 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toxic gas removal – metal–organic frameworks for the capture and degradation of toxic gases and vapours
Elisa
Barea
,
Carmen
Montoro
and
Jorge A. R.
Navarro
*
Departamento de Química Inorgánica, Universidad de Granada, Av. Fuentenueva, S/N, 18071, Granada, Spain. E-mail: jarn@ugr.es; Web: http://www.ugr.es/local/jarn Tel: +34 958 248 093
First published on 7th April 2014
Abstract
The release of anthropogenic toxic pollutants into the atmosphere is a worldwide threat of growing concern. In this regard, it is possible to take advantage of the high versatility of MOFs materials in order to develop new technologies for environmental remediation purposes. Consequently, one of the main scientific challenges to be achieved in the field of MOF research should be to maximize the performance of these solids towards the sensing, capture and catalytic degradation of harmful gases and vapors by means of a rational control of size and reactivity of the pore walls that are directly accessible to guest molecules.
 Elisa Barea (left), Carmen Montoro (middle) and Jorge A. R. Navarro (right) | Elisa Barea obtained her PhD in 2004 at the University of Granada. After a postdoctoral stage in the University of Milan (2006–2008), she got a research contract (Ramon y Cajal Grant) to join the University of Granada. Since April 2011, she is an Associate Professor in the Department of Inorganic Chemistry. Her current research focuses on the study of porous coordination polymers with potential applications in air purification and drug-delivery. The quality of her research has been recognized by the Excellence Research Award of the University of Granada (2011) and the Young Researcher Award of the Spanish Royal Society of Chemistry (2012). |
Carmen Montoro born in 1983 graduated in Chemical Engineering in 2008, and received her PhD in Chemistry at the University of Granada in 2013 under the supervision of Prof. Jorge A. R. Navarro and Dr Elisa Barea. Her research focuses on novel porous materials for gas separation and storage and air purification. |
Jorge A. R. Navarro born in 1969 is Full Professor of Inorganic Chemistry at the University of Granada since 2010. His research is focused on the synthesis and applications of discrete and extended polygonal coordination assemblies with molecular recognition properties suitable for applications in the fields of environmental and biomedical chemistry. He is the author of ca. 75 publications and the quality of his research has been recognised by the Excellence Research Award of the University of Granada (2002, 2009) and the RSEQ-Sigma-Aldrich Young Researcher Award (2006). He is also a member of the editorial board of ICA Journal. |
Key learning pointsThe reader will learn:About the major anthropogenic toxic gases and vapors emitted to the atmosphere together with their toxicity levels. About the performance of metal–organic frameworks in environmental remediation processes. About the key features of the porous structure and functionality of MOFs that determine the capture of major atmospheric pollutants. About the relevance of catalytically active MOFs in the degradation of toxic gases and vapors into harmless substances. About the importance of creating hybrid composites containing MOFs in order to further improve the performance of MOF materials for the capture and degradation of toxic gases and vapors. |
Introduction
The release of anthropogenic toxic pollutants into the atmosphere, which include products of combustion/chemical reactions, leaks of harmful industrial gases and vapors as well as the deliberate emission of chemical warfare agents, is a worldwide risk of growing concern. Common hazardous compounds such as NOx, SOx, CO, H2S, NH3, other nitrogen (e.g. hydrogen cyanide) or sulfur-containing compounds (e.g. organothiols), hydrocarbons, volatile organic compounds (benzene, toluene, methanol, etc.) are of major concern for environmental air pollution. The main sources of these gases are anthropogenic. For example, emissions of SO2, NO2 and CO are mainly due to the burning of fossil fuels that cover the current energy demand. SOx and NOx are involved in the formation of photochemical smog and acid rain, which are a major threat to the environment and health. H2S is another poisonous, corrosive and odorous gas. It is naturally occurring in both natural gas and biogas. The purification of these gases prior to their use implies the recovery of this acid contaminant. Moreover, its removal from reformate gas is of great interest for the production of ultrapure hydrogen for fuel cells as H2S damages fuel cell catalysts. NH3 is another contaminant widely used in pharmaceutical and chemical industries for various purposes, such as a fertilizer, cleaner, fermentation agent, antimicrobial agent, refrigerant, precursor of most N-containing compounds, etc.Volatile organic compounds (VOCs) are also considered a major group of air pollutants. The European Union defines them as chemicals with a vapor pressure greater than 10 Pa at 293 K, which potentially lead to photochemical smog, carcinogenesis, teratogenesis and mutagenesis. VOCs are present in indoor/outdoor air, as a consequence of the emissions from chemical process industries, building materials, cosmetics, pesticides, detergents, etc. A particularly harmful class of VOCs are the Chemical Warfare Agents, although banned since 1997 by the Chemical Weapons Convention, after the 65th country deposited its instrument of ratification, they are easily available to terrorist groups or unscrupulous governments and consequently are a major worldwide threat.
The properties of some of these harmful gases and vapors as well as the concentration levels that are likely to cause severe health effects are listed in Table 1.
| Adsorbate | BPb (K) | Comments | IDLH (ppm) |
|---|---|---|---|
| a NIOSH Pocket Guide to Chemical Hazards. Cincinnati, OH: U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Institute for Occupational Safety and Health, DHHS (NIOSH). 2005, publication no. 2005-149. b Boiling point. c 129I and 131I are common radioisotopes of iodine with respective half-life periods of ∼107 years and 8 days. When iodine radioisotopes are present in high levels in the environment from radioactive fallout, they can be absorbed through contaminated food, and will accumulate in the thyroid. As it decays, it may cause damage to the thyroid with concomitant occurrence of radiogenic thyroid cancer in later life. d CWA = Chemical Warfare Agent. | |||
| NO | 121.4 | σ-Donor/π-acceptor | 100 |
| NO2 (N2O4) | 302.2 | σ-Donor | 20 |
| N2O | 184.7 | Redox active | — |
| CO | 81.7 | σ-Donor/π-acceptor | 1200 |
| CS2 | 319.4 | Acidic | 500 |
| COS | 222.7 | Acidic | — |
| SO2 | 263.1 | σ-Donor/π-acceptor | 100 |
| H2S | 212.8 | Acidic redox active | 100 |
| (CH3)2S | 310.5 | σ-Donor | 7 |
| NH3 | 239.8 | Basic σ-donor H-bonding | 300 |
| HCN | 299.5 | Acidic | 50 |
| ClCN | 287 | — | 0.3 |
| PH3 | 185.0 | σ-Donor/π-acceptor | 50 |
| AsH3 | 211 | σ-Donor/π-acceptor | 3 |
| c-C6H12 | 353.9 | — | 1300 |
| C6H6 | 353.2 | π-Stacking | 500 |
| Ni(CO)4 | 316.2 | — | 30 |
| B2H6 | 180.7 | — | 15 |
| F2 | 85.0 | Oxidant | 25 |
| Cl2 | 239.1 | Oxidant | 10 |
| Br2 | 332 | Oxidant | 3 |
| I2 | 457.4 | Nuclear wastec | 2 |
| COCl2 | 281 | Reagent/CWAd | 2 |
| S(C2H4Cl)2 | 490.2 | CWAd | 0.7 |
| POFCH3(OC3H7) | 431.2 | CWAd | 0.1 |
| C11H26NO2PS | 571.2 | CWAd | 0.003 |
In view of all the above, the effective sensing, capture and eventually the degradation of these harmful chemicals is of great importance both for the protection of the environment and for health issues. Noteworthy, porous materials are at the forefront of minimizing the undesired effects of human activity by means of increasing the efficiency of industrial and remediation processes.1 In this regard, a new class of crystalline nanoporous materials known as metal–organic frameworks (MOFs) has emerged, as an alternative to zeolites, as a consequence of their fascinating potential applications in adsorption, sensing and catalysis.2
Consequently, one of the main scientific challenges to be achieved in the field of MOF research should be to maximize the performance of these solids towards the sensing, capture and degradation of harmful gases and vapors by means of a rational control of size and reactivity of the pore walls that are directly accessible to guest molecules.
In this review, the features of classical MOFs as well as hybrid materials based on MOFs suitable for the selective sensing, capture and catalytic degradation of toxic gases and vapors will be discussed.
1. Adsorption of toxic gases and harmful volatile organic compounds
The effective capture of harmful chemicals is of great importance both for the protection of the environment and for those who are at risk of being exposed to such substances. In this context, the use of MOFs with adequate pore size/shape is not enough for an efficient capture of unsafe gases/vapors and other more specific interactions between the hazardous adsorbates and the host are desirable. For example, the presence of open metal sites (coordinatively unsaturated metal centres) or certain functionalizations on the pore surface may enhance the adsorption selectivity/efficiency of MOFs towards certain toxic compounds via coordination bonds, acid–base/electrostatic interactions, π-complex/H-bonding formation, etc.3Adsorptive removal of harmful gases
The possible use of MOFs for the removal of gaseous sulfur compounds has been recently investigated. The presence of open metal sites and H-bonding donor–acceptor groups in these adsorbents have been proposed as suitable features for the efficient removal of S-compounds. In this context, two different classes of MIL systems (MIL stands for Materials of the Institute Lavoisier), the series MIL-53(Al, Cr) and MIL-47(V) [M(X)(bdc)] (bdc = 1,4-terephthalate; X = O for M = V4+, OH for M = Al3+, Cr3+) and the series MIL-100(Cr) [Cr3F(H2O)2O(btc)2] (btc = 1,3,5-tricarboxylate) and MIL-101(Cr) [Cr3F(H2O)2O(bdc)3] have been explored for the adsorption of H2S.4,5 Small-pore MIL-47(V) and MIL-53(Al, Cr) exhibit reversible adsorption behaviour under H2S pressure. Moreover, IR measurements and molecular simulations reveal that the MIL-47(V) framework remains rigid upon H2S adsorption up to a pressure of 1.8 MPa, while MIL-53(Cr) shows a flexible behaviour undertaking two structural transitions: the first one from a large pore form (LP) to its narrow pore version (NP) at very low pressure and the second one from the NP to the LP form at higher pressure. These structural transitions explain the two-step shape of the adsorption isotherm of MIL-53(Cr). Moreover, the arrangements of H2S molecules within the pores of these MIL species have been investigated. At an initial stage, H2S molecules form hydrogen bonds, either as a hydrogen donor (in MIL-47(V)) or hydrogen acceptor (in MIL-53(Cr)) with the μ2-O and μ2-OH groups, respectively, at the pore surface. In the case of MIL-47(V), the interaction of acidic H2S is also reinforced through other basic centers of the porous matrix, such as the oxygens of the carboxylate groups and the π electrons of the benzene rings. At higher pressures (1.8 MPa), the adsorbates arrange within the channel to form dimers and the strength of the hydrogen bonds becomes weaker. It should be noted that the strength of H2S–adsorbent interactions follow the sequence: MIL-53(Cr) NP > MIL-47(V) > MIL-53(Cr) LP. This means that MIL-47(V) would be the most promising candidate for purification of gases containing H2S as a pollutant as H2S is more easily desorbed from MIL-47(V) than from MIL-53(Cr) NP. Preliminary simulations performed on MIL-47(V) suggest a high selectivity for H2S over CH4 in the range of pressures relevant for Pressure Swing Adsorption (PSA). On the other hand, large-pore MIL-100(Cr) and MIL-101(Cr) also show a high uptake of H2S exhibiting type-I adsorption isotherms (16.7 and 38.4 mmol g−1 at 2 MPa, respectively). However, the desorption appears to be irreversible, either because of a partial destruction of the framework or because of the strong interactions between the host and the H2S molecules. Their lack of ability to regenerate hampers the use of these mesoporous MOFs for the removal of H2S from a practical application point of view.M-CPO-27 also known as M-MOF-74 [M2(2,5-dhbdc)(H2O)2], (2,5-dhbdc = 2,5-dihydroxyterephthalate M = Ni2+, Zn2+) can bind H2S relatively strongly, allowing the storage of the gas for several months.6 It should be noted that in spite of the rather aggressive chemical nature of H2S, Ni-CPO-27 only shows slight loss in crystallinity after storage and release whereas Zn-CPO-27 was amorphized by prolonged storage over a year. The study of the interaction of Ni-CPO-27 with H2S using powder X-ray diffraction and X-ray pair distribution function analysis reveals that H2S is clearly coordinated to coordinatively unsaturated metal sites. The release of H2S from these MOFs has also been studied for biological applications taking into account the signal properties of this molecule. It has been demonstrated that a proportion of the stored hydrogen sulfide can be released by exposing these materials to moisture as the water molecules replace the adsorbate at the metal site. Thus, the contact of the H2S loaded materials with a moist atmosphere gives rise to the release of around one third of the chemisorbed H2S from Ni-CPO-27 (1.8 mmol g−1) and Zn-CPO-27 (0.5 mmol g−1) after 1 h. The lower adsorptive and delivery potential of Zn-CPO-27 has been attributed to difficulties in the full activation of the sample7 and to the softer nature of Zn metal centres which gives rise to stronger and irreversible sulfur binding.
FMOF-2 [Zn2OH(2,2′-bis(4-carboxyphenyl)hexafluoropropane)1.5] is a breathing MOF, which has been tested for potential acid gas removal applications.8 Indeed, it remains structurally stable upon adsorption of SO2 and H2S reaching weight capacities of 14% and 8.3%, respectively, at 1 bar. Moreover, both gas adsorption–desorption isotherms show hysteresis confirming the breathing motion of FMOF-2 after gas adsorption–desorption.
Six isoreticular MOFs, namely MOF-5 ([Zn4O(bdc)3], bdc = 1,4-terephthalate), IRMOF-3 ([Zn4O(abdc)3], abdc = 2-amino-1,4-benzene dicarboxylate), MOF-177 ([Zn4O(btb)3], btb = 1,3,5-benzenetribenzoate), IRMOF-62 ([Zn4O(bdb)3], bdb = butadiynedibenzoate), Zn-CPO-27, and [Cu3(btc)2] (btc = 1,3,5-tricarboxylate), prove to be very effective in removing gas and vapor contaminants, such as sulfur dioxide, ammonia, chlorine tetrahydrothiophene, benzene, dichloromethane and ethylene oxide (Fig. 1).9 The behaviour of each MOF has been compared to that of BPL carbon. In this section, only the behaviour related to the adsorption of toxic gases will be discussed (for the discussion of the capture of volatile organic compounds, please see below). Noteworthy, it has been demonstrated that the presence of coordinatively unsaturated metal sites (i.e. Zn-CPO-27 and [Cu3(btc)2]) and an amino functionality in IRMOF-3 prove to be effective in adsorbing ammonia and sulfur dioxide highly outperforming the behaviour of BPL carbon by a factor of at least 59 in the case of ammonia or performing equally well, in the case of sulfur dioxide (Fig. 1).
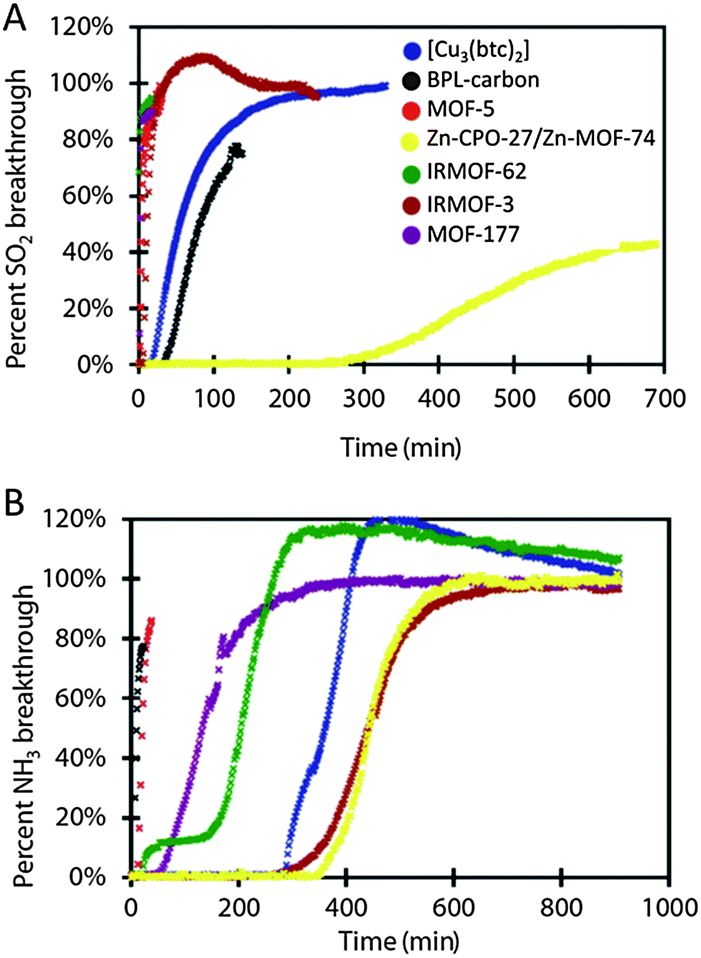 | ||
| Fig. 1 Kinetic breakthrough curves of SO2 (A) and NH3 (B) contaminants over various MOFs. Reproduced from ref. 9. | ||
The improvement of ammonia adsorption in Zn-CPO-27 and [Cu3(btc)2] is explained because of the presence of Zn2+ and Cu2+ open metal sites, which behave as Lewis acids to coordinate NH3 Lewis bases, which is not possible in the activated carbon. Deeper studies on the interaction of NH3 with [Cu3(btc)2] have postulated that [Cu3(btc)2] reacts with ammonia to form a presumed diammine–copper(II) complex, under dry conditions.10 Under humid conditions, initially, water enhances NH3 adsorption on [Cu3(btc)2] via dissolution of NH3 in the water film11 but in a latter step Cu(OH)2 and (NH4)3BTC species are formed thereby inducing an irreversible loss of structure and porosity reflected in a significant decrease in available sites and capacity over multiple dynamic adsorption cycles of breakthrough curves.10 In the case of IRMOF-3, amine groups constitute reactive electron-rich groups available for hydrogen bonding with NH3 molecules, improving the adsorption behavior in comparison with MOF-5 and BPL carbon. MOF-5, MOF-177 and IRMOF-62 lack any reactive functionality and show worse dynamic adsorption capacity for ammonia than the previously mentioned functionalized MOFs (Fig. 1). In spite of this, these MOFs show a better performance than BPL carbon towards this gas. However, they show little or no capacity for SO2.
Regarding chlorine adsorption, open metal sites in [Cu3(btc)2] are ineffective in this case as Cl2 does not typically act as a ligand. However, halogen molecules can give rise to oxidative addition reactions to Pt redox active metal centers as demonstrated in the spin cross-over (SCO) {Fe(pyz)[PtX2n(CN)4]} (n = 0 to 1) materials as reported by Kitagawa, Real and colleagues.12 Moreover, the halogen addition reaction gives rise to a precise control of the SCO transition temperature of the resulting {Fe(pyz)[PtX2n(CN)4]} materials. On the other hand, IRMOF-3 and IRMOF-62 have demonstrated considerable capacity for chlorine adsorption. Indeed, IRMOF-3 outperforms BPL carbon by a factor of 1.76 and the adsorption capacity of IRMOF-62 is in the same order of magnitude than the activated carbon. These results outline that despite the high capacities for thermodynamic gas adsorption, MOFs lacking reactive adsorption sites are ineffective in kinetic gas adsorption. In contrast, it should be noted that, in the case of MOFs with open metal sites (OMS) the presence of humidity may hamper the adsorption of the adsorbate of interest as a consequence of the irreversible coordination of water molecules to the open metal sites (see above). This feature is a main drawback for the application of MOFs containing OMS in competition with classical hydrophobic adsorbents like activated carbons (see also the section on the capture of volatile organic compounds).
Mavrandonakis and colleagues13 have recently carried out a computational study of the interaction of a series of toxic gases CO, OCS, SO2, NO, NO2, N2O, NH3, PH3 with the open metal sites of [Cu3(btc)2] (Fig. 2). The calculated adsorption energies of the toxic gases and common adsorbate molecules follow the trend NH3 > H2O > PH3 > H2S > SO2 > CO ∼ OCS ∼ CO2 ∼ NyOx > N2 > O2. Only ammonia is adsorbed more strongly on the metal sites than water, exceeding the water–framework interaction by 30 kJ mol−1. Noteworthy, although hydrogen sulfide and phosphane are less strongly bound by 10 kJ mol−1 compared to water, the interactions are significantly stronger than physisorption falling in the regime of chemisorption. These findings are in agreement with the experimental observations that ammonia, water and hydrogen sulfide decompose the framework.10
 | ||
| Fig. 2 Optimized structures of the interaction of small gases (including highly toxic ones) with the open metal sites of [Cu3(btc)2]: (a) CO, (b) CO2, (c) O2, (d) OCS(O), (e) OCS(S), (f) PH3, (g) NH3, (h) N2, (i) NO, (j) N2O(O), (k) N2O(N), (l) NO2. Reproduced from ref. 13. | ||
On the other hand, it has been demonstrated that it is possible to improve the performance of the adsorption capacities of toxic gases (NH3, H2S and NO2) using composites of MOFs (MOF-5, [Cu3(btc)2] and MIL-100(Fe)) with graphitic compounds (graphite or graphite oxide, GO) (Fig. 3).14 In the case of MOF@GO composites, the results are indicative of the strong coordination of the GO oxygen groups to the MOFs' metallic centres, which in the case of MOF-5@GO and [Cu3(btc)2]@GO gives rise to the formation of a new pore space in the interface between the carbon layers and the MOF units. This new structural feature enhances the physical adsorption capacity of these composites towards toxic gases compared to the parent MOFs. For example, an increase of 12% (for NH3), 50% (for H2S) and 4% (for NO2) is observed in the case of [Cu3(btc)2]@GO in comparison with the original [Cu3(btc)2]. However, in addition to physisorption, reactive adsorption mechanisms of NH3, H2S and NO2 also take place for these composite materials as well as for their parent MOFs (Fig. 3). In the case of the [Cu3(btc)2] based composites, the coordination of these toxic gases to the unsaturated Cu centers takes place and the formation of diammine–copper(II) complexes, CuS or Cu(NO3)2 is proposed, depending on the adsorbate. These processes are accompanied by the decomposition of the framework with a significant loss of porosity. However, for the MIL-100(Fe) and MOF-5 composites, which do not possess open metal sites, besides physisorption, acid–base reactions and hydrogen bonding take place, respectively, with the frameworks retaining their original porosity. The impact of the presence of water on the adsorption of NH3 and NO2 was also studied for these composites. As observed for the parent MOFs, water enhances NH3 adsorption in Cu-based composites and MOF-5@GO. However, in the latter case, this fact is counterbalanced by the collapse of the MOF structure caused by the formation of hydrogen bonds between the water molecules and the zinc oxide tetrahedra. However, competitive adsorption of water and NH3 is observed on MIL-100(Fe)@GO. The same competitive behavior is observed for water and NO2 in [Cu3(btc)2]@GO.
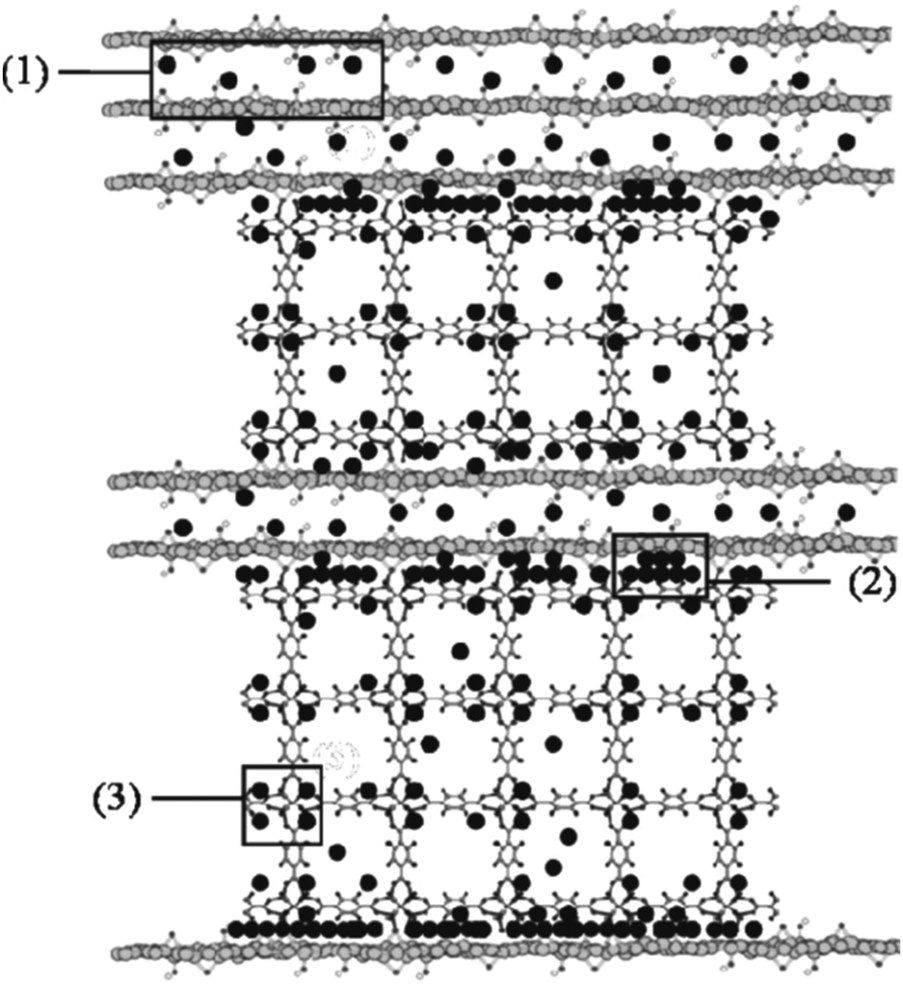 | ||
| Fig. 3 Scheme of the mechanisms of ammonia (black circle) adsorption in the MOF-5@GO composites including: (1) intercalation between the graphene layers, (2) adsorption at the interface between the graphene layers and the MOF-5 segments, (3) hydrogen bonding with the oxygen atoms of the zinc oxide in MOF-5. Reproduced from ref. 14. | ||
The M-CPO-27 (M-MOF-74) (M = Zn2+, Co2+, Ni2+, Mg2+) series have been tested for removing NH3, CNCl and SO2 from air in dry and humid conditions and dynamic breakthrough capacities were compared to 13X zeolite and BPL activated carbon.15 The results reveal that in the presence of a humid atmosphere at 25 °C CPO-27 analogues adsorb an appreciable amount of water accounting at least for 0.1 g of water per gram of adsorbent. In dry conditions, the dynamic loadings of NH3 on all the CPO-27 analogues are generally near to or exceed the maximum loadings of traditional materials, such as 13X zeolite, which adsorbs 2.86 mol kg−1 under the same conditions, and BPL activated carbon, which has no capacity for dry ammonia. The best performing material, Mg-CPO-27 loads three times the capacity of 13X zeolite and the worst performing analog, Ni-CPO-27 loads 80% of the capacity of 13X. It should be noted that when ammonia is loaded in dry conditions, a significant portion of the adsorbate is retained when the MOF materials are exposed to dry air during desorption. However, the 13X zeolite did not retain any of the adsorbed ammonia under the same breakthrough conditions. As expected, the adsorptive capacity of these materials is reduced in a humid gas stream, but they maintain significant adsorption capacity for NH3. Indeed, Mg-CPO-27, the worst performing material under humid conditions, shows 6 times the capacity of BPL activated carbon. On the other hand, CPO-27 structures adsorb dry cyanogen chloride in the same order of magnitude as classical materials, reaching 70% of the retention by Co-CPO-27 even after desorption. However, under humid conditions, none of these MOFs adsorb an appreciable quantity of CNCl resembling the behaviour of 13X zeolite in both dry and humid conditions. Regarding SO2 removal, only Mg-CPO-27 loads this acid gas. In general, Co and Mg analogues show better adsorption of the four gases in both dry and humid conditions. Furthermore, it is clear that the presence of water diminished the adsorption capacity of these MOFs towards the studied gases as a consequence of the competition of the water molecules with the toxic gases for the adsorption at the metal centres leading to diminished breakthrough times and total capacities.
The presence of open metal sites is also highly beneficial for the capture of NO. A series of MOFs have been assayed for the capture and release of NO for biological applications. This is the case for [Cu3(btc)2] and CPO-27 structures,16 which adsorb up to 3 and 8 mmol of NO per gram of MOF, respectively, considerably more than other similar nanoporous solids, such as zeolites. However, the adsorption of NO on [Cu3(btc)2] can be considered nearly irreversible as only 1 μmol of NO per gram of material is released upon exposure to moisture while CPO-27 materials completely desorb all the stored NO under similar conditions. Other biodegradable materials, such as the Fe-based MIL-88A [Fe3O(CH3CO2−)(fumarate)3], MIL-88B [Fe3OX(1,4-bdc)3], 1,4-bdc = 1,4-benzenedicarboxylate (X = F, Cl, OH), MIL-88B-NO2 [Fe3OX(2-NO2-bdc)3], 2-NO2-bdc = 2-nitroterephthalate (X = F, Cl, OH) and MIL-88B-2OH [Fe3OX(2,5-dhbdc)3], 2,5-dhbdc = 2,5-dihydroxyterephthalate (X = F, Cl, OH), show loading capacities lying within the 1–2.5 mmol g−1 range.17 In this case, the low accessibility to the pores is due to the absence of a breathing mechanism of the structure with the pores remaining closed and preventing the easy diffusion of NO to bind the accessible iron metal sites. The released amount of NO from these structures in the presence of a wet gas is even lower, a few tenths of mmol g−1, which is attributed to the desorption of the physisorbed NO prior to the desorption tests, but it is still enough to ensure a significant release at the biological level over prolonged periods of time. A similar behaviour is observed in BioMIL-3 [Ca2(azbz-TC)(H2O)(DMF)], azbz-TC = 3,3′,5,5′-azobenzenetetracarboxylate, which adsorbs 0.8 mmol g−1 of NO at 1 atm, a quantity which is much lower than the theoretical unsaturated Ca2+ metal sites.18 The absence of a plateau in the NO adsorption branch suggests a possible higher adsorption at higher pressures. However, residual coordinated DMF, steric hindrance and diffusion constraints make coordination on the unsaturated metal sites more difficult. These factors also contribute to the slow and low delivery of NO from this material (5 μmol g−1 overnight). Other methodologies have been developed to store NO in MOFs. This is the case for the postsynthetic functionalization of the open metal sites in [Cu3(btc)2] with the bifunctional 4-(methylamino)-pyridine to produce secondary amines, which are able to react with NO to yield the diazen-1-ium-1,2-diolate (NONOate) derivative.19 A similar strategy consists in the functionalization of the organic linkers with amines available to interact with NO yielding NONOate compounds.
CO adsorption has also been studied in MOFs with open-metal sites. It has been demonstrated that [Cu3(btc)2] preferentially adsorbs CO towards H2 and N2 at 298 K. This can be explained because of the electrostatic interactions between the partial charge of the Cu2+ sites and the CO dipole.20 On the other hand, Ni-CPO-27 strongly coordinates CO at room temperature.21 Most of the Ni2+ sites are involved in the interaction (about 80%), forming 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 linear adducts whose interaction enthalpy is slightly above 50 kJ mol−1. However, the adsorption is completely reversible at room temperature after prolonged evacuation. As well as for NO, the biocompatible series MIL-88B and MIL-88B-NH2 [Fe3OX(2-NO2-bdc)3], 2-NH2-bdc = 2-aminoterephthalate (X = F, Cl, OH), have been studied for the capture and release of CO for therapeutic applications showing that CO binding occurs to unsaturated FeII/FeIII coordination sites generated by the activation procedure.22
:1 linear adducts whose interaction enthalpy is slightly above 50 kJ mol−1. However, the adsorption is completely reversible at room temperature after prolonged evacuation. As well as for NO, the biocompatible series MIL-88B and MIL-88B-NH2 [Fe3OX(2-NO2-bdc)3], 2-NH2-bdc = 2-aminoterephthalate (X = F, Cl, OH), have been studied for the capture and release of CO for therapeutic applications showing that CO binding occurs to unsaturated FeII/FeIII coordination sites generated by the activation procedure.22
Finally, noteworthy, the cages of ZIF-8 [Zn(2-MeIm)2] (2-MeImH = 2-methylimidazole) bind iodine 4 times more strongly than activated carbon, the traditional high capacity iodine adsorbent.23 The latter result is of relevance in order to capture radioactive iodine isotopes produced in nuclear processes.
Capture of volatile organic compounds
The characteristics of adsorbent materials for the capture of volatile organic compounds can be significantly different from the ones for gases. In this regard, the diffusion kinetics of vapor molecules might be very slow in narrow pore materials and, on the other hand, the presence of open metal sites can be sometimes disadvantageous for air/gas purification purposes in the presence of moisture.As mentioned in the previous section, six isoreticular MOFs, namely MOF-5, IRMOF-3, MOF-177, IRMOF-62, Zn-CPO-27, and [Cu3(btc)2] are shown as selective adsorbents of tetrahydrothiophene, benzene, dichloromethane and ethylene oxide.9 In the same line as the results of breakthrough experiments on gaseous contaminants, MOF-5 and MOF-177 do not perform well as kinetic adsorption media. IRMOF-62 is largely outclassed by BPL carbon except in the case of ethylene oxide adsorption, for which both materials are equally ineffective. IRMOF-3 is a poor adsorbent for the vapors chosen, because none of them behave as good Lewis acids. MOFs containing open metal sites are found to be the most effective in removing vapors from the gas stream. Zn-CPO-27 and [Cu3(btc)2] outperform BPL carbon by an order of magnitude in ethylene oxide adsorption. However, Zn-CPO-27 is not effective against the entire range of vapors, whereas [Cu3(btc)2] is effective. [Cu3(btc)2] outperforms BPL carbon in tetrahydrothiophene adsorption by a factor of 3 although there is essentially no difference in performance between the activated carbon and [Cu3(btc)2] in dichloromethane and benzene adsorption. It should be noted, however, that the presence of moisture can significantly diminish the performance of [Cu3(btc)2] as a consequence of the blockage of the open metal sites by water molecules (see below).
MIL-101 ([Cr3F(H2O)2O(bdc)]) has proved to be effective in the adsorption of a wide range of VOCs with various functional groups and polarities, i.e. acetone, benzene, toluene, ethylbenzene, xylenes, n-hexane, methanol, butanone, dichloromethane and n-butylamine. Yan et al. demonstrated that MIL-101 has higher affinity for VOCs containing heteroatoms or aromatic rings, especially the amines.24 In addition, MIL-101 shows higher adsorption capacities for acetone, toluene, ethylbenzene and xylenes than other commonly used adsorbents, such as zeolites, resins, activated carbon together with their derivatives.25 For example, MIL-101 adsorbs 16.7 mmol g−1 of benzene at 288 K and 56 mbar, which is almost twice the values for zeolites (silicalite-1 and SBA-15) and 3–5 times larger than that of activated carbons (Ajax and ACF). In this case, the profile of the thermally programmed desorption curve shows two separated peaks corresponding to two major sites for benzene adsorption. The strong interactions would occur between the Cr3+ metal centers and the adsorbate molecules and the slightly weaker interactions take place between the space of the pores and the adsorbate. Consecutive cycles of adsorption–desorption show that fast desorption kinetics, high desorption efficiency and stable adsorption capacity over five cycles are available. The efficiency of benzene desorption can reach 97% exhibiting a high reversibility of benzene adsorption–desorption.26
On the other hand, the modulation of the porous network of the anionic MOF NH4[Cu3(OH)(capz)] (capz = 4-carboxypyrazolato) by means of ion-exchange processes of the extra-framework NH4+ cations leads to profound changes in the textural properties of its porous surface and in the adsorption selectivity towards benzene–cyclohexane mixtures.27 Indeed, exposure of the A[Cu3(OH)(capz)] materials (A = NH4+, Li+, Na+, K+, Me3NH+, Et3NH+) to benzene–cyclohexane 1:1 vapor mixtures shows significant enrichment of the adsorbate phase in the benzene component. This behaviour is explained taking into account that although the increasing bulk of the exchanged cations still permits access of molecules to the porous structure, a concomitant increase in size-exclusion selectivity takes place.
The flexible MIL-53(Al) framework adsorbs xylene isomers and ethylbenzene.28 Adsorption isotherms at 110 °C show two well-defined steps and hysteresis, corresponding to the opening or breathing of the framework, as induced by the presence of adsorbing molecules. At low pressures, adsorption leads to framework contraction. In the limited space of the closed form of the MIL-53(Al) framework, only a single-file arrangement of molecules can be accommodated, adsorbed along the length of the pores. At much higher pressures, the pores are reopened. In the open form, there is space enough for the xylene isomers to be adsorbed in pairs along the length of the pores, leading to a doubling of the amount adsorbed (Fig. 4). Due to differences in the efficiencies with which the different isomers can be packed inside the pores, the adsorption capacity is highly affected. This phenomenon is known as commensurate adsorption and occurs when the molecular size and shape of the adsorbate lead to an orientation and adsorbed amount that is compatible and self-consistent with the crystal symmetry and pore structure of the adsorbent.29
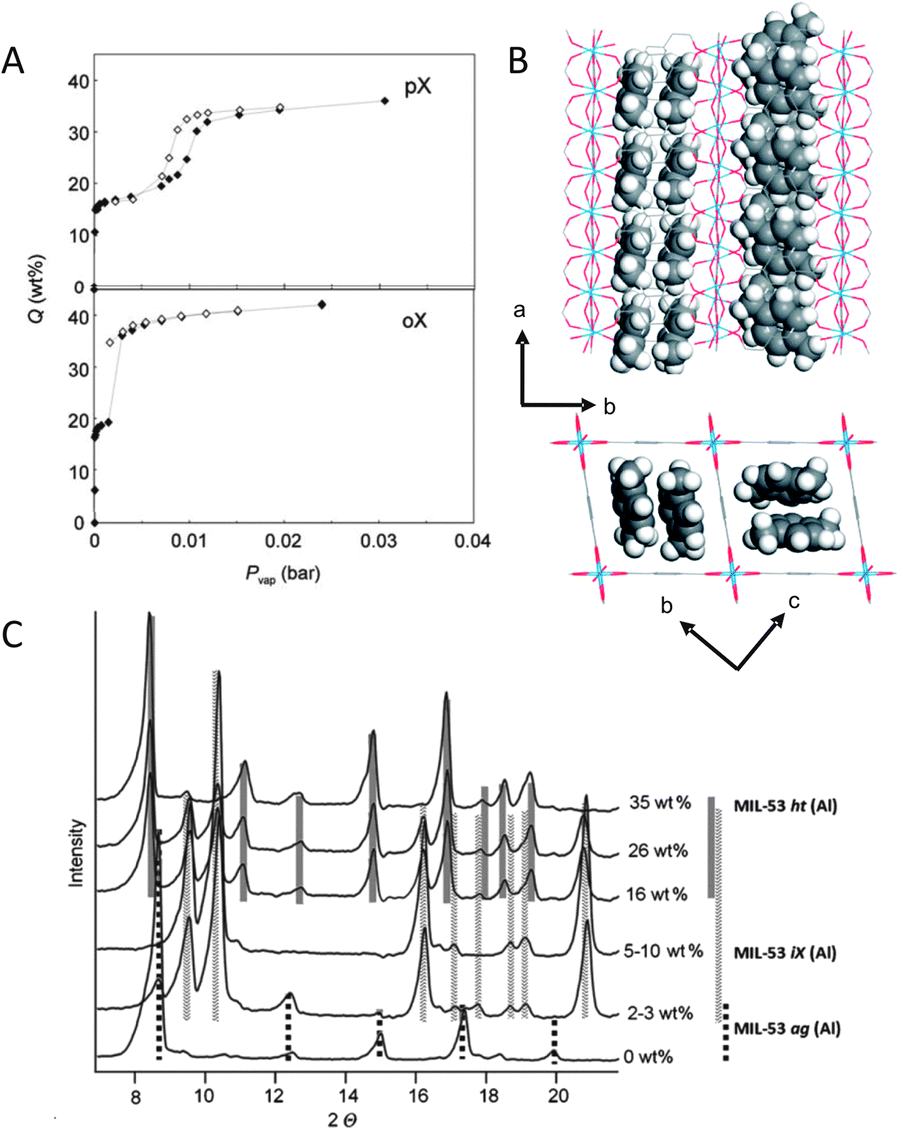 | ||
| Fig. 4 (A) Adsorption (solid symbols) and desorption (open symbols) isotherms for o-xylene (oX) and p-xylene (pX) measured at 110 °C on MIL-53(Al); (B) p-xylene at 110 °C in the pores of MIL-53(Al); (C) X-ray diffractograms of MIL-53(Al) with different oX loadings. Adapted from ref. 28 and 29. | ||
A modified MOF, CuCl2-loaded MIL-47(V) shows remarkable adsorption capacity of benzothiophene.30 MIL-47(V) shows an unexpected reduction ability to form CuI ions from loaded CuII ions probably because of the presence of VIII in the MOF. The obtained CuI ions show a beneficial effect on the adsorption of benzothiophene probably through π-complexation. It has been demonstrated that the adsorption capacity increases with increasing CuCl2 loading up to a specific content (Cu/V = 0.05 mol mol−1) and decreases with further increase in the CuCl2 content, showing that there is an optimum CuCl2 concentration. This is due to the contribution of both the porosity/acidity of MIL-47(V) and a CuI site derived from CuCl2.
On the other hand, the development of sensor materials is also a very active field of research. In this regard, [Zn2(bdc)2(dpNDI)]n (H2bdc = benzene-1,4-dicarboxylic acid; dpNDI = N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide) gives rise to strong stacking interactions with VOC molecules with a concomitant strong change in the emission color sensitive to the ionization potential of each molecule (Fig. 5).31
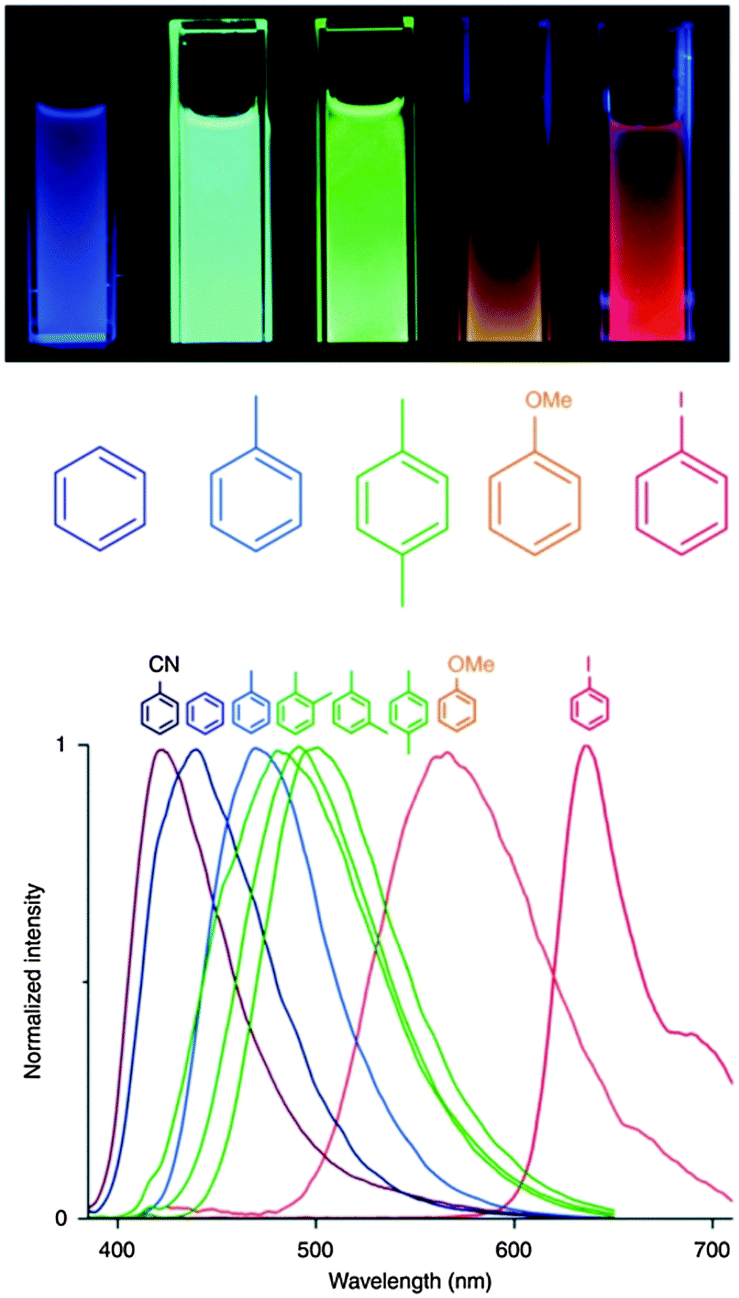 | ||
| Fig. 5 (top) Luminescence of the powdered [Zn2(bdc)2(dpNDI)]n material suspended in the liquid exposed to different VOC molecules after excitation at 365 nm using a commercial ultraviolet lamp. (bottom) Collected normalized spectra of the [Zn2(bdc)2(dpNDI)]n@VOC compounds excited at 370 nm. Adapted from ref. 31. | ||
In this context, the {Fe(pyz)[Pt(CN)4]} system can incorporate a variety of vapor guest molecules, namely benzene and CS2, which modify the pore size and optical and magnetic properties of the materials and might be useful for sensing purposes.32
As mentioned before, MOFs with coordinatively unsaturated metal sites, such as [Cu3(btc)2], are ineffective for the capture of VOCs in the presence of ambient moisture. With the aim of overcoming this problem, we have reported a novel flexible MOF, namely [Ni(bpb)]33 (H2bpb = 1,4-(4-bispyrazolyl)benzene), which is efficient for the adsorption of benzene and cyclohexane. In addition, this material captures tetrahydrothiophene (up to 0.34 g of tetrahydrothiophene per gram of material) from CH4–CO2 mixtures in dynamic conditions even in the presence of humidity (60%), thus overcoming the problems raised by prototypic MOF-5 and [Cu3(btc)2] MOF materials in practical applications.
As previously mentioned in the introduction, a special family of VOCs is the warfare agents. The search for efficient adsorbents of these harmful molecules in practical applications is of great interest. It should be noted that in real conditions, humidity will always be present and water molecules will constitute an important competitor in the adsorption process of these toxic molecules. In this context, we have demonstrated that the use of highly hydrophobic MOFs for the capture of warfare agents is an adequate strategy for the selective adsorption of these molecules even in extreme humid conditions. The basis of this strategy remains in the high affinity of the porous matrix towards the non-polar warfare agents in contrast with its low affinity for polar water molecules. For example, the MOF-5 analog [Zn4O(dmcapz)3] (dmcapz = 3,5-dimethyl-4-carboxypyrazolato) is suitable for the capture of diisopropylfluorophosphate (DIFP, model of sarin nerve gas) and diethylsulfide (DES, model of vesicant mustard gas).34 This compound shows remarkable chemical, mechanical and thermal stability granted by the nature of the M–N,O(carboxypyrazolato) coordinative bonds. In addition, it is highly hydrophobic, which results in large VOC–H2O partition coefficients. Under ambient conditions, [Zn4O(dmcapz)3] clearly outperforms the behavior of [Cu3(btc)2] which, when hydrated, does not retain either DES or DIFP. However, the performance of [Zn4O(dmcapz)3] approaches that of molecular sieve activated carbon Carboxen suggesting similar adsorption processes dominated by the small size and apolar nature of the pores in both materials. Then, it has been clearly demonstrated the necessity of stabilizing MOFs against ambient humidity to turn these porous materials more suitable for specialized and industrial applications. In this context, pre- or postsynthetic modification approaches have been explored in order to enhance the hydrophobicity of MOFs. One of the most successful approaches is the introduction of alkane or fluoroalkane35 residues in the organic linkers. In this context, Omary and colleagues36 reported the synthesis of totally fluorinated MOFs, coined FMOF-1 and FMOF-2, from the combination of Ag+ with 3,5-bis(trifluoromethyl)-1,2,4-triazolate. The hydrophobic character of FMOF-1 is mainly responsible for the observed efficient selective adsorption ability for aliphatic and aromatic oil components (benzene, toluene, p-xylene, cyclohexane and n-hexane), while preventing the entrance of water molecules into their pores. Likewise, the isoreticular series [Ni8(OH)4(H2O)2(L)6] (H2L1 = 1H-pyrazole-4-carboxylic acid; H2L2 = 4-(1H-pyrazol-4-yl)benzoic acid, H2L3 = 4,4′-benzene-1,4-diylbis(1H-pyrazole), H2L4 = 4,4′-buta-1,3-diyne-1,4-diylbis(1H-pyrazole), H2L5 = 4,4′-(benzene-1,4-diyldiethyne-2,1-diyl)bis(1H-pyrazole), H2L5-R (R = methyl, trifluoromethyl)) proves that the use of metal–azolate coordinative bonds gives rise to materials with enhanced stability towards hydrolysis.35 Moreover, the length and functionalization of the linkers impact on the pore size as well as on the surface polarity (Fig. 6). The most significant results regarding the capture of warfare agents were achieved under a strongly competitive moist atmosphere (up to 80% RH) and have been compared with the hydrophobic active carbon Blücher-101408, which is the active phase of Saratoga filtering systems. Indeed, 1H NMR analysis of the adsorbate phase after the adsorption process of DES in 80% RH moist streams reveals that only [Ni8(OH)4(H2O)2(L5-CF3)6] and Blücher-101408 efficiently capture DES under these conditions, witnessing that the adsorption of DES in this MOF is not affected by the presence of humidity. This isoreticular series might be considered as the first step towards the rational design of MOFs for the capture of harmful VOCs under highly demanding environmental conditions.
 | ||
| Fig. 6 (A) Crystal structure of highly hydrophobic [Ni8(OH)4(H2O)2(L5)6] (H2L5 = 4,4′-(benzene-2,5-trifluoromethyl-1,4-diyldiethyne-2,1-diyl)bis(1H-pyrazole)). (B) Impact of benzene functionalization with hydrophobic residues on the water adsorption isotherms at 298 K. Reproduced from ref. 35. | ||
2. Catalytic degradation of harmful gases and volatile organic compounds
The catalytic degradation of harmful gases and vapors into non-toxic substances should be regarded as the ideal case for gas/air purification processes since it overcomes the problems related to adsorbent saturation or its behavior as secondary emitters. Noteworthy, the development of tailored heterogeneous catalysts is one of the areas of major current interest in MOFs research, since these materials are amenable for finely tuning their catalytic properties.37 However, there are still few reports in which the catalytic activity of MOF materials has been applied for the removal of harmful substances from gas/air streams.Catalytic decomposition of trace gases and vapors
CO oxidation to CO2 can be achieved by gold nanoparticles (NPs) deposited on zeolite-type metal–organic framework ZIF-8 [Zn(2-MeIm)2] by a simple solid grinding method. The catalytic activity of the resulting Au@ZIF-8 material for CO oxidation is improved along with increasing Au loadings with the highest catalytic activity being obtained for 5.0 wt% Au@ZIF-8, which presents half conversion of CO at ∼170 °C. Gold NPs are close to being monodispersed and do not give rise to aggregation during catalytic reactions over multiple cycles.38 This reaction might be useful for hydrogen purification purposes for fuel cell applications. The 3D [Yb(OH)(2,6-AQDS)(H2O)] (AQDS: anthraquinone-2,6-disulfonate)39 polymeric framework acts as a good catalyst in hydrodesulfurization reactions of thiophene under mild conditions (7 bar of H2 and 70 °C) as a consequence of the low coordination of the Yb atom and/or lability of coordinated ligands. The enhancement of SO2 trapping has also been achieved by impregnating [Cu3(btc)2] with Ba2+ salts (BaCl2, Ba(NO3)2, Ba(CH3COO)2). Indeed, the BaCl2–[Cu3(btc)2] composite is able to capture SO2 in an oxidative atmosphere in the form of sulfates outperforming the reference material BaCO3/Al2O3/Pt,40 which is the first generation storage reduction catalyst known to store SO2. The formation of small microcrystals of Ba(CH3COO)2 and Ba(NO3)2 in the pores of [Cu3(btc)2] is observed, while the MOF structure is maintained. However, after impregnation with BaCl2, the parent MOF loses crystallinity and the BaCl2 appears highly dispersed in the material. The SO2 uptake of all Ba–[Cu3(btc)2] samples exceeded the stoichiometric uptake capacity based on the Ba2+ concentration, which is indicative that a fraction of SOx is bound to Cu cations. At low temperatures, when the MOF still maintains its structural integrity, the SOx storage is due to the concomitant adsorption of SO2 on Cu2+ and the formation of Ba-sulfates. However, at high temperatures [Cu3(btc)2] decomposes leading to isolated Cu species that react with SOx to form Cu-sulfates. This process is of interest in the elimination of traces of sulfur compounds in fuels prior to the NOx storage-reduction (NOx-SR) concept. However, irreversible SOx adsorption is the main drawback of the above described materials.[M3OF(bdc)2] MIL-101 (M = Cr, Fe) have also been recently found to be active for solvent-free selective oxidation of cyclohexane with molecular oxygen and/or tert-butyl hydroperoxide under mild conditions.41 Similarly, the zeomimetic NH4[Cu3(OH)(capz)] system is also able to catalyse the oxidation of cyclohexane and cyclohexene with tert-butyl hydroperoxide under mild conditions.27
Polyoxometalates (POMs) are regarded as active acid and oxidation catalysts in the homogeneous phase. Noteworthy, POMs can be incorporated in the structure of MOFs either as building blocks or encapsulated in the pore structure of MOFs thereby enhancing their stability and giving rise to advanced heterogeneous catalysts.42 In this regard, Hill and co-workers have shown that the redox active POM [CuPW11O39]5− encapsulated in [Cu3(btc)2] might be useful for the catalytic air based oxidative degradation of SH2 and mercaptane molecules (Fig. 7).43 Indeed, the [Cu3(btc)2]@[CuPW11O39] hybrid material catalyzes the rapid chemo- and shape-selective oxidation of thiols into disulfides and, more significantly, the rapid and sustained removal of toxic H2S via H2S + 1/2O2 → 1/8S8 + H2O (4000 turnovers in <20 h) while the POM or the MOF alone is catalytically slow or inactive. Similarly, the redox active POM [CoPW11O39]5− incorporated inside [Cr3OF(bdc)2] MIL-101(Cr) voids behaves as an active heterogeneous catalyst for the molecular oxygen allylic oxidation of α-pinene, which can be used as a flavoring substance and drug precursor.44
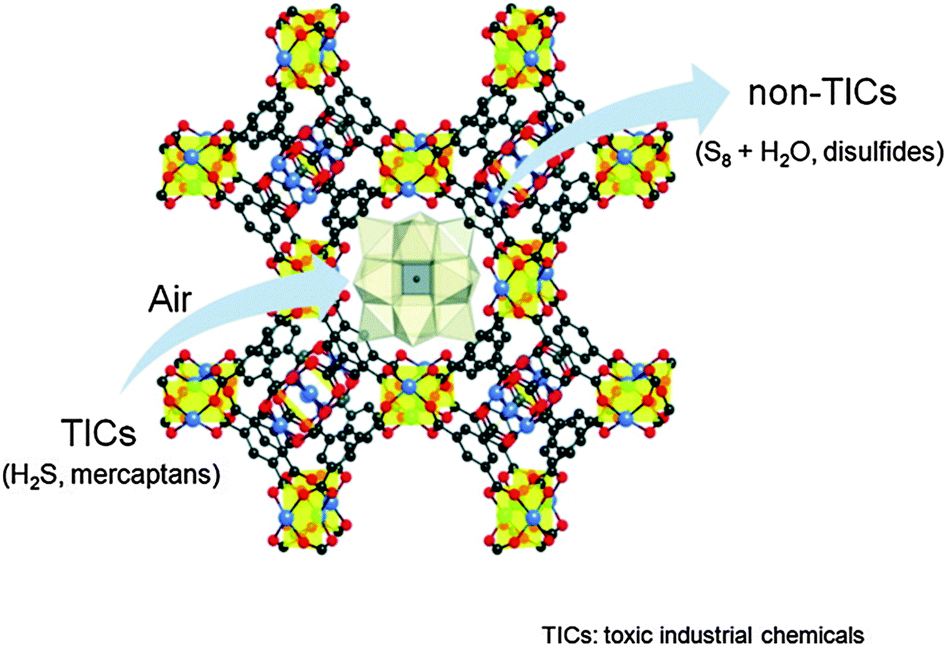 | ||
| Fig. 7 Schematic representation of the selective air oxidation of thiols to disulfides and the oxidation of toxic H2S to S8 by [Cu3(btc)2]@[CuPW11O39]. Reproduced from ref. 43. | ||
Lin and co-workers45 have recently shown that it is possible to obtain oxidation and photoactive catalysts by doping of [Zr6O4(OH)4(bpdc)6] (UiO-67), (bpdc = para-biphenyldicarboxylic acid), with IrIII or RuII metal fragments attached to 2,2′-bipyridine-5,5′-dicarboxylato spacers (Fig. 8). The resulting modified MOF-6 containing the phosphorescent [RuII(bpy)2(dcbpy)]Cl2(H2L6) (bpy = 2,2′-bipyridine) behaves as an efficient aerobic oxidation catalyst of thioanisole (Fig. 8).
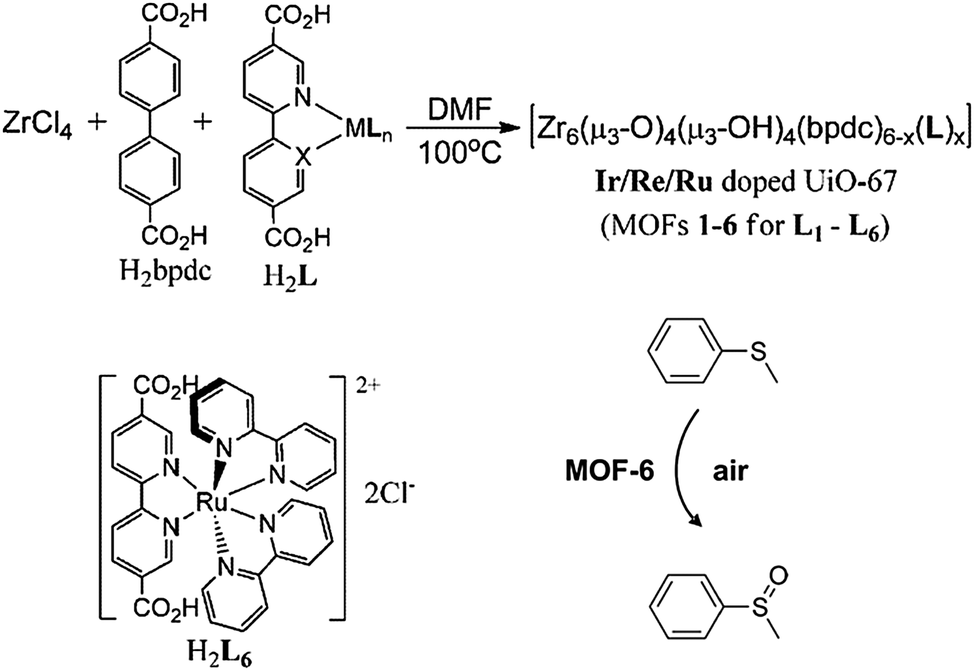 | ||
| Fig. 8 Synthesis of IrIII and RuII doped [Zr6O4(OH)4(bpdc)6] (UiO-67) systems and the resulting aerobic catalytic oxidation of thioanisole. Adapted from ref. 45. | ||
The use of MOF materials with intrinsic photochemical properties is also an advantageous feature for the possible degradation of harmful substances. In this regard, [Ti8O8(OH)4(bdc)6] (MIL-125)46 under UV-irradiation is able to oxidize adsorbed alcohols giving rise to a photochromic behaviour associated with the formation of stable mixed valence titanium-oxo compounds (Fig. 9). The titanium oxo clusters are back oxidized in the presence of oxygen closing the catalytic cycle. The photogenerated electrons are trapped as TiIII centers, while a concomitant oxidation of the adsorbed alcohol molecules occurs. Noteworthy, the photocatalytic properties of [Ti8O8(OH)4(bdc)6] (MIL-125) can be tuned by replacing bdc by 2-aminoterephthalate. The NH2-MIL-125 system behaves as an active photocatalyst under visible light irradiation in the reduction of carbon dioxide to the formate anion with TEOA (triethanolamine) acting as an electron donor and a base.47
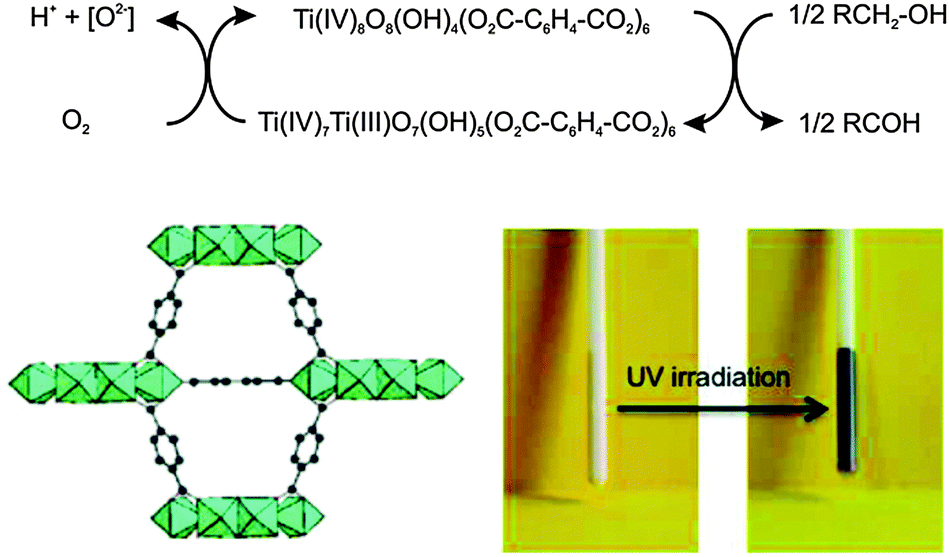 | ||
| Fig. 9 (top) Proposed mechanism for the photochemical oxidation of alcohols by [Ti8O8(OH)4(bdc)6] (MIL-125) under UV radiation. (bottom) Photochromic behavior of MIL-125 before and after UV irradiation in the presence of adsorbed benzyl alcohol. Adapted from ref. 46. | ||
Catalytic degradation of chemical warfare agents
The catalytic degradation of the highly toxic chemical warfare agents is of high interest, since it avoids the problems related to the manipulation of the contaminated materials after exposure to such substances. In this regard, the usual degradation pathways of such substances involve oxidation as well as hydrolysis routes (Scheme 1). This fact has led to the use of redox and/or acid–base active MOFs as catalysts for the remediation systems after exposure to these extremely harmful molecules.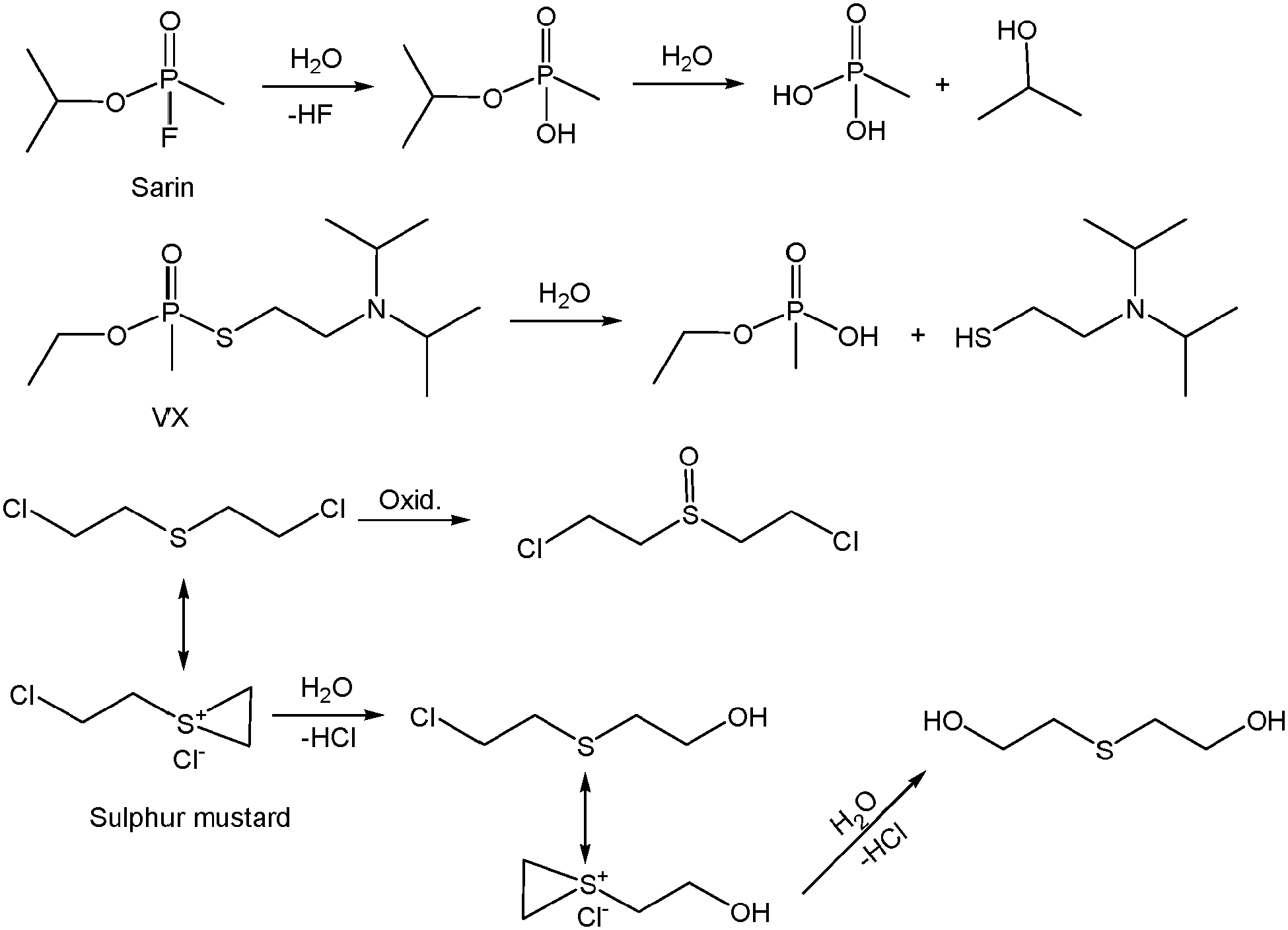 | ||
| Scheme 1 Common oxidative and hydrolysis degradation pathways of selected chemical warfare agents. | ||
In this regard, the presence of Cu open metal sites with Lewis acidic nature in [Cu3(btc)2] favors the hydrolysis, although slow, of both sulfur mustard and organophosphate nerve agents.48
The catalytic activity in hydrolysis reactions of the [Cu3(btc)2] type systems can be improved by the incorporation of acid polyoxometalates in their pore structure. Thus, Su, Liu and colleagues have taken advantage of the improved hydrolytic activity of the sodalite type H3[(Cu4Cl)3(btc)8]2[PW12O40]3(C4H12N)6 (NENU-11) system for the capture and posterior hydrolytic degradation of the model nerve agent dimethylmethylphosphonate (DMMP).49
Hatton and colleagues50 have reported the incorporation of highly nucleophilic 4-methylaminopyridine residues by means of covalent coupling of the amino residues of aluminium aminoterephthalate MOF systems to give rise to [Al3OF(4-MAP-NH-bdc)2] (NH2-MIL-101(Al)) and [AlOH(4-MAP-NH-bdc)] (4-MAP-NH-MIL-53(Al)) active materials (Fig. 10). Noteworthy, this MOF can be incorporated onto protective rubber glove surfaces prior to curing giving rise to self-detoxifying surfaces exhibiting a high catalytic activity in the hydrolytic degradation of chemical warfare simulant diisopropylfluorophosphate (DIFP) as proven by 31P-MAS NMR studies.
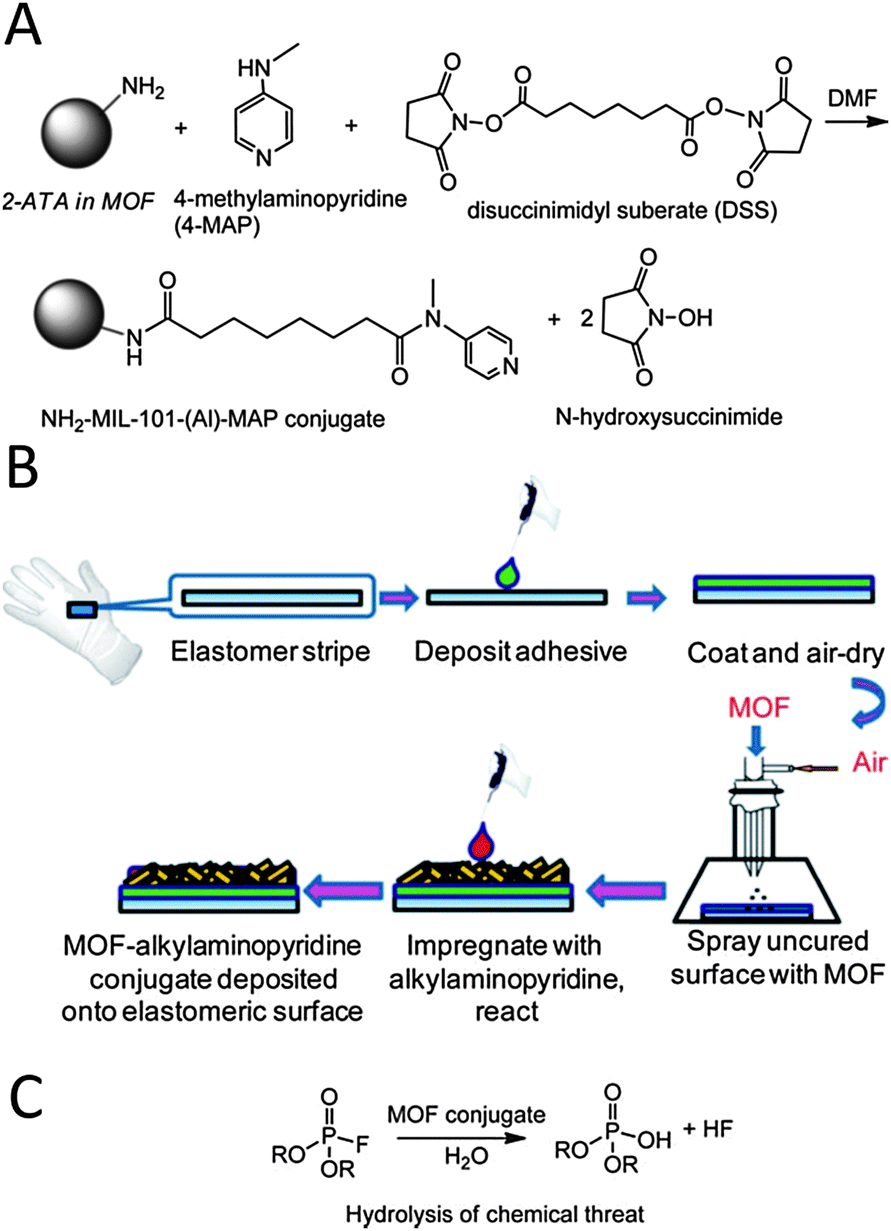 | ||
| Fig. 10 (A) Functionalization of MOF pore surface with nucleophilic 4-methylaminopyridine (4-MAP). (B) MOF@elastomeric films deposited onto the outer surfaces of the Guardian butyl rubber gloves. (C) Catalytic degradation of DIFP sarin simulant. Adapted from ref. 50. | ||
Conclusions
The use of MOFs with adequate pore size/shape is not enough for an efficient capture of hazardous gases/vapors and other more specific interactions between the harmful adsorbates and the host are desirable. For example, the presence of open metal sites (coordinatively unsaturated metal centres) or certain functionalizations on the pore surface may enhance the adsorption selectivity/efficiency of MOFs towards certain toxic compounds via coordination bonds, acid–base/electrostatic interactions, π-complex/H-bonding formation, etc. Thus, an effective strategy for the synthesis of materials that strongly retain one gas in preference to another is to target a particular surface chemistry instead of optimizing the surface area alone. Adsorption selectivity can, however, become a drawback for protective applications (i.e. fire rescuers or army applications). In this regard, the fabrication of multicomponent composite materials (i.e. core shell adsorbents, hybrid textiles and thin film coatings of MOFs on various substrates) is becoming a common practice, which provides a way to enhance the performance of the materials towards the generation of multi-target adsorbents. Moreover, the design of chemically, thermally and mechanically robust MOF materials able to stand the typical working conditions of practical applications (i.e. moisture, strong oxidising/reducing agents, acidic/basic environments, high temperature/pressure…) needs to be addressed by means of the right choice of metal–ligand combination, synthetic and postsynthetic conditions, formation of composite materials, etc.Noteworthy, the amenable properties of MOFs offers a way towards the development of advanced catalysts suitable for the degradation of harmful gases and vapors into non-toxic substances. This strategy should be regarded as the ideal case for gas/air purification processes since it overcomes the problems related to adsorbent saturation or its behavior as secondary emitters. Finally, the integration of these catalysts into composite materials may lead to advanced filters, textiles and surfaces with self-cleaning properties.
Acknowledgements
The authors are grateful for the generous support by the Spanish Ministries of Economy (project: CTQ2011-22787) and Defense (COINCIDENTE Program) as well as Junta de Andalucia (P09-FQM-4981).Notes and references
- S. Aguado, A. C. Polo, M. P. Bernal, J. Coronas and J. Santamaría, J. Membr. Sci., 2004, 240, 159 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244, 444 CrossRef PubMed.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. De Weireld, J. Am. Chem. Soc., 2009, 131, 8775 CrossRef CAS PubMed.
- L. Hamon, H. Leclerc, A. Ghoufi, L. Oliveiro, A. Travert, J.-C. Lavalley, T. Devic, C. Serre, G. Férey, G. De Weireld, A. Vimont and G. Maurin, J. Phys. Chem. C, 2011, 115, 2047 CAS.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. De Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060 RSC.
- P. D. C. Dietzel, R. E. Johnsen, H. Fjellvag, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 5125 RSC.
- C. A. Fernandez, P. K. Thallapally, R. K. Motkuri, S. K. Nune, J. C. Sumrak, J. Tian and J. Liu, Cryst. Growth Des., 2010, 10, 1037 CAS.
- D. Britt, D. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623 CrossRef CAS PubMed.
- G. W. Peterson, G. W. Wagner, A. Balboa, J. Mahle, T. Sewell and C. J. Karwacki, J. Phys. Chem. C, 2009, 113, 13906 CAS.
- C. Petit, B. Mendoza, D. O'Donnell and T. J. Bandosz, Langmuir, 2011, 27, 10234 CrossRef CAS PubMed.
- G. Agustí, R. Ohtani, K. Yoneda, A. B. Gaspar, M. Ohba, J. F. Sánchez-Royo, M. C. Muñoz, S. Kitagawa and J. A. Real, Angew. Chem., Int. Ed., 2009, 48, 8944 CrossRef PubMed.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2013, 117, 14570 CAS.
- C. Petit and T. Bandosz, Adv. Funct. Mater., 2010, 20, 111 CrossRef CAS PubMed; C. Petit and T. Bandosz, Dalton Trans., 2012, 41, 4027 RSC.
- T. G. Glover, G. W. Peterson, B. J. Schindler, D. Britt and O. Yaghi, Chem. Eng. Sci., 2011, 66, 163 CrossRef PubMed.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS PubMed , and references therein.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturin, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592 CrossRef CAS.
- S. R. Miller, E. Alvarez, L. Fradxourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773 RSC.
- M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986 CrossRef CAS PubMed.
- J. R. Karra and K. S. Walton, Langmuir, 2008, 24, 8620 CrossRef CAS PubMed.
- S. Chavan, J. G. Vitillo, E. Groppo, F. Bonino, C. Lamberti, P. D. C. Dietzel and S. Bordiga, J. Phys. Chem. C, 2009, 113, 3292 CAS.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill, M. Muhler, R. A. Fischer, Y. Wang, U. Schatzschneider and N. Metzler-Nolte, Chem. – Eur. J., 2013, 19, 6785 CrossRef CAS PubMed.
- J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256 CrossRef CAS PubMed.
- C.-Y. Huang, M. Song, Z.-Y. Gu, H.-F. Wang and X.-P. Yan, Environ. Sci. Technol., 2011, 45, 4490 CrossRef CAS PubMed.
- K. Yang, Q. Sun, F. Xue and D. Lin, J. Hazard. Mater., 2011, 195, 124 CrossRef CAS PubMed.
- Z. Zhao, X. Li, S. Huang, Q. Xia and Z. Li, Ind. Eng. Chem. Res., 2011, 50, 2254 CrossRef CAS.
- E. Quartapelle Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS PubMed.
- V. Finsy, C. E. A. Kirschhock, G. Vedts, M. Maes, L. Alaerts, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Chem. – Eur. J., 2009, 15, 7724 CrossRef CAS PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed.
- N. A. Khan and S. H. Jhung, Angew. Chem., Int. Ed., 2012, 51, 1198 CrossRef CAS PubMed.
- Y. Takashima, V. Martinez Martinez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767 CrossRef CAS PubMed.
- S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-García, I. Fernández-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664 CrossRef CAS.
- C. Montoro, F. Linares, E. Quartapelle Procopio, I. Senkovska, S. Kaskel, S. Galli, N. Masciocchi, E. Barea and J. A. R. Navarro, J. Am. Chem. Soc., 2011, 133, 11888 CrossRef CAS PubMed.
- N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290 CrossRef CAS PubMed.
- C. Yang, U. Kaipa, Q. Z. Mather, X. P. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed.
- H.-L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302 CrossRef CAS PubMed.
- F. Gandara, E. Gutierrez-Puebla, M. Iglesias, D. Proserpio, N. Snejko and M. A. Monge, Chem. Mater., 2009, 21, 655 CrossRef CAS.
- H. Dathe, A. Jentys and J. A. Lercher, Phys. Chem. Chem. Phys., 2005, 7, 1283 RSC.
- N. V. Maksimchuk, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Chem. Commun., 2012, 48, 6812 RSC.
- B. Nohra, H. El Moll, L. M. Rodriguez-Albelo, P. Mialane, J. Marrot, C. Mellot-Draznieks, M. O'Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2011, 133, 13363 CrossRef CAS PubMed.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839 CrossRef CAS PubMed.
- N. V. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. Shmakov, Y. A. Chesalov, D. N. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364 CrossRef CAS PubMed.
- G. W. Peterson and G. W. Wagner, J. Porous Mater., 2014, 21, 121 CrossRef CAS.
- F.-J. Ma, S.-X. Liu, C.-Y. Sun, D.-D. Liang, G.-J. Ren, F. Wei, Y.-G. Chen and Z.-M. Su, J. Am. Chem. Soc., 2011, 133, 4178 CrossRef CAS PubMed.
- L. Bromberg, Y. Klichko, E. P. Chang, S. Speakman, C. M. Straut, E. Wilusz and T. A. Hatton, ACS Appl. Mater. Interfaces, 2012, 4, 4595 CAS.
| This journal is © The Royal Society of Chemistry 2014 |