 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in zinc–air batteries
Yanguang
Li
*a and
Hongjie
Dai
*b
aInstitute of Functional Nano & Soft Materials (FUNSOM) & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou 215123, China. E-mail: yanguang@suda.edu.cn
bDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: hdai@stanford.edu
First published on 13th June 2014
Abstract
Zinc–air is a century-old battery technology but has attracted revived interest recently. With larger storage capacity at a fraction of the cost compared to lithium-ion, zinc–air batteries clearly represent one of the most viable future options to powering electric vehicles. However, some technical problems associated with them have yet to be resolved. In this review, we present the fundamentals, challenges and latest exciting advances related to zinc–air research. Detailed discussion will be organized around the individual components of the system – from zinc electrodes, electrolytes, and separators to air electrodes and oxygen electrocatalysts in sequential order for both primary and electrically/mechanically rechargeable types. The detrimental effect of CO2 on battery performance is also emphasized, and possible solutions summarized. Finally, other metal–air batteries are briefly overviewed and compared in favor of zinc–air.
 Yanguang Li | Yanguang Li is a professor in the Institute of Functional Nano & Soft Materials (FUNSOM) at Soochow University, China. He received his BS in Chemistry from Fudan University, China in 2005; and completed his PhD in 2010 under Professor Yiying Wu in Chemistry at Ohio State University. He then moved to Stanford University and completed his post-doctoral training in the Department of Chemistry under the supervision of Professor Hongjie Dai. His research focuses on nanostructured functional materials for energy applications, particularly in the realm of electrocatalysis, advanced batteries and photo water splitting. |
 Hongjie Dai | Hongjie Dai is the J. G. Jackson and C. J. Wood Professor of Chemistry at Stanford University. His research interfaces chemistry, physics, materials science and the biological and medical sciences. Thus far, his group has made advances in the basic science of carbon nanotubes and graphene and potential applications in the areas of nanoelectronics, nanobiotechnology, nanomedicine, energy storage and catalysis. He has published more than 200 journal papers that have been cited over 50 |
1. Introduction
Our society has been in transition from a fossil fuel based economy to a clean energy economy. This gradual but inevitable process is being accelerated by recent active research worldwide on sustainable energy harvesting, conversion and storage. Batteries have long been recognized for their capacity to efficiently convert and store electrical energy.1–10 They now find use in a myriad of applications extending from portable electronic devices, grid-scale energy storage to electric vehicles. Of the many different types of batteries marketed so far, lithium-ion technology has dominated the consumer market since its advent by virtue of its high specific energy and power density.1–10In the last five years or so, there has been a strong global incentive to develop electric vehicles (EVs) – starting from hybrid EVs to plug-in EVs and ultimately to pure EVs – powered by state-of-the-art lithium-ion batteries, as a move to reduce foreign oil dependence and mitigate green gas emission.11 Unfortunately EVs have achieved little market penetration so far. In 2012, EV sales in the United States market accounted for only 0.1% of over 14 million sold U. S. vehicles.12 This was to a great extent due to the high cost and insufficient energy density of current EV batteries. At 400–800 $ kW−1 h−1 currently,13 a lithium-ion battery pack alone would cost over $30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for a 240 mile range passenger vehicle, equal to the cost of an entire gasoline-powered car. Tremendous research efforts have been dedicated to increasing the energy density and lowering the cost of EV batteries. Recently, the US Department of Energy launched the “EV Everywhere Grand Challenge” as an initiative for improved batteries with dramatically reduced cost and weight, aimed at producing EVs that are as affordable as today's gasoline-powered vehicles.14
000 for a 240 mile range passenger vehicle, equal to the cost of an entire gasoline-powered car. Tremendous research efforts have been dedicated to increasing the energy density and lowering the cost of EV batteries. Recently, the US Department of Energy launched the “EV Everywhere Grand Challenge” as an initiative for improved batteries with dramatically reduced cost and weight, aimed at producing EVs that are as affordable as today's gasoline-powered vehicles.14
As one of the proposed post lithium-ion technologies, metal–air batteries have received revived interest recently. These systems feature the electrochemical coupling of a metal negative electrode to an air-breathing positive electrode through a suitable electrolyte.1 Metal–air batteries are between traditional batteries and fuel cells. They have the design features of traditional batteries in which a metal is used as the negative electrode. They also have similarities to conventional fuel cells in that their porous positive electrode structure requires a continuous and inexhaustible oxygen supply from the surrounding air as the reactant, making possible very high theoretical energy densities – about 2–10 folds higher than those of lithium-ion batteries.1
Among the different types of metal–air batteries, aqueous zinc–air is a relatively mature technology and holds the greatest promise for future energy applications. Its primary batteries have been known to the scientific community since the late nineteenth century.15 Commercial products started to emerge in the 1930s.1 Zinc–air batteries have a high theoretical energy density of 1086 Wh kg−1 (including oxygen), about five times higher than the current lithium-ion technology. They can potentially be manufactured at very low cost (<10 $ kW−1 h−1 estimated,16 about two orders of magnitude lower than lithium-ion). For many applications, zinc–air batteries offer the highest available energy density of any primary battery systems.1 They have frequently been advocated as the most viable option, both technically and economically, to replace lithium-ion batteries for future EV applications.17 Despite their early start and great potential, the development of zinc–air batteries has been impeded by problems associated with the metal electrode and air catalyst. So far, primary zinc–air batteries have been most successfully implemented for medical and telecommunication applications.1 They are noted for their high energy densities but low power output capability (<10 mW for hearing aid button cells) due to the inefficiency of air catalysts available. In addition, the development of electrically rechargeable zinc–air batteries with an extended cycle life has been troubled by non-uniform zinc dissolution and deposition, and the lack of satisfactory bifunctional air catalysts.
Some previous papers have provided excellent reviews on zinc–air batteries, covering either individual components or the whole system.18–22 Nevertheless, in light of the latest increased research activity, especially on rechargeable batteries, an up-to-date account of the current status and challenges of zinc–air technology has become highly necessary. In this review, we focus on the more recent progress and technical issues with respect to the battery components, electrode materials and air catalysts of zinc–air batteries. We start with a brief introduction of the zinc–air battery configuration and operation principle, followed by a sequential discussion of the zinc electrode, electrolyte, separator and air electrode for both primary and rechargeable types. Further, mechanically rechargeable zinc–air batteries and their regeneration using renewable solar energy are reviewed. The detrimental effect of atmospheric CO2 on the air electrode and battery performance is also emphasized, and possible solutions summarized. Finally, other types of metal–air batteries are briefly introduced and compared before the concluding remarks on the alluring promise of zinc–air technology. With these, we aim to provide readers with a timely snapshot of this rapidly developing area.
2. Battery configuration and operation principle
Fig. 1 schematically illustrates the basic structure of a primary zinc–air battery. It is comprised of a negative zinc electrode, a membrane separator and a positive air electrode assembled together in an alkaline electrolyte. Upon battery discharge, the oxidation of zinc occurs, giving rise to soluble zincate ions (i.e. Zn(OH)42−).1,18 This process usually proceeds until they are supersaturated in the electrolyte, after which the zincate ions decompose to insoluble zinc oxide as shown by the equations below.| Negative electrode: Zn + 4OH− → Zn(OH)42− + 2e− |
| Zn(OH)42− → ZnO + H2O + 2OH− |
| Positive electrode: O2 + 4e− + 2H2O → 4OH− |
| Overall reaction: 2Zn + O2 → 2ZnO |
| Parasitic reaction: Zn + 2H2O → Zn(OH)2 + H2 |
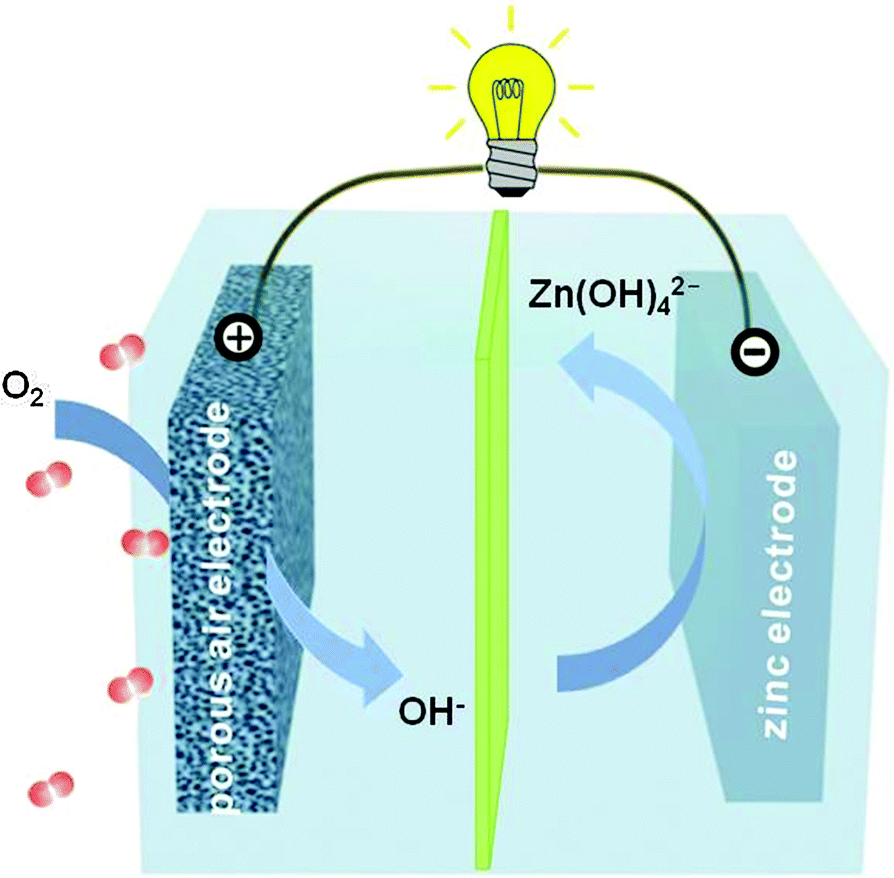 | ||
| Fig. 1 Schematic principle of operation for zinc–air batteries. | ||
In electrically rechargeable zinc–air batteries, the aforementioned electrochemical reactions are reversed during recharge with zinc metals plated at the negative electrode and oxygen evolving at the positive electrode. Zinc is the most active metal that can be plated from an aqueous electrolyte.1 However, its cyclability is typically poor because of the high solubility of its discharge product (i.e. zincate) in alkaline electrolytes and its escape from the negative electrode vicinity. Upon recharge, the reluctance of zincate to fully return to the same location at the electrode surface triggers electrode shape change or dendritic growth, which gradually degrade the battery performance, or even more seriously, short out the battery.1,21 Furthermore, electrically rechargeable zinc–air batteries rely on bifunctional air electrodes that are capable of both oxygen reduction and evolution electrocatalysis.19,20,22 The requirement of bifunctionality imposes strong criteria on the selection of catalyst materials. So far few candidates have been able to meet the stringent demands of both high activity and long durability.
Zinc–air batteries have a standard potential of 1.65 V. In practice, their working voltages are markedly lower, typically <1.2 V in order to get considerable discharge current densities. For rechargeable batteries, electrochemical reactions can't be reverted until a large charging voltage of 2.0 V or higher is applied. Significant deviation of both charge and discharge voltages from the equilibrium value are mostly contributed by the substantial overpotentials of oxygen electrocatalysis at the positive electrode.18 As a result, electrically rechargeable zinc–air batteries usually have a low round-trip energy efficiency of <60%.
Besides challenges with positive and negative electrode materials, a major operating constraint to zinc–air batteries as well as to alkaline fuel cells is their sensitivity to the CO2 concentration in the feed gas stream. The reaction of CO2 with electrolyte leads to the formation of carbonates by the following reactions, which decreases the electrolyte conductivity. Precipitation of carbonates on the air electrode also clogs pores, negatively affecting the performance of the air electrodes and batteries. This issue is discussed in detail in Section 8.
| 2KOH + CO2 → K2CO3 + H2O and/or KOH + CO2 → KHCO3 |
3. Zinc electrode
Since the invention of the first battery by Volta in 1796, metallic zinc has been the negative electrode material of choice for many primary systems such as zinc–carbon, zinc–manganese dioxide, zinc–nickel and zinc–air.1 It possesses a unique set of attributes including low equivalent weight, reversibility, high specific energy density, abundance and low toxicity, and is the most electropositive metal that is relatively stable in aqueous and alkaline media without significant corrosion.1,25 In many commercial zinc batteries, zinc electrode materials are generally a gelled mixture of granulated zinc powders in the range of 50–200 mesh with some additives.26,27 The shape or morphology of the zinc granules has been found to be of great importance in achieving better inter-particle contact and lowering internal electrical resistance in the negative electrode pack.1 In principle, high surface area zinc particles are preferred for better electrochemical performance. Durkot and co-workers reported that a zinc electrode with a large proportion of fine zinc powders (>200 mesh) had increased high-rate discharge characteristics.26 Oyama and co-workers favored a well-balanced combination of coarse and fine particles as a tradeoff between high-rate performance and self-corrosion.28 In addition to powders, other high-surface-area zinc electrode materials such as spheres, flakes, ribbons, fibers, dendrites and foams have been explored.29–35 Zhang developed a spin cast method to produce zinc fibers, which can be further processed into different physical forms like sheets, rods, bars and plates with various thickness and lengths (Fig. 2).31,32 The fibrous electrodes are advantageous for large-sized alkaline or zinc–air battery systems due to their good electrical conductivity, mechanical stability and design flexibility for controlling mass distribution, porosity and effective surface area. A zinc–air battery using the fibrous zinc electrode provided ∼40% more capacity, ∼50% more energy and ∼30% more active material utilization at high discharging currents than a battery using a gelled powder electrode (Fig. 2d).31 Drillet and coworkers prepared zinc foams by consolidation of emulsified zinc suspensions.35 The open porosity was adjusted via controlling the dispersed phase concentration in the emulsions. Using these zinc foams as the metallic negative electrode, they demonstrated an energy density of 300–500 Wh kgZn−1.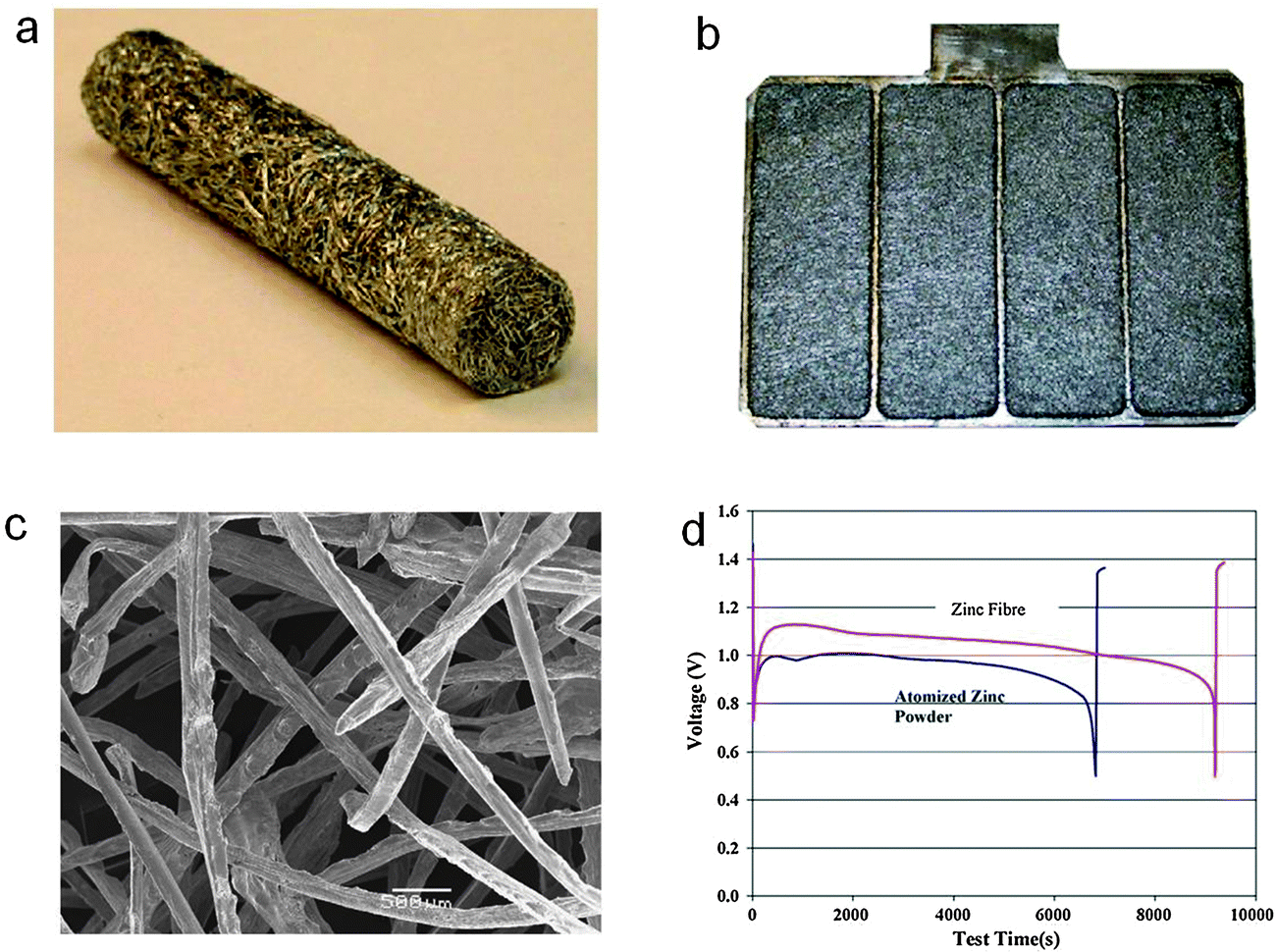 | ||
| Fig. 2 Fibrous zinc electrode for zinc–air batteries. (a) A rod form for an AA battery and (b) a plate form for a large mechanically rechargeable zinc–air battery; (c) scanning electron microscopy (SEM) image of the zinc fibers; (d) discharge curves of zinc–air batteries using a gelled atomized zinc powder electrode and a fibrous zinc electrode in a laboratory test cell at a current density of 100 mA cm−2. Reprinted with permission from ref. 31, copyright 2006, Elsevier. | ||
However, as the electrode surface area increases, the corrosion rate of the zinc electrode generally becomes more significant. This side reaction consumes electrolyte, lowers the utilization efficiency of the zinc electrode, and eventually shortens battery lifetime.21 Many efforts have been made to slow down or suppress self-corrosion. In the early days, a partial remedy was amalgamation of the zinc.36,37 With the addition of mercury, a new phase of zinc-rich amalgam was formed at the initiation of hydrogen evolution. Baugh and co-workers observed that when the mercury level exceeded 100 μg cm−2, corrosion of the zinc was inhibited to less than 40% of unamalgamated zinc.37 However, because of concerns over its high toxicity and negative environmental impact, the use of mercury nowadays is restricted in many products including batteries. Alternative solutions have been pursued (Table 1). Alloying zinc with other metals (e.g. lead, cadmium, bismuth, tin and indium) was found to stabilize the zinc electrode.38–41 Introducing additives such as silicates, surfactants and polymers could also alter the electrochemical properties of the zinc and suppress hydrogen gas generation to different extents.42–46 In addition, coating the zinc metal with other materials offered an effective strategy for improving the comprehensive properties of the negative electrode.47,48 Cho and co-workers treated the zinc surface with 0.1 wt% lithium boron oxide, and observed increased discharge capacity, reduced hydrogen generation and self-discharge.47 Lee and coworkers reported that coating zinc particles with an Al2O3 layer could also minimize the corrosion rate, and 0.25 wt% Al2O3 coated zinc electrode provided 50% longer discharging time over pristine zinc in 9 M KOH.48
| Modifications to the Zn electrode | Effects | Ref. |
|---|---|---|
| Alloys with Pb, Cd, Bi, Sn, In, Mg, Al or Ni | Suppress H2 generation, reduce dendrite formation, improve cycling reversibility | 39–41 |
| Surface coating with Al2O3 or lithium boron oxide | Suppress H2 generation and self-discharge | 47 and 48 |
| Inorganic additives: Ca(OH)2, Bi2O3, Tl2O3, Ga2O3, In2O3, In(OH)3, HgO, PbO, CdO or silicates | Suppress H2 evolution, reduce dendrite formation, improve discharge performance and cycling reversibility | 38, 43 and 52–60 |
| Polymer additives: ionomers, PEG, PMMA, polypyrrole, polyaniline, poly(vinyl acetate) or polycarbonate | Restrict the dissolution of discharge product, reduce dendrite formation and shape change | 45, 46, 49, 50, 51 and 68 |
| Surfactant additives: perfluorosurfactants, CTAB, TBABr, tetra-alkyl ammonium hydroxides, triethanolamine or lignosulfonate | Suppress H2 generation and electrode corrosion, reduce dendrite formation | 44 and 63–67 |
Making electrically rechargeable zinc–air batteries necessitates the development of cyclable zinc electrodes. The electrochemistry of zinc in alkaline electrolytes is easily reversible, but its non-uniform dissolution and deposition usually results in electrode shape change or dendritic growth during extensive charge–discharge cycling, which is detrimental to battery performance and cycle life.1,21 Different approaches have been attempted to mitigate these problems (Table 1). First of all, modifications to the electrode or electrolyte have been carried out so as to better retain the discharge product. Physically, these can be done through coating zinc electrodes with surface trapping layers.49–51 Vatsalarani and co-workers found that a fibrous network of polyaniline coating on a porous zinc electrode allowed the movement of hydroxide ions but restricted the diffusion of zincate ions.51 After 100 cycles, the coated electrode exhibited a uniform surface morphology, smoother than the untreated zinc electrode. The solubility of the zinc discharge products can also be reduced with chemical additives. Addition of calcium hydroxide to the zinc electrode or electrolyte has long been recognized as effective.52–54 It forms an insoluble compound with the zincate ions, thereby maintaining much of the zinc in a solid form in the proximity of the zinc electrode and allowing the interconversion between Zn(0) and Zn(II) at sufficiently high rates during battery charge–discharge cycles. Importantly, calcium hydroxide is insoluble in alkaline electrolytes, and therefore retains a uniform distribution as the battery is cycled. Calcium zincate itself has also been tested as a battery electrode material with much success.55–57 Other alkaline-earth metal hydroxides such as barium hydroxide and magnesium hydroxide as well as citrate, carbonate, borate, chromate, fluoride and silicate compounds have also been used to trap the discharge products.25
Furthermore, in order to suppress dendritic growth, McBreen and co-worker investigated Bi2O3, Tl2O3, Ga2O3, In2O3, HgO, PbO, CdO and In(OH)3 as electrode additives.58–60 It was proposed and proven by in situ XRD experiments that these oxides–hydroxides were reduced and formed an electronic network at the nanometer scale before zinc deposition, which could in turn enhance the electronic conductivity and polarizability of the electrode, improve the current distribution, and promote the formation of compact, thin zinc deposits.61,62 They also had high hydrogen overpotentials, and could inhibit electrochemical hydrogen evolution on the zinc electrode. Other than metal oxides–hydroxides, organic additives to the electrode or electrolyte have been investigated for suppressing dendritic initiation and propagation.63–67 These usually adsorb at the sites of rapid growth, and can ease the irregularity of the electrode surface. Banik and coworker observed that over a wide concentration range (100–10000 ppm), polyethylene glycol (PEG) in the electrolyte reduced the zinc electrodeposition kinetics and suppressed dendrite formation (Fig. 3).68 This was in accordance with their electrochemical modeling, which predicted an order of magnitude reduction in the zinc dendrite growth rates in the presence of a high concentration of PEG. More often, a combination of different types of additives is used so as to achieve the best cycling performance of the zinc electrode. Recently, S.C.P.S. of France developed a cyclable zinc electrode composed of a copper foam current collector filled with a mixture of zinc, ceramic electronic conductor (TiN) and a polymer binder.16 The ceramic conductor helps to retain zincate ions, allowing a uniform zinc deposition on charge. The resulting zinc electrode is capable of 800–1500 cycles without degradation.16
 | ||
| Fig. 3 Optical microscope images of zinc dendrites deposited on the tip of wire electrodes from 0.1 M ZnCl2 electrolyte with various concentrations of PEG. Experiments were performed potentiostatically at two operating potentials versus Ag/AgCl as indicated. Reprinted with permission from ref. 68, copyright 2013, The Electrochemical Society. | ||
4. Electrolyte
Except for a few scattered reports,69 zinc–air batteries mostly operate in alkaline media, such as KOH and NaOH, for the sake of higher activity of both the zinc electrode and air electrode. KOH is usually preferred over NaOH because of its better ionic conductivity, higher oxygen diffusion coefficients, and lower viscosity.70 Moreover, its reaction products with atmospheric CO2 – K2CO3 or KHCO3 – have higher solubilities than their sodium counterparts, and can therefore alleviate the carbonate precipitation problem which is a serious challenge for zinc–air batteries (see discussion in Section 8). Most commonly, 7 M or 30 wt% KOH solution is employed for its maximum electric conductivity.For open systems like zinc–air batteries, water loss from the liquid electrolytes is an important cause of performance degradation. They usually require regular topping up with water. It has been found that gelling of the electrolyte can help minimize water loss, and enhance battery performance and life. Hydroponics gel has the capacity to store solution 20–100 times its weight. It was first investigated as a gelling agent to immobilize KOH electrolyte for zinc–air batteries by Othman and coworkers.71,72 In a follow-up study, Mohamad demonstrated that a battery using 6 M KOH/hydroponics gel had an improved specific capacity of 657.5 mA h g−1 (789 W kg−1).73 Yang and coworker prepared a polymer gel electrolyte comprising of KOH dissolved in a polyethylene oxide (PEO)—polyvinyl alcohol (PVA) polymer matrix.74 It exhibited high ionic conductivity, mechanical strength and electrochemical stability suitable for solid-state zinc–air batteries. Zhu and coworkers solution polymerized acrylate-KOH–H2O at room temperature and derived an alkaline polymer gel electrolyte with a high specific conductivity of 0.288 S cm−1.75 Laboratory zinc–air, zinc–MnO2, and Ni–Cd batteries using this polymer gel electrolyte had almost the same performance characteristics as those with aqueous alkaline solution.
Recently, the viability of aprotic electrolytes, especially ionic liquids, for zinc–air batteries has been proposed and evaluated. These electrolytes are beneficial to the cyclability of the zinc electrodes. With them, dendrite-free zinc deposition has been demonstrated. They are also able to suppress the self-corrosion of zinc, slow down the drying-out of the electrolyte and eliminate its carbonation. Simons and co-workers investigated the electrodeposition and dissolution of Zn2+ in 1-ethyl-3-methylimidazolium dicyanamide ([emim][dca]) ionic liquid.76 They concluded that deposition from Zn(dca)2 in [emim][dca] containing 3 wt% H2O resulted in uniform, non-dendritic morphologies. The system had a high current density and efficiency appropriate for use in secondary zinc batteries. Xu and co-workers examined different ionic liquids, and observed a small overpotential for zinc redox chemistry in imidazolium cation and dicyanamide anion based ionic liquids.77 The exchange current densities derived from Tafel analysis were about 10−2 mA cm−2 in imidazolium based ionic liquids.
Despite being amenable to zinc electrochemistry, aprotic electrolytes in general do not work well with the current air electrodes designed specifically for aqueous solutions as will be discussed in more detail under Section 6.1. The oxygen electrocatalysis in aprotic electrolytes is drastically different from that in aqueous media. Studies have shown that cations in aprotic electrolytes strongly influence the reduction mechanism.78 Larger cations such as tetrabutylammonium (TBA) salts favor the reversible O2/O2− reaction, whereas smaller cations cause the irreversible reduction of oxygen, forming insoluble metal peroxides or superoxides.78 In non-aqueous lithium–air or sodium–air batteries, the precipitation of the discharge products at the air electrode is known to clog pores and thereby gradually shut off the reaction.79 Air electrodes also have distinct wettability in aprotic electrolytes. Harting and co-workers observed that ionic liquids were too viscous to effectively wet the gas-diffusing electrode, giving rise to a quick voltage decrease in the zinc–air battery during discharge. Using pure 1-butyl-3-methylimidazolium dicyanamide ([BMIM][dca]) as the electrolyte, they only obtained a discharge current density of 0.2 mA cm−2 at 0.8 V.80 So far, the performance of zinc air batteries using KOH as the electrolyte could not yet be approached by any aprotic electrolyte.
5. Separator
The separator in batteries physically keeps apart the positive and negative electrodes. It is an electrochemically inactive component but has a direct impact on key battery parameters. Separators for zinc–air batteries must have a low ionic resistance and high electrical resistance. They should have a high adsorption capacity of the alkaline electrolyte but display sufficient chemical resistance against the corrosive electrolyte and oxidation. Moreover, in light of the possible dendritic growth of zinc deposits in rechargeable batteries, separators should also have strong structural resistance to the perforation of zinc dendrites for the sake of safety and long-term reliability.Nonwoven polymeric separators made of polyethylene (PE), polypropylene (PP), polyvinyl alcohol (PVA) and polyamide have been widely employed in traditional alkaline and lithium batteries as well as metal–air batteries as reviewed by some excellent works.81,82 Their fibrous structure offers a high porosity (up to 75% pore volume) necessary for high electrolyte retention and low ionic resistance. Commercial zinc–air batteries typically use laminated nonwoven separators such as Celgard® 5550.81,82 They have a trilayer structure (PP/PE/PP) where the PP layer is designed to maintain the integrity of the separator, and the PE core is intended to shut down the battery if it overheats (Fig. 4). These separator membranes are often produced by a dry process in which a polyolefin resin is melted, thermally annealed to increase the size and amount of lamella crystallites, and then precisely stretched to form tightly ordered micropores.81,82 They are usually coated with surfactants for rapid electrolyte wetting. Recently, Wu and coworkers reported that sulfonation of nonwoven PE/PP separators could improve their hydrophilicity, and consequently double the ionic conductivity in alkaline electrolytes.83,84 Zinc–air batteries assembled from sulfonated nonwoven separators exhibited an improved peak power density of 27–38 mW cm−2.
 | ||
| Fig. 4 Laminated nonwoven separator membranes (Celgard® 5550) used in zinc–air batteries. (a) A cross-section image and (b) a top-view image of the membrane. Reproduced with permission from Celgard, LLC and ref. 82, copyright 2007, The Electrochemical Society. | ||
Saputra and coworkers investigated an inorganic microporous MCM-41 membrane as an alternative separator material.85 Compared to organic polymers, inorganic separators have the advantage of better thermal stability. The authors dip-coated a zinc electrode with a 5 μm MCM-41 membrane, and assembled it with a commercial air electrode in KOH. The full battery delivered a peak power density of 32 mW cm−2 and a volumetric energy density of 300 Wh L−1, comparable to commercial zinc–air button cells of equivalent size.85
One drawback of porous separators is that their open structure allows the easy permeation of soluble zincate ions from the negative electrode, giving rise to an increased polarization and decreased battery cycling efficiency. It would be beneficial to develop and employ anion-exchange membranes that are only selective to the passage of hydroxide ions. Dewi and coworkers prepared a poly(methylsufonio-1,4-phenylenethio-1,4-phenylene triflate) membrane with high anion selectivity.86 When used in zinc–air batteries, the polysulfonium separator permitted no detectable crossover of zinc species and effectively increased the discharge capacity by six times as compared to commercial Celgard separators. However, the use of anion-exchange membranes in zinc–air batteries is generally plagued by their insufficient long-term stability at high pHs – a problem similarly restricting their application in alkaline fuel cells.87 Regular anion-exchange membranes typically show a 10% loss in performance after only 1000 h, far from satisfactory for meeting necessary battery shelf life and cycle life.
6. Air electrode
6.1 Gas diffusion architecture
ORR mainly occurs at the triple phase boundary where the electrode is in intimate contact with the ion-conducting electrolyte and the gas phase. Oxygen diffusion through the gas phase is significantly faster than through the electrolyte since most solvents possess low oxygen solubility and diffusivity. The conventional electrode architectures cannot provide sufficient triple phase boundary to sustain continuous, high-current-density ORR, and therefore are rather ineffective as air electrodes. One way to increase the electrocatalytic current density is through the preparation of porous gas diffusion electrodes. When the earliest zinc–air battery was introduced to the public around 1878, it used a porous platinized carbon air electrode.1,15 In 1932, Heise and Schumacher of the National Carbon Company constructed alkaline zinc–air batteries with the carbon electrodes treated with wax to prevent flooding.1,88 This design has been used almost unchanged since then. Modern gas diffusion electrodes consist of several polytetrafluoroethylene (PTFE)-bonded carbon layers with two interpenetrating subsystems of hydrophilic and hydrophobic microchannels on current collecting materials (usually nickel or carbon).1,89–92 Their hydrophilic microchannels are properly wetted for providing access to the liquid electrolyte. The hydrophobic counterparts are designed to provide a barrier to prevent electrolyte penetration, and to facilitate fast oxygen diffusion from the atmosphere to the catalytic sites (Fig. 5). Zinc–air batteries have close interactions with the surrounding environment. Too low or high humidity may lead to the gradual drying-out of the electrolyte or flooding of the air electrode, either of which is detrimental to battery performance. Well balanced hydrophobicity and hydrophilicity in the air electrode can help slow down water evaporation loss or resist flooding under extreme conditions. They can be tuned by varying the carbon supports used, the carbon to PTFE ratio and the fabrication conditions.93,94 A successful example of a commercial GDE design is the Toray carbon paper first developed by Toray Industries, Inc in 1980. In this material, carbon fibers are bound together by layers of graphitized carbon to ensure high porosity and electrical conductivity, and then treated with PTFE as the wet-proofing agent for varying research purposes (Fig. 5b). In order to maximize the oxygen gas permeability, GDEs usually have to be as thin as possible. Zhu and co-workers reported a unique design of an ultrathin air electrode by entrapping carbon particles in a sinter-locked network of metal fibers. The electrode had a highly open porous structure with a thickness of 0.13–0.33 mm, 30–75% thinner than commercial products. As a result, it exhibited improved rate and pulse performance.95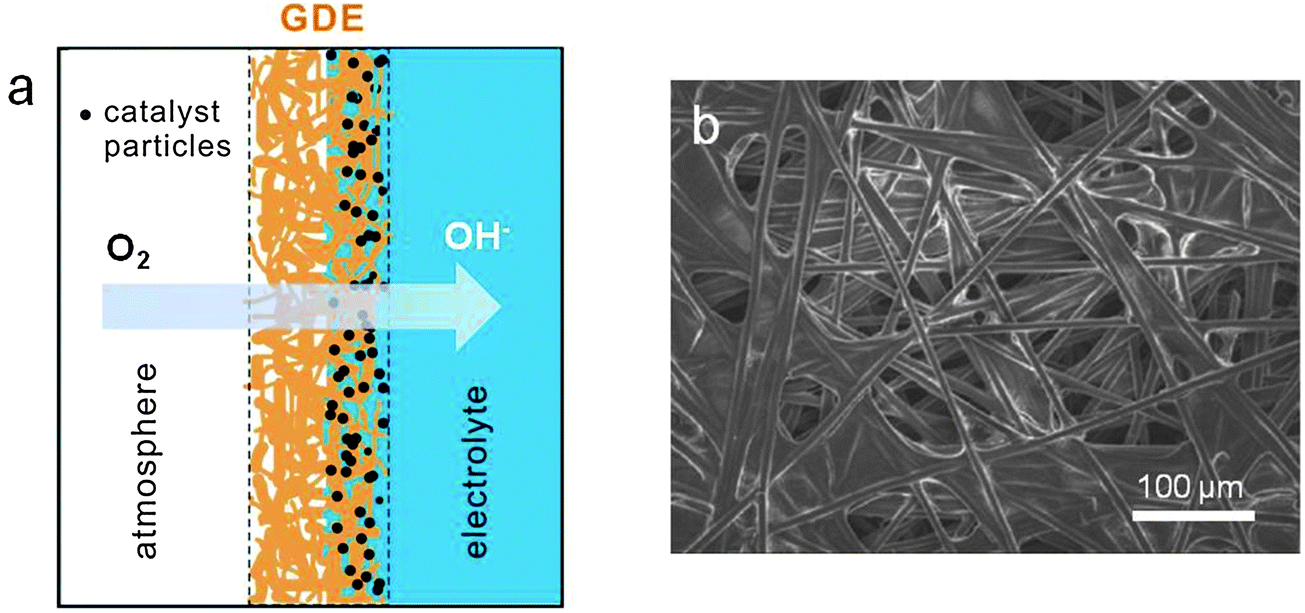 | ||
| Fig. 5 The structure of a gas diffusion electrode. (a) Schematic of a catalyst-loaded gas diffusion electrode in contact with the liquid electrolyte. It allows the easy permeation of oxygen from the surrounding atmosphere and its subsequent reduction on supported electrocatalysts. (b) SEM image of the commercial Toray carbon paper treated with hydrophobic PTFE. | ||
These PTFE-bonded multilayered GDEs work efficiently for most aqueous electrolytes. However they may not be optimal for aprotic or ionic liquid electrolytes used in non-aqueous metal–air batteries. Little research has been done so far to study the properties of conventional GDEs in non-aqueous media.96,97 While ionic liquids are generally too viscous to properly wet the air electrode,80 most organic solvents (e.g. acetonitrile) easily flood PTFE and the carbon electrode pores, pushing out the gas. Both circumstances lead to the greatly diminished triple phase boundary essential to ORR electrocatalysis. It is suggested that in non-aqueous lithium–air batteries, an electrode–electrolyte “two-phase reaction zone” is in effect. In this model, the air electrode is completely wetted with the liquid electrolyte, and only oxygen dissolved in the electrolyte actually participates in the charge-transfer process (Fig. 6b).97,98 This may partly explain the much smaller observed current density (about three orders of magnitude) non-aqueous metal–air batteries are able to deliver compared to aqueous versions. To tackle this problem, Balaish and co-workers impregnated an air electrode with perfluorocarbons as the special oxygen carriers, which are immiscible with aprotic electrolytes used in lithium–air batteries (Fig. 6c and d).98 Thus a formed artificial three phase boundary substantially increased the discharge capacity by over 50%. The same principle may also be applicable to the non-aqueous zinc–air batteries being proposed. Future research on GDEs must be directed toward the optimization of electrode architecture and properties together with electrolyte formulation and electrocatalysts.
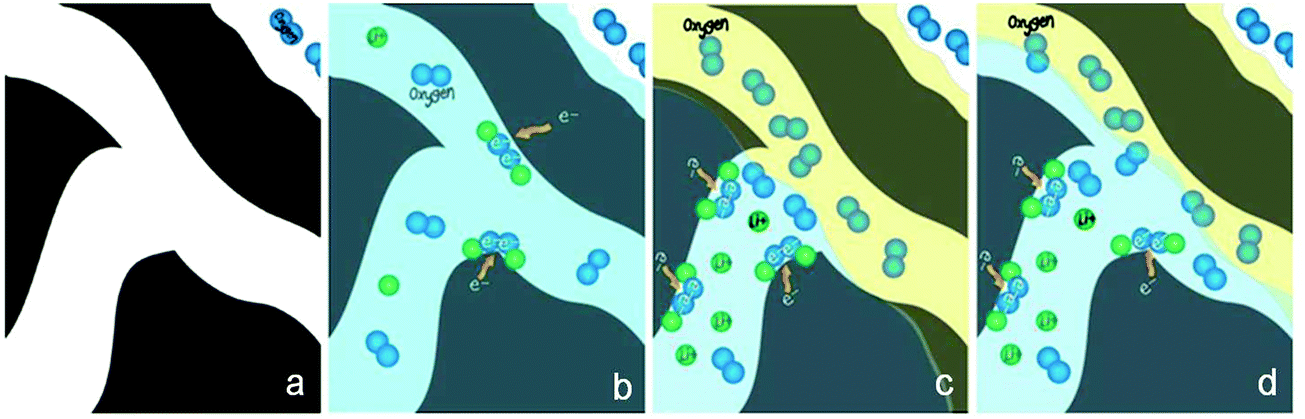 | ||
| Fig. 6 Proposed mechanism for the formation of an artificial three-phase reaction zone in a non-aqueous metal–air positive electrode. (a) Channels inside the pristine porous carbon; (b) channels are flooded with an organic electrolyte (in blue) thus, only dissolved oxygen is participating in the reduction reaction; (c and d) different possibilities of two distinct subsystem channels, formed as a result of the perfluorocarbon treatment. Reprinted with permission from ref. 98, copyright 2014, Wiley-VCH. | ||
6.2 Air catalysts
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (498 kJ mol−1), which is extremely hard to break electrochemically. This usually requires the assistance of electrocatalysts for the bond activation and cleavage. ORR electrocatalysis has been a heated topic of research and debate in the fields of fuel cells, metal–air batteries and chlor-alkali electrolysis.19,20,22–24 For metal–air batteries, ORR electrocatalysts have a decisive impact on battery power density, energy efficiency and lifetime. Many efforts have been invested in finding proper electrocatalysts to reduce ORR overpotential and enhance battery discharge performance (Table 2).19,20,22 Based on their functional ingredients, ORR catalysts can be grouped into the categories of precious metals, metal oxides or carbonaceous materials.
O bond (498 kJ mol−1), which is extremely hard to break electrochemically. This usually requires the assistance of electrocatalysts for the bond activation and cleavage. ORR electrocatalysis has been a heated topic of research and debate in the fields of fuel cells, metal–air batteries and chlor-alkali electrolysis.19,20,22–24 For metal–air batteries, ORR electrocatalysts have a decisive impact on battery power density, energy efficiency and lifetime. Many efforts have been invested in finding proper electrocatalysts to reduce ORR overpotential and enhance battery discharge performance (Table 2).19,20,22 Based on their functional ingredients, ORR catalysts can be grouped into the categories of precious metals, metal oxides or carbonaceous materials.
| Air catalysts | Electrolytes | Battery performancea | Ref. |
|---|---|---|---|
| a Polarization refers to the difference between charge and discharge voltages at the same working current density. | |||
| Unifunctional catalysts for primary batteries | |||
| Ag/C | 6.5 M KOH | Peak power density: 34 mW cm−2 at 35 °C and 72 mW cm−2 at 80 °C | 107 |
| Mn3O4/rGO-IL | Current density @ 1 V: 70 mA cm−2; peak power density: 120 mW cm−2 | 118 | |
| Mn3O4/ketjenblack | 6 M KOH | Current density @ 1 V: 120 mA cm−2; peak power density: ∼190 mW cm−2 | 119 |
| N-doped CNTs | 6 M KOH | Current density @ 1 V: 50 mA cm−2; peak power density: 69.5 mW cm−2 | 142 |
| N-doped porous carbon nanofibers | 6 M KOH | Current density @ 1 V: 150 mA cm−2; peak power density: 194 mW cm−2 | 143 |
| B,N-codoped nanodiamond | 6 M KOH | Current density @ 1 V: ∼15 mA cm−2; peak power density: 25 mW cm−2 | 141 |
| Pyrolyzed CoTMPP (Co–N–C) | 6 M KOH | Current density @ 1 V: 120 mA cm−2 | 157 |
| Pyrolyzed FeCo–EDA (FeCo–N–C) | 6 M KOH | Current density @ 1 V: 150 mA cm−2; peak power density: 232 mW cm−2 | 158 |
| Bifunctional catalysts for rechargeable batteries | |||
| MnO2–NCNT | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼1.5 V; cyclability: charged–discharged at ∼8 mA cm−2 with 300 s per step for 50 cycles, polarization increased ∼0.4 V at the end. | 167 |
| MnO2/Co3O4 | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼1.4 V; cyclability: charged–discharged at 15 mA cm−2 with 7 min per step for 60 cycles, polarization increased ∼0.3 V at the end. | 168 |
| CoMn2O4/N-rGO | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼0.7 V; cyclability: charged–discharged at 20 mA cm−2 with 300 s per step for 100 cycles, polarization increased ∼0.2 V at the end. | 170 |
| NiCo2O4 | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼0.7 V; cyclability: charged–discharged at 20 mA cm−2 with 20 min per step for 50 cycles, polarization increased ∼0.2 V at the end. | 169 |
| LaNiO3/NCNT | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼1.2 V; cyclability: charged–discharged at ∼17.6 mA cm−2 with 300 s per step for 75 cycles, polarization increased 0.1∼0.2 V at the end. | 179 |
| La2NiO4 | 6 M KOH | Voltage polarization @ j = 20 mA cm−2: ∼0.8 V; cyclability: charged–discharged at ∼25 mA cm−2 with 150 s per step for 20 cycles, polarization increased 0.4 V at the end. | 180 |
| Catalysts for three-electrode configuration | |||
| MnO2 (ORR) + stainless steel (OER) | 7 M KOH | Voltage polarization @ j = 20 mA cm−2: >0.9 V; cyclability: charged–discharged at 5–15 mA cm−2 with 12–15 h per step for ∼120 h, negligible voltage change at the end. | 16 |
| CoO/NCNT (ORR) + NiFe-LDH/CNT (OER) | 6 M KOH | Peak power density: ∼265 mW cm−2; voltage polarization @ j = 20 mA cm−2: 0.75 V; cyclability: charged–discharged at 20 mA cm−2 with 10 h per step for ∼200 h, negligible voltage change at the end. | 183 |
In the early days of zinc–air research, precious metal air catalysts were widely used.99–102 Platinum was the choice in the first zinc–air battery due to its high activity.15 It remains the benchmark in the evaluation of new alternative electrocatalysts.23 In addition to platinum, other precious metals such as palladium, silver, gold and their alloys have received considerable attention. Silver is particularly attractive due to its relatively low cost (only 1% of the price of platinum), decent activity (overpotential 50–100 mV larger than platinum) and better long-term stability.24,103–105 At very high base concentrations relevant to zinc–air batteries (e.g. 30 wt% KOH), silver was reported to even outperform platinum catalysts.106 This was attributed to the electronic structure of silver with a completely filled d band giving rise to a much less oxophilic surface than platinum,106 whereas a higher coverage of OH on the platinum surface is known to suppress its ORR kinetics with increasing pH.24 Accordingly, silver catalysts are usually favored over platinum for alkaline fuel cells and metal–air batteries.24,101,102,107 Using a Ag-amalgam electrode, Chireau from Yardney Electric Corporation (now Yardney Technical Products) demonstrated an energy density of 155 W h kg−1 for a 24 V, 20 Ah battery at a 3 h rate discharge.102 A more recent study showed that zinc–air batteries with a 10 wt% Ag/C air electrocatalyst delivered a peak power density of 34 mW cm−2 at 35 °C.107
Precious metal ORR catalysts have high electrocatalytic activities. However their widespread use is prohibited by their scarcity.23,24 Compared with precious metals, non-precious metal oxides are more desired as air catalysts due to their lower cost. A plethora of binary and ternary oxides in the form of spinel, perovskite or other structures have been extensively investigated (Table 2).108–115 Among them, manganese oxide (MnOx) is a particularly interesting candidate due to its rich oxidation states, chemical compositions and crystal structures. In fact, MnO2 is the most common ORR electrocatalyst in commercial zinc–air batteries. For example, during the fabrication of Duracell hearing-aid batteries, γ-MnO2 particles were intensely milled together with carbon until they were <10 μm in size, and then blended with PTFE. A uniform layer of the mixture was applied and pressed onto a metal grid to form the positive electrode sheet.116,117 Such batteries have high energy densities up to >400 Wh kg−1. Lee and co-workers devoted considerable efforts to fabricating air electrodes comprising of MnOx supported on conductive nanocarbon matrices.118,119 Using a polyol method, they synthesized amorphous MnOx nanowires on Ketjenblack (Fig. 7). A laboratory zinc–air battery based on this composite air electrode exhibited a peak power density of 190 mW cm−2 and a discharge capacity of 300 mA h g−1 (normalized to the mass of zinc), which was comparable to batteries equipped with the platinum catalyst (Fig. 7).119
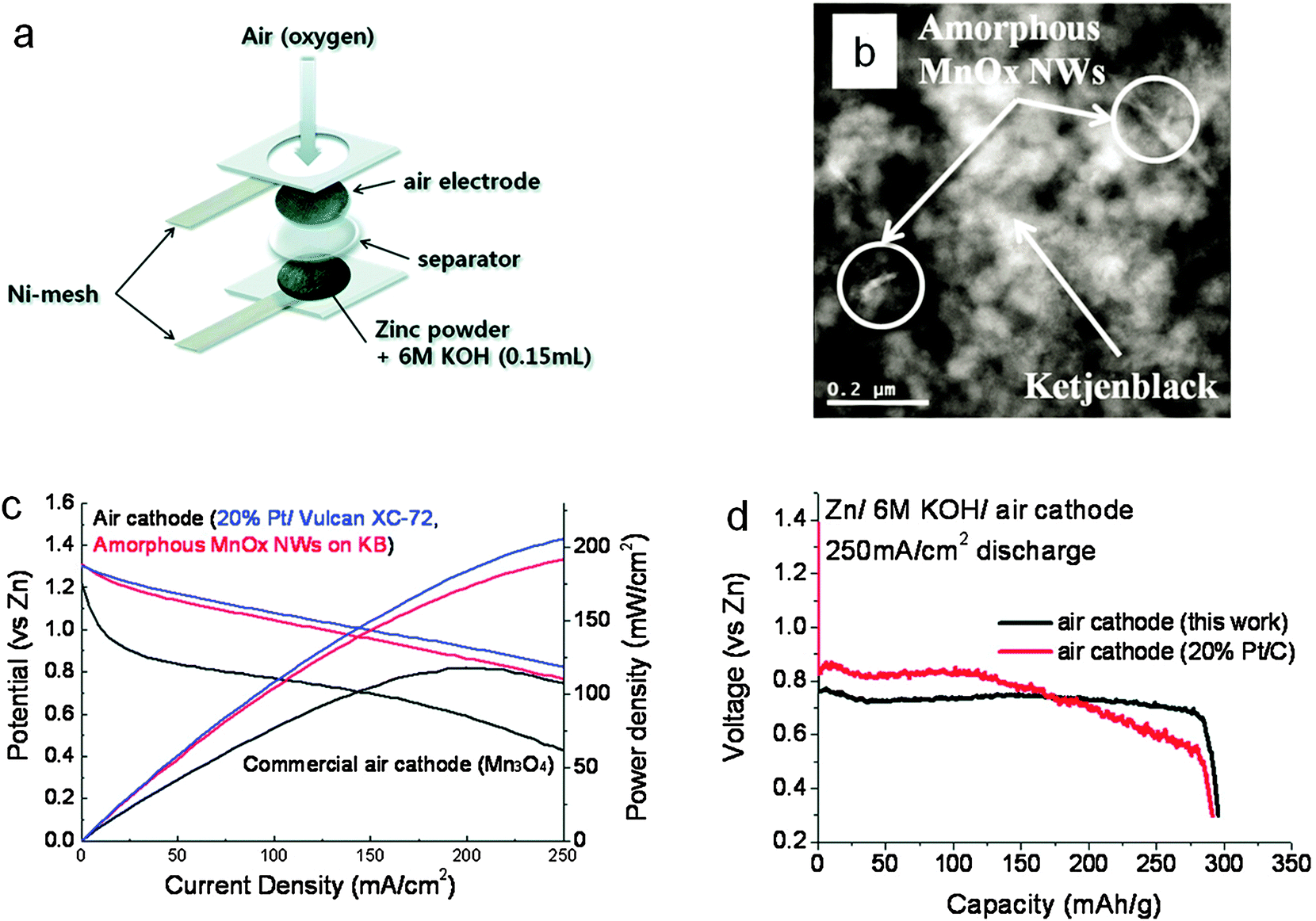 | ||
| Fig. 7 Amorphous manganese oxides supported on Ketjenblack for zinc–air batteries. (a) Schematic configuration of the battery; (b) transmission electron microscopy (TEM) image of amorphous MnOx nanowires on Ketjenback composites; (c) polarization curves and (d) discharge curves at 250 mA cm−2 of zinc–air batteries in comparison with the standard 20% Pt/C. Reprinted with permission from ref. 119, copyright 2011, American Chemical Society. | ||
Besides manganese oxide, perovskite oxides have also received tremendous attention for ORR electrocatalysis in alkaline media. Interestingly, it has been noted that their activity is mainly determined by the B-site cations whereas the A-site cations play a minor role.120,121 In a recent study, Suntivich and co-workers quantitatively correlated the intrinsic ORR activity with the σ*-filling of the B-site cations, and observed a volcano-shaped trend with eg electron number among the 15 different compositions they investigated.121 Perovskite-type oxides, particularly La0.6Ca0.4CoO3, have been seriously pursued by several different groups as air electrocatalysts for zinc–air batteries.122–124 Yamazoe and co-worker prepared large-surface-area La0.6Ca0.4CoO3 from an amorphous citrate precursor.122 Using this active catalyst, they constructed a zinc–air battery with a power density of ∼260 mW cm−2 at 290 mA cm−2.
Carbon based ORR electrocatalysts have also been the subject of recent scrutiny.125 In aqueous solutions, pristine carbon materials have poor inherent ORR activity with a predominant two-electron pathway to form peroxides.126–128 Chemical modification of the carbon surface, such as heteroatom doping, can enhance their ORR electrocatalytic activity via increasing the structural disorder or forming heteroatom functionalities.125,128 Metal-free nitrogen-doped carbon materials, including carbon black, mesoporous carbon, graphene sheets, nanofibers and nanotubes, have been intensively explored for ORR electrocatalysis,128–141 some of which have been applied to zinc–air batteries (Table 2).142,143 Nitrogen is introduced to these carbonaceous materials in pyridinic, pyrrolic or graphitic form by reacting with appropriate nitrogen precursors at elevated temperatures.125 The amount of each species depends on the detailed synthetic conditions such as the nitrogen precursors used and reaction temperatures.144–146
Metal-free nitrogen-doped carbon materials have respectable ORR activity in alkaline media, however they are still considerably distant from the platinum benchmark. A more active type of carbon-based ORR catalyst consists of metal–nitrogen–carbon (M–N–C) materials. These are usually synthesized via pyrolyzing metal (most commonly iron or cobalt) and nitrogen precursors on a carbon support at 800–1000 °C.147–152 Although the exact nature of the active sites in this type of catalyst remains elusive, there has been mounting evidence in support of the hypothesis that metal cations coordinated by nitrogen are responsible for the electrocatalytic activity.153–156 To design high performing catalysts requires a careful and creative choice of carbon support, metal and nitrogen precursors and synthetic conditions. Zhu and co-workers reported that pyrolyzing a tetramethoxyphenyl porphyrin cobalt complex on carbon at a mild temperature yielded a highly active Co–N–C type ORR electrocatalyst.157 A zinc–air battery loaded with a small amount (0.08 mg cm−2) of this catalyst delivered a current density of 120 mA cm−2 at 1 V. Similarly, a FeCo–N–C type catalyst was prepared by pyrolyzing ethylenediamine chelated iron and cobalt, and evaluated in zinc–air batteries.158 The resulting battery exhibited a peak power density of 232 mW cm−2.
There are several bifunctional catalysts that have been reported in the past.159,162 The electrocatalytic activity of metal oxide is associated with the ability of the cations to adopt different valency states, particularly when they form redox couples at the potential of oxygen reduction or evolution.163 Jaramillo and coworkers electrodeposited a nanostructured MnOx thin film on a glassy carbon substrate followed by heat treatment in air. This simple yet effective approach yielded a catalyst with remarkable bifunctional activity close or comparable to the best known precious metals.164 Co3O4 has long been known for its OER capability, but its ORR activity was generally poor. Liang and coworkers grew Co3O4 nanoparticles on graphene sheets and found the hybrid material exhibited a surprising ORR activity due to the synergistic effect between the two components while the OER activity of Co3O4 was further enhanced.112 This is one of the best bifunctional oxygen catalysts available currently. Other bifunctional spinel oxides such as CoxMn3−xO4, MnCo2O4, CuxCo3−xO4 and NiCo2O4 have also been investigated.114,115,165,166 NiCo2O4 nanopowders, CoMn2O4/N-doped graphene composite, MnO2 nanotubes/N-doped carbon nanotube composite, Co3O4 nanoparticle-modified MnO2 nanotubes, and so on, have been tested in full zinc–air batteries with decent performance (Table 2).167–170 Among the perovskite oxides, research has primarily focused on the composition La1−xAxMO3−δ (where A = Ca or Sr, and M = Co, Ni and Mn).171 It was found that La0.6Ca0.4CoO3 showed the most promising bifunctional catalytic activity.172,173 The influence of heat treatment, synthetic conditions and carbon support on the catalytic activity has been carefully studied.174–177 La0.6Ca0.4CoO3 is also one of the earliest bifunctional electrocatalysts developed and evaluated for electrically rechargeable zinc–air batteries.123,172,173,178 Muller and co-workers initiated serious efforts to build zinc–air batteries of different sizes using this catalyst in the 1990s.123,178 With a 200 cm2 zinc electrode and air electrode, they achieved a cycle life of about 1250 h in C/9 charge and discharge cycles. A battery module of ten such cells connected in series exhibited a practical working voltage of 20 V and specific energy density of 95 Wh kg−1.123 More recently, Chen and co-workers derived a core–corona structured bifunctional catalyst comprising of LaNiO3 particles supporting nitrogen-doped carbon nanotubes (Fig. 8).179 In this material, the core and corona were designed to catalyze OER and ORR, respectively, with high efficiency. A zinc air battery based on the catalyst displayed a discharge voltage of around 0.9 V and charge voltage of around 2.2 V at a current density of ∼17.6 mA cm−2 (or 24.5 A g−1 when normalized to the mass of the catalyst), and improved cycling performance compared with both Pt/C and LaNiO3 (Fig. 8d and e). Jung and co-workers fabricated a rechargeable zinc–air battery using La1.7Sr0.3NiO4 as the bifunctional catalyst.180 At 25 mA cm−2, the battery had an initial discharge voltage of around 1.2 V and charge voltage of around 2.0 V with a fair cycling stability.
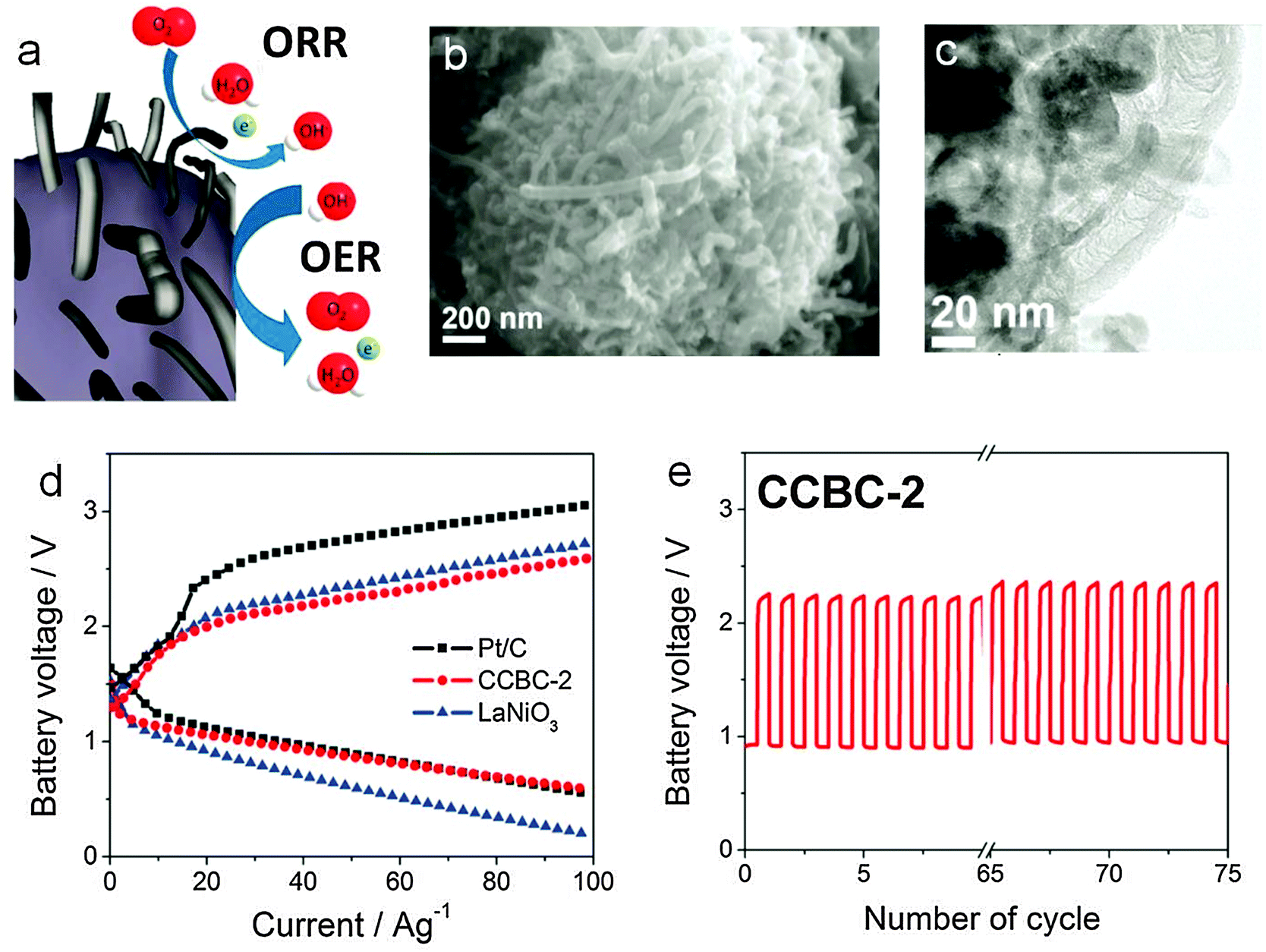 | ||
| Fig. 8 Core-corona structured bifunctional catalyst (CCBC) for rechargeable zinc–air batteries. (a) Schematic of the core–corona structure consisting of LaNiO3 centers supporting nitrogen-doped carbon nanotubes; (b) SEM and (c) TEM images of the CCBC; (d) charge and discharge polarization curves of Pt/C, CCBC-2 and LaNiO3; (e) charge–discharge cycling of CCBC-2 at 17.6 mA cm−2 and 10 min per cycle. Reprinted with permission from ref. 179, copyright 2012, American Chemical Society. | ||
Despite all this progress, the cycling stability of bifunctional electrodes in general is far from satisfactory. Degradation usually starts within a few numbers of cycles.167,168,179,180 It is believed that alternating reductive and oxidative environments during ORR–OER cycles may cause gradual but unrecoverable damage to catalyst materials, leading to deteriorating activities. For example, even though MnOx is often deemed as the most popular bifunctional catalyst, it has a strong propensity to get oxidized to MnO4− at OER potentials.16 Carbon as the catalyst support is also susceptible to electrochemical corrosion. Continuous efforts are therefore needed to design and prepare highly efficient and robust bifunctional electrocatalysts and electrodes for electrically rechargeable zinc–air batteries.
6.3 Three-electrode configuration
One way to bypass the cycling stability issue is to adopt a three-electrode configuration. In this design, a pair of air electrodes is employed for charge and discharge separately. The zinc electrode as the third electrode is located between them and connects to the ORR electrode for discharge, and switches to the OER electrode for charge. Consequently, exposure of ORR (or OER) electrocatalysts to the oxidative (or reductive) potentials is avoided, which dramatically improves their lifetime. Even though this configuration unavoidably increases the cell volume and decreases the corresponding volumetric energy and power density, it physically decouples ORR and OER, allowing easier electrocatalyst optimization and manipulation, and ultimately enhances the battery cycling stability.181A few examples of three-electrode zinc–air batteries are available in the literature (Table 2). Zhong developed a three-electrode battery consisting of an ORR electrode formed from cobalt tetramethoxylphenyl porphyrin (CoTMPP) and an OER electrode composed of 30 wt% Ag2O and 70 wt% LaNiO3.182 Evaluation suggested that such a 6 Ah battery maintained a stable performance for at least 30 cycles. Toussaint and co-workers used MnO2 mixed with carbon as the ORR electrode and a stainless steel grid as the OER electrode in a three-electrode rechargeable zinc–air battery.16 They demonstrated a 9 Ah cell with a specific energy density of 130 Wh kg−1. Remarkably, more than 5000 h of operation and 200 cycles were accomplished. Building on their latest breakthroughs in hybrid oxygen electrocatalysts, Li and co-workers employed cobalt oxide nanocrystals grown on nitrogen-doped carbon nanotubes (CoO/NCNT) as the ORR catalyst and nickel iron layered double hydroxide grown on carbon nanotubes (NiFe–LDH/CNT) as the OER catalyst for zinc–air batteries (Fig. 9a–c).183 In 6 M KOH, these two hybrid catalysts were superior to precious metal benchmarks (Pt/C and IrO2/C for ORR and OER, respectively) in both activity and durability. Primary batteries incorporating CoO/NCNT exhibited a peak power density of around 265 mW cm−2 and a large specific energy density of >700 Wh kgZn−1 at room temperature.183 Rechargeable batteries in the three-electrode configuration had a high round trip efficiency of 60–65%. When repeatedly charged and discharged at 20–50 mA cm−2 for a total of 240 h, the battery showed excellent cycling stability (Fig. 9d), much improved over the conventional two-electrode configuration in which the same air electrode was used for both ORR and OER.167,168,179,180,183
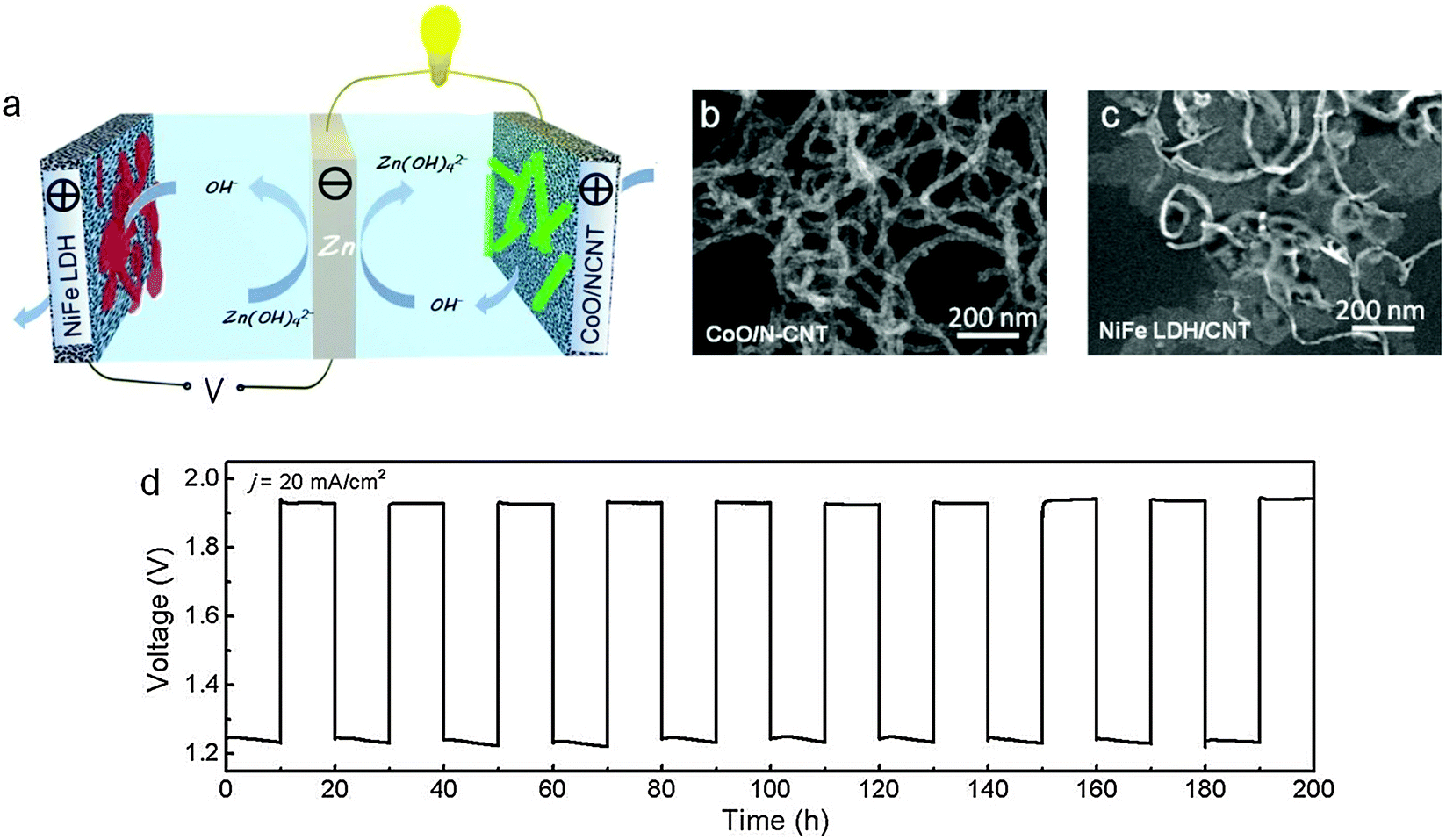 | ||
| Fig. 9 Electrically rechargeable zinc–air batteries based on high-performance hybrid electrocatalysts. (a) Schematic of a rechargeable battery in the three-electrode configuration; (b and c) TEM images of CoO/NCNT and NiFe LDH/CNT, respectively; (d) charge and discharge cycling performance of the battery at 20 mA cm−2 and a 20 h cycle period. Reprinted with permission from ref. 183, copyright 2013, Nature Publishing Group. | ||
7. Mechanically rechargeable zinc–air batteries
Another approach to overcome problems with electrical recharging is mechanically rechargeable batteries, where the spent zinc electrode and electrolyte are physically removed and replaced.1 Mechanically rechargeable batteries are refurbishable primary batteries, so they can use relatively simple unifunctional catalysts that need to operate only in a discharge mode, and can avoid the problematic dendritic zinc deposition most electrically rechargeable batteries encounter.In the late 1960s, mechanically rechargeable zinc–air batteries were considered for powering portable military electronic equipment by virtue of their high energy density and ease of recharging.1 Since the 1990s, there have been serious efforts to develop EVs powered by mechanically rechargeable zinc–air batteries.184–187 Cooper and co-workers in Lawrence Livermore National Laboratory conducted on-vehicle tests of nine 12-cell modules, and achieved a specific energy density of 130 Wh kg−1 and a power density of 65 W kg−1. A self-feeding feature was designed by them to add zinc pellets and the alkaline electrolyte, which helped reduce the refueling time to less than ten minutes for a 15 kW, 55 kW h battery unit.184 Electric Fuel Limited of Israel (now Arotech), on the other hand, employed mechanically replaceable zinc cassettes. Spent cassettes could be replaced in 30 s. This technology was evaluated in a four-passenger mini-jeep using a 32-cell module connected in series. The battery unit delivered a specific energy density of 191 Wh kg−1 and a power density of over 80 W kg−1.185
Based on these preliminary data, it seems that EVs powered by mechanically rechargeable zinc–air batteries are able to compete with conventional vehicles in price, performance, safety and quick refueling. However, the commercial success of a zinc–air based transportation system relies on the establishment of two other system elements: public refueling stations to replenish the negative electrode materials and electrolytes, as well as zinc regeneration facilities for centralized recycling of the discharge products (Fig. 10).187 The collected discharged fuel can be electrochemically regenerated by a zinc electrowinning process using off-peak electricity.187,188 Alternatively, zinc oxide can be reduced to zinc through its direct thermal dissociation or carbothermic reduction in a high-temperature chemical reactor heated by concentrated solar energy, as shown in Fig. 10.189 Carbothermic reduction of zinc oxide with a solid or gaseous (e.g. methane) carbon source is fundamentally less challenging, and can proceed at low temperatures of 1100–1200 °C. The product gases of zinc can be easily condensed out of the off-gas mixture containing zinc.189 Several reactor prototypes have been built and evaluated. In Europe, a pilot plant with a concentrated solar power input of approximately 300 kW has been set up. It successfully operates with zinc production rates of up to 50 kg h−1.189 The use of solar energy offers a sustainable option for the recharge of spent zinc–air batteries.
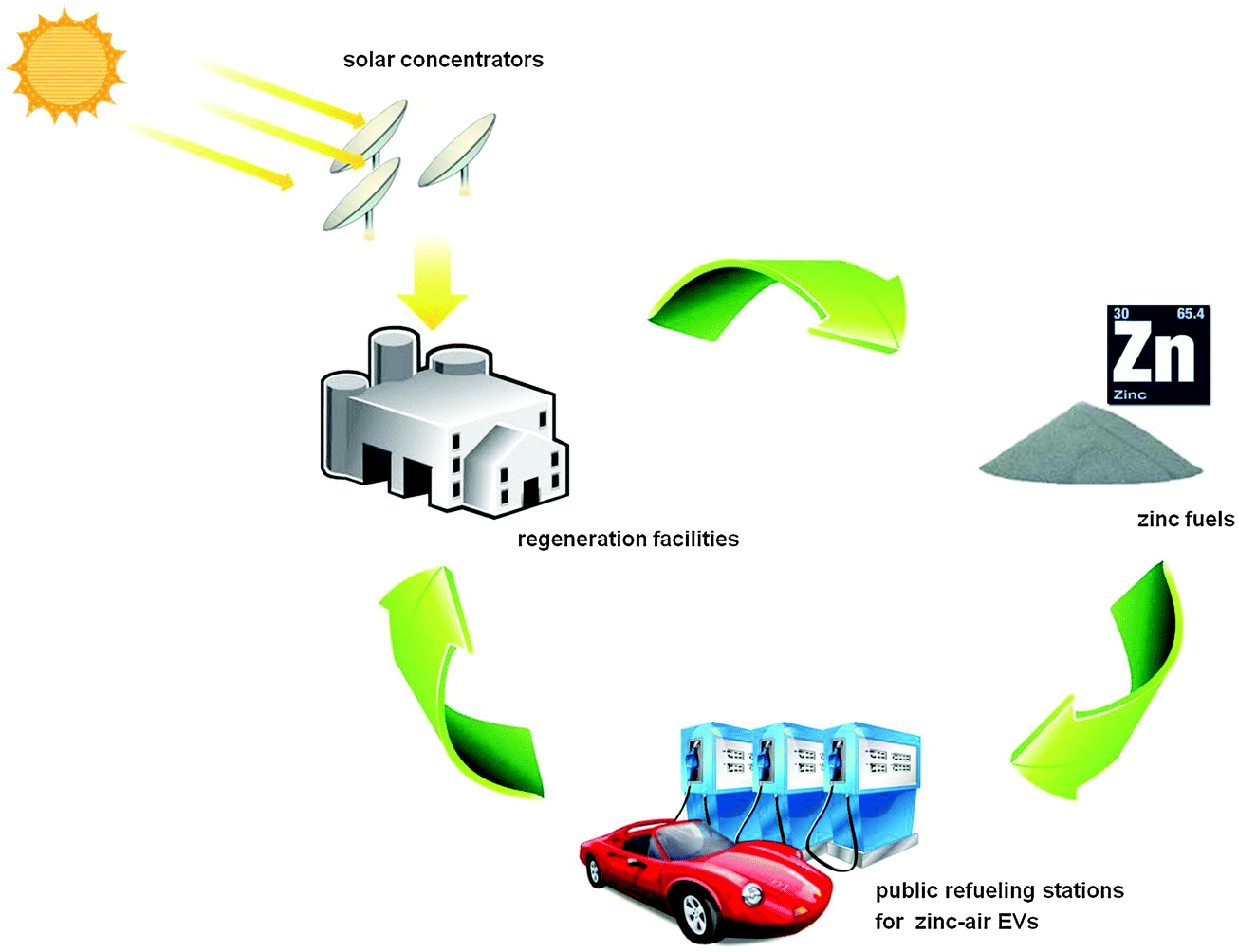 | ||
| Fig. 10 Schematic illustration of the Zn–ZnO cycle for an electrified transportation system. The system requires the development of three basic elements: zinc–air electric vehicles, public refueling stations and regeneration facilities. It is possible to regenerate discharge products via carbothermic reduction using the heat from concentrated solar radiation. | ||
8. CO2 management
Since zinc–air batteries are open systems, their electrolytes are very sensitive to atmospheric CO2, which can react to form carbonates. This reduces electrolyte conductivity.1 Crystallization of carbonates in the air electrode also clogs pores and decreases its activity.1,18 This is a predominant threat to the operation of both alkaline metal–air batteries and alkaline fuel cells.92,190 Drillet and co-workers compared the stability of bifunctional La0.6Ca0.4CoO3 oxygen electrodes as a function of the CO2 concentration in the feed gas.191 They observed that when pure oxygen was used, the OER electrode was life-limiting and stable up to 2500 h. However, with air containing 409–1000 ppm CO2, the ORR became life limiting and was only able to run for about 270 h due to pore clogging. Al-Saleh and coworkers observed that 0.03% CO2 in the oxygen stream did not affect the performance of the Ag/PTFE ORR electrode at all temperatures for a period of 200 h, whereas 1% CO2 in oxygen caused a drop in electrode current density with time.192 Analysis revealed the presence of 7% carbonate on the final electrode.It is evident that proper CO2 management is essential to the endurance of zinc–air batteries. CO2 can be effectively removed by passing the inlet air through a “scrubber” of inexpensive hydroxides (e.g. soda lime and Ca(OH)2) or amines (e.g. monoethanolamine).191,193 It is estimated that one kilogram of soda lime can clean 1000 m3 of air, taking the CO2 concentration from 0.03% to 0.001%.194 If this efficiency can be readily achieved, our calculations indicate approximately 1200 kWh of clean air per kilogram of soda lime with a cost of ∼ 0.1 $ kg−1. One alternative strategy for CO2 management involves the synergistic possibility of changing electrolyte to remove any accumulated carbonate. Gulzow suggested that in a 3.5 kW alkaline fuel cell system, changing electrolyte every 800 h ensured no carbonate precipitation and also maintained the electrolyte concentration.195 In addition, operating zinc–air batteries at higher temperatures would increase the solubility of carbonates and therefore retard its precipitation. There seems to be no obvious technical hurdle toward high-temperature (e.g. 60–80 °C) zinc–air batteries if the self-corrosion of zinc and water loss can be properly managed. Other possible approaches include employing CO2-tolerant electrolytes such as ionic liquids.196 But more often, these options result in much inferior battery performance as a sacrifice.
9. Comparison between zinc–air and other metal–air batteries
Besides zinc–air batteries, other aqueous metal–air systems such as iron–air, aluminum–air and magnesium–air have also been considered, but are not as favored as zinc–air.197–199 These batteries were first invented in the 1960s and 1970s (Table 3).200–202 Among them, iron–air is the only one that can be electrically recharged. Practical iron–air batteries are capable of a long cycle life (>1000 cycles).197 However, their energy density is not high, typically in the range of 60–80 Wh kg−1, which falls short of the desired target set for EV applications. Iron–air batteries are mostly intended for grid-scale energy storage because of their low-cost (<$ 100 kW−1h−1) and long cycle life.197 Aluminum–air and magnesium–air batteries have high theoretical energy densities and working voltages (Table 3). Unfortunately, practically attainable values are much lower due to the parasitic corrosion reaction evolving hydrogen gas at the metallic negative electrode.198,199 Aluminum–air and magnesium–air are not electrically rechargeable since the electrodeposition of aluminum and magnesium is not thermodynamically feasible in aqueous electrolytes. Efforts have been directed towards mechanically rechargeable designs. Aluminum–air and magnesium–air batteries are candidates for ocean power supplies (e.g. ocean buoys) and underwater vehicle propulsion using oxygen present in the ocean.1 They have also been proposed for electric vehicle propulsion.203–206 Yet few products have penetrated the alternative power markets.| Battery systems | Fe–air | Zn–air | Al–air | Mg–air | Na–air | K–air | Li–air |
|---|---|---|---|---|---|---|---|
| a Data source: http://www.metalprices.com. b Oxygen inclusive. c Reported values in literature were normalized to the mass of catalysts. | |||||||
| Year invented | 1968 | 1878 | 1962 | 1966 | 2012 | 2013 | 1996 |
| Cost of metals ($ kg−1)a | 0.40 | 1.85 | 1.75 | 2.75 | 1.7 | ∼20 | 68 |
| Theoretical voltage (V) | 1.28 | 1.65 | 2.71 | 3.09 | 2.27 | 2.48 | 2.96 |
| Theoretical energy density (Wh kg−1)b | 763 | 1086 | 2796 | 2840 | 1106 | 935 | 3458 |
| Electrolyte for practical batteries | Alkaline | Alkaline | Alkaline or saline | Saline | Aprotic | Aprotic | Aprotic |
| Practical voltage (V) | ∼1.0 | 1.0–1.2 | 1.1–1.4 | 1.2–1.4 | ∼2.2 | ∼2.4 | ∼2.6 |
| Practical energy density (Wh kg−1) | 60–80 | 350–500 | 300–500 | 400–700 | Unclearc | Unclearc | Unclearc |
| Primary (P) or electrically recharge-able (R) | R | R | P | P | R | R | R |
Nonaqueous metal–air batteries such as lithium–air, sodium–air and potassium–air were introduced to the public more recently, however they have gained rapidly increasing attention recently.79,207–212 Lithium–air is particularly appealing due to its very high theoretical energy density (3458 Wh kg−1, Table 3). Nonaqueous metal–air batteries have a starkly different battery electrochemistry from their aqueous counterparts. ORR in organic solvents proceeds at a rate orders of magnitude slower than in aqueous electrolytes.19 This leads to the formation of insoluble metal peroxide or superoxide particles, the accumulation of which at the air electrode blocks oxygen diffusion, and gradually shuts off battery reactions.19,79,211,212 Unlike zinc–air batteries, the real capacity that a nonaqueous metal–air battery can achieve is determined by the air electrode – especially by its surface area and pore volume available for the deposition of discharge products – rather than by the metal electrode.79,211,212 This characteristic essentially eliminates the feasibility of mechanical recharging in nonaqueous metal–air batteries unless a method to dissolve the discharge product can be identified. In many studies on lithium–air, the battery capacity is normalized to the mass of air catalysts instead of the metal electrode, resulting in encouraging numbers on the order of thousands or even tens of thousands of mA h g−1. These results are misleading, and should not be taken as an indication that current lithium–air batteries surpass zinc–air batteries in performance. The absolute capacity that a nonaqueous metal–air battery is able to deliver is usually a very small fraction of what a zinc–air battery is capable of. To make it more complicated, recent studies suggested that observed battery currents might be due to the irreversible decomposition and oxidation of its electrolytes.210,213,214 Without doubt, nonaqueous metal–air batteries have tremendous potential. However, they are plagued by intrinsic performance limitations (low power capability and poor cyclability), and might not be able to rival zinc–air batteries at least in the near future.
10. Conclusion and outlook
This review highlights recent advances in zinc–air battery research. Zinc–air is one of the most promising candidates for future energy storage beyond lithium-ion technology. It has been known to the scientific community for over a century. Traditional zinc–air batteries are non-rechargeable and famed for their high energy density. However, they only penetrate a niche market (such as hearing aids and buoys) as a result of their limited power density. ORR electrocatalysis is the performance-limiting reaction in zinc–air batteries. In the last five years, many high-performance non-precious metal based ORR electrocatalysts have been developed through engineering their chemical compositions, structures or their interaction with the carbon support. When incorporated in zinc–air batteries, these materials greatly improve the power performance. It is now possible to achieve a peak power density of over 200 mW cm−2 at room temperature from small-scale laboratory tests. Moreover, progress has been made toward electrically rechargeable zinc–air batteries even though some challenges remain. The shape change of the zinc electrode is a long known problem, but can be alleviated in part with the addition of chemical additives and corrosion inhibitors to the electrode or electrolyte. Cyclable zinc electrodes stable for over 1000 cycles have been successfully demonstrated with little capacity loss. As for the air electrode, serious efforts have been invested in bifunctional oxygen electrocatalysts for both ORR and OER. Several metal oxide and nanocarbon hybrid materials are particularly attractive for their high electrocatalytic activity close or comparable to precious metal benchmarks. Unfortunately, bifunctional catalysts in general suffer from poor cycling stability. It is believed that alternating oxidative and reductive environments at the air electrode during charge–discharge cycles accelerate the catalyst failure. In light of this, a three-electrode configuration has been devised, in which a pair of air electrodes are designed for ORR and OER separately. Compared to the conventional two-electrode configuration, this new battery configuration warrants better cycling performance as confirmed by increasing numbers of studies. In addition, zinc–air batteries can also be mechanically recharged via physically replacing the zinc electrode and electrolyte. It is proposed that the discharge products can be regenerated off-site using renewable energy such as the heat from concentrated solar radiation. Another serious challenge to zinc–air research or alkaline metal–air research in general is CO2 management. Fortunately, there is increasing evidence showing that this can be readily achieved by passing the inlet air through a scrubber of inexpensive materials such as soda lime.Among the different types of metal–air systems – aqueous or non-aqueous, zinc–air by far represents the only viable contender to replace lithium-ion for EV applications. With recent improvements in power capability and lifetime, zinc–air batteries nowadays have most performance parameters on a par with or exceeding those of lithium-ion, not to mention their inherent safety and much lower cost. Even though electrically rechargeable zinc–air batteries are still not mature, there is no major technical hurdle toward zinc–air EVs with mechanical recharging. Many field trials have been carried out in US, Europe and China to assess this technology. It allows fast refueling in a few minutes just like conventional gasoline vehicles. Although it has been reported that some novel lithium-ion technologies are capable of fast recharging in 10 minutes or less, in practice such fast recharging requires extremely high power and may not be feasible at most residential sites. Moreover, zinc–air batteries can be combined with other high-power rechargeable batteries such as lead-acid or even supercapacitors for EVs. In such a hybrid configuration, high energy zinc–air batteries can be used as the primary energy source during periods of light load while high power batteries or supercapacitors handle the peak power requirements. Future research on zinc–air should be placed on the continuous optimization of battery design, electrolyte and electrode materials.
Acknowledgements
Y. L. acknowledges startup support from Soochow University, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Program for Jiangsu Specially-Appointed Professors. H. D. acknowledges support from Stanford Precourt Institute for Energy (PIE) and Global Climate and Energy Project (GCEP).References
- D. Linden and T. B. Reddy, Handbooks of Batteries, McGraw-Hill, 2001 Search PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- J. Chen and F. Cheng, Acc. Chem. Res., 2009, 42, 713 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- J. H. Williams, A. DeBenedictis, R. Ghanadan, A. Mahone, J. Moore, W. R. Morrow, S. Price and M. S. Torn, Science, 2012, 335, 53 CrossRef CAS PubMed.
- Electric Drive Transportation Association, (EDTA), Electric Drive Vehicle Sales Figures (U.S. Market) – EV Sales, http://www.electricdrive.org/index.php?ht=d/sp/i/20952/pid/20952, 2013.
- S. Sun, Energy Smart Technologies – Energy – Research Notes, Bloomberg New Energy Finance, 2012 Search PubMed.
- US DOE EERE, EV Everywhere Grand Challenge, http://www1.eere.energy.gov/vehiclesandfuels/electric_vehicles/index.html.
- L. Maiche, French Pat., 127069, 1878 Search PubMed.
- G. Toussaint, P. Stevens, L. Akrour, R. Rouget and F. Fourgeot, ECS Trans., 2010, 28, 25 CAS.
- Policy white paper, http://www.meridian-int-res.com/Projects/The_Zinc_Air_Solution.pdf.
- J. S. Lee, S. T. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34 CrossRef CAS.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- R. Cao, J. S. Lee, M. L. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816 CrossRef CAS.
- H. Kim, G. Jeong, Y.-U. Kim, J.-H. Kim, C.-M. Park and H.-J. Sohn, Chem. Soc. Rev., 2013, 42, 9011 RSC.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014 10.1039/C3CS60248F.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557 CrossRef CAS PubMed.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- F. R. Mclarnon and E. J. Cairns, J. Electrochem. Soc., 1991, 138, 645 CrossRef CAS PubMed.
- R. E. Durkot, L. Lin and P. B. Harris, US Pat., 6284410, 2001 Search PubMed.
- H. Chang and I. Chi, US Pat., 6593023, 2003 Search PubMed.
- A. Oyama, T. Odahara, S. Fuchino, M. Shinoda and H. Shimomura, US Pat., 6746509, 2004 Search PubMed.
- H. Ma, C. S. Li, Y. Su and J. Chen, J. Mater. Chem., 2007, 17, 684 RSC.
- C. C. Yang and S. J. Lin, J. Power Sources, 2002, 112, 174 CrossRef CAS.
- X. G. Zhang, J. Power Sources, 2006, 163, 591 CrossRef CAS PubMed.
- X. G. Zhang, US Pat., 7291186, 2004 Search PubMed.
- N. C. Tang, US Pat., 6221527, 2001 Search PubMed.
- L. F. Urry, US Pat., 6022639, 2000 Search PubMed.
- J.-F. Drillet, M. Adam, S. Barg, A. Herter, D. Koch, V. M. Schmidt and M. Wilhelm, ECS Trans., 2010, 28, 13 CAS.
- L. Z. Vorkapic, D. M. Drazic and A. R. Despic, J. Electrochem. Soc., 1974, 121, 1385 CrossRef CAS PubMed.
- L. M. Baugh, F. L. Tye and N. C. White, J. Appl. Electrochem., 1983, 13, 623 CrossRef CAS.
- Y. Sato, M. Takahashi, H. Asakura, T. Yoshida, K. Tada, K. Kobayakawa, N. Chiba and K. Yoshida, J. Power Sources, 1992, 38, 317 CrossRef CAS.
- D. P. Bhatt and R. Udhayan, J. Power Sources, 1994, 47, 177 CrossRef CAS.
- A. R. S. Kannan, S. Muralidharan, K. B. Sarangapani, V. Balaramachandran and V. Kapali, J. Power Sources, 1995, 57, 93 CrossRef.
- C. W. Lee, K. Sathiyanarayanan, S. W. Eom and M. S. Yun, J. Power Sources, 2006, 160, 1436 CrossRef CAS PubMed.
- T. Keily and T. J. Sinclair, J. Power Sources, 1981, 6, 47 CrossRef.
- J.-Y. Huot, J. Appl. Electrochem., 1992, 22, 443 CrossRef CAS.
- J. L. Zhu, Y. H. Zhou and C. Q. Gao, J. Power Sources, 1998, 72, 231 CrossRef CAS.
- Y. Ein-Eli, M. Auinat and D. Starosvetsky, J. Power Sources, 2003, 114, 330 CrossRef CAS.
- J. Vatsalarani, S. Geetha, D. C. Trivedi and P. C. Warrier, J. Power Sources, 2006, 158, 1484 CrossRef CAS PubMed.
- Y. D. Cho and G. T. K. Fey, J. Power Sources, 2008, 184, 610 CrossRef CAS PubMed.
- S.-M. Lee, Y.-J. Kim, S.-W. Eom, N.-S. Choi, K.-W. Kim and S.-B. Cho, J. Power Sources, 2013, 227, 177 CrossRef CAS PubMed.
- N. A. Hampson and A. J. S. Mcneil, J. Power Sources, 1985, 15, 261 CrossRef CAS.
- J. L. Zhu and Y. H. Zhou, J. Power Sources, 1998, 73, 266 CrossRef CAS.
- J. Vatsalarani, D. C. Trivedi, K. Ragavendran and P. C. Warrier, J. Electrochem. Soc., 2005, 152, A1974 CrossRef CAS PubMed.
- R. Jain, T. C. Adler, F. R. Mclarnon and E. J. Cairns, J. Appl. Electrochem., 1992, 22, 1039 CrossRef CAS.
- Y. M. Wang and G. Wainwright, J. Electrochem. Soc., 1986, 133, 1869 CrossRef CAS PubMed.
- J. S. Chen and L. F. Wang, J. Appl. Electrochem., 1996, 26, 227 CrossRef CAS.
- C. Zhang, J. M. Wang, L. Zhang, J. Q. Zhang and C. N. Cao, J. Appl. Electrochem., 2001, 31, 1049 CrossRef CAS.
- X. M. Zhu, H. X. Yang, X. P. Ai, J. X. Yu and Y. L. Cao, J. Appl. Electrochem., 2003, 33, 607 CrossRef CAS.
- C.-C. Yang, W.-C. Chien, P.-W. Chen and C.-Y. Wu, J. Appl. Electrochem., 2009, 39, 39 CrossRef CAS.
- J. McBreen and E. Gannon, J. Power Sources, 1985, 15, 169 CrossRef CAS.
- J. McBreen and E. Gannon, Electrochim. Acta, 1981, 26, 1439 CrossRef CAS.
- J. McBreen and E. Gannon, J. Electrochem. Soc., 1983, 130, 1980 CrossRef CAS PubMed.
- K. Bass, P. J. Mitchell, G. D. Wilcox and J. Smith, J. Power Sources, 1991, 35, 333 CrossRef CAS.
- F. Moser, F. Fourgeot, R. Rouget, O. Crosnier and T. Brousse, Electrochim. Acta, 2013, 109, 110 CrossRef CAS PubMed.
- J. M. Wang, L. Zhang, C. Zhang and J. Q. Zhang, J. Power Sources, 2001, 102, 139 CrossRef CAS.
- C. J. Lan, C. Y. Lee and T. S. Chin, Electrochim. Acta, 2007, 52, 5407 CrossRef CAS PubMed.
- Y. Sharma, M. Aziz, J. Yusof and K. Kordesch, J. Power Sources, 2001, 94, 129 CrossRef CAS.
- Z. Tan, Z. Yang, X. Ni, H. Chen and R. Wen, Electrochim. Acta, 2012, 85, 554 CrossRef CAS PubMed.
- Y. Wen, T. Wang, J. Cheng, J. Pan, G. Cao and Y. Yang, Electrochim. Acta, 2012, 59, 64 CrossRef CAS PubMed.
- S. J. Banik and R. Akolkar, J. Electrochem. Soc., 2013, 160, D519 CrossRef CAS PubMed.
- J. Jindra, J. Mrha and M. Musilova, J. Appl. Electrochem., 1973, 3, 297 CrossRef CAS.
- D. M. See and R. E. White, J. Chem. Eng. Data, 1997, 42, 1266 CrossRef CAS.
- R. Othman, W. J. Basirun, A. H. Yahaya and A. K. Arof, J. Power Sources, 2001, 103, 34 CrossRef CAS.
- R. Othman, A. H. Yahaya and A. K. Arof, J. New Mater. Electrochem. Syst., 2002, 5, 177 CAS.
- A. A. Mohamad, J. Power Sources, 2006, 159, 752 CrossRef CAS PubMed.
- C. C. Yang and S. J. Lin, J. Power Sources, 2002, 112, 497 CrossRef CAS.
- X. Zhu, H. Yang, Y. Cao and X. Ai, Electrochim. Acta, 2004, 49, 2533 CrossRef CAS PubMed.
- T. J. Simons, A. A. J. Torriero, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochem. Commun., 2012, 18, 119 CrossRef CAS PubMed.
- M. Xu, D. G. Ivey, Z. Xie and W. Qu, Electrochim. Acta, 2013, 89, 756 CrossRef CAS PubMed.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2009, 113, 20127 CAS.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193 CrossRef CAS.
- K. Harting, U. Kunz and T. Turek, Z. Phys. Chem., 2012, 226, 151 CrossRef CAS.
- P. Arora and Z. M. Zhang, Chem. Rev., 2004, 104, 4419 CrossRef CAS.
- P. Kritzer and J. A. Cook, J. Electrochem. Soc., 2007, 154, A481 CrossRef CAS PubMed.
- G. M. Wu, S. J. Lin, J. H. You and C. C. Yang, Mater. Chem. Phys., 2008, 112, 798 CrossRef CAS PubMed.
- G. M. Wu, S. J. Lin and C. C. Yang, J. Membr. Sci., 2006, 284, 120 CrossRef CAS PubMed.
- H. Saputra, R. Othman, A. G. E. Sutjipto and R. Muhida, J. Membr. Sci., 2011, 367, 152 CrossRef CAS PubMed.
- E. L. Dewi, K. Oyaizu, H. Nishide and E. Tsuchida, J. Power Sources, 2003, 115, 149 CrossRef CAS.
- Y. J. Wang, J. L. Qiao, R. Baker and J. J. Zhang, Chem. Soc. Rev., 2013, 42, 5768 RSC.
- G. W. Heise, US Pat., 1899615, 1933 Search PubMed.
- J. Chottiner, US Pat., 4152489, 1979 Search PubMed.
- H. F. Gibbard, US Pat., 4333993, 1982 Search PubMed.
- C.-L. Lou and Y.-K. Shun, US Pat., 6127061, 2000 Search PubMed.
- F. Bidault, D. J. L. Brett, P. H. Middleton and N. P. Brandon, J. Power Sources, 2009, 187, 39 CrossRef CAS PubMed.
- M. Maja, C. Orecchia, M. Strano, P. Tosco and M. Vanni, Electrochim. Acta, 2000, 46, 423 CrossRef CAS.
- K. Kordesch, S. Jahangir and M. Schautz, Electrochim. Acta, 1984, 29, 1589 CrossRef CAS.
- W. H. Zhu, B. A. Poole, D. R. Cahela and B. J. Tatarchuk, J. Appl. Electrochem., 2003, 33, 29 CrossRef CAS.
- C. Tran, X. Q. Yang and D. Y. Qu, J. Power Sources, 2010, 195, 2057 CrossRef CAS PubMed.
- S. S. Zhang, D. Foster and J. Read, J. Power Sources, 2010, 195, 1235 CrossRef CAS PubMed.
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, ChemElectroChem, 2014, 1, 90 CrossRef.
- P. C. Foller, J. Appl. Electrochem., 1986, 16, 527 CrossRef CAS.
- R. Thacker, Energy Convers., 1972, 12, 17 CrossRef CAS.
- O. C. Wagner, Secondary zinc-air cell investigations, Army Electron. Command, Fort Monmouth, NJ, USA, 1972 Search PubMed.
- R. F. Chireau, The performance of silver-amalgam cathodes as oxygen electrodes in high power metal-air batteries, Proc. Int. Symp., 9th edn, 1975 Search PubMed.
- N. Wagner, M. Schulze and E. Gulzow, J. Power Sources, 2004, 127, 264 CrossRef CAS PubMed.
- C. Coutanceau, L. Demarconnay, C. Lamy and J. M. Leger, J. Power Sources, 2006, 156, 14 CrossRef CAS PubMed.
- H. Meng and P. K. Shen, Electrochem. Commun., 2006, 8, 588 CrossRef CAS PubMed.
- M. Chatenet, L. Genies-Bultel, M. Aurousseau, R. Durand and F. Andolfatto, J. Appl. Electrochem., 2002, 32, 1131 CrossRef CAS.
- J.-J. Han, N. Li and T.-Y. Zhang, J. Power Sources, 2009, 193, 885 CrossRef CAS PubMed.
- F. Y. Cheng, Y. Su, J. Liang, Z. L. Tao and J. Chen, Chem. Mater., 2010, 22, 898 CrossRef CAS.
- Y. Gorlin, C. J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2, 2687 CrossRef CAS.
- F. Y. Cheng, T. R. Zhang, Y. Zhang, J. Du, X. P. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474 CrossRef CAS PubMed.
- J. Feng, Y. Liang, H. Wang, Y. Li, B. Zhang, J. Zhou, J. Wang, T. Regier and H. Dai, Nano Res., 2012, 5, 718 CrossRef CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 2013 CrossRef CAS PubMed.
- Y. Y. Liang, H. L. Wang, J. G. Zhou, Y. G. Li, J. Wang, T. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517 CrossRef CAS PubMed.
- F. Y. Cheng, J. A. Shen, B. Peng, Y. D. Pan, Z. L. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- A. Borbely and J. Molla, US Pat., 4894296A, 1987 Search PubMed.
- K. Buckle, US Pat., 7001439 B2, 2003 Search PubMed.
- J. S. Lee, T. Lee, H. K. Song, J. Cho and B. S. Kim, Energy Environ. Sci., 2011, 4, 4148 CAS.
- J. S. Lee, G. S. Park, H. I. Lee, S. T. Kim, R. G. Cao, M. L. Liu and J. Cho, Nano Lett., 2011, 11, 5362 CrossRef CAS PubMed.
- H. Tanaka and M. Misono, Curr. Opin. Solid State Mater. Sci., 2001, 5, 381 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 647 CrossRef CAS.
- N. Yamazoe, N. Miura, Y. Shimizu and K. Uemura, Prog. Batteries Sol. Cells, 1989, 8, 276 CAS.
- S. Muller, F. Holzer and O. Haas, Proc. – Electrochem. Soc., 1997, 97(18), 859 Search PubMed.
- X. Wang, P. J. Sebastian, M. A. Smit, H. Yang and S. A. Gamboa, J. Power Sources, 2003, 124, 278 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. G. Jin and S. Z. Qiao, Small, 2012, 8, 3550 CrossRef CAS PubMed.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527 CrossRef CAS.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787 CrossRef CAS PubMed.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707 CrossRef CAS PubMed.
- Y. Tang, B. L. Allen, D. R. Kauffman and A. Star, J. Am. Chem. Soc., 2009, 131, 13200 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Muellen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- H. Jin, H. Zhang, H. Zhong and J. Zhang, Energy Environ. Sci., 2011, 4, 3389 CAS.
- D. S. Geng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 CAS.
- S. Shanmugam and T. Osaka, Chem. Commun., 2011, 47, 4463 RSC.
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206 CrossRef CAS PubMed.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. Lu, Adv. Mater., 2013, 25, 998 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350 CrossRef CAS PubMed.
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293 CrossRef CAS PubMed.
- Y. Sun, C. Li and G. Shi, J. Mater. Chem., 2012, 22, 12810 RSC.
- S. M. Zhu, Z. Chen, B. Li, D. Higgins, H. J. Wang, H. Li and Z. W. Chen, Electrochim. Acta, 2011, 56, 5080 CrossRef CAS PubMed.
- G. S. Park, J. S. Lee, S. T. Kim, S. Park and J. Cho, J. Power Sources, 2013, 243, 267 CrossRef CAS PubMed.
- G. Q. Jian, Y. Zhao, Q. Wu, L. J. Yang, X. Z. Wang and Z. Hu, J. Phys. Chem. C, 2013, 117, 7811 CAS.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939 CrossRef CAS PubMed.
- K. Stanczyk, R. Dziembaj, Z. Piwowarska and S. Witkowski, Carbon, 1995, 33, 1383 CrossRef CAS.
- C. W. B. Bezerra, L. Zhang, K. C. Lee, H. S. Liu, A. L. B. Marques, E. P. Marques, H. J. Wang and J. J. Zhang, Electrochim. Acta, 2008, 53, 4937 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878 CrossRef CAS PubMed.
- M. Lefevre and J. P. Dodelet, Electrochim. Acta, 2008, 53, 8269 CrossRef CAS PubMed.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394 CrossRef CAS PubMed.
- D. A. Scherson, S. B. Yao, E. B. Yeager, J. Eldridge, M. E. Kordesch and R. W. Hoffman, J. Phys. Chem., 1983, 87, 932 CrossRef CAS.
- M. S. Thorum, J. M. Hankett and A. A. Gewirth, J. Phys. Chem. Lett., 2011, 2, 295 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705 CrossRef CAS.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001 CAS.
- A. L. Zhu, H. J. Wang, W. Qu, X. X. Li, Z. Jong and H. Li, J. Power Sources, 2010, 195, 5587 CrossRef CAS PubMed.
- Z. Chen, J. Y. Choi, H. J. Wang, H. Li and Z. W. Chen, J. Power Sources, 2011, 196, 3673 CrossRef CAS PubMed.
- L. Jorissen, J. Power Sources, 2006, 155, 23 CrossRef PubMed.
- E. S. Buzzet, US Pat., 3977901, 1976 Search PubMed.
- J. F. Jackovitz and C.-T. Liu, US Pat., 5318862, 1994 Search PubMed.
- V. Neburchilov, H. J. Wang, J. J. Martin and W. Qu, J. Power Sources, 2010, 195, 1271 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 1991, 36, 225 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- V. Nikolova, P. Iliev, K. Petrov, T. Vitanov, E. Zhecheva, R. Stoyanova, I. Valov and D. Stoychev, J. Power Sources, 2008, 185, 727 CrossRef CAS PubMed.
- D. U. Lee, B. J. Kim and Z. W. Chen, J. Mater. Chem. A, 2013, 1, 4754 CAS.
- Z. Chen, A. Yu, R. Ahmed, H. Wang, H. Li and Z. Chen, Electrochim. Acta, 2012, 69, 295 CrossRef CAS PubMed.
- G. Du, X. Liu, Y. Zong, T. S. A. Hor, A. Yu and Z. Liu, Nanoscale, 2013, 5, 4657 RSC.
- M. Prabu, K. Ketpang and S. Shanmugam, Nanoscale, 2014, 6, 3173 RSC.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59 CrossRef CAS PubMed.
- H. M. Zhang, Y. Teraoka and N. Yamazoe, Chem. Lett., 1987, 665 CrossRef CAS.
- Y. Shimizu, K. Uemura, H. Matsuda, N. Miura and N. Yamazoe, J. Electrochem. Soc., 1990, 137, 3430 CrossRef CAS PubMed.
- S. Mueller, K. Striebel and O. Haas, Electrochim. Acta, 1994, 39, 1661 CrossRef CAS.
- C. K. Lee, K. A. Striebel, F. R. McLarnon and E. J. Cairns, J. Electrochem. Soc., 1997, 144, 3801 CrossRef CAS PubMed.
- N. L. Wu, W. R. Liu and S. J. Su, Electrochim. Acta, 2003, 48, 1567 CrossRef CAS.
- S. Malkhandi, B. Yang, A. K. Manohar, A. Manivannan, G. K. S. Prakash and S. R. Narayanan, J. Phys. Chem. Lett., 2012, 3, 967 CrossRef CAS.
- H. Arai, S. Muller and O. Haas, J. Electrochem. Soc., 2000, 147, 3584 CrossRef CAS PubMed.
- S. Muller, F. Holzer, O. Haas, C. Schlatter and C. Comninellis, Chimia, 1995, 49, 27 Search PubMed.
- Z. Chen, A. Yu, D. Higgins, H. Li, H. Wang and Z. Chen, Nano Lett., 2012, 12, 1946 CrossRef CAS PubMed.
- K.-N. Jung, J.-H. Jung, W. B. Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902 CAS.
- T. Tsai and A. Vartek, Patent, WO2003061057 A1, Evionyx Inc, 2003 Search PubMed.
- Z. M. Zhong, US Pat., 6383675, 2002 Search PubMed.
- Y. G. Li, M. Gong, Y. Y. Liang, J. Feng, J. E. Kim, H. L. Wang, G. S. Hong, B. Zhang and H. J. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- J. F. Cooper, D. Fleming, L. Keene, A. Maimoni, K. Peterman and R. Koopman, Demonstration of zinc/air fuel battery to enhance the range and mission of fleet electric vehicles: preliminary results in the refueling of a multicell module, Lawrence Livermore National Laboratory, 1994 Search PubMed.
- J. R. Goldstein and B. Koretz, Proc. Intersoc. Energy Convers. Eng. Conf., 1993, 28, 2.279 Search PubMed.
- J. Noring, S. Gordon, A. Maimoni, M. Spragge and J. F. Cooper, Proc. – Electrochem. Soc., 1993, 93(8), 235 Search PubMed.
- J. Goldstein, I. Brown and B. Koretz, J. Power Sources, 1999, 80, 171 CrossRef CAS.
- J. R. Goldstein, N. Lapidot, M. Aguf, I. Gektin and M. Givon, Proc. – Electrochem. Soc., 1997, 97(18), 881 Search PubMed.
- C. Wieckert, M. Epstein, G. Olalde, S. Santen and A. Steinfeld, in Zinc Electrodes: Solar Thermal Production, ed. J. Garche, et al., Encyclopedia of Electrochemical Power Sources, 2009 Search PubMed.
- G. F. McLean, T. Niet, S. Prince-Richard and N. Djilali, Int. J. Hydrogen Energy, 2002, 27, 507 CrossRef CAS.
- J. F. Drillet, F. Holzer, T. Kallis, S. Muller and V. M. Schmidt, Phys. Chem. Chem. Phys., 2001, 3, 368 RSC.
- M. A. Al-Saleh, S. Gultekin, A. S. Alzakri and H. Celiker, J. Appl. Electrochem., 1994, 24, 575 CrossRef CAS.
- H. H. Cheng and C. S. Tan, J. Power Sources, 2006, 162, 1431 CrossRef CAS PubMed.
- A. J. Appleby and F. R. Foulkes, Fuel Cell Handbook, Krieger Publishing Company, Florida, 1993 Search PubMed.
- E. Gulzow, J. Power Sources, 1996, 61, 99 CrossRef.
- E. B. Fox, H. R. Colon-Mercado, Y. Chen and W. S. W. Ho, ACS Symp. Ser., 2012, 1117, 129 CrossRef CAS PubMed.
- S. R. Narayanan, G. K. S. Prakash, A. Manohar, B. Yang, S. Malkhandi and A. Kindler, Solid State Ionics, 2012, 216, 105 CrossRef CAS PubMed.
- D. R. Egan, C. P. de Leon, R. J. K. Wood, R. L. Jones, K. R. Stokes and F. C. Walsh, J. Power Sources, 2013, 236, 293 CrossRef CAS PubMed.
- T. R. Zhang, Z. L. Tao and J. Chen, Mater. Horiz., 2014, 1, 196 RSC.
- L. Ojefors and L. Carlsson, J. Power Sources, 1978, 2, 287 CrossRef CAS.
- S. Zaromb, J. Electrochem. Soc., 1962, 109, 1125 CrossRef CAS PubMed.
- W. N. Carson and C. E. Kent, in The Magnesium-Air Cell, ed. D. H. Collins, Power Sources, 1966 Search PubMed.
- A. S. Homa, E. J. Rudd, Proc. Intersoc. Energy Convers. Eng. Conf., 1989, 24th, 1331.
- D. J. Levy, R. P. Hollandsworth, E. M. Gonzales, E. L. Littauer, Proc. Intersoc. Energy Convers. Eng. Conf., 1983, 18th, 1635.
- S. Yang and H. Knickle, J. Power Sources, 2002, 112, 162 CrossRef CAS.
- E. J. Rudd, S. Lott, The development of aluminum-air batteries for application in electric vehicles: final report, Res. Dev. Cent., ELTECH Systems Corp., Fairport Harbor, OH, USA, 1990.
- P. Hartmann, C. L. Bender, M. Vracar, A. K. Durr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky, R. Hadar, H. Mazor, M. Goor and L. Burstein, J. Power Sources, 2013, 244, 771 CrossRef CAS PubMed.
- X. D. Ren and Y. Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, J. Power Sources, 2011, 196, 886 CrossRef CAS PubMed.
- S. A. Freunberger, Y. H. Chen, Z. Q. Peng, J. M. Griffin, L. J. Hardwick, F. Barde, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040 CrossRef CAS PubMed.
- Z. Q. Peng, S. A. Freunberger, Y. H. Chen and P. G. Bruce, Science, 2012, 337, 563 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |