 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation exchange at the secondary building units of metal–organic frameworks
C. K.
Brozek
and
M.
Dincă
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: mdinca@mit.edu
First published on 16th May 2014
Abstract
Cation exchange is an emerging synthetic route for modifying the secondary building units (SBUs) of metal–organic frameworks (MOFs). This technique has been used extensively to enhance the properties of nanocrystals and molecules, but the extent of its applications for MOFs is still expanding. To harness cation exchange as a rational tool, we need to elucidate its governing factors. Not nearly enough experimental observations exist for drawing these conclusions, so we provide a conceptual framework for approaching this task. We address which SBUs undergo exchange, why certain ions replace others, how the framework influences the process, the role of the solvent, and current applications. Using these guidelines, certain trends emerge from the available data and missing experiments become obvious. If future studies follow this framework, then a more comprehensive body of observations will furnish a deeper understanding of cation exchange and inspire future applications.
 C. K. Brozek | Carl K. Brozek was born in Hinsdale, IL, in 1988. He earned his S.B. from the University of Chicago in 2010, while working for Prof. Gregory L. Hillhouse on the synthesis of nickel-containing compounds bearing disulfide ligands. His current work at the Massachusetts Institute of Technology with Prof. Mircea Dincă involves showing that a new realm of coordination chemistry is only possible with the bizarre ligand fields of MOF SBUs. |
 M. Dincă | Mircea Dincă was born in Făgăraş, Romania, in 1980. He received a Bachelor's Degree in Chemistry from Princeton University in 2003 and a PhD in Inorganic Chemistry from the University of California, Berkeley, in 2008. Following postdoctoral work at MIT, he joined the faculty at MIT in 2010. His research is concerned with the study of crystalline extended solids as unusual platforms for redox reactivity, small molecule activation, electrochemistry, and emerging electronic phenomena. |
Key learning points(1) The secondary building units (SBUs) that undergo cation exchange often contain metal sites that are coordinatively unsaturated, are coordinated by at least one solvent molecule, or are capable of higher coordination numbers than suggested by the crystal structures of the respective MOFs.(2) Metal sites that are coordinatively saturated by the MOF framework/ligands can still undergo cation exchange if the ligands in the framework form a weak field ligand environment at the SBU. (3) Although periodic trends of cation exchange are not fully established yet, Cu2+ ions tend to replace most other second row transition metals, but Pb2+, Mn2+, and Cd2+ exchange faster than Cu2+. (4) The structure of the MOF may influence the extent of cation exchange; the primary reason for this may be the limited distortion allowed by any given lattice during the exchange process. (5) Applications of cation exchange in MOFs are just emerging, but the technique has already enabled the formation of previously unknown molecular species, highlighting MOFs as new platforms for coordination chemistry and small molecule reactivity. |
Introduction
Cation exchange is a powerful tool for designing new materials. Broadly defined, it is the partial or complete substitution of a metal ion at the site of another. This process offers an alternative, typically milder, route for accessing materials when conventional synthesis at high temperature fails. For decades, it has been employed to tailor the composition of zeolites and, more recently, nanocrystals. Metal–organic frameworks (MOFs) emerged decades ago, but cation exchange was only first demonstrated with them in 2007.1 In these materials, the exchange occurs at the inorganic clusters, often called the metal nodes or secondary-building units (SBUs). Although these clusters are integral to the MOF structure, the metal ions can be replaced, sometimes entirely and in a matter of hours, without compromising the structure. The details of this fascinating transformation are unknown and the bounty of MOF structures that undergo metal ion substitution present a host of curiosities to be explained.Geochemists have long known cation exchange as diadochy.2 Minerals are rarely pure phases because minor amounts of foreign ions of similar charge and size often incorporate into the structure. The replacement of an ion for another at a particular crystalline lattice position is a diadochic transformation, and often requires high temperatures and pressures. For instance, the volcanic rocks known as the olivine series, (Mg2+, Fe2+)SiO4, differ by their relative composition of Mg2+ or Fe2+, which result from diadochic transformations in magma.3 Meanwhile, the substitution of Na+ into porous leucite, KAlSi2O6, occurs at temperatures as low as 150 °C, illustrating the role of porosity in facilitating the exchange process.4 V. M. Goldschmidt developed a set of rules to explain the mutual replacement of ions in magmatic minerals.5 This contends that ions undergo diadochy if they possess similar charge and radii. Ions with greater charge or smaller radii are incorporated to a great degree because they form stronger, more ionic bonds. To account for the covalent components of these bonds, Ringwood's rule states that ions with similar electronegativity replace each other.6 The ion with the lower value will be exchanged more because it will form bonds with greater ionic character. These trends are useful for assessing the cation exchange behavior of MOFs, though they derive from observations with minerals, which are typically densely packed structures.
Cation exchange is also employed with nanocrystals to fine-tune their band structures by inserting specific ions into well-defined environments.7 Unlike in bulk CdSe, Cu2S, or similar extended materials, cation exchange in nanocrystals occurs at room temperature at sub-second rates due to enhanced surface area and low atomic counts. The small size of these particles also facilitates atomic reorganization and diminishes lattice strain. This technique enables the synthesis of metastable phases that are not achievable by conventional “hot injection” synthesis, such as Cu2S particles with turn-on plasmon resonance.8 Cation exchange also enables complexity to be engineered into a nanocrystal device. For instance, templating CdSe on PbSe nanorods for fixed amounts of time generates CdSe–PbSe core–shell heterostructures so that electron and hole carriers are confined within the lower band-gap PbSe core, resulting in high quantum yield excitonic emission.9
In solution, metallo-cluster compounds and mononuclear complexes are also known to substitute for other cations. For decades, transmetallation has been used to replace cations in mononuclear compounds featuring multidentate ligands. The mechanism of these exchanges often involves the transfer of a ligand to a new metal ion.10 Cation substitution at a molecular cluster that left the anionic framework intact was first documented in 1982 for the adamantane-like cage compounds, [M4−n, Mn′, (SC6H5)]2− (M, M′ = Fe2+, Co2+, Zn2+, Cd2+).11 Metal exchange in these compounds was believed to involve free ions exiting the cage before the inserting species associated. However, mechanistic studies of the simpler case of Co2+ incorporating into [M4(SPh)10]2− (M = Zn or Fe) revealed a process that was quite complex.12 Few other reports have attempted to understand cation exchange in molecules, though metallothioneins are thought to mediate detoxification of trace metals through some version of metal ion substitution.13
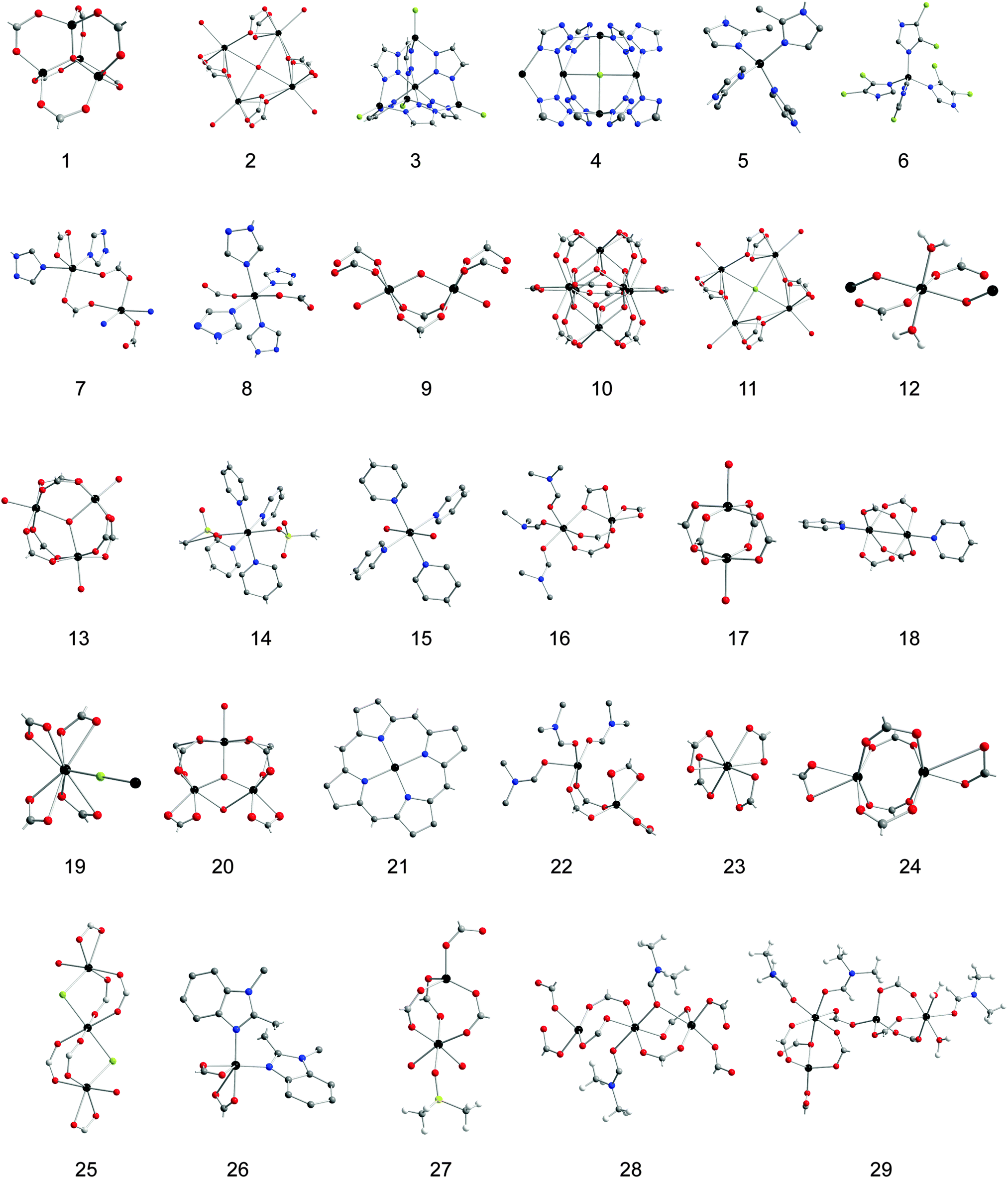
This article outlines the available observations of cation exchange at MOF SBUs so that general trends and future studies can be sketched. We organize data around questions that need to be answered to endow this technique with predictive capabilities. All known examples of metal ion substitution at MOF SBUs and relevant details are listed in Table 1 with pictorial representations of the SBUs in Table 2. We also note that we confined our discussion to substitution that occurs at SBUs and not in the pores or when metal ions are part of the ligands, in the so-called metalloligands. More general reviews of cation exchange in MOFs have been published elsewhere.14,15 Cation exchange has already yielded some surprising results and new materials that have not been accessible otherwise, but the extent of its use for designing new MOFs in a systematic and predictive manner depends on understanding its mechanism. This tutorial review is intended to provide a blueprint towards this goal.
| Molecular formula | SBU | Common name | Inserted cation | Extent | Conditions | Characterization | Ref. |
|---|---|---|---|---|---|---|---|
| Al(OH)(BDC-Br) | 9 | MIL-53(Al)-Br | Fe3+ | Undetermined | H2O, 85 °C, 5 d | PXRD | 34 |
| Co(H3O)[(Co4Cl)3(HMTT)8] | 11 | POST-65(Co) | Mn2+ | Complete | DMF, RT, 1 mo | SXRD, BET | 26 |
| Co 6(BTB)4(BP)3 | 18 | Ni2+, Cu2+, Zn2+ | Complete | DMF, RT,1 d, 1 d, 2 d | BET, PXRD | 38 | |
| [Co2(BTX)2(BDC)2(H2O)2]n | 8 | Cd2+ | Complete | H2O, RT, 7 d | IR, PXRD | 39 | |
| [Co3+2Co2+O(BTB)2(H2O)x(DMF)y]·zDMF·nH2O | 20 | Co3+ | Complete | DMF-EtOH RT, 1–3 wk | SXRD | 28 | |
| [Zn2.5Co1.5(DCPP)2(DMF)3(H2O)2]n | 29 | Zn2+ | Complete | MeCN, 80 °C, 3 h | PXRD, optical photos | 53 | |
| Zn3.9Co2.1(BTB)4(BP)3 | 18 | SUMOF-1-(Co:2Zn) | Zn2+ | Complete | DMF, RT, 7 d | SXRD, PXRD | 37 |
| Cd 1.5(H3O)3[(Cd4O)3(HMTT)8]·6H2O | 2 | Pb2+, Dy3+, Nd3+ | Complete | H2O, RT, 7 d | SXRD, PXRD | 25 | |
| [Cd(BTX)2Cl2]n | Unknown | Cu2+ | Complete | H2O, RT, 7 d | IR, PXRD | 39 | |
| {[Cd2(BTX)2(BDC)2]H2O}n | 7 | Cu2+ | Complete | H2O, RT, 7 d | IR PXRD | 39 | |
| Cd 3[(Cd4Cl)3(BTT)8]2 | 4 | CdCd-BTT | Co2+, Ni2+not Mn2+, Fe2+, Cu2+, or Zn2+ | Complete | MeOH, 80 °C, 30 d | IR, SXRD, BET | 21 |
| {[Cd(BTTN)2(H2O)2]·2(PF6)·pyrene·2(H2O)}n | 15 | Cu2+ | Complete | MeOH, RT, 8 h | SXRD | 42 | |
| {[Cd(BP)2(O3SFcSO3)]·(CH3OH)4}n | 14 | Cu2+ | 50% | MeOH, RT, 30 d | SXRD | 41 | |
| [(CH3)2NH2]15[(Cd2Cl)3(TATPT)4]·12DMF·18H2O | 19 | Cu2+, Co2+, Ni2+, Zn2+ | Cu2+ 7.7%, Co2+ 6.2%, Ni2+ 4.3%, Zn2+ 4.6% | DMF, RT, 5 d | PXRD | 33 | |
| C156H60Cd11N12O51 | 21 | MMPF-5(Cd) | Co2+ | Complete at porphyrin node untouched | DMSO, 85 °C, 2 d | SXRD | 48 |
| [Cd4(BPT)4]·[Cd(C44H36N8)S]·S (S = MeOH, H2O) | 24 | Porph@MOM-11-Cd | Cu2+ | Complete | MeOH, RT, 10 d | SXRD | 36 |
| [Cd6(BPT)4Cl4(H2O)4]·[C44H36N8CdCl]·[H3O] | 25 | Porph@MOM-10 | Mn2+, Cu2+ | Mn complete, Cu 76% | MeOH, RT, 1 mo | SXRD, BET | 32 |
| Na0.25[(CH3)2NH2]1.75[Cd(HMBM)2]·xsolvent | 23 | Cu2+not Ni2+or Co2+ | 96% | MeOH, RT, 7 d | PXRD | 35 | |
| Cr 3F(H2O)2O(BDC)3 | 13 | MIL-101(Cr) | Al3+, Fe3+ | Al3+ 10%, Fe3+ 5.8% | H2O, 100 °C, 3 d | BET, PXRD | 27 |
| Cu 2/3Mn1/3(H3O)[(Cu8/3Mn4/3Cl)3(HMTT)8] | 11 | POST-65(Cu) | Mn2+ | 34% | DMF, RT, 1 mo | SXRD, BET | 26 |
| {[Cu(BTTN)2(H2O)2]·2(PF6)·pyrene·2(H2O)}n | 15 | Cd2+ | Complete | MeOH, RT, 10 d | SXRD | 42 | |
| Cu 6(BTB)4(BP)3 | 18 | SUMOF-1-Cu | Zn2+ | 38% | DMF, RT, 3 mo | SXRD, PXRD | 37 |
| Cu 8(BIM)16 | 26 | Zn2+, Co2+ | Zn 20.81%, Co 14.97% | H2O, RT, 5 d | PXRD | 44 | |
| [Cu4(DCPP)2(DMF)3(H2O)2]n | 29 | Zn2+ | Complete | MeCN, 80 °C, 6 h | PXRD, optical photos | 53 | |
| Fe(OH)(BDC-Br) | 9 | MIL-53(Fe)-Br | Al3+ | Undetermined | H2O, 85 °C, 5 d | PXRD | 34 |
| Mn 3[(Mn4Cl)3BTT8]2 | 4 | MnMn-BTT | Li+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+ | Li+ 21%, Fe2+ 20%, Co2+ 20%, Ni2+ 18%, Cu2+ 92%, Zn2+ 80% | MeOH, RT, 1 mo | PXRD, SXRD, MAD, BET | 1, 20, 45 |
| Mn(H3O)[(Mn4Cl)3(HMTT)8] | 11 | POST-65(Mn) | Fe3+, Co2+, Ni2+, Cu2+ | Fe3+ complete, Co2+ complete, Ni2+ complete, Cu2+ 66% | DMF, RT, 12 d | SXRD, BET | 26 |
| Ni 6(BTB)4(BP)3 | 18 | Cu2+ | Complete | DMF, RT, 15 d | BET, PXRD | 38 | |
| Zn3.72Ni2.28(BTB)4(BP)3 | 18 | SUMOF-1-(Ni:2Zn) | Zn2+ | Complete | DMF, RT, 7 d | SXRD, PXRD | 37 |
| Ni(H3O)[(Ni4Cl)3(HMTT)8] | 11 | POST-65(Ni) | Mn2+ | Complete | DMF, RT, 1 mo | SXRD, BET | 26 |
| [Zn2Ni2(DCPP)2(DMF)3(H2O)2]n | 29 | Zn2+ | Complete | MeCN, 80 °C, 4 h | PXRD, optical photos | 53 | |
| Pb 1.5(H3O)3[(Pb4O)3(HMTT)8]·6H2O | 2 | Cd2+ | Complete | H2O, RT, 3 wk | SXRD, PXRD | 25 | |
| {[(ZnOOCClH3C6Fc)2(H2O)3](H2O)}n | 12 | Pb2+, Cd2+, Cu2+, Ni2+, Co2+, Mn2+, Cr2+ | Pb2+ complete, Cd2+ 92%, Cu2+ 76% | H2O, RT, 5 d | SXRD, PXRD, IR | 31 | |
| [Zn(4,4′-BP)2-(FcphSO3)2]n | 14 | Cd2+, Pb2+, Cu2+ | Cd2+ 40%, Pb2+ 75%, Cu2+ 50% | MeOH, RT, 30 d | SXRD | 40 | |
| {[Zn(BTTN)2(H2O)2]·2(PF6)·pyrene·2(H2O)}n | 15 | Cu2+ | Complete | MeOH, RT, 6 h | SXRD | 42 | |
| {[Zn2(BDCPPI)(DMF)3]·7DMF·5H2O}n | 16 | Cu2+not Co2+, Ni2+, Cd2+ | 97% | MeOH, RT, 4 d | PXRD, IR | 30 | |
| Zn 3BTC2(H2O)3 | 17 | Zn-HKUST-1 | Cu2+ | 53% | MeOH, RT, 3 mo | PXRD | 22 |
| Zn 24TDCPEB8(H2O)12 | 17 | PMOF-2 | Cu2+ | Complete | MeOH, RT, 3 d | Optical photos, PXRD | 22 |
| Zn 6(BTB)4(BP)3 | 18 | Ni2+, Cu2+, Co2+ | Complete | DMF, RT, 2 d | BET, PXRD | 38 | |
| [Zn2(TADYDI)(DMF)3]n | 22 | NTU-101-Zn | Cu2+ | 80% | DMF, RT, 14 d | IR, PXRD | 29 |
| Zn 4(ETTB)·4DMF·xsolvent | 17 | PCN-921 | Cu2+ | Complete | DMF, RT, 4 d | SXRD | 23 |
| Zn 6(BTB)4(BP)3 | 18 | SUMOF-1-Zn | Cu2+, Co2+, Ni2+ | Cu complete, Co 35%, Ni 38% | DMF, RT, 3 mo | SXRD, PXRD | 37 |
| [Zn7(PPBOTCDITC)3(H2O)7]n[Zn5(PPBOTCDITC)3(H2O)5]n·xsolvent | 17 | Cu2+not Ni2+or Co2+ | 87% | MeOH, RT, 7 d | PXRD | 24 | |
| [Zn4(TIAPy)·(H2O)4·(EGME)2] | 27 | JUC-118 | Cu2+ | 98.8% | 2-Methoxyethanol, RT, 3 d | SXRD, optical photos | 52 |
| [Zn3(CBAI)2(DMF)2]·2DMF | 28 | Cu2+, not Co2+or Ni2+ | Complete | DMF–H2O, RT, 5 d | PXRD, FT-IR, SEM | 54 | |
| [Zn4(DCPP)2(DMF)3(H2O)2]n | 29 | Cu2+, Co2+, Ni2+ | Cu2+ Complete, Co2+ and Ni2+ only 6-coordinate sites | MeCN, 80 °C, 4 h | PXRD, optical photos | 53 | |
| ZnZn4Cl4(BTDD6) | 3 | MFU-4l | Co2+ | 80% (all but central Zn2) | DMF, 140 °C, 20 h | SXRD | 19 |
| Zn-(MeIm) | 5 | ZIF-8 | Mn2+ | 12% | MeOH, 55 °C, 24 h | PXRD, BET | 18 |
| Zn-(Cl2Im) | 6 | ZIF-71 | Mn2+ | 10% | MeOH, 55 °C, 24 h | PXRD, BET | 18 |
| Zn 4O(BDC)3 | 1 | MOF-5 | Ti3+, V3+, V2+, Cr3+, Cr2+, Mn2+, Fe2+, Ni2+ | Ti3+ 2.3%, V3+ 5%, V2+ 4.3%, Cr3+ 35%, Cr2+ 24%, Mn2+ 11%, Fe2+ 24%, Ni2+ 25% | DMF, RT, 7 d | PXRD, BET, IR | 16, 17 |
| Zr 6O4(OH)4(BDC)12 | 10 | UiO-66 | Ti4+, Hf4+ | Ti4+ 94%, Hf4+ 18% | DMF, 85 °C, 5 d | PXRD, BET | 34 |
|
Which SBUs undergo cation exchange?
If we can predict which MOFs are susceptible to cation exchange, it will become a rational tool for synthesizing new materials with intended properties. After elucidating the factors that make an SBU exchangeable, specific materials could be selected for cation exchange from among the thousands of reported MOFs, and their exact compositions could be designed beforehand. These factors are yet unknown, but surveying the reported examples of cation exchange in MOFs reveals several common features among their SBUs.A foremost observation is that the exchangeable metal ions in an SBU are often capable of higher coordination numbers than those observed in the X-ray crystal structures. For example, the series of materials known as (Cl)M-MOF-5 arise from Ti3+, V3+, V2+, Cr3+, Cr2+, Mn2+, Fe2+, or Ni2+ replacing a four-coordinate Zn2+ cation in each cluster of MOF-5 (Zn4O(BDC)3) (see the Abbreviations section below).16,17 Similarly, the tetrahedral Zn2+ sites in ZIF-8 (Zn-(MeIm)) and ZIF-71 (Zn-(Cl2Im)) can be replaced by Mn2+ ions,18 while the four-coordinate Zn2+ sites in MFU-4l (ZnZn4Cl4(BTDD6)) can be replaced by Co2+ ions.19
In several examples, the exchangeable metal ions contain open sites when fully evacuated, but become partially solvated when immersed in solution. The family of MOFs known as MM-BTT, M3[(M4Cl)3BTT8]2, begin with a two-coordinate Cs-symmetric Mn2+ site and five-coordinate Mn2+ site with C4v symmetry.20 When in methanol, the latter gains a solvent ligand to become six-coordinate, while the former becomes fully solvated in the cavities of the structure. Either the fully solvated or both Mn2+ sites exchange for Fe2+, Co2+, Ni2+, Cu2+, or Zn2+.1 An isostructural material known as Cd3[(Cd4Cl)3BTT8]2 contains Cd2+ that demonstrates similar coordinative changes upon solvation and replaces for Co2+ or Ni2+.21
Not all structures can be desolvated as MM-BTT, but the metal sites in many other SBUs typically feature bound solvent molecules. The materials known as Zn-HKUST-1 ([Zn3BTC2(H2O)3]),22 P-MOF-2 (Zn24TDCPEB8(H2O)12),22 PCN-921 (Zn4(ETTB)·4DMF·xSolvent),23 and [Zn7(PPBOTCDITC)3(H2O)7]n[Zn5((PPBOTCDITC))3(H2O)5]n·xSolvent24 contain SBUs with “paddlewheel” structures. Each of the metal sites in these clusters is bound to four carboxylates from the framework and one solvent molecule at the axial position. Cd1.5(H3O)3[(Cd4O)3(HMTT)8]2·6H2O25 and POST-65(Mn) (Mn(H3O)[(Mn4Cl)3(HMTT)8]2)26 have the sodalite topology, like MM-BTT, with similar partially solvated SBUs. The metal sites in the planar Cd4O clusters of Cd1.5(H3O)3[(Cd4O)3(HMTT)8]·6H2O are each bound to a solvent molecule and exchange for Pb2+. The Mn4Cl clusters of POST-65(Mn) are partially solvated, as in MM-BTT, and can be replaced by Fe3+, Co2+, Ni2+, Cu2+. In the case of Fe3+ exchange, the {M4Cl}7+ SBU transforms into {Fe4OH}11+, with two μ2-O providing additional charge balance. Similar to the metal sites in the “paddlewheel” and the planar MCl/O clusters, the exchangeable Cr3+ sites in MIL-101(Cr)27 (Cr3F(H2O)2O(BDC)3) would be coordinatively unsaturated if not for a pendent solvent ligand in the axial position. Similarly, the SBU of the series [Co3+2Co2+O(BTB)2(H2O)x(DMF)y]·zDMF·nH2O (x = y = 1, z = 7.5, n = 12; x = 2, y = 0, z = 8.5, n = 8; x = 2, y = 1, z = 7, n = 8) contains a cobalt site with a bound solvent molecule and all three Co2+ sites exchange to form an entirely new structure.28 In another case of partial solvation, the exchangeable di-zinc sites in NTU-101-Zn29 [Zn2(TADYDI)(DMF)3]n and {[Zn2(BDCPPI)(DMF)3]·7DMF·5H2O}n contain a Zn2+ ion held to the framework by only three bonds, with its remaining coordination sphere filled by three solvent molecules.30 The material {[Zn(OOCClH3C6Fc)2(H2O)3](H2O)}n features [–Zn2+–O2−–Zn2+–]∞ chains with each Zn2+ site bound to two bridging carboxylates that are oriented trans from each other.31 These otherwise four-coordinate Zn2+ ions include two ligated water molecules and can be replaced by Pb2+, Cd2+, Cu2+, Ni2+, Co2+, Mn2+, or Cr2+.
Conversely, SBUs with metal sites that are octahedrally coordinated by the framework ligands and have no terminal solvent species typically do not undergo cation exchange. For instance, of the two crystallographically distinct Zn2+ sites in MFU-4l, the ion attached through six bonds to the framework does not exchange for Co2+. In the MOF known as porph@MOM-10-Cd ([Cd6(BPT)4Cl4(H2O)4]·[C44H36N8CdCl]·[H3O]), one Cd2+ is coordinatively saturated in octahedral fashion by framework ligands, while the other site contains a solvent ligand.32 Cu2+ only exchanges the latter completely. Unlike the previous two examples where the extent of cation exchange could be compared between two types of coordination environments within the same MOF, we do not have this vantage point for analysing [(CH3)2NH2]15[(Cd2Cl)3(TATPT)4]·12DMF·18H2O, where a single nine-coordinate Cd2+ ion is present in the asymmetric unit.33 Consistent with the generally small degree of exchange for more highly coordinated ions, Cd2+ centers in this structure exchange with Cu2+, Co2+, Ni2+, and Zn2+, but only to a small degree. Finally, the MOFs known as UiO-6634 (Zr6O4(OH)4(BDC)12) and MIL-53(Al)-Br34 (Al(OH)(BDC-Br)) also contain SBUs with metals bound to the framework in high coordination and do not exchange for other ions completely. Given that Zr4+ and Al3+ form some of the strongest metal–oxygen bonds among the metals incorporated into MOFs, it is remarkable that they undergo any extent of cation exchange.
Metal sites that are coordinately saturated by the framework and undergo complete cation exchange might do so because their weak field ligands dissociate readily. A ligand field analysis of Ni-MOF-5 indicates that the MOF-5 framework is a stronger ligand than halides, but is significantly weaker than coordinating solvents such as DMSO or DMF. Considering that in MOF-5 the ligand field is weak despite the presence of an O2− in the coordination sphere, this study suggests that SBUs comprised of only carboxylates form weak bonds with late transition metal ions. For example, the metal sites in both Na0.25[(CH3)2NH2]1.75[M(HMBM)2]·xSolvent35 (M = Cd2+ or Cu2+) and porph@MOM-11-Cd36 ([Cd4(BPT)4]·[Cd(C44H36N8)S]·S) (S = MeOH, H2O) are bound to six carboxylate ligands, yet exchange for Cu2+ at 96% of the sites, virtually quantitatively. Here, the weak field carboxylates might dissociate and permit cation exchange despite the metal sites being octahedrally coordinated. The almost complete exchange of seemingly coordinatively saturated ions is also observed with ligands other than carboxylates. Unlike [(CH3)2NH2]15[(Cd2Cl)3(TATPT)4]·12DMF·18H2O or MIL-53(Al)-Br, which exchange partially, the environments of these SBUs typically do not contain single atom μ2 ligands, such as O2− or Cl−. The “paddlewheel” SBUs of PCN-921, SUMOF-1-Zn (Zn6(BTB)4(BP)3),37 and M6(BTB)4(BP)3 (M = Co, Cu, Ni) contain 4,4′-bipyridine bridging to an adjacent SBU, rather than a solvent molecule at the axial position.38 Despite lacking solvent ligands, the metal sites in these materials exchange for Cu2+ completely. Metal ions in the SBUs of [Zn(4,4′-BP)2-(FcphSO3)2]n,40 {[Cd(BP)2(FcphSO3)]·(CH3OH)4}n,41 and {[M(BTTN)2(H2O)2]·2(PF6)·pyrene·2(H2O)}n (ref. 42) (M = Cd2+, Zn2+) can be entirely replaced by Cu2+, despite being bound to four 4,4′-bipyridine ligands and two carboxylates. Similarly, the six-coordinate metal sites in {[Cd2(BTX)2(BDC)2]H2O}n and [Co3(BTX)4(BDC)3(H2O)4]n (ref. 39) can be replaced by Cu2+, even though they are bound to bridging carboxylates and triazole ligands. None of these examples contain chains bridged by single atom μ2 ligands, and undergo complete exchange despite being coordinatively saturated by framework ligands. Importantly, the family of MOFs known as M-MOF-74 feature SBUs with [–M2+–O2−–M2+–]∞ chains and is conspicuously absent from the known examples of cation exchange.
Taken together, these observations begin to reveal the factors that enable cation exchange at certain SBUs. The pervasiveness of partially solvated SBUs among these examples and the coordination changes that MM-BTT undergoes upon solvation call into question whether the metal sites in MOF-5, ZIF-8, and MFU-4l are indeed unsaturated when surrounded by a solvent. If geometric flexibility and the ability of metal sites to interact with the solvent are requisites for cation exchange, then we can begin to sketch a mechanism for this process (see Scheme 1). Perhaps the metal ion does not readily leave the cluster as a dissociated cation. Instead, solvent molecules might associate step-wise to the exiting metal ion as it remains partially bound to the cluster. Furthermore, since cation exchange occurs in “paddlewheel” structures with either a solvent or 4,4′-bipyridine at the axial position of the metal site, the clusters must be flexible enough to accommodate the inserting metal ions or, alternatively, the carboxylates and 4,4′-bipyridine must readily dissociate without compromising the framework. Alternatively, we may construct a model where the MOF ligands dynamically dissociate from metal sites in the presence of coordinating solvents and thereby enable cation exchange. The ability of coordinatively saturated metal sites to exchange when surrounded by weak field carboxylates, but not bridging O2− ligands, suggests that cation exchange might become a predictable tool by quantifying the interaction of the SBU with the metal ions. If future studies measured the ligand field strength of the exchangeable SBUs, then general trends might emerge and aid our understanding of the cation exchange process. This might be achieved by UV-vis spectroscopy, for instance, in a manner analogous to classic solution studies of homoleptic complexes.43
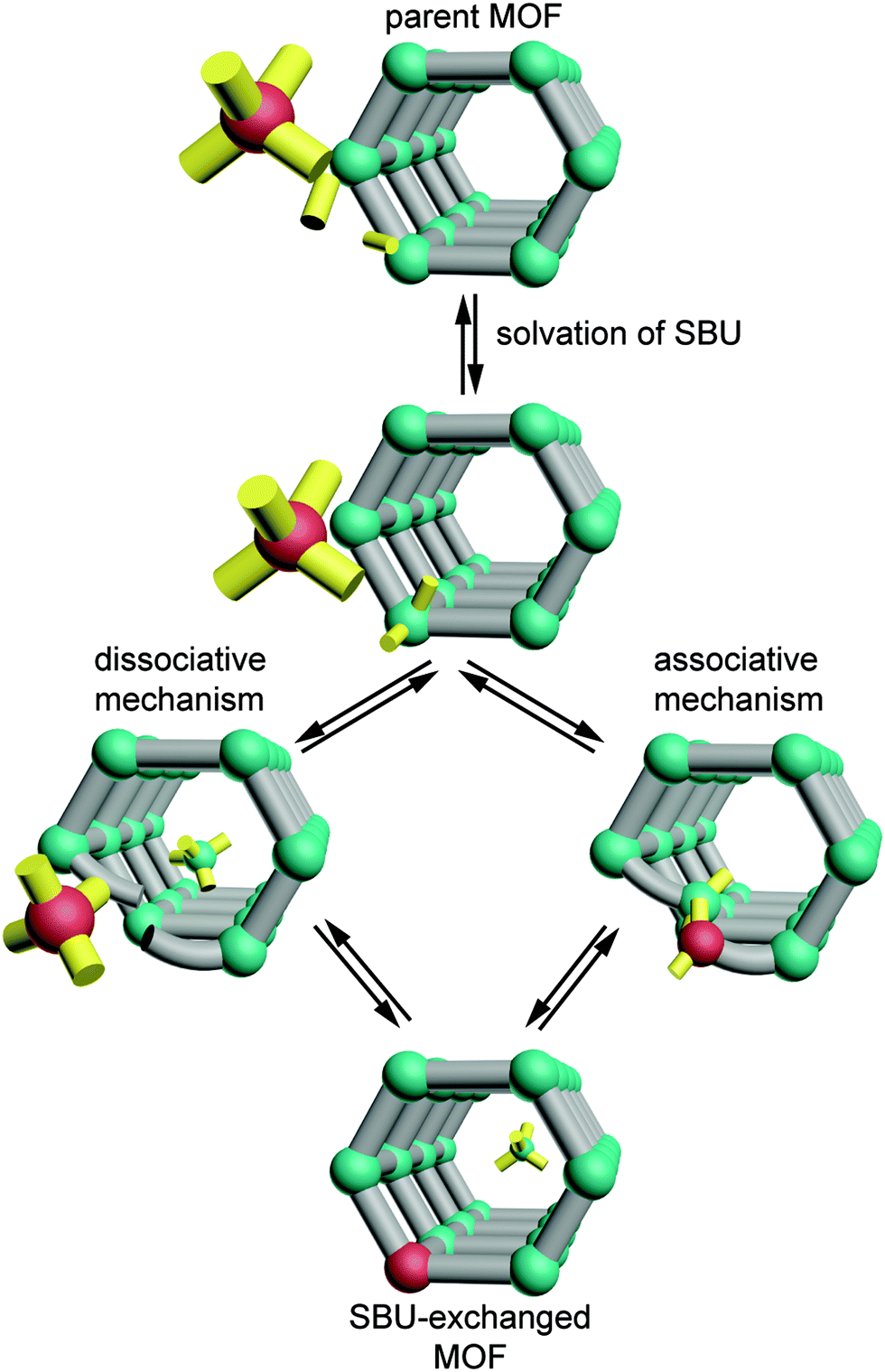 | ||
| Scheme 1 Simplified mechanistic pathways for cation exchange at MOF SBUs. Green and red spheres represent exiting and inserting metal ions, respectively. Organic linkers are shown in gray and solvent is depicted in yellow. | ||
Which ions exchange into SBUs?
To program physical properties into a SBU through cation exchange, we must be able to predict whether a particular cation will replace another and to what extent. By controlling the initial concentration of the inserting cation solution, the thermodynamic equilibria of the exchange processes could be controlled to furnish heterometallic SBUs for specific catalytic applications. Clusters with unusual magnetic and electronic properties could be assembled through judicious cation exchange that might be otherwise impossible through direct synthesis. Attaining this depth of understanding can be achieved by comparing how a wide variety of cations replace SBUs in a particular MOF structure. Unfortunately, few studies report the results of more than one exchange and almost none report unsuccessful attempts, which in the context of mechanistic investigations can be equally informative.Most examples of cation exchange at SBUs involve Cu2+ replacing Zn2+ or Cd2+. The Zn2+ ions in porph@MOM-11-Zn, PCN-921, NTU-101-Zn, and PMOF-2 are known to undergo a high degree of substitution for Cu2+, with no reported attempts to exchange with other ions.22,23,29,36 Similarly, the Cd2+ ions in {[Cd2(BTX)2(BDC)2]H2O}n and [Cd(BTX)2Cl2]n can be totally replaced by Cu2+, but their exchange with other ions is unknown.39 In the isostructural variants of {[M2(BDCPPI)(DMF)3]·7DMF·5H2O}n (M = Cd2+ or Zn2+) both Cd2+ and Zn2+ are fully replaced by Cu2+.30 The Zn2+ ions in Zn-HKUST-122 and Zn2+ or Cd2+ ion in {[M(BP)2(FcphSO3)]·(CH3OH)4}n (M = Zn2+ or Cd2+) both exchange for Cu2+,40,41 though not to completion. These reports do not always test whether the cation exchange is reversible, but the reversibility of a process lends insight into the relative thermodynamic stability of the exchanged variants. We do know, however, that reversible Zn2+ exchange into NTU-101-Cu29 or Cu-PMOF-222 is impossible, while Zn2+ can partially replace Cu2+ in the framework of porph@MOM-11-Cu, but not at the porphyrin metalloligand.36
When information is available for Cu2+ as well as other transition metals exchanging in the same host structure, Cu2+ typically inserts to the greatest extent and is the least reversible. In {[Zn2(BDCPPI)(DMF)3]·7DMF·5H2O}n, 97% of the Zn2+ sites are exchangeable for Cu2+, but none can be replaced by Ni2+, Co2+, or Cd2+.30 Similarly, Cu2+ exchanges Zn2+ in [Zn7((PPBOTCDITC))3(H2O)7]n [Zn5((PPBOTCDITC))3(H2O)5]n·xSolvent24 and Cd2+ in Na0.25[(CH3)2NH2]1.75[Cd(HMBM)2]·xSolvent,35 but Ni2+ or Co2+ do not. Cu2+, Co2+, and Ni2+ replace Zn2+ in SUMOF-1-Zn, but only Cu2+ replaces all the sites, while Co2+ replaces 35% and Ni replaces 38% after an identical number of times.37 In the reverse process, the all-Zn2+ material can be regenerated from the Co2+ or Ni2+ variants after 7 days, but Zn2+ can replace only 38% of the Cu2+ sites in SUMOF-1-Cu. Furthermore, the Co2+, Ni2+, and Zn2+ materials are all interchangeable through reversible cation exchange, while their replacement for Cu2+ is irreversible. Similarly, the isostructural series M6(BTB)4(BP)3 (M = Zn2+, Co2+, or Ni2+) generate a Cu2+ analogue through irreversible cation exchange, while the Co2+ and Zn2+ variants are completely interchangeable.38 Despite the overall low degree of cation exchange in [(CH3)2NH2]15[(Cd2Cl)3(TATPT)4]·12DMF·18H2O, Cu2+ still replaced Cd2+ more than Co2+, Ni2+, or Zn2+ did.33 Perhaps most tellingly, there is only one instance in which Cu2+ is replaced by other transition metal ions: Zn2+ and Co2+ both exchange the Cu2+ sites in Cu8(BIM)16, albeit only 21% and 15% of the Cu2+ sites are replaced, respectively.44
SBUs in which a variety of cations are exchanged but are not fully exchangeable by Cu2+ still demonstrate preference for Cu2+. All the Mn2+ sites of POST-65(Mn) can be replaced by Co2+, and Ni2+ but not Cu2+. Nevertheless, Mn2+ can replace only 34% of the Cu2+, whereas the Co2+ and Ni2+ processes are fully reversible.26 The Mn2+ ions in the SBU of the material known as MnMn-BTT are exchangeable for Cu2+ and Zn2+, with Cu2+ replacing Mn2+ to the fullest extent.45 A notable exception to the apparent dominance of Cu2+ is porph@MOM-10-Cd, where Mn2+ replaces all Cd2+ sites, while Cu2+ replaces 76%.32
Outside the first transition series, Pb2+ and Cd2+ tend to exchange preferentially into SBUs over Cu2+ and other transition metals. The extent that Zn2+ sites can be exchanged in {[Zn(OOCClH3C6Fc)2(H2O)3](H2O)}n follows the order Pb2+ > Cd2+ > Cu2+ > Mn2+ > Ni2+ > Co2+ > Cr2+.31 In a related system, Pb2+ replaces 75% of the Zn2+ sites of [Zn(4,4′-BP)2-(FcphSO3)2]n, whereas Cu2+ replaces just 50%.40
Although little rigorous work has been done to interrogate the kinetics of cation exchange in MOFs, the present studies indicate that the rate of substitution into a particular SBU depends on the identity of the metal ions. For MOF-5, Ni2+ requires up to a year to replace 25% of the original Zn2+ sites, whereas Cr2+ and Fe2+ reach that extent in a week. Furthermore, the exchange with Mn2+ is so rapid at room temperature that the process destroys the crystals and only proceeds in a controlled fashion when conducted at −35 °C.17 Though the resulting materials are isostructural, Cu2+ fully exchanges Zn6(BTB)4(BP)3 in 2 days, Co6(BTB)4(BP)3 in 1 day, and Ni6(BTB)4(BP)3 in 15 days.38 Pb2+ replaces Cd2+ in 7 days for Cd1.5(H3O)3[(Cd4O)3(HMTT)8]·6H2O, yet Co2+, Ni2+, and Cu2+ require 12 days to replace Mn2+ in a similar structure.25
The dominance of Cu2+ among these examples and the preference for Cd2+ and Pb2+ over Cu2+ might be explained by differences in electronegativity. Calculations suggest that Pb2+ has the lowest electronegativity among the cations that undergo exchange, followed by Mn2+ and Cd2+. Cu2+, on the other hand, has the highest electronegativity.46 Perhaps Pb2+, Mn2+, and Cd2+ ions form labile ionic bonds, allowing them to perform cation exchange faster. This kinetic argument might explain why Pb2+ and Cd2+ exchange more sites than Cu2+ in {[Zn(OOCClH3C6Fc)2(H2O)3](H2O)}n (ref. 31) and why Mn2+ replaces more sites than Cu2+ in porph@MOM-10-Cd.32 If these experiments were allowed to go on longer, Cu2+ might have been exchanged completely. The high electronegativity of Cu2+ would enable it to form bonds that are more covalent and thermodynamically stable. A greater thermodynamic driving force would be consistent with the irreversibility and high degree of substitution of Cu2+ exchanges. This trend in electronegativity is also consistent with cation preference following the Irving–Williams series,47 since labile Mn2+ species and thermodynamically stable Cu2+ bonds constitute either end of the series.
Even with the general trends exposed above, we cannot yet predict whether a particular cation will replace another and to what extent. In the absence of more experimental and empirical evidence, quantum chemical calculations could prove useful in predicting which cations form more thermodynamically stable complexes in a given SBU. With the computed energy values, thermodynamic equilibria could be manipulated to engineer SBUs with certain mixed-metal compositions. The mechanism of cation exchange, on the other hand, will need to be studied on a case-by-case basis. With a more detailed understanding of how the process depends on the identity of the cation, one might control the kinetics and harness cation exchange as a synthetic tool.
How does the framework influence the exchange?
To rationalize how cation exchange occurs at SBUs, one must remember that SBUs are embedded in the lattice of a MOF. Although they often resemble molecular clusters, they do not possess the degrees of freedom of molecules in solution. Thus, the lattice limits the geometrical distortions available to an SBU. We must also understand that the cation exchange process must occur in the MOF pores. The process is therefore likely influenced by diffusion and pore size effects. We do not know how these intrinsic features of MOFs impact cation exchange, but any mechanistic understanding must account for them. The scant observations already suggest the MOF lattice impacts the cation exchange and vice versa.An important evidence for this co-dependency is that certain cation exchanges can compromise the structural integrity of a framework. For instance, after Cu2+ replaces the Zn2+ sites in the materials known as {[Zn2(BDCPPI)(DMF)3]·7DMF·5H2O}n (ref. 30) and {[Zn(BTTN)2(H2O)2]·2(PF6)·pyrene·2(H2O)}n,42 the resulting crystals are known to crack. As mentioned above, especially fast exchanges into MOF-5, such as Mn2+, also cause deterioration of the crystals, which is evidenced both optically and especially through surface area measurements. For example, synthesizing Fe-MOF-5 using a solution of anhydrous FeCl2 is rapid and leads to inferior quality powder, whereas the exchange from Fe(BF4)2·xH2O is slow and gives a superior material.17 Among the cations that substitute into MOF-5, Ni2+ is the slowest and has the highest apparent surface area. Similarly, after Co2+ replaces Cd2+ in MMPF-5(Cd), the surface area decreases, possibly due to collapsed pores.48
Observations suggest that the framework itself limits the extent of cation exchange. The replacement of Zn2+ by Co2+ in Zn6(BTB)4(BP)3 occurs initially at the exterior of the crystals and replaces the interior sites after approximately a day. The authors contend that this time dependence is the result of the lattice being more flexible at the exterior, not of diffusion limitations in the framework pores.38 When rationalizing why Cu2+ exchanges 53% of the Zn2+ sites in Zn-HKUST-1 but all sites in PMOF-2, the authors invoked a similar argument: the longer linkers in PMOF-2 endow the lattice with greater flexibility, even though its SBUs are the same as in Zn-HKUST-1.22 Perhaps this reasoning might explain why the extent of cation exchange in Zn(4,4′-BP)2-(FcphSO3)2 is lower for a powder material than for single crystals.40 Larger particles might better accommodate distortions and defects introduced by the exchange process than a small one. In perhaps the most surprising case of homogeneous exchange limited by a MOF lattice, the substitution of cations in the SBU of MOF-5 is almost universally capped at 25% (i.e. only one Zn2+ ion in every Zn4O cluster). In fact, it may be surprising that the MOF-5 lattice, which has seemingly saturated pseudo-tetrahedral Zn2+ ions, enables cation exchange at all. Attempting to substitute Ni2+ into basic zinc acetate, a molecular analogue of the MOF-5 SBUs, is not possible with retention of the cluster geometry.16 Perhaps the M-MOF-74 class of materials do not undergo cation exchange because any distortion to the [–M2+–O2−–M2+–]∞ SBUs would require a large activation energy imposed by the lattice.
Predicting how a MOF framework influences the cation exchange process will become a general tool by first proceeding on a case-by-case basis. Still, knowing how a lattice inhibits or enables substitution at a SBU would allow us to design the composition of a material with greater precision.
What role does the solvent play in cation exchange?
Solvents differ along a wide variety of parameters that might be relevant to the mechanism of cation exchange at SBUs. The dielectric constant of solvent, HOMO level, molecular size, or ligand field strength might dictate how substitution occurs. When we develop a deeper understanding of this process, careful selection of the solvent might become a powerful handle for studying the rate and extent of cation exchange. Studies on the effect of employing different solvents are rarest for cation exchange in MOFs, but the available observations are still useful.{[Zn2(BDCPPI)(DMF)3]·7DMF·5H2O}n is the only exchangeable material to be tested against several solvents. Though perhaps expected because of intra-pore diffusion limitations, the results suggest that the size of the solvation sphere impacts the rate of substitution. While the exchange is fast in methanol, it is slow in acetone and does not occur in larger solvents such as DMF or 1-pentanol.30 However, solvents appear to play a mechanistic role aside from shuttling solvated cations through pores. Given that most SBUs feature coordinatively unsaturated metal sites or solvent ligands, it is significant that all exchanges involve coordinating solvents. Most use methanol, DMF, or H2O – all of which are strongly donating ligands with relatively high ligand field strengths. The Cu2+ substitution into Zn-HKUST-1 occurs more slowly in DMF than in the stronger field ligand methanol.22 Perhaps the Co2+ exchange into MMPF-5(Cd) does not go to completion because the weak field solvent, DMSO, is used.48 Based on the ligand field analysis of Ni-MOF-5,16 the lattice is a far weaker ligand than solvents used for cation exchange. If solvents act as ligands during the exchange mechanism, then they might associate with SBUs and weaken the bonds between the exiting metal ion and the framework. They might also stabilize reactive intermediates or dictate the rate at which the inserting metal ion desolvates and subsequently enters the SBU.
Systematic studies will be needed to elucidate how solvents influence the mechanistic details. Future reports should attempt their synthesis procedures with multiple solvents and plot the extent of exchange versus relevant solvent parameters. Finding a single parameter that correlates well with exchange rate would shed light on the crucial steps of the exchange process. For an example, if substitution rate in a particular MOF correlates with the dielectric constant, then perhaps the role of the solvent is to stabilize an intermediate with a large dipole moment. Each system will need to be studied individually, but with many thorough solvent investigations we could learn about the cation exchange mechanism in general.
Applications
As a research direction, cation exchange at MOF SBUs is still in its infancy, but the exchange process already has applications that are impossible to achieve through conventional synthetic routes. Most of the materials covered in this review can only be made through cation exchange. Isolating Ni-MOF-5 is possible by solvothermal synthesis, but all other variants in the (Cl)M-MOF-5 family are not. M-HKUST-1 (M = Zn2+ or Cu2+), M-PMOF-2 (M = Zn2+ or Cu2+), MIL-53(Fe)-Br, MIL-53(Al)-Br, MIL-101(Fe), MIL-101(Al), and the class of MOFs known as M6(BTB)4(BP)3 (M = Co2+, Ni2+, Cu2+, or Zn2+) are accessible through direct synthesis, but the mixed-metal derivatives have only been accessed by cation exchange. The Mn2+,20 Fe2+,49 and Cu2+ (ref. 50) variants of MM-BTT can be made directly, but cation exchange remains the only route to the Zn2+, Co2+, Ni2+-based materials.1,21The most common application for cation exchanged-MOFs is in gas storage. Installing cations with open coordination sites and open shell electronic structures enhances the adsorption interaction between the SBU and guest molecule to increase the overall gas uptake. Whether starting from CdCd-BTT21 or MnMn-BTT,1 altering the cation identity leads to tunable apparent surface areas, H2 uptake, and H2 adsorption enthalpies. So far accessible only by cation exchange, the partially exchanged Co2+ derivative exhibited an unprecedented initial enthalpy of adsorption, ΔH, of 10.5 kJ mol−1. Calculations suggest that ZnZn-BTT should exhibit the largest enthalpy of adsorption. Although only a partially substituted Zn analogue has been reported, the all-Zn material may be accessible through cation exchange.51 Soaking POST-65(Mn) in a solution of Fe2+, Co2+, Ni2+, or Cu2+ leads to isostructural analogues with enhanced H2 uptake when measured in mol mol−1. Most variants show greater ΔH than the initial 5.21 kJ mol−1 of POST-65(Mn), with POST-65(Fe) displaying a ΔH of 6.60 kJ mol−1. Each variant also displays distinct magnetic properties, with the Co2+, Ni2+, and Cu2+ materials showing antiferromagnetic coupling while the Fe2+ version exhibits ferromagnetic coupling.26 The Zn2+-variants of HKUST-1 and PMOF-2 do not show appreciable gas uptake since they are not stable to complete desolvation. The Cu2+ analogue of HKUST-1 is, on the other hand, stable to desolvation, and greater amounts of Cu2+ substitution into the Zn2+ parent material lead to significant N2 uptake indicative of greater porosity and stability.22 Similarly, the ability of M6(BTB)4(BP)3 (M = Co2+, Ni2+, or Zn2+) to adsorb N2 can be tailored by altering the ratio of any two of these cations in the structure.38 Finally, while NTU-101-Zn exhibits a BET surface area of just 37 m2 g−1, the Cu2+ variant adsorbs significant amounts of H2, CO2, and N2 to give a BET value of 2017 m2 g−1.29
The most exciting potential application of cation exchange lies in the area of small molecule reactivity and catalysis, yet catalysis at SBUs altered through cation exchange is only just emerging. Even in these examples, most reports focus on simply demonstrating reactivity or catalysis; it is unfortunately not yet common practice to show how the new SBUs compare with the state-of-the-art (heterogeneous) catalysts for a given transformation. For instance, after replacing the Cd2+ ions in porph@MOM-10-Cd with Mn2+ or Cu2+, the MOFs are capable of catalysing the oxidation of trans-stilbene to stilbene oxide and benzaldehyde in the presence of tert-butyl hydroperoxide.32 Here, the conversion and turnover number compare well to molecular Mn3+TMPyP under similar conditions. The Cu2+, Zn2+, and Co2+ variants of the helical framework known as Cu8(BIM)16 catalyse the self-coupling of 2,6-di-tert-butylphenol under ambient conditions to afford 3,3′,5,5′-tetra-tert-butyl-4,4′-diphenoquinone.44 After replacing the four exterior Zn2+ sites in the SBU of MFU-4l with Co2+, Co-MFU-4l becomes catalytically active in oxidizing CO to CO2.
Cation exchange builds a fundamentally new platform for reactivity studies because the resultant metal clusters of SBUs are often unusual coordination motifs that are difficult or impossible to achieve as solution-phase molecules. For example, no molecule is known to stabilize Ni2+ or Co2+ in the two-coordinate environment conferred by MM-BTT. The metal species in the (Cl)M-MOF-5 family are without a precedent in both materials and molecules because of the unusual all-oxygen, dianionic, and tripodal ligand field in the MOF-5 SBU. These sites are some of the few examples of divalent metal ions in three-fold symmetric tetradentate environments. A ligand field analysis of Ni-MOF-5 indicates that MOF-5 is by far the strongest ligand to stabilize Ni2+ in a pseudo-tetrahedral geometry, which is remarkable because ligand fields of similar strength coerce Ni2+ to assume a square planar configuration. Preliminary studies demonstrate that these unusual species perform small molecule activation without compromising the integrity of the lattice. The Fe2+ centers in Fe-MOF-5 react with NO to generate an unusual ferric nitrosyl, which is the only example of electron transfer to NO in a MOF and the only example of a ferric nitrosyl in an all-oxygen environment.
Viewing the cation exchanged SBUs as molecular entities will be a useful perspective for conceiving new applications in reactivity and catalysis. Reimagining SBUs as coordination pockets for various transition metal ions constructs an entirely new platform for coordination and redox chemistry. SBUs will act as superior catalysts only by treating them as an unusual ligand environment. This viewpoint inspired the use of open coordination and open shell metal ions to enhance H2 uptake. Novel porous magnets might result from installing particular metal ions into desirable molecular entities. Only a few reports have investigated the applications of cation exchange, but the ability to insert reactive metal ions into specific geometries should enable chemistry that is otherwise impossible to achieve.
Outlook
Being able to substitute specific metal ions into predefined environments is a level of control uncommon to solid state synthetic chemistry. Cation exchange into the SBUs of MOFs is already unlocking materials with unprecedented properties that cannot be achieved otherwise. However, harnessing this process as a predictive synthetic tool will require understanding its mechanistic details. The available experimental observations are insufficient to draw meaningful conclusions about how the process transpires in even a particular material. Future studies, including those we proposed here, will uncover trends that will make this technique predictive. We recommend that if a MOF appears active for cation exchange, then the substitution should be attempted for a variety of metal species and solvents to tease out trends. The rate and extent of exchange under these different conditions could be compared against various chemical properties of the metal ions and solvents to find parameters that are most relevant to the mechanism. Future studies should also report exchange conditions that did not work along with those that did. Such detailed, seemingly obscure, observations might prove critical in uncovering a deeper understanding of cation exchange.Discovering how SBUs undergo cation exchange will teach us about MOF chemistry and dynamics in general. For example, if coordinating solvents enable the exchange process by binding to metal sites in SBUs, perhaps this will reveal that MOFs dynamically interact with solvents and are not as rigid as commonly assumed or as portrayed by X-ray crystal structures. Elucidating these sorts of fundamentals about MOFs will have profound consequences for any of their applications. Understanding how the lattice flexibility or the symmetry of the SBU limits the geometrical distortions of the metal site will shape future catalytic studies of MOFs. The reactivity of metal sites could be controlled with the fine level of control we enjoy with molecular catalysts, but with the unexplored solid-state ligand environment of MOFs. Cation exchange at the SBUs of MOFs promises a new landscape of materials chemistry and our investigations have only just begun.
Addendum
During the preparation and review of this manuscript, several relevant reports were published that reinforce the trends stated above. Consistent with our comments on the types of SBUs that undergo exchange, these new examples feature metal sites that are capable of higher coordination numbers. Thus, one of the two replaceable Zn2+ sites in JUC-118 ([Zn4(TIAPy)·(H2O)4·(EGME)2]) is 4-coordinate,52 as are two of the four unique Zn2+ sites in [Zn4(DCPP)2(DMF)3(H2O)2]n,53 while one of the two exchangeable Zn2+ sites in [Zn3(CBAI)2(DMF)2]·2DMF is 5-coordinate.54 In addition, the SBUs in these new examples contain metal sites with bound solvent molecules. DMSO occupies a coordination site of the 6-coordinate Zn2+ in JUC-118, two DMF molecules are bound to a Zn2+ atom in the asymmetric unit of [Zn3(CBAI)2(DMF)2]·2DMF, and two of the four Zn2+ sites in [Zn4(DCPP)2(DMF)3(H2O)2]n are ligated by DMF or H2O. In one of the most complete mechanistic reports of cation exchange in a MOF, magnetic measurements revealed that the first Zn2+ to exchange in [Zn4(DCPP)2(DMF)3(H2O)2]n is the one with most bound solvent. Even though some of these exchangeable metal sites are coordinatively saturated, the substitution presumably happens only because the ligands are weak-field carboxylates, as explained before. Like most examples of cation exchange, Cu2+ replaces the Zn2+ atoms in these MOFs completely and, in the case of JUC-118 and [Zn3(CBAI)2(DMF)2]·2DMF, does so irreversibly. While Ni2+ and Co2+ do not exchange at all into [Zn3(CBAI)2(DMF)2]·2DMF, they insert only into the 6-coordinate sites of [Zn4-(DCPP)2(DMF)3(H2O)2]n, due to the preference for these geometries. The new reports also offer insight into the role of the solvent. For instance, Cu2+ inserts into ([Zn4(TIAPy)·(H2O)4·(EGME)2]) in the presence of 2-methoxyethanol but not common solvents such as DMF, MeOH, or acetone. Since 2-methoxyethanol also induces a single crystal-to-single crystal transformation, perhaps it allows cation exchange by facilitating bond rupture between the Zn2+ and carboxylate ligands in the MOF. A recent report also investigated the solvent dependence of Co2+ exchanging into MFU-4l and Ni2+ into MOF-5. By plotting the rates of exchange against a variety of solvent parameters, the exchange of Co2+ into MFU-4l appeared to be limited by the ability of the solvents to solvate the exiting Zn2+ ions, while the Ni2+ exchange into MOF-5 was limited by the ability of the solvent to desolvate the inserting Ni2+ ions.55 The publication of these reports in just the past few months speaks of the burgeoning interest in this field, while offering observations that reinforce the trends we propose in this review.Abbreviations
| BDC | 1,4-p-Benzenedicarboxylate |
| BDCPPI | N,N′-Bis(3,5-dicarboxyphenyl)pyromellitic diimide |
| BIM | 4′-[4-Methyl-6-(1-methyl-benzimidazolyl-2-group)-2-n-propyl-benzimidazolyl methyl] |
| BP | 4,4′-Bipyridine |
| BPT | Biphenyl-3,4′,5-tricarboxylate |
| BTB | 1,3,5-Benzenetribenzoate |
| BTC | 1,3,5-Benzenetricarboxylate |
| BTDD | Bis(1,2,3-triazolo-[4,5-b],[4′,5′-i])dibenzo-[1,4]-dioxin |
| BTT | 1,3,5-Tris(tetrazol-5-yl)benzene |
| BTTN | Benzene-1,3,5-triyltriisonicotinate |
| BTX | 1,4-Bis(triazol-1-ylmethyl)benzene |
| CBAI | 5-(4-Carboxybenzoylamino)-isophthalate |
| Cl2Im | Dichloroimidazolate |
| DCPP | 4,5-Bis(4′-carboxylphenyl)-phthalate |
| EGME | 2-Methoxyethanol |
| ETTB | 4′,4′′′,4′′′′′,4′′′′′′′-Ethene-1,1,2,2-tetrayltetrakis[1,1′-biphenyl]-3,5-dicarboxylate |
| FcphSO3 | m-Ferrocenyl benzenesulfonate |
| HMTT | 5,5′,10,10′,15,15′-Hexamethyltruxene-2,7,12-tricarboxylate |
| HMBM | 2-Hydroxymethyl-4,6-bi(2′-methoxyl-4′-(2′′-1′′-carboxyl)-ethylene)-1,3,5-mesitylene |
| MeIm | 2-Methylimidazolate |
| O3SFcSO3 | Ferrocene-1,1′-disulfonate |
| PPBOTCDITC | N-Phenyl-N′-phenyl bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxydiimide tetracarboxylate |
| TADYDI | 5,5′-(1,2,3-Triazole-1,4-diyl)-diisophthalate |
| TATPT | 2,4,6-Tris(2,5-dicarboxylphenyl-amino)-1,3,5-triazine |
| TDCPEB | 1,3,5-Tris(3,5-dicarboxylphenylethynyl)benzene |
| TIAPy | 1,3,6,8-Tetrakis(3,5-isophthalate)pyrene (H8TIAPy) |
Acknowledgements
Our work on cation exchange in MOFs is partially supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0006937. MD thanks 3M, the Sloan Foundation, and the Research Corporation for Science Advancement (Cottrell Scholars) for non-tenured faculty funds. CKB is partially supported through an NSF Graduate Research Fellowship Program through Grant 1122374. We also thank the MIT-Hayashi (MISTI) Seed Fund for travel funds.Notes and references
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef PubMed.
- K. A. Misra, Introduction to Geochemistry: Principles and Applications, Wiley, West Sussex, 2012 Search PubMed.
- H. Jaffe, Crystal Chemistry and Refractivity, Dover, 1988 Search PubMed.
- A. Putnis, Mineral. Mag., 2002, 66, 689–708 CrossRef CAS PubMed.
- V. M. Goldschmidt, J. Chem. Soc., 1937, 655–673 RSC.
- A. E. Ringwood, Geochim. Cosmochim. Acta, 1955, 7, 189–202 CrossRef CAS.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS PubMed.
- M. Eigen and R. Wilkins, Mechanisms of Inorganic Reactions, American Chemical Society, Washington, D.C., 1965, vol. 49 Search PubMed.
- K. S. Hagen, D. W. Stephan and R. H. Holm, Inorg. Chem., 1982, 21, 3928–3936 CrossRef CAS.
- V. Autissier and R. A. Henderson, Inorg. Chem., 2008, 47, 6393–6403 CrossRef CAS PubMed.
- H. Li and J. D. Otvos, Biochemistry, 1996, 35, 13929–13936 CrossRef CAS PubMed.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5468 CAS.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014 10.1039/c4cs00076e.
- C. K. Brozek and M. Dincă, Chem. Sci., 2012, 3, 2110–2113 RSC.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- D. Denysenko, T. Werner, M. Grzywa, A. Puls, V. Hagen, G. Eickerling, J. Jelic, K. Reuter and D. Volkmer, Chem. Commun., 2012, 48, 1236–1238 RSC.
- M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef PubMed.
- J.-H. Liao, W.-T. Chen, C.-S. Tsai and C.-C. Wang, CrystEngComm, 2013, 3377–3384 RSC.
- X. Song, S. Jeong, D. Kim and M. S. Lah, CrystEngComm, 2012, 14, 5753–5756 RSC.
- Z. Wei, W. Lu, H.-L. Jiang and H.-C. Zhou, Inorg. Chem., 2013, 52, 1164–1166 CrossRef CAS PubMed.
- Z.-J. Zhang, W. Shi, Z. Niu, H.-H. Li, B. Zhao, P. Cheng, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2011, 47, 6425–6427 RSC.
- S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed.
- Y. Kim, S. Das, S. Bhattacharya, S. Hong, M. G. Kim, M. Yoon, S. Natarajan and K. Kim, Chem. – Eur. J., 2012, 18, 16642–16648 CrossRef CAS PubMed.
- P. Á. Szilágyi, P. Serra-Crespo, I. Dugulan, J. Gascon, H. Geerlings and B. Dam, CrystEngComm, 2013, 15, 10175–10178 RSC.
- J. Li, P. Huang, X. Wu, J. Tao, R.-B. Huang and L. Zheng, Chem. Sci., 2013, 4, 3232–3236 RSC.
- X.-J. Wang, P.-Z. Li, L. Liu, Q. Zhang, P. Borah, J. D. Wong, X. X. Chan, G. Rakesh, Y. Li and Y. Zhao, Chem. Commun., 2012, 48, 10286–10288 RSC.
- T. K. Prasad, D. H. Hong and M. P. Suh, Chem. – Eur. J., 2010, 16, 14043–14050 CrossRef CAS PubMed.
- J. Li, L. Li, H. Hou and Y. Fan, Cryst. Growth Des., 2009, 9, 4504–4513 CAS.
- Z. Zhang, L. Zhang, L. Wojtas, P. Nugent, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 924–927 CrossRef CAS PubMed.
- C.-Y. Sun, X.-L. Wang, X. Zhang, C. Qin, P. Li, Z.-M. Su, D.-X. Zhu, G.-G. Shan, K.-Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717–2724 Search PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- H.-M. Zhang, J. Yang, Y.-C. He and J.-F. Ma, Chem. – Asian J., 2013, 8, 2787–2791 CrossRef CAS PubMed.
- Z. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 5982–5985 CrossRef CAS PubMed.
- Q. Yao, J. Sun, K. Li, J. Su, M. V. Peskov and X. Zou, Dalton Trans., 2012, 41, 3953–3955 RSC.
- X. Song, T. K. Kim, H. Kim, D. Kim, S. Jeong, H. R. Moon and M. S. Lah, Chem. Mater., 2012, 24, 3065–3073 CrossRef CAS.
- S. Huang, X. Li, X. Shi, H. Hou and Y. Fan, J. Mater. Chem., 2010, 20, 5695–5699 RSC.
- L. Mi, H. Hou, Z. Song, H. Han and Y. Fan, Chem. – Eur. J., 2008, 14, 1814–1821 CrossRef CAS PubMed.
- L. Mi, H. Hou, Z. Song, H. Han, H. Xu, Y. Fan and S. Ng, Cryst. Growth Des., 2007, 7, 2553–2561 CAS.
- G. Mukherjee and K. Biradha, Chem. Commun., 2012, 48, 4293–4295 RSC.
- A. B. P. Lever and S. T. Metal, J. Chem. Educ., 1968, 45, 711–712 CrossRef CAS.
- J. Zhao, L. Mi, J. Hu, H. Hou and Y. Fan, J. Am. Chem. Soc., 2008, 130, 15222–15223 CrossRef CAS PubMed.
- C. K. Brozek, A. F. Cozzolino, S. J. Teat, Y. Chen and M. Dincă, Chem. Mater., 2013, 25, 2998–3002 CrossRef CAS.
- K. Li and D. Xue, J. Phys. Chem. A, 2006, 110, 11332–11337 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- X.-S. Wang, M. Chrzanowski, L. Wojtas, Y.-S. Chen and S. Ma, Chem. – Eur. J., 2013, 19, 3297–3301 CrossRef CAS PubMed.
- K. Sumida, S. Horike, S. S. Kaye, Z. R. Herm, W. L. Queen, C. M. Brown, F. Grandjean, G. J. Long, A. Dailly and J. R. Long, Chem. Sci., 2010, 1, 184–191 RSC.
- M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419–1422 CrossRef PubMed.
- K. Sumida, D. Stück, L. Mino, J.-D. Chai, E. D. Bloch, O. Zavorotynska, L. J. Murray, M. Dincă, S. Chavan, S. Bordiga, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2013, 135, 1083–1091 CrossRef CAS PubMed.
- N. Zhao, F. Sun, H. He, J. Jia and G. Zhu, Cryst. Growth Des., 2014, 14, 1738–1743 CAS.
- W. Meng, H. Li, Z. Xu, S. Du, Y. Li, Y. Zhu, Y. Han, H. Hou, Y. Fan and M. Tang, Chem. – Eur. J., 2014, 20, 2945–2952 CrossRef CAS PubMed.
- Y.-F. Niu, W. Zhao, J. Han and X.-L. Zhao, CrystEngComm, 2014, 16, 2344–2347 RSC.
- C. K. Brozek, L. Bellarosa, T. Soejima, T. V. Clark, N. López and M. Dincă, Chem. – Eur. J., 2014, 20 DOI:10.1002/chem.201402682.
| This journal is © The Royal Society of Chemistry 2014 |