 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal complexes designed to bind to amyloid-β for the diagnosis and treatment of Alzheimer's disease
David J.
Hayne
,
SinChun
Lim
and
Paul S.
Donnelly
*
School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Melbourne, 3010, Australia. E-mail: pauld@unimelb.edu.au
First published on 27th March 2014
Abstract
Alzheimer's disease is the most common form of age-related neurodegenerative dementia. The disease is characterised by the presence of plaques in the cerebral cortex. The major constituent of these plaques is aggregated amyloid-β peptide. This review focuses on the molecular aspects of metal complexes designed to bind to amyloid-β. The development of radioactive metal-based complexes of copper and technetium designed as diagnostic imaging agents to detect amyloid burden in the brain is discussed. Separate sections of the review discuss the use of luminescent metal complexes to act as non-conventional probes of amyloid formation and recent research into the use of metal complexes as inhibitors of amyloid formation and toxicity.
1 Brief introduction to the pathology of Alzheimer's disease
Alzheimer's disease (AD) is a progressive neurodegenerative condition that results in synaptic failure and neuronal death. These symptoms initially manifest as mild forgetfulness but lead to complete loss of cognition.1 Characteristic pathological hallmarks in the brains of those suffering with the disease include the presence of extracellular senile plaques, intracellular neurofibrillary tangles and altered levels of neurotransmitters. Amyloid plaques are composed of an insoluble aggregated peptide called amyloid-β (Aβ).2,3 This peptide is 39–43 residues in length and derived from the amyloid-β precursor protein (APP). The amyloid plaque burden in subjects does not consistently correlate with cognitive impairment and some argue that smaller soluble oligomeric species are the toxic species responsible for neuronal death.4–8 However, oligomers and plaques are thought to be in equilibrium. Although the exact role of amyloid plaques in the onset of dementia is controversial, what is certain is that histopathological studies show extensive cortical Aβ deposition in post-mortem analysis of AD subjects.1 Neurofibrillary Tangles (NFT) consist of a hyper phosphorylated form of a microtubule-associated protein called tau. NFT initiate with the formation of bundles of paired helical filaments that accumulate in the neuronal cytoplasm. The hyper-phosphorylation of tau results in its detachment from microtubules that consequently lose structural integrity with concomitant impaired axonal transport and compromised synaptic function.9 Another prominent and consistent feature of AD is deficits to the acetylcholine (cholinergic) neurotransmitter system. Post-mortem studies have revealed cholinergic neuronal loss and reduced acetylcholine levels. This correlates with a decrease in the concentration of acetylcholine esterase, an enzyme responsible for the hydrolysis of acetylcholine in the brain.10The focus of this review is metal complexes designed to bind to either Aβ fibrils or Aβ plaques as distinct from metal-binding “free” ligands designed to displace either copper or zinc or iron ions that are thought to be bound to Aβ plaques. The approaches that are discussed in this review include: the potential of radioactive copper and technetium complexes to be used as diagnostic imaging agents to elucidate plaque burden in living patients; the use of luminescent metal complexes as non-conventional probes of amyloid formation; and the use of platinum, ruthenium, iridium and rhodium metal complexes to inhibit the aggregation and toxicity of Aβ. Several elegant and sophisticated approaches that use chelating ligands designed to attenuate CuII/ZnII–Aβ interactions and redistribute metal ions bound to Aβ plaques that are of significant interest as potential therapeutics will not be discussed in this review but selected leading references11–26 and reviews27–32 are available.
2 The amyloid-β peptide and plaques
The amyloid precursor protein (APP) is encoded by a single gene on chromosome 21 and resembles a cell-surface receptor having a single transmembrane domain, a large extracellular domain and a short cytoplasmic tail. The transmembrane domain extends into the intracellular region and contains the C terminus while the N terminus resides within the extracellular domain.30 Two pathways exist for the processing of APP, one being non-amyloidogenic whilst the other pathway is amyloidogenic (Fig. 1). Non-amyloidogenic metabolism involves cleavage of APP by α-secretase, a membrane anchored secretase, releasing the soluble N terminus fragment, sAPPα, to the extracellular space. The remaining transmembrane fragment is cleaved by γ-secretase within the transmembrane domain liberating non-amyloidogenic P3 (Aβ17–40 or Aβ17–42) to the extracellular space. The amyloidogenic proteolysis of APP follows a similar series of events but is cleavage with β-secretase (also known as β-APP cleaving enzyme or BACE-1) followed by γ-secretase releases sAPPβ and Aβ. Aβ ranges in length from 39–43 amino acids depending where γ-secretase cleavage occurs giving variability at the hydrophobic C-termini connected to the hydrophilic N-terminal domain.33 The two main peptides produced are 40 (Aβ40) and 42 (Aβ42) amino acid residues in length; Aβ42 shows the greater propensity for aggregation in vivo and is often considered the more toxic.34,35 As monomers, the peptides show unfolded random coil conformation with some α-helical and β-sheet structure. An imbalance between the production and clearance of Aβ peptide results in its accumulation as oligomers, protofibrils, fibrils, and then extracellular Aβ plaque deposits in the brain parenchyma. The amyloid hypothesis argues that Aβ plaques and/or their precursors trigger a cascade of events leading to synaptic dysfunction, microgliosis, and neuronal loss.1,36,37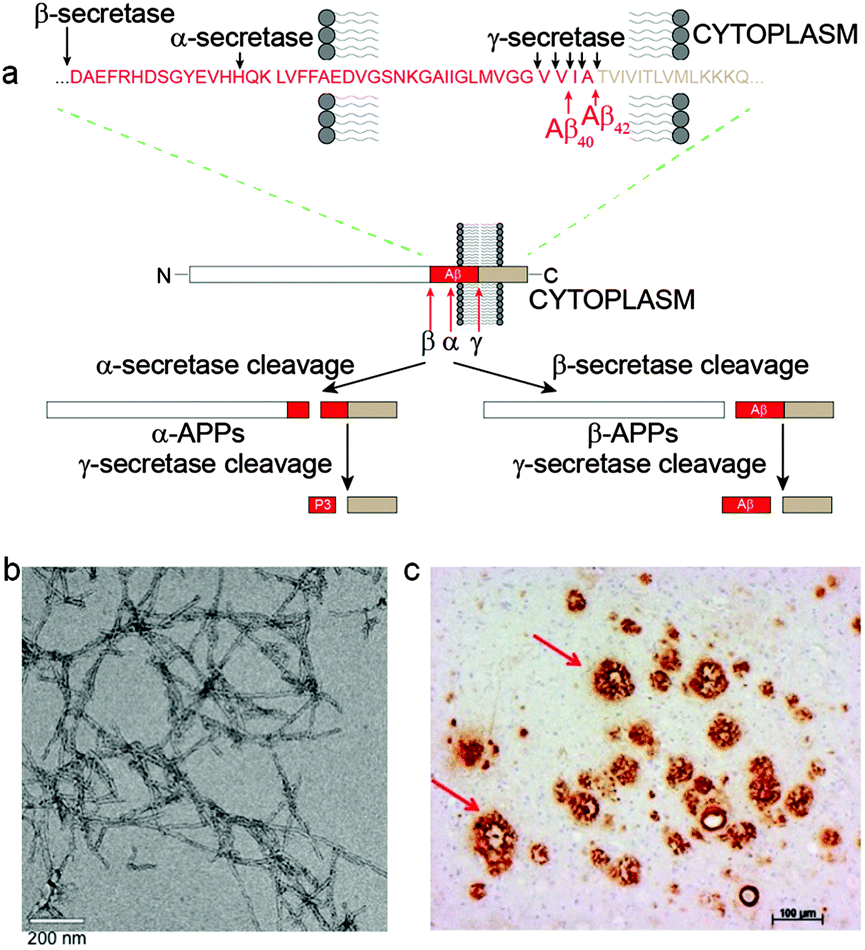 | ||
| Fig. 1 (a) Schematic representation of the non-amyloidogenic (α-secretase cleavage) and amyloidogenic (β-secretase cleavage) proteolytic pathways of APP. The specific enzyme cleavage sites on the peptide sequences are as shown by the black arrows.38,39 (b) TEM image of Aβ42 fibrils negatively stained with uranyl acetate. (c) A section of AD affected human brain tissue treated with an Aβ antibody, 1E8, showing extensive Aβ plaque deposition. | ||
Amyloid is a general term used to describe peptides or proteins that form β-sheets which accumulate as highly ordered fibrillar aggregates.40 One of the earliest methods to identify amyloids was the apple green birefringence histological dye Congo Red (Fig. 5) displayed once bound to amyloid when observed under polarized light leading to the first definition criterion of amyloid.41–43 The second definition criterion for amyloid is to possess fibrillar morphology.43,44 Negatively stained thin tissue sections containing amyloid imaged with electron microscopy (EM) reveal bundles of straight, rigid fibrils. Mature amyloid fibrils are estimated to be about 75–100 Å in width. Assembly involves two to four thin protofilaments twisting together parallel to the fibril axis. An individual protofilament consists of several β-sheets composed of peptide chains bound together by hydrogen bonds. The β-strands (monomeric Aβ) are aligned perpendicular to the fibril axis (cross-β structure) (Fig. 2).45,46 Electron microscopy and X-ray diffraction analysis of the Aβ protofilament reveals the inter-peptide chain spacing to be about 4.8 Å and inter-sheet spacing to be about 10 Å, characteristic of the cross-β structure.42,47,48
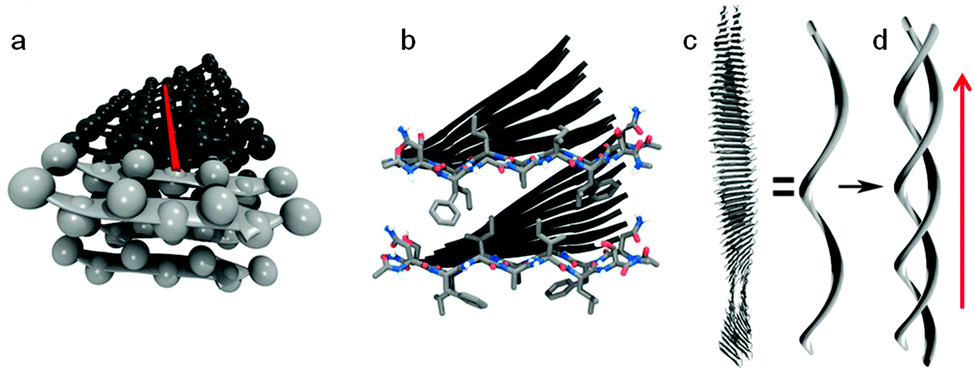 | ||
| Fig. 2 Schematic representation of amyloid fibril. (a) β-Strands are arranged perpendicular to the fibril axis (axis shown in red, side-chains shown as spheres). (b) Two sheets are paired by the interlocking of side-chains, the two lighter coloured peptides in each sheet highlight this and correspond with the lighter coloured peptides in (a). (c) β-Sheets twist together to form a protofibril which aggregates to form the mature fibril (d) (direction of fibril axis shown with red arrow). Reproduced with permission from K. K. Skeby, J. Soerensen and B. Schioett, J. Am. Chem. Soc., 2013, 135, 15114. Copyright 2013 American Chemical Society. | ||
Amyloid fibrils are not crystalline and generally insoluble in aqueous solvents. This precludes analysis by conventional solution NMR. Nevertheless, recent advances in cryoelectron microscopy and solid-state NMR have provided structural information on the inter- and intra-molecular interactions that bind the cross-β structural motifs of Aβ fibrils.45,48–53 Structural studies using fibrils formed from 35MoxAβ42 peptides (containing a methionine sulfoxide at position 35) have given insight into the 3D structure of Aβ42 fibrils (Fig. 3). The methionine is oxidised during the production of the monomeric peptide but maintains similar properties to the non-oxidised Aβ42.54 A single 35MoxAβ42 protofilament consists of two β-sheets (β1 and β2) that run along the fibril axis forming a parallel β-sandwich stabilized by inter-β-sheet side-chain interactions. Residues 17–26 define sheet β1 and residues 31–42 form sheet β2 with a connecting loop containing residues 27–30. Intermolecular interactions between amino acid residues on β1- and β2-sheets involve ionic interactions (Asp23 and Lys28), hydrophobic interactions (Leu17, Phe19, Ala21, Gly38 and Val36) and backbone hydrogen bonds. The loop region and β1 interact by salt bridge formation between the side-chains of residues Asp23 and Lys28. Residue Lys28 also forms close contacts with residues Ile32 and Leu34 of β2. The intermolecular hydrophobic interaction formed between the odd-numbered residues of the β1-strands (Leu17, Phe19 and Ala21) and the even-numbered residues (Gly38 and Val36) of neighbouring β2-strands establishes the hydrophobic core of Aβ42 fibrils. The extension and growth of the Aβ42 protofilament is unidirectional; an incoming nth Aβ42 monomer can initiate contact with the fibrillar end by means of hydrophobic interactions between its β1-strand and the β2-strand of the (n − 1)th monomer. The ionic interactions between the residues in the loop regions and backbone hydrogen bonds further strengthen the intermolecular association. In order to prevent the dissociation of the nth monomer, another Aβ42 monomer is required to interact with the β2-strand of the nth monomer to stabilize it. This suggests that the sequence-selective and cooperative mechanism of Aβ42 fibril growth is a first-order kinetic process.46
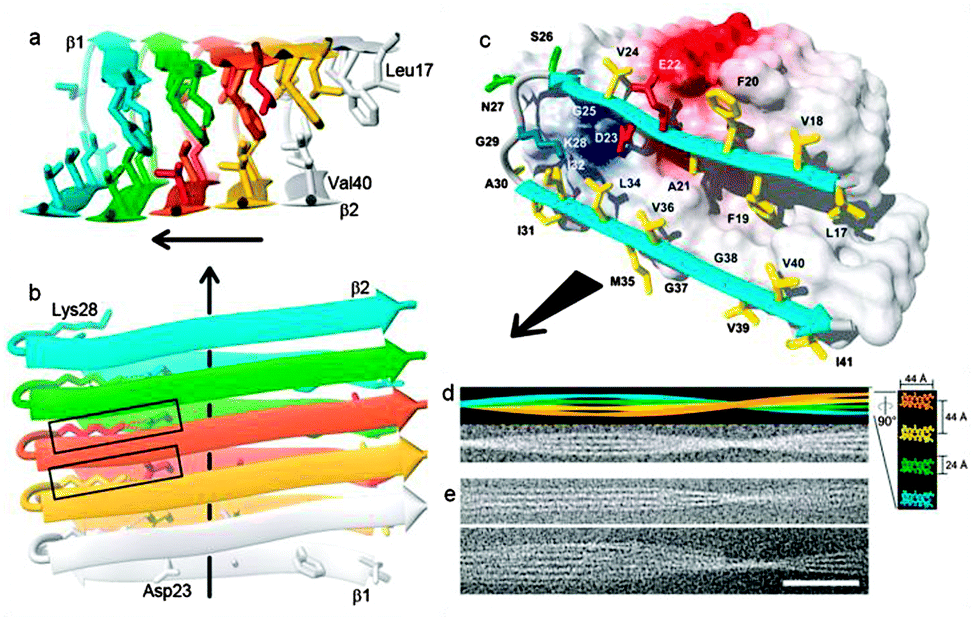 | ||
| Fig. 3 (a and b) Ribbon diagrams of the protofibril core structure (residues 17–42) illustrating the intermolecular nature of the β-strand interactions. Aβ monomers are individually colored. The β-strands are indicated by arrows, non-regular secondary structure is indicated by spline curves through the Cα atom coordinates of the corresponding residues. The bonds of side chains that constitute the core of the protofilament are shown. (b) Dotted lines indicate ion pair interactions forming intermolecular salt bridges between residues D23 and K28, two salt bridges formed by the central Aβ42 molecule are highlighted by rectangles. (c) Van der Waals contact surface polarity with a ribbon diagram at the odd end of the 35MoxAβ42 protofilament showing residues 17–42. The colours of side chains indicate if they are hydrophobic (yellow), polar (green), negatively charged (red), or positively charged (blue). Positively and negatively charged surface patches are shown in blue and red, respectively, and all others are shown in white. (d) (upper) Simulation of a 35MoxAβ42 fibril that consists of four protofilaments colored individually. Lower shows the same fibril in a noisy gray-scale image, which has been blurred corresponding to a resolution of 2 nm. In right, a ×5-magnified cross section perpendicular to the fibril axis is shown, using the same color code. Dimensions are indicated. To match the experimental twist of the protofilament of the fibril shown in (e), a twist angle of 0.45° per molecule was used. (e) Two examples of cryoelectron micrographs of single 35MoxAβ42 fibrils. The direction of the fibril axis is indicated by the direction of the black arrows (scale bar, 50 nm). Adapted with permission from T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342. Copyright 2005 National Academy of Sciences, U. S. A. | ||
3 Structural morphology of amyloid plaques
The relationship between soluble Aβ monomers, oligomers Aβ, protofibrils, fibrils and plaques is complicated and several aspects of the dynamics remain uncertain.55,56 Amyloid-β plaques are classified into two morphological types: (i) neuritic and (ii) diffuse (“pre-amyloid”) plaques (Fig. 4). Neuritic plaques, observed after staining, have a dense compact spherical appearance with a diameter ranging from 10 μm to greater than 120 μm. Neuritic plaques can be further sub-classified into ‘fibrillar’ or ‘dense-cored’ plaque forms. Fibrillar plaques show dense accumulations of Aβ throughout the plaque structure (Fig. 4B) whereas cored plaques have a distinct central core of Aβ encircled by a void or clearing surrounded by an outer spherical rim of Aβ (Fig. 4A). These Aβ deposits are referred to as neuritic plaques as they are spatially localized with dystrophic neuritis (clusters of abnormal neuronal processes) both inside and immediately surrounding the deposits.57–59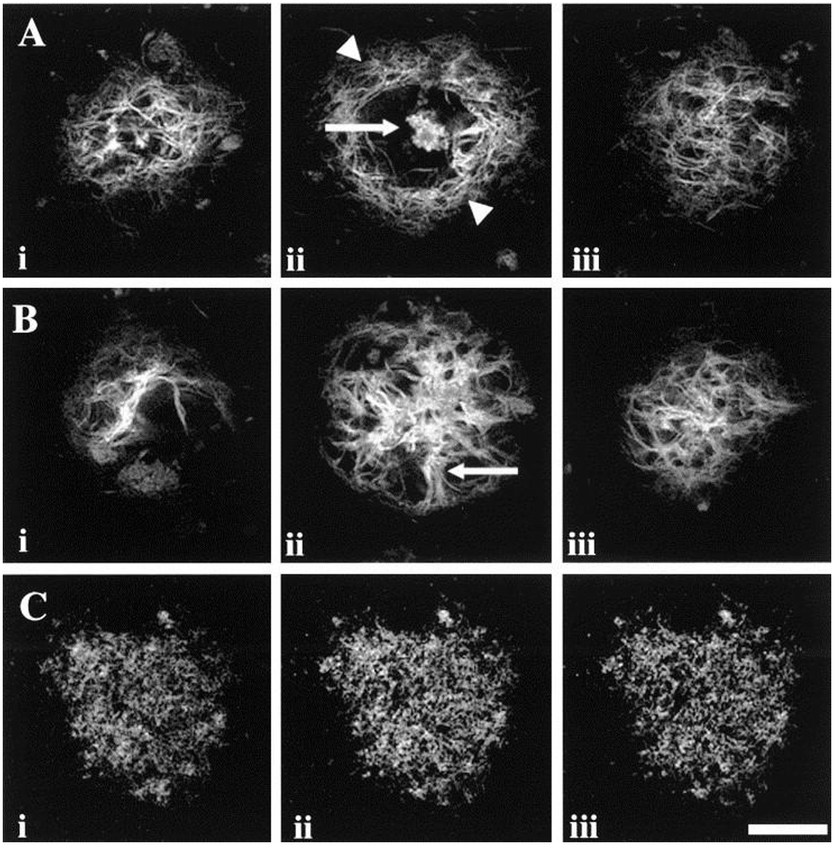 | ||
| Fig. 4 Serial sections of human brain tissue displaying Aβ plaques of different morphologies. (A, i–iii) Optical sections of a dense core plaque, observed after staining with a histological dye for amyloid. Images were captured at ∼14.5 μm intervals and show the dense Aβ core of the plaque (arrow) (A, ii) surrounded by a void defined by an outer rim of Aβ (arrow heads). (B, i–iii) Similar images of a fibrillar plaque with images captured at intervals of ∼17.5 μm. Spoke-like Aβ accumulations (arrow) radiate from the dense central accumulation. (C, i–iii) Confocal images of a diffuse plaque after treatment with an Aβ antibody. The images were captured at intervals of ∼2.5 μm and show the punctate accumulation of Aβ giving a granular pattern. Scale bar = 20 μm (A and B), 50 μm (C). Reproduced with permission from T. C. Dickson and J. C. Vickers, Neuroscience, 2001, 105, 99–107. Copyright 2001 Elsevier. | ||
Following staining, diffuse plaques display a finely granular pattern of amorphous shape that lacks a fibrillar, compacted centre (Fig. 4C). These non-fibrillar plaques are the only Aβ deposits found in regions of the brain not clearly associated with the typical symptoms of AD. They also form within the same regions of the brain as neuritic plaques, however, very little or no detectable dystrophic neurites are associated with diffuse plaques. Studies in a transgenic AD mouse model, expressing mutant human APP, show that the mice develop diffuse plaques before fibrillar plaques supporting the hypothesis that diffuse plaques are immature or precursor lesions to neuritic plaques and are therefore also termed “pre-amyloid” plaques.57
4 Molecules that bind to Aβ fibrils and plaques
The histopathological dyes Congo Red and Thioflavin T (ThT) (Fig. 5) have provided the structural inspiration for the development of many radiolabeled tracers aimed at detection of amyloid in living patients. Both dyes are relatively planar, hydrophobic aromatic molecules that interact with and bind to the cross-β sheet structure found in all amyloid fibrils. They are unable to cross the blood–brain barrier at least in part due to their ionic charge.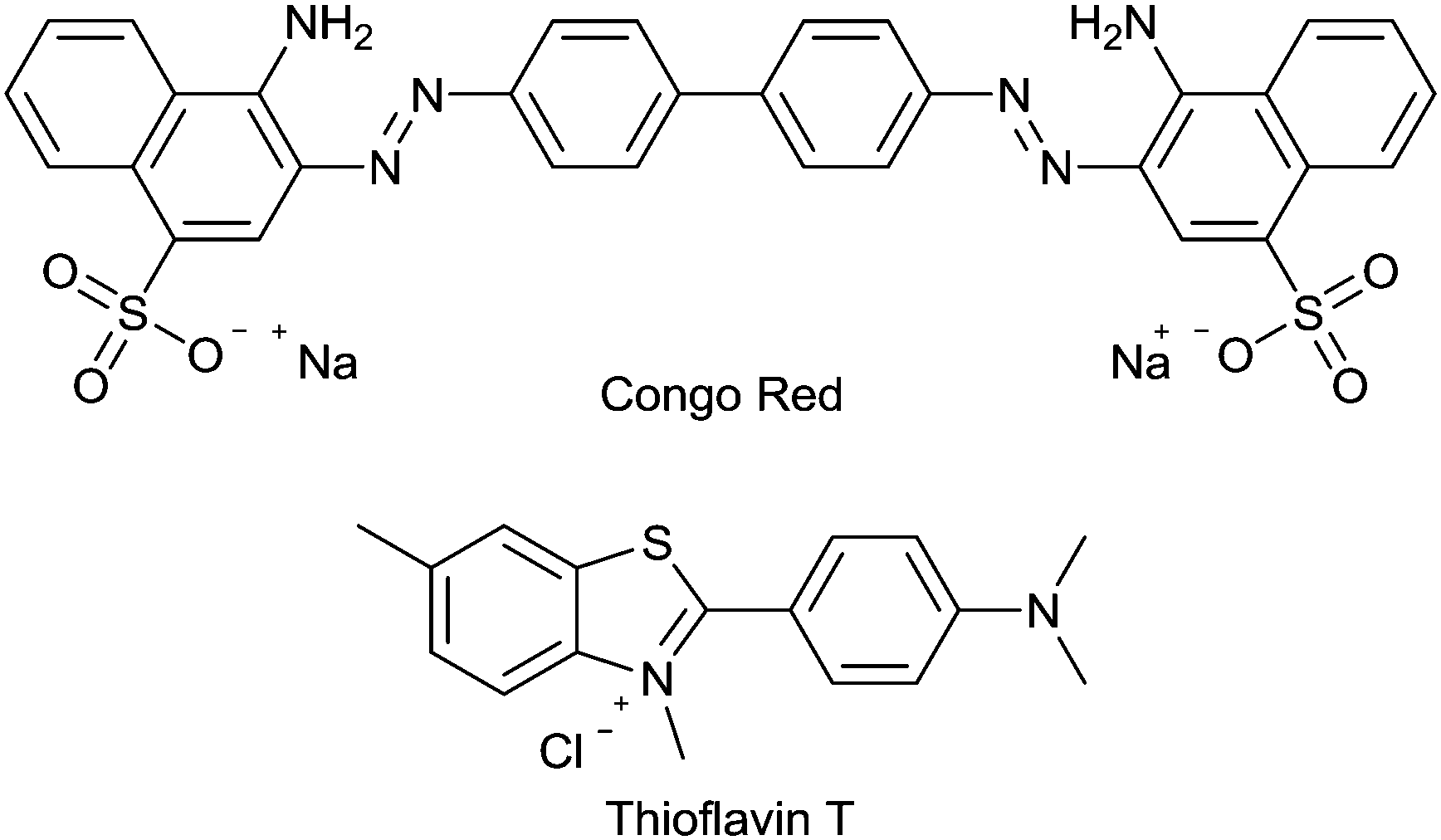 | ||
| Fig. 5 The structure of the histological dyes, Congo Red and Thioflavin T. | ||
Over the last decade there has been significant progress in the development of positron emission tomography (PET) imaging agents to characterise Aβ plaque burden. PET is a non-invasive molecular imaging technique that relies on the detection of radioactivity emitted from a positron-emitting isotope that is incorporated into a molecular tracer or imaging agent. The emitted positron annihilates releasing two gamma photons travelling in opposite directions. Detection of the emitted photons allows the generation of an image with a spatial resolution of 3–5 mm with high sensitivity. An analogue of ThT, the 2-phenylbenzothiazole derivative 2-(4(methylamino)phenyl)benzothiazol-6-ol (11C-PiB) (Fig. 6), contains the positron emitting isotope carbon-11, is able to cross the blood brain barrier, and labels Aβ plaques in vivo. It has been used in pioneering clinical studies that have demonstrated the tremendous potential of PET imaging in assisting AD diagnosis.60 The longer half-life of fluorine-18 when compared to carbon-11 is more practical and fluorine-18 is currently the most widely used radionuclide for PET imaging. Benzothiazole derivatives and an aromatic fluoro-substituted pyridinyl benzofuran radiolabeled with fluorine-18 are in the late stages of clinical development. The pyridinyl benzofuran compound, AZD4694 (Fig. 6), has completed phase III clinical trials and compares favourably to PiB displaying relatively low non-specific binding when compared to other 18F radiotracers.61 Flutemetamol (Fig. 6), a benzothiazole derivative, has recently gained FDA approval to estimate beta amyloid neuritic plaque density.62 Another pharmacophore that has shown considerable potential in binding to Aβ plaques contain diaryl alkenes such as stilbenes and styrylpyridines. The development of the 18F labelled stilbenes and styrylpyridines is presented in a ‘perspective’ article63 and the research in this area has led to the recent FDA approval of 18F-AV45 (florbetapir) (Fig. 6) to detect the presence of amyloid.64
 | ||
| Fig. 6 Structures of AZD4694, PiB, flutemetamol, and florbetapir. | ||
The use of the benzothiazole dye ThT as a fluorescent probe for the detection of amyloid-like structures was first reported in 1959.65 In protic solvents ThT absorbs at a wavelength of 340 nm with a relatively weak emission at 445 nm but when bound to amyloid fibrils the dye undergoes a 115 nm red shift in excitation profile (λex = 445 nm) with enhanced emission at a wavelength of 485 nm. In aqueous solutions rapid rotation about the C–C bond linking the benzothiazole group to the N,N-dimethylaniline portion of the molecule provides a pathway for non-emissive decay of the excited state. This occurs by a twisted internal charged-transfer process (TICT) from a locally excited state (LE) by increasing the torsion angle from 37° to 90°. The interaction of ThT with amyloid fibrils restricts rotation about the C–C bond and is partially responsible for the increased emission (Fig. 7).66–70 It is likely that there are three different binding sites for ThT on Aβ40/42 fibrils.71,72 The first and second binding sites (BS1 and BS2) have higher binding affinities than the third (BS3) and are located closely and/or overlapping to each other. BS1 occurs every ∼35 Aβ40 monomers while BS2 occurs every ∼4 Aβ40 monomers and low capacity BS3 occurs every ∼300 Aβ40 monomers.72 Hydrophobic grooves are formed on the β-sheet surface by the side-chains of amino acids, particularly Phe and Val. These hydrophobic grooves propagate along the “twisting” Aβ fibril forming BS1 and BS2. Interaction of ThT with the side-chains of these grooves is thought to be primarily via π–π stacking. The low affinity and less abundant BS3 is present on the β-sheet extremities of the fibril.73
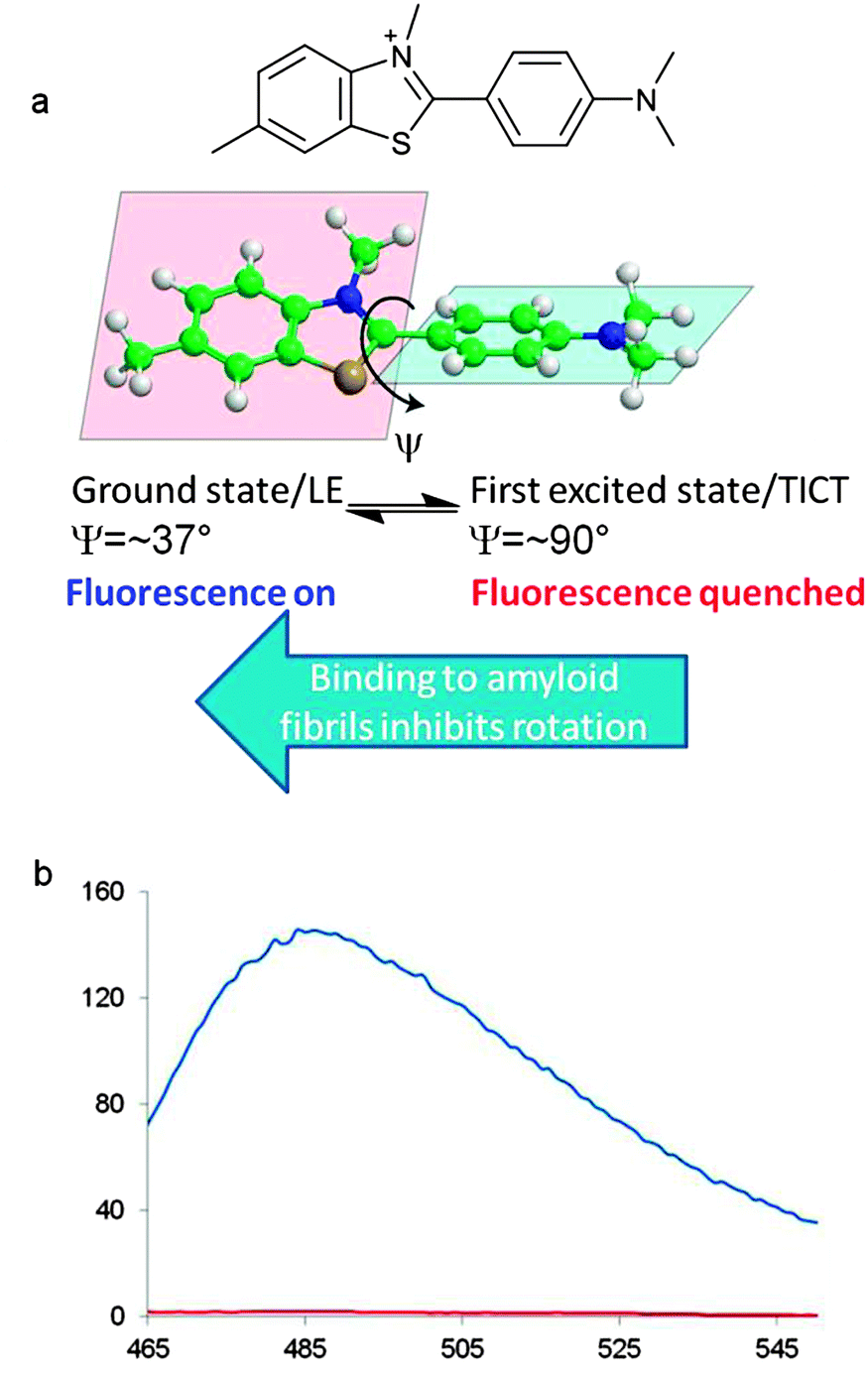 | ||
| Fig. 7 Photophysical properties of ThT. (a) The two planar groups within ThT rotate about the C–C bond with an angle of torsion, ψ. (b) Fluorescence emission of ThT (red) and ThT with Aβ42 in phosphate buffer (λex = 444 nm). M. Biancalana and S. Koide, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412. Copyright 2010 Elsevier. | ||
Simulated molecular dynamics support the proposed groove binding of histological dyes and small-molecule imaging agents to amyloid. The Asp-Phe-Gly-Ala-Ile-Leu-Ser peptide sequence, found within human islet amyloid polypeptide, acts as a useful model for binding simulations of small molecule imaging agents and dyes, including ThT and the related radiotracer 11C-PiB. The simulated fibril construct consists of a double layer of β-sheets, each containing 10 in-register antiparallel β-strands. The amino acid side chains decorate the large faces of the fibril to form grooves on both faces (Fig. 8a).
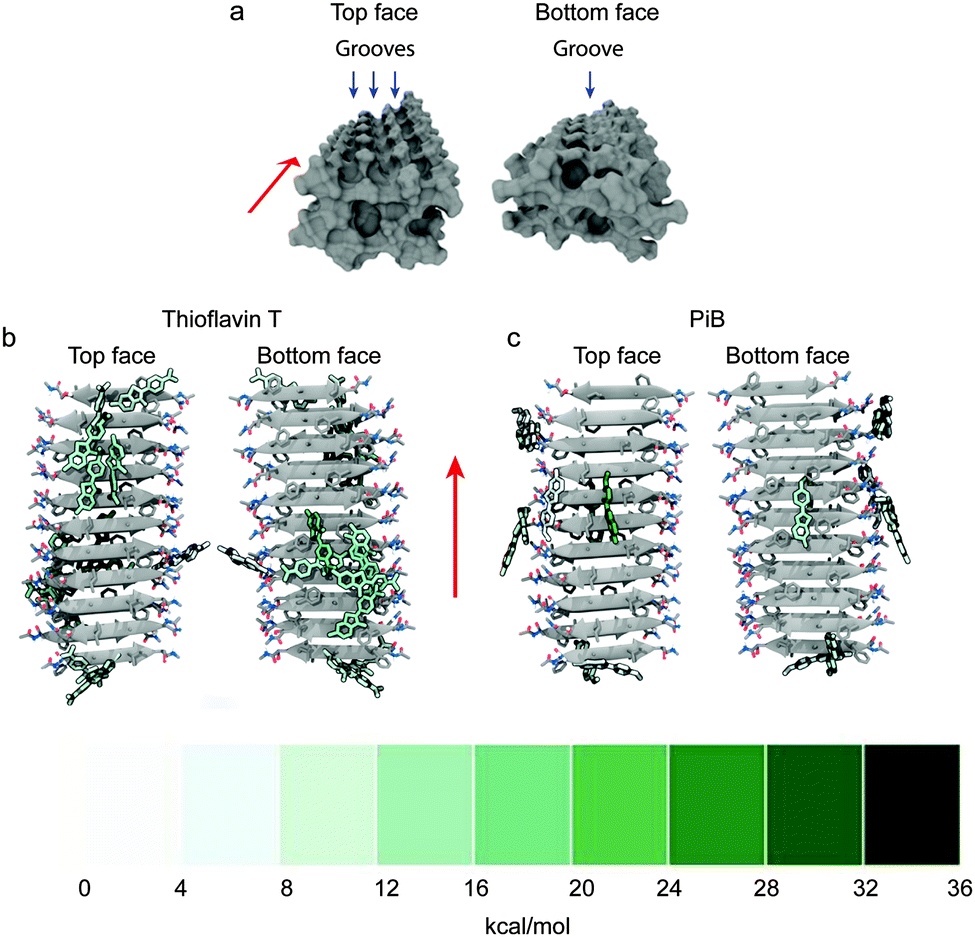 | ||
| Fig. 8 (a) Surface representation of the top face and bottom face of the simulated Asp-Phe-Gly-Ala-Ile-Leu-Ser fibril construct, blue arrows show three grooves on the top face and single groove on the bottom face with red arrows showing the direction of the fibril axis. Representative structures for ThT (b) and PiB (c) clusters are coloured by their free energies of binding to the simulated fibril. A large binding free energy value represents a favourable contribution to binding energy. Clusters are defined by the positions of heavy atoms within the compound with respect to the Cα atoms of the fibril, the representative is chosen as the structure with the lowest RMSD to the average structure of each respective cluster. Adapted with permission from K. K. Skeby, J. Soerensen and B. Schioett, J. Am. Chem. Soc., 2013, 135, 15114. Copyright 2013 American Chemical Society. | ||
The top face of the fibril has a central groove consisting of isoleucine residues and two neighbouring minor grooves formed with isoleucine on one side and alternating arginine and serine residues on the other. Although the bottom face of the fibril has potential for two grooves, phenylalanine residues leaning toward the alanine residues restrict access to one groove but the small alanine residues allow room for a single small hydrophobic central groove. Amongst the multiple potential binding sites available, the two binding modes with the highest binding affinities are in the hydrophobic grooves of the top and bottom faces. The absolute energetic contributions of binding can be classified as polar or non-polar. The non-polar interactions provided the largest contributions to the interactions and a major proportion can be speculatively assigned to π–π interactions. Comparison of the charged ThT and its neutral analogue PiB reveals greater insertion into the hydrophobic grooves for PiB corresponding to a more energetically favourable binding to the fibril (Fig. 8b and c). It is reasonable to expect this to translate to fibrils formed with other peptides, such as Aβ, due to the similarity across amyloid fibril structure.55
5 Toward diagnostic imaging of Alzheimer's disease with copper radiopharmaceuticals
The positron-emitting isotopes of copper offer considerable potential for diagnostic PET imaging.74,75 There are two comparatively short-lived isotopes, copper-60 (t1/2 = 20 min) which is relatively easy to produce with a small medical cyclotron and copper-62 (t1/2 = 20 min), available from a convenient generator. Two longer half-life isotopes, copper-61 (t1/2 = 3.4 h) and copper-64 (t1/2 = 12.7 h) can both be produced in low energy hospital cyclotrons.74 The low energy of the positron-emission from copper-64 is coupled to a lack of interfering gamma emissions so high quality images are obtained that are comparable to those obtained with fluorine-18.76 In principle, the rapid and simple incorporation of a radioactive copper isotope into a specifically designed targeting ligand is an attractive alternative to the sometimes challenging covalent incorporation of carbon-11 or fluorine-18 into plaque binding tracers. Such a system needs to be carefully designed as factors such as complexation kinetics, thermodynamic stability and biodistribution need to be considered. A single ligand framework is suitable for the full range of copper isotopes and would serve as a valuable platform to develop versatile Aβ plaque imaging agents and improve the number of clinical centers that are capable of assessing Aβ plaque burden in patients.A family of ligands known as bis(thiosemicarbazones) (btsc), derived from 1,2-diones, can be used as delivery vehicles for radioactive copper isotopes as they form stable (Ka = 1018) and neutral membrane permeable copper complexes.77–81 The copper complex [64CuII(atsm)] (Fig. 9) is currently undergoing clinical trials in humans for imaging hypoxia in head and neck cancers.82–85 It is thought that the hypoxia selectivity of the complex involves the metal ion being reduced from CuII to CuI. The copper(II) bis(thiosemicarbazonato) complex with a single methyl substituent on the backbone of the ligand, [CuII(ptsm)] (Fig. 9), has been used to image blood perfusion in humans and over 5% of the injected dose reaches the brain highlighting its ability to cross the blood brain barrier.86,87
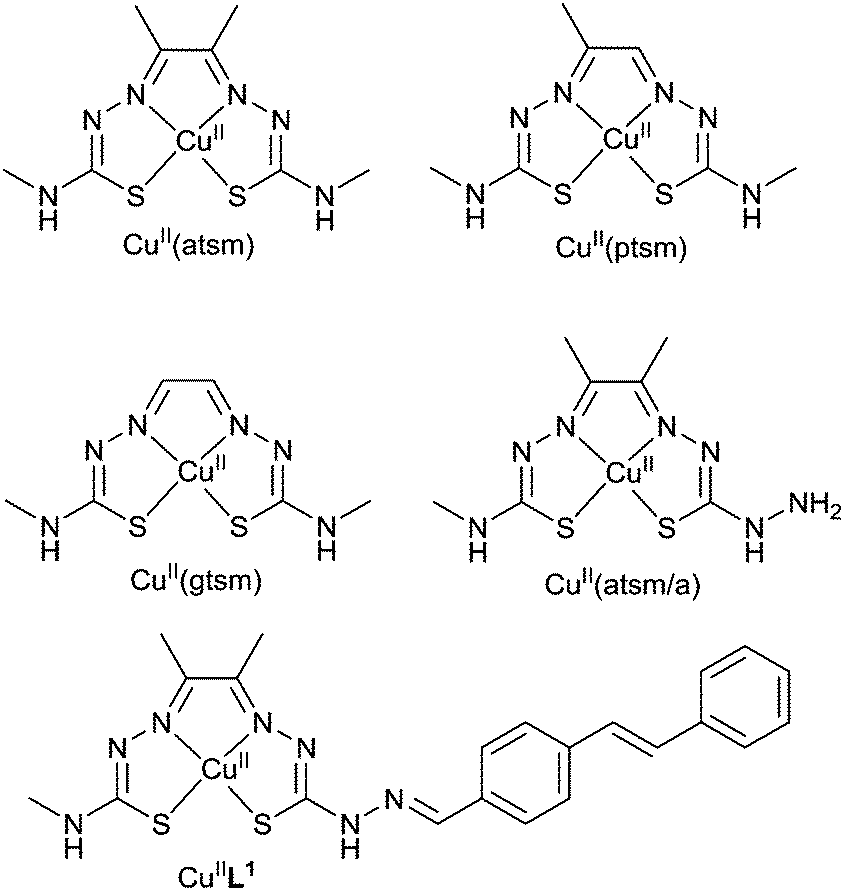 | ||
| Fig. 9 Structures of bis(thiosemicarbazonato) copper(II) complexes. | ||
[CuII(atsm)] is also able cross the blood–brain barrier and the uptake of radiolabeled [CuII(atsm)] in the brain has been used to probe cellular redox status in the brains of patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and to evaluate striatal oxidative stress in patients with Parkinson's disease. The CuII/CuI redox potential of [CuII(btsc)] complexes is dependent on the substituents on the diimine-like backbone of the ligand. For example, the CuII/CuI reduction potential in [CuII(atsm)] is about 160 mV more negative than the complex with two hydrogen atoms on the backbone, [CuII(gtsm)]. This difference in redox chemistry has been used to investigate differences in copper metabolism in a mouse model of amyloid pathology. The Aβ42 peptide binds copper and amyloid plaques have elevated levels of copper (as well as zinc and iron) and it has been suggested that the transmembrane amyloid precursor protein may play a role in the control of nutrient copper. Both [64CuII(atsm)] and [64CuII(gtsm)] cross the blood–brain barrier but the brain uptake of [64CuII(gtsm)] in APP/PS1 model of amyloid pathology mice is higher than [64CuII(atsm)]. The uptake of [64CuII(gtsm)] in the brain (expressed as % of the injected dose per gram (body weight), %ID per g) is significantly higher (p = 0.01) in APP/PS1 mice when compared to control animals (3.0 ± 0.25% ID per g in the APP/PS1 mice compared to 1.58 ± 0.14% ID per g in wild type control animals). This difference is presumably a result of changes to copper metabolism and is not a measure of plaque burden.81 The ability to alter both the CuII/CuI reduction potential and the biodistribution of [CuII(btsc)] complexes by altering the substituents on the ligand means they have potential to investigate both redox status and altered copper metabolism in other neurological conditions such as Wilson's and Menkes disease. The potential of [CuII(gtsm)] and related complexes to probe molecular changes in Alzheimer's disease and other neurological diseases warrants more detailed investigations.
As [CuII(atsm)] has sufficient stability for brain imaging it appeared to be a useful starting framework to develop bifunctional chelates with the potential to be used as Aβ plaque imaging agents. The CuII/CuI reduction potential in [CuII(atsm)] is such that it is thought, at least to some extent, reduction is only likely to occur intracellularly in hypoxic cells so reduction to CuI would not be expected when targeting extracellular Aβ plaques. A new bis(thiosemicarbazone) ligand with an appended plaque targeting trans-stilbene functional group, H2L1 was prepared by a condensation reaction between trans-stilbene aldehyde and the bifunctional chelate atsm/a (Fig. 9). The copper complex, [CuIIL1], interacts with synthetic Aβ42 fibrils in vitro and binds selectively to Aβ plaques in post-mortem samples of human brains from AD subjects (Fig. 10). The binding of [CuIIL1] to Aβ plaques was demonstrated in serial sections (7 μm) of frontal cortex tissue treated with a solution of [CuIIL1]. The complex is fluorescent due to the presence of the stilbene functional group allowing examination by epifluorescence microscopy and comparison to the contiguous section that was immuno-stained with an Aβ antibody (1E8) to identify plaques. Aβ plaques are typically 40–60 μm in diameter, indicating that 7 μm serial sections would comprise the same Aβ plaques. The [64CuL1] complex crosses the blood–brain barrier and displays increased uptake in the APP/PS1 model of amyloid pathology when compared to age-matched control animals. The brain uptake was significantly higher (p = 0.005) in the APP/PS1 transgenic mice when compared with wild-type (2.5% ± 0.6 compared to 1.7% ± 0.6, %ID per g) at 7 minutes after the injection.88
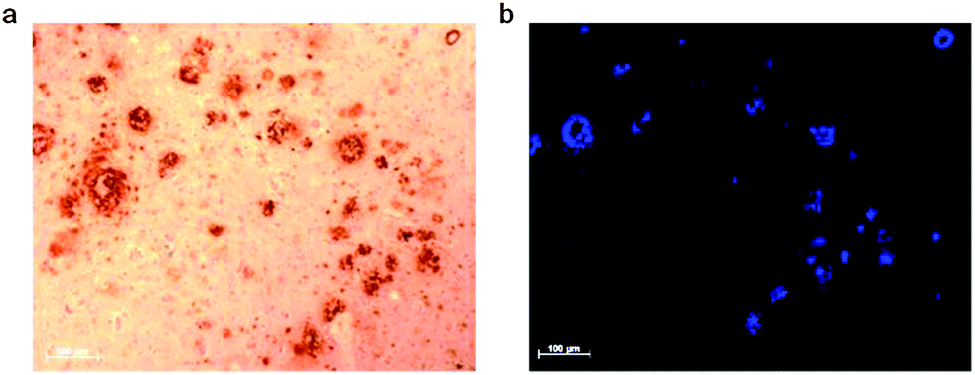 | ||
| Fig. 10 Serial sections of human brain tissue from AD affected subjects. (a) Sample immuno-stained with 1E8 antibody and (b) sample treated with [CuIIL1]. | ||
To reduce the size and molecular weight of the ligand hybrid thiosemicarbazone–benzothiazole and thiosemicarbazone–styrylpyridine tetradentate ligands H2L2–4 were prepared. All three ligands form stable complexes with CuII and the monocations [CuIIHL2]+ and [CuIIHL3]+, where the hydrazine limb of the ligand remains protonated, were characterised by single crystal X-ray crystallography (Fig. 11b). In the solid state the CuII is 5-coordinate square pyramidal with the ligand acting as a quadridentate N3S donor and weaker axial interactions with the sulfur atom from an adjacent molecule to give a dimer. Electronic spectroscopy showed that all the complexes were charge-neutral in aqueous buffer at pH 7.4. The ligands form stable complexes with CuII and for [CuIIL4] the conditional dissociation constant (KD) is 5.8(4) × 10−18 M at pH 7.4. The CuII complexes that feature the styrylpyridine functional group, [CuIIL3] and [CuIIL4], have a quasi reversible reduction at about −0.68 V (vs. SCE) attributed to a CuII/CuI couple, 80 mV more negative the CuII/CuI couple for [CuII(atsm)]. The complexes [CuIIL3–4] bind to Aβ plaques in AD affected human brain tissue, with plaques identified by staining the contiguous section with 1E8 antibody (Fig. 11c), even though the pyridyl nitrogen atom within the styrylpyridine is coordinated to CuII. Both H2L3 and H2L4 formed complexes with copper-64 at room temperature, [64CuIIL3] and [64CuIIL4], and the distribution coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D) values of 1.90 and 1.87 are comparable to [CuII(atsm)] (1.85). The brain uptake of [64CuIIL3] was investigated by micro-PET imaging in wild-type mice and revealed essentially no radioactivity evident in the brain five minutes post injection. The images obtained following administration of [64CuIIL4] were more promising with significant radioactivity observable in the brain. A separate biodistribution study that revealed that, at five minutes post injection, 1.11 (0.20) %ID per g of [CuIIL4] had accumulated in the brain, and in wild-type mice without Aβ plaques, the radioactivity cleared to 0.38 (0.09) %ID per g 30 min post injection. The addition of a –(CH2)2N(CH3)2 functional group to form [CuIIL4] increases the molecular weight and has little effect on the logD but increases the ability to cross the blood–brain barrier when compared to [CuIIL3]. This functional group is capable of forming an intramolecular hydrogen bond and may alter the solvation properties of the complex thus modifying membrane permeability. It is also possible that this functional group produces other complicating interactions such as those with serum proteins or the p-glycoprotein efflux system. Further studies on these systems are warranted including investigation in transgenic models of amyloid and more detailed characterisation of their biodistribution and metabolism.89
D) values of 1.90 and 1.87 are comparable to [CuII(atsm)] (1.85). The brain uptake of [64CuIIL3] was investigated by micro-PET imaging in wild-type mice and revealed essentially no radioactivity evident in the brain five minutes post injection. The images obtained following administration of [64CuIIL4] were more promising with significant radioactivity observable in the brain. A separate biodistribution study that revealed that, at five minutes post injection, 1.11 (0.20) %ID per g of [CuIIL4] had accumulated in the brain, and in wild-type mice without Aβ plaques, the radioactivity cleared to 0.38 (0.09) %ID per g 30 min post injection. The addition of a –(CH2)2N(CH3)2 functional group to form [CuIIL4] increases the molecular weight and has little effect on the logD but increases the ability to cross the blood–brain barrier when compared to [CuIIL3]. This functional group is capable of forming an intramolecular hydrogen bond and may alter the solvation properties of the complex thus modifying membrane permeability. It is also possible that this functional group produces other complicating interactions such as those with serum proteins or the p-glycoprotein efflux system. Further studies on these systems are warranted including investigation in transgenic models of amyloid and more detailed characterisation of their biodistribution and metabolism.89
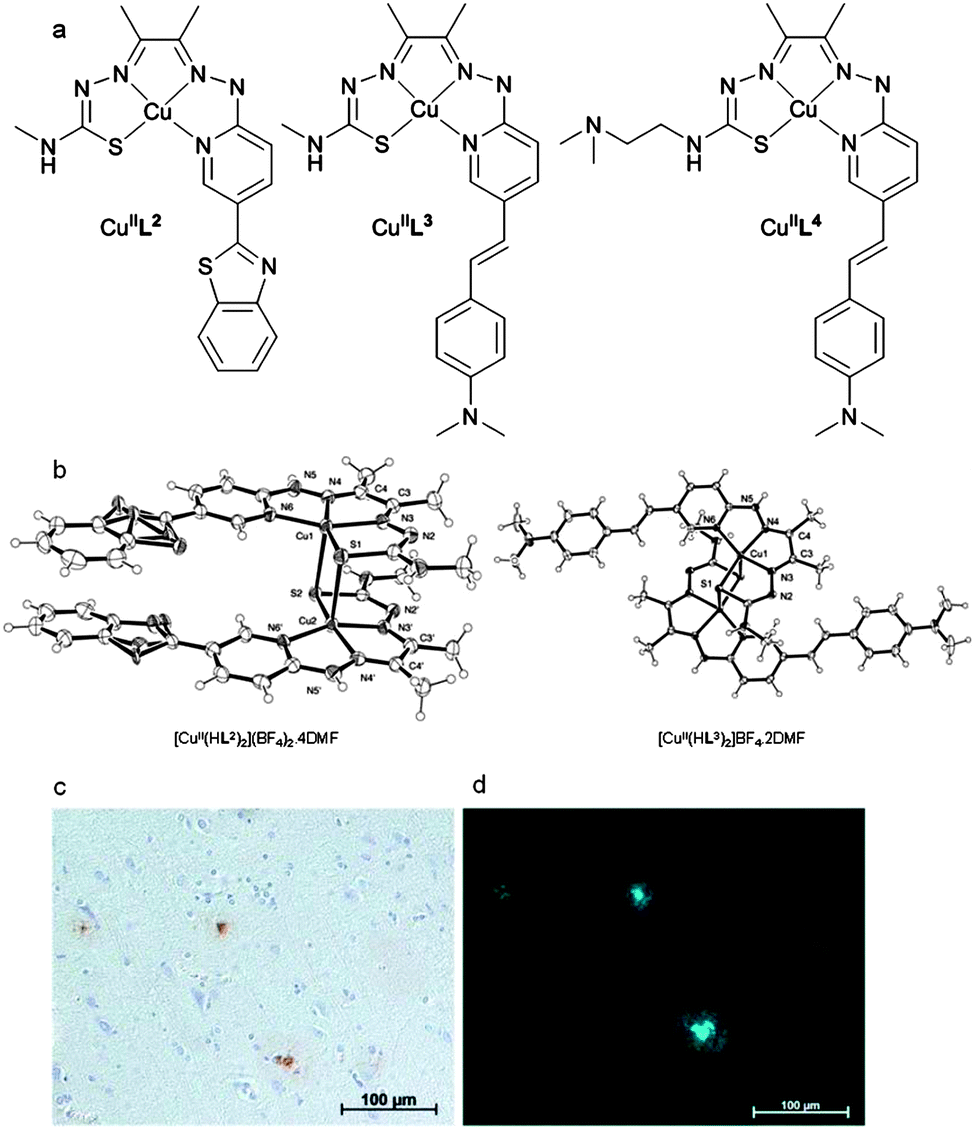 | ||
| Fig. 11 (a) Structure of complexes [CuL2], [CuL3], and [CuL4]. (b) ORTEP (ellipsoids at 40% probability) representations of [CuII(HL2)]2(BF4)2·4DMF and [CuII(H2L3)]BF4·2DMF with solvent molecules and anions omitted for clarity. The dication dimer, [CuII(HL2)]2(BF4)2·4DMF contains benzothiazole rings that are disordered over 2 sites (occupancy 0.5). (c) AD human brain sections (×20 magnification) with 1E8 antibody stain and (d) epi-fluoresence microscopy of CuL4 binding to Aβ plagues. | ||
Specifically designed ligands to probe Aβ plaque burden such as the systems presented above could be incorporated into ‘kit’ formulations in a similar manner to successful technetium-99m based radiopharmaceuticals. In principle, these ligands would be for suitable each of the positron-emitting isotopes of copper offering incredible versatility and flexibility from a single platform.
6 Technetium-99m radiotracers designed to bind to amyloid-β
Although the use of the imaging technique single photon emission computed tomography (SPECT) is routine and widespread, progress toward a SPECT compatible molecular probe has been slower than that of PET.90 The most commonly used radioisotope for SPECT imaging is technetium-99m. This γ-emitting isotope has a half-life of six hours and is available from convenient generator systems. A 99mTc-based radiotracer for determining plaque burden would be of considerable clinical utility and increase the number of centres that are able to perform diagnostic scans for AD based on both cost and availability of the necessary infrastructure. As there are no non-radioactive isotopes of technetium, it is common practice to use the relatively stable isotopes of rhenium to form analogues of 99mTc complexes for comprehensive characterisation and preliminary in vitro assessment.90–92 Detailed reviews on the chemistry of technetium are available93–95 and earlier detailed reviews on diagnostic imaging of AD with 99mTc complexes were published in 2011 and early 2012 (see Fig. 12 for selected examples).90,92 The significant progress towards a 99mTc complex for Aβ plaque imaging published in articles from 2012 onwards are highlighted in the following section.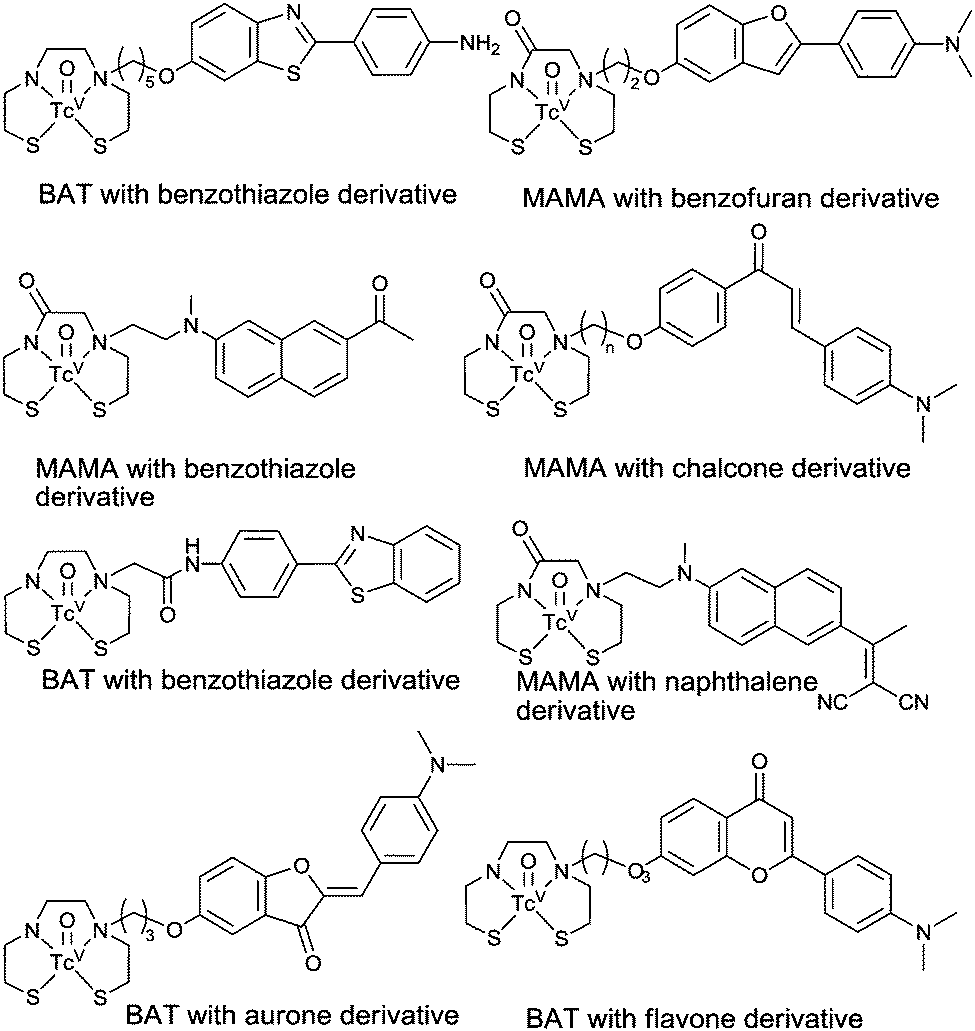 | ||
| Fig. 12 Selected examples of technetium complexes designed to bind to Aβ plaques. For recent reviews see ref. 90 and 92. | ||
A N2S2 donor often forms neutral complexes with the [MVO]3+ (M = Re or Tc) core, particularly the ligands derived from bis(aminoethanethiol) (BAT) and monoamine–monoamide dithiol (MAMA). A comparison between [TcOBAT] and [TcOMAMA] complexes tethered to a phenylbenzoxazole plaque targeting group revealed that the [TcOBAT] complexes displayed higher brain uptake.96 Amino, monomethylamino, or dimethylamino substituted pyridyl benzofuran functional groups have also been used as the basis for targeted binding of Aβ plaques in a bifunctional chelate formed by N-alkylation of BAT (Fig. 13).97 Tetradentate N2S2 ligands with N-alkylation of the ligand leads to the possibility for syn and anti isomers with respect to the position of the pendant group in relation to the apical oxo ligand. However, in the case of H3L5–7 a single radioactive peak was observed by radio-high performance liquid chromatography upon radiolabeling with 99mTc. The octanol water partition coefficient (P) is used to give an indication of the biodistribution of compounds. LogP values were 0.68, 1.35, and 2.09 for [99mTcOL5–7] respectively. Brain uptake in ‘normal’ mice 2 min post injection was 1.59 (0.21) %ID per g and 1.80 (0.16) %ID per g for [99mTcOL5] and [99mTcOL6] respectively while [99mTcOL7] has a brain uptake of 1.64 (0.27) %ID per g at 10 min with all three showing good clearance from the brain. The highest brain uptake was shown by [99mTcOL6] (1.80% ID per g) but, of the rhenium analogues, [ReOL7] displayed the highest binding affinity toward Aβ42 aggregates in vitro (Ki 13.6 nM). The compound that displayed the highest uptake in the brain, [99mTcOL6], was evaluated in Tg2576 mice which display high Aβ plaque deposition in the brain. The murine brain samples examined by ex vivo autoradiography demonstrated that the tracer accumulated in the same deposits as Thioflavin S.97
 | ||
| Fig. 13 General structure of complexes (represented in text as MOLx) containing BAT chelate conjugated to pyridyl benzofuran derived Aβ binding group; where M = ReV or 99mTcV; L5, R = NH2; L6, R = NHMe; L7, R = NMe2. | ||
In an alternative approach a dibenzylideneacetone derivative, containing a dimethylamino group, was conjugated to both BAT and MAMA (Fig. 14). Once again, rhenium complexes were used as non-radioactive surrogates to assess the binding affinity of complexes toward aggregated Aβ. The binding affinities of MAMA rhenium complexes, [ReOL8–9], to Aβ42 aggregates were moderate (Ki = 121 and 60 nm for [ReOL8–9] respectively) increasing to high affinity for BAT rhenium complexes (Ki = 25 and 14 nm for [ReOL10–11] respectively). All complexes, [ReOL8–11], bind in vitro to Aβ plaques within brain samples from transgenic model mice (APP/PS1). Complexes [99mTcOL9–11] display relatively high lipophilicity, with logD values ranging from, 3.17 to 3.53. Brain uptake of the complexes in wild-type mice is relatively low with no significant difference between BAT containing complexes, [99mTcOL8–9], and the MAMA containing complex [99mTcOL10], having an uptake ranging from 0.47 to 0.49% ID per g 2 min post injection. An even lower brain uptake of 0.31% ID per g 2 min post injection was observed for [99mTcOL11]. Compounds [99mTcOL8–10] cleared the brain over time (brain2min/brain60min = 6.13, 3.92 and 5.33 for [99mTcOL8–10] respectively) while a significant amount of activity was recorded in the brain 60 min post injection for [99mTcOL11] (brain2min/brain60min = 2). The brain clearance is important for radiotracers as fast brain clearance is necessary to remove unbound radiotracer, giving a high signal to noise ratio affording clear images.98
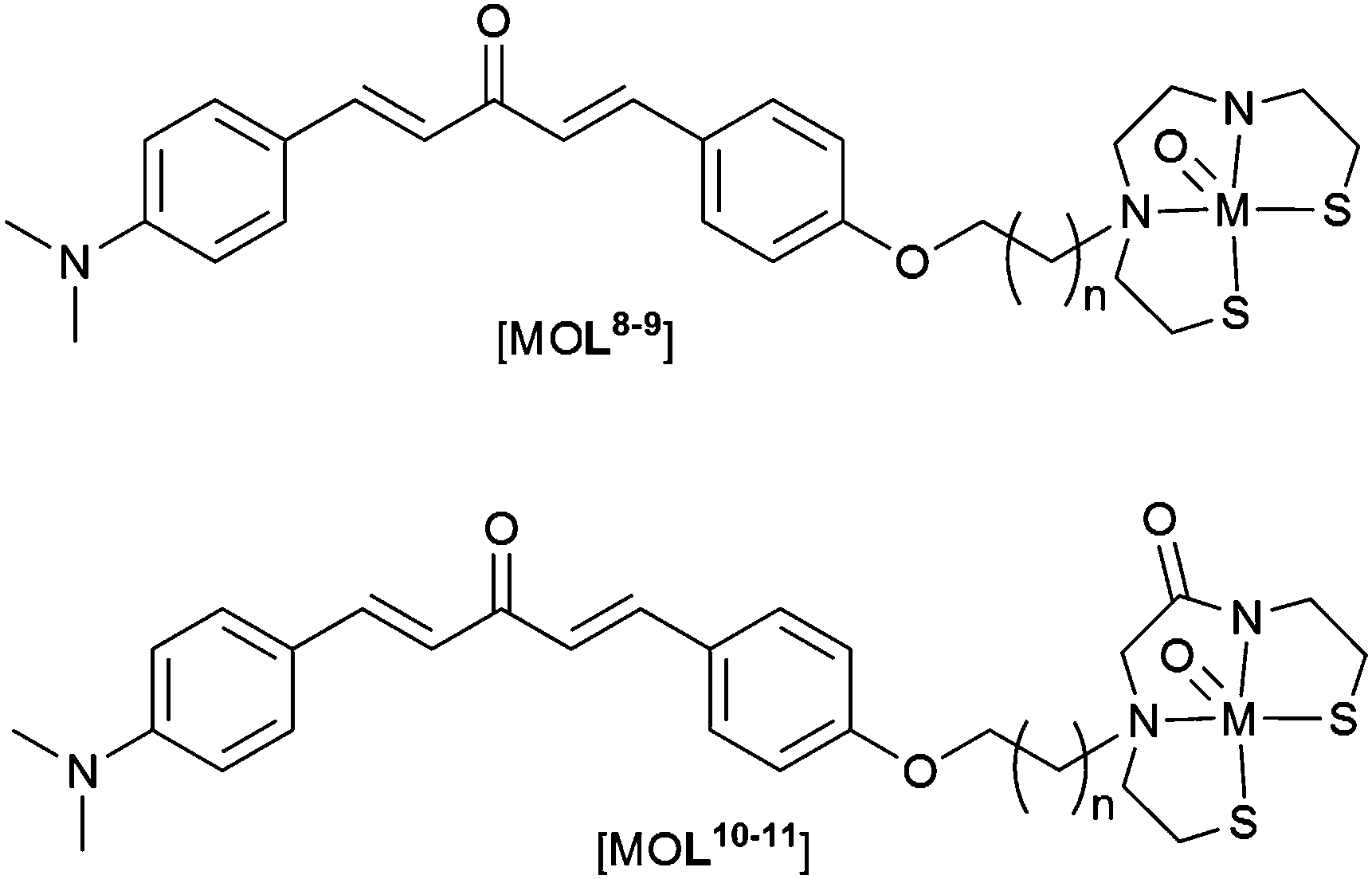 | ||
| Fig. 14 General structure of complexes containing MAMA and BAT ligands conjugated to dibenzylideneacetone derived Aβ binding group; M = ReV or 99mTcV; L8, n = 2; L9, n = 4; L10, n = 2; L11, n = 4. | ||
An integrated approach similar to the tetradentate hybrid thiosemicarbazone–styrylpyridine systems ([CuL3] and [CuL4]), whereby the binding motif forms part of the chelate backbone, reduces the molecular weight of compounds and potentially aid blood–brain barrier permeability. Examples of such ligands have been synthesised incorporating 2-arylbenzothiazole (2-ABT) derivatives as the targeting motif (Fig. 15). The binding affinity of rhenium complexes, [ReOL12–19], was assessed according to their binding affinity to Aβ40 fibrils and logPC18 (PC18 = estimation of P by a reverse phase HPLC method). This revealed that both the type of chelator and substitution positions about 2-ABT (see Fig. 15 ThT for numbering) influences the affinity of complexes toward Aβ fibrils. Addition of an electron donating group or halogen to 2-ABT at 6- or 4′-positions, in general, increases binding affinity of these complexes toward Aβ40 fibrils as does incorporating 2-ABT into the ligand chelator via its phenyl ring. The two rhenium complexes that show the most promise of this set of compounds are [ReOL15] (R = OMe) and ReL18 (R = F). Both contain semi-rigid chelates that integrate the 2-ABT group through the phenyl ring with the amino group at the 4′-position. Although the binding affinities of these complexes (Ki ∼ 30 nM) is inferior to that of most clinical Aβ imaging agents (<10 nM) the integrated approach using other Aβ binding motifs may prove more successful.99
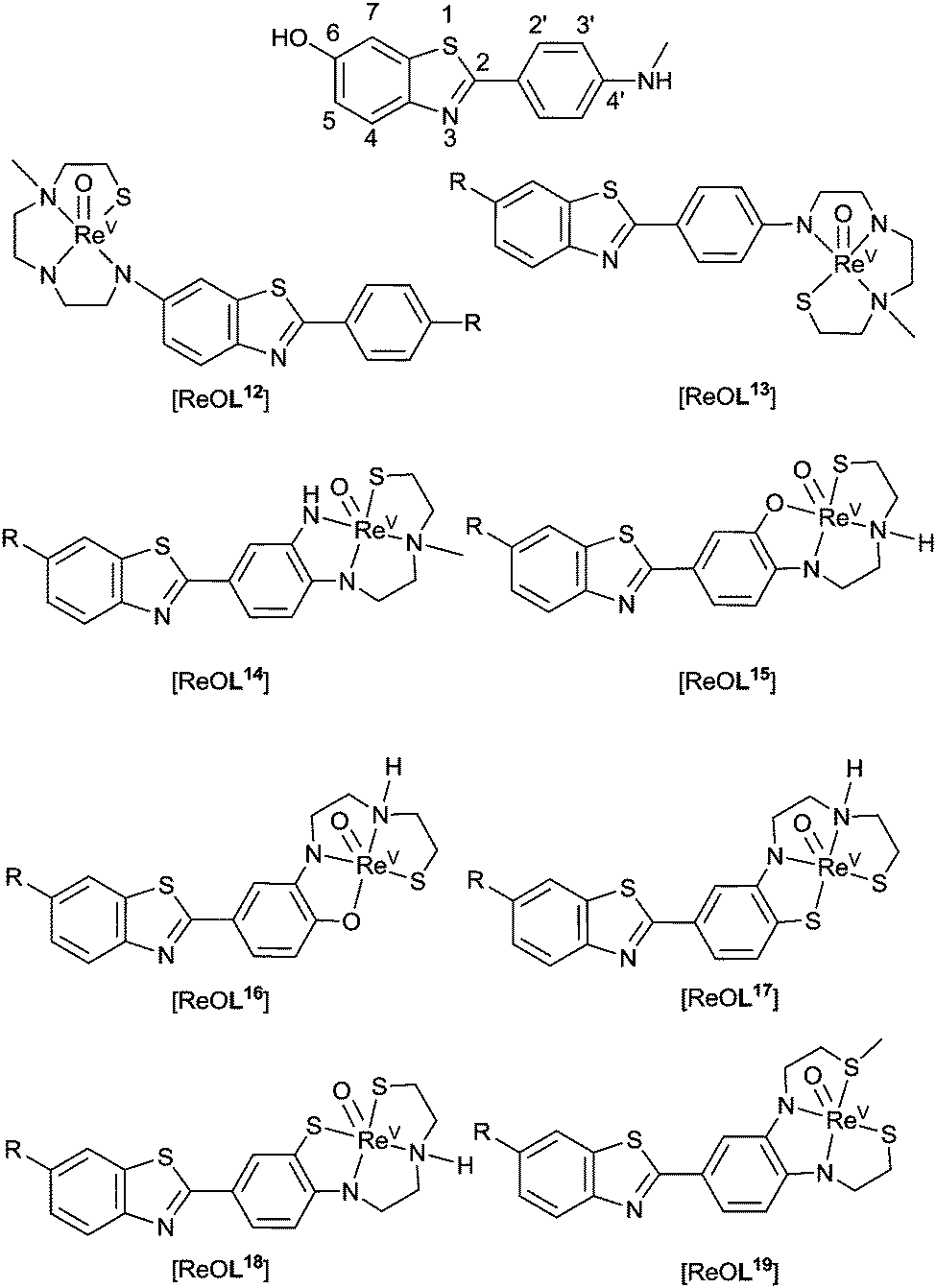 | ||
| Fig. 15 General structure of complexes containing 2-arylbenzothiazoles for Aβ binding; R = H, F, or OMe. | ||
The low valent oxidation state of technetium(I) can be stabilized by carbonyl ligands. Pioneering method development has led to the synthesis of fac-[99mTcI(CO)3(H2O)3]+ from [99mTcVIIO4]− using sodium boranocarbonate as both reducing agent and an in situ source of carbon monoxide allowing for the convenience of a kit formulation.100–102 These low spin d6 systems offer high kinetic stability. The stable M–C bonds favour exchange of the water ligands allowing the introduction of mono-, bi- or tridentate ligands and the possibility for [2+1] mixed ligand complexes by coordination of both a mono and bidentate ligand in the one complex.
A benzothiazole pendant group conjugated to a picolylamine monoacetate chelate ligand coordinates in a tridentate manner with [MI(CO)3]+ (M = Re or 99mTc) to give stable, neutral complexes [M(CO)3L20] (Fig. 16). Human brain tissue, collected from subjects diagnosed with AD, when treated with [Re(CO)3L20] shows binding of the complex to Aβ plaques. Similarly, [99Tc(CO)3L20] also binds to Aβ plaques but no binding is detected when a shorter acetamide linker is used to connect the benzothiazole group with the chelate. Brain uptake of [99mTc(CO)3L20] administered through tail vein injection to Swiss Albino mice showed moderate brain uptake at 1 minute post injection (0.69% ID per g) combined with fast clearance (0.05% ID per g 15 min post injection; 0.02% ID per g 60 min post injection).103
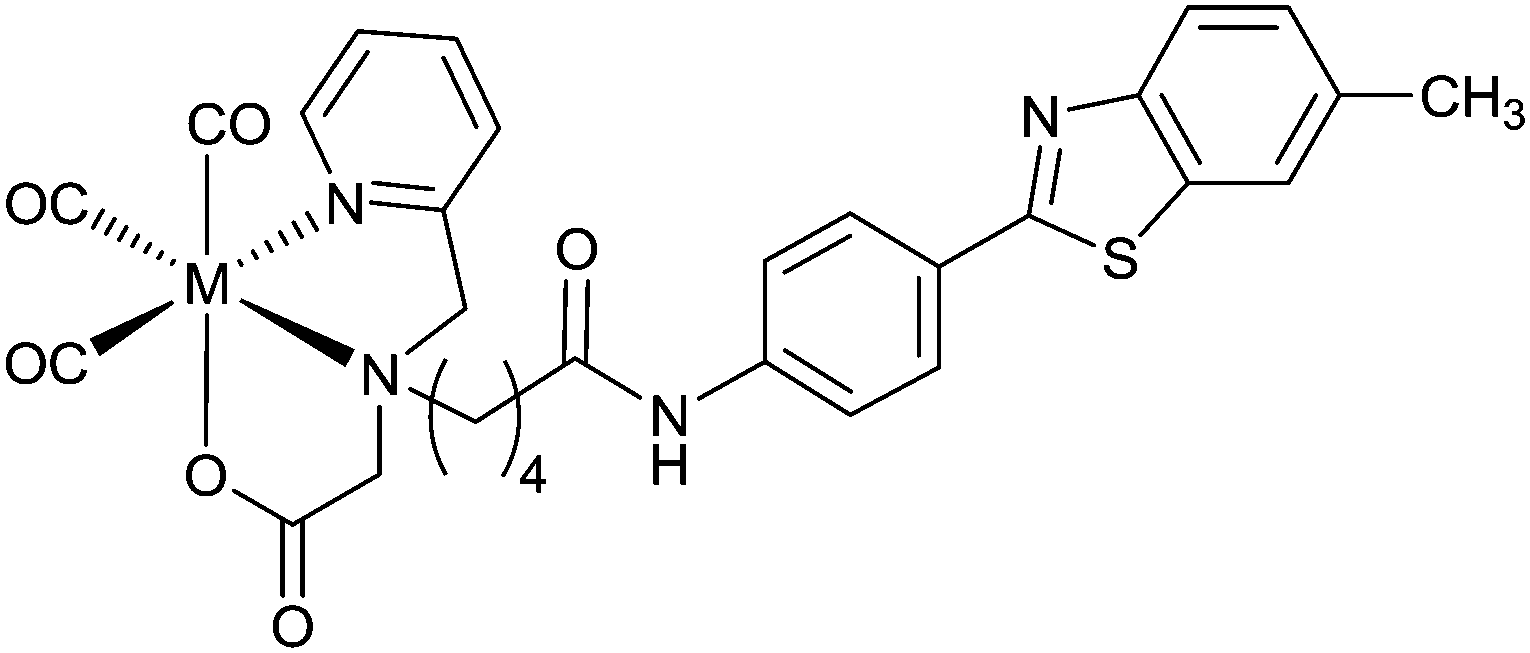 | ||
| Fig. 16 Structure of ML20 containing benzothiazole based pendant group conjugated to a tridentate chelate; M = ReI, 99TcI or 99mTcI. | ||
Selected substituted chalcones have a conjugated planar structure and bind to Aβ plaques. Replacement of a phenyl ring with a cyclopentadienyl functional group allows the synthesis of “piano stool” type complexes [CpM(CO)3] (where Cp = cyclopentadienyl and M = ReI or TcI). Rhenium complexes were synthesised by acetylation of (cyclopentadienyl)tricarbonylrhenium using acetyl chloride.91 The product was then reacted via base catalysed Claisen condensation with the chosen aromatic aldehyde enabling variation in the length of the alkenyl group within complexes (Fig. 17). The synthesis of 99mTc analogues, [99mTc(CO)3L21–23], can be accomplished by a two-step process: first, [CH3COCp99mTc(CO)3] is synthesised from pertechnetate by a double ligand transfer (DLT) using acetylferrocene and [Mn(CO)5Br]; the resultant complex is then reacted via a base catalysed Claisen condensation with the selected aromatic aldehydes to afford complexes in approximately 25% radiochemical yield over the two steps.
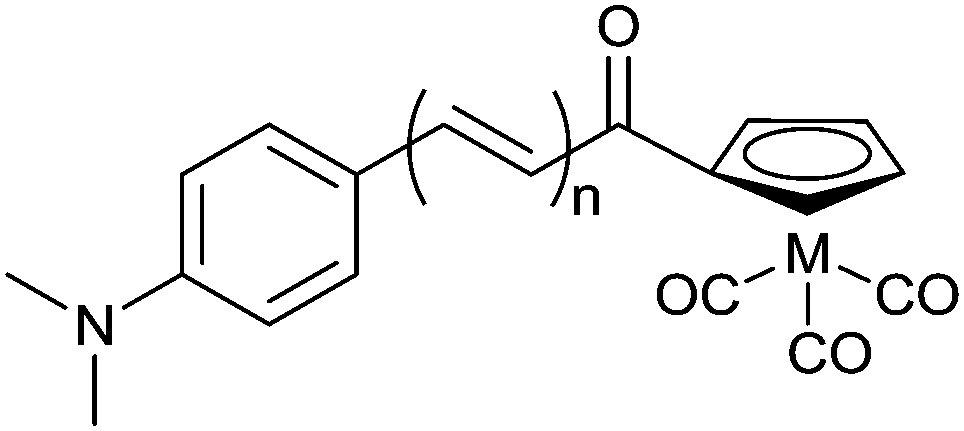 | ||
| Fig. 17 General structure of complexes containing chalcone inspired cyclopentadienyl ligand; M = ReI or 99mTcI; L21n = 1, L22n = 2, L23n = 3. | ||
The rhenium surrogates, [Re(CO)3L21–23], show moderate binding affinities toward Aβ42 aggregates. Binding affinity towards Aβ42 improves with increasing length of the alkenyl group to be at a maximum for [Re(CO)3L23] (Ki = 108 ± 16 nM). This trend is repeated for the binding of complexes [99mTc(CO)3L21–23] to Aβ42 aggregates, however, an inverse relationship between the length of alkenyl chain and brain uptake in normal ICR mice results in 99mTcL21 showing the highest uptake at 2 min post injection (4.10 ± 0.38% ID per g) and good clearance from the brain (brain2min/brain60min = 8.20).91
A mixed ligand system, whereby bidentate and monodentate ligands are used to complete the coordination sphere about fac-[M(CO)3]+ producing a “2+1” system, allows for the possibility of altering pharmacokinetics without modifying the binding motif. The α,β-unsaturated β-diketone curcumin (HL24) binds to Aβ plaques and is able to act as a bidentate ligand when reacted with fac-[M(CO)3(H2O)3]+ to form fac-[M(CO)3L24(H2O)] (M = Re or 99mTc). The remaining aqua ligand can be exchanged for imidazole, isocyanocyclohexane, and triphenylphosphine (Fig. 18).104,105 The large trans effect exerted by triphenylphosphine can lead to further substitution and formation of [Re(CO)2(PPh3)2L24]. The substitution chemistry is similar for 99mTc. The aqua ligand of the intermediate, [99mTc(CO)3L24(H2O)], is replaced by a phosphine ligand at room temperature to give [99mTc(CO)3L24(PPh3)] in 80% radiochemical yield, with 20% remaining as the intermediate complex. When incubated at 60 °C for 20 min, the reaction progresses to form cis–trans-[99mTc(CO)2(PPh3)2L24]. Both fac-[Re(CO)3L24(PPh3)] and cis–trans-[Re(CO)2(PPh3)2L24] bind to Aβ plaques in post-mortem human brain tissue.105
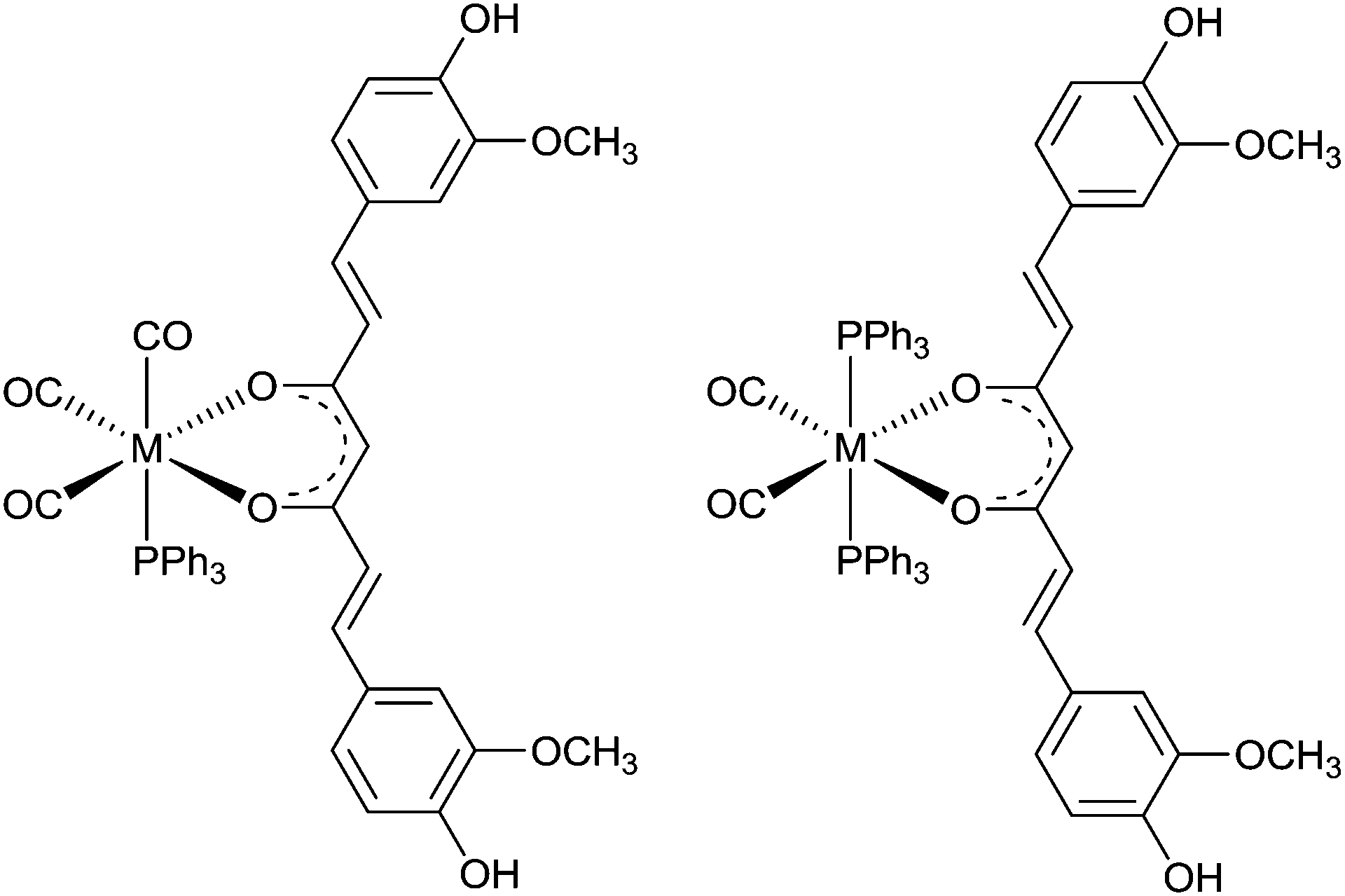 | ||
| Fig. 18 General structure of complexes containing curcumin (HL24); left: fac-[Re(CO)3 (PPh3) L24] a “2+1” complex, right cis–trans-[Re(CO)2(PPh3)2L24] “2+1+1” complex, M = ReI or 99mTcI. | ||
7 The interaction of phosphorescent ruthenium polypyridyl complexes with Aβ fibrils
Specifically designed phosphorescent metal complexes are an interesting alternative to conventional organic fluorescent probes of fibril formation due to their long-lived fluorescent life times due to the spin–orbit coupling promoted by heavy metal ions. The ruthenium polypyridyl complex [RuII(bpy)2(dppz)]2+ (where bpy = 2,2-bipyridine and dppz = dipyridophenazine) (Fig. 19a) binds to DNA which results in an enhancement of luminescence attributed to a metal–ligand charge-transfer (MLCT) from the ruthenium atom to a primarily ligand (dppz) based π* orbital.106 This 1MLCT excited state decays via intersystem crossing to a 3MLCT excited state primarily localised on the phenazine nitrogen atoms. When the complex is in aqueous environments water quenches this excited state by hydrogen bonding with the phenazine nitrogen atoms but when the dppz is shielded from water the complex is luminescent.107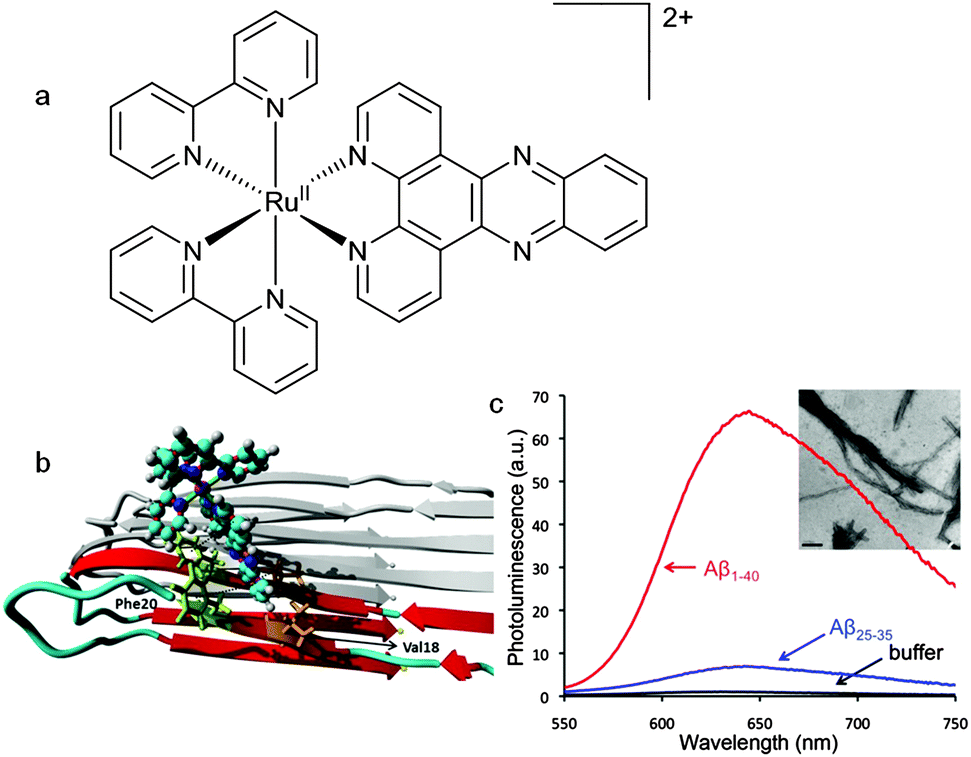 | ||
| Fig. 19 (a) Structure of the ruthenium polypyridyl complex, [Ru(bpy)2(dppz)]2+. (b) Representation of a molecular dynamics simulation showing binding of [Ru(bpy)2(dppz)]2+ to an Aβ fibril. The simulation predicts that binding occurs in a hydrophobic cleft formed between Val18 and Phe20. (c) Photoluminescence spectra of [Ru(bpy)2(dppz)]2+ incubated with Aβ40 (red), Aβ25–35 (blue), and buffer (black). Inset shows TEM image of Aβ25–35 fibrils (scale bar = 200 nm). Images adapted with permission from N. P. Cook, M. Ozbil, C. Katsampes, R. Prabhakar and A. A. Marti, J. Am. Chem. Soc., 2013, 135, 10810–10816. Copyright 2013 American Chemical Society. | ||
In similar fashion to its interaction with DNA the interaction of [Ru(bpy)2(dppz)]2+ with Aβ fibrils also results in a change in the polarity of the microenvironment resulting in enhanced luminescence (Fig. 19c).108 The use of a metal based luminescent probe with a long-lived excited state has potential advantages over more conventional dyes such as ThT. The relatively long photoluminescent lifetime of [Ru(bpy)2(dppz)]2+ bound to Aβ fibrils (185 ns) can be used to differentiate between the competitive binding of other molecules with shorter fluorescent lifetimes. The dissociation constant for the binding of [Ru(bpy)2dppz]2+ to Aβ fibrils is 2.1 μM with a binding stoichiometry of 2.6 Aβ monomers per [Ru(bpy)2dppz]2+. Computational methods that combines molecular docking and all atom molecular dynamics simulation predicted that a hydrophobic cleft between Val18 and Phe20 was a likely binding site with the side chains of these two amino acid residues interacting with the dppz functional group via CH–π and π–π interactions (Fig. 19b).109 The versatility and potential of this family of ruthenium complexes as unconventional probes of protein aggregation is highlighted by the use of the 1,10-phenanthroline analogue, [Ru(phen)2dppz]2+ (where phen = 1,10-phenanthroline), a probe to monitor the fibrilisation of α-syn, a protein associated with Parkinson's disease, in neuroglioma cells.110
8 Metal complexes to inhibit aggregation and toxicity of amyloid-β (Aβ)
Amyloid-β peptides possess moderate affinity for copper(II), copper(I) and zinc(II). The interaction of Aβ with CuII involves coordination of the metal ion to imidazole residues on His6, His13, and His14 and alters the aggregation and toxicity profiles of the peptide.28,31,33,111–115 The resulting CuII–Aβ complexes are redox active and lead to the production of toxic reactive oxygen species.116 Modification of the CuII and ZnII binding site of Aβ has the potential to mitigate toxicity and inhibit amyloid formation. An innovative approach of the metal binding properties of Aβ is to use metal complexes with kinetically inert metal ions to form stable coordinate bonds with the histidine residues that are involved in coordination to CuII and ZnII. The challenge is to achieve selectivity for the histidine residues and this has, in part, been achieved by using PtII complexes with hydrophobic phenanthroline ligands and two monodentate co-ligands poised for substitution reactions with the N-donors of the imidazole residues of histidine.The aggregation and neurotoxicity of Aβ is modulated by the addition of [PtIICl2(phen)] (where phen = 1,10-phenanthroline) and similar complexes (Fig. 20).117 These PtII complexes target the metal binding site of Aβ by exchange of the monodentate chloride ligands followed by coordination of the PtII ion to the imidazole functional group in the histidine residues to give [PtII(phen)–Aβ]2+ adducts. It is thought that the complexes target the N-terminal domain of Aβ through π–π interactions between the aromatic phen ligand and the aromatic residues of the peptide (Phe4, Tyr10 and Phe19) that span the CuII binding site. The coordination of the [Pt(phen)]2+ complexes to Aβ inhibits the aggregation of the peptide and leads to precipitation of amorphous aggregates rather than amyloid fibrils. The formation of the [PtII(phen)–Aβ]2+ adducts results inhibition of the neurotoxicity of Aβ and protection against Aβ induced synaptotoxicity in mouse hippocampal tissue.117 The non-covalent interactions between the aromatic phen ligand and aromatic residues of amino acids in the Aβ peptide lead to an acceleration in the rate of reaction as demonstrated by the reaction of [PtCl2(phen)] with the shorter variant Aβ16 (t1/2 ≈ 4 min) when compared to the platination of comparable N-donor ligands.118 The formation of [PtII(phen)–Aβ16]2+ adducts alters the CuII and ZnII binding properties of Aβ16 and thus changes the aggregation and toxicity profiles of the peptide.119 Following the reaction of Aβ42 with a water soluble sulfonated analogue of [PtCl2(phen)], [PtCl2(4,7-diphenyl-[1,10]phenanthroline disulfonate)], several products containing a ratio of 1:1 platinum to Aβ42 were identified by mass spectrometry. X-ray absorption spectroscopy in combination with theoretical calculations (density functional theory) suggested the major component was a square planar PtII complex, [PtII((4,7-diphenyl-[1,10]-phenanthroline disulfonate))(hist)Cl]+, and the EXAFS refinement gave bond lengths of Pt–N(phen), 1.993(5) Å, Pt–N(imidazole), 2.03(1) Å and Pt–Cl and 2.235(7) Å. In contrast to [PtCl2(phen)] and its analogues, the reaction of cis-[Pt(NH3)2Cl2] with Aβ42 results in a mixture of products that primarily involve coordination to the sulfur atom of Met35. Similar mixed RuII and PtII complexes also prevent the aggregation of Aβ42 as does a cyclometalated PtII complex, [Pt(Cl)(dimethylsulfoxide)(2-phenyl-5-methylpyridine)], which binds to Aβ28.120,121 The binding of the platinum complex to the peptide does not preclude the coordination of CuII to Aβ28 but does result in a complex where the CuII is in a different coordination environment.122 The interaction of this organometallic complex with this shorter form of the Aβ peptide also inhibits ZnII induced aggregation.123 A strategy of combining the Aβ binding ability of platinum complexes containing aromatic ligands with the copper(II) and zinc(II) binding properties of the tetraazamacrocyclic chelator cyclen has led to the synthesis of cyclen–Pt(bpy) conjugates (where bpy = 2,2-bipyridine). Upon interaction with Aβ40 the PtII ion of these conjugates coordinates to His14/13 and the macrocyclic ligand is able to sequester CuII and ZnII. The simultaneous metal chelation and coordinate bond modification of the peptide alter the aggregation of Aβ40.124
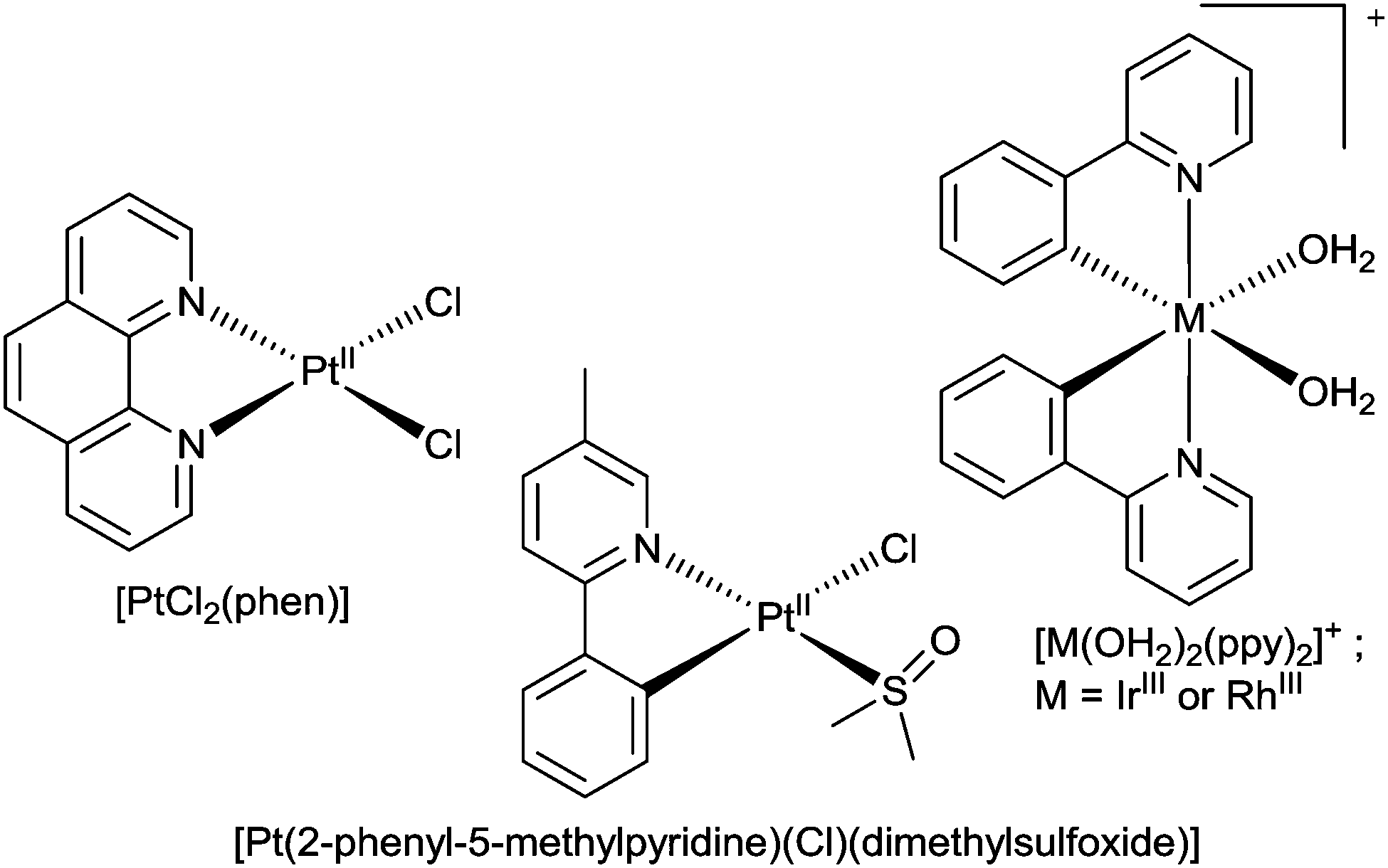 | ||
| Fig. 20 The structures of [PtCl2(phen)], [PtCl(dimethylsulfoxide)(2-phenyl-5-methylpyridine)] and [M(ppy)2(OH2)2]+ (where M = RhIII or IrIII). | ||
Each of these platinum(II) complexes are likely to have poor bioavailability and blood–brain barrier penetration and this has the potential to compromise their clinical development for the treatment of neurodegeneration. The desire to provide orally bioavailable platinum(II) complexes with the potential to alter Aβ aggregation led to the synthesis of a platinum(IV) complex with a specifically designed benzoimidazole quinoline ligand (L25) (Fig. 21). The idea of using a PtIV complex as a pro-drug activated by in vivo reduction to a PtII species was inspired by the similar approach used to make orally bio-available and less toxic platinum complexes for the treatment of cancer (Satraplatin). Complex [PtIVCl4L25] was administered orally to the APP/PS1 mouse model of amyloid pathology. Analysis of the brain tissue following treatment revealed a 40% reduction in Aβ42 levels and a reduction in Aβ plaques when compared to untreated animals (Fig. 21).125
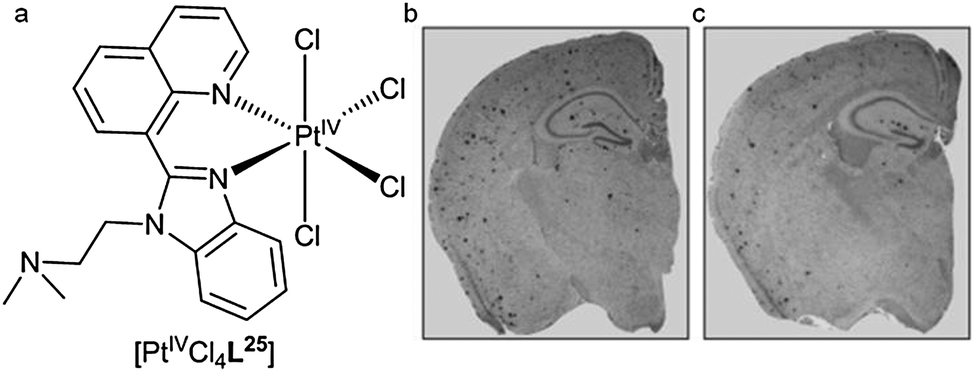 | ||
| Fig. 21 (a) Structure of [PtIVCl4L25]. Representative examples of brain tissue from transgenic APP/PS1 mice immunohistochemical stained for Aβ when (b) untreated or (c) treated with [PtIVCl4L25]. Images adapted with permission from V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378. Copyright 2013 John Wiley and Sons. | ||
The potential of metal complexes with kinetically inert d6 complexes to inhibit Aβ amyloid has been extended to IrIII, RhIII, and RuII complexes. Iridium(III) and rhodium(III) complexes with orthometallated phenylpyridine (ppy) ligands, [M(ppy)2(OH2)2]+ (where M = RhIII or IrIII) (Fig. 20), inhibit the aggregation of Aβ40 with [Rh(ppy)2(OH2)2]+ the most effective, inhibiting aggregation at concentrations of 5 μM. It is likely that exchange of the aqua ligands is followed by coordination of the metal ion to the imidazole side chains of the histidine residues in Aβ40. The complexes formed from [Ir(ppy)2(OH2)2]+ in the presence of Aβ40 are luminescent and the emission intensity (λem = 491 nm) is higher for aggregated [Aβ40–Ir(ppy)2]+ complexes when compared to the monomeric form suggesting this system could be used as an alternative to conventional fluorescent dyes for monitoring fibrilisation.126 A series of ruthenium(II) complexes also interact with Aβ peptides and pioneering studies have identified some interesting substitution chemistry. The reaction of fac-[Ru(CO)3Cl2(1,3-thiazole)] (Fig. 22) with Aβ28 results in a mixture where the predominant product identified by electrospray mass spectrometry is of the composition [Ru(CO)3–Aβ28]2+ and similar complexes were identified when longer Aβ42 replaced Aβ28. The coordination of the [Ru(CO)3]2+ fragment to Aβ28 results in dramatic changes to the 1H NMR spectrum with significant broadening of signals attributed to the three histidine residues and tyrosine-10 implicating the histidine residues in coordination to the RuII.127 A ruthenium(III) monoanionic complex with two 2-aminothiazole N-bound ligands and four chloride ligands (PMRU20) (Fig. 22) was found to offer protection against Aβ induced toxicity in rat primary cortical neurons. Interestingly two other structurally related RuIII complexes, NAMI A and KP1019 (Fig. 22), offered little to no protection. The difference in reactivity between these complexes is intriguing and warrants further investigation.128 The reaction of selected ruthenium complexes with proteins involved in the pathology of type II diabetes (human islet amyloid polypeptide) and prion diseases (PrP106–126) also results in significant changes to the aggregation of the respective proteins. This further highlights the potential of coordinate bond formation with kinetically inert metal ions to act as novel inhibitors of amyloid formation.129,130
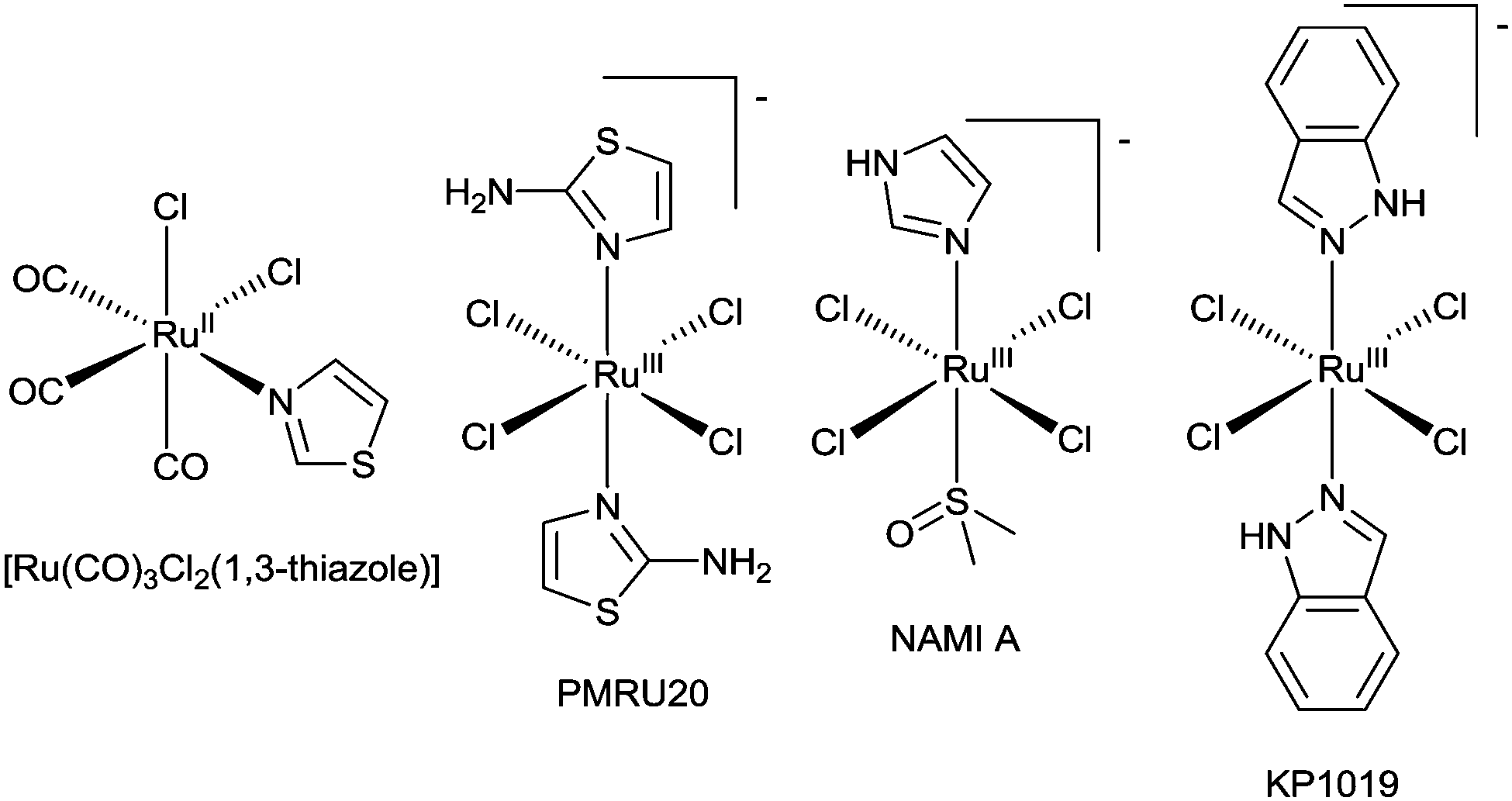 | ||
| Fig. 22 Structure of ruthenium complexes whose reaction with Aβ peptides have been investigated. | ||
9 Conclusions
The full details of the role Aβ plaques, oligomers, tau and neurofibrillary tangles play in cognitive impairment in Alzheimer's disease remains a mystery. What is known about the extracellular amyloid deposits evident in the cortex of subjects with the disease is that they contain relatively high concentrations of aggregated amyloid-β protein, a 39–43 amino acid fragment derived from the amyloid precursor protein. Our understanding of the structure and actual function of both Aβ and APP remains incomplete. It is possible that there is still much to learn about the generation, clearance and toxicity of Aβ as well as the molecular dynamics involving Aβ monomers, oligomers and plaques. Significant progress in the last decade has led to the development of molecular tracers that can offer insight into the deposition of amyloid in the brain of living patients using non-invasive PET imaging. The results of pioneering multi-centre studies using PET tracers to detect amyloid in large patient cohorts are emerging and providing valuable insights into the relationship between amyloid and cognition.131–133There are several radioactive isotopes of copper that have the potential to be of use in PET imaging and technetium-99m continues to be the radioisotope of choice for SPECT imaging. In principle, ligands designed to bind to amyloid plaques and form stable copper or technetium complexes have the potential to extend the capabilities of amyloid imaging. A new ligand based on a bis(thiosemicarbazone) framework with appended plaque targeting functional group and hybrid thiosemicarbazone–styrylpyridine ligands have shown some promise as ligands for copper radiopharmaceuticals. A wide range of Tc complexes designed to bind to Aβ plaques is discussed in the review. The “piano stool” complex, [99mTcIL21] (Fig. 21), shows significant potential and warrants further investigation.
Luminescent ruthenium and iridium metal complexes with long fluorescent lifetimes that undergo significant changes to their electronic spectra upon binding to Aβ fibrils have potential as useful probes to gain increased insight into the molecular aspects of amyloid formation and aggregation. The interaction of the hydrophobic complex [Ru(bpy)2(dppz)]2+ with Aβ40 results in a dramatic change in the photoluminescence of the complex and the long fluorescent life-time could be of use in competitive binding assays with other small molecules. An innovative approach at altering the metal-binding properties, toxicity and aggregation profiles of Aβ is to use metal complexes of kinetically inert metal ions to form coordinate bonds to the histidine residues in the metal-binding domain of Aβ. Complexes with the kinetically inert ions, PtII, RuII, IrIII and RhIII all interact with Aβ peptides of varying length to give adducts that have altered toxicity and propensity to aggregate. The viability of metal complexes to be therapeutically useful inhibitors of amyloid formation and Aβ toxicity will be dependent on them satisfying the stringent and challenging requirements of therapeutic agents.134
Acknowledgements
Australian Research Council and National Health and Medical Research Council for financial support. Timothy Connell and Stacey Rudd (both University of Melbourne) for expert assistance in the preparation of this manuscript.References
- C. L. Masters, R. Cappai, K. J. Barnham and V. L. Villemagne, J. Neurochem., 2006, 97, 1700 CrossRef CAS PubMed.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245 CrossRef CAS.
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 120, 885 CrossRef CAS PubMed.
- D. J. Selkoe, Nat. Med., 2011, 17, 1060 CrossRef CAS PubMed.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535 CrossRef CAS PubMed.
- D. B. Teplow, N. D. Lazo, G. Bitan, S. Bernstein, T. Wyttenbach, M. T. Bowers, A. Baumketner, J.-E. Shea, B. Urbanc, L. Cruz, J. Borreguero and H. E. Stanley, Acc. Chem. Res., 2006, 39, 635 CrossRef CAS PubMed.
- D. M. Walsh and D. B. Teplow, Prog. Mol. Biol. Transl. Sci., 2012, 107, 101 CAS.
- R. Roychaudhuri, M. Yang, M. M. Hoshi and D. B. Teplow, J. Biol. Chem., 2009, 284, 4749 CrossRef CAS PubMed.
- J. Marx, Science, 2007, 316, 1416 CrossRef CAS PubMed.
- N. Okamura, M. T. Fodero-Tavoletti, Y. Kudo, C. C. Rowe, S. Furumoto, H. Arai, C. L. Masters, K. Yanai and V. L. Villemagne, Expert Opin. Med. Diagn., 2009, 3, 705 CrossRef CAS PubMed.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y.-S. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665 CrossRef CAS PubMed.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43 CrossRef CAS PubMed.
- L. R. Perez and K. J. Franz, Dalton Trans., 2010, 39, 2177 RSC.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, J. Neurochem., 2011, 119, 220 CrossRef CAS PubMed.
- H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, K. H. Thompson and C. Orvig, Angew. Chem., Int. Ed., 2007, 46, 1716 CrossRef CAS PubMed.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, J. Am. Chem. Soc., 2007, 129, 7453 CrossRef CAS PubMed.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663 CrossRef CAS PubMed.
- J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990 CrossRef CAS PubMed.
- M. R. Jones, E. L. Service, J. R. Thompson, M. C. P. Wang, I. J. Kimsey, A. S. DeToma, A. Ramamoorthy, M. H. Lim and T. Storr, Metallomics, 2012, 4, 910 RSC.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625 CrossRef CAS PubMed.
- A. Kochi, T. J. Eckroat, K. D. Green, A. S. Mayhoub, M. H. Lim and S. Garneau-Tsodikova, Chem. Sci., 2013, 4, 4137 RSC.
- Y. Liu, A. Kochi, A. S. Pithadia, S. Lee, Y. Nam, M. W. Beck, X. He, D. Lee and M. H. Lim, Inorg. Chem., 2013, 52, 8121 CrossRef CAS PubMed.
- T. Storr, L. E. Scott, M. L. Bowen, D. E. Green, K. H. Thompson, H. J. Schugar and C. Orvig, Dalton Trans., 2009, 3034 RSC.
- L. E. Scott, M. Telpoukhovskaia, C. Rodriguez-Rodriguez, M. Merkel, M. L. Bowen, B. D. G. Page, D. E. Green, T. Storr, F. Thomas, D. D. Allen, P. R. Lockman, B. O. Patrick, M. J. Adam and C. Orvig, Chem. Sci., 2011, 2, 642 RSC.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299 CrossRef CAS PubMed.
- D. S. Folk and K. J. Franz, J. Am. Chem. Soc., 2010, 132, 4994 CrossRef CAS PubMed.
- P. J. Crouch and K. J. Barnham, Acc. Chem. Res., 2012, 45, 1604 CrossRef CAS PubMed.
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 16, 67 CrossRef CAS PubMed.
- C. Rodriguez-Rodriguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308 CrossRef CAS.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856 CrossRef CAS PubMed.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836 RSC.
- K. J. Franz, Curr. Opin. Chem. Biol., 2013, 17, 143 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193 CrossRef CAS PubMed.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101 CrossRef CAS PubMed.
- C. L. Masters and K. Beyreuther, BMJ, 1998, 316, 446 CrossRef CAS PubMed.
- J. Hardy and J. Selkoe Dennis, Science, 2002, 297, 353 CrossRef CAS PubMed.
- M. T. Fodero-Tavoletti, V. L. Villemagne, C. C. Rowe, C. L. Masters, K. J. Barnham and R. Cappai, Int. J. Biochem. Cell Biol., 2011, 43, 1247 CrossRef CAS PubMed.
- S. S. Sisodia and P. H. St George-Hyslop, Nat. Rev. Neurosci., 2002, 3, 281 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333 CrossRef CAS PubMed.
- A. S. Cohen and E. Calkins, Nature, 1959, 183, 1202 CrossRef CAS PubMed.
- J. D. Sipe and A. S. Cohen, J. Struct. Biol., 2000, 130, 88 CrossRef CAS PubMed.
- G. G. Glenner, N. Engl. J. Med., 1980, 302, 1283 CrossRef CAS PubMed.
- T. Shirahama and A. S. Cohen, J. Cell Biol., 1967, 33, 679 CrossRef CAS PubMed.
- R. Tycko, Q. Rev. Biophys., 2006, 39, 1 CrossRef CAS PubMed.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342 CrossRef CAS PubMed.
- D. A. Kirschner, C. Abraham and D. J. Selkoe, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 503 CrossRef CAS.
- L. C. Serpell and J. M. Smith, J. Mol. Biol., 2000, 299, 225 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742 CrossRef CAS PubMed.
- O. N. Antzutkin, J. J. Balbach, R. D. Leapman, N. W. Rizzo, J. Reed and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13045 CrossRef CAS PubMed.
- J. J. Balbach, A. T. Petkova, N. A. Oyler, O. N. Antzutkin, D. J. Gordon, S. C. Meredith and R. Tycko, Biophys. J., 2002, 83, 1205 CrossRef CAS PubMed.
- O. N. Antzutkin, J. J. Balbach and R. Tycko, Biophys. J., 2003, 84, 3326 CrossRef CAS PubMed.
- A. T. Petkova, W.-M. Yau and R. Tycko, Biochemistry, 2006, 45, 498 CrossRef CAS PubMed.
- H. Dobeli, N. Draeger, G. Huber, P. Jakob, D. Schmidt, B. Seilheimer, D. Stuber, B. Wipf and M. Zulauf, Biotechnology, 1995, 13, 988 CrossRef CAS PubMed.
- K. K. Skeby, J. Soerensen and B. Schioett, J. Am. Chem. Soc., 2013, 135, 15114 CrossRef CAS PubMed.
- C. L. Masters and D. J. Selkoe, Cold Spring Harbor Perspect. Med., 2012, 2, a006262 Search PubMed.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741 CAS.
- J. C. Vickers, T. C. Dickson, P. A. Adlard, H. L. Saunders, C. E. King and G. McCormack, Prog. Neurobiol., 2000, 60, 139 CrossRef CAS PubMed.
- T. C. Dickson and J. C. Vickers, Neuroscience, 2001, 105, 99 CrossRef CAS PubMed.
- W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergstrom, I. Savitcheva, G.-f. Huang, S. Estrada, B. Ausen, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Langstrom, Ann. Neurol., 2004, 55, 306 CrossRef CAS PubMed.
- C. C. Rowe, S. Pejoska, R. S. Mulligan, G. Jones, J. G. Chan, S. Svensson, Z. Cselenyi, C. L. Masters and V. L. Villemagne, J. Nucl. Med., 2013, 54, 880 CrossRef CAS PubMed.
- U. S. F. a. D. Adminsitration, News & Events, U.S. Food and Drug Adminsitration, 2013 Search PubMed.
- H. F. Kung, S. R. Choi, W. Qu, W. Zhang and D. Skovronsky, J. Med. Chem., 2010, 53, 933 CrossRef CAS PubMed.
- N. S. Mason, C. A. Mathis and W. E. Klunk, J. Labelled Compd. Radiopharm., 2013, 56, 89 CrossRef CAS PubMed.
- P. S. Vassar and C. F. A. Culling, AMA Arch. Pathol., 1959, 68, 487 CAS.
- V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, K. K. Turoverov and I. M. Kuznetsova, J. Phys. Chem., 2007, 111, 4829 CrossRef CAS PubMed.
- E. S. Voropai, M. P. Samtsov, K. N. Kaplevskii, A. A. Maskevich, V. I. Stepuro, O. I. Povarova, I. M. Kuznetsova, K. K. Turoverov, A. L. Fink and V. N. Uverskii, J. Appl. Spectrosc., 2003, 70, 868 CrossRef CAS.
- W. Dzwolak and M. Pecul, FEBS Lett., 2005, 579, 6601 CrossRef CAS PubMed.
- A. Srivastava, P. K. Singh, M. Kumbhakar, T. Mukherjee, S. Chattopadyay, H. Pal and S. Nath, Eur. J. Chem., 2010, 16, 9257 CrossRef CAS PubMed.
- A. I. Sulatskaya, A. A. Maskevich, I. M. Kuznetsova, V. N. Uversky and K. K. Turoverov, PLoS One, 2010, 5, 1 Search PubMed.
- H. LeVine, Amyloid, 2005, 12, 5 CrossRef CAS PubMed.
- A. Lockhart, L. Ye, D. B. Judd, A. T. Merritt, P. N. Lowe, J. L. Morgenstern, G. Hong, A. D. Gee and J. Brown, J. Biol. Chem., 2005, 280, 7677 CrossRef CAS PubMed.
- C. Wu, Z. Wang, H. Lei, Y. Duan, M. T. Bowers and J. Shea, J. Mol. Biol., 2008, 384, 718 CrossRef CAS PubMed.
- P. J. Blower, J. S. Lewis and J. Zweit, Nucl. Med. Biol., 1996, 23, 957 CrossRef CAS PubMed.
- S. V. Smith, J. Inorg. Biochem., 2004, 98, 1874 CrossRef CAS PubMed.
- Production and Selection of Metal PET Radioisotopes for Molecular Imaging, ed. S. V. Smith, M. Jones and V. Holmes, InTech, 2011 Search PubMed.
- D. H. Petering, Biochem. Pharmacol., 1974, 23, 567 CrossRef CAS PubMed.
- M. A. Green, D. L. Klippenstein and J. R. Tennison, J. Nucl. Med., 1988, 29, 1549 CAS.
- K. A. Price, P. J. Crouch, I. Volitakis, B. M. Paterson, S. Lim, P. S. Donnelly and A. R. White, Inorg. Chem., 2011, 50, 9594 CrossRef CAS PubMed.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40, 3005 RSC.
- M. T. Fodero-Tavoletti, V. L. Villemagne, B. M. Paterson, A. R. White, Q.-X. Li, J. Camakaris, G. O'Keefe, R. Cappai, K. J. Barnham and P. S. Donnelly, J. Alzheimer's Dis., 2010, 20, 49 CAS.
- Y. Fujibayashi, H. Taniuchi, Y. Yonekura, H. Ohtani, J. Konishi and A. Yokoyama, J. Nucl. Med., 1997, 38, 1155 CAS.
- J. L. J. Dearling, J. S. Lewis, D. W. McCarthy, M. J. Welch and P. J. Blower, J. Chem. Soc., Chem. Commun., 1998, 2531 RSC.
- A. L. Vavere and J. S. Lewis, Dalton Trans., 2007, 4893 RSC.
- A. R. Cowley, J. R. Dilworth, P. S. Donnelly, E. Labisbal and A. Sousa, J. Am. Chem. Soc., 2002, 124, 5270 CrossRef CAS PubMed.
- T. R. Wallhaus, J. Lacy, J. Whang, M. A. Green, R. J. Nickles and C. K. Stone, J. Nucl. Med., 1998, 39, 1958 CAS.
- M. A. Green, C. J. Mathias, M. J. Welch, A. H. McGuire, D. Perry, F. Fernandez-Rubio, J. S. Perlmutter, M. E. Raichle and S. R. Bergmann, J. Nucl. Med., 1990, 31, 1989 CAS.
- S. Lim, B. M. Paterson, M. T. Fodero-Tavoletti, G. J. O'Keefe, R. Cappai, K. J. Barnham, V. L. Villemagne and P. S. Donnelly, Chem. Commun., 2010, 46, 5437 RSC.
- J. L. Hickey, S. C. Lim, D. J. Hayne, B. M. Paterson, J. M. White, V. L. Villemagne, P. Roselt, D. Binns, C. Cullinane, C. M. Jeffery, R. I. Price, K. J. Barnham and P. S. Donnelly, J. Am. Chem. Soc., 2013, 135, 16120 CrossRef CAS PubMed.
- M. Ono and H. Saji, Int. J. Mol. Imaging, 2011, 543267 Search PubMed.
- Z. Li, M. Cui, J. Dai, X. Wang, P. Yu, Y. Yang, J. Jia, H. Fu, M. Ono, H. Jia, H. Saji and B. Liu, J. Med. Chem., 2013, 56, 471 CrossRef CAS PubMed.
- J. L. Hickey and P. S. Donnelly, Coord. Chem. Rev., 2012, 256, 2367 CrossRef CAS.
- G. Bandoli, F. Tisato, A. Dolmella and S. Agostini, Coord. Chem. Rev., 2006, 250, 561 CrossRef CAS.
- G. Bandoli, A. Dolmella, M. Porchia, F. Refosco and F. Tisato, Coord. Chem. Rev., 2001, 214, 43 CrossRef CAS.
- F. Tisato, F. Refosco and G. Bandoli, Coord. Chem. Rev., 1994, 135/136, 325 CrossRef CAS.
- X. Wang, M. Cui, P. Yu, Z. Li, Y. Yang, H. Jia and B. Liu, Bioorg. Med. Chem. Lett., 2012, 22, 4327 CrossRef CAS PubMed.
- Y. Cheng, M. Ono, H. Kimura, M. Ueda and H. Saji, J. Med. Chem., 2012, 55, 2279 CrossRef CAS PubMed.
- Y. Yang, M. Cui, B. Jin, X. Wang, Z. Li, P. Yu, J. Jia, H. Fu, H. Jia and B. Liu, Eur. J. Med. Chem., 2013, 64, 90 CrossRef CAS PubMed.
- J. Pan, N. S. Mason, M. L. Debnath, C. A. Mathis, W. E. Klunk and K.-S. Lin, Bioorg. Med. Chem. Lett., 2013, 23, 1720 CrossRef CAS PubMed.
- R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram and T. A. Kaden, J. Am. Chem. Soc., 1998, 120, 7987 CrossRef CAS.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135 CrossRef CAS PubMed.
- G. R. Morais, A. Paulo and I. Santos, Organometallics, 2012, 31, 5693 CrossRef CAS.
- M. Sagnou, S. Tzanopoulou, C. P. Raptopoulou, V. Psycharis, H. Braband, R. Alberto, I. C. Pirmettis, M. Papadopoulos and M. Pelecanou, Eur. J. Inorg. Chem., 2012, 4279 CrossRef CAS.
- M. Sagnou, D. Benaki, C. Triantis, T. Tsotakos, V. Psycharis, C. P. Raptopoulou, I. Pirmettis, M. Papadopoulos and M. Pelecanou, Inorg. Chem., 2011, 50, 1295 CrossRef CAS PubMed.
- C. Triantis, T. Tsotakos, C. Tsoukalas, M. Sagnou, C. Raptopoulou, A. Terzis, V. Psycharis, M. Pelecanou, I. Pirmettis and M. Papadopoulos, Inorg. Chem., 2013, 52, 12995 CrossRef CAS PubMed.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS.
- C. Turro, S. H. Bossmann, Y. Jenkins, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 9026 CrossRef CAS.
- N. P. Cook, V. Torres, D. Jain and A. A. Marti, J. Am. Chem. Soc., 2011, 133, 11121 CrossRef CAS PubMed.
- N. P. Cook, M. Ozbil, C. Katsampes, R. Prabhakar and A. A. Marti, J. Am. Chem. Soc., 2013, 135, 10810 CrossRef CAS PubMed.
- N. P. Cook, K. Kilpatrick, L. Segatori and A. A. Marti, J. Am. Chem. Soc., 2012, 134, 20776 CrossRef CAS PubMed.
- Z.-G. Xiao and A. G. Wedd, Nat. Prod. Rep., 2010, 27, 768 RSC.
- S. C. Drew and K. J. Barnham, Acc. Chem. Res., 2011, 44, 1146 CrossRef CAS PubMed.
- C. Hureau, Coord. Chem. Rev., 2012, 256, 2164 CrossRef CAS.
- I. Zawisza, M. Rozga and W. Bal, Coord. Chem. Rev., 2012, 256, 2297 CrossRef CAS.
- B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, J. Biol. Chem., 2002, 277, 40302 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813 CrossRef CAS PubMed.
- G. Ma, F. Huang, X. Pu, L. Jia, T. Jiang, L. Li and Y. Liu, Chem.–Eur. J., 2011, 17, 11657 CrossRef CAS PubMed.
- G. Ma, E. Wang, H. Wei, K. Wei, P. Zhu and Y. Liu, Metallomics, 2013, 5, 879 RSC.
- V. A. Streltsov, V. Chandana Epa, S. A. James, Q. I. Churches, J. M. Caine, V. B. Kenche and K. J. Barnham, Chem. Commun., 2013, 49, 11364 RSC.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691 CrossRef CAS PubMed.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404 RSC.
- F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130 RSC.
- X. Wang, X. Wang, C. Zhang, Y. Jiao and Z. Guo, Chem. Sci., 2012, 3, 1304 RSC.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374 CrossRef CAS PubMed.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917 RSC.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720 CrossRef CAS PubMed.
- L. Messori, M. Camarri, T. Ferraro, C. Gabbiani and D. Franceschini, ACS Med. Chem. Lett., 2013, 4, 329 CrossRef CAS PubMed.
- L. He, X. Wang, C. Zhao, H. Wang and W. Du, Metallomics, 2013, 5, 1599 RSC.
- X. Wang, B. Zhang, C. Zhao, Y. Wang, L. He, M. Cui, X. Zhu and W. Du, J. Inorg. Biochem., 2013, 128, 1 CrossRef CAS PubMed.
- V. L. Villemagne, S. Burnham, P. Bourgeat, B. Brown, K. A. Ellis, O. Salvado, C. Szoeke, S. L. Macaulay, R. Martins, P. Maruff, D. Ames, C. C. Rowe and C. L. Masters, Lancet Neurol., 2013, 12, 357 CrossRef CAS PubMed.
- R. J. Bateman, C. Xiong, T. L. S. Benzinger, A. M. Fagan, A. Goate, N. C. Fox, D. S. Marcus, N. J. Cairns, X. Xie, T. M. Blazey, D. M. Holtzman, A. Santacruz, V. Buckles, A. Oliver, K. Moulder, P. S. Aisen, B. Ghetti, W. E. Klunk, E. McDade, R. N. Martins, C. L. Masters, R. Mayeux, J. M. Ringman, M. N. Rossor, P. R. Schofield, R. A. Sperling, S. Salloway and J. C. Morris, N. Engl. J. Med., 2012, 367, 795 CrossRef CAS PubMed.
- C. R. Jack, Jr., H. J. Wiste, T. G. Lesnick, S. D. Weigand, D. S. Knopman, P. Vemuri, V. S. Pankratz, M. L. Senjem, J. L. Gunter, M. M. Mielke, V. J. Lowe, B. F. Boeve and R. C. Petersen, Neurology, 2013, 80, 890 CrossRef PubMed.
- D. J. Selkoe, Ann. Neurol., 2013, 74, 328 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |