 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoporous materials for clean energy technologies
Noemi
Linares
a,
Ana M.
Silvestre-Albero
b,
Elena
Serrano
a,
Joaquín
Silvestre-Albero
*b and
Javier
García-Martínez
*a
aLaboratorio de Nanotecnología Molecular, Departamento de Química Inorgánica, Universidad de Alicante, Ap. 99, E-03080 Alicante, Spain. E-mail: j.garcia@ua.es; Fax: +34965903454; Tel: +34965903400x2372
bLaboratorio de Materiales Avanzados, Departamento de Química Inorgánica-Instituto Universitario de Materiales, Universidad de Alicante, Ap. 99, E-03080 Alicante, Spain. E-mail: joaquin.silvestre@ua.es; Fax: +34965903454; Tel: +34965909350x2226
First published on 4th April 2014
Abstract
Alternative energy technologies are greatly hindered by significant limitations in materials science. From low activity to poor stability, and from mineral scarcity to high cost, the current materials are not able to cope with the significant challenges of clean energy technologies. However, recent advances in the preparation of nanomaterials, porous solids, and nanostructured solids are providing hope in the race for a better, cleaner energy production. The present contribution critically reviews the development and role of mesoporosity in a wide range of technologies, as this provides for critical improvements in accessibility, the dispersion of the active phase and a higher surface area. Relevant examples of the development of mesoporosity by a wide range of techniques are provided, including the preparation of hierarchical structures with pore systems in different scale ranges. Mesoporosity plays a significant role in catalysis, especially in the most challenging processes where bulky molecules, like those obtained from biomass or highly unreactive species, such as CO2 should be transformed into most valuable products. Furthermore, mesoporous materials also play a significant role as electrodes in fuel and solar cells and in thermoelectric devices, technologies which are benefiting from improved accessibility and a better dispersion of materials with controlled porosity.
 Noemi Linares | Noemi Linares obtained her PhD Degree in 2010 at the Molecular Nanotechnology Lab in the University of Alicante (Spain), under the supervision of Prof. García-Martínez. After her dissertation, she moved to Florence (Italy) for a postdoctoral fellow at the ICCOM-CNR, where she was working under the ITN NANO-HOST on the immobilization of homogeneous catalysts in nanostructured materials, mainly for the synthesis of fine-chemicals under flow conditions. In May 2013, she came back to the Molecular Nanotechnology Lab to work on the synthesis of nanostructured solids with different functionalities incorporated in their structure, mainly for energetic applications (H2 storage, water-gas shift reaction, biomass valorization). |
 Ana M. Silvestre-Albero | Ana M. Silvestre Albero obtained her PhD Degree in 2010 working on the preparation and characterization of Pt catalysts supported on micro-mesoporous materials to ethanol combustion reaction in the group of Prof. Rodríguez-Reinoso. Since 2012, she has been working as a researcher at the University of Alicante. Her research interests include the preparation and characterization of micro-mesopores materials, environmental pollution control, CO2 storage, the catalytic epoxidation reaction and catalytic combustion of VOCs. She is the co-author of more than 20 peer-reviewed manuscripts. |
 Elena Serrano | Elena Serrano obtained her PhD in 2006 at the University of Basque Country (Spain) working on the nanostructuration of functional materials. She carried out her post-doctoral activity in collaboration with Arkema at the National Institute of Applied Sciences (INSA) at Lyon (France). In 2009, she joined the Molecular Nanotechnology Lab at the University of Alicante as a Research Fellow. Her current research interests are in the area of new synthetic pathways to prepare heterogeneous catalysts by the self-assembly of functional materials (metal nanoparticles, metal complexes, etc.), based on silica and titania materials for photocatalytic applications, where she has published extensively. |
 Joaquín Silvestre-Albero | Joaquín Silvestre-Albero, born 1976, studied chemistry in Alicante, Spain, where he completed his PhD thesis in 2003 in the group of Prof. Rodríguez-Reinoso. Then, he spent one-year in the group of Prof. Corma at ITQ, Valencia (Spain), and two more years at the Fritz-Haber-Institute, Berlín (Germany), in the group of Prof. Freund. In 2006, he moved back to the University of Alicante where he is now Associate Professor for Inorganic Chemistry. His interests range from materials science, adsorption processes, nanotechnology, heterogeneous catalysis and nanomedicine. His scientific work has been published in more than 75 peer-reviewed manuscripts, 4 book chapters and several patent applications have been filled in the last few years. Joaquín Silvestre-Albero has received several awards including the Alexander von Humboldt Stiftung. He has been Visiting Professor at the Vienna University of Technology (Austria), Universidad de los Andes (Colombia) and Universidad Nacional de San Luis (Argentina). |
 Javier García-Martínez | Javier García-Martínez is the Director of the Molecular Nanotechnology Lab at the University of Alicante, Spain. He has published extensively in the areas of nanomaterials, catalysis and energy and is the inventor of more than 25 patents. His latest books are Nanotechnology for the Energy Challenge (Wiley, 2013) and The Chemical Element (Wiley, 2011). He is the founder and chief scientist of Rive Technology, Inc. (Boston, MA), a venture capital-funded Massachusetts Institute of Technology (MIT) spin-off commercializing nanostructured catalysts for energy applications. He has received the Europe Medal in 2005, the Silver Medal of the European Young Chemist Award in 2006, and the TR 35 Award from MIT's Technology Review magazine; in 2009, he was selected as a Young Global Leader. Since 2010, he has been a member of the World Economic Forum Council on Emerging Technologies. He is a Fellow of the Royal Society of Chemistry, a member of the Global Young Academy and a Bureau member of the IUPAC. |
1. Introduction
Renewable energy technologies hold the promise to meet the increasing energy demands of a 7 billion people planet without depleting our natural resources and compromising the quality of our environment.1 Despite the significant growth of clean energy technologies observed in recent years, they are still far from reaching their full potential as in many cases they suffer from low yields, high cost, material scarcity, poor robustness, or challenging scalability.Many of these challenges are being overcome thanks to the ingenuity and creativity of chemists, materials scientists and nanotechnologists, who are developing new materials and reinventing old ones by modifying their structure, size, morphology, surface chemistry and porosity.2–4 Among the material science contributions which are providing some of the most significant advances in energy applications, we are covering here the development and role of mesoporosity, highlighting both new techniques to introduce controlled mesoporosity in solids and their application in a wide range of energy technologies.
Porous solids are ubiquitous due to their many advantages such as a large surface area, an enhanced accessibility and the ability to anchor different chemical functionalities on their surface. The use of molecular and supramolecular templates, especially surfactants, has been one of the most successful strategies for the production of materials with a controlled porosity.5 Since the discovery of MCM-41, a myriad of surfactant-templated solids has populated the research efforts of many groups, dealing with their synthesis, characterization and application. Some of the main advantages of this methodology are: their versatility, robustness, simplicity and ability to produce very complex and interconnected porous structures. Sol–gel chemistry techniques are typically used in combination with different surfactants to produce a wide variety of porous metal oxides. Both soft templates, such as surfactant and polymers and hard templates such as carbon and metal oxides and carbonates which can be burned-off or easily dissolved at a certain pH, have being extensively used to introduce controlled mesoporosity in a wide variety of solids. The combination of these strategies has yielded new hierarchical materials whose unique porous structures provide significant advantages, many of them described in this review.6
The control and/or modification of the surface chemistry of porous materials is a critical step in the synthesis of advanced materials for energy applications.7 The right surface polarity is needed for the good wettability of the electrode with the electrolyte solution, and for the good contact between the catalyst and the reaction solution. In other cases, more complex surface modification is needed. For example, the introduction of complex chemical species, such as dyes, on the surface of nanoparticles, is typically carried out in the preparation of dye-sensitized solar cells.
More recently, a novel strategy has been described to introduce a wide variety of chemical functionalities in porous solids, ranging from metal complexes to nanoparticles, clusters and homogenous catalysts. This has been possible thanks to the combination of surfactant-template and sol–gel chemistry techniques.8 Ranging from gains in accessibility, a higher surface area and enhanced yields to improved selectivity, the precise control over the pore structure and surface chemistry of mesoporous materials is having a major impact on the performance of a wide range of energy technologies, as described in some detail in this review.
2. New mesoporous materials with unique properties
2.1. Nanostructured mesoporous materials
Twenty years after the pioneering work of Mobil's research on the synthesis and characterization of a new class of porous materials, the M41S family,9 the literature about mesoporous materials has proliferated explosively. Since the early nineties, when the first Kresge et al.9a article was published, the investigations into mesoporous materials have continuously grown resulting in a new independent research area, closely related to zeolites and microporous materials. Proof of the interest that nanostructured mesoporous materials now generate can be found in the number of publications in this field: 6000 papers included the words mesoporous materials in the ‘Web of Science’ database in the year 2012, besides the numerous special issues, conferences, books and patents, that every year add to the accumulated knowledge of the subject. For all these reasons, it is clear that in this section we can only give a short summary of mesoporous materials, emphasizing the benefit that these materials can bring to renewable related technologies. For a more in-depth examination of the synthesis of mesoporous materials, readers are kindly directed to ref. 10–16 and to the themed issue about Mesoporous Materials that Chemical Society Reviews recently published to coincide with the 20th anniversary of this new material family.17According to IUPAC, mesoporous materials are those possessing pore dimensions between 2 and 50 nm. That is, between micropores and macropores in size (the Greek prefix meso- means ‘in between’).18 Initially, the aim of developing these materials was to overcome the ‘1 nm restriction’ that the use of zeolites imposes.5 Theoretically, using materials with similar features to zeolites (acidity, regular pore size, high BET surface area) but with larger pores could open the door to processing large molecules that are unable to enter into a zeolite porous framework.
The synthesis of nanostructured mesoporous solids is based on the supramolecular templating approach, where long chain organic surfactants are used as structure-directing agents (SDA) or templates. The assembly of these surfactant molecules in the presence of a silica precursor leads to a composite mesostructure during the condensation of the silica network. The subsequent removal of the surfactant gives a mesoporous material with porous systems replicating the surfactant's assembly, see Fig. 1. This method generates solids with narrow pore size distributions, high BET surface areas (∼1000 m2 g−1) and a tuneable pore size (depending on the surfactant used). Moreover, it can be applied to the synthesis of a great number of different solids, inorganic, organic–inorganic hybrids and organic solids.10
 | ||
| Fig. 1 Different structures of the M41S family: (top) surfactant supramolecular assemblies and (bottom) TEM images of the final materials. Reproduced from ref. 5. Copyright 2013, RSC. | ||
The M41S family was originally prepared employing cationic surfactants (S+) at high pH, where the silicate species are negatively charged (I−), and the electrostatic interaction (S+I−) is responsible for the silica structuration. Since the discovery of this approach, other synthetic pathways have been employed to prepare mesoporous materials in a wide range of pHs, temperatures and surfactant types (anionic (S−), neutral (S0/N0)). Moreover, the mesoporous materials family has been extended from the initial silica and silica/alumina solids to a huge number of other inorganic oxides. Some examples of these types of material, along with the synthetic pathways employed for their structuration, are shown in Table 1.
| Surfactant type | Interaction type | Inorganic solids | Ref. |
|---|---|---|---|
| Cationic S+ | S+I− | SiO2: M41S | 9 |
| WO3 | 19 | ||
| Sb2O5 | 19a | ||
| SnS2 | 19a, 20 | ||
| S+X−I+ | SiO2: SBA-1, SBA-2, SBA-3 | 19a, 21 | |
| SnO2 | 22 | ||
| ZnPO4 | 19a | ||
| Anionic S− | S−I+ | Mg, Al, Ga, Mn, Fe, Co, Ni, Zn, Pb oxides | 19a |
| S−M+I− | Silica: AMS (using a co-structure directing agent) | 23 | |
| ZnO, Al2O3 | 19a | ||
| Neutral N0/S0 | S0I0 | HMS | 24 |
| ZrO2 | 25 | ||
| SnO2 | 26 | ||
| CdS, CdSe, SnS2, Sb2S3 | 27 | ||
| (S0H+)(X−I+) | SiO2: SBA-15 | 28 | |
| TiO2, ZrO2, Al2O3, Nb2O5, Ta2O5, WO3, HfO2, SnO2, and mixed oxides | 29 | ||
| N0I0 | SiO2: MSU-X | 30 | |
| γ-Al2O3 | 31 |
Even if these materials did not accomplish the task for which they were initially conceived, which was to replace zeolites in different applications (mainly in petrochemistry), since they do not have the high catalytic activity of zeolites or their hydrothermal stability, the applications in which mesoporous materials are currently used have become countless. They are used in catalysis (as catalysts or supports), adsorption, pollutant remediation, sensors, drug delivery systems and, more related to the present review topic, photocatalysis, solar cells, fuel cells and batteries.10 The flexibility of the templating methods permits the synthesis of materials with a controlled pore size and structure, controlled wall compositions and highly interconnected surface areas, all of which allow the optimisation of the material for the specific application required.
In energy related devices, nanostructured materials have attracted much attention because of their unique properties compared to bulk materials.10 Especially relevant is the case of mesoporous transition metal oxides, which can confine d-electrons to the walls between the pores, endowing such materials with unusual magnetic, electrochemical and optical properties.15,32 For example, mesoporous SnO2 materials show a drastic reduction of dielectrics compared to bulk SnO2 and these unique optical and electrical properties can extend their applications to gas sensors, optoelectronics and as electrodes in solid-state ionic devices.33 Mesoporous TiO2 presents a much higher photocatalytic activity due to its higher surface area;34 mesoporous Nb2O5·nH2O shows improved efficiency for the hydrolysis of cellobiose than supermicroporous and bulk Nb2O5·nH2O;35 mesoporous versions of α-Fe2O3 exhibit enhanced lithium-ion storage capabilities and excellent cycling stabilities.36 These are not isolated cases, there are many other examples where the incorporation of mesoporosity in an inorganic solid brings new (or improved) properties that can expand the traditional applications of the material and make them, in many cases, ideal for energy technologies.7 This will be thoroughly discussed in the following sections.
2.2. Hierarchical materials
Hierarchical porous materials contain porous systems at two or three different scales in an ordered structure with interconnectivity between the pores. Interest in this kind of materials is growing rapidly due to their practical potential in different areas from nanoscience to catalysis, separation, electronics, optics, optoelectronics, energy, environmental and life science.37In the catalytic field, these materials can improve the performance of materials with only one type of porosity. For example, the incorporation of mesoporosity in zeolites, usually with micropores <1 nm, improves the diffusion of compounds within their pores and the size of molecules that can be catalyzed.38 This is a subject of enormous interest, mainly due to the improvements in fluid catalytic cracking (FCC) that can be obtained from the incorporation of mesoporosity into zeolites, while maintaining their other features. Different reviews in this topic can be found elsewhere.39 In summary, two main approaches have been employed to incorporate mesopores into zeolites: (1) top-down techniques that involve the removal of one of the two main components of zeolites, i.e. silica (e.g. via desilication by treatment in alkaline solutions)40 or alumina (e.g. via dealumination by steaming at high temperatures or by chemical treatments),41 and (2) bottom-up procedures that utilize soft42,43 or hard44 templates (e.g. surfactants, inorganic sacrificial materials, etc.). Fig. 2 shows a scheme of the different synthetic approaches to prepare mesoporous zeolites.
 | ||
| Fig. 2 Different synthetic approaches to prepare hierarchical zeolites. Reproduced with permission from ref. 48. Copyright 2013, Wiley-VCH. | ||
Regardless of the preparation method, the improved diffusion and accessibility to active sites of mesoporous zeolites results in higher catalytic activities and longer lifetimes than traditional microporous zeolites.39a Moreover, the incorporation of mesoporosity increases the selectivity of primary products, due to the shortening of contact time. Ying and García-Martínez have patented,45 published46 and more recently commercialized,47,48 the first method to introduce mesoporosity into Y zeolites with low Si/Al ratios, which are relevant to catalytic cracking, based on a new top-down approach using surfactant-templated crystal rearrangement, see Fig. 2. The mesostructured zeolite Y demonstrated excellent hydrothermal stability, which is critical to such applications. The testing of FCC catalysts made from mesostructured zeolite Y showed a significantly improved selectivity in product yields (more transportation fuels, i.e. gasoline and light cycle oil (LCO), and less coke, dry gases and uncracked residues).
With regard to renewable energy, these hierarchical zeolites can be very useful in bio-oil upgrading. Bio-oils produced from the pyrolysis of biomass are very inexpensive renewable liquid fuels. However, the fuel quality of the bio-oils is inferior to that of petroleum-based fuels and, in order to be used as replacements or supplements for fossil diesel or gasoline, they require upgrading treatment.49 This is mainly due to the high oxygen and unsaturated content that these fuels present which, consequently, causes a low stability over time and a low heating value.50
The hydro-processing of bio-oil reduces both the oxygen and unsaturated content, making hydrodeoxygenation a promising method for bio-oil upgrading.51 As in FCC, the upgrading of bio-oil in zeolites can be limited by the zeolite restricted pore size (<1 nm) since the process involves bulky reactants products.52 With this in mind, Fang and co-workers53 recently prepared a mesoporous ZSM-5 with wormhole like intracrystalline mesopore channels by a bottom-up approach using a dual template. That is, the typical microporous template for ZSM-5, tetrapropylammonium hydroxide (TPAOH), and a mixture of mesoporous templates such as, octyldimethyl-ammonium chloride (TPOAC) and cetyl trimethylammonium bromide (CTAB). The mesoporous zeolite was used to support Pt and its catalytic performance was evaluated in a dibenzofuran hydrodeoxygenation reaction, as a model bio-oil compound. This hierarchical material showed a better catalytic performance than Pt/ZSM-5 and Pt/Al2O3 due to the combination of a high acidity and easily accessible mesopore channels.
On another scale, the incorporation of macropores into mesoporous architectures also minimises diffusion barriers and may enhance the distribution of the active sites during catalyst preparation.39a The synthetic procedures of macro/mesoporous materials involve: (1) the combination of a supermolecular assembly of amphiphilic polymers or surfactants with second surfactant systems or with macrotemplates such as solid particles, liquid drops and air bubbles or (2) a single templating method combined with a supplementary chemical or physical method (chemical etching, chemical modification, physical deposition, and physical leaching, etc.).54
There are multiple instances where the use of macro/mesoporous materials enhances the catalytic properties of the material. Certain organic transformations of hierarchical materials result in more active and selective processes than their mesoporous counterparts. Some representative examples include single-site Ti-containing hierarchical macro/mesoporous silica, which displays higher catalytic activities for the epoxidation of linear α-olefins compared to Ti-containing mesoporous silica without macropores (the reaction rate is enhanced with increasing alkyl chain length of linear α-olefins).55 Pd supported on macro/mesoporous TiO2 monolith (7 nm mesopores and 2.5 μm macropores) exhibits better productivity (twice as good) and excellent durability in catalytic hydrogenation reactions under continuous flow conditions than its mesoporous counterpart (7 nm mesoporous). This is probably due to a smaller pressure drop, a higher mass transfer and more uniform residence time for reactants throughout the hierarchical material.56 Finally, SAPO-34, a microporous alumina phosphate silicate zeolite, can be synthesised in a monolith form with 4 nm mesopores and bimodal 1 and 6 μm macropores, and shows a higher catalytic activity and stability than the conventional SAPO-34 in the catalytic conversion of methanol to light olefins.57
This enhancement in the performance of hierarchical materials holds true for other properties as well. For example, since macropores have comparable dimensions to the wavelength of visible and UV light, their incorporation into porous materials can aid light scattering within them.39a As a result, greater light scattering and harvesting can amplify the photocatalytic efficiency, a fact which has been proven in different studies. For instance, hierarchical materials with flower-like morphologies (see Fig. 3), made up of mesoporous located within the nanosheets, and larger macro/mesoporous in the spacing between consecutive nanosheets, have been synthesized for photocatalytic applications. These solids have demonstrated superior photocatalytic activities, when compared with homologous mesoporous materials.
 | ||
| Fig. 3 SEM images of bismuth subcarbonate with a flower-like morphology. Reproduced from ref. 58b with permission from The Royal Society of Chemistry. | ||
In Fig. 3, a 3D in situ N-doped (BiO)2CO3 flower-like architecture is shown. This material was proved to be an efficient photocatalyst in the removal of NO in indoor air under both visible light and UV irradiation, with a higher activity than the material in particulate form.58 Other examples of the photocatalytic enhancement in hierarchical materials are, a TiO2 with a chrysanthemum-like morphology which has recently been reported to have a higher photoactivity for the degradation of methylene blue than the same material after grinding,59 and macro/mesoporous ZnO double-pyramids synthesised by an ultrasound-assisted approach that present a better photocatalytic activity in the degradation of organic dyes than irregular ZnO.60
Another application in which hierarchical materials have shown a superior performance over other morphologies is in energy storage technologies. Low density, ultraporous 3D nanoarchitectures combine a high surface area for heterogeneous reactions with a continuous and hierarchical porous network for rapid molecular flux. They therefore present the appropriate electronic, ionic, and electrochemical requirements for, among other uses, Li-ions batteries, supercapacitors and solar thermal storage systems. Excellent reviews about hierarchical materials for energy conversion and storage61 and specifically for lithium batteries,62 have recently been published.
Regarding Li-ion batteries (LIBs), their desired performance characteristics can have opposing requirements with the micro/nano-structure of electrodes. This problem can be solved using hierarchically designed electrodes, tailored to satisfy these conflicting requirements. For instance, novel porous NiO hollow microspheres prepared by an ultrasound-assisted template-free route and composed of loosely packed nanoparticles with diameters around 30–80 nm, see Fig. 4, showed an enhanced electrochemical performance when evaluated as an anode material for LIBs. Their good lithium-storage performance can be attributed to their unique porous architecture, which provides the structural flexibility for volume change and the routes for fast Li+ diffusion.63
 | ||
| Fig. 4 The SEM images of porous NiO hollow microspheres at different magnifications. Reprinted with permission from ref. 63. Copyright 2013, Elsevier. | ||
Many other hierarchical materials have been used in energy storage applications; indeed it is one of the most interesting applications of such materials. Transition-metal oxides have exhibited a high capacity for reversible lithium storage64 while structured carbon materials show an excellent performance as supercapacitor electrodes.65 The impressive potential of hierarchical materials in energy storage devices will be carefully reviewed later on.
2.3. Nanocasted mesoporous materials
Whilst their attractions are obvious, there are some limitations to the use of soft templates for the synthesis of transition metal mesoporous oxides. The hydrolysis and condensation of transition metal alkoxides is not as easy to control as that of silicon alkoxides. For this reason, non-siliceous mesoporous materials prepared via direct templating show, in most cases, low order, low crystallinity and poor stability.66 One strategy that can overcome these limitations is nanocasting. Nanocasting is a procedure where nanoscale moulds are used to prepare solids replicating those moulds. Fig. 5 shows a schematic representation of the nanocasting procedure. A 3D-interconnected porous solid is used as a hard template and the desired material is synthesised in its porous system using a suitable precursor and the necessary chemistry. Once the solid is prepared, the removal of the hard template gives the required porous material as a negative replica of the initial template. For a more comprehensive revision of the synthetic methods and applications of these nanocasted solids, readers are directed to ref. 67.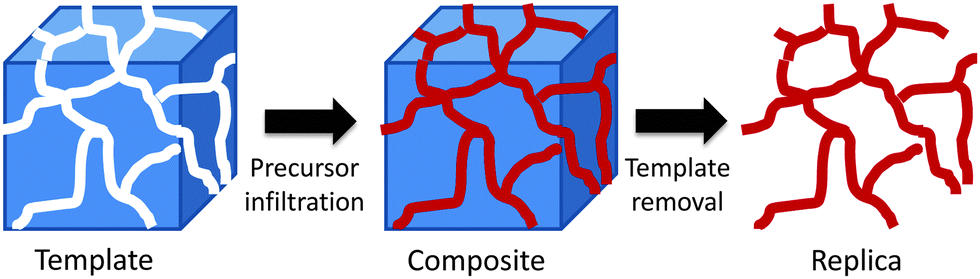 | ||
| Fig. 5 A schematic representation of the nanocasting pathway using a 3D-interconnected hard template. | ||
Following this approach, and with the appropriate experimental conditions, the nanocasting of carbons and metal oxides into meso- and macro/mesoporous silicas has been extensively studied, while nanocasting of metals is less common.68 Nanocasting offers a different way to prepare mesoporous and hierarchical materials, but the applications of the resulting materials are the same as have been mentioned above. Next some examples of nanocasted mesoporous solids used for energy applications are shown.
As has been already mentioned, structured porous carbons are one of the most interesting materials for energy storage. Their excellent chemical, mechanical and thermal stability, coupled with good conductivity and a high surface area, makes them ideal electrode materials for supercapacitors or batteries. A typical application of hierarchically porous carbon in LIBs was shown by Stein and co-workers (Fig. 6).69 They used a two-step procedure to synthesise hierarchically structured carbon monoliths. Firstly, a hierarchical silica monolith was prepared using poly(methyl methacrylate) (PMMA) colloids as templates for the macropores and silicate/poly(oxyethylene) surfactant solutions as precursors for the mesoporous walls. Once calcined, the macro/mesoporous silica monolith was used as a silica template to produce carbon monoliths with similar structural hierarchy by a nanocasting procedure. The resulting carbon monoliths maintained the open, interconnected macropore structure of the preform and the mesoporosity of the skeleton, which provided a high surface area >1200 m2 g−1 to the material. Next, more graphitic, nitrogen-doped carbon was introduced into the mesopores via CVD, producing a monolithic nanocomposite material, 3DOM/m C/C (not shown in Fig. 6). This composite material was more resistant to forming a solid–electrolyte interface layer and had a greater lithium capacity at high charge and discharge rates, when compared to the same material without template mesopores and walls consisting only of amorphous carbon.
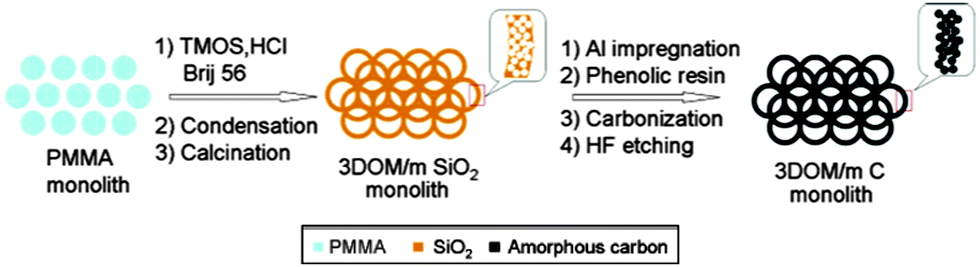 | ||
| Fig. 6 A diagram of the synthesis procedure of the 3D ordered carbon monolith. Reprinted with permission from ref. 69. Copyright 2006, American Chemical Society. | ||
Regarding transition metal oxides, among the different oxides studied, cobalt oxide has demonstrated an excellent electrochemical performance in terms of specific capacity and cyclability.64 Using two different hard templates, KIT-6 cubic mesoporous silica and SBA-15 hexagonal mesoporous silica, Wang et al. have recently prepared two highly ordered mesoporous Co3O4 materials replicating the previous cubic and hexagonal silica structures. These two solids showed ferromagnetic ordering even at room temperature, due to the geometric confinement of antiferromagnetic order within nanoparticles. The KIT-6 mesoporous Co3O4 electrode delivered a lithium storage capacity of 1760 mA h g−1 at a current rate of 50 mA h g−1, which is similar to what has been previously reported for porous Co3O4 nanotubes and nanoneedles.70 During 100 cycles, the electrode maintained a reversible capacity above 1100 mA h g−1, which is higher than the theoretical capacity of Co3O4 (890 mA h g−1). It can be attributed to the high surface area and interconnected porous structures, providing extra active sites for Li+ storage.71 Differing from other morphologies like powders, where the specific capacity decreases when the current density is increased, these mesoporous electrodes exhibited almost the same specific capacity, 300 mA h g−1, at high and low current densities.
The extraordinary performance (long-term cyclability and high-current performance) of these mesoporous Co3O4 materials should be attributed to the highly ordered mesoporous structure (see Fig. 7), in which the electrolyte can easily diffuse into the mesopores.72
 | ||
| Fig. 7 TEM images at low (a) and high (b) magnifications of KIT-6 mesoporous Co3O4, showing highly ordered mesopores. The inset is the corresponding SAED pattern of the circled zone. Reprinted with permission from ref. 72. Copyright 2010, Wiley-VCH. | ||
2.4. Biomimetic mesoporous materials
Over the past decade, bioentity immobilisation has opened up a new way to explore the potential energy applications for the integration of biological systems into electronic devices: chiefly in biofuel and the mitigation of CO2 for environmental purposes. For an intensive study of this approach, the readers' attention is drawn to the outstanding review published by the group of Prof. Meunier in 2011.73The term ‘biomimetic’, coined by Otto H. Schmitt in 1969, refers to the process of comprehending and applying biological principles to man-made design.74 Later, IUPAC defined ‘biomimetic’ as ‘a compound that mimics a biological material in its structure or function,’ and additionally, as ‘a laboratory procedure designed to imitate a natural chemical process’.75
There are numerous examples of the potential of biomimetic materials in different applications, including biology, medicine, aerospace, energy, etc. As far as energy conversion, capture and storage applications are concerned, biotemplated materials, mostly with hierarchical structures (see Section 2.2), show great promise. Su et al. recently reviewed the state of the art of applications of hierarchically structured porous materials in energy conversion and storage.61 In this section, characteristic examples of biomimetic mesoporous materials for renewable energy will be put forward.
Much effort has been devoted to extending the use of TiO2 under sunlight irradiation, including by the biomimetic route. Accordingly, hierarchical macro-mesoporous titania obtained from biotemplates such as plant leaves or butterfly wings has been reported to enhance light harvesting and photocatalytic hydrogen production, as well as showing promising properties as a photoanode for solar cells and dye sensitised solar cells (DSSCs).61,76–79 An example of this was recently reported by Zhang et al., who synthesised a new photoanode-titania based structure for DSSCs, inspired by butterfly wing scales (Fig. 8).76
Continuing with titania and DSSCs, diatoms are also promising biotemplates for the biomimetic fabrication of nanostructured materials and devices. Diatoms produce different regular 3D silica structures with nanometre to micrometre dimensions. These structures have been used as biosupports, generating new hierarchical materials by coating them with titania layers giving an enhanced electrical output of experimental DSSCs.80,81
 | ||
| Fig. 8 Nature picture and low-magnification optical microscopy, low-magnification FE-SEM images and high-resolution FE-SEM images showing the wing of the blue male (a–c) and black male (d–f). The insets in the lower left-hand (c, f) corner show the two-dimensional, logarithmic Fourier power spectra of square areas selected from the images. Reprinted with permission from ref. 77. Copyright 2009, American Chemical Society. | ||
But the potential applications of hierarchical biotemplated materials are not restricted to the DSSC field. Hierarchical macro–mesoporous wood-templated NiO,82 Fe3O4,83 manganese oxide,84 chromium oxides,85 alumina;86 as well as diatomaceous earth-templated carbon87 and mesoporous biocarbon-coated Li3V2(PO4)88 have all been synthesised as promising candidates for carbon electrodes in LIBs.
Besides hierarchical biotemplated materials, metal–organic framework materials (MOFs), which will be discussed in the following section, are ideal candidates for building biomimetic systems due to their extremely high surface area and chemical tuneability. Zhou et al.89 recently published an excellent review on MOFs as biomimetic catalysts. Their review focuses on implanting biomimetic active sites into stable MOFs. Maspoch et al.90 highlighted the advances in the synthesis of metal–biomolecule frameworks (MBioFs). In any case, these approaches are focused far away from the concept which is discussed herein, of ‘biomimetic’ materials as sacrificial templates for energy applications.
2.5. Hybrid mesoporous materials
Previous sections are devoted to the synthetic methods and properties of mesoporous/nanostructured/hierarchical materials for potential applications in clean energy technology. In general they owe their promise to (i) their high surface area, needed for efficient electrochemical reactions in fuel cells and Li-ion batteries, as well as for attaching a broad spectrum of sensitizers, both organic and inorganic to DSSC and quantum-dot sensitized solar cells; and (ii) their improved diffusion, i.e. a fast electrolyte-surface diffusion of the electrochemically active materials in solar cells, fuel cells and batteries, and a shorter solid state diffusion path for the Li ions in batteries.7Usually, the incorporation of active sites into mesoporous materials is essential to their application and thus the success of the synthetic route will determine the efficiency of the solid when used in a device. The role of the incorporation of functionality into mesoporous solids, and of mesoporosity itself, in device performance in fuel cells is highlighted in Section 4.1. Additionally, the use of mesoporous hybrid TiO2, SiO2, SnO2 and ZnO-based materials for clean photocatalytic energy technologies and for mesoporous advanced solar cells will be carefully discussed in Sections 3.7 and 4.2, respectively. This section, on the other hand, will seek to summarise existing synthetic strategies for obtaining hybrid mesoporous solids, for the aforementioned applications. Examples of materials which have already found uses in these areas are given as an illustration.
Most of the techniques for the incorporation of functionality in mesoporous solids are based on post-synthetic treatments of preformed solids, which usually lead to a partial blocking of the mesoporosity and a poor control of the location and geometry of the active phase. Traditionally, supported oxide catalysts have been prepared by ion exchange, wet impregnation, deposition–precipitation or chemical vapour deposition. More efficient post-synthetic techniques are atomic layer deposition and grafting. The latter technique also includes so-called organometallic grafting, using LnM[O2P(OtBu)2]m or LnM[OSi(OtBu)3]m (Ln = alkoxide, amide, alkyl ligands) as organometallic precursors that, having been cleaved thermolytically, are able to react with hydroxyl groups of the surface of the oxide support.91,92
More recently, a new strategy has been developed to introduce a wide range of functionalities in mesoporous materials: the so-called ‘sol–gel coordination chemistry’ (Fig. 9).8
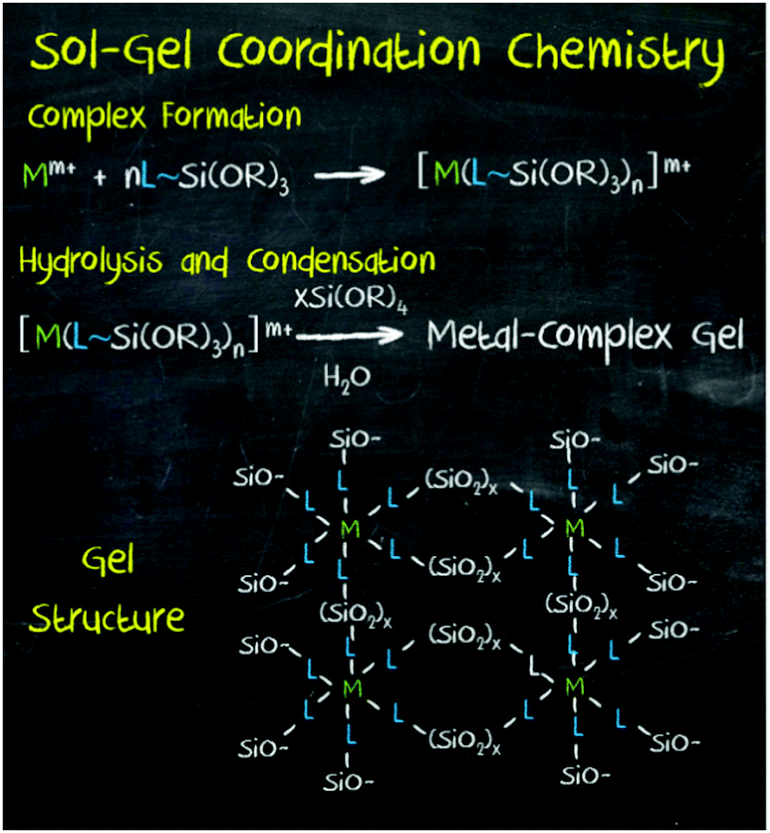 | ||
| Fig. 9 Sol–gel coordination chemistry, an approach developed for the synthesis of hybrid mesoporous materials through the in situ incorporation of chemical functionalities into the framework of silica and organosilica porous materials. Reprinted with permission from ref. 8. Copyright 2013, Wiley-VCH. | ||
Inspired by periodic mesoporous organosilica (PMO) hybrid materials, this approach is based on the in situ incorporation of chemical functionality through its co-condensation with the metal oxide precursor during the sol–gel process. The corresponding metal complex or nanoparticle is modified with terminal trialkoxysilane groups for its co-condensation with a silica source and in the presence of an adequate surfactant. In this manner, mesoporous hybrid materials containing a wide variety of functionalities, including metal nanoparticles, metal complexes, clusters, organic molecules and dyes, as well as other homogeneous catalysts have been synthesised. In addition to silica, other mesoporous solids such as PMO and TiO2 have been used as supports. In all cases, it has been proved that higher surface areas and enhanced yields are possible with this method, as well as precise control over the pore structure and surface chemistry of mesoporous materials.93,94
As an example of a hybrid mesoporous material with applications in energy-related technologies, mesoporous titania is the most common material used as the working electrode (WE) in DSCCs, and it is usually prepared by post-synthetic methods; the TiO2 is synthesised by using hard-template or soft-template approaches, and the dye is usually incorporated by the impregnation and/or immersion of TiO2 in a solution of the complex to incorporate, by interactions between the carboxylic groups of the metal complex with the TiO2 surface.7,34 Different examples of the above hybrid porous titania and its performance are given in Sections 3.7 and 4.3.
As far as the sol–gel coordination chemistry approach is concerned, Rico-Santacruz et al.93 recently reported the synthesis of a novel hybrid mesoporous titania–N3 dye with high organic content (up to 50%) and a surface area of ca. 250 m2 g−1, without the need of surfactants (and calcinations), using only water and ethanol as a solvents. This method allows for overcoming the drawbacks of traditional titania, keeping the textural properties of unmodified titanias but with a considerable improvement of the photocatalytic properties in comparison with both the unmodified one and the one prepared by the grafting of the dye on the pre-existing mesoporous titania.93b
The combination of their gas storage/capture performance at room temperature with the simplicity of their synthesis makes MOFs ideal candidates for clean energy technologies. The most relevant parameter for those applications is the surface area per volume ratio as well as their stability.96–98 Non-mesoporous MOF-5 has one of the highest reported values of surface area per volume ratio, ca. 2200 m2 cm−3.98 Despite such high surface to volume ratios, pores larger than 2 nm are required to expand the application of MOFs, to areas such as macromolecular catalysis or separation. Thus, the expansion of these structures into the mesoporous range could herald the MOFs' coming of age in the industry.96,97,99
Mesoporous metal–organic framework is a term denoting MOFs with pores ranging between 2 and 50 nm, as defined by IUPAC for other mesoporous materials. However, the pores of these novel solids encompass not only channels but also the cavities of the material. Thus few mesoporous MOFs have a type IV isotherm.97 Adsorption in these materials is governed by the size of the accessible channel into the pore, i.e. micropores with the typical type I isotherm. A second uptake then takes place in which microporous windows open to mesoporous cages, leading to type I isotherms with a second uptake in most cases, as shown in Table 2.97
| Family | Name | Pore apertures (nm) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Type N2 isothermb | Structure typeb,c | Ref. | |
|---|---|---|---|---|---|---|---|---|
| BET | Langmuir | |||||||
| a STP units. b Data from ref. 3. c Data from ref. 1. | ||||||||
| IRMOF | IRMOF-12/14 | 2.45 | — | 1750/1936 | 0.61/0.69 | 3D channel | 100 | |
| IRMOF-16 | 2.9 | 1910 | — | 100 | ||||
| IRMOF-74-IX | 9.8 | 1760 | — | 3.30 | Type IV | 112 | ||
| MesoMOF-1-HX | 2.2 × 2.6, 3.85 | 685 | — | 500–730a | Type IV | 3D channel | 101 | |
| MIL | MIL-100 | 1.2, 2.5 × 2.9 | — | 3100 | 1.16 | Mainly type I | Cage | 104 |
| MIL-101 | 1.4 × 1.6, 2.9 × 3.4 | 4100 | 5900 | 2.01 | Type I with secondary uptakes | 105a | ||
| MIL-101_NDC | 1.8 × 2.0, 3.9, 4.6 | 2100 | — | — | Type I with secondary uptakes | 105b | ||
| ZIF | ZIF-95 | 0.37, 2.5 × 1.4 | 1050 | 1240 | 0.43 | Mainly type I | Cage | 109 |
| ZIF-100 | 0.35, 3.56 | 600 | 770 | 0.37 | ||||
| UMCM | UMCM-1 | 1.4 × 1.7, 2.4 × 2.9 | 4160 | — | Type I with secondary uptakesc/type IVb | 1D channel | 103 | |
| UMCM-2 | 2.6 × 3.2 | 5200 | 6060 | 2.32 | Cage | 108 | ||
| PCN | PCN-61 | 2.3, 1.5, 1.3 | 3000 | 3500 | 1.36 | Type I with secondary uptakes | Cage | 111 |
| PCN-66 | 2.6, 1.6, 1.3 | 4000 | 4000 | 1.63 | ||||
| JUC | JUC-48 | 2.5 × 2.8 | 629 | 880 | 0.19 | Mainly type I | 1D channel | 102 |
| MOF | MOF-210 | 2.69 × 4.83 | 6240 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
3.60 | Type IV | Cage | 115 |
| NU | NU-109E | 7010 | — | 3.75 | Type IV | Cage | 113 | |
| NU-100 | 1.34, 1.54, 2.74 | 6143 | — | 2.82 | 114 | |||
| NU-110E | 7140 | — | 4.40 | Type IV | 113 | |||
In 2012 Cui et al.95 published a tutorial review which covers new advances in the field of mesoporous metal–organic framework (mesoMOFs). The review covers the design and synthesis of mesoMOFs, their porosity activation and surface modification, as well as their potential applications in storage and separation, catalysis, drug delivery and imaging. It is accepted that novel mesoMOF materials can be synthesised following different strategies, although the solvothermal technique is the most common.
As an example, Fig. 10 shows the synthesis of a mesoporous MOF through surfactant-assisted synthesis, as an efficient soft-templating approach to mesoporous silicates.
 | ||
| Fig. 10 A schematic representation of the surfactant-assisted synthesis of mesoporous MOF particles from microporous MOFs. Reproduced from ref. 95. Copyright 2012, RSC. | ||
Zhou et al. classified mesoporous MOFs as five different types: those with 3D channels; those with 1D channels; mesoporous ZIFs (from zeolite imidazolate frameworks) with large cavities; mesoporous MOFs based on supramolecular templates and lastly, mesoporous MOFs with large cavities, the final group being the most common type of MOFs found.97a Cui et al.95 further reduced this classification to four types: cage-type mesoMOFs (including ZIFs), channel-type mesoMOFs (including 1D and 3D MOFs), chiral mesoMOFs and mesoporous particles of MOFs. For a more in-depth examination of the synthesis, properties and applications of these novel materials, readers are directed to ref. 95–97 and 99. Herein, we will show just a few examples of mesoMOFs with potential applications in clean energy technologies.
The first mesoporous MOF was reported in 2002 by Yaghi et al.100 Named IRMOF-16, it is a 3D MOF which belongs the IRMOF family (isoreticular MOF-5), whose structure consists of ZnO4 tetrahedra and a long organic linker ([1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylic acid, TPDC).
IRMOF-16 exhibited a high capacity for methane storage (240 cm3 STP per g at 298 K and 36 atm.) and an extremely low density, 0.21 g cm−3. It possesses a percentage free volume of 91.1%, much higher than some of the most open zeolites, such as faujasite, and similar to silica xerogel and aerogels.97–100 Since then, numerous mesoporous MOFs have been synthesised,101–115 the most representative of which are summarised in Table 2.
For example, Zhou et al.101 reported the first example of a mesoporous MOF exhibiting a type IV adsorption–desorption isotherm, mesoMOF-1, by using TATAB (4,4′,4′′-s-triazine-1,3,5-triyltri-p-aminobenzoate) as an organic linker and acids (HX, X = F, Cl, Br) to stabilise the meso-channels (mesoMOF-HX). Yaghi et al.109 synthesised ZIF-95 and ZIF-100, two mesoporous MOFs that can selectively capture CO2 from several different gas mixtures with methane, CO or nitrogen, at room temperature.
Recently, the same laboratory expanded the original phenylene unit of MOF-74 to prepare a series of mesoporous materials, namely IRMOF-74-I to IX, with pore apertures ranging from 1.4 to 9.8 nm, the latter figure being the highest ever reported in the literature.112 Farha et al.113 synthesised two mesoporous MOFs, NU-109 and NU-110 (Northwestern University), with BET areas ca. 7010–7140 m2 g−1. These BET values are the highest of any porous materials reported to date, even superseding the area of the previously synthesised NU-100114 and MOF-210 (see Fig. 11).115
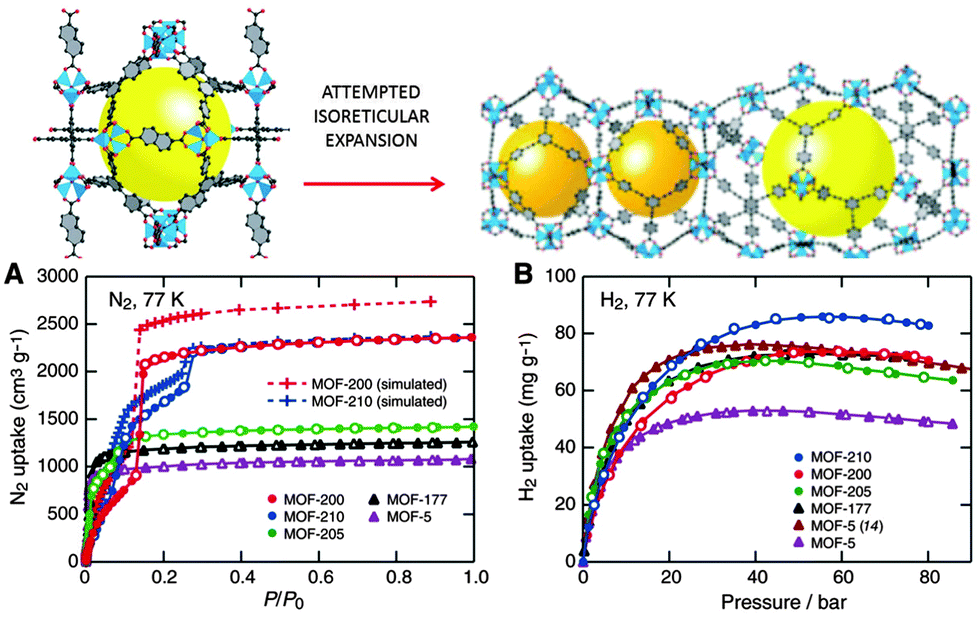 | ||
| Fig. 11 (Up) The attempted isoreticular expansion of MOF-205 leads to MOF-210, and (bottom) the low-pressure N2 isotherms (A) and high-pressure H2 isotherms at 77 K (B) of MOF-5, -177, -200, -205, and -210. Reprinted with permission from ref. 115. Copyright 2009. American Association for the Advancement of Science. | ||
The surface area of a football pitch is about 7000 square metres, which means that every gram of the last four mesoporous MOFs has almost the same surface area as a football pitch. MOF-210 and NU-100 exhibited hydrogen adsorption as high as 7.9 to 9.0 wt% at 56 bar. The former is increased up to 15 wt% at 80 bar (Fig. 11)114,115. These values are higher than the targets of the U.S. Department of Energy (DOE) for hydrogen adsorption −5.5 wt% and 40 g l−1 of volume capacity at a operating temperature of −40 °C to 60 °C under a maximum delivery pressure of 100 atm.116 Mercedes-Benz has already deployed MOF hydrogen fuel tanks in a fuel cell-powered demonstration model, the F125.117
These are just a few examples of the possible application of these mesoporous materials in clean energy technologies, as shown in Fig. 12. The use of these novel materials in clean energy technologies, including catalysis, will be discussed in the following sections.
 | ||
| Fig. 12 The illustration of the potential applications of porous materials in energy conversion and storage. Adapted from ref. 61. Copyright 2012, Wiley-VCH. | ||
3. Mesoporous catalysts for clean energy technologies
3.1. Biofuels production and upgrading with mesoporous catalysts
From a plethora of biofuels, bioethanol and biodiesel alone have been commercialised worldwide for transportation uses. The Directive 2003/30/EC on the promotion of the use of biofuels or other renewable fuels established for state members of the European Union a goal of 5.75% share of renewable energy in the transport sector for 2010, this share rising to a minimum of 10% in every member state in 2020 (Directive 2009/28/EC). Biofuels must contribute to CO2 reductions of 50% by 2017. Other countries, including the U.S., Canada, Japan and Australia have adopted similar policies for the use of biofuels over the next few years. At this point it is important to highlight that the EU is the world leader in the production of biodiesel (European demand in biodiesel could increase up to 10 million tons per year by 2010) whereas the US is the biggest ethanol producer worldwide.
Among the different biomass sources, edible biomass feedstocks (e.g. sugars, starches and vegetable oils) were initially proposed for the large scale production of biofuels or biochemical compounds, the so-called “first generation” biofuels. However, important concerns arise about the impact of edible feedstock on biodiversity and land use (possible deforestation), as well as their high production cost and competition with food crops (there is a strong argument that increases in food prices are due in part to this competition between food and fuel).118 Newer biofuels, the so-called “second generation” biofuels, offer the potential to avoid the aforementioned drawbacks of edible biomass resources. Non-edible biomass resources (e.g. lignocellulosic feedstocks) could achieve a sustainable production of fuels and chemicals without affecting food supplies or forcing extensive changes in land use. Additionally, second generation biofuels are expected to be superior in terms of energy balance, land requirement and competition with food crops, and greenhouse gas emission reduction.
In order to evaluate the possible use of biomass derivatives in the production of biofuels in a biorefinery, the US DOE has selected so-called platform molecules, which include organic acids (e.g. succinic acid, lactic acid, etc.), polyols (e.g. glycerol, sorbitol, etc.) and others (e.g. bioethanol, furans, etc.). The processing of these renewable biomass derivatives in a biorefinery requires the design of new catalytic strategies taking into account the fact that these biomass derivatives exhibit a rich surface chemistry (high degree of oxygenated groups), are water-soluble, highly reactive and have low volatility, when compared to conventional petrochemical feeds (these systems must be designed for aqueous-phase processing in mild conditions). The processing of biomass feedstocks in biorefineries is seen as a challenging procedure, with multiple steps, because of the complex chemical composition of the feedstocks. Chemical processes using inorganic acid and base catalysts play a crucial role, for example the trans-esterification of vegetable oils with methanol in the production of biodiesel, or the hydrolysis of cellulose to fermentable sugars, etc. However, homogenous acid or base catalysts have significant drawbacks, mainly associated with the neutralisation and separation of catalysts from the reaction products. In this sense, the design of new solid catalysts with a proper porous structure can be envisaged as a promising alternative not only in terms of the processing and production cost (recovery and reuse of the solid catalyst at the end of the reaction) but also for the control of the reaction pathways in the production of the new biofuels from biomass.
In the following sections we will focus on some mesostructured materials which are envisaged to be promising candidates for the biomass conversion processes, both for first and second generation biofuels production, as well as the conversion of platform molecules to value-added products. The presence of complex reaction intermediates in biorefineries will make mesoporous materials extremely important as catalyst supports, able to control the preferred reaction pathways via pore diameter.
3.1.2.1. Bioethanol processing. Ethanol is usually converted by the fermentation of edible-biomass aqueous sugars. Bioethanol produced from biomass is included as an additive in the formulation of gasoline in low concentrations (e.g. E5–15) due to the limitations of current spark-ignition engines. High-concentration blends (e.g. E85) will require newly designed engines, designed as flexible-fuel vehicles (FFVs). Besides being used as an additive or as a fuel, bioethanol has enormous potential as a chemical feedstock to produce renewable hydrogen via steam reforming or chemicals (e.g. ethylene, acetaldehyde, acetic acid, etc.) A number of reactions, dehydration, dehydrogenation, oxidation and so on, might be used in this case. These processes require the design of proper catalysts with porous structures, acid–base properties and thermal/catalytic stability under harsh temperature conditions.
One of the main conversion processes for bioethanol concerns the production of C3 derivatives such as propene oxide, acrylonitrile and polypropene. Conventional catalysts like zeolites and metal oxides come with the dual caveat of a low catalytic activity and lifetime. Although the shape selectivity in acidic zeolites favour the formation of C3 and C4 derivatives, coke formation gives rise to strong deactivation processes.119–121 Recent studies from Iwamoto et al. using Ni-MCM41 materials have shown that mesoporous silica materials exhibit an improved stability towards coke formation together with a higher water resistance under reaction conditions compared to conventional zeolites.122 Although the selectivity to C3 and C4 derivatives is low on these mesoporous systems, the selectivity increases for low space-velocity. A similar low selectivity towards higher hydrocarbons (C5–C12 derivatives) has been reported for H-ZSM-5 nanocrystals deposited on Al-MCM-41.123 While conventional ZSM-5 exhibits a significant selectivity towards liquid hydrocarbons, mesoporous H-ZSM-5/Al-MCM-41 led to the formation of ethylene with a 100% ethanol conversion.
Another important reaction involving ethanol is steam reforming to carbon dioxide and hydrogen. Traditionally this reaction has been performed using noble metal catalysts (e.g. Pt, Rh, Pd) and transition metals (Co and Ni) supported on oxide supports (Al2O3, MgO, SiO2, CeO2, etc.).124–127 Unfortunately, these catalysts have two important problems, their deactivation owing to coke formation, and the low selectivity to hydrogen (the main reaction products being unwanted carbon monoxide, acetaldehyde, acetone and methane). An improved performance in terms of the catalytic activity and selectivity to hydrogen has been observed in mesostructured materials. Their controllable pore size allows the accommodation of small size metal nanoparticles (large metal ensembles are related with coke deposition and nanofibers growth). In this manner, Gac and co-workers described an improved catalytic behaviour using a Co3O4/MCM-48 catalyst synthesised by the nanocasting approach.128 Mesoporous Co–Si catalysts exhibit an ethanol conversion of 100% at 420 °C with a hydrogen selectivity higher than 80%. A high selectivity for hydrogen (above 70%) was reported by Carrero and co-workers on Cu–Ni/SBA-15 catalysts although at a slightly higher reaction temperature (600 °C).129 The metal content, metal particle size, Ni/Cu ratio and the preparation conditions (Cu/Ni framework species or deposited metal nanoparticles) have been proposed to define the final hydrogen selectivity. Interestingly, both the hydrogen selectivity and catalyst lifetime can be highly improved in mesoporous Cu–Ni/SBA-15 catalysts by incorporating basic promoters such Ca and Mg (0–20 wt%) that inhibit ethanol dehydration to ethylene and subsequent coke formation.130 More examples of ethanol steam reforming with mesoporous catalysts can be found later on, in the hydrogen production section.
3.1.2.2. Catalytic strategies to produce biodiesel. It is hoped that in the near future, biofuels will begin to mitigate the large consumption (and side-effects) of petroleum-based fuels for transportation. Biodiesel represents a major candidate for achieving this end. Biodiesel is a mixture of fatty acid methyl esters (FAME) produced by the trans-esterification of triglycerides with an alcohol (e.g. methanol), using a homogeneous acid or base catalyst. Different heterogeneous catalysts (basic zeolites, alkaline earth metal oxides and hydrotalcites) have been evaluated for the trans-esterification of vegetable oils as replacements for the current homogenous base catalysts.131 CaO has been the most widely tested base catalyst for the trans-esterification of various vegetable oils such as soybean, rapeseed and sunflower oils.132,133
Microporous solid catalysts (titanosilicate ETS and zeolite β) were unable to carry out mass transfer adequately; giving rise to their poor catalytic performance compared to non-porous solid catalysts. Indeed non porous examples, such as Amberlyst-15 and Nafion NR50, exhibit a promising catalytic activity compared to homogenous catalysts. Similarly, Suppes and co-workers studied the trans-esterification of soybean oil with methanol using a series of zeolites and metal catalysts.134 The catalytic measurements show that zeolite ETS-10 gives a higher conversion than the zeolite-X catalysts, the increase being attributed to the higher basicity and improved intra-crystalline diffusion afforded by ETS-10's larger pore diameter. In general, zeolites performed better than metal oxides and also homogenous catalysts in the trans-esterification of soybean oil.
Taking into account the aforementioned diffusional limitations when dealing with high molecular weight reactants, mesoporous silica materials could have an advantage over microporous zeolites in the production of biodiesel because the mass transfer resistance and diffusion limitations can be significantly reduced. Indeed, the deposition of calcium oxide on different mesoporous silicas (SBA-15 and MCM-41) gave rise to improved catalysts for the trans-esterification of sunflower oil and castor oil with methanol.135 A sample containing 14 wt% CaO on SBA-15 was the most active catalyst, reaching a conversion as high as 95% with sunflower oil (after 5 h of reaction) and 65% for castor oil (after 1 h). Interestingly, these catalysts showed no lixiviation of the active phase under the reaction conditions, in contrast to commercial CaO.
Li and co-workers investigated mesoporous silicas (MCM-41, KIT-6 and SBA-15) loaded with MgO as a base catalyst, for the transesterification of a blended vegetable oil with ethanol to produce biodiesel.136 Among all catalysts, MgO-impregnated SBA-15 exhibited the highest catalytic activity for the production of biodiesel, achieving a conversion as high as 96% within 5 h. Similarly, Abdullah and co-workers achieved very active catalysts by the deposition of potassium on SBA-15 using conventional impregnation methods. The K/SBA-15 catalyst attained an optimum biodiesel yield of ca. 87% at 70 °C. The excellent performance of these systems was attributed to a high surface area, tuneable mesopore size and high thermal stability.137 Corma and co-workers performed the glycerolysis of triolein and rapeseed oil using different base catalysts such as mesoporous Cs-MCM41, Cs-sepiolite and hydrotalcites. The final catalytic activity was defined by the strength of the basic sites and not by the porous structure.138 The final conversion achieved was as high as 92% for hydrotalcite, versus 26% on mesoporous Cs-MCM41, with a high selectivity to monoolein (78% and 46%, respectively).
Despite the excellent results reported in the literature for heterogeneous acidic or basic catalysts in biodiesel production processes, their catalytic performance is still far from that of the conventional catalysts (NaOH, H2SO4, etc.). Furthermore, heterogeneous base catalysts are still very sensitive to the free fatty acid (FFA) content in the oil. Consequently, the design of new porous catalysts is required, bearing in mind not only the acid or basic site strength but also the available active surface and tolerance to high FFA content oil at mild reaction conditions. Another method in biodiesel processing where mesoporous catalysts may be desirable is the use of coproduced glycerol for the synthesis of oxygenated fuel components. This is described in the next section.
3.1.2.3. Glycerol conversion. One of the main by-products of biodiesel production by the trans-esterification of vegetable oils (and residual oils and fats), is glycerol. The valorisation of this highly versatile molecule into valuable chemicals, using greener catalytic processes, presents several alternatives: (i) glycerol esterification with acetic acid or trans-esterification with methyl acetate to obtain glycerol triacetate (triacetin),139–142 (ii) glycerol etherification with different aryl/alkyl alcohols to produce a wide variety of substitute ethers,139 (iii) glycerol oxidation with an oxygen donor substrate to produce glyceric and glycolic acids as major products,139,143,144 and (iv) glycerol dehydration on solid acid catalysts to give acrolein and hydroxyacetone.145
Where glycerol esterification and etherification are concerned, the number of studies dealing with the effect of mesoporosity is rather scarce. These reactions are traditionally performed using solid acid catalysts, and the selectivity towards the different reaction products (mono-, di- and tri-esters or the corresponding ethers) is highly sensitive to the reaction conditions (substrate/acid or alcohol ratio, reaction time, amount of catalysts, etc.), the acid strength of the support and the presence of microwave radiation.134–137 Di- and tri- derivatives are valuable petrol fuel additives which give enhanced viscosity properties when blended with diesel fuel or antiknocking properties when added to gasoline. Interestingly, Luque and co-workers confirmed the considerable effect of the pore size not only on the final conversion but also on the selectivity towards different reaction products.139 As can be observed in Fig. 13, the mesoporous Starbon acid catalyst exhibits an improved catalytic activity compared to traditional solid acid catalysts (e.g. sulfated zirconia) with an unprecedented high selectivity to the triacetylglycerol. A similar improvement in terms of the conversion values (95%) and selectivity was observed for mesoporous Starbon acid carbon in the etherification of glycerol with a high selectivity to the monoether in the 1-position compared to the monoether in the 2-position (5/1).139 Using sulfonic-acid functionalised mesostructured materials (SBA-15), Melero and co-workers found that glycerol conversion in the esterification of glycerol, to yield acetylated derivates, was higher with acetic acid than conventional acid catalysts. The total conversion reached by mesoporous materials was ca. 90% with over 80% selectivity to diacetylglycerol and triacetylglycerol. Significantly, the reaction activity was highly sensitive to the acid strength of the sulfonic acid groups: propylsulfonic < arenesulfonic < fluorosulfonic.142
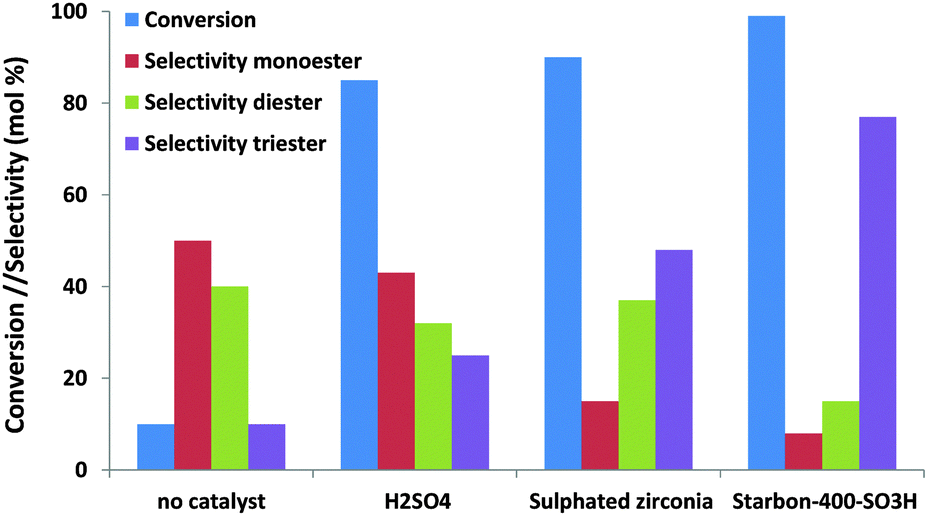 | ||
| Fig. 13 The catalytic activity and selectivity of different solid acids in the esterification of glycerol with acetic acid. | ||
For the selective liquid-phase oxidation of glycerol, noble-metal supported catalysts have been much investigated for their catalytic activity and selectivity. Using molecular oxygen as an oxidant, carbon-supported Pt and Pd nanoparticles have demonstrated a good catalytic activity, as well as selectivity for glyceric acid.146,147 The nature of the metal nanoparticles (Pd or Pt), the pH of the reaction media and the presence of secondary species (e.g. Bi) exhibit a large influence not only on the catalytic activity but also on the final selectivity. The oxidation of the secondary hydroxyl group of glyceric acid to yield dihydroxyacetone was preferred under acidic conditions and after the incorporation of bismuth on platinum nanoparticles (Scheme 1).
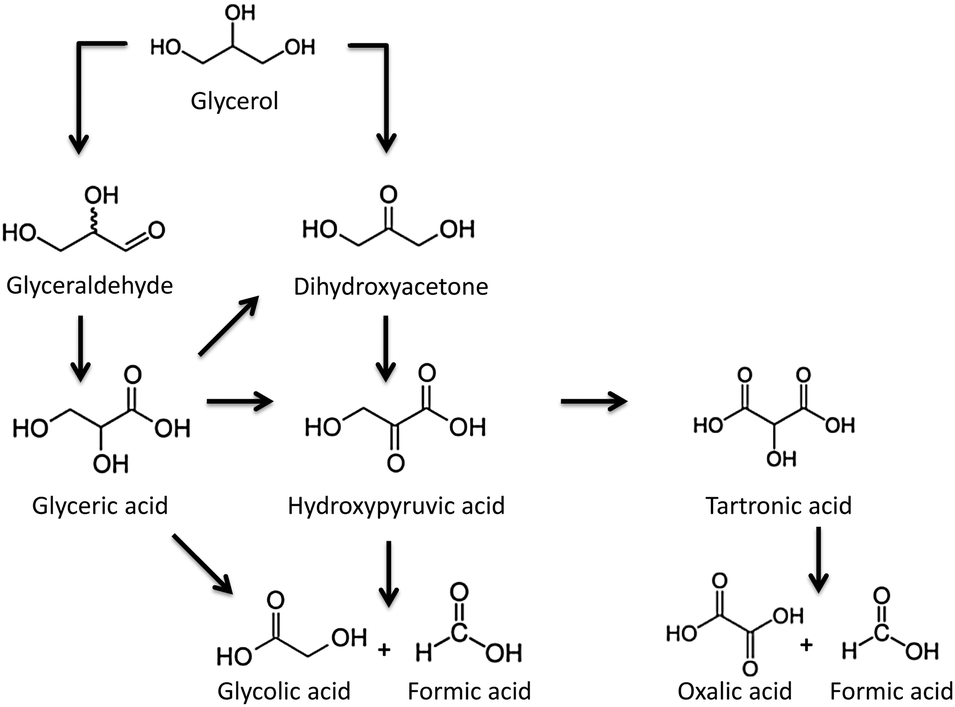 | ||
| Scheme 1 Different reaction paths for the selective oxidation of glycerol. | ||
While various studies have demonstrated the influence of the noble-metal particle size on the catalytic activity, selectivity does not seem to correlate with the size of the nanoparticles.148–150 Indeed, Villa and co-workers confirmed that Au particle size had a negligible effect on the final selectivity when using MgAl2O4 spinels as a support and molecular oxygen as the oxidant.151 By using spinels with a different surface compositions (Al/Mg ratio) and apparent surface areas, these authors anticipated the important role of the textural and chemical properties of the support in the selectivity of the oxidation reaction.
This point was further confirmed by McMorn and co-workers143 and Rodrigues and co-workers.144 Apparently, wide mesopores favour the formation of partial-oxidation products (glyceric acid and glyceraldehyde) while narrow pores can lead to an easier over-oxidation of the primary products (oxalic acid and formic acid), independently of the catalyst and reaction conditions used. Indeed, the partial-oxidation products (mainly glyceraldehyde, dihydroxyacetone and glyceric acid) are the main reaction products when titania–silica materials with a large pore size (up to 15 nm) are used. It would seem that narrow micropores favour slow diffusion of the reaction products, giving rise to over-oxidation reactions, while an increase in the pore size decreases diffusion limitations, and partially oxidised products are observed.
The effect of the pore size on the final selectivity is clearly illustrated in Fig. 14 using Au nanoparticles supported in different carbon xerogels.144 Whereas carbon xerogels with a narrow pore size favour the formation of highly oxidized products (mainly glyceric acid (GLYCEA), glycolic acid and tartronic acid), an increase in the pore size diameter from 5 to 20 nm leads to a progressive shift in the product selectivity towards partially-oxidized dihydroxyacetone (DIHA).
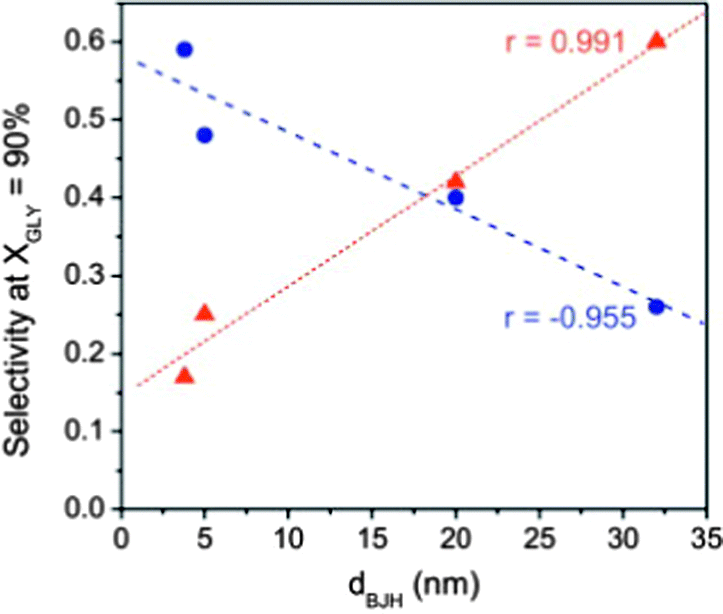 | ||
Fig. 14 Correlations between selectivities to dihydroxyacetone (DIHA –  ) and glyceric acid (GLYCEA – ) and glyceric acid (GLYCEA –  ) and the main pores size (calculated by the BJH method) for Au nanoparticles supported on mesoporous carbon supports. Reprinted with permission from ref. 144. Copyright 2012, Elsevier. ) and the main pores size (calculated by the BJH method) for Au nanoparticles supported on mesoporous carbon supports. Reprinted with permission from ref. 144. Copyright 2012, Elsevier. | ||
Besides diffusion limitations, the higher selectivity towards DIHA of larger pores has been explained by the requirement of a larger void space inside the nanocavities (e.g. adsorption mode of the glycerol molecule). A similar observation on selectivity towards DIHA was described for Au nanoparticles supported on MWCNTs, which contained a larger proportion of mesopores than similar nanoparticles supported on a microporous activated carbon.152
In summary, it is clear that the liquid-phase oxidation of glycerol is highly influenced by diffusional limitations, with mesoporous materials being preferred in order to avoid undesired over-oxidation reactions.
Another reaction where the role of mesoporosity is crucial is the dehydration of glycerol to hydroxyacetone and acrolein in the presence of acidic catalysts. Using mesoporous aluminophosphates which incorporated transition metal-ions (e.g. Cr(III), Cu(II) and La(III)), Liu and co-workers observed the good performance of these systems in terms of the glycerol conversion (as high as 99%) and selectivity to acrolein (ca. 80%).145 Interestingly, the selectivity and conversion of glycerol was sensitive to the reaction temperature, the textural characteristics of the support and the Cr/Cu molar ratio.
3.2. Mesoporous materials in gas-to-liquid conversion
Natural gas is considered as an alternative to petroleum for the production of chemicals and as clean(er) liquid fuel. Natural gas reserves are more plentiful than those of petroleum, making the conversion of natural gas to value-added chemicals (gas to liquid (GTL) technology) an attractive proposition. There are two approaches for achieving that conversion, the direct one, from natural gas itself, and the indirectly route, via synthesis gas (syngas) or methanol. Of these approaches, the indirect route is the more commercialised technology. One of the main steps in the conversion of natural gas is the activation of the methane molecule using heterogeneous catalysts.The direct conversion of natural gas involves multiple reactions, including pyrolysis – to produce hydrogen and carbon nanotubes, methane dehydro-aromatization – giving higher hydrocarbons (benzene, toluene, etc.), and direct oxidative conversions – to ethane, methanol and formaldehyde.153 Concerning methane dehydroaromatization (MDA), Su and co-workers found an improvement in the conversion of methane to aromatics using Mo catalysts supported on modified HZSM-5.154a The unique selectivity to aromatics and the stability of the catalysts, which were derived from alkali-treated ZSM5, was attributed to the coexistence of mesopores which enhance the diffusion of the reaction products (aromatics) and inherent micropores in the zeolite that are the active sites for aromatics formation. Similar results were obtained by Chu and co-workers using an unusual hierarchical ZSM5 developed by the assembly of many French fry-like crystals using SBA-15 as the silica source.154b Mo nanoparticles supported on modified ZSM5 resulted in a higher methane conversion and a stronger coking-resistance compared to the conventional Mo/ZSM5 catalyst. The higher resistance to deactivation by coke formation is due, according to Martinez and co-workers, to the presence of intracrystalline mesopores that allow more coke to be formed while keeping the active sites (acidic sites) in the 10-ring channels of the zeolite active for aromatization.155 Similarly, hierarchical mesoporous Mo/HMCM-22-HS catalysts, prepared by hard templating with carbon black, exhibit exceptional behaviour in terms of methane conversion, benzene yield and catalyst lifetime.156
Direct oxidative conversion of methane to methanol and formaldehyde has been widely investigated in mesoporous materials (e.g. MCM-41 or SBA-15) using oxide nanoparticles (MoOx, VOx and FePO4) as the active species.157 Compared to crystalline oxides, Wang and co-workers demonstrated that the confinement of FePO4 species in ordered mesoporous silicas gives an improvement in the catalytic activity and selectivity towards HCHO. The catalytic behaviour is further improved after the incorporation of phosphorous species and the subsequent formation of the FePO4 active sites.158a Apparently, the reducibility of the FePO4 cluster was improved when embedded in mesoporous silicas, as this gave rise to a larger proportion of the lattice oxygen required for the selective oxidation reaction. Similar improvements were described by Li and co-workers with copper nanoparticles attached to mesoporous SBA-15.158b Using these catalysts the oxidation of methane produces formaldehyde as the main reaction product (5.6 mol HCHO mol Cu−1 s−1) together with a small amount of carbon dioxide.
The indirect conversion of methane involves a first step to syngas followed by conversion of this syngas to the desired product (e.g. methanol) or the further conversion of methanol to value-added products.
For the methane reforming reaction to syngas, mesoporous catalysts have been mainly applied to control the size of the supported active species by the diameter of the pores. In this way, Ni nanoparticles supported on mesoporous nanocrystalline ZrO2 showed a high catalytic activity and an improved lifetime under low temperatures and high carbon dioxide to methane ratios.159
The synthesised syngas can be converted into higher hydrocarbons or oxygenates with predominantly straight carbon chains via Fischer–Tropsch (FT) synthesis. The formation of hydrocarbons via FTS is a surface-catalysed polymerisation reaction which proceeds via a modified carbene mechanism on bifunctional catalysts, i.e. active species (Fe, Co, etc.) for FTS supported on acidic zeolites. However, the lack of sufficient external surface area makes the use of mesoporous materials very attractive due to the high metal loading required. Mesoporous materials allow an optimum dispersion of the active species together with improved transport properties which increase the selectivity towards liquid hydrocarbons.160a–c
Sartini and co-workers compared the catalytic behaviour of Co/mesoH-ZSM5 catalysts in FTS with conventional Co/SiO2.160d After 140 h of reaction, the former was twice as active and three times more selective to the C5–C11 fraction with a large content of unsaturated hydrocarbons.
With regard to the synthesis of methanol via the hydrogenation of carbon monoxide, van den Berg et al. compared the catalytic performance of a supported copper catalyst, Cu/ZnO/MCM48, with the typical copper catalyst, the former having a fourfold higher surface area and a fivefold higher copper content. According to these authors, the formation of confined nanocrystalline ZnO species on the mesoporous channels of MCM-48 prior to copper impregnation is of major importance to achieve a large catalytic activity.161a,b
Methanol dehydration to dimethyl ether (DME), as a new synthetic fuel, is another interesting process to substitute diesel or liquefied petroleum gas. Traditionally, methanol dehydration has been performed over acidic catalysts such as zeolites. However, the microporous nature of zeolites does not allow a proper diffusion of DME causing a fast loss of activity and selectivity. Tang et al. reported a micro-mesoporous ZSM-5/MCM-41 composite molecular sieve prepared by combining a microporous zeolite silica source with nano self-assembly methods for the catalytic dehydration of methanol to DME.162a The synthesised catalyst showed an excellent catalytic behaviour, with a methanol conversion of 86.6%, a selectivity to DME of 100% and a long catalyst lifetime better than the parent ZSM-5 catalyst. Similarly, Cho and co-workers reported a higher methanol conversion, lower selectivity to hydrocarbons and a slower deactivation for mesoporous LTA zeolites compared to the conventional CaA microporous zeolite.162b Unfortunately, the selectivity to DME was larger in the conventional CaA zeolite.
Another important process for methanol concerns its conversion to hydrocarbons and gasoline (methanol to hydrocarbon MTH and methanol to gasoline MTG). These processes play an important role in the conversion of biomass, coal, natural gas and CO2 to liquid hydrocarbon fuels which can be tuned to produce gasoline-rich or olefin-rich products depending on the catalyst and the reaction conditions. The reactions are catalysed by shape-selective solid acid catalysts, such as zeolites. Due to the limited growth of the hydrocarbon chains inside the zeolite cavities as a result of steric hindrance, nanozeolites have been proposed as a potential replacement to avoid the aforementioned drawbacks. In this sense, nanoscale HZSM5 crystals have shown a higher activity, a lower coke content and a better stability in the conversion of methanol to hydrocarbons compared to conventional, larger zeolitic crystals.163a
Another approach to minimize kinetic limitations is the use of mesoporous catalysts. Bjorgen and co-workers studied the methanol to gasoline reaction on HZSM5 zeolite chemically treated with NaOH to develop mesoporosity.163b The catalyst lifetime increased by a factor of 3.3 on the desilicated zeolites and the selectivity towards the gasoline fraction (C5+) was increased by 1.7. It seems that the hydrogen transfer reaction was faster in the modified samples and led to more aromatic and paraffinic compounds in the product. A similar improvement in the catalyst lifetime was reported by Kim and co-workers using a series of mesoporous MFI zeolites during the methanol to hydrocarbon conversion.163c While the catalyst's activity is related to the acid site density, the catalyst's stability (deactivation rate) correlates with its mesoporosity.163d,e Mesoporous catalysts exhibit an improved stability due to the faster removal of products with a shorter diffusion path-length and lower coke formation in the conversion of methanol to gasoline-range hydrocarbons.
3.3. The catalytic production of hydrogen
Interest in hydrogen production has grown inexorably in recent times. Some reviews have been published on this topic.164–167 As it is known, hydrogen has emerged as a strategic clean energy carrier for both transportation and stationary applications. It is considered to be one of the best alternative fuels due to its abundance and non-polluting nature when used in fuel cells. In fact, hydrogen conversion in fuel cells efficiently generates energy, producing water as the only residue.168,169 Hydrogen is the most abundant element in the universe and it is also the major constituent of Earth. However, it does not occur in a significant amount as a free H2 molecule, but it is mainly found in the form of water or biomass. Nowadays, nearly 95% of the total hydrogen supply is produced from fossil fuels, mainly by methane steam reforming. However, applications of hydrogen in the transportation sector or to regulate the generation of renewable electricity are still a hot research topic, because of the techno-economical limitations and competition with other technologies. Catalytic technologies are expected to play an essential role in enhancing the yield and the energy efficiency. Ordered mesoporous materials are starting to play a significant role in the development of new types of catalyst for the production of clean hydrogen from different sources and through a variety of routes. The production of hydrogen can be achieved following 4 methods: (i) steam reforming of biomass, (ii) catalytic decomposition of methane, (iii) catalytic decomposition of ammonia and (iv) photocatalytic process. This review will show the most relevant articles related to the effect of mesoporosity on the different technologies. For more detailed information readers are referred to a recent review by Serrano et al.167Transition metal-based catalysts (Ru, Rh, Ni, Ir, Pd, Pt, Co and Fe) have been used since the 1970s in steam reforming.171,172 Ni-based catalysts have been widely used due to their low cost and high catalytic activity. However, Ni-based catalysts show high carbon deposition related to the deactivation of the catalyst. Additional efforts have been made to increase the stability of Ni-based catalysts and, using mesoporous materials as a support, a strong resistance to catalyst deactivation has been achieved.173–175 The high surface area and mesopore uniformity of these systems seem to be the key factors behind the superior catalytic behaviour compared to microporous catalysts.
Bang and co-workers prepared a series of mesoporous nickel–alumina xerogel catalysts by a single-step carbon-templating sol–gel method to study the steam reforming of liquefied natural gas (LNG).176,177 They concluded that the crystallite size of metallic nickel served as an important factor in determining the catalytic activity. Later on, Bang and co-workers studied the same reaction but with a series of mesoporous alkaline earth metal-promoted nickel–alumina xerogel (M = Mg, Ca, Sr and Ba) catalysts, these were prepared by a single-step epoxide-driven sol–gel method and a subsequent incipient wetness impregnation method.178 It was demonstrated that the exposed Ni surface area of the reduced catalysts decreased in the order of Mg > Sr > Ca > Ba, and the hydrogen production is correlated with the exposed surface area of nickel. Highly exposed nickel gives rise to higher LNG conversion and hydrogen yield. It has also been reported that high Ni exposure provides more stable catalysts towards deactivation.179
Recently, another steam reforming process based on ethanol has been studied for the production of hydrogen. Ethanol can be considered as a renewable source since it is produced by the fermentation of a relatively large variety of biomass types with a high carbohydrate content. As with methane reforming, Ni-based catalysts have been employed in the case of ethanol. For instance, a series of Ni catalysts supported on Al-SBA-15 mesoporous materials were studied in the context of ethanol steam reforming by Lindo and co-workers.180 They demonstrated that the Ni/Al-SBA-15 catalysts produce larger amounts of ethylene and coke, with a slightly lower hydrogen selectivity than the Ni/SBA-15 catalysts. This result was explained by the ethanol dehydration taking place in the Ni/Al-SBA-15 acid sites, while an ethanol dehydrogenation mechanism predominates in the Ni/SBA-15 catalyst. Thus it was concluded that for steam ethanol reforming, Ni supported onto pure silica SBA-15 was a promising catalyst.
Han and co-workers used mesoporous Ni–Al2O3–ZrO2 catalysts prepared using sol–gel chemistry and the results confirmed the importance of a high nickel surface area being exposed.181–183 At the same time, Serra et al. studied bioethanol steam reforming on Ni-modified mordenite to see the effect of mesoporosity and the addition of alkaline metals.184 Ni nanoparticles deposited on mesoporous mordenite show a high activity and selectivity and are resistant against coke deposition. The mesoporosity seems to be the main reason for the good performance while the addition of alkaline metals (Na) improves significantly the selectivity to hydrogen while decreasing coke deposition.
In a typical catalytic reforming reaction, hydrogen evolves at a high temperature over a Ni or noble metal-based catalysts. In order to reduce the maximum temperature for high hydrogen generation, Sn was used by Lee and co-workers185 as a promoter for steam ethanol reforming. It was demonstrated that the addition of Sn reduces the temperature for the steam ethanol reforming with high hydrogen yield.
In addition to methane and ethanol, ethylene glycol and glycerol are considered interesting renewable sources for hydrogen production. In this case, recent work has shown the benefits of employing different types of ordered mesoporous materials as supports for the preparation of steam ethylene glycol or glycerol catalysts. Serrano and co-workers167 analysed the effect of Pt-MCM-41 materials or Ni nanoclusters embedded in multicomponent mesoporous metal oxides. Both catalysts favour a higher hydrogen production because of their good metal dispersion. For instance, the ethylene glycol steam reforming reaction was studied with Pt nanoparticles supported on ordered mesoporous carbon by Kim and co-workers.186 The Pt catalyst with a 3D pore structure shows a higher catalytic performance which is due to the fact that there is little metal sintering during the reaction process and more favourable transport and diffusion of the reactants and products.
Metal-based catalysts have been used as an active phase for methane decomposition. However, important deactivation effects have been encountered due to coke formation. In order to mitigate these drawbacks, mesoporous materials have been proposed as promising candidates for supporting active phases in the catalytic decomposition of methane.167 For instance, Ni dispersed within an ordered mesoporous material, consisting of partially Ce-substituted MCM-41 silica showed a higher methane conversion, as well as total selectivity for hydrogen and a high resistance to deactivation, compared with other metal-based catalysts.
Botas and co-workers have used a new class of carbonaceous materials (CMK-3 and CMK-5), which have an ordered mesoporous structure.187,188 Their results showed that CMK-5 is an active catalyst for hydrogen production with a high stability. Carbon deposition from methane decomposition may leave the CMK-5 pores, growing towards the outside part of the catalyst particles, which avoids the total blockage of the mesopores. Recently, Jin and co-workers prepared Fe–Al2O3 supported on carbon materials to study the methane decomposition.189 It has been demonstrated that the introduction of Fe and Al2O3 into the activated carbon decreases the surface area and pore volume, but mesopores with an average pore size around 4.5 nm are formed as an activation process. The resultant catalyst, with appropriate Fe/Al2O3 loading, exhibits good methane conversion and stability. Apparently, mesopores and larger pore volumes are beneficial to achieving a good dispersion of the active Fe species, giving rise to a higher catalytic activity than microporous carbon.
The use of ordered mesoporous materials can provide significant advantages over conventional photocatalysts. Thus, well-defined and uniform pores are ideal to disperse the photoactive component, in the case of the as supported catalysts, and metal doping. Furthermore, with a pure mesostructured semiconductor, both a high light absorption and favourable interaction with the reagents take place simultaneously. Nevertheless, the crystallinity is important because the amorphous components are inactive in photocatalysis.167
TiO2 modified with WS2 was reported by Jing and co-workers showing that the mesoporous structure of the substrate was beneficial for a larger amount of WS2 loading. They demonstrated the effectiveness of using mesoporous TiO2 as the substrate and WS2 as a sensitiser to construct an efficient and stable heterogeneous photocatalyst system responding to visible light.196 Later on, TiO2 modified with ZrO2 was prepared as a mesoporous structured phase using the block co-polymer surfactants method, and was tested in the aforementioned reaction.197 The most important results are shown in Fig. 15.
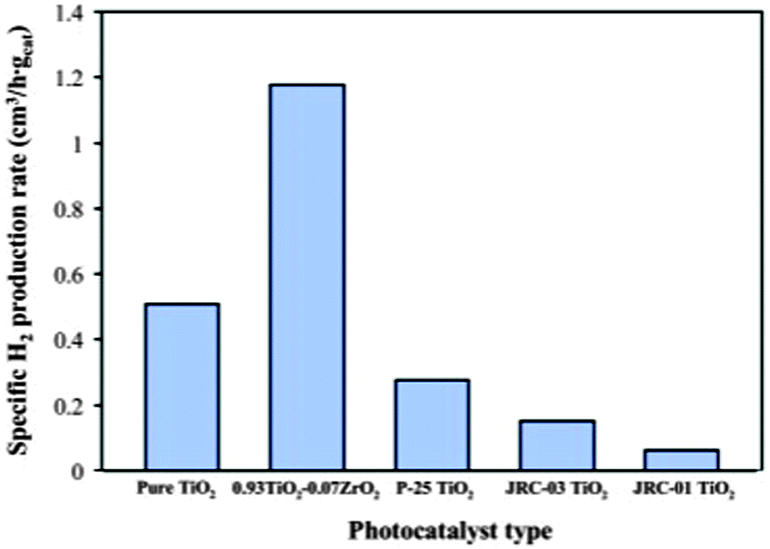 | ||
| Fig. 15 The effect of the photocatalyst type on the specific H2 production rate over the synthesised mesoporous-assembled pure TiO2 and 0.93TiO2–0.07ZrO2 mixed oxide photocatalysts calcined at 500 °C when compared to commercial TiO2 photocatalysts (P25, JRC-01 Ishihara Sangyo Co., JRC-03 Ishihara Sangyo Co). | ||
Comparing the specific hydrogen production rate shows that the synthesised mesoporous-assembled TiO2–ZrO2 mixed oxide photocatalyst with a specific Ti/Zr ratio provide the highest photocatalytic activity for hydrogen production compared to commercial TiO2 photocatalysts. The mesoporous-assembled structure, with a pore diameter of around 5.6 nm, is considered to be a prime factor in increasing the photocatalytic hydrogen production. All the investigated commercial TiO2 photocatalysts possess undesired non-mesoporous characteristics and this may lead to their comparatively low photocatalytic hydrogen production activities, because the photoactive species is less accessible to the reactant.
Dang and co-workers198 have studied photocatalytic hydrogen production with Cu(OH)2/TNTs catalysts prepared by the hydrothermal precipitation process. TNTs loaded with 8 wt% Cu(OH)2 exhibit a remarkably improved activity due to the higher dispersion of Cu(OH)2 onto the TNT surface compared with pure TNTs.
Another alternative to improve the photocatalytic activity is the incorporation of transition metal oxides into the mesoporous silica frameworks in a one-pot synthesis process. The photocatalytic mechanism in this case is based on a metal–metal charge transfer excitation, where incorporated metals act as chromophores, absorbing UV and/or visible light which leads to the formation of an excited state of charge transfer. Moreover, the high dispersion of these isolated species can lead to an increase in the conversion per centre in the case of bulk catalysts.
ZrO2 is a promising light harvesting agent because its conduction band is located at a much higher energy level. For this reason it has been used to prepare Zr-MCM-41 to study the photocatalytic activity in water splitting. The experimental results show that the enhancements in the photocatalytic activity afforded by the Zr-MCM-41 photocatalysts could be due to the high dispersion of ZrO2 in the amorphous wall of MCM-41.199a Similar results were reported using W supported on MCM-48.199b
Recently, Peng et al.200 have prepared two sets of titania, one containing MCM-41 and the other MCM-48, as catalysts for water splitting. The photocatalytic H2 evolution results indicate that the cubic phase MCM-48 exhibits a higher activity as the host photocatalyst than hexagonal MCM-41 materials. Moreover, it was also confirmed that tetrahedrally coordinated Ti species possess a much higher photocatalytic efficiency than the octahedral ones and that the cubic phase is a better support than the hexagonal mesoporous silica support.
Ordered mesoporous materials have significant potential for improving a variety of catalytic routes for the production of hydrogen. Some additional results related to the importance of the mesoporosity in the H2 production can be found elsewhere.167
3.4. Catalytic valorisation of CO2
Nowadays CO2 emissions to the atmosphere are growing, owing to the higher demand for fossil fuels caused by increases in population and living standards. The European Union has set an ambitious target to drastically reduce greenhouse gas emissions over the next few decades. In 2020 the reduction of greenhouse gas emissions should be 20% and in 2050 around 50% in Spain.201 The need to mitigate the accumulation of CO2 in the atmosphere requires technologies able to reduce CO2 emission. Among the different technologies hoping to lower such emissions, the utilization of CO2 as a building block may represent an interesting approach to obtain valuable chemicals and materials.202–206 The most difficult task related to CO2 conversion into organic compounds is the stability of the CO2 molecule. In order to activate the CO2 molecule a high quantity of energy is necessary to weaken the C–O bond. The valorisation process of CO2 must be partnered with a renewable energy like solar energy, to supply the energy involved in the process.207Traditionally, CO2 was limited to a few processes like the synthesis of urea, salicylic acid and polycarbonates. However, nowadays the versatility of CO2 to produce chemical products is being developed constantly, as shown in Fig. 16. Carbon dioxide reacts with hydrogen, alcohols, acetals, epoxides, amines, carbon–carbon unsaturated compounds and oxetanes in the presence of metal compounds as catalysts.204–209
 | ||
| Fig. 16 The chemical transformation of CO2. Reproduced with permission from ref. 209. Copyright 2001, American Chemical Society. | ||
Intensive research efforts are necessary to find new ways of functionalising carbon dioxide and to improve the selectivity of the products by new catalysts. Although most organic compounds are synthesised by homogeneous catalysis, the most important obstacle for large-scale production using this approach is the removal of the catalyst and the purity of the final products. Heterogeneous catalysis is more realistically applicable to large-scale production because it is possible to remove the catalysts and, consequently, the purity of the products is higher.
Therefore, it is vital to develop heterogeneous catalysts if CO2 is to be converted into valuable molecules for different applications.210
CO2 valorisation includes different processes like the hydrogenation of CO2, methanol or ethanol reforming, the conversion of alkane to alkenes and the fixation of CO2 into organic molecules.
Over the last two decades, the hydrogenation of CO2 to CH3OH has been studied in order to develop effective catalysts. At the beginning, supported precious metals (Pt or Pd) were used to study CO2 hydrogenation. It is well established that both the support and the active phase can modify the activity and selectivity.213–215
Additionally, the activity can be increased by the addition of promoters like Cr2O3, Ga2O3 and ZrO2.206,216,217 Recently, it has been observed that the uniform pore structure of mesoporous materials may provide nano-sized uniform reaction environments which enable the stabilisation of small metal nanoparticles inside the mesopores. In this connection, it is worth mentioning that a so-called bi-functional mechanism has been proposed for CH3OH formation from CO2, over metal oxide-promoted Pd catalysts, where the metal oxide promoters stabilise adsorbed formate (or CO2) species and metallic Pd-dissociated H2 molecules simultaneously.218 Thus, a better catalytic activity for CH3OH formation would be expected when small Pd and metal oxide nanoparticles are incorporated inside mesocavities. In addition, if the size of the Pd nanoparticles can be controlled systematically by mesoporous materials with different pore diameters, these catalysts could also become an interesting model catalyst.
Although some research groups have already shown the use of mesoporous silica with a high activity for the hydrodesulfuration of dibenzothiophene,219 the Fischer–Tropsch synthesis,220 and H2O2 synthesis221 as a support, its utility in the hydrogenation of CO2 to methanol is still very limited. In 2006 Yu and co-workers studied the effect of a Ru catalyst supported on functionalised MCM-41.222 In this study they combined the benefits of heterogeneous catalysis with the benefits of homogeneous catalysis, immobilising Ru complexes on the functionalized mesoporous MCM-41. Their results showed that MCM-41-immobilised ruthenium complexes exhibit a promising catalytic performance as heterogeneous catalysts for the synthesis of formate from CO2 and H2 in CO2-expanded solvent. Later on, Kosizumi and co-workers studied the effect of nano-structured mesoporous supports and the effect of the addition of alkali/alkaline earth metal oxides on the CO2 hydrogenation reaction.223
As shown in Fig. 17, mesoporosity does not guarantee good performance in CO2 hydrogenation. This paper demonstrates that the small mesopores of MCM-41 and SBA-15 work as a template for the formation of small Pd0 nanoparticles, leading to a higher activity for methanol formation compared to the conventional amorphous silica. Indeed, the importance of mesoporosity is related to the development of specific Pd nanoparticles rather than diffusional problems. It seems that the overall reaction for methanol formation is governed by the surface reaction rather than diffusion on the reactants, probably due to the low reaction temperature used.
 | ||
| Fig. 17 The dependency of the rate of CH3OH formation over Ca(0.4)/Pd(6) catalysts on the average pore diameter. Adapted from ref. 223. Copyright 2012, Elsevier. | ||
This reaction has been extensively studied using mesoporous materials as a support for the active phase. Rh-based catalysts are known to have a high activity, stability and low coke formation in the CO2 reforming of methane and the reforming of ethanol reactions.224 Wu and Kawi studied the effect of Rh/Ce-SBA-15 as a catalyst for the CO2 reforming of ethanol.225 They observed that the mobility of the surface oxygen species and the Rh dispersion over the Ce/SBA-15 catalyst support were important parameters in achieving a high catalytic performance. It was demonstrated that hexagonal mesoporous SBA-15 remains intact after the CO2–ethanol reforming reaction for 24 h at 600, 650, 700 and 750 °C. So, the 1%Rh/Ce-SBA15 catalyst is a potential commercial catalyst for CO2 reforming with ethanol to produce syngas and hydrogen.
One important point about CO2 reforming is the inhibition of the catalysts by carbon deposition. This coke deposition is related to the active metals and the support. Ni-based catalysts have been reported to easily deactivate due to the formation of coke. The low cost of Ni has encouraged the development of new Ni-based catalysts with an improved stability. Xu and co-workers studied the effect of an ordered mesoporous NiO–Al2O3 composite in the CO2 reforming of methane.226 It was demonstrated that ordered mesostructured catalysts can offer a high activity and long stability, by supplying much more accessible Ni active sites to the reactant and stabilising the Ni nanoparticles via the “confinement effect” during the reaction. Ni nanoparticles exist among the framework of the material, which contribute to suppressing the coke deposition.
Later on, the incorporation of MgO was evaluated in order to produce mesoporous NiO–MgO–Al2O3via a one pot evaporation induced self-assembly (EISA) strategy.227 The benefits of the material NiO–MgO–Al2O3 over NiO–Al2O3 were found to be a large specific surface area, a large pore volume and a favourable thermal stability. The addition of moderate amounts of Mg promotes the catalytic properties. These materials were tested over a 100 h reaction time with no loss in catalytic activity.
Furthermore, it is well known that the nature of the supports greatly affected the catalytic performance of Ni based catalysts for the catalytic reforming of methanol (CRM). Various materials such as MgO, Al2O3, SiO2, CeO2 and ZrO2 have been investigated as a support for Ni based catalysts. Currently, different metal oxides have been developed in order to obtain materials with improved thermal stability.
In order to compare the effect of the support, Sarkar and co-workers studied the CO2 reforming to methane, catalysed by Ni supported on SiO2/Al2O3 and ZSM-5.228 They demonstrated that mesoporous ZSM-5 as a support was more active than mesoporous SiO2/Al2O3 due to the different Ni dispersion on the supports. Ni-ZSM with 5% of Ni showed 96.2% methane conversion at 800 °C in the used conditions. Consequently, mesoporous materials are important to synthesise active phase nanoparticles with good metal dispersion.
After studying the effect of Ni and Ni–Mg mesoporous materials on Al2O3, Xu and co-workers studied CO2 reforming by using Ni–Ce/Zr mesoporous solids.229 It was the mesoporous framework that endowed the mesoporous catalyst with more activity. Specifically, predominant textural properties provided more “accessible” Ni active centres for the gaseous reactants accounting for the higher activity. The “confinement effect” of the mesopores imbued the Ni nanoparticles with a higher stability. As noble metals commonly excel in coke resistance and thermal sintering, Takahashi and co-workers analysed the effect of a Pt/ZrO2 mesoporous catalyst in the CRM reaction.230 As previously mentioned, the presence of a mesoporous support is important to achieve good metal dispersion and to improve the resistance to coking.
Recently, the CRM reaction has been studied on different mesoporous catalysts, like Ni-SBA-15 prepared with β-cyclodextrine.231 β-Cyclodextrine was added to increase Ni dispersion and the catalysts prepared with β-cyclodextrine were more active than their NiSBA-15 counterparts. The presence of mesopores was required in order to favour the intimate contact between the cyclodextrine and the Ni particles, thus preventing Ni sintering effects. It is known that Y can act as a promoter for catalytic reforming reactions. With this in mind, Li and co-workers studied the effect of Ni/Y-SBA15 mesoporous materials in the CRM reaction.232 Y is a promoter of the CRM reaction by enhancing the reduction of NiO, which was attributed to the oxygen vacancies on the surface of the catalyst and the high mobility and activity of the surface oxygen species. The presence of a mesoporous support is important to achieve a good Y dispersion around the structure.
As mentioned before, CeO2 is a good support for catalysts due to its redox properties. However, its thermal stability must be improved. Zeng and co-workers described the influence of pore distribution on the catalytic performance of an inverse CeO2/Co3O4 catalyst for the CMR reaction.233 It was demonstrated that the CeO2/Co3O4 catalyst with double pore distribution could give more active sites as well as better gas circulation channels, reducing the internal diffusion resistance and improving the catalytic performance for CH4/CO2 reforming. This work is in agreement with the results obtained using bimodal silica.234 Finally, one of the latest works related to the CRM reaction has been done by Guo and co-workers, who have studied the catalytic properties and stability of cubic mesoporous LaxNiyOz/KIT-6 catalysts.235 La2NiO4/KIT-6, showing the highest activity for CMR reaction at 800 °C, still retains the mesoporous structure and high surface area, which is favourable for the diffusion or adsorption of reactant molecules. During the reforming reaction, the formation of La2O2CO3 species can maintain the balance between carbon deposition and elimination. The thermal stability of the La2NiO4/KIT-6 catalyst is significantly higher than that of La2NiO4/SBA-15 due to the stable cubic structure of the KIT-6 support.
Taking into account the promoting effect of Cr2O3, Rao and co-workers studied the effect of Cr addition on different siliceous materials derived from MCM-41.239,240 The results showed that the ethane dehydrogenation reaction depends on the chromia loading and the catalyst preparation. Cr/MCM-41 catalysts were more active than bulk Cr2O3, this effect being attributed to the better metal dispersion on the mesoporous material supports.
Another important process is the oxidative dehydrogenation (ODH) of propane to propene. It is known that a limited propene selectivity at higher propane conversions is linked with the propene adsorption on acid sites and their subsequent oxidation to carbon oxides. Thus, it is challenging to develop new efficient catalyst systems which can allow the production of propene with a high selectivity at a higher propane conversion. Cr catalysts were studied in the propane dehydrogenation reaction by Michorczyk and co-workers using mesoporous materials as the support.241 In dehydrogenation reactions the higher the dispersion of the active phase, the higher the activity.
Recently, Zhang and co-workers studied the effect of ZSM-5-supported chromium oxide catalysts prepared by the incipient wetness method in the dehydrogenation of propane.242 This study demonstrates that chromium oxide supported on an Na-type ZSM-5 with a smaller crystal size (ca. 400 nm) is an attractive new catalyst applicable for propane dehydrogenation to propylene with CO2. This catalyst exhibits a substantially higher activity than chromium oxide supported on ZSM-5 with a larger crystal size (ca. 2 μm).
Finally, it is noteworthy to mention that the number of studies dealing with large molecules is rather scarce. As an example, Wang and co-workers prepared a series of mesoporous silica SBA-15 supported chromia catalysts and examined their catalytic properties for the ODH of ethane, propane, n-butane and isobutane.243 All catalysts showed an excellent catalytic activity and the results could be explained by a combination of factors: (1) the good redox behaviour of the Cr species, (2) the formation of highly disperse mono- and poly-chromate domains, and (3) the formation of well-ordered mesostructured banana-like SBA-15 rods that provide a local environment favourable for the adsorption, desorption and diffusion of the reactant or product molecules. One important process is the catalytic dehydrogenation of ethylbenzene (EB) to obtain styrene. Styrene is one of the most important monomers in the petrochemical industry because of its use in the production of various polymers such as polystyrene, styrene-acrylonitrile (SAN) copolymer, styrene-butadiene rubber (SBR) and acrylonitrile-butadiene-styrene (ABS) tert-polymer.
Contrary to the process that uses oxygen as a strong oxidant, the dehydrogenation of ethylbenzene with CO2 produces styrene with a much higher selectivity. Qiao and co-workers studied VOx/MCM-41 catalysts for the EB reaction.244 They concluded that catalysts prepared using a mesoporous support showed a higher EB conversion, which must be attributed to the better metal dispersion on the support.
The functionalisation of well-ordered mesoporous solids by post-grafting techniques has been postulated as a more efficient synthetic route since it increases the efficiency for the utilisation of large pore materials. Hybrid inorganic–organic mesoporous materials based on the co-condensation of siloxane and organosiloxane precursors, in the presence of a templating surfactant solution are of interest for producing a regular and ordered architecture. Udayakumar and co-workers studied the fixation of CO2 to allyl glycidyl ether over an ionic liquid immobilisation on MS41.245 The results showed that the modified MS41 materials were generated with a large number of active sites, uniform pores and high surface area. The excellent activity of these materials is owed to the large number of active sites in the host system. Recently, Nelson and Adam studied the synthesis of styrene carbonate via the cycloaddition of CO2 using ordered mesoporous MCM-41-Imi/Br catalysts.246 This material promises to be a heterogeneous, environmentally friendly and active catalyst for the production of cyclic carbonates from styrene oxide. The fixation of CO2 into three-member heterocyclic rings such as aziridine for their transformation into five-member heterocyclic rings such as oxazolidinones is important for their versatile intermediates. MCM-41 grafted with amine groups has been reported to catalyse many organic reactions. These catalysts have a good mesoporous structure ordering, a large BET surface area and a high porosity. All these qualities are important to permit the diffusion of the molecules into the catalyst. Recently, Nale and co-workers studied the regioselective synthesis of 5-aryl-2-oxazolidinones from CO2 and aziridines by amine functionalised MCM-41.247 The results make it a promising material for the cyclic carbonates reaction from styrene oxide.
The efficient CO2 valorisation process requires mesoporous catalysts containing uniformly distributed small nanoparticles. For the big size of the molecules involved in this process, mesoporous materials also help to avoid diffusional problems.
3.5. Photocatalytic energy technologies
By adsorption of light with greater photonic energy, semiconductors can generate electron–hole pairs, which initiate simultaneously oxidative and reductive reactions with surface species, before recombination. Catalysts under light irradiation, called photocatalysts, are attracting a great deal of attention, both for fundamental science and practical applications.248 This technology can be divided into the degradation of molecules into CO2 or the utilization of CO2 to produce organic molecules.During the last few years, a lot of research has been done to activate mesoporous materials with titanium oxide in order to obtain large pore catalysts allowing high diffusion rates and therefore an improved photocatalytic activity for bulky molecules.93a,251,252 The preparation conditions such as the synthesis time and calcination temperature significantly influence the photocatalytic activity of meso–macroporous TiO2, illustrating the role of porosity in light harvesting photocatalysis253,254
For instance, Saadoun and co-workers255 tested the activity of a titania, synthesised using an ammonium salt as a hydrolysis control agent, in the oxidation reaction of formaldehyde. The results indicated that the diffusion of the reactants or products within a porous catalyst could be a rate-limiting factor. TiO2 catalysts prepared hydrothermally showed a significantly higher efficiency than the commercial titania, Degussa P25.
Later on, Wang and co-workers256 prepared bimodal meso–macroporous TiO2 by a self-formation phenomenon process in the presence of surfactants. Ethylene photodegradation in the gas-phase was chosen as a probe reaction to study the photocatalytic activity. The results showed that catalysts calcined at 350 °C possess an intact macro/mesoporous structure and higher photoactivity (60% higher than of commercial P25 titania). When the calcination temperature was increased above 600 °C, the macro/mesoporous structure was destroyed and a loss in the photocatalytic activity was observed. The high photocatalytic performance of the low-temperature-calcined macro/mesoporous TiO2 may be explained by the existence of macrochannels that increase the photoabsorption efficiency and allow the efficient diffusion of gaseous molecules. Similar results were obtained by Yu and co-workers.257 These authors found that the hierarchical macro–mesoporous TiO2 calcined at 300 °C had the maximum photocatalytic activity for the oxidation of acetone in the gas phase (around twice that of P25). The activity then decreased as the calcination temperature increased, due to the destruction of the macroporous structure and the decrease in the surface area.
The incorporation of supported titania within silica materials has been also proposed for improving the surface area of these materials. Hamdy and co-workers studied the incorporation of TiO2 in a silica mesoporous material (TUD-1), to evaluate the selective light-activated oxidation of propane to acetone.258 The results showed that nanoparticle-containing TiO2-TUD-1 can be more selective in the photooxidation of propene to acetone than commercially available microcrystalline anatase.
Later on, Busuioc and co-workers259 reported a post-synthesis deposition method to form crystalline titania plugs inside the mesopores of SBA-15. Anatase nanoparticles with different sizes were deposited into the SBA-15 channels, influencing the adsorption properties and photocatalytic activity. These materials exhibit an improved catalytic activity for the destruction of Rhodamine 6G under UV light irradiation.
More recently, Tasbihi and co-workers260 studied the photocatalytic degradation of isopropanol in the gaseous medium. They catalysed the reaction with titania, incorporated in ordered (SBA-15) and disordered (KIL-2) mesoporous silica via the sol–gel impregnation method. The results showed that the photocatalytic activity towards the formation of acetone depends on the accessibility and number of titania nanoparticles. In the STi/SBA-15 (1/1) sample, prepared using an aqueous crystalline anatase sol with a Ti/Si ratio of 1, the loading of titania was not too high to decrease accessibility and high enough to ensure a sufficient quantity of active nanoparticles for the reaction. Therefore among all the investigated samples the photocatalytic activity (acetone formation rate) of STi/SBA-15 (1/1) was the highest. The results revealed the benefits of a mesoporous support when removing an organic contaminant from the gaseous phase.
Later on, Dong and co-workers261 prepared 2-D hexagonal mesoporous TiO2–SiO2 nanocomposites consisting of anatase TiO2 nanocrystals and amorphous SiO2 nanoparticles, with large mesochannels and high specific surface areas. The photocatalytic degradation of different organic dyes like Methylene Blue, Safranin O, Crystal Violet, Brilliant Green, Basic Suchsin, Rhodamine-6G, Acid Fuchsin, Orange II, Reactive Brilliant Red X3B, Acid Red 1 and Microcystin-LR was evaluated in these systems. The results showed that the samples prepared by TiO2–SiO2 nanocomposites exhibited an excellent degradation activity for all the contaminants, much higher than that of the P25 photocatalyst. The dyes were not only decolourised promptly but degrade readily as well. It was thus demonstrated that these catalysts had promising applications in the fast and highly efficient degradation of various organic pollutants. Recently, Rico et al. synthesised a mesoporous titania–organosilica material with a high surface area, (ca. 300 m2 g−1) with excellent activity in the degradation of a commercial dye under UV light.93a
As far as the morphology of TiO2 is concerned, Zhang et al.262 employed highly crystalline mesoporous TiO2 synthesised via the surfactant sulfuric acid carbonisation method for the degradation of Rhodamine B (RhB) under UV light irradiation. A higher photocatalytic activity was achieved than from the commercial P25, which can be attributed to the higher surface area of the ordered mesoporous TiO2. At the same time, Zhu and co-workers263 prepared titanium oxides with hierarchical structures using biotemplates by the sonochemical method. The calcination temperature has a strong effect on the structural reproducibility and photocatalytic activity of the replicas. It was demonstrated that a calcination temperature of 450 °C results in the best structural replication, the highest surface area, (ca. 58.4 m2 g−1), and hence the best photocatalytic properties. TiO2-450 showed better photocatalytic properties than P25, which could be attributed to its hierarchical interconnected structures with open and accessible pores, as well as the fine grain size and large surface area of the TiO2 replica. Finally, He and co-workers264 have studied the photocatalytic activity in the degradation of gaseous benzene with mesoporous TiO2. Mesoporous anatase TiO2 samples were prepared via a solvothermal method, using TBT (tetrabutyl titanate) as the Ti source and acetic acid as the solvent. The materials were studied for the degradation of gaseous benzene under UV irradiation. It was demonstrated that nano-sized TiO2 particles prepared under optimal conditions had the largest specific surface area (two times higher than Degussa P25) and a significantly higher efficiency for benzene degradation compared to commercial P25.
Up to now, the importance of the development of mesoporous TiO2 for photocatalytic degradation has been demonstrated. To further improve the photocatalytic activity of hierarchical porous TiO2, several strategies based on chemical and physical concepts have been adopted. Metal doping of porous TiO2 structures has been thought to be a good way to enhance the photocatalytic activity, while the coupling of TiO2 with another semiconductor is another widely used approach.192,248
For instance, Zhou and co-workers265 studied the effect of Fe doping on mesoporous TiO2. The samples were prepared by the ultrasonic-induced hydrolysis reaction of tetrabutyl titanate in a ferric nitrate aqueous solution without using any template. The photocatalytic activity of the Fe-doped TiO2 powders calcined at 400 °C was more than twice that of Degussa P25 (at an optimal atomic ratio of Fe to Ti of 0.25) for the photodegradation of acetone. The high activity of the Fe-doped TiO2 powder can be attributed to the results of the synergetic effect of Fe-doping, the large BET specific surface area and the small crystallite size.
Recently, the doping of different metals has been used in different photocatalytic degradation reactions. As an example, Ismail266 reported a synthesis of mesoporous PdO–TiO2 nanocomposites, through a simple one-step sol–gel reaction, for the photooxidation of methanol in aqueous suspensions. Comparing these catalysts with Pd/aeroxide TiO2–P25, the PdO–TiO2 nanocomposites showed more effective and efficient photocatalytic activities for methanol oxidation to formaldehyde (4 and 2 times more than PdO–TiO2 and Pd/TiO2-P25, respectively). The photocatalytic results indicated that these PdO–TiO2 nanocomposites with a mesoporous structure enable the high flux and rapid diffusion of methanol.
Additionally, Ismail267 reported the synthesis of mesoporous Ag/TiO2 films, where the nanoparticles are highly dispersed into TiO2 thin films in a simple one-pot reaction, with the P123 triblock copolymer as a template. The obtained mesoporous Ag/TiO2 films were evaluated for their photocatalytic degradation of 2-chlorophenols showing that these catalysts were more photoactive (8 times more) than the nonporous commercial photocatalyst Pilkington Glass Activ. Furthermore, the Ag/TiO2 films were quite stable during the reaction with no significant loss of activity after 10 runs. Recently, Choina and co-workers268 have studied the effect of Zr-doped anatase titania prepared by sol–gel and chemical vapour deposition methods in the oxidative decomposition of Ibuprofen (IBP). The obtained photocatalytic material shows a high initial activity and exhibits improved adsorption properties in aqueous solution. Both of these properties make the catalyst interesting for the decomposition of hazardous compounds like IBP, especially in a low concentration aqueous solution.
Since doping both with transition metals and lanthanide ions each has its respective advantages, it can be assumed that doping with two dopants can show a synergetic effect with an improved photocatalytic activity. For instance, Nesic and co-workers269 prepared titanium dioxide photocatalysts co-doped with lanthanum and vanadium using a facile microwave-assisted hydrothermal method. The co-doping of vanadium contributes to the extension of absorption into the visible region. The photocatalytic activity of the samples was evaluated by the decolourisation of the textile dye Blue 52 in aqueous solutions under sun-like radiation. Compared with La singly doped TiO2, the co-doped catalysts showed a significant improvement in photoactivity, even higher than commercially available titania P25. Although gold was recognised to be poorly active as a catalyst, when Au nanoparticles were highly dispersed on semiconductor metal oxides or hydroxides, they exhibited a good catalytic activity. Furthermore, it was expected that the encapsulation of gold nanoparticles in the MCM-41 or TiMCM-41 surface would tailor the photoresponsiveness into the visible region.
With this in mind, Kumar and co-workers270 prepared Au/TiMCM-41 photocatalysts to study the photodegradation of methyl orange. It was demonstrated that Au/TiMCM-41 had a higher photocatalytic activity than TiMCM-41. Recently, Zhan and co-workers271 have studied the photocatalytic degradation of methyl orange by using Mo/TiO2/SiO2 materials. A Mo-doped, TiO2-fumed SiO2 composite was prepared by a feasible sol–gel method. The prepared catalysts exhibit a good photocatalytic activity under both ultraviolet and visible light irradiation for methyl orange degradation. The high surface area and high active site concentration on the composite surface are considered the key factors in this.
Yan et al.272 have studied the effect of WO3 in the photodegradation of Rhodamine B dye and phenol under simulated solar light irradiation. It is worth noting that WO3, with a narrow band gap, is one of the most important photocatalysts. For that reason, Yan prepared a highly ordered mesoporous WO3–TiO2 composite material by a surfactant template-assisted evaporation-induced self-assembly process. The presence of a large quantity of mesopores in the composite system allows the diffusion of organic compounds inside the bulk material, shortening the migration distance of photogenerated charge carriers and reducing the opportunity for electron–hole recombination. The results show that both the surface area and crystallinity degree are important parameters in the photodegradation reaction.
Similar to metals, non-metals doped or deposited on TiO2 can effectively improve its photoactivity and selectivity under visible light irradiations. The most commonly used non-metals so far investigated, in a number of studies, are C, N, F, I and S. For instance, Hu and co-workers273 studied the effect of N doping on mesoporous TiO2. Non-metal doping is one of the typical chemical modifications able to enhance the visible light photocatalytic activity of oxides. N-doping has been proved to be a simple and effective way to increase the visible light adsorption. N-doped mesoporous TiO2 spheres were prepared by a dual-templating synthesis. The ordered arrays of N-doped mesoporous TiO2 spheres showed an enhanced visible light photocatalytic activity, which could be attributed to the N doping effect, abundant ordered mesopores and a unique opal structure. Such TiO2 sphere arrays may be promising for practical applications in the fields of environmental purification, water photoelectrolysis and dye sensitized solar cells.
Non-semiconducting metal oxides, like alumina and silica, have frequently been used for supporting photocatalysts, in order to control the product selectivity or to shift the reaction to visible light irradiation conditions. For instance, Cr–SiO2 materials containing highly dispersed chromate species have been proved to catalyse the photo-oxidation of different olefins under visible light irradiation, with a high selectivity to partially oxidized products; while TiO2 promoted complete decomposition.274 Mo, Mn, Cr, Cu, Co and Ag have all been studied as metal doping photocatalysts. Rodriguez and co-workers275 compared the photoactivity of Degussa P25 with Co–Al-MCM41 materials in the degradation of acetaldehyde using visible light and UV-visible light. The results showed that Co–Al-MCM41 with an optimum Co/Al ratio effected good photocatalytic degradation in visible light, but did not work as well as P25 in UV-visible light conditions. Similar results were obtained using Cr–Al-MCM-41 and AgBr/Al-MCM-41 catalysts.276,277 These are thus representative examples which prove that mesoporous materials as a support have many advantages due to their large surface area, uniform pore size and accessible open frameworks.
Recently, the effect of mesoporosity in artificial photosynthesis has been developed by many research groups. For instance, Frei and co-workers283 studied the formation of formic acid as the main product over a Ti silicalite molecular sieve (TS-1), under UV light and using methanol as an electron donor. On studying the mechanism of the reaction with FTIR spectroscopy measurements they found that CO2 splits to CO and O2 at the excited metal-to-metal charge-transfer sites.284 Recently, Tahir et al.285 have studied photocatalytic CO2 reduction with H2O in the gaseous phase, using TiO2 and indium doped TiO2 nanoparticles in a microchannel monolith photoreactor. The results show that this photoreactor possesses a larger illuminated surface areas, higher light utilisation, an efficient adsorption–desorption process and a higher catalyst interparticle mesoporosity. All of these are key factors in improving the yield rates in a monolithic photo-reactor. They have demonstrated that well-developed mesopores, a larger surface area and a higher pore volume can enhance the molecular transportation rates of reactants and products to increase CO2 conversion efficiency.
Another study using mesoporous silica supported Cu–TiO2 nanocomposites and prepared by a one-pot sol–gel method shows that the high surface area mesoporous silica substrate greatly enhanced CO2 photoreduction due to improved TiO2 dispersion and the increased adsorption of CO2 and H2O on the catalyst.286
Until now, a large number of studies have been reported for the increased CO2 photocatalytic conversion efficiency using various TiO2-based photocatalysts. However, because of the wide band gap and relatively slow carrier transport of titania, the activity of TiO2-based photocatalysts in the reduction of CO2 with H2O is not high enough for practical uses, especially under visible or solar light irradiation. CeO2 is an n-type semiconductor and therefore has been used as a dopant to improve titania-based catalysts. Wang and co-workers287 have studied the reduction of CO2 with H2O under simulated solar irradiation using ordered mesoporous CeO2–TiO2 composites synthesised through a nanocasting route, with ordered mesoporous SBA-15 as the template. The results are shown in Fig. 18.
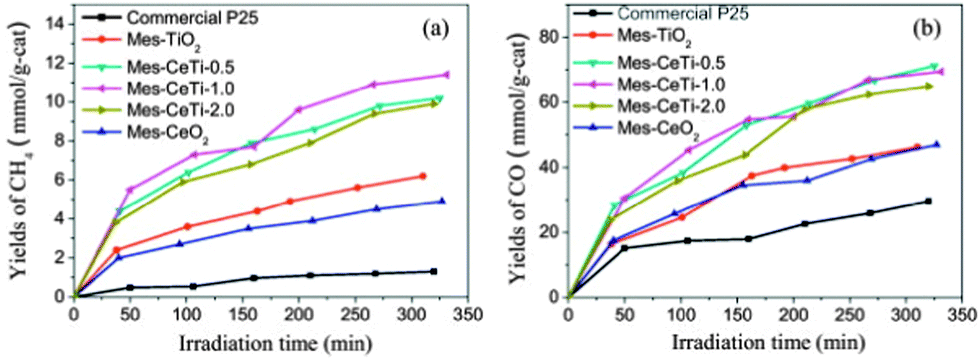 | ||
| Fig. 18 The yield of CH4 (a) and CO (b) as a function of the irradiation time over all photocatalysts. Reproduced with permission from ref. 287. Copyright 2013, Elsevier. | ||
It is demonstrated that the ordered mesoporous CeO2–TiO2 composites exhibit a higher photocatalytic activity in the photoreduction of CO2 with H2O under simulated solar irradiation than commercial P25. The results show that CeO2 addition can enhance the photocatalytic efficiency of pure mesoporous TiO2. The enhanced photocatalytic performance for these CeO2–TiO2 composites can be ascribed to their unique structure, which confers a variety of favourable properties. Firstly, ordered mesoporous architecture with a large surface area and 2D open-pore system makes the reactant diffusion into the bulk of the catalyst easy, and hence provides fast intraparticle molecular transfer. Secondly, the introduction of CeO2 species into composites can effectively extend the spectral response from the UV to the visible area owing to the CeO2-photosensitisation. Later on, Wang's group prepared ordered mesoporous Fe-doped CeO2 catalysts by nanocasting, using ordered mesoporous SBA-15 as the template.288 These catalysts exhibit a good photocatalytic performance in the reduction of CO2 under simulated solar irradiation.
Another alternative to CO2 photoreduction is the use of surface-anchored molecular catalysts that combine the advantages of homogeneous and heterogeneous catalysis.289 For instance, Dubois et al.290 have studied the photocatalytic reduction of CO2 using Re-bby immobilised on mesoporous silica. The results show that covalent attachment through simple organic linkages is a promising strategy for immobilising molecular CO2-reduction photocatalysts on solid-state surfaces.
4. Beyond catalysis
4.1. Novel mesoporous electrodes for fuel cell technologies
In a fuel cell, the chemical energy of a fuel and an oxidant is converted into electrochemical energy. The process involves electron transfer during the oxidation and reduction reactions with an essentially invariant electrode–electrolytic system.291 Nowadays, there are several types of fuel cell (FC) differing, mainly, in the nature of the electrolyte. However, the basic operating principle of all types of FC is the same; see the schematic representation in Fig. 19. A fuel, i.e. hydrogen, is oxidised at the anode into protons and electrons, while at the cathode, oxygen is reduced to oxide species, and these then react to form water. Depending on the electrolyte, protons or oxide ions are transported through the electrolyte, which should be ion conducting but electronically insulating. Finally, electrons travel round an external circuit delivering electric power.292 FC are classified based on the choice of fuel and electrolyte into 6 major groups:293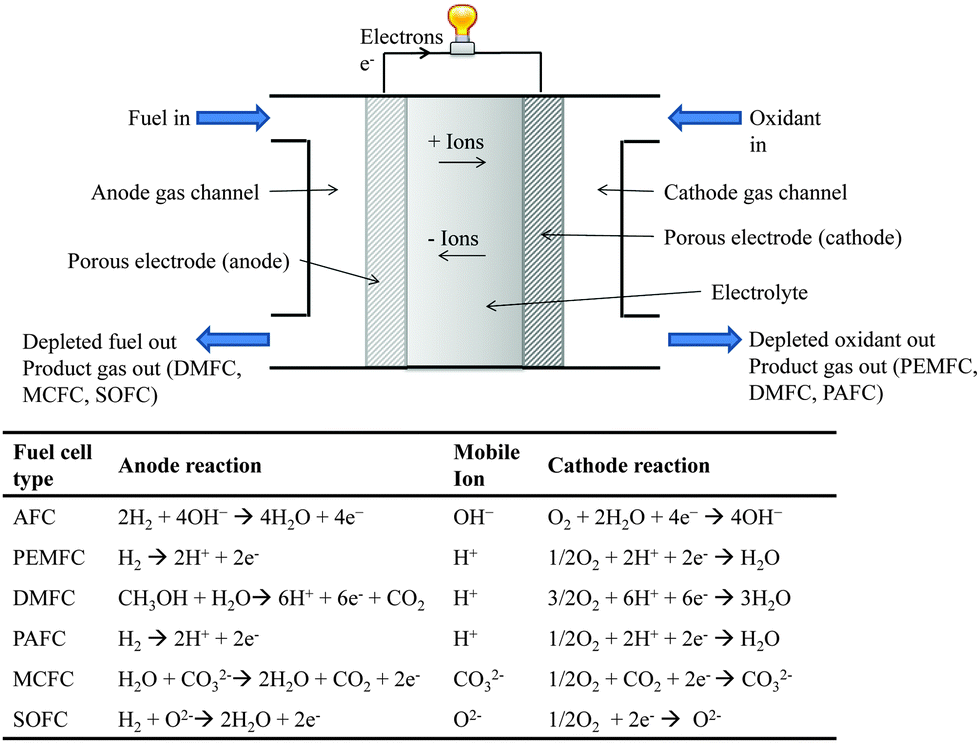 | ||
| Fig. 19 A schematic representation showing the general operating principles of a fuel cell and the principle anode and cathode reactions, as well as the mobile ions associated with the most common fuel cell types. Adapted from ref. 296. Copyright 2011, IEEE. | ||
– alkaline fuel cells (AFC) utilise an aqueous potassium hydroxide (KOH) solution as the alkaline electrolyte;
– proton exchange membrane fuel cells (PEMFC) and direct methanol fuel cells (DMFC) use a polymer membrane as the electrolyte;
– phosphoric acid fuel cells (PAFC) use pure phosphoric acid as the electrolyte;
– molten carbonate fuel cells (MCFC) use a molten mixture of lithium, sodium, and potassium carbonates as the electrolyte;
– solid oxide fuel cells (SOFC) use a ceramic material as the electrolyte.
As the process is not governed by Carnot's law, fuel cells do not need high pressure or thermal gradients to obtain good efficiency, having much higher theoretical efficiencies (35–55%) than traditional combustion-based thermal energy conversion. Furthermore, they present very low levels of pollutant emission due to the absence of high pressure, high temperature a combustion chamber and the fact that air and fuel are separated.294 Hence, FCs are cleaner and more efficient energy conversion technologies than traditional systems. However, it is necessary to point out that FCs are only classed as renewable energy systems if the fuel used is renewable (biofuels, hydrogen from wind or solar conversion of water).295
Regarding the use of porous materials in FCs, anodes and cathodes have to be porous in order to allow the diffusion of the fuel and the products and, moreover, the use of mesoporous supports in the active catalyst region would increase the dispersion and stability of the catalyst (typically Pt and Pt based alloys297).
As reported by Sharma and Pollet,298 an ideal support for the electrocatalyst should have: (1) a good electrical conductivity, (2) good catalyst–support interaction, (3) a large surface area, (4) a mesoporous structure enabling the ionomer and polymer electrolyte to bring the catalyst nanoparticles close to the reactants, i.e. to maximise the triple-phase boundary (TPB), (5) a good water handling capability to avoid flooding, (6) good corrosion resistance, and (7) easy recovery of the catalyst.
Usually, electrocatalysts in FCs have been supported in highly conductive porous carbons and different reviews have recently been published dealing with this topic.299 Mesoporous carbons show very good properties as FC catalyst supports: high surface area, high electrical conductivity and a relatively good stability. However, carbon has the issue of its corrosion during the reaction, leading to catalyst loss and dramatically affecting the performance of the FC. For this reason, lately, non-carbonaceous-based materials have been extensively studied as electrocatalyst supports. An interesting review about non-carbon support materials for PEMFC electrocatalysts can be found elsewhere.300
Due to the large body of reports dealing with mesoporous supports for FC catalysts, and as the topic has been extensively reviewed in the lasts years,291,298,301 here we only discuss the most relevant contributions published in the last year on mesoporous electrodes for PEMFC and DMFC, because they are two of the most promising systems. An extensive summary of the application of mesoporous materials in FC technology, showing last year's contributions, is shown in Table 3.
| Materials | Structure | Preparation method | Properties and applications | Ref. |
|---|---|---|---|---|
| a M = transition metal. | ||||
| Pt/C, PtRu/C | Mesoporous | Non-ionic surfactant | Enhancement of the activity in DAFC and PEMFC | 310–312 |
| Dual soft/hard templating | 313 | |||
| SBA-15 as hard template | 302–304, 314 | |||
| Hierarchical meso/macroporous | Hard template and adjusting of the synthetic conditions | 315 | ||
| Mesoporous | SBA-15 as hard template | Acid treatments to improve catalytic activity in DAFC | 315 | |
| Micro/mesoporous | Xerogel | 316 | ||
| Mesoporous | Alkylamine surfactants | Different studies/treatments to enhance the ORR activity | 317 | |
| SBA-15 as hard template | 318 | |||
| Micro/mesoporous | Derived from Mo2C | 319 | ||
| Pt/MxOy/C,a Pt/TiOxC | Mesoporous | Non-ionic surfactant | Studies of the dopant influence into the electrocatalysis | 320 |
| SBA-15 as hard template | 321 | |||
| Pt/C doped with other C structures | Mesoporous | Silicas as hard template | Enhanced electrochemical performance and long-term stability | 322 |
| PtRuIrNi/C | Hierarchical meso/macroporous | Silicas as hard template | Enhancement in activity and better electrochemical stability in DAFC | 323 |
| Pt/CrN, Pt/TiN, Pt/Ti0.5Nb0.5N | Micro/mesoporous | Ammonolysis | High tolerance to corrosion, excellent long-term stability. | 307 |
| Pt/(SiO2/RuO2), Pt/(TiO2/RuO2) | Mesoporous | Cationic surfactant | Better stability results in electrooxidation | 308 |
| Pt/MxOya | Mesoporous | Different procedures | Alternative electrodes to the C based ones | 309 |
Concerning carbon based electrodes, a recent work published by Lazaro and co-workers shows the influence of different morphologies of carbon materials as electrocatalyst supports on the catalysts' performance in DAFCs.302 Platinum catalysts supported on carbon nanofibres (CNFs), carbon nanocoils (CNCs) and ordered mesoporous carbons (CMK-3) were evaluated in the alcohol oxidation reaction, and compared with a platinum catalyst supported on Vulcan XC-72R (by the same method), and with the commercial Pt/C catalyst from E-TEK. The support material's characteristics were shown to have an influence on both the physicochemical and electrochemical properties of electrocatalysts. The highest current densities, both in methanol and ethanol oxidations, were reached with the mesoporous Pt/CMK-3 catalyst. This could be rationalised by proposing that the high electrical conductivity and ordered porous structure of the support allow better electron transfer and diffusion of the reactants and by-products, during the electrochemical reactions. In the same vein, other very recent works also demonstrate the superior performances of mesoporous catalysts over other kinds of support.299a,303,304
Without doubt, the most employed and studied electrodes nowadays are the ones based on carbon, see Table 3. However, as has been previously mentioned, there is a key drawback associated with the use of carbon electrodes, their corrosion with use under high potentials. This can be overcome using non-carbon catalyst supports possessing the good properties of carbon (especially high surface area and high electrical conductivity), but also being corrosion-resistant. Some of the materials developed lately include porous metal and alloys,306 nitrides,307 different mixed oxides,308 as well as metal oxides.309 As an example, DiSalvo and co-workers have developed a methodology to prepare mesoporous nitrides of Ti, Cr and Ti/Nb by ammonolysis of different oxides of those metals.307 These nitrides showed excellent electronic conductivity and high surface area, performing very well as a result in methanol electrooxidation and the ORR in acid and alkaline media. Furthermore, they present in every case, a higher electrocatalytic stability than commercial Pt/C. Their high tolerance to corrosion makes them very promising candidates to replace carbon black as supports for fuel cell catalysts.
Finally, it is necessary to mention that, even with the great potential that FC systems show as clean energy conversion systems, there are some bottlenecks which inhibit FCs from finding a wide range of applications. Two of these bottlenecks are the poor efficiency of the oxygen reduction reaction (ORR) and the use of noble metals (mainly Pt) in the electrocatalysts, which makes the process unsustainable and/or expensive. With this in mind, Asefa and co-workers have recently published the synthesis of a metal-free electrocatalyst based on N- and O-doped mesoporous carbon for the ORR.305 The synthesis of this novel carbon catalyst was carried out using SBA-15 as a hard template, see the schematic representation of the synthetic procedure in Fig. 20. The aforementioned N- and O-doped mesoporous carbon, was produced in two broad steps. First, polymerised mesoporous silica-supported polyaniline (PANI) was carbonised in situ, before the mesoporous silica template was etched away. This synthetic method also permitted the immobilisation of non-noble metals, Fe and Co, into the system. All the resulting materials showed excellent electrocatalytic activity toward the ORR. However, the metal-free, PANI-derived mesoporous carbon exhibited the highest activity, challenging conventional paradigms. This unprecedented activity of the metal-free catalyst towards the ORR can be attributed to the synergetic activities of the nitrogen and oxygen (or hydroxyl) species that were implanted in it by PANI/mesoporous silica during pyrolysis. Other very recent reports also show the possibility of using metal free catalysts in the ORR which can definitively help reduce costs in FC, avoiding the use of expensive Pt.324
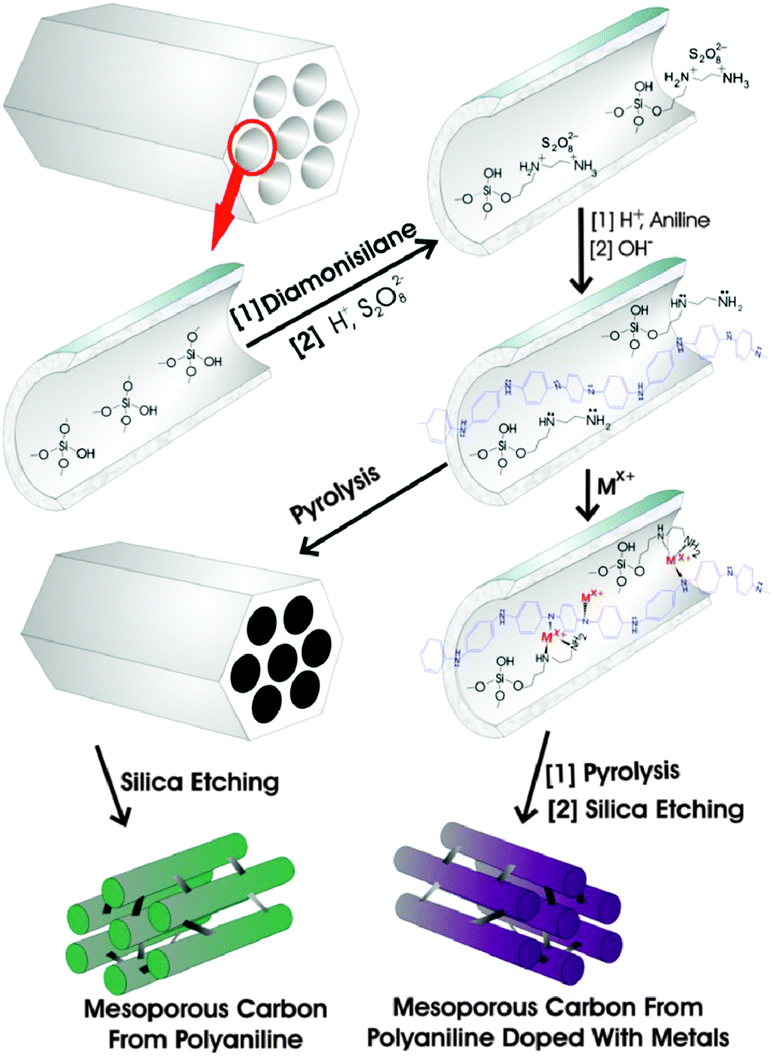 | ||
| Fig. 20 The synthetic procedure of N- and O-doped mesoporous carbons with or without metal dopants by the carbonization of polymerized mesoporous silica (SBA-15)-supported polyaniline, and the subsequent etching of the SBA-15. Reprinted with permission from ref. 305. Copyright 2013, American Chemical Society. | ||
To conclude, it is true that much work has been done over recent years on FC systems, indeed, all the references cited here were published only in 2013. However, while the results are encouraging, a lot remains to be done in order to provide materials solutions to the main deficiencies that FC technology still suffers from such as the high-cost (mainly due to the catalyst), and durability issues.
4.2. Mesoporous materials in advanced solar cells
A dye sensitised solar cell (DSSC) is a photoelectrochemical cell which generates electricity using solar power. It consists of three parts: the working electrode (WE) which contains the sensitising dye, the electrolyte, and the counter electrode (CE), see Fig. 21.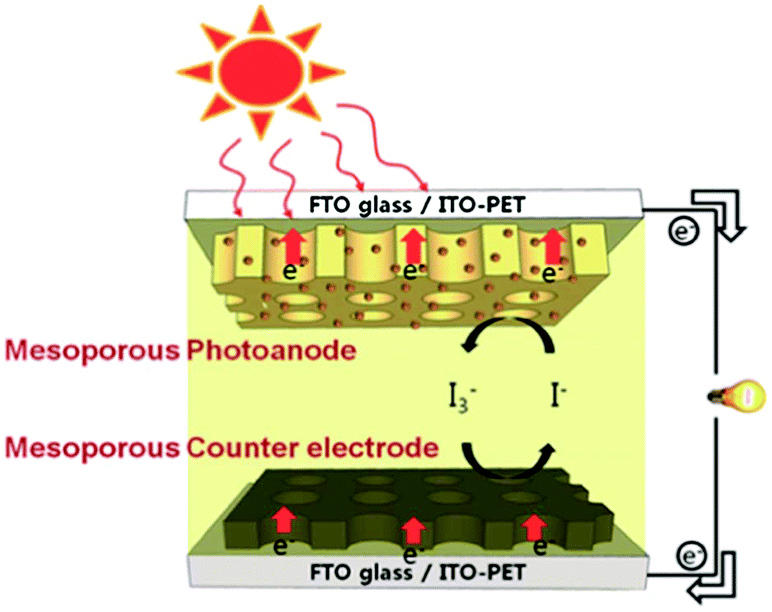 | ||
| Fig. 21 A schematic representation of a dye sensitized solar cell (DSSC) showing the mesoporous working electrode and counter electrode. Reproduced from ref. 7. Copyright 2013, RSC. | ||
Since O'Regan and Grätzel reported the first high-efficiency DSSC in 1991,325 this class of solar cells has emerged as a promising alternative to conventional silicon solar cells. A lot of effort has been made to increase the efficiency of the cell since that first report, reaching a top reported efficiency of 12.3% in 2011 for liquid DSC, again by the Grätzel group.326,327 DSSCs use wide band-gap semiconductors, while the sensitisation to the solar spectrum is performed by the dye. Different materials have been tested in DSCs including SnO2, rutile TiO2, ZnO, anatase TiO2, Nb2O5 and SrTiO3. As pointed out in Section 2.5, the most commonly used material, and the one offering the best performance, is anatase TiO2.328 Regarding the morphology of the electrodes, mesoporous nanostructures offer interesting advantages which can improve the characteristics of both the WE and CE. Next, some selected examples will be discussed to indicate those ideal features for mesoporous WE and CE; for a broader description, readers are kindly addressed to ref. 7, 34 and 248.
The overall conversion efficiency of the WE increases with the following factors: (1) a higher dye loading which will produce a higher light harvest, (2) an efficient transport of the injected electrons from the dye molecules to the external circuit in order to decrease the back reaction with oxidised species in the electrolyte and the holes in the dye, (3) a higher porosity to improve the binding of the dye molecules and the diffusion of the electrolyte and, (4) scattering functions in the WE, which can lengthen the light pathway and therefore improve light harvesting. All these factors can be enhanced using a mesoporous electrode as the WE.7,34,248,326,329 Herein, the two highest DSSC efficiencies reported using mesoporous hybrid titania as the WE, will be highlighted. Grätzel et al.326 reported mesoscopic solar cells prepared using a mesoporous hybrid titania as the WE, a zinc porphyrin dye used as a sensitiser and a Co(II/III)tris(bipyridyl) complex as the electrolyte. The WE was made by immersing a 10 μm mesoporous TiO2 film (5 μm thick transparent mesoporous layer + 5 μm thick scattering layer) in a solution of the dye used as the sensitiser. With this configuration, the efficiency values for these DSSC based on liquid junctions of 12.3% was achieved. More recently, the same group reported the fabrication of a solid-state mesoscopic perovskite-sensitised solar cell with an efficiency of 15% and a stability comparable to that of today's best thin-film solar cells. The cell used mesoporous anatase TiO2 as the WE.329
Similarly in the CE, different studies show better performances when using mesoporous carbon (MC), with a well-developed nanostructure and high surface area, as the CE than with graphite and activated carbon.330–335 The good performances of these inexpensive and abundant carbonaceous materials, added to their high corrosion resistance and good catalytic activity, make them very promising alternative materials for the CE. That is, replacements for the commonly used Pt catalyst, which is very active but scarce and expensive, inhibiting its large scale applications in the future.336
As examples, two works of Ramasamy and Lee are explained. They developed two different mesoporous carbon CEs using hard332 and soft333 templating approaches. An ordered mesoporous carbon (OMC) with a high surface area (∼1575 m2 g−1) and bimodal mesoporosity (2.5 and 6.1 nm) was synthesised using a triblock copolymer, namely F127, as a structure directing agent. The DSC prepared with this OMC based CE offered an energy conversion efficiency of 7.46%, which represents a 42% enhancement over a commercial Vulcan based CE. Electrochemical impedance spectroscopy analysis reveals decreased charge transfer resistance at the OMC counter electrode–electrolyte interface, and thus an improved fill factor and energy conversion efficiency.332 Simultaneously, the same group published a CE comprised of large-pore sized mesoporous carbon. The synthesis of this new large-pore MC involved the use of a mesocellular silica, MSU-F, as a hard template. Large-pore sized mesopores with an interconnected pore structure of sub-micron size MSU-F–C facilitated redox electrolyte penetration, and consequently most of the active surface area of the MSU-F–C carbon CE participates in the I3− reduction reaction. As a result, an 8.18% solar to electric energy conversion efficiency was obtained, a 21% increase in device performance compared with CMK-3 carbon (SBA-15 templated). Fig. 22 shows the characteristics of the MSU-F–C and the Nyquist plots displaying the similarity of its behaviour to Pt-based CEs.333
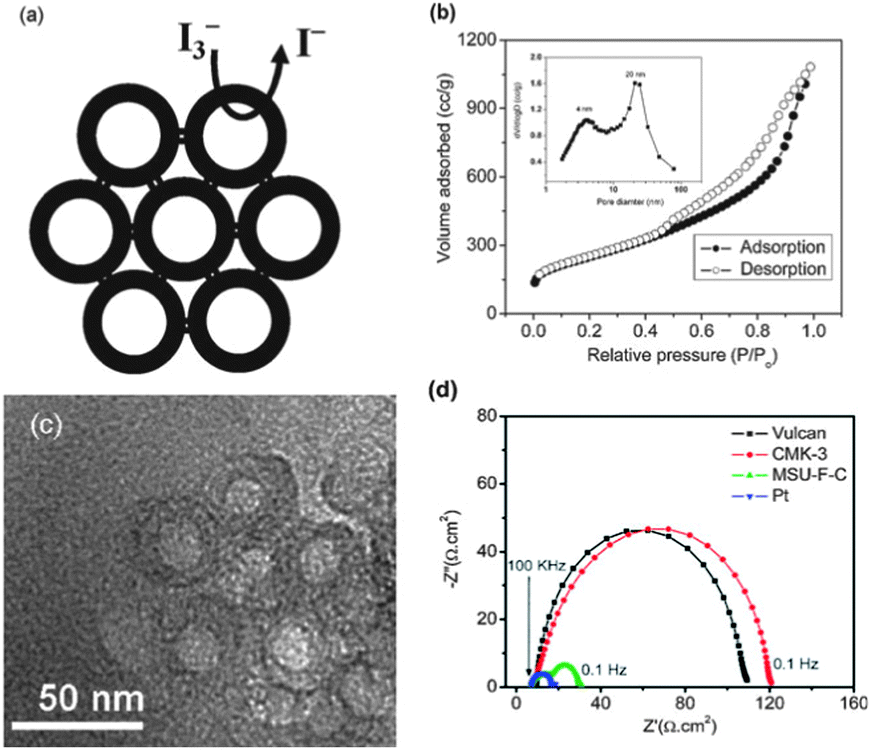 | ||
| Fig. 22 (a) A schematic illustration of the facile diffusion and reduction of I3− ions in MSU-F–C carbon. (b) The N2 isotherm and pore size distribution (inset) of MSU-F–C carbon. (c) High-resolution TEM image of microtomed MSU-F–C carbon. (d) Nyquist plots of various carbon electrodes and conventional Pt electrodes in a thin layer symmetric cell configuration. Reproduced from ref. 333. Copyright 2010, RSC. | ||
4.3. Mesoporous thermoelectric materials
The thermoelectric effect is a phenomenon involving a direct energy conversion from heat into electricity (or vice versa). Materials showing this effect, that is, capable of directly converting temperature gradients to electric voltage and vice versa, are considered thermoelectric (TE) materials. These materials offer the possibility of directly converting waste thermal energy back to electricity, as well as to cool atmosphere without using harmful chemicals like CFC and/or moving parts.337 A huge amount of waste heat is produced in both automotive and industry operations, and many of mankind's activities may be converted into useful energy using TE. As an illustration, in 2008 the US Department of Energy (DOE) reported that somewhere between 20 to 50% of industrial energy input in the US is lost as waste heat.338 Hence, capturing and reusing this lost heat would be a valuable approach to improve the overall energy efficiency. For this reason, TE materials have been receiving increasing attention in the last few years.339The energy-conversion efficiency of a TE material is evaluated by a dimensionless quantity called the figure of merit (zT), defined as zT = S2σT/κ; where S is the Seebeck coefficient (thermoelectric power, the change in voltage per unit temperature difference in a material), σ is the electrical conductivity, κ is the thermal conductivity, and T is the average temperature between the hot and the cold sides. A high electrical conductivity, a high Seebeck coefficient, and a low thermal conductivity are essential characteristics of thermoelectric materials. However, it is very difficult to control these factors individually, because the electrical conductivity and the Seebeck coefficient have an inverse relationship. Insulators, for example, tend to have very high Seebeck coefficients, while metals usually present low values in the order of 1–10 μV K−1. In this sense, semiconductors with Seebeck coefficients in the order of 102 to 103 μV K−1, are ideal thermoelectric devices. Heavily doped semiconductors were found to have a comparatively good zT. G. A. Slack340 proposed that in order to optimise the figure of merit, an ideal material should have a low lattice thermal conductivity as in a glass, and a high electrical conductivity as in a crystal, which is known as ‘electron crystal, phonon glass’ (ECPG). Consequently, thermoelectric materials comprise a huge family, including different materials: semimetals, semiconductors and ceramics; different crystalline forms: monocrystals, polycrystals, nanocomposites; and covering varying dimensions: from bulk to film, wire to cluster.337 Some polymers are also showing interesting thermoelectric material properties.341
In this sense, nanocrystals are expected to reduce thermal conductivity because of the higher presence of boundaries than in bulk materials, and mesopores are likely to further reduce the thermal conductivity via crystalline wall-pore interfaces. In addition, a continuous mesostructure nanocrystalline framework can maintain a high electrical conductivity.342 However, there are limited publications studying such porous TE materials and most of them are based on theoretical calculations.343
Regarding experimental studies, Park and co-workers are one of the groups that have been most active in analysing how mesoporosity and the synthetic parameters of mesoporous solids influence their thermoelectric properties. They have reported the synthesis of TiO2344–347 and ZnO348,349 mesoporous films using a triblock copolymer, Pluronic-123, and a neutral surfactant, Brij-76, as porogens, respectively. In the case of TiO2, the resistivity of mesoporous TiO2 films increases when the surfactant concentration increases because the porosity induces a reduction in the electrical path for carriers.347 This resistivity affects the Seebeck coefficient, which is higher for the higher concentration of surfactant. The group concludes that both porosity and pore arrangement could have an effect on the electrical conductivity of the mesoporous film. On further investigating the porosity of TiO2 films, at different surfactant/precursor ratios and different annealing temperatures, they consolidated their previous conclusions, as well as the idea that a disordered structure also has a positive effect on the thermoelectric property of the film.344 In the case of mesoporous ZnO films, the same trend was found, by increasing the porosity of the ZnO films, their conductivity ratio and Seebeck coefficients increased.348 Thus, the higher the porosity of the ZnO mesoporous films, the better their thermoelectric properties. However, even if these works show interesting results about the relationship between the porosity and the enhancement of the TE properties and the growing interest that the TE properties of transition metal oxides are lately attracting for their potential future prospects,350 the prototype TE materials in the low and high temperature ranges nowadays are Bi2Te3 and Si.342 In this regard, and following on from this study, two very recent and interesting works exploring the possibilities of mesoporous Bi2Te3 and Si are reviewed.
Stucky and co-workers reported in 2012 the first mesoporous monolith for TE applications.342 The monolith was prepared by a two-step procedure. First, mesoporous n-type Bi2Te3 powder was synthesised, using colloidal silica nanospheres (LUDOX) as a hard template. A mesoporous solid with 28 m2 g−1 of BET surface area, 0.17 cm3 g−1 of pore volume and an average pore size of 15 nm (consistent with the particle size of the silica nanospheres, 14 nm) was obtained. In the second step, the raw powder was hot-pressed into a mechanically stable monolith; most of the porosity in the final 3D monolith came from the synthetic mesopores of the initial Bi2Te3 material. The authors demonstrate that a continuous mesoporous nanocrystalline framework is highly effective for phonon scattering. The generated interfaces and boundaries which are a result of the mesoporosity, scatter phonons with a high efficiency leading to a substantially reduced thermal conductivity. When comparing with the control sample prepared without porosity, the thermal conductivities of the meso-Bi2Te3 are reduced by more than 50%. This huge reduction compensates for the loss of electrical conductivity caused by the mesopores and finally, the porous monolith presents a higher zT in the entire temperature range (300–500 K) then the fully condensed control sample. The maximum enhancement in the figure of merit (zT) is achieved in the temperature range from 400 to 500 K being over 45% higher in the porous monolith. As the authors finally conclude being ‘the first reported mesoporous monolith, the mesopores in the n-type Bi2Te3 monolith suggest a new viable avenue for the heterostructured synthesis of efficient TE materials’.
Similar results were obtained for Fang et al. when studying the thermal conductivity of ordered mesoporous nanocrystalline silicon thin films.351 In this case, two silica films were synthesised using two different diblock copolymers, namely, PB-PEO [poly(butadiene)-b-poly(ethylene oxide)] and PEP-PEO [poly(ethylene-propylene)-b-poly(ethylene oxide)]. Once the ordered mesoporous silica films are produced, they were subsequently reduced by magnesium vapor under an inert atmosphere, giving as a result a silicon film replicating the silica structure. Thermal conductivity measurements for the ordered mesoporous nanocrystalline silicon thin films from 25 to 315 K, resulted in a 3 to 5 orders of magnitude smaller thermal conductivity than that of the bulk single crystal silicon, depending on the temperature range examined. In addition, thin films templated by the PB-PEO copolymer had a smaller thermal conductivity than those templated by the PEP-PEO copolymer due to their smaller pores and increased disorder. Moreover, good agreement was found between the measured data and the model predictions.
Even if the literature regarding mesoporous materials for thermoelectric applications is scarce, the majority of the cases reported show an enhancement in the thermoelectric properties mostly coming from the reduction in thermal conductivities for the mesoporous solids regarding their dense counterparts. However, a reported model for polycrystallized SixGe1−x alloys with a nanosized (≈20 nm) interparticle porosity, shows a significant reduction in zT.352 Consequently, it seems evident that the porosity can have a positive or negative effect on zT depending on the material composition, the effective mass of the carriers, the pore size distributions and the type of pores (inter or intraparticulate). Therefore, a precise synthetic design is required if one wishes to improve zT by the incorporation of mesopores into a condensed matrix.
5. Conclusions
Recent advances in the synthesis of highly tuneable and complex porous structures have unlocked a wide range of opportunities in clean energy technologies, providing an improved accessibility, a higher surface area and a better dispersion of the active species. In addition to the development of controlled mesoporosity, the modification of the surface chemistry of porous materials using a wide range of techniques, from chemical grafting to the oxidation of the material surface has allowed further improvement in their performance when dealing with molecules of different polarity.New approaches to introducing mesoporosity, as well as revisited traditional techniques, have both been reviewed highlighting the possibility of combining different approaches to produce hierarchical structures with a controlled porosity at different scales. In this regard, a wide range of nanotechnologies from surfactant-templating to nanocasting have been successfully applied in the last decades, yielding novel materials with unprecedented control over their architecture.
In many cases, mesoporous materials are used as catalysts for the transformation of clean energy sources into electricity, fuels or valuable molecules. Herein we have provided some relevant examples of the role of controlled mesoporosity in increasing the activity, but mainly the selectivity of some of the most challenging processes, including the transformation of biomass to high quality fuels, the efficient production of hydrogen or the valorisation of CO2. However, mesoporous materials are widely used beyond catalysis, mainly as electrodes in fuel and solar cells and in thermoelectric devices. In these cases, the presence of mesoporosity significantly enhances the dispersion of the active phase and the accessibility of the electrolyte, which suggests that new advances in mesoporous materials will play a significant role in the clean production of energy in the future.
Acknowledgements
We specially thank Daniel Johnson for his invaluable help with the editing of this paper. The authors wish to thank the Spanish MINECO (Project CTQ2011-28954-C02-01) for financial support. E.S. acknowledges financial support from UA (Project GRE12-39).Notes and references
- (a) http://www.iea.org/publications/freepublications/publication/EnergyTechnologyInitiatives_2013.pdf, accessed on 15th November 2013; (b) http://www3.weforum.org/docs/WEF_EN_EnergyVision Report_2013.pdf, accessed on 15th November 2013; (c) http://www.un.org/wcm/webdav/site/sustainableenergyforall/shared/Documents/SEFA-Action%20Agenda-Final.pdf, accessed on 15th November 2013.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- E. Serrano, K. Li, G. Rus and J. García-Martínez, in Nanotechnology for the Energy Challenge, ed. J. García-Martínez, Wiley-VCH, Weinheim, Germany, 2nd edn, 2013 Search PubMed.
- E. Serrano, G. Rus and J. García-Martínez, Renewable Sustainable Energy Rev., 2009, 13, 2373–2384 CrossRef CAS PubMed.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663–3670 RSC.
- J. García-Martínez, in Tomorrow's Chemistry Today, ed. B. Pignataro, Wiley-VCH, Weinheim, Germany, 2008, ch. 3 Search PubMed.
- Y. Ye, C. Jo, I. Jeong and J. Lee, Nanoscale, 2013, 5, 4584–4605 RSC.
- E. Serrano, N. Linares, J. R. Berenguer and J. García-Martínez, ChemCatChem, 2013, 5, 844–860 CrossRef CAS PubMed.
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS; (b) C. T. Kresge, M. E. Leonowicz, W. J. Roth and J. C. Vartuli, Synthetic Mesoporous Crystalline Material, US Pat., 5,098,684, 1992 Search PubMed; (c) C. T. Kresge, M. E. Leonowicz, W. J. Roth and J. C. Vartuli, Synthetic Porous Crystalline Material, Its Synthesis, US Pat., 5,102,643, 1992 Search PubMed.
- N. Pal and A. Bhaumik, Adv. Colloid Interface Sci., 2013, 189–190, 21–41 CrossRef CAS PubMed.
- W. Li and D. Zhao, Chem. Commun., 2013, 49, 943–946 RSC.
- Introduction to Zeolite Science and Practice, Stud. Surf. Sci. and Catal., ed. J. Cějka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, Amsterdam, 2007, vol. 168 Search PubMed.
- R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, Chemistry of Zeolites and Related Porous Materials, Wiley, Singapore, 2007 Search PubMed.
- F. Di Renzo, A. Galarneau, P. Trens and F. Fajula, in Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, Wiley-VCH, Weinheim, 2002, pp. 1311–1395 Search PubMed.
- G. J. de, A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS PubMed.
- Mesoporous Materials [Themed collection], Chem. Soc. Rev., ed. B. Lebeau, A. Galarneau and M. Linden, 2013, vol. 42(9) Search PubMed.
- P. Behrens, Adv. Mater., 1993, 5, 127–132 CrossRef CAS PubMed.
- (a) Q. Huo, D. I. Margolese, U. Ciesla, D. K. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi, B. F. Chmelka, F. Schüth and G. D. Stucky, Chem. Mater., 1994, 6, 1176–1991 CrossRef CAS; (b) A. Sayari, Stud. Surf. Sci. Catal., 1996, 102, 1–46 CrossRef CAS; (c) N. D. Hoa, N. V. Duy and N. V. Hieu, Mater. Res. Bull., 2013, 48, 440–448 CrossRef CAS PubMed.
- J. Li, L. Delmotte and H. Kessler, Chem. Commun., 1996, 1023–1024 RSC.
- Q. Huo, R. Leon, P. M. Petroff and G. D. Stucky, Science, 1995, 268, 1324–1327 CAS.
- Y. Wang, C. Ma, X. Sun and H. Li, Microporous Mesoporous Mater., 2001, 49, 171–178 CrossRef CAS.
- S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801–805 CrossRef CAS PubMed.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS PubMed.
- N. Ulagappan, Neeraj, B. V. N. Raju and C. N. R. Rao, Chem. Commun., 1996, 2243–2244 RSC.
- K. G. Severin, T. M. Abdel-Fattah and T. J. Pinnavaia, Chem. Commun., 1998, 1471–1472 RSC.
- Neeraj and C. N. R. Rao, J. Mater. Chem., 1998, 8, 1631–1634 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- (a) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS PubMed; (b) C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe, J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 1128–1132 CrossRef CAS PubMed.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef PubMed.
- Z. Zhang, R. W. Hicks, T. R. Pauly and T. J. Pinnavaia, J. Am. Chem. Soc., 2002, 124, 1592–1593 CrossRef CAS PubMed.
- (a) P. Yang, T. Deng, D. Zhao, P. Feng, D. Pine, B. F. Chmelka, G. M. Whitesides and G. D. Stucky, Science, 1998, 282, 2244–2246 CrossRef CAS; (b) X. He and D. Antonelli, Angew. Chem., Int. Ed., 2002, 41, 214–229 CrossRef CAS; (c) S. Yuan, Z. Zho and G. Li, CrystEngComm, 2011, 13, 4709–4713 RSC.
- D. Chandra, N. Mukherjee, A. Mondal and A. Bhaumik, J. Phys. Chem. C, 2008, 112, 8668–8674 CAS.
- J. L. Vivero-Escoto, Y.-D. Chiang, K. C.-W. Wu and Y. Yamauchi, Sci. Technol. Adv. Mater., 2012, 13, 013003 CrossRef.
- K. Nakajima, T. Fukui, H. Kato, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, Chem. Mater., 2010, 22, 3332 CrossRef CAS.
- K. Brezesinski, J. Haetge, J. Wang, S. Mascotto, C. Reitz, A. Rein, S. H. Tolbert, J. Perlich, B. Dunn and T. Brezesinski, Small, 2011, 7, 407–414 CrossRef CAS PubMed.
- B. L. Su, C. Sanchez and X.-Y. Yang, Hierarchically structured porous materials: from nanoscience to catalysis, biomedicine, optics and energy, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- J. Cějka, A. Corma and S. I. Zones, Zeolites and Catalysis: Synthesis, Reactions and Applications, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- (a) C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC; (b) K. Na, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2013, 166, 3–19 CrossRef CAS PubMed; (c) L.-H. Chen, X.-Y. Li, J. C. Rooke, Y.-H. Zhang, X.-Y. Yang, Y. Tang, F.-S. Xiao and B.-L. Su, J. Mater. Chem., 2012, 22, 17381–17403 RSC; (d) S. Lopez-Orozco, A. Inayat, A. Schwab, T. Selvam and W. Schwieger, Adv. Mater., 2011, 23, 2602–2615 CrossRef CAS PubMed; (e) J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC; (f) K. Egeblad, C. H. Christensen, M. Kustova and C. H. Christensen, Chem. Mater., 2008, 20, 946–960 CrossRef CAS.
- (a) K. P. de Jong, J. Zecevic, H. Friedrich, P. E. de Jongh, M. Bulut, S. van Donk, R. Kenmogen, A. Finiels, V. Hulea and F. Fajula, Angew. Chem., Int. Ed., 2010, 49, 10074–10078 CrossRef CAS PubMed; (b) J. Pérez-Ramírez, D. Verboekend, A. Bonilla and S. Abelló, Adv. Funct. Mater., 2009, 19, 3972–3979 CrossRef PubMed; (c) J. C. Groen, T. Bach, U. Ziese, A. M. Paulaime-van Donk, K. P. de Jong, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2005, 127, 10792–10793 CrossRef CAS PubMed; (d) J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérez-Ramírez, Chem. - Eur. J., 2005, 11, 4983–4994 CrossRef CAS PubMed; (e) M. Ogura, S. Y. Shinomiya, J. Tateno, Y. Nara, E. Kikuchi and H. Matsukata, Chem. Lett., 2000, 882–883 CrossRef CAS.
- (a) C.-Y. Chen and S. I. Zones, in Zeolites and Catalysis: Synthesis, Reactions and Application, ed. J. Cějka, A. Corma and S. I. Zones, Wiley-VCH, Weinheim, Germany, 2010, pp. 155–170 Search PubMed; (b) S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev. Sci. Eng., 2003, 45, 297–319 CrossRef CAS PubMed; (c) C. S. Triantafillidis, A. G. Vlessidis and N. P. Evmiridis, Ind. Eng. Chem. Res., 2000, 39, 307–319 CrossRef CAS.
- (a) B. Liu, C. Li, Y. Ren, Y. Tan, H. Xi and Y. Qian, Chem. Eng. J., 2012, 210, 96–102 CrossRef CAS PubMed; (b) K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS PubMed; (c) F.-S. Xiao, L. Wang, C. Yin, Y. Di, J. Li, R. Xu, D. S. Su, R. Schlogl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef CAS PubMed; (d) H. Wang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2006, 45, 7603–7606 CrossRef CAS PubMed; (e) M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CAS.
- D. H. Park, S. S. Kim, H. Wang, T. J. Pinnavaia, M. C. Papapetrou, A. A. Lappas and K. S. Triantafyllidis, Angew. Chem., Int. Ed., 2009, 48, 7645–7648 CrossRef CAS PubMed.
- (a) J.-B. Koo, N. Jiang, S. Saravanamurugan, M. Bejblová, Z. Musilová, J. Cějka and S.-E. Park, J. Catal., 2010, 276, 327–334 CrossRef CAS PubMed; (b) W. Fan, M. A. Snyder, S. Kumar, P.-S. Lee, W. C. Yoo, A. V. McCormick, R. L. Penn, A. Stein and M. Tsapatsis, Nat. Mater., 2008, 7, 984–991 CrossRef CAS PubMed; (c) C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116–7117 CrossRef CAS; (d) H. Zhu, Z. Liu, Y. Wang, D. Kong, X. Yuan and Z. Xie, Chem. Mater., 2008, 20, 1134–1139 CrossRef CAS; (e) C. H. Christensen, I. Schmidt, A. Carlsson, K. Johannsen and K. Herbst, J. Am. Chem. Soc., 2005, 127, 8098–8102 CrossRef CAS PubMed; (f) Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- J. Y. Ying and J. García-Martínez, US Pat., US 2005/0239634 A1, 2004 Search PubMed.
- J. García-Martínez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987–994 Search PubMed.
- J. García-Martínez, K. Li and G. Krishnaiah, Chem. Commun., 2012, 48, 11841–11843 RSC.
- K. Li, J. Valla and J. García-Martínez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS PubMed.
- S. Xiu and A. Shahbazi, Renewable Sustainable Energy Rev., 2012, 16, 4406–4414 CrossRef CAS PubMed.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS PubMed.
- K. Jacobson, K. C. Maheria and A. K. Dalai, Renewable Sustainable Energy Rev., 2013, 23, 91–106 CrossRef CAS PubMed.
- Y. Wang, Y. Fang, T. He, H. Hu and J. Wu, Catal. Commun., 2011, 12, 1201–1205 CrossRef CAS PubMed.
- For a review of synthetic procedures see: X.-Y. Yang, Y. Li, A. Lemaire, J.-G. Yu and B.-L. Su, Pure Appl. Chem., 2009, 81, 2265–2307 CrossRef CAS.
- T. Kamegawa, N. Suzuki, M. Che and H. Yamashita, Langmuir, 2011, 27, 2873–2879 CrossRef CAS PubMed.
- N. Linares, S. Hartmann, A. Galarneau and P. Barbaro, ACS Catal., 2012, 2, 2194–2198 CrossRef CAS.
- H. Yang, Z. Liu, H. Gao and Z. Xie, J. Mater. Chem., 2010, 20, 3227–3231 RSC.
- (a) F. Dong, S. C. Lee, Z. Wu, Y. Huang, M. Fu, W.-K. Ho, S. Zou and B. Wang, J. Hazard. Mater., 2011, 195, 346–354 CrossRef CAS PubMed; (b) F. Dong, Y. Sun, W. K. Ho and Z. Wu, Dalton Trans., 2012, 41, 8270–8284 RSC.
- P.-C. Chen, M.-C. Tsai, M.-H. Yang, T.-T. Chen, H.-C. Chen, I.-C. Changa, Y.-C. Chang, Y.-L. Chen, I.-N. Lin, H.-T. Chiu and C.-Y. Lee, Appl. Catal., B, 2013, 142–143, 752–760 CrossRef CAS PubMed.
- D. Xie, L. Chang, F. Wang, G. Du and B. Xu, J. Alloys Compd., 2012, 545, 176–181 CrossRef CAS PubMed.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS PubMed.
- J. Xiao, J. Zheng, X. Li, Y. Shao and J.-G. Zhang, Nanotechnology, 2013, 24, 424004 CrossRef PubMed.
- D. Xie, W. Yuan, Z. Dong, Q. Su, J. Zhang and G. Du, Electrochim. Acta, 2013, 92, 87–92 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- (a) X. Li and B. Wei, Nano Energy, 2013, 2, 159–173 CrossRef CAS PubMed; (b) Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098–4140 RSC.
- A.-H. Lu, D. Zhao and Y. Wan, Nanocasting: A Versatile Strategy for Creating Nanostructured Porous Materials, RSC Publishing, Cambridge, UK, 2009 Search PubMed.
- (a) Y.-S. Hu, P. Adelhelm, B. M. Smarsly, S. Hore, M. Antonietti and J. Maier, Adv. Funct. Mater., 2007, 17, 1873–1878 CrossRef CAS PubMed; (b) L. Yu, N. Brun, K. Sakaushi, J. Eckert and M. M. Titirici, Carbon, 2013, 61, 245–253 CrossRef CAS PubMed; (c) S. Wang, Z. Yang, H. Zhang, H. Tan, J. Yu and J. Wu, Electrochim. Acta, 2013, 106, 307–311 CrossRef CAS PubMed.
- Z. Wang, F. Li, N. S. Ergang and A. Stein, Chem. Mater., 2006, 18, 5543–5553 CrossRef CAS.
- (a) N. Du, H. Zhang, B. D. Chen, J. B. Wu, X. Y. Ma, Z. H. Liu, Y. Q. Zhang, D. Yang, X. H. Huang and J. P. Tu, Adv. Mater., 2007, 19, 4505–4509 CrossRef CAS PubMed; (b) X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS PubMed; (c) X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, J. Mater. Chem., 2008, 18, 4397–4401 RSC.
- L. Tian, H. L. Zou, J. X. Fu, X. F. Yang, Y. Wang, H. L. Guo, X. H. Fu, C. L. Liang, M. M. Wu, P. K. Shen and M. Q. Gao, Adv. Funct. Mater., 2010, 20, 617–623 CrossRef CAS PubMed.
- G. Wang, H. Liu, J. Horvat, B. Wang, S. Qiao, J. Park and H. Ahn, Chem. - Eur. J., 2010, 16, 11020–11027 CrossRef CAS PubMed.
- C. F. Meunier, X. Y. Yang, J. C. Rooke and B. L. Su, ChemCatChem, 2011, 3, 476–488 CrossRef CAS PubMed.
- O. H. Schmitt, Some interesting and useful biomimetic transforms, Proc. 3rd Int. Biophysics Congress, 1969, p. 297.
- Compendium of Chemical Terminology, 2nd ed; (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford (1997), XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins, ISBN 0967855098, DOI: 10.1351/goldbook, accessed on October 2013.
- H. Zhou, X. Li, T. Fan, F. E. Osterloh, J. Ding, E. M. Sabio, D. Zhang and Q. Guo, Adv. Mater., 2010, 22, 951–956 CrossRef CAS PubMed.
- W. Zhang, D. Zhang, T. J. Fan, J. J. Gu, J. Ding, H. Wang, Q. X. Guo and H. Ogawa, Chem. Mater., 2009, 21, 33–40 CrossRef.
- X. F. Li, T. X. Fan, H. Zhou, S. K. Chow, W. Zhang, D. Zhang, Q. X. Guo and H. Ogawa, Adv. Funct. Mater., 2009, 19, 45–56 CrossRef CAS PubMed.
- Z. Schnepp, W. Yang, M. Antonietti and C. Giordano, Angew. Chem., Int. Ed., 2010, 49, 6564–6566 CrossRef CAS PubMed.
- D. Losic, P. J. Evans, A. Atanacio, J. G. Mitchell and N. H. Voelcker, J. Mater. Chem., 2006, 16, 4029–4034 RSC.
- (a) C. Jeffryes, T. Gutu, J. Jiao and G. L. Rorrer, ACS Nano, 2008, 2, 2103–2112 CrossRef CAS PubMed; (b) C. Jeffryes, T. Gutu, J. Jiao and G. L. Rorrer, J. Mater. Res., 2008, 23, 3255–3262 CrossRef CAS.
- Z. Liu, T. Fan and D. Zhang, J. Am. Ceram. Soc., 2006, 89, 662–665 CrossRef CAS.
- Z. Liu, T. Fan, W. Zhang and D. Zhang, Microporous Mesoporous Mater., 2005, 85, 82–88 CrossRef CAS PubMed.
- X. Li, T. Fan, Z. Liu, J. Ding, Q. Guo and D. Zhang, J. Eur. Ceram. Soc., 2006, 26, 3657–3664 CrossRef CAS PubMed.
- T. Fan, X. Li, Z. Liu, J. Gu, D. Zhang and Q. Guo, J. Am. Ceram. Soc., 2006, 89, 3511–3515 CrossRef CAS.
- J. Cao and H. Sieber, J. Porous Mater., 2004, 11, 163–172 CrossRef CAS.
- S. M. Holmes, B. E. Graniel-Garcia, P. Foran, P. Hill, E. P. L. Roberts, B. H. Sakakini and J. M. Newton, Chem. Commun., 2006, 2662–2663 RSC.
- W. He, X. Zhang, X. Du, Y. Zhang, Y. Yue, J. Shen and M. Li, Electrochim. Acta, 2013, 112, 295–303 CrossRef CAS PubMed.
- Z.-Y. Gu, J. Park, A. Raiff, Z. Wei and H.-C. Zhou, ChemCatChem, 2014, 6, 67–75 CrossRef CAS PubMed.
- I. Imaz, M. Rubio-Martínez, J. An, I. Solé-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC.
- S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS PubMed.
- K. L. Fujdala and T. D. Tilley, J. Catal., 2003, 216, 265–275 CrossRef CAS.
- (a) M. Rico-Santacruz, A. E. Sepúlveda, E. Serrano, J. R. Berenguer, E. Lalinde and J. García-Martínez, Spanish Pat., ref. 20130535, 2013 Search PubMed; (b) M. Rico-Santacruz, A. E. Sepúlveda, E. Serrano, J. R. Berenguer, E. Lalinde, J. García-Martínez, unpublished results.
- (a) E. Coronado, A. Ribera, J. García-Martínez, N. Linares and L. M. Liz-Marzán, J. Mater. Chem., 2008, 18, 5682–5688 RSC; (b) J. García-Martínez, N. Linares, S. Sinibaldi, E. Coronado and A. Ribera, Microporous Mesoporous Mater., 2009, 117, 170–177 CrossRef PubMed; (c) N. Linares, A. E. Sepulveda, M. C. Pacheco, J. Berenguer, E. Lalinde, C. Nájera and J. García-Martínez, New J. Chem., 2011, 35, 225–234 RSC; (d) N. Linares, A. E. Sepulveda, J. Berenguer, E. Lalinde and J. García-Martínez, Microporous Mesoporous Mater., 2012, 158, 300–308 CrossRef CAS PubMed; (e) A. I. Carrillo, J. García-Martínez, R. Llusar, E. Serrano, I. Sorribes, C. Vicente and J. A. Vidal-Moya, Microporous Mesoporous Mater., 2012, 151, 380–389 CrossRef CAS PubMed; (f) M. Rico, A. E. Sepulveda, E. Serrano, S. Ruiz, J. García-Martínez, J. R. Berenguer and E. Lalinde, Chem. Commun., 2012, 48, 8883–8885 RSC; (g) N. Linares, E. Serrano, A. Carrillo and J. García-Martínez, Mater. Lett., 2013, 95, 93–96 CrossRef CAS PubMed.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- (a) Q. R. Fang, T. A. Makal, M. D. Young and H. C. Zhou, Comments Inorg. Chem., 2010, 31, 165–195 CrossRef CAS; (b) Metal-organic frameworks: design and applications, ed. L. R. MacGillivray, Wiley-VCH, 2010, ISBN 978-0-470-19556-7 Search PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- X. S. Wang, S. Q. Ma, D. F. Sun, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 16474–16475 CrossRef CAS PubMed.
- Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638–6642 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677–680 CrossRef CAS PubMed.
- G. Ferey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef CAS PubMed.
- (a) G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed; (b) A. Sonnauer, F. Hoffmann, M. Froba, L. Kienle, V. Duppel, M. Thommes, C. Serre, G. Ferey and N. Stock, Angew. Chem., Int. Ed., 2009, 48, 3791–3794 CrossRef CAS PubMed.
- (a) J. Juan-Alcaniz, M. G. Goesten, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, New J. Chem., 2012, 36, 977–987 RSC; (b) M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056–6058 RSC.
- Y. K. Park, S. B. Choi, H. Kim, K. Kim, B. H. Won, K. Choi, J. S. Choi, W. S. Ahn, N. Won, S. Kim, D. H. Jung, S. H. Choi, G. H. Kim, S. S. Cha, Y. H. Jhon, J. K. Yang and J. Kim, Angew. Chem., Int. Ed., 2007, 46, 8230–8233 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 4184–4185 CrossRef CAS PubMed.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- L.-G. Qiu, T. Xu, Z.-Q. Li, W. Wang, Y. Wu, X. Jiang, X.-Y. Tian and L.-D. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9487–9491 CrossRef CAS PubMed.
- D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186–9188 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydın, R. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- O. K. Farha, A. O. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- Mercedes-Benz F125, www.emercedesbenz.com/autos/mercedes-benz/concept-vehicles/mercedes-benz-f125-research-vehicle-technology, accessed on 20th October 2013.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS PubMed.
- A. G. Gayudo, A. Alonso, B. Valle, A. T. Aguayo, M. Olazar and J. Bilbao, Chem. Eng. J., 2011, 167, 262–277 CrossRef PubMed.
- M. Inaba, K. Murata, M. Saito and I. Takahara, Green Chem., 2007, 9, 638–646 RSC.
- H. Oikawa, Y. Shibata and T. Baba, Appl. Catal., A, 2006, 312, 181–185 CrossRef CAS PubMed.
- M. Iwamoto, K. Kasai and T. Haishi, ChemSusChem, 2011, 4, 1055–1058 CrossRef CAS PubMed.
- A. M. Varvarin, K. N. Khomenko and V. V. Brei, Theor. Exp. Chem., 2011, 47, 36–40 CrossRef CAS.
- P. D. Vaidya and A. E. Rodrigues, Chem. Eng. J., 2006, 117, 39–49 CrossRef CAS PubMed.
- S. Sá, H. Silva, L. Brandao, J. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43–57 CrossRef PubMed.
- L. P. R. Profeti, E. A. Ticianelli and E. M. Assaf, Appl. Catal., A, 2009, 360, 17–25 CrossRef CAS PubMed.
- C. B. Wang, C. C. Lee, J. L. Bi, J. Y. Siang, J. Y. Liu and C. T. Yeh, Catal. Today, 2009, 146, 76–81 CrossRef CAS PubMed.
- W. Gac, W. Zawadzki and B. Tomaszewska, Catal. Today, 2011, 176, 97–102 CrossRef CAS PubMed.
- A. Carrero, J. A. Calles and A. J. Vizcaíno, Appl. Catal., A, 2007, 327, 82–94 CrossRef CAS PubMed.
- A. J. Vizcaíno, A. Carrero and J. A. Calles, Catal. Today, 2009, 146, 63–70 CrossRef PubMed.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS PubMed.
- S. Yan, M. Kim, S. O. Salley and K. Y. Simon Ng, Appl. Catal., A, 2009, 360, 163–170 CrossRef CAS PubMed.
- D. E. López, J. G. Goodwin Jr., D. A. Bruce and E. Lotero, Appl. Catal., A, 2005, 295, 97–105 CrossRef PubMed.
- G. J. Suppes, M. A. Dasari, E. J. Doskocil, P. J. Mankidy and M. J. Goff, Appl. Catal., A, 2004, 257, 213–223 CrossRef CAS PubMed.
- M. C. G. Albuquerque, I. Jiménez-Urbistondo, J. Santamaría-González, J. M. Mérida-Robles, R. Moreno-Tost, E. Rodríguez-Castellón, A. Jiménez-López, D. C. S. Azevedo, C. L. Cavalcante Jr. and P. Maireles-Torres, Appl. Catal., A, 2008, 334, 35–43 CrossRef CAS PubMed.
- E. Li and V. Rudolph, Energy Fuels, 2008, 22, 145–149 CrossRef CAS.
- A. Z. Abdullah, N. Razali and K. T. Lee, Fuel Process. Technol., 2009, 90, 958–964 CrossRef CAS PubMed.
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1998, 173, 315–321 CrossRef CAS.
- R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Appl. Catal., B, 2008, 82, 157–162 CrossRef CAS PubMed.
- J. Delgado, SP Pat., 2,201,894, 2002 Search PubMed.
- R. Wessendorf, Erdoel Kohle, Erdgas, Petrochem., 1995, 48, 138–143 CAS.
- J. A. Melero, R. van Grieken, G. Morales and M. Paniagua, Energy Fuels, 2007, 21, 1782–1791 CrossRef CAS.
- P. McMorn, G. Roberts and G. J. Hutchings, Catal. Lett., 1999, 63, 193–197 CrossRef CAS.
- E. G. Rodrigues, M. F. R. Pereira and J. J. M. Orfao, Appl. Catal., B, 2012, 115–116, 1–6 CrossRef CAS PubMed.
- S.-Y. Liu, C.-J. Zhou, Q. Liu, G.-C. Liu, C.-J. Huang and Z.-S. Chao, ChemSusChem, 2008, 1, 575–578 CrossRef PubMed.
- H. Kimura, K. Tsuto, T. Wakisaka, Y. Kazumi and Y. Inaya, Appl. Catal., A, 1993, 96, 217–228 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- N. Dimitratos, A. Villa, C. L. Bianchi, L. Prati and M. Makkee, Appl. Catal., A, 2006, 311, 185–192 CrossRef CAS PubMed.
- S. Demirel-Gülen, M. Lucas and P. Claus, Catal. Today, 2005, 102–103, 166–172 CrossRef PubMed.
- W. C. Ketchie, Y.-L. Fang, M. S. Wong, M. Murayana and R. J. Davis, J. Catal., 2007, 250, 94–101 CrossRef CAS PubMed.
- A. Villa, A. Gaiassi, I. Rossetti, C. L. Bianchi, L. Prati, K. van Benthem, G. M. Veith and L. Prati, J. Catal., 2010, 275, 108–116 CrossRef CAS PubMed.
- E. G. Rodrigues, M. F. R. Pereira, J. J. Delgado, X. Chen and J. J. M. Orfao, Catal. Commun., 2011, 16, 64–69 CrossRef CAS PubMed.
- M. Gharabi, F. T. Zangeneh, F. Yaripour and S. Sahebdelfar, Appl. Catal., A, 2012, 443–444, 8–26 CrossRef PubMed.
- (a) L. Su, L. Liu, J. Zhuang, H. Wang, Y. Li, W. Shen, Y. Xu and X. Bao, Catal. Lett., 2003, 91, 155–167 CrossRef CAS; (b) N. Chu, J. Yang, C. Li, J. Cui, Q. Zhao, X. Yin, J. Lu and J. Wang, Microporous Mesoporous Mater., 2009, 118, 169–175 CrossRef CAS PubMed.
- A. Martinez, E. Peris, M. Derewinski and A. Burkat-Dulak, Catal. Today, 2011, 169, 75–84 CrossRef CAS PubMed.
- N. B. Chu, J. Q. Wang, Y. Zhang, J. H. Yang, J. M. Lu and D. H. Yin, Chem. Mater., 2010, 22, 2757–2763 CrossRef CAS.
- Y. Wang, X. Wang, Z. Su, Q. Guo, Q. Tang, Q. Zhang and H. Wan, Catal. Today, 2004, 93–95, 155–161 CrossRef CAS PubMed.
- (a) J. He, Y. Li, D. An, Q. Zhang and Y. Wang, J. Nat. Gas Chem., 2009, 18, 288–294 CrossRef CAS; (b) Y. Li, D. An, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2008, 112, 13700–13708 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, J. Nat. Gas Chem., 2008, 17, 278–282 CrossRef CAS.
- (a) J. Kang, K. Cheng, L. Zhang, Q. Zhang, J. Ding, W. Hua, Y. Lou, Q. Zhai and Y. Wang, Angew. Chem., 2011, 123, 5306–5309 CrossRef PubMed; (b) S. Sartipi, K. Parashar, M. Makkee, J. Gascon and F. Kapteijn, Catal. Sci. Technol., 2013, 3, 572–575 RSC; (c) S. Sarpiti, M. Alberts, M. J. Meijerink, T. C. Keller, J. Pérez-Ramirez, J. Gascon and F. Kapteijn, ChemSusChem, 2013, 1646–1650 Search PubMed; (d) S. Sartini, M. Alberts, V. P. Santos, M. Nasalevich, J. Gascon and F. Kapteijn, ChemCatChem, 2014, 6, 142–151 CrossRef PubMed.
- (a) M. W. E. van den Berg, S. Polarz, O. P. Tkachenko, K. V. Klementiev, M. Bandyopadhyay, L. Khodeir, H. Gies, M. Muhler and W. Grünert, J. Catal., 2006, 241, 446–455 CrossRef CAS PubMed; (b) M. W. E. van den Berg, S. Polarz, O. P. Tkachenko, K. Kahler, M. Muhler and W. Grünert, Catal. Lett., 2009, 128, 49–56 CrossRef.
- (a) Q. Tang, H. Xu, Y. Zheng, J. Wang, H. Li and J. Zhang, Appl. Catal., A, 2012, 413–414, 36–42 CrossRef CAS PubMed; (b) K. Cho, H. S. Cho, L. C. de Menorval and R. Ryoo, Chem. Mater., 2009, 21, 5664–5673 CrossRef CAS.
- (a) M. Sugimoto, H. Katsuno, K. Takatsu and N. Kawata, Zeolites, 1987, 7, 503–507 CrossRef CAS; (b) M. Bjorgen, F. Joensen, M. S. Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef PubMed; (c) J. Kim, M. Choi and R. Ryoo, J. Catal., 2010, 269, 219–228 CrossRef CAS PubMed; (d) A. A. Rownaghi, F. Rezaei and J. Hedlund, Microporous Mesoporous Mater., 2012, 151, 26–33 CrossRef CAS PubMed; (e) P. N. R. Vennestrom, M. Grill, M. Kustova, K. Egeblad, L. F. Lundegaard, F. Joensen, C. H. Christensen and P. Beato, Catal. Today, 2011, 168, 71–79 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- J. Tollefson, Nature, 2010, 464, 1262–1264 CrossRef CAS PubMed.
- H. F. Abbas and W. M. A. W. Daud, Int. J. Hydrogen Energy, 2010, 35, 1160–1190 CrossRef CAS PubMed.
- D. P. Serrano, J. M. Coronado, V. A. de la Peña O'Shea, P. Pizarro and J. A. Botas, J. Mater. Chem. A, 2013, 1, 12016–12027 CAS.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- Hydrogen and Syngas, Production and Purifcation Technologies, ed. K. Lui, C. Song and V. Subramani, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- J. R. R. Nielsen, J. Catal., 1973, 31, 173–199 CrossRef.
- G. Jones, J. G. Jakobsen, S. S. Shim, J. Kleis, M. P. Andersson, J. Rossmeisl, F. A. Pedersen, T. Bligaard, S. Helveg, B. Hinnemann, J. R. R-Nielsne, I. Chorkendorff, J. Sehested and J. N. Norskov, J. Catal., 2008, 259, 147–160 CrossRef CAS PubMed.
- J. H. Kim, D. J. Suh, T. J. Park and K. L. Kim, Appl. Catal., A, 2000, 197, 191–200 CrossRef CAS.
- G. Li, L. Hu and J. M. Hill, Appl. Catal., A, 2006, 301, 16–24 CrossRef CAS PubMed.
- J. G. Seo, M. H. Youn, S. Park, J. C. Jung, P. Kim, J. S. Chung and I. K. Song, J. Power Sources, 2009, 186, 178–184 CrossRef CAS PubMed.
- Y. Bang, J. Lee, S. J. Han, J. G. Seo, M. H. Youn, J. H. Song and I. K. Song, Int. J. Hydrogen Energy, 2012, 37, 11208–11217 CrossRef CAS PubMed.
- Y. Bang, S. J. Han, J. G. Seo, M. H. Youn, J. H. Song and I. K. Song, Int. J. Hydrogen Energy, 2012, 37, 17967–17977 CrossRef CAS PubMed.
- J. Yoo, Y. Bang, S. J. Han, T. H. Kang, J. Lee and I. K. Song, J. Mol. Catal. A: Chem., 2013, 380, 28–33 CrossRef CAS PubMed.
- N. Wang, W. Chu, T. Zhang and X. S. Zhao, Int. J. Hydrogen Energy, 2012, 37, 19–30 CrossRef CAS PubMed.
- M. Lindo, A. J. Vizcaíno, J. A. Calles and A. Carrero, Int. J. Hydrogen Energy, 2010, 35, 5895–5901 CrossRef CAS PubMed.
- S. J. Han, Y. Bang, J. Yoo, J. G. Seo and I. K. Song, Int. J. Hydrogen Energy, 2013, 38, 8285–8292 CrossRef CAS PubMed.
- S. J. Han, Y. Bang, J. Yoo, K. H. Kang, J. H. Song, J. G. Seo and I. K. Song, Int. J. Hydrogen Energy, 2013, 38, 15119–15127 CrossRef CAS PubMed.
- Y. Bang, S. J. Han, J. Yoo, J. H. Choi, K. H. Kang, J. H. Song, J. G. Seo, J. C. Jung and I. K. Song, Int. J. Hydrogen Energy, 2013, 38, 8751–8758 CrossRef CAS PubMed.
- J. F. C. Serra, M. T. Navarro, F. Rey and A. Chica, Int. J. Hydrogen Energy, 2012, 37, 7101–7108 CrossRef PubMed.
- J. S. Lee, G. B. Han and M. Kang, Energy, 2012, 44, 248–256 CrossRef CAS PubMed.
- H. D. Kim, T. W. Kim, H. J. Park, K. E. Jeong, H. J. Chae, S. Y. Jeong, C. H. Lee and C. U. Kim, Int. J. Hydrogen Energy, 2012, 37, 12187–12197 CrossRef CAS PubMed.
- J. A. Botas, D. P. Serrano, R. G. López, P. Pizarro and G. Gómez, Int. J. Hydrogen Energy, 2010, 35, 9788–9794 CrossRef CAS PubMed.
- D. P. Serrano, J. A. Botas, P. Pizarro and G. Gómez, Int. J. Hydrogen Energy, 2013, 38, 5671–5683 CrossRef CAS PubMed.
- L. Jin, H. Si, J. Zhang, P. Lin, Z. Hu, B. Qiu and H. Hu, Int. J. Hydrogen Energy, 2013, 38, 10373–10380 CrossRef CAS PubMed.
- H. Tan, K. Li, S. Sioud, D. Cha, M. H. Amad, M. N. Hedhili and Z. A. A. Talla, Catal. Commun., 2012, 26, 248–252 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- M. D. H. Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 Search PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- R. M. N. Yerga, M. C. A. Galván, F. Vaquero, J. Arenales and J. L. G. Fierro, Renewable Hydrogen Technol.: Prod., Purif., Storage, Appl. Saf., 2013, 43–61 CAS.
- H. Yang, L. Guo, W. Yan and H. Liu, J. Power Sources, 2006, 159, 1305–1309 CrossRef CAS PubMed.
- D. Jing and L. Guo, Catal. Commun., 2007, 8, 795–799 CrossRef CAS PubMed.
- S. Onsuratoom, S. Chavadej and T. Sreethawong, Int. J. Hydrogen Energy, 2011, 36, 5246–5261 CrossRef CAS PubMed.
- H. Dang, X. Dong, Y. Dong, Y. Zhang and S. Hampshire, Int. J. Hydrogen Energy, 2013, 38, 2126–2135 CrossRef CAS PubMed.
- (a) S. H. Liu and H. P. Wang, Int. J. Hydrogen Energy, 2002, 27, 859–862 CrossRef CAS; (b) D. Zhao, A. Rodriguez, N. M. Dimitrijevic, T. Rajh and R. T. Koodall, J. Phys. Chem. C, 2010, 114, 15728–15734 CrossRef CAS.
- R. Peng, D. Zhao, M. M. Dimitrijevic, T. Rajh and R. T. Koodall, J. Phys. Chem. C, 2012, 116, 1605–1613 CAS.
- IEA, CO2 emissions from fuel combustion 2012.
- G. Centi and S. Perathoner, Stud. Surf. Sci. Catal., 2004, 153, 1–8 CrossRef CAS.
- M. Aresta, A. Dibenedetto, F. Nocita, C. Pastore, A. M. Venezia, F. Chirikalova, V. I. Kononenko, V. G. Shevchenko and I. A. Chupova, Catal. Today, 2006, 115, 117–123 CrossRef PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS PubMed.
- P. G. Jessop, E. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. R. Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- L. Lloyd, Handbook of Industrial Catalysts, Springer, New York, USA, 2011 Search PubMed.
- J. L. G. Fierro, Metal Oxides: Chemistry and Applications, CRC Taylor & Francis Pub., Boca Raton, FL, USA, 2006, ch. 18, p. 569 Search PubMed.
- H. Sano, Energy Convers. Manage., 1995, 36, 895–898 CrossRef CAS.
- L. Fan and K. Fujimoto, J. Catal., 1994, 150, 217–220 CrossRef CAS.
- X. L. Liang, X. Dong, G. D. Lin and H. B. Zhang, Appl. Catal., B, 2009, 88, 315–322 CrossRef CAS PubMed.
- K. P. Yu, W. Y. Yu, M. C. Kuo, Y. C. Liou and S. H. Chien, Appl. Catal., B, 2008, 84, 112–118 CrossRef CAS PubMed.
- T. Fujitani, M. Saito, Y. Kanai, Y. Watanabe, J. Nakamura and T. Uchijima, Appl. Catal., A, 1995, 125, L199–L202 CrossRef CAS.
- J. Stoczyniskia, R. Grabowskia, P. Olszewskia, A. Kozlowskaa, J. Stoch and M. Lachowskab, Appl. Catal., A, 2006, 310, 127–137 CrossRef PubMed.
- S. E. Collins, D. L. Chiavassa, A. L. Bonivardi and M. A. Baltanás, Catal. Lett., 2005, 103, 83–88 CrossRef CAS.
- C. S. Song and K. M. Reddy, Appl. Catal., A, 1999, 176, 1–10 CrossRef CAS.
- Y. Ohtsuka, T. Arai, S. Takasaki and N. Tsubouchi, Energy Fuels, 2003, 17, 804–809 CrossRef CAS.
- E. Ghedini, F. Menegazzo, M. Signoretto, M. Manzoli, F. Pinna and G. Strukul, J. Catal., 2010, 273, 266–273 CrossRef CAS PubMed.
- Y. M. Yu, J. H. Fei, Y. P. Zhang and X. M. Zheng, J. Fuel Chem. Technol., 2006, 34, 700–705 CrossRef CAS.
- N. Kosizumi, X. Jiang, J. Kugai and C. Song, Catal. Today, 2012, 194, 16–24 CrossRef PubMed.
- M. Maestri, D. G. Vlachos, A. Beratta, G. Groppi and E. Tronconi, J. Catal., 2008, 259, 211–222 CrossRef CAS PubMed.
- X. Wu and S. Kawi, Catal. Today, 2009, 148, 251–259 CrossRef CAS PubMed.
- L. Xu, H. Song and L. Chou, Catal. Sci. Technol., 2011, 1, 1032–1042 CAS.
- L. Xu, H. Song and L. Chou, Appl. Catal., B, 2011, 108–109, 177–190 CrossRef CAS PubMed.
- B. Sarkar, R. Tiwari, R. K. Singha, S. Suman, S. Ghosh, S. S. Acharyya, K. Mantri, L. N. S. Konathala, C. Penden and R. Bal, Catal. Today, 2012, 198, 209–214 CrossRef CAS PubMed.
- L. Xu, H. Song and L. Chou, Int. J. Hydrogen Energy, 2012, 37, 18001–18020 CrossRef CAS PubMed.
- Y. Takahashi and T. Yamazaki, Fuel, 2012, 102, 239–246 CrossRef CAS PubMed.
- H. Liu, Y. Li, H. Wu, T. Miyake and D. He, Int. J. Hydrogen Energy, 2013, 38, 15200–15209 CrossRef CAS PubMed.
- B. Li and S. Zhang, Int. J. Hydrogen Energy, 2013, 38, 14250–14260 CrossRef CAS PubMed.
- S. Zeng, X. Fu, T. Zhou, X. Wang and H. Su, Fuel Process. Technol., 2013, 114, 69–74 CrossRef CAS PubMed.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- Y. H. Guo, C. Xia and B. S. Liu, Chem. Eng. J., 2014, 237, 421–429 CrossRef CAS PubMed.
- K. Nakagawa, M. Okamura, N. Ikenaga, T. Suzuki and T. Kobayashi, Chem. Commun., 1998, 1025–1026 RSC.
- I. Takahara and M. Saito, Chem. Lett., 1996, 973–974 CrossRef CAS.
- K. T. Leth, A. K. Rovik, M. S. Holm, M. Brorson, H. J. Jakobsen, J. Skibsted and C. H. Christensen, Appl. Catal., A, 2008, 348, 257–265 CrossRef CAS PubMed.
- T. V. M. Rao, Y. Yang and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 152–158 CrossRef CAS PubMed.
- T. V. M. Rao, E. M. Zahidi and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 159–165 CrossRef CAS PubMed.
- P. Michorczyk, J. Ogonowski, P. Kustrowski and L. Chmielarz, Appl. Catal., A, 2008, 349, 62–69 CrossRef CAS PubMed.
- F. Zhang, R. Xu, Y. Yue, W. Yang, S. Gu, C. Miao, W. Hua and Z. Gao, Microporous Mesoporous Mater., 2011, 145, 194–199 CrossRef CAS PubMed.
- G. Wang, L. Zhang, J. Deng, H. Dai, H. He and C. T. Au, Appl. Catal., A, 2009, 355, 192–201 CrossRef CAS PubMed.
- Y. Qiao, C. Miao, Y. Yue, Z. Xie, W. Yang, W. Hua and Z. Gao, Microporous Mesoporous Mater., 2009, 119, 150–157 CrossRef CAS PubMed.
- S. Udayakumar, M. K. Lee, H. L. Shim and D. W. Park, Appl. Catal., A, 2009, 365, 88–95 CrossRef CAS PubMed.
- J. N. Appaturi and F. Adam, Appl. Catal., B, 2013, 136–137, 150–159 CrossRef CAS PubMed.
- D. B. Nale, S. Rana, K. Parida and B. M. Bhanage, Appl. Catal., A, 2014, 469, 340–349 CrossRef CAS PubMed.
- R. Zhang, A. A. Elzatahry, S. S. Al-Deyab and D. Zhao, Nano Today, 2012, 7, 344–366 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- S. Wang, H. M. Ang and M. O. Tade, Environ. Int., 2007, 33, 694–705 CrossRef CAS PubMed.
- P. Van Der Voort, P. I. Ravikovitch, K. P. De Jong, A. V. Neimark, A. H. Janssen, M. Benjelloum, E. Van Bavel, P. Cool, B. M. Wechuysen and E. F. Vansant, Chem. Commun., 2002, 1010–1011 RSC.
- M. Kruk, M. Jaroniec, S. H. Joo and R. Ryoo, J. Phys. Chem. B, 2003, 107, 2205–2213 CrossRef CAS.
- F. Bosc, P. L. Desmazes and A. Ayral, J. Colloid Interface Sci., 2006, 304, 545–548 CrossRef CAS PubMed.
- J. I. L. Chen, G. von Freymann, V. Kitaev and G. A. Ozin, J. Am. Chem. Soc., 2007, 129, 1196–1202 CrossRef CAS PubMed.
- L. Saadoun, J. A. Ayllon, J. Jiménez-Becerril, J. Peral, X. Domènech and R. Rodríguez-Clemente, Mater. Res. Bull., 2000, 35, 193–202 CrossRef CAS.
- X. Wang, J. C. Yu, C. Ho, Y. Hou and X. Fu, Langmuir, 2005, 21, 2552–2559 CrossRef CAS PubMed.
- J. Yu, Y. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984–1990 CrossRef CAS PubMed.
- M. S. Hamdy, O. Berg, J. C. Jansen, T. Maschmeyer, J. A. Moulijm and G. Mul, Chem. - Eur. J., 2006, 12, 620–628 CrossRef PubMed.
- A. M. Busuioc, V. Meynen, E. Beyers, P. Cool, N. Bilba and E. F. Vansant, Catal. Commun., 2007, 8, 527–530 CrossRef CAS PubMed.
- M. Tasbihi, U. L. Stangar, A. Ristic, V. Kaucic and N. N. Tusar, J. Photochem. Photobiol., A, 2010, 216, 167–178 CrossRef CAS PubMed.
- W. Dong, Y. Sun, Q. Ma, L. Zhu, W. Hua, X. Lu, G. Zhuang, S. Zhang, Z. Guo and D. Zhao, J. Hazard. Mater., 2012, 229–230, 307–320 CrossRef CAS PubMed.
- R. Y. Zhang, B. Tu and D. Y. Zhao, Chem. - Eur. J., 2010, 16, 9977–9981 CrossRef CAS PubMed.
- S. Zhu, D. Zhang, Z. Chen, G. Zhou, H. Jiang and J. Li, J. Nanopart. Res., 2010, 12, 2445–2456 CrossRef CAS.
- F. He, J. Li, T. Li and G. Li, Chem. Eng. J., 2014, 237, 312–321 CrossRef CAS PubMed.
- M. Zhou, J. Yu and B. Cheng, J. Hazard. Mater., 2006, 137, 1838–1847 CrossRef CAS PubMed.
- A. A. Ismail, Appl. Catal., B, 2012, 117–118, 67–72 CrossRef CAS PubMed.
- A. A. Ismail, Microporous Mesoporous Mater., 2012, 149, 69–75 CrossRef CAS PubMed.
- J. Choina, Ch. Fischer, G. U. Flechsig, H. Kosslick, V. A. Tuan, N. D. Tuyen, N. A. Tuyen and A. Schulz, J. Photochem. Photobiol., A, 2014, 274, 108–116 CrossRef CAS PubMed.
- J. Nesic, D. D. Manojlovic, I. Andelkovic, B. P. Dojcinovic, P. J. Vulic, J. Krstic and G. M. Roglic, J. Mol. Catal. A: Chem., 2013, 378, 67–75 CrossRef CAS PubMed.
- P. S. S. Kumar, M. R. Raj and S. Anandan, Sol. Energy Mater. Sol. Cells, 2010, 94, 1783–1789 CrossRef PubMed.
- C. Zhan, F. Chen, H. Dai, J. Yang and M. Zhong, Chem. Eng. J., 2013, 225, 695–703 CrossRef CAS PubMed.
- X. Yan, X. Zong, G. Q. Lu and L. Wang, Prog. Nat. Sci., 2012, 22, 654–660 CrossRef PubMed.
- Z. Hu, L. Xu and J. Chen, Mater. Lett., 2013, 106, 421–424 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Teshima and T. Hirai, J. Phys. Chem. B, 2006, 110, 6257–6263 CrossRef CAS PubMed.
- S. Rodriguez, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Photochem. Photobiol., A, 2004, 165, 51–58 CrossRef PubMed.
- S. Rodríguez, K. T. Ranjit, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Catal., 2005, 230, 158–165 CrossRef PubMed.
- S. Rodríguez, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Catal., 2005, 233, 405–410 CrossRef PubMed.
- E. C. C. Baly, I. M. Heilbron and W. F. Barker, J. Chem. Soc. Trans., 1921, 119, 1025–1035 RSC.
- M. Tahir and N. A. S. Amin, Renewable Sustainable Energy Rev., 2013, 25, 560–579 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- J. S. Hwang, J. S. Chang, S. E. Park, K. Ikeue and M. Anpo, Top. Catal., 2005, 35, 311–319 CrossRef CAS PubMed.
- N. Ulagappan and H. Frei, J. Phys. Chem. A, 2000, 104, 7834–7839 CrossRef CAS.
- W. Lin and F. Frei, J. Am. Chem. Soc., 2005, 127, 1610–1611 CrossRef CAS PubMed.
- M. Tahir and N. A. S. Amin, Appl. Catal., A, 2013, 467, 483–496 CrossRef CAS PubMed.
- Y. Li, W. N. Wang, Z. Zhan, M. H. Woo, C. Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS PubMed.
- Y. Wang, B. Li, C. Zhang, L. Cui, S. Kang, X. Li and L. Zhou, Appl. Catal., B, 2013, 130–131, 277–284 CrossRef CAS PubMed.
- Y. Wang, F. Wang, Y. Chen, D. Zhang, B. Li, S. Kang, X. Li and L. Cui, Appl. Catal., B, 2014, 147, 602–609 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. P. Saing-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- K. D. Dubois, H. He, C. Liu, A. S. Vorushilov and G. Li, J. Mol. Catal. A: Chem., 2012, 363–364, 208–213 CrossRef CAS PubMed.
- S. B. Yoon, B. Fang, M. Kim, J. Ho Kim and J.-S. Yu, in Frontiers of Nanoscience, Nanostructured Materials, ed. G. Wilde, Elsevier, Amsterdam, the Netherlands, 2009, vol. 1, pp. 173–231 Search PubMed.
- R. M. Ormerod, Chem. Soc. Rev., 2003, 32, 17–28 RSC.
- A comparison between the different fuel cell types can be found in: S. Mekhilef, R. Saidurb and A. Safaria, Renewable Sustainable Energy Rev., 2012, 16, 981–989 CrossRef CAS PubMed.
- S. Giddey, S. P. S. Badwal, A. Kulkarni and C. Munnings, Prog. Energy Combust. Sci., 2012, 38, 360–399 CrossRef CAS PubMed.
- J.-J. Hwang, Renewable Sustainable Energy Rev., 2013, 19, 220–229 CrossRef CAS PubMed.
- M. W. Ellis, M. R. Von Spakovsky and D. J. Nelson, Proc. IEEE, 2001, 89, 1808–1818 CrossRef CAS.
- N. V. Long, Y. Yang, C. M. Thi, N. V. Minh, Y. Cao and M. Nogami, Nano Energy, 2013, 2, 636–676 CrossRef PubMed.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS PubMed.
- (a) J. B. Xu and T. S. Zhao, RSC Adv., 2013, 3, 16–24 RSC; (b) J. Liang, S. Z. Qiao, G. Q. Lu and D. Hulicova-Jurcakova, in Novel Carbon Adsorbents, ed. Juan. M. D. Tascón, Elsevier, Oxford, 2012, pp. 549–581 Search PubMed; (c) E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS PubMed.
- Y.-J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- S. Zhang, Y. Shao, G. Yin and Y. Lin, J. Mater. Chem. A, 2013, 1, 4631–4641 CAS.
- L. Calvillo, V. Celorrio, R. Moliner, A. B. Garcia, I. Caméan and M. J. Lazaro, Electrochim. Acta, 2013, 102, 19–27 CrossRef CAS PubMed.
- J. R. C. Salgado, V. A. Paganin, E. R. Gonzalez, M. F. Montemor, I. Tacchini, A. Ansón, M. A. Salvador, P. Ferreira, F. M. L. Figueiredo and M. G. S. Ferreira, Int. J. Hydrogen Energy, 2013, 38, 910–920 CrossRef CAS PubMed.
- J. Cao, Z. Chen, J. Xu, W. Wang and Z. Chen, Electrochim. Acta, 2013, 88, 184–192 CrossRef CAS PubMed.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- R. Li, H. Mao, J. Zhang, T. Huang and A. Yu, J. Power Sources, 2013, 241, 660–667 CrossRef CAS PubMed.
- (a) M. Yang, R. Guarecuco and F. J. DiSalvo, Chem. Mater., 2013, 25, 1783–1787 CrossRef CAS; (b) M. Yang, Z. Cui and F. J. DiSalvo, Phys. Chem. Chem. Phys., 2013, 15, 7041–7044 RSC; (c) M. Yang, Z. Cui and F. J. DiSalvo, Phys. Chem. Chem. Phys., 2013, 15, 1088–1092 RSC; (d) Z. Cui, R. G. Burns and F. J. DiSalvo, Chem. Mater., 2013, 25, 3782–3784 CrossRef CAS.
- (a) A. Kumar and V. K. Ramani, Appl. Catal., B, 2013, 138–139, 43–50 CrossRef CAS PubMed; (b) C.-P. Lo, G. Wang, A. Kumar and V. Ramani, Appl. Catal., B, 2013, 140–141, 133–140 CrossRef CAS PubMed.
- (a) Q. Wang, G. Wang, K. Sasaki, T. Takeguchi, T. Yamanaka, M. Sadakane and W. Ueda, J. Power Sources, 2013, 241, 728–735 CrossRef CAS PubMed; (b) J. Zhang, J.-p. Tu, G.-h. Du, Z.-m. Dong, Q.-m. Su, D. Xie and X.-l. Wang, Electrochim. Acta, 2013, 88, 107–111 CrossRef CAS PubMed; (c) H. Hua, C. Hu, Z. Zhao, H. Liu, X. Xie and Y. Xi, Electrochim. Acta, 2013, 105, 130–136 CrossRef CAS PubMed; (d) M. P. Gurrola, M. Guerra-Balcázar, L. Álvarez-Contreras, R. Nava, J. Ledesma-García and L. G. Arriaga, J. Power Sources, 2013, 243, 826–830 CrossRef CAS PubMed.
- R. Zolfaghari, F.-R. Ahmadun, M. R. Othman, W. R. W. Daud and M. Ismail, Mater. Chem. Phys., 2013, 139, 262–269 CrossRef CAS PubMed.
- K. K. Tintula, A. Jalajakshi, A. K. Sahu, S. Pitchumani, P. Sridhar and A. K. Shukla, Fuel Cells, 2013, 13, 158–166 CrossRef CAS PubMed.
- M. M. Bruno, M. A. Petruccelli, F. A. Viva and H. R. Corti, Int. J. Hydrogen Energy, 2013, 38, 4116–4123 CrossRef CAS PubMed.
- S. Song, K. Wang, Y. Liu, C. He, Y. Liang, R. Fu, D. Wu and Y. Wang, Int. J. Hydrogen Energy, 2013, 38, 1405–1412 CrossRef CAS PubMed.
- (a) F. Li, K.-Y. Chan, H. Yung, C. Yang and S. W. Ting, Phys. Chem. Chem. Phys., 2013, 15, 13570–13577 RSC; (b) S.-J. Park, B.-J. Kim and S.-Y. Lee, J. Colloid Interface Sci., 2013, 405, 150–156 CrossRef CAS PubMed.
- (a) A. M. Baena-Moncada, G. A. Planes, M. S. Moreno and C. A. Barbero, J. Power Sources, 2013, 221, 42–48 CrossRef CAS PubMed; (b) Q.-X. Li, H.-M. Mao, J.-G. Li and Q.-J. Xu, J. Fuel Cell Sci. Technol., 2013, 10, 051006 CrossRef; (c) C. Y. Wong, S.-K. Chen, A.-Y. Lo, C.-M. Tseng, C.-Y. Lin and S.-B. Liu, Int. J. Hydrogen Energy, 2013, 38, 12984–12990 CrossRef CAS PubMed.
- C. Alegre, M. E. Gálvez, E. Baquedano, R. Moliner, E. Pastor and M. J. Lázaro, J. Phys. Chem. C, 2013, 117, 13045–13058 CAS.
- D. Banham, F. Feng, K. Pei, S. Ye and V. Birss, J. Mater. Chem. A, 2013, 1, 2812–2820 CAS.
- S. Shrestha, S. Asheghi, J. Timbro and W. E. Mustain, Appl. Catal., A, 2013, 464–465, 233–242 CrossRef CAS PubMed.
- (a) K. Vaarmets, J. Nerut, E. Härk and E. Lust, Electrochim. Acta, 2013, 104, 216–227 CrossRef CAS PubMed; (b) K. Vaarmets, S. Sepp, J. Nerut, E. Härk, I. Tallo and E. Lust, J. Solid State Electrochem., 2013, 17, 1729–1741 CrossRef CAS.
- (a) J. Tang, T. Wang, X. Sun, Y. Guo, H. Xue, H. Guo, M. Liu, X. Zhang and J. He, Microporous Mesoporous Mater., 2013, 177, 105–112 CrossRef CAS PubMed; (b) M. Lei, T. Z. Yang, W. J. Wang, K. Huang, R. Zhang, X. L. Fu, H. J. Yang, Y. G. Wang and W. H. Tang, Int. J. Hydrogen Energy, 2013, 38, 205–211 CrossRef CAS PubMed; (c) C. Rüdiger, J. Brumbarov, F. Wiesinger, S. Leonardi, O. Paschos, C. Valero Vidal and J. Kunze-Liebhäuser, ChemCatChem, 2013, 5, 3219–3223 CrossRef PubMed.
- (a) J. Zeng, C. Francia, C. Gerbaldi, V. Baglio, S. Specchia, A. S. Aricò and P. Spinelli, Electrochim. Acta, 2013, 94, 80–91 CrossRef CAS PubMed; (b) L. Samiee, F. Shoghi and A. Vinu, Appl. Surf. Sci., 2013, 265, 214–221 CrossRef CAS PubMed.
- (a) J. Y. Cheon, C. Ahn, D. J. You, C. Pak, S. H. Hur, J. Kim and S. H. Joo, J. Mater. Chem. A, 2013, 1, 1270–1283 RSC; (b) X. Bo and L. Guo, Electrochim. Acta, 2013, 90, 283–290 CrossRef CAS PubMed; (c) J. Tang, T. Wang, X. Sun, Y. Hu, Q. Xie, Y. Guo, H. Xue and J. He, Electrochim. Acta, 2013, 90, 53–62 CrossRef CAS PubMed.
- J. H. Kim, S. Y. Kwon, D. Bhattacharjya, G. S. Chai and J.-S. Yu, J. Catal., 2013, 306, 133–145 CrossRef CAS PubMed.
- (a) J. Yan, H. Meng, F. Xie, X. Yuan, W. Yu, W. Lin, W. Ouyang and D. Yuan, J. Power Sources, 2014, 245, 772–778 CrossRef CAS PubMed; (b) J.-e. Park, Y. J. Jang, Y. J. Kim, M.-s. Song, S. Yoon, D. H. Kim and S.-J. Kim, Phys. Chem. Chem. Phys., 2014, 16, 103–109 RSC; (c) J.-S. Lee, K. Jo, T. Lee, T. Yun, J. Cho and B.-S. Kim, J. Mater. Chem. A, 2013, 1, 9603–9607 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, Md. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- An overview of the state-of-the-art conversion efficiencies in DSCs can be found in: H. M. Upadhyaya, S. Senthilarasu, M.-H. Hsu and D. K. Kumar, Sol. Energy Mater. Sol. Cells, 2013, 119, 291–295 CrossRef CAS PubMed.
- G. Hodes and A. Zaban, in Frontiers of Nanoscience, Nanostructured Materials, ed. G. Wilde, Elsevier, Amsterdam, the Netherlands, 2009, vol. 1, pp. 232–269 Search PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. l. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- T. Peng, W. Sun, X. Sun, N. Huang, Y. Liu, C. Bu, S. Guo and X.-Z. Zhao, Nanoscale, 2013, 5, 337–341 RSC.
- G. Wang, S. Kuang, D. Wang and S. Zhuo, Electrochim. Acta, 2013, 113, 346–353 CrossRef CAS PubMed.
- S.-j. Xu, Y.-f. Luo, W. Zhong and Z.-h. Xiao, New Carbon Mater., 2013, 28, 254–261 CrossRef CAS.
- E. Ramasamy and J. Lee, Chem. Commun., 2010, 46, 2136–2138 RSC.
- E. Ramasamy, J. Chun and J. Lee, Carbon, 2010, 48, 4563–4565 CrossRef CAS PubMed.
- P. Srinivasu, S. P. Singh, A. Islam and L. Han, Int. J. Photoenergy, 2011, 617439 Search PubMed.
- (a) A. Kay and M. Grätzel, Sol. Energy Mater. Sol. Cells, 1996, 44, 99–117 CrossRef CAS; (b) K. Imoto, K. Takahashi, T. Yamaguchi, T. Komura, J. Nakamura and K. Murata, Sol. Energy Mater. Sol. Cells, 2003, 79, 459–469 CrossRef CAS; (c) T. N. Murakami, S. Ito, Q. Wang, M. K. Nazeeruddin, T. Bessho, I. Cesar, P. Liska, R. H. Bakar, P. Comte, P. Pechy and M. Grätzel, J. Electrochem. Soc., 2006, 153, A2255–A2261 CrossRef CAS PubMed.
- H. Alama and S. Ramakrishna, Nano Energy, 2013, 2, 190–212 CrossRef PubMed.
- Waste Heat Recovery: Technologies and Opportunities in U.S. Industry, U.S. Department of Energy, 2008 Search PubMed.
- W. Liu, X. Yan, G. Chen and Z. Ren, Nano Energy, 2012, 1, 42–56 CrossRef CAS PubMed.
- G. A. Slack, in CRC Handbook of Thermoelectrics, ed. M. Rowe, CRC, Boca Raton, FL, 1995, pp. 407–440 Search PubMed.
- A. Shakouri and S. Li, Proceedings of International Conference on Thermoelectrics, Baltimore, September 1999.
- Y. Zhang, T. Day, M. L. Snedaker, H. Wang, S. Krämer, C. S. Birkel, X. Ji, D. Liu, G. J. Snyder and G. D. Stucky, Adv. Mater., 2012, 24, 5065–5070 CrossRef CAS PubMed.
- (a) B. Qiu and X. Ruan, Appl. Phys. Lett., 2010, 9, 183103–183107 Search PubMed; (b) J.-H. Lee, G. A. Galli and J. C. Grossman, Nano Lett., 2008, 8, 3750–3754 CrossRef CAS PubMed; (c) Y. He, D. Donadio, J.-H. Lee, J. C. Grossman and G. Galli, ACS Nano, 2011, 5, 1839–1844 CrossRef CAS PubMed.
- M.-H. Hong, S.-Y. Jung, T.-J. Ha, W.-S. Seo, Y. S. Lim, S. Shin, H. H. Cho and H.-H. Park, Surf. Coat. Technol., 2013, 231, 370–373 CrossRef CAS PubMed.
- S.-Y. Jung, T.-J. Ha, C.-S. Park, W.-S. Seo, Y. S. Lim, S. Shin, H. H. Cho and H.-H. Park, Thin Solid Films, 2013, 529, 94–97 CrossRef CAS PubMed.
- S.-Y. Jung, T.-J. Ha, W.-S. Seo, Y. S. Lim, S. Shin, H. H. Cho and H.-H. Park, J. Electron. Mater., 2011, 40, 652–656 CrossRef CAS.
- T.-J. Ha, H.-H. Park, S.-Y. Jung, S.-J. Yoon, J.-S. Kim and H. W. Jang, Thin Solid Films, 2010, 518, 7196–7198 CrossRef CAS PubMed.
- M.-H. Hong, C.-S. Park, S. Shin, H. H. Cho, W.-S. Seo, Y. S. Lim, J.-K. Lee and H.-H. Park, J. Nanomater., 2013, 172504 CAS.
- M.-H. Hong, C.-S. Park, W.-S. Seo, Y. S. Lim, J.-K. Lee and H.-H. Park, J. Nanomater., 2013, 131537 CAS.
- S. Walia, S. Balendhran, H. Nili, S. Zhuiykov, G. Rosengarten, Q. H. Wang, M. Bhaskaran, S. Sriram, M. S. Strano and K. Kalantar-zadeh, Prog. Mater. Sci., 2013, 58, 1443–1489 CrossRef CAS PubMed.
- J. Fang, C. B. Kang, Y. Huang, S. H. Tolbert and L. Pilon, J. Phys. Chem. C, 2012, 116, 12926–12933 CAS.
- H. Lee, D. Vashaee, D. Z. Wang, M. S. Dresselhaus, Z. F. Ren and G. Chen, J. Appl. Phys., 2010, 107, 094308 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |