Matching chelators to radiometals for radiopharmaceuticals
Eric W.
Price
*ab and
Chris
Orvig
*a
aMedicinal Inorganic Chemistry Group, Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia, Canada V6T 1Z1. E-mail: ericwprice@gmail.com; orvig@chem.ubc.ca
bTRIUMF, 4004 Wesbrook Mall, Vancouver, British Columbia, Canada V6T 2A3
First published on 30th October 2013
Abstract
Radiometals comprise many useful radioactive isotopes of various metallic elements. When properly harnessed, these have valuable emission properties that can be used for diagnostic imaging techniques, such as single photon emission computed tomography (SPECT, e.g.67Ga, 99mTc, 111In, 177Lu) and positron emission tomography (PET, e.g.68Ga, 64Cu, 44Sc, 86Y, 89Zr), as well as therapeutic applications (e.g.47Sc, 114mIn, 177Lu, 90Y, 212/213Bi, 212Pb, 225Ac, 186/188Re). A fundamental critical component of a radiometal-based radiopharmaceutical is the chelator, the ligand system that binds the radiometal ion in a tight stable coordination complex so that it can be properly directed to a desirable molecular target in vivo. This article is a guide for selecting the optimal match between chelator and radiometal for use in these systems. The article briefly introduces a selection of relevant and high impact radiometals, and their potential utility to the fields of radiochemistry, nuclear medicine, and molecular imaging. A description of radiometal-based radiopharmaceuticals is provided, and several key design considerations are discussed. The experimental methods by which chelators are assessed for their suitability with a variety of radiometal ions is explained, and a large selection of the most common and most promising chelators are evaluated and discussed for their potential use with a variety of radiometals. Comprehensive tables have been assembled to provide a convenient and accessible overview of the field of radiometal chelating agents.
 Eric W. Price and Chris Orvig | Eric Price received his undergraduate education at the University of Victoria in Victoria, British Columbia, Canada, where he completed his honours BSc (2009) in Chemistry as a part of the CO-OP program, performing research both in industry and academia, including work at TRIUMF and an NSERC USRA research term with Dr Matthew Moffitt. In 2009 he began his PhD studies as an NSERC scholar in a joint project between Dr Chris Orvig at the University of British Columbia and Dr Michael J. Adam at TRIUMF, partially in collaboration with Nordion, researching new radiometal-based radiopharmaceuticals of 67/68Ga, 64Cu, 111In, 177Lu, 89Zr, and 225Ac. He initiated, and has been pivotal in, an international research collaboration with Dr Jason S. Lewis and Dr Brian M. Zeglis, traveling to Memorial Sloan-Kettering Cancer Center in New York, USA, and has also collaborated with the BC Cancer Agency in Vancouver, as part of his PhD work. In 2013 he won the Berson–Yalow award from the Society of Nuclear Medicine and Molecular Imaging. |
Chris Orvig earned his Hons. BSc from McGill and his PhD as an NSERC of Canada scholar at MIT with Alan Davison, FRS. After postdoctoral fellowships with Kenneth N. Raymond at the University of California, Berkeley, and the late Colin J. L. Lock at McMaster University, he joined the University of British Columbia in 1984, where he is now Professor of Chemistry and Pharmaceutical Sciences. Orvig, a Fellow of the Royal Society of Canada, has received various teaching, research and service awards, and published more than 200 research papers. He is a co-inventor on many issued patents, and a certified ski instructor. |
1. Introduction
Radiometals are radioactive isotopes that can be harnessed for applications in medical diagnosis, as well as for cancer therapy. In order to apply these isotopes to specific biological applications, the “free” radiometal ions must be sequestered from aqueous solution using chelators (ligands) to obviate transchelation and hydrolysis. Chelators used for this application are typically covalently linked to a biologically active targeting molecule, making an active radiopharmaceutical agent. The chelator is used to tightly bind a radiometal ion so that when injected into a patient, the targeting molecule can deliver the isotope without any radiometal loss from the radiopharmaceutical, effectively supplying a site-specific radioactive source in vivo for imaging or therapy. A rapidly expanding number of radiometals are routinely produced, with a broad variety of half-lives, emission types, energies, and branching ratios (Table 1).1–4 The availability of a wide range of radiometal ions makes it possible to carefully pick the specific nuclear properties that are needed for a vast number of different applications. Some examples of radiometals that can be used for positron emission tomography (PET) imaging are 68Ga, 64Cu, 86Y, 89Zr, and 44Sc, with PET imaging providing sensitive, quantitative, and non-invasive images of a variety of molecular processes and targets. Single photon emission computed tomography (SPECT) is an older and more ubiquitous imaging modality than PET, and, since its inception in the 1960s, 99mTc has been the workhorse isotope of SPECT. More recently, the radiometals 67Ga, 111In, and 177Lu have been increasingly used for SPECT imaging in chelator-based radiopharmaceuticals. For therapy applications, particle emitters such as 111In (Auger electron emitter), 90Y and 177Lu (β−), and 225Ac, 212Pb, and 213Bi (α), are being heavily investigated, typically in conjunction with antibody vectors (immunoconjugates) or peptides. Each radiometal ion has unique aqueous coordination chemistry properties; these must be properly attended to if these isotopes are to be safely harnessed for medical applications and use in vivo.| Isotope | t 1/2 (h) | Decay mode | E (keV) | Production method |
|---|---|---|---|---|
| 60Cu | 0.4 | β+ (93%) | β+, 3920, 3000, 2000 | Cyclotron, 60Ni(p,n)60Cu |
| EC (7%) | ||||
| 61Cu | 3.3 | β+ (62%) | β+, 1220, 1150, 940, 560 | Cyclotron, 61Ni(p,n)61Cu |
| EC (38%) | ||||
| 62Cu | 0.16 | β+ (98%) | β+, 2910 | 62Zn/62Cu generator |
| EC (2%) | ||||
| 64Cu | 12.7 | β+ (19%) | β+, 656 | Cyclotron, 64Ni(p,n)64Cu |
| EC (41%) | ||||
| β− (40%) | ||||
| 66Ga | 9.5 | β+ (56%) | β+, 4150, 935 | Cyclotron, 63Cu(α,nγ)66Ga |
| EC (44%) | ||||
| 67Ga | 78.2 | EC (100%) | γ, 93, 184, 300 | Cyclotron, 68Zn(p,2n)67Ga |
| 68Ga | 1.1 | β+ (90%) | β+, 1880 | 68Ge/68Ga generator |
| EC (10%) | ||||
| 44Sc | 3.9 | β+ (94%) | γ, 1157 | 44Ti/44Sc generator |
| EC (6%) | β+, 1474 | |||
| 47Sc | 80.2 | β− (100%) | γ, 159 | 47Ti(n,p)47Sc |
| β−, 441, 600 | ||||
| 111In | 67.2 | EC (100%) | γ, 245, 172 | Cyclotron, 111Cd(p,n)111m,gIn |
| 114mIn | 49.5 d | EC (100%) | γ, 190 | Cyclotron, 114Cd(p,n)114mIn or 116Cd(p,3n)114mIn |
| 114In (daughter) | 73 s | β− (100%) | β−, 1989 | |
| 177Lu | 159.4 | β− (100%) | γ, 112, 208 | 176Lu(n,γ)177Lu |
| β−, 177, 385, 498 | ||||
| 86Y | 14.7 | β+ (33%) | β+, 1221 | Cyclotron, 86Sr(p,n)86Y |
| EC (66%) | ||||
| 90Y | 64.1 | β− (100%) | β−, 2280 | 90Zr(n,p)90Y |
| 89Zr | 78.5 | β+ (23%) | β+, 897 | Cyclotron, 89Y(p,n)89Zr |
| EC (77%) | ||||
| 212Bi | 1.1 | α (36%) | α, 6050 | 228Pb/212Pb generator |
| β− (64%) | β−, 6089 | |||
| 213Bi | 0.76 | α (2.2%) | α, 5549 | 228Th/212Pb generator |
| β− (97.8%) | β−, 5869 | |||
| 212Pb (daughter is 212Bi) | 10.6 | β− (100%) | α, 570 | 224Ra/212Pb generator |
| 225Ac | 240 | α (100%) | α, 5600–5830 (6) | 226Ra(p,2n)225Ac |
| n-Capture of 232Th → 233U → 225Ac | ||||
The major difference between radioactive (“hot”) and non-radioactive (“cold”) metal ion chemistry is that radiochemistry is typically performed under extremely dilute conditions, with radiometal ions typically being utilized at nM to pM concentrations.5 It is also important to note that several of the elements being discussed have multiple radioactive isotopes that are useful for diagnostic or therapeutic purposes (e.g.86/90Y, 67/68Ga, 44/47Sc, 60/61/62/64Cu), and all isotopes of a given element have identical chemistry.6–11 This means that a single radiopharmaceutical agent can be radiolabeled with different isotopes of the same element (e.g.86/90Y), and provide the same charge and physical properties, and therefore the same biological behavior and distribution in vivo.6–11 This class of radiopharmaceutical that utilizes two isotopes of the same element, such as 86Y for PET imaging and 90Y for therapy, has been referred to as a theranostic agent.12,13 An interesting note about 90Y is that as a β− emitter it is possible to perform biodistribution, imaging, and dosimetry studies with its bremsstrahlung X-rays, but because the spatial resolution and image quality obtained is very poor an imaging surrogate is typically used (e.g.111In or 86Y).14 Alternatively, 90Y is unique and in some circumstances can be used directly for PET imaging because it emits a very low abundance of positrons (0.003%), which can be used to collect imaging data superior to that obtained by 90Y bremsstrahlung imaging.14
2. Radiometal-based radiopharmaceutical design
Ligands that are typically used to construct radiometal-based radiopharmaceuticals (not always with 99mTc) are bifunctional chelators (BFCs), which are simply chelators with reactive functional groups that can be covalently coupled (conjugated) to targeting vectors (e.g. peptides, nucleotides, antibodies, nanoparticles). Common bioconjugation techniques utilize functional groups such as carboxylic acids or activated esters (e.g. N-hydroxysuccinimide NHS-ester, tetrafluorophenyl TFP-ester) for amide couplings, isothiocyanates for thiourea couplings, and maleimides for thiol couplings (Fig. 1).17,18 Click chemistry is gaining popularity in bioconjugate chemistry, with both the traditional copper(I) catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition “click” reaction (forming a 1,2,3-triazole-ring linkage), or newer copper-free reactions like strain-promoted azide–alkyne cycloadditions (e.g. dibenzocyclooctyne/azide reaction) and Diels–Alder click reactions (e.g. transcyclooctene/1,2,4,5-tetrazine) (Fig. 1).19 It is interesting to note that the transcyclooctene/1,2,4,5-tetrazine copper-free click coupling displays remarkably fast reaction kinetics, allowing for novel applications like in vivo pre-targeting, where the click reaction can occur in vivo at very dilute concentrations.20–24 The modular design of BFC systems allows for a theoretically limitless number of different vectors to be conjugated, providing molecular targeting to a constantly increasing number of biological targets.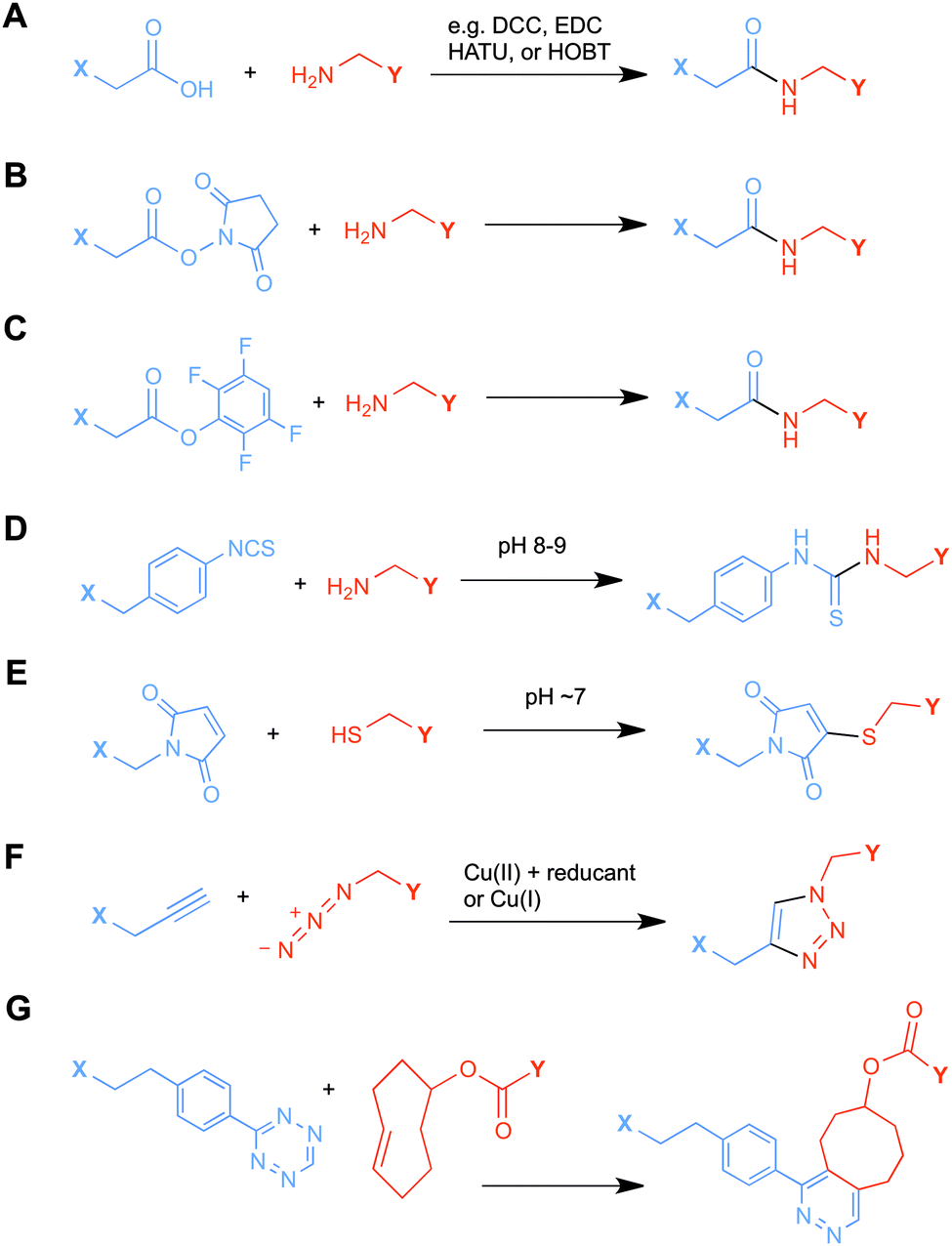 | ||
| Fig. 1 Examples of bioconjugation reactions: (A) standard peptide coupling reaction between a carboxylic acid and a primary amine with a coupling reagent; (B and C) peptide coupling reactions between activated esters of tetrafluorophenyl (TFP) or N-hydroxysuccinimide (NHS) and a primary amine; (D) thiourea bond formation between an isothiocyanate and a primary amine; (E) thioether bond formation between a maleimide and thiol; (F) standard Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition (“click” reaction) between an azide and an alkyne; and (G) strain-promoted Diels–Alder “click” reaction between a tetrazine and transcyclooctene. | ||
The structure and physical properties of the radiometal–chelate complex have a large impact on the overall pharmacokinetic properties of a radiopharmaceutical, with many radiometal complexes being very hydrophilic and subsequently leading to rapid renal excretion when attached to small vectors like peptides and nucleotides (less prominent with large ∼150 kDa antibodies).6–11,179 It has been observed in peptide-conjugates that keeping the radiometal ion and peptide constant, and changing only the chelator can have drastic effects on biodistributions.25 Radiometal-based radiopharmaceuticals contain many synthetically exchangeable components, which can be separated into different modules: the radiometal, which changes the radioactive emission properties and half-life (γ for SPECT, β+ for PET, and β− or α particles, or Auger electrons, for therapy); the chelator, which must be carefully matched with the radiometal for optimal stability; the BFC–vector conjugation method, for different types of bioconjugation reactions and linkages; and the vector/targeting moiety, which allows for the selection of any known molecular target for site-specific delivery of the radioactive “payload” (Fig. 2).
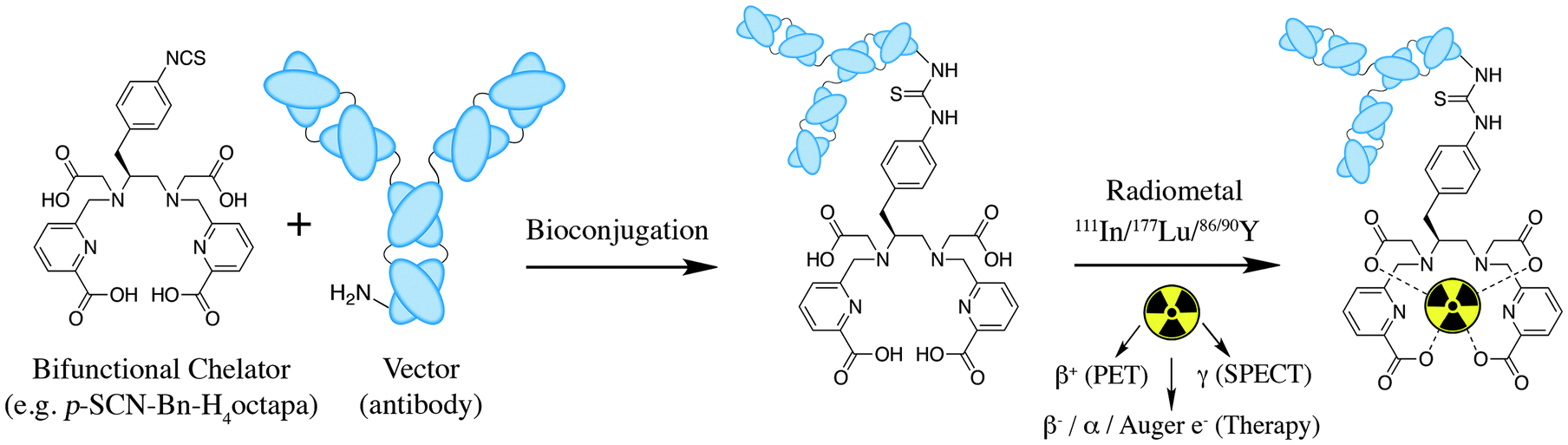 | ||
| Fig. 2 Illustration of an archetypal radiometal-based radiopharmaceutical agent containing a bifunctional chelator (BFC) conjugated to a targeting vector (e.g. antibody, peptide, nanoparticle) using a variety of conjugation methods (e.g. isothiocyanate–amine coupling, peptide coupling, maleimide–thiol coupling, activated ester amide coupling, click-coupling) and then radiolabeled with a radiometal ion (e.g.111In3+/177Lu3+/86/90Y3+). | ||
Each radiometal ion has different chemical demands, including ligand donor atom preferences (e.g. N, O, S, hard/soft), coordination number, and coordination geometry; however, there are many key design considerations that can be applied universally.26 Ligand synthesis should be relatively simple and avoid stereoisomers and non-enantio/diastereospecific reactions, modularity should be incorporated in BFC synthesis as much as possible to allow for incorporation of different bioconjugation handles, and modular synthesis also should allow for tuning of denticity and physical properties by changing the polarity and charge of the chelate (the degree of polarity can be assessed by octanol–water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P)), so that biodistribution properties can be adjusted.
P)), so that biodistribution properties can be adjusted.
The intention of this article is to act as a guide for researchers to find the optimal match of chelator with radiometal for radiopharmaceutical applications. The methods by which chelators are evaluated with different radiometals will be discussed, and the current “gold standard” chelators for each relevant radiometal ion will be identified. A large number of review articles with broader scopes have been written that discuss the application of radiometals in radiopharmaceuticals, covering topics ranging from chelators, coordination chemistry, synthetic methodologies, radiometal production, radiochemistry, bioconjugation strategies, automation, molecular targeting groups (vectors), imaging modalities, and therapeutics.3,13,15,17,18,26–51 Technetium chemistry is not covered here, because the chemistry of technetium, and the types of ligands used with it, differ drastically from those for the radiometals discussed here.26,35,52
2.1 Macrocyclic versus acyclic chelators
When designing new chelators, a historical glance at previous work reveals that macrocycles are generally more kinetically inert than acyclic chelators, even if their thermodynamic stabilities have been determined to be very similar.53–57 Macrocyclic chelators require minimal physical manipulation during metal ion coordination, as they possess inherently constrained geometries and partially pre-organized metal ion binding sites, thereby decreasing the entropic loss experienced upon metal ion coordination.58 To contrast this, acyclic chelators must undergo a more drastic change in physical orientation and geometry in solution in order to arrange donor atoms to coordinate with a metal ion, and subsequently they suffer a more significant decrease in entropy than do macrocycles (thermodynamically unfavorable). The thermodynamic driving force towards complex formation is therefore greater for macrocycles in general, a phenomenon referred to as the macrocycle effect.58 A crucial property where most acyclic chelators excel and most macrocycles suffer is in the coordination kinetics and radiolabeling efficiency. The ability to quantitatively radiolabel/coordinate a radiometal in less than 15 minutes at room temperature is a common property of acyclic chelators, whereas macrocycles often require heating to 60–95 °C for extended times (30–90 minutes).59–61 Fast room temperature radiolabeling becomes a crucial property when working with BFC-conjugates of heat sensitive molecules such as antibodies and their derivatives, or when working with short half-life isotopes such as 68Ga, 212/213Bi, 44Sc, and 62Cu.2.2 Matching chelators with radiometals – how are chelators evaluated?
When a new chelator is synthesized for the purpose of radiometal ion sequestration, or an old chelator is repurposed for use with a new radiometal ion, initial screening experiments are usually done by simple radiolabeling to determine a number of factors: whether the chelator can bind the radiometal ion and effectively radiolabel in high yields (quantitative is best), what temperature is required (ambient temperature is best), and what reaction time is required (faster is better). Short half-life isotopes like 68Ga are ideally matched with chelators that can radiolabel rapidly (fast radiolabeling kinetics). Longer half-life isotopes such as 111In and 177Lu allow for extended reaction times, but even if the half-life allows for long reaction times, completing the radiolabeling portion of radiopharmaceutical preparation is most convenient if finished in less than 10 or 15 minutes. As previously mentioned, room temperature radiolabeling is crucial for sensitive antibody vectors, which are degraded at elevated temperatures. DOTA inconveniently requires elevated temperatures for radiolabeling with essentially all radiometals (e.g.44Sc, 111In, 177Lu, 86/90Y, 225Ac) but its abundant application in radiochemistry for decades, its exceptional in vivo stability, and the commercial availability of many different bifunctional DOTA derivatives and vector conjugates means that it is likely the most commonly used chelator to this day. Moving forward to the design and testing of new chelators, fast room temperature radiolabeling kinetics should be a priority; however, fast kinetics of radiometal incorporation (on-rate) and consequently low energetic barriers to radiometal–chelate complexation can also mean fast radiometal decorporation (off-rates) and low energetic barriers to radiometal release (Fig. 3). An arduous balancing act is required to obtain the best set of chelate properties for each application and radiometal ion, requiring the study and availability of a broad selection of different chelators with a variety of properties from which to choose.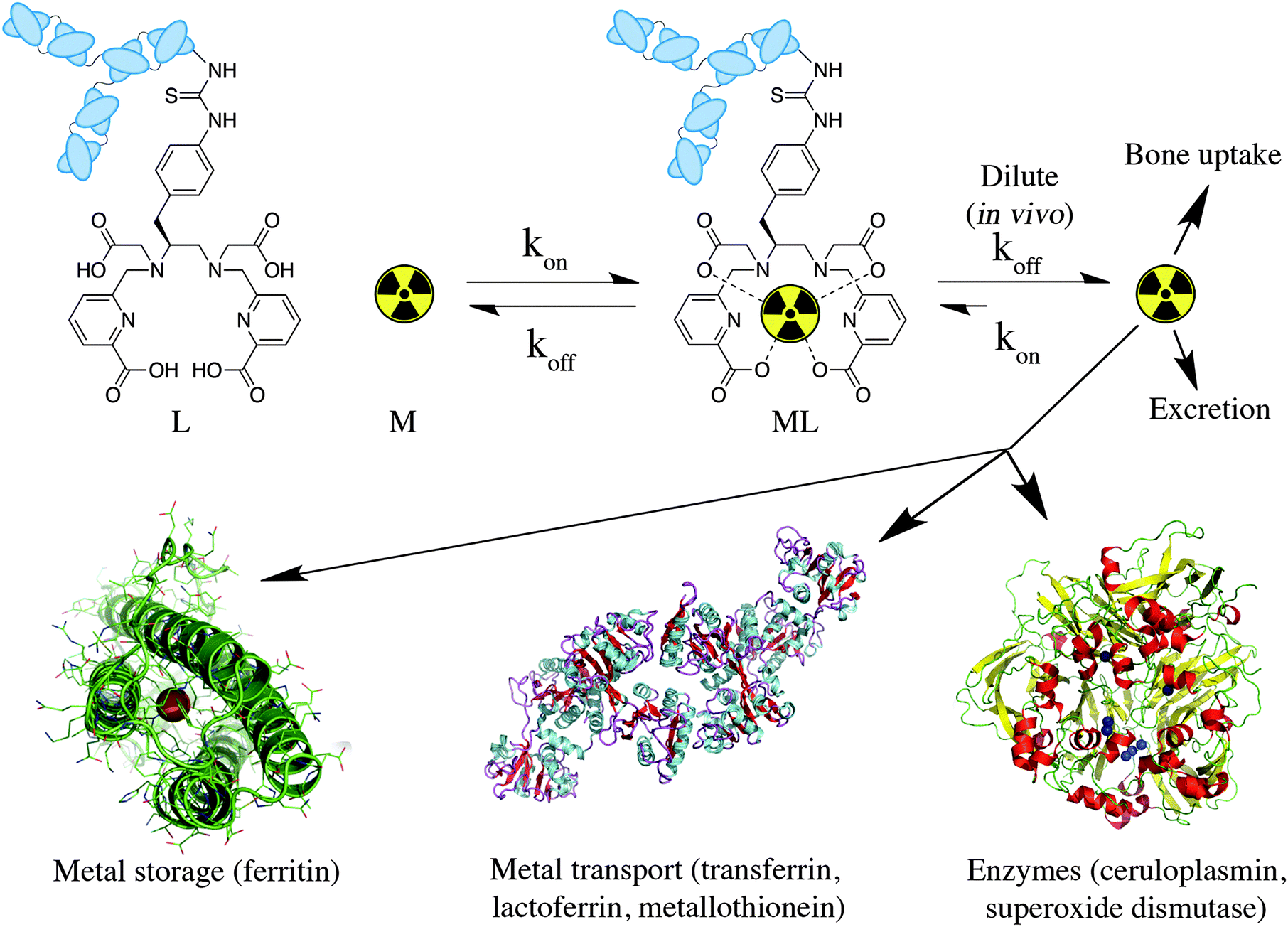 | ||
| Fig. 3 Cartoon depiction of metal ion coordination kinetics, enhanced off-rate kinetics in vivo (extremely dilute conditions), and possible routes of radiometal ion loss in vivo (solid-state structures of ferritin H-chain homopolymer PDB file 1FHA, ceruloplasmin PDB file 2J5W, and apo-transferrin PDB file 2HAV shown). | ||
When a chelator is identified through early screening to possess radiolabeling properties that are suitable for use with a particular radiometal ion, it must then be experimentally determined to be highly stable and inert. Further experiments are performed with the specific radiometal–chelate complex under conditions relevant to in vivo translation to judge its potential as the core component of a radiometal-based radiopharmaceutical. The result of radiometal loss from a radiopharmaceutical in vivo is the non-targeted distribution of the “free” radiometal ion in the body, and its exact fate and distribution in the body depends on the properties and biological behavior of the specific radiometal ion in question (Fig. 3). For example, 89Zr and 68Ga are known to accumulate in the bone when released from a BFC, where 64Cu is known to accumulate in the liver. The fate of these radiometal ions can be tracked using PET/SPECT imaging in the living animal, and/or biodistribution experiments where animals are euthanized at predetermined time points, their organs harvested, and the distribution of radioactivity measured and calculated for the percentage of injected dose of radioactivity per gram of tissue (%ID/g). Each metal ion has its own unique properties that must be considered when constructing a radiometal-based imaging/therapeutic agent, such as its aqueous hydrolysis chemistry, redox chemistry, and affinity for native biological chelators.
2.3 Thermodynamic stability
When evaluating and selecting a chelator to match with a specific radiometal ion for use in a radiopharmaceutical, kinetic inertness in vivo is ultimately the most crucial consideration, even beyond that of the absolute thermodynamic stability of the metal–chelate complex. Thermodynamic stability/formation constants (KML = [ML]/[M][L]) are usually experimentally determined by potentiometric and/or spectrophotometric titrations, but evaluating kinetic inertness under conditions relevant to in vivo applications can be much more problematic. Thermodynamic stability constants can be a useful metric for preliminary comparisons of various chelators with a particular metal ion, but they do not predict in vivo stability with any level of competence.62,63 A thermodynamic parameter that provides more biologically relevant information than KML values is the pM value (−log[M]Free).64–67 The pM value is the negative log of the concentration of free metal ion uncomplexed by a given chelator under specific conditions. The pM value is a condition-dependent value that is calculated from the standard thermodynamic stability constant (logKML), accounting for variables and conditions such as ligand basicity, metal ion hydrolysis, (physiological) pH, and ligand:metal ratio.
Stability constants and pM values provide a number for the direction and magnitude of the equilibrium in a metal–chelate coordination reaction under specific conditions, but give no kinetic information (e.g. off-rates for dissociation).68,69 This is a very important factor because the rate of dissociation in vivo is what governs the kinetic inertness of a radiometal complex, regardless of thermodynamic stability, and these off-rates are greatly influenced by the high dilution encountered in vivo when a tiny quantity of radiopharmaceutical (micro- or nano-grams) is diluted into the blood pool (Fig. 3). Even more complicating is the abundant presence in the body of many strong native biological chelators and competing ions that can transchelate radiometals from BFC-conjugates. These are often present in higher concentrations than is the radiopharmaceutical, and include transport proteins such as transferrin, ceruloplasmin, and metallothionein, storage proteins like ferritin, and metal containing enzymes such as superoxide dismutase (Fig. 3). This wide range of complicating factors means that a single in vitro assay is typically not accurate in predicting in vivo stability.
2.4 Kinetics – acid dissociation and competitive radiolabeling
Acid dissociation experiments can be used to measure and assess the relative kinetic inertness of a metal complex to acidic conditions. Most complexes are found to de-ligate fairly quickly below pH 2.0,70 and experiments evaluating the rate of dissociation at a constant pH (e.g. 2.0) have been performed to compare and evaluate chelators with a particular metal/radiometal ion.55,70,71 In these acid dissociation experiments, decomplexation can be observed over time with techniques such as high-performance liquid chromatography (HPLC), thin-layer chromatography (TLC), and nuclear magnetic resonance (NMR) for diamagnetic metal complexes. With the exception of copper chelates,68,72 acid dissociation experiments are not commonly performed as they do not provide an accurate prediction of in vivo kinetic inertness, because low pH is not encountered in the blood or most organs (except the stomach) and radiometal dissociation typically occurs via transchelation to serum proteins and enzymes, and is not acid-mediated.69,73–77Competitive radiolabeling experiments can be performed by adding to a radiolabeling mixture an excess of non-radioactive ions, such as Na+, K+, Ca2+, Mg2+, Cu2+, Zn2+, or Fe3+, followed by addition of a chelator to evaluate the impact of these competing ions on radiolabeling yields.55,78–81 These experiments can reveal the radiolabeling selectivity of a chelator for a specific radiometal ion in the presence of other biologically relevant competing ions. Alternatively, if the radiometal complex is preformed under standard metal-free radiolabeling conditions, and is then added to a mixture containing these competing ions, stability to transchelation can be assessed. Because chelate-based radiopharmaceuticals are typically radiolabeled in strictly metal-free conditions (deionized ultrapure water, often passed through a metal-scavenging chelex resin), these experiments may not appear completely relevant for predicting in vivo stability and utility. Depending on the method of radiometal production and purification, the specific activity of radiometal ions can vary greatly, as can the concentrations of impurity metals ions.5 Radiometals are used in very small quantities and under extremely dilute conditions; therefore, any impurity metal ions present (even if only a few nanomoles) may actually be in excess of the radiometal ion concentration, and competitive binding could be problematic.5,82 The presence and quantity of metal ion impurities in radiometal mixtures depends on the source of the radiometal ion, method of production, and purification.5,82
2.5 In vitro and in vivo stability
Experiments that are more pertinent to in vivo translation are metal-exchange competitions with biologically relevant mixtures, including blood serum, apo-transferrin, superoxide dismutase, and hydroxyapatite (bone).18,83–88 By incubating radiometallated chelators with these different competition mixtures, the quantity of radiometal that is transchelated from chelator to serum proteins/enzymes can be evaluated over time using size exclusion HPLC, iTLC, or disposable PD10 size exclusion columns.18,83–88 These experiments provide a directly relevant measure of stability and kinetic inertness by competition with the most likely transchelation culprits in vivo; additionally, in these in vitro assays the concentrations of these biological reagents can often be elevated above normal physiological levels to provide a more stringent challenge.Ultimately the most relevant and practical test of radiopharmaceutical stability is in vivo. Biodistribution studies in healthy mice can be performed on “naked” radiometal–chelate complexes (no conjugated vectors) to assess clearance and uptake profiles and ensure no abnormal organ distribution or critical instability occurs. If a radiometal–chelate complex is very stable in vivo, the complex is typically cleared quickly from the animal through the kidneys/bladder/urine or digestive system/liver/feces depending on polarity and metabolism.89 Unstable complexes often demonstrate persistent uptake in organs and tissues where the non-bound radiometal is known to associate (e.g. Zr4+ and lanthanides in the bone, Cu2+ in the liver).89 The major drawback to this type of experiment with “naked” chelate complexes is that highly polar and charged radiometal–chelate complexes are typically cleared very quickly from the body, and therefore do not persist in vivo for long enough to encounter any significant challenge to their structural integrity.84,85 Conversely, highly lipophilic complexes tend to accumulate in the liver and digestive tract, regardless of stability.84,85
To evaluate properly the stability and kinetic inertness of a chelate in vivo, a suitable vector must be attached (e.g. peptide, antibody, nanoparticle) so that the radiometal–chelate complex is made to persist in blood circulation for a substantial period of time, and can be monitored over several hours or days (depending on isotope half-life and the subsequent imaging/therapy window).90 An additional concern with in vivo experiments is the significant normal variance between animals that introduces error; for example, 10 mice of the same sex and breed, procured from the same supplier, will often show significant differences in biodistribution of the same radiopharmaceutical. Additionally, the specific experimental techniques and methods (e.g. radiopharmaceutical preparation, injection, animal dissection, organ counting) used for these biodistribution experiments can cause large variability between data sets. Variables such as specific activity of the source radiometal, specific activity of the radiolabeled agent, radiolabeling temperature, purity of the final radiopharmaceutical preparation, injected mass of radiopharmaceutical, and injected volume can make large differences in biodistribution profiles. The same radiopharmaceutical used at different institutions may offer drastically different tumor uptake and organ distribution values, if the above variables are not tightly controlled, and even then differences between animals and between experimental techniques can introduce large variances. For these reasons it is crucial to include internal control experiments for every study; for example, evaluation of a new chelator in vivo should be done in parallel with an existing and established “gold standard” chelator so that a direct comparison can be made because comparison to previously performed studies (even in the exact same animal model and/or cell line) is often not reliable due to the above mentioned complications.79,83,85,91
3. A selection of chelators and their most suitable radiometal companions
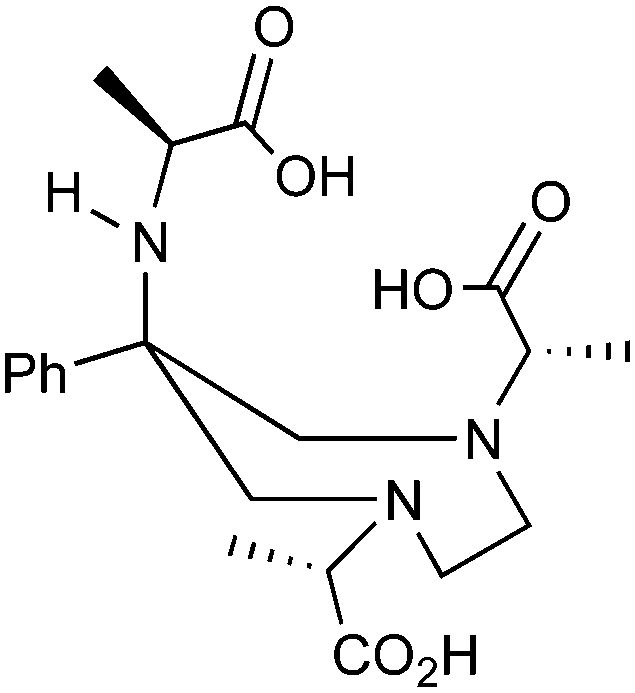
A survey of radiometal chelators has been undertaken, with the most relevant and promising examples being discussed. The most suitable chelate–radiometal matches have been identified, and detailed tables included, with the aim of providing a quick reference for those looking to find the optimal match between chelator and radiometal. A color-coding scheme has been used, and justification for the assignment of good (green), fair (orange), and poor (red) matches between chelators and radiometals can be found in the text and the provided references. The color code indicates the authors' opinions on the general suitability of a chelator for use with a specific radiometal, accounting for factors such as radiolabeling conditions and in vitro/in vivo stability. An assignment of green may suggest that either a chelator is currently the “gold standard” for use with a given radiometal, or that early work with a new chelator looks very promising and in vivo studies have shown that it works comparably to the current “gold standards”. Assignment of orange to a chelator–radiometal pair may be made if the combination has been used in vivo successfully, but perhaps in recent years has been surpassed by a new and superior chelator and is no longer the best choice. Also, an assignment of orange could mean that a new chelator–radiometal ion pair looks very promising, but perhaps a bifunctional derivative has yet to be synthesized, or only preliminary in vivo work has been done and more study is required (e.g. no study of bioconjugates, or no direct comparisons to existing “gold standards” for internal reference). An assignment of red indicates that the highlighted chelator–radiometal ion pair is an unsuitable match, and generally should not be used due to severe instability or radiolabeling problems. These color “ratings” were made using the referenced studies and reflect the current trends in radiochemistry and nuclear medicine, with the goal of acting as a quick guide for those seeking to find an optimal chelate–radiometal ion pairing.3.1 DOTA
DOTA is one of the primary workhorse chelators for radiometal chemistry, and is one of the current “gold standards” for a number of isotopes, including 111In, 177Lu, 86/90Y, 225Ac, and 44/47Sc. DOTA has been extensively used with 67/68Ga, but is widely accepted to be less stable than its more petite macrocyclic counterpart NOTA (Table 2). 68Ga-DOTATOC has been shown to exhibit superior in vivo properties to 111In-DOTATOC (Octreoscan) despite non-optimal stability.92 Even though a chelator may be more stable and inert with a given radiometal (e.g. NOTA vs. DOTA for 68Ga), it may not be the optimal choice for a certain application (e.g. a specific peptide vector). For example, NOTA is widely accepted to form a more stable complex with 68Ga than does DOTA, but due to differences in charge and physical properties (e.g. neutral vs. charged complex), DOTA may provide superior in vivo properties with certain vectors.25,79 This example highlights the complex set of variables one must consider when constructing radiometal-based radiopharmaceuticals.KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
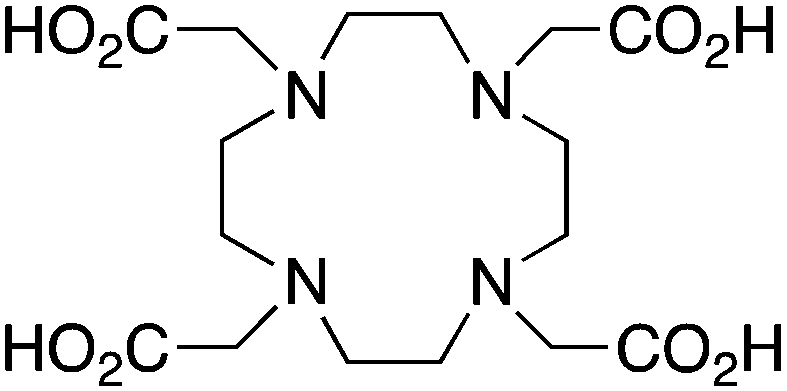
DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, maximum CN = 8, donor set N4O4 |
64Cu2+ |

|
25–90 °C, 30–60 min, pH 5.5–6.5 | 22.2, 22.7 | Distorted octahedron | 84, 87, 90, 104–110 |
| 67/68Ga3+ |

|
37–90 °C, 10–30 min, pH 4.0–5.5 | 21.3 (pM 15.2, 18.5) | Distorted octahedron | 64, 92, 110–118 | |
| 44/47Sc3+ |

|
95 °C, 20–30 min, pH 4.0 | 27.0 (pM 26.5) | Square antiprism | 96–98 | |
| 111In3+ |

|
37–100 °C, 15–60 min, pH 4.0–6.0 | 23.9 (pM 17.8–18.8) | Square antiprism? | 64, 85, 93, 114, 117, 119–124, 179 | |
| 177Lu3+ |

|
25–100 °C, 15–90 min, pH 4.0–6.0 | 23.5, 21.6 (pM 17.1) | Square antiprism | 124–130 | |
| 86/90Y3+ |

|
25–100 °C, 15–90 min, pH 4.0–6.0 | 24.3–24.9 | Square antiprism | 55, 70, 93, 124, 127, 130–132 | |
| 213Bi3+ |

|
95–100 °C,133 5 min, pH 6.0–8.7 | — | Square antiprism | 134, 135 | |
| 212Pb2+ |

|
25–75 °C, 30–60 min, pH 4.0–5.5 | — | Square antiprism | 127, 136–138 | |
| 225Ac3+ |

|
37–60 °C, 30–120 min, pH 6.0 | — | Square antiprism? | 100, 101 | |
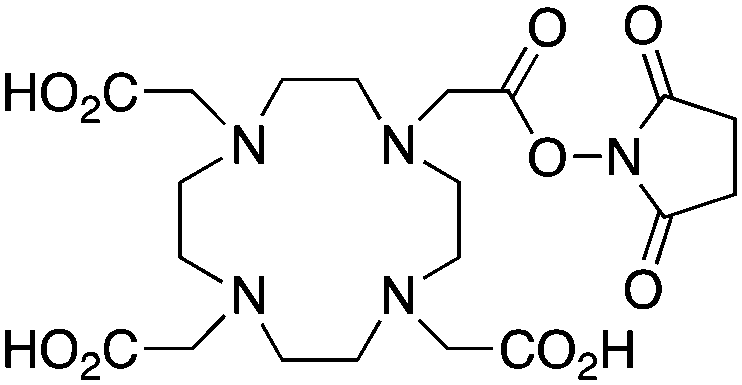
DOTA-NHS-ester139 |
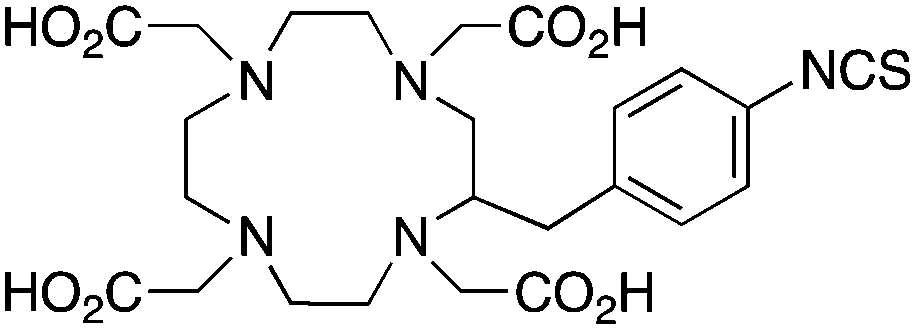 p-SCN-Bn-DOTA (C-DOTA)95,140,141
p-SCN-Bn-DOTA (C-DOTA)95,140,141
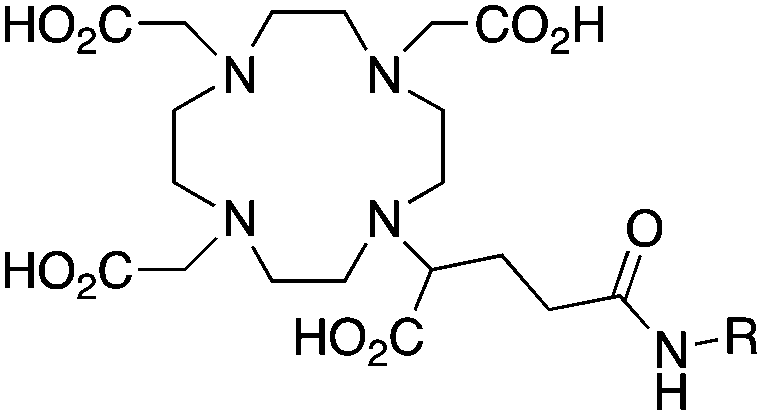
|
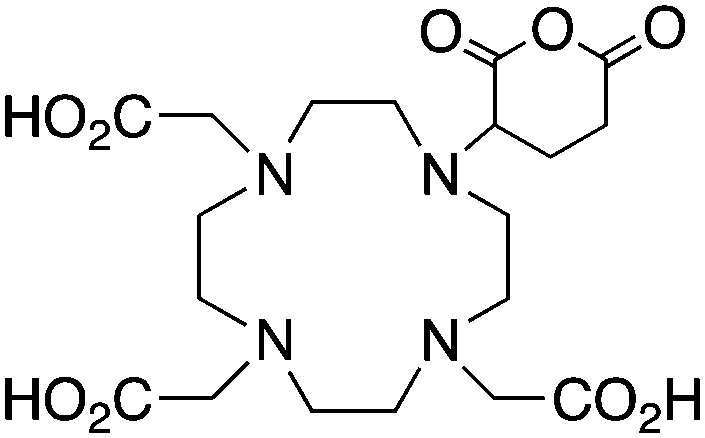
DOTAGA, R = amide, NHS ester142 |
DOTAGA-anhydride143 |
|||
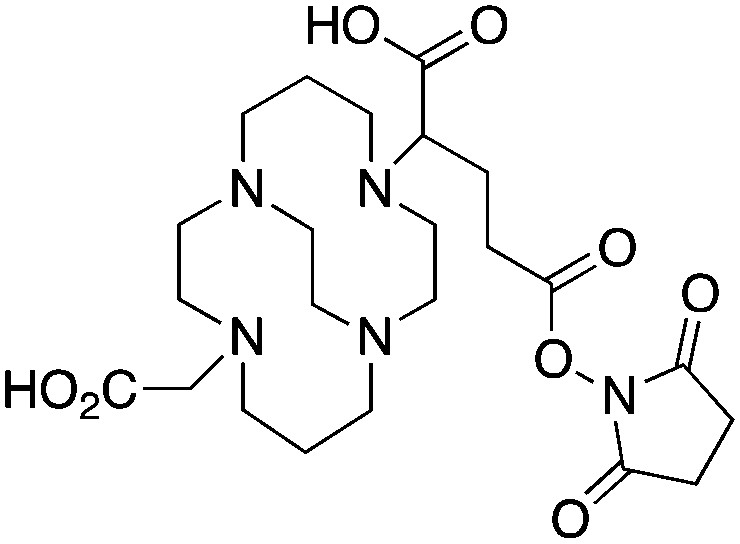
When designing new BFCs or modifying existing chelators to make BFC derivatives, care should be taken not to disrupt the original coordination sphere of the chelator. Some common bifunctional DOTA derivatives utilize one of the carboxylic acid arms for the site of vector conjugation, effectively blocking one of the coordinating carboxylate arms (amide carbonyl groups may still coordinate, albeit weakly) (Table 2).59 DOTAGA, DOTASA, and various isothiocyanate derivatives of DOTA have been synthesized and solve this problem by conjugating to vectors through the carbon backbone and side-arm functionalization (Table 2).59,93–95 These bifunctional DOTA derivatives retain their maximum potential denticity (octadentate) as well as the same thermodynamic stability and kinetic inertness as unadulterated DOTA.59,93,94
Despite its slow radiolabeling kinetics and required heating, DOTA is currently the “gold standard” chelator for use with 111In, 177Lu, and 86/90Y (Table 2). The coordination chemistry and properties of Y3+ and Lu3+ are very similar; they both preferentially form 8–9 coordinate complexes in square antiprismatic or monocapped square antiprismatic geometries, and are hard metal ions with a preference for hard ligand donors such as carboxylate-oxygens and amine-nitrogens. In3+ has similar properties to Y3+ and Lu3+, but being smaller forms 7–8 coordinate complexes, typically with square antiprismatic geometry. According to acid dissociation experiments performed at pH 2.0 with the 90Y complexes of DOTA, DTPA (vide infra), and CHX-A′′-DTPA, DOTA is the most kinetically inert to acid dissociation by a significant margin, with CHX-A′′-DTPA showing moderate stability, and DTPA decomposing almost immediately.55 The validity of acid dissociation experiments is questionable, as conditions such as these are never encountered in vivo (except for the stomach), but they are a quantifiable metric for measuring off-rate kinetics and further substantiates the place of DOTA as the current “gold standard” chelator for 111In, 177Lu, and 86/90Y.55
Although very little investigation of this isotope has been performed, DOTA is also the current chelator of choice for 44Sc, although the requisite high temperature (90 °C) radiolabeling conditions are not optimal.96–98 As 44Sc has only recently become more popular in the literature, little in vivo work has been performed, and new chelators (preferably with ambient temperature radiolabeling properties) are of strong interest. Another isotope of recent interest for use with DOTA is the α-emitter 225Ac, which is a large actinide isotope that possesses no non-radioactive isotopologue, making study of its coordination chemistry difficult. The large macrocycles HEHA and PEPA (vide infra) had previously been used most extensively with 225Ac, but recently DOTA has been shown to have superior in vivo properties.99 The long half-life of 225Ac (10.0 days) lends well to radioimmunotherapy (RIT), and so the high temperatures required for 225Ac radiolabeling with DOTA are very unfavorable for sensitive antibodies. The current method used for synthesizing 225Ac–DOTA–antibody conjugates is to radiolabel the BFC p-SCN-Bn-DOTA at 60 °C with 225Ac first, followed by antibody conjugation (thiourea coupling) and purification.100,101 It is important to realize that the isothiocyanate functionality in the BFC p-SCN-Bn-DOTA is very sensitive and degrades rapidly under these conditions. A study comparing the therapeutic effects of 225Ac, 213Bi, and 90Y using an antibody vector demonstrated 225Ac to be the most effective, with renal toxicity from the daughters 221Fr and 213Bi being the most significant concern.102 A recent clinical trial in humans with 225Ac-DOTA-HuM195 (humanized anti-CD33 monoclonal antibody) has shown promise, with DOTA being the current “gold standard” for 225Ac.100,103 A high denticity acyclic chelator (CN = 8–10) that could match the in vivo stability of DOTA and radiolabel at room temperature would be of great utility and would offer a more streamlined radiopharmaceutical preparation than the one outlined above for p-SCN-Bn-DOTA.
3.2 DOTA derivatives: CB-DO2A, 3p-C-DEPA, TCMC, and Oxo-DO3A
CB-DO2A has only been investigated with the radiometal 64Cu for radiochemical use (Table 3).68,144 An X-ray crystal structure of CB-DO2A with Ga3+ is available and shows a distorted octahedral coordination geometry, and acid stability experiments have been performed with Ga3+, revealing impressive acid inertness.145 Although very little work has been done with CB-DO2A, studies with the non-bifunctional 64Cu(CB-DO2A) have suggested that it had superior in vivo stability to DOTA and TETA, but inferior stability to CB-TE2A.139 Further comparision of CB-DO2A to CB-TE2A, DOTA, and TETA revealed that CB-TE2A possessed superior acid and electrochemical inertness, which explains why DOTA and its cross-bridged derivative CB-DO2A have been replaced by CB-TE2A and are no longer a prominent subject of current research with 64Cu.56 Another interesting DOTA derivative is Oxo-DO3A, which is a heptadentate N3O4 macrocycle that has shown improved radiolabeling kinetics and stability with 64Cu and 67/68Ga compared to DOTA (Table 3). The p-NH2-Bn-Oxo-DO3A derivative is shown in Table 3, which can be directly coupled to a peptide via standard peptide coupling reactions, or transformed to a benzyl-isothiocyanate.KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
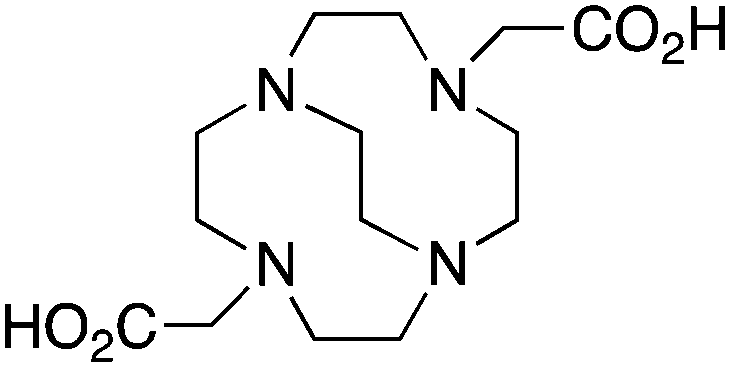
CB-DO2A, 4,10-bis(carboxymethyl)-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane, N4O2, CN = 6 |
64Cu2+ |

|
80 °C, 30–60 min, pH 5–7 | — | Distorted octahedron | 68, 144 |
| 67/68Ga3+ |

|
— | — | Distorted octahedron | 145 | |
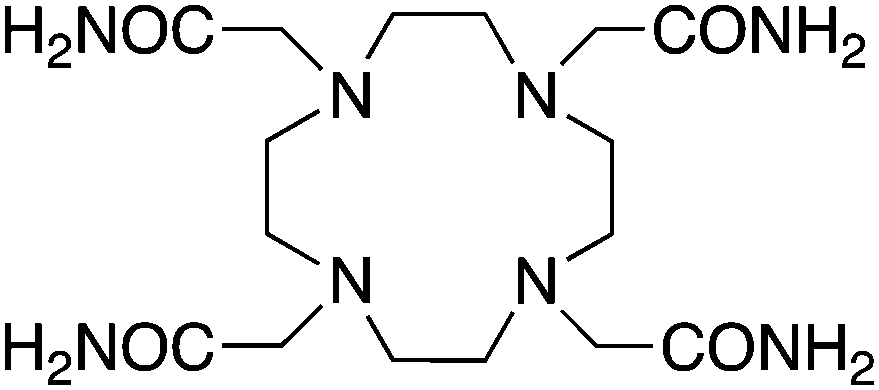
TCMC, 1,4,7,10-tetrakis(carbamoylmethyl)-l,4,7,10-tetraazacyclododecane, N4O4, CN = 8 |
212Pb2+ |

|
37 °C, 30–60 min, pH 5–6.5 | >19 | Square antiprismatic | 138, 147, 148, 150, 153, 154 |
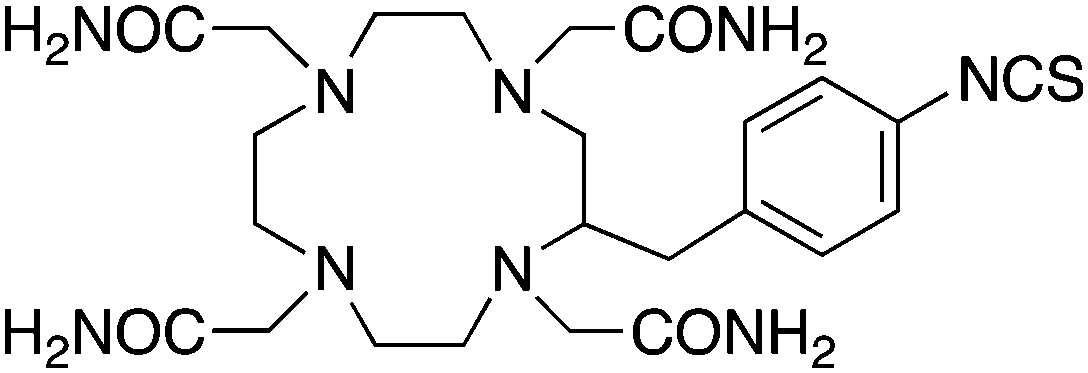
p-SCN-Bn-TCMC150 |
||||||
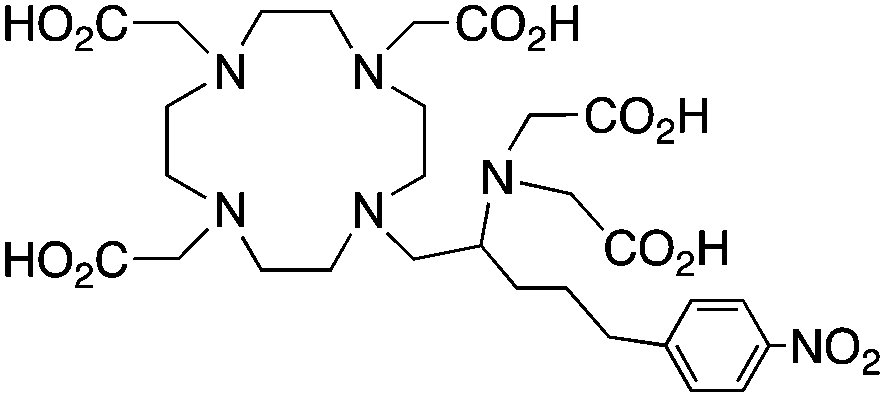
3p-C-DEPA, N5O5, CN = 10 |
212/213Bi |

|
25 °C, 5–10 min, pH 5.5 | — | Square antiprismatic | 152 |
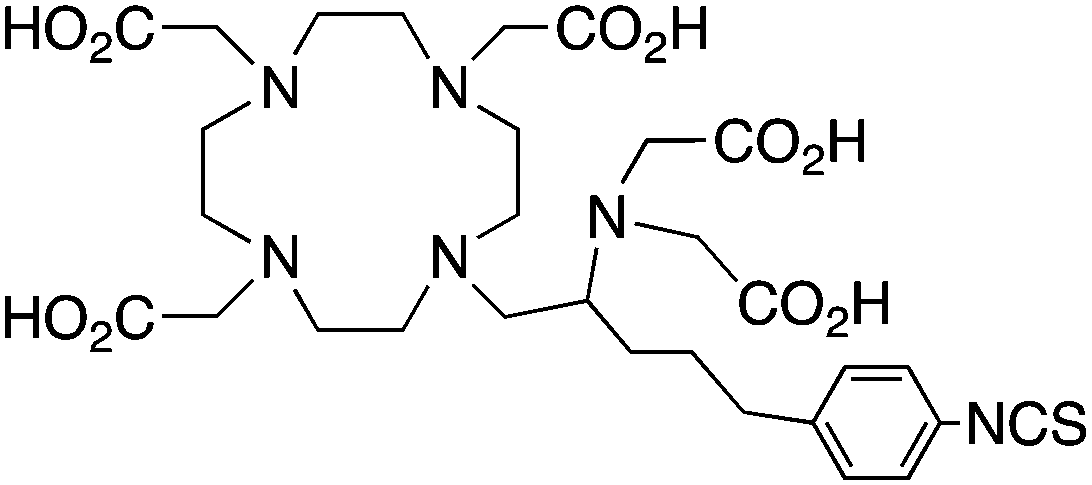
3p-C-DEPA-NCS152 |
3p-C-DEPA = 2-[(carboxymethyl)][5-(4-nitrophenyl-1-[4,7,10-tris-(carboxymethyl)-1,4,7,10-tetraazacyclododecan-1-yl]pentan-2-yl)-amino]acetic acid | |||||
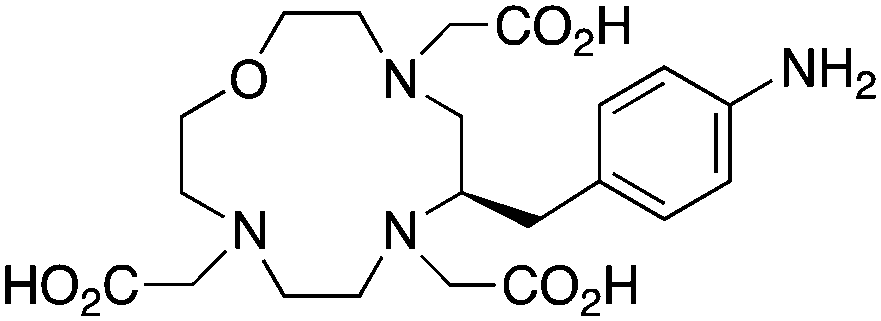
p-NH2-Bn-Oxo-DO3A, N3O4, CN = 7 |
64Cu2+ |

|
25 °C, 10 min, pH 5.5 | — | Distorted octahedron? | 86, 87, 155, 156 |
| 67/68Ga3+ |

|
25 °C, 10 min, pH 4–5 | — | Distorted octahedron? | 86, 157, 158 | |
Although DOTA has been used successfully with 212Pb, its slow radiolabeling kinetics and stability properties were not ideal.146 Replacement of the carboxylic acid donor arms of DOTA with amide arms has produced the chelator TCMC (Table 3), which to date is the best chelator available for 212Pb.147–149 Chappell et al. have shown TCMC to be superior to C-DOTA (Table 2) with 212Pb due to improved radiolabeling kinetics and in vivo stability.150,151 Although not optimal for 212Pb, 3p-C-DEPA (Table 3) has been shown to be an excellent chelator for 212/213Bi, with superior properties to C-DOTA.152 The DOTA derivative 3p-C-DEPA has demonstrated greatly improved radiolabeling kinetics with 212/213Bi compared to DOTA, with comparable in vivo stability.152 3p-C-DEPA can be considered to be one of the current “gold standards” for 212/213Bi; however, the NOTA analogue 3p-C-NETA (vide infra) also looks very promising and has not been directly compared with 3p-C-DEPA, and so the superior of the two has yet to be identified.
3.3 TETA
TETA is an octadentate N4O4 macrocyclic chelator that has only been investigated with 64Cu for radiopharmaceutical applications (Table 4). TETA is generally not used anymore, and has been superseded by newer generation chelators such as NOTA, CB-TE2A, and CB-TE1A1P/CB-TE2P (Table 4) that are more stable and kinetically inert in vivo. Copper is a difficult metal ion to harness, as it is quite labile, has active redox chemistry between Cu1+/Cu2+in vivo, and has a fast water exchange rate of 2 × 108 s−1.37,159 Copper(II) is a first row transition metal with a d9 electron configuration, is best used with hexadentate chelators that saturate its coordination sphere, and has a preference for borderline soft ligand donor atoms such as amines, imines, and thiols.15,37 Despite being made obsolete in recent years, TETA has previously been used successfully for 64Cu imaging with vectors like octreotide.160,161 TETA can radiolabel with 64Cu at room temperature, but it lacks kinetic inertness and overall stability in vivo. Surprisingly, the TETA derivative TE2A (Table 4) shows enhanced kinetic inertness, despite the fact that TE2A is merely a hexadentate derivative of TETA that is missing two carboxylic acid arms.162 This effect is enhanced even further when the two secondary amines of TE2A are methylated or cross-bridged by an ethylene unit (Table 4).144,163 Unlike DOTA (cyclen-based), TETA (cyclam-based) has not been heavily investigated with any radiometals other than 64Cu; however, some structural and physical data is available with other metal ions (Table 4).KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
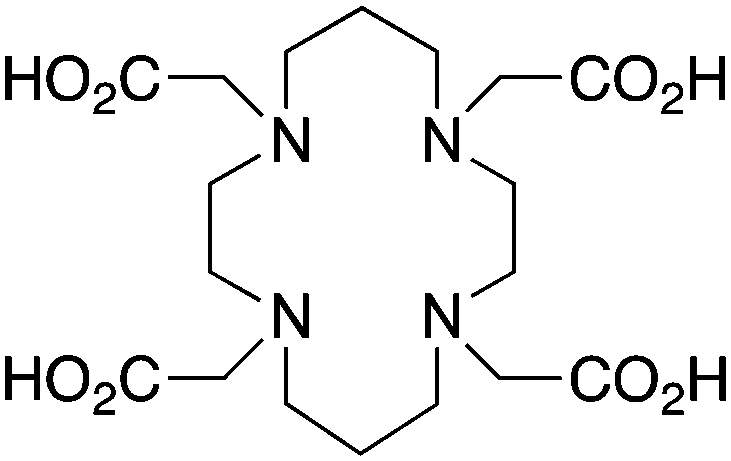
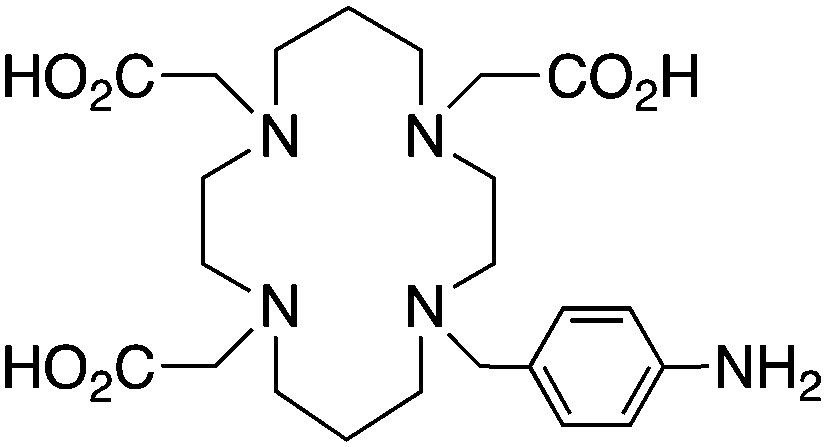
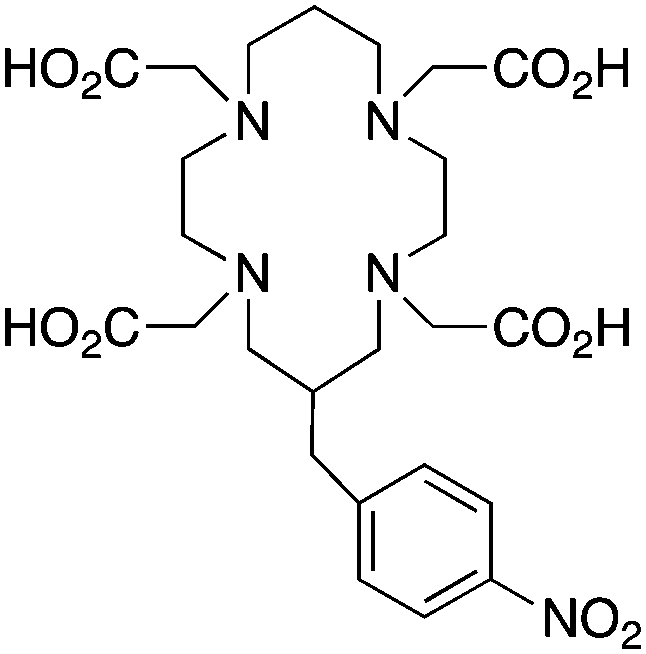
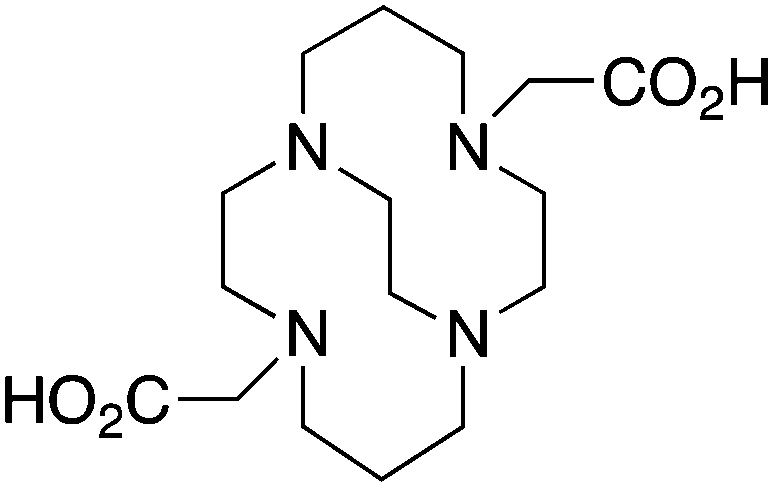
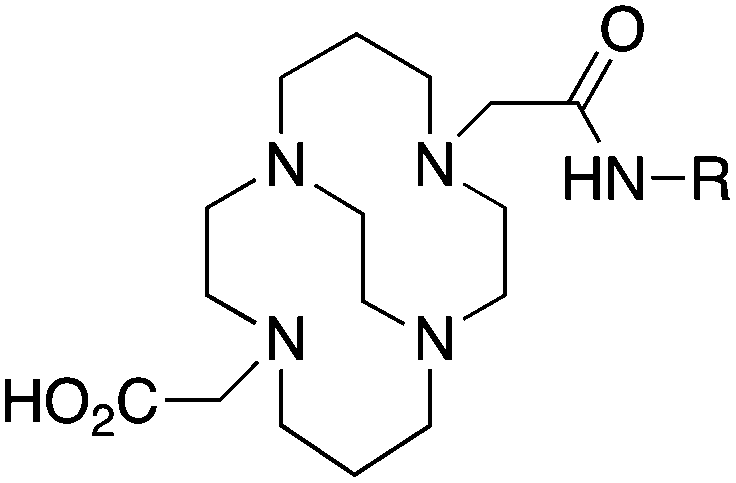
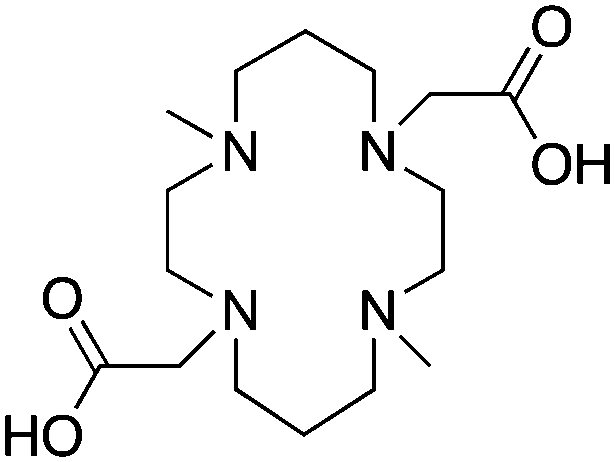
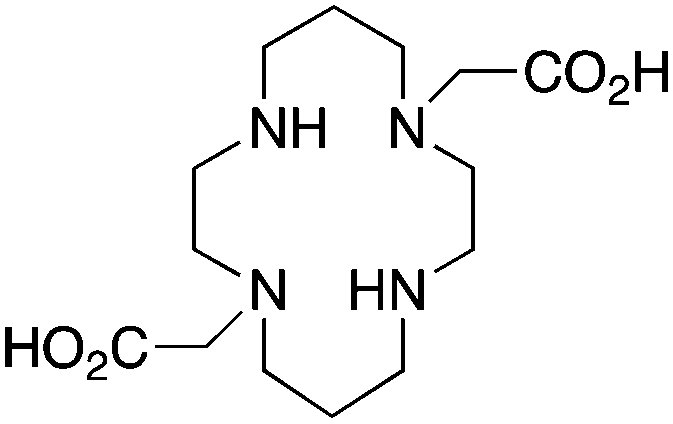
TETA, 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid, N4O4 CN = 8 |
64Cu2+ |

|
25 °C, 60 min, pH 5–7 | 21.9, 21.6 | Distorted octahedron | 160, 169 |
| 67/68Ga3+ |

|
— | 19.74 (pM 14.1) | Distorted octahedron | 64 | |
| 111In3+ |

|
— | 21.9 (pM 16.3) | Distorted octahedron? | 64 | |
| 177Lu3+ |

|
— | 15.3 | Distorted dodecahedron? | 170 | |
| 86/90Y3+ |

|
— | 14.8 | Distorted dodecahedron? | 170 | |
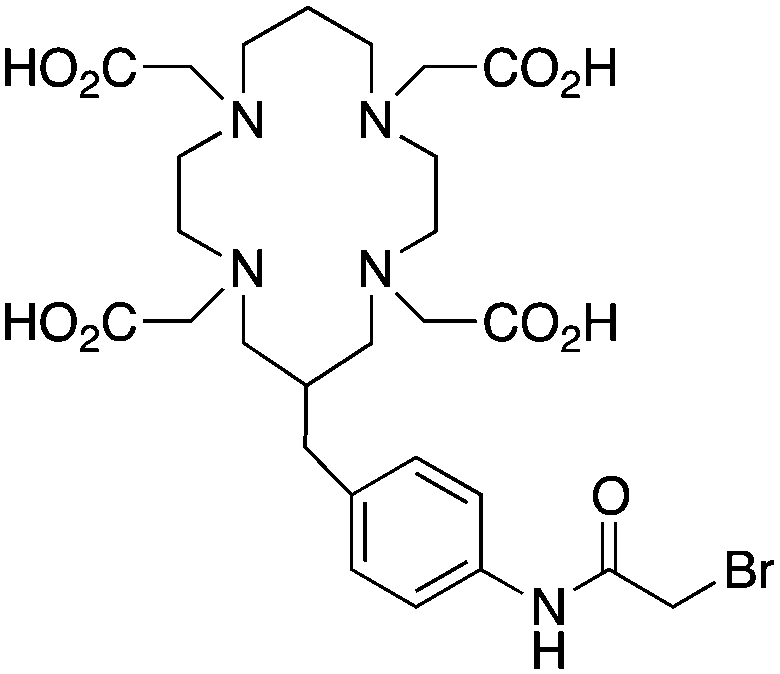
BAT (C-TETA derivative)171 |
 p-NH2-Bn-TE3A172
p-NH2-Bn-TE3A172
|
 C-TETA (p-NO2-Bn-TETA)173
C-TETA (p-NO2-Bn-TETA)173
|
||||
CB-TE2A, 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane, N4O2 CN = 6 |
64Cu2+ |

|
95 °C, 60 min, pH 6–7 | — | Distorted octahedron | 68, 90, 106, 108, 164, 166–168, 174, 175 |
|
 176
176
|
|||||
CB-TE1A1P164,177 |
 CB-TE2P164,177
CB-TE2P164,177
|
 MM-TE2A163
MM-TE2A163
|
 DM-TE2A163
DM-TE2A163
|
 TE2A162
TE2A162
|
||
3.4 TE2A, CB-TE2A, CB-TE1A1P, CB-TE2P, MM-TE2A, DM-TE2A
The cyclam-based macrocycle TETA has spawned a large number of successful derivatives (Table 4). Despite the requisite harsh radiolabeling conditions (95 °C), CB-TE2A has become one of the premier 64Cu chelators, having superior in vivo stability to TETA, TE2A, DOTA, NOTA, and CB-DO2A.144 The stability gains from adding an ethylene cross bridge to TETA/TE2A are significant, despite the removal of two coordinating carboxylic acid arms, and a subsequent decrease in denticity from 8 to 6. In order to surmount the harsh radiolabeling conditions required for CB-TE2A, several derivatives of CB-TE2A have been synthesized. Improved radiolabeling kinetics have been achieved by replacement of one (CB-TE1A1P) or both (CB-TE2P) carboxylic acid arms with methylenephosphonate groups, with in vitro and in vivo stability being retained or enhanced compared to the native CB-TE2A (Table 4).144,164–168 Most importantly, the methylenephosphonate derivatives CB-TE1A1P and CB-TE2P can be radiolabeled with 64Cu at room temperature, although relatively slowly. Another noteworthy set of derivatives were made by methylating one (MM-TE2A) or two (DM-TE2A) nitrogen atoms of TETA/TE2A, instead of linking them together with an ethylene bridge (Table 4).163 Surprisingly, these methylated TE2A derivatives possessed similar radiolabeling kinetics and stability to CB-TE2A.163 This observation suggests that the high stability of CB-TE2A is not a result of steric protection by the cage-like structure created by the cross-bridged ethylene unit, but instead by changing the electronic properties of the nitrogen atoms (secondary to tertiary amines) and their donor properties. Although CB-TE2A has been the most heavily investigated ligand of the above mentioned TETA derivatives, the new methylenephosphonate derivatives CB-TE1A1P and CB-TE2P show the most promise, as they appear to retain the stability of CB-TE2A, while having greatly enhanced radiolabeling kinetics.3.5 Diamsar and derivatives
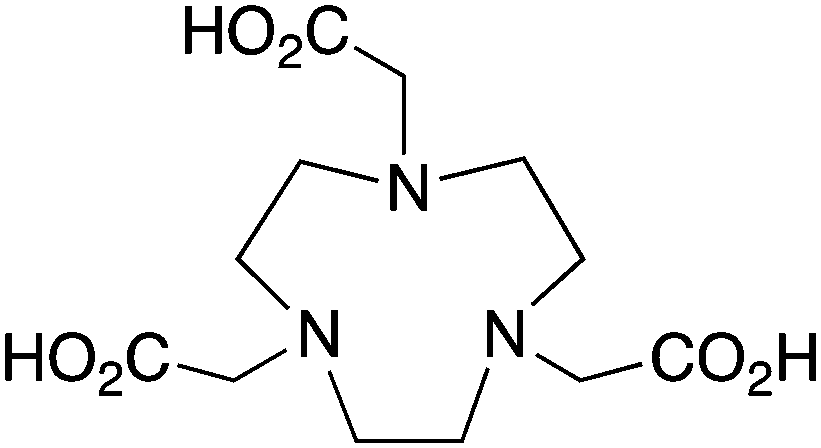
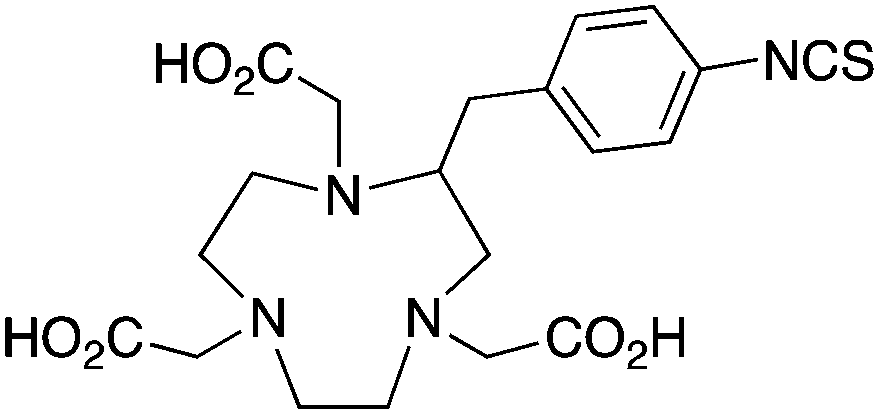
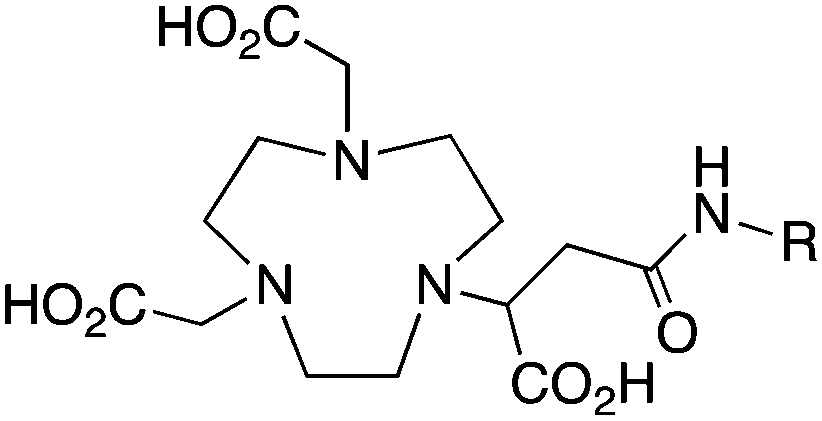
The radiometal ion 64Cu2+, similar to 68Ga3+, has been extremely popular in recent years due to its favorable positron emission (β+) properties for PET imaging and its intermediate half-life (12.7 hours), which is suitable for use with peptides, antibody fragments, and even whole antibodies. Unlike 68Ga, 64Cu also undergoes β− emission, making it useful for therapy in addition to PET imaging. It follows, therefore, that there has been a strong interest in developing new and improved bifunctional chelators to optimize radiolabeling procedures and in vivo performance with isotopes of copper. In addition to the ubiquitous chelator NOTA (vide infra), new entrants such as CB-TE2A, CB-TE1A1P, CB-TE2P, MM-TE2A, and DM-TE2A have recently been determined to be excellent 64Cu chelators (vide supra). The Sar family of chelators (hexamine sarcophagines) are hexaazamacrobicyclic cage type ligands, and include the ligands SarAr, SarAr-NCS, diamSar, AmBaSar, and BaBaSar (Table 5). One of the major benefits of these nitrogen rich chelators is that they can radiolabel with 64Cu in only a few minutes (5–10 min) at room temperature, which is faster even than the recently devised ligands CB-TE1A1P and CB-TE2P (Table 4).37,178–180 Of all the chelators discussed for 64Cu, Sar derivatives have the fastest room temperature radiolabeling kinetics. In addition to fast kinetics, they demonstrate excellent in vivo stability, and strong resistance to acid dissociation in vitro.178–181 One drawback to nitrogen rich N6 chelators such as the Sar family is lipophilicity, as they contain little or no acid functionality to add negative charge, and as a result form cationic or neutral complexes with 64Cu (depending on the Sar derivative). Because 64Cu pairs exceptionally well with peptides, a lipophilic radiometal–chelator complex can have deleterious affects on biodistribution and tumor targeting properties by directing activity to the liver and digestive tract (depending on hydrophilicity of the selected peptide). The Sar derivative BaBaSar adds two carboxylic acid functional groups to the Sar scaffold, thereby augmenting hydrophilicity. Animal studies have been promising with the Sar family of chelators, but more work is required for further validation; furthermore, limited commercial availability and challenging synthesis may be a reason why widespread adoption has yet to occur.37,178–181KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
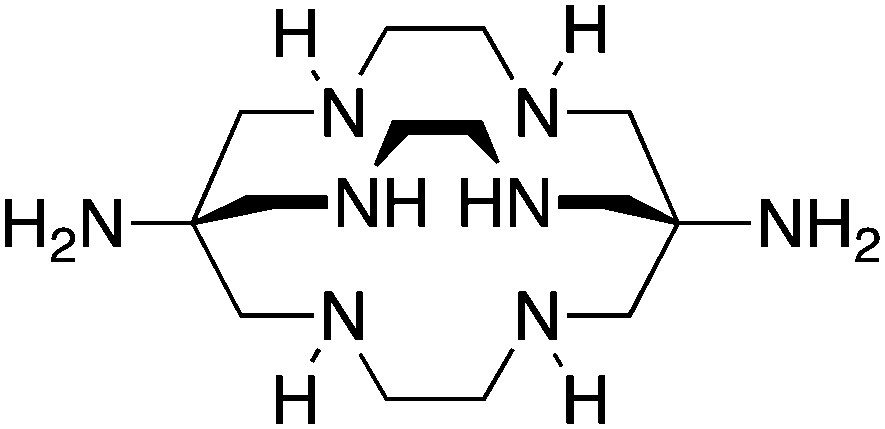
Diamsar, N6, CN = 6 |
64Cu2+ |

|
25 °C, 5–30 min, pH 5.5 | Distorted octahedron or trigonal prism | 178–181 | |
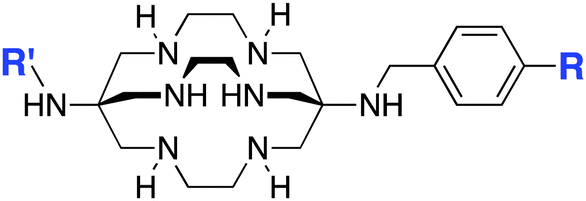
|
1-N-(4-Aminobenzyl)-3,6,10,13,16,19-hexaazabicyclo[6.6.6]-eicosane-1,8-diamine (SarAr)178–182
|
|||||
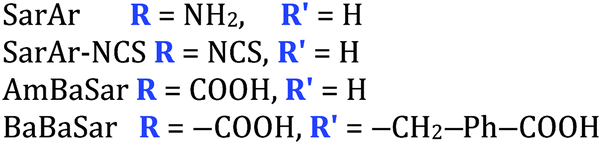
3.6 NOTA, NETA, and TACN-TM
NOTA is a hexadentate N3O3 chelator, and is one of the oldest and most successful chelators for use with 67/68Ga and 64Cu (Table 6). NOTA is generally considered to be the “gold standard” for Ga3+ chelation, possessing favorable radiolabeling conditions (RT, 30–60 minutes) and excellent in vivo stability.110,117,183,184 Of all the isotopes discussed in this review, 68Ga is probably the most popular in recent years. 68Ga has a very favorable positron emission (1880 keV, 90%) for PET imaging, and a short half-life (68 min) suitably matched for imaging with peptide vectors. The recent development of several 68Ge/68Ga-generator systems with shelf-lives of ∼9–12 months has propelled this radiometal into the spotlight of the international research scene.18567Ga is a longer-lived isotope of gallium that is suitable for SPECT imaging. Ga3+ is a hard acidic metal ion (pKa 2.6) with an ionic radius of 62 pm (CN = 6), a preference for amine and oxygen donor atoms, and is ultimately a challenging metal ion to chelate because it has a strong affinity for hydroxide anions causing precipitation of gallium hydroxide (Ga(OH)3) between pH 3–7.62,159,186KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
NOTA, 1,4,7-triazacyclononane-1,4,7-triacetic acid, CN = 6, N3O3 |
64Cu2+ |

|
25 °C, 30–60 min, pH 5.5–6.5 | 21.6 | Distorted trigonal prism | 187, 196 |
| 67/68Ga3+ |

|
25 °C, 30–60 min, pH 4.0–5.5 | 31.0 (pM 26.4, 27.9) | Distorted octahedron | 110, 117, 118, 183, 184, 197, 198 | |
| 44/47Sc3+ |

|
95 °C, 20–30 min, pH 4.0 | 16.5 (pM 19.2) | Distorted octahedron | 98 | |
| 111In3+ |

|
60–95 °C, 20–30 min, pH 4.0–5.0 | 26.2 (pM 21.6) | Distorted octahedron | 70, 117, 184, 199–201 | |
p-SCN-Bn-NOTA(C-NOTA)141,199 |
 NODASA, R = NHS ester, amide202
NODASA, R = NHS ester, amide202
|
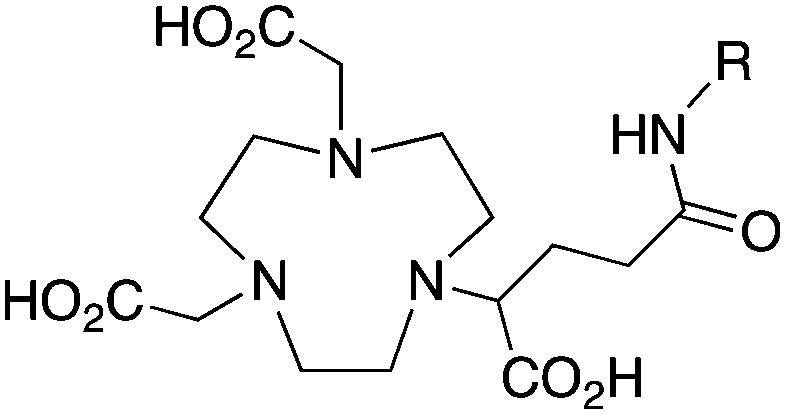 NODAGA, R = NHS ester, amide200
NODAGA, R = NHS ester, amide200
|
||||
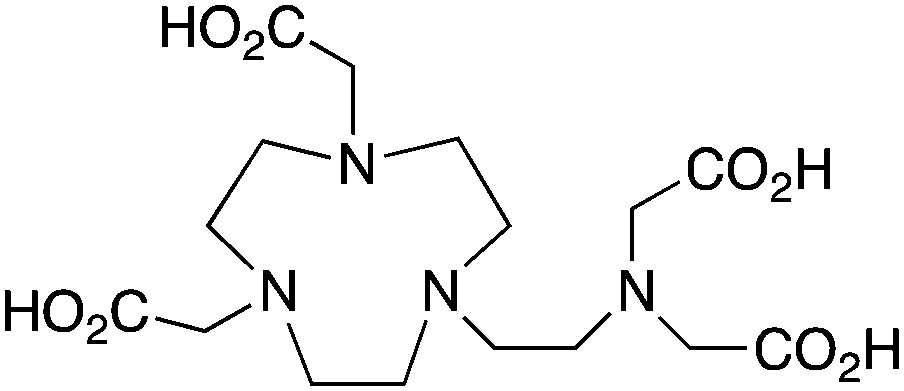
NETA, {4-[2-(bis-carboxymethylamino)-ethyl]-7-carboxymethyl-[1,4,7]triazonan-1-yl}-acetic acid, N4O4, CN = 8 |
177Lu3+ |

|
25 °C, 5 min, pH 4.5 | — | Square antiprism? | 127, 149, 190, 191 |
| 86/90Y3+ |

|
25 °C, 5 min, pH 4.0 | — | Square antiprism? | 127, 189, 191 | |
| 212/213Bi3+ |

|
25 °C, 5 min, pH 4.0 | — | Square antiprism? | 149, 190, 191, 194 | |
| 212Pb2+ |

|
25 °C, 5 min, pH 4.0 | — | Square antiprism? | 191 | |
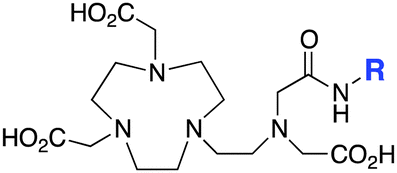
NETA-monoamide189 |
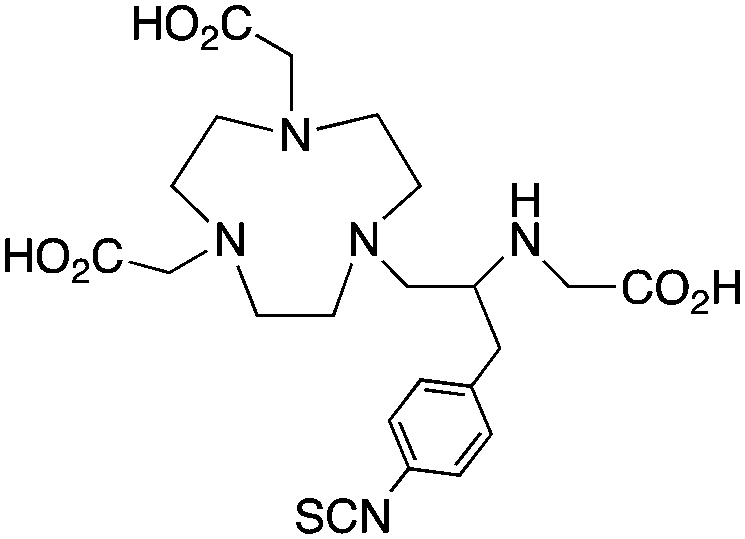 C-NE3TA-NCS191
C-NE3TA-NCS191
|
 C-NETA-NCS191
C-NETA-NCS191
|
 3p-C-NETA127,193,194
3p-C-NETA127,193,194
|
|||
TACN-TM, N,N′,N′′, tris(2-mercaptoethyl)1,4,7-triazacyclononane, N3S3 CN = 6 |
67/68Ga3+ |

|
25 °C, 10 min, degassed ethanol | 34.2 | Distorted octahedron | 110, 195, 203, 204 |
| 111In3+ |

|
25 °C, 10 min, degassed ethanol | 36.1 | Distorted octahedron | 99, 176, 177, 183 | |
Because of its potential clinical utility, many new gallium chelators have been published in the last ∼5 years; however, most have not yet amassed the same amount of in vivo data as NOTA. NOTA has been shown to have superior radiolabeling properties and stability with 64Cu when compared to common chelators such as DOTA, EDTA, DTPA, and TETA.155,187 NOTA can radiolabel with 64Cu at room temperature in 30–60 minutes, making it compatible with heat sensitive antibody vectors.188 Although many new chelators like TRAP, AAZTA (DATA), H2dedpa, CP256, and PCTA (vide infra) show great promise for use with 67/68Ga in early experiments, due to commerical availability of BFC derivatives and widespread acceptance, NOTA is still currently the “gold standard” for 67/68Ga. A similar story can be told for 64Cu, where a plethora of new chelators, including the Sar family (e.g. SarAr, DiamSar), CB-TE2A, and various CB-TE2A derivatives (e.g. CB-TE1A1P, CB-TE2P, MM/DM-TE2A) appear to possess many superior properties, but NOTA is still the practical “gold standard” for 64Cu.
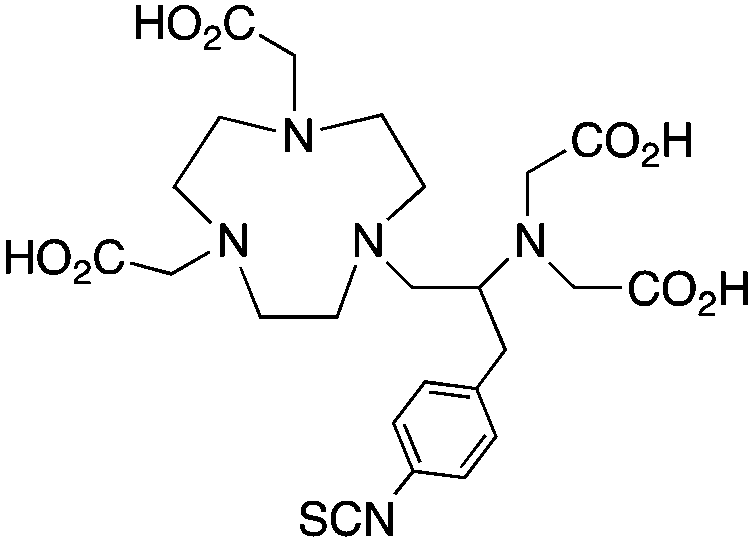
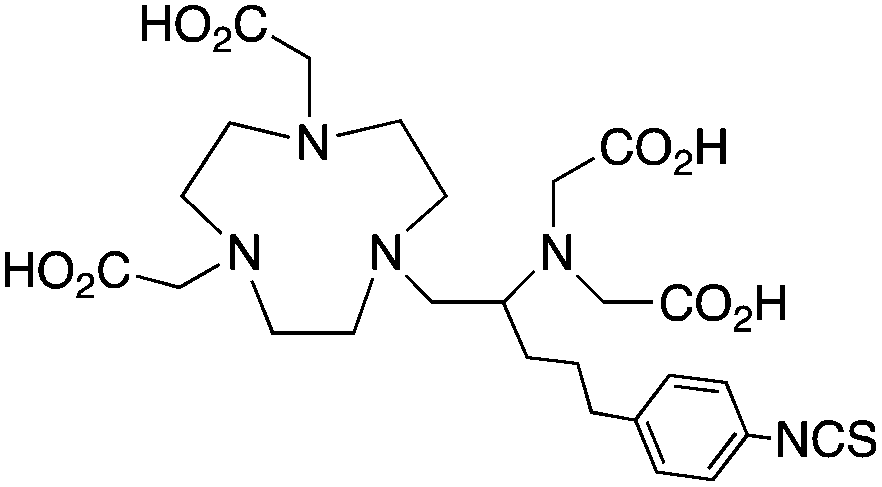
The modified NOTA chelator NETA (Table 6) is an interesting derivative that shows promise with 86/90Y, possessing fast radiolabeling kinetics similar to those of acyclic chelators (5–10 minutes, RT), as well as a high degree of stability and rigidity imparted by the macrocyclic framework.189 A direct comparison between NETA and DOTA has demonstrated greatly enhanced radiolabeling kinetics for NETA with Y3+.189 Biodistribution experiments performed with the 86Y complexes of the non-bifunctional chelators NETA and DOTA have suggested that NETA had comparable clearance and stability properties to DOTA, even showing lower bone accumulation than DOTA.189 NETA has been evaluated with other therapeutic isotopes for RIT, including 203/212Pb, 212/213Bi, and 177Lu.149,190,191 NETA and bifunctional derivatives C-NETA and C-NE3TA (Table 6) were found to be unstable with 203/212Pb isotopes, but looked very stable with 212/213Bi, 90Y, and 177Lu during a period of 11 days in blood serum.191–193 Biodistribution studies of C-NETA complexes in healthy mice demonstrated excellent clearance and stability of the 212/213Bi, 90Y, and 177Lu complexes, with the exception of high kidney uptake with the 212/213Bi-C-NETA complex.191 Recent experiments with an isothiocyanate bearing derivative of C-NETA, 3p-C-NETA (Table 6) have demonstrated that 90Y and 177Lu radiolabeled immunoconjugates (trastuzumab) had very rapid room temperature radiolabeling kinetics, and excellent in vivo performance, with the potential to surpass DOTA and also DTPA analogues such as CHX-A′′-DTPA.127 3p-C-NETA has been further evaluated with 212/213Bi for RIT applications, and trastuzumab immunoconjugates have been shown to exhibit rapid radiolabeling kinetics and excellent in vitro and in vivo stability, suggesting improved stability over C-NETA, which showed high kidney accumulation of 212/213Bi.191,194 It is interesting to note that these studies were actually performed with 205/206Bi, as the longer half-lives (15.3/6.2 days, respectively) of these 212/213Bi isotopologues are more amenable to extensive in vitro and in vivo study than are the short half-lives of 212/213Bi.194 Currently it is not clear if 3p-C-DEPA (Table 3) is superior to 3p-C-NETA (Table 6) for use with 212/213Bi, and a direct comparison would be timely.
TACN-TM (Table 6) is an interesting thiol-based chelator, and shows extremely high thermodynamic stability constants (logKML) with Ga3+ and In3+ of 34.2 and 36.1, respectively.99,176,177,183 Another interesting thiol TACN derivative is TACN-HSB, which substitutes alkyl-thiols for thiophenol groups.195 Despite the high thermodynamic stabilities, radiolabeling experiments required the use of degassed ethanol due to instability of the thiol groups of TACN-TM/TACN-HSB to air, and ultimately these ligands could not be used for in vivo applications.195 For these reasons, no bifunctional derivatives have been synthesized of these thiol-based TACN/NOTA derivatives, and further study has not been pursued.
3.7 DTPA, 1B4M-DTPA, and CHX-A′′-DTPA
DTPA is one of the oldest and most pervasive acyclic chelators used in radiochemistry, and like most acyclic chelators it can be radiolabeled with many radiometal ions at room temperature in a matter of minutes (Table 7). As a first generation radiometal chelator, it suffers from stability issues in vivo with many radiometal ions and is universally not as stable as the macrocycles DOTA and NOTA, making its use obsolete in recent years.56 DTPA has been successfully used as the BFC in the FDA-approved SPECT agent OctreoScanTM (111In-DTPA-octreotide), a somatostatin-targeting peptide-conjugate used for imaging neuroendocrine tumors.205,206 DTPA has also been successfully used with radiometals such as 64Cu, 111In, 177Lu, and 86/90Y, but has been made redundant by more stable new chelators like DOTA, NOTA, and CHX-A′′-DTPA (Table 7).56,161 The shortcomings of DTPA have been improved through design of novel derivatives, such as the DTPA derivatives 1B4M-DTPA (Tiuxetan) and CHX-A′′-DTPA.56,207 1B4M-DTPA is a bifunctional DTPA derivative that contains a single methyl group on one of its ethylene backbones, and has been successfully incorporated into the FDA-approved 90Y therapeutic immunoconjugate Zevalin (Table 7).207–209,210KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
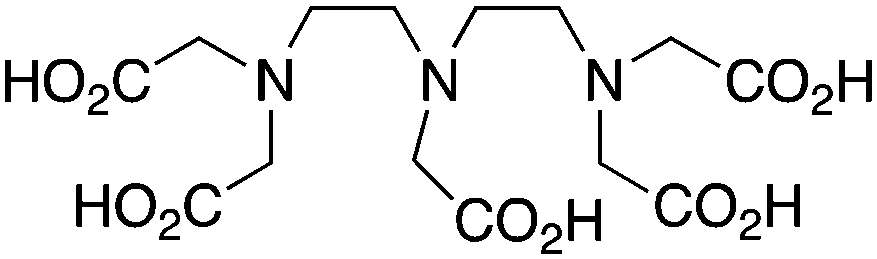
DTPA, diethylenetriaminepentaacetic acid, N3O5, CN = 8 |
64Cu2+ |

|
40 °C, 60 min, pH 6.5 | 21.4 | Distorted octahedron? | 119, 234 |
| 67/68Ga3+ |

|
25 °C, 30 min, pH 3.5 | 24.3, 25.5 (pM 20.2) | Distorted octahedron? | 117, 119, 235 | |
| 44/47Sc3+ |

|
25 °C, 10 min, pH 6.0 | — | Square antiprism? | 16 | |
| 111In3+ |

|
25 °C, 5–10 min, pH 4.5–5.5 | 29.0 (pM 24.9) | Pentagonal bipyramid or square antiprism | 117, 119, 236–238 | |
| 177Lu3+ |

|
25 °C, 10–20 min, pH 5.5 | 22.6 | Square antiprism? | 119, 131, 239 | |
| 86/90Y3+ |

|
25 °C, 10–20 min, pH 5.5 | 21.2, 22.0, 22.5 | Monocapped square antiprism | 55, 131, 132, 239 | |
| 89Zr4+ |

|
25 °C, 60 min, pH 7 (<0.1% yield) | 35.8–36.9 | Distorted dodecahedron | 119, 240 | |
| 212/213Bi3+ |

|
25 °C, 10–20 min, pH 5 | 35.6 | Square antiprism | 119, 230–232 | |
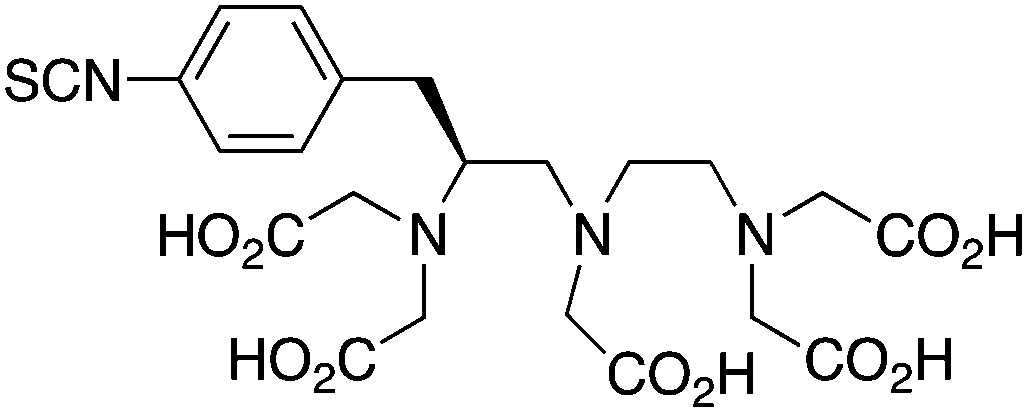
p-SCN-Bn-1B-DTPA211,241 |
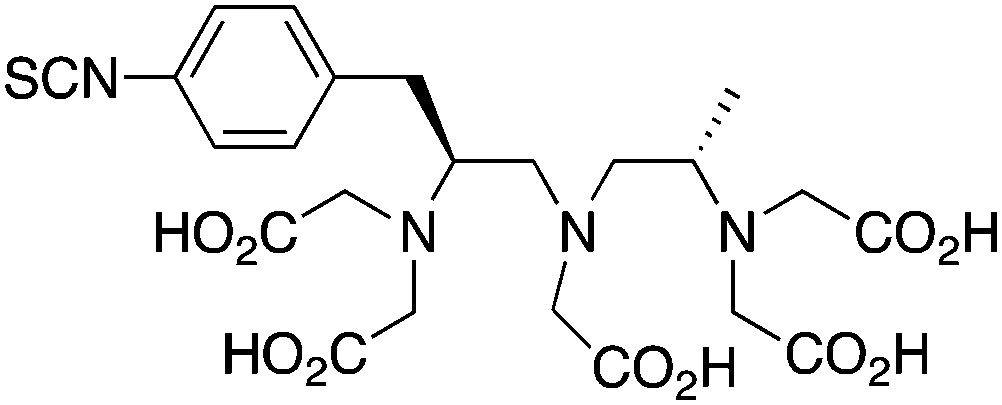 p-SCN-Bn-1B4M-DTPA224, 242, 243
p-SCN-Bn-1B4M-DTPA224, 242, 243
|
|||||
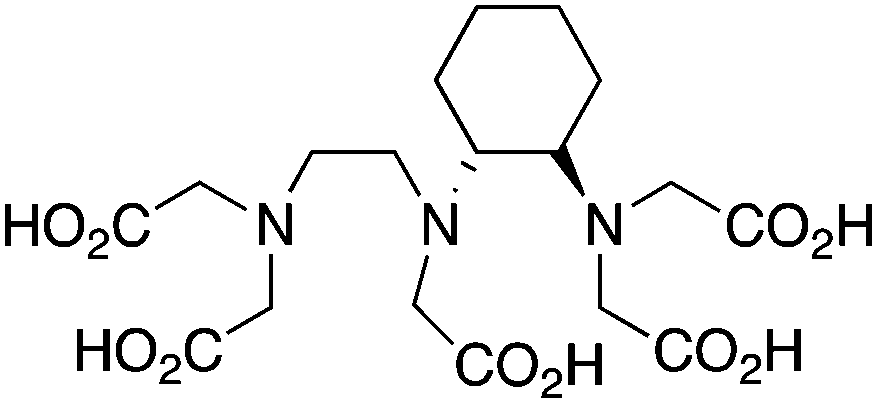
CHX-A′′-DTPA, 2-(p-isothiocyanatobenzyl)-cyclohexyldiethylenetriaminepentaacetic acid, N3O5, CN = 8 |
67/68Ga3+ |

|
85 °C, 20 min, pH 5.5 | — | Distorted octahedron? | 217 |
| 111In3+ |

|
25–60 °C, 30–60 min, pH 5.5 | — | Square antiprism | 217, 218, 224–229 | |
| 177Lu3+ |

|
37–75 °C, 30–60 min, pH 5–5.5 | — | Square antiprism | 54, 126, 128 | |
| 86/90Y3+ |

|
37–75 °C, 30–60 min, pH 5–5.5 | — | Square antiprism | 6, 7, 55, 56, 212–220 | |
| 213Bi3+ |

|
25 °C, 10–20 min, pH 5 | — | Square antiprism | 201, 221–223 | |
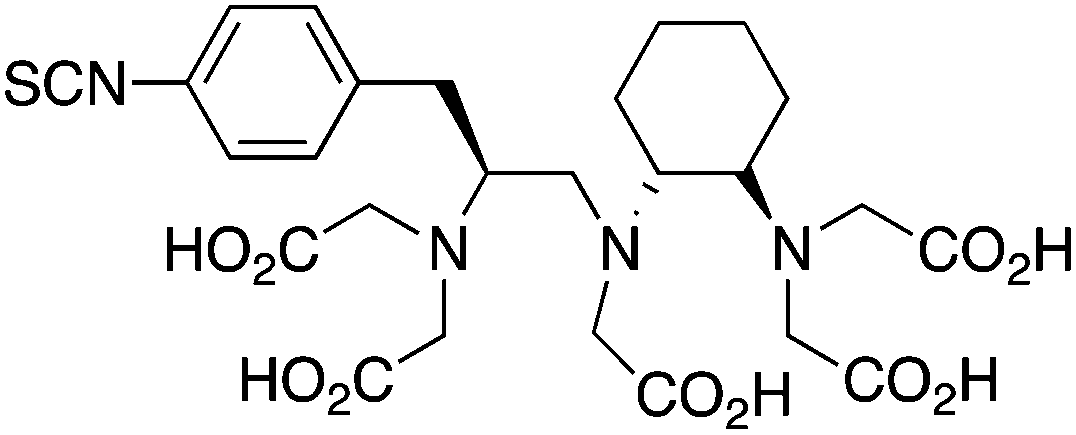
p-SCN-Bn-CHX-A’’-DTPA56,212,244 |
||||||
Although DTPA and DOTA are the most commonly investigated chelate systems for Y3+, the chelator CHX-A′′-DTPA (Table 7) shows significantly improved stability over DTPA; however, it is still generally considered to be less stable than DOTA.56 The cyclohexyl backbone of CHX-A′′-DTPA makes the chelator more rigid and imposes a degree of preorganization on the metal ion binding site, enhancing kinetic inertness, but retarding radiolabeling kinetics compared to those of DTPA.56,211 Due to the success of antibody vectors, developing acyclic chelators with fast radiometal ion coordination kinetics, as well as high stability and kinetic inertness comparable to that of macrocycles such as DOTA is an important goal. The work done towards developing CHX-A′′-DTPA was particularly interesting because of the 4 possible isomers of this chelator. The CHX-B′′-DTPA isomer was found to be much less stable than the CHX-A′′-DTPA isomer in vivo with 90Y, despite in vitro stability assays suggesting they were very similar.56,212 If CHX-DTPA were to be used as a racemic mixture it would result in significant decomposition of the less stable isomers and would lead to radiodemetallation in vivo, highlighting the importance of enantiopurity.56,212 Different isomers of a BFC–radiometal complex can have different stabilities and off-rates (kinetic inertness), potentially leading to differential biodistributions.42,59
Although CHX-A′′-DTPA has enhanced radiolabeling kinetics when compared to DOTA, both are still much slower than typical acyclic chelators such as DTPA and H4octapa (Table 9), and often require mild heating (∼37–60 °C) and reaction times of 30–60 minutes to achieve reasonable yields. CHX-A′′-DTPA has been heavily investigated for use with many radiometals, including 86/90Y,6,7,55,56,212–220177Lu,54,126,128 and 212/213Bi,201,221–223 but work with 111In has comparatively been limited.217,218,224–229 Studies that have been performed with 111In(CHX-A′′-DTPA) conjugates have shown good in vivo results, but to our knowledge little work has been done comparing it with the “gold standard” 111In chelator DOTA.228 A study comparing the in vivo behavior of the affibody conjugate 114mIn-CHX-A′′-DTPA-ABD-(ZHER2:342)2 to 111In-DOTA-ABD-(ZHER2:342)2 suggests that both chelators have comparable stability and performance, with CHX-A′′-DTPA exhibiting slightly higher tumor uptake, but also slightly higher non-target organ uptake (e.g. kidneys, liver, bone).228114mIn is a notable γ-emitting isotope because it has identical chemistry to 111In, but a longer 49.5-day half-life and interesting decay scheme (Table 1).228 When 114mIn decays it emits a γ-ray for SPECT imaging, but the daughter nuclide 114In has a half-life of 72 seconds and emits high-energy (1989 keV) β− particles useful for therapy.228 When the parent 114mIn radiolabeled affibody internalizes into cells, it acts as an in vivo generator of 114In for intracellular delivery of therapeutic radioactivity (β− particles) to cancer cells.228
Investigations with 212/213Bi have found DTPA to possess fast radiolabeling kinetics, but poor in vivo stability, ultimately rendering it unusable.119,230–232 CHX-A′′-DTPA has been shown to possess enhanced stability with 212/213Bi when compared to DTPA and 1B4M-DTPA, and even comparable stability to DOTA, while additionally possessing greatly enhanced radiolabeling kinetics (important for the short half-life of 212/213Bi).201,221–223,231,233 The solid-state structure of the [Bi(CHX-A′′-DTPA)]2− complex suggests that the partially pre-organized binding site offered by the rigid cyclohexyl backbone may contribute to the enhanced stability of the complex (also suggested as the cause for enhanced stability with other isotopes such as 86/90Y and 177Lu).221 Although comparable to DOTA, CHX-A′′-DTPA is still considered to be inferior to 3p-C-NETA and 3p-C-DEPA for use with 212/213Bi.
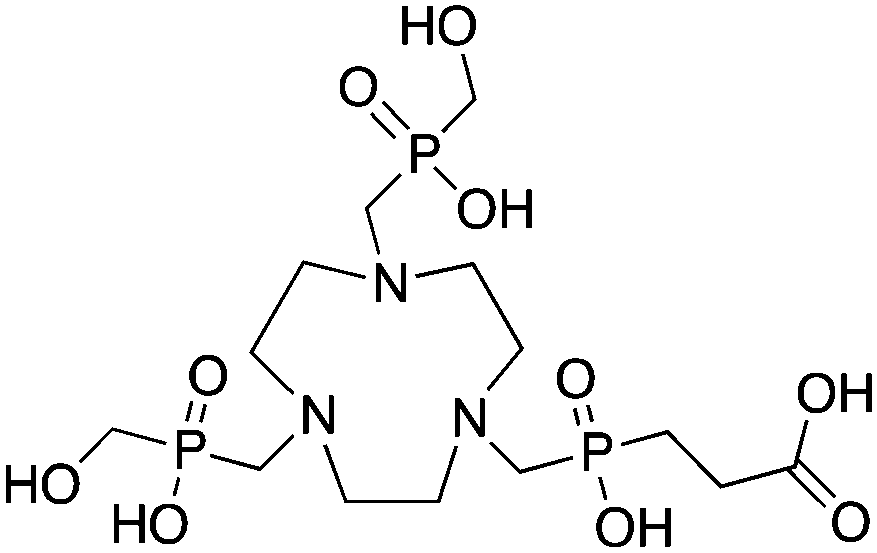
3.8 TRAP (PRP9) and NOPO
TRAP (PRP9) is a derivative of the macrocycle NOTA, wherein the traditional carboxylic acid arms have been replaced with phosphinic acid groups (Table 8).79–81,245–249 The most successful TRAP derivative, TRAP-Pr, contains an additional ethyl-carboxylate moiety extending distally from the phosphinic acid groups (Table 8).79–81,245–249 The most interesting property of the TRAP-Pr ligand is its improved apparent specificity for Ga3+ when compared to NOTA.78,81 This phenomenon is expressed by the difference in thermodynamic stability constants (logKML) of TRAP and NOTA for competing metal ions such as Mg2+, Ca2+, Cu2+, and Zn2+,81 and by competitive radiolabeling experiments done in the presence of an excess of these other ions.78 As a general rule, radiolabeling experiments are performed in strictly metal-free water and so the selectivity of TRAP-Pr for Ga3+ may not appear to be totally relevant, but it does suggest that transchelation by these competing metal ions in vivo should occur to a lesser extent than with NOTA. Additionally, as discussed in the introduction, the enhanced specificity of TRAP-Pr for 68Ga over NOTA may be of benefit if trace-impurities are present in the stock radiometal solution (e.g.68Ge/68Ga generator eluent, or commercially supplied 64Cu). A similar derivative to TRAP is NOPO, which is a triazacyclononane-triphospinate ligand that has a similar structure to TRAP-Pr, but with two terminal alcohol groups in place of carboxylic acids.78,248,250 Like TRAP, NOPO has shown promising properties for 64Cu and 68Ga radiolabeling.78,248,250
KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
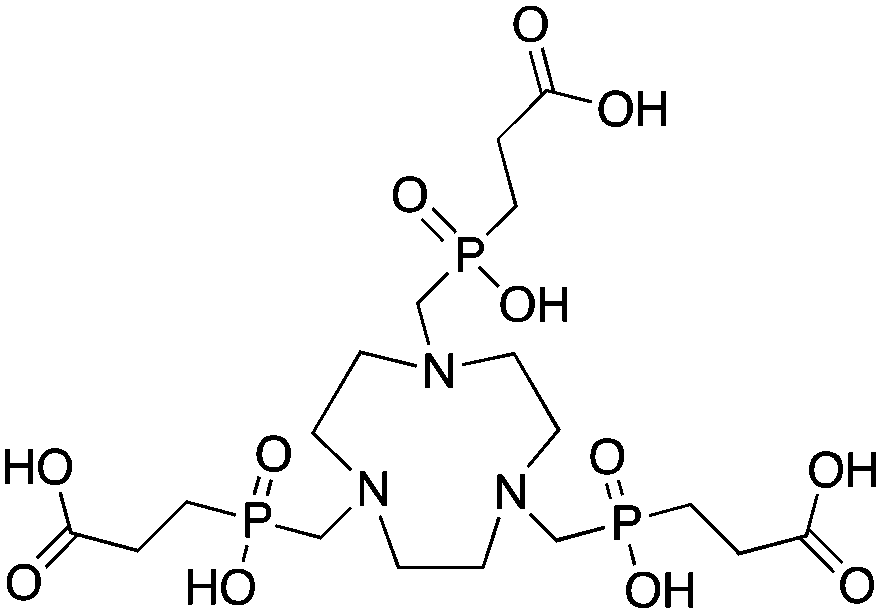
TRAP (PRP9, TRAP-Pr), 1,4,7-triazacyclononane-1,4,7-tris[methyl(2-carboxyethyl)phosphinic acid], N3O3 CN = 6 (+ distal carboxylates CN = 9?) |
67/68Ga3+ |

|
95 °C, 5 min, pH 3.2 | 26.24 | Distorted octahedron | 79–81, 91, 245–249 |
|
L = Linker, e.g. PEG8, PEG4, Glu, AHX, N3AHX.
P = Peptide, e.g. RGD, DRG.
NOPO 78,248,257 |
|||||
AAZTA, and novel DATA derivatives, 1,4-bis (hydroxycarbonyl methyl)-6-[bis(hydroxylcarbonyl methyl)] amino-6-methyl perhydro-1,4-diazepine, N2O4 or N3O3, CN = 6 |
67/68Ga3+ |

|
25 °C, 1–5 min, pH 4–6.8 | — | Distorted octahedron | 253–256 |
H3L1 |
 H3L4
H3L4
|
|||||
The peptide conjugate TRAP(RGD)3 was radiolabeled with 68Ga in 5 minutes at 95 °C (HEPEs, pH 3.2), and despite the high reaction temperature was able to radiolabel at lower ligand concentrations than the corresponding DOTA or NOTA conjugates.14,15 RGD (arginine–glycine–aspartic acid tripeptide) is a popular cyclic peptide vector that targets overexpression of integrin αvβ3 receptors on various types of cancer, and is commonly used as a proof-of-principle vector for studying new isotopes and new BFC.14,15,30,91,250,25168Ga-TRAP(RGD)3 demonstrated good in vivo tumor targeting properties, showing great promise for use in 68Ga-based PET imaging agents;14,15 however, a recent comparative biodistribution study between 68Ga-TRAP(RGD)3 and 68Ga-NODAGA-RGD revealed higher uptake in several non-target organs for the TRAP-based agent.79,91,246 Despite these apparent shortcomings, TRAP-Pr is a very promising 68Ga chelator, which is a claim substantiated by the work of Dr Richard P. Baum at Zentralklinik Bad Berka in Germany, who has performed imaging experiments in human patients using 68Ga-TRAP-peptide conjugates.252
3.9 AAZTA and derivatives (DATA)
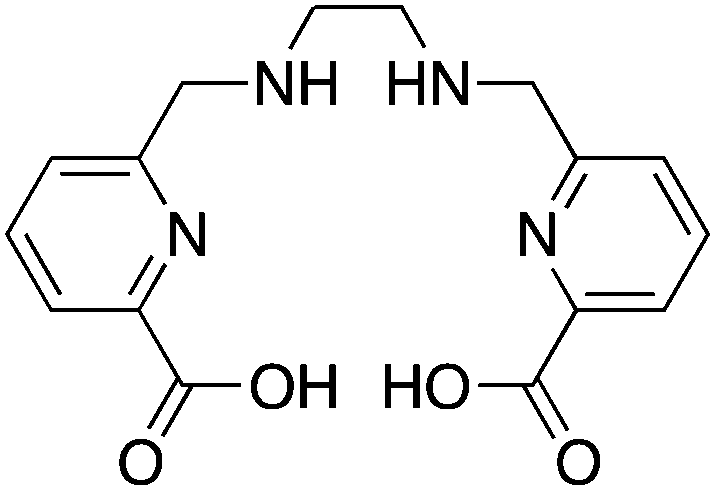
The novel AAZTA derivatives H3L1 and H3L4 are new 67/68Ga-chelators that have shown improved radiolabeling and serum stability properties over the parent AAZTA chelator (Table 8). The parent AAZTA chelator was found to form multiple isomers with Ga3+, and so the derivatives H3L1 and H3L4 were crafted such that one major isomer of the Ga3+ complex was kinetically trapped by attachment of bulky alkyl/aryl groups.253–256 These AAZTA derivatives (now called DATA) are promising new Ga3+ chelators with extremely fast radiolabeling kinetics (1–5 minutes at RT) and good preliminary in vitro and in vivo stability results (as non-bifunctional chelators). These chelators are very recent, as most of this work has just been published in 2013, and so more extensive in vitro and in vivo experimentation will likely follow in the coming years. Additionally, no bifunctional derivatives (e.g. peptide conjugates) have been published to date, which will be required for proper evaluation and comparison in vivo to existing “gold standards” like NOTA.3.10 H2dedpa, H4octapa, H2azapa, and H5decapa
This family of ligands has only been published over the previous 3 years and has been referred to as the “pa family”, as they are all crafted around central picolinic acid (“pa”) binding moieties (Table 9). This work originated in chelators made by Rodriguez-Blas and coworkers, originally purposed as contrast agents for magnetic resonance imaging (MRI) and luminescence.258–267 H2dedpa was the first chelator in this family to be investigated, with its acyclic hexadentate scaffold being ideal for 67/68Ga, radiolabeling in less than 10 minutes at room temperature and forming a very symmetrical hexedentate coordination geometry (determined by solid-state X-ray structure).118,262,264,265,267In vitro analysis of the [67/68Ga(dedpa)]+ complex demonstrated excellent stability and resistance to transchelation by apo-transferrin and blood serum.118 The bifunctional derivative p-SCN-Bn-H2dedpa has been synthesized and conjugated to the cyclic peptide RGD, radiolabeled with 68Ga and 64Cu, and has shown promising results for in vivo tumor targeting and PET imaging.268,269 The H2dedpa chelator has also been functionalized with lipophilic moieties, and the resulting cationic 68Ga complexes have been investigated for cardiac perfusion imaging.270 Although preliminary work looks very promising for H2dedpa, especially towards the production of kit-formulations with its very fast room temperature radiolabeling kinetics, more in vivo validation is required. As is the case with all new bifunctional chelators, the largest barrier to widespread adoption is commercial availability, as the synthesis and purification of most nitrogen and oxygen rich chelators are typically tedious and challenging.KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
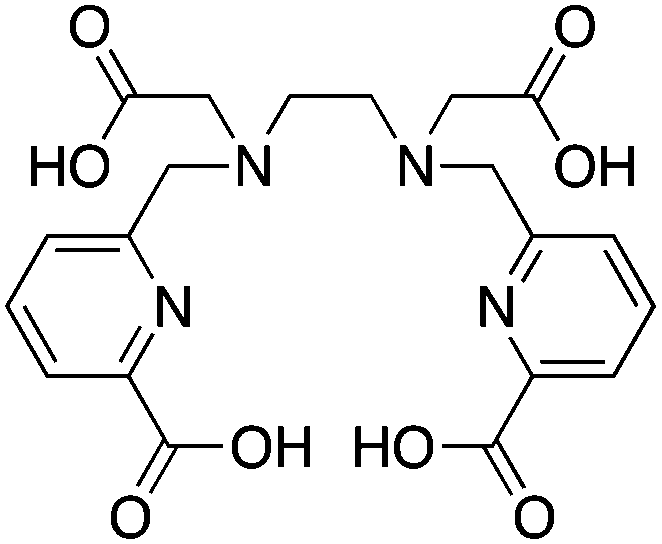
H2dedpa, 1,2-[[6-(carboxy)-pyridin-2-yl]methylamino]ethane, N4O2 CN = 6 |
64Cu2+ |

|
25 °C, 5–10 min, pH 5.5 | 19.2 (pM = 18.5) | Distorted octahedron | 269 |
| 67/68Ga3+ |

|
25 °C, 5–10 min, pH 4.5 | 28.1 (pM = 27.4) | Distorted octahedron | 118, 268, 270 | |
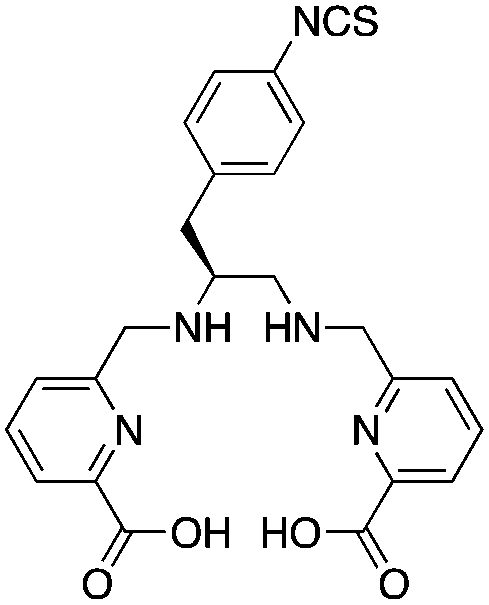
p-SCN-Bn-H2dedpa118,268 |
||||||
H4octapa, N,N′-bis(6-carboxy-2-pyridylmethyl)ethylenediamine-N,N′-diacetic acid, N4O4 CN = 8 |
111In3+ |

|
25 °C, 5–10 min, pH 4.5 | 26.8 (pM = 26.5) | Square antiprism | 83, 85 |
| 177Lu3+ |

|
25 °C, 5–10 min, pH 4.5 | 20.1 (pM = 19.8) | Square antiprism | 83 | |
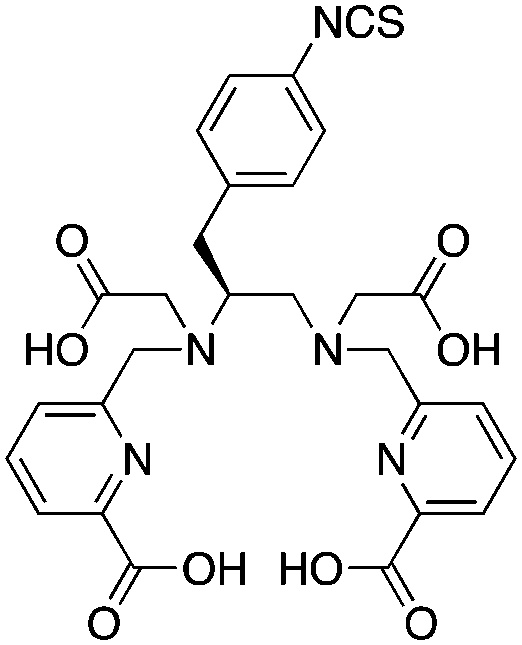
p-SCN-Bn-H4octapa83 |
||||||
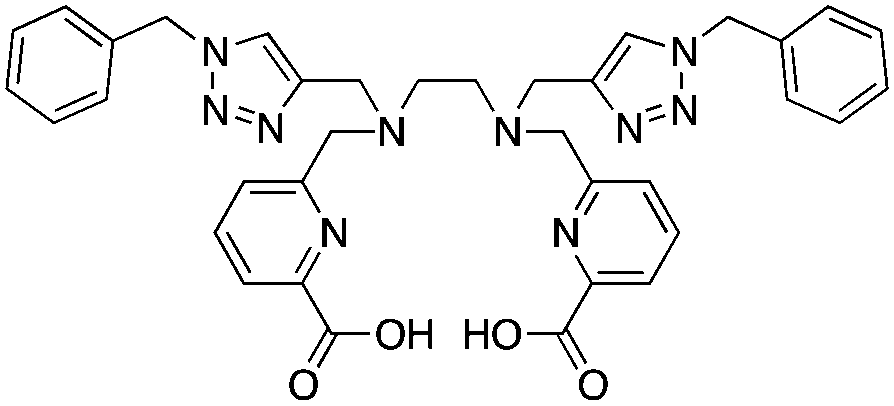
H2azapa, N,N′-[1-benzyl-1,2,3-triazole-4-yl]methyl-N,N′-[6-(carboxy)pyridin-2-yl]-1,2-diaminoethane, N6O2 CN = 8 (benzyl groups as vector placeholder) |
64Cu2+ |

|
25 °C, 5–10 min, pH 5.5 | — | Distorted octahedron? | 84 |
| 67/68Ga3+ |

|
25 °C, 5–10 min, pH 4–4.5 | — | Distorted octahedron? | 84 | |
| 111In3+ |

|
25 °C, 5–10 min, pH 4.5 | — | Square antiprism? | 84 | |
| 177Lu3+ |

|
25 °C, 5–10 min, pH 4.5 | — | Square antiprism? | 84 | |
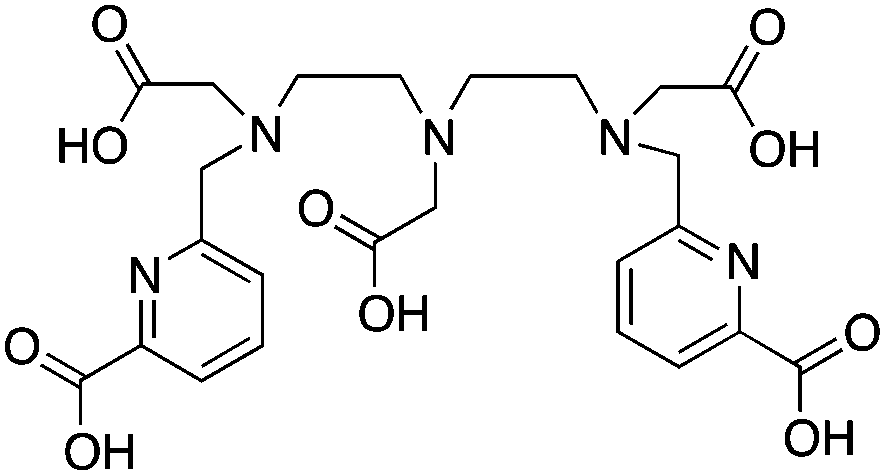
H5decapa, N,N′′-[[6-(carboxy)pyridin-2-yl]methyl]diethylenetriamine-N,N′,N′′-triacetic Acid, N5O5 CN = 10 |
111In3+ |

|
25 °C, 5–10 min, pH 4.5 | 27.56 (pM = 23.1) | Square antiprism | 83, 85 |
H2azapa was built on a variation of the click-to-chelate approach, where the peptide vector is conjugated to the chelator by click-chemistry, and the resultant triazole-rings are uniquely positioned so that they are capable of coordinating to radiometal ions in the inner-coordination sphere.32,84,271 The triazole-containing H2dedpa/H4octapa derivative, H2azapa, has been synthesized and evaluated with 64Cu, 68Ga, 111In, and 177Lu (Table 9).84 Preliminary results were promising with 64Cu, but in vitro stability assays suggest that complexes with 68Ga, 111In, and 177Lu were unstable.84 Due to the very lipophilic character of the neutral [Cu(azapa)] complex, in vivo evaluation revealed high liver and digestive tract uptake as expected, therefore more hydrophilic conjugates (e.g. peptide conjugates) must be synthesized and evaluated in the future.84 The large decadentate derivative of H4octapa, H5decapa, was evaluated with 111In in vitro and in vivo, and was found to be significantly less stable than were H4octapa and DOTA (Table 9).83,85
H4octapa is a derivative of H2dedpa, with an additional two carboxylic acid arms increasing the maximum denticity from 6 to 8 (Table 9).83,85,262,267 H4octapa has recently been found to exhibit ideal properties for 111In and 177Lu radiochemistry, radiolabeling in quantitative yields at room temperature in less than 10–15 minutes.83,85 Direct comparisons between H4octapa and DOTA with both 111In and 177Lu have demonstrated similar in vivo properties and stability, with H4octapa having greatly enhanced radiolabeling kinetics (significantly faster even than CHX-A′′-DTPA), suggesting that it may be a suitable alternative and improvement to DOTA.83,85 Based on its excellent properties with 177Lu, H4octapa may also be a suitable match for the similar isotopes 86/90Y, although no results have been published towards this goal thus far. Further preclinical work must be performed to validate H4octapa as a suitable alternative to DOTA, although a direct comparison has been made with 111In and 177Lu in vitro and in vivo.83,85
It appears that CHX-A′′-DTPA, C-NETA/3p-C-NETA, and H4octapa are the best current alternatives to DOTA for fast radiolabeling kinetics and excellent in vivo stability with 111In for SPECT imaging and dosimetry, and 177Lu and 86/90Y for therapeutic applications.83,85,127 It is currently not clear how C-NETA/3p-C-NETA perform with 111In because, to our knowledge, no studies have been published, and work with 111In and CHX-A′′-DTPA is limited. If a SPECT imaging surrogate isotope such as 111In is required for pre-therapy imaging and dosimetry, it appears that currently CHX-A′′-DTPA and H4octapa are the most promising alternatives to DOTA.83,85 A direct comparison between all of these chelators with 111In, 177Lu, and 86/90Y in the same animal model at the same time would be of significant value.
3.11 HBED and SHBED
HBED and its derivative SHBED present an interesting example of chelator development, as the addition of an aromatic para-sulfonate group in SHBED to the phenol functionality of HBED serves to decrease the pKa of the phenolic protons and alter their hard/soft metal-donor properties (Table 10). The original purpose of adding this aryl-sulfonate group to HBED was to increase its negative charge and hydrophilicity/solubility (fully deprotonated SHBED has a 6− charge vs. 4− for HBED), but the effect of changing the donor properties of the attached phenolic oxygen highlights an interesting option for tuning other chelators that contain aromatic donor groups.272 Decreasing the pKa of acidic functional groups in ligands can also help to improve radiolabeling efficiency: in this example by making the phenolic protons of SHBED more acidic and easier to deprotonate, and subsequently making SHBED a more effective ligand at lower pH. The bifunctional derivative HBED-CC has been conjugated to antibodies and was found to radiolabel much faster than NOTA, achieving a radiochemical yield (RCY) of 97% in under 5 minutes at room temperature, while also showing comparable in vivo performance to NOTA.273,274 Although HBED has been around for several decades, limited work with 68Ga has been performed, and other isotopes such as 64Cu and 111In have not shown optimal properties.117,273–276 HBED demonstrates that phenol and catechol donor groups are effective for Ga3+, but interestingly recent Ga3+ chelators such as H2dedpa, TRAP, PCTA, AAZTA (DATA), and CP256 have not utilized them. Although an effective gallium chelator, HBED has received limited clinical use and has been supplanted in recent years by a plethora of new ligands.KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
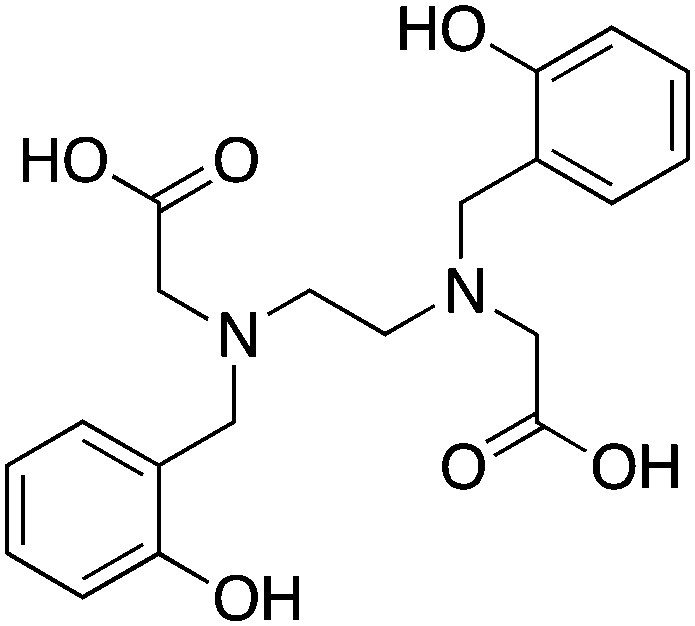
HBED, N,N′-bis(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid, N2O4, CN = 6 |
67/68Ga3+ |

|
25 °C, 10–20 min, pH 4–4.5 | 38.5 (pM 28.6) | Distorted octahedron? | 117, 273–276 |
| 44/47Sc3+ |

|
25 °C, 10 min, pH 6.0 | — | Square antiprism? | 16 | |
| 111In3+ |

|
25 °C, 10–20 min, pH 4–7 | 27.9 (pM 17.9) | Distorted octahedron? | 117, 275, 276 | |
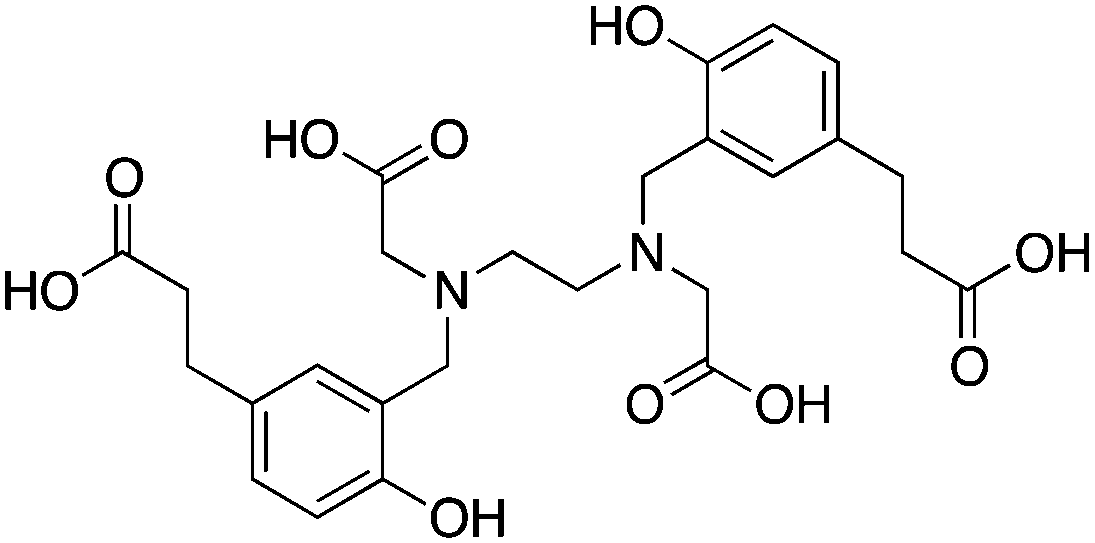
HBED-CC274,279 |
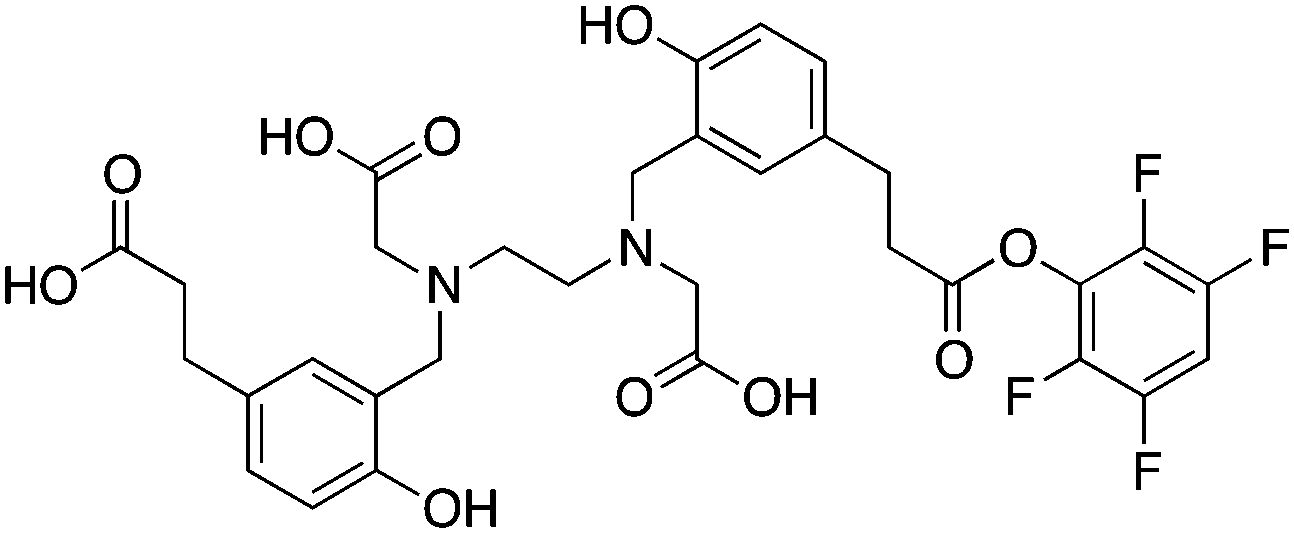
(HBED-CC)TFP274 |
|||||
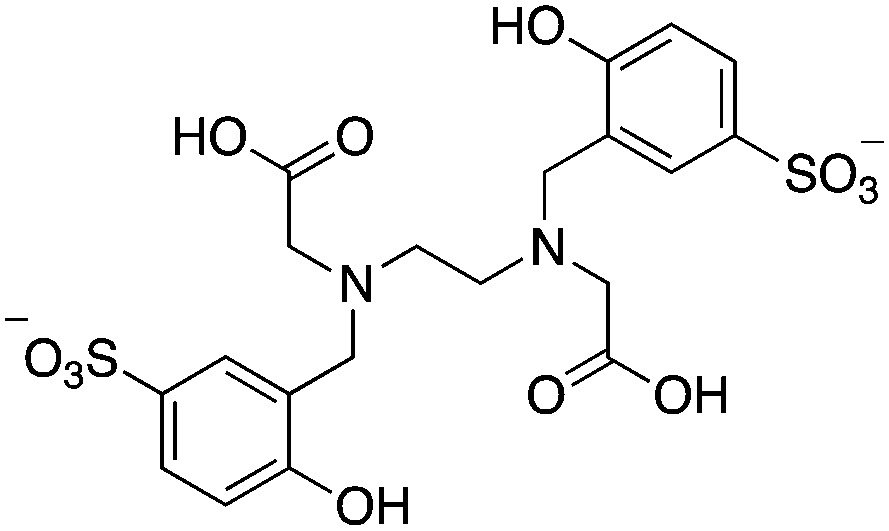
SHBED, N,N′-bis(2-hydroxy-5-sulfobenzyl)ethylenediamine-N,N′-diacetic acid, N2O4, CN = 6 |
67/68Ga3+ |

|
25 °C, 10–20 min, pH 4–4.5 | 37.5 (pM 28.3) | Distorted octahedron? | 117, 272, 276 |
| 111In3+ |

|
25 °C, 10–20 min, pH 4–7 | 29.4 (pM 20.6) | Distorted octahedron? | 117, 272, 276 | |
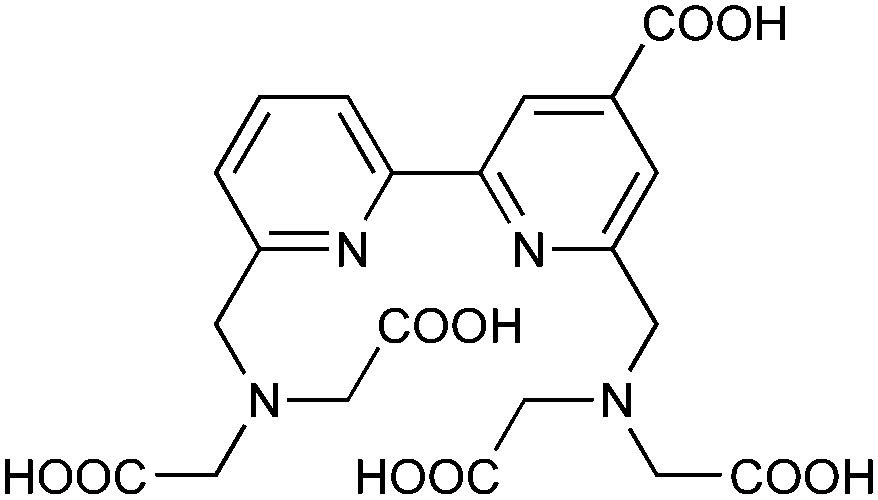
BPCA, N4O4, CN = 8 |
111In3+ |

|
25 °C, 60 min, pH 5 | — | Square antiprism? | 277, 278 |
3.12 BPCA
BPCA is an interesting chelator that was published in 2010, which is based on a 2,2′-bipyridine backbone, and provides an octadentate N4O4 donor set (Table 10).277,278 BPCA was conjugated to a novel cholecystokinin C-terminal tetrapeptide (CCK4), which targets tumors expressing the cholecystokinin receptor subtype 2 (CCK2R), and radiolabeled with 111In in high RCY after 1 hour at room temperature. The novel peptide-conjugate BPCA–(Ahx)2–CCK4 was evaluated in vitro by serum stability transchelation assays, and in vivo by biodistribution and planar scintigraphy experiments. The same (Ahx)2–CCK4 conjugate was made with CHX-A′′-DTPA as an internal reference, and it was demonstrated that BPCA possessed superior stability to CHX-A′′-DTPA in serum (89% vs. 45% stable after 2.5 hours, respectively). Biodistribution and planar scintigraphic imaging studies showed that BPCA-based conjugates had higher tumor uptake, lower background organ uptake, and higher tumor/muscle ratios than CHX-A′′-DTPA, suggesting that BPCA is a very promising new chelator for 111In. A study evaluating BPCA with 177Lu and 86/90Y would be of great interest.3.13 CP256
CP256 and its bifunctional derivative YM103 are acyclic tripodal tris(hydroxypyridinone) ligands that can rapidly radiolabel with 68Ga in 5 minutes at room temperature (Table 11).280 The maleimide derivative YM103 was conjugated to cysteine residues of the protein C2Ac, and in vivo experiments in normal healthy mice (no tumor models) showed rapid clearance through the kidneys with no observable decomposition after 1.5 hours.280 This new chelator was published less than 2 years ago, and further studies in tumor bearing mice will help to establish its potential utility. CP256 has an interesting acyclic tripodal design, similar to L3 (hydroxamate-based, Fig. 3), and most interestingly both chelators are oxygen-rich with O6 donor sets, suggesting a potential untapped application as 89Zr chelators (yet to be investigated, to our knowledge).KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
CP256, O6 CN = 6 |
67/68Ga3+ |

|
25 °C, 5 min, pH 6.5 | — | Distorted octahedron | 280 |
Bifunctional derivative YM103280 |
CP256 = 4-acetylamino-4-[2-[(3-hydroxy-1,6-dimethyl-4-oxo-1,4-dihydro-pyridin-2-ylmethyl)-carbamoyl]-ethyl]-heptanedioic acid bis-[(3-hydroxy-1,6-dimethyl-4-oxo-1,4-dihydro-pyridin-2-ylmethyl)-amide] | |||||
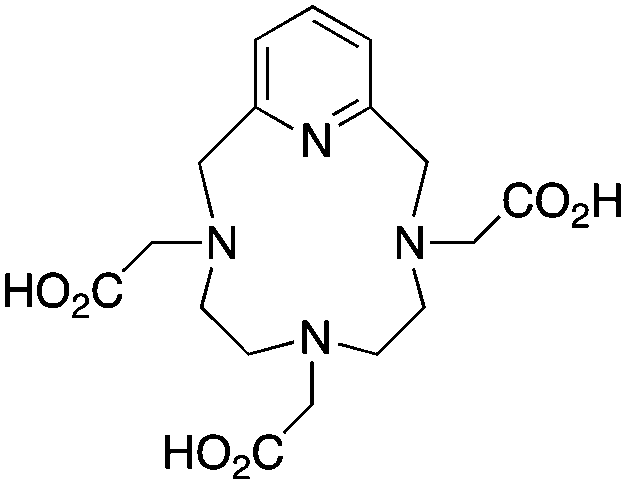
PCTA, 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,6,9,-triacetic acid, N4O3 CN = 7 |
64Cu2+ |

|
25 °C, 5 min, pH 5.5 | 19.1 | Distorted octahedron | 87, 90, 214, 290 |
| 67/68Ga3+ |

|
25 °C, 5–10 min, pH 4–5 | — | Distorted octahedron | 86, 290–292 | |
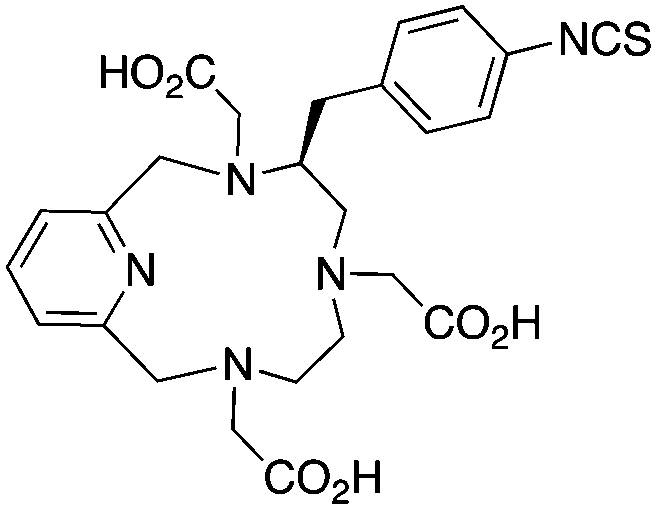
p-SCN-Bn-PCTA214 |
||||||
DFO, desferrioxamine B, O6, CN = 6 |
67/68Ga3+ |

|
25 °C, 30 min, pH 3.5 | 28.6 | Distorted octahedron? | 235, 293 |
| 89Zr4+ |

|
25 °C, 60 min, pH 7–7.3 | — | Distorted octahedron? | 51, 130, 240, 281, 283–288, 294–300 | |
p-SCN-Bn-DFO296 |
 Bifunctional DFO derivatives297,301
Bifunctional DFO derivatives297,301
|
|||||
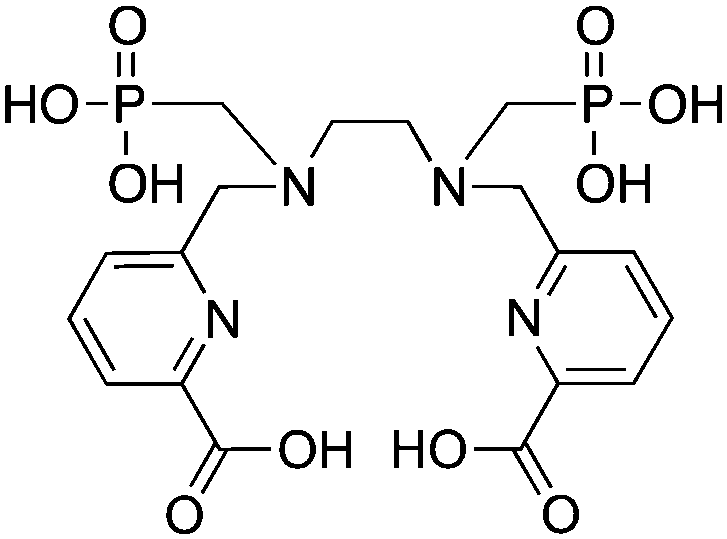
H6phospa, N,N′-(methylenephosphonate)-N,N′-[6-(methoxycarbonyl)pyridin-2-yl]methyl-1,2-diaminoethane, N4O4 CN = 8 |
89Zr4+ |

|
25 °C, 60 min, pH 7.4 | — | Square antiprism? | 289 |
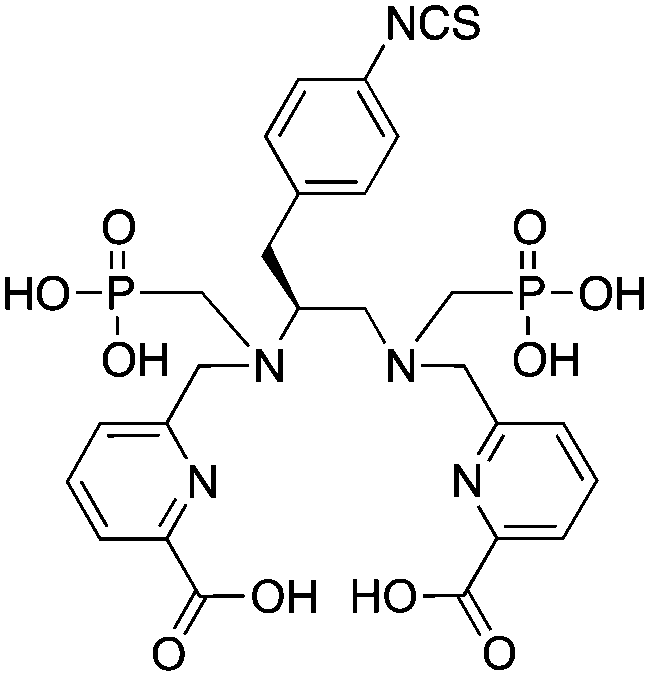
Bifunctional derivative p-SCN-Bn-H6phospa |
||||||
3.14 Desferrioxamine (DFO)
Desferrioxamine B (DFO) is a bacterial siderophore that natively binds Fe3+, and has also been used extensively with isotopes of gallium and zirconium (Table 11). DFO is the only competent 89Zr chelator available for radiolabeling and in vivo applications.281 Zr4+ is a highly charged, very hard metal ion with a relatively small ionic radius (84 and 89 pm for CN = 8 and 9, respectively),186 that is prone to forming insoluble polynuclear hydroxide species in aqueous solution under non-acidic conditions, and subsequently is difficult to radiolabel effectively. DFO is thought to bind 89Zr with its three hydroxamate groups in a hexadentate fashion, although no crystal structures have been obtained.282 For many years the next best chelator for 89Zr has been DTPA, which forms a thermodynamically stable complex with Zr4+ (logKML = 35.8–36.9), but studies have shown that radiolabeling with 89Zr is inefficient and in vivo stability is exceptionally poor.119 A DTPA–antibody conjugate of Zevalin was radiolabeled with 89Zr, and radiochemical yields obtained were <0.1% after 1 hour at room temperature;240 in contrast, DFO can radiolabel with 89Zr in quantitative yields (>99%) after 1 hour. The new chelator H6phospa has recently been studied with 89Zr, and has shown greatly improved radiolabeling performance relative to DTPA, but still inferior to DFO (vide infra).
No FDA-approved radiopharmaceuticals currently utilize 89Zr, although a number of 89Zr–DFO–antibody conjugates are in clinical trials.20889Zr has a long half life (78.5 hours), making it ideally paired with antibody vectors, as the biological half-life of an antibody is on the order of 2–3 weeks.3889Zr–DFO–Zevalin was the first 89Zr antibody conjugate imaged in humans, and was shown to be a suitable PET surrogate for 90Y-Zevalin dosimetry.283 A number of other studies have been performed using 89Zr-DFO-antibody conjugates with success, such as 89Zr-DFO-U36 (anti-CD446 chimeric mAb),208,28489Zr-DFO-bevacizumab,208,28589Zr-DFO-J591,28189Zr-DFO-TRC105,286 and 89Zr-DFO-trastuzumab.208,287,288
The high 4+ charge makes 89Zr challenging to incorporate into BFC systems while still retaining bioequivalence with other common 3+ cationic metal ions (e.g. In3+, Ga3+, Y3+, Lu3+), because the chelate–radiometal charge and polarity is different. Evaluating the stability of a potential BFC for 89Zr can be done in vivo, because 89Zr that is lost from a BFC typically localizes in bone, as demonstrated by the highly unstable 89Zr-DOTA- and 89Zr-DTPA-based antibody conjugates with cetuximab that showed significant 89Zr accretion in the thighbone 72 hours post injection.13089Zr-chloride and 89Zr-oxalate have also been injected directly into mice, with 89Zr-chloride forming colloids and accumulating in the liver, and the weakly chelated 89Zr-oxalate demonstrating rapid bone uptake.281 Although DFO is an excellent 89Zr chelator, some decomposition can be observed over time in vivo as 89Zr slowly accumulates in bone.27,51 The design of novel chelators for 89Zr with improved solubility, in vivo stability, and chelation properties would be timely, considering there is currently only one option (DFO). Based on the success of DFO, it would appear that oxygen-rich donors are most suitable for 89Zr chelation, with O6–O8 coordination being preferred, and donor groups including hydroxamates, carboxylates, carbonyls, catechols, and hydroxypyridinones being logical choices.
3.15 H6phospa
Developing new chelating agents for 89Zr is currently of great interest, as the only chelator available that can radiolabel 89Zr with any level of proficiency is DFO. As discussed in Section 3.14, the best alternative chelator to DFO for 89Zr radiolabeling is DTPA; however, with meager radiochemical yields of <0.1% after 1 hour at room temperature and poor in vivo stability, DTPA is not a viable alternative to DFO.240 DFO can radiolabel with 89Zr in quantitative yields (>99%) after 1 hour at room temperature, and demonstrates imperfect but acceptable stability in vivo, and no acceptable alternatives have been published to date. The acyclic chelator H6phospa (Table 11),264,265 and the bifunctional derivative p-SCN-Bn-H6phospa are methylenephosphonate derivatives of H4octapa (Table 9) that have been recently studied with 89Zr.289 The antibody conjugate H6phospa-trastuzumab was able to achieve radiochemical yields of ∼8% with 89Zr after 1 hour at room temperature (PBS, pH 7.4).289 This work was compared to previous attempts to radiolabel H4octapa-trastuzumab with 89Zr, which performed similarly to DTPA and exhibited essentially no radiolabeling after 1 hour.289 These results demonstrate that replacing the carboxylic acid arms of H4octapa with methylenephosphonate arms in H6phospa improved radiolabeling kinetics with 89Zr, suggesting that methylenephosphonate groups are more suitable than carboxylic acid groups for chelating 89Zr.289 Although a RCY of ∼8% is insufficient to supplant DFO as the “gold standard” chelator for radiolabeling with 89Zr, it is a significant improvement over DTPA and H4octapa, and to our knowledge is the highest 89Zr radiolabeling yield behind DFO to be published.3.16 PCTA
The heptadentate macrocyclic chelator PCTA was originally synthesized by Sherry and coworkers as a potential MRI contrast agent (Table 11).290 PCTA has been recently re-purposed and evaluated with 68Ga and 64Cu, and has been shown to possess much faster radiolabeling kinetics than DOTA (5–10 min, RT).86 The serum stability of the 67/68Ga–PCTA complex was found to be superior to DOTA, and comparable to NOTA.86 Additionally, the biodistribution profile of the non-bifunctional 68Ga–PCTA revealed lower kidney retention than 68Ga–NOTA, showing potential improvement over NOTA for use in peptide conjugates.86 Comparisons of RGD conjugates of the bifunctional derivatives of both PCTA and NOTA have demonstrated excellent in vivo stability and comparable biodistribution profiles, with the PCTA-based agent having lower kidney uptake than its NOTA counterpart.291 PCTA has also been investigated with 64Cu, showing greater radiolabeling kinetics and yields to DOTA, and superior in vitro and in vivo stability.75 PCTA has only recently been investigated, but further work may show it to be an excellent chelator for copper and gallium-based radiopharmaceuticals.3.17 HEHA and PEPA
HEHA is a large 18-membered, 12 coordinate, macrocyclic chelator, and PEPA is a structurally similar 10 coordinate derivative (Table 12). Both HEHA and PEPA have primarily been investigated for 225Ac complexation and RIT; preliminary radiolabeling and in vitro/in vivo stability experiments with 225Ac had suggested that HEHA was superior to DOTA and PEPA.224 The antibody conjugate HEHA-MAb-201B was radiolabeled with 225Ac and used to treat lung tumors in mice, and although effective at delivering large doses of 225Ac to tumors, high radiotoxicity from decay daughter isotopes resulted in death.302 More recently, using different in vitro experiments (e.g. cation exchange resin challenge) and different in vivo targets, DOTA has been shown to be the most suitable 225Ac chelator.101,103,303 A recent clinical trial with 225Ac-DOTA-HuM195 (humanized anti-CD33 monoclonal antibody) in humans has shown promise.100,103 PEPA has also been investigated for use with 212/213Bi for therapy applications, but was found to be unsuitable.304 α-emitting isotopes are currently of very high interest and potential impact, due to their effectiveness at killing cancer cells when they are tightly chelate-bound and safely delivered to their targets.3,134,149,151,210,303 α-particles travel a very short distance (μm range, compared to mm range for β− particles) and have high linear energy transfer values (LET), meaning they deposit huge amounts of energy over a very short distance, making them very effective at treating various cancers when properly directed, but also potentially very toxic.3,134,149,151,210,303 Development of new chelators for use with α-emitting isotopes is currently of high impact in the field of BFC.KML), coordination geometry, and color-coded ranking
| Chelator and common bifunctional derivatives | Radiometal ion | Radiolabeling conditions | logKML |
Proposed geometry | Ref. | |
|---|---|---|---|---|---|---|
| a Green “✓” = good/best match, orange “∼” = suitable match, or requires more evaluation but shows potential, red “✗” = poor/unstable match. | ||||||
HEHA, 1,4,7,10,13,16-hexaazacyclohexadecane-N,N′,N′′,N′′′,N′′′′,N′′′′′-hexaacetic acid, N6O6, CN = 12 |
225Ac3+ |

|
37–40 °C, 30 min, pH 5.8–7 | — | ? | 99, 101, 103, 303–305 |
p-SCN-Bn-HEHA (C-HEHA)99 |
||||||
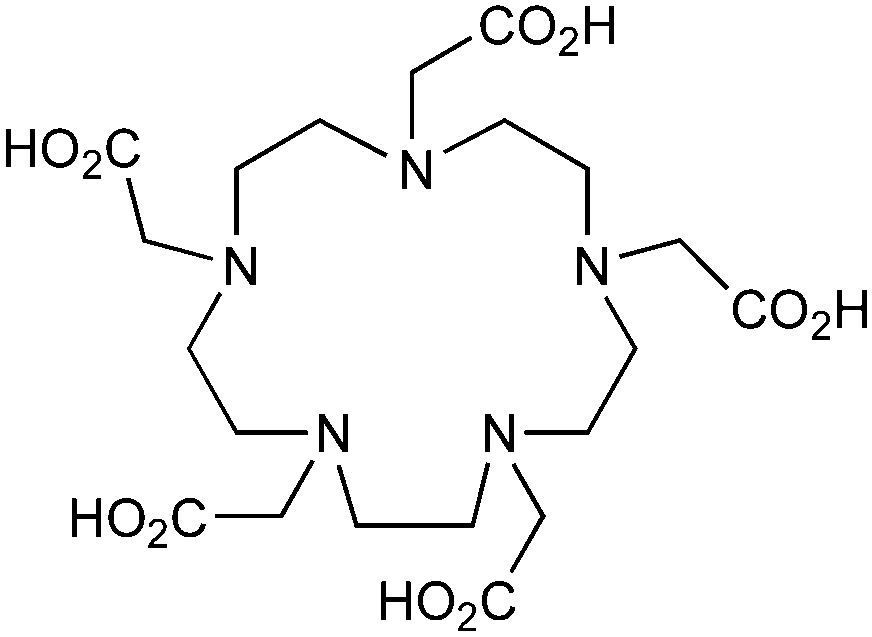
PEPA, 1,4,7,10,13-pentaazacyclopentadecane-N,N′,N′′,N′′′,N′′′′-pentaacetic acid, N5O5, CN = 10 |
212/213Bi3+ |

|
25 °C, 15 min, pH 5–5.5 | — | ? | 304 |
| 225Ac3+ |

|
37–40 °C, 30-60 min, pH 5.8–7 | — | ? | 99 | |
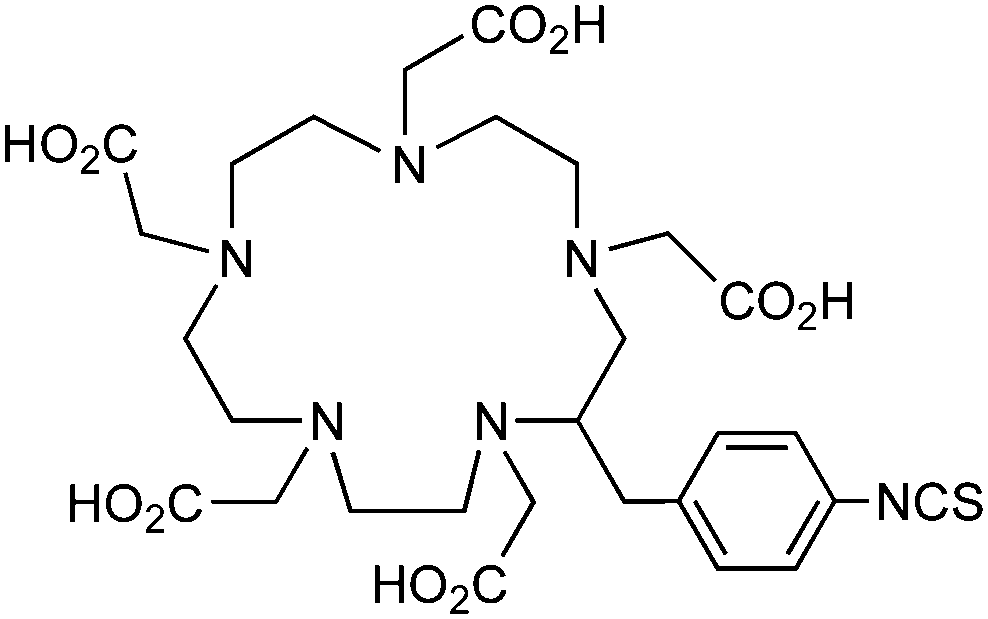
p-SCN-Bn-PEPA (C-PEPA)304 |
||||||
4. A brief look at less-developed chelators
A large number of chelators have been used transiently over the years for study and application with a variety of radiometals, but have not garnered very much interest from the radiochemistry community, have not been sufficiently developed to date, or have lacked a required level of proficiency (e.g. reaction kinetics, RCY, stability) for mainstream use. Additionally, a chelator may have shown great promise for use with a radiometal, but a suitable synthetic route to a bifunctional derivative may never have been published. Shown in Fig. 4 is a selection of chelators that have not been discussed here, but still may be of interest or of use to some researchers. These chelators may not have been found to be a suitable match to any of the current gamut of available radiometal ions discussed here, but production of more exotic radiometals in the future may bring them into relevance. Inspiration may also be drawn from the chemical structures, donor-atom types, connectivity, or geometries of these chelators, and potentially used towards new and novel chelators. | ||
| Fig. 4 A selection of less-discussed chelators with potential applications in radiometal chemistry, including NEC-SP,306 L1 (TACN-HSB, tris(2-mercaptobenzyl)amine),307 BAT-TM,308,309 EC,117,310 SBAD,311 BAPEN,312–314 TACHPYR,315–317 L2 (9-hydroxy-2,4-dipyridin-2-yl-3,7-bis(pyridin-2-ylmethyl)-3,7-diazabicyclo-[3.3.1]nonane-1,5-dicarboxylic acid),318 DiP-LICAM,319,320 and the hydroxamate-based L3.321 | ||
4.1 Conclusions
After examining the large number of highly competent radiometal chelators displayed in this review, some researchers may be daunted or even disheartened at the idea of creating new scaffolds with the existence of so many chelators that already work “well enough”. Radiometal chemistry teaches us that the intricate relationships amongst radiometal ion, chelator, linker, biological vector (e.g. peptide, antibody, antibody fragment, nanoparticle), and animal/tumor models are surprisingly complicated and only superficially understood. In order to optimally tune the properties of radiometal-based radiopharmaceuticals, a large variety of different tools (e.g. chelators, linkers, vectors) are crucial to have available so that the physical properties of an agent can be easily modified. Factors such as the charge, polarity, denticity, donor atom type (e.g. N, O, S), and kinetics (e.g. macrocyclic vs. acyclic) must be modular in order to optimize in vivo behavior of a radiopharmaceutical, and this requires the availability of many different bifunctional chelators. The synthetic canon of radiopharmaceutical components will always have space for new innovation and curiosity so that radioactive imaging agents and therapeutics can be continually improved.Current directions in radiochemistry and nuclear medicine highlight the importance of PET imaging, and subsequently the significance of optimal utilization of a growing number of β+ emitting radiometal ions. Promising new PET isotopes such as 44Sc and 89Zr currently have poorly elaborated chelate chemistry, and moving forward the development of fast-radiolabeling and exceptionally stable chelators for these isotopes will be important. 68Ga is perhaps the most promising of all isotopes discussed here, primarily due to its highly portable and long-lived generator system. To properly utilize 68Ga and translate it to the clinic, the wide-range of excellent 68Ga chelators that are currently under investigation must be transformed into viable imaging agents to provide useful diagnostic tools to the medical community. α-emitting isotopes are also currently of strong interest, as they are extremely effective, when targeted, at site-specifically delivering therapeutic doses of radioactivity to a variety of cancers. The caveat of α-emitters is their potentially fatal toxicity, and therefore robust chelate chemistry is of utmost importance for the continued development of therapeutic agents based on isotopes such as 225Ac, 212/213Bi, and 212Pb.
Acknowledgements
We acknowledge NSERC for CGS-M/CGS-D fellowships (E.W.P.) and many years of research support (C.O.), as well as the University of British Columbia for a 4YF fellowship (E.W.P.). C.O. acknowledges the Canada Council for the Arts for a Killam Research Fellowship (2011–2013), the University of Canterbury for a Visiting Erskine Fellowship (2013), and the Alexander von Humboldt Foundation for a Research Award, as well as Prof. Dr Peter Comba and his research group in Heidelberg for hospitality and interesting discussions. We also thank Caterina F. Ramogida for thoughtful editing.References
- S. Y. F. Chu, L. P. Ekström and R. B. Firestone, WWW table of radioactive isotopes, database version 2/28/1999, fromhttp://nucleardata.nuclear.lu.se/nucleardata/toi/, 1999.
- C. M. Lederer and V. S. Shirley, Table of Isotopes, John Wiley & Sons, New York, 7th edn, 1978 Search PubMed.
- J. P. Holland, M. J. Williamson and J. S. Lewis, Mol. Imaging, 2010, 9, 1–20 CAS.
- S. M. Qaim, Q. J. Nucl. Med. Mol. Imaging, 2008, 52, 111–120 CAS.
- D. Zeng and C. J. Anderson, Chem. Commun., 2013, 49, 2697–2699 RSC.
- T. K. Nayak, K. Garmestani, D. E. Milenic, K. E. Baidoo and M. W. Brechbiel, PLoS One, 2011, 6, e18198 CAS.
- T. K. Nayak, K. Garmestani, K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, Int. J. Cancer, 2010, 128, 920–926 CrossRef PubMed.
- S. Palm, R. M. Enmon, C. Matei, K. S. Kolbert, S. Xu, P. B. Zanzonico, R. L. Finn, J. A. Koutcher, S. M. Larson and G. Sgouros, J. Nucl. Med., 2003, 44, 1148–1155 CAS.
- L. I. Gordon, T. E. Witzig, G. A. Wiseman, I. W. Flinn, S. S. Spies, D. H. Silverman, C. Emmanuolides, L. Cripe, M. Saleh, M. S. Czuczman, T. Olejnik, C. A. White and A. J. Grillo-López, Semin. Oncol., 2002, 29, 87–92 CrossRef CAS PubMed.
- M. F. Giblin, B. Veerendra and C. J. Smith, In Vivo, 2005, 19, 9–29 CAS.
- H. Herzog, F. Rösch, G. Stöcklin, C. Lueders, S. M. Qaim and L. E. Feinendegen, J. Nucl. Med., 1993, 34, 2222–2226 CAS.
- F. Rösch and R. P. Baum, Dalton Trans., 2011, 40, 6104–6111 RSC.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- S. Walrand, G. Flux, M. Konijnenberg, R. Valkema, E. Krenning, R. Lhommel, S. Pauwels and F. Jamar, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 57–68 CrossRef CAS PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- M. Połosak, A. Piotrowska, S. Krajewski and A. Bilewicz, J. Radioanal. Nucl. Chem., 2013, 295, 1867–1872 CrossRef PubMed.
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC.
- B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168–6195 RSC.
- D. Zeng, B. M. Zeglis, J. S. Lewis and C. J. Anderson, J. Nucl. Med., 2013, 54, 829–832 CrossRef CAS PubMed.
- B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D. Carlin, P. Zanzonico, R. Weissleder and J. S. Lewis, J. Nucl. Med., 2013, 54, 1389–1396 CrossRef CAS PubMed.
- T. Reiner, E. J. Keliher, S. Earley, B. Marinelli and R. Weissleder, Angew. Chem., Int. Ed., 2011, 50, 1922–1925 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920–922 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- T. Reiner, J. Lacy, E. Keliher, K. S. Yang, A. Ullal, R. H. Kohler, C. Vinegoni and R. Weissleder, Neoplasia, 2012, 14, 169–177 CrossRef CAS PubMed.
- M. Lin, M. Welch and S. Lapi, Mol. Imaging Biol., 2013, 1–8 Search PubMed.
- M. D. Bartholoma, A. S. Louie, J. F. Valliant and J. Zubieta, Chem. Rev., 2010, 110, 2903–2920 CrossRef CAS PubMed.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, Nucl. Med. Biol., 2013, 40, 3–14 CrossRef CAS PubMed.
- M. D. Bartholomä, Inorg. Chim. Acta, 2012, 389, 36–51 CrossRef.
- W. A. P. Breeman, E. de Blois, H. Sze Chan, M. Konijnenberg, D. J. Kwekkeboom and E. P. Krenning, Semin. Nucl. Med., 2011, 41, 314–321 CrossRef PubMed.
- S. L. Rice, C. A. Roney, P. Daumar and J. S. Lewis, Semin. Nucl. Med., 2011, 41, 265–282 CrossRef PubMed.
- Z. Li and P. S. Conti, Adv. Drug Delivery Rev., 2010, 62, 1031–1051 CrossRef CAS PubMed.
- M. Glaser and E. G. Robins, J. Labelled Compd. Radiopharm., 2009, 52, 407–414 CrossRef CAS.
- D. S. Urch, Annu. Rep. Prog. Chem., Sect. A, 2009, 105, 460–476 RSC.
- S. Vallabhajosula, Chemistry of Metal Radionuclides (Rb, Ga, In, Y, Cu and Tc), Molecular Imaging, Springer, Berlin Heidelberg, 2009, pp. 179–196 Search PubMed.
- S. Liu, Adv. Drug Delivery Rev., 2008, 60, 1347–1370 CrossRef CAS PubMed.
- J. S. Lewis and C. J. Anderson, in Peptide Characterization and Application Protocols, ed. G. B. Fields, Humana Press, Inc., New York, 2007, vol. 386, pp. 227–240 Search PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Curr. Pharm. Des., 2007, 13, 3–16 CrossRef CAS PubMed.
- C. A. Boswell and M. W. Brechbiel, Nucl. Med. Biol., 2007, 34, 757–778 CrossRef CAS PubMed.
- G. A. M. S. van Dongen, G. W. M. Visser, M. N. Lub-de Hooge, E. G. de Vries and L. R. Perk, Oncologist, 2007, 12, 1379–1389 CrossRef CAS PubMed.
- D. S. Urch and M. J. Welch, Annu. Rep. Prog. Chem., Sect. A, 2006, 102, 542–563 RSC.
- S. Liu, ChemInform, 2005, 36, 445–461 Search PubMed.
- S. Liu and D. Edwards, in Contrast agents II: optical, ultrasound, x-ray and radiopharmaceutical imaging, ed. W. Krause, Springer-Verlag, Berlin Heidelberg, 2002, vol. 222, pp. 259–278 Search PubMed.
- C. J. Anderson and M. J. Welch, Chem. Rev., 1999, 99, 2219–2234 CrossRef CAS PubMed.
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2012, 113, 858–883 CrossRef PubMed.
- L. Koehler, K. Gagnon, S. McQuarrie and F. Wuest, Molecules, 2010, 15, 2686–2718 CrossRef CAS PubMed.
- M. W. Brechbiel, Q. J. Nucl. Med. Mol. Imaging, 2008, 52, 166–173 CAS.
- M. Pagani, S. Stone-Elander and S. A. Larsson, Eur. J. Nucl. Med. Mol. Imaging, 1997, 24, 1301–1327 CrossRef CAS.
- M. Fani, J. P. André and H. R. Maecke, Contrast Media Mol. Imaging, 2008, 3, 53–63 CrossRef CAS PubMed.
- S. Hassfjell and M. W. Brechbiel, Chem. Rev., 2001, 101, 2019–2036 CrossRef CAS PubMed.
- J. D. G. Correia, A. Paulo, P. D. Raposinho and I. Santos, Dalton Trans., 2011, 40, 6144–6167 RSC.
- B. M. Zeglis, P. Mohindra, G. I. Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder and J. S. Lewis, Bioconjugate Chem., 2011, 22, 2048–2059 CrossRef CAS PubMed.
- S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205–2218 CrossRef CAS PubMed.
- J. Byegård, G. Skarnemark and M. Skålberg, J. Radioanal. Nucl. Chem., 1999, 241, 281–290 CrossRef.
- J. B. Stimmel and F. C. Kull Jr, Nucl. Med. Biol., 1998, 25, 117–125 CrossRef CAS PubMed.
- J. B. Stimmel, M. E. Stockstill and F. C. Kull, Bioconjugate Chem., 1995, 6, 219–225 CrossRef CAS PubMed.
- L. Camera, S. Kinuya, K. Garmestani, C. Wu, M. W. Brechbiel, L. H. Pai, T. J. McMurry, O. A. Gansow, I. Pastan, C. H. Paik and J. A. Carrasquillo, J. Nucl. Med., 1994, 35, 882–889 CAS.
- A. Harrison, C. A. Walker, D. Parker, K. J. Jankowski, J. P. L. Cox, A. S. Craig, J. M. Sansom, N. R. A. Beeley, R. A. Boyce, L. Chaplin, M. A. W. Eaton, A. P. H. Farnsworth, K. Millar, A. T. Millican, A. M. Randall, S. K. Rhind, D. S. Secher and A. Turner, Int. J. Radiat. Appl. Instrum., Part B, 1991, 18, 469–476 CrossRef CAS.
- R. D. Hancock, J. Chem. Educ., 1992, 69, 615–621 CrossRef CAS.
- S. Liu and D. S. Edwards, Bioconjugate Chem., 2000, 12, 7–34 CrossRef.
- D. L. Kukis, S. J. DeNardo, G. L. DeNardo, R. T. O'Donnell and C. F. Meares, J. Nucl. Med., 1998, 39, 2105–2110 CAS.
- Y. H. Jang, M. Blanco, S. Dasgupta, D. A. Keire, J. E. Shively and W. A. Goddard, J. Am. Chem. Soc., 1999, 121, 6142–6151 CrossRef CAS.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Vol. 3: Other Organic Ligands, Plenum Press, New York, 1977 Search PubMed.
- R. M. Smith and A. E. Martell, Sci. Total Environ., 1987, 64, 125–147 CrossRef CAS.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 190, 37–46 CrossRef CAS.
- W. R. Harris and V. L. Pecoraro, Biochemistry, 1983, 22, 292–299 CrossRef CAS PubMed.
- K. N. Raymond, G. Müller and B. Matzanke, in Top. Curr. Chem., ed. F. L. Boschke, Springer-Verlag, Berlin, Heidelberg, 1984, vol. 123, pp. 49–102 Search PubMed.
- M. J. Kappel and K. N. Raymond, Inorg. Chem., 1982, 21, 3437–3442 CrossRef CAS.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 4829–4833 CrossRef CAS.
- J.-F. Morfin and É. Tóth, Inorg. Chem., 2011, 50, 10371–10378 CrossRef CAS PubMed.
- C. J. Broan, J. P. L. Cox, A. S. Craig, R. Kataky, D. Parker, A. Harrison, A. M. Randall and G. Ferguson, J. Chem. Soc., Perkin Trans. 2, 1991, 87–99 RSC.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, É. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960–10969 CrossRef PubMed.
- X. Sun, M. Wuest, G. R. Weisman, E. H. Wong, D. P. Reed, C. A. Boswell, R. Motekaitis, A. E. Martell, M. J. Welch and C. J. Anderson, J. Med. Chem., 2001, 45, 469–477 CrossRef.
- L. A. Bass, M. Wang, M. J. Welch and C. J. Anderson, Bioconjugate Chem., 2000, 11, 527–532 CrossRef CAS PubMed.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O'Halloran, Science, 1999, 284, 805–808 CrossRef CAS PubMed.
- W. R. Harris and L. Messori, Coord. Chem. Rev., 2002, 228, 237–262 CrossRef CAS.
- H. Sun, H. Li and P. J. Sadler, Chem. Rev., 1999, 99, 2817–2842 CrossRef CAS PubMed.
- W. Harris, in Less Common Metals in Proteins and Nucleic Acid Probes, ed. M. J. Clarke, Springer-Verlag, Berlin Heidelberg, 1998, vol. 92, pp. 121–162 Search PubMed.
- J. Šimeček, P. Hermann, H.-J. Wester and J. Notni, ChemMedChem, 2013, 8, 95–103 CrossRef PubMed.
- J. Notni, K. Pohle and H.-J. Wester, EJNMMI Res., 2012, 2, 1–5 CrossRef PubMed.
- J. Notni, J. Šimeček, P. Hermann and H.-J. Wester, Chem.–Eur. J., 2011, 17, 14718–14722 CrossRef CAS PubMed.
- J. Šimeček, M. Schulz, J. Notni, J. Plutnar, V. Kubíček, J. Havlíčková and P. Hermann, Inorg. Chem., 2011, 51, 577–590 CrossRef PubMed.
- D. Zeng and C. J. Anderson, Solvent Extr. Ion Exch., 2013, 31, 337–344 CrossRef CAS.
- E. W. Price, B. M. Zeglis, J. F. Cawthray, C. F. Ramogida, N. Ramos, J. S. Lewis, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2013, 135, 12707–12721 CrossRef CAS PubMed.
- G. A. Bailey, E. W. Price, B. M. Zeglis, C. L. Ferreira, E. Boros, M. J. Lacasse, B. O. Patrick, J. S. Lewis, M. J. Adam and C. Orvig, Inorg. Chem., 2012, 51, 12575–12589 CrossRef CAS PubMed.
- E. W. Price, J. F. Cawthray, G. A. Bailey, C. L. Ferreira, E. Boros, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2012, 134, 8670–8683 CrossRef CAS PubMed.
- C. L. Ferreira, E. Lamsa, M. Woods, Y. Duan, P. Fernando, C. Bensimon, M. Kordos, K. Guenther, P. Jurek and G. E. Kiefer, Bioconjugate Chem., 2010, 21, 531–536 CrossRef CAS PubMed.
- C. L. Ferreira, D. T. Yapp, E. Lamsa, M. Gleave, C. Bensimon, P. Jurek and G. E. Kiefer, Nucl. Med. Biol., 2008, 35, 875–882 CrossRef CAS PubMed.
- W. P. Li, D. S. Ma, C. Higginbotham, T. Hoffman, A. R. Ketring, C. S. Cutler and S. S. Jurisson, Nucl. Med. Biol., 2001, 28, 145–154 CrossRef CAS PubMed.
- A. Ando, I. Ando, T. Hiraki and K. Hisada, Int. J. Radiat. Appl. Instrum., Part B, 1989, 16, 57–80 CrossRef CAS.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- I. Laitinen, J. Notni, K. Pohle, M. Rudelius, E. Farrell, S. Nekolla, G. Henriksen, S. Neubauer, H. Kessler and H.-J. Wester, EJNMMI Res., 2013, 3, 1–9 CrossRef PubMed.
- M. H. Hofmann, H. M. Maecke, A. Börner, E. W. Weckesser, P. Schöffski, M. O. Oei, J. S. Schumacher, M. H. Henze, A. H. Heppeler, G. M. Meyer and W. K. Knapp, Eur. J. Nucl. Med. Mol. Imaging, 2001, 28, 1751–1757 CrossRef CAS PubMed.
- S. Liu, J. Pietryka, C. E. Ellars and D. S. Edwards, Bioconjugate Chem., 2002, 13, 902–913 CrossRef CAS PubMed.
- J. Fichna and A. Janecka, Bioconjugate Chem., 2003, 14, 3–17 CrossRef CAS PubMed.
- L. L. Chappell, B. E. Rogers, M. B. Khazaeli, M. S. Mayo, D. J. Buchsbaum and M. W. Brechbiel, Bioorg. Med. Chem., 1999, 7, 2313–2320 CrossRef CAS PubMed.
- S. Eigner, D. R. B. Vera, M. Fellner, N. S. Loktionova, M. Piel, O. Lebeda, F. Rösch, T. L. Roß and K. E. Henke, Mol. Imaging Biol., 2013, 15, 79–86 CrossRef PubMed.
- D. V. Filosofov, N. S. Loktionova and F. Rösch, Radiochim. Acta, 2010, 98, 149–156 CrossRef CAS.
- A. Majkowska-Pilip and A. Bilewicz, J. Inorg. Biochem., 2011, 105, 313–320 CrossRef CAS PubMed.
- L. L. Chappell, K. A. Deal, E. Dadachova and M. W. Brechbiel, Bioconjugate Chem., 2000, 11, 510–519 CrossRef CAS PubMed.
- Å. M. Ballangrud, W.-H. Yang, S. Palm, R. Enmon, P. E. Borchardt, V. A. Pellegrini, M. R. McDevitt, D. A. Scheinberg and G. Sgouros, Clin. Cancer Res., 2004, 10, 4489–4497 CrossRef PubMed.
- M. R. McDevitt, D. Ma, J. Simon, R. K. Frank and D. A. Scheinberg, Appl. Radiat. Isot., 2002, 57, 841–847 CrossRef CAS PubMed.
- H. Song, R. F. Hobbs, R. Vajravelu, D. L. Huso, C. Esaias, C. Apostolidis, A. Morgenstern and G. Sgouros, Cancer Res., 2009, 69, 8941–8948 CrossRef CAS PubMed.
- M. Miederer, M. R. McDevitt, G. Sgouros, K. Kramer, N.-K. V. Cheung and D. A. Scheinberg, J. Nucl. Med., 2004, 45, 129–137 CAS.
- S. Chaves, R. Delgado and J. J. R. F. Da Silva, Talanta, 1992, 39, 249–254 CrossRef CAS PubMed.
- R. Delgado and J. J. R. F. da Silva, Talanta, 1982, 29, 815–822 CrossRef CAS PubMed.
- J. C. Garrison, T. L. Rold, G. L. Sieckman, S. D. Figueroa, W. A. Volkert, S. S. Jurisson and T. J. Hoffman, J. Nucl. Med., 2007, 48, 1327–1337 CrossRef CAS PubMed.
- P. McQuade, Y. Miao, J. Yoo, T. P. Quinn, M. J. Welch and J. S. Lewis, J. Med. Chem., 2005, 48, 2985–2992 CrossRef CAS PubMed.
- S. H. Hausner, D. L. Kukis, M. K. J. Gagnon, C. E. Stanecki, R. Ferdani, J. F. Marshall, C. J. Anderson and J. L. Sutcliffe, Mol. Imaging, 2009, 8, 111–121 CAS.
- G. B. Biddlecombe, B. E. Rogers, M. de Visser, J. J. Parry, M. de Jong, J. L. Erion and J. S. Lewis, Bioconjugate Chem., 2007, 18, 724–730 CrossRef CAS PubMed.
- A. E. Martell, R. J. Motekaitis, E. T. Clarke, R. Delgado, Y. Sun and R. Ma, Supramol. Chem., 1996, 6, 353–363 CrossRef CAS.
- I. Velikyan, Å. L. Sundberg, Ö. Lindhe, A. U. Höglund, O. Eriksson, E. Werner, J. Carlsson, M. Bergström, B. Långström and V. Tolmachev, J. Nucl. Med., 2005, 46, 1881–1888 CAS.
- A. Dimitrakopoulou-Strauss, P. Hohenberger, U. Haberkorn, H. R. Mäcke, M. Eisenhut and L. G. Strauss, J. Nucl. Med., 2007, 48, 1245–1250 CrossRef CAS PubMed.
- G. Kramer-Marek, N. Shenoy, J. Seidel, G. Griffiths, P. Choyke and J. Capala, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 1967–1976 CrossRef CAS PubMed.
- C. Decristoforo, I. Hernandez Gonzalez, J. Carlsen, M. Rupprich, M. Huisman, I. Virgolini, H.-J. Wester and R. Haubner, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 1507–1515 CrossRef PubMed.
- M. Gabriel, C. Decristoforo, D. Kendler, G. Dobrozemsky, D. Heute, C. Uprimny, P. Kovacs, E. Von Guggenberg, R. Bale and I. J. Virgolini, J. Nucl. Med., 2007, 48, 508–518 CrossRef CAS PubMed.
- L. Wei, Y. Miao, F. Gallazzi, T. P. Quinn, M. J. Welch, A. L. Vavere and J. S. Lewis, Nucl. Med. Biol., 2007, 34, 945–953 CrossRef CAS PubMed.
- Y. Sun, C. J. Anderson, T. S. Pajeau, D. E. Reichert, R. D. Hancock, R. J. Motekaitis, A. E. Martell and M. J. Welch, J. Med. Chem., 1996, 39, 458–470 CrossRef CAS PubMed.
- E. Boros, C. L. Ferreira, J. F. Cawthray, E. W. Price, B. O. Patrick, D. W. Wester, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2010, 132, 15726–15733 CrossRef CAS PubMed.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Plenum Press, New York, 1974–1989, vol. 1–6 Search PubMed.
- S. Liu, Z. He, W.-Y. Hsieh and P. E. Fanwick, Inorg. Chem., 2003, 42, 8831–8837 CrossRef CAS PubMed.
- L. Li, J. Bading, P. J. Yazaki, A. H. Ahuja, D. Crow, D. Colcher, L. E. Williams, J. Y. C. Wong, A. Raubitschek and J. E. Shively, Bioconjugate Chem., 2007, 19, 89–96 CrossRef PubMed.
- S. Chakraborty, J. Shi, Y.-S. Kim, Y. Zhou, B. Jia, F. Wang and S. Liu, Bioconjugate Chem., 2010, 21, 969–978 CrossRef CAS PubMed.
- A. Tatsi, T. Maina, R. Cescato, B. Waser, E. Krenning, M. de Jong, P. Cordopatis, J. Reubi and B. Nock, EJNMMI Res., 2012, 2, 1–13 CrossRef PubMed.
- W. A. P. Breeman, M. de Jong, T. J. Visser, J. L. Erion and E. P. Krenning, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 917–920 CrossRef CAS PubMed.
- Ė. Tóth and E. Brücher, Inorg. Chim. Acta, 1994, 221, 165–167 CrossRef.
- M. Hens, G. Vaidyanathan, X.-G. Zhao, D. D. Bigner and M. R. Zalutsky, Nucl. Med. Biol., 2010, 37, 741–750 CrossRef CAS PubMed.
- C. S. Kang, X. Sun, F. Jia, H. A. Song, Y. Chen, M. Lewis and H.-S. Chong, Bioconjugate Chem., 2012, 23, 1775–1782 CrossRef CAS PubMed.
- D. E. Milenic, K. Garmestani, L. L. Chappell, E. Dadachova, A. Yordanov, D. Ma, J. Schlom and M. W. Brechbiel, Nucl. Med. Biol., 2002, 29, 431–442 CrossRef CAS PubMed.
- S. Rasaneh, H. Rajabi, M. H. Babaei and F. Johari Daha, J. Labelled Compd. Radiopharm., 2010, 53, 575–579 CrossRef CAS.
- L. R. Perk, G. W. M. Visser, M. J. W. D. Vosjan, M. Stigter-van Walsum, B. M. Tijink, C. R. Leemans and G. A. M. S. van Dongen, J. Nucl. Med., 2005, 46, 1898–1906 CAS.
- K. Kumar, C. A. Chang, L. C. Francesconi, D. D. Dischino, M. F. Malley, J. Z. Gougoutas and M. F. Tweedle, Inorg. Chem., 1994, 33, 3567–3575 CrossRef CAS.
- M. Koudelková, H. Vinšová and V. Jedináková-Křížová, J. Chromatogr., A, 2003, 990, 311–316 CrossRef.
- J. P. Norenberg, B. J. Krenning, I. R. H. M. Konings, D. F. Kusewitt, T. K. Nayak, T. L. Anderson, M. de Jong, K. Garmestani, M. W. Brechbiel and L. K. Kvols, Clin. Cancer Res., 2006, 12, 897–903 CrossRef CAS PubMed.
- D. Cordier, F. Forrer, F. Bruchertseifer, A. Morgenstern, C. Apostolidis, S. Good, J. Müller-Brand, H. Mäcke, J. C. Reubi and A. Merlo, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1335–1344 CrossRef CAS PubMed.
- D. Wild, M. Frischknecht, H. Zhang, A. Morgenstern, F. Bruchertseifer, J. Boisclair, A. Provencher-Bolliger, J.-C. Reubi and H. R. Maecke, Cancer Res., 2011, 71, 1009–1018 CrossRef CAS PubMed.
- E. Horak, F. Hartmann, K. Garmestani, C. Wu, M. Brechbiel, O. A. Gansow, N. F. Landolfi and T. A. Waldmann, J. Nucl. Med., 1997, 38, 1944–1950 CAS.
- G. Ruble, C. Wu, R. A. Squire, O. A. Gansow and M. Strand, Int. J. Radiat. Oncol., Biol., Phys., 1996, 34, 609–616 CrossRef CAS.
- K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, Nucl. Med. Biol., 2013, 40, 592–599 CrossRef CAS PubMed.
- W. Mier, J. Hoffend, S. Krämer, J. Schuhmacher, W. E. Hull, M. Eisenhut and U. Haberkorn, Bioconjugate Chem., 2004, 16, 237–240 CrossRef PubMed.
- L. L. Chappell, D. Ma, D. E. Milenic, K. Garmestani, V. Venditto, M. P. Beitzel and M. W. Brechbiel, Nucl. Med. Biol., 2003, 30, 581–595 CrossRef CAS PubMed.
- T. J. McMurry, M. Brechbiel, K. Kumar and O. A. Gansow, Bioconjugate Chem., 1992, 3, 108–117 CrossRef CAS PubMed.
- K.-P. Eisenwiener, P. Powell and H. R. Mäcke, Bioorg. Med. Chem. Lett., 2000, 10, 2133–2135 CrossRef CAS PubMed.
- C. Bernhard, M. Moreau, D. Lhenry, C. Goze, F. Boschetti, Y. Rousselin, F. Brunotte and F. Denat, Chem.–Eur. J., 2012, 18, 7834–7841 CrossRef CAS PubMed.
- C. A. Boswell, X. Sun, W. Niu, G. R. Weisman, E. H. Wong, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2004, 47, 1465–1474 CrossRef CAS PubMed.
- W. Niu, E. H. Wong, G. R. Weisman, Y. Peng, C. J. Anderson, L. N. Zakharov, J. A. Golen and A. L. Rheingold, Eur. J. Inorg. Chem., 2006, 676–677 CrossRef CAS.
- B. Bartoś, K. Lyczko, A. Kasperek, S. Krajewski and A. Bilewicz, J. Radioanal. Nucl. Chem., 2013, 295, 205–209 CrossRef PubMed.
- K. J. Yong, D. E. Milenic, K. E. Baidoo and M. W. Brechbiel, Br. J. Cancer, 2013, 108, 2013–2020 CrossRef CAS PubMed.
- K. J. Yong, D. E. Milenic, K. E. Baidoo and M. W. Brechbiel, Int. J. Radiat. Oncol., Biol., Phys., 2013, 85, 1119–1126 CrossRef CAS PubMed.
- H.-S. Chong, D. E. Milenic, K. Garmestani, E. D. Brady, H. Arora, C. Pfiester and M. W. Brechbiel, Nucl. Med. Biol., 2006, 33, 459–467 CrossRef CAS PubMed.
- L. L. Chappell, E. Dadachova, D. E. Milenic, K. Garmestani, C. Wu and M. W. Brechbiel, Nucl. Med. Biol., 2000, 27, 93–100 CrossRef CAS PubMed.
- K. Yong and M. W. Brechbiel, Dalton Trans., 2011, 40, 6068–6076 RSC.
- H. A. Song, C. S. Kang, K. E. Baidoo, D. E. Milenic, Y. Chen, A. Dai, M. W. Brechbiel and H.-S. Chong, Bioconjugate Chem., 2011, 22, 1128–1135 CrossRef CAS PubMed.
- L. Carlton, R. D. Hancock, H. Maumela and K. P. Wainwright, J. Chem. Soc., Chem. Commun., 1994, 1007–1008 RSC.
- H. Maumela, R. D. Hancock, L. Carlton, J. H. Reibenspies and K. P. Wainwright, J. Am. Chem. Soc., 1995, 117, 6698–6707 CrossRef CAS.
- S. Ait-Mohand, P. Fournier, V. R. Dumulon-Perreault, G. E. Kiefer, P. Jurek, C. L. Ferreira, F. O. Bénard and B. Guérin, Bioconjugate Chem., 2011, 22, 1729–1735 CrossRef CAS PubMed.
- D. T. Yapp, C. L. Ferreira, R. K. Gill, E. Boros, M. Q. Wong, D. Mandel, P. Jurek and G. E. Kiefer, Mol. Imaging, 2013, 12, 263–272 CAS.
- P. A. Knetsch, M. Petrik, C. Rangger, G. Seidel, H.-J. Pietzsch, I. Virgolini, C. Decristoforo and R. Haubner, Nucl. Med. Biol., 2013, 40, 65–72 CrossRef CAS PubMed.
- P. A. Knetsch, M. Petrik, C. Rangger, G. Seidel, H. J. Pietzsch, I. Virgolini, C. Decristoforo and R. Haubner, Nucl. Med. Biol., 2013, 40, 65–72 CrossRef CAS PubMed.
- C. F. Baes Jr and R. E. Mesmer, The Hydrolysis of Cations, Wiley-Interscience, New York, 1976 Search PubMed.
- C. J. Anderson, F. Dehdashti, P. D. Cutler, S. W. Schwarz, R. Laforest, L. A. Bass, J. S. Lewis and D. W. McCarthy, J. Nucl. Med., 2001, 42, 213–221 CAS.
- P. J. Blower, J. S. Lewis and J. Zweit, Nucl. Med. Biol., 1996, 23, 957–980 CrossRef CAS PubMed.
- D. N. Pandya, J. Y. Kim, J. C. Park, H. Lee, P. B. Phapale, W. Kwak, T. H. Choi, G. J. Cheon, Y.-R. Yoon and J. Yoo, Chem. Commun., 2010, 46, 3517–3519 RSC.
- A. V. Dale, D. N. Pandya, J. Y. Kim, H. Lee, Y. S. Ha, N. Bhatt, J. Kim, J. J. Seo, W. Lee, S. Kim, Y.-R. Yoon, G. I. An and J. Yoo, ACS Med. Chem. Lett., 2013, 10, 927–931 CrossRef PubMed.
- R. Ferdani, D. J. Stigers, A. L. Fiamengo, L. Wei, B. T. Y. Li, J. A. Golen, A. L. Rheingold, G. R. Weisman, E. H. Wong and C. J. Anderson, Dalton Trans., 2012, 41, 1938–1950 RSC.
- K. Suzuki, M. Satake, J. Suwada, S. Oshikiri, H. Ashino, H. Dozono, A. Hino, H. Kasahara and T. Minamizawa, Nucl. Med. Biol., 2011, 38, 1011–1018 CrossRef CAS PubMed.
- R. A. Dumont, F. Deininger, R. Haubner, H. R. Maecke, W. A. Weber and M. Fani, J. Nucl. Med., 2011, 52, 1276–1284 CrossRef CAS PubMed.
- M. Fani, L. Del Pozzo, K. Abiraj, R. Mansi, M. L. Tamma, R. Cescato, B. Waser, W. A. Weber, J. C. Reubi and H. R. Maecke, J. Nucl. Med., 2011, 52, 1110–1118 CrossRef CAS PubMed.
- J. E. Sprague, Y. Peng, A. L. Fiamengo, K. S. Woodin, E. A. Southwick, G. R. Weisman, E. H. Wong, J. A. Golen, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2007, 50, 2527–2535 CrossRef CAS PubMed.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 190, 27–36 CrossRef CAS.
- M. Kodama, T. Koike, A. B. Mahatma and E. Kimura, Inorg. Chem., 1991, 30, 1270–1273 CrossRef CAS.
- M. R. Lewis, C. A. Boswell, R. Laforest, T. L. Buettner, D. Ye, J. M. Connett and C. J. Anderson, Cancer Biother. Radiopharm., 2001, 16, 483–494 CrossRef CAS PubMed.
- M. Maillet, C. S. Kwok, A. A. Noujaim and V. Snieckus, Tetrahedron Lett., 1998, 39, 2659–2662 CrossRef CAS.
- S. V. Deshpande, S. J. DeNardo, C. F. Meares, M. J. McCall, G. P. Adams, M. K. Moi and G. L. DeNardo, J. Nucl. Med., 1988, 29, 217–225 CAS.
- L. Wei, Y. Ye, T. J. Wadas, J. S. Lewis, M. J. Welch, S. Achilefu and C. J. Anderson, Nucl. Med. Biol., 2009, 36, 277–285 CrossRef CAS PubMed.
- M. Eiblmaier, R. Andrews, R. Laforest, B. E. Rogers and C. J. Anderson, J. Nucl. Med., 2007, 48, 1390–1396 CrossRef CAS PubMed.
- C. A. Boswell, C. A. S. Regino, K. E. Baidoo, K. J. Wong, A. Bumb, H. Xu, D. E. Milenic, J. A. Kelley, C. C. Lai and M. W. Brechbiel, Bioconjugate Chem., 2008, 19, 1476–1484 CrossRef CAS PubMed.
- D. J. Stigers, R. Ferdani, G. R. Weisman, E. H. Wong, C. J. Anderson, J. A. Golen, C. Moore and A. L. Rheingold, Dalton Trans., 2010, 39, 1699–1701 RSC.
- K. A. Lears, R. Ferdani, K. Liang, A. Zheleznyak, R. Andrews, C. D. Sherman, S. Achilefu, C. J. Anderson and B. E. Rogers, J. Nucl. Med., 2011, 52, 470–477 CrossRef CAS PubMed.
- N. Di Bartolo, A. M. Sargeson and S. V. Smith, Org. Biomol. Chem., 2006, 4, 3350–3357 CAS.
- N. M. Di Bartolo, A. M. Sargeson, T. M. Donlevy and S. V. Smith, Dalton Trans., 2001, 2303–2309 RSC.
- S. Liu, Z. Li, L.-P. Yap, C.-W. Huang, R. Park and P. S. Conti, Chem.–Eur. J., 2011, 17, 10222–10225 CrossRef CAS PubMed.
- E. Mume, A. Asad, N. M. Di Bartolo, L. Kong, C. Smith, A. M. Sargeson, R. Price and S. V. Smith, Dalton Trans., 2013, 42, 14402–14410 RSC.
- I. Velikyan, H. Maecke and B. Langstrom, Bioconjugate Chem., 2008, 19, 569–573 CrossRef CAS PubMed.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 181, 273–280 CrossRef CAS.
- M. Lin, D. Ranganathan, S. Lapi and M. Welch, J. Nucl. Med. Meeting Abstracts, 2012, 53, 1457 Search PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Y. Zhang, H. Hong, J. W. Engle, J. Bean, Y. Yang, B. R. Leigh, T. E. Barnhart and W. Cai, PLoS One, 2011, 6, e28005 CAS.
- A. J. Chang, R. Sohn, Z. H. Lu, J. M. Arbeit and S. E. Lapi, PLoS One, 2013, 8, e58949 CAS.
- H.-S. Chong, K. Garmestani, D. Ma, D. E. Milenic, T. Overstreet and M. W. Brechbiel, J. Med. Chem., 2002, 45, 3458–3464 CrossRef CAS PubMed.
- H.-S. Chong, H. A. Song, N. Birch, T. Le, S. Lim and X. Ma, Bioorg. Med. Chem. Lett., 2008, 18, 3436–3439 CrossRef CAS PubMed.
- H.-S. Chong, H. A. Song, X. Ma, D. E. Milenic, E. D. Brady, S. Lim, H. Lee, K. Baidoo, D. Cheng and M. W. Brechbiel, Bioconjugate Chem., 2008, 19, 1439–1447 CrossRef CAS PubMed.
- H.-S. Chong, X. Ma, H. Lee, P. Bui, H. A. Song and N. Birch, J. Med. Chem., 2008, 51, 2208–2215 CrossRef CAS PubMed.
- H.-S. Chong, H. A. Song, C. S. Kang, T. Le, X. Sun, M. Dadwal, H. Lee, X. Lan, Y. Chen and A. Dai, Chem. Commun., 2011, 47, 5584–5586 RSC.
- C. S. Kang, H. A. Song, D. E. Milenic, K. E. Baidoo, M. W. Brechbiel and H.-S. Chong, Nucl. Med. Biol., 2013, 40, 600–605 CrossRef CAS PubMed.
- Y. Sun, C. S. Cutler, A. E. Martell and M. J. Welch, Tetrahedron, 1999, 55, 5733–5740 CrossRef CAS.
- A. Bevilacqua, R. I. Gelb, W. B. Hebard and L. J. Zompa, Inorg. Chem., 1987, 26, 2699–2706 CrossRef CAS.
- J. Strand, H. Honarvar, A. Perols, A. Orlova, R. K. Selvaraju, A. E. Karlström and V. Tolmachev, PLoS One, 2013, 8, e70028 CAS.
- A. Fuchs, I. Greguric and G. Roe, Nucl. Med. Biol., 2010, 37, 692 CrossRef.
- T. J. McMurry, M. Brechbiel, C. Wu and O. A. Gansow, Bioconjugate Chem., 1993, 4, 236–245 CrossRef CAS PubMed.
- K.-P. Eisenwiener, M. I. M. Prata, I. Buschmann, H.-W. Zhang, A. C. Santos, S. Wenger, J. C. Reubi and H. R. Maecke, Bioconjugate Chem., 2002, 13, 530–541 CrossRef CAS PubMed.
- V. Tolmachev, M. Altai, M. Sandström, A. Perols, A. E. Karlström, F. Boschetti and A. Orlova, Bioconjugate Chem., 2011, 22, 894–902 CrossRef CAS PubMed.
- J. P. André, H. R. Maecke, M. Zehnder, L. Macko and K. G. Akyel, Chem. Commun., 1998, 1301–1302 RSC.
- R. Ma, M. J. Welch, J. Reibenspies and A. E. Martell, Inorg. Chim. Acta, 1995, 236, 75–82 CrossRef CAS.
- Y. Sun, A. E. Martell, R. J. Motekaitis and M. J. Welch, Tetrahedron, 1998, 54, 4203–4210 CrossRef CAS.
- P. Aschoff, M. Öksüz, B. Kemke, K. Zhernosekov, M. Jennewein, F. Rösch and H. Bihl, Nuklearmedizin, 2005, 44, A58(V144) Search PubMed.
- P. Aschoff, B. Kemke, M. Öksüz, K. Zhernosekov, M. Jennewein, F. Rösch and H. Bihl, Nuklearmedizin, 2005, 44, A59(V146) Search PubMed.
- K. Hohloch, P. L. Zinzani, W. Linkesch, W. Jurczak, A. Deptala, M. Lorsbach, C. Windemuth-Kiesselbach, G. G. Wulf and L. H. Truemper, Bone Marrow Transplant., 2010, 46, 901–903 CrossRef PubMed.
- http://www.clinicaltrials.gov/, accessed July 29, 2013.
- J. A. Carrasquillo, J. D. White, C. H. Paik, A. Raubitschek, N. Le, M. Rotman, M. W. Brechbiel, O. A. Gansow, L. E. Top, P. Perentesis, J. C. Reynolds, D. L. Nelson and T. A. Waldmann, J. Nucl. Med., 1999, 40, 268–276 CAS.
- R. M. Sharkey, J. Burton and D. M. Goldenberg, Expert Rev. Clin. Immunol., 2005, 1, 47–62 CrossRef CAS PubMed.
- M. W. Brechbiel, O. A. Gansow, R. W. Atcher, J. Schlom, J. Esteban, D. Simpson and D. Colcher, Inorg. Chem., 1986, 25, 2772–2781 CrossRef CAS.
- C. Wu, H. Kobayashi, B. Sun, T. M. Yoo, C. H. Paik, O. A. Gansow, J. A. Carrasquillo, I. Pastan and M. W. Brechbiel, Bioorg. Med. Chem., 1997, 5, 1925–1934 CrossRef CAS PubMed.
- T. J. McMurry, C. G. Pippin, C. Wu, K. A. Deal, M. W. Brechbiel, S. Mirzadeh and O. A. Gansow, J. Med. Chem., 1998, 41, 3546–3549 CrossRef CAS PubMed.
- T. K. Nayak, K. Garmestani, K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, Int. J. Cancer, 2011, 128, 920–926 CrossRef CAS PubMed.
- T. K. Nayak, K. Garmestani, K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, J. Nucl. Med., 2010, 51, 942–950 CrossRef CAS PubMed.
- T. Nayak, C. Regino, K. Wong, D. Milenic, K. Garmestani, K. Baidoo, L. Szajek and M. Brechbiel, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1368–1376 CrossRef CAS PubMed.
- L. Wei, X. Zhang, F. Gallazzi, Y. Miao, X. Jin, M. W. Brechbiel, H. Xu, T. Clifford, M. J. Welch, J. S. Lewis and T. P. Quinn, Nucl. Med. Biol., 2009, 36, 345–354 CrossRef CAS PubMed.
- D. W. Schneider, T. Heitner, B. Alicke, D. R. Light, K. McLean, N. Satozawa, G. Parry, J. Yoo, J. S. Lewis and R. Parry, J. Nucl. Med., 2009, 50, 435–443 CrossRef CAS PubMed.
- T. Clifford, C. A. Boswell, G. B. Biddlecombe, J. S. Lewis and M. W. Brechbiel, J. Med. Chem., 2006, 49, 4297–4304 CrossRef CAS PubMed.
- F. T. Lee, A. J. Mountain, M. P. Kelly, C. Hall, A. Rigopoulos, T. G. Johns, F. E. Smyth, M. W. Brechbiel, E. C. Nice, A. W. Burgess and A. M. Scott, Clin. Cancer Res., 2005, 11, 7080s–7086s CrossRef CAS PubMed.
- M. W. Brechbiel, O. A. Gansow, C. G. Pippin, R. D. Rogers and R. P. Planalp, Inorg. Chem., 1996, 35, 6343–6348 CrossRef CAS.
- T. K. Nikula, M. R. McDevitt, R. D. Finn, C. Wu, R. W. Kozak, K. Garmestani, M. W. Brechbiel, M. J. Curcio, C. G. Pippin, L. Tiffany-Jones, M. W. Geerlings, C. Apostolidis, R. Molinet, O. A. Gansow and D. A. Scheinberg, J. Nucl. Med., 1999, 40, 166–176 CAS.
- M. R. McDevitt, R. D. Finn, D. Ma, S. M. Larson and D. A. Scheinberg, J. Nucl. Med., 1999, 40, 1722–1727 CAS.
- L. Camera, S. Kinuya, K. Garmestani, L. H. Pai, M. W. Brechbiel, O. A. Gansow, C. H. Paik, I. Pastan and J. A. Carrasquillo, Nucl. Med. Biol., 1993, 20, 955–962 CrossRef CAS PubMed.
- M. J. Blend, J. J. Stastny, S. M. Swanson and M. W. Brechbiel, Cancer Biother. Radiopharm., 2003, 18, 355–363 CrossRef CAS PubMed.
- M. P. Borgman, T. Coleman, R. B. Kolhatkar, S. Geyser-Stoops, B. R. Line and H. Ghandehari, J. Controlled Release, 2008, 132, 193–199 CrossRef CAS PubMed.
- J. Malmberg, V. Tolmachev and A. Orlova, Exp. Ther. Med., 2011, 2, 523–528 Search PubMed.
- V. Tolmachev, H. Wållberg, K. Andersson, A. Wennborg, H. Lundqvist and A. Orlova, Eur. J. Nucl. Med. Mol. Imaging, 2009, 36, 1460–1468 CrossRef PubMed.
- S. J. Hosseinimehr, A. Orlova and V. Tolmachev, Hum. Antibodies, 2010, 19, 107–111 CAS.
- R. W. Kozak, T. A. Waldmann, R. W. Atcher and O. A. Gansow, Trends Biotechnol., 1986, 4, 259–264 CrossRef CAS.
- D. E. Milenic, M. Roselli, S. Mirzadeh, C. G. Pippin, O. A. Gansow, D. Colcher, M. W. Brechbiel and J. Schlom, Cancer Biother. Radiopharm., 2001, 16, 133–146 CrossRef CAS PubMed.
- C. L. Ruegg, W. T. Anderson-Berg, M. W. Brechbiel, S. Mirzadeh, O. A. Gansow and M. Strand, Cancer Res., 1990, 50, 4221–4226 CAS.
- O. A. Gansow, Int. J. Radiat. Appl. Instrum., Part B, 1991, 18, 369–381 CrossRef CAS.
- P. Panizzi, M. Nahrendorf, J.-L. Figueiredo, J. Panizzi, B. Marinelli, Y. Iwamoto, E. Keliher, A. A. Maddur, P. Waterman, H. K. Kroh, F. Leuschner, E. Aikawa, F. K. Swirski, M. J. Pittet, T. M. Hackeng, P. Fuentes-Prior, O. Schneewind, P. E. Bock and R. Weissleder, Nat. Med., 2011, 17, 1142–1146 CrossRef CAS PubMed.
- S. Kojima and M. Jay, Eur. J. Nucl. Med., 1987, 13, 366–370 CAS.
- W. H. Bakker, R. Albert, C. Bruns, W. A. P. Breeman, L. J. Hofland, P. Marbach, J. Pless, D. Pralet, B. Stolz, J. W. Koper, S. W. J. Lamberts, T. J. Visser and E. P. Krenning, Life Sci., 1991, 49, 1583–1591 CrossRef CAS PubMed.
- M. N. Lub-de Hooge, J. G. W. Kosterink, P. J. Perik, H. Nijnuis, L. Tran, J. Bart, A. J. H. Suurmeijer, S. de Jong, P. L. Jager and E. G. E. de Vries, Br. J. Pharmacol., 2004, 143, 99–106 CrossRef CAS PubMed.
- S. Liu and D. S. Edwards, Bioconjugate Chem., 2001, 12, 630–634 CrossRef CAS PubMed.
- M. J. Koppe, R. P. Bleichrodt, A. C. Soede, A. A. Verhofstad, D. M. Goldenberg, W. J. G. Oyen and O. C. Boerman, J. Nucl. Med., 2004, 45, 1224–1232 CAS.
- L. R. Perk, O. J. Visser, M. Stigter-van Walsum, M. J. W. D. Vosjan, G. W. M. Visser, J. M. Zijlstra, P. C. Huijgens and G. A. M. S. Dongen, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1337–1345 CrossRef CAS PubMed.
- M. W. Brechbiel and O. A. Gansow, Bioconjugate Chem., 1991, 2, 187–194 CrossRef CAS PubMed.
- L. Camera, S. Kinuya, K. Garmestani, M. Brechbiel, C. Wu, L. Pai, T. McMurry, O. Gansow, I. Pastan, C. Paik and J. Carrasquillo, Eur. J. Nucl. Med., 1994, 21, 640–646 CrossRef CAS PubMed.
- M. Roselli, J. Schlom, O. A. Gansow, M. W. Brechbiel, S. Mirzadeh, C. G. Pippin, D. E. Milenic and D. Colcher, Int. J. Radiat. Appl. Instrum., Part B, 1991, 18, 389–394 CrossRef CAS.
- M. W. Brechbiel and O. A. Gansow, J. Chem. Soc., Perkin Trans. 1, 1992, 1173–1178 RSC.
- J. Šimeček, P. Hermann, J. Havlíčková, E. Herdtweck, T. G. Kapp, N. Engelbogen, H. Kessler, H.-J. Wester and J. Notni, Chem.–Eur. J., 2013, 19, 7748–7757 CrossRef PubMed.
- J. Notni, K. Pohle and H.-J. Wester, Nucl. Med. Biol., 2013, 40, 33–41 CrossRef CAS PubMed.
- J. Notni, J. Plutnar and H.-J. Wester, EJNMMI Res., 2012, 2, 1–4 CrossRef PubMed.
- J. Šimeček, O. Zemek, P. Hermann, H.-J. Wester and J. Notni, ChemMedChem, 2012, 7, 1375–1378 CrossRef PubMed.
- J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem.–Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed.
- R. H. Kimura, Z. Cheng, S. S. Gambhir and J. R. Cochran, Cancer Res., 2009, 69, 2435–2442 CrossRef CAS PubMed.
- R. Haubner, H.-J. Wester, F. Burkhart, R. Senekowitsch-Schmidtke, W. Weber, S. L. Goodman, H. Kessler and M. Schwaiger, J. Nucl. Med., 2001, 42, 326–336 CAS.
- R. P. Baum and H. R. Kulkarni, Theranostics, 2012, 2, 437–447 CrossRef CAS PubMed.
- Z. Baranyai, F. Uggeri, G. B. Giovenzana, A. Bényei, E. Brücher and S. Aime, Chem.–Eur. J., 2009, 15, 1696–1705 CrossRef CAS PubMed.
- B. P. Waldron, D. Parker, C. Burchardt, D. S. Yufit, M. Zimny and F. Roesch, Chem. Commun., 2013, 49, 579–581 RSC.
- D. Parker and B. P. Waldron, Org. Biomol. Chem., 2013, 11, 2827–2838 CAS.
- D. Parker, B. P. Waldron and D. S. Yufit, Dalton Trans., 2013, 42, 8001–8008 RSC.
- J. Simecek, H.-J. Wester and J. Notni, Dalton Trans., 2012, 41, 13803–13806 RSC.
- R. Ferreiros-Martinez, D. Esteban-Gomez, C. Platas-Iglesias, A. de Blas and T. Rodriguez-Blas, Inorg. Chem., 2009, 48, 10976–10987 CrossRef CAS PubMed.
- R. Ferreiros-Martinez, D. Esteban-Gomez, A. de Blas, C. Platas-Iglesias and T. Rodriguez-Blas, Inorg. Chem., 2009, 48, 11821–11831 CrossRef CAS PubMed.
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gomez, E. Toth, A. de Blas, C. Platas-Iglesias and T. Rodriguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS PubMed.
- L. Vaiana, D. Esteban-Gómez, M. Mato-Iglesias, C. Platas-Iglesias, A. de Blas and T. Rodríguez-Blas, Eur. J. Inorg. Chem., 2009, 400–411 CrossRef CAS.
- R. Ferreiros-Martinez, D. Esteban-Gomez, C. Platas-Iglesias, A. de Blas and T. Rodriguez-Blas, Dalton Trans., 2008, 5754–5765 RSC.
- M. Mato-Iglesias, A. Roca-Sabio, Z. Palinkas, D. Esteban-Gomez, C. Platas-Iglesias, E. Toth, A. de Blas and T. Rodriguez-Blas, Inorg. Chem., 2008, 47, 7840–7851 CrossRef CAS PubMed.
- E. Balogh, M. Mato-Iglesias, C. Platas-Iglesias, E. Toth, K. Djanashvili, J. A. Peters, A. de Blas and T. Rodriguez-Blas, Inorg. Chem., 2006, 45, 8719–8728 CrossRef CAS PubMed.
- M. Mato-Iglesias, E. Balogh, C. Platas-Iglesias, E. Toth, A. de Blas and T. R. Blas, Dalton Trans., 2006, 5404–5415 RSC.
- M. Mato-Iglesias, C. Platas-Iglesias, K. Djanashvili, J. A. Peters, E. Toth, E. Balogh, R. N. Muller, L. V. Elst, A. de Blas and T. Rodriguez-Blas, Chem. Commun., 2005, 4729–4731 RSC.
- C. Platas-Iglesias, M. Mato-Iglesias, K. Djanashvili, R. N. Muller, L. V. Elst, J. A. Peters, A. de Blas and T. Rodríguez-Blas, Chem.–Eur. J., 2004, 10, 3579–3590 CrossRef CAS PubMed.
- E. Boros, C. L. Ferreira, D. T. T. Yapp, R. K. Gill, E. W. Price, M. J. Adam and C. Orvig, Nucl. Med. Biol., 2012, 39, 785–794 CrossRef CAS PubMed.
- E. Boros, J. F. Cawthray, C. L. Ferreira, B. O. Patrick, M. J. Adam and C. Orvig, Inorg. Chem., 2012, 51, 6279–6284 CrossRef CAS PubMed.
- E. Boros, C. L. Ferreira, B. O. Patrick, M. J. Adam and C. Orvig, Nucl. Med. Biol., 2011, 38, 1165–1174 CrossRef CAS PubMed.
- H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, Chem.–Eur. J., 2008, 14, 6173–6183 CrossRef CAS PubMed.
- R. J. Motekaitis, Y. Sun and A. E. Martell, Inorg. Chim. Acta, 1989, 159, 29–39 CrossRef CAS.
- M. Eder, A. V. Krivoshein, M. Backer, J. M. Backer, U. Haberkorn and M. Eisenhut, Nucl. Med. Biol., 2010, 37, 405–412 CrossRef CAS PubMed.
- M. Eder, B. Wängler, S. Knackmuss, F. LeGall, M. Little, U. Haberkorn, W. Mier and M. Eisenhut, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 1878–1886 CrossRef CAS PubMed.
- R. Ma, R. J. Motekaitis and A. E. Martell, Inorg. Chim. Acta, 1994, 224, 151–155 CrossRef CAS.
- C. J. Mathias, Y. Sun, M. J. Welch, M. A. Green, J. A. Thomas, K. R. Wade and A. E. Martell, Int. J. Radiat. Appl. Instrum., Part B, 1988, 15, 69–81 CrossRef CAS.
- S. Brillouet, S. Dorbes, F. Courbon, C. Picard, J. P. Delord, E. Benoist, M. Poirot, B. Mestre-Voegtlé and S. Silvente-Poirot, Bioorg. Med. Chem., 2010, 18, 5400–5412 CrossRef CAS PubMed.
- F. Havas, M. Danel, C. Galaup, P. Tisnès and C. Picard, Tetrahedron Lett., 2007, 48, 999–1002 CrossRef CAS.
- J. Schuhmacher, S. Kaul, G. Klivényi, H. Junkermann, A. Magener, M. Henze, J. Doll, U. Haberkorn, F. Amelung and G. Bastert, Cancer Res., 2001, 61, 3712–3717 CAS.
- D. J. Berry, Y. Ma, J. R. Ballinger, R. Tavare, A. Koers, K. Sunassee, T. Zhou, S. Nawaz, G. E. D. Mullen, R. C. Hider and P. J. Blower, Chem. Commun., 2011, 47, 7068–7070 RSC.
- J. P. Holland, V. Divilov, N. H. Bander, P. M. Smith-Jones, S. M. Larson and J. S. Lewis, J. Nucl. Med., 2010, 51, 1293–1300 CrossRef CAS PubMed.
- W. E. Meijs, J. D. M. Herscheid, H. J. Haisma and H. M. Pinedo, Int. J. Radiat. Appl. Instrum., Part A, 1992, 43, 1443–1447 CrossRef CAS.
- L. Perk, O. Visser, M. Stigter-van Walsum, M. Vosjan, G. Visser, J. Zijlstra, P. Huijgens and G. van Dongen, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1337–1345 CrossRef CAS PubMed.
- P. K. E. Börjesson, Y. W. S. Jauw, R. Boellaard, R. de Bree, E. F. I. Comans, J. C. Roos, J. A. Castelijns, M. J. W. D. Vosjan, J. A. Kummer, C. R. Leemans, A. A. Lammertsma and G. A. M. S. van Dongen, Clin. Cancer Res., 2006, 12, 2133–2140 CrossRef PubMed.
- W. B. Nagengast, E. G. de Vries, G. A. Hospers, N. H. Mulder, J. R. de Jong, H. Hollema, A. H. Brouwers, G. A. van Dongen, L. R. Perk and M. N. Lub-de Hooge, J. Nucl. Med., 2007, 48, 1313–1319 CrossRef CAS PubMed.
- H. Hong, G. Severin, Y. Yang, J. Engle, Y. Zhang, T. Barnhart, G. Liu, B. Leigh, R. Nickles and W. Cai, Eur. J. Nucl. Med. Mol. Imaging, 2011, 39, 138–148 CrossRef PubMed.
- J. P. Holland, E. Caldas-Lopes, V. Divilov, V. A. Longo, T. Taldone, D. Zatorska, G. Chiosis and J. S. Lewis, PLoS One, 2010, 5, e8859 Search PubMed.
- A. J. Chang, R. DeSilva, S. Jain, K. Lears, B. Rogers and S. Lapi, Pharmaceuticals, 2012, 5, 79–93 CrossRef CAS PubMed.
- E. W. Price, B. M. Zeglis, J. S. Lewis, M. J. Adam and C. Orvig, Dalton Trans., 2013 10.1039/C3DT51940F.
- W. D. Kim, G. E. Kiefer, F. Maton, K. McMillan, R. N. Muller and A. D. Sherry, Inorg. Chem., 1995, 34, 2233–2243 CrossRef CAS.
- C. L. Ferreira, D. T. T. Yapp, D. Mandel, R. K. Gill, E. Boros, M. Q. Wong, P. Jurek and G. E. Kiefer, Bioconjugate Chem., 2012, 23, 2239–2246 CrossRef CAS PubMed.
- G. Tircsó, E. T. Benyó, E. H. Suh, P. Jurek, G. E. Kiefer, A. D. Sherry and Z. Kovács, Bioconjugate Chem., 2009, 20, 565–575 CrossRef PubMed.
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Inorg. Chem., 1989, 28, 2189–2195 CrossRef CAS.
- I. Verel, G. W. M. Visser, R. Boellaard, M. Stigter-van Walsum, G. B. Snow and G. A. M. S. van Dongen, J. Nucl. Med., 2003, 44, 1271–1281 CAS.
- M. J. W. D. Vosjan, L. R. Perk, G. W. M. Visser, M. Budde, P. Jurek, G. E. Kiefer and G. A. M. S. van Dongen, Nat. Protocols, 2010, 5, 739–743 CAS.
- L. Perk, M. Vosjan, G. Visser, M. Budde, P. Jurek, G. Kiefer and G. van Dongen, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 250–259 CrossRef CAS PubMed.
- J. N. Tinianow, H. S. Gill, A. Ogasawara, J. E. Flores, A. N. Vanderbilt, E. Luis, R. Vandlen, M. Darwish, J. R. Junutula, S. P. Williams and J. Marik, Nucl. Med. Biol., 37, 289–297 CrossRef CAS PubMed.
- C. Heneweer, J. P. Holland, V. Divilov, S. Carlin and J. S. Lewis, J. Nucl. Med., 2011, 52, 625–633 CrossRef CAS PubMed.
- J. P. Holland, Y. Sheh and J. S. Lewis, Nucl. Med. Biol., 2009, 36, 729–739 CrossRef CAS PubMed.
- E. C. F. Dijkers, J. G. W. Kosterink, A. P. Rademaker, L. R. Perk, G. A. M. S. van Dongen, J. Bart, J. R. de Jong, E. G. E. de Vries and M. N. Lub-de Hooge, J. Nucl. Med., 2009, 50, 974–981 CrossRef CAS PubMed.
- W. E. Meijs, H. J. Haisma, R. P. Klok, F. B. van Gog, E. Kievit, H. M. Pinedo and J. D. M. Herscheid, J. Nucl. Med., 1997, 38, 112–118 CAS.
- S. J. Kennel, L. L. Chappell, K. Dadachova, M. W. Brechbiel, T. K. Lankford, I. A. Davis, M. Stabin and S. Mirzadeh, Cancer Biother. Radiopharm., 2000, 15, 235–244 CrossRef CAS PubMed.
- J. Schwartz, J. S. Jaggi, J. A. O'Donoghue, S. Ruan, M. McDevitt, S. M. Larson, D. A. Scheinberg and J. L. Humm, Phys. Med. Biol., 2011, 56, 721 CrossRef CAS PubMed.
- K. Garmestani, Z. Yao, M. Zhang, K. Wong, C. W. Park, I. Pastan, J. A. Carrasquillo and M. W. Brechbiel, Nucl. Med. Biol., 2001, 28, 409–418 CrossRef CAS PubMed.
- K. A. Deal, I. A. Davis, S. Mirzadeh, S. J. Kennel and M. W. Brechbiel, J. Med. Chem., 1999, 42, 2988–2992 CrossRef CAS PubMed.
- G. Papini, S. Alidori, J. S. Lewis, D. E. Reichert, M. Pellei, G. Gioia Lobbia, G. B. Biddlecombe, C. J. Anderson and C. Santini, Dalton Trans., 2009, 177–184 RSC.
- R. J. Motekaitis, A. E. Martell, S. A. Koch, J. Hwang, D. A. Quarless, Jr. and M. J. Welch, Inorg. Chem., 1998, 37, 5902–5911 CrossRef CAS.
- L. C. Francesconi, B.-L. Liu, J. J. Billings, P. J. Carroll, G. Graczyk and H. F. Kung, J. Chem. Soc., Chem. Commun., 1991, 94–95 RSC.
- Y. Y. Zheng, S. Saluja, G. P. A. Yap, M. Blumenstein, A. L. Rheingold and L. C. Francesconi, Inorg. Chem., 1996, 35, 6656–6666 CrossRef CAS PubMed.
- Y. Li, A. E. Martell, R. D. Hancock, J. H. Reibenspies, C. J. Anderson and M. J. Welch, Inorg. Chem., 1996, 35, 404–414 CrossRef CAS PubMed.
- E. Wong, P. Caravan, S. Liu, S. J. Rettig and C. Orvig, Inorg. Chem., 1996, 35, 715–724 CrossRef CAS.
- B. W. Tsang, C. J. Mathias, P. E. Fanwick and M. A. Green, J. Med. Chem., 1994, 37, 4400–4406 CrossRef CAS PubMed.
- V. Sharma, A. Beatty, S. P. Wey, J. Dahlheimer, C. M. Pica, C. L. Crankshaw, L. Bass, M. A. Green, M. J. Welch and D. Piwnica-Worms, Chem. Biol., 2000, 7, 335–343 CrossRef CAS PubMed.
- Y.-M. Hsiao, C. J. Mathias, S.-P. Wey, P. E. Fanwick and M. A. Green, Nucl. Med. Biol., 2009, 36, 39–45 CrossRef CAS PubMed.
- G. Park, E. Dadachova, A. Przyborowska, S.-J. Lai, D. Ma, G. Broker, R. D. Rogers, R. P. Planalp and M. W. Brechbiel, Polyhedron, 2001, 20, 3155–3163 CrossRef CAS.
- D. Ma, F. Lu, T. Overstreet, D. E. Milenic and M. W. Brechbiel, Nucl. Med. Biol., 2002, 29, 91–105 CrossRef CAS PubMed.
- T. Bowen, R. P. Planalp and M. W. Brechbiel, Bioorg. Med. Chem. Lett., 1996, 6, 807–810 CrossRef CAS.
- S. Juran, M. Walther, H. Stephan, R. Bergmann, J. Steinbach, W. Kraus, F. Emmerling and P. Comba, Bioconjugate Chem., 2009, 20, 347–359 CrossRef CAS PubMed.
- A. Riesen, T. A. Kaden, W. Ritter and H. R. Maecke, J. Chem. Soc., Chem. Commun., 1989, 460–462 RSC.
- S. M. Moerlein and M. J. Welch, Int. J. Nucl. Med. Biol., 1981, 8, 277–287 CrossRef CAS PubMed.
- R. J. Motekaitis, Y. Sun and A. E. Martell, Inorg. Chem., 1991, 30, 1554–1556 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |