 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled coordination cages based on banana-shaped ligands
Muxin
Han
,
David M.
Engelhard
and
Guido H.
Clever
*
Institute for Inorganic Chemistry, Georg-August University Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: gclever@gwdg.de; Web: http://www.clever-lab.de
First published on 6th February 2014
Abstract
The combination of pyridyl ligands and square-planar Pd(II) or Pt(II) cations has proven to be a very reliable recipe for the realization of supramolecular self-assemblies. This tutorial review deals with the design, synthesis and host–guest chemistry of discrete coordination cages built according to this strategy. The focus is set on structures obeying the formula [PdnL2n] (n = 2–4). The most discussed ligands are bent, bis-monodentate bridges having their two donor sites pointing in the same direction. The structures of the resulting cages range from simple globules over intertwined knots to interpenetrated dimers featuring three small pockets instead of one large cavity. The cages have large openings that allow small guest molecules to enter and leave the cavities. Most structures are cationic and thus favour the uptake of anionic guests. Some examples of host–guest complexes are discussed with emphasis on coencapsulation and allosteric binding phenomena. Aside from cages in which the ligands have only a structural role, some examples of functional ligands based on photo- and redox-active backbones are presented.
 David M. Engelhard, Muxin Han and Guido H. Clever | Muxin Han was born in Tianjin, China. She studied chemistry at the University of Göttingen where she received her Diploma degree under the supervision of Carola Schulzke in 2009. In 2010, she joined the group of Guido H. Clever as a fellow of the CaSuS doctoral programme. Her research is focused on the rational design, synthesis and host–guest chemistry of low-symmetric and light-switchable coordination cages. |
David M. Engelhard studied chemistry at the University of Göttingen, where he received his BSc in 2009. He obtained his master's degree in chemistry in 2011 concluding with a thesis on the coordination chemistry of light-switchable ligands. In 2011 he started his PhD within the IRTG program “Metal Sites in Biomolecules” under the supervision of Guido H. Clever, investigating coordination cages and DNA G-quadruplex–metal interactions. |
Guido H. Clever is a Professor of Inorganic Chemistry at the University of Göttingen. He studied chemistry in Heidelberg and received his PhD from LMU Munich under the supervision of Thomas Carell. From 2007 to 2009 he was a JSPS postdoctoral researcher in the group of Mitsuhiko Shionoya at The University of Tokyo. In 2009, he was appointed as an Assistant Professor at the same institution. In 2010 he became a Junior-Professor at the University of Göttingen where he was appointed as a Professor in 2013. He was awarded the ADUC prize 2012 for young investigators. His research focuses on the redox-, photo- and host–guest chemistry of functionalized coordination cages. Further interests include metal-mediated DNA architectures. |
Key learning points(1) Concave bis-monodentate organic ligands self-assemble with square-planar metal cations into discrete cage structures.(2) The Pd(pyridine)4 motif is a very popular and easy to handle coordination environment. (3) Besides simple [M2L4] cages, tubular [M3L6] and [M4L8] assemblies, knots and interpenetrated dimers are sometimes formed. (4) The formation of the assembly product is a function of the ligand geometry, flexibility and chemical makeup, the solvent and the counter anions. (5) One or multiple guests can be incorporated into the cages. (6) Dynamic features and switchable functions allow for the control of guest binding affinity and selectivity. |
1. Introduction
The metal-mediated self-assembly of organic ligands into discrete nanoscopic objects1 such as rings, helicates, cages and capsules2 as well as knots and links3 has afforded a large number of amazing structures over the last few decades. Some of these assemblies were only synthesised due to their aesthetic appeal or in order to demonstrate the capabilities of rational design in supramolecular chemistry. Other structures were prepared with the aim of encapsulating certain guest compounds with high selectivity, often motivated by the search for new receptors, sensors and molecular transporters.4 Also the stabilization of reactive compounds5 was shown to be feasible inside self-assembled cages. Moreover, the number of chemical reactions carried out inside the confined environments of such artificial nanoscopic cavities is steadily increasing. Nevertheless, the field of these “molecular flasks” is still in its infancy. Mentionable examples include the rate enhancement of Diels–Alder reactions in Rebek's organic capsules, cycloadditions with unusual regioselectivities in Fujita's octahedral cage and Nazarov cyclisations inside Raymond's anionic Ga4-tetrahedron.6 Other fields in which self-assembled coordination architectures are playing a role include molecular machinery7 and membrane-spanning channels.8Most of the cages reported to date have very high symmetries, often resembling the Platonic or Archimedean solids in shape. A recent development is the rational design of cage structures of lower symmetry. This can be achieved by using more than one kind of metal centre, e.g. in heterobimetallic assemblies.9 Also, a controlled arrangement of different ligands around the metal centres can be realized, when a heteroleptic coordination environment is favoured due to electronic10 or steric constraints.11 Alternatively, rational ligand design can offer the possibility to generate a single cage architecture of reduced symmetry comprising several non-equal coordination environments. This has been even shown in systems comprising only one kind of metal cation and one type of donor site. Examples include the square-cuboid box reported by Clever and coworkers12 and Fujita's [M18L24] stellated cuboctahedron.13
Besides all design efforts invested in the construction of such self-assembled cages it is fair to say, however, that more than just a few of these structures were the products of serendipity. This was also the case for some of the structures discussed in this review. In particular, the self-assemblies showing a complex topology such as those with interwoven ligands or catenated subunits were quite often obtained by chance. Exceptions include Stoddart's Borromean Rings14 and Leigh's pentafoil knot15 which were the products of a rational design approach.
The main part of this review deals with cage structures containing a rather low number of metal centres (nuclearity, n = 2–4), in which all four coordination sites of the square-planar metal centres are occupied by pyridine-based bridging ligands. The bis-monodentate ligands have a concave shape resembling a banana. They can adopt conformations in which the bonding vectors of the two pyridyl donors are collinear. Furthermore, only structures in which pairs of metal centres share at least two bridging ligands are addressed (Fig. 1a–e). In this sense, the family of giant spheres [PdnL2n] composed of 6, 12 or 24 Pd(II) centres and bis-pyridyl ligands having a wide angle between their nitrogen donors will not be treated. The interested reader is referred to a recent review by Fujita and coworkers.16 Likewise, self-assemblies containing cis-blocked square-planar Pd(II) centres that allow for the binding of two further ligands are only touched at the periphery.17
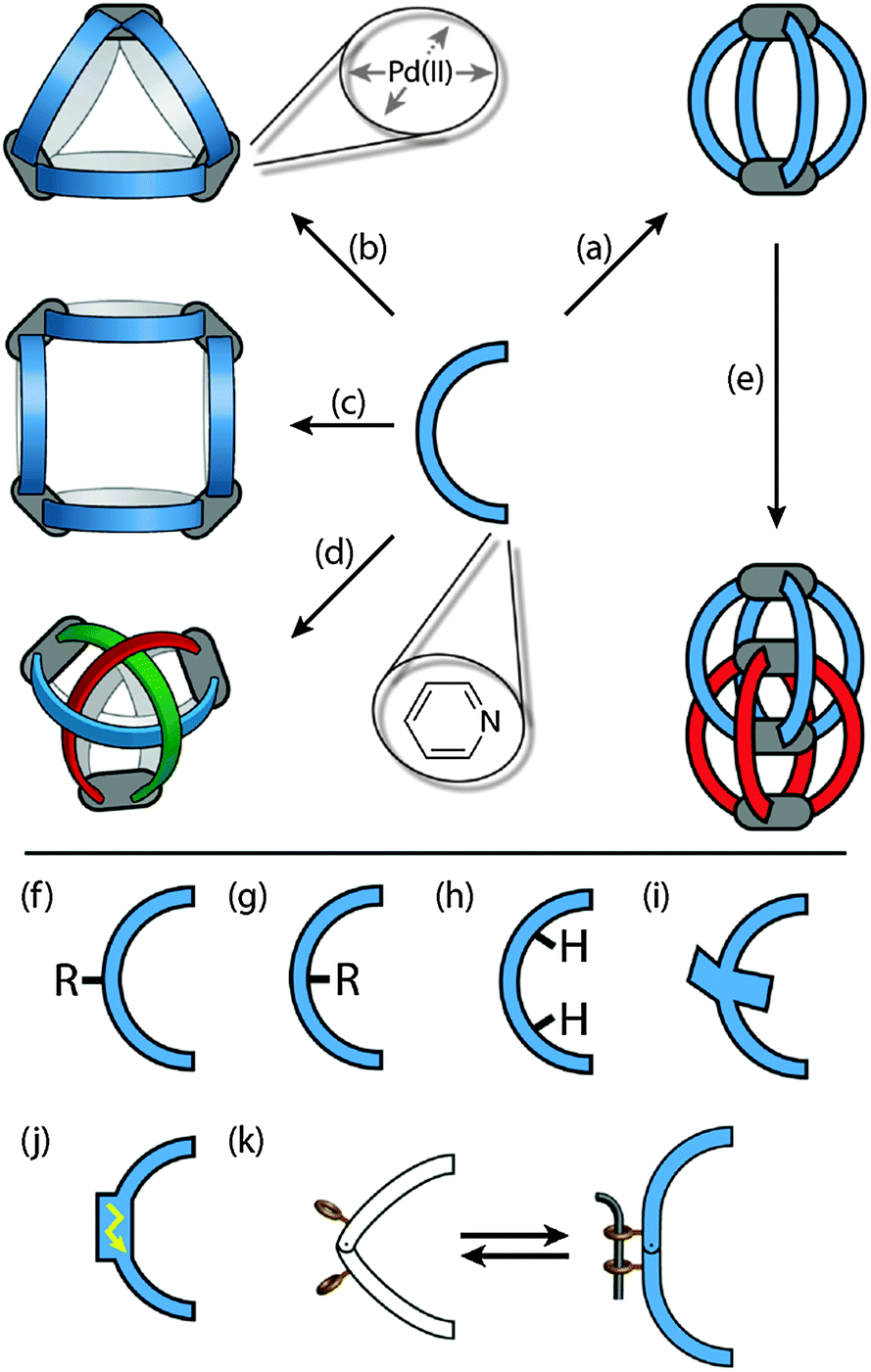 | ||
| Fig. 1 Overview of discussed [PdnL2n] (n = 2–4) cages including (a) simple [Pd2L4] cages, (b) [Pd3L6] and (c) [Pd4L8] rings, (d) a [Pd3L6] double-trefoil knot with intertwined ligands, and (e) interpenetrating [Pd4L8] cage dimers. Ligand functionalisations include substitution in (f) exterior or (g) endohedral positions, (h) the placement of hydrogen-bond donors inside the cavity, (i) the introduction of steric bulk around the ligand backbone, and the implementation of (j) redox activity or (k) photo-switchability into the ligand backbones. | ||
A further recent development in the area of coordination cages is the implementation of functionality other than the shape-forming structure into the ligand backbones. In particular, redox-active or photoswitchable ligands are currently studied as integral components of cage assemblies resulting in compounds that can undergo multi-electron reductions/oxidations or light-triggered structural changes, respectively. Fig. 1f–k schematically compares some ligand designs. Apart from their structural role, the ligands may carry exterior (Fig. 1f) or endohedral (Fig. 1g) substituents, inward pointing hydrogen-bond donors (Fig. 1h), sterically demanding backbones (Fig. 1i), redox activity (Fig. 1j) and light-switchable functionalities (Fig. 1k).
This tutorial review will not only provide information about the design and preparation of a selection of coordination cages but it will also give some examples of informative host–guest complexes. In particular, the implication of dynamic features and ligand-centred functions on guest encapsulation processes is covered. A focus is set on the binding of anions18 and strategies to control the cavity size and shape. Where appropriate (stacked guest assemblies and interpenetrated double-cages), related structures are also discussed, even when the cages they are composed of do not contain banana-shaped bis-monodentate ligands.
2. Assembly principles
Most of the structures discussed in this review are formed by following a few rules. As depicted in Fig. 1, the ligands have a concave, banana-like shape with two pyridyl donors at their ends. Most of the structures are composed of aromatic rings connected either directly via single bonds, through linear alkyne struts or via flexible sp3 or amide linkers. To allow for the formation of the rather small architectures that are shown in Fig. 1a–e, the bonding vectors of the two donor atoms must be able to point in a common direction. As a precursor for the Pd(II) cations, a metal salt containing non-coordinating counter anions such as BF4− is used. Furthermore, the assembly is usually carried out in polar solvents such as acetonitrile, acetone, DMSO or even water. The cationic reaction products are generally better soluble in these solvents than the ligands. Depending on the ligand length, bending angle, other structural features as well as the solvent, metal source and counter anion, a variety of discrete structures may be formed. The assemblies shown in Fig. 1 range from simple [Pd2L4] cages (Fig. 1a), over [Pd3L6] and [Pd4L8] rings (Fig. 1b and c) to advanced topologies such as a [Pd3L6] double-trefoil knot (Fig. 1d) and [Pd4L8] interpenetrated double-cages (Fig. 1e).3. Assembly and anion binding of [M2L4] cages
The small [M2L4] assemblies represent one of the most simple but important structural classes among the various self-assembled macrocycles, helicates, cages and capsules with different metal–ligand stoichiometries.2 Compared to the densely packed helicates, the cavity-containing cages and capsules provide more possibilities for the application in specific guest recognition, transport of molecular cargo and catalysis. Back in 1998, Atwood and co-workers have synthesised the first example of a [M2L4] assembly in which two octahedral Cu(II) ions are connected by four bis(amidomethyl)pyridyl ligands 1 (Fig. 2a).19 The pyridyl groups of the ligands were found to coordinate to the four equatorial positions of each Cu centre while water molecules occupied the axial positions in [Cu214(H2O)4]4+. Consequently, two water molecules take positions inside the cavity while the other two aquo ligands are situated at the cage's periphery. Bis-monodentate benzimidazole ligands were used by Su, Kaim and Zur Loye to prepare [M2L4] cages using octahedral {NiCl2} fragments carrying the two chloro substituents in a trans relationship.20 On the other hand, Amouri et al. prepared [Co224]4+ cages based on benzimidazole ligands 2 and Co(II) cations (Fig. 2b).21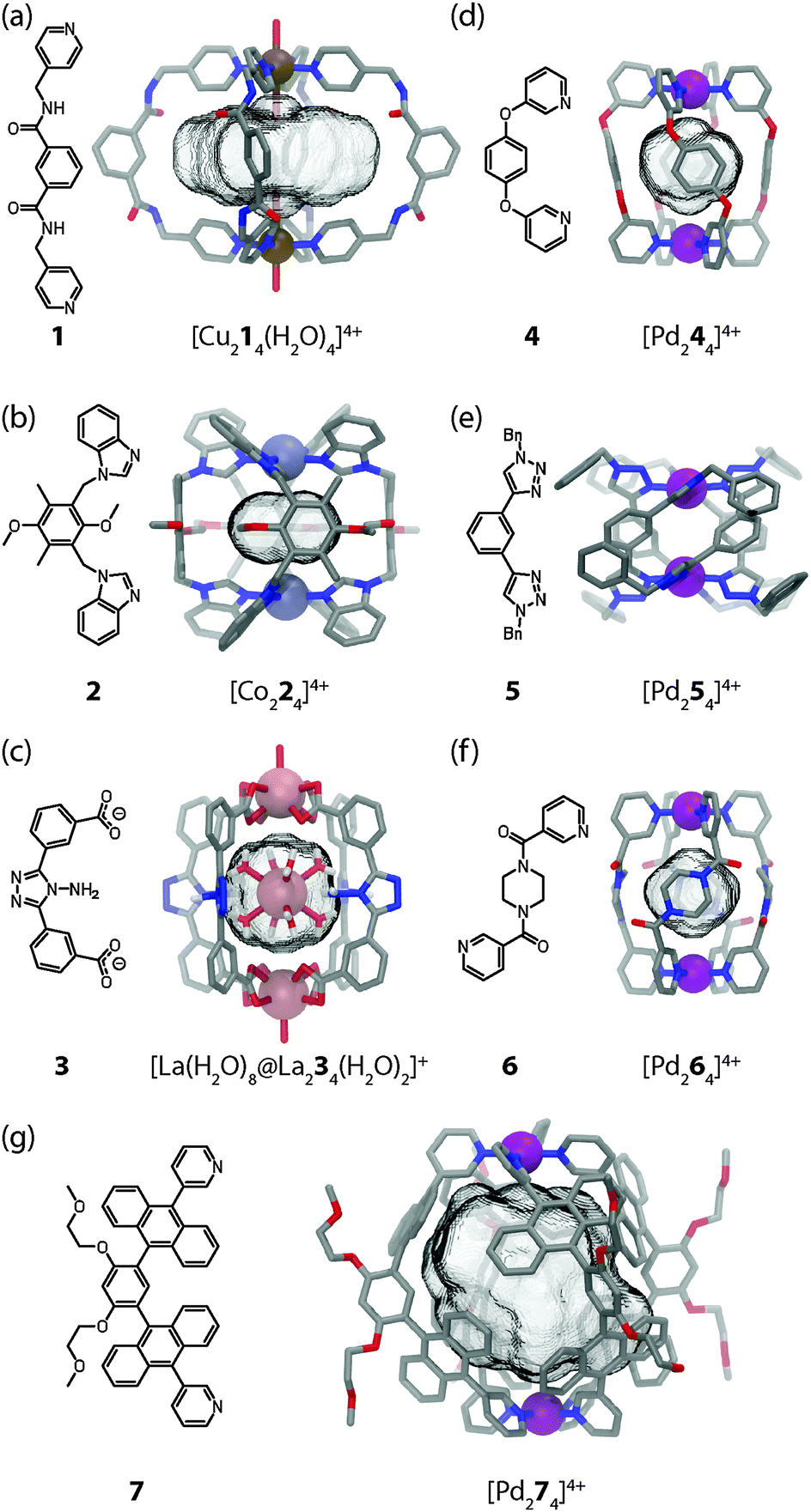 | ||
| Fig. 2 X-ray structures of cages (a) [Cu214(H2O)4]4+, (b) [Co224]4+, (c) [[Ln(H2O)8]@Ln234(H2O)2]+, (d) [Pd244]4+, (e) [Pd254]4+, (f) [Pd264]4+ and (g) [Pd274]4+. For all figures applies: hydrogens, solvent molecules and counter anions were omitted for clarity; C: grey, O: red; N: blue; Cu: brown; Pd: purple; Co: light blue; and La: light red; metal cations are represented as spheres; cavities are depicted as translucent grey potatoids. | ||
Dong et al. reported a very interesting family of luminescent, isostructural host–guest complexes [M′@M234]+ composed of four banana-shaped bis-carboxylate ligands 3 coordinating two M = {Ln(H2O)} fragments (Ln = La, Ce, Sm, Eu or Tb; all metals in oxidation state III and coordinated by nine oxygen donors) and one central M′ = [Ln(H2O)8]3+ cation inside the bis-anionic cage (Fig. 2c).22 For further examples of [M2L4] coordination cages and related structures the reader is referred to a recent review by Debata et al.23
Among the metals used in the construction of [M2L4] cages, square-planar palladium(II) ions take a prominent role due to their predictable coordination sphere, convenient ligand exchange kinetics and diamagnetism. The latter property facilitates the examination of the solution behaviour by various NMR techniques, a fact that has contributed a tremendous body of information to the field. Steel and co-workers marked the first milestone in the self-assembly of a [Pd244]4+ cage in 1998 (Fig. 2d).24 The compound was prepared by mixing the subcomponents 1,4-bis(3-pyridyloxy)-benzene 4 and [PdI2(py)2] in acetonitrile together with silver triflate in order to remove the halide anions. The cage was shown to encapsulate one PF6− anion when NH4PF6 was present during the reaction.
Fujita's group illustrated that the quantitative formation of a [Pd2L4] cage can be monitored by using 1H NMR spectroscopy.25 A combination of the ligand 1,3-bis(3-pyridylphenyl)benzene and the salt Pd(NO3)2 was used to form the cage in DMSO. Since no strongly coordinating halides were introduced with the metal precursor, the addition of a silver(I) salt was not required. The X-ray structure revealed that one of the nitrate anions is encapsulated inside the cavity by ionic interactions.
Puddephatt and co-workers reported a [Pd2L4] cage that was formed by combining a bis(amidopyridyl) ligand with Pd(CF3COO)2 in a CH2Cl2–DMF solution.26 Since the eight amide bonds (two per ligand) comprising the cage scaffold were found to be able to adopt different conformations, the self-assembled structure is highly flexible. In its different conformations, the cage was shown to encapsulate neutral molecules, cations or anions and thus termed an “amphitopic” receptor.
Using click chemistry, Crowley and co-workers27 have synthesized a number of bis-monodentate ligands (for example 5 as shown in Fig. 2e) containing two 1,2,3-triazole donor sites. Their self-assembly using Pd(II) ions yielded quadruply stranded cages [Pd254]4+ whose backbone structures can be widely varied owing to the modularity of the click approach.
Another example of a [Pd2L4] cage was reported shortly afterwards by Sahoo and Chand (Fig. 2f).28 The N,N′-bis(3-pyridylformyl)piperazine ligand 6 can adopt two different conformers (syn and anti) in solution. Reaction of 6 with Pd(NO3)2 in acetonitrile leads to the formation of a dinuclear cage [Pd264]4+ in which all four ligands were shown to exist in the anti conformation by X-ray analysis. One nitrate anion was found to be encapsulated inside the cavity.
Meanwhile Hooley and co-workers have created a self-assembled “paddle-wheel” [Pd2L4] cage, which forms upon addition of Pd(II) ions to a solution of a simple 1,3-bis(3-pyridylethynyl)benzene ligand.29 The uptake and binding affinity of a series of neutral aromatic nitriles were described with respect to their size and electronic complementarity.
Yoshizawa and co-workers reported the preparation of a [Pd274]4+ molecular capsule from four bis-anthracene ligands 7 and two Pd(II) ions (Fig. 2g).30 The resulting architecture encloses a large hydrophobic cavity. The ability to encapsulate compact guests such as [2,2]paracyclophane through hydrophobic interactions was shown. Furthermore, C60 was selectively recognized due to its complementary size and shape from a mixture of fullerenes.
Shionoya and Clever reported the synthesis of [M2L4] cages (M = Pd(II) or Pt(II)) from rigid concave ligands 8a based on an annelated norbornene backbone that was obtained by a series of cycloaddition reactions (Fig. 3a).31 The ribbon-shaped backbone does not contain any rotatable bonds except for the attachment sites of the pyridine donors, which is a quite unique structural feature among the ligands used in such self-assemblies.
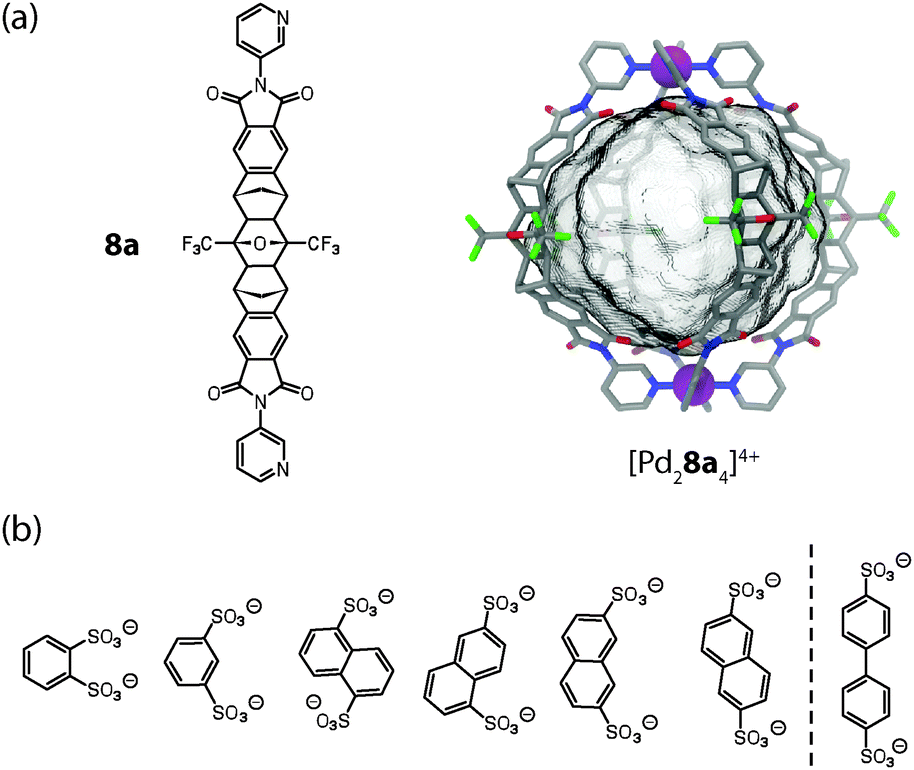 | ||
| Fig. 3 (a) Ligand 8a and X-ray structure of cage [Pd28a4]4+; (b) series of bis-sulfonates that were tested as guest molecules for the uptake in [Pd28a4]4+. Only the first six fit inside the cage. | ||
The globular structure of cage [Pd28a4]4+ was revealed by an X-ray structure. It is maintained in solution as evidenced by a diffusion ordered spectroscopy (DOSY) NMR experiment. The two Pd(II) cations are spaced 1.7 nm apart and act as electrostatic anchors for the quantitative encapsulation of a series of bis-anionic guest compounds. The encapsulation was monitored by 1H NMR titrations, DOSY experiments, ESI mass spectrometry and – in the case of a redox-active guest based on ferrocene – cyclic voltammetry.31 By using carefully selected aromatic bis-sulfonate anions of different size (with regard to their S–S distance), it was shown that the relative binding affinity is a function of the length match between the anionic guests and the separated cationic sites of the cage (Fig. 3b).32 Longer bis-sulfonates were found to replace shorter ones from the inside of the cage in competition experiments. This effect was observed, however, only up to an optimal length (reached with the guest 2,6-naphthyl bis-sulfonate) and longer guests (such as 4,4′-bipyridyl bis-sulfonate) were found to be too large to be encapsulated. Interestingly, the addition of such oversized guests to the cage resulted in the precipitation of an insoluble salt composed of the cages joined by the bis-anions via the outer sites of the Pd(pyridine)4-planes. The same effect was observed when a solution of host–guest complexes (“full cages”) was treated with excess amounts of a bis-anionic guest. In the latter case, however, the size of the guest was not a determining factor.
Both observations formed the basis for experiments with the light-switchable guest 4,4′-azobenzene bis-sulfonate (Fig. 4a).33 Whereas the cis-isomer was found to be perfectly accommodated inside the cavity, the trans-isomer is too large and thus mediates the aggregation of the cages through ionic interactions via their outer sides. The use of a highly soluble PEGylated cage derivative allowed for the NMR monitoring of this process by alternating irradiation with UV light (365 nm, generating the cis-isomer) and white light (producing the trans-isomer). The situation became even more interesting when the soluble host–guest complex of the unsubstituted cage derivative [Pd28a4]4+ including the cis-isomer was subjected to irradiation with white light (Fig. 4b). The resulting aggregates of the cage with the released trans-isomer of the guest happened to have such a low solubility that single crystals separated from the sample within 20 minutes after the onset of the irradiation. An X-ray structure analysis revealed that the expelled trans-isomeric guests have linked the cages into chain-like assemblies {[cage][guest]}n by ionic interactions. The chains are aligned in parallel and further cross-linked by additional bis-sulfonate guests (Fig. 4c).
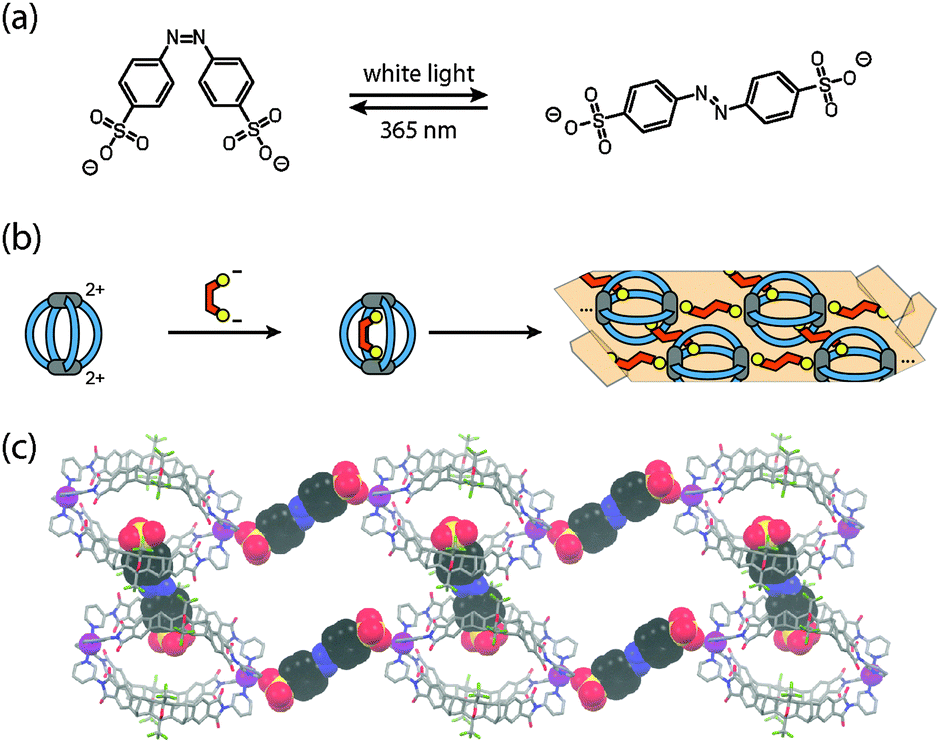 | ||
| Fig. 4 (a) Structure of a light-switchable bis-sulfonate guest based on azobenzene; (b) encapsulation of the cis-guest into [Pd28a4]4+ followed by light-triggered crystallization; (c) X-ray structure of the cage network linked by the trans-configured guests. | ||
In a following work, the mechanism of guest exchange was studied based on four (pseudo)rotaxanes prepared from the isostructural Pd(II) or Pt(II) centred cages and two rod-like bis-sulfonate threads with small and large stoppers, respectively (see Fig. 5a for a model of the small stoppered derivative).34 By a combination of NMR spectroscopic and mass spectrometric experiments it was shown that the small stoppered thread slips into the cage within seconds, regardless of the kind of metal in the cage structure. In contrast, the uptake of the large stoppered thread was found to be not only much slower per se but also dependent on the kind of metal composing the cage. With the Pd(II) cage, uptake was complete within 90 min at room temperature. Uptake into the Pt(II) cage, however, required heating the sample to 80 °C for several days. This indicates a change of the rotaxanation mechanism from slipping to clipping in the case of the large-stoppered guest as the ligand exchange kinetics around the cage's metal centres become the determining factor.
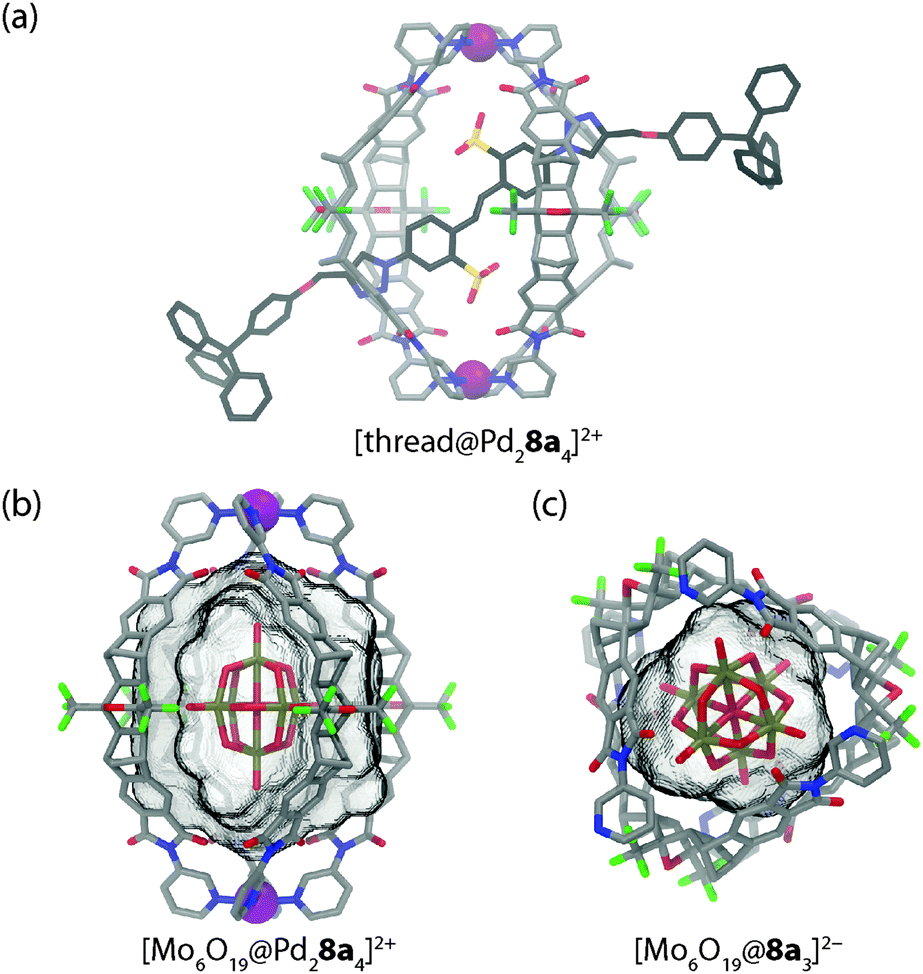 | ||
| Fig. 5 (a) Molecular model of a pseudorotaxane based on cage [Pd28a4]4+ and a rod-like bis-sulfonate thread; (b) PM6 model of inclusion complex [Mo6O19@Pd28a4]2+ and (c) X-ray structure of non-covalent aggregate [Mo6O19@8a3]2−. | ||
In a further work, Clever and co-workers have shown that anion encapsulation in [Pd28a4]4+ is not limited to aromatic sulfonate guests. Also the large globular anion hexamolybdate [Mo6O19]2− can be quantitatively encapsulated inside the cage as evidenced by 1H NMR titrations and ESI mass spectrometry (Fig. 5b).35 Comparable to the aforementioned observations, the addition of excess amounts of the polyoxometallate guest led to the formation of a precipitate. A surprising result was obtained when this microcrystalline solid was heated and subjected to slow evaporation in acetonitrile. The X-ray structure analysis of the formed single crystals indicated a breakdown of the initial host–guest complex furnishing a structure in which three ligands 8a are wrapped around one hexamolybdate dianion in a chiral, cyclic arrangement in the absence of any Pd(II) ions (Fig. 5c). To compensate for the charge of the anionic guest, two pyridine sites are protonated.
4. Encapsulation of stacked metal complexes
With regard to host–guest chemistry, a further degree of complexity is introduced in systems capable of coencapsulating more than one guest molecule. On the one hand, this feature offers the possibility to run bimolecular chemical reactions inside the self-assembled cages.6 On the other hand, it allows for the study of close interactions between discrete metal complexes inside the constrained environment of a molecular cavity. For example, coordination cage [Pt28a4]4+ has been shown by Shionoya and Clever to coencapsulate two equivalents of [PtX4]2− (X = Cl− or Br−) and one equivalent of [Pt(NH2C4H9)4]2+ or [Pt(pyridine)4]2+. The guests are sandwiched between the cage's two integral [Pt(pyridine-ligand)4]2+ sites, hence forming a soluble Pt5-array of alternating cationic and anionic square-planar platinum(II) complexes.36 The discrete supramolecular assembly thus represents a monodisperse cut-out from a parental polymeric structure related to the long known Magnus' salt {[Pt(NH3)4][PtCl4]}n. The self-assembly of the three guests inside the cavity proceeds with positive cooperativity, as evidenced by NMR titrations and ESI mass spectrometric measurements. Furthermore, the single-crystal X-ray data revealed that all five involved platinum centres are linearly aligned along the cage's fourfold axis (Fig. 6a). Owing to the substantial size of the pyridine donor sites, the observed Pt–Pt distances within the Pt5-array were found to lie between 3.81 Å and 4.15 Å, thus significantly exceeding the sum of the van der Waals radii of two Pt(II) ions (3.50 Å) as well as the Pt–Pt distances found in Magnus' Green salt (3.25 Å). Consequently, no UV-Vis spectroscopic indications of direct orbital interactions between the linearly aligned Pt-centres were found in the described system.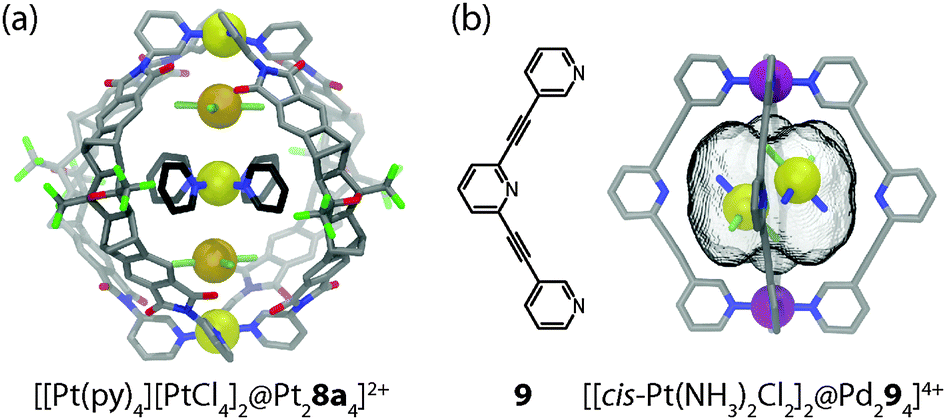 | ||
| Fig. 6 (a) X-ray structure of the quaternary host–guest complex [[Pt(py)4][PtCl4]2@Pt28a4]2+ and (b) X-ray structure of the ternary host–guest complex [cis-[Pt(NH3)2Cl2]2@Pd294]4+ including two molecules of the anticancer drug cisplatin. | ||
In a separate study, Crowley and co-workers reported a molecular cage [Pd294]4+ based on four tripyridyl ligands 9 and two Pd(II) ions, in which two neutral molecules of the anticancer drug cisplatin were encapsulated in an aqueous environment (Fig. 6b).37 In contrast to the previous example, the guest dimer does not form a linear stack together with the cage's [Pd(pyridine-ligand)4]2+ sites. Instead, the line through its Pt-centres is inclined by 62.5° with respect to the cage's fourfold Pd2-axis. The reason for this difference can be explained by the dissimilarity of the interactions that are responsible for the guest uptake: alternating electrostatic attractions in the former example as opposed to a combination of hydrophobic effects and hydrogen bonds between the amines of the cisplatin molecules and the central, non-metal-bound pyridyl moieties in the latter case. The cage itself shows a stimuli-responsive behaviour and can be reversibly disassembled/reassembled upon the addition/removal of suitable competing ligands. This feature of controlled release of a molecular cargo promises to play a role in future medical applications.
A number of further examples of coordination cage based metal stacks are worth to be mentioned in this context, although their ligand and/or cage structures deviate from the main topic of this article. Fujita and co-workers have reported a set of trigonal prisms synthesized by the combination of a cis-capped Pd(II) acceptor, a planar tripyridyl-triazine donor (10), and a linear bipyridyl pillar (11a or 11b) in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 molar ratio (Fig. 7). The size of the hydrophobic cavity of the cage can be adjusted by altering the length of the pillar ligands. The prisms with larger cavities were able to encapsulate some neutral flat metal complexes in pairs or triples, presumably by a combination of hydrophobic effects, π-stacking contacts between their ligand moieties and d–d orbital interactions between their metal centres.38 For example, two neutral [M(acac)2] (M = Pt, Pd or Cu; acac = acetylacetonato) complexes could be inserted in the form of a dimeric stack into the prismatic cage [Pd610211a3]12+ (Fig. 7).39 Strong metal–metal interactions were confirmed by powder UV-Vis measurements in the solid state in the case of the encapsulated Pt and Pd complexes. The X-ray analysis of the [Pt(acac)2] containing cage revealed a short Pt–Pt distance of 3.32 Å.
:2:3 molar ratio (Fig. 7). The size of the hydrophobic cavity of the cage can be adjusted by altering the length of the pillar ligands. The prisms with larger cavities were able to encapsulate some neutral flat metal complexes in pairs or triples, presumably by a combination of hydrophobic effects, π-stacking contacts between their ligand moieties and d–d orbital interactions between their metal centres.38 For example, two neutral [M(acac)2] (M = Pt, Pd or Cu; acac = acetylacetonato) complexes could be inserted in the form of a dimeric stack into the prismatic cage [Pd610211a3]12+ (Fig. 7).39 Strong metal–metal interactions were confirmed by powder UV-Vis measurements in the solid state in the case of the encapsulated Pt and Pd complexes. The X-ray analysis of the [Pt(acac)2] containing cage revealed a short Pt–Pt distance of 3.32 Å.
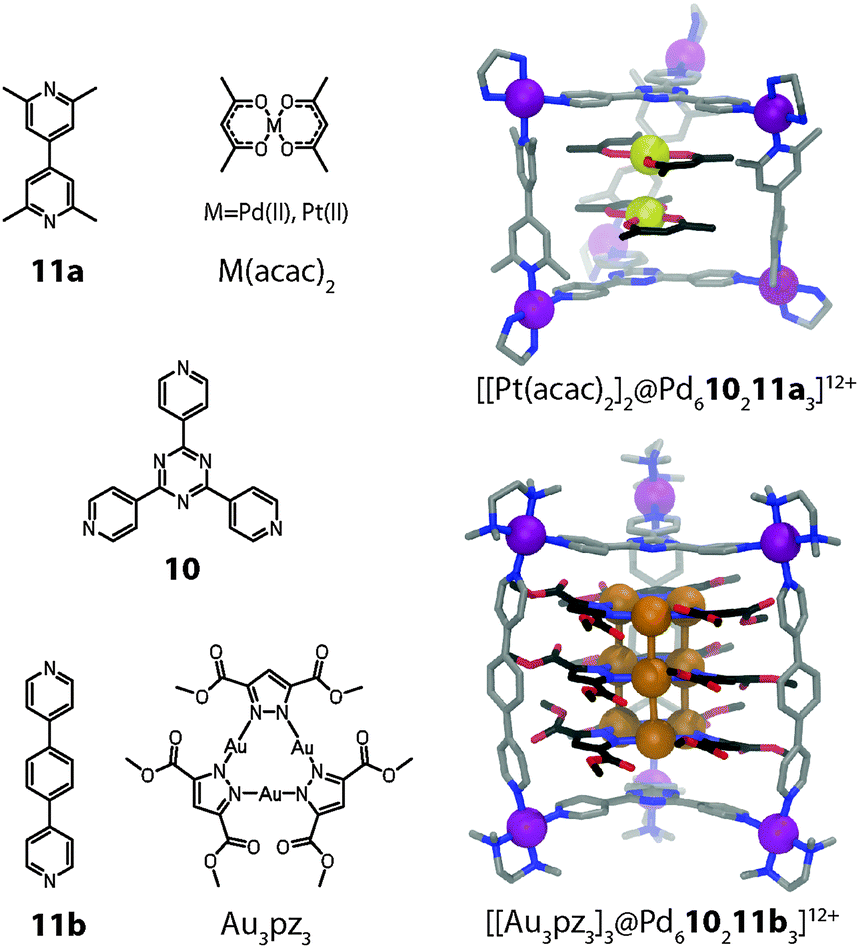 | ||
| Fig. 7 X-ray structures of ternary host–guest complexes [[Pt(acac)2]2@Pd610211a3]12+ and [[Au3(pz)3]3@Pd610211b3]12+. | ||
A strong spin–spin interaction within an encapsulated dimer of [Cu(acac)2] complexes, on the other hand, was found by EPR analysis of the corresponding host–guest complex. The ferromagnetic coupling (as confirmed by observing the Δms= 2 transition of the triplet state) between the two Cu(II) centres was attributed to the twisted orientation of the two stacked Cu(II) complexes relative to each other.
In a related study, the same group demonstrated that up to three stacked trinuclear pyrazolato-bridged Au(I) clusters can be incorporated inside a series of structurally related prismatic cages (e.g. [Pd610211b3] depicted in Fig. 7).40 The cage height thereby controls the number of stacked structures in the encapsulated array. Intermolecular aurophilic interactions were observed in the X-ray structures of the assemblies. The intermolecular Au–Au distances of 3.23 Å are significantly shorter than the sum of the van der Waals radii (3.60 Å). In summary, this approach allows for the precise 3D arrangement of metal centres with possible applications in the fields of catalysis, tuneable redox systems and luminescent materials.
5. Assemblies with M3L6 and M4L8 stoichiometry
The combination of two metal ions with four banana-shaped ligands is a straightforward way to produce cage compounds [M2L4], representing the smallest possible architectures with a defined inner cavity. Under certain circumstances, however, these assemblies can be thermodynamically disfavoured and coordination compounds with a higher nuclearity and a more complex topology are formed instead. Important members of the latter category include entanglements, knots, and catenated structures.3A general approach towards the design of coordination cages with such unusual geometries does not exist. A desired topology can often be influenced, however, by a careful ligand design, the coordination environment of the metal ions, solvent effects as well as the presence of templating molecules and counter ions. The most important features of the ligand design include the number and kind of the donor functionalities, the angle between the donor sites, the flexibility and steric demand of the backbone structure. Furthermore, the presence of functional groups and sites for non-covalent interactions can be determining for the outcome of the self-assembly. Solvent molecules and counter anions may influence the topology due to their size, dipole moment or charge. Most often they act as templates and define the size and nature of the cavity.
Two examples from the recent literature illustrate the different contributions, which are able to direct the shape of a supramolecular assembly. In a system reported by Fujita and co-workers,41 the self-assembly of ligand 12 with Pd(II) cations was found to either yield tubular trigonal or tetragonal prismatic coordination cages (Fig. 8a and b). In contrast to the [M2L4] cage forming ligands discussed above, the fixed donor angle of approximately 60° renders ligand 12 incapable of forming [M2L4] cages. Instead, the system escapes the situation of unbearable strain in the ligand backbones or the square-planar coordination spheres by forming structures of higher nuclearity. Both the trigonal and the tetragonal prismatic geometries seem to possess comparable strain energies, although the formation of [Pd3126]6+ should be entropically favoured. Interestingly, the choice of solvent turned out to be a decisive trigger to drive the exclusive formation of one geometry or the other. In DMSO-d6 the tetragonal shape is favoured, whereas in CD3CN (and other solvents) the trigonal prismatic cage is formed. Analysing both crystal structures, this effect can be attributed to the two different cavity sizes, which are filled by counter anions and solvent molecules. In [Pd3126]6+ the cavity is small and filled by one water and two acetonitrile molecules. In the case of [Pd4128]8+ in DMSO, however, the cavity contains a favourable aggregate of two nitrate anions and two DMSO molecules, which compensates for the entropical disadvantage. This example demonstrates that despite a careful ligand design, all the different contributions to a thermodynamically stable assembly have to be considered. Quite often, this limits the predictability for the formation of a desired structure.
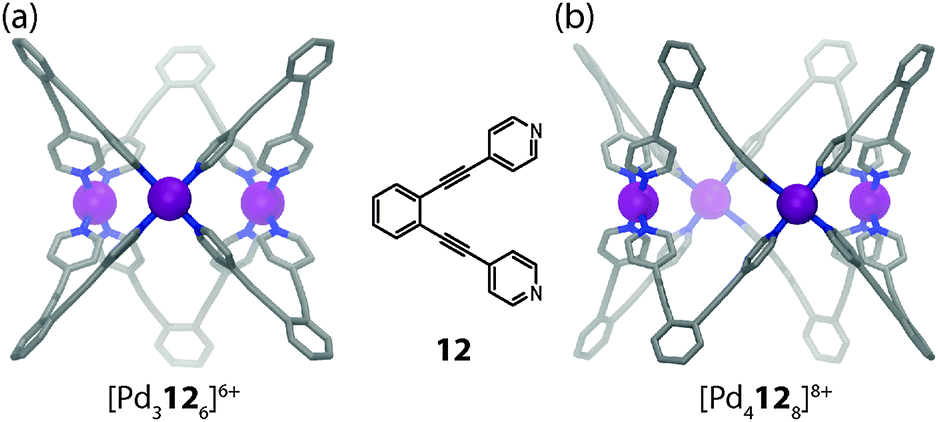 | ||
| Fig. 8 X-ray structures of (a) tubular trigonal prismatic cage [Pd3126]6+ and (b) tubular tetragonal prismatic cage [Pd4128]8+. | ||
This also holds true for the outcome of the self-assembly of another bidentate pyridine-based ligand 8c with palladium cations.42 Ligand 8c is a slightly altered version of ligand 8a, which was already described above in detail. In order to increase the cavity size with respect to cage [Pd28a4]4+, the two isomeric ligands 8b and 8c were synthesized carrying 1,3- or 1,4-phenylene spacers, respectively, between the backbone and the pyridyl donors (Fig. 9). As expected, the reaction of ligand 8b with Pd(II) cations led to the formation of a [Pd28b4]4+ cage that is widened in the directions perpendicular to the Pd2-axis. The isomeric ligand 8c, intended to elongate the cage along the Pd2-axis, however, was found to form a more complicated assembly obeying the formula [Pd38c6]6+ as evidenced by ESI mass spectrometry. Surprisingly, the 1H-NMR spectrum of the cage assembly [Pd38c6]6+ shows not only a signal shift due to the metal coordination, but also tremendous signal splittings. The observed splitting pattern indicates a low symmetric surrounding of the ribbon-shaped backbones of every ligand 8c in the cage as compared to the free ligand. A simple trigonal prismatic tube shape as found for [Pd3126]6+ can be immediately ruled out. Devoid of suitable crystals for an X-ray analysis, 2D NMR techniques together with molecular modelling were used to determine the 3D arrangement of the ligands. NOESY (Nuclear Overhauser Effect SpectroscopY) data allowed us to calculate a series of unique interligand proton distances, which in turn led to the identification of the correct topology (Fig. 10).
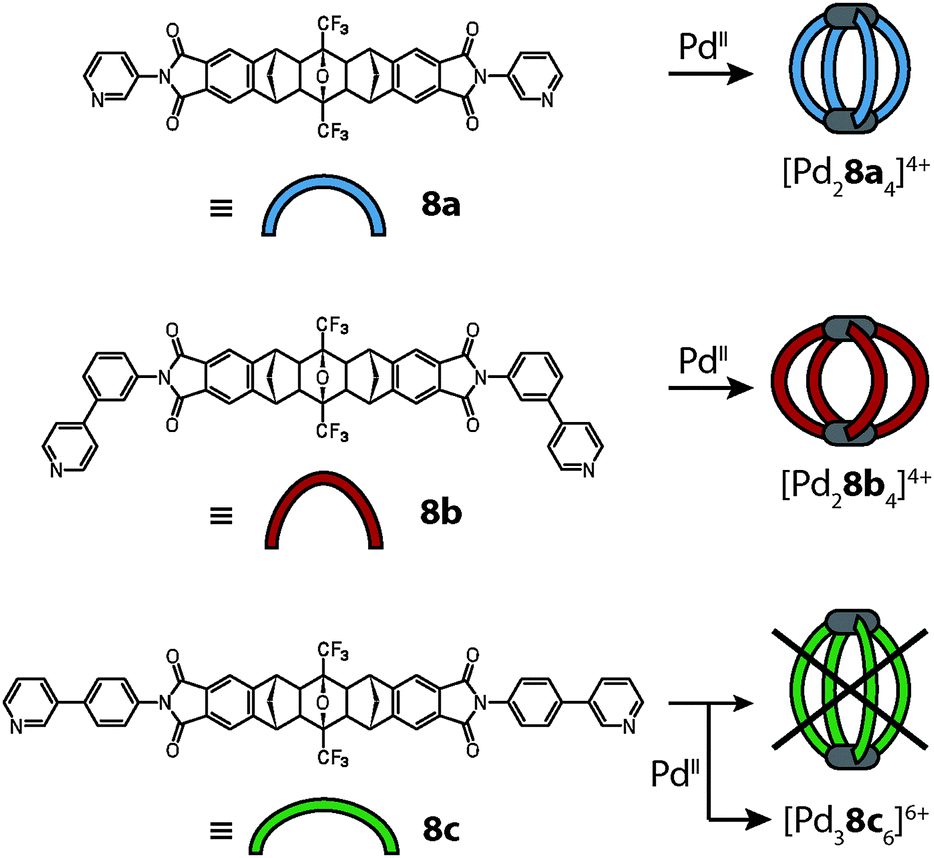 | ||
| Fig. 9 Assembly scheme for the Pd(II)-mediated assembly of the structurally related ligands 8a, 8b and 8c to give the anticipated cage structures [Pd28a4]4+ and [Pd28b4]4+ and the serendipitous double-trefoil knot [Pd38a6]6+ as the exclusive reaction products, respectively. | ||
 | ||
| Fig. 10 (a) Side view, (b) top view and (c) space-filling representation of the double-trefoil knot cage [Pd38c6]6+; (d) schematic depiction of the underlying topology with the two hemispheres separated. Figure in parts reproduced from ref. 42. | ||
As seen in Fig. 10 the cage is composed of two hemispheres, held together by the three metal ions. Each half resembles a trefoil knot. Although the ligand backbone is locked in the banana-shaped geometry, the pyridine and phenylene units exhibit a degree of freedom for rotation. Thus, a fourfold cage symmetry as in [Pd28a4]4+ is theoretically possible, but thermodynamically disfavoured, most probably due to attractive interactions within the intertwined double-trefoil knot structure. On top of the metal–ligand interactions, additional stabilization arises through inter-ligand hydrogen bonding and extended π-stacking between the aromatic units of the ligands. Due to the dense packing within the structure, the size of the cavity is 1300 Å3, nearly of the same size as for the parental structure [Pd28a4]4+ (V = 1380 Å3).
Each of the two trefoil-knotted substructures is chiral, therefore two combinations for the hemispheres of the cage are possible. One combination leads to a racemic mixture of a chiral cage, the second leads to an achiral meso-cage possessing a mirror plane defined by the three metal cations. The latter case was confirmed to be the most probable structure with the help of an encapsulation experiment using chiral camphorsulfonates as guest molecules.
6. Interpenetrated double-cages
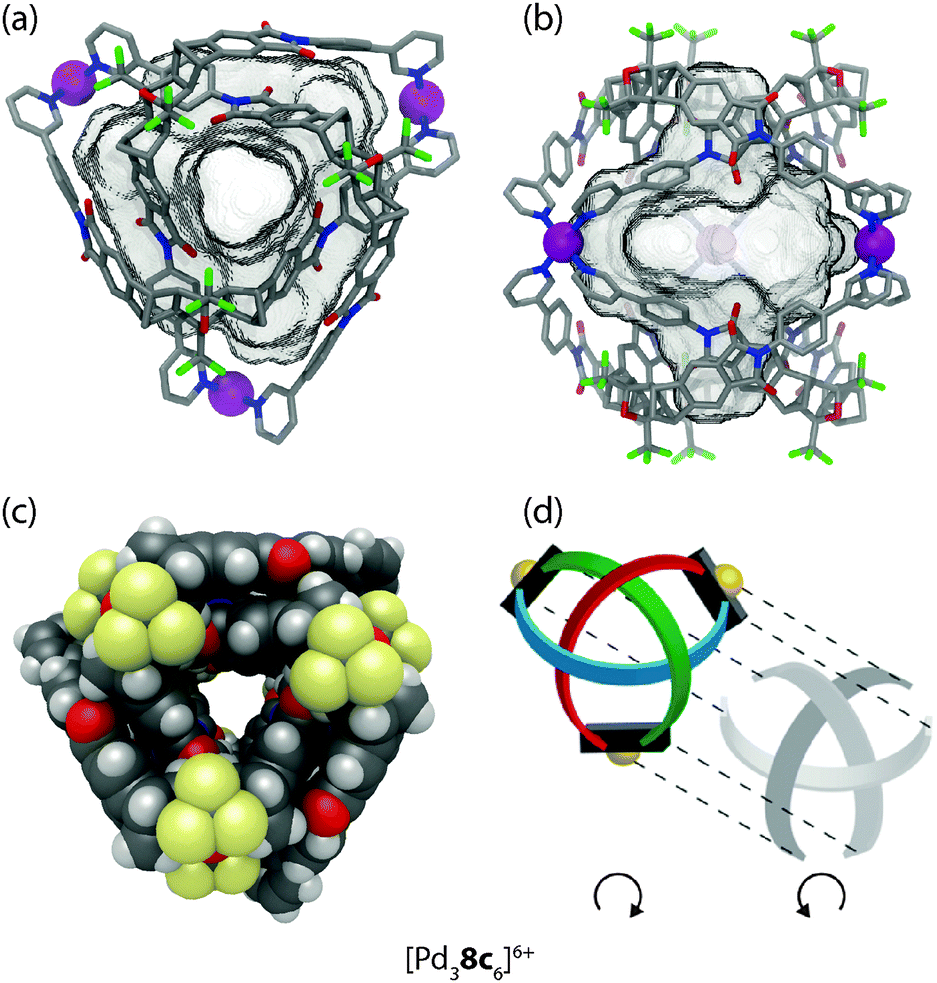
A special case of molecular entanglement is interpenetration, where two or more discrete species are interlocked without direct bonding connections between each other. In the field of discrete 3-dimensional coordination cages, the first example was reported in 1999 by the group of Fujita.43 They showed that a combination of two different tridentate ligands with three cis-blocked square-planar metal cations (Pd(II) or Pt(II)) yields interlocked dimers of coordination cages (Fig. 11).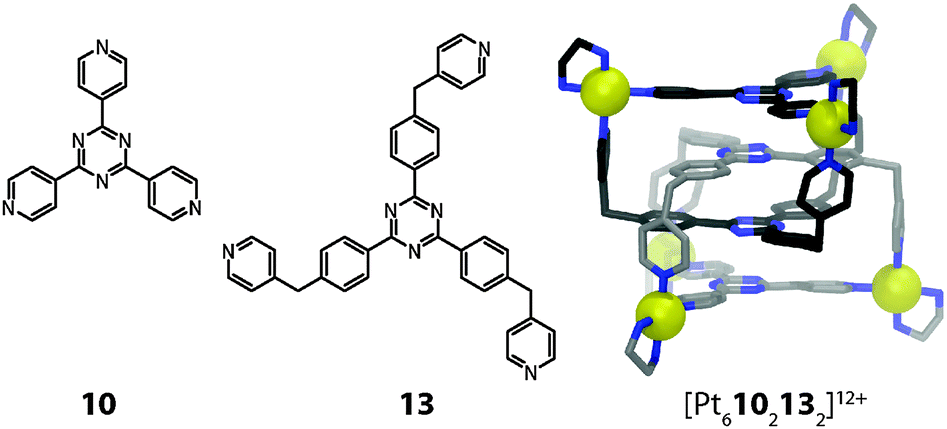 | ||
| Fig. 11 X-ray structure of the interpenetrated double-cage [Pt6102132]12+. | ||
Ligand 10 is planar and rigid, whereas ligand 13 has flexible methylene bridges. Constrained by the cis-capped metal centres, this restricts the outcome of the assembly to the formation of individual cages containing both ligands with the formula [M3(10)(13)]6+ (M = Pd(II) and Pt(II)). The driving force for the interpenetration leading to [M6102132]12+ is the attractive π–π stacking between the planar, aromatic ligand backbones. In addition, an entropic contribution arises as a consequence of the interpenetration: in contrast to the solvent filled cavity of the single cage, the interpenetrated cage assembly shows no cavity due to the dense packing. During the self-assembly process, cavity-confined molecules are released, giving rise to a favourable increase in entropy.
In 2008 Hardie reported another type of dimeric interpenetrated coordination cage.44 Whereas the aforementioned cage [M6102132]12+ is composed of two different ligands, each of Hardie's systems contains only one type of ligand. In the case of [M6144(NO3)6]6+ (M = Zn(II) or Co(II)) the ligand is composed of a bowl-shaped cyclotriveratrylene (Fig. 12). The conical geometry of the central macrocycle preorganises the attached 2,2′-bipyridine units on the same side, thus promoting the formation of a discrete self-assembly. Two of these ligands form a trigonal bipyramidal cage together with Zn(II) or Co(II) ions (one nitrate anion fills the remaining two coordination sites of each metal cation) and two of these cages dimerise to give the interpenetrated structure. The bowl-shape of the ligands gives rise to a cavity (V = 240 Å3), which has three openings to the outside. The interpenetration does not occur due to π–π stacking, as the distance between the aromatic units is too large (d = 4.77 Å). Instead, two kinds of weak hydrogen bonding interactions hold the two individual cages in place.
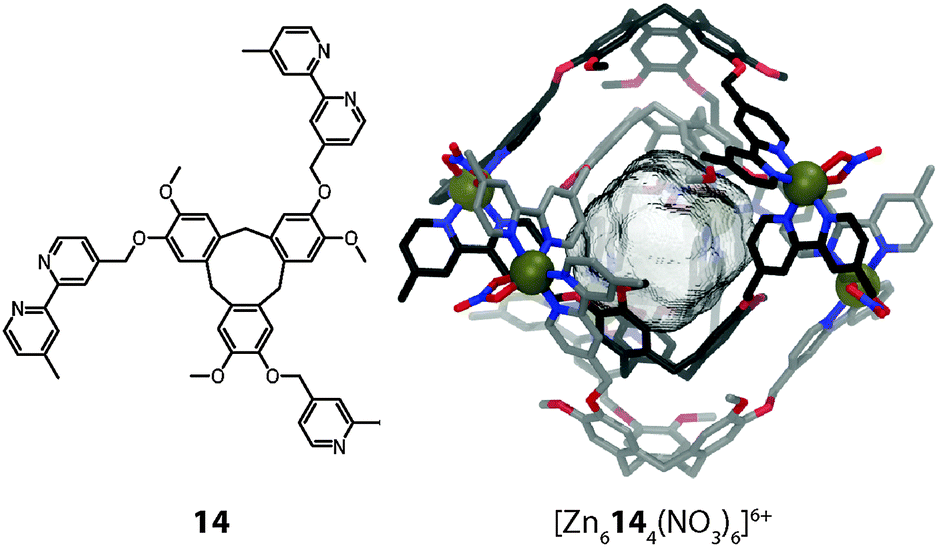 | ||
| Fig. 12 X-ray structure of the cyclotriveratrylene-based interpenetrated double-cage [M6144(NO3)6]6+. | ||
In both previous examples the involved metal ions are located in two parallel planes perpendicular to the axis of highest symmetry (C3). Within the interpenetrated cage dimer [Pd4158]8+ reported by Kuroda and co-workers the metal ions are all located on one single fourfold axis (C4) instead (Fig. 13).45 This mode of interpenetration leads to the formation of three separate cavities, possibly occupied by counter anions or solvent molecules although none could be resolved in the crystal structure.
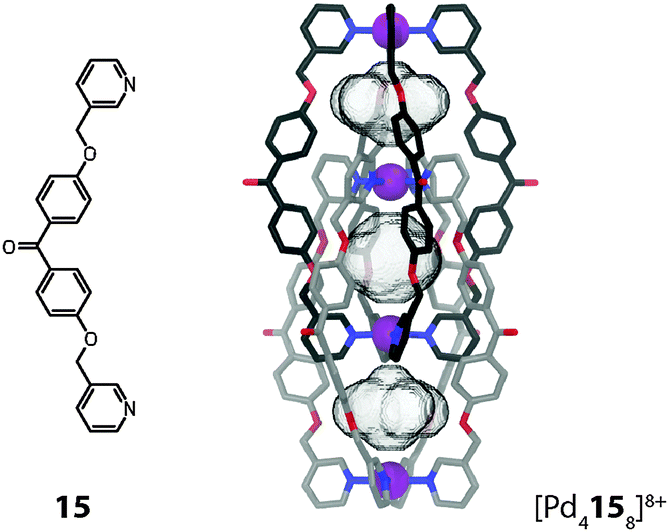 | ||
| Fig. 13 X-ray structure of the interpenetrated double-cage [Pd4158]8+. | ||
As compared to the previous example, this arrangement of the metal ions leads to distinct differences in the properties of the cavities: within [M6144(NO3)6]6+ the octahedrally coordinated metal ions are shielded from the cavity by the organic parts of the donor sites. In [Pd4158]8+, however, the square-planar coordinated metal ions are part of the atoms directly lining the cavity. The exposed positive charges facilitate the incorporation of anionic guests, which is indeed observed in these systems.
The binding of anions inside the pockets of [Pd4L8] assemblies was studied in great detail by Clever et al. on a series of structurally related interpenetrated coordination cages. First, three closely related structures based on the redox-active heterocycle phenothiazine will be discussed (Fig. 14).46 The ligands feature a sulphur atom at their concave side which is either not carrying any substituents (16a) or one (16b; sulfoxide) or two (16c; sulfone) oxygen attachments. Upon reaction with Pd(II), monomeric cages [Pd2164]4+ are quickly formed as reaction intermediates but shortly after consumed in the quantitative formation of the interpenetrated double-cages [Pd4168]8+. All three cage derivatives share a common topology and structurally resemble the aforementioned cage dimer [Pd4158]8+. The three pockets of the cages [Pd4168]8+ were found to be filled with tetrafluoroborate counter anions, both in the solid state (by X-ray analysis) and in solution (by 19F NMR spectroscopy). Phenothiazines show a rich redox chemistry undergoing one- and two-electron oxidations. In the presence of water, oxidation (and disproportionation) leads to the formation of the sulfoxide derivative. Under harsher conditions, the sulfoxides can be further oxidized to sulfones.
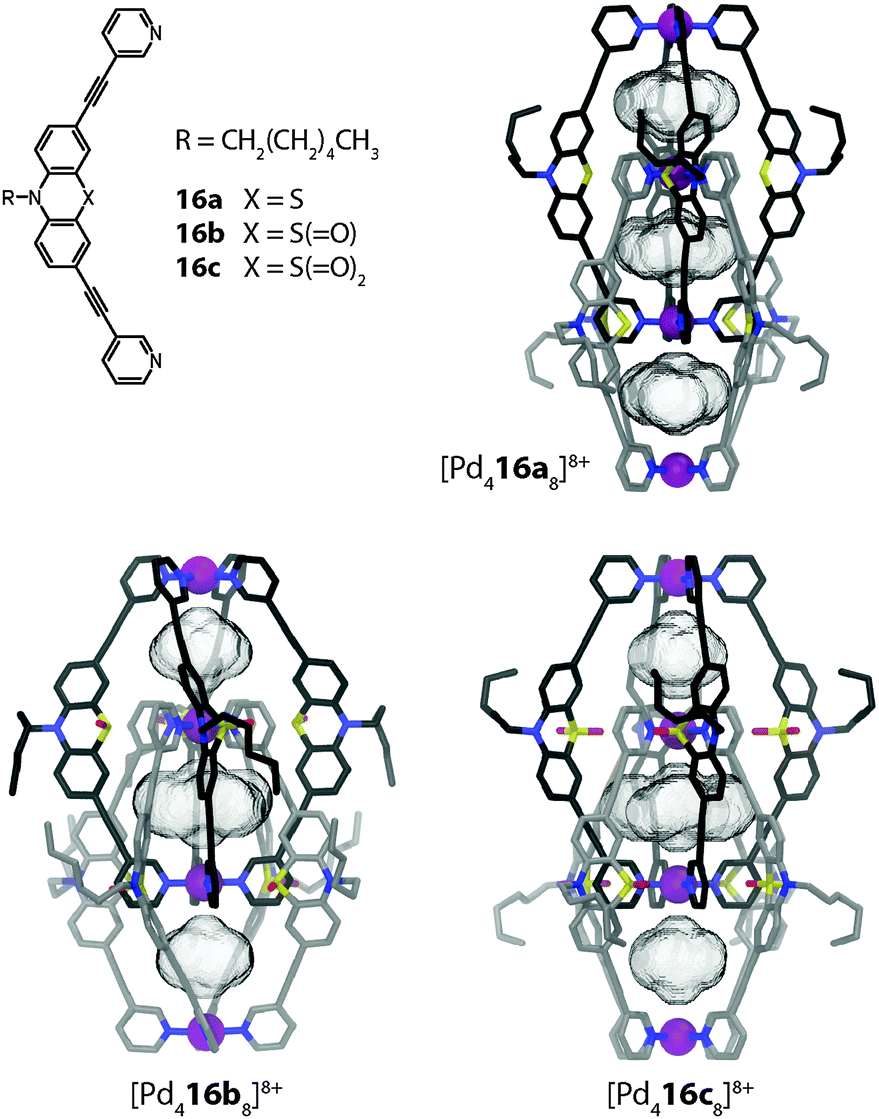 | ||
| Fig. 14 X-ray structures of three related interpenetrated double-cages [Pd416a–c8]8+ based on phenothiazine and its mono- and di-oxygenated derivatives. | ||
Indeed, the eightfold monooxygenated double-cage [Pd416b8]8+ was first obtained as the oxidation product of crystals of the parental structure [Pd416a8]8+ upon long-time exposure to air. Alternatively, the cage could be prepared by oxygenation of the ligand and subsequent addition of Pd(II). The sulfone cage [Pd416c8]8+ was assembled in a similar way. All three cages were structurally characterized by single crystal analysis (Fig. 14).46 From the structures, the Pd–Pd distances as well as the cavity sizes were obtained. Since all three cages bind halide anions (in particular chloride and bromide) with high but slightly different affinities in their outer two pockets, a correlation of their structural features with the relative anion binding strengths and selectivities was assumed. It turned out that the non-oxygenated cage, featuring the largest pockets, binds the halides weaker than the oxygenated variants, which showed comparable affinities. All cages bind chloride stronger than bromide, an observation that was also supported by theoretical calculations.47
An even stronger binder for halide anions was realized by the Clever group on the basis of a dibenzosuberone backbone containing a 7-membered ring carrying an endohedrally oriented carbonyl group ([Pd4178]8+, Fig. 15a).48 As for the phenothiazine cages, halide binding was found to proceed with strong positive cooperativity according to an allosteric mechanism, thus accompanied by a structural change. In particular chloride was bound with such a high affinity (Knet ≈ 1020 M−2) that solid AgCl could be dissolved by the cage in acetonitrile.49 A diffusion ordered spectroscopy (DOSY) NMR experiment clearly showed that the double-cage species undergoes a significant shrinking upon chloride binding. A deeper investigation based on NOESY NMR experiments and electronic structure calculations revealed that this reduction in size is caused by a compression of the cigar-shaped structure along the Pd4-axis by 3.3% accompanied by a relative rotation of the monomeric subunits by 8° (Fig. 15b). This said, the halide-binding induced movement resembles a screw motion which is no surprise regarding the cage's helical conformation. One and two-dimensional 19F NMR spectroscopic methods were deployed in order to study the dynamics of the tetrafluoroborate anions in the three pockets of the double-cage. The BF4− anions in the outer pockets were found to be in fast exchange with the anions in the free solution and this effect shows a strong solvent dependence.49 In contrast, the templating BF4− anion in the central pocket is locked in place due to the densely packed ligand surrounding. The compression of the double-cage structure upon replacement of the outer BF4− anions with chloride, however, leads to an increase in the size of the central cavity, which was observed to affect the nuclear spin relaxation kinetics of the encapsulated anion.49
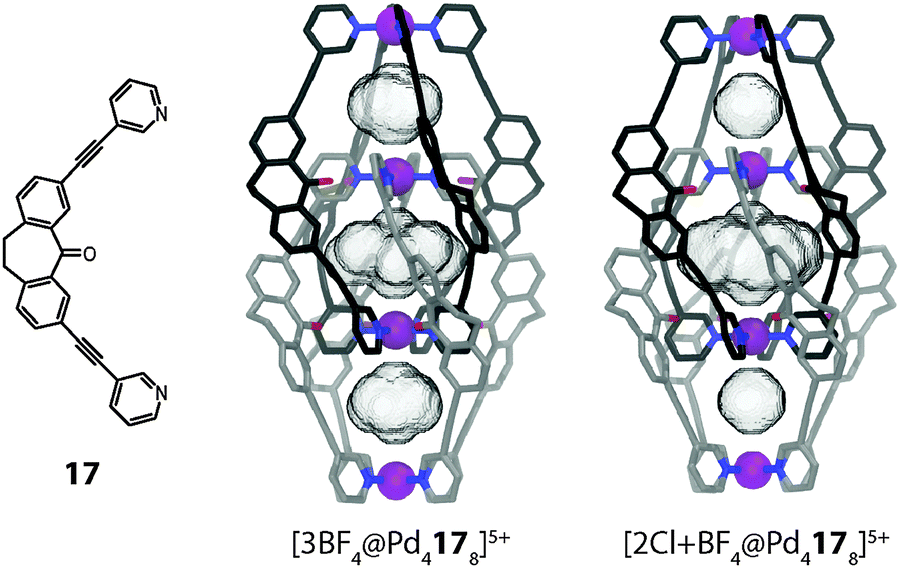 | ||
| Fig. 15 (a) X-ray structure of interpenetrated double-cage [Pd4178]8+ carrying three tetrafluoroborate anions in its two outer and one central pockets; (b) calculated structure of the double-cage after the two BF4− anions in the outer pockets have been replaced by chloride anions (instead of the anions, the cavity volumes are shown). | ||
The system was further modified by subjecting the carbonyl group of the dibenzosuberone backbone to the addition of various aromatic substituents by a Grignard reaction.50 The resulting ligand derivative carrying an anisyl substituent is shown in Fig. 16. Interestingly, addition of the [Pd(CH3CN)4](BF4)2 precursor to this bulky structure led to the clean formation of a monomeric cage [Pd2184]8+ in which the aryl substituents occupy positions near all four openings of the cage. Apparently, the rather large BF4− anion is not able to act as a template for the dimerization of this cage derivative since this would lead to a sterically overcrowded double-cage. Most surprisingly, however, double-cage formation could be induced by the addition of chloride, acting as a small template for the central pocket, since this allowed the two intertwined subunits to substantially move away from each other. The aryl substituents are then able to adopt positions in which they do not clash with the interpenetrating parts of the other cage subunit, as can be seen in the X-ray structure of the resulting double-cage (Fig. 16). The comparison of the pocket sizes of [Pd4188]8+ with the previously discussed cage dimers [Pd4168]8+ and [Pd4178]8+ reveals that the template-directed minification of the inner cavity leads to a substantial increase of the Pd–Pd distances and hence volumes of the outer pockets. It was shown that this structural alteration has a significant effect on the anion binding selectivity of the system. Whereas the previous double-cages containing the large BF4− template were found to bind small halide anions, double-cage [Pd4188]8+ strongly favours the binding of large oxoanions such as perrhenate ReO4− in its outer pockets.
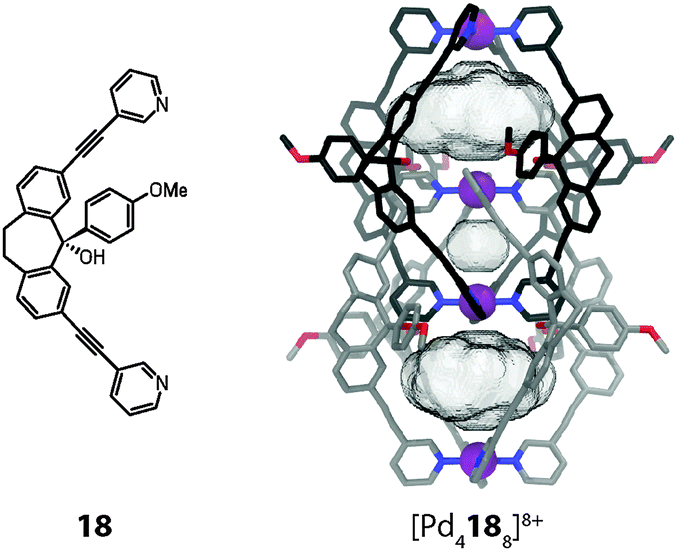 | ||
| Fig. 16 X-ray structure of the chloride-templated double-cage [Pd4188] whose voluminous outer pockets can encapsulate large anions such as ReO4− (instead of the anions, the cavity volumes are shown). | ||
7. Light-switchable cages
Over the past decades much attention has been devoted to molecular switching processes triggered by external stimuli such as irradiation with light.51 Light is considered as an ideal stimulus because of the ease of controlling irradiation time and location, precise regulation of the wavelength and the tuneable intensity. Molecular photoswitches were introduced into supramolecular structures such as host–guest systems early on. For example, Ueno described in 1979 the combination of an azobenzene-bridged β-cyclodextrin whose guest binding ability is modulated by photoisomerization of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond.52 Fujita and co-workers attached multiple azobenzenes to the concave side of a spherical cage, thereby enabling to switch the interior's hydrophobicity by a reversible photoisomerization.53
N double bond.52 Fujita and co-workers attached multiple azobenzenes to the concave side of a spherical cage, thereby enabling to switch the interior's hydrophobicity by a reversible photoisomerization.53
Recently, Clever and coworkers reported a photochromic coordination cage [Pd2194]4+ composed of four bis-monodentate pyridyl ligands 19 based on a light-switchable dithienylethene (DTE) backbone and two square-planar coordinated Pd(II) ions (Fig. 17).54 The DTE photoswitch can exist in a colourless “open-ring” photoisomeric form which contains rotatable single bonds and a deep blue “closed ring” form in which the scaffold is stiffened as the consequence of an electrocyclic ring closure. Irradiation of the flexible “open-ring” form o-19 of the ligand with UV light resulted in a quantitative interconversion into the structurally rigid “closed-ring” form c-19. The photoreaction was found to be fully reversible upon irradiation with white light. Interestingly, the assembled cage compound is amenable to a similar reversible interconversion process. As revealed by DOSY NMR experiments, the “open form” cage [Pd2(o-19)4]4+ has a higher structural flexibility and a smaller hydrodynamic radius than the stretched “close ring” form [Pd2(c-19)4]4+. The ability of both cage isomers to encapsulate anionic guests in their interior differs significantly. The affinity of the spherical closo-borate anion [B12F12]2− to the cavity of [Pd2(o-19)4]4+ was found to be about 100 times higher than observed for the [Pd2(c-19)4]4+ photoisomer. Furthermore, irradiation of the host–guest complexes allowed the light-controlled uptake and release of the guest compound. This effect might find application in supramolecular catalysis, drug delivery (for example in boron neutron-capture therapy) and molecular machinery.
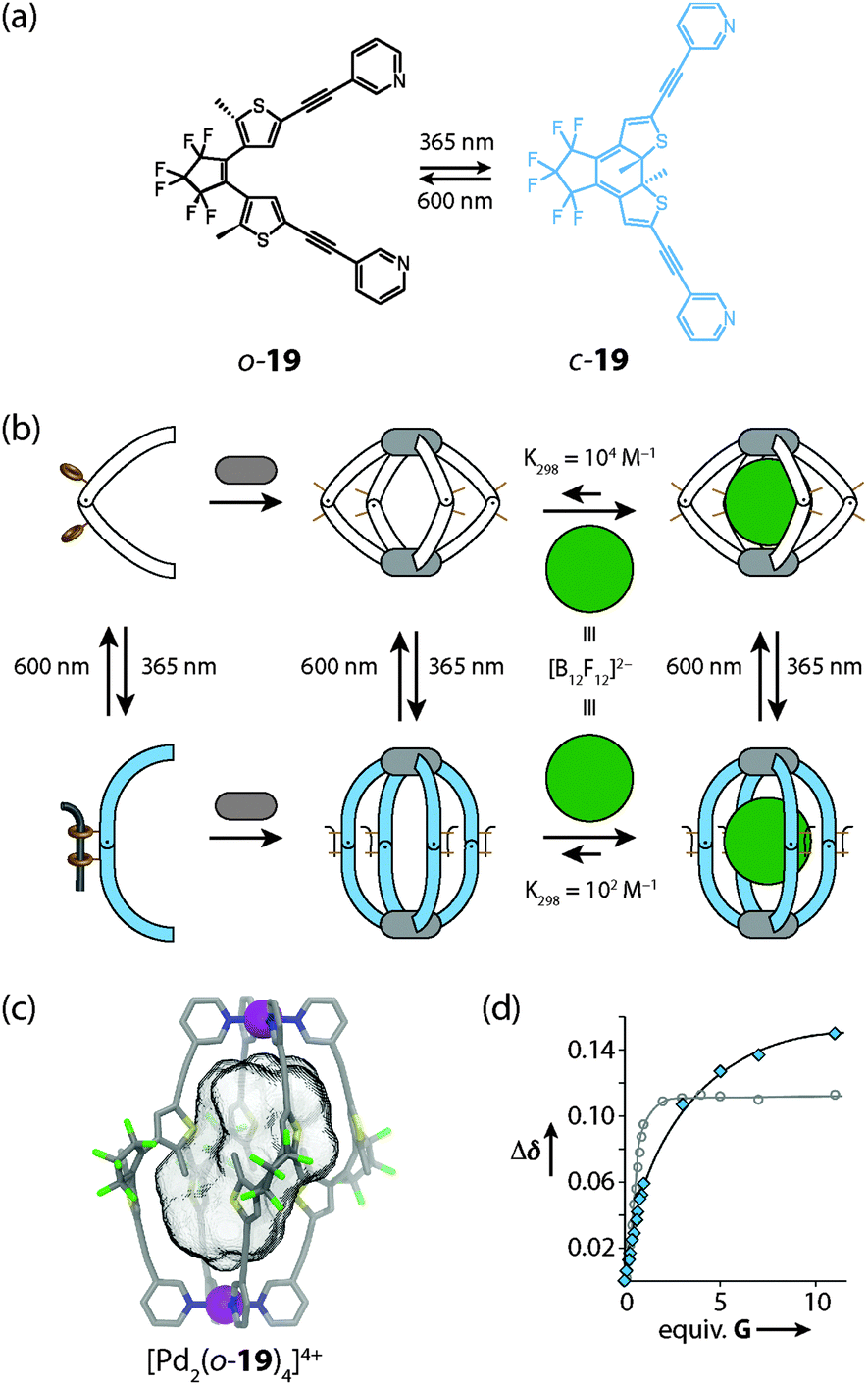 | ||
| Fig. 17 (a) Light-induced interconversion between the photoisomeric ligands o-19 and c-19, (b) schematic representation of the cage assembly and guest uptake/release processes; (c) X-ray structure of the [Pd2(o-19)4]4+ photoisomer of the cage; (d) comparison of binding isotherms for the uptake of the guest [B12F12]2− into the cavity of [Pd2(o-19)4]4+ (white dots) and [Pd2(c-19)4]4+ (blue dots), respectively. Figure in parts reproduced from ref. 54. | ||
8. Conclusion and outlook
The self-assembly of coordination cages has emerged into a vibrant area within the field of supramolecular chemistry. Beyond aesthetic appeal, functions associated with the nanoscopic cavities inside the cages promise to find applications in molecular diagnostics, separation techniques, catalysis and material science. Various strategies for gaining control over the size, shape, structural flexibility and topology of the cages are currently under development. Among the plethora of architectural principles and coordination environments that have been used to generate discrete coordination cages and capsules, this review has mainly focused on the combination of concave (“banana-shaped”) bis-monodentate pyridyl ligands and square-planar d8 metal cations (Pd(II) or Pt(II)). We showed that although the [M2L4] stoichiometry is the most commonly found structural motif, assemblies of higher nuclearity (such as [M3L6] and [M4L8]) may be formed under certain circumstances. Furthermore, compounds exhibiting complex topologies based on ligand entanglement or catenation are sometimes formed as the exclusive reaction products. The presented examples include a double-trefoil knot and a variety of interpenetrated double-cages.The factors deciding the formation of either simple topologies or more complicated, entangled structures are not yet fully understood. The ligand shape and flexibility play a role as do non-covalent interaction sites (e.g. for hydrogen bonding or π-stacking) included in the ligand structures. Also, counter anions, templating guest molecules and solvent effects can determine the outcome of an assembly reaction. In the case of the [Pd4L8]8+ interpenetrated double-cages, the understanding of the assembly principles has grown up to the point where the design of new interpenetrated cage derivatives became predictable.
Most of the discussed coordination cages show rich host–guest chemistry. Many cationic cages bind anions with high affinities and selectivities, depending on the size and shape of their cavities. On the other hand, water-soluble self-assemblies may be able to bind uncharged molecules by hydrogen bonding or π-stacking interactions, assisted by hydrophobic effects. Of particular interest are phenomena of coencapsulation, which enable guest–guest interactions. These are of importance in the fields of molecular magnetism and reactions inside confined cavities.
The anionic guests occupying the three pockets of the interpenetrated cages [Pd4L8]8+ were shown to communicate via a structural relay mechanism involving a relative movement of the two monomeric cage fragments along the Pd4-axis. As a result, the anion binding in the outer pockets proceeds with positive cooperativity and its selectivity is controlled by the size of the template in the central pocket.
The implementation of ligand-centred functions such as redox activity and photoswitchability opens opportunities for the use of self-assembled coordination cages in stimuli-responsive receptors, molecular transporters and smart materials. In future, the combination of the discussed subtopics (rational design of structure and topology, host–guest chemistry and implementation of functionalities) promises to lead to increasingly complex supramolecules with applications in molecular machinery and artificial enzyme mimicry.
Acknowledgements
M. H. thanks the CaSuS program of Lower Saxony for a PhD fellowship. D. M. E. thanks the Fonds der Chemischen Industrie for a PhD fellowship. We thank the DFG (CL 489/2-1 and SFB 1073) for financial support.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, 2nd edn, 2009 Search PubMed.
- (a) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC; (b) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS PubMed; (c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (e) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- (a) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed; (b) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- (a) J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC; (b) S. Kubik, Top. Curr. Chem., 2012, 319, 1–34 CrossRef CAS; (c) L. Palmer and J. Rebek, Org. Biomol. Chem., 2004, 2, 3051–3059 RSC.
- (a) M. Ziegler, J. Brumaghim and K. N. Raymond, Angew. Chem., Int. Ed., 2000, 39, 4119–4121 CrossRef CAS; (b) M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558–6559 CrossRef CAS PubMed.
- (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (b) D. Vriezema, M. Aragones, J. Elemans, J. Cornelissen, A. Rowan and R. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed; (c) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- M. Jung, H. Kim, K. Baek and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 5755–5757 CrossRef CAS PubMed.
- S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058–10059 CrossRef CAS PubMed.
- S. Mirtschin, A. Slabon-Turski, R. Scopelliti, A. H. Velders and K. Severin, J. Am. Chem. Soc., 2010, 132, 14004–14005 CrossRef CAS PubMed.
- M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383–5388 CrossRef CAS PubMed.
- M. Han, R. Michel and G. H. Clever, Chem.–Eur. J., 2014 DOI:10.1002/chem.201303181.
- Q.-F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330–333 CrossRef CAS PubMed.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed.
- J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2011, 4, 15–20 CrossRef PubMed.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- (a) J. L. Sessler, P. Gale, W.-S. Cho and S. J. Rowan, Anion Receptor Chemistry, Monographs in Supramolecular Chemistry, Royal Society of Chemistry, 1st edn, 2006 Search PubMed; (b) Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley-VCH, New York, 1997 Search PubMed; (c) P. D. Beer and S. Bayly, Top. Curr. Chem., 2005, 255, 125–162 CAS.
- L. J. Barbour, G. W. Orr and J. L. Atwood, Nature, 1998, 393, 671–673 CrossRef CAS PubMed.
- C.-Y. Su, Y.-P. Cai, C.-L. Chen, M. D. Smith, W. Kaim and H.-C. zur Loye, J. Am. Chem. Soc., 2003, 125, 8595–8613 CrossRef CAS PubMed.
- H. Amouri, L. Mimassi, M. N. Rager, B. E. Mann, C. Guyard-Duhayon and L. Raehm, Angew. Chem., Int. Ed., 2005, 44, 4543–4546 CrossRef CAS PubMed.
- Y.-B. Dong, P. Wang, J.-P. Ma, X.-X. Zhao, H.-Y. Wang, B. Tang and R.-Q. Huang, J. Am. Chem. Soc., 2007, 129, 4872–4873 CrossRef CAS PubMed.
- N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS PubMed.
- D. McMorran and P. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295–3297 CrossRef CAS.
- D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1652–1653 RSC.
- N. Yue, Z. Qin, M. C. Jennings, D. J. Eisler and R. J. Puddephatt, Inorg. Chem. Commun., 2003, 6, 1269–1271 CrossRef CAS.
- J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035–4037 RSC.
- H. S. Sahoo and D. K. Chand, Dalton Trans., 2010, 39, 7223–7225 RSC.
- P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef CAS PubMed.
- G. H. Clever, W. Kawamura and M. Shionoya, Inorg. Chem., 2011, 50, 4689–4691 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS PubMed.
- G. H. Clever and M. Shionoya, Chem.–Eur. J., 2010, 16, 11792–11796 CrossRef CAS PubMed.
- M. Han, J. Hey, W. Kawamura, D. Stalke, M. Shionoya and G. H. Clever, Inorg. Chem., 2012, 51, 9574–9576 CrossRef CAS PubMed.
- G. H. Clever, W. Kawamura, S. Tashiro, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 2606–2609 CrossRef CAS PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714–1725 RSC.
- M. Yoshizawa, K. Ono, K. Kumazawa, T. Kato and M. Fujita, J. Am. Chem. Soc., 2005, 127, 10800–10801 CrossRef CAS PubMed.
- T. Osuga, T. Murase, K. Ono, Y. Yamauchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 15553–15555 CrossRef CAS PubMed.
- K. Suzuki, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 2819–2822 CrossRef CAS PubMed.
- D. M. Engelhard, S. Freye, K. Grohe, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 4747–4750 CrossRef CAS PubMed.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS PubMed.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950–2951 CrossRef CAS PubMed.
- M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 706–710 CrossRef CAS PubMed.
- (a) M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102–10106 CrossRef CAS PubMed; (b) M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014 10.1039/C3DT53243G.
- M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906–15910 RSC.
- S. Freye, J. Hey, A. Torras Galán, D. Stalke, R. Herbst Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed.
- S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem.–Eur. J., 2013, 2114–2121 CrossRef CAS PubMed.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 2–34 Search PubMed.
- A. Ueno, H. Yoshimura, R. Saka and T. Osa, J. Am. Chem. Soc., 1979, 101, 2779–2780 CrossRef CAS.
- T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133–5136 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |