 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral metal nanoparticle-catalyzed asymmetric C–C bond formation reactions
Tomohiro
Yasukawa
,
Hiroyuki
Miyamura
and
Shū
Kobayashi
*
Department of Chemistry, School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shu_kobayashi@chem.s.u-tokyo.ac.jp; Fax: +81-3-5684-0634
First published on 5th December 2013
Abstract
Chiral ligand-modified metal nanoparticles possess an attractive potential for application in asymmetric synthesis. This article focuses on chiral-nanoparticle-catalyzed asymmetric C–C bond formation reactions and discusses the nature of the active species.
 Tomohiro Yasukawa | Tomohiro Yasukawa was born in Kanagawa, Japan, in 1987 and studied at the University of Tokyo, receiving his MSc degree in 2012 working under the direction of Prof. Shū Kobayashi. At present, he is a PhD student in the group of Prof. Shū Kobayashi. His research interests include development of chiral metal nanoparticle catalysts, polymer immobilized catalysts and environmentally benign organic synthesis. |
 Hiroyuki Miyamura | Hiroyuki Miyamura was born in 1982 in Saitama, Japan. He received his BS degree in pharmaceutical sciences in 2004 from The University of Tokyo. He received his MSc and PhD degrees at the same University in 2006 and 2009 under the supervision of Professor Shū Kobayashi. In 2009 just after receiving PhD, he was appointed assistant professor in the Department of chemistry at The University of Tokyo. In 2010, he received the winner of The Reaxys PhD Prize. His research interest includes metal nanocluster catalysis and reaction integration such as tandem reaction and flow synthesis. |
 Shū Kobayashi | Shū Kobayashi studied at the University of Tokyo, receiving his PhD in 1988 working under the direction of Professor T. Mukaiyama. Following an initial period as assistant professor, he was promoted to lecturer then associate professor at Science University of Tokyo (SUT). In 1998, he moved to the Graduate School of Pharmaceutical Sciences, the University of Tokyo, as full professor. In 2007, he was appointed to his current position as professor of organic chemistry in the Department of Chemistry, Faculty of Science, The University of Tokyo. He has held various visiting professorships, including the Universite Louis Pasteur, Strasbourg (1993), Kyoto University (1995), Nijmegen University (1996), Philipps-University of Marburg (1997), Paris-Sud (2010). Professor Kobayashi has wide-ranging research interests that include the development of new synthetic methods and novel catalysts, organic reactions in water, solid-phase synthesis, total synthesis of biologically interesting compounds, and organometallic chemistry. |
1. Introduction
Rapid progress has been made in the field of homogeneous chiral metal complexes and many types of difficult transformations have been realized, including asymmetric C–C bond formation, to construct complicated molecular structures with high selectivity.1,2 However, the application of these homogeneous catalysts to large-scale industrial processes is still limited.3–9 This is because industry generally prefers heterogeneous catalysts because of their great advantages, such as low cost, reusability, avoidance of metal contamination of products, and the availability for reaction integration such as in continuous flow systems and tandem reactions.10–14 One of the common strategies to convert homogeneous catalysts to heterogeneous catalysts is immobilization of chiral ligands on inorganic or organic supports followed by introduction of metal salts to form “immobilized chiral metal complexes.”15–19 While this strategy has already been investigated for several reactions, these systems have drawbacks, such as complicated preparation of monomeric ligands and individual fabrication of heterogeneous polymers. In addition, these catalysts themselves are not robust because of metal leaching and the instability of immobilized ligands, and it is difficult to revive their activity when the catalysts are deactivated.Metal nanoparticles (NPs) in catalysts are of great interest in both academia and industry, not only due to their very large surface areas, producing high catalytic activity, but also due to their quantum size effect that sometimes shows high activity and unique selectivity.20–23 Recently, several reports have shown that homogeneous metal complex-catalyzed reactions can be conducted by using immobilized metal NP catalytic systems, with the advantages of the heterogeneous catalysts mentioned above.24 It might indicate that metal NPs possess the potential for application in asymmetric synthesis, namely, the use of chiral-ligand-modified metal NPs (chiral NPs) as asymmetric catalysts (Fig. 1).
 | ||
| Fig. 1 Functionalized chiral NP catalysts. | ||
Chiral NPs themselves have attracted the attention of many researchers in various fields as they show characteristic physical properties25–28 such as circular dichroism (CD) and metal-based electronic transitions. Furthermore, it may be possible to use them not only for asymmetric catalysis, but also as chiral nematic liquid crystals,29–32 in chiral recognition,33–36etc.
Chiral NPs have been mainly prepared by two types of methods:28 direct synthesis of NPs in the presence of a chiral modifier as a stabilizer (Fig. 2a) or ligand exchange from stabilized NPs (Fig. 2b). In the latter method, it is possible that chiral NPs form in situ simply by combining chiral modifiers and metal NPs in the reaction medium and then using them directly as chiral catalysts.
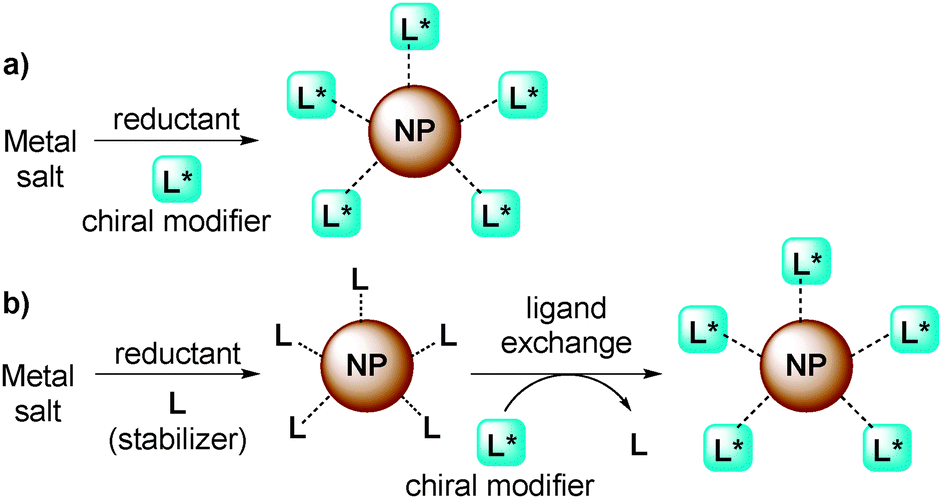 | ||
| Fig. 2 (a) Direct synthesis of chiral NPs; (b) synthesis of chiral NPs via ligand exchange. | ||
The first example of heterogeneous asymmetric catalysis using this concept was reported by Orito and co-workers in 1979, demonstrating asymmetric hydrogenation of methyl pyruvate or methyl benzoylformate catalyzed by cinchonidine-modified Pt on carbon (Scheme 1).37,38 Although the first Orito-type reaction was reported more than 30 years ago, this type of reaction is still being studied.39–43 Recently, studies were carried out to improve the catalyst system44–61 and to reveal its detailed mechanism.62–67 It is generally accepted that the chemisorption layer, through the formation of individual 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric complexes from substrates and chiral modifiers, is the key to inducing chirality in this reaction.62 Very recently, Maeda, Baiker and co-workers proposed a possible intermediate and complex between a substrate and a modifier formed by multiple hydrogen bonds.64 Based on this chiral NP system, other types of chiral NP catalysts for asymmetric hydrogenation reactions, for example nickel-boride NPs,68 Pd NPs,69–85 Ru NPs,86–90 Rh NPs,88,91–96 Ir NPs,88,97–99 and Fe NPs,100 have been widely investigated. Several reviews illustrate examples of heterogeneous asymmetric hydrogenation reactions.17,101–104
:1 diastereomeric complexes from substrates and chiral modifiers, is the key to inducing chirality in this reaction.62 Very recently, Maeda, Baiker and co-workers proposed a possible intermediate and complex between a substrate and a modifier formed by multiple hydrogen bonds.64 Based on this chiral NP system, other types of chiral NP catalysts for asymmetric hydrogenation reactions, for example nickel-boride NPs,68 Pd NPs,69–85 Ru NPs,86–90 Rh NPs,88,91–96 Ir NPs,88,97–99 and Fe NPs,100 have been widely investigated. Several reviews illustrate examples of heterogeneous asymmetric hydrogenation reactions.17,101–104
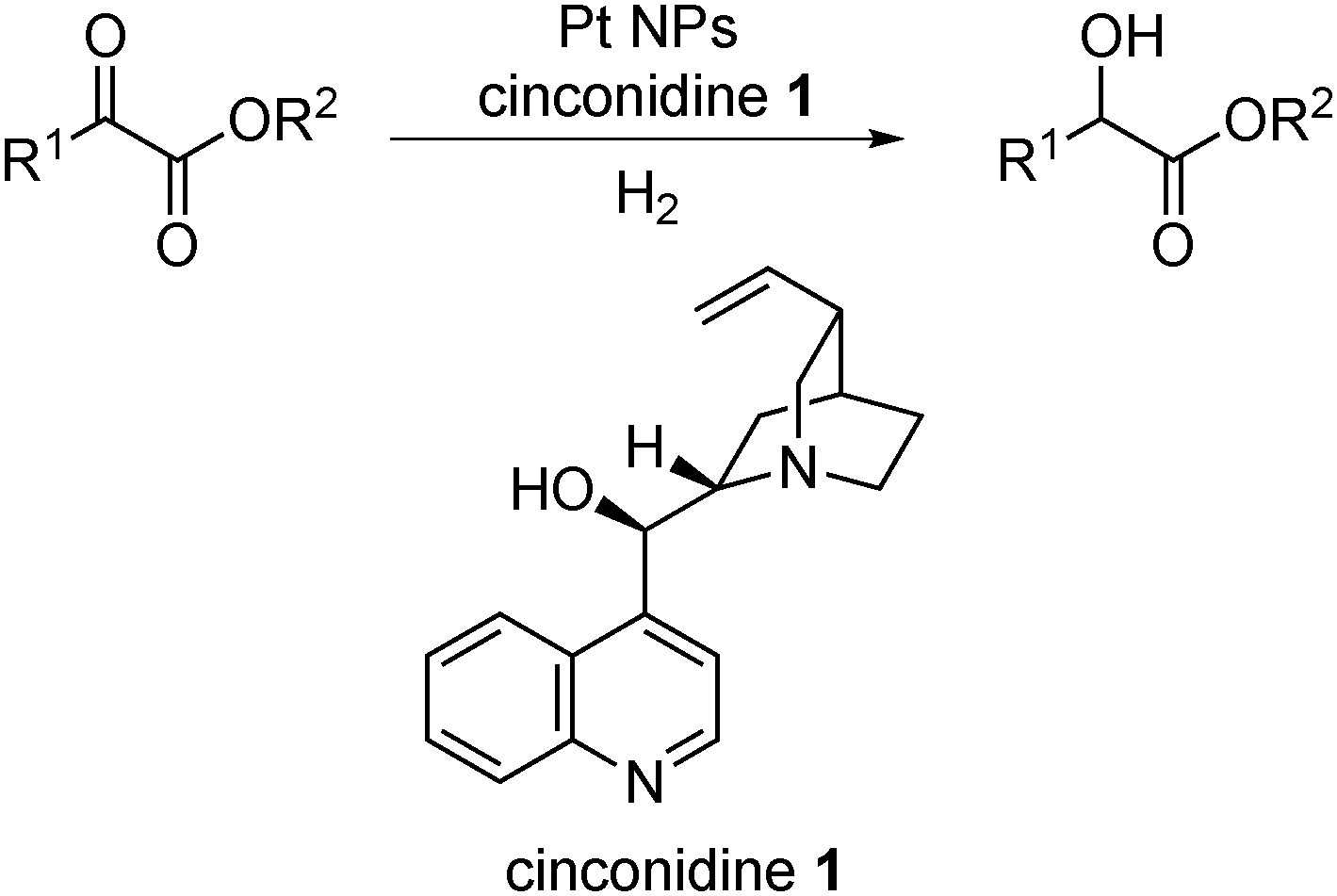 | ||
| Scheme 1 Orito-type reactions. | ||
On the other hand, asymmetric C–C bond formation reactions catalyzed by chiral NPs are a challenging topic and less well developed, judging from the mechanism of the corresponding homogeneous metal complex system. The mechanism of chiral induction is more complicated and multicoordination of both chiral modifier and substrates to NPs is required in transition states. Moreover, this type of multicoordination may easily cause the metal to leach from supports or the decomposition of NPs to form molecular complexes. For these reasons, the nature of the active species should be examined carefully because it is also possible that such a homogeneous molecular complex is a real active species.105–108 However, lack of fair observation and discussion about heterogeneity is sometimes found. Widely accepted tests to confirm heterogeneity, for example, the mercury-poisoning test, hot filtration test (filtrate transfer test), and three-phase test, are usually conducted.105
In this review, we focus on chiral NP-catalyzed asymmetric C–C bond formation. Although the detailed mechanism and active species are still unclear, some reports describe the specific nature of chiral NPs catalysts, which showed different catalyst behavior from the corresponding homogeneous molecular complex catalysts.
2. Palladium nanoparticles
2.1 Asymmetric allylic alkylation
Palladium-catalyzed asymmetric allylic alkylation reactions are one of the most powerful tools to construct asymmetric C–C bonds and have been widely investigated in homogeneous Pd complex systems.109,110 Several groups have reported that chiral palladium NPs catalyzed this reaction using various chiral modifiers and, interestingly, the assumed nature of the active species was different for each of the catalyst systems.In general, stabilization methods for metal NPs are crucial to develop efficient catalysts. In this context, Kobayashi and co-workers developed microencapsulated (MC) catalysts.111–114 Microcapsules have been used for coating and isolating substances until their activity is required, and their applications in medicine and pharmacy have been extensively studied.115 This microencapsulation technique was applied to the immobilization of metal NP catalysts into polymers. That is, catalysts would be physically enveloped by polymer backbones and, simultaneously, immobilized by the interaction between the π electrons of the benzene rings of the polystyrene used as a polymer support and vacant orbitals of the catalysts (metal NPs) (Fig. 3).
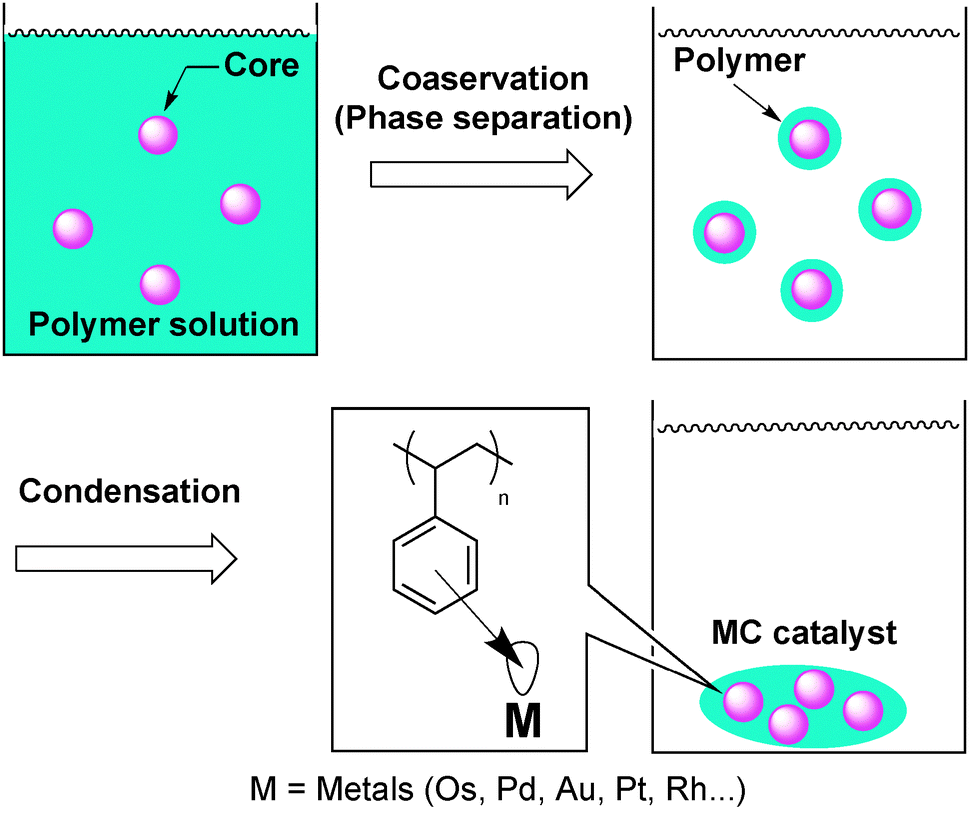 | ||
| Fig. 3 Microencapsulation technique. | ||
In 2002, Kobayashi et al. stabilized Pd(0) by the microencapsulation technique, and the thus prepared MC Pd(0) (MC Pd(PPh3)) was applied to a catalytic asymmetric allylation reaction (Scheme 2).116 The reaction of 1,3-diphenyl-2-propen-1-yl ethyl carbonate (1.0 equiv.) with dimethyl malonate (3.0 equiv.) was performed in the presence of MC Pd(PPh3) (20 mol%), chiral phosphine-oxazoline 2 (20 mol%), bis(trimethylsilyl)acetamide (BSA) (3.0 equiv.), and potassium acetate (0.10 equiv.) under reflux conditions in acetonitrile. The allylation adduct (R)-3 was obtained in 87% yield with 83% ee.
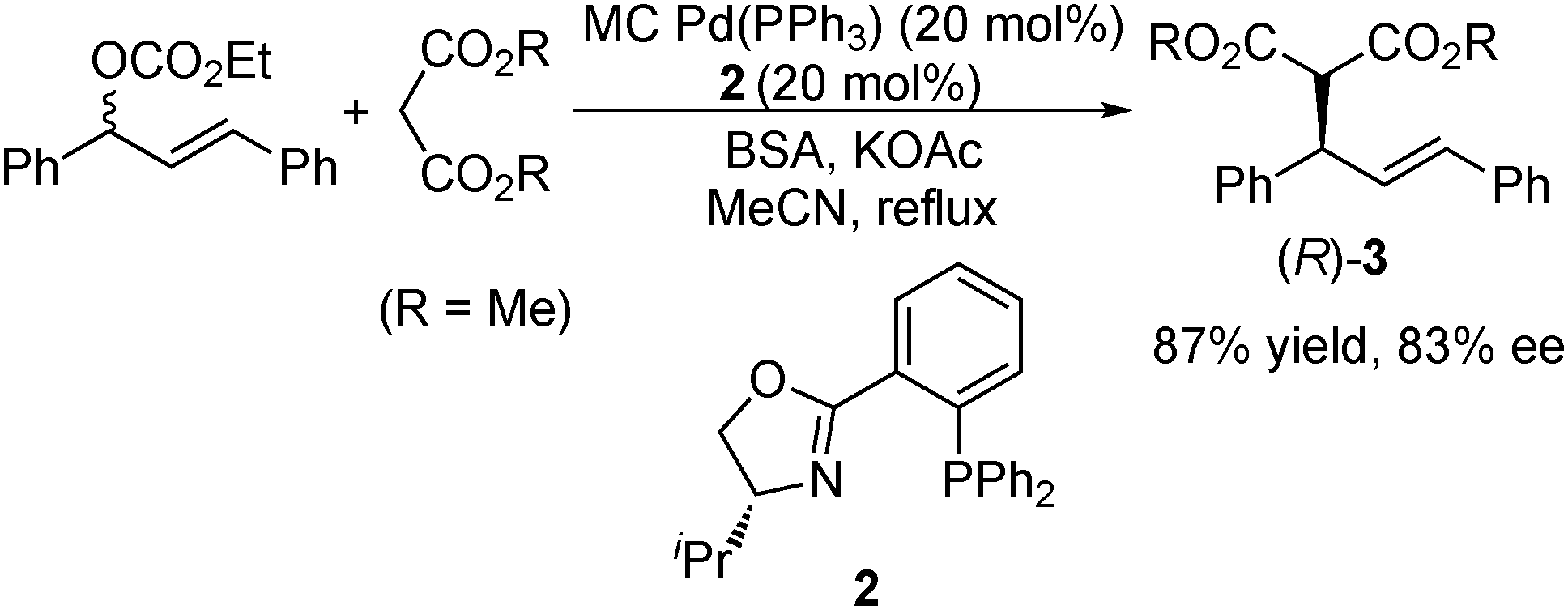 | ||
| Scheme 2 MC Pd-catalyzed asymmetric allylation. | ||
In 2004, Gómez, Philippot, Chaudret and co-workers reported asymmetric allylic alkylation reactions catalyzed by chiral diphosphite 4 with xylose-backbone-modified colloidal Pd NPs (Schemes 3 and 4).117 The reactivity of a molecular Pd complex (referred to as Mol. 4), which was generated in situ by the reaction of [Pd(C3H5)Cl]2 with 4, was compared with that of NP catalysts. A significant difference was observed (Table 1). In the presence of a NP catalyst (Pd4), the reaction mainly proceeded with one enantiomer of the racemic 8 and, consequently, a very high degree of kinetic resolution was demonstrated, while no kinetic resolution was observed in the presence of Mol. 4. The relative rates of alkylation of the two enantiomers of 8 in two catalyst systems were studied. The kinetic preference for the R-8 was only a factor of 2 in the Mol. 4 system, whereas it was clearly higher (12–20) in the Pd4 system.
 | ||
| Scheme 3 Synthesis of chiral Pd NPs. | ||
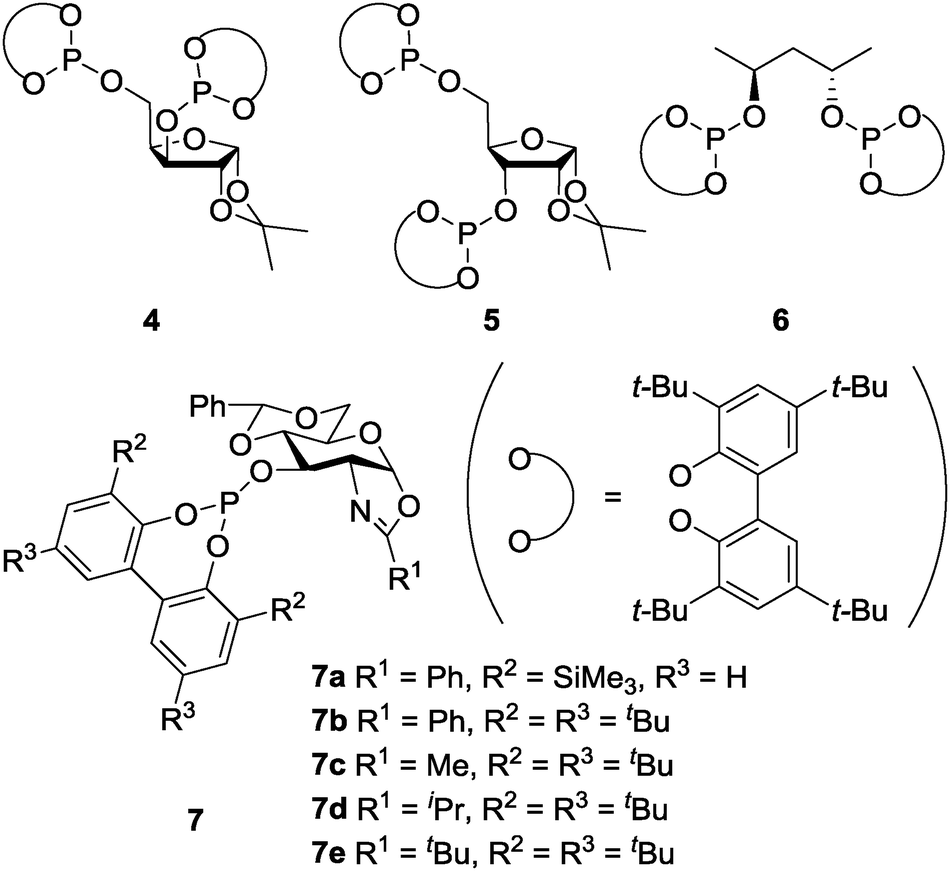 | ||
| Scheme 4 Chiral diphosphite ligands. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Pd/L* | Time (h) | Conv. (%) | ee of 3 (%) | ee of 8 (%) |
| 1 | Pd4 | 1/0.2 | 24 | 56 | 97 | 89 |
| 2 | Pd4 | 1/0.2 | 168 | 59 | 97 | 89 |
| 3 | Pd4 | 1/1.05 | 168 | 61 | 97 | 89 |
| 4 | Mol. 4 | 1/1 | 1.5 | 95 | 90 | 0 |
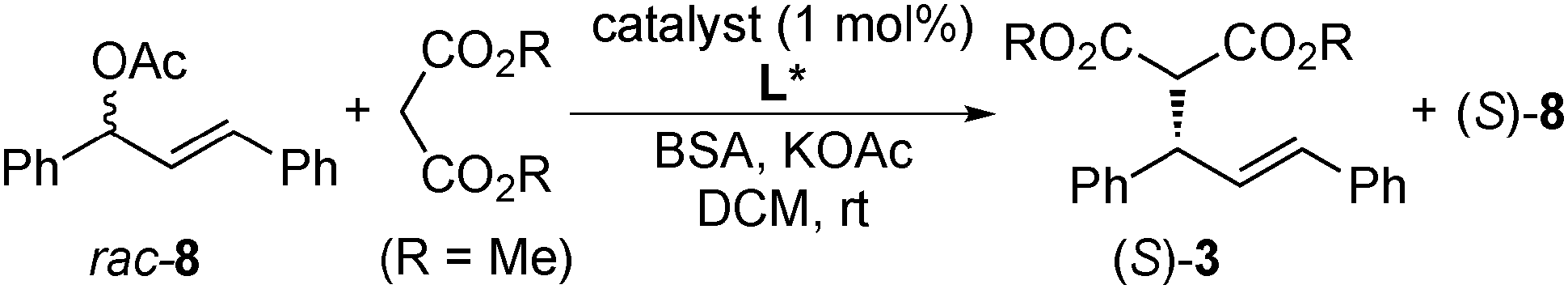
In 2007, the same group reported detailed studies of this reaction and showed higher sensitivity of NP catalyst systems upon adjustment of the metal, the chiral modifier and the substrate than that of molecular catalysts.118 For example: (i) Pd5 showed a remarkable difference in stability and decomposed into molecular species, while Pd4 did not, and (ii) alkyl-substituted allyl acetates were not alkylated by using Pd4 or Pd6, while the corresponding molecular catalyst systems gave high conversions. In 2008, Diéguez, Gómez, Leeuwen and co-workers reported modified Pd NPs systems with chiral oxazolinyl-phosphite ligands 7a–7e for the same reaction.119 The nature of the active species was carefully studied by continuous-flow membrane reactor experiments, transmission electron microscopy observations, classical poisoning experiments, and kinetic measurements. The results proved the molecular nature of the true catalysts, which were leached, as measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) analysis.
In 2005, Felpin and Landais reported a heterogeneous Pd catalyst with a chiral modifier that promoted asymmetric allylic alkylation.120 Pd/C and (R)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) 9 were used as catalysts for the reaction of racemic 8 with diethylmalonate in water, and high enantioselectivity (80% ee) was achieved in spite of low yield (21%). Subsequently, in 2007, Baiker and co-workers reported 9 modified Pd/Al2O3 catalyst systems for the reaction of racemic 8 with the sodium salt of dimethyl malonate (Scheme 5).121 Reductive treatment of Pd/Al2O3 in hydrogen before the reaction was required for obtaining reproducibility and, as a result, good chemoselectivity (94%) and moderate enantioselectivity (∼60% ee) were obtained. The enantioselectivity of this system was independent of the reaction temperature (60 and 120 °C), while the corresponding homogeneous complex system afforded poor enantioselectivity to give the opposite enantiomer of the product (R-3) at 60 °C or above. In a report from the same group in 2008, the enantioselectivity of this system was improved (82% ee) by changing the chiral modifier to a ferrocenyl phosphine ligand 10.122 As in the case of the Pd/Al2O3–BINAP system, no dependence of enantioselectivity on temperature was observed (60 and 120 °C), while the enantioselectivity was decreased by increasing the reaction temperature from 85% ee at 60 °C to 55% ee at 120 °C in the corresponding homogeneous complex system. Here, the same enantiomer of the product (S-3) was produced. To confirm the metal leaching in the reaction mixture, the catalytic activity in the filtrate with additional substrates was examined after the first reaction was conducted at 120 °C for 6 h and the catalyst was removed by centrifugation. The reaction was continued for a further 6 h and gave the additional product in 19% yield, while the yield in the blank experiment, which was conducted in the absence of Pd/Al2O3, was 10%.
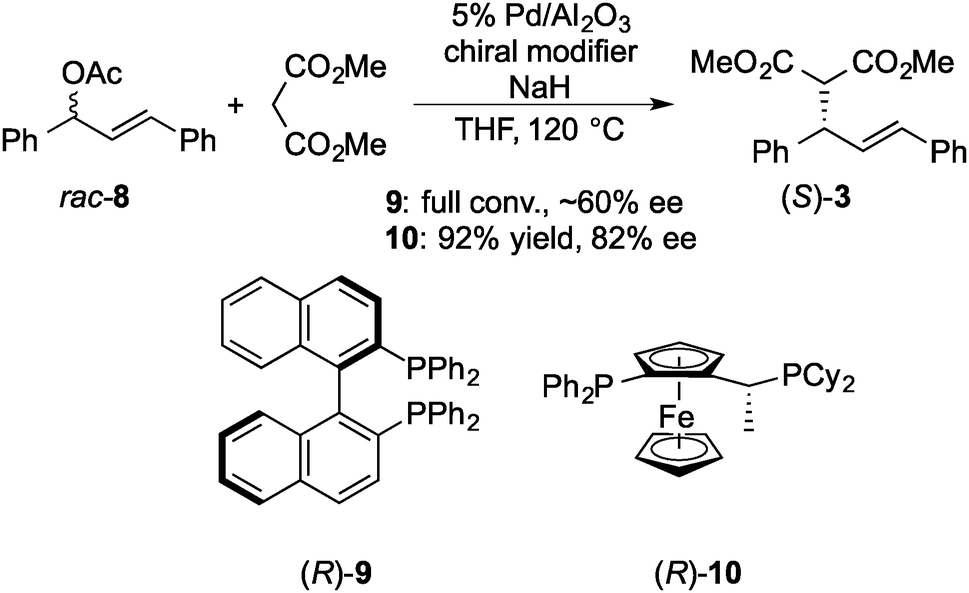 | ||
| Scheme 5 Asymmetric allylic alkylation of rac-8 with dimethyl malonate by Pd catalyst. | ||
In 2010, the same group reported a detailed study of the nature of the active species in the Pd/Al2O3–BINAP system and emphasized heterogeneity.123 The oxidation state of Pd was investigated by in situ X-ray absorption near-edge structure analysis of the reaction mixture. They did not observe oxidized Pd in the supernatant solution and hence proposed that the molecules in the reaction mixture, such as the solvent and sodium dimethyl malonate, were able to reduce Pd atoms on the surface of Pd NPs and to maintain the reduced state during the reaction. They also pointed out that addition of a chlorinated solvent, such as chloroform, favored dissolution of metallic Pd to form homogeneous catalysis. After the first reaction at 120 °C for 3 h, the reaction mixture was cooled. The catalyst was removed by centrifugation and the supernatant with additional sodium malonate did not give further conversion at room temperature over 18 h in the case of pure THF as solvent, while a small conversion was observed in a similar control experiment in the presence of chloroform.
2.2 Asymmetric cross-coupling reactions
Palladium-catalyzed cross-coupling reactions are one of the biggest breakthroughs in transition metal-catalyzed C–C bond formation reactions.124 Not only have homogeneous catalysts been developed, but also many heterogeneous catalyst systems.107,108,125 Among these systems, a couple of examples of asymmetric cross-coupling systems were reported recently.In 2008, Fujihara and co-workers succeeded in preparing the optically active mono- and bisphosphine-modified chiral Pd NPs and applying these NPs to asymmetric Suzuki–Miyaura coupling reactions (Scheme 6).126 The zero oxidation state of the chiral Pd NPs was confirmed by X-ray photoelectron spectroscopy (XPS) analysis, and CD spectra of the chiral Pd NPs showed negative Cotton effects. It was demonstrated that (S)-BINAP-modified chiral Pd NPs could catalyze the coupling reactions with naphthyl halides and arylboronic acids to afford axially chiral biaryl compounds in good enantioselectivity at low temperature (−7 to 25 °C), although reported syntheses of binaphthalenes catalyzed by a chiral Pd–bisphosphine complex required high temperatures to obtain acceptable yields. The latter showed low enantioselectivity.
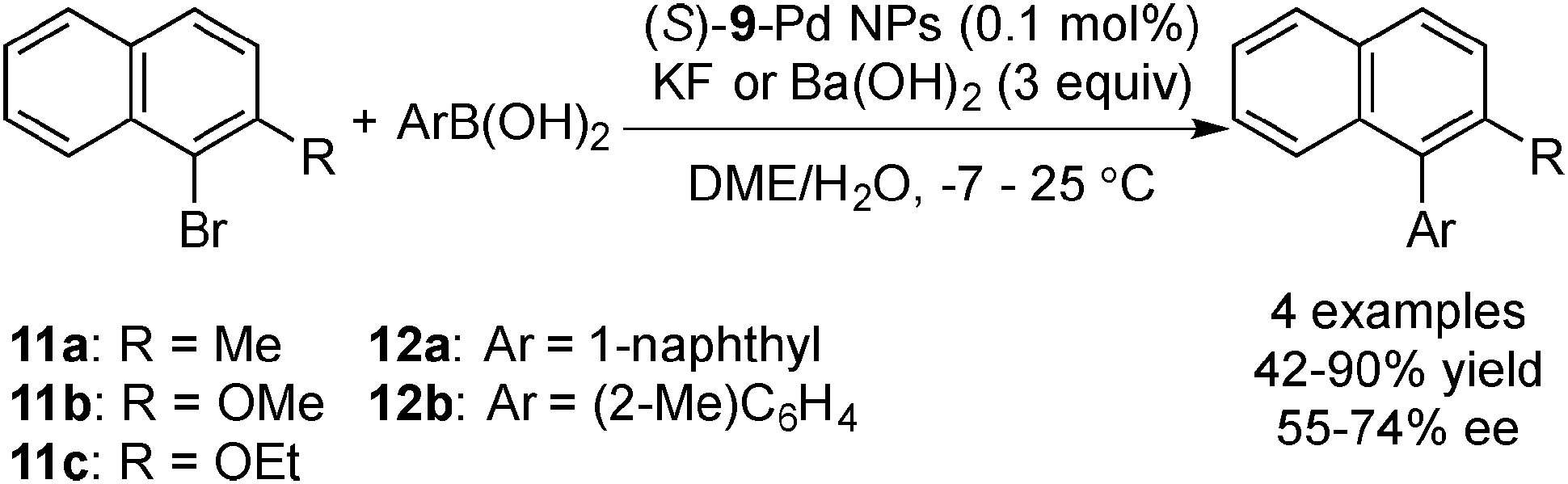 | ||
| Scheme 6 Asymmetric Suzuki–Miyaura coupling reactions catalyze by chiral Pd NPs. | ||
In 2009, Yamashita and co-workers reported the BINAP-modified core–shell structured bimetallic Fe/Pd NP-catalyzed asymmetric Suzuki–Miyaura coupling reaction.127 The Fe/Pd NPs with an Fe-rich core and a Pd-rich shell were synthesized by thermal decomposition of Fe(CO)5 and subsequent reduction of Pd(acac)2 in the presence of oleic acid and oleylamine, and BINAP-modified NPs were formed through a simple ligand exchange procedure. X-ray absorption measurements revealed that most Pd atoms existed in a metallic form while Fe atoms existed in an oxidized form. The optically active BINAP-modified Fe/Pd NPs showed catalytic activity for the coupling reaction between halide 11b and boronic acid 12a with moderate enantioselectivity (up to 48% ee). The NPs were recovered easily by applying an external magnet and could be reused, while maintaining identical enantioselectivity without an external addition of chiral modifier. A hot filtration test was conducted and the reaction hardly occurred in the filtrate after removal of the catalyst.
In 2010, Glorius and co-workers reported the first successful application of chiral N-heterocyclic carbene (NHC) as a chiral modifier of magnetite supported Pd NPs (Scheme 7) in the asymmetric α-arylation of ketones with aryl halides (Scheme 8).128 Chiral Pd NPs were prepared by addition of a NHC precursor and a base in the presence of Fe3O4/Pd. A zero-oxidation state of Pd was confirmed, even after surface modification, by XPS analysis. The presence of a NHC-modified surface was supported by differences between spectra of the free salt 13 and that of Fe3O4/Pd/13 in attenuated total reflection infrared spectroscopy analysis. Various ketones and aryl halides could be employed for asymmetric α-arylation reactions, including intramolecular reactions, to afford the corresponding α-arylated ketones in moderate-to-good enantioselectivity. The paramagnetic catalyst could be recovered using a magnet and then reused five times without significant loss of activity and selectivity. The heterogeneous nature of active species was indicated by several experiments, such as the hot filtration test, mercury-poisoning test and trace metal analysis. Moreover, dramatically low selectivity was exhibited by several homogeneous complex systems, which afforded the racemic α-arylated product, biphenyl, and other side products. The same research group also developed a homogeneous Pd catalyst system with quinine for this asymmetric α-arylation reaction and achieved excellent substrate generality and enantioselectivity.129 A combination of Fe3O4/Pd and quinine as a chiral modifier was also examined. The heterogeneous nature of the real active species was indicated by the mercury-poisoning test and hot filtration test. The results emphasized the important role of chiral NHC for both stability and activity of the Pd NPs as catalysts.
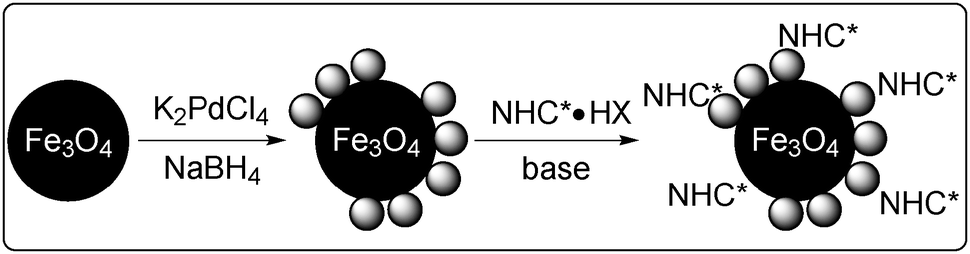 | ||
| Scheme 7 Preparation of Fe3O4/Pd NPs modified by chiral NHC 13. | ||
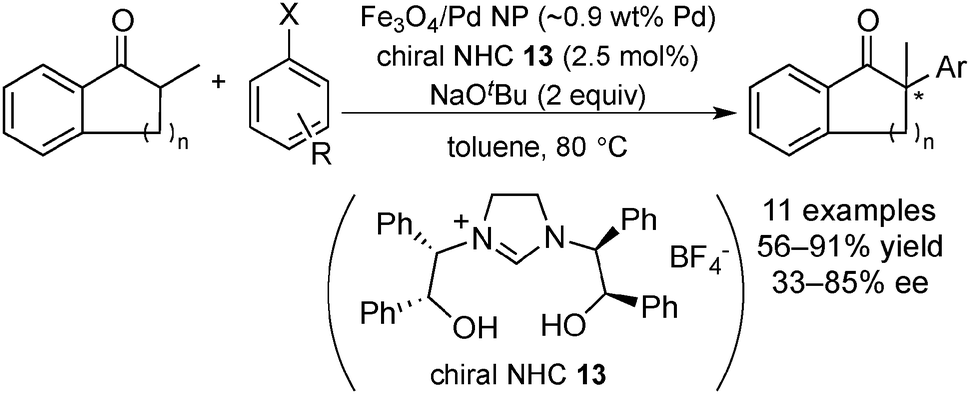 | ||
| Scheme 8 The asymmetric α-arylation of ketones with aryl halides. | ||
In 2011, Glorius and co-workers further developed immobilized Pd catalysts with NHC.130 From magnetite-supported NHC 14, a molecular Pd catalyst 15 was prepared with Pd(OAc)2, while a Pd NP catalyst 16 was also prepared by the reduction of K2PdCl4 (Scheme 9). These catalysts were characterized by XPS analysis to confirm the formation of a Pd complex or Pd(0), respectively, and the catalytic activities of these two Pd catalysts were tested in the asymmetric allylation of 4-nitrobenzaldehyde with allyltributyltin (Scheme 10). The NP catalyst 16 provided improved allylation results for both yield and enantioselectivity, and, surprisingly, the product was obtained with the opposite absolute configuration to the one formed with the molecular catalyst 15.
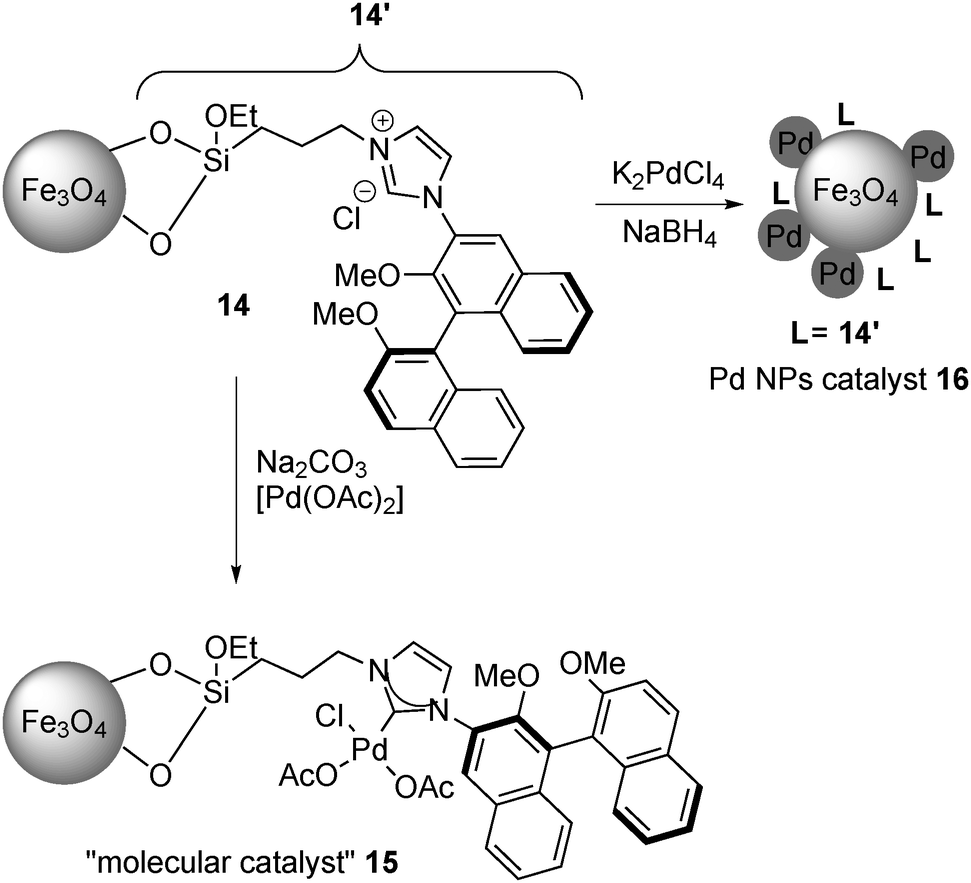 | ||
| Scheme 9 Preparation of a molecular catalyst and a Pd NP catalyst on Fe3O4. | ||
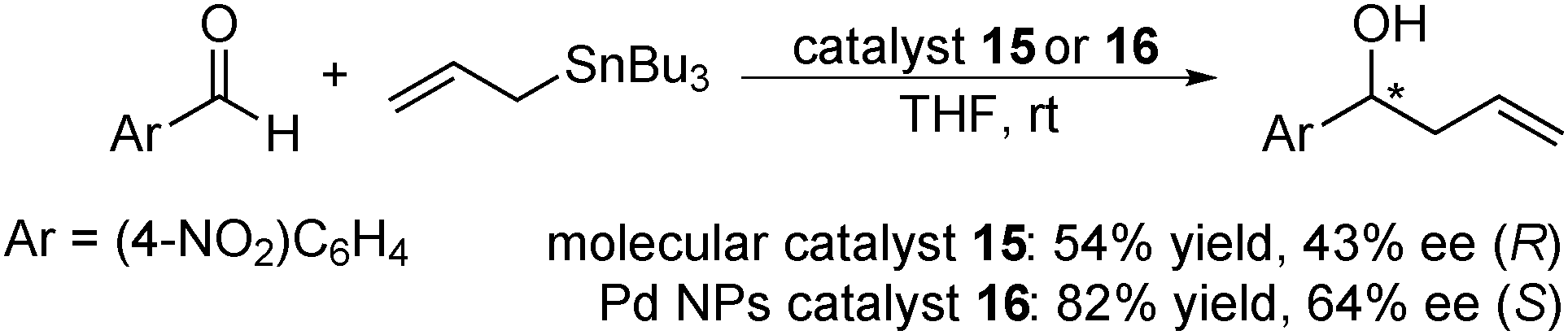 | ||
| Scheme 10 Asymmetric allylation of 4-nitrobenzaldehyde with allyltributyltin. | ||
3. Rhodium nanoparticles
3.1 Hydroformylation
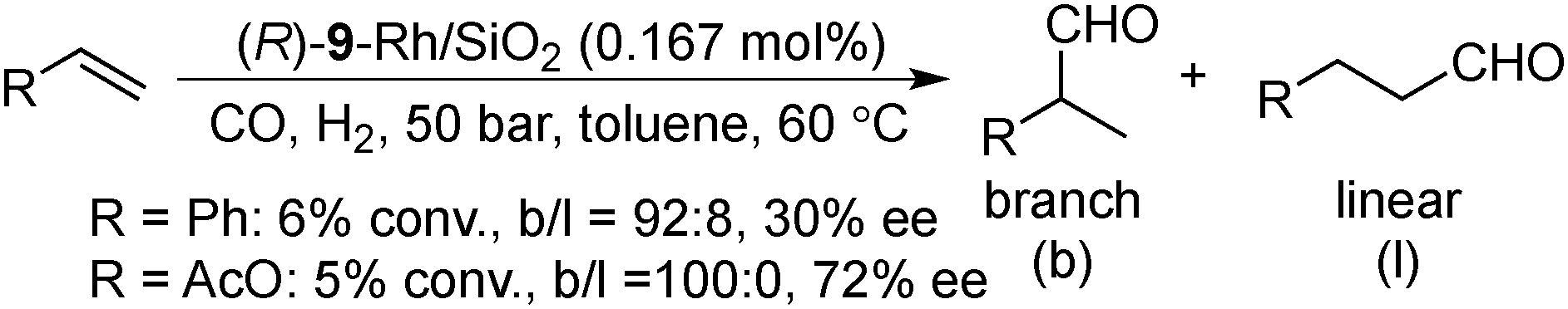
Asymmetric hydroformylation of olefins using chiral Rh complexes in the presence of syngas is a useful method to synthesize optically active aldehydes with excellent regioselectivity and enantioselectivity.131 Despite extensive development of homogeneous catalysis and recent advances in Rh NP catalysts,132 heterogeneous asymmetric hydroformylation is still a challenging topic.133In 2000, Anderson and co-workers reported styrene hydroformylation over chiral-ligand-modified Rh/SiO2·Al2O3. Chiral induction was achieved by using DIOP 17 or chiraphos 18 (Scheme 11) as a chiral modifier although selectivity was very low (up to 9% ee).134 In 2006, Li and co-workers reported the asymmetric hydroformylation of styrene or vinyl acetate catalyzed by BINAP-modified Rh/SiO2 (Scheme 12).135 To achieve chiral induction, an optimized ratio of Rh and modifier was close to 1.0 and, as a result, up to 72% ee and 100% selectivity of a branched aldehyde for the hydroformylation was obtained, although conversion was very low (<10%). It was noted that the DIOP 17-modified Rh/SiO2 catalyst showed higher turnover frequency (TOF) for the hydroformylation of vinyl acetate (128 h−1) than that of unmodified Rh/SiO2 (90 h−1), while other diphosphine ligands decreased the catalytic activity. 31P magic angle spinning nuclear magnetic resonance (MAS NMR) analysis and IR spectra of adsorbed CO indicated the existence of a strong interaction between the P atoms of the chiral modifier and the surface Rh sites. In 2008, the same group reported a different method to prepare modified Rh NPs, by one-pot chemical reduction of aqueous rhodium chloride dispersed in toluene in the presence of amphiphilic tetraoctylammonium bromide and (R)-BINAP.136 These BINAP-modified Rh NPs could also be immobilized on SiO2 in a vacuum by adsorption; however, no significant improvement of catalytic performance was observed.
 | ||
| Scheme 11 Chiral diphosphine ligands. | ||
 | ||
| Scheme 12 Asymmetric hydroformylation of olefins by BINAP-Rh/SiO2. | ||
In 2008, Axet, Claver and Philippot reported that carbohydrate-derived diphosphite-ligand-modified colloidal Rh NPs (Scheme 13) catalyzed the asymmetric hydroformylation of styrene.137 With the addition of an excess amount of the ligand, high conversion, high regioselectivity, and moderate enantioselectivity were observed in the presence of Rh19 NPs (Scheme 14). The catalytic activities of molecular complexes, which were prepared from [Rh(acac)(CO)2] and the corresponding chiral ligands, were examined. The enantioselectivities in the molecular systems were slightly lower (∼10% ee difference) than those in the respective colloidal systems. As results of experiments carried out with dilute solutions and poisoning tests were not conclusive, in situ high-pressure NMR spectroscopic studies under catalytic conditions in the presence of Rh4 NPs were conducted and the well-known hydridorhodium diphosphite complex [RhH(CO)2(4)] was detected after a reaction time of 45 h. The results indicated that molecular species generated from NPs are real active species. Given the difference in enantioselectivity between the molecular complex system and the NPs system, the possibility that some activity comes from NPs cannot be excluded.
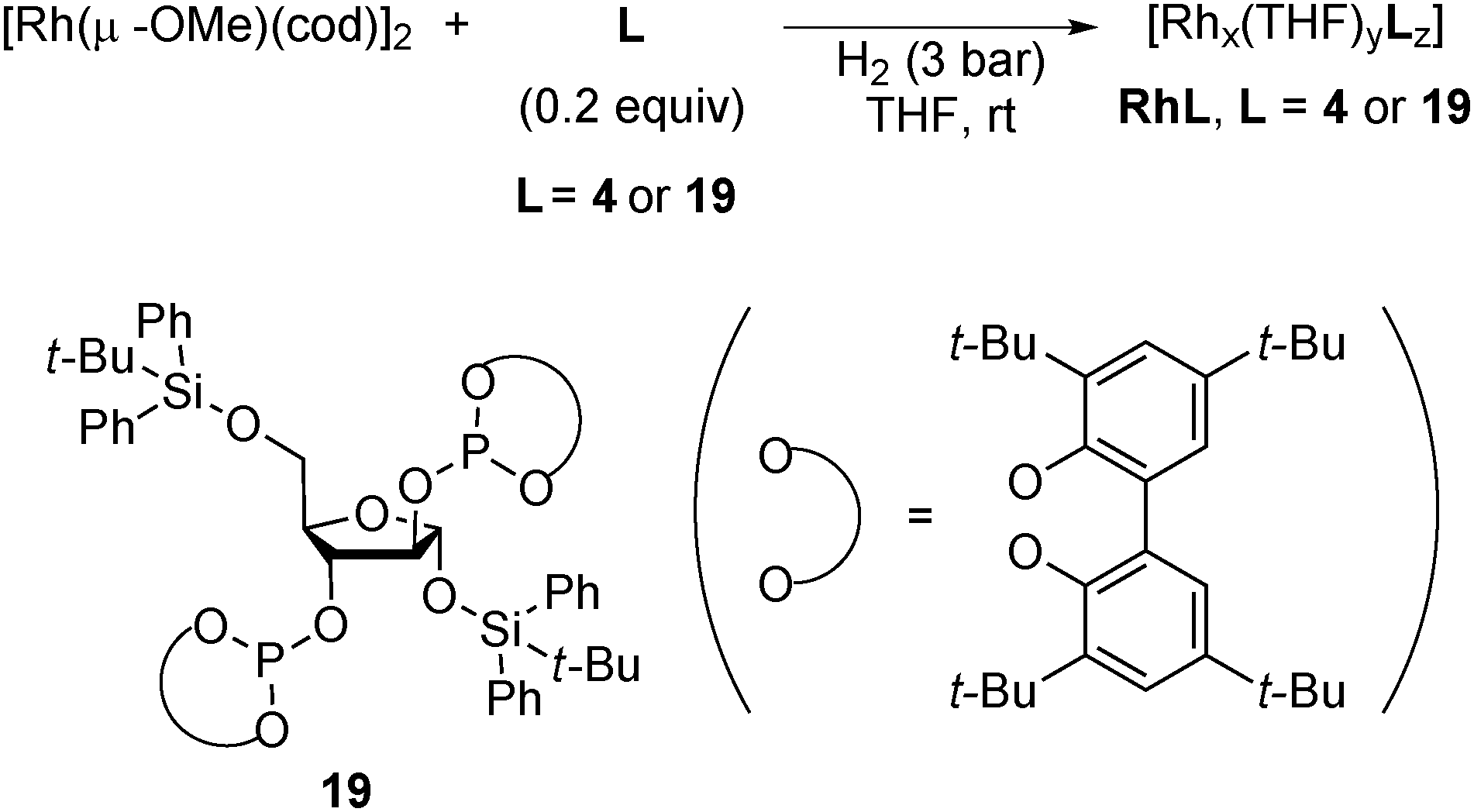 | ||
| Scheme 13 Synthesis of chiral Rh NPs 4 and 19. | ||
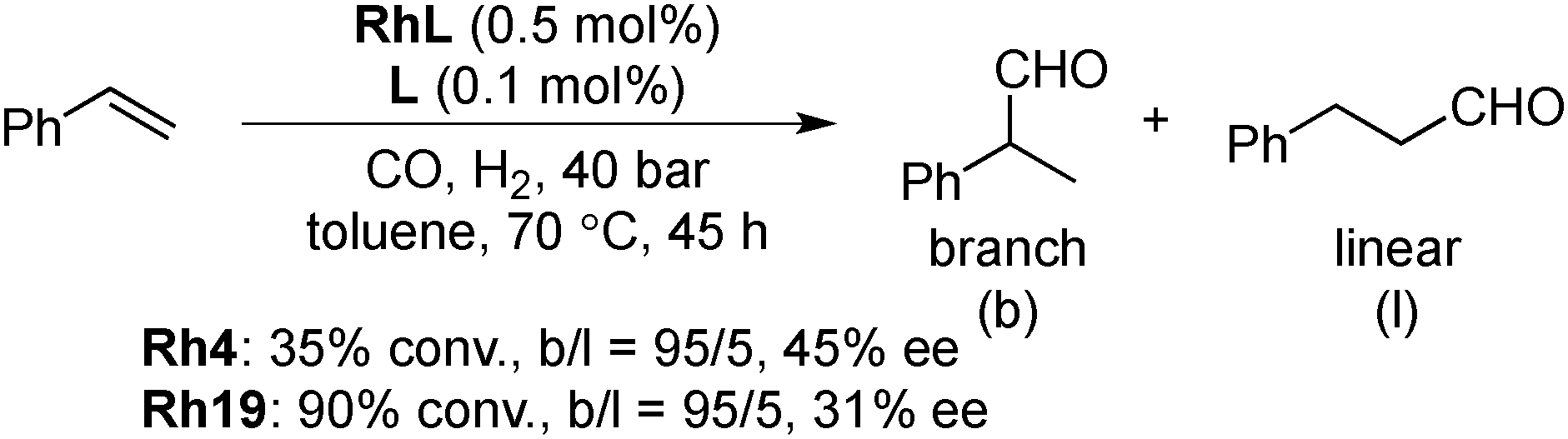 | ||
| Scheme 14 Asymmetric hydroformylation of styrene by Rh NPs. | ||
3.2 Pauson–Khand reaction
The Pauson–Khand reaction is a [2+2+1] cycloaddition of an alkyne, an alkene, and carbon monoxide to construct a cyclopentanone skeleton. In early studies of this reaction, a dicobalt octacarbonyl complex was used as the catalyst,138,139 but further developments revealed that several other transition metal complexes138,140 or cobalt NPs141 could be used for this reaction. Moreover, the use of heterobimetallic NP catalysts has attracted more attention due to their high catalytic activity.141,142 In 2005, Park and Chung reported charcoal-immobilized Co/Rh heterobimetallic NP-catalyzed asymmetric Pauson–Khand-type reactions with a chiral modifier.143 In the presence of the Co/Rh NP catalyst and chiral diphosphine modifier 20, the reactions of several enynes with crotonaldehyde as an alternative CO source proceeded, and the corresponding bicyclic cyclopentanones were obtained in high yields with moderate-to-good enantioselectivities (Scheme 15). The catalyst could be recovered by filtration and then reused five times with the addition of a new chiral modifier without significant loss of yield and enantioselectivity. Although elemental analysis of the reaction mixture by ICP-AES after three cycles detected 3.9 ppm of Co and 0.3 ppm of Rh species, results of the mercury-poisoning test supported a heterogeneous nature of the active species as Hg(0) completely eliminated further catalysis after the addition of Hg(0) to the reaction mixture.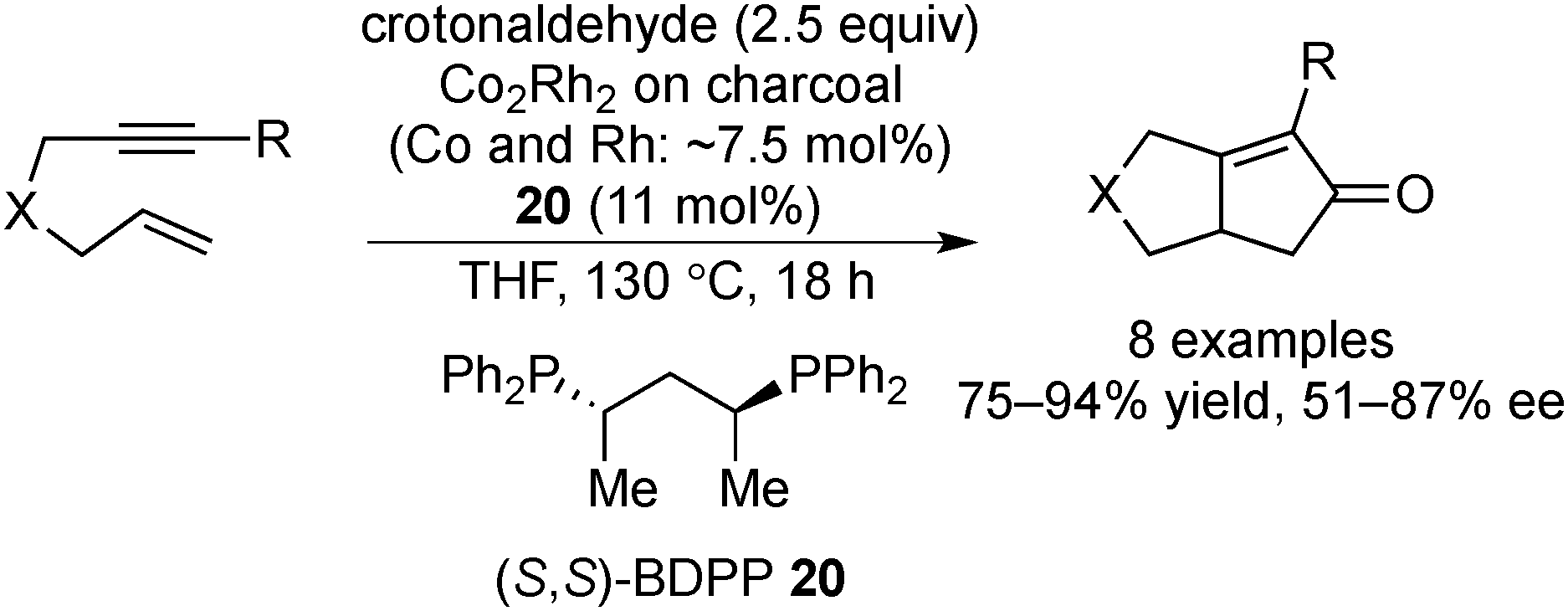 | ||
| Scheme 15 Co/Rh NP-catalyzed asymmetric Pauson–Khand type reactions. | ||
3.3 1,4-Addition reaction
Since the development of the rhodium-catalyzed asymmetric 1,4-addition of boron compounds to α,β-unsaturated carbonyl compounds by Hayashi and Miyaura,144 both academic research145–147 and large-scale processes148–150 (the largest example being 20 kg) of this reaction have been widely demonstrated; however, due to the high cost of Rh, reusable and robust chiral heterogeneous Rh catalysts are required.In 2012, Kobayashi and co-workers developed chiral Rh NPs and Rh/Ag bimetallic NPs, which were immobilized on a mixture of polystyrene-based copolymer with a cross-linking moiety and carbon black (PI/CB Rh and PI/CB Rh/Ag), and used them in the asymmetric 1,4-addition of boronic acids to α,β-unsaturated ketones (Scheme 16).151 In the presence of PI/CB Rh and (S)-BINAP as a chiral modifier, a significant amount of Rh leaching was observed during the reaction between a cyclic substrate and phenylboronic acid. When changing the chiral modifier to optically active chiral diene 21, Rh leaching in a crude mixture was dramatically suppressed to the detection limit of ICP-AES, and the desired product was obtained in high yield and excellent enantioselectivity. The low catalytic activity of PI/CB Rh towards acyclic substrates was improved after the formation of bimetallic NPs with Ag; various substrates were applicable and the desired β-arylated ketones were obtained in high yields and high enantioselectivities by using PI/CB Rh/Ag and chiral modifiers 21 or 22, without leaching of the metals. Scanning transmission electron microscopy (STEM) analysis and energy dispersive X-ray spectroscopy (EDS) mapping showed that Ag formed alloy NPs with Rh that could prevent the aggregation of Rh NPs, to enhance the catalytic activity. PI/CB Rh/Ag could be recovered by filtration and then reused. High yields and high enantioselectivities could be maintained for eight reactions. The deactivated catalyst, after repeated use, could be restored fully simply by heating. Fourteen cycles were demonstrated. Furthermore, a hot filtration test confirmed that after the reaction the filtrate did not catalyze the 1,4-addition reaction, indicating that the reaction is not catalyzed by homogeneous leached species. A one-pot aerobic oxidation-asymmetric 1,4-addition reaction of an allyl alcohol and an arylboronic acid was also demonstrated by combining with PI/CB Au as an aerobic oxidation catalyst.
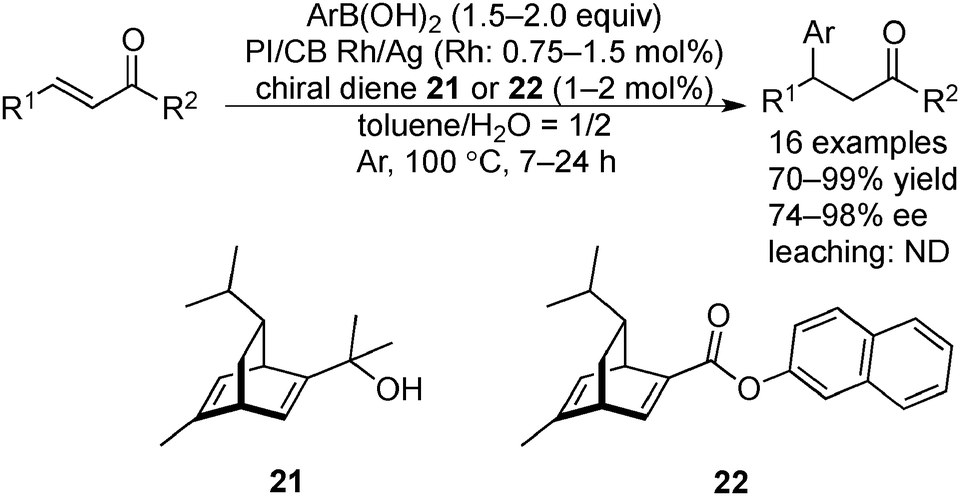 | ||
| Scheme 16 Rh/Ag NP-catalyzed asymmetric 1,4-addition reactions. | ||
4. Other metal nanoparticles
4.1 Nanocrystalline magnesium oxide
Examples of asymmetric catalysis by chiral modifiers and nontransition metal NPs are rather limited. In 2005, Choudary et al. reported several asymmetric reactions using aerogel-prepared nanocrystalline MgO (NAP–MgO), which is commercially available and possesses a large surface area (590 m2 g−1) and a three-dimensional polyhedral structure.152 The NAP–MgO was used for asymmetric Henry reactions with aldehydes or α-ketoesters in the presence of (S)-BINOL 23 (Scheme 17) and asymmetric Michael reactions with nitroalkanes in the presence of optically active chiral diamine 24 (Scheme 18). In both reactions, high yields and high enantioselectivities were achieved for several substrates, and NAP–MgO could be reused five times after heating at 250 °C for 1 h without loss of activity and enantioselectivity. The control experiments using silylated MgO or protected hydroxyls of 23 indicated that the hydrogen bond interaction between the hydroxy groups of 23 and the hydroxy groups of partially hydrated MgO are essential for obtaining high enantioselectivity. In 2006, the same group reported that NAP–MgO systems modified with chiral diamine 24 were applicable for asymmetric aldol reactions with moderate yields and enantioselectivities (Scheme 19).153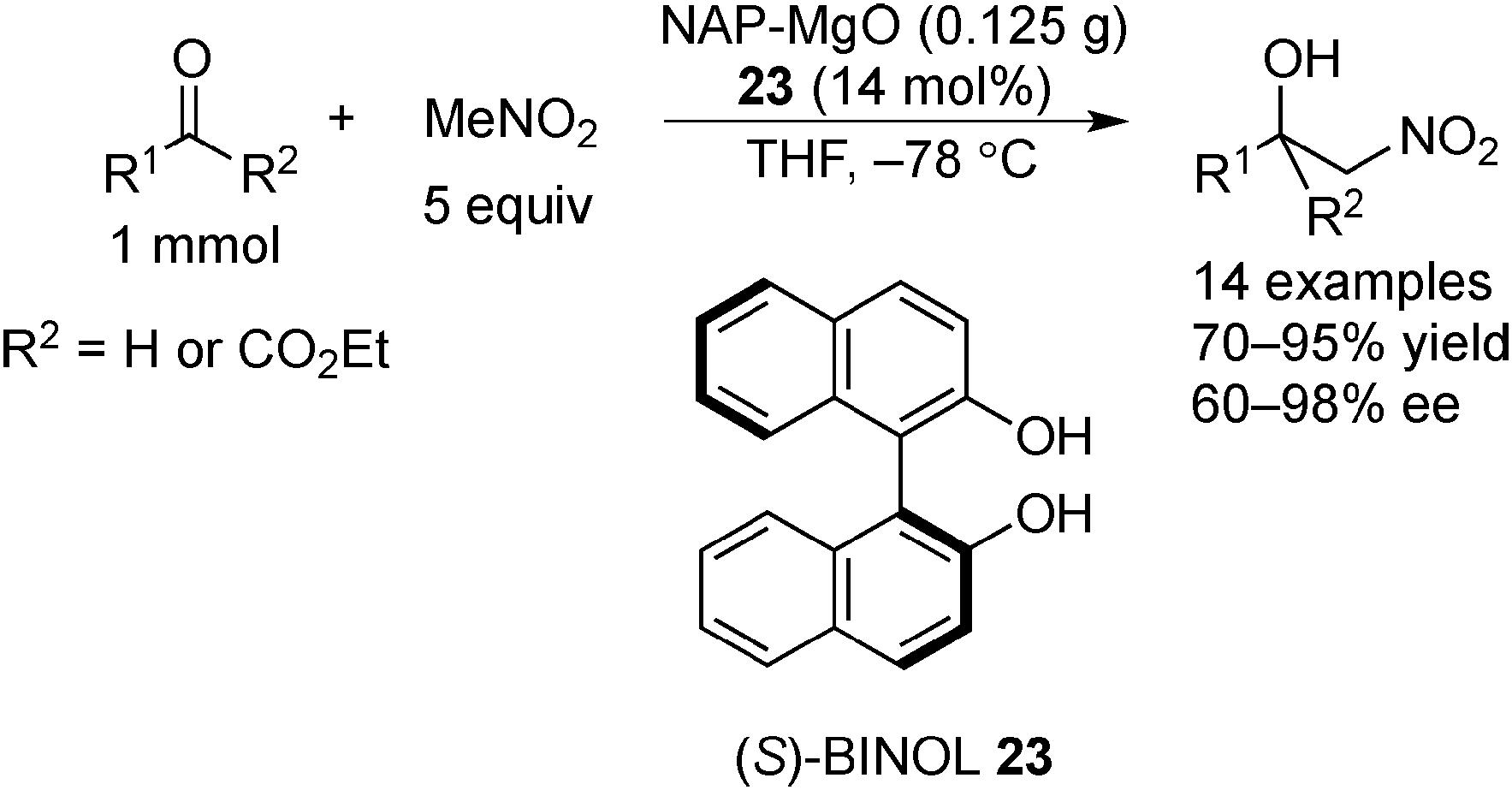 | ||
| Scheme 17 NAP–MgO-catalyzed asymmetric Henry reactions. | ||
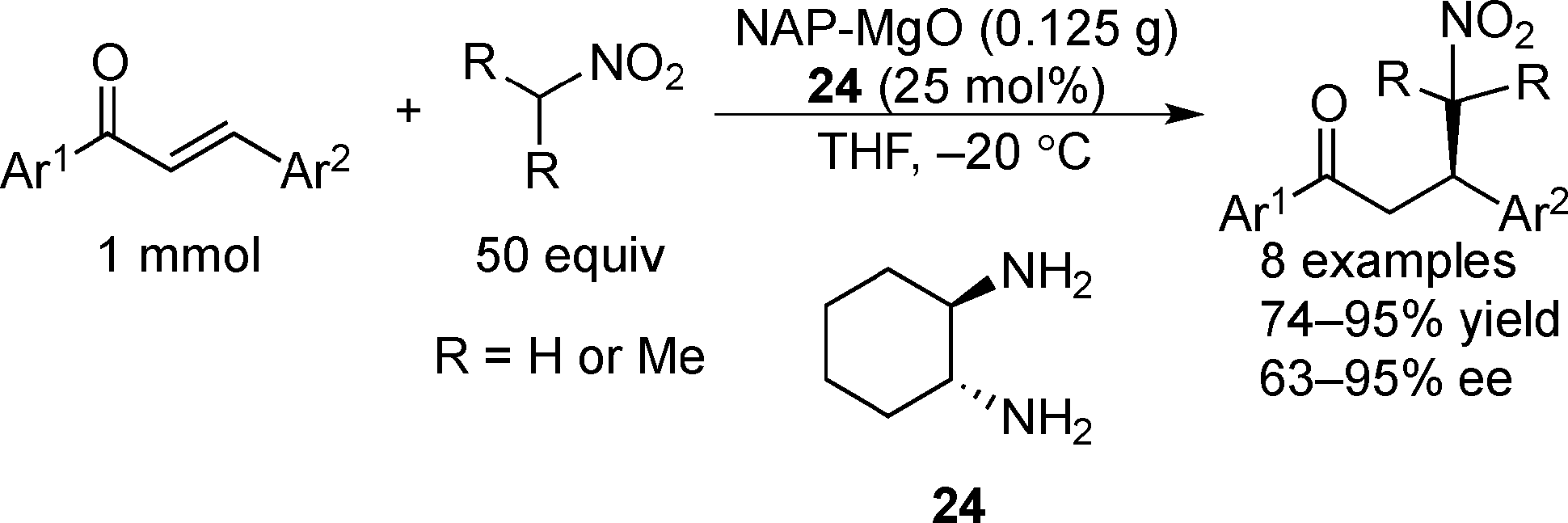 | ||
| Scheme 18 NAP–MgO-catalyzed asymmetric Michael reactions with nitroalkanes. | ||
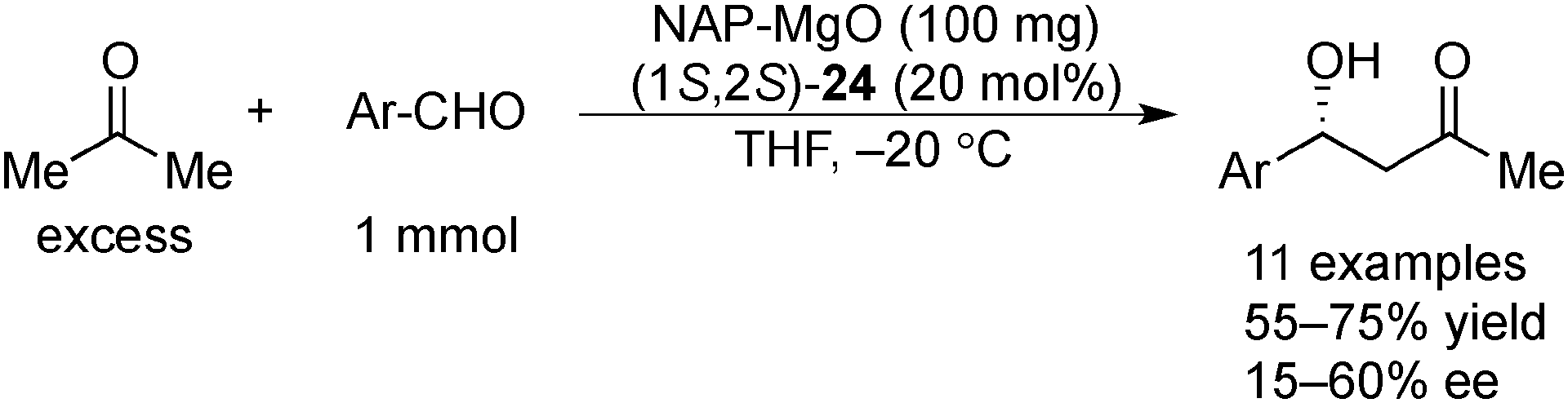 | ||
| Scheme 19 NAP–MgO-catalyzed asymmetric aldol reactions. | ||
Kantam and co-workers also investigated the NAP–MgO-mediated asymmetric Michael reactions with malonates. The desired products were obtained in high yields and high enantioselectivities in the presence of optically active chiral diamine 25 (Scheme 20).154
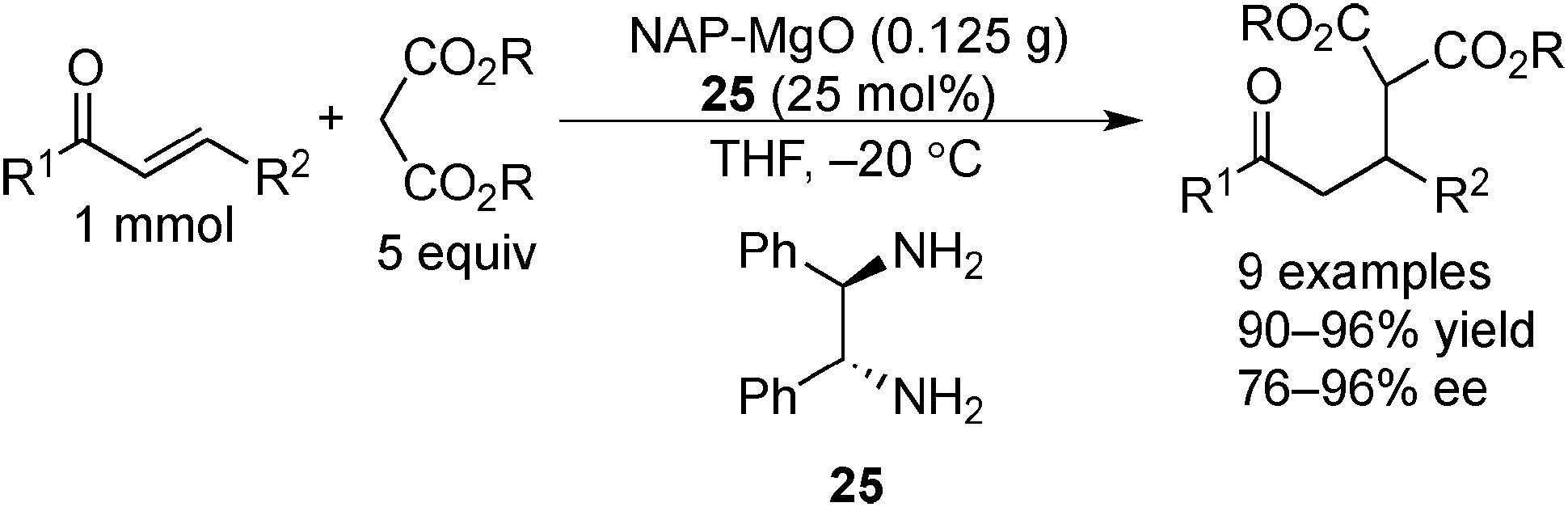 | ||
| Scheme 20 NAP–MgO-catalyzed asymmetric Michael reactions with malonates. | ||
4.2 Copper nanoparticles
In 2008, Kantam and co-workers found that nanocrystalline CuO (nano-CuO) could also be utilized for the asymmetric aldol reactions of acetone with arylaldehydes in the presence of chiral diamine 25 as a chiral modifier (Scheme 21).155 Nano-CuO had a higher large surface area (136 m2 g−1) and smaller crystal size (7–9 nm) and showed higher activity and enantioselectivity compared with commercial CuO. Nano-CuO could be reused for four cycles without significant loss of activity, and no structure or morphology change was observed after the reaction, as confirmed by X-ray diffraction (XRD) analysis. The filtrate, which was obtained after removal of the solids, did not promote the reaction, and the absence of copper in the filtrate was confirmed by atomic absorption spectrometry studies.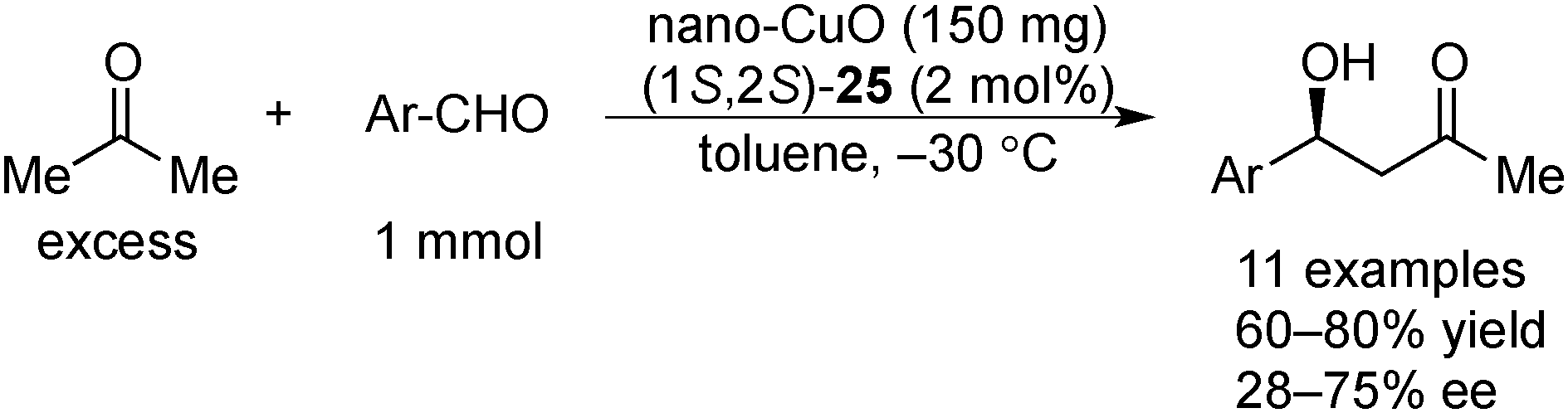 | ||
| Scheme 21 Nano-CuO-catalyzed asymmetric aldol reactions. | ||
4.3 Gold nanoparticles
Although gold NPs as catalysts, such as in aerobic oxidation, have been extensively studied,156–160 asymmetric catalysis with gold NPs had not been reported until very recently. In 2013, Toste, Somorjai and co-workers investigated enantioselective heterogeneous Au NP catalysts for the asymmetric cyclopropanation reaction by immobilization of a chiral self-assembled monolayer (SAM) on the surface of a mesoporous silica support.161 After amino acids, as chiral modifier, were attached to the surface of mesoporous SiO2 (MCF-17) through the OH terminal, Au ions were introduced, and the encapsulated Au ions were reduced to NPs by exposure to H2 (Fig. 4).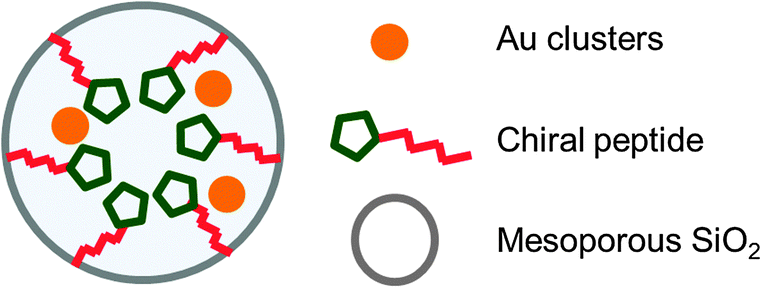 | ||
| Fig. 4 Au NPs encapsulated in chiral SAM/mesoporous MCF-17 support. | ||
The obtained Au NPs catalyst (Au@SAM/MCF) was used for the intermolecular cyclopropanation reaction of styrene with propargyl pivalate in the presence of an oxidizer (Scheme 22). The desired product 26 was obtained in good diastereoselectivity and moderate enantioselectivity, when peptide 28 was immobilized on the surface of mesoporous SiO2. The same catalyst could also be used for the asymmetric intramolecular cyclopropanation reaction although, there, moderate conversion and moderate enantioselectivity were observed (Scheme 23). The corresponding homogeneous system using AuCl3 and unsupported diproline in the intermolecular reaction produced low yield and no enantioselection. While ICP analysis showed no leaching of Au ions to the solution phase due to the repulsion forces between hydrophilic catalyst and hydrophobic solvent, in situ near-edge X-ray adsorption fine structure measurements carried out under reaction conditions proved that the formation of Au(III) ions, which were generated from Au(0) NPs by oxidation with PhICl2, were the active species. Spectroscopic measurements revealed that the enantioselectivity was correlated with the stability of SAM, by the formation of a hydrogen bonding network. Although, in this study, the active species was proved to be not the NPs themselves, the advantages of the surrounding chiral SAM for the formation of a mesoscale enantioselective catalyst were noted.
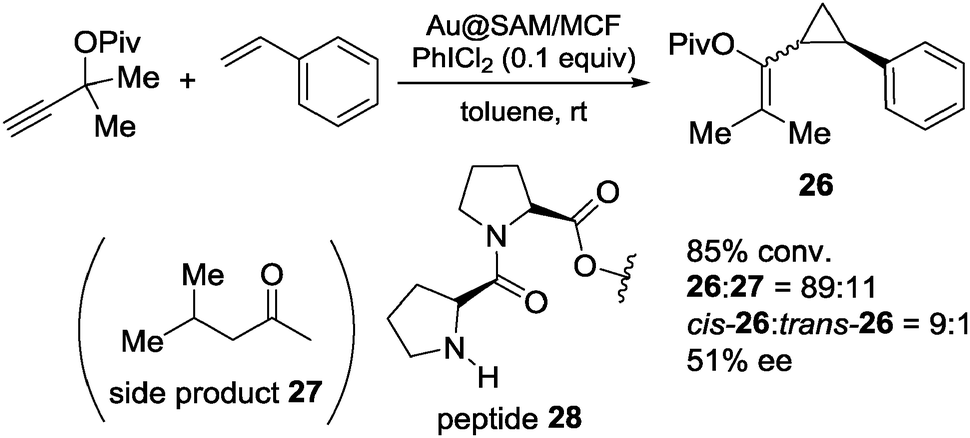 | ||
| Scheme 22 Au@SAM/MCF-17-catalyzed asymmetric intermolecular cyclopropanation. | ||
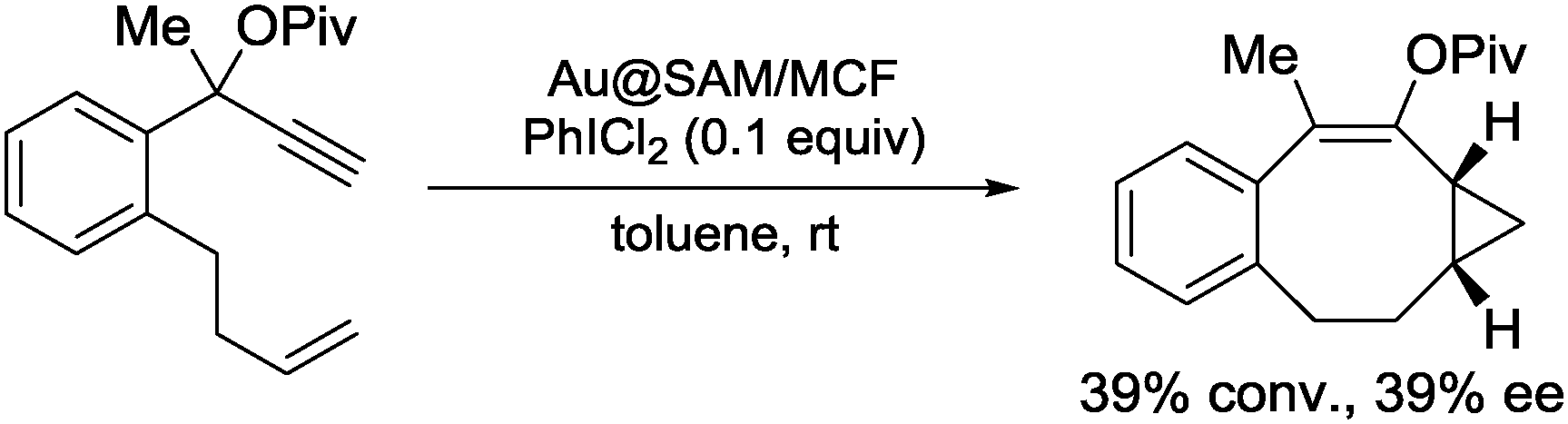 | ||
| Scheme 23 Au@SAM/MCF-17-catalyzed asymmetric intramolecular cyclopropanation. | ||
5. Conclusion
The recent reports that we reviewed clearly showed the potential of chiral NP catalysts not only for asymmetric hydrogenation, but also for various types of asymmetric C–C bond formation reactions. Chiral NPs can be easily generated from metal precursors and chiral modifiers. The obtained NPs can be immobilized on several insoluble supports, such as metal oxides, polymers and magnetic NPs, to form heterogeneous catalysts. It was demonstrated that such catalysts could be recovered by simple operations and reused several times, without significant loss of activities and selectivities. The enantioselectivities in chiral NP catalyst systems were comparable to those in the corresponding homogeneous catalytic systems, and some chiral NP catalyst systems showed different selectivities compared with homogeneous catalytic systems. These facts indicate that chiral NP catalysts are not just “immobilized homogeneous catalysts;” they should be considered as distinct active species. The choice of the chiral modifier is a critical key not only to control enantioselectivity, but also to stabilize the NP catalyst itself. A wrong choice of chiral modifier can lead to the decomposition of NP catalyst to a corresponding homogeneous catalyst.Given these aspects, we should be careful when discussing the real active species. Although the heterogeneity was confirmed by several traditional experiments, such as the poisoning test, hot filtration test, three-phase test, and ICP analysis in a reaction mixture, it is still unclear how the chiral environment is constructed around the NPs and how chiral modifiers and substrates interact with the surface of NPs or any other active species.
It is important to clarify the chiral active species and the mechanism of specific selectivities in chiral NP-catalyzed reactions. This is an emerging area of research. Further studies should lead to achievement of more efficient and unique chiral NP-catalyzed reactions.
Acknowledgements
Our work written in this article was partially supported by a Grant-in-Aid for Science Research from the Japan Society for the Promotion of Science (JSPS), Global COE Program, The University of Tokyo, MEXT Japan, NEDO, and Japan Science Technology Agency (JST). T.Y. thanks the JSPS for a Research Fellowship for Young Scientists.References
- Asymmetric Synthesis More Methods and Applications, ed. M. Christmann and S. Bräse, Wiley-VCH John Wiley & Sons, Hoboken, 2012 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley, Hoboken, N.J., 3rd edn, 2010 Search PubMed.
- Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, ed. H. U. Blaser and H.-J. Federsel, Wiley-VCH, Weinheim, 2010 Search PubMed.
- G. P. Howell, Org. Process Res. Dev., 2012, 16, 1258–1272 CrossRef CAS.
- C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS.
- N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291–1299 CrossRef CAS PubMed.
- H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385–1393 CrossRef CAS PubMed.
- V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS PubMed.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- P. Barbaro, F. Liguori, N. Linares and C. M. Marrodan, Eur. J. Inorg. Chem., 2012, 3807–3823 CrossRef CAS.
- C. Lucarelli and A. Vaccari, Green Chem., 2011, 13, 1941–1949 RSC.
- B. Pugin and H.-U. Blaser, Top. Catal., 2010, 53, 953–962 CrossRef CAS.
- L. Yin and J. Liebscher, Chem. Rev., 2006, 107, 133–173 CrossRef PubMed.
- M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J.-P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002–3027 CrossRef CAS PubMed.
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC.
- S. Bräse, F. Lauterwasser and R. E. Ziegert, Adv. Synth. Catal., 2003, 345, 869–929 CrossRef.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS PubMed.
- Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008 Search PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- D. L. Fedlheim and C. A. Foss, Metal Nanoparticles: Synthesis, Characterization, and Applications, CRC Press, 2001 Search PubMed.
- H. Cong and J. A. Porco, ACS Catal., 2012, 2, 65–70 CrossRef CAS PubMed.
- Y. Xia, Y. Zhou and Z. Tang, Nanoscale, 2011, 3, 1374–1382 RSC.
- A. Guerrero-Martínez, J. Lorenzo Alonso-Gómez, B. Auguié, M. Magdalena Cid and L. M. Liz-Marzán, Nano Today, 2011, 6, 381–400 CrossRef PubMed.
- C. Noguez and I. L. Garzón, Chem. Soc. Rev., 2009, 38, 757–771 RSC.
- C. Gautier and T. Bürgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS PubMed.
- U. Shivakumar, J. Mirzaei, X. Feng, A. Sharma, P. Moreira and T. Hegmann, Liq. Cryst., 2011, 38, 1495–1514 CrossRef CAS.
- W. Hu, H. Zhao, L. Shan, L. Song, H. Cao, Z. Yang, Z. Cheng, C. Yan, S. Li, H. Yang and L. Guo, Liq. Cryst., 2010, 37, 563–569 CrossRef CAS.
- H. Qi, J. O'Neil and T. Hegmann, J. Mater. Chem., 2008, 18, 374–380 RSC.
- H. Qi and T. Hegmann, J. Mater. Chem., 2006, 16, 4197–4205 RSC.
- Y. Sun, L. Zhang and H. Li, New J. Chem., 2012, 36, 1442–1444 RSC.
- M. Zhang and B.-C. Ye, Anal. Chem., 2011, 83, 1504–1509 CrossRef CAS PubMed.
- C.-C. You, S. S. Agasti and V. M. Rotello, Chem.–Eur. J., 2008, 14, 143–150 CrossRef CAS PubMed.
- Y. X. Wang, X. L. Yin, M. H. Shi, W. Li, L. Zhang and J. L. Kong, Talanta, 2006, 69, 1240–1245 CrossRef CAS PubMed.
- Y. Orito, S. Imai and S. Niwa, Nippon Kagaku Kaishi, 1979, 1118–1120 CrossRef CAS.
- Y. Orito, S. Imai, S. Niwa and G. H. Nguyen, J. Synth. Org. Chem., Jpn., 1979, 37, 173–174 CrossRef CAS.
- G. Kyriakou, S. K. Beaumont and R. M. Lambert, Langmuir, 2011, 27, 9687–9695 CrossRef CAS PubMed.
- F. Zaera, J. Phys. Chem. C, 2008, 112, 16196–16203 CAS.
- H.-U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed.
- M. Studer, H. U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS.
- M. von Arx, T. Mallat and A. Baiker, Top. Catal., 2002, 19, 75–87 CrossRef CAS.
- G. Szőllősi, Z. Makra, M. Fekete, F. Fülöp and M. Bartók, Catal. Lett., 2012, 142, 889–894 CrossRef.
- E. Schmidt, C. Bucher, G. Santarossa, T. Mallat, R. Gilmour and A. Baiker, J. Catal., 2012, 289, 238–248 CrossRef CAS PubMed.
- S. Sano, M. J. Beier, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 2012, 357, 117–124 CrossRef CAS PubMed.
- B. Li, X. Li, Y. Ding and P. Wu, Catal. Lett., 2012, 142, 1033–1039 CrossRef CAS.
- A. Indra and G. K. Lahiri, Chem.–Eur. J., 2012, 18, 6742–6745 CrossRef CAS PubMed.
- M. J. Beier, J.-M. Andanson, T. Mallat, F. Krumeich and A. Baiker, ACS Catal., 2012, 2, 337–340 CrossRef CAS.
- B. Török, A. Kulkarni, R. DeSousa, K. Satuluri, M. Török and G. K. S. Prakash, Catal. Lett., 2011, 141, 1435–1441 CrossRef.
- G. Szőllősi, Z. Makra, F. Fueloep and M. Bartók, Catal. Lett., 2011, 141, 1616–1620 CrossRef.
- B. Li, X. Li, H. Wang and P. Wu, J. Mol. Catal. A: Chem., 2011, 345, 81–89 CrossRef CAS PubMed.
- N. Erathodiyil, H. Gu, H. Shao, J. Jiang and J. Y. Ying, Green Chem., 2011, 13, 3070–3074 RSC.
- Z. Chen, Z. Guan, M. Li, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2011, 50, 4913–4917 CrossRef CAS PubMed.
- S. Böttcher, C. Hoffmann, K. Räuchle and W. Reschetilowski, ChemCatChem, 2011, 3, 741–748 CrossRef.
- M. U. Azmat, Y. Guo, Y. Guo, Y. Wang and G. Lu, J. Mol. Catal. A: Chem., 2011, 336, 42–50 CrossRef CAS PubMed.
- H. Wang, X. Li, Y. M. Wang and P. Wu, ChemCatChem, 2010, 2, 1303–1311 CrossRef CAS.
- S. Nowag, X.-S. Wang, J. Keilitz, A. Thomas and R. Haag, ChemCatChem, 2010, 2, 807–811 CrossRef CAS.
- C. Mondelli, C. Bucher, A. Baiker and R. Gilmour, J. Mol. Catal. A: Chem., 2010, 327, 87–91 CrossRef CAS PubMed.
- J. Keilitz, S. Nowag, J.-D. Marty and R. Haag, Adv. Synth. Catal., 2010, 352, 1503–1511 CrossRef CAS.
- S. Cserényi, G. Szőllősi, K. Szöri, F. Fülöp and M. Bartók, Catal. Commun., 2010, 12, 14–19 CrossRef PubMed.
- G. Goubert and P. H. McBreen, ChemCatChem, 2013, 5, 683–685 CrossRef CAS.
- F. Meemken, N. Maeda, K. Hungerbühler and A. Baiker, ACS Catal., 2012, 2, 464–467 CrossRef CAS.
- F. Meemken, N. Maeda, K. Hungerbühler and A. Baiker, Angew. Chem., Int. Ed., 2012, 51, 8212–8216 CrossRef CAS PubMed.
- N. Maeda, S. Sano, T. Mallat, K. Hungerbühler and A. Baiker, J. Phys. Chem. C, 2012, 116, 4182–4188 CAS.
- N. Maeda, K. Hungerbühler and A. Baiker, J. Am. Chem. Soc., 2011, 133, 19567–19569 CrossRef CAS PubMed.
- E. Schmidt, A. Vargas, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 12358–12367 CrossRef CAS PubMed.
- K. Molvinger, M. Lopez and J. Court, Tetrahedron Lett., 1999, 40, 8375–8378 CrossRef CAS.
- Z. Makra, G. Szőllősi and M. Bartók, Catal. Today, 2012, 181, 56–61 CrossRef CAS PubMed.
- N. Győrffy and A. Tungler, J. Mol. Catal. A: Chem., 2011, 336, 72–77 CrossRef PubMed.
- T. Sugimura and H. Ogawa, Chem. Lett., 2010, 232–233 CrossRef CAS.
- T. Y. Kim and T. Sugimura, J. Mol. Catal. A: Chem., 2010, 327, 58–62 CrossRef CAS PubMed.
- N. Győrffy, A. Tungler and M. Fodor, J. Catal., 2010, 270, 2–8 CrossRef PubMed.
- S. K. Beaumont, G. Kyriakou, D. J. Watson, O. P. H. Vaughan, A. C. Papageorgiou and R. M. Lambert, J. Phys. Chem. C, 2010, 114, 15075–15077 CAS.
- D. J. Watson, R. J. B. R. J. Jesudason, S. K. Beaumont, G. Kyriakou, J. W. Burton and R. M. Lambert, J. Am. Chem. Soc., 2009, 131, 14584–14589 CrossRef CAS PubMed.
- G. Szőllősi, Z. Nemeth, K. Hernadi and M. Bartók, Catal. Lett., 2009, 132, 370–376 CrossRef.
- S. Li, C. Chen, E. Zhan, S.-B. Liu and W. Shen, J. Mol. Catal. A: Chem., 2009, 304, 88–94 CrossRef CAS PubMed.
- S. C. Mhadgut, M. Török, S. Dasgupta and B. Török, Catal. Lett., 2008, 123, 156–163 CrossRef CAS.
- S. Li, E. Zhan, Y. Li, Y. Xu and W. Shen, Catal. Today, 2008, 131, 347–352 CrossRef CAS PubMed.
- E. Zhan, S. Li, Y. Xu and W. Shen, Catal. Commun., 2007, 8, 1239–1243 CrossRef CAS PubMed.
- A. I. McIntosh, D. J. Watson and R. M. Lambert, Langmuir, 2007, 23, 6113–6118 CrossRef CAS PubMed.
- A. I. McIntosh, D. J. Watson, J. W. Burton and R. M. Lambert, J. Am. Chem. Soc., 2006, 128, 7329–7334 CrossRef CAS PubMed.
- R. Strobel, F. Krumeich, W. J. Stark, S. E. Pratsinis and A. Baiker, J. Catal., 2004, 222, 307–314 CrossRef CAS PubMed.
- A. Tungler, Y. Nitta, K. Fodor, G. Farkas and T. Mathe, J. Mol. Catal. A: Chem., 1999, 149, 135–140 CrossRef CAS.
- A. Tungler, T. Mathe, T. Tarnai, K. Fodor, G. Toth, J. Kajtar, I. Kolossvary, B. Herenyi and R. A. Sheldon, Tetrahedron: Asymmetry, 1995, 6, 2395–2402 CrossRef CAS.
- L. Ye, H. Lin, H. Zhou and Y. Yuan, J. Phys. Chem. C, 2010, 114, 19752–19760 CAS.
- H.-y. Jiang, H. Chen and R.-x. Li, Catal. Commun., 2010, 11, 584–587 CrossRef CAS PubMed.
- A. Gual, C. Godard, K. Philippot, B. Chaudret, A. Denicourt-Nowicki, A. Roucoux, S. Castillón and C. Claver, ChemSusChem, 2009, 2, 769–779 CrossRef CAS PubMed.
- J. Wang, H. Feng, R. Qin, H. Fu, M. Yuan, H. Chen and X. Li, Tetrahedron: Asymmetry, 2007, 18, 1643–1647 CrossRef CAS PubMed.
- S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gómez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 115–122 RSC.
- E. G. Bilé, E. Cortelazzo-Polisini, A. Denicourt-Nowicki, R. Sassine, F. Launay and A. Roucoux, ChemSusChem, 2012, 5, 91–101 CrossRef PubMed.
- Y. Huang, S. Xu and V. S. Y. Lin, ChemCatChem, 2011, 3, 690–694 CrossRef CAS.
- E. G. Bilé, A. Denicourt-Nowicki, R. Sassine, P. Beaunier, F. Launay and A. Roucoux, ChemSusChem, 2010, 3, 1276–1279 CrossRef PubMed.
- F. Hoxha, N. van Vegten, A. Urakawa, F. Krurneich, T. Mallat and A. Baiker, J. Catal., 2009, 261, 224–231 CrossRef CAS PubMed.
- F. Hoxha, T. Mallat and A. Baiker, J. Catal., 2007, 248, 11–19 CrossRef CAS PubMed.
- O. J. Sonderegger, G. M. W. Ho, T. Burgi and A. Baiker, J. Catal., 2005, 230, 499–506 CrossRef CAS PubMed.
- H.-y. Jiang, B. Sun, X.-x. Zheng and H. Chen, Appl. Catal., A, 2012, 421, 86–90 CrossRef PubMed.
- C. Yang, H. Jiang, J. Feng, H. Fu, R. Li, H. Chen and X. Li, J. Mol. Catal. A: Chem., 2009, 300, 98–102 CrossRef CAS PubMed.
- H.-y. Jiang, C.-f. Yang, C. Li, H.-y. Fu, H. Chen, R.-x. Li and X.-j. Li, Angew. Chem., Int. Ed., 2008, 47, 9240–9244 CrossRef CAS PubMed.
- J. F. Sonnenberg, N. Coombs, P. A. Dube and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 5893–5899 CrossRef CAS PubMed.
- K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13–22 CAS.
- P. Barbaro, V. Dal Santo and F. Liguori, Dalton Trans., 2010, 39, 8391–8402 RSC.
- S. Roy and M. A. Pericas, Org. Biomol. Chem., 2009, 7, 2669–2677 CAS.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2011, 112, 1536–1554 CrossRef PubMed.
- L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Beland and P. D. Cara, ChemCatChem, 2012, 4, 432–445 CrossRef CAS.
- J. Durand, E. Teuma and M. Gómez, Eur. J. Inorg. Chem., 2008, 3577–3586 CrossRef CAS.
- B. M. Trost, Org. Process Res. Dev., 2012, 16, 185–194 CrossRef CAS PubMed.
- B. M. Trost and D. L. VanVranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed.
- S. Kobayashi and S. Nagayama, J. Am. Chem. Soc., 1998, 120, 2985–2986 CrossRef CAS.
- R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS PubMed.
- S. Kobayashi and H. Miyamura, Chem. Rec., 2010, 10, 271–290 CrossRef CAS PubMed.
- S. Kobayashi and H. Miyamura, Aldrichimica Acta, 2013, 46, 3–19 CAS.
- M. Donbrow, Microcapsules and Nanoparticles in Medicine and Pharmacy, CRC Press, Boca Raton, FL, 1991 Search PubMed.
- R. Akiyama and S. Kobayashi, Angew. Chem., Int. Ed., 2001, 40, 3469–3471 CrossRef CAS.
- S. Jansat, M. Gómez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillón and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592–1593 CrossRef CAS PubMed.
- I. Favier, M. Gómez, G. Muller, M. R. Axet, S. Castillon, C. Claver, S. Jansat, B. Chaudret and K. Philippot, Adv. Synth. Catal., 2007, 349, 2459–2469 CrossRef CAS.
- M. Diéguez, O. Pamies, Y. Mata, E. Teuma, M. Gómez, F. Ribaudo and P. W. N. M. van Leeuwen, Adv. Synth. Catal., 2008, 350, 2583–2598 CrossRef.
- F. X. Felpin and Y. Landais, J. Org. Chem., 2005, 70, 6441–6446 CrossRef CAS PubMed.
- S. Reimann, T. Mallat and A. Baiker, J. Catal., 2007, 252, 30–38 CrossRef CAS PubMed.
- S. Reimann, T. Mallat and A. Baiker, J. Catal., 2008, 254, 79–83 CrossRef CAS PubMed.
- S. Reimann, J.-D. Grunwaldt, T. Mallat and A. Baiker, Chem.–Eur. J., 2010, 16, 9658–9668 CrossRef CAS PubMed.
- Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments, ed. Á. Molnár, Wiley-VCH, Weinheim, 2013 Search PubMed.
- M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS PubMed.
- K. Mori, Y. Kondo and H. Yamashita, Phys. Chem. Chem. Phys., 2009, 11, 8949–8954 RSC.
- K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS PubMed.
- C. Richter, K. V. S. Ranganath and F. Glorius, Adv. Synth. Catal., 2012, 354, 377–382 CrossRef CAS.
- K. V. S. Ranganath, A. H. Schaefer and F. Glorius, ChemCatChem, 2011, 3, 1889–1891 CrossRef CAS.
- J. Klosin and C. R. Landis, Acc. Chem. Res., 2007, 40, 1251–1259 CrossRef CAS PubMed.
- Y. Yuan, N. Yan and P. J. Dyson, ACS Catal., 2012, 2, 1057–1069 CrossRef CAS.
- A. C. B. Neves, M. J. F. Calvete, T. M. V. D. Pinho e Melo and M. M. Pereira, Eur. J. Org. Chem., 2012, 6309–6320 CrossRef CAS.
- J. M. Coronado, F. Coloma and J. A. Anderson, J. Mol. Catal. A: Chem., 2000, 154, 143–154 CrossRef CAS.
- D. Han, X. Li, H. Zhang, Z. Liu, J. Li and C. Li, J. Catal., 2006, 243, 318–328 CrossRef CAS PubMed.
- D. Han, X. Li, H. Zhang, Z. Liu, G. Hu and C. Li, J. Mol. Catal. A: Chem., 2008, 283, 15–22 CrossRef CAS PubMed.
- M. R. Axet, S. Castillon, C. Claver, K. Philippot, P. Lecante and B. Chaudret, Eur. J. Inorg. Chem., 2008, 3460–3466 CrossRef CAS.
- J. Blanco-Urgoiti, L. Anorbe, L. Perez-Serrano, G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC.
- S. T. Ingate and J. Marco-Contelles, Org. Prep. Proced. Int., 1998, 30, 121–143 CrossRef CAS.
- H.-W. Lee and F.-Y. Kwong, Eur. J. Org. Chem., 2010, 789–811 CrossRef CAS.
- J. H. Park, K.-M. Chang and Y. K. Chung, Coord. Chem. Rev., 2009, 253, 2461–2480 CrossRef CAS PubMed.
- J. H. Park and Y. K. Chung, Dalton Trans., 2008, 2369–2378 RSC.
- K. H. Park and Y. K. Chung, Adv. Synth. Catal., 2005, 347, 854–866 CrossRef CAS.
- Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579–5580 CrossRef CAS.
- P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS.
- H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC.
- T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed.
- G. P. Howell, Org. Process Res. Dev., 2012, 16, 1258–1272 CrossRef CAS.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- S. Brock, D. Hose, J. Moseley, A. Parker, I. Patel and A. Williams, Org. Process Res. Dev., 2008, 12, 496–502 CrossRef CAS.
- T. Yasukawa, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 16963–16966 CrossRef CAS PubMed.
- B. M. Choudary, K. V. S. Ranganath, U. Pal, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 13167–13171 CrossRef CAS PubMed.
- B. M. Choudary, L. Chakrapani, T. Ramani, K. V. Kumar and M. L. Kantam, Tetrahedron, 2006, 62, 9571–9576 CrossRef CAS PubMed.
- M. L. Kantam, K. V. S. Ranganath, K. Mahendar, L. Chakrapani and B. M. Choudary, Tetrahedron Lett., 2007, 48, 7646–7649 CrossRef CAS PubMed.
- M. L. Kantam, T. Ramani, L. Chakrapani and K. V. Kumar, Tetrahedron Lett., 2008, 49, 1498–1501 CrossRef CAS PubMed.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- T. Tsukuda, H. Tsunoyama and H. Sakurai, Chem.–Asian J., 2011, 6, 736–748 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- A. Haruta, Chem. Rec., 2003, 3, 75–87 CrossRef PubMed.
- E. Gross, J. H. Liu, S. Alayoglu, M. A. Marcus, S. C. Fakra, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 3881–3886 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |