz, P. P. Speiser and H. J. Bisson, Z. Naturforsch., C: J. Biosci., 1977, 32, 133–134 Search PubMed.
- W. Xu, R. Schmidt, M. Whaley, J. N. Demas, B. A. DeGraff, E. K. Karikari and B. A. Famer, Anal. Chem., 1995, 67, 3172–3180 CrossRef CAS.
- R. Badia and M. E. D. Garcia, Anal. Lett., 2000, 33, 307–322 CrossRef CAS.
- M. A. Chan, J. L. Lawless, S. K. Lam and D. Lo, Anal. Chim. Acta, 2000, 408, 33–37 CrossRef CAS.
- J. I. Peterson and R. V. Fitzgerald, Rev. Sci. Instrum., 1980, 51, 670–671 CrossRef CAS PubMed.
- J. N. Demas, E. W. Harris and R. P. McBride, J. Am. Chem. Soc., 1977, 99, 3547–3551 CrossRef CAS.
- M. Z. Ongun, O. Oter, G. Sabancı, K. Ertekin and E. Celik, Sens. Actuators, B, 2013, 183, 11–19 CrossRef CAS PubMed.
- L. Li and D. R. Walt, Anal. Chem., 1995, 67, 3746–3752 CrossRef CAS.
- G. Kuncová and M. Fialová, Biotechnol. Tech., 1995, 9, 175–178 CrossRef.
- J. F. Gouin, F. Baros, D. Birot and J. C. André, Sens. Actuators, B, 1997, 39, 401–406 CrossRef CAS.
- Y. Amao and I. Okura, Sens. Actuators, B, 2003, 88, 162–167 CrossRef CAS.
- L. Liu, B. Li, R. F. Qin, H. F. Zhao, X. G. Ren and Z. M. Su, Nanotechnology, 2010, 21, 285701 CrossRef PubMed.
- M. V. Rigo, R. Olsson and P. Geissinger, Sens. Transducers J., 2010, 113, 18–32 CAS.
- G. Liebsch, I. Klimant and O. S. Wolfbeis, Adv. Mater., 1999, 11, 1296–1299 CrossRef CAS.
- M. E. Köse, B. F. Carroll and K. S. Schanze, Langmuir, 2005, 21, 9121–9129 CrossRef PubMed.
- A. S. Kocincova, S. M. Borisov, C. Krause and O. S. Wolfbeis, Anal. Chem., 2007, 79, 8486–8493 CrossRef CAS PubMed.
- H. Xu, J. W. Aylott, R. Kopelman, T. J. Miller and M. A. Philbert, Anal. Chem., 2001, 73, 4124–4133 CrossRef CAS.
- Z. J. Fuller, W. D. Bare, K. A. Kneas, W. Y. Xu, J. N. Demas and B. A. DeGraff, Anal. Chem., 2003, 75, 2670–2677 CrossRef CAS.
- I. Klimant and O. S. Wolfbeis, Anal. Chem., 1995, 67, 3160–3166 CrossRef CAS.
- F. J. Mingoarranz, M. C. Moreno-Bondi, D. García-Fresnadillo, C. de Dios and G. Orellana, Mikrochim. Acta, 1995, 121, 107–118 CrossRef CAS.
- J. F. Fernández-Sánchez, T. Roth, R. Cannas, M. K. Nazeeruddin, S. Spichiger, M. Graetzel and U. E. Spichiger-Keller, Talanta, 2007, 71, 242–250 CrossRef PubMed.
- I. Klimant, P. Belser and O. S. Wolfbeis, Talanta, 1994, 41, 985–991 CrossRef CAS.
- K. P. McNamara, X. Li, A. D. Stull and Z. Rosenzweig, Anal. Chim. Acta, 1998, 361, 73–83 CrossRef CAS.
- M. P. Xavier, D. García-Fresnadillo, M. C. Moreno-Bondi and G. Orellana, Anal. Chem., 1998, 70, 5184–5189 CrossRef CAS.
- C. Malins, H. G. Glever, B. D. MacCraith, S. Fanni and J. G. Vos, Anal. Commun., 1999, 36, 3–4 RSC.
- E. Holder, D. Oelkrug, H.-J. Egelhaaf, H. A. Mayer and E. Lindner, J. Fluoresc., 2002, 12, 383–395 CrossRef CAS.
- Z. Wang, A. R. McWilliams, C. E. B. Evans, X. Lu, S. Chung, M. A. Winnik and I. Manners, Adv. Funct. Mater., 2002, 12, 415–419 CrossRef CAS.
- P. Hartmann, M. J. P. Leiner and M. E. Lippitsch, Sens. Actuators, B, 1995, 29, 251–257 CrossRef CAS.
- S. J. Payne, G. L. Fiore, C. L. Fraser and J. N. Demas, Anal. Chem., 2010, 82, 917–921 CrossRef CAS PubMed.
- K. A. McGee, D. J. Veltkamp, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2007, 129, 15092–15093 CrossRef CAS PubMed.
- K. A. McGee, B. J. Marquardt and K. R. Mann, Inorg. Chem., 2008, 47, 9143–9145 CrossRef CAS PubMed.
- K. A. McGee and K. R. Mann, J. Am. Chem. Soc., 2009, 131, 1896–1902 CrossRef CAS PubMed.
- X.-d. Wang, D. E. Achatz, C. Hupf, M. Sperber, J. Wegener, S. Bange, J. M. Lupton and O. S. Wolfbeis, Sens. Actuators, B, 2013, 188, 257–262 CrossRef CAS PubMed.
- D. Xiao, Y. Mo and M. M. F. Choi, Meas. Sci. Technol., 2003, 14, 862 CrossRef CAS.
- D. S. Daivasagaya, L. Yao, K. Y. Yung, M. Hajj-Hassan, M. C. Cheung, V. P. Chodavarapu and F. V. Bright, Sens. Actuators, B, 2011, 157, 408–416 CrossRef CAS PubMed.
- C. Preininger, I. Klimant and O. S. Wolfbeis, Anal. Chem., 1994, 66, 1841–1846 CrossRef CAS.
- Z. Rosenzweig and R. Kopelman, Anal. Chem., 1995, 67, 2650–2654 CrossRef CAS.
- A. Mills and M. Thomas, Analyst, 1997, 122, 63–68 RSC.
- Y. Amao and I. Okura, Polym. J., 2000, 32, 452–455 CrossRef CAS.
- A. M. Morin, W. Xu, J. N. Demas and B. A. DeGraff, J. Fluoresc., 2000, 10, 7–12 CrossRef CAS.
- H. He, R. J. Fraatz, M. J. P. Leiner, M. M. Rehn and J. K. Tusa, Sens. Actuators, B, 1995, 29, 246–250 CrossRef CAS.
- C.-M. Chan, M.-Y. Chan, M. Zhang, W. Lo and K.-Y. Wong, Analyst, 1999, 124, 691–694 RSC.
- M. M. F. Choi and D. Xiao, Anal. Chim. Acta, 1999, 387, 197–205 CrossRef CAS.
- A. K. McEvoy, C. McDonagh and B. D. MacCraith, J. Sol–Gel Sci. Technol., 1997, 8, 1121–1125 CAS.
- B. D. MacCraith, C. M. McDonagh, G. O'Keeffe, E. T. Keyes, J. G. Vos, B. O'Kelly and J. F. McGilp, Analyst, 1993, 118, 385–388 RSC.
- C. McDonagh, C. Kolle, A. K. McEvoy, D. L. Dowling, A. A. Cafolla, S. J. Cullen and B. D. MacCraith, Sens. Actuators, B, 2001, 74, 124–130 CrossRef CAS.
- Y. Tang, E. C. Tehan, Z. Tao and F. V. Bright, Anal. Chem., 2003, 75, 2407–2413 CrossRef CAS.
- I. Klimant, F. Ruckruh, G. Liebsch, C. Stangelmayer and O. S. Wolfbeis, Mikrochim. Acta, 1999, 131, 35–46 CrossRef CAS.
- X. Chen, Z. Zhong, Z. Li, Y. Jiang, X. Wang and K. Wong, Sens. Actuators, B, 2002, 87, 233–238 CrossRef CAS.
- A. Apostolidis, I. Klimant, D. Andrzejewski and O. S. Wolfbeis, J. Comb. Chem., 2004, 6, 325–331 CrossRef CAS PubMed.
- K. Ertekin, S. Kocak, M. Sabih Ozer, S. Aycan and B. Cetinkaya, Talanta, 2003, 61, 573–579 CrossRef CAS.
- G. Di Marco, M. Lanza and S. Campagna, Adv. Mater., 1995, 7, 468–471 CrossRef CAS.
- B. Meier, T. Werner, I. Klimant and O. S. Wolfbeis, Sens. Actuators, B, 1995, 29, 240–245 CrossRef CAS.
- P. Zhang, J. Guo, Y. Wang and W. Pang, Mater. Lett., 2002, 53, 400–405 CrossRef CAS.
- M. Ahmad, N. Mohammad and J. Abdullah, J. Non-Cryst. Solids, 2001, 290, 86–91 CrossRef CAS.
- D. García-Fresnadillo, M. D. Marazuela, M. C. Moreno-Bondi and G. Orellana, Langmuir, 1999, 15, 6451–6459 CrossRef.
- N. Velasco-Garcia, M. J. Valencia-Gonzalez and M. E. Diaz-Garcia, Analyst, 1997, 122, 1405–1410 RSC.
- J. Ji, N. Rosenzweig, I. Jones and Z. Rosenzweig, J. Biomed. Opt., 2002, 7, 404–409 CrossRef CAS PubMed.
- J. Ji, N. Rosenzweig, I. Jones and Z. Rosenzweig, Anal. Chem., 2001, 73, 3521–3527 CrossRef CAS.
- G. Di Marco, M. Lanza, M. Pieruccini and S. Campagna, Adv. Mater., 1996, 8, 576–580 CrossRef CAS.
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265–287 RSC.
- M.-L. Ho, J.-C. Wang, T.-Y. Wang, C.-Y. Lin, J. F. Zhu, Y.-A. Chen and T.-C. Chen, Sens. Actuators, B, 2014, 190, 479–485 CrossRef CAS PubMed.
- G. Di Marco, M. Lanza, A. Mamo, I. Stefio, C. Di Pietro, G. Romeo and S. Campagna, Anal. Chem., 1998, 70, 5019–5023 CrossRef CAS PubMed.
- Y. Amao, Y. Ishikawa and I. Okura, Anal. Chim. Acta, 2001, 445, 177–182 CrossRef CAS.
- L. Huynh, Z. Wang, J. Yang, V. Stoeva, A. Lough, I. Manners and M. A. Winnik, Chem. Mater., 2005, 17, 4765–4773 CrossRef CAS.
- P. Steunenberg, A. Ruggi, N. S. van den Berg, T. Buckle, J. Kuil, F. W. B. van Leeuwen and A. H. Velders, Inorg. Chem., 2012, 51, 2105–2114 CrossRef CAS PubMed.
- C. S. K. Mak, D. Pentlehner, M. Stich, O. S. Wolfbeis, W. K. Chan and H. Yersin, Chem. Mater., 2009, 21, 2173–2175 CrossRef CAS.
- S. M. Borisov and I. Klimant, Anal. Chem., 2007, 79, 7501–7509 CrossRef CAS PubMed.
- B. H. Leavens, C. Trindle, M. Sabat, Z. Altun, J. Demas and B. A. DeGraff, J. Fluoresc., 2012, 22, 163–174 CrossRef CAS PubMed.
- P. Jin, Z. Guo, J. Chu, J. Tan, S. Zhang and W. Zhu, Ind. Eng. Chem. Res., 2013, 52, 3980–3987 CrossRef CAS.
- S. M. Borisov and I. Klimant, Microchim. Acta, 2009, 164, 7–15 CrossRef CAS.
- A. L. Medina-Castillo, J. F. Fernandez-Sanchez, C. Klein, M. K. Nazeeruddin, A. Segura-Carretero, A. Fernandez-Gutierrez, M. Graetzel and U. E. Spichiger-Keller, Analyst, 2007, 132, 929–936 RSC.
- B. Carlson, B. E. Eichinger, W. Kaminsky and G. D. Phelan, Sens. Actuators, B, 2010, 145, 278–284 CrossRef CAS PubMed.
- A. Ruggi, M. Berenguel Alonso, D. N. Reinhoudt and A. H. Velders, Chem. Commun., 2010, 46, 6726–6728 RSC.
- M. C. DeRosa, D. J. Hodgson, G. D. Enright, B. Dawson, C. E. B. Evans and R. J. Crutchley, J. Am. Chem. Soc., 2004, 126, 7619–7626 CrossRef CAS PubMed.
- A. Habibagahi, Y. Mébarki, Y. Sultan, G. P. A. Yap and R. J. Crutchley, ACS Appl. Mater. Interfaces, 2009, 1, 1785–1792 CAS.
- Y.-K. Peng, C.-W. Lai, C.-L. Liu, H.-C. Chen, Y.-H. Hsiao, W.-L. Liu, K.-C. Tang, Y. Chi, J.-K. Hsiao, K.-E. Lim, H.-E. Liao, J.-J. Shyue and P.-T. Chou, ACS Nano, 2011, 5, 4177–4187 CrossRef CAS PubMed.
- M. C. DeRosa, P. J. Mosher, G. P. A. Yap, K. S. Focsaneanu, R. J. Crutchley and C. E. B. Evans, Inorg. Chem., 2003, 42, 4864–4872 CrossRef CAS PubMed.
- M.-L. Ho, Y.-A. Chen, T.-C. Chen, P.-J. Chang, Y.-P. Yu, K.-Y. Cheng, C.-H. Shih, G.-H. Lee and H.-S. Sheu, Dalton Trans., 2012, 41, 2592–2600 RSC.
- M. Marín-Suárez, B. F. E. Curchod, I. Tavernelli, U. Rothlisberger, R. Scopelliti, I. Jung, D. Di Censo, M. Grätzel, J. F. Fernández-Sánchez, A. Fernández-Gutiérrez, M. K. Nazeeruddin and E. Baranoff, Chem. Mater., 2012, 24, 2330–2338 CrossRef.
- L. H. Fischer, M. I. J. Stich, O. S. Wolfbeis, N. Tian, E. Holder and M. Schaferling, Chem.–Eur. J., 2009, 15, 10857–10863 CrossRef CAS PubMed.
- E. M. Kober, J. L. Marshall, W. J. Dressick, B. P. Sullivan, J. V. Caspar and T. J. Meyer, Inorg. Chem., 1985, 24, 2755–2763 CrossRef CAS.
- S. B. Bambot, G. Rao, M. Romauld, G. M. Carter, J. Sipior, E. Terpetchnig and J. R. Lakowicz, Biosens. Bioelectron., 1995, 10, 643–652 CrossRef CAS.
- W. Xu, K. A. Kneas, J. N. Demas and B. A. DeGraff, Anal. Chem., 1996, 68, 2605–2609 CrossRef CAS PubMed.
- L. Sacksteder, J. N. Demas and B. A. DeGraff, Anal. Chem., 1993, 65, 3480–3483 CrossRef CAS.
- L. Sacksteder, M. Lee, J. N. Demas and B. A. DeGraff, J. Am. Chem. Soc., 1993, 115, 8230–8238 CrossRef CAS.
- K. A. Kneas, W. Xu, J. N. Demas, B. A. DeGraff and A. P. Zipp, J. Fluoresc., 1998, 8, 295–300 CrossRef CAS.
- Y. Shen, B. P. Maliwal and J. R. Lakowicz, J. Fluoresc., 2001, 11, 315–318 CrossRef CAS.
- T. M. McLean, J. L. Moody, M. R. Waterland and S. G. Telfer, Inorg. Chem., 2012, 51, 446–455 CrossRef CAS PubMed.
- C.-E. Wang, J. Lumin., 2014, 145, 531–538 CrossRef CAS PubMed.
- H. Hong, L. Zhu, A. Wang and H. Lu, Spectrochim. Acta, Part A, 2012, 98, 466–473 CrossRef CAS PubMed.
- G. Xu, M. Lu, C. Huang, Y. Wang and S. Ge, Spectrochim. Acta, Part A, 2014, 123, 369–375 CrossRef CAS PubMed.
- Y. Amao, I. Okura and T. Miyashita, Bull. Chem. Soc. Jpn., 2000, 73, 2663–2668 CrossRef CAS.
- Y. Amao, I. Okura and T. Miyashita, Chem. Lett., 2000, 29, 934–935 CrossRef.
- Y. Li, Spectrochim. Acta, Part A, 2011, 79, 356–360 CrossRef CAS PubMed.
- Q. Zuo, B. Li, L. Zhang, Y. Wang, Y. Liu, J. Zhang, Y. Chen and L. Guo, J. Solid State Chem., 2010, 183, 1715–1720 CrossRef CAS PubMed.
- S. Li and X. Zhao, Synth. Met., 2011, 161, 737–742 CrossRef CAS PubMed.
- N. Feng, J. Xie and D. Zhang, Spectrochim. Acta, Part A, 2010, 77, 292–296 CrossRef PubMed.
- Y. Amao, I. Okura and T. Miyashita, Chem. Lett., 2000, 29, 1286–1287 CrossRef.
- Y. Amao, Y. Ishikawa, I. Okura and T. Miyashita, Bull. Chem. Soc. Jpn., 2001, 74, 2445–2449 CrossRef CAS.
- G. L. Law, R. Pal, L. O. Palsson, D. Parker and K. L. Wong, Chem. Commun., 2009, 7321–7323 RSC.
- S. Borisov and I. Klimant, Anal. Bioanal. Chem., 2012, 404, 2797–2806 CrossRef CAS PubMed.
- Y. Wang, B. Li, L. Zhang, Q. Zuo, P. Li, J. Zhang and Z. Su, ChemPhysChem, 2011, 12, 349–355 CrossRef CAS PubMed.
- O. Burkhard and W. K. R. Barnikol, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 1719 Search PubMed.
- B. W. Atwater, J. Fluoresc., 1992, 2, 237–246 CrossRef CAS PubMed.
- D. Eastwood and M. Gouterman, J. Mol. Spectrosc., 1970, 35, 359–375 CrossRef CAS.
- W. Rumsey, J. Vanderkooi and D. Wilson, Science, 1988, 241, 1649–1651 CAS.
- R. B. Beswick and C. W. Pitt, Chem. Phys. Lett., 1988, 143, 589–592 CrossRef CAS.
- J. Kavandi, J. Callis, M. Gouterman, G. Khalil, D. Wright, E. Green, D. Burns and B. McLachlan, Rev. Sci. Instrum., 1990, 61, 3340–3347 CrossRef PubMed.
- A. K. Bansal, W. Holzer, A. Penzkofer and T. Tsuboi, Chem. Phys., 2006, 330, 118–129 CrossRef CAS PubMed.
- W. W.-S. Lee, K.-Y. Wong, X.-M. Li, Y.-B. Leung, C.-S. Chan and K. S. Chan, J. Mater. Chem., 1993, 3, 1031–1035 RSC.
- S.-K. Lee and I. Okura, Anal. Commun., 1997, 34, 185–188 RSC.
- Y. Amao, T. Miyashita and I. Okura, J. Fluorine Chem., 2001, 107, 101–106 CrossRef CAS.
- T. Mayr, S. M. Borisov, T. Abel, B. Enko, K. Waich, G. Mistlberger and I. Klimant, Anal. Chem., 2009, 81, 6541–6545 CrossRef CAS.
- S. M. Borisov, P. Lehner and I. Klimant, Anal. Chim. Acta, 2011, 690, 108–115 CrossRef CAS PubMed.
- B. Zelelow, G. E. Khalil, G. Phelan, B. Carlson, M. Gouterman, J. B. Callis and L. R. Dalton, Sens. Actuators, B, 2003, 96, 304–314 CrossRef CAS.
- G. E. Khalil, C. Costin, J. Crafton, G. Jones, S. Grenoble, M. Gouterman, J. B. Callis and L. R. Dalton, Sens. Actuators, B, 2004, 97, 13–21 CrossRef CAS.
- G. Martin, C. James, D. Larry, K. Gamal, M. Youssef, R. C. Kevin and G. Michel, Meas. Sci. Technol., 2004, 15, 1986 CrossRef.
- G. Khalil, M. Gouterman, S. Ching, C. Costin, L. Coyle, S. Gouin, E. Green, M. Sadilek, R. Wan and J. Yearyean, J. Porphyrins Phthalocyanines, 2002, 6, 135–145 CrossRef CAS.
- S. M. Borisov, G. Nuss and I. Klimant, Anal. Chem., 2008, 80, 9435–9442 CrossRef CAS PubMed.
- S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Klimant, J. Photochem. Photobiol., A, 2009, 201, 128–135 CrossRef CAS PubMed.
- S. M. Borisov, D. B. Papkovsky, G. V. Ponomarev, A. S. DeToma, R. Saf and I. Klimant, J. Photochem. Photobiol., A, 2009, 206, 87–92 CrossRef CAS PubMed.
- D. B. Papkovsky, G. V. Ponomarev, W. Trettnak and P. O'Leary, Anal. Chem., 1995, 67, 4112–4117 CrossRef CAS.
- D. B. Papkovsky, Sens. Actuators, B, 1995, 29, 213–218 CrossRef CAS.
- S. M. Borisov, G. Zenkl and I. Klimant, ACS Appl. Mater. Interfaces, 2010, 2, 366–374 CAS.
- O. S. Finikova, A. V. Cheprakov and S. A. Vinogradov, J. Org. Chem., 2005, 70, 9562–9572 CrossRef CAS PubMed.
- F. Niedermair, S. M. Borisov, G. Zenkl, O. T. Hofmann, H. r. Weber, R. Saf and I. Klimant, Inorg. Chem., 2010, 49, 9333–9342 CrossRef CAS PubMed.
- W. Wu, W. Wu, S. Ji, H. Guo, X. Wang and J. Zhao, Dyes Pigm., 2011, 89, 199–211 CrossRef CAS PubMed.
- K. Koren, S. M. Borisov, R. Saf and I. Klimant, Eur. J. Inorg. Chem., 2011, 1531–1534 CrossRef CAS PubMed.
- K. Koren, R. I. Dmitriev, S. M. Borisov, D. B. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184–1190 CrossRef CAS PubMed.
- V. V. Vasil'ev and S. M. Borisov, Sens. Actuators, B, 2002, 82, 272–276 CrossRef CAS.
- P. Gewehr and D. Delpy, Med. Biol. Eng. Comput., 1993, 31, 11–21 CAS.
- T. C. O'Riordan, A. V. Zhdanov, G. V. Ponomarev and D. B. Papkovsky, Anal. Chem., 2007, 79, 9414–9419 CrossRef CAS PubMed.
- T. C. O'Riordan, K. Fitzgerald, G. V. Ponomarev, J. Mackrill, J. Hynes, C. Taylor and D. B. Papkovsky, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2007, 292, R1613–R1620 CrossRef CAS PubMed.
- Y. Will, J. Hynes, V. I. Ogurtsov and D. B. Papkovsky, Nat. Protoc., 2006, 1, 2563–2572 CrossRef CAS PubMed.
- R. D. Shonat, K. N. Richmond and P. C. Johnson, Rev. Sci. Instrum., 1995, 66, 5075–5084 CrossRef CAS PubMed.
- L.-W. Lo, C. J. Koch and D. F. Wilson, Anal. Biochem., 1996, 236, 153–160 CrossRef CAS PubMed.
- T. Saito, N. Asakura, T. Kamachi and I. Okura, J. Porphyrins Phthalocyanines, 2007, 11, 160–164 CrossRef CAS.
- Y. Amao, K. Asai and I. Okura, Anal. Commun., 1999, 36, 179–180 RSC.
- Y. Amao and I. Okura, Analyst, 2000, 125, 1601–1604 RSC.
- Y. Amao, K. Asai and I. Okura, J. Porphyrins Phthalocyanines, 2000, 4, 179–184 CrossRef CAS.
- A. Fercher, G. V. Ponomarev, D. Yashunski and D. Papkovsky, Anal. Bioanal. Chem., 2010, 396, 1793–1803 CrossRef CAS PubMed.
- R. I. Dmitriev, A. V. Zhdanov, G. V. Ponomarev, D. V. Yashunski and D. B. Papkovsky, Anal. Biochem., 2010, 398, 24–33 CrossRef CAS PubMed.
- M. Obata, Y. Tanaka, N. Araki, S. Hirohara, S. Yano, K. Mitsuo, K. Asai, M. Harada, T. Kakuchi and C. Ohtsuki, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2997–3006 CrossRef CAS.
- Y. Q. Tian, B. R. Shumway, A. C. Youngbull, Y. Z. Li, A. K. Y. Jen, R. H. Johnson and D. R. Meldrum, Sens. Actuators, B, 2010, 147, 714–722 CrossRef CAS PubMed.
- Y. Tian, B. R. Shumway and D. R. Meldrum, Chem. Mater., 2010, 22, 2069–2078 CrossRef CAS PubMed.
- H. Lu, Y. Jin, Y. Tian, W. Zhang, M. R. Holl and D. R. Meldrum, J. Mater. Chem., 2011, 21, 19293–19301 RSC.
- Y. Tian, B. R. Shumway, W. Gao, C. Youngbull, M. R. Holl, R. H. Johnson and D. R. Meldrum, Sens. Actuators, B, 2010, 150, 579–587 CrossRef CAS PubMed.
- K. Koren, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2012, 169, 173–181 CrossRef CAS PubMed.
- R. P. Briñas, T. Troxler, R. M. Hochstrasser and S. A. Vinogradov, J. Am. Chem. Soc., 2005, 127, 11851–11862 CrossRef PubMed.
- S. Sakadzic, E. Roussakis, M. A. Yaseen, E. T. Mandeville, V. J. Srinivasan, K. Arai, S. Ruvinskaya, A. Devor, E. H. Lo, S. A. Vinogradov and D. A. Boas, Nat. Methods, 2010, 7, 755–759 CrossRef CAS PubMed.
- O. S. Finikova, A. Y. Lebedev, A. Aprelev, T. Troxler, F. Gao, C. Garnacho, S. Muro, R. M. Hochstrasser and S. A. Vinogradov, ChemPhysChem, 2008, 9, 1673–1679 CrossRef CAS PubMed.
- O. S. Finikova, T. Troxler, A. Senes, W. F. DeGrado, R. M. Hochstrasser and S. A. Vinogradov, J. Phys. Chem. A, 2007, 111, 6977–6990 CrossRef CAS PubMed.
- J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen, S. A. Vinogradov and S. Charpak, Nat. Med., 2011, 17, 893–898 CrossRef CAS PubMed.
- R. C. Evans, P. Douglas, J. A. G. Williams and D. L. Rochester, J. Fluoresc., 2006, 16, 201–206 CrossRef CAS PubMed.
- S. R. Ricketts and P. Douglas, Sens. Actuators, B, 2008, 135, 46–51 CrossRef CAS PubMed.
- X. D. Wang, X. Chen, Z. X. Xie and X. R. Wang, Angew. Chem., Int. Ed., 2008, 47, 7450–7453 CrossRef CAS PubMed.
- X. D. Wang, R. J. Meier, M. Link and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2010, 49, 4907–4909 CrossRef CAS PubMed.
- R. A. Potyrailo and G. M. Hieftje, Anal. Chim. Acta, 1998, 370, 1–8 CrossRef CAS.
- S.-K. Lee and I. Okura, Anal. Chim. Acta, 1997, 342, 181–188 CrossRef CAS.
- S. K. Lee and I. Okura, Analyst, 1997, 122, 81–84 RSC.
- I. Klimant, M. Kühl, R. N. Glud and G. Holst, Sens. Actuators, B, 1997, 38, 29–37 CrossRef CAS.
- Y. Amao, T. Miyashita and I. Okura, React. Funct. Polym., 2001, 47, 49–54 CrossRef CAS.
- Y. Amao, K. Asai, T. Miyashita and I. Okura, Polym. Adv. Technol., 2000, 11, 705–709 CrossRef CAS.
- Y. Amao, T. Miyashita and I. Okura, Analyst, 2000, 125, 871–875 RSC.
- Y. Amao, K. Asai, I. Okura, H. Shinohara and H. Nishide, Analyst, 2000, 125, 1911–1914 RSC.
- R. N. Gillanders, M. C. Tedford, P. J. Crilly and R. T. Bailey, Anal. Chim. Acta, 2004, 502, 1–6 CrossRef CAS PubMed.
- G. DiMarco and M. Lanza, Sens. Actuators, B, 2000, 63, 42–48 CrossRef CAS.
- P. Douglas and K. Eaton, Sens. Actuators, B, 2002, 82, 200–208 CrossRef CAS.
- X. Lu, I. Manners and M. A. Winnik, Macromolecules, 2001, 34, 1917–1927 CrossRef CAS.
- Y. E. L. Koo, Y. F. Cao, R. Kopelman, S. M. Koo, M. Brasuel and M. A. Philbert, Anal. Chem., 2004, 76, 2498–2505 CrossRef CAS PubMed.
- Y. Amao, Y. Tabuchi, Y. Yamashita and K. Kimura, Eur. Polym. J., 2002, 38, 675–681 CrossRef CAS.
- A. Sheila Holmes-Smith, A. Hamill, M. Campbell and M. Uttamlal, Analyst, 1999, 124, 1463–1466 RSC.
- A. Mills and A. Lepre, Anal. Chem., 1997, 69, 4653–4659 CrossRef CAS.
- Z. Pang, X. Gu, A. Yekta, Z. Masoumi, J. B. Coll, M. A. Winnik and I. Manners, Adv. Mater., 1996, 8, 768–771 CrossRef CAS.
- N. B. Borchert, G. V. Ponomarev, J. P. Kerry and D. B. Papkovsky, Anal. Chem., 2010, 83, 18–22 CrossRef PubMed.
- P. Hartmann and W. Trettnak, Anal. Chem., 1996, 68, 2615–2620 CrossRef CAS PubMed.
- C. Kolle, W. Gruber, W. Trettnak, K. Biebernik, C. Dolezal, F. Reininger and P. O'Leary, Sens. Actuators, B, 1997, 38, 141–149 CrossRef CAS.
- M. Trinkel, W. Trettnak and C. Kolle, Quim. Anal., 2000, 19, 112–117 CAS.
- K. Õige, T. Avarmaa, A. Suisalu and R. Jaaniso, Sens. Actuators, B, 2005, 106, 424–430 CrossRef PubMed.
- T.-S. Yeh, C.-S. Chu and Y.-L. Lo, Sens. Actuators, B, 2006, 119, 701–707 CrossRef CAS PubMed.
- C.-S. Chu, Y.-L. Lo and T.-W. Sung, Talanta, 2010, 82, 1044–1051 CrossRef CAS PubMed.
- C.-S. Chu and Y.-L. Lo, Sens. Actuators, B, 2007, 124, 376–382 CrossRef CAS PubMed.
- C. S. Chu and Y. L. Lo, Sens. Actuators, B, 2011, 155, 53–57 CrossRef CAS PubMed.
- Y. Amao, T. Miyashita and I. Okura, J. Porphyrins Phthalocyanines, 2001, 5, 433–438 CrossRef CAS.
- Y. Amao, K. Asai, K. Miyakawa and I. Okura, J. Porphyrins Phthalocyanines, 2000, 4, 19–22 CrossRef CAS.
- Y. Amao, K. Miyakawa and I. Okura, J. Mater. Chem., 2000, 10, 305–308 RSC.
- A. J. Twarowski and L. Good, J. Phys. Chem., 1987, 91, 5252–5257 CrossRef CAS.
- D. B. Papkovsky, J. Olah, I. V. Troyanovsky, N. A. Sadovsky, V. D. Rumyantseva, A. F. Mironov, A. I. Yaropolov and A. P. Savitsky, Biosens. Bioelectron., 1992, 7, 199–206 CrossRef.
- M. Mitsuishi, H. Tanaka, M. Obata and T. Miyashita, Langmuir, 2010, 26, 15117–15120 CrossRef CAS PubMed.
- Y. Amao, K. Asai and I. Okura, Anal. Chim. Acta, 2000, 407, 41–44 CrossRef CAS.
- Y. M. Liu, R. Pereiro-Garcia, M. J. Valencia-Gonzalez, M. E. Diaz-Garcia and A. Sanz-Medel, Anal. Chem., 1994, 66, 836–840 CrossRef CAS.
- F. Alava-Moreno, M. J. Valencia-González, A. Sanz-Medel and M. E. Díaz-García, Analyst, 1997, 122, 807–810 RSC.
- J. M. Costa-Fernández, M. E. Diaz-García and A. Sanz-Medel, Anal. Chim. Acta, 1998, 360, 17–26 CrossRef.
- J. M. Costa-Fernández, N. Bordel, J. C. Campo, F. J. Ferrero, M. A. Pérez and A. Sanz-Medel, Mikrochim. Acta, 2000, 134, 145–152 CrossRef.
- J. Díaz-García, J. M. Costa-Fernández, N. Bordel-García and A. Sanz-Medel, Anal. Chim. Acta, 2001, 429, 55–64 CrossRef.
- A. Mills, A. Lepre, B. R. C. Theobald, E. Slade and B. A. Murrer, Anal. Chem., 1997, 69, 2842–2847 CrossRef CAS.
- A. Mills, C. Tommons, R. T. Bailey, P. Crilly and M. C. Tedford, Anal. Chim. Acta, 2011, 702, 269–273 CrossRef CAS PubMed.
- H. Jiang, H. Yang, F. Liu and Y. Li, J. Lumin., 2012, 132, 198–204 CrossRef PubMed.
- C. S. Smith and K. R. Mann, Chem. Mater., 2009, 21, 5042–5049 CrossRef CAS.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079–14085 CrossRef CAS PubMed.
- C. Wen, G. Tao, X. Xu, X. Feng and R. Luo, Spectrochim. Acta, Part A, 2011, 79, 1345–1351 CrossRef CAS PubMed.
- C. S. Smith and K. R. Mann, J. Am. Chem. Soc., 2012, 134, 8786–8789 CrossRef CAS PubMed.
- J. Jia, Y. Tian and Z. Li, Synth. Met., 2011, 161, 1377–1382 CrossRef CAS PubMed.
- W. W. S. Lee, K. Y. Wong and X. M. Li, Anal. Chem., 1993, 65, 255–258 CrossRef CAS.
- C. Liu, X. Song, X. Rao, Y. Xing, Z. Wang, J. Zhao and J. Qiu, Dyes Pigm., 2014, 101, 85–92 CrossRef CAS PubMed.
- A. Vogler and H. Kunkely, in Top. Curr. Chem., ed. H. Yersin, Springer, Berlin/Heidelberg, 2001, vol. 213, pp. 143–182 Search PubMed.
- M. T. Miller and T. B. Karpishin, Sens. Actuators, B, 1999, 61, 222–224 CrossRef CAS.
- E. Vander Donckt, B. Camerman, R. Herne and R. Vandeloise, Sens. Actuators, B, 1996, 32, 121–127 CrossRef CAS.
- X.-M. Li and K.-Y. Wong, Anal. Chim. Acta, 1992, 262, 27–32 CrossRef CAS.
- Y. S. Xia, T. L. Zhang, X. L. Diao and C. Q. Zhu, Chem. Lett., 2007, 36, 242–243 CrossRef CAS.
- E. D. Lee, T. C. Werner and W. R. Seitz, Anal. Chem., 1987, 59, 279–283 CrossRef CAS.
- Y. Kostov, P. Harms, R. S. Pilato and G. Rao, Analyst, 2000, 125, 1175–1178 RSC.
- Y. Kostov and G. Rao, Sens. Actuators, B, 2003, 90, 139–142 CrossRef CAS.
- Y. Kostov, Z. Gryczynski and G. Rao, Anal. Chem., 2002, 74, 2167–2171 CrossRef CAS.
- Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775–9786 RSC.
- X. Liu, W. Sun, L. Zou, Z. Xie, X. Li, C. Lu, L. Wang and Y. Cheng, Dalton Trans., 2012, 41, 1312–1319 RSC.
- C. A. Parker and W. J. Barnes, Analyst, 1957, 82, 606–618 RSC.
- A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS PubMed.
- H. Xiang, L. Zhou, Y. Feng, J. Cheng, D. Wu and X. Zhou, Inorg. Chem., 2012, 51, 5208–5212 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- H. D. Hendricks, Mol. Phys., 1971, 20, 189–191 CrossRef.
- M. Anni and R. Rella, J. Phys. Chem. B, 2009, 114, 1559–1561 CrossRef PubMed.
- M. R. Ayers and A. J. Hunt, J. Non-Cryst. Solids, 1998, 225, 343–347 CrossRef CAS.
- Y. Zhang and D. T. Johnson, Anal. Chim. Acta, 2004, 511, 333–337 CrossRef CAS PubMed.
- R. Ghosh, Appl. Phys. Lett., 1999, 75, 2885 CrossRef CAS PubMed.
- R. N. Ghosh, D. Osborn and G. L. Baker, Proc. IEEE Sens. 2003, IEEE Int. Conf. Sens., 2nd, 2003, 2, 807–808 CAS.
- R. N. Ghosh, P. A. Askeland, S. Kramer and R. Loloee, Appl. Phys. Lett., 2011, 98, 221103 CrossRef PubMed.
- J. T. Remillard, B. D. Poindexter and W. H. Weber, Appl. Opt., 1997, 36, 3699–3707 CrossRef CAS.
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699–766 CrossRef CAS PubMed.
- G. Korotcenkov, Handbook of Gas Sensor Materials, Springer, New York, 2014, pp. 209–222 Search PubMed.
- M. I. J. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102–3114 RSC.
- M. Cox and B. Dunn, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 621–636 CrossRef CAS.
- M. E. Cox and B. Dunn, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2395–2400 CrossRef CAS.
- G. Shaw, Trans. Faraday Soc., 1967, 63, 2181–2189 RSC.
- J. W. Parker and M. E. Cox, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1179–1188 CrossRef CAS.
- M. Kubin and P. Spacek, Coll. Czechpsloy. Chem. Commun., 1965, 30, 3294 CAS.
- D. Badocco and P. Pastore, Anal. Chem., 2008, 80, 2091–2096 CrossRef CAS PubMed.
- D. Badocco, A. Mondin and P. Pastore, Sens. Actuators, B, 2012, 163, 165–170 CrossRef CAS PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer handbook, Wiley, New York, 1999, vol. 1 and 2 Search PubMed.
- G. J. Mohr, T. Werner, I. Oehme, C. Preininger, I. Klimant, B. Kovacs and O. S. Wolfbeis, Adv. Mater., 1997, 9, 1108–1113 CrossRef CAS.
- E. R. Carraway, J. N. Demas and B. A. DeGraff, Langmuir, 1991, 7, 2991–2998 CrossRef CAS.
- S. B. Bambot, R. Holavanahali, J. R. Lakowicz, G. M. Carter and G. Rao, Biotechnol. Bioeng., 1994, 43, 1139–1145 CrossRef CAS PubMed.
- M. E. Köse, R. J. Crutchley, M. C. DeRosa, N. Ananthakrishnan, J. R. Reynolds and K. S. Schanze, Langmuir, 2005, 21, 8255–8262 CrossRef PubMed.
- K. Eaton and P. Douglas, Sens. Actuators, B, 2002, 82, 94–104 CrossRef CAS.
- X. Yang, L. Peng, L. Yuan, P. Teng, F. Tian, L. Li and S. Luo, Opt. Commun., 2011, 284, 3462–3466 CrossRef CAS PubMed.
- A. Mills and M. D. Thomas, Analyst, 1998, 123, 1135–1140 RSC.
- J. Brandup, E. Immergut and E. Grulke, Polymer handbook, John Wiley & Sons, New York, 1989, pp. 435–449 Search PubMed.
- D. B. Papkovsky, G. J. Mohr and O. S. Wolfbeis, Anal. Chim. Acta, 1997, 337, 201–205 CrossRef CAS.
- G. Lashkov and A. Kavtrev, Polym. Sci. U.S.S.R., 1986, 28, 1885–1891 CrossRef.
- K. A. Kneas, W. Xu, J. N. Demas and B. A. Degraff, Appl. Spectrosc., 1997, 51, 1346–1351 CrossRef CAS.
- K. Koren, L. Hutter, B. Enko, A. Pein, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2013, 176, 344–350 CrossRef CAS PubMed.
- W. Trettnak, C. Kolle, F. Reininger, C. Dolezal and P. O'Leary, Sens. Actuators, B, 1996, 36, 506–512 CrossRef CAS.
- R. Ruffolo, C. E. B. Evans, X.-H. Liu, Y. Ni, Z. Pang, P. Park, A. R. McWilliams, X. Gu, X. Lu, A. Yekta, M. A. Winnik and I. Manners, Anal. Chem., 2000, 72, 1894–1904 CrossRef CAS.
- C. Kelly, C. Toncelli, J. Kerry and D. B. Papkovsky, J. Mater. Chem. C, 2014 10.1039/C3TC32529F.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- D. Badocco, A. Mondin, P. Pastore, S. Voltolina and S. Gross, Anal. Chim. Acta, 2008, 627, 239–246 CrossRef CAS PubMed.
- S. M. Borisov and O. S. Wolfbeis, Anal. Chem., 2006, 78, 5094–5101 CrossRef CAS PubMed.
- S. M. Borisov, A. S. Vasylevska, C. Krause and O. S. Wolfbeis, Adv. Funct. Mater., 2006, 16, 1536–1542 CrossRef CAS.
- M. I. J. Stich, M. Schaeferling and O. S. Wolfbeis, Adv. Mater., 2009, 21, 2216–2220 CrossRef CAS.
- R. Shah, S. Margerum and M. Gold, Proc. SPIE, 1988, 906, 65–73 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, P. Käuper, H. van den Bergh and G. Wagnières, Chimia, 2011, 65, 691–695, DOI:10.2533/chimia.2011.691.
- K. Pospiskova, I. Safarik, M. Sebela and G. Kuncova, Microchim. Acta, 2013, 180, 311–318 CrossRef CAS.
- P. Schrenkhammer and O. S. Wolfbeis, Biosens. Bioelectron., 2008, 24, 994–999 CrossRef CAS PubMed.
- L. H. Fischer, C. Karakus, R. J. Meier, N. Risch, O. S. Wolfbeis, E. Holder and M. Schäferling, Chem.–Eur. J., 2012, 18, 15706–15713 CrossRef CAS PubMed.
- A. S. Kocincová, S. Nagl, S. Arain, C. Krause, S. M. Borisov, M. Arnold and O. S. Wolfbeis, Biotechnol. Bioeng., 2008, 100, 430–438 CrossRef PubMed.
- G. Zhan, D. B. Henthorn and K. Chang-Soo, IEEE Sens. J., 2010, 79–82, DOI:10.1109/ICSENS.2010.5690839.
- D. P. O'Neal, M. A. Meledeo, J. R. Davis, B. L. Ibey, V. A. Gant, M. V. Pishko and G. L. Cote, IEEE Sens. J., 2004, 4, 728–734 CrossRef CAS.
- J. F. Lovell, A. Roxin, K. K. Ng, Q. Qi, J. D. McMullen, R. S. DaCosta and G. Zheng, Biomacromolecules, 2011, 12, 3115–3118 CrossRef CAS PubMed.
- X.-D. Wang, T.-Y. Zhou, X. Chen, K.-Y. Wong and X.-R. Wang, Sens. Actuators, B, 2008, 129, 866–873 CrossRef CAS PubMed.
- A. Habibagahi, Y. Mébarki, Y. Sultan and R. J. Crutchley, J. Photochem. Photobiol., A, 2011, 225, 88–94 CrossRef CAS PubMed.
- M. Marín-Suárez, M. Arias-Martos, T. Galeano-Díaz, J. Fernández-Sánchez and A. Fernández-Gutiérrez, Microchim. Acta, 2013, 180, 1201–1209 CrossRef.
- Y. Amao, K. Asai, T. Miyashita and I. Okura, Anal. Commun., 1999, 36, 367–369 RSC.
- Y. Amao, T. Miyashita and I. Okura, Anal. Chim. Acta, 2000, 421, 167–174 CrossRef CAS.
- E. Puklin, B. Carlson, S. Gouin, C. Costin, E. Green, S. Ponomarev, H. Tanji and M. Gouterman, J. Appl. Polym. Sci., 2000, 77, 2795–2804 CrossRef CAS.
- O. Oter, K. Ertekin, O. Dayan and B. Cetinkaya, J. Fluoresc., 2008, 18, 269–276 CrossRef CAS PubMed.
- C. Hirel, O. Oter, A. G. Gürek, K. Ertekin, E. Önal and S. Z. Topal, J. Porphyrins Phthalocyanines, 2013, 17, 431–439 CrossRef.
- M. Kaneko and S. Hayakawa, J. Macromol. Sci., Chem., 1988, 25, 1255–1261 CrossRef.
- T. Ishiji, K. Kudo and Kaneko, Sens. Actuators, B, 1994, 22, 205–210 CrossRef CAS.
- K. Stubenrauch, M. Sandholzer, F. Niedermair, K. Waich, T. Mayr, I. Klimant, G. Trimmel and C. Slugovc, Eur. Polym. J., 2008, 44, 2558–2566 CrossRef CAS PubMed.
- L. S. Egan, M. A. Winnik and M. D. Croucher, Langmuir, 1988, 4, 438–445 CrossRef CAS.
- X.-L. Qi, S.-Y. Liu, R.-B. Lin, P.-Q. Liao, J.-W. Ye, Z. Lai, Y. Guan, X.-N. Cheng, J.-P. Zhang and X.-M. Chen, Chem. Commun., 2013, 49, 6864–6866 RSC.
- G. Liebsch, I. Klimant, B. Frank, G. Holst and O. S. Wolfbeis, Appl. Spectrosc., 2000, 54, 548–559 CrossRef CAS.
- E. Schmälzlin, J. T. van Dongen, I. Klimant, B. Marmodee, M. Steup, J. Fisahn, P. Geigenberger and H. G. Lohmannsröben, Biophys. J., 2005, 89, 1339–1345 CrossRef PubMed.
- A. Bizzarri, H. Koehler, M. Cajlakovic, A. Pasic, L. Schaupp, I. Klimant and V. Ribitsch, Anal. Chim. Acta, 2006, 573, 48–56 CrossRef PubMed.
- B. P. H. Schaffar and O. S. Wolfbeis, Proc. SPIE, 1989, 990, 122–129 CrossRef CAS PubMed.
- P. S. Grant and M. J. McShane, IEEE Sens. J., 2003, 3, 139–146 CrossRef CAS.
- D. A. Chang-Yen, A. Badardeen and B. K. Gale, Sens. Actuators, B, 2007, 120, 426–433 CrossRef CAS PubMed.
- K. B. Guice, M. E. Caldorera and M. J. McShane, J. Biomed. Opt., 2005, 10, 064031 CrossRef PubMed.
- R. N. Gillanders, O. V. Arzhakova, A. Hempel, A. Dolgova, J. P. Kerry, L. M. Yarysheva, N. F. Bakeev, A. L. Volynskii and D. B. Papkovsky, Anal. Chem., 2010, 82, 466–468 CrossRef CAS PubMed.
- P. C. A. Jerónimo, A. N. Araújo, M. Conceiçã and B. S. M. Montenegro, Talanta, 2007, 72, 13–27 CrossRef PubMed.
- G. T. John, I. Klimant, C. Wittmann and E. Heinzle, Biotechnol. Bioeng., 2003, 81, 829–836 CrossRef CAS PubMed.
- P. Hartmann, M. J. P. Leiner and M. E. Lippitsch, J. Fluoresc., 1994, 4, 327–330 CrossRef CAS PubMed.
- G. A. Holst, T. Köster, E. Voges and D. W. Lübbers, Sens. Actuators, B, 1995, 29, 231–239 CrossRef CAS.
- M. Ogawa, T. Nakamura, J.-i. Mori and K. Kuroda, J. Phys. Chem. B, 2000, 104, 8554–8556 CrossRef CAS.
- H. Zhang, Y. Sun, K. Ye, P. Zhang and Y. Wang, J. Mater. Chem., 2005, 15, 3181–3186 RSC.
- M. Fang, Y. Wang, P. Zhang, S. Li and R. Xu, J. Lumin., 2000, 91, 67–70 CrossRef CAS.
- B. Wang, L. Zhang, B. Li, Y. Li, Y. Shi and T. Shi, Sens. Actuators, B, 2014, 190, 93–100 CrossRef CAS PubMed.
- Y. Wang, B. Li, L. Zhang and H. Song, Langmuir, 2013, 29, 1273–1279 CrossRef CAS PubMed.
- A. Čeklovský and S. Takagi, Cent. Eur. J. Chem., 2013, 11, 1132–1136 CrossRef.
- S. Z. Topal, K. Ertekin, D. Topkaya, S. Alp and B. Yenigul, Microchim. Acta, 2008, 161, 209–216 CrossRef CAS.
- O. Oter, K. Ertekin and S. Derinkuyu, Mater. Chem. Phys., 2009, 113, 322–328 CrossRef CAS PubMed.
- A. Lobnik and O. S. Wolfbeis, Analyst, 1998, 123, 2247–2250 RSC.
- C. McDonagh, B. D. MacCraith and A. K. McEvoy, Anal. Chem., 1998, 70, 45–50 CrossRef CAS PubMed.
- M. T. Murtagh, M. R. Shahriari and M. Krihak, Chem. Mater., 1998, 10, 3862–3869 CrossRef CAS.
- B. J. Basu, Sens. Actuators, B, 2007, 123, 568–577 CrossRef CAS PubMed.
- O. S. Wolfbeis, I. Klimant, T. Werner, C. Huber, U. Kosch, C. Krause, G. Neurauter and A. Dürkop, Sens. Actuators, B, 1998, 51, 17–24 CrossRef CAS.
- Z. Tao, E. C. Tehan, Y. Tang and F. V. Bright, Anal. Chem., 2006, 78, 1939–1945 CrossRef CAS PubMed.
- C.-S. Chu, J. Lumin., 2013, 135, 5–9 CrossRef CAS PubMed.
- R. M. Bukowski, R. Ciriminna, M. Pagliaro and F. V. Bright, Anal. Chem., 2005, 77, 2670–2672 CrossRef CAS PubMed.
- R. Ciriminna and M. Pagliaro, Analyst, 2009, 134, 1531–1535 RSC.
- Y. Xiong, J. Xu, D.-q. Zhu, C.-f. Duan and Y.-f. Guan, J. Sol–Gel Sci. Technol., 2010, 53, 441–447 CrossRef CAS PubMed.
- Y. Xiong, D. Zhu, S. Chen, H. Peng and Y. Guan, J. Fluoresc., 2010, 20, 269–274 CrossRef CAS PubMed.
- Y. Xiong, Z. Ye, J. Xu, Y. Zhu, C. Chen and Y. Guan, Analyst, 2013, 138, 1819–1827 RSC.
- V. S. Tripathi, G. Lakshminarayana and M. Nogami, Sens. Actuators, B, 2010, 147, 741–747 CrossRef CAS PubMed.
- J. Estella, D. Wencel, J. P. Moore, M. Sourdaine and C. McDonagh, Anal. Chim. Acta, 2010, 666, 83–90 CrossRef CAS PubMed.
- B. Lei, B. Li, H. Zhang, S. Lu, Z. Zheng, W. Li and Y. Wang, Adv. Funct. Mater., 2006, 16, 1883–1891 CrossRef CAS.
- X. Lu and M. A. Winnik, Chem. Mater., 2001, 13, 3449–3463 CrossRef CAS.
- G. S. Vasylevska, S. M. Borisov, C. Krause and O. S. Wolfbeis, Chem. Mater., 2006, 18, 4609–4616 CrossRef CAS.
- A. Mills, A. Graham and C. O'Rourke, Sens. Actuators, B, 2014, 190, 907–912 CrossRef CAS PubMed.
- N. Opitz and D. W. Lübbers, Adv. Exp. Med. Biol., 1984, 169, 899–905 CrossRef CAS.
- S. M. Borisov, T. Mayr, G. Mistlberger, K. Waich, K. Koren, P. Chojnacki and I. Klimant, Talanta, 2009, 79, 1322–1330 CrossRef CAS PubMed.
- J. M. Kürner, I. Klimant, C. Krause, H. Preu, W. Kunz and O. S. Wolfbeis, Bioconjugate Chem., 2001, 12, 883–889 CrossRef PubMed.
- A. Fercher, S. M. Borisov, A. V. Zhdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499–5508 CrossRef CAS PubMed.
- E. Schmälzlin, B. Walz, I. Klimant, B. Schewe and H. G. Lohmannsroben, Sens. Actuators, B, 2006, 119, 251–254 CrossRef PubMed.
- X. D. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. M. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901–906 RSC.
- S. M. Borisov, T. Mayr and I. Klimant, Anal. Chem., 2008, 80, 573–582 CrossRef CAS PubMed.
- P. J. Cywinski, A. J. Moro, S. E. Stanca, C. Biskup and G. J. Mohr, Sens. Actuators, B, 2009, 135, 472–477 CrossRef CAS PubMed.
- S. H. Im, G. E. Khalil, J. Callis, B. H. Ahn, M. Gouterman and Y. Xia, Talanta, 2005, 67, 492–497 CrossRef CAS PubMed.
- X. D. Wang, T. Y. Zhou, X. H. Song, Y. Jiang, C. J. Yang and X. Chen, J. Mater. Chem., 2011, 21, 17651–17653 RSC.
- H. A. Clark, M. Hoyer, M. A. Philbert and R. Kopelman, Anal. Chem., 1999, 71, 4831–4836 CrossRef CAS.
- Y. F. Cao, Y. E. L. Koo and R. Kopelman, Analyst, 2004, 129, 745–750 RSC.
- Y. E. Koo Lee, E. E. Ulbrich, G. Kim, H. Hah, C. Strollo, W. Fan, R. Gurjar, S. Koo and R. Kopelman, Anal. Chem., 2010, 82, 8446–8455 CrossRef CAS PubMed.
- M. P. Coogan, J. B. Court, V. L. Gray, A. J. Hayes, S. H. Lloyd, C. O. Millet, S. J. A. Pope and D. Lloyd, Photochem. Photobiol. Sci., 2010, 9, 103–109 CAS.
- H. Liu, H. Yang, X. Hao, H. Xu, Y. Lv, D. Xiao, H. Wang and Z. Tian, Small, 2013, 9, 2639–2648 CrossRef CAS PubMed.
- T. C. O'Riordan, A. E. Soini and D. B. Papkovsky, Anal. Chem., 2002, 74, 5845–5850 CrossRef CAS.
- M. Burke, P. J. O'Sullivan, A. E. Soini, H. Berney and D. B. Papkovsky, Anal. Biochem., 2003, 320, 273–280 CrossRef CAS.
- K. P. McNamara and Z. Rosenzweig, Anal. Chem., 1998, 70, 4853–4859 CrossRef CAS.
- Z. Cheng and C. A. Aspinwall, Analyst, 2006, 131, 236–243 RSC.
- P. Chojnacki, G. Mistlberger and I. Klimant, Angew. Chem., Int. Ed., 2007, 46, 8850–8853 CrossRef CAS PubMed.
- S. H. Cheng, C. H. Lee, C. S. Yang, F. G. Tseng, C. Y. Mou and L. W. Lo, J. Mater. Chem., 2009, 19, 1252–1257 RSC.
- B. B. Collier, S. Singh and M. McShane, Analyst, 2011, 136, 962–967 RSC.
- C. M. Lemon, P. N. Curtin, R. C. Somers, A. B. Greytak, R. M. Lanning, R. K. Jain, M. G. Bawendi and D. G. Nocera, Inorg. Chem., 2013, 52, 10394–10406 CrossRef CAS PubMed.
- M. A. Acosta, P. Ymele-Leki, Y. V. Kostov and J. B. Leach, Biomaterials, 2009, 30, 3068–3074 CrossRef CAS PubMed.
- H. E. Posch and O. S. Wolfbeis, Sens. Actuators, 1988, 15, 77–83 CrossRef CAS.
- F. Navarro-Villoslada, G. Orellana, M. C. Moreno-Bondi, T. Vick, M. Driver, G. Hildebrand and K. Liefeith, Anal. Chem., 2001, 73, 5150–5156 CrossRef CAS.
- O. S. Wolfbeis, B. Kovács, K. Goswami and S. M. Klainer, Mikrochim. Acta, 1998, 129, 181–188 CrossRef CAS.
- D. B. Papkovsky, A. N. Ovchinnikov, V. I. Ogurtsov, G. V. Ponomarev and T. Korpela, Sens. Actuators, B, 1998, 51, 137–145 CrossRef CAS.
- M. I. Gutiérrez, C. G. Martínez, D. García-Fresnadillo, A. M. Castro, G. Orellana, A. M. Braun and E. Oliveros, J. Phys. Chem. A, 2003, 107, 3397–3403 CrossRef.
- P. Ceroni, A. Y. Lebedev, E. Marchi, M. Yuan, T. V. Esipova, G. Bergamini, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Photochem. Photobiol. Sci., 2011, 10, 1056–1065 CAS.
- R. S. Atkinson, D. R. G. Brimage, R. S. Davidson and E. Gray, J. Chem. Soc., Perkin Trans. 1, 1973, 960–964 RSC.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- I. B. C. Matheson and J. Lee, J. Am. Chem. Soc., 1972, 94, 3310–3313 CrossRef CAS.
- R. A. Ackerman, I. Rosenthal and J. N. Pitts, J. Chem. Phys., 1971, 54, 4960–4961 CrossRef CAS PubMed.
- J. P. Guillory and C. F. Cook, J. Polym. Sci., Part A: Polym. Chem., 1973, 11, 1927–1937 CrossRef CAS.
- C. S. Foote, T. T. Fujimoto and Y. C. Chang, Tetrahedron Lett., 1972, 13, 45–48 CrossRef.
- N. Hasty, P. B. Merkel, P. Radlick and D. R. Kearns, Tetrahedron Lett., 1972, 13, 49–52 CrossRef.
- B. Enko, S. M. Borisov, J. Regensburger, W. Bäumler, G. Gescheidt and I. Klimant, J. Phys. Chem. A, 2013, 117, 8873–8882 CrossRef CAS PubMed.
- O. S. Wolfbeis, L. J. Weis, M. J. P. Leiner and W. E. Ziegler, Anal. Chem., 1988, 60, 2028–2030 CrossRef CAS.
- M. C. Moreno-Bondi, O. S. Wolfbeis, M. J. P. Leiner and B. P. H. Schaffar, Anal. Chem., 1990, 62, 2377–2380 CrossRef CAS.
- T. Mayr, I. Klimant, O. S. Wolfbeis and T. Werner, Anal. Chim. Acta, 2002, 462, 1–10 CrossRef CAS.
- G. O'Keeffe, B. D. MacCraith, A. K. McEvoy, C. M. McDonagh and J. F. McGilp, Sens. Actuators, B, 1995, 29, 226–230 CrossRef CAS.
- V. I. Ogurtsov and D. B. Papkovsky, Sens. Actuators, B, 1998, 51, 377–381 CrossRef CAS.
- W. Feng, N. Zhou, L. Chen and B. Li, J. Opt., 2013, 15, 055502 CrossRef.
- B. D. MacCraith, G. O'keeffe, C. McDonagh and A. McEvoy, Electron. Lett., 1994, 30, 888–889 CrossRef.
- G. Holst, R. N. Glud, M. Kühl and I. Klimant, Sens. Actuators, B, 1997, 38, 122–129 CrossRef CAS.
- K. W. Berndt and J. R. Lakowicz, Anal. Biochem., 1992, 201, 319–325 CrossRef CAS.
- H. Malak, J. W. Dobrucki, M. M. Malak and H. M. Swartz, Biophys. J., 1998, 74, A189 Search PubMed.
- S. A. Vinogradov, M. A. Fernandez-Searra, B. W. Dugan and D. F. Wilson, Rev. Sci. Instrum., 2001, 72, 3396–3406 CrossRef CAS PubMed.
- R. J. Woods, S. Scypinski, L. J. C. Love and H. A. Ashworth, Anal. Chem., 1984, 56, 1395–1400 CrossRef CAS.
- R. M. Ballew and J. N. Demas, Anal. Chem., 1989, 61, 30–33 CrossRef CAS.
- S. J. Payne, J. N. Demas and B. A. DeGraff, Appl. Spectrosc., 2009, 63, 437–441 CrossRef CAS PubMed.
- A. Sharma and O. S. Wolfbeis, Appl. Spectrosc., 1988, 42, 1009–1011 CrossRef CAS.
- E. J. McLaurin, A. B. Greytak, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 12994–13001 CrossRef CAS PubMed.
- C. M. Lemon, E. Karnas, M. G. Bawendi and D. G. Nocera, Inorg. Chem., 2013, 52, 10394–10406 CrossRef CAS PubMed.
- D. E. Achatz, R. J. Meier, L. H. Fischer and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2011, 50, 260–263 CrossRef CAS PubMed.
- I. Gryczynski, Z. Gryczynski, J. R. Lakowicz and G. Rao, Analyst, 1999, 124, 1041–1044 RSC.
- L. Shen, M. Ratterman, D. Klotzkin and I. Papautsky, Sens. Actuators, B, 2011, 155, 430–435 CrossRef CAS PubMed.
- O. S. Finikova, P. Chen, Z. Ou, K. M. Kadish and S. A. Vinogradov, J. Photochem. Photobiol., A, 2008, 198, 75–84 CrossRef CAS PubMed.
- A. V. Kondrashina, R. I. Dmitriev, S. M. Borisov, I. Klimant, I. O'Brien, Y. M. Nolan, A. V. Zhdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931–4939 CrossRef CAS.
- C. M. Lemon, E. Karnas, M. G. Bawendi and D. G. Nocera, Inorg. Chem., 2013, 52, 10394–10406 CrossRef CAS PubMed.
- H. Y. Aboul-Enein, R.-I. Stefan, J. F. van Staden, X. R. Zhang, A. M. Garcia-Campana and W. R. G. Baeyens, Crit. Rev. Anal. Chem., 2000, 30, 271–289 CrossRef CAS.
- A. Burr and D. Mauzerall, Biochim. Biophys. Acta, Bioenerg., 1968, 153, 614–624 CrossRef CAS.
- J. W. Hastings, J. Cell. Comp. Physiol., 1952, 39, 1–30 CrossRef CAS.
- R. Oshino, N. Oshino, M. Tamura, L. Kobilinsky and B. Chance, Biochim. Biophys. Acta, Gen. Subj., 1972, 273, 5–17 CrossRef CAS.
- B. Chance and R. Oshino, Methods Enzymol., 1978, 57, 223–226 CAS.
- D. Lloyd, K. James, J. Williams and N. Williams, Anal. Biochem., 1981, 116, 17–21 CrossRef CAS.
- M. Beijerinck, K. Ned. Akad. Wet., Proc., Ser. B: Phys. Sci., 1902, 4, 45–49 Search PubMed.
- G. E. Collins and S. L. Rose-Pehrsson, Anal. Chem., 1995, 67, 2224–2230 CrossRef CAS.
- R.-J. Zheng, Y.-M. Fang, S.-F. Qin, J. Song, A.-H. Wu and J.-J. Sun, Sens. Actuators, B, 2011, 157, 488–493 CrossRef CAS PubMed.
- S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360–4368 CrossRef CAS.
- I.-S. Shin, T. Hirsch, B. Ehrl, D.-H. Jang, O. S. Wolfbeis and J.-I. Hong, Anal. Chem., 2012, 84, 9163–9168 CAS.
- D. S. Smith, Y. Kostov and G. Rao, Sens. Actuators, B, 2007, 127, 432–440 CrossRef CAS PubMed.
- S. Baluschev, F. Yu, T. Miteva, S. Ahl, A. Yasuda, G. Nelles, W. Knoll and G. Wegner, Nano Lett., 2005, 5, 2482–2484 CrossRef CAS PubMed.
- O. S. Wolfbeis, Proc. SPIE, 1990, 1368, 218–222 CrossRef PubMed.
- O. S. Wolfbeis and E. Urbano, Anal. Chem., 1983, 55, 1904–1906 CrossRef CAS.
- M. I. J. Stich, S. Nagl, O. S. Wolfbeis, U. Henne and M. Schaeferling, Adv. Funct. Mater., 2008, 18, 1399–1406 CrossRef CAS.
- L. H. Fischer, S. M. Borisov, M. Schaeferling, I. Klimant and O. S. Wolfbeis, Analyst, 2010, 135, 1224–1229 RSC.
- S. Nagl, M. I. J. Stich, M. Schaferling and O. S. Wolfbeis, Anal. Bioanal. Chem., 2009, 393, 1199–1207 CrossRef CAS PubMed.
- S. M. Borisov, C. Krause, S. Arain and O. S. Wolfbeis, Adv. Mater., 2006, 18, 1511–1516 CrossRef CAS.
- C. R. Schroder, L. Polerecky and I. Klimant, Anal. Chem., 2007, 79, 60–70 CrossRef PubMed.
- S. Borisov, R. Seifner and I. Klimant, Anal. Bioanal. Chem., 2011, 400, 2463–2474 CrossRef CAS PubMed.
- R. Liu, T. Xiao, W. Cui, J. Shinar and R. Shinar, Anal. Chim. Acta, 2013, 778, 70–78 CrossRef CAS PubMed.
- T. Kameya, Y. Matsuda, Y. Egami, H. Yamaguchi and T. Niimi, Sens. Actuators, B, 2014, 190, 70–77 CrossRef CAS PubMed.
- J. Hradil, C. Davis, K. Mongey, C. McDonagh and B. D. MacCraith, Meas. Sci. Technol., 2002, 13, 1552 CrossRef CAS.
- L. M. Coyle and M. Gouterman, Sens. Actuators, B, 1999, 61, 92–99 CrossRef CAS.
- T.-W. Sung and Y.-L. Lo, Sens. Actuators, B, 2012, 173, 406–413 CrossRef CAS PubMed.
- H. Lam, G. Rao, J. Loureiro and L. Tolosa, Talanta, 2011, 84, 65–70 CrossRef CAS PubMed.
- R. J. Meier, S. Schreml, X.-d. Wang, M. Landthaler, P. Babilas and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2011, 50, 10893–10896 CrossRef CAS PubMed.
- X.-d. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS PubMed.
- C. R. Schroeder, G. Neurauter and I. Klimant, Microchim. Acta, 2007, 158, 205–218 CrossRef CAS.
- L. Zhang, F. Su, S. Buizer, H. Lu, W. Gao, Y. Tian and D. Meldrum, Biomaterials, 2013, 34, 9779–9788 CrossRef CAS PubMed.
- M. Schaeferling, A. Duerkop and U. Resch-Genger, in Springer series on fluorescence, ed. U. Resch-Genger, Springer, Berlin, 2008, vol. 5, pp. 373–414 Search PubMed.
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885–4901 RSC.
- D. W. Lübbers, US Pat., 4,306,877, 1981 Search PubMed.
- E. J. Park, K. R. Reid, W. Tang, R. T. Kennedy and R. Kopelman, J. Mater. Chem., 2005, 15, 2913–2919 RSC.
- C. F. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741–2745 CrossRef CAS PubMed.
- P. A. S. Jorge, M. Mayeh, R. Benrashid, P. Caldas, J. L. Santos and F. Farahi, Appl. Opt., 2006, 45, 3760–3767 CrossRef CAS.
- M. Amelia, A. Lavie-Cambot, N. D. McClenaghan and A. Credi, Chem. Commun., 2011, 47, 325–327 RSC.
- C.-S. Chu and Y.-L. Lo, Sens. Actuators, B, 2008, 134, 711–717 CrossRef CAS PubMed.
- Y. Kostov, K. A. Van Houten, P. Harms, R. S. Pilato and G. Rao, Appl. Spectrosc., 2000, 54, 864–868 CrossRef CAS.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., 2012, 124, 4224–4227 CrossRef.
- T. Doussineau, A. Schulz, A. Lapresta-Fernandez, A. Moro, S. Körsten, S. Trupp and G. J. Mohr, Chem.–Eur. J., 2010, 16, 10290–10299 CrossRef CAS PubMed.
- S. J. Payne, G. Zhang, J. N. Demas, C. L. Fraser and B. A. Degraff, Appl. Spectrosc., 2011, 65, 1321–1324 CrossRef CAS PubMed.
- D. P. Saini S. M. Klainer and S. L. Coulter, US Pat., 5,737,457, 1998.
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef PubMed.
- P. Hartmann and W. Ziegler, Anal. Chem., 1996, 68, 4512–4514 CrossRef CAS.
- www.presens.de/references/application-notes.html?tx_presensapplicationnotes_pi1[product_group] = 16; accessed in January 2014.
- D. H. Song, H. D. Kim and K. C. Kim, Opt. Laser. Eng., 2012, 50, 74–81 CrossRef PubMed.
- M. I. J. Stich, S. M. Borisov, U. Henne and M. Schäferling, Sens. Actuators, B, 2009, 139, 204–207 CrossRef CAS PubMed.
- B. Ungerböck, V. Charwat, P. Ertl and T. Mayr, Lab Chip, 2013, 13, 1593–1601 RSC.
- S. Park, S. G. Achanta, J. Yang and C.-S. Kim, Sens. Actuators, B, 2012, 164, 101–108 CrossRef CAS PubMed.
- S. Achanta, P. Sanghan and K. Chang-Soo, IEEE Sens. J., 2012, 1–4 Search PubMed.
- J. Park, W. Hong and C.-S. Kim, IEEE Sens. J., 2010, 10, 1855–1862 CrossRef CAS.
- K. Kellner, G. Liebsch, I. Klimant, O. S. Wolfbeis, T. Blunk, M. B. Schulz and A. Gopferich, Biotechnol. Bioeng., 2002, 80, 73–83 CrossRef CAS PubMed.
- P. Babilas, G. Liebsch, V. Schacht, I. Klimant, O. S. Wolfbeis, R. M. Szeimies and C. Abels, Microcirculation, 2005, 12, 477–487 CrossRef CAS PubMed.
- P. Babilas, P. Lamby, L. Prantl, S. Schreml, E. M. Jung, G. Liebsch, O. S. Wolfbeis, M. Landthaler, R. M. Szeimies and C. Abels, Skin Res. Technol., 2008, 14, 304–311 CrossRef PubMed.
- R. J. Meier, L. H. Fischer, O. S. Wolfbeis and M. Schäferling, Sens. Actuators, B, 2013, 177, 500–506 CrossRef CAS PubMed.
- J. Hofmann, R. J. Meier, A. Mahnke, V. Schatz, F. Brackmann, R. Trollmann, C. Bogdan, G. Liebsch, X. Wang, O. S. Wolfbeis and J. Jantsch, Methods Appl. Fluoresc., 2013, 1, 045002 CrossRef.
- G. Holst, O. Kohls, I. Klimant, B. Konig, M. Kuhl and T. Richter, Sens. Actuators, B, 1998, 51, 163–170 CrossRef CAS.
- X. F. Wang, T. Uchida, D. M. Coleman and S. Minami, Appl. Spectrosc., 1991, 45, 360–366 CrossRef CAS.
- G. Holst and B. Grunwald, Sens. Actuators, B, 2001, 74, 78–90 CrossRef CAS.
- J. R. Lakowicz and K. W. Berndt, Rev. Sci. Instrum., 1991, 62, 1727–1734 CrossRef CAS PubMed.
- C.-S. Chu and Y.-L. Lo, Sens. Actuators, B, 2010, 147, 310–315 CrossRef CAS PubMed.
- O. S. Wolfbeis and W. Trettnak, Spectrochim. Acta, Part A, 1987, 43, 405–408 CrossRef.
- A. Mondin, D. Badocco and P. Pastore, Sens. Actuators, B, 2014, 190, 775–781 CrossRef CAS PubMed.
- D. Badocco, A. Mondin and P. Pastore, Sens. Actuators, B, 2011, 158, 54–61 CrossRef CAS PubMed.
- D. Badocco, A. Mondin and P. Pastore, Sens. Actuators, B, 2013, 181, 943–948 CrossRef CAS PubMed.
- D. Badocco, A. Mondin and P. Pastore, Sens. Actuators, B, 2013, 181, 949–954 CrossRef CAS PubMed.
- X.-d. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- H. Kroneis, PhD thesis, TU Graz, 1983.
- E. Reynolds, J. N. Demas and B. A. DeGraff, J. Fluoresc., 2013, 23, 237–241 CrossRef CAS PubMed.
- W. Miller, M. Yafuso, C. Yan, H. Hui and S. Arick, Clin. Chem., 1987, 33, 1538–1542 CAS.
- C.-S. Chu, Y.-L. Lo and T.-W. Sung, Photonic Sens., 2011, 1, 234–250 CrossRef PubMed.
- I. A. Zakharov and T. I. Grishaeva, Zh. Anal. Khim., 1981, 36, 112 CAS.
- B.-H. Han, M. A. Winnik, A. B. Bourlinos and E. P. Giannelis, Chem. Mater., 2005, 17, 4001–4009 CrossRef CAS.
- O. S. Wolfbeis, Anal. Chem., 2000, 72, 81–90 CrossRef CAS.
- O. S. Wolfbeis, Anal. Chem., 2002, 74, 2663–2678 CrossRef CAS.
- O. S. Wolfbeis, Anal. Chem., 2004, 76, 3269–3284 CrossRef CAS PubMed.
- O. S. Wolfbeis, Anal. Chem., 2006, 78, 3859–3874 CrossRef CAS PubMed.
- O. S. Wolfbeis, Anal. Chem., 2008, 80, 4269–4283 CrossRef CAS PubMed.
- X.-D. Wang and O. S. Wolfbeis, Anal. Chem., 2012, 85, 487–508 CrossRef PubMed.
- B. A. Shapiro, C. K. Mahutte, R. D. Cane and I. J. Gilmour, Crit. Care Med., 1993, 21, 487–494 CrossRef CAS PubMed.
- E. J. Fogt, Clin. Chem., 1990, 36, 1573–1580 CAS.
- D. W. Lübbers, Acta Anaesthesiol. Scand., 1995, 39, 37–54 CrossRef.
- M. J. P. Leiner, Sens. Actuators, B, 1995, 29, 169–173 CrossRef CAS.
- W. Barnikol, T. Gaertner, N. Weiler and O. Burkhard, Rev. Sci. Instrum., 1988, 59, 1204–1208 CrossRef CAS PubMed.
- R. Chen, A. D. Farmery, A. Obeid and C. E. W. Hahn, IEEE Sens. J., 2012, 12, 71–75 CrossRef CAS.
- P. A. S. Jorge, P. Caldas, C. C. Rosa, A. G. Oliva and J. L. Santos, Sens. Actuators, B, 2004, 103, 290–299 CrossRef CAS PubMed.
- C. A. Browne, D. H. Tarrant, M. S. Olteanu, J. W. Mullens and E. L. Chronister, Anal. Chem., 1996, 68, 2289–2295 CrossRef CAS.
- M. V. Rigo and P. Geissinger, J. Nanomater., 2010, 2010, 1–11 CrossRef PubMed.
- M. V. Rigo and P. Geissinger, J. Sens., 2012, 2012, 1–10 CrossRef PubMed.
- C.-S. Chu, T.-W. Sung and Y.-L. Lo, Sens. Actuators, B, 2013, 185, 287–292 CrossRef CAS PubMed.
- E. E. Hardy, D. J. David, N. S. Kapany and F. C. Unterleitner, Nature, 1975, 257, 666–667 CrossRef CAS.
- Ó. Esteban and C. Pulido, Proc. SPIE, 2013, 8794, 879410 CrossRef PubMed.
- X. Yang, Y. Liu, F. Tian, L. Yuan, Z. Liu, S. Luo and E. Zhao, Opt. Lett., 2012, 37, 2115–2117 CrossRef CAS PubMed.
- R. Xue, P. Behera, J. Xu, M. S. Viapiano and J. J. Lannutti, Sens. Actuators, B, 2014, 192, 697–707 CrossRef CAS PubMed.
- B. A. A. Dremel, R. D. Schmid and O. S. Wolfbeis, Anal. Chim. Acta, 1991, 248, 351–359 CrossRef CAS.
- W. Trettnak and O. S. Wolfbeis, Anal. Lett., 1989, 22, 2191–2197 CrossRef CAS.
- B. Lamprecht, A. Tschepp, M. Cajlakovic, M. Sagmeister, V. Ribitsch and S. Köstler, Analyst, 2013, 138, 5875–5878 RSC.
- V. G. Bordo and H.-G. Rubahn, Optics and spectroscopy at surfaces and interfaces, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Z.-M. Qi, S. Xia and N. Matsuda, Anal. Biochem., 2008, 374, 196–202 CrossRef CAS PubMed.
- B. W. Dodson, Proc. SPIE, 1984, 497, 91 CrossRef PubMed.
- R. C. Murray and S. M. Lefkowitz, Eur. Pat., 190,830, 1986 Search PubMed.
- Y. Xiong, J. Xu, J.-W. Wang and Y.-F. Guan, Anal. Bioanal. Chem., 2009, 394, 919–923 CrossRef CAS PubMed.
- E. Singer, G. L. Duveneck, M. Ehrat and H. M. Widmer, Sens. Actuators, A, 1994, 42, 542–546 CrossRef CAS.
- W. Cao and Y. Duan, Sens. Actuators, B, 2006, 119, 363–369 CrossRef CAS PubMed.
- D. Wales, R. Parker, J. Gates, M. Grossel and P. Smith, Integrated planar Bragg grating oxygen sensor, 2010, http://eprints.soton.ac.uk/340796/340791/344630.pdf. Visited in January 2014 Search PubMed.
- J. Luo, X. Zhuang and J. Yao, Proc. Inst. Mech. Eng., Part N, 2012, 226, 39–43 CAS.
- C. R. Lavers, K. Itoh, S. C. Wu, M. Murabayashi, I. Mauchline, G. Stewart and T. Stout, Sens. Actuators, B, 2000, 69, 85–95 CrossRef CAS.
- O. S. Wolfbeis, TrAC, Trends Anal. Chem., 1996, 15, 225–232 CAS.
- C. Monat, P. Domachuk and B. J. Eggleton, Nat. Photon., 2007, 1, 106–114 CrossRef CAS.
- R. A. Potyrailo, S. E. Hobbs and G. M. Hieftje, Fresenius' J. Anal. Chem., 1998, 362, 349–373 CrossRef CAS.
- D. P. Saini, US Pat., 5,439,647, 1995 Search PubMed.
- Z. Wang, S. Y. Al-Raqa, I. Chambrier, D. A. Russell and M. J. Cook, J. Porphyrins Phthalocyanines, 2012, 16, 1186–1195 CrossRef CAS.
- C. S. Burke, O. McGaughey, J.-M. Sabattie, H. Barry, A. K. McEvoy, C. McDonagh and B. D. MacCraith, Analyst, 2005, 130, 41–45 RSC.
- D. A. Chang-Yen and B. K. Gale, Lab Chip, 2003, 3, 297–301 RSC.
- P. J. R. Roche, M. C. K. Cheung, K. Y. Yung, A. G. Kirk, V. P. Chodavarpu and F. V. Bright, Sens. Actuators, B, 2010, 147, 581–586 CrossRef CAS PubMed.
- B. J. Prince, A. W. Schwabacher and P. Geissinger, Anal. Chem., 2001, 73, 1007–1015 CrossRef CAS.
- S. Personick, Bell Syst. Tech. J, 1977, 56, 355–366 CrossRef.
- S. Eich, E. Schmälzlin and H.-G. Löhmannsröben, Sensors, 2013, 13, 7170–7183 CrossRef CAS PubMed.
- R. A. Potyrailo and G. M. Hieftje, Anal. Chem., 1998, 70, 3407–3412 CrossRef CAS.
- S. Eich, E. Schmälzlin and H.-G. Löhmannsröben, Proc. SPIE, 2010, 77260A CrossRef PubMed.
- B. J. Prince, N. T. Kaltcheva, A. W. Schwabacher and P. Geissinger, Appl. Spectrosc., 2001, 55, 1018–1024 CrossRef CAS.
- P. E. Henning and P. Geissinger, Meas. Sci. Technol., 2012, 23, 045104 CrossRef.
- P. E. Henning, A. Benko, A. W. Schwabacher, P. Geissinger and R. J. Olsson, Rev. Sci. Instrum., 2005, 76, 062220 CrossRef PubMed.
- G. Mistlberger, K. Koren, S. M. Borisov and I. Klimant, Anal. Chem., 2010, 82, 2124–2128 CrossRef CAS PubMed.
- E. W. Stein, P. S. Grant, H. Zhu and M. J. McShane, Anal. Chem., 2007, 79, 1339–1348 CrossRef CAS PubMed.
- E. W. Stein, S. Singh and M. J. McShane, Anal. Chem., 2008, 80, 1408–1417 CrossRef CAS PubMed.
- S. Singh and M. McShane, Biosens. Bioelectron., 2011, 26, 2478–2483 CrossRef CAS PubMed.
- J. Prasad, A. Joshi, R. D. Jayant and R. Srivastava, Biotechnol. Bioeng., 2011, 108, 2011–2021 CrossRef CAS PubMed.
- J. Q. Brown, R. Srivastava and M. J. McShane, Biosens. Bioelectron., 2005, 21, 212–216 CrossRef CAS PubMed.
- K. Nagamine, S. Ito, M. Takeda, S. Otani and M. Nishizawa, Electrochemistry, 2012, 80, 318–320 CrossRef CAS PubMed.
- J. López-Gejo, A. Arranz, Á. Navarro, C. Palacio, E. Muñoz and G. Orellana, J. Am. Chem. Soc., 2010, 132, 1746–1747 CrossRef PubMed.
- J. López-Gejo, Á. Navarro-Tobar, A. Arranz, C. Palacio, E. Muñoz and G. Orellana, ACS Appl. Mater. Interfaces, 2011, 3, 3846–3854 Search PubMed.
- S. Lee, B. L. Ibey, G. L. Coté and M. V. Pishko, Sens. Actuators, B, 2008, 128, 388–398 CrossRef CAS PubMed.
- H. Zhu, X. Zhou, F. Su, Y. Tian, S. Ashili, M. R. Holl and D. R. Meldrum, Sens. Actuators, B, 2012, 173, 817–823 CrossRef CAS PubMed.
- V. Nock, M. Alkaisi and R. J. Blaikie, Microelectron. Eng., 2010, 87, 814–816 CrossRef CAS PubMed.
- J. R. Etzkorn, W.-C. Wu, Z. Tian, P. Kim, S.-H. Jang, D. R. Meldrum, A. K. Y. Jen and B. A. Parviz, J. Micromech. Microeng., 2010, 20, 095017 CrossRef.
- R. Ambekar, P. Jongwon, D. B. Henthorn and K. Chang-Soo, IEEE Sens. J., 2009, 9, 169–175 CrossRef CAS PubMed.
- A. Martínez-Olmos, J. Fernández-Salmerón, N. Lopez-Ruiz, A. Rivadeneyra Torres, L. F. Capitan-Vallvey and A. J. Palma, Anal. Chem., 2013, 85, 11098–11105 CrossRef PubMed.
- M. Marín-Suárez, S. Medina-Rodríguez, O. Ergeneman, S. Pané, J. F. Fernández-Sánchez, B. J. Nelson and A. Fernández-Gutiérrez, Nanoscale, 2014, 6, 263–271 RSC.
- I. Dunphy, S. A. Vinogradov and D. F. Wilson, Anal. Biochem., 2002, 310, 191–198 CrossRef CAS.
- I. B. Rietveld, E. Kim and S. A. Vinogradov, Tetrahedron, 2003, 59, 3821–3831 CrossRef CAS.
- D. F. Wilson, W. M. F. Lee, S. Makonnen, O. Finikova, S. Apreleva and S. A. Vinogradov, J. Appl. Physiol., 2006, 101, 1648–1656 CrossRef CAS PubMed.
- A. Y. Lebedev, A. V. Cheprakov, S. Sakadžić, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292–1304 CAS.
- L. S. Ziemer, W. M. F. Lee, S. A. Vinogradov, C. Sehgal and D. F. Wilson, J. Appl. Physiol., 2005, 98, 1503–1510 CrossRef CAS PubMed.
- L.-W. Lo, S. H.-Y. Huang, C.-H. Chang, W.-Y. Chen, P.-J. Tsai and C.-S. Yang, J. Med. Biol. Eng., 2003, 23, 19–28 Search PubMed.
- T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756–8765 CrossRef CAS PubMed.
- A. Y. Lebedev, T. Troxler and S. A. Vinogradov, J. Porphyrins Phthalocyanines, 2008, 12, 1261–1269 CrossRef CAS PubMed.
- S. V. Apreleva, D. F. Wilson and S. A. Vinogradov, Appl. Opt., 2006, 45, 8547–8559 CrossRef CAS.
- M. A. Yaseen, V. J. Srinivasan, S. Sakadi, W. Wu, S. Ruvinskaya, S. A. Vinogradov and D. A. Boas, Opt. Express, 2009, 17, 22341–22350 CrossRef CAS PubMed.
- Y.-E. K. Lee, R. Smith and R. Kopelman, Annu. Rev. Anal. Chem., 2009, 2, 57–76 CrossRef PubMed.
- Y. Kuang and D. R. Walt, Biotechnol. Bioeng., 2007, 96, 318–325 CrossRef CAS PubMed.
- X.-H. Wang, H.-S. Peng, Z. Chang, L.-L. Hou, F.-T. You, F. Teng, H.-W. Song and B. Dong, Microchim. Acta, 2012, 178, 147–152 CrossRef CAS.
- X.-H. Wang, H.-S. Peng, H. Ding, F.-T. You, S.-H. Huang, F. Teng, B. Dong and H.-W. Song, J. Mater. Chem., 2012, 22, 16066–16071 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cs00039k |
| ‡ We use the common term “oxygen” for the more correct but less usual term “dioxygen”. |
| This journal is © The Royal Society of Chemistry 2014 |