 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent extraction: the coordination chemistry behind extractive metallurgy†
A. Matthew
Wilson
a,
Phillip J.
Bailey
a,
Peter A.
Tasker
*a,
Jennifer R.
Turkington
a,
Richard A.
Grant
b and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: jason.love@ed.ac.uk; peter.tasker@ed.ac.uk; Fax: +44 (0)131 6504743; Tel: +44 (0)131 6504762
bJohnson Matthey Technology Centre, Sonning Common, Reading, RG4 9NH, UK
First published on 3rd October 2013
Abstract
The modes of action of the commercial solvent extractants used in extractive hydrometallurgy are classified according to whether the recovery process involves the transport of metal cations, Mn+, metalate anions, MXxn−, or metal salts, MXx into a water-immiscible solvent. Well-established principles of coordination chemistry provide an explanation for the remarkable strengths and selectivities shown by most of these extractants. Reagents which achieve high selectivity when transporting metal cations or metal salts into a water-immiscible solvent usually operate in the inner coordination sphere of the metal and provide donor atom types or dispositions which favour the formation of particularly stable neutral complexes that have high solubility in the hydrocarbons commonly used in recovery processes. In the extraction of metalates, the structures of the neutral assemblies formed in the water-immiscible phase are usually not well defined and the cationic reagents can be assumed to operate in the outer coordination spheres. The formation of secondary bonds in the outer sphere using, for example, electrostatic or H-bonding interactions are favoured by the low polarity of the water-immiscible solvents.
Key learning points(1) Metals are critical resources in the 21st century due to technological advances and societal needs.(2) Extractive metallurgy is an efficient method for metal refining and is underpinned by coordination chemistry. (3) Reagents used commercially in the selective separation of metals by solvent extraction can be classified according to their modes of operation. (4) Extractants (either singly or in mixtures) interact with metal species through the inner-sphere, outer-sphere, or combinations of both. (5) An understanding of the fundamental coordination chemistry allied to solvent extraction processes can form the basis of new reagent designs for more efficient and sustainable metal refining. |
Introduction
Although the techniques for the recovery of metals have advanced steadily since the bronze and iron ages,1,2 it can be argued that the need to accelerate the pace of development of new technologies for extractive metallurgy has never been greater if we are to respond to the rapidly increasing demand for metals, the diminishing supplies of high grade ores, and the pressures to recycle.3,4 Governments worldwide are proposing policies to ensure that new technologies are developed in order to maintain supplies of critical metals, in particular those, such as the rare earth elements, that are becoming essential for communications and greener energy production.5–8This tutorial review describes the coordination chemistry of commercial solvent extraction processes that are used to achieve the separation and concentration of the metals of value in extractive hydrometallurgy. Hydrometallurgy, which is underpinned by coordination chemistry, is often favoured over traditional pyrometallurgy by industry as it can lead to excellent materials and energy balances because reagents are recycled. Also, hydrometallurgical plants are smaller than those for pyrometallurgical processing so can be located close to the mining operations, and are often environmentally favoured due to lack of SO2 emission that is frequently associated with pyrometallurgy. The technology is well suited to metal recovery from low grade, mixed metal, ores and can be adapted to the recovery of metals from secondary sources, for example catalytic converters and electrical goods.3 Even with these advantages, the embodied energy associated with the production of metals is not necessarily higher for metals obtained via smelting or blast furnaces.9 As such, the choice between technologies is not always clear cut and in some cases, for example in the extraction of precious metals, a combination of pyro- and hydrometallurgical processes is preferred.
A hydrometallurgical flow sheet usually involves the operations shown in Fig. 1. After dissolution of the ore into an aqueous medium (leaching), the metal to be recovered can be separated and concentrated using solvent extraction and the resulting, high purity, aqueous solution is reduced to generate the metal.10
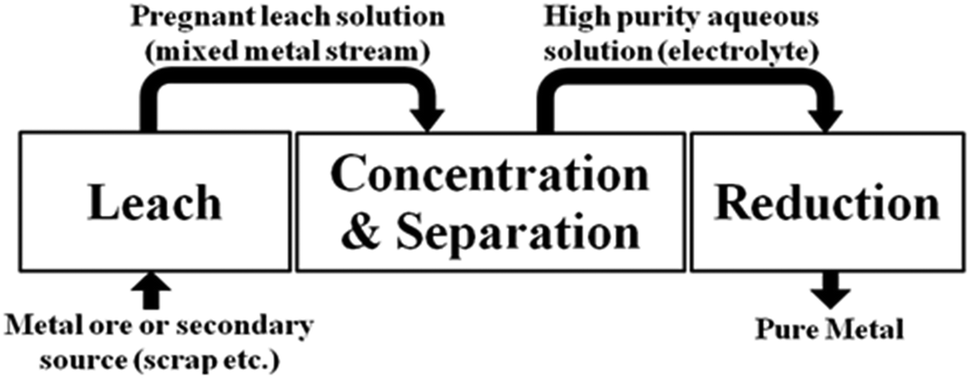 | ||
| Fig. 1 The key operations in a hydrometallurgical process to recover metals from minerals or from secondary sources. | ||
Most metals are obtained from oxidic or sulfidic ores and leaching processes can involve a wide range of conditions.11 A lot of development work has been undertaken recently on pressure and oxidative leaching of base metal sulfides to generate elemental sulfur as the major by-product and sulfate solutions for downstream processing. Chloride media are favoured for the recovery of precious metals, whilst nitrate, chloride and sulfate streams are involved in the extraction and purification of f-block elements.12 While a number of methods can be used to achieve the concentration and separation operations shown in Fig. 1, this review will only consider solvent extraction. This technology, first developed for large scale operations for uranium production in the Manhattan Project,10 has proven robust and can be used in continuous, rather than batch processes.
An idealised representation of a flow sheet using solvent extraction to achieve the concentration and separation of a desired metal is shown in Fig. 2. At the front end of such a system, the aqueous solution from leaching (the “pregnant leach solution”) is contacted with the solvent extractant in a mixer-settler (Fig. 3) which selectively transfers the desired metal to a water-immiscible phase. After phase separation, the aqueous solution (the raffinate) is recycled back to leaching and the water-immiscible phase (the “organic”) is moved to another mixer-settler where it is contacted with an aqueous ‘stripping’ solution to create a concentrated, pure aqueous solution of the metal for reduction. The stripped water-immiscible phase (the “organic”) is reused and the aqueous solution from electrolysis, the “spent electrolyte” is reused in stripping.
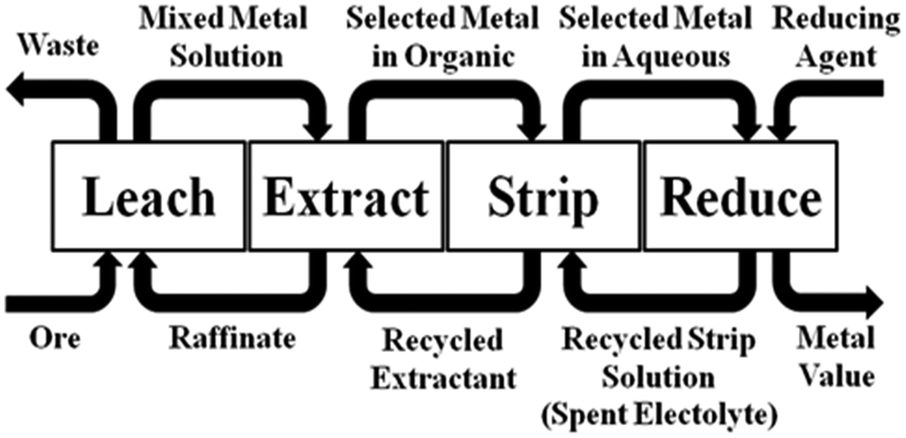 | ||
| Fig. 2 The incorporation of solvent extraction to achieve metal separation and concentration in a hydrometallurgical circuit which involves recycling of all reagents. | ||
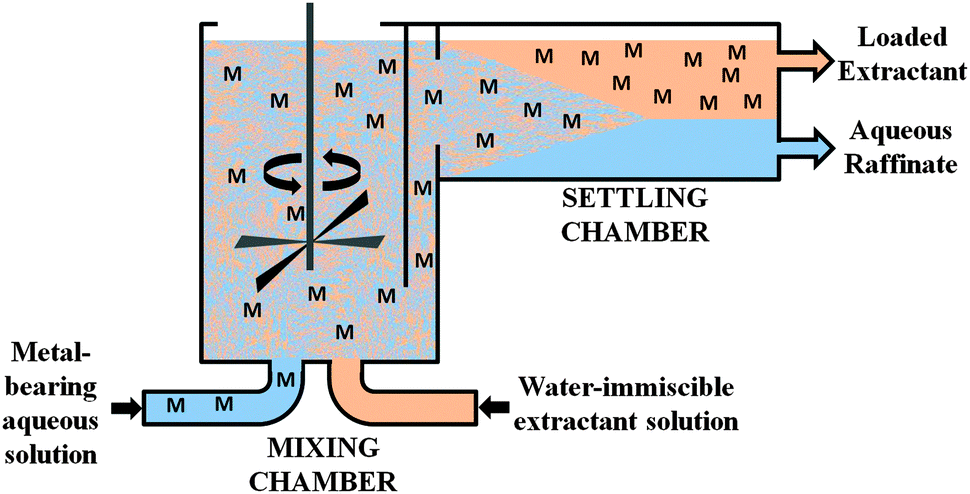 | ||
| Fig. 3 Schematic representation of a mixer-settler for continuous operation of solvent extraction. | ||
The nature of the counter anion present in the pregnant leach solution has a major influence on the selection of the metal-extracting reagent used in downstream processing. As chloride is a good inner-sphere ligand it is possible to generate extractable, charge-neutral, complexes [MClx(L)n] by using a neutral reagent L which effectively “solvates” the metal salt, MClx, as in eqn (1) (“Metal Salt Extractants” section below). Such solvating reagents may be present in the inner or outer coordination sphere, or both.
| nL(org) + MClx ⇌ [MClx(L)n](org) | (1) |
At high chloride concentrations, chloridometalates MClxn− are more likely to be present in the aqueous solution. These can be transferred to the water-immiscible solvent by formation of outer sphere assemblies or ion pairs, [(LH)nMClx], with cationic extractants, LH+, for example as in eqn (2) (see “Metalate Extractants” below).
| nL(org) + nH+ + MClxn− ⇌ [(LH)nMClx](org) | (2) |
A different strategy is needed for metal removal from sulfate streams because sulfate is a weak inner sphere ligand,11 and the most effective approach is to generate a charge-neutral complex in the organic phase, [M(L)n], by combining anionic ligands L− with the metal cation Mn+ as in eqn (3) (see “Metal Cation Extractants”).
| nLH(org) + Mn+ ⇌ [M(L)n](org) + nH+ | (3) |
Good materials balances in the flow sheet outlined in Fig. 2 depend on the retention of reagents in the three closed loops. Consequently, it is very important that a solvent extractant and its metal complexes have a very low solubility in water and a high solubility in the water-immiscible phase, usually a high boiling hydrocarbon on the grounds of safety and cost. The low polarity of such kerosene diluents favours the formation of H-bonds and other forms of secondary bonding between extractant molecules. These interactions in the outer coordination sphere of a complexed metal ion can contribute significantly to the strength and selectivity of an extractant and can be used in conjunction with more conventional aspects of ligand design which influence the stability of the inner coordination sphere, to develop reagents which meet the requirements of a particular circuit.12
In addition to showing the appropriate strength and selectivity of extraction any new reagent should:
• Facilitate fast metal transfer between the two liquid phases in both loading and stripping and rapid phase disengagement, otherwise the throughput of metal in the overall process will be low;
• Show high chemical stability under the conditions of operation to reduce make-up costs and minimise the transfer of degradation products to other parts of the circuit or to the environment.
Commercial extractants
Only a small percentage of the solvent extractants described in the literature has been extensively tested or used in commercial operations.13 Most frequently, the reagents used in commercial operations are formulations which contain the extractant blended with modifiers that enhance solubility, phase disengagement properties, and metal transfer. These formulations are frequently denoted by trade numbers which often make it difficult for a non-expert to identify the active components. Also, reports on the performance of the most commercially viable reagents have often been written by metallurgists and engineers and consequently relatively little attention has been paid to the fundamental chemistry involved in the processes, in particular the structures of the assemblies formed in the water-immiscible phase.This article reviews the coordination chemistry that underpins the use of different classes of commercial solvent extractants. We focus on examples where this chemistry is well understood and where the structures of the assemblies formed in the water-immiscible phase are best defined. An understanding of the coordination chemistry, which often involves interactions in the outer- as well as in the inner-coordination spheres can usually account for the strength and selectivity of metal transfer to the organic phase and can aid the design of new reagents and processes.
Metal cation extraction
These reagents transfer metal cations, Mn+, from the aqueous phase by an exchange process in which the metal cation usually substitutes ionisable protons on the extractant, as shown in eqn (3).As the equilibrium in eqn (3) is pH dependent, the “strengths” of the extractants can be compared by recording the pH values at which they are 50%-loaded with metal (the “pH1/2 values”), see for example Fig. 4. Strong extractants for metal cations have low pH1/2 values, indicating that they are capable of recovering metals from highly acidic feed solutions. Conversely, a weak extractant for metal cations is only capable of recovering metals at high pH. The pH-dependence of the equilibrium shown in eqn (3) makes it possible to control the loading and stripping of the metal in the organic phase by varying the pH of the aqueous phase with which it is in contact.
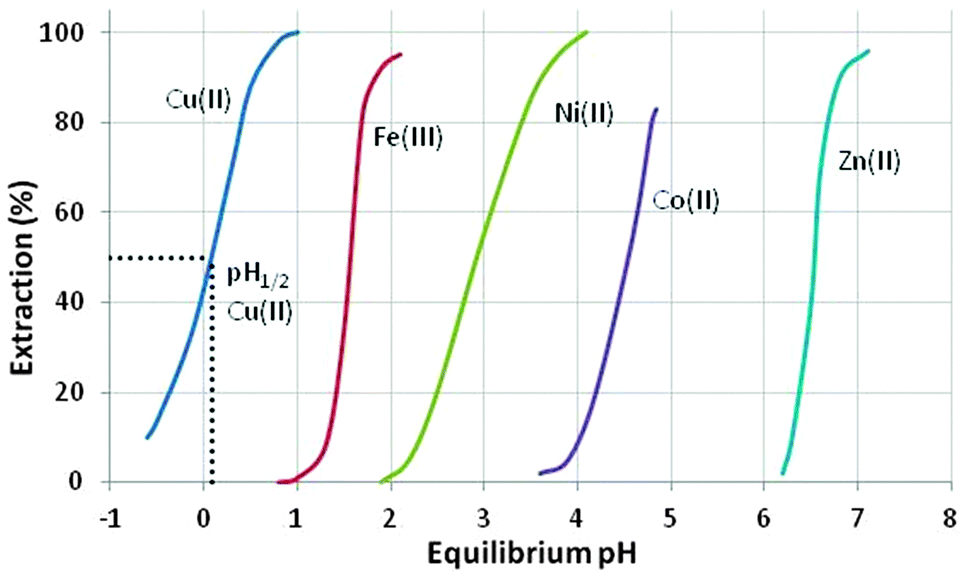 | ||
| Fig. 4 The pH dependence of metal cation loading by AcorgaP50.‡ | ||
In some cases weak extractants have to be first converted to their alkali metal or ammonium salts to ensure that loading of the desired metal cation is successful. The cation exchange process in eqn (4) is facilitated by the sodium ion having a higher hydration energy than the Mn+ cation, favouring its transfer to the aqueous phase, and by the [M(L)n] complex being more thermodynamically stable than the sodium salt.
| n[Na(L)](org) + Mn+ ⇌ [M(L)n](org) + nNa+ | (4) |
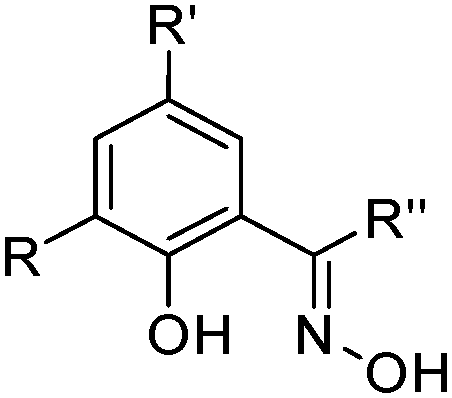
Oxime extractants
Phenolic oximes of the types shown in Table 1,§ are used extensively in copper recovery,11 accounting for ca. 25% of world production.14 The sequence of reactions outlined in Scheme 1 ensures that the leachant, the extractant and the stripping solution are recycled and leads to an excellent materials balance.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11
11
| Phenolic oximes | |||
|---|---|---|---|
|
|
|||
| Substituents | Commercial name(s) | ||
| R | R′ | R′′ | |
| H | C9H19 | H | Acorga P50 |
| H | C9H19 | Me | Acorga K2000, LIX 84-I, SME 529 |
| H | C9H19 | Phenyl | LIX 65N |
| H | C12H25 | Phenyl | LIX 64 |
| Cl | C9H19 | Phenyl | LIX 70 |
| H | C12H25 | H | LIX 860, LIX 622 |
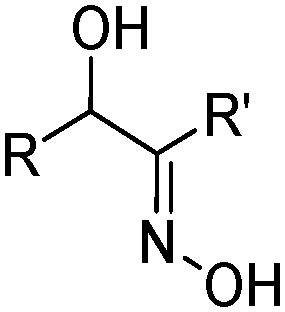
| α-Hydroxyoximes | ||
|---|---|---|
|
|
||
| Substituents | Commercial name(s) | |
| R = R′ | n-C4H9(C2H5)CHCH2 | LIX 63 |
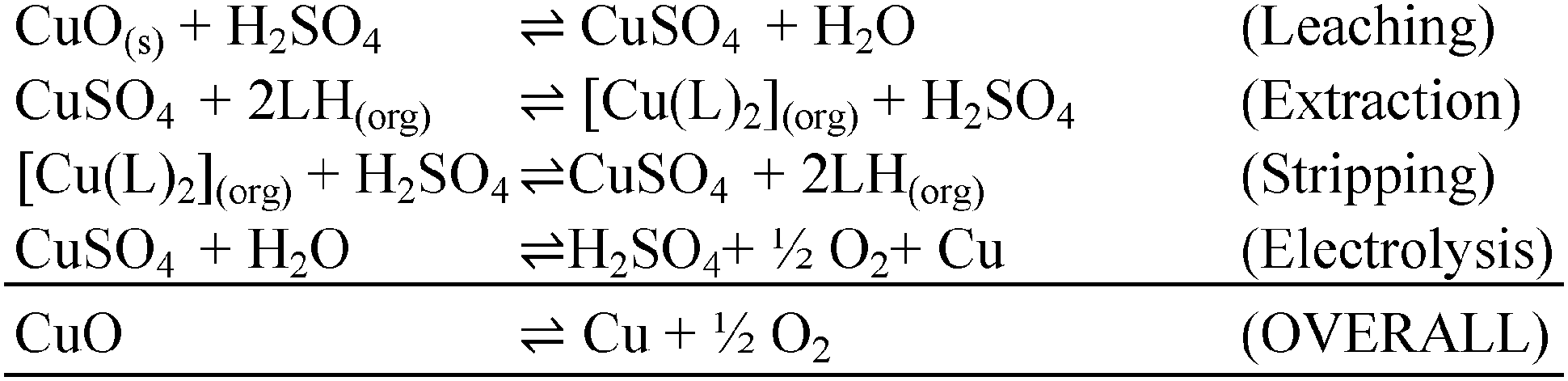 | ||
| Scheme 1 The sequence of reactions and overall materials balance for copper recovery from oxide ores by the leach/solvent extraction/electrowinning process.12 | ||
The suitability of the phenolic oxime reagents in these operations arises from their coordination chemistry. They are strong enough extractants (pH1/2 < 1.0, see Fig. 4) to load copper from aqueous leach solutions that are sufficiently acidic to ensure that iron(III) remains in the aqueous phase.¶ This strength and the selectivity for Cu2+ over other base-metal cations arises in part from the pseudo-macrocyclic structure of the copper complex (Scheme 2). The strong interligand H-bonds between the oxime hydrogen and the phenolic oxygen atoms define a planar donor set and a cavity which is a particularly good fit for Cu(II).12
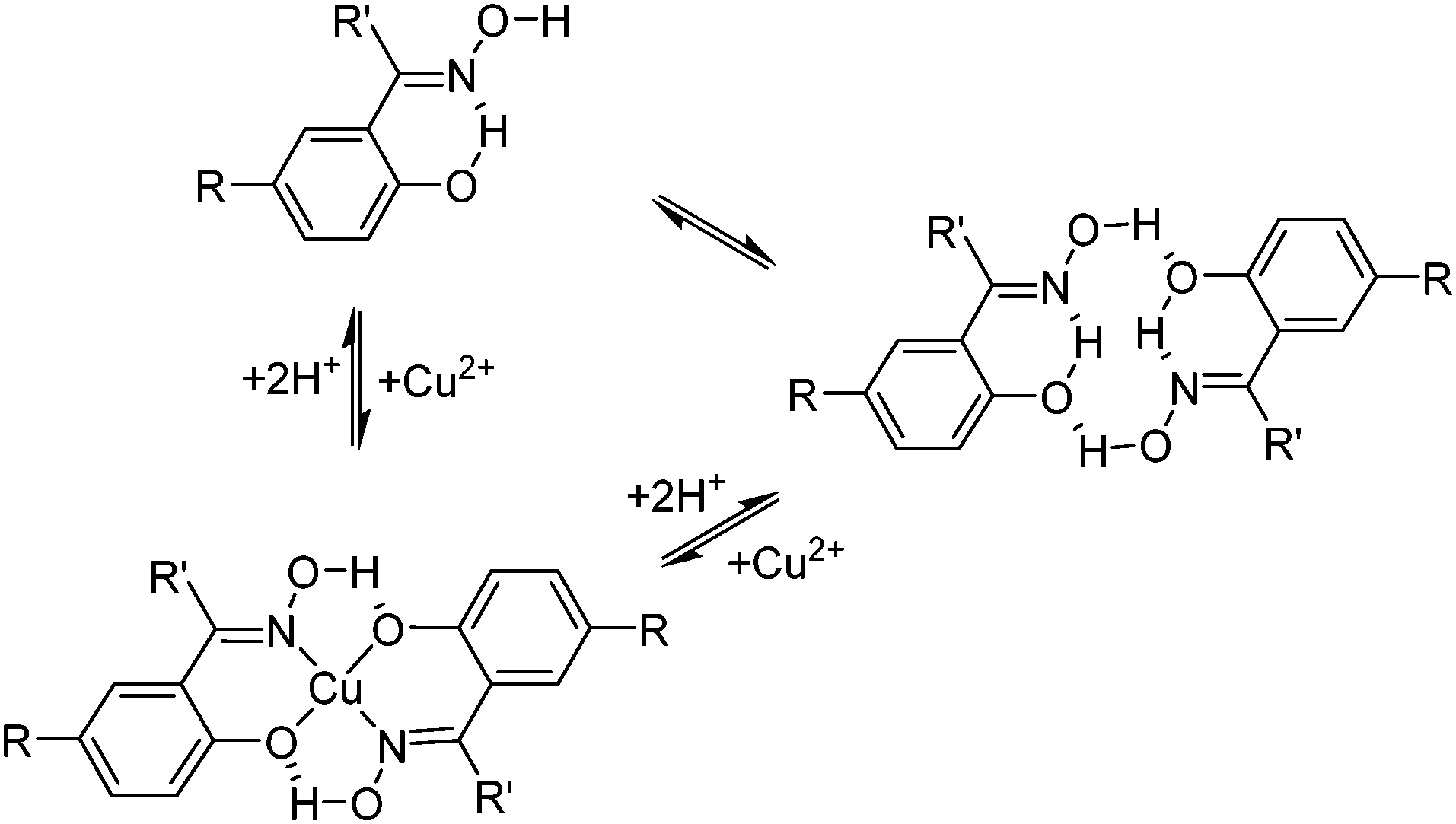 | ||
| Scheme 2 Pre-organisation of phenolic oxime pro-ligands and the pseudo-macrocyclic structures of their Cu(II) complexes. | ||
The selectivity of metal extraction in reactions of the type shown in eqn (3) is revealed by the difference in the pH1/2 values for the various metals. It is clear from the S-curve plots shown in Fig. 4 that Acorga P50 will reject Ni(II) and Zn(II) when Cu(II) is present. The efficacy of extraction of the metals shown in Fig. 4 is roughly that which would be predicted by the Irving–Williams order but with those metals which have a preference for the planar geometry of the pseudo-macrocyclic donor set, Ni(II), and Cu(II), being favoured.
Oximic reagents are susceptible to hydrolysis under acidic conditions.15 However, in two phase systems the imine bond can show remarkable resistance to hydrolysis, depending on the hydrophobicity of the reagent. Ketoximes appear to be more stable than aldoximes and the phenolic oximes are more stable than the aliphatic α-hydroxyoximes such as LIX 63.
Whilst used mainly in Cu recovery, phenolic oximes can be used to separate Pd(II) and Pt(IV). In this case, the former is extracted as a 2:1 complex, [Pd(L)n], at pH ≥ 3.0 whilst the kinetic inertness of Pt(IV) ensures that it is only loaded very slowly and so remains in the aqueous phase.16
Extraction of Mo(VI) and U(VI) by the aliphatic reagent, 5,8-diethyl-7-hydroxydodecan-6-one oxime (LIX 63), has been shown to involve the complexation of the metal cation MO22+, forming the neutral complex [MO2(LIX63-H)2] in which two reagent molecules are deprotonated.17 Eu(III) and Th(IV) can be recovered using 3,5-di-tert-butyl-2-hydroxy-benzaldehyde oxime, which is structurally similar to commercially available salicylaldoximes. The stoichiometry of [EuCl2(L)] was inferred from plots of logD against the log [L] (where D = distribution coefficient = [M]org/[M]aq), whereas, under the same conditions, Th(IV) forms [ThCl(L)3] in the water-immiscible phase.18 The presence of both the extractant anion (L−) and an anion from the aqueous feed solution (chloride) indicates that these latter extractions do not proceed by the simple cation exchange process shown in eqn (3), but display characteristics of the metal salt extraction processes (see below).
Stripping of loaded hydroxyoximes is usually achieved by contacting the loaded organic phase with an acidic aqueous solution (see Scheme 1). Controlling the pH can allow selective stripping of phenolic oximes which have been loaded with more than one metal, e.g. the more weakly bound Ni(II) can be completely transferred from a Cu(II)/Ni(II)-loaded solution of LIX 84 at pH 3, whereas the copper remains in the organic phase until the pH of the aqueous phase is lowered further.19
Phosphinic, phosphonic and phosphoric acid extractants
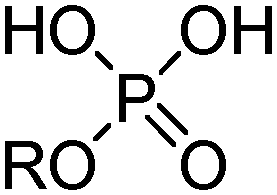
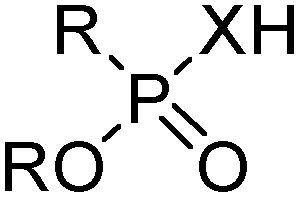
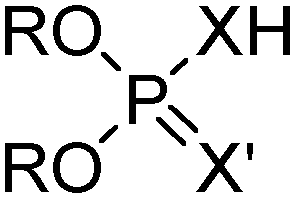
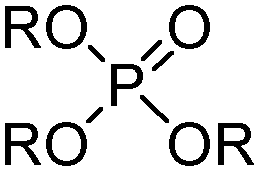
Commercial extractants based on phosphinic, phosphonic acids and phosphoric acids are listed in Table 2.§ These have one ionisable proton, with the exception of the phosphate monoesters, which have two. Their metal donor atoms (X = O or S) can be varied to suit the hard–soft properties of different metals.11
| Monoalkylphosphoric acids | ||
|---|---|---|
|
|
||
| Substituents | Commercial names | |
| R | n-C4H9(C2H5)CHCH2 | MEHPA |
| Dialkylphosphonic acids | ||
|---|---|---|
|
|
||
| Substituents | Commercial names | |
| R | n-C4H9(C2H5)CHCH2 | PC 88A, P507, Ionquest 801 |
| Dialkylphosphinic acids and derivatives | ||
|---|---|---|
|
|
||
| Substituents | Commercial names | |
| R | t-C4H9CH2(CH3)CHCH2 | Cyanex 272 |
| X = X′ | O | |
| R | t-C4H9CH2(CH3)CHCH2 | Cyanex 302 |
| X | O | |
| X′ | S | |
| R | t-C4H9CH2(CH3)CHCH2 | Cyanex 301 |
| X = X′ | S | |
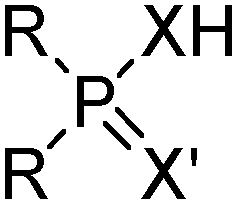
| Dialkylphosphoric acids and derivatives | ||
|---|---|---|
|
|
||
| Substituents | Commercial names | |
| R | n-C4H9(C2H5)CHCH2 | D2EHPA, DEHPA, P204, Baysolvex DEDP |
| X = X′ | O | |
| R | n-C4H9(C2H5)CHCH2 | Hoe F 3787 |
| X | O | |
| X′ | S | |
| R | n-C4H9(C2H5)CHCH2 | DEHTPA |
| X = X′ | S | |
A characteristic of the phosphorus(V) acid extractants is that they form stable dimeric structures in non-polar solvents (see Fig. 5), similar to those of carboxylic acids (see below). When the extractant is present in excess, formation of metal complexes involves the retention of one of the hydrogen bonds in the dimer, whilst the other is broken to release a proton which is replaced by the metal cation; this leads to the formation of an 8-membered pseudo-chelate (see Fig. 5). Extraction of M2+ cations of the base metals by the commercial reagent Cyanex 272 and related phosphoric, phosphonic and phosphinic acids (see Table 2), usually gives complexes with 4:1 ligand:metal stoichiometry, [M(L)2(LH)2]. The 8-membered, hydrogen-bonded rings in these complexes are able to subtend O–M–O angles greater than 90° and may contribute to these reagents showing selectivity for base metals which adopt tetrahedral coordination geometries. The ease of extraction of M2+ cations of the first transition series reflects this, and follows the sequence Zn > Cu > Mn > Co > Ni (see Fig. 6) which is not the Irving–Williams order of stability. The high affinity for zinc by the phosphoric acid D2EHPA is the basis for its application in a recently commissioned plant for the recovery of zinc on a 150000 tonne p.a. scale.20
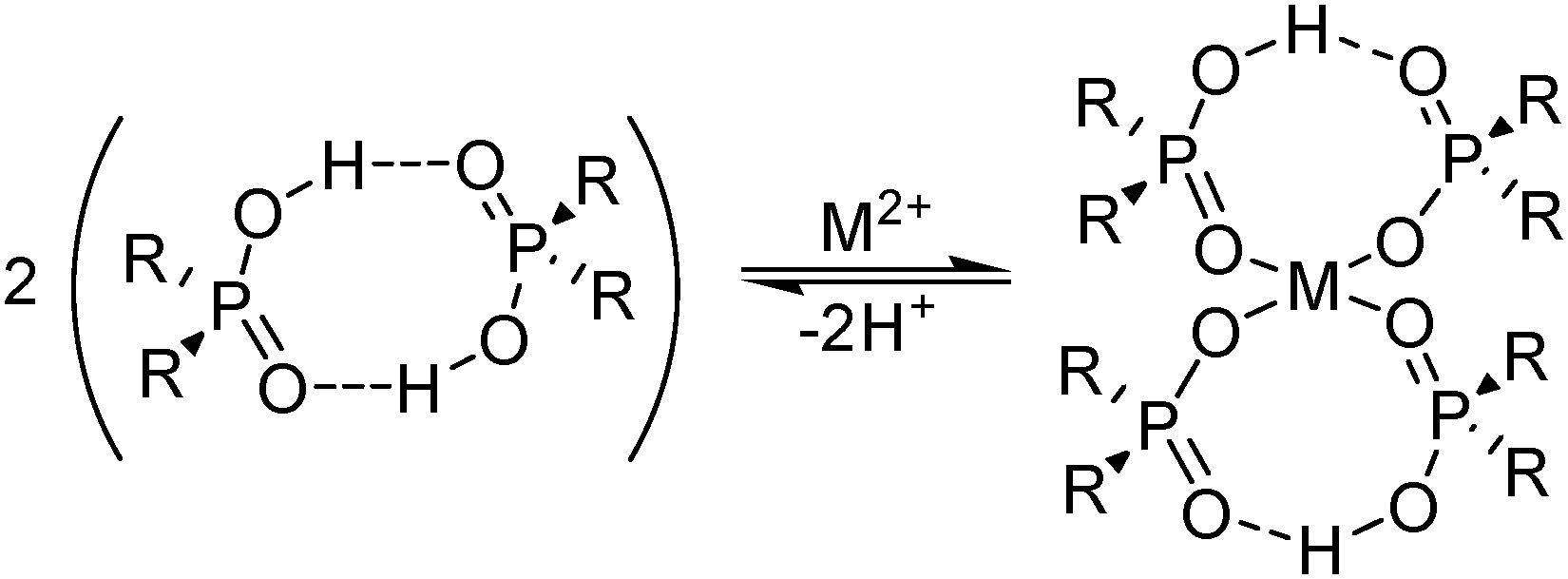 | ||
| Fig. 5 Complex formation equilibrium involving phosphinic acids. | ||
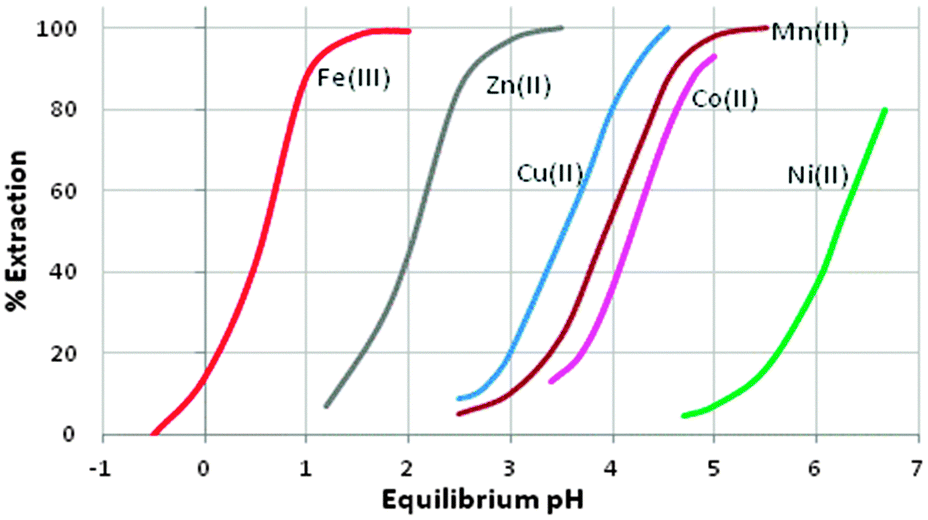 | ||
| Fig. 6 The pH-dependence of metal cation loading by Cyanex 272; data are adapted from ref. 22. | ||
The 8-membered pseudo-chelate motif has been identified in X-ray crystal structure determinations, e.g. in the complex [Ni(Ph2PO2)2(Ph2PO2H)2(DMF)2] in which diphenylphosphinic acid is used as a model for Cyanex 272 (Fig. 7).21
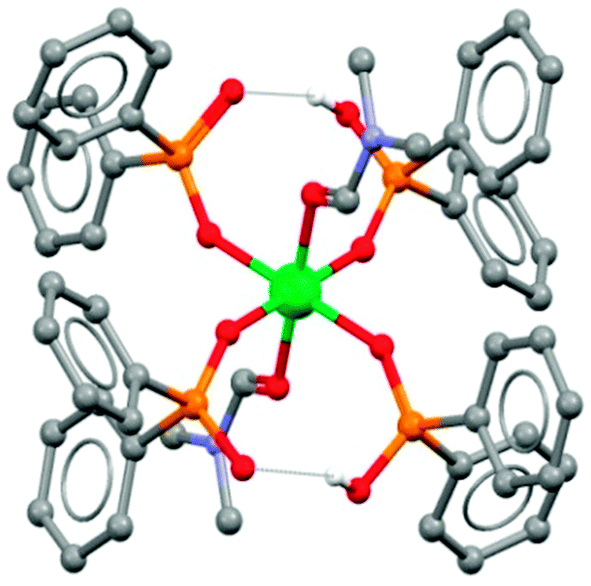 | ||
| Fig. 7 The structure of [Ni(Ph2PO2)2(Ph2PO2H)2(DMF)2] from single crystal X-ray diffraction.21 | ||
Phosphorus(V) acids have been shown to be effective in the recovery of a wide range of metals, particularly d- and f-block elements, the latter often in synergistic mixtures (see below). D2EHPA extracts MoO22+ in a 4:1 L:M ratio, possibly forming a complex similar to that in Fig. 5 with the Mo(VI) atom retaining two cis-oxo groups in a six coordinate structure. Cyanex 600, a variant of Cyanex 272 (Table 2) has been shown to extract Mo from acidic solutions that contain Cu, Fe and Al cations.23
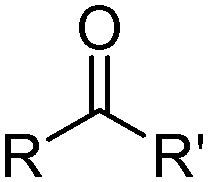
Carboxylic and other organic acids
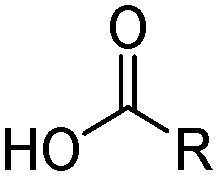
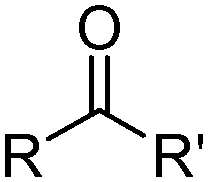
Carboxylic acids, like the phosphorus(V) acids have a propensity to associate in low polarity solvents but in this case this leads to more complicated speciation than would be inferred from the equilibrium shown in eqn (3). The carboxylic acid dimer, [RCO2H]2, shown in Scheme 3, is particularly stable in hydrocarbon solvents. One or both of the protons can be replaced by metal cations to generate mono- or dinuclear complexes and remaining coordination sites can then can accommodate neutral carboxylic acid or water molecules.11 Consequently, a large number of species of the formula [M(RCO2)n(RCO2H)m(H2O)p]x can coexist in the water-immiscible phase, with the charge on the extracted action, Mn+, being counterbalanced by an appropriate number of carboxylate anions.24 These variations of speciation contribute to the carboxylic acids showing poor selectivity as metal extractants and they are much weaker pH swing extractants than the phosphorus(V) acids. Like the hydroxamic acids (Table 3), they cannot be used to process aqueous feed solutions of base metals without first precipitating all of the iron(III) because they form very stable complexes of this cation. Hydrophobic, straight-chain carboxylic acids are good surfactants and their propensity to form soaps leads to problems in phase disengagement in solvent extraction processes. Reagents which are used in metal recovery, e.g. Versatic acid (Table 3,§) have branched alkyl chains to overcome this problem.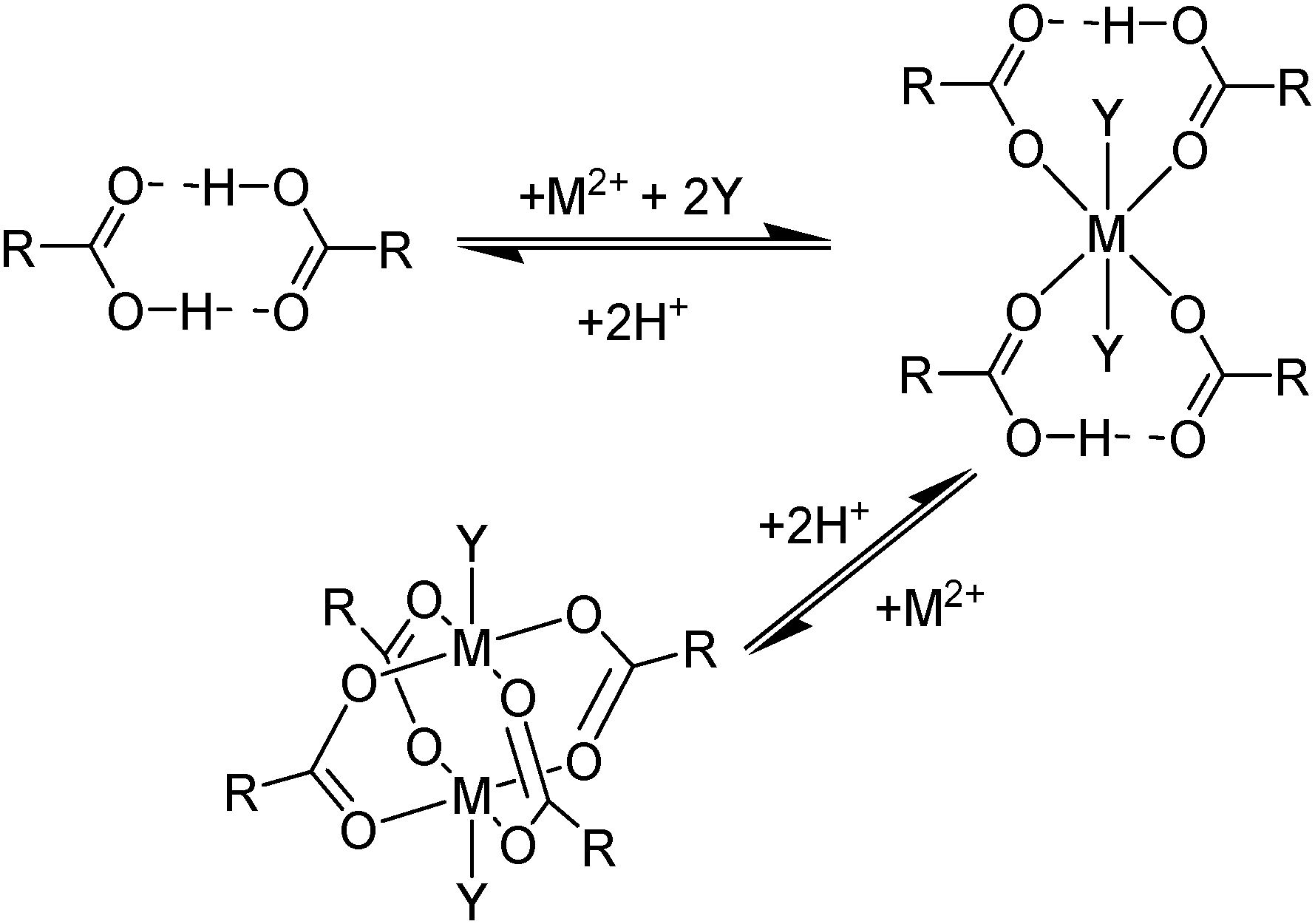 | ||
| Scheme 3 Some of the neutral complexes formed on extraction of M2+ cations by hydrophobic carboxylic acids (Y = H2O or RCO2H). | ||
11
| Substituents | Commercial name(s) | ||
|---|---|---|---|
a Gravimetric reagents containing 8-hydroxyquinoline are often referred to as oxines. These should not be confused with oximes which contain a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH unit (see Table 1). N–OH unit (see Table 1).
|
|||
|
R | Branched aliphatic groups (C9H19) | Versatic acid 10 |
|
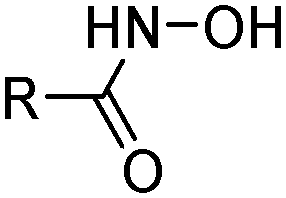
R | Branched aliphatic groups | LIX 1104 |
|
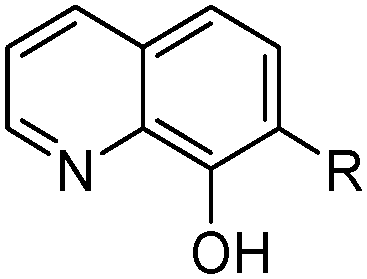
R | i-C11H23 | Kelex 100a |
The hydroxamic acid LIX 1104 (Table 3) has been used to extract Fe, Co, Ni, Cu, Zn and Cd, loading at pH 3–7;25 Cu(II) can be stripped with ca. 1 M sulfuric acid, whilst Fe(III) requires 4 M acid. Deprotonation to give the hydroxamate generates an O2− chelating donor set which is known to form extremely stable Fe(III) complexes similar to the highly stable, iron-chelating siderophores found in nature.
8-Hydroxyquinolines (LH) have a longstanding record of use in gravimetric and colorimetric analysis, yielding stable neutral complexes [M(L)n] with metal di-, tri-, and tetracations when solutions are buffered to favour the deprotonation of the hydroxyl group. The hydrocarbon-soluble reagent Kelex 100 (Table 3) can be used to recover gallium from spent Bayer liquor in which it is present in concentrations of 100–300 mg L−1.26 The extraction is undertaken on the alkaline liquor after alumina has been precipitated and involves the reaction shown in eqn (5). A significant amount of aluminium and sodium are co-extracted, but these are selectively stripped by contacting with 6 M HCl leaving only gallium in the organic phase as [(LH2)GaCl4], a neutral assembly of tetrachloridogallium(III) and monoprotonated Kelex 100. Reagents specifically designed to extract metalate anions are discussed below.
| [Ga(OH)4]− + 3LH(org) ⇌ [M(L)3](org) + OH− + 3H2O | (5) |
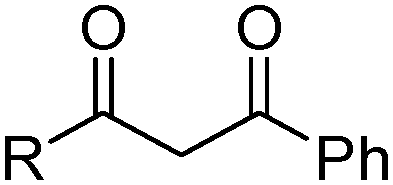
β-Diketones (Table 4,§) are weak extractants which operate under basic conditions that promote the formation of the bidentate monoanionic ligands as in Fig. 8.27,28
11
| β-Diketones | ||
|---|---|---|
|
|
||
| Substituents | Commercial name(s) | |
| R | i-C7H15 | LIX 54 |
| R | C(CH3)(C2H5)C3H7 | XI-N54 |
| R | CH(C2H5)C3H7 | XI-55 |
| R | C(CH3)2C6H13 | XI-57 |
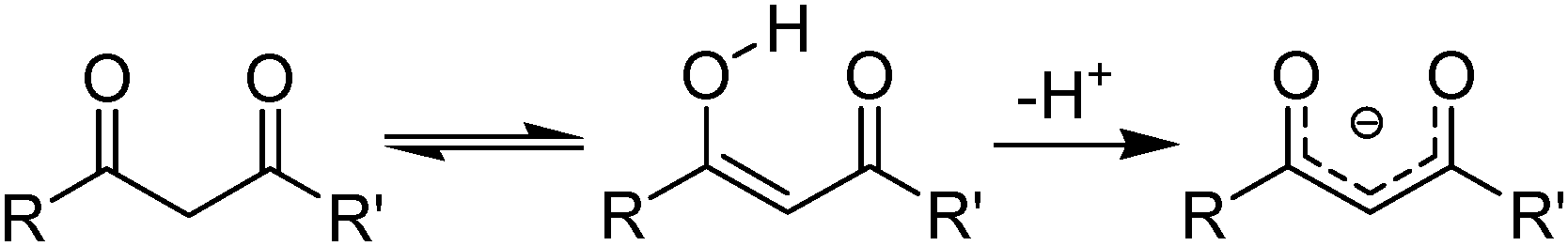 | ||
| Fig. 8 Keto and enol tautomers of 1,3-diketones which can be deprotonated to form monoanionic ligands. | ||
LIX 54 was introduced as a commercial reagent for the recovery of copper from ammoniacal solutions.29 The 2:1 complex in the extraction process shown in eqn (6) is assumed to have a planar structure analogous to that of the acetylacetonate complex shown in Fig. 9.30 The aqueous raffinate contains a mixture of ammonium ions and ammonia which is recycled back to the leaching of chalcocite, Cu2S (eqn (7)) to generate the ammine complex and enriched covellite (CuS) that is further processed in a conventional smelter.
| [Cu(NH3)4]2+ + 2LH(org) ⇌ [Cu(L)2](org) + 2[NH4]+ + 2NH3 | (6) |
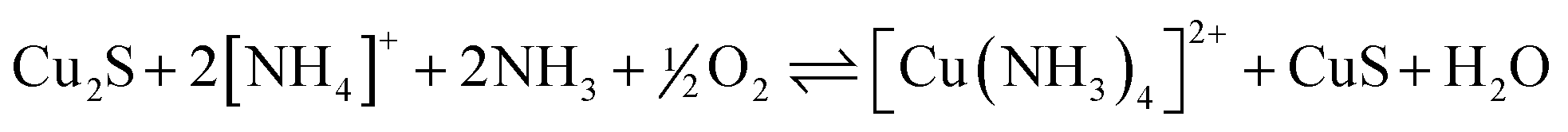 | (7) |
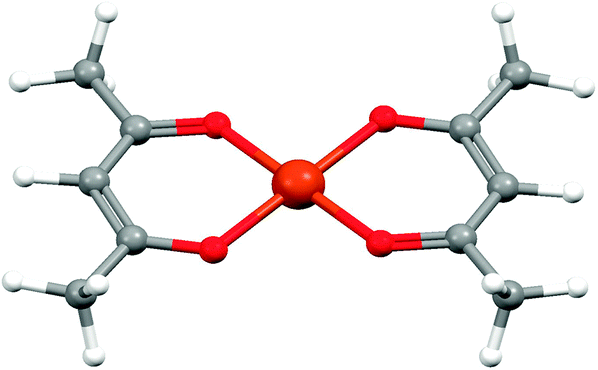 | ||
| Fig. 9 The structure of bis(acetylacetonato)copper(II) from single crystal X-ray diffraction.30 | ||
Metalate anion extraction
A number of leaching processes generate solutions in which the metals are present as metalate anions, MXxn−. This is particularly the case for the chloridometalates of the platinum group metals for which oxidative leaching in acidic chloride media is the most practicable approach to dissolution.22 Also, the efficiency of recently developed chloride-leaching processes for base metals12 makes it practicable to recover these metals from acidic chloride streams, despite these being more corrosive than the more common sulfate-based streams.The use of solvent extraction to concentrate and separate the metal values which are present as metalate anions requires the formation of neutral assemblies (often referred to as “ion pairs”) in the water immiscible phase. This can be achieved in two ways. Mixing a solution of a neutral extractant with an acidic aqueous solution can lead to protonation of the extractant and the “pH-swing” process shown in eqn (2) in which loading is favoured by lowering the pH of the aqueous phase and stripping by raising pH.12 Amines, ketones and phosphine oxides (Table 5,§) have been used commercially in this way. Alternatively, an extractant that carries a permanent positive charge can be employed in an anion exchange process, eqn (8). In this case, loading and stripping are influenced by variation of the concentration of the counteranion Y− in an “anion-swing” process.12
| n(LY)(org) + MClxn− ⇌ [(L)nMClx](org) + nY− | (8) |
11
| Primary amines (H2NR) | |
|---|---|
| Substituent(s), R | Commercial name(s) |
| C16–22H34–46 (mixture of isomers) | Primene JM-T |
| C12–14H26–30 (mixture of isomers) | Primene 81-R |
| Secondary amines (HNRR′) | ||
|---|---|---|
| R = R′ | C13H27 | Ditridecylamine, HOE 2652 |
| R | C12H25 | Amberlite LA-2 |
| R′ | C12–14H25–29 | |
| R = R′ | C8H17 (mixture of isomers) | Adogen 283 |
| Tertiary amines (NR3) | |
|---|---|
| C8H17 | TOA, Alamine 300 |
| C8H17 and C10H21 (2:1) |
Alamine 336 |
| C8H17 (mixture of isomers) | Alamine 308 |
| C8H17 (2-ethylhexyl) | TEHA |
| C10H21 (mixture of isomers) | Alamine 310 |
| C12H25 | Alamine 304 |
| Quaternary amines (NR3R′X) | ||
|---|---|---|
| R | C8H17 and C10H21 (2:1) |
Aliquat 336 |
| R′ | CH3 | |
| X | Counter-ion | Cl− |
| Phosphoric acid esters and phosphine oxides | |||
|---|---|---|---|
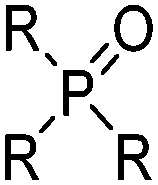
|
R | O-n-C4H9 | TBP |
| R | C8H17 | TOPO | |
| Ketones and ethers | |||
|---|---|---|---|
|
R | Me | MIBK |
| R′ | i-C4H9 | ||
| R = R′ | i-C4H9 | DIKB | |

|
DBC | ||
Designing organic reagents to give high selectivity of extraction of metalate anions is challenging because the extractants do not enter the inner coordination sphere and consequently it is not possible to exploit a metal's preference for a particular coordination geometry or donor atom type. As transfer of a metalate anion into a non-polar, water-immiscible, solvent requires removal of most or all of the hydration sphere, it is generally more difficult to extract small, highly charged, anions. This phenomenon is well documented and has been referred to as the Hofmeister bias because it is related to the Hofmeister Series that was established in 1888 to rank the efficacy of inorganic anions in the coagulation of proteins.31
Solvent extraction processes to separate metalate anions on the basis of their charge are easier to implement in practice when the metals are kinetically inert because they do not lose coordinated anions to generate lower charged, more readily extractable species during the contact time in a mixer settler. An example is the use of tri-n-butylphosphate (TBP, Table 5) to recover Pt(IV) selectively as its dianion PtCl62− from acidic chloride streams which contain Ir(III) and Rh(III) as their trianions IrCl63− and RhCl63−. In these processes, it is important to maintain the oxidation potential of the feed solution22 to ensure that the Ir(III) is not converted to the more readily extracted Ir(IV) complex, IrCl62−. Hydrophobic amines (Table 5) are very efficient extractants for PGM chloridometalates, but their high basicity makes it more difficult to strip the loaded solution; this latter process can be accompanied by the irreversible formation of inner sphere complexes as in eqn (9).
Formation of such inner sphere complexes is less of a problem when tertiary amines of the Alamine type (Table 5) are used as extractants, presumably as a consequence of steric hindrance.22 Much more weakly basic alkyl amides have been used for the extraction of Pt(IV) from acidic chloride streams and have the advantage of being much more readily stripped. Tertiary amines in the Alamine® series, particularly Alamine 336, have been shown to be useful in the separation and extraction of uranium as (R3NH)4UO2(SO4)3 from sulfate leach solutions.20,32 This provides an alternative to conventional uranium extraction from nitrate streams by tributyl phosphate (TBP) in the PUREX process, which is best described as neutral metal-salt extraction.
| [LH]2[PtCl6](org) + Na2CO3 → [(L)2PtCl4](org) + CO2 + H2O + 2NaCl | (9) |
The Hofmeister bias underpins the efficient recovery of gold as its monoanion, AuCl4−, from mixed-metal chloride streams and several of the reagents listed in Table 5 have been used in gold recovery, including the triether dibutylcarbitol (DBC) which has been exploited by a number of Platinum Group Metal (PGM) refiners. Whilst this gives good separation from most of the precious metals, it is not very selective over some of the metalloid elements (see Fig. 10), particularly at high HCl concentrations, presumably because under these conditions other monoanionic chloridometalates are present.22 DBC and methylisobutyl ketone (MIBK, Table 5), which is used extensively in gold recovery from chloride solutions, are fairly soluble in aqueous HCl solutions presenting problems of loss of reagent in continuous operations.
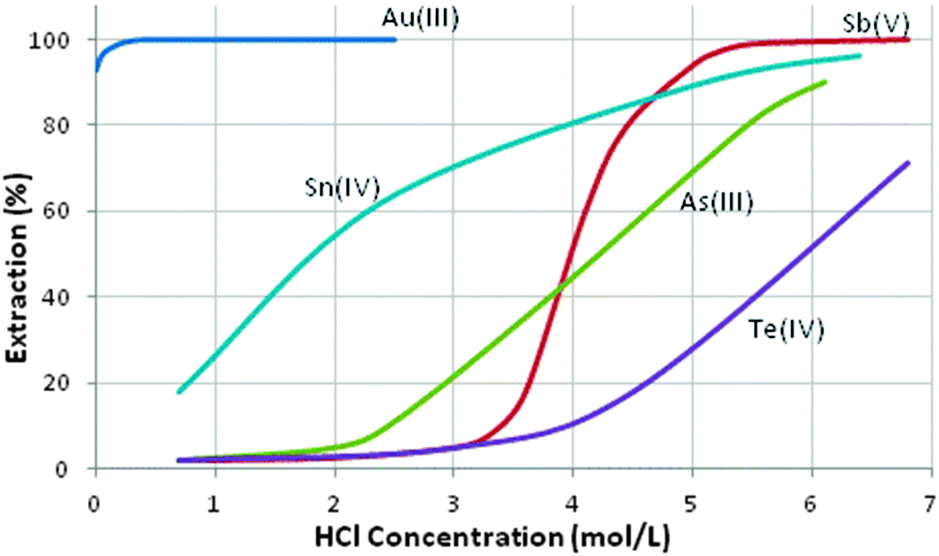 | ||
| Fig. 10 Selectivity of gold extraction with dibutylcarbitol.22 | ||
Because metalate extraction involves the transfer of preformed complexes from the aqueous phase, conventional (inner sphere) coordination chemistry does not play a part determining the selectivity of binding of the metalate anions to the organic cations in the assemblies (ion pairs) that form in the organic phase. However, the relative stability of the metalates formed initially in the aqueous phase does contribute to the distribution coefficients for extraction. This is exploited to great effect in the separation of Co(II) from Ni(II) in the Outokumpu process.33 Dissolution of a Co/Ni matte in chloride solutions generates CoCl42− which is significantly more stable than its nickel analogue, NiCl42−, and consequently a clear separation can be effected using Alamine reagents because the overall process represented by eqn (10) and (11) is more favourable when M = Co.
| MCl2 + 2Cl− ⇌ [MCl4]2− | (10) |
| [MCl4]2− + 2H+ + 2R3N(org) ⇌ [(R3NH)2MCl4](org) | (11) |
In general, the structures of assemblies formed in commercial metalate extraction processes are not well understood. They involve electrostatic, hydrogen bonding, and other supramolecular interactions which have been discussed in a recent Feature Article.12 New types of metalate extractants have been developed to exploit the concepts of anion recognition and can overcome the Hofmeister bias, providing the possibility of opening up new flow sheets for metal recovery.12 For example, tris-2-ethylhexylamine (TEHA) is a model for the trialkylamine reagents Alamine 330 etc., and shows the expected selectivity for FeCl4− over ZnCl42− (Fig. 11). In contrast, the monoamido derivative L1 in Fig. 11 is a much stronger extractant for Zn(II), requiring a lower concentration of chloride to effect 100% recovery of Zn(II) compared to Fe(III). Such a reagent will allow the selective recovery of zinc from the oxidative chloride leaching of sulfidic ores or from pickling liquors from galvanising processes, both of which involve aqueous solutions with an excess of iron.
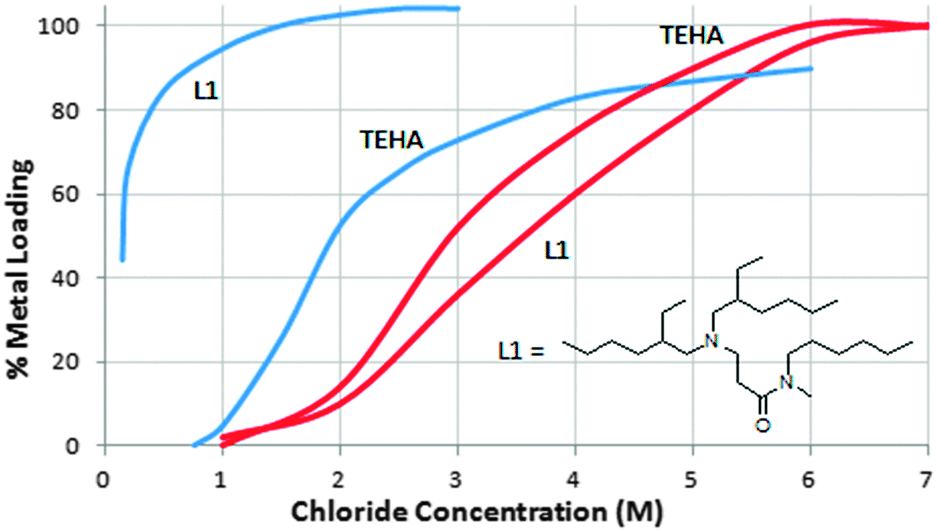 | ||
| Fig. 11 Loadings of [ZnCl4]2− (blue lines) and [FeCl4]2− (red lines) by TEHA and a closely related monoamido derivative (L1) as a function of chloride concentration in the aqueous feed. | ||
An alternative approach to the recovery of base metals from oxidative chloride leaching of sulfidic ores is to transfer the chloride salt to the water-immiscible phase, and is discussed in the following section.
Metal-salt extractants
These reagents transport the metal cation and its attendant anion(s) from aqueous solution as a salt, MXy, (eqn (1)) and, because they appear to make the salt soluble in the water immiscible phase, they are frequently referred to by metallurgists as “solvating extractants”.34 This historically useful terminology can be misleading because the reagent usually does not merely interact with the outer sphere of a neutral metal salt, MXy, to generate a hydrophobic species but also functions as an inner-sphere ligand for metal, as in the CUPREX and PUREX processes described below.35,36Attention also needs to be paid to the conditions under which the extractants listed in Tables 6 and 7§ are used. When contacted with highly acidic solutions of metal salts, the oxygen atom in the phosphate esters (e.g. TBP), the ketones (e.g. MIBK) or the phosphine oxides (e.g. Cyanex 923) can be protonated and the resulting cations can form ion-pair assemblies with metalate anions, transferring them to the water-immiscible phase by the mechanisms discussed above.
11
| Substituents | Commercial name(s) | ||
|---|---|---|---|
|
R | C10H21 | Acorga CLX50 |
|
R | COOC13H27 | Acorga ZNX50 |
|
R | n-C4H9 | TBP |
|
R | Me | MIBK |
| R′ | i-C4H9 | ||
| R = R′ | i-C4H9 | DIBK | |
11,37,38
| Phosphine | ||
|---|---|---|
|
|
||
| Substituents | Commercial name(s) | |
| R = R′ = R′′ | i-C4H9 | Cyanex 471 |
| X | S | |
| R = R′ = R′′ | CH3(CH2)7 | Cyanex 921, TOPO |
| X | O | |
| R = R′ = R′′ | i-CH3(CH2)7 | Cyanex 925 |
| X | O | |
| R = R′ = R′′ | CH3(CH2)5 and CH3(CH2)7 | Cyanex 923 |
| X | O | |
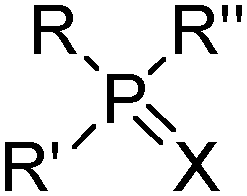
In the PUREX process, first developed for the recovery of plutonium and uranium as part of the Manhattan Project,10 two molecules of tri-n-butylphosphate (TBP, Table 6) bind to the uranyl cation in the equilibrium shown in eqn (12). This operates on a “nitrate-swing” in which the loading and stripping are controlled by variation of the concentration of nitric acid in the aqueous phase. Because it has a low viscosity and is fairly resistant to radiation damage, TBP can be used as the diluent, (the water-immiscible phase), and consequently TBP molecules are also present in the outer/solvation sphere.
| UO22+ + 2NO3− + 2TBP(org) ⇌ [UO2(NO3)2(TBP)2](org) | (12) |
The equatorial plane of the uranyl dication is defined by two bidentate nitrate ions and by two PO groups in the X-ray crystal structure shown in Fig. 12 of a model compound that contains the tri-iso-butyl analogue of TBP.39
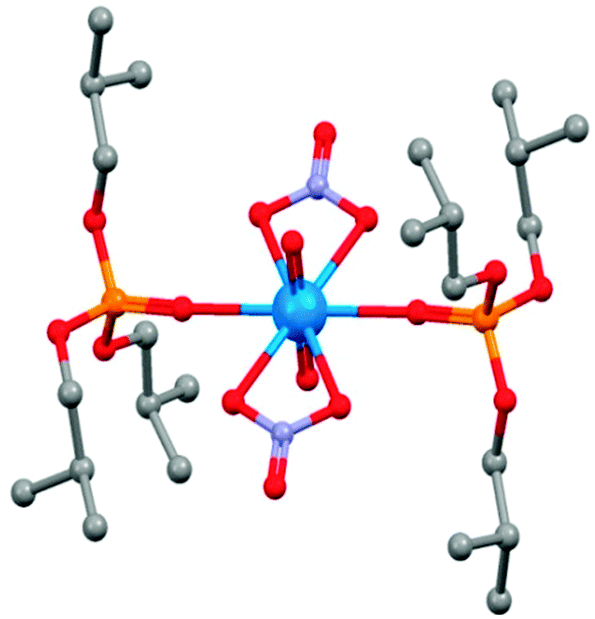 | ||
| Fig. 12 The structure of [UO2(NO3)2(iso-TBP)2] from single crystal X-ray diffraction.39 | ||
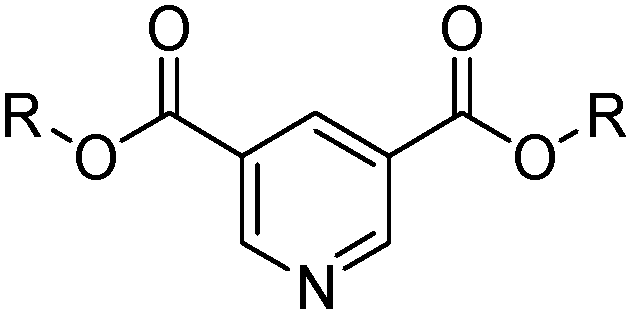
The CUPREX process was developed to recover copper from chloride solutions.35 It uses a neutral ligand, didecyl pyridine-3,5-dicarboxylate, CLX50 (Table 6) to generate the neutral, kerosene-soluble, four-coordinate copper complex [CuCl2(L)2] according to the reaction in eqn (13). Variation of chloride concentration and temperature are used to control copper loading and stripping.35
| Cu2+ + 2L(org) + 2Cl− ⇌ [CuCl2(L)2](org) | (13) |
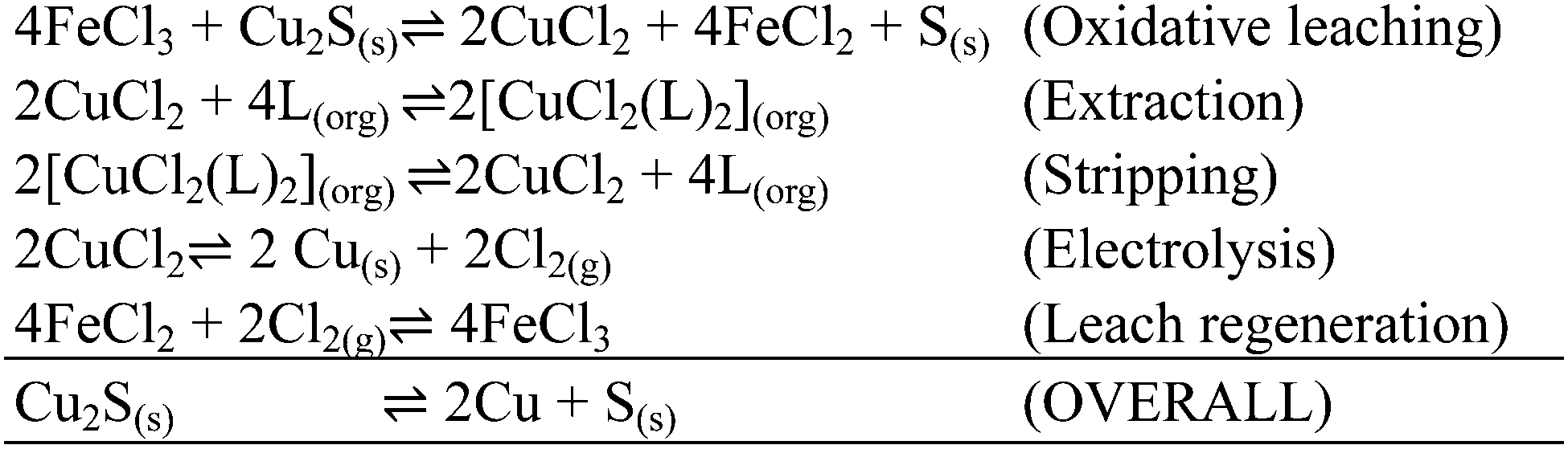 | ||
| Scheme 4 The equilibria and overall materials balance for the CUPREX process.35 | ||
The trialkylphosphine oxide reagent, Cyanex 923 (Table 7), extracts La(III) and Sm(III) from nitric acid solutions. Analysis of the distribution ratio data indicates that two molecules of Cyanex 923 are involved in the extracted species with the charge on the Ln(III) cations satisfied by the coordination of three nitrate anions.38
The best understood metal salt reagents (as in the examples above) operate with reagent molecules being bound in the inner coordination sphere of the metal cation and with the counter anions acting as ligands or contact ion pairs. This mechanism does not work well for extraction of sulfate salts because sulfate is a weak inner sphere ligand and is intrinsically difficult to transport from an aqueous solution because of its high hydration energy. To overcome these problems, some ditopic reagents have been designed to give separated ions pairs M2+ and SO42− with the reagent providing appropriate binding sites for each. The ditopic reagent HNT (Fig. 13) is a strong nickel sulfate extractant, operating in a zwitterionic form with a trianionic binding site for Ni2+ provided by the three salicylaldiminate units and a tricationic site for the SO42− provided by the protonated pendant amine groups. Nickel can be stripped almost quantitatively by contacting the loaded organic phase with dilute sulfuric acid giving a 98% recovery in a circuit comprising two extraction and one stripping stage.40 Commercial exploitation of this reagent for the recovery of nickel from pressure leaching of lateritic ores failed because the imine linkages are sensitive to hydrolysis under the acidic conditions preferred for stripping. Also, the reagent is not sufficiently selective for sulfate over chloride to generate an NiSO4 electrolyte of the purity required to operate a conventional sulfate electrowinning plant for nickel.40
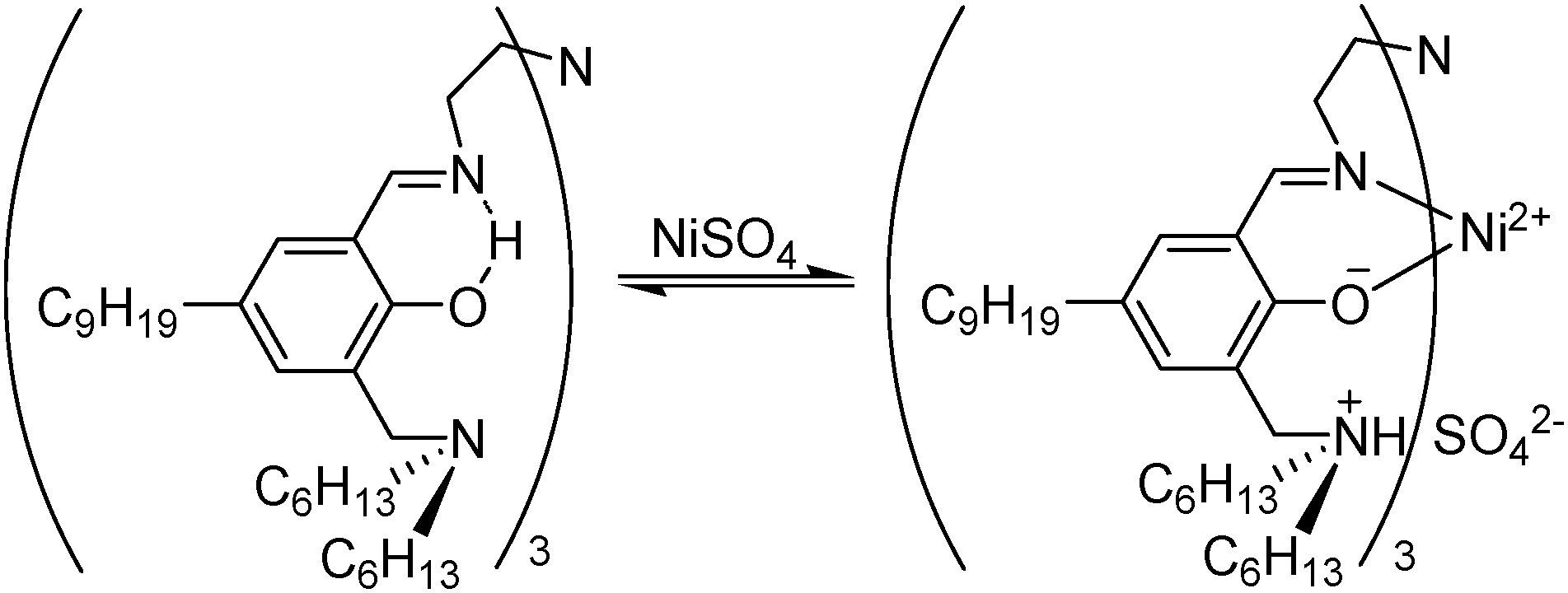 | ||
| Fig. 13 Uptake of NiSO4 by HNT to give a neutral complex [Ni(HNT)SO4] containing a zwitterionic form of the ligand. | ||
Synergistic and other extractants
In the metal-recovery processes discussed above, the modes of action of the extractants and the coordination chemistry involved in forming charge-neutral assemblies are well defined. The processes involve the transport of either a metal cation, M2+, a metalate anion, MYxn−, or a metal salt, MYx, into the water-immiscible phase. Some of the extractants, e.g. phosphine oxides and trialkylphosphates in Tables 6 and 7, can perform more than one of these roles, transporting metalate anions when they are in a protonated form and metal salts when they are in their neutral form. However, a tactic frequently used in extractive hydrometallurgy to achieve efficient separation and concentration of metals is to employ mixtures of solvent extractants.41 Most of these synergistic systems depend on the complementarity of reagents that show different modes of action to enhance extraction.Both the α-hydroxyoxime LIX 63 and the neodecanoic acid, Versatic 10 (see Tables 1 and 3), are weak Ni extractants (see Fig. 14), requiring several stages of extraction with interstage pH-adjustment to recover Ni effectively from sulfate streams. In contrast, a blend of these reagents is much stronger, recovering Ni at a pH1/2 value more than two units lower than with the individual reagents.42 In general, the origin of the synergism in blends of extractants is not well defined. In the case of the LIX63/Versatic 10, early work suggested that it was the hydroxy-oxime which was deprotonated with the carboxylic acid functioning as a neutral solvating extractant. More recently, on the basis of the structures of model systems (see below), deprotonation of the Versatic acid to give a complex such as [Ni(RCOO·LIX63)2] is thought to be more likely.
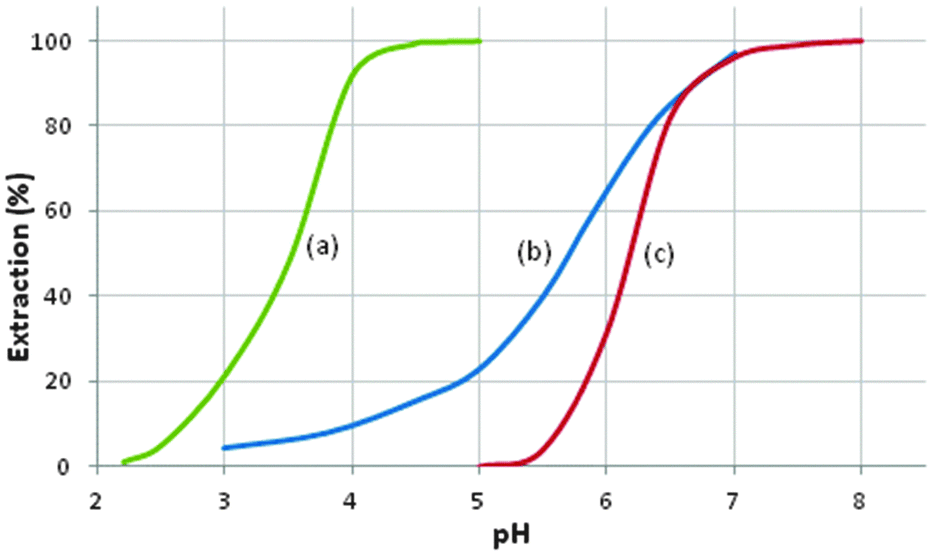 | ||
| Fig. 14 The pH dependence of Ni-loading from a solution typifying a laterite pregnant leach solution by: (a) a blend of Versatic 10 and LIX 63; (b) Versatic 10; (c) LIX 63.42 | ||
X-ray structure determinations of Ni(II) complexes formed by model carboxylic acids and α-hydroxyoximes that have aromatic or shorter aliphatic groups to enhance the formation of crystalline forms, support structures in which it is the carboxylic acid rather than the oxime that is deprotonated to balance the charge on the nickel dication (Fig. 15).43 Inter-ligand hydrogen bonding generates meridional tridentate [NO2]− donor sets with 5- and 7-membered chelate rings resulting from the N–OH group of the oxime unit acting as the hydrogen bond donor to the uncoordinated oxygen atom of the carboxylate. The importance of such H-bonding interactions in the outer coordination spheres of metal complexes has been recognised and can be incorporated into ligand design to tune the strength and selectivity of extractants.12
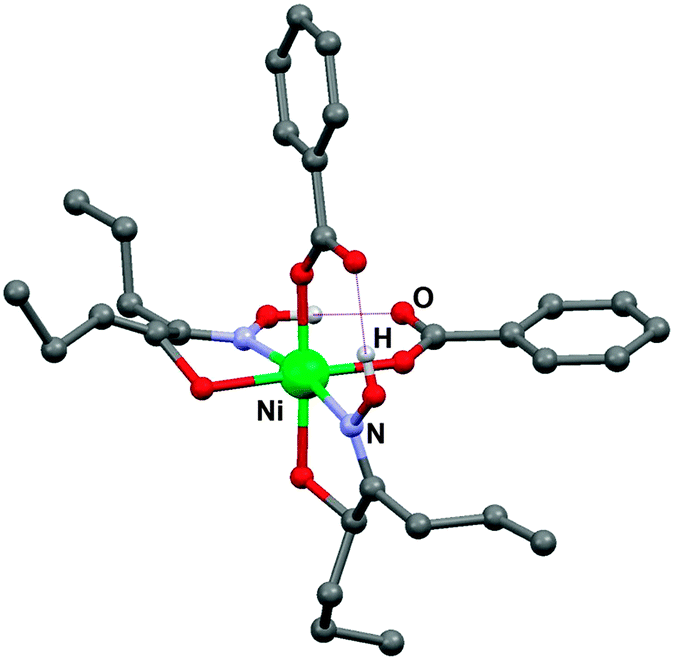 | ||
| Fig. 15 Molecular structure of a Ni(II) complex containing a di-n-propyl analogue of LIX63 (R = CH3CH2CH2–) and benzoic acid instead of Versatic acid from single crystal X-ray diffraction.43 | ||
There are many examples of synergistic systems being used in the extraction of f-block elements, particularly those involving phosphorus(V) acids.44,45 Light rare earth elements can be separated using blends of extractants such as PC 88A, Cyanex 302 and D2EHPA, and while there is some information on the stoichiometries of the assemblies formed, their structures are not as well defined as the Ni(II) systems described above.46
In some cases the principal role of the extractant is not as a ligand but as a surfactant, stabilising reverse micelles in which the metal is found in the “water pool”. Extraction of metalate anions by this mechanism is shown schematically in Fig. 16. Recent work on the extraction of lanthanides by malonamides provides important information on the composition and structures of the reverse micelles formed.47 An understanding of the supramolecular chemistry involved in forming such large assemblies is key to defining the origin of the chemical potential which favours the transfer of the metal-containing species from a homogenous aqueous phase into the water pool in the reverse micelle.48
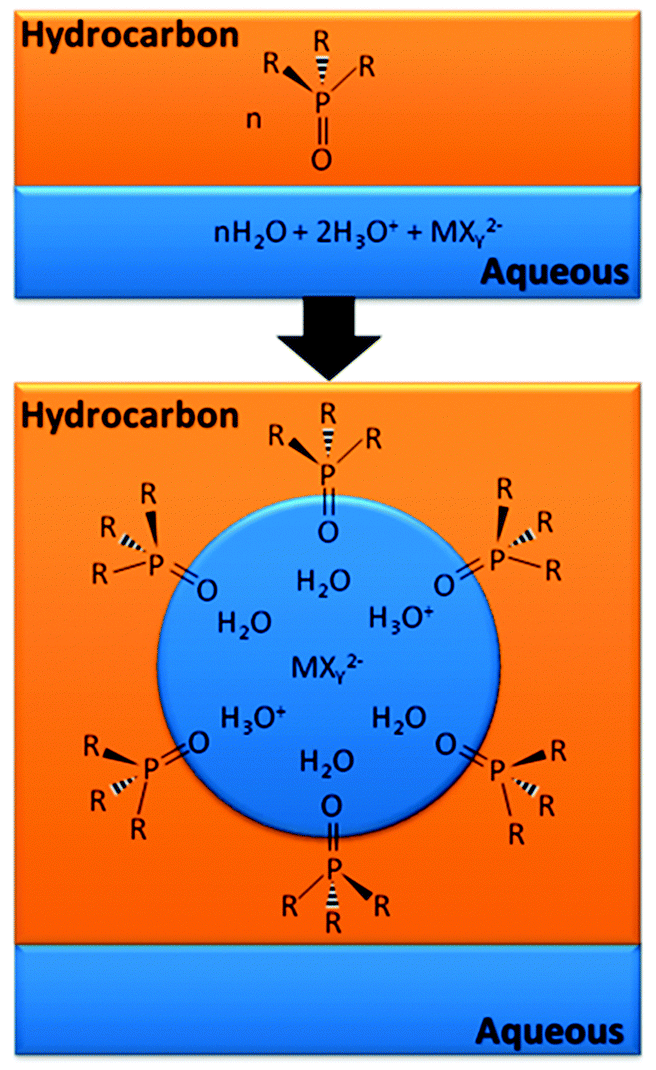 | ||
| Fig. 16 Reverse micelle formation by protonated and neutral phosphine oxide extractants with a metalate anion in the acidic “water pool”.49 | ||
Conclusions
Solvent extraction of d- and f-block metals provides many examples where ligand design can be used to great effect to achieve the separation and concentration operations in extractive hydrometallurgy. This is particularly true when the extractants operate in the inner coordination sphere of the metal ion, and high strength and selectivity of complex formation can be controlled by ensuring that the donor atom types and dispositions meet the requirements of the targeted metal. It has also been recognised that interactions between ligands in the outer sphere can be used to tune the strength and selectivity of extraction. The importance of these secondary bonding interactions arises because the water-immiscible solvents usually have very low polarities which favour formation of outer-sphere interactions such hydrogen bonds. Indeed, when the extractants transport metalate anions, MYxn−, the inner coordination sphere usually remains unchanged and it is only variations in the effectiveness of the secondary bonding which can give rise to selectivity and strength of recovery.Whilst this review has focused on the role of coordination chemistry in extractant design, many of the processes in use and which operate on kilotonne scales also depend on supra-molecular chemistry to ensure that the rates of transport between liquid phases are high and that phase disengagement is efficient. It is challenging to determine the structures of assemblies which are responsible for these processes at interfaces, and there is often also ambiguity concerning their structure in the bulk of the water-immiscible phase. As a consequence, it should be recognised that whilst the classification of extractants into the three types that we have used above is convenient for analysing the function of reagents, it does not preclude extractants functioning in more than one role, nor does it deal with systems where synergistic mixtures of extractants are used. For information on more detailed subclassification of modes of extraction, reviews in more specialist publications are recommended.31
Acknowledgements
We thank the EPSRC (UK), Cytec Industries Inc., (studentship JRT), Johnson Matthey and Anglo American (studentship AMW) for funding, Mary Healy and Kirsty MacRuary for helpful discussions and Kathryn Sole, Adam Fischmann and Violina Cocalia for advice and for providing data for Fig. 4, 6 and 10.Notes and references
- A history of metallurgy, ed. I. Weibull, 2nd edn, The Institute of Materials, 1993 Search PubMed.
- D. Killick and T. Fenn, Annu. Rev. Anthropol., 2012, 41, 559–575 CrossRef PubMed.
- P. Gramatyka, R. Nowosielski and P. Sakiewicz, J. Achiev. Mater. Manuf. Eng., 2007, 20, 535–538 Search PubMed.
- T. E. Graedel, J. Allwood, J.-P. Birat, M. Buchert, C. Hageluken, B. K. Reck, S. F. Sibley and G. Sonnemann, J. Ind. Ecol., 2011, 15, 355–366 CrossRef CAS.
- A. Tajani, Critical Raw Materials for the EU, http://ec.europa.eu/enterprise/policies/raw-materials/files/docs/report-b_en.pdf, accessed 22 July, 2013.
- DEFRA, Resource security action plan: making the most of valuable materials, 2012.
- D. Bauer, D. Diamond, J. Li, D. Sandalow, P. Telleen and B. Wanner, Critical Minerals Strategy, U.S. Department of Energy, 2010 Search PubMed.
- RSC, Resources that Don't Cost the Earth, 2011.
- J. Rankin, 4th High Temperature Processing Symposium, Swinburne University of Technology, Melborne, Australia, 2012.
- F. Habashi, Hydrometallurgy, 2005, 79, 15–22 CrossRef CAS PubMed.
- Comprehensive Coordination Chemistry II, ed. P. A. Tasker, P. G. Plieger and L. C. West, Elsevier Ltd, Oxford, 2004 Search PubMed.
- J. R. Turkington, P. J. Bailey, J. B. Love, A. M. Wilson and P. A. Tasker, Chem. Commun., 2013, 49, 1891–1899 RSC.
- D. S. Flett, J. Chem. Technol. Biotechnol., 1999, 74, 99–105 CrossRef CAS.
- G. A. Kordosky, 18th International Solvent Extraction Conference, Cape Town, South Africa, 2002.
- S. F. Woollam and R. A. Grant, Solvent Extr.: Fundam. Ind. Appl., Proc. ISEC 2008, Int. Solvent Extr. Conf., Tuscon, Arizona, USA, 2008.
- M. V. Rane and V. Venugopal, Hydrometallurgy, 2006, 84, 54–59 CrossRef CAS PubMed.
- M. H. H. Mahmoud, S. Nakamura and K. Akiba, Anal. Sci., 1997, 13, 149–152 CrossRef CAS.
- N. Sehati, Z. Shiri-Yekta, A. A. Zamani, M. R. Yaftian and N. Noshiranzadeh, Sep. Sci. Technol., 2012, 47, 670–676 CrossRef CAS.
- G. A. Kordosky, W. H. Champion, J. R. Dolegowski, S. M. Olafson and W. S. Jensen, Miner. Eng., 1981, 33, 291–299 CAS.
- K. C. Sole, A. M. Feather and P. M. Cole, Hydrometallurgy, 2005, 78, 52–78 CrossRef CAS PubMed.
- J. Langer, H. Görls, G. Gillies and D. Walther, Z. Anorg. Allg. Chem., 2005, 631, 2719–2726 CrossRef CAS.
- K. C. Sole, in Solvent extraction and liquid membranes: fundamentals and applications in new materials, ed. M. Aguilar and J. L. Cortina, Taylor & Francis, 2008, pp. 141–200 Search PubMed.
- V. Cocalia, T. Bednarski, D. J. Harris, H. Yáñez, A. Soto, M. Soderstrom and E. Kamenetzky, 19th International Solvent Extraction Conference, Santiago, Chile, 2011.
- H. Yamada and M. Tanaka, Adv. Inorg. Chem. Radiochem., 1985, 29, 143–168 CrossRef CAS.
- B. K. Tait, K. E. Mdlalose and I. Taljaard, Hydrometallurgy, 1995, 38, 1–6 CrossRef CAS.
- Z. Zhao, Y. Yang, Y. Xiao and Y. Fan, Hydrometallurgy, 2012, 125–126, 115–124 CrossRef CAS PubMed.
- F. J. Alguacil and A. Cobo, Hydrometallurgy, 1998, 48, 291–299 CrossRef CAS.
- G. A. Kordosky, M. J. Virnig and P. Mattison, 18th International Solvent Extraction Conference, Cape Town, South Africa, 2002.
- G. Kyuchoukov, M. B. Bogacki and J. Szymanowski, Ind. Eng. Chem. Res., 1998, 37, 4084–4089 CrossRef CAS.
- P. C. Lebrun, W. D. Lyon and H. A. Kuska, J. Crystallogr. Spectrosc. Res., 1986, 16, 889–893 CrossRef CAS.
- B. A. Moyer, P. V. Bonnesen, R. Custelcean, L. H. Delmau and B. P. Hay, Kem. Ind., 2005, 54, 65–87 CAS.
- G. Ramadevi, T. Sreenivas, A. S. Navale and N. P. H. Padmanabhan, J. Radioanal. Nucl. Chem., 2012, 294, 13–18 CrossRef CAS.
- O. Hyvarinen, M. Hamalainen and R. Leimala, 32nd Annual Hydrometallurgy Meeting, Montreal, Canada, 2002, p. 609.
- V. K. Manchanda, P. N. Pathak and P. K. Mohapatra, in Ion Exchange and Solvent Extraction, ed. B. A. Moyer, CRC Press, Boca Raton, 2010, vol. 19, pp. 69–71 Search PubMed.
- R. Dalton, G. Diaz, R. Price and A. Zunkel, JOM, 1991, 43, 51–56 CrossRef CAS.
- A. P. Paiva and P. Malik, J. Radioanal. Nucl. Chem., 2004, 261, 485–496 CrossRef CAS.
- E. Dziwinski and J. Szymanowski, Solvent Extr. Ion Exch., 1999, 17, 1515–1522 CrossRef CAS.
- Y. A. El-Nadi, N. E. El-Hefny and J. A. Daoud, Solvent Extr. Ion Exch., 2007, 25, 225–240 CrossRef CAS.
- J. H. Burns, G. M. Brown and R. R. Ryan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1446–1448 CrossRef.
- R. A. Coxall, L. F. Lindoy, H. A. Miller, A. Parkin, S. Parsons, P. A. Tasker and D. J. White, Dalton Trans., 2003, 55–64 RSC.
- G. M. Ritcey, Tsinghua Sci. Technol., 2006, 11, 137–152 CrossRef CAS.
- C. Y. Cheng, Hydrometallurgy, 2006, 84, 109–117 CrossRef CAS PubMed.
- K. R. Barnard, G. L. Nealon, M. I. Ogden and B. W. Skelton, Solvent Extr. Ion Exch., 2010, 28, 778–792 CrossRef CAS.
- R. Banda, H. Jeon and M. Lee, Sep. Purif. Technol., 2012, 98, 481–487 CrossRef CAS PubMed.
- J. M. Reddy, T. J. Sudhavani, S. N. Reddy, K. K. S. V. Rao and K. L. Reddy, Int. J. Res. Chem. Environ., 2012, 2, 158–163 CAS.
- H. Chang, M. Li, Z. Liu, Y. Hu and F. Zhang, J. Rare Earths, 2010, 28, 116–119 CrossRef.
- R. J. Ellis, M. Audras and M. R. Antonio, Langmuir, 2012, 28, 15498–15504 CrossRef CAS PubMed.
- R. J. Ellis, Y. Meridiano, R. Chiarizia, L. Berthon, J. Muller, L. Couston and M. R. Antonio, Chem.–Eur. J., 2013, 19, 2663–2675 CrossRef CAS PubMed.
- K. Huang, J. Qi, X.-X. Liu, Y.-F. Liu, W.-H. Li, Z.-L. Yang, S.-F. Weng, Y.-Z. Xu and J.-G. Wu, Guangpuxue Yu Guangpu Fenxi, 2008, 28, 2038–2043 CAS.
Footnotes |
| † The School of Chemistry at Edinburgh celebrates 300 years of chemistry research and teaching in 2013. |
| ‡ Data provided by Cytec Industries Inc. |
| § The active component in commercial reagents often has the same molecular formula but contains different ratios of structural isomers and/or alternative phase modifiers. |
| ¶ Fe(III) is present in high concentrations in most pregnant leach solutions containing base metals and separates from aqueous solutions as an oxyhydroxide at pH > 2.3. Formation of solid materials presents major operating problems in mixer-settlers. Consequently, acidic feeds and strong extractants are preferred for the circuit shown in Scheme 1. |
| This journal is © The Royal Society of Chemistry 2014 |