 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
About supramolecular systems for dynamically probing cells
Jenny
Brinkmann
,
Emanuela
Cavatorta
,
Shrikrishnan
Sankaran
,
Bettina
Schmidt
,
Jasper
van Weerd
and
Pascal
Jonkheijm
*
MESA+ Institute for Nanotechnology and Department of Science and Technology, Laboratory Group of Bioinspired Molecular Engineering, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: p.jonkheijm@utwente.nl; Tel: +31 534892987 Web: www.jonkheijm.org
First published on 31st March 2014
Abstract
This article reviews the state of the art in the development of strategies for generating supramolecular systems for dynamic cell studies. Dynamic systems are crucial to further our understanding of cell biology and are consequently at the heart of many medical applications. Increasing interest has therefore been focused recently on rendering systems bioactive and dynamic that can subsequently be employed to engage with cells. Different approaches using supramolecular chemistry are reviewed with particular emphasis on their application in cell studies. We conclude with an outlook on future challenges for dynamic cell research and applications.
 Jenny Brinkmann (far left), Emanuela Cavatorta (second from left), Shrikrishnan Sankaran (third from left), Pascal Jonkheijm (middle), Bettina Schmidt (second from right) and Jasper van Weerd (far right) | Jenny Brinkmann is currently doing her PhD focusing on the development of dynamic bioactive surfaces for cell studies making use of supramolecular host–guest interactions. She received her BSc in Biomedical Engineering in 2009, a program offered jointly by the Technical University of Denmark and the Faculty of Health Sciences at Copenhagen University. In 2011 she graduated with an MSc in Biomedical Engineering with a focus on Molecular Bioengineering, from the Federal Swiss Institute of Technology Zurich (Switzerland). |
Emanuela Cavatorta received her MSc degree in Chemistry in 2012 from the University of Parma (Italy). During her master’s assignment she worked on surface-immobilized peptide nucleic acids for sensory systems under the supervision of Prof. R. Corradini. The same year she started her PhD research in the group of Pascal Jonkheijm on work that focuses on supramolecular functionalization of cell membranes. |
Shrikrishnan Sankaran obtained his BE (Hons) in Electronics and Instrumentation and MSc (Hons) in Biological Sciences from the Birla Institute of Technology and Science (BITS), Pilani (India). He did his master’s thesis at the Raman Research Institute, Bangalore, under Prof. V. Lakshminarayanan on Electrochemical Analysis of Biomolecular Interactions. He is currently pursuing his PhD at the University of Twente, working on developing dynamic bacterial systems using supramolecular interactions. |
Bettina Schmidt studied chemistry at the Technische Universität Dresden (Germany), where she received her MSc degree in 2011. She conducted the research for her master’s thesis under the direction of Prof. K. Gloe working on the synthesis and characterization of flavonoid-based ligands and the study of their transition metal complexes. Since October 2011 she is a PhD candidate at the University of Twente under the supervision of Pascal Jonkheijm. The aim of her project is the development of porphyrin-based supramolecular assemblies and their functionalization with biomolecules for targeting studies under physiological conditions. |
Jasper van Weerd received his BA in Biology and Medical Laboratory Research from the Saxion University of Applied Science in 2007 (Netherlands). Afterwards, he obtained his MSc in Biomedical Engineering from the University of Twente in 2010, focusing on molecular, cellular and tissue engineering. From 2008 till 2009 he was working as a part-time researcher for ZebraBioscience BV on modulation-encoded multiplex DNA detection in lab-on-a-chip devices. Since 2010 he has been pursuing his PhD at the University of Twente on supported lipid bilayers for directing cell fate, membrane electrophoresis and surface gradient formation. |
Pascal Jonkheijm earned his PhD in macromolecular chemistry from the University of Eindhoven (Netherlands) with Prof. E. W. Meijer and Prof. A. P. H. J. Schenning and he performed his post-doctoral studies at the Max Planck Institute of Molecular Physiology in Dortmund (Germany) with Prof. H. Waldmann. He has recently been appointed at the MESA+ Institute for Nanotechnology and the University of Twente as Adjunct Professor and is heading research in the so-called Bioinspired Molecular Engineering. He received an NWO Innovation VENI and VIDI Grant, the Young Investigator Award of the Biomedical Materials Program and a Starting ERC Grant. His group aims to develop dynamic chemical strategies to understand, direct and manipulate cellular processes with temporal and spatial control (densities, specificities, separation). Insight into the mechanisms that direct and regulate cellular function will be used to make a new generation of smart biomaterials, to fabricate multifunctional biochips and to renew synthetic biology. |
1. Introduction
Cellular plasma membranes possess spatially organized arrays of receptors serving as fingerprints. These receptor fingerprints regulate information transfer into the cell thereby initiating a plethora of intracellular signaling processes.1–3 Also when cells interact with other cells or materials, these fingerprints regulate a number of cellular processes. Although receptor clustering has been recognized to play an important role in cell function, it remains largely unknown how the collective interaction between populations of different receptors and ligands in two opposed fluid membranes occurs, how it is maintained and regulated. Most intriguing is the potential association of these receptors to lipid platforms, known as rafts, to supposedly assist the formation of larger functional complexes.4–6 Often different ligands activate exactly the same receptors of a certain signal chain. However, temporal variability of protein activities is not the only factor that allows cells to manage many vital processes with comparatively few components. Even the spatial distribution of the proteins within the cells plays a major role.7 Insight into the mechanisms that control and regulate cell function will be gained by assembling synthetic architectures decorated with ligands and using those architectures to engage with receptor fingerprints, mimic and modulate the interaction thereof. So far, the organization of receptors and their mutual interaction have been difficult to address in living cells. Equally challenging is the manipulation of this well-defined organization and monitoring with sufficient spatial and temporal resolution the functional consequences of an altered distribution of receptors. Tailored supramolecular systems with biological ligands will provide an extremely flexible platform (in terms of ligand density, different ligands, separation, etc.). Such systems would allow for analyzing the complexity of the cell membrane and for correlating organization and structure with biological function.In this review the focus lies on surveying different strategies that have been used to fabricate supramolecular bioactive systems for exploring their interaction with cells. Often integrated strategies combining supramolecular assembly with fabrication techniques, modern molecular biology strategies and imaging techniques are employed. We strongly believe that supramolecular and adaptive chemistry combined with fabrication methods provide excellent tools to construct dynamic biological systems, which are to be employed for cell manipulation experiments. Making use of reversible chemical strategies is a rewarding task in developing functional materials and devices.8,9 Knowing the limitations involved in ordering peptides and proteins at different length scales will surely hasten the development of future applications, e.g. tissue engineering and chemical biology.10,11 Self-assembly is an attractive tool and an efficient bottom-up strategy to govern thermodynamic control aiming to position ligands at predefined locations and the modulation thereof. Dynamic bioactive systems open exciting possibilities in fundamental and applied research, and eventually supramolecular cell manipulation.
We start with a review of polymer systems in which bioactive ligands can be dynamically presented through hydrogen-bonded and host–guest driven supramolecular interactions and their properties are illustrated in the context of cell studies. Reorganization of polymers or hydrogels can also be accomplished by real-time switching by e.g. light, temperature, pH, electric field, hydrolysis or enzymolysis methods for which the reader is referred to elsewhere.12 Then, we continue with the description of work in which various peptide based systems, such as α-helices, β-sheets and defined protein scaffolds, and nucleotide based systems are employed in cellular environments. We carry on reviewing a range of amphiphilic systems, which are formed through hydrophilic and lipophilic properties, that dynamically present bioactive ligands and are evaluated in cellular studies. Finally we review dynamic self-assembled monolayers that have been used in a cellular context focusing on real-time switching by external triggers.
1.1 Interfacing cells with hydrogen-bonded and host–guest driven supramolecular polymers
Hydrogen-bonded biopolymers are promising candidates for construction of cell-interactive matrices. An excellent example exploiting the directionality and reversibility of hydrogen bonding is given by 2-ureido-4[1H]-pyrimidinone (UPy) quadruple hydrogen bonding units.13 The reversible nature of these hydrogen-bonding interactions (with lifetimes between 0.1 and 1 s) creates responsive materials and allows for a modular approach. For example, Meijer and coworkers attached this UPy unit to either end of oligocaprolactam or oligo(trimethylene carbonate) and also to one end of a bioactive peptide such as cell-adhesive RGD peptides. RGD is a fibrinogen derived peptide that binds with integrins and so promotes cell adhesion, making it an ideal candidate to probe integrin mediated cell adhesion. When both Upy building blocks are mixed together, a bioactive supramolecular scaffold was fabricated through Upy–Upy interactions while electrospinning (Fig. 1A).13In vitro studies showed good cell adhesion and spreading when the Upy-materials contained the Upy–RGD peptides.13 The in vivo behaviour was studied by the subcutaneous implantation of solution-cast supramolecular polymer films in rats. It was observed that the peptide bearing polymer was able to induce signalling of cells and the physiological process resulting in the growth of new blood vessels from pre-existing ones.13 An important consideration in using hydrogen bonding in aqueous media is the competitive environment. Hydrogen bonds are much weaker in water than in the bulk; however the hydrophobic shielding of this bond in the upper layer of the polymer film makes this binding strong but dynamic.13 Additional hydrogen bonding in the lateral direction can be introduced by incorporating urea (U) moieties adjacent to both Upy-moieties on either end of oligocaprolactone.14 When Upy-functionalized extracellular matrix (ECM) derived peptides were mixed into these Upy-U-biomaterials, such bioactive biomaterials induced human primary tubular epithelial cells to form tight monolayers, which was not seen on the biomaterials lacking these Upy-ECM peptides.14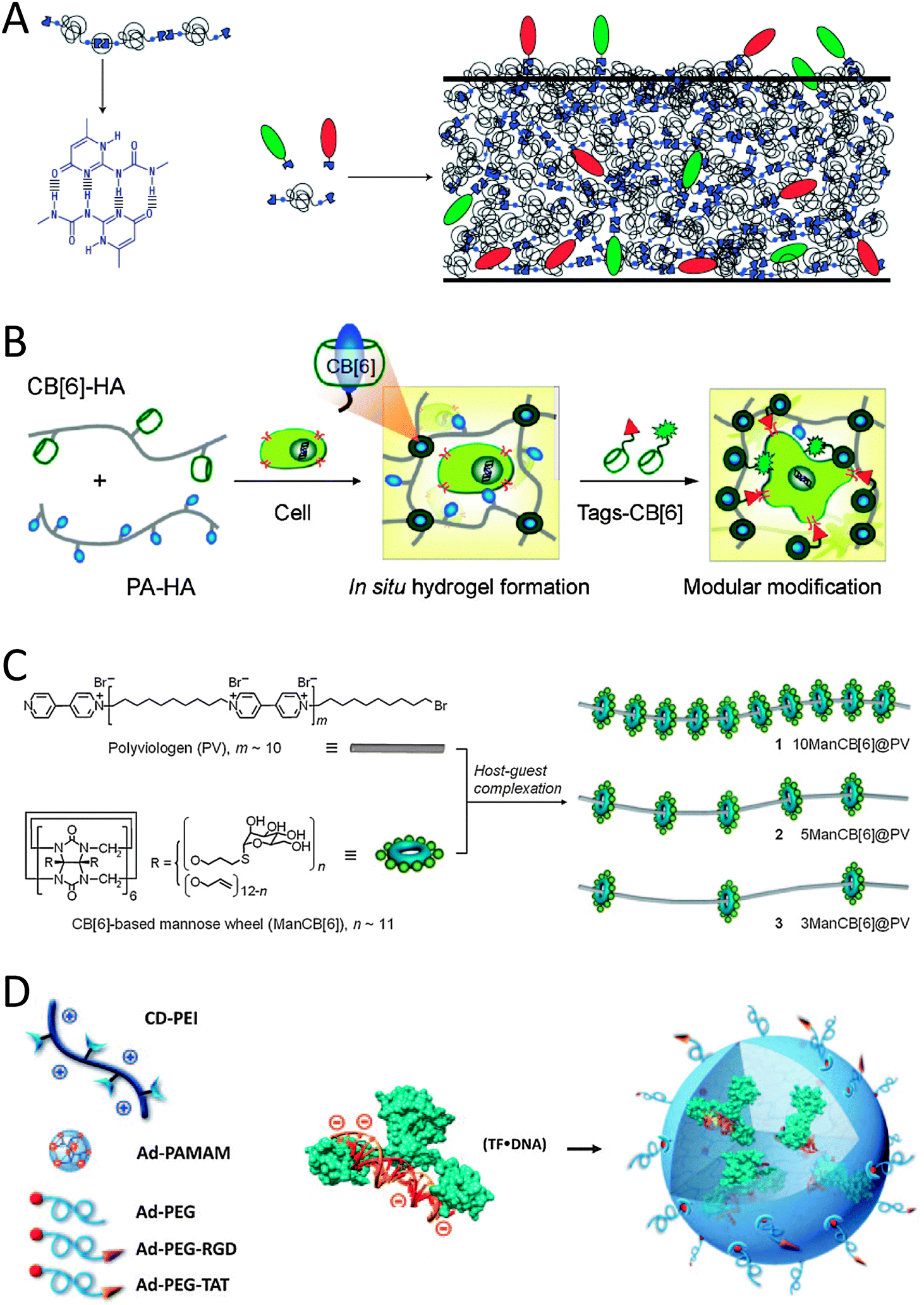 | ||
| Fig. 1 Schematic representations of (A) the self-complementary hydrogen-bonding Upy moiety in a supramolecular polymer that allows for the incorporation of different Upy-functionalized bioactive ligands (green and red moieties) by simply mixing the components (reproduced with permission from ref. 13 provided by the Nature Publishing Group, 2014) and (B) the host–guest complexation between host CB[6]-conjugated biopolymers and guest polyamine-conjugated biopolymers to form a supramolecular hydrogel that allows simply mixing in of various bio-ligand-tagged CB[6] (reproduced with permission from ref. 17 provided by the American Chemical Society, 2014). (C) Glyco-pseudopolyrotaxanes composed of various densities of CB[6]-based mannose wheels threaded on polyviologen showed the highest inhibitory potency for E. coli ORN178-induced hemagglutination relative to monomeric mannoside studies in the case of the lowest density of wheels per chain (reproduced with permission from ref. 23 provided by John Wiley and Sons, 2014). (D) Schematic representation of the host–guest self-assembly approach for the preparation of supramolecular nanoparticles by simply mixing host CD-functionalized polymers with complex guest Ad-functionalized dendrimers and Ad-functionalized bioactive ligands. In the mixture pre-complexed Tf with DNA is present and can be included in the SNP via electrostatic interactions (reproduced with permission from ref. 28 provided by John Wiley and Sons, 2014). | ||
An alternative to supramolecularly end-cap polymeric chains with bioactive ligands is to supramolecularly graft bioactive ligands onto linear co-polymers. Scherman and coworkers illustrated this concept using cucurbit[8]uril (CB[8]) to connect mannose-functionalized viologen to pendant naphthol-moieties on a methacrylate polymer through ternary complexation.15 Alternatively, simply mixing CB[6]-grafted hyaluronic acid (CB[6]-HA) with spermidine-functionalized bioactive peptides results in the formation of a stable in vitro and in vivo supramolecular host–guest system as described by Kim and coworkers (Fig. 1B).16 For example, when formyl peptide receptor like 1 (FPRL1) specific peptide WKYMV was mixed in, its therapeutic signal transduction with elevated Ca2+ and phosphor-extracellular signal-regulated kinase (pERK) levels in FPRL1-expressing human breast adenocarcinoma cells was observed.16 An interesting hydrogel system was made after mixing CB[6]-HA with HA carrying 1,6-diaminohexane or spermine as pendant moieties (in their protonated forms) to make ultrastable host–guest complexes between the two HA-polymers (Fig. 1B).17 The hydrogel could be further modularly modified with for example a bioactive peptide-tagged CB[6], which can be anchored to (residual) diaminohexane moieties in the hydrogel.17 When an RGD-tagged CB[6] was incorporated into the hydrogel, human fibroblast cells entrapped in the hydrogel proliferated approximately 5-fold in 14 days and showed a spread morphology.17 The results were thought to match well with the characteristic cell adhesion and proliferation behaviours in the RGD environment.17 In contrast, when hydrogels lacked the RGD-tagged CB[6], cell proliferation within the hydrogel network was relatively low and the cells retained a round morphology showing poor adhesion.16 Stupp and coworkers reported the complexation of RGD containing adamantane guest molecules to the culture medium on β-cyclodextrin (βCD) host engrafted alginate gels.18 Such supramolecular host–guest gels induced focal adhesion formation and cell spreading.18 Similarly, Cooper-White and coworkers spatially assembled βCD-modified peptides onto adamantane-terminated polystyrene–polyethyleneglycol films.19 Depending on the density of the surface bound peptide, human mesenchymal stem cells showed increased adhesion and variations in morphology ranging from rounded to highly spread, with associated changes in cytoskeletal organization from a disorganized actin cytoskeleton to well-defined and highly aligned stress fibers.19 In another contribution from Kim and coworkers, a covalent polymerized network of side chain functionalized CB[6] was used as a host template.20,21 Through host–guest interactions of two spermidine conjugates, a typical polyamine guest of CB[6], bearing galactose20 or folate21 as targeting ligands, has been introduced to the polymeric spherical nanonetwork of CBs for receptor mediated endocytosis.
Another strategy to create supramolecular polymers decorated with bioactive ligands makes use of threading ligand-functionalized host molecules on various polymers. For example Stoddart and coworkers threaded lactoside-functionalized αCD22 and Kim and coworkers threaded mannose-functionalized CB[6] onto the decamethylene segments of a linear chain of polyviologen.23 The self-assembled lactoside-pseudorotaxanes were investigated for their ability to inhibit galectin-1-mediated T-cell agglutination.22 The lactoside-pseudopolyrotaxane exhibited a valency-corrected 10-fold enhancement over native lactose in the agglutination assay, which was greater than the enhancements observed for lactoside-bearing trivalent glycoclusters and a lactoside-bearing chitosan polymer tested using the same assay.22 The self-assembled mannose-pseudopolyrotaxanes not only effectively induce bacterial aggregation, but also exhibit high inhibitory activity against bacterial binding to host urinary epithelial cells (Fig. 1C).23 The most potent inhibitor was the mannose-pseudorotaxane threaded with only three mannose-CB[6] showing 300 times higher inhibitory potency as compared to free mannose, indicating that the density of bioactive ligands along the rotaxane is key for optimizing interactions.23 The CD-based pseudorotaxane will show appreciable dethreading due to weaker interactions with the polyviologen–decamethylene copolymer when compared to the CB-based pseudorotaxane. A polyrotaxane architecture would prevent unwanted dethreading from occurring. For example, Yui and coworkers showed that the mobility of cationic αCDs along a polyethylene glycol (PEG) chain in a polyrotaxane architecture is favorable for efficient polyplex formation with anionic DNA as expected.24 Also, it is expected that this polyrotaxane will show sufficient cleavage of the disulfide linkages under reducing conditions, because the introduction of only two disulfide linkages avoids the overstabilization of the polyplex. The cleavage of the disulfide bond will trigger the pDNA release through the slow dissociation of the non-covalent linkages between αCDs and PEG as was studied in vitro using dithiothreitol.24 Also in vivo release of the pDNA was followed by microscopy.24 The polyplex was completely removed from endosomes and/or lysosomes 90 min after transfection of fibroblast cells.24 Yui and coworkers also made polyrotaxanes with RGD-functionalized αCDs on linear guest polyethyleneglycol.25 As a result, a faster initial recognition process was observed by quartz crystal microbalance-dissipation (QCM-D) measurements when RGD peptides were residing on the polyrotaxane architecture in comparison to covalently immobilized RGD systems.25 However, despite this faster initial recognition process stable focal adhesion formation that is accompanied by actin polymerization was suppressed.25
Host–guest characteristics can also be used to create supramolecular polymer nanoparticles (SNP). Tseng and coworkers developed a molecular recognition system based on an adamantane guest and a βCD host to achieve self-assembly of SNPs (Fig. 1D). Three basic molecular building blocks were used to form the SNPs: adamantane-grafted polyamidoamine dendrimer (Ad-PAMAM), βCD-grafted branched polyethylenimine (CD-PEI) and adamantane-functionalized PEG (Ad-PEG).26,27 Ad-PEG functions as a capping reagent and solvates to improve the water solubility, structural stability and non-adhesiveness.26 When Ad-PEG functionalized with cell-adhesive RGD or cell-penetrating TAT peptides was blended in during the one-pot mixing process, SNPs are formed that can engage in specific cell-integrin interactions and subsequently can be internalized into cells.27 When a transcription factor (Tf) pre-complexed with a luciferase reporter vector (DNA) was also blended in during the one-pot mixing process, uniform SNPs of 50 nm diameter were found that were internalized by HeLa cells after 12 h (Fig. 1D).28 The bioactivity of Tf was quantified by measuring the bioluminescence intensity from the DNA reporter.28 The results show that these SNP systems provide a method for manipulating cellular behaviour.
1.2 Employing α-helices for cell studies
One widely studied naturally occurring structural motif that has been exploited to dynamically interface with cells is the α-helix. An α-helix is a chain of amino acids forming a right-handed coil stabilized by hydrogen bonds. Coiled-coils consist of two or more α-helices wrapped around each other like strands of a rope. This is achieved by a patterned arrangement of hydrophobic and charged residues along the polypeptide chain of each α-helix. The most explored pattern is that of leucine zippers that contain seven-residue repeats (heptads) (Fig. 2A).29 Having high binding affinities (low nM range), these leucine zippers prove to be effective, non-toxic labels to study cellular processes at the membrane. Leucine zippers have been extensively used in vivo and in vitro to immobilize various biomolecules in close proximity to each other by fusing complementary coils to them. For example, Matsuzaki and coworkers engineered them to selectively label receptors on the cell membrane.30 In this work the protein receptors were genetically fused with one coil and expressed in mammalian cells while the complementary coil was conjugated with a fluorescent dye and mixed with the cells.30 Labeling of the receptors on the membrane was witnessed within minutes. Receptor functions like internalization of the receptor and calcium influx as a response to certain chemical stimuli were not impaired and were monitored.30 Cholesterol-modified coiled-coil forming peptides E and K were successfully employed by Kros and coworkers for insertion in the membranes of Chinese hamster ovary cells and the skin of zebrafish embryos.31 Fluorescent microscopy was used to confirm the specific supramolecular coiled-coil formation on the membranes.31 Futaki and coworkers used the heterodimeric coiled-coil formation between E and K peptides for the artificial activation of epidermal growth factor receptor (EGFR) by supramolecular dimerization.32 Whereas Famulok and coworkers previously employed extracellular constitutively dimerized (disulfide bridged) leucine-zipper EGFR fusion to analyze the mechanism of an intracellular EGFR activating factor,33 in the report of Futaki and coworkers it is the addition of dimeric K-coil peptides that triggered dimerization of fusions of the E-coil peptide with EGFR receptors on plasma membranes and thereby activated the receptor (Fig. 2B).32 Autophosphorylation of EGFR was confirmed by Western blot using a phosphorylated Y1173-specific antibody.32 | ||
| Fig. 2 (A) Helical-wheel representation of a parallel dimer with a heptad repeat of amino acids present in assemblies of coiled-coil peptide motifs. The heptad repeat positions are labeled from a to g and the α-helices propagate into the page. The a, d surface is predominantly hydrophobic, and residues at positions e and g are often charged (reproduced with permission from ref. 29b provided by Springer, 2014). (B) Scheme of artificial activation of transmembrane receptors by a helical peptide through coiled-coil formation (reproduced with permission from ref. 32 provided by John Wiley and Sons, 2014). (C) Schematic diagram showing the self-assembly of keratins in the presence of extracellular matrix protein domains (reproduced with permission from ref. 36 provided by the American Chemical Society, 2014). (D) Illustration of the molecular design of bioactive surfaces capable of dynamically and reversibly regulating immobilized ligands using the leucine zipper assembly (reproduced with permission from ref. 37 provided by the American Chemical Society, 2014). | ||
The self-assembling property of leucine zippers has enabled researchers to further assemble them into microstructures.34 Microscopic fibers have been assembled using two synthetic complementary α-helical coils and some of the side chains have been functionalized with azides, alkenes and thiols by click reactions in water.34 This was visualized by conjugating biotin to the fibers that were then allowed to bind with gold or rhodamine labeled streptavidin (SAv).34 Through binding of biotin-functionalized biomolecules to SAv, these fibers can thus potentially be used to develop multi-component functionalized systems. Similarly, fibrils have been constructed from synthetic leucine zippers with RGD peptides fused to their N-terminal ends.35 The presence of numerous RGD peptides at the surface of these fibrils promoted multivalent interactions with the integrins at the surface of cells.35 These fibrils were shown to bind with much higher affinities than short RGD-containing peptides in solution by cell-adhesion inhibition assays. Also, by immobilizing these fibrils on the surface, substrates promoting cell adhesion comparable to that achieved with the commonly used fibronectin and vitronectin were designed.35 Going one step further, keratin based α-helices have been used to fabricate biocompatible hydrogels through lateral association of the coiled-coils as reported by Iwata and coworkers (Fig. 2C).36 By integrating a laminin domain, which interacts with various types of integrins, the hydrogel was developed to promote cell adhesion and proliferation.36 The hydrogels were shown to be a suitable platform for the adhesion and proliferation of neural progenitor cells.36 Since these hydrogels are biocompatible and biodegradable, they can be implanted into brain tissue to potentially combat neurodegenerative disorders like Parkinson's disease.
The reversible nature of binding between complementary coils has also been used to produce functional solid supports for cell interactions (Fig. 2D). Substrates for reversible cell adhesion have been designed by immobilizing leucine zipper coils on a surface.37 The coils on the surface (coil-A) have a RGD peptide fused to their C-terminal end, accessible to cell receptors in solution.37 These surfaces were made cell repellant by adding a high affinity complementary coil with a PEG linker (B) to the solution.37 The coils bind and the PEG linkers mask the RGD peptides preventing cells from adhering to the surface. This situation was then reversed by adding coil-A into solution that binds competitively to coil-B, thus releasing it from the surface and unmasking the RGD peptide, allowing the cells to attach to it again.37 Surface immobilization of coiled-coils has enabled the generation of cell surface interactive gradients as studied by Tirrell and coworkers.38 A glass surface was modified by generating a monolayer of an α-helical coil (ZR) on it and fabricating a polydimethylsiloxane (PDMS) microchannel to enclose it.38 The complementary coil (ZE) was fused to a fibronectin protein domain which promotes cell adhesion. By passing unmodified ZE and fibronectin-fused ZE through a microfluidic gradient generator and flowing this mixture through the ZR immobilized microchannel, a gradient of surface immobilized fibronectin was produced.38 Cell adhesion assays on these surfaces revealed that the cell density increased with the increasing concentration of fibronectin.38
1.3 Employing β-sheets for cell studies
Self-assembled nanostructures can also be fabricated using synthetic β-sheet forming peptides. Alternate placement of positively charged, hydrophobic and negatively charged residues gives rise to electrostatic and solvophobic interactions between the peptides and directs the β-sheet formation. Attaching various groups to the terminals of the peptides provides functionalization of these β-sheets and also promotes the formation of particular supramolecular structures like tapes, ribbons, fibrils and fibers. By attaching carbohydrate conjugated coils of different geometries to these peptides, the effect of functional groups on the length of assembled nanostructures was studied by Lee and coworkers (Fig. 3A).39 Short linear coils resulted in micrometer scaled ribbons but long dendritic coils generated ribbons only about 150 nm long due to steric crowding effects.39 The carbohydrates were selected to bind with receptors on pathogenic bacteria and it was shown that both nano-assemblies were effective in immobilizing particular bacterial strains.39 At high concentrations, the long ribbons caused bacterial clustering whereas the shorter structures did not, thus showing that the size of these assemblies elicits different responses from the living entities binding to them.39 | ||
| Fig. 3 (A) Supramolecular building blocks representing β-sheet peptides with attached coils and carbohydrates. Negatively stained transmission electron micrographs show nanostructures from the peptides. GP1-peptides, with a small and linear coil, form nanostructures of several micrometers long, whereas GP2-peptides, with a high-volume-fraction dendritic coil, form nanostructures of only about 150 nm long. Both nanostructures could immobilize bacteria to a similar degree; however, only the long nanostructures were shown to induce the formation of bacterial clusters (reproduced with permission from ref. 39 provided by the American Chemical Society, 2014). (B) Representation of the nanoribbon self-assembly of a peptide consisting of a random coil block (Tat CPP), a flexible-linker block (GSGG) and a β-sheet assembly block. After encapsulation of hydrophobic guest molecules (Nile red), the nanoribbons can be internalized in cells as observed from the overlaid confocal laser scanning microscopy (CLSM) images (reproduced with permission from ref. 41 provided by John Wiley and Sons, 2014). | ||
Another approach involved using triblock constructs made of a carbohydrate, a PEG linker and a β-sheet forming peptide.40 By modifying the length of the PEG linker, the stability of the nanoribbons formed was modified and the most stable structure for bacterial motility inhibition and agglutination was identified.40 Using mannose as the carbohydrate, the ribbons were designed to be specific towards a particular strain of pathogenic bacteria.40 The ribbons were further modified by encapsulating the fluorescent probe, Nile red, into the hydrophobic interface formed by the bilayer of β-tapes.40 This enabled the ribbons to be used for the fluorescent detection of clustered pathogenic bacteria. These techniques can be extended to clinical screening and isolation of pathogens in food and water. Encapsulating dyes in the hydrophobic domain of these structures potentially enables therapeutic drugs to be transported into human cells.41 β-Sheet forming peptides were fused with cationic cell penetrating peptides borrowed from the HIV-1 Tat protein.41 A green dye was conjugated to the peptides and Nile red was incorporated into the nanoribbons.41 These assemblies were shown to penetrate the cell membrane of HeLa cells and stain the cytoplasm whereas the peptide conjugated dye entered the nucleus (Fig. 3B).41 This was speculated to be due to the disassembly and transport of different components of the structure within the cell due to various cytoplasmic entities. This could facilitate multiple drug delivery into different regions of the cell.
1.4 Interfacing cells with protein scaffolds
One of the key aspects in developing supramolecular systems that interact with cells is identifying ligands that can specifically bind with particular receptors on the membrane. Amino acids confer a wide range of structural and functional properties to the peptides they form. This enables researchers to identify even short peptides that bind strongly with most proteins. Scaffolding proteins have been developed as an effective tool for identifying and displaying these peptide sequences. These proteins usually consist of an underlying stable structural element, termed the scaffold. A flexible and variable loop containing the required peptide extends from this scaffold and is exposed to the solvent. Nature's most successful protein scaffolds for molecular recognition are antibodies, which can bind to countless biomedical targets with high specificity and affinity. However, non-antibody protein scaffolds have generated much interest because of limitations of antibodies, which generally include expensive recombinant production in mammalian cells and the inability to site-specifically incorporate chemical tags. Alternative protein scaffolds have been developed such as affibodies, ankyrin repeat proteins, kunitz domains, PDZ-domains, knottins and anticalins, which have been reviewed elsewhere.42 Most of these have been employed to display libraries containing millions of random peptide sequences on viral phages, bacterial or yeast cells for selecting sequences that bind specifically to particular proteins. Peptides that bind to various receptors present on the surface of mammalian cells have been identified using this technique primarily for imaging, diagnostic and potentially therapeutic purposes. High affinity integrin binding peptides were discovered by Cochran and coworkers using the Ecballium elaterium trypsin inhibitor (EETI-II) knottin scaffold in yeast-display libraries.43 Knottins are short peptides (∼3 kDa) structurally stabilized by at least 3 disulfide bridges forming a cystine knot structure from which their name is derived. One or more of the loops present between these disulfide bridges can usually be extensively modified without disrupting their three-dimensional fold thus enabling knottins to be exploited as scaffold proteins. To develop high affinity binding peptides for integrin, the 6 amino acid trypsin binding loop of EETI-II was replaced by a randomized 11 amino acid peptide library with the RGD motif located at different locations.43 The knottins were expressed in yeast cells and displayed on the surface.43 Integrin binding was detected using a labeled anti-integrin antibody and fluorescence activated cell sorting (FACS).43 Since these knottins are short peptides, they were artificially synthesized and shown to bind with multiple integrins specific to cancerous glioblastoma tumor cells at low nanomolar affinities consequently inhibiting these cells from adhering to the extracellular matrix.43 Thus it is possible to identify peptides that interact with receptors of targeted cell types with extremely high specificity and binding affinity (Fig. 4). | ||
| Fig. 4 An engineered ∼3.5 kDa, conformationally constrained cystine knot peptide that binds with high affinity and specificity to αvβ3, αvβ5, and α5β1 integrins. The blue star indicates the location of N-terminal modification with e.g. dye AF680, and red indicates an engineered integrin-binding loop, which replaces the native trypsin-binding loop. Sequence and disulfide bond connectivity for the engineered knottin is given (integrin-binding loop sequence shown in red). AF680-labeled integrin engineered knottin illuminates mouse medulloblastoma in vivo imaged 2 h after tail vein injection (mouse with (left) and without (right) tumor) (reproduced with permission from ref. 43 provided by the National Academy of Sciences, 2014). | ||
1.5. Interfacing cells with nanostructures based on DNA
Well-defined Watson–Crick base-pairing between nucleotides and advances in single stranded DNA oligomer synthesis have enabled researchers to design DNA strands that organize into various nanostructures. By placing complementary sequences of nucleotides at different locations on multiple DNA strands, numerous structures have been self-assembled for various nanotechnological applications,44 including the fabrication of tunable ECM.45 In the latter case from Way and coworkers, five DNA strands that form a four helix ribbon with biotin terminal overhangs were designed (Fig. 5A).45 The synthetic protein consisted of a fibronectin domain fused to a monomeric SAv protein that would bind to the biotin on the DNA ribbons. Together the two components formed a cell interacting ECM scaffold to which HeLa cells could strongly adhere to.45 By modifying the DNA strands to contain unhybridized single stranded regions, the stiffness and persistence lengths of the scaffolds could be fine-tuned.45 This was utilized to modulate cytoskeletal organization and shape of the cells, the status of particular signal transduction proteins and localization of an intracellular transcription factor.45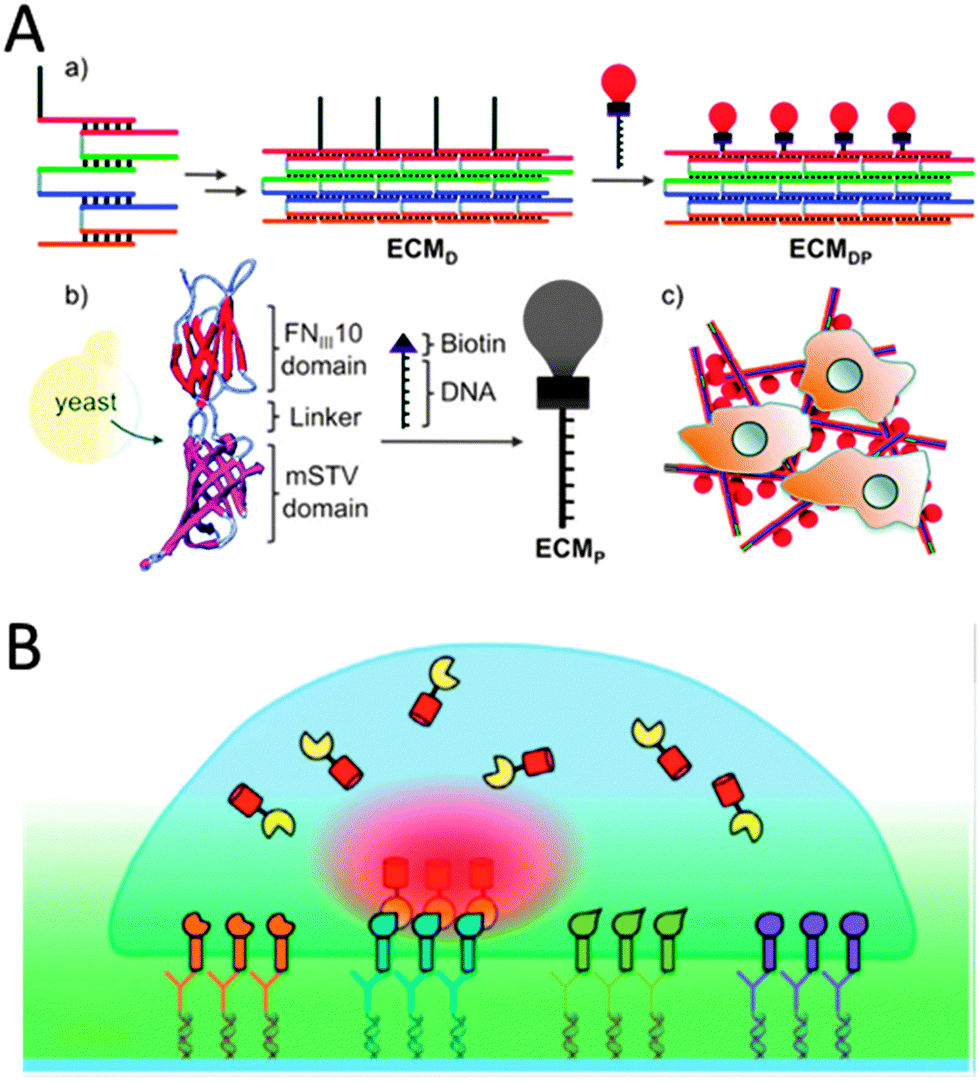 | ||
| Fig. 5 (A) A DNA/protein-based matrix (ECMDP) was constructed from a DNA ribbon (ECMD) surface-functionalized with proteins (ECMP) containing the RGD domain of human fibronectin (reproduced with permission from ref. 45 provided by the American Chemical Society, 2014). (B) Schematic representation of a protein-array inside living cells using surface-bound ligand patterns, DDI and bait-presenting artificial receptor constructs (bait-proteins, orange, blue, green and purple surface bound ligands via DDI). Interaction analysis of a cytosolic prey protein (yellow) fused to a fluorescent protein (red) can be imaged by total-internal reflection fluorescence microscopy (reproduced with permission from ref. 53 provided by John Wiley and Sons, 2014). | ||
Another way to employ the specificity of Watson–Crick base pairing is the DNA-directed immobilization (DDI) of proteins for generating microstructured patterns of proteins on surfaces (Fig. 5B).46 This method requires conjugates of the protein-of-interest functionalized with short single-stranded oligonucleotides (ssDNA), e.g. via simple biotin–SAv conjugation, and surface substrates functionalized with capture oligonucleotides complementary to the DNA–protein conjugates employing various modern microstructuring techniques.47 For example, Heath and coworkers installed biotin on cell-surface proteins via incubation with N-hydroxysuccinimide–biotin. The biotinylated human primary neurons and astrocyte cells were then encoded with ssDNA–SAv conjugates.48 The DNA-tagged cells were immobilized on the corresponding spots bearing the complementary DNA.48 Various methods are available for the cell-surface attachment of ssDNA tags,46 for example membrane anchoring of oligonucleotides or the Staudinger ligation of phosphine-modified ssDNA to azide groups that are installed on the cell surface through metabolic labeling.49,50 DDI of cells on glass surfaces has been used to study cell sorting, cell adhesion and intercellular interactions or to grow small populations of cells which could be utilized for screening purposes.50–52 Niemeyer and Dehmelt and coworkers fabricated protein ligand arrays with subcellular dimensions (Fig. 5B).53,54 To this end, a DDI array was fabricated by dip-pen lithography to generate micrometer-sized patterns, typically 45 μm with a pitch of 12 μm.53 The DDI arrays represented either biotinylated epidermal growth factor for culturing with carcinoma cells or different antibodies with binding specificity for peptide epitopes on transmembrane receptor constructs such as the regulatory unit II-β.54 The latter recognition led to specific recruitment and concentration of the transmembrane proteins which was visualized by total internal reflection microscopy (TIRF).54 TIRF studies were used to further visualize the recruitment of the cytoplasmic catalytic subunit cat-α of protein kinase A to the DDI-areas.54
1.6. Employing peptide amphiphile nanostructures for cell studies
When peptides are substituted with amphiphilic moieties, they become interesting building blocks to create self-assembling biofunctional nanostructures.55,56 Such peptide amphiphiles (PA) self-assemble in aqueous conditions into 1D nanostructures that possess a hydrophobic core and a hydrophilic shell. These nanostructures could easily be used to encapsulate small hydrophobic molecules for drug delivery. Moreover, the fibrous character of the nanostructures allows their employment in 3D networks as bioactive scaffolds. The structure of the PAs that is required to create such high-aspect-ratio nanofibers is shown in Fig. 6A. One part of the PAs is a hydrophobic block, usually an alkyl chain the length of which can be tuned. The hydrophobic part of the molecule is conjugated to a β-sheet forming segment coupled to a sequence with charged amino acids. The final element in the structure is a bioactive peptide sequence. The charged amino acids guarantee good solubility under physiological conditions. Between them there is electrostatic repulsion which pushes the molecules apart. The balance of all three forces, hydrophobic (alkyl chain), hydrogen bonding (β-sheet forming segment) and electrostatic repulsion (charged amino acids), determines the size and shape of the final cylindrical assembly (Fig. 6B).55 Due to their composition the PAs can be considered biodegradable since they can be metabolized into amino acids and lipids.57 Essential to the biofunctionality of the nanostructures are the epitopes that are introduced that can interact with cells or proteins. This part is presented on the periphery of the assembled nanostructures. Depending on the bioactive peptide sequence a variety of biological functions can be addressed (Fig. 6C).55 For example, bundles of PA nanofibers displaying RGD epitopes have been used as ECM-mimicking scaffolds by Stupp and coworkers.58 Best results in the so-called focal adhesion and cell migration studies were achieved when the synthesized PAs displayed the RGD epitope in branched ways. In a similar approach, a laminin derived epitope IKVAV was displayed on PA structures following the same procedure (Fig. 6C).59 The use of IKVAV displaying PA nanofibers in mice with spinal cord injuries led to functional recovery after 9 weeks post treatment.57 The PA nanofibers were able to promote regeneration of motor and sensory axons. The axons entered and sometimes even crossed the lesion that was induced. Furthermore, the PA nanofibers were not only involved in cell signalling, but also provided structural support to neuronal cells. A comparison with only the pure IKVAV peptide showed that it did not promote any functional recovery indicating the role of structural templating. Due to the charged nature of PAs their self-assembly is largely influenced by the ionic conditions and pH of the biological environment.59 This might prove beneficial for medical applications enabling PA delivery via injection and triggered in vivo self-assembly. Injectable therapies are desired due to their non-invasive nature compared to traditional surgical procedures, for example in the case of articular cartilage regeneration, which, if left untreated, can lead to osteoarthritis.57 To treat spinal cord injuries in mice a peptide domain with binding affinity to the transforming growth factor β1 (TGF-β1), known to enhance cartilage repair, is used.57 A self-assembling bioactive hydrogel prepared by using two different PAs, a non-bioactive and the TGF-binding one, was used to treat full thickness articular cartilage defects in rabbits. The TGF-binding hydrogel induced de novo cartilage formation quite similar to the surrounding tissue. Histological evaluation showed excellent tissue integration virtually undistinguishable from native cartilage. The binding epitopes for TGF-β allowed for slow and prolonged release of the growth factor and are suspected to be essential for the observed tissue regeneration. It is postulated that the epitopes also protect the TGF-β from proteolytic degradation. The great advantage of this approach is that there is no need for exogenous growth factors or transplanted cells. | ||
| Fig. 6 (A) Molecular structure of a representative PA with four rationally designed chemical entities. (B) Molecular graphics illustration of an IKVAV-containing PA molecule and its self-assembly into nanofibers (reproduced with permission from ref. 55 provided by John Wiley and Sons, 2014). (C) Scanning electron micrograph of the IKVAV nanofiber network formed by adding cell media to the PA aqueous solution (reproduced with permission from ref. 55 provided by John Wiley and Sons, 2014). | ||
The hydrophobic and hydrophilic regions that are key to the self-assembly of PAs can also be combined using supramolecular host–guest chemistry. Scherman and coworkers made use of the CB[8] to form a stable ternary complex in aqueous solution between an electron-poor and an electron-rich guest inside the CB[8] cavity.60 A pyrene (electron-rich)-labeled peptide and a viologen (electron-poor)-capped long alkyl chain were coupled via the CB[8] cavity to form a supramolecular PA.60 These supramolecular PA building blocks can subsequently self-assemble further into nanostructures. After internalization in HeLa cells the vesicular structures could trigger cell death.60 By addition of adamantane, a guest that binds strongly to CB[8], both viologen and pyrene guests are released from the CB[8] cavity thereby resulting in a loss of the nanostructures.60 The release of pyrene was confirmed by the increase in fluorescence, while uncomplexed viologen is cytotoxic to the cells.60 No cell death, but an increase of pyrene fluorescence was observed upon addition of electron-rich naphthol, which replaces only pyrene, to form a more stable ternary complex with CB[8] and viologen.60
1.7 Interfacing cells with aromatic rod- and disc-shaped amphiphile nanostructures
It is well known that when hydrophobic rod-like segments are connected to hydrophilic head groups, molecular building blocks with amphiphilic characteristics are created that drive the systems to self-assemble into organized nanostructures such as micelles, cylinders and hollow vesicles in aqueous solutions.61 For example, Lee and coworkers have synthesized amphiphiles consisting of rigid aromatic hydrophobic tetra(p-phenylene)s as rod segments and hydrophilic mannose functionalized poly(ethylene oxide) as coil segments (Fig. 7A). Stable carbohydrate-coated nanocapsules were formed in aqueous solution.62 Another molecule consisted of a rigid aromatic alkyl-substituted oligo(phenylene) segment connected to a flexible dendritic branch. These amphiphiles self-assemble in aqueous conditions into various nanostructures depending on the degree of branching in the dendron. Hydrophobic dye Nile Red can be encapsulated in those cylinders enabling the observation of the nanostructures by fluorescence microscopy.63 Investigation of the interactions of the carbohydrate-coated nanostructures with E. coli cells showed that both nano-objects could immobilize bacterial cells.62,63 Disc-shaped aromatic molecules have been used by Brunsveld and coworkers for fabricating nanostructures that can be used for interaction with bacteria (Fig. 7B).64 Their discotics consist of an extended aromatic core of three 2,2′-bipyridine-3,3′-diamines linked to a central benzene-1,3,5-tricarbonyl unit.64 These discotics can self-assemble in water to form an auto-fluorescent columnar polymer.64 The peripheral hydrophilic ethylene glycol chains provide water-solubility and shield the hydrophobic core to promote hydrogen bonding and π–π stacking between adjacent discotics. Functionalization of the peripheral ethylene oxide tails with mannose groups led to selective binding of the supramolecular polymers to E. coli bacteria.64 The same discotic molecules have been adopted as cellular uptake carriers by Brunsveld and coworkers.65 π–π-Stacked assemblies of discotic monomers that feature peripheral amine groups are easily taken up by the cells via endocytosis to the cytoplasm.65 As expected, when these peripheral amine groups are absent, the supramolecular polymers are not taken up.65 Interestingly when non-cell permeable monomers, which are functionalized with biotinylated ligands, are blended in supramolecular polymers of amine-functionalized monomers, successful uptake of the supramolecular polymer was observed through staining with a fluorescently labelled anti-biotin antibody after fixation of the cells.65 | ||
| Fig. 7 (A) A triblock rigid aromatic-flexible dendritic block molecule consisting of a hydrophobic alkyl chain (n = 21), a rigid aromatic segment and flexible carbohydrate conjugate dendrons. The carbohydrate-coated nanostructures can immobilize E. coli cells. The degree of immobilization was significantly dependent on the shape of the nanostructure (reproduced with permission from ref. 63 provided by the American Chemical Society, 2014). (B) Discotic compounds functionalized with mannose ligands (R2) and inert glycol side chains (R1) form columnar architectures. Binding of these systems to bacteria result in their clustering, which was visualized using fluorescence microscopy (reproduced with permission from ref. 64 provided by John Wiley and Sons, 2014). | ||
1.8 Interfacing cells with lipid bilayer architectures
The early notion that the cell membrane, which consists of a lipid bilayer with associated proteins and carbohydrates, acts as a liquid in which its constituents can move freely is considered nowadays an oversimplification of reality. This so-called ‘fluid mosaic model’ has been updated to include variable patchiness, variable thickness and higher protein occupancy than was previously considered. Studying membrane associated processes forced scientists to conceive model lipid bilayers that mimicked the amphiphilic lipid architecture ensuring that bilayer properties are comparable to those of natural cell membranes. Such model membranes can be subdivided into suspended, so-called black lipid membranes (BLMs), and solid supported lipid bilayers (SLBs) and lipid vesicles (Fig. 8). Among the broad class of self-assembled amphiphilic lipids that are generally termed as vesicles, liposomes are unilamellar vesicles where the aqueous interior of each vesicle is bound by a single lipid bilayer.66–69 Besides the aforementioned examples other lipid inspired structures have also been studied like lipid monolayers and lipid derived polymers. Common, either natural or synthetic, lipid building blocks can be categorized according to their structure, e.g. fatty acids, phospholipids, glycerophospholipids, triacylglycerols, eicosanoids, waxes, sphingolipids, as well as steroids, isoprenoids and terpenes. Composition and protocol of preparation can be tailored to tune the physicochemical properties of the lipid system, either vesicular or supported, e.g. surface charge, bilayer fluidity and lamellarity (i.e. number of lipid bilayers). Fluidity, which refers to the lateral diffusion of the membrane constituents, is affected by the gel–liquid transition temperature of the lipids, which is determined by the alkyl tails and is modulated in vivo by cholesterol. The lipid systems can be further modified by direct covalent coupling of ligands to lipid bilayers, non-covalent interaction using chelating lipids and via insertion of natural or synthetic lipid anchors. A detailed overview of ligand-bilayer immobilization via covalent and non-covalent chemistry is reviewed elsewhere.68 Modifications of lipid bilayer structures can improve the limited stability in certain environments, such as in air or in vivo.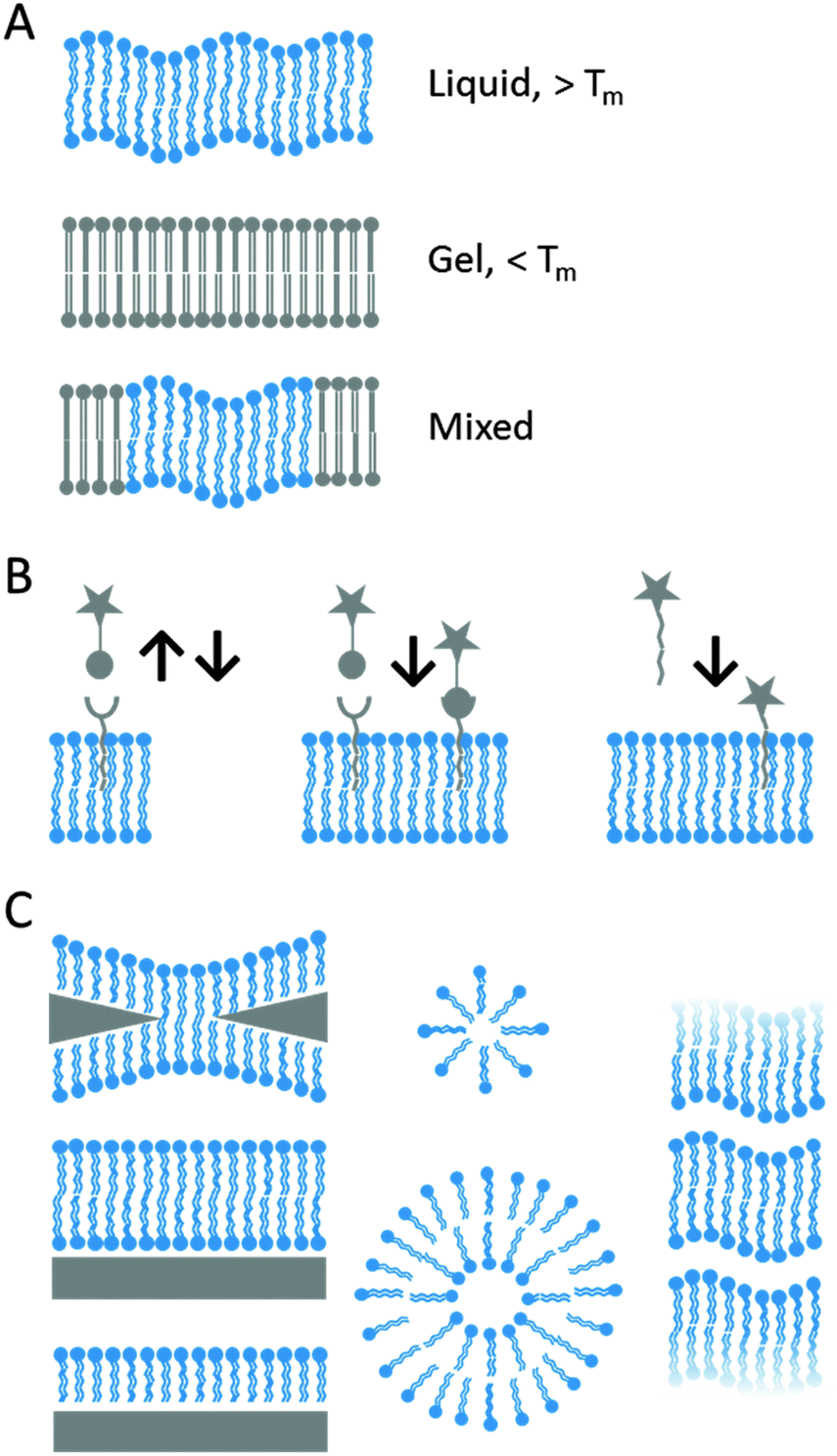 | ||
| Fig. 8 An overview of artificial lipid bilayers and a few parameters are given that can be fine-tuned such as (A) phase transition temperature resulting in mobile (liquid) or immobile (gel) bilayers, (B) ligand immobilization through e.g. non-covalent interactions (left), covalent reactions (middle) and post-insertion of ligand-modified lipids (right). (C) Various types of lipid-based solid-supported and solution architectures. | ||
Liposomes and liposome-based structures have matured for the intracellular delivery of drugs and imaging agents over the last decades and have found clinical application.69 The delivery takes place by uptake of the particle and its cargo by the cell. The internalization can be mediated by non-specific electrostatic recognition between positively charged vesicles and the negatively charged lipids of cell membranes,71 as extensively demonstrated in the field of gene transfection where typically DNA and cationic lipids are combined in the so-called lipoplexes.71 Anderson and coworkers have adopted the use of lipidoids to facilitate non-viral delivery of small interfering RNA to endothelial cells (Fig. 9A).72–74 Michael addition chemistry between alkylacrylate and acrylamide materials and amines was utilized to create a structurally diverse library of lipid-like molecules termed lipidoids, which were analyzed for their ability to transfect cells both in vitro and in vivo.72,73 The library allowed the study of the role of the length and number of alkyl chains, the charge and the substitution of the amines as well as the amide bond. The lead candidates facilitate sequence-specific knockdown in a variety of cellular targets and animal species.72 Typically these lipidoids form cationic 200 nm spherical lipid-like particles.73 Improvement in delivery efficacy has been achieved through synthesizing lipidoids following ring-opening of alkyl-substituted epoxides by amine substrates.74 With these lipid-like particles siRNA-directed liver gene silencing at therapeutically relevant doses was possible.74
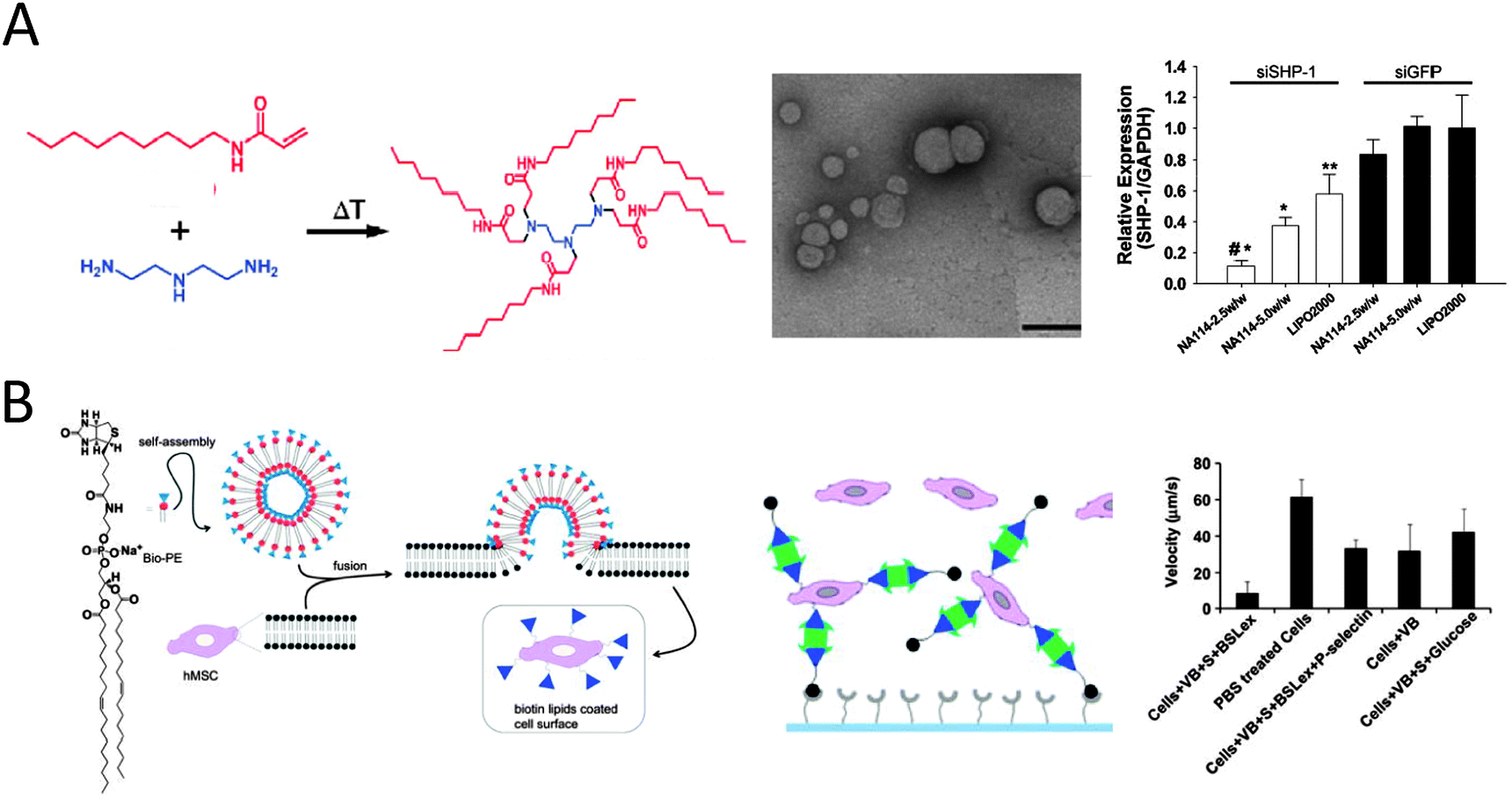 | ||
| Fig. 9 (A) An example of the synthesis of lipidoids through the conjugate addition of amines to acrylamides. TEM images of lipidoid nanoparticles complexed with siRNA (scale bar 200 nm). Successful silencing of SHP-1 by siSHP-1 delivery in HUVECs (10% serum) as concluded from quantitative real-time PCR measurements to determine the SHP-1 expression in HUVECs at 2 days after siRNA transfection using the synthesized lipidoids (NA114, two doses) and compared with lipofectamine 2000 (LIPO2000) and all siGFP-transfected groups (reproduced with permission from ref. 73 provided by John Wiley and Sons, 2014). (B) Schematic showing formation of vesicles from biotinylated lipid and modification of cells with biotinylated lipid vesicles. The vesicle modified MSCs are further modified with sialyl Lewis X using SAv and biotinylated SLeX. Rolling of SLeX modified hMSCs on a P-selectin coated surface was significantly improved (8 μm s−1 as compared to 61 μm s−1 for unmodified MSCs) from flow chamber assays (0.5 dyn cm−2) (reproduced with permission from ref. 84 provided by Elsevier, 2014). | ||
Endocytotic pathways can also be exploited to enter the vesicle in the cytosol in the form of an endosome. To this end, vesicles can be decorated with specific ligands that recognize complementary cell receptors to enhance their uptake.75 The lateral diffusion of such ligands on the surface of the vesicle allows their pre-organization and thereby creates an array of multiple binding sites for cell receptors, which leads to a more favorable interaction according to the concept of multivalency.76 However, in addition to a thermodynamically favorable binding of the carrier vesicle to the membrane, the carrier vesicle has to release its cargo to elicit cell responses. Moreover, especially when considering in vivo applicability, vesicle features such as persistence, clearance or accumulation of the vesicles in the body have to be balanced by enzyme degradability into non-toxic byproducts after the release of the load. Typically a decoration with hydrophilic polymers such as polyethylene glycol improves the in vitro and in vivo stability of lipid vesicles. Vesicles based on an amphiphilic CB[6] derivatized with EG6 at its periphery allow for incorporation of spermine-modified targeting ligands and imaging probes through specific host–guest interactions between spermidine and the CB[6] host.77 When these supramolecular lipid vesicles were loaded with doxorubicin, internalization into cells by receptor mediated endocytosis and release of entrapped drugs was confirmed.77
Further developments have been demonstrated employing a silicate porous framework to decorate the surface of porphyrin-conjugated vesicles.78 These particles circulated in blood for prolonged time and reached the cytosol by endocytosis. Upon irradiation of the porphyrin units by light inside the cells, the singlet oxygen caused cell death of cancer cells.78 Enhanced stability could also be achieved by using interbilayer-crosslinked multilamellar vesicles.79 Crosslinking was achieved by the maleimide modified head-groups of opposing bilayers using a dithiol linker. Residual maleimides were capped by reaction with thiolated PEG and yielded an inert vesicular surface. Interbilayer-crosslinked vesicles stably carried antigenic proteins in the hydrophilic vesicle core and apolar immunostimulatory agents in the hydrophobic vesicle wall. Endosomal lipases catalyzed the selective rupture and release of the cargo.79
Alternatively, fusion of vesicles to the cellular membrane can install chemical functionalities on the cell surface.80 This fusion leads to cell membranes that are transiently doped with artificial lipids without altering the cellular machinery.80 This method serves as an alternative to e.g. antibody recognition,81 metabolic labeling82 and covalent modification83 methods. Transient chemically modified membranes are of interest as they would potentially transform cells in therapeutic agents as tumor vaccines or building blocks for tissue regeneration. The surface of stem cells (hMSC) was chemically engineered by incubation with liposomes integrally composed of an amphiphilic biotinylated lipid.84 The artificial lipids were observed as rafts, i.e. locally clustered on the membrane. The exposed biotin groups were accessible for binding to SAv and subsequently to biotinylated homing ligand sialyl Lewis X (Fig. 9B).84 The engineered hMSCs with sialyl Lewis X showed improved rolling behavior on P-selectin surfaces under flow conditions while not changing the cell phenotype. These results indicate that the transitory modification of cell surfaces with lipid vesicles can be used to efficiently immobilize adhesion ligands and potentially target systemically administered cells to the site of inflammation.84 Again, size, charge and overall composition of the vesicle can be tailored, as well as the individual lipids in terms of their gel–liquid transition temperature and fusogenicity (i.e. propensity to fuse).67 Chemoselective cell-surface engineering based on electrostatic interaction between the positively charged liposomes and negatively charged lipids of the cellular membrane led to the display of bio-orthogonal functional groups at cellular membranes.85 This strategy, as reported by Yousaf and coworkers, allowed for the display of ketone or oxyamine groups to different populations of cells for subsequent cell assembly via oxime ligation.85 The authors demonstrated several applications including the selective fluorescent labeling of the cell surface, the formation of small spheroid cell assemblies that are yet able to differentiate, and the generation of large and dense, 3D multilayered tissue-like structures for tissue engineering applications.85 In another report thiol-reactive maleimide headgroups of the lipid bilayer surface of drug-loaded uni- or multilamellar liposomes were reacted with the plasma membrane of lymphocytes.86 This strategy enabled continuous autocrine stimulation of donor cells in vivo.86 From these examples it is evident that lipid vesicles can be employed to change native cell membranes, either by the tunable delivery of active agents or by cell membrane functionalization, into reactive surfaces for new biotechnological applications such as tissue regeneration and cell-based therapies.
 | ||
| Fig. 10 (A) Schematic of a T-cell on a substrate patterned with diffusion barriers (reproduced with permission from ref. 92 provided by Elsevier, 2014). (B) Schematic of the method used to create peptide RGD-functionalized SLB glass surfaces for studying neural stem cell adhesion. Phase contrast images of NSCs after incubation on RGD-SLBs (FGF2-containing media, 5 days, scale bar: 100 μm) (reproduced with permission from ref. 96 provided by Elsevier, 2014). (C) Targeting and fusogenic peptides are chemically conjugated to maleimide–lipids and mixed in at 1–5 wt%. The vesicles composed of either fluid (DOPC) or non-fluid (DPPC) zwitterionic phosphatidylcholine lipids with 30 wt% cholesterol are further modified with 5 wt% PEG-2000 PE to enhance colloidal stability and decrease nonspecific interactions. The vesicles are loaded with cargo (chemotherapeutics (doxorubicin, 5-fluorouracil, cisplatin)), q-dots for imaging, diphtheria toxin A-chain and siRNA. The vesicles (1) bind to human hepatocellular carcinoma cells with high affinity owing to recruitment of the targeting peptides (magenta) to the cell surface, (2) become internalized by receptor-mediated endocytosis and (3) release their cargo into the cytosol upon endosome acidification and protonation of the fusogenic peptide (blue). (4) Cargos modified with a nuclear localization sequence (NLS) are transported through the nuclear pore complex and become concentrated in the nucleus (reproduced with permission from ref. 104 provided by the Nature Publishing Group, 2014). | ||
Owing to the lipid bilayers' inherent non-fouling nature with respect to protein adsorption and cell adhesion, specific cell responses can be observed. Tirrell and coworkers designed highly tunable cell culture surfaces using SLBs. One study showed the potential of SLBs to culture neuronal cells.96 SLBs were doped with cell adhesive RGD peptide amphiphiles consisting of a hydrophobic anchor and a hydrophilic spacer conjugated to the peptide sequence (Fig. 10B).96 Having an accessible RGD motif, cell responses could be fine-tuned by varying the hydrophilic spacer. A similar approach was adopted by Gold and coworkers that utilized SLBs decorated with an IKVAV peptide instead.97 This peptide sequence is derived from the ECM glycoprotein laminin-1 that is known to modulate cell adhesion and neurite outgrowth activity. Instead of using peptide amphiphiles, incorporated during bilayer formation, they demonstrated in situ modification of the SLBs.97 During bilayer formation thiol-reactive maleimide-functionalized lipids were included that at a later stage react with cysteine modified IKVAV peptides.64 Model neuronal cells (PC12) only adhered to the peptide modified SLBs. In a follow-up study they were able to elucidate a positive, nonlinear correlation between the number of attached rat neuronal cells (AHP) and the density of IKVAV peptides tethered to the SLB. Also, a threshold of ligand concentration was found, required for cell binding.98 Adopting a similar in situ modification strategy, SLBs were prepared serving as a carrier for ECM components. Huang and coworkers demonstrated that an SLB consisting of part of a carboxylic acid modified lipid could easily be modified to express collagen type 1 or fibronectin.99 In their set-up, collagen and the underlying lipid bilayer are a better representation of the cellular environment where lipid–collagen interactions are important. The system allowed for sustained A10 smooth muscle cell culture only on collagen modified SLBs. Not only the presentation of certain bioactive ligands on a SLB dictate cellular responses but also their mobility as was demonstrated by Reimhult and coworkers.100 Depending on the phase transition temperature, the lateral mobility of lipids was greatly affected. In a liquid state system diffusion coefficients in the order of μm2 per second are found while gel state SLBs show negligible diffusion.100 This in turn influences the mobility of cadherin tethered to biotin-modified lipids. The bilayer state influenced cell response dramatically; a more spread cell morphology was observed on gel state SLBs compared to liquid state ones.100 Within the rich field of planar lipid bilayer research, others chose to focus on the incorporation of functionally active proteins into such model systems and to ensure proper function, often involving electrical signaling. Therefore lipid bilayer systems should meet certain electrical standards compatible with the use of electrochemical techniques. For instance, means of lipid bilayer tethering must be considered. An overview of design criteria involved and past results are nicely presented in the review by Naumann and coworkers.101 Another aspect that deserves attention is the lack of stability of SLBs when exposed to air. Bilayer stabilization can be achieved by the use of photo-cross-linkable lipids that bear apolar tails containing diacetylene bonds.102 These lipids readily crosslink under UV light to yield a solvent resistant and air-stable SLB that is also applicable to prepare polymerized lipid vesicles. However, such strategies do not yield mobile SLBs. Several attempts in that direction have been made; for instance sterol tethering of SLB and the use of protein layers on top of SLB imparted air-stability while retaining lateral motion of lipids.103
Both liposomes, as discussed in the previous section, and SLBs are powerful systems for cell research due to the high degree of surface tunability and their multipurpose, dynamic nature and are proven bio-mimetic systems. Ashley and coworkers designed a multicomponent particle-supported lipid bilayer combining both systems.104 These so-called protocells consist of a carrier synthesized by liposome fusion onto spherical nanoporous silica particles, followed by modification of the resulting supported lipid bilayer with PEG, targeting peptides and fusogenic peptides (Fig. 10C).104 Therapeutic and imaging agents could be simultaneously loaded into the silica core with high capacity due to the nano-sized pores which increase the surface area. The rate of the cargo release could be optimized by affecting the particle dissolution in the acidic endosomal environment through modifying silica composition and the protonation of the fusogenic peptide. In their study the protocell displayed a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold higher affinity for human hepatocellular carcinoma compared to hepatocytes, endothelial or immune cells, demonstrated the delivery of encapsulated cargo upon cellular uptake and showed enhanced stability over traditional liposomes.104
000 fold higher affinity for human hepatocellular carcinoma compared to hepatocytes, endothelial or immune cells, demonstrated the delivery of encapsulated cargo upon cellular uptake and showed enhanced stability over traditional liposomes.104
1.9 Interfacing cells with dynamic self-assembled monolayers
The highly complex composition of the ECM and its interaction with molecular fingerprints on the cell membrane make it a highly challenging task to create model systems that allow us to mimic cells' spatial as well as temporal organization. Due to their advantage in being well-ordered, permitting high interfacial control of specific bioactive ligands and allowing great flexibility in the creation of complex substrates with spatial precision, self-assembled monolayers (SAMs) have been a particularly attractive strategy since their discovery in the early 80s. Analogous to self-assembling systems in solution, they are highly ordered nanostructures that spontaneously organize on surfaces. More recently, SAMs are also actively investigated as model substrates for dynamic cell studies. This is desirable, as they not only allow the spatial control of bioactive ligands, but additionally permit control of their presence on the substrate in time. Dynamic SAMs can change their surface properties in response to external stimuli such as a change in pH, temperature, light or electrochemical stimuli. However, since cells require steady physiological conditions, like pH and temperature, conversion of substrate properties in response to light or electrochemical stimuli has been used favourably for cell studies. To date, a number of different types of substrate materials and chemical strategies to immobilize bioactive ligands have been exploited for use as dynamic substrates. Nevertheless, the majority of systems use SAMs of alkanethiols on gold for a number of advantages: e.g. being bio-inert, allowing simple and well-ordered functionalization and being available to various substrate analysis techniques. Alkanethiolates form densely packed well-ordered monolayers on gold where the thiol head group binds with high affinity to the underlying gold substrate. They can furthermore be simply adapted to hold several different functional groups in order to modify the substrate with specific bioactive ligands. Conductivity of the gold substrate makes it suitable for even more analytical techniques to characterize interfacial interactions. We will here describe selected examples from the literature to illustrate the various strategies using dynamic SAMs for cell studies.In a recent example, thiol interaction with gold acted as anchoring points for RGD peptides to achieve sub-cellular control by functionalizing a gold electrode array.107 Modification of the separating glass regions with PEG to make them cell-repellent resulted in selective cell-adhesion to the RGD-functionalized electrodes. Spatial design of sub-cellular distances between adjacent electrodes allowed cells to spread over multiple electrodes. When Searson and coworkers applied a negative voltage pulse on an individual gold electrode, selective release of the thiols was triggered and consequently retraction of that very region of the attached cell could be visualized by monitoring fluorescently labelled actin with fluorescence microscopy.107 Modulating cell detachment in this manner opens a new road to gain significant insights into cellular pathway signalling. Flexibility in alkanethiol functionalization makes them a useful tool for creation of multi-purpose cell culture surfaces with modular cell-repellent and cell-adhesive regions. The combination of micropatterning technology with self-assembly of differentially terminated thiols presents a powerful approach to generate spatially resolved micro-confinements to create multifunctional SAM profiles on the substrate.47,108–110 Recent studies have already demonstrated great potential of such patterned systems for studying cell motility as well as cell–cell interactions.47 With the appropriate design of the SAM properties, such as spatial definition alongside chemical functionality, an electrical stimulus can for instance be used to trigger the release of a particular section on the surface and allow cells to migrate into newly available areas.108 This design makes it possible to study cell migration in a non-invasive manner. However, since coupling alkanethiols to a desired biomolecule can be laborious, other coupling strategies taking place directly at the interface of a pre-assembled monolayer have been explored. Some of the frequently employed organic strategies include redox-active hydroquinone (HQ) monolayers in combination with Diels–Alder conjugation,111,112 oxime reaction113–117 and Cu(I)-catalysed Huisgen cycloaddition.118,119 The electrochemical conversion of HQ-terminated monolayers on gold has been a successfully employed strategy to chemically regulate interfacial cell–substrate interactions. HQ can undergo oxidation upon an electrochemical stimulus and convert to benzoquinone (BQ), which, in turn, can react with a number of functional groups.111 To ensure specific interactions, selective to the bioactive ligand of choice, a critical step is to block the background to unspecific binding of proteins present in biological media. Short ethylene glycol linkers have proven themselves as a very efficient choice for that purpose, commonly used at 1:99% ligand to ethylene glycol (EG) ratios. When HQ groups, present in a cell-inert background, are oxidized to BQ, they can readily form a covalent adduct via a Diels–Alder reaction with a cyclopentadiene (Fig. 11A).111 When such a cyclopentadiene is functionalized with RGD a reversible substrate is obtained that allows for controlling cell-repellent and cell-adhesive areas by changing the redox-potentials as described by Mrksich and coworkers.111 For spatial control of cell adhesion and migration this strategy was extended using micropatterns. Patterns of hexadecanethiolate on gold were reacted with fibronectin (Fn), onto which cells were allowed to adhere.111 The backfilling of the non-patterned regions with a 1:99% HQ to EG mixture made this at first an area unavailable for fibroblast cells.111 Upon electrically stimulated oxidation of the HQs to BQ and following Diels–Alder mediated RGD binding, these cells could consequently migrate out of their patterns into the surrounding areas.111 Instead of starting with a cell-inert HQ layer, Mrksich and coworkers used a silyl ether conjugated to HQ to create an O-silyl HQ functionality further coupled to an RGD.112 Upon electrical activation HQ converts to BQ and causes hydrolysis of the silyl ether, causing the release of the RGD. The resulting BQ functionality could as in the previous example again react with a diene-coupled RGD ligand via a Diels–Alder reaction, creating a dual-functional cell-adhesive or repellent surface. In a similar fashion, the redox-active HQ-terminated SAMs can, when oxidized to the resulting BQ, be coupled to aminooxy-terminated ligands by an oxime reaction as described by Yousaf and coworkers.113–117 Oxyamine reacts covalently with the ketone groups on BQ in high yield under physiological conditions to form a stable oxyamine linkage. The reacting aminooxy groups can furthermore be introduced in most biomolecules by using standard synthetic procedures. Complex surfaces with well-defined spatial control were generated based on this approach by improving the system with microarray or micropatterning techniques.113,114 In the latter case, the patterned areas were cell-adhesive and non-patterned areas at first cell-repellent. In this example, photo-induced conversion as well as electrochemical activation was combined to selectively convert the non-patterned areas.114 These regions initially presented a 1:99% mixture of HQ groups terminated with a photo-sensitive protection group and cell-repellent EG4.114 UV illumination through a gradient photomask could then selectively deprotect the HQ groups, which, followed by electrochemical conversion to BQ, could bind an aminooxy-terminated RGD ligand via an oxime conjugate (Fig. 11B).114 This caused the fibroblasts (adhering to the μCP areas) to spread to the photo-activated RGD-gradient. By applying a reductive potential these aminooxy-RGD could then be selectively released. Consequently cells adhering to these non-patterned functionalized areas were released, while cells still remained attached in the μCP areas.
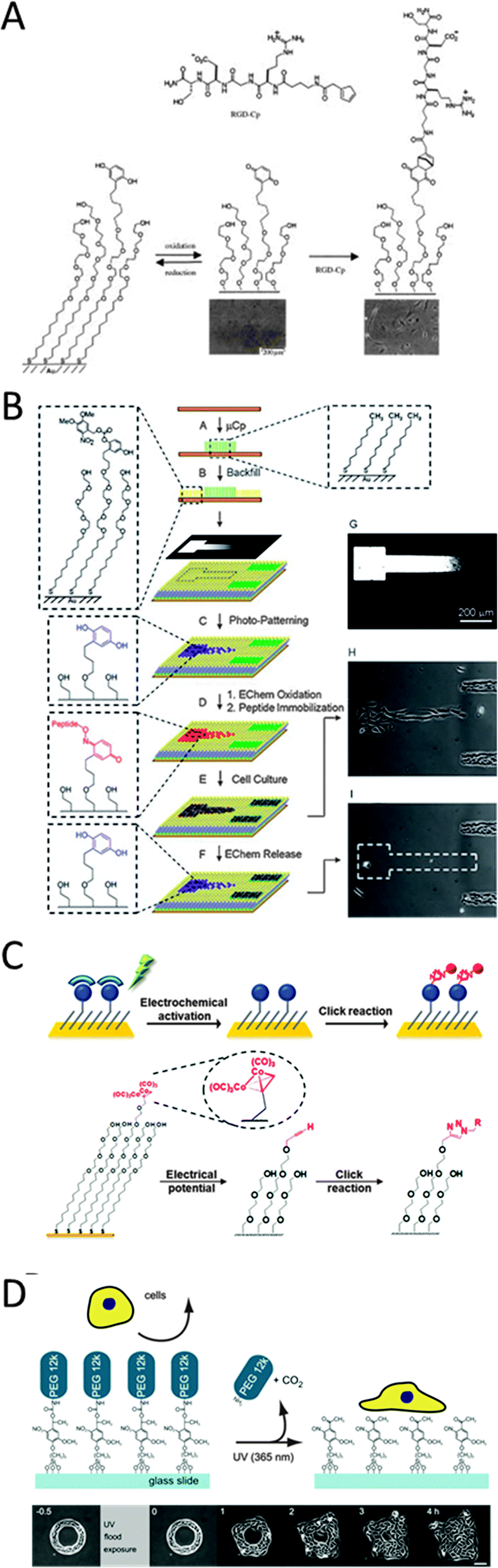 | ||
| Fig. 11 (A) HQ immobilized to gold substrates oxidizes to BQ and undergoes Diels–Alder reaction with RGD–cyclopentadiene (Cp). Attachment of RGD changes the substrate from cell-repellent to cell adhesive (reproduced with permission from ref. 111 provided by John Wiley and Sons, 2014). (B) Combination of μCP and electrochemical as well as photo-induced activation for complex cell substrates (reproduced with permission from ref. 114 provided by John Wiley and Sons, 2014).114 (C) Electrochemical stimulated oxidative degradation of dicobalt hexacarbonyl complexes to reveal alkynes on the monolayer and subsequent reaction with azides coupled to biomolecules via Cu(I)-catalyzed Huisgen cycloaddition reaction (reproduced with permission from ref. 118 provided by the American Chemical Society, 2014). (D) Glass modified with PEG moieties featuring a photocleavable linker. Upon light exposure these photocleavable groups undergo intermolecular oxidation and thus cause PEG to be released. Whilst PEG is inert to cell adhesion, the photocleavage allows cells to spread to the newly available cell-adhesive areas (reproduced with permission from ref. 119 provided by Elsevier, 2014). | ||
In regenerative medicine a challenging and rewarding task is to understand the underlying mechanisms driving stem cell differentiation. Combining electroactive SAMs with microarray technology was demonstrated to be an elegant and promising strategy to immobilize a library of bioactive ligands on a substrate to screen for optimal conditions.110,115 As in the examples above, alkanethiol-gold substrates terminated with HQ groups or EG4 were used as the basic building blocks. Using microarray printing and mixing different ratios of HQ to EG4-terminated moieties, patterns of various ligand densities were obtained. A solution of alkanethiol-EG4 was then used to block the remaining areas around the patterned areas. Creating a library of different oxyamine-tethered ligands that could react with electrically oxidized HQ, BQ, arrays of precisely controlled spatial resolution and varying densities could be tailored on the substrate. With this versatile and complex platform at hand it was possible to screen for the effect of ligand density and composition on the rate of stem cell (hMSC) differentiation.115 A similar platform has been used for studies of single cell polarization.117 Cu(I)-catalyzed Huisgen cycloaddition between azides and alkynes is a chemoselective reaction that proceeds under mild reaction conditions and therefore makes it an attractive approach for fabricating bioactive dynamic SAMs. Recently it was applied in combination with the oxime reaction as an in situ “hide-and-reveal” strategy of small biomolecules to create a dual chemoselective SAM, displaying switchable (HQ) as well as non-switchable azide functional groups.116 Bifunctional bioactive moieties were created by co-coupling the peptides with an oxyamine as well as an alkyne end group, which are the respective chemical partners for the BQ and azide. When the moieties were switched from presenting linear to cyclic RGD, by application of a mild electrochemical potential, they could monitor real-time changes in cell behaviour such as changes in cell adhesion and migration.116
Cu(I)-catalyzed Huisgen alkyne–azide cycloaddition has also been reported by Yeo and coworkers as a direct strategy for electroactive substrate dynamicity without use of the HQ linkage (Fig. 11C).118 Alkyne-terminated monolayers were masked with dicobalt hexacarbonyl complexes, making them inert to react with azides.118 Upon electrochemical activation the dicobalt hexacarbonyl complex was oxidatively degraded and the thus exposed alkyne groups were available for reaction with azides.118 Coupling azides to a cell-adhesive peptide resulted in substrates that were turned from cell-repellent to cell-adhesive upon reaction with alkynes.118
Besides electrochemical stimulated release systems, the use of photo-induced activation has been explored.119,120 Silicon oxide substrates presenting free amine groups could provide a reactive substrate for photo-cleavable linkers via carbamate bonding as described by del Campo and coworkers.120 These linkers contained an intercalated photolabile group, the o-nitrobenzyl derivative (NVOC), which upon UV irradiation underwent an intermolecular redox reaction, and hence, underwent cleavage from the substrate.120 Tethering this linker with a bio-ligand created a cell-adhesive substrate, that, upon light exposure, could be reversed to be cell-repellent. Similarly, a photo-cleavable 2-nitrobenzyl-linked PEG monolayer formed on glass could make the substrate cell repellent, and upon light exposure cause PEG-release and feature a cell-adhesive substrate (Fig. 11D).119 Spatial control of these systems can easily be achieved by a photomask, thus regulating the desired areas of illumination. Yousaf and coworkers integrated a bioactive surface strategy with photoelectroactive surface strategy to generate dynamic ligand surface gradients for controlling cell adhesion, tissue shape morphing and cell migration.121 To this end an NVOC-protected HQ terminated alkanethiol was mixed in with the hydrophilic parts of a SAM that was otherwise hydrophobic.121 Irradiation revealed the HQ group and fibroblasts were allowed to adhere to the hydrophobic regions of the SAM.121 Only when the HQ was converted to the quinone group and oxyamine–RGD was coupled to the photo-deprotected regions, cells started to migrate into the RGD regions.121 Jiang and coworkers used SAMs of photoresponsive azobenzenes conjugated to RGD on a background of EG6 to control cell adhesion reversibly. Switching of azobenzenes between E isomer and Z isomer by UV light rendered the surface to be either adhesive by presenting the RGD or repellent by shielding the RGD within the inert EG6 layer.122
Since natural interfaces between the cell and its interacting ligands build on non-covalent interactions, chemical systems relying on non-covalent binding such as hydrophobic, van der Waals, ion-dipole or hydrogen bonds are being explored and are promising to come yet a step closer to mimic natural cell–ECM interactions. A novel strategy to reversibly attach and release cells is the use of supramolecular host–guest chemistry. On a quartz substrate a host monolayer was formed by silane-terminated α-CD (Fig. 12A).123 α-CD can recognize multiple different guests, such as naphthalene, stilbene and azobenzene, and therefore holds potential as a multifunctional template for immobilizing different biological ligands to one substrate. In this particular study azobenzene was used as a guest to form an inclusion complex with α-CD when present in the trans isomer. To make the substrate available for cell adhesion, a RGD-peptide was coupled to azobenzene. UV irradiation of 365 nm triggered trans-to-cis isomerization of azobenzene which leads to the release of the azobenzene complex and thus also the release of the cells from the substrate.123 Jonkheijm and coworkers recently reported the use of a supramolecular CB[8]-host as a versatile system for fabricating dynamic SAMs (Fig. 12B).124,125 Electroactive viologen was used as a first guest to link the CB[8] to the gold substrate by modifying it with a thiol. Cell-adhesive RGD could act as a second guest when terminated with the aromatic amino acid tryptophan to bind to CB[8] with high affinity. When this ternary complex was assembled on gold substrates to form a monolayer, cells were able to spread on the available RGD epitopes.125 Electrochemical activation of the ternary complex led to reduction of viologen and caused a release of RGD, thus causing cellular release.125 This principle was furthermore applied to gold microelectrode arrays to achieve release with sub-cellular resolution. In another example supramolecular control of adhesion of cells is demonstrated using synthetic RGD–ferrocene conjugates that were immobilized via host–guest chemistry onto cucurbit[7]uril coated gold surfaces.126 CB[7] spontaneously adsorbs onto gold surfaces and forms a stable SAM.127 While the ferrocene ring system fills the lipophilic CB[7] cavity, the protonated amino functionality protrudes from the cavity, where it makes stabilizing electrostatic interactions with the polar carbonyl groups located at the CB[7] rim. The binding constant is not determined on surfaces; however binding constants in the case of such systems in solutions are found to be in the range of 10−11 M−1 and these systems are much more stable than the ternary complexation motif that was discussed above.
 | ||
| Fig. 12 Supramolecular host–guest assemblies of a (A) photo-responsive α-CD-host and azobenzene–RGD–guest (reproduced with permission from ref. 123 provided by the American Chemical Society, 2014) and (B) electrochemically responsive CB[8]-host with two guests, i.e. surface bound viologen and RGD-peptide functionalized tryptophan (reproduced with permission from ref. 125 provided by John Wiley and Sons, 2014). | ||
2. Conclusions and outlook
From the literature survey it becomes evident that many diverse strategies have been described to fabricate dynamic bioactive systems to explore interactions with living cells and bacteria. Often integrated strategies combining synthetic and supramolecular assembly with fabrication techniques, modern molecular biology strategies, surface chemistry and imaging techniques are employed to assess the biofunctionality of interfacing artificial structures with cells. Supramolecular assembly offers the ability to decorate the generated architectures with bioactive ligands to engage in applications ranging from imaging to diagnostics, and from drug delivery to tissue engineering. Essential characteristics of a dynamic bioactive system are the control over architectural shape, ligand composition, density, mobility and diversity and the architectures can be found in solution or at solid supports or their interface. Further essential components of a dynamic bioactive system are a stimulus that can be arbitrarily applied to the system, either a chemical compound or any external trigger, and a system that is designed to transduce the stimulus into a signal, ideally reversibly and precisely. While the understanding of fabricating such multifaceted dynamic bioactive architectures has matured over the last decade, nonetheless their dynamics, stability and susceptibility to changes by addition of chemical compounds, electronic and/or optical control need to be further integrated. We strongly believe that each of these synthetic bioactive systems provides excellent chemical tools to gain insight into biological mechanisms that control and regulate cell function as these synthetic systems are decorated with bioactive ligands and are used to engage with receptor fingerprints, mimic and modulate the interaction thereof fully exploiting the dynamic properties of the designed systems. Living cells are inherently dynamic and continuously adapt to their environment in the relentless pursuit of homeostasis. Many of the kinetic aspects of for example cytoskeleton responses are difficult to interpret when using static synthetic systems. Such processes may include adhesion, spreading and migration. While the use of dynamic systems, materials and interfaces can mirror the intrinsic dynamic behavior of living cells, seeding cells in the presence of such systems that present time-invariant bioactive signals, i.e. static cues, leads to a convolution of nascent adhesion and spreading with other types of cellular responses that may be of interest.Acknowledgements
The authors acknowledge discussions with and contributions of former and current colleagues. The European Research Council is acknowledged for funding through Starters Grant 310105 and P4.02 Superdices of the research program of the BioMedical Materials institute co-funded by the Dutch Ministry of Economic Affairs, Agriculture and Innovation.References
- K. Jacobson, E. D. Sheets and R. Simson, Science, 1995, 268, 1441–1442 CAS.
- C. R. F. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky and A. Kupfer, Nature, 1998, 395, 82–88 CrossRef CAS PubMed.
- F. R. Maxfield, Curr. Opin. Cell Biol., 2002, 14, 483–487 CrossRef CAS PubMed.
- J. F. Hancock, Nat. Rev. Mol. Cell Biol., 2006, 7, 456–462 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- D. M. Engelman, Nature, 2005, 438, 578–580 CrossRef CAS PubMed.
- E. Zamir and P. I. H. Bastiaens, Nat. Chem. Biol., 2008, 4, 643–647 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- (a) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (b) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- T. Fenske, H.-G. Korth, A. Mohr and C. Schmuck, Chem. – Eur. J., 2012, 18, 738–755 CrossRef CAS PubMed.
- (a) M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS PubMed; (b) C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC; (c) C. Alexander, Nat. Mater., 2008, 7, 767–768 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- P. Y. W. Dankers, J. M. Boomker, A. Huizinga-van der Vlag, E. Wisse, W. P. J. Appel, F. M. M. Smedts, M. C. Harmsen, A. W. Bosman, E. W. Meijer and M. J. A. van Luyn, Biomaterials, 2011, 32, 723–733 CrossRef CAS PubMed.
- J. Geng, F. Biedermann, J. M. Zayed, F. Tian and O. A. Scherman, Macromolecules, 2011, 44, 4276–4281 CrossRef CAS.
- K. S. Kim, S. J. Park, J. A. Yang, J. H. Jeon, S. H. Bhang, B. S. Kim and K. Kim, Biomaterials, 2011, 32, 7687–7694 CrossRef PubMed.
- K. M. Park, J.-A. Yang, H. Jung, J. Yeom, J. S. Park, K.-H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS PubMed.
- J. Boekhoven, C. M. Rubert Pérez, S. Sur, A. Worthy and S. I. Stupp, Angew. Chem., Int. Ed., 2013, 52, 12077–12080 CrossRef CAS PubMed.
- H. Li, J. Frith and J. J. Cooper-White, Biomacromolecules, 2014, 15, 43–52 CrossRef CAS PubMed.
- E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405–4408 CrossRef CAS PubMed.
- D. Kim, E. Kim, J. Kim, K. Park, K. Baek, M. Jung, Y. Ko, W. Sung, H. Kim, J. Suh, C. Park, O. Na, D.-k. Lee, K. Lee, S. Han and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 3471–3474 CrossRef CAS PubMed.
- A. Nelson, J. M. Belitsky, S. Vidal, C. S. Joiner, L. G. Baum and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 11914–11922 CrossRef CAS PubMed.
- J. Kim, Y. Ahn, K. M. Park, D.-W. Lee and K. Kim, Chem. – Eur. J., 2010, 16, 12168–12173 CrossRef CAS PubMed.
- N. Yui and T. Ooya, Chem. – Eur. J., 2006, 12, 6730–6737 CrossRef CAS PubMed.
- J.-H. Seo, S. Kakinoki, Y. Inoue, T. Yamaoka, K. Ishihara and N. Yui, J. Am. Chem. Soc., 2013, 135, 5513–5516 CrossRef CAS PubMed.
- H. Wang, S. Wang, H. Su, K.-J. Chen, A. Armijo, W.-Y. Lin, Y. Wang, J. Sun, K.-i. Kamei, J. Czernin, C. G. Radu and H.-R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 4344–4348 CrossRef CAS PubMed.
- H. Wang, K. Liu, K.-J. Chen, Y. Lu, S. Wang, W.-Y. Lin, F. Guo, K. Kamei, Y.-C. Chen, M. Ohashi, M. Wang, X.-Z. Zhao, C. K.-F. Shen and H.-R. Tseng, ACS Nano, 2010, 4, 6235–6243 CrossRef CAS PubMed.
- Y. Liu, H. Wang, K.-i. Kamei, M. Yan, K.-J. Chen, Q. Yuan, L. Shi, Y. Lu and H.-R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3058–3062 CrossRef CAS PubMed.
- (a) H. Robson Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef CAS PubMed; (b) J. Mason, K. Müller and K. Arndt, in Protein Engineering Protocols, ed. K. Arndt and K. Müller, Humana Press, 2007, vol. 352, pp. 35–70 Search PubMed.
- Y. Yano, A. Yano, S. Oishi, Y. Sugimoto, G. Tsujimoto, N. Fujii and K. Matsuzaki, ACS Chem. Biol., 2008, 3, 341–345 CrossRef CAS PubMed.
- H. R. Zope, F. Versluis, A. Ordas, J. Voskuhl, H. P. Spaink and A. Kros, Angew. Chem., Int. Ed., 2013, 52, 14247–14251 CrossRef CAS PubMed.
- I. Nakase, S. Okumura, G. Tanaka, K. Osaki, M. Imanishi and S. Futaki, Angew. Chem., Int. Ed., 2012, 51, 7464–7467 CrossRef CAS PubMed.
- A. Bill, A. Schmitz, B. Albertoni, J.-N. Song, L. C. Heukamp, D. Walrafen, F. Thorwith, P. J. Verveer, S. Zimmer, L. Meffert, A. Schreiber, S. Chatterjee, R. K. Thomas, R. T. Ullrich, T. Lang and M. Famulok, Cell, 2010, 143, 201–211 CrossRef CAS PubMed.
- Z. N. Mahmoud, S. B. Gunnoo, A. R. Thomson, J. M. Fletcher and D. N. Woolfson, Biomaterials, 2011, 32, 3712–3720 CrossRef CAS PubMed.
- V. Villard, O. Kalyuzhniy, O. Riccio, S. Potekhin, T. N. Melnik, A. V. Kajava, C. Rüegg and G. Corradin, J. Pept. Sci., 2006, 12, 206–212 CrossRef CAS PubMed.
- T. Nakaji-Hirabayashi, K. Kato and H. Iwata, Biomacromolecules, 2008, 9, 1411–1416 CrossRef CAS PubMed.
- B. Liu, Y. Liu, J. J. Riesberg and W. Shen, J. Am. Chem. Soc., 2010, 132, 13630–13632 CrossRef CAS PubMed.
- K. Zhang, A. Sugawara and D. A. Tirrell, ChemBioChem, 2009, 10, 2617–2619 CrossRef CAS PubMed.
- Y.-b. Lim, S. Park, E. Lee, H. Jeong, J.-H. Ryu, M. S. Lee and M. Lee, Biomacromolecules, 2007, 8, 1404–1408 CrossRef CAS PubMed.
- Y.-b. Lim, S. Park, E. Lee, J.-H. Ryu, Y.-R. Yoon, T.-H. Kim and M. Lee, Chem.–Asian J., 2007, 2, 1363–1369 CrossRef CAS PubMed.
- Y.-b. Lim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 3475–3478 CrossRef CAS PubMed.
- R. J. Hosse, A. Rothe and B. E. Power, Protein Sci., 2006, 15, 14–27 CrossRef CAS PubMed.
- (a) R. H. Kimura, A. M. Levin, F. V. Cochran and J. R. Cochran, Proteins, 2009, 77, 359–369 CrossRef CAS PubMed; (b) S. J. Moore, M. G. Hayden Gephart, J. M. Bergen, Y. S. Su, H. Rayburn, M. P. Scott and J. R. Cochran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14598–14603 CrossRef CAS PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- F. A. Aldaye, W. T. Senapedis, P. A. Silver and J. C. Way, J. Am. Chem. Soc., 2010, 132, 14727–14729 CrossRef CAS PubMed.
- R. Meyer, S. Giselbrecht, B. E. Rapp, M. Hirtz and C. M. Niemeyer, Curr. Opin. Chem. Biol., 2014, 18, 8–15 CrossRef CAS PubMed.
- J. Voskuhl, J. Brinkmann and P. Jonkheijm, Curr. Opin. Chem. Biol., 2014, 18, 1–7 CrossRef CAS PubMed.
- U. Vermesh, O. Vermesh, J. Wang, G. A. Kwong, C. Ma, K. Hwang and J. R. Heath, Angew. Chem., Int. Ed., 2011, 50, 7378–7380 CrossRef CAS PubMed.
- N. S. Selden, M. E. Todhunter, N. Y. Jee, J. S. Liu, K. E. Broaders and Z. J. Gartner, J. Am. Chem. Soc., 2012, 134, 765–768 CrossRef CAS PubMed.
- R. A. Chandra, E. S. Douglas, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 896–901 CrossRef CAS PubMed.
- H. Schroeder, B. Ellinger, C. F. W. Becker, H. Waldmann and C. M. Niemeyer, Angew. Chem., Int. Ed., 2007, 46, 4180–4183 CrossRef CAS PubMed.
- H. Onoe, S. C. Hsiao, E. S. Douglas, Z. J. Gartner, C. R. Bertozzi, M. B. Francis and R. A. Matthies, Langmuir, 2010, 28, 8120–8130 CrossRef PubMed.
- S. Gandor, S. Reisewitz, M. Venkatachalapahty, G. Arrabito, M. Reibner, H. Schroeder, K. Ruf, C. M. Niemeyer, P. I. Bastiaens and L. Dehmelt, Angew. Chem., Int. Ed., 2013, 52, 4790–4794 CrossRef CAS PubMed.
- G. Arrabito, S. Reisewitz, L. Dehmelt, P. I. Bastiaens, B. Pignataro, H. Schroeder and C. M. Niemeyer, Small, 2013, 9, 4243–4249 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- R. N. Shah, N. A. Shah, M. M. Del Rosario Lim, C. Hsieha, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS PubMed.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS PubMed.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS PubMed.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T. C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- J.-H. Ryu, D.-J. Hong and M. Lee, Chem. Commun., 2008, 1043–1054 RSC.
- B.-S. Kim, W.-Y. Yang, J.-H. Ryu, Y.-S. Yoo and M. Lee, Chem. Commun., 2005, 2035–2037 RSC.
- J.-H. Ryu, E. Lee, Y.-b. Lim and M. Lee, J. Am. Chem. Soc., 2007, 129, 4808–4814 CrossRef CAS PubMed.
- M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921–2924 CrossRef PubMed.
- K. Petkau-Milroy, M. H. Sonntag, A. H. A. M. van Onzen and L. Brunsveld, J. Am. Chem. Soc., 2012, 134, 8086–8089 CrossRef CAS PubMed.
- L. Huang and R. E. Pagano, J. Cell Biol., 1975, 67, 38–48 CrossRef CAS PubMed.
- H. R. Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- C. H. Yu and J. T. Groves, Med. Biol. Eng. Comput., 2010, 48, 955–963 Search PubMed.
- D. S. Watson, A. N. Endsley and L. Huang, Vaccine, 2012, 30, 2256–2272 CrossRef CAS PubMed.
- J. Nguyen and F. C. Szoka, Acc. Chem. Res., 2012, 45, 1153–1162 CrossRef CAS PubMed.
- B. Yang, S.-Y. Geng, X.-M. Liu, J.-T. Wang, Y.-K. Chen, Y.-L. Wang and J.-Y. Wang, Soft Matter, 2012, 8, 518–525 RSC.
- A. Akinc, A. Zumbuehl, M. Goldberg, E. S. Leshchiner, V. Busini, N. Hossain, S. A. Bacallado, D. N. Nguyen, J. Fuller, R. Alvarez, A. Borodovsky, T. Borland, R. Constien, A. de Fougerolles, J. R. Dorkin, K. N. Jayaprakash, M. Jayaraman, M. John, V. Koteliansky, M. Manoharan, L. Nechev, J. Qin, T. Racie, D. Raitcheva, K. G. Rajeev, D. W. Y. Sah, J. Soutschek, I. Toudjarska, H.-P. Vornlocher, T. S. Zimmermann, R. Langer and D. G. Anderson, Nat. Biotechnol., 2008, 26, 561–569 CrossRef CAS PubMed.
- S.-W. Cho, M. Goldberg, S. M. Son, Q. Xu, F. Yang, Y. Mei, S. Bogatyrev, R. Langer and D. G. Anderson, Adv. Funct. Mater., 2009, 19, 3112–3118 CrossRef CAS PubMed.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1864–1869 CrossRef CAS PubMed.
- E. Kluza, D. W. J. van der Schaft, P. A. Hautvast, W. J. Mulder, K. H. Mayo, A. W. Griffioen, G. J. Strijkers and K. Nicolay, Nano Lett., 2010, 10, 52–58 CrossRef CAS PubMed.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, J. Am. Chem. Soc., 2009, 131, 2408–2415 CrossRef CAS PubMed.
- K. M. Park, D.-W. Lee, B. Sarkar, H. Jung, J. Kim, Y. H. Ko, K. E. Lee, H. Jeon and K. Kim, Small, 2010, 6, 1430–1441 CrossRef CAS PubMed.
- X. Liang, X. Li, X. Yue and Z. Dai, Angew. Chem., Int. Ed., 2011, 50, 11622–11627 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. H. Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243–251 CrossRef CAS PubMed.
- D. Dutta, A. Pulsipher, W. Luo, H. Mak and M. N. Yousaf, Bioconjugate Chem., 2011, 22, 2423–2433 CrossRef CAS PubMed.
- S. S. Agasti, M. Liong, C. Tassa, H. J. Chung, S. Y. Shaw, H. Lee and R. Weissleder, Angew. Chem., Int. Ed., 2012, 51, 450–454 CrossRef CAS PubMed.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS PubMed.
- S. C. Hsiao, B. J. Shum, H. Onoe, E. S. Douglas, Z. J. Gartner, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Langmuir, 2009, 25, 6985–6991 CrossRef CAS PubMed.
- D. Sarkar, P. K. Vemula, W. Zhao, A. Gupta, R. Karnik and J. M. Karp, Biomaterials, 2010, 31, 5266–5274 CrossRef CAS PubMed.
- D. Dutta, A. Pulsipher, W. Luo and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 8704–8713 CrossRef CAS PubMed.
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bersthteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- D. Afanasenkau and A. Offenhausser, Langmuir, 2012, 28, 13387–13394 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS.
- J. van Weerd, S. O. Krabbenborg, J. C. T. Eijkel, H. B. J. Karperien, J. Huskens and P. Jonkheijm, J. Am. Chem. Soc., 2014, 136, 100–103 CrossRef CAS PubMed.
- J. M. Nam, P. M. Nair, R. M. Neve, J. W. Gray and J. T. Groves, ChemBioChem, 2006, 7, 436–440 CrossRef CAS PubMed.
- N. C. Hartman and J. T. Groves, Curr. Opin. Cell Biol., 2011, 23, 370–376 CrossRef CAS PubMed.
- N. C. Hartman, J. A. Nye and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12729–12734 CrossRef PubMed.
- B. N. Manz, B. L. Jackson, R. S. Petit, M. L. Dustin and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9089–9094 CrossRef CAS PubMed.
- M. Charnley, R. Kroschewski and M. Textor, Integr. Biol., 2012, 4, 1059–1071 RSC.
- B. Ananthanarayanan, L. Little, D. V. Schaffer, K. E. Healy and M. Tirrell, Biomaterials, 2010, 31, 8706–8715 CrossRef CAS PubMed.
- S. Svedhem, D. Dahlborg, J. Ekeroth, J. Kelly, F. Höök and J. Gold, Langmuir, 2003, 19, 6730–6736 CrossRef CAS.
- D. Thid, K. Holm, P. S. Eriksson, J. Ekeroth, B. Kasemo and J. Gold, J. Biomed. Mater. Res., Part A, 2008, 84A, 940–953 CrossRef CAS PubMed.
- C.-J. Huang, N.-J. Cho, C.-J. Hsu, P.-Y. Tseng, C. W. Frank and Y.-C. Chang, Biomacromolecules, 2010, 11, 1231–1240 CrossRef CAS PubMed.
- M. Andreasson-Ochsner, G. Romano, M. Håkanson, M. L. Smith, D. E. Leckband, M. Textor and E. Reimhult, Lab Chip, 2011, 11, 2876–2883 RSC.
- R. L. C. Naumann, C. Nowak and W. Knoll, Soft Matter, 2011, 7, 9535–9548 RSC.
- K. Morigaki, H. Schönherr and T. Okazaki, Langmuir, 2007, 23, 12254–12260 CrossRef CAS PubMed.
- Y. Deng, Y. Wang, B. Holtz, J. Li, N. Traaseth, G. Veglia, B. J. Stottrup, R. Elde, D. Pei, A. Guo and X.-Y. Zhu, J. Am. Chem. Soc., 2008, 130, 6267–6271 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. Jeffrey Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- S. Yamaguchi, S. Yamahira, K. Kikuchi, K. Sumaru, T. Kanamori and T. Nagamune, Angew. Chem., 2012, 124, 132–135 CrossRef.
- K. Ishihara and M. Takai, J. R. Soc., Interface, 2009, 6, S279–S291 CrossRef CAS PubMed.
- B. Wildt, D. Wirtz and P. C. Searson, Nat. Methods, 2009, 6, 211–213 CrossRef CAS PubMed.
- S. Raghavan, R. A. Desai, Y. Kwon, M. Mrksich and C. S. Chen, Langmuir, 2010, 26, 17733–17738 CrossRef CAS PubMed.
- Z. Chen, Y. Li, W. Liu, D. Zhang, Y. Zhao, B. Yuan and X. Jiang, Angew. Chem., Int. Ed., 2009, 48, 8303–8305 CrossRef CAS PubMed.
- X. Jiang, R. Ferrigno, M. Mrksich and G. M. Whitesides, J. Am. Chem. Soc., 2003, 125, 2366–2367 CrossRef CAS PubMed.
- M. N. Yousaf, B. T. Houseman and M. Mrksich, Angew. Chem., Int. Ed., 2001, 40, 1093–1096 CrossRef CAS.
- W. S. Yeo, M. N. Yousaf and M. Mrksich, J. Am. Chem. Soc., 2003, 125, 14994–14995 CrossRef CAS PubMed.
- E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542–15546 CrossRef CAS PubMed.
- E. W. L. Chan, S. Park and M. N. Yousaf, Angew. Chem., Int. Ed., 2008, 47, 6267–6271 CrossRef CAS PubMed.
- W. Luo, E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2010, 132, 2614–2621 CrossRef CAS PubMed.
- B. M. Lamb and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 8870–8873 CrossRef CAS PubMed.
- D. K. Hoover, E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2008, 130, 3280–3281 CrossRef CAS PubMed.
- I. Choi, Y. K. Kim, D. H. Min, S. W. Lee and W. S. Yeo, J. Am. Chem. Soc., 2011, 133, 16718–16721 CrossRef CAS PubMed.
- C. G. Rolli, H. Nakayama, K. Yamaguchi, J. P. Spatz, R. Kemkemer and J. Nakanishi, Biomaterials, 2011, 33, 2409–2418 CrossRef PubMed.
- M. Wirkner, J. M. Alonso, V. Maus, M. Salierno, T. T. Lee, A. J. García and A. del Campo, Adv. Mater., 2011, 23, 3907–3911 CrossRef CAS PubMed.
- W. Luo and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 10780–10783 CrossRef CAS PubMed.
- D. Liu, Y. Xie, H. Shao and X. Jiang, Angew. Chem., Int. Ed., 2009, 48, 4406–4408 CrossRef CAS PubMed.
- Y.-H. Gong, C. Li, J. Yang, H.-Y. Wang, R.-X. Zhuo and X.-Z. Zhang, Macromolecules, 2011, 44, 7499–7502 CrossRef CAS.
- A. González-Campo, M. Brasch, D. Uhlenheuer, A. Gómez-Casado, L. Yang, L. Brunsveld, J. Huskens and P. Jonkheijm, Langmuir, 2012, 28, 16364–16371 CrossRef PubMed.
- A. Qi, J. Brinkmann, J. Huskens, S. Krabbenborg, J. de Boer and P. Jonkheijm, Angew. Chem., Int. Ed., 2012, 51, 12233–12337 CrossRef PubMed.
- P. Neirynck, J. Brinkmann, Q. An, D. van der Schaft, L. Gustav Milroy, P. Jonkheijm and L. Brunsveld, Chem. Commun., 2013, 49, 3679–3681 RSC.
- J. Young, H. D. Nguyen, L. Yang, J. Huskens, P. Jonkheijm and L. Brunsveld, ChemBioChem, 2010, 11, 180–183 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |