The gold–hydrogen bond, Au–H, and the hydrogen bond to gold, Au⋯H–X
Hubert
Schmidbaur
ab,
Helgard G.
Raubenheimer
c and
Liliana
Dobrzańska
d
aDepartment Chemie, Technische Universität München, Garching, Germany. E-mail: H.Schmidbaur@lrz.tum.de
bChemistry Department, King Abdulaziz University, Jeddah, Saudi Arabia
cDepartment of Chemistry and Polymer Science, University of Stellenbosch, Matieland, South Africa. E-mail: hgr@sun.ac.za
dDepartment of Chemistry, KU Leuven, Heverlee, Belgium. E-mail: lianger@chem.kuleuven.be
First published on 2nd September 2013
Abstract
In the first part of this review, the characteristics of Au–H bonds in gold hydrides are reviewed including the data of recently prepared stable organometallic complexes with gold(I) and gold(III) centers. In the second part, the reports are summarized where authors have tried to provide evidence for hydrogen bonds to gold of the type Au⋯H–X. Such interactions have been proposed for gold atoms in the Au(−I), Au(0), Au(I), and Au(III) oxidation states as hydrogen bonding acceptors and H–X units with X = O, N, C as donors, based on both experimental and quantum chemistry studies. To complement these findings, the literature was screened for examples with similar molecular geometries, for which such bonding has not yet been considered. In the discussion of the results, the recently issued IUPAC definitions of hydrogen bonding and the currently accepted description of agostic interactions have been used as guidelines to rank the Au⋯H–X interactions in this broad range of weak chemical bonding. From the available data it appears that all the intra- and intermolecular Au⋯H–X contacts are associated with very low binding energies and non-specific directionality. To date, the energetics have not been estimated, because there are no thermochemical and very limited IR/Raman and temperature-dependent NMR data that can be used as reliable references. Where conspicuous structural or spectroscopic effects have been observed, explanations other than hydrogen bonding Au⋯H–X can also be advanced in most cases. Although numerous examples of short Au⋯H–X contacts exist in the literature, it seems, at this stage, that these probably make only very minor contributions to the energy of a given system and have only a marginal influence on molecular conformations which so far have most often attracted researchers to this topic. Further, more dedicated investigations will be necessary before well founded conclusions can be drawn.
1. Introduction
The chemistry of compounds with one or more discrete gold–hydrogen bonds, Au–H, has proved to be a challenging field, as many reported – and probably many more unreported – preparative endeavors met with complete failure.1–3 For a long time, the use of the standard general methods for the preparation of transition metal hydrides employed with a wide variety of conventional auxiliary ligands did not lead to any stable or metastable gold hydrides even under very mild conditions. However, this situation has changed: at the end of the last decade significant progress has been made when the arsenal of ligands was complemented by novel systems that now allow a clear-cut synthesis of hydride complexes of gold in both the Au(I) and Au(III) oxidation states.4–7 These preparations gave access to the first structural and spectroscopic data, and have provided highly valuable benchmarks for gold hydrogen chemistry. Surprisingly, a stable phosphine complex (R3P)AuH has not yet been prepared.Prior to this new phase of progress, fundamental information on the nature of Au–H bonds was obtained only from data collected for highly reactive species in the gas phase or in low temperature matrices by spectroscopic techniques.8,9 These molecules with simple formulae like AuH or AuH3 were also the prime subject of quantum chemical calculations which gave an idea of some basic thermodynamic and structural characteristics, for which relativistic effects were recognized to be particularly relevant.10–14 Surface chemistry of gold did not contribute much to the understanding of the bonding of hydrogen to gold metal,15–18 because a pristine surface of bulk pure gold with a minimum of defects does not show any affinity to gaseous molecular hydrogen (H2). Chemisorption of hydrogen is only observed when there is a significant number of active centers present on surfaces or on similarly activated gold (nano)particles. Hydrogen radicals H˙ prepared by predissociation of H2 become attached to cold gold surfaces (<150 K), but upon warming to 216 K molecular hydrogen is released.19–21 Solubility of hydrogen gas in metallic gold is extremely low and endothermic.22
With this as a background, the following question has been asked for quite some time: would gold, instead of forming conventional Au–H bonds, rather entertain hydrogen bonds of the types Au⋯H–X, Au⋯H⋯Au or Au–H⋯X?23–27 This idea has quite a reasonable basis when considering several peculiar atomic characteristics of gold atoms and ions:
(i) Gold is one of the most electronegative elements among the metals, mainly due to relativistic effects.10–12 Therefore, in its compounds it may be negatively polarized, similar to the situation in bonds of most elements to a halogen.
(ii) The radii of gold anions Au−, radicals Au˙ and cations Au+ are subject to relativistic contraction which has a significant influence on the interactions with approaching substrates.10–12 In recent DFT calculations of models for hydrogen bonding at gold atoms, the radius of the auride Au− has been calculated to be similar to that of the bromide anion Br− and smaller than that of the iodide anion I−.28
(iii) Gold atoms (radicals) Au˙ have a high electron affinity and thus readily form auride anions Au−. These anions resemble halide anions (like e.g. I−) with an – albeit different – closed shell of electrons (5d106s2 for Au− instead of 5s25p6 for the I− example).
(iv) Univalent gold (Au+) has the lowest known coordination number (CN 2) found for metal atoms, with the two ligands in a linear array. Virtually any substrate therefore has open access to the metal atom, and this situation could favor the formation of even very weak interactions. Neither steric hindrance nor major entropy deficits (requiring other ligands to move from their equilibrium positions) would oppose the approach of a potential hydrogen bonding candidate H–X.
(v) The affinity of bulk gold can be manipulated by an electrical charge. Anodic gold is known to add a molecule of water with one of the hydrogen atoms as the contact atom, whereas cathodic gold instead has the usual contact with the oxygen atom.29 This flexibility in the approach of molecules indicates that hydrogen bonding at gold atoms, clusters, nanoparticles, and surfaces may be realized in many systems even though the associated energy changes may be very small.
Because of the increasing importance of gold clusters and nanoparticles in medical applications, for diagnostics and therapy, several research groups have recently become interested in potential hydrogen bonding interactions of gold atoms (Au) and ions (Aum±), or clusters (Aun) and the corresponding ions (Aunm±), with peptides, pteridines, uracils, and other DNA bases.23,24 These substrates are classical multiple hydrogen bond donors (with O–H or N–H functions) and acceptors (with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN– and other functions), and may thus exhibit specific docking properties via e.g. Au–OC and/or Au⋯H–X bonding. However, there is still a lack of experimental structural or thermodynamic data for these systems; most of the research carried out to date has been limited to quantum chemical studies of model systems.
O, CN– and other functions), and may thus exhibit specific docking properties via e.g. Au–OC and/or Au⋯H–X bonding. However, there is still a lack of experimental structural or thermodynamic data for these systems; most of the research carried out to date has been limited to quantum chemical studies of model systems.
Obviously, any contacts of the type Au⋯H–X with X = carbon must be considered in the context of “agostic”30,31 interactions as defined by Brookhart and Green of the general type M⋯H–C observed in or inferred for many organometallic compounds with the metal atoms M taken mainly from the early transition metal series with nd0–nd6 electronic configurations. For M⋯H–C interactions of late transition elements, Lippard has later proposed the term “anagostic”,32 while Venanzi et al. introduced the term “pre-agostic”,33 but the border of course is not clearly defined. One way of describing the transition is the move from three center-two electron bonding to three center-four electron bonding, meaning that in “agostic” interactions the metal atom is assumed to be electron-deficient, while in “anagostic” interactions the metal atom is to be taken as more electron-rich. In a more recent description of the former class of M⋯H–C interactions, the systems with late transition elements M are referred to as “electron-rich agostic complexes”.34 In the typical examples for this type of interaction, for instance with an ethyl group attached to a dn metal center, M–CαH2–CβH3, one of the H–Cβ units becomes involved in a σ-complex through side-on coordination to the metal atom, similar to the situation in dihydrogen complexes. By contrast, in agostic interactions with the early transition metals the H–C bonds are actually less affected and the major changes appear in the M–Cα–Cβ bonding.34
It is worth remembering that crystallographers had been puzzled by the observation of short M⋯H–C contacts in a variety of coordination compounds already in the 1960s, including the groups of Ibers (Ru⋯H),35 Mason and Shaw (Pd⋯H),36 Vrieze (Pt⋯H),37 and Maitlis (Pd⋯H).38 In a second round in the 1970s, the groups of Trofimenko,39 Echols,40 and Cotton have presented numerous examples of Mo⋯H interactions, and in this work the X-ray diffraction studies were complemented by variable temperature NMR investigations.41,42 Referring to these early contributions, Cotton in 1989 expressed the view that the term “agostic” proposed in 198343 was “a curious word” for a phenomenon “not in any essential way different” from the previous findings, meaning that there was no fundamental difference between the various forms of close M⋯H–X contacts.44
While work on M⋯H–C interactions in early transition metals, mainly with the d0 examples Ti and Zr, continued to advance on a broad front in the 1980s, and later, owing to the relevance to e.g. olefin polymerization catalysis,30,31,34 the investigations of related systems with late transition metals were more limited. It was mainly the group of Albinati and Pregosin which continued to focus on compounds with Pt⋯H contacts.45–48 In extended NMR studies it was shown that Pt⋯H–C contacts could be best diagnosed by monitoring J(195Pt,1H) or J(195Pt13C) coupling constants due to the fortuitously high abundance and large magnetic moment of the 195Pt (s = ½) nucleus. Again it was felt that these interactions were set apart from “agostic” interactions with the early transition metals, and therefore the neutral term “three center interactions” was used exclusively. However, as also pointed out by Cotton, this distinction was supposed to be likely “one of degree as opposed to type”.48 It should also be noted that the observation of spin–spin couplings and of nuclear Overhauser effects (NOE) in the platinum (and also rhodium) compounds does only indicate spin communication between electrons and nuclei of atoms in close contact, which however may not be associated with any significant (hydrogen) bonding.
In all this early work, the compounds of coinage metals in general and of gold in particular were not considered. It should be mentioned that gold has no isotope with a suitable magnetic moment and nuclear spin to allow a direct observation of e.g.197Au–1H/13C spin–spin couplings or δ(197Au) chemical shifts which makes it more difficult to identify Au⋯H–X contacts.
Au⋯H–X interactions may of course include not only the closed-shell auride anions Au−, but also the gold cations Au+ coordinatively saturated in common compounds of the types L–Au–X, [X–Au–X]−, [L–Au–L]+, or others. Au⋯H–X contacts to Au3+ with its 5d8 configuration as the central metal ion have their parallels in the corresponding interactions observed at isoelectronic Pt2+. The various forms of Au⋯H–X contacts could play a role in gold catalysis, for instance by lowering the energy of significant transition states, as it has long been assumed for the Pt2+ analogs. A very recent paper discussing various pathways of the gold-catalyzed hydrogenation of olefins is a very good example. Not only gold(I) hydrides, but also weaker gold–hydrogen interactions such as agostic bonding have been considered for intermediates and transition states.49
In the present review, the currently available information on compounds with gold–hydrogen bonds, Au–H, and with hydrogen bonding to gold, Au⋯H–X, is summarized based upon explicit pertinent considerations in the literature, and also on results of a new search for hitherto unnoticed potential evidence for related phenomena. For the latter, solely the simple term “hydrogen bonding” is used, thus avoiding the recently introduced terms “unconventional” or “non-conventional hydrogen bonding”,23,24 or others32,34,48 for which there is a certain danger of confusion. Following the original definition,31 the term “agostic” is also excluded, since its use should best be restricted to H–C donor functions engaged in bonding to distinctly electron-deficient metal centers, regardless in which way this bonding may be described.34 The proximity of a gold and a hydrogen atom in a given system (Au⋯H–X) is called a “contact” not necessarily implying any significant “interaction”, which may be attractive or repulsive.
2. Gold hydrides
Experimental data on compounds with discrete Au–H bonds are still rare.1–3,8,50 They include benchmark values on the molecule Au–H in the gas phase and in inert gas matrices at low temperature, including its adducts with dihydrogen, and results collected from crystals and solutions of its very recently prepared complexes (NHC)AuH, where NHC represents an N-heterocyclic carbene ligand.4,5 The same sort of ligands enabled the preparation of hydrogen-bridged dinuclear gold complexes [μ-HAu2(NHC)2]+.4 With ligands other than NHCs, such as tertiary phosphines, only the hydrogen-bridged dinuclear gold(I) species were observed.51–53 In addition, there is one unique case of a crystalline gold(III) hydride with a dianionic tridentate ligand.6 Earlier attempts to obtain gold trihydride AuH3 or its dimer Au2H6 and tetrahydridoaurates(III) M[AuH4] had been unsuccessful.54For several decades, hydrides of gold in its various oxidation states have also been the subject of quantum chemical calculations.10–15
2.1 Gold(I) hydride Au–H and its dihydrogen adducts
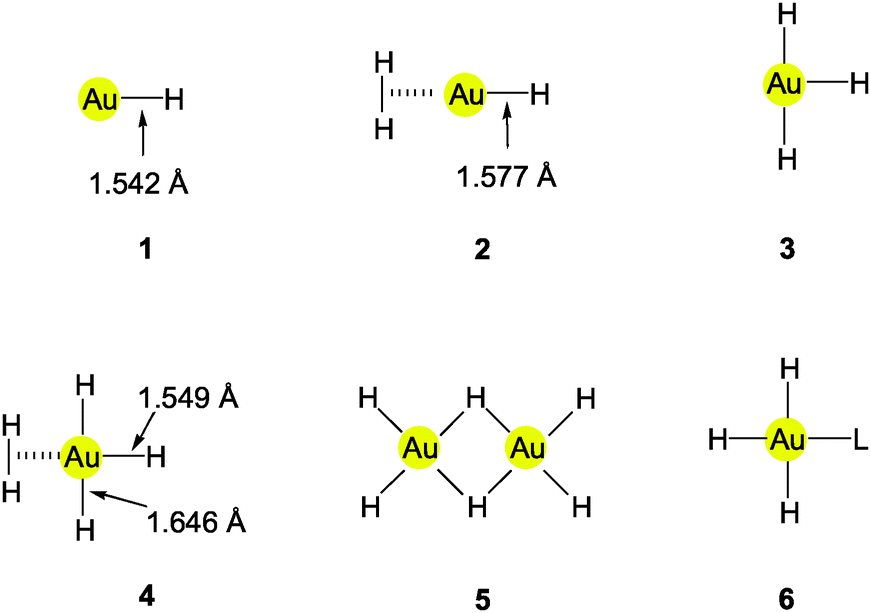
The AuH molecule (1) has been observed in the gas phase as a product of the reaction of gold vapor and hydrogen radicals.9,55,56 From spectrometric observations, the dissociation energy DE of Au–H was calculated to be 74.3 ± 2.6 kcal mol−1.55,57,58 The enthalpy of formation from atomic gold and atomic hydrogen is 69.9 ± 1.9 kcal mol−1.9 Similar data were obtained from the thermodynamics of reactions of gold cations [Au]+ with protic species like alcohols, [Au]+ + RR′CHOH → Au–H + [RR′C(OH)]+, or of gold anions [Au]− with weak acids in the gas phase. From the gas phase acidity constants for the reactions [Au]− + X–H → Au–H + [X]− a DE value of 70.6 kcal mol−1 has been calculated.59The AuH molecules produced from Laser-ablated gold vapor and hydrogen in an excess of argon and trapped in solid argon show a ν(Au–H) band at 2226.6 cm−1 (1597.2 cm−1 for Au–D).58,60 The corresponding gas phase values for Au–H/D obtained from electronic band spectra are 2218.8/1591.7 cm−1. The AuH molecule has a dipole moment of −2.70 D.9 If excess H2 (or D2) is given access to matrix-isolated AuH molecules, a further shift of ν(Au–H) to 2173.6/1559.2 cm−1 is observed which is ascribed to the formation of HAu(H2)/DAu(D2) adducts (2 or 3).61 In solid hydrogen, there is evidence of the existence of another adduct with hydrogen (H2)AuH3 (4).60
From the experimental data, the Au–H distance in the AuH molecule (1, C∞v symmetry) was calculated as d(Au–H) = 1.542 Å. This distance is elongated to 1.577 Å in (H2)AuH (2). However, in the (H2)AuH3 adduct (4), one of the Au–H bonds is short, 1.549 Å, while the other two are long, 1.646 Å.61 Calculated values for d(Au–H) approach this benchmark quite closely upon inclusion of relativistic effects.10,14 Gold atoms in the 2P excited state undergo insertion into the H2 molecule to form AuH2 (2B2) with a significant barrier to the decomposition into Au and H2. Cluster hydrides like Au2H and Au2H− are also formed in this system.60
Of the two structural alternatives discussed for AuH3, the T-shaped molecule (3) with well separated hydrogen atoms62 and the Y-shaped dihydrogen complex (2) (both with C2v symmetry), the latter is clearly favored by the matrix-isolation data.63 This result has been confirmed by several quantum chemical calculations which revealed that the T-shaped molecule was about 30 kcal mol−1 higher in energy. The addition of H2 to AuH is only mildly exothermic (13.44 kcal mol−1). However, the dimerization of AuH3 (2, 3) to afford Au2H6 (5) would contribute about 20 kcal mol−1 to the stability of the latter, and monomeric AuH3 would also be stabilized through L donors in complexes (L)AuH3 (6).64–68 In the recent paper on a proposed catalytic cycle of gold-catalyzed hydrogenation of olefins, a dihydrogen complex of the composition Me3PAu(H2)Cl has been considered as a transition state (see Section 3.5).49
2.2 Gold hydride complexes (L)Au–H
Complexes of AuH with L representing an NHC have been prepared from the corresponding chloride (NHC)AuCl with LiBEt3H,4 or from the t-butoxide (NHC)AuOtBu or the hydroxide (NHC)AuOH with (MeO)3SiH.4,5 The example with NHC = IPr {1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene} has been crystallized and its structure determined by X-ray diffraction (7). However, it was not possible to refine the position of the Au–H hydrogen atom, and therefore the Au–H distance is not accurately known. Surprisingly, the signal of this proton could not be localized in the 1H-NMR spectrum, but for the deuterated compound a signal was detected in the 2D-NMR spectrum. In the IR spectrum, the ν(Au–H) vibration was assigned to the band at 1976 cm−1 [1407 cm−1 for ν(Au–D)]. (IPr)AuH was used for the preparation of several new complexes (IPr)AuX with X = tBuO, C(N2)COOEt, C[C(O)OMe]CH(CO)OMe, etc.4
The structure of (IPr)AuH was the subject of several quantum chemical calculations. At the B3LYP/cc-pVCZ level, the latest results gave an Au–H distance of 1.618 Å.69
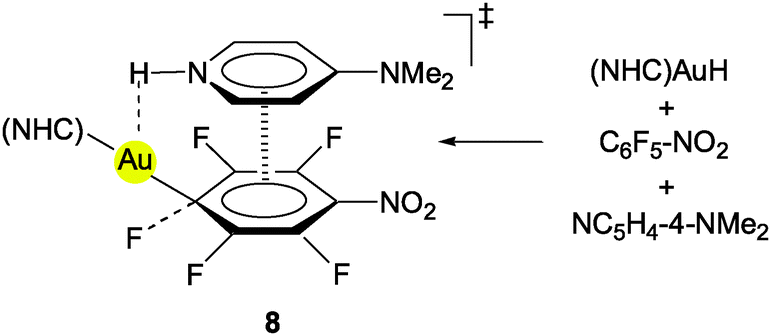
(NHC)AuH complexes have recently been employed as stoichiometric and catalytic reagents for the hydrodefluorations of fluoroarenes. The catalytic version has used triorganosilanes R3SiH as the hydride source, the formation of the corresponding fluorosilanes R3SiF providing the driving force for the process. In the mechanisms proposed for the possible catalytic cycles, a gold(III) hydride species which is formed via an oxidative addition of the fluoroarene to gold(I) has been considered. Alternatively, the approach of an auxiliary base (4-dimethylaminopyridine) at the Au–H function could establish an Au⋯H–N contact in a π–π assisted intermediate which induces the hydrogen transfer (8, see also below)7 and C–F bond cleavage.
Recent attempts to obtain (IPr)AuH by β-elimination of ethene from (IPr)AuEt have failed. The activation barrier for this thermal extrusion is too high (ca. 50 kcal mol−1), and thus the temperature required (>120 °C) exceeds the range of stability of the product. Attempts to insert ethene into the Au–H bond (8 bar at 25 °C or 1 bar at 80 °C) were equally unsuccessful.69 Experimental and theoretical studies involving (L)AuEt69 and (L)AuH49 systems are discussed below.
Careful examination of the spectral patterns of the products of the same reaction carried out with different excitation energies has provided evidence for an additional species (2052, 2050, 1726, 814, 800, and 781 cm−1 for [AuH4]−; 1452, 1450, 1230, 567, 571, 556, and 555 cm−1 for [AuD4]−) which was identified by comparison with data calculated for the square planar (D4h symmetry) anions [AuH/D4]− and for mixed H/D species (10). It features the distance d(Au–H) = 1.653 Å. The addition of a H2 molecule to the [H–Au–H]− anion has been calculated to be endothermic by ca. 10 kcal mol−1.66

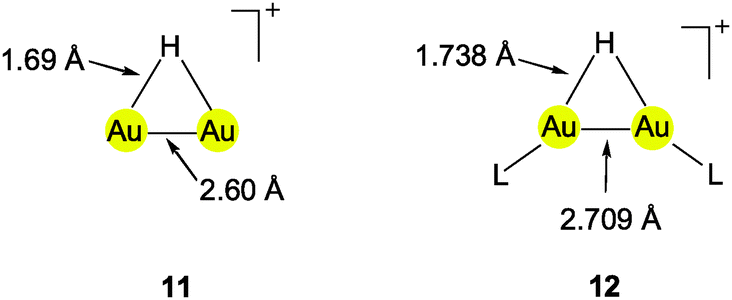
Using (R3P)AuCl compounds as substrates, the cations [(R3P)2Au2H]+ have been identified with R = Me, Ph, and a model structure has been calculated by quantum chemical methods for R = H.72 Distances d(Au–H) and d(Au⋯Au) were calculated at 1.738 and 2.907 Å, respectively, significantly longer than for the ligand-free species.70 With bidentate Xantphos ligands, the same core unit [Au2H]+ becomes trapped between four phosphorus atoms with each Au atom chelated by one ligand (Fig. 1). The triflate salt of this cation (which has C2 symmetry) has been structurally characterized by X-ray diffraction.73
 | ||
| Fig. 1 Structure of the cation [{(Xantphos)Au}2H]+ in a crystal of the CF3SO3− salt; irrelevant H atoms are omitted for clarity; the dashed line indicates the aurophilic interaction.73 | ||
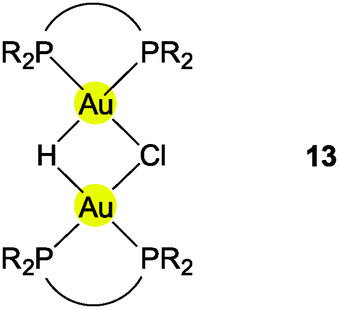
The distances d(Au–H) and d(Au⋯Au) are 1.70(3) and 2.7542(3) Å, respectively, rather close to the distances within the ligand-free core unit.70 In a related work with similar ligands, the Au–H–Au bridge was found to be further supported by a chlorine atom (13, R2P∧PR2 = Xantphos). The bridging hydrogen atom was identified by its 1H-NMR quintet signal due to 2J(PH) couplings to four phosphorus atoms,74,75 and by its contribution to the mass of the molecular ion in the mass spectrum.
Reaction of (IPr)AuOTf with (IPr)AuH yields the salt [{(IPr)Au}2H]+ OTf − which also features the [Au2H]+ core (12, L = NHC). The crystal structure has been determined, but again, the bridging hydrogen atom could not be localized. The d(Au⋯Au) distance of 2.709 Å is in good agreement with the data for the complexes with phosphine ligands (above). In the 1H-NMR spectrum, the signal of the hydrogen atom [Au2H]+ was detected at δ 0.42 ppm. No IR bands have been assigned to the [Au2H]+ unit.4,5

As noted above, in studies of the hydrodefluorination of perfluoroarenes using (NHC)AuH complexes, an interaction between the gold hydride, the substrate and the auxiliary base (4-dimethylamino-pyridine) has been proposed for a transition state which involves Au⋯H–N contacts (8). In the oxidative addition, the oxidation state of the gold atom is increased from (I) to (III), and in the following reductive elimination the original state is restored.7 On the basis of DFT calculations, the tri-component transition state has been assumed to be supported significantly by π–π interactions between the two aromatic six-membered rings. A contribution from Au⋯H–C bridging to the energetics of the process is uncertain.
3. Hydrogen bonds to gold atoms
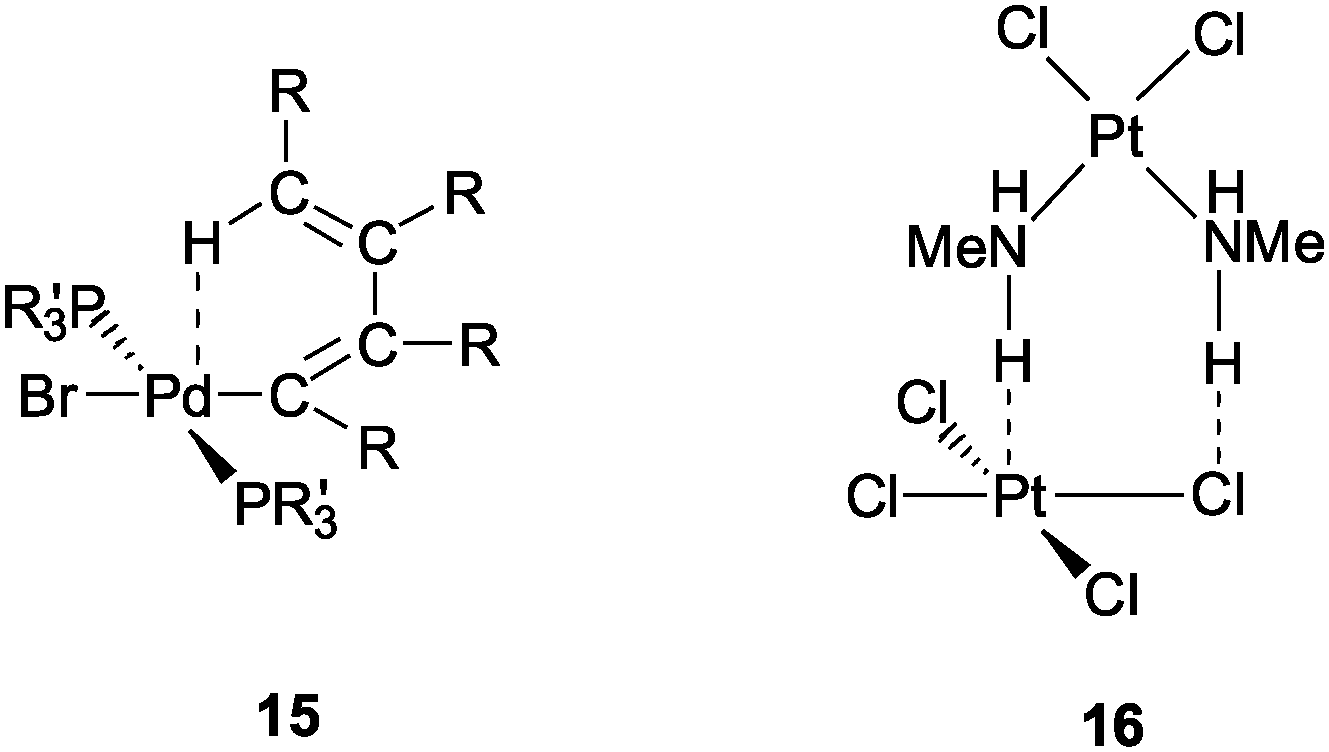
As mentioned in the Introduction, intramolecular contacts M⋯H–C in organometallic complexes have been the subject of extensive considerations.30–48 In single crystal X-ray diffraction studies of many coordination compounds certain molecular conformations were observed which suggested an orientation of a flexible ligand towards a transition metal atom with any of the possible d0–d8 electron configurations establishing rather short M⋯H distances. Formula 15 (R = COOMe, R′ = Ph) is an early illustrative example (Pd⋯H 2.3, C–H 1.08 Å; to be compared with van der Waals radii for Pd and H at 1.9 and 1.2 Å, respectively). The idea of a specific Pd⋯H–C interaction was also supported by the unusual down-field chemical shift and two-bond coupling of the proton concerned in the NMR spectra.38 Early examples have also included intermolecular contacts of the type Pt⋯H–N between the components of the compound [NnPr4]2[PtCl4][cis-PtCl2(NH2Me)2] for which the hydrogen atoms were localized by neutron diffraction experiments: Pt⋯H 2.261, N–H 1.032 Å, N–H⋯Pt 167.1° (16).76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) †
†
While the experimental evidence for similar intra- and intermolecular M⋯H–X contacts was growing as summarized in several general reviews on both “hydrogen bonding” and “agostic interactions”,30,31,78–99 the increasing number of known examples became also an incentive for several theoretical approaches to the subject.34,100–104 With this rapid growth of experimentally and theoretically obtained data, it has become more and more important to revise or extend existing concepts of the bonding concerned. More generally, as many kinds of “weak interactions” between molecular and ionic components of supramolecular aggregates are currently in the focus of research activities, there has been a great demand for clear-cut definitions of the criteria that determine the nature of the interactions under consideration.
An authoritative IUPAC Technical Report87 and the corresponding IUPAC Recommendation 201188,89 briefly summarize the following essentials that should be met by experimental or theoretical observations for a system X–H⋯Y–Z (where X–H is taken as the donor part and Y–Z as the acceptor part) in order to characterize it as a “hydrogen bond”.‡
(i) The forces involved may have electrostatic, charge transfer (covalent) and dispersion (van der Waals) components.
(ii) The X–H bond should be polarized owing to a difference in the electronegativity of H and X, with X as the more electronegative partner.
(iii) The angle of the X–H⋯Y connection should ideally be close to 180° and preferentially not drop below 110°.
(iv–vi) There should be some significant structural or spectroscopic evidence for the interaction, e.g. changes of bond lengths (including X–H and Y–Z), of vibrational modes (mainly for X–H) or of 1H-NMR chemical shifts.
(vii) The Gibbs free energy of the interaction should provide a minimum of thermal stability to the bond concerned.
From criterion (ii) of this definition it follows that, in addition to F–H, O–H and N–H functions, C–H and S–H units are acceptable as hydrogen bond donors, even though the difference in electronegativity between H(δ+) and C(δ−) or S(δ−) may be very small, but B–H units are excluded owing to an inverse polarization of the bond (B(δ+)–H(δ−)).
Within the given IUPAC formalism, gold atoms (Y = Au) in their various oxidation states and molecular environments (Z) are considered here as hydrogen bond acceptors in systems X–H⋯Au–Z. As already mentioned above, this approach appears to be straightforward for the closed-shell auride anions Au−, which are analogs not only of halide anions, but also of the rare gases.8 Hydrogen bonds to halide anions are well established, with the binding energies increasing from I− to F−. Moreover, hydrogen bonding of water molecules to xenon atoms with an almost linear Xe⋯H–O(H) contact has been observed in molecular beam and photoelectron spectroscopy (PES) experiments.90,91 Au− therefore should clearly qualify as a hydrogen bond acceptor.
With AuI or AuIII metal centres and for Au0 atoms (mainly in gold clusters), the hydrogen bond acceptor properties are less obvious and very much dependent on the ligand part Z. Strong hydrogen bonds are generally not expected, but medium, weak or very weak interactions may become discernible if suitable donor parts X–H are involved. In the case of the latter category (very weak), generally an upper threshold of the binding energies of 17 kJ mol−1 (4 kcal mol−1) has been set, with an open lower end as the values approach those of standard dispersion forces.87
Regarding criterion (iii) (above) it will be shown further down that Au⋯H–X angles with only few exceptions (viz. the bonding to auride anions Au−) are smaller than the proposed 180° optimum for “IUPAC hydrogen bonding” which indicates that the contacts concerned are far from this standard hydrogen bonding model. By contrast, for idealized agostic bonding much smaller angles (less than 120 °C) are the rule.30,31,34,97 Au⋯H–X angles are thus falling in a range in between these two models, similar to the situation described earlier for M⋯H–N/C contacts with M representing a d8 metal center in its typical square planar environment.86
As already mentioned in the Introduction, for this intermediate range the bonding was considered in the literature as “very similar” or at least “substantially similar” to “classical” hydrogen bonding,84 as “anagostic” bonding,32 “pre(a)gostic bonding”,33 “pseudoagostic bonding”,93 “non-conventional hydrogen bonding”,85 or – as mentioned above – “electron-rich agostic”.34 Realizing that all these interactions are associated with small energy contributions, it has been noted that both charge-assisted intermolecular contacts (charge-assisted hydrogen bonds) and multiple intermolecular M⋯H–X contacts (multicenter hetero-acceptor hydrogen bonding) may be the key to observe more stable prototypes, the latter being described as the “Gulliver effect” where intermolecular contacts are concerned.93
The formulae used to represent such interactions are not uniform in the literature. It has been proposed that the direction of the electron flow should be indicated by using two alternatives (X–H⋯M or M⋯H–X) where the electrons are meant to flow always from right to left.93 While for Au− with its electronic configuration 5s106s2 this flow can be considered as X–H⋯Au in standard cases, for AuI and AuIII with their electronic configurations 5d106s0 and 5d86s0, respectively, and with different sets of ligands the situation is less clear. Therefore a uniform representation as Au⋯H–X has been chosen.
Under criteria (iv–vi) (above), particular attention should be paid to cases where parts of the molecular structure show unexpected conformations which may be caused by the influence of the hydrogen bonding acceptor (in the present context by the influence of the gold centre) on the ligand conformation. However, it may be very difficult in most cases to distinguish this influence from for example intraligand resonance effects, ligand–counterion interactions or most efficient molecular packing in the crystal.
Even though there has been an enormous growth in gold chemistry in recent years, hydrogen bonding at gold centers was not considered systematically and comprehensively in most reports describing the progress made in experimental and theoretical studies.1–3,34,84–86,95 In the following sections pertinent observations are summarized and analyzed.
3.1 Experimental evidence for Au⋯H–X hydrogen bonding at auride anions Au−
Auride anions Au− were first recognized as components of alkali salts such as Cs+Au− in the 1940s.105 A large family of binary and ternary alkali and alkaline earth aurides has subsequently been investigated, including also some metal auride oxides.106–108 In solutions of these metal aurides in solvents like liquid ammonia and amines, the anions may be engaged in Au⋯H–N bonding, but no details are available.109 However, complexation of the alkali cations by crown ethers in liquid ammonia provided crystalline materials in which there is clear evidence for Au⋯H–N bonding.25 In the compound [Rb([18]crown-6)(NH3)3]Au·NH3 the auride anion is surrounded by five ammonia molecules in a distorted square pyramidal geometry. The positions of the hydrogen atoms have been located, and the shortest (apical) distance Au⋯H is 2.58(1) Å with an angle N–H⋯Au of 158(1)°. The remainder Au⋯H distances are in the range of 2.835–3.015 Å with Au⋯H–N angles between 162.8 and 166.7° (Fig. 2). (In an evaluation of the data for the present review, a minor error in this paper has been noted and corrected.) The structural parameters compare well with those found for conventional hydrogen bonds Br/I⋯H in the corresponding halides.25 | ||
| Fig. 2 Environment of the auride anion Au− in a crystal of [Rb([18]crown-6)(NH3)3]Au·NH3; Au⋯H–N contacts are shown as orange dashed lines, H atoms of the [18]crown-6 have been omitted for clarity; the shortest contact has been labelled.25 | ||
In a related compound, [Cs([18]crown-6)]Au·8NH3, two different auride anions are surrounded by 7 ammonia molecules with Au–N distances in the range of 3.67–3.90 Å (Fig. 3).26 Previous studies of the quaternary ammonium salt [NMe4]Au (CsCl-type) have revealed a large set of not less than 20 C–H⋯Au contacts between the auride anion Au− and the methyl groups of surrounding NMe4+ cations. In four of these contacts the Au⋯H and Au–C distances are 2.92(10) and 3.849(1) Å, respectively, and the Au⋯H–C angles are close to 170° (Fig. 4).27 The structure is isomorphous with that of the bromide [NMe4]Br, and similar hydrogen bonding between cations and anions can be assumed. 197Au Mössbauer spectra have confirmed the influence of hydrogen bonding on the isomeric shift values of Au− in [NMe4]+Au− as compared to Cs+Au−.27
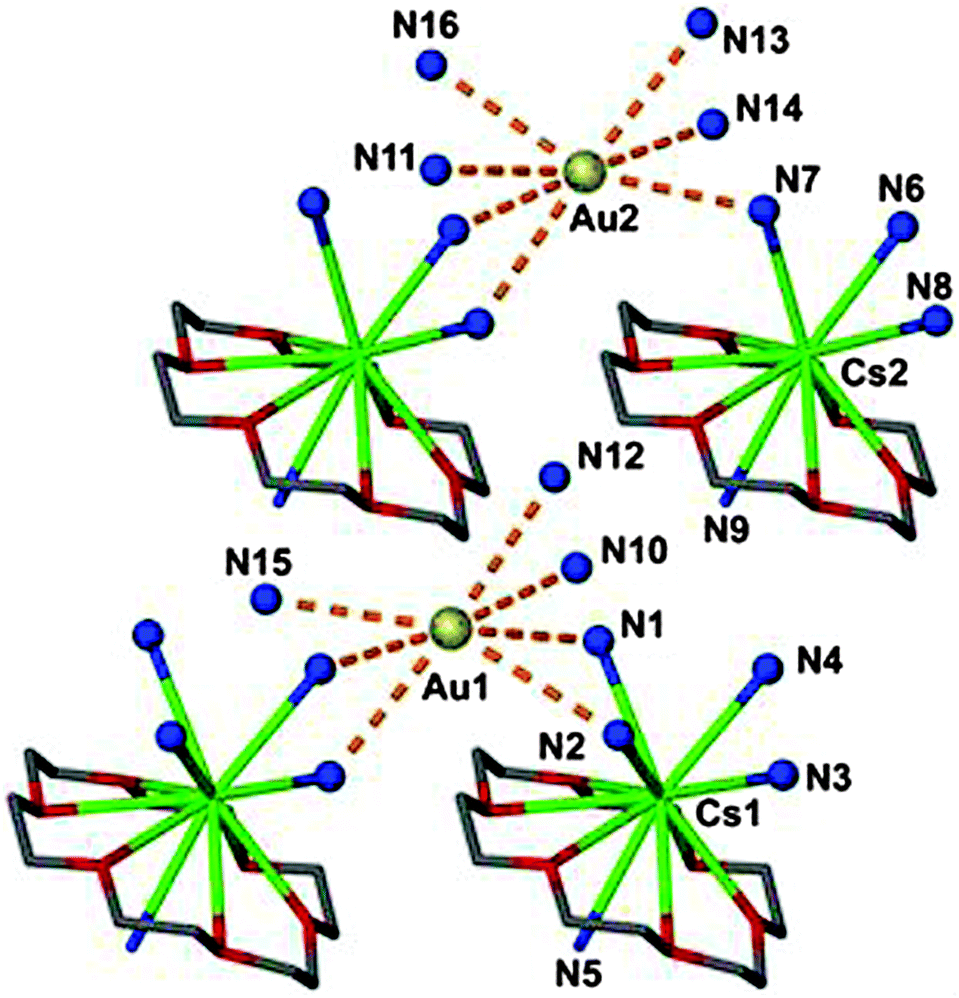 | ||
| Fig. 3 (Au−)⋯N connectivity in a crystal of [Cs([18]crown-6)]Au·8NH3 (dashed orange lines); H atoms are not shown; the two crystallographically independent auride anions Au− present in the asymmetric unit show different environments.26 | ||
 | ||
| Fig. 4 The shortest (Au−)⋯H–C contacts (Au⋯C = 3.849(1) Å) present in a crystal of [(CH3)4N]Au, indicated as dashed green lines.27 | ||
This brief summary of experimental evidence for hydrogen bonding at Au− shows that so far only Au⋯H–N and Au⋯H–C contacts have been observed in the condensed phase. Interestingly, the strongest hydrogen bond donor units H–O and H–F have not yet been encountered as partners of Au− in any crystalline systems. This is probably due to the fact that these groups choose other acceptor units where they can become engaged in stronger bonding. However, simple adducts of auride anions with the classical hydrogen bond donor molecules HF, H2O and NH3, and with amines and even N-heterocycles have been the targets of extended quantum-chemical calculations which gave valuable information on the molecular and electronic structures of these models (17–22) (see also below).28,110 (Analysis of the results from such calculations indicates electron transfer from gold to the hydrogen in X–H units and a dashed arrow is used to indicate this for structures obtained theoretically.)
In parallel to these theoretical studies, the hydrates of auride anions and auride clusters [(H2O)nAum]− (as isolated species) became also the subject of experimental investigations. Predissociation infrared spectral observations of these anions in the range of 3700–3100 cm−1 are clearly reminiscent of the patterns observed for hydrates of the halide anions which supports the halide-auride analogy.111
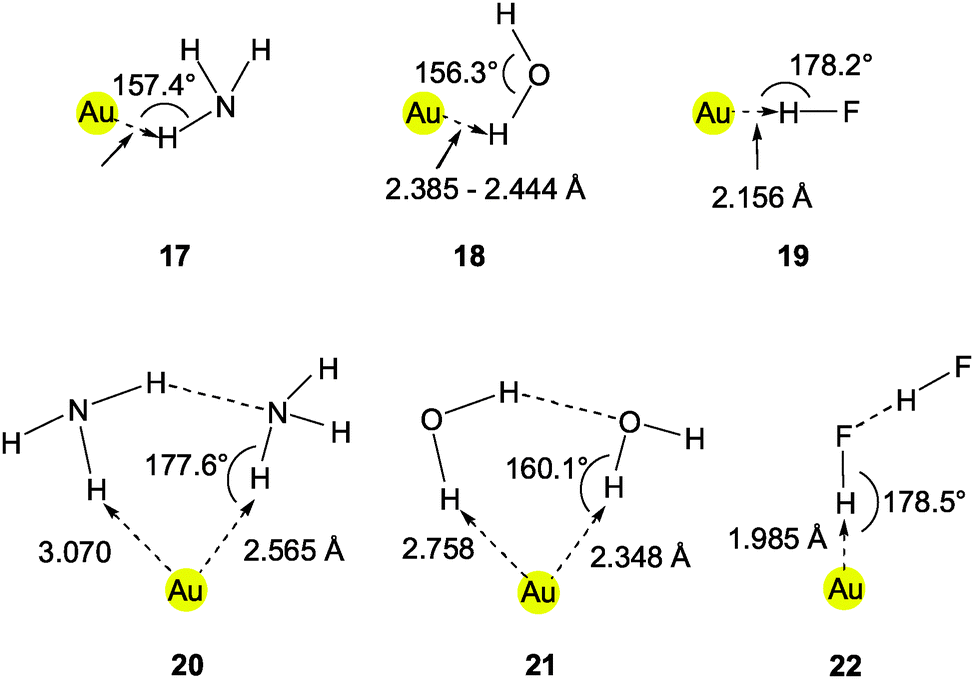
The anions [(H2O)Au]− (18) and [(H2O)2Au]− (21) were also studied by photoelectron spectroscopy (PES). The vertical electron detachment energy (VDE) of the anion Au− (the electron affinity of the gold atom) is shifted from 2.31 to 2.76 eV upon addition of the first water molecule. The second water molecule causes a further shift to 3.20 eV. These values differ markedly from those of the hydrates of Cu− mainly owing to relativistic effects which cause the gold atom to have a higher electron affinity. The hydrates of the dinuclear anion radical [Au2]− were also included and showed similar effects.112 In a subsequent photoelectron spectroscopic investigation the vertical detachment energy of the anion [(H2O)Au]− of 2.76 eV was confirmed and supported by ab initio calculations (2.82 eV).91
The emerging physical data show significant changes in the vibrational and rotational dynamics of donor molecules like water and changes in the electronic configuration of the acceptor Au− as demonstrated by photoelectron spectroscopy. The binding energies appear to be well above those of common dispersion forces, and there is a clear analogy to the behavior of large halide anions as hydrogen bond acceptors. Therefore there is not really a need to set this type of interaction apart from standard hydrogen bonding by using terms like “non-conventional” or “unconventional” hydrogen bonding.113,114
The results of theoretical studies on model systems with auride anions Au− or gold cluster anions [Aun]− as nucleation units for the addition of HF, H2O, NH3 and even for larger components like DNA bases are summarized in Section 3.6.
3.2 Experimental evidence for Au⋯H–X hydrogen bonding at Au+ cations and in their complexes
Owing to their positive charge, ligand-free gold cations Au+ are not expected to undergo interactions with potential hydrogen bond donor functions like N–H, O–H or F–H to give adducts with hydrogen bonding characteristics. In agreement with simple rules of Coulomb interactions, dipolar donors like ammonia or water will form complexes such as [Au(NH3)n]+ and [Au(OH2)n]+, respectively, exclusively via Au–N and Au–O bonding (23, 24 for n = 2). This has been confirmed in experimental studies at least for ammonia,115 while there is less information on the aquo complexes which are unstable to disproportionation.50 Theoretical studies on model systems with ammonia and water,116–119 or alcohols58 and (enolizable) acetone120 also gave the expected results. Only in multicomponent systems with several water molecules engaged in mutual hydrogen bonding, or with an auxiliary ligand, Au⋯H–O contacts may contribute to the overall stability of the aggregates.118
The situation may be different for complexes of AuI where the formal charge of the metal atom can be distributed over a ligand system and where there is still ready access to the metal atom for any kind of substrates owing to the notorious linear two-coordination of gold(I). Especially well-tailored ate-complexes [Y–Au–Y]− with Y bearing a substituent with a hydrogen atom close to the metal atom should be good candidates for hydrogen bonding. However, in spite of this possibly favorable potential there is still very limited evidence for inter- or intramolecular contacts with typical O–H or N–H donors which might lead to Au⋯H–O or Au⋯H–N interactions. The examples summarized in the following have all been selected from the Cambridge Structural Database (CSD system 2013, up till the update of February 2013) with distances and angle cut-offs set at Au⋯H 1.2–3.0, Au⋯X 2.2–4.0 Å and Au⋯H–X 90–180°, respectively, as suggested in the literature.79,93 The bond lengths were normalized during the search (see histograms), but the data given in this review are not normalized.
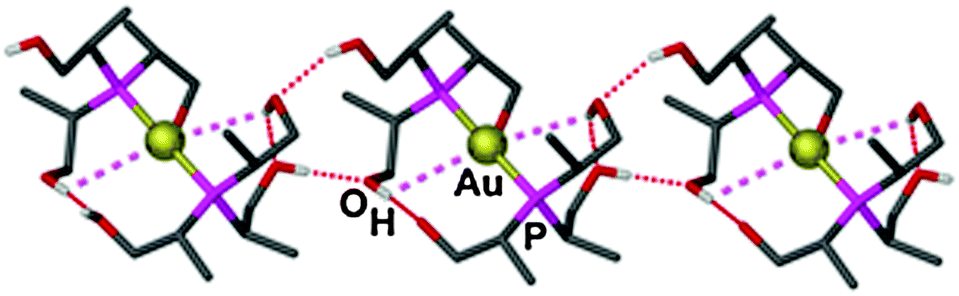 | ||
| Fig. 5 Chain structure of the cation [{(R,R,R)-(HOCH2CH(CH3))3P}2Au]+ in the CF3SO3− salt. Carbon-bound hydrogen atoms have been omitted for clarity; Au⋯O–H contacts and O–H⋯O hydrogen bonds are shown as purple and red dashed lines, respectively.121 | ||
A similar situation is observed in the gold(I) complex with diphenyl(hydroxymethyl)phosphine ligands Ph2PCH2OH (1:3). One of the hydroxyl groups in 25 located near the gold center with Au⋯H–O characteristics of 2.881, 3.624 Å and 126.04° is simultaneously involved in much stronger intra- and intermolecular O–H⋯O interactions.122
In the crystal structures of dinuclear complexes of ligands of the general formula (Ph2PCH2)2N(R), where R carries OH and/or COOH groups in various positions, comparable combinations of contacts are encountered. While for the example shown in 26 intramolecular O–H⋯O contacts are established, in crystals of examples with the C(O)OH and OH groups not in adjacent positions (2-carboxy, 6-hydroxy; 5-carboxy, 2-hydroxy), the C(O)OH group in the former is engaged in hydrogen bonding to the chloride ligands of neighboring molecules in a complex array (not shown) and the OH group comes close to the gold atom (Au⋯H–O characteristics of 2.722, 3.375 Å, 135.75°). The C(O)OH group in the latter is involved in strong intermolecular O–H⋯O interactions and the OH group participates in intermolecular O–H⋯Cl bonding, thereby closely approaching the gold atom from a neighboring molecule (Au⋯H–O characteristics of 2.896, 3.394 Å, 119.89°). No significance has been attributed to these secondary contacts.123

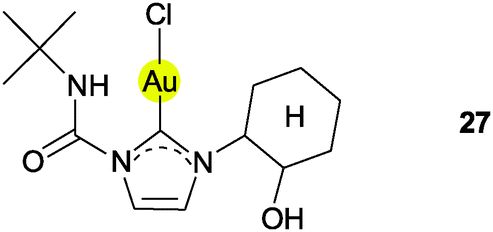
In a structural study of (NHC)AuCl complexes with ligands bearing a 2-hydroxy-cyclohexyl group at one of the N-atoms (27) no unusual intramolecular attraction of the OH groups towards the gold atoms was observed, even though this substituent should be free to rotate into a suitable conformation. For two selected structures, short intermolecular contacts between the OH groups and gold atoms from adjacent molecules can be observed, namely for 27 (2.974, 3.607 Å, 135.92°)124 and for an analog with a CH2Ph substituent on the imidazolium ring (2.935, 3.502 Å, 128.13°).125 However, in both molecules the orientation of the OH unit is rather influenced by intermolecular O–H⋯Cl interactions (2.585, 3.358 Å, 158°; 2.615, 3.402 Å, 161.22°). For a third related compound (with a CH2C(O)Ph group), no Au⋯H contact was observed.
Regarding gold(I) complexes which crystallize with OH-functionalized solvate molecules, the compounds of gold carboxylates with bis(diphenylphosphino)methane (dppm) or -ethane (dppe) can be mentioned as examples. In the former case, the MeOH solvate molecule approaches the gold(I) center with the Au⋯H–O parameters 3.011, 3.414 Å and 112.32°, whereas in the latter the corresponding data are 2.936, 3.378 Å and 115.53°. The position of the solute molecule may be largely determined by the size and the form of the void in which it is accommodated.126
It is obvious from this small selection of representative potential candidates for AuI⋯H–O hydrogen bonding that the influence of this type of interaction on the structural characteristics of gold(I) complexes has so far proved to be largely negligible. However, the dinuclear cationic complex of 1-thioglycerol with a diaurated sulfur center (28) is somewhat exceptional in that therein a surprisingly short intramolecular contact was observed. In the crystals of the tetrafluoroborate salt, the (disordered) hydroxyl group in the 2-position is clearly orientated towards one of the gold atoms to establish distances Au⋯H and Au⋯O of 2.393 and 3.114 Å, respectively, and an angle Au⋯H–O of 142.7°. These are the shortest distances in the present collection which together with the rather wide angle may be indicative of a significant interaction. It should be noted that the two geminal gold atoms of the [SAu2]+ unit are tied together by a strong aurophilic interaction which may significantly alter the acceptor properties of the metal atoms, clearly a feature for further attention.127

To summarize, even though there are many gold(I) compounds which offer O–H donor and AuI acceptor functions that could lead to close Au⋯H–O intramolecular contacts without conformational constraints, or to similarly unforced intermolecular contacts, there appears to be no particular preference for establishing such interactions in the solid state. Studies on solutions aiming at any evidence for Au⋯H–O interactions have not been published.
In the present context, the crystals of [(NH3)2Au]Cl·4NH3 have recently been investigated as a simple example. The gold atoms were found to be pseudo-hexacoordinated by two N-atoms through standard donor–acceptor bonding, two gold atoms through aurophilic interactions and two H atoms of ammonia molecules. The latter have Au⋯H distances of 2.95 Å and small Au⋯H–N angles of 122°, but are also engaged in N–H⋯Cl contacts making the interactions bifurcated (not shown in 29).128

An early pertinent example was presented by Laguna et al. in a study of 2-(ferrocenylmethylamino)pyridine complexes (Fig. 6). In the crystals of one of the compounds a rather close intramolecular Au⋯H–N contact with an H⋯Au distance of 2.55 Å and an angle Au⋯H–N of 117° was observed. The planes of the pyridine and the pentafluorophenyl groups are close to parallel which places one of the fluorine atoms next to the NH hydrogen atom. The gold atom is further engaged in two aurophilic contacts established through the stacking of the monomers. There is no spectroscopic evidence of an effect of these Au⋯H–N and N–H⋯F interactions on the molecular dynamics (e.g. restricted rotation of substituents to be noticed in NMR spectra of solutions).129
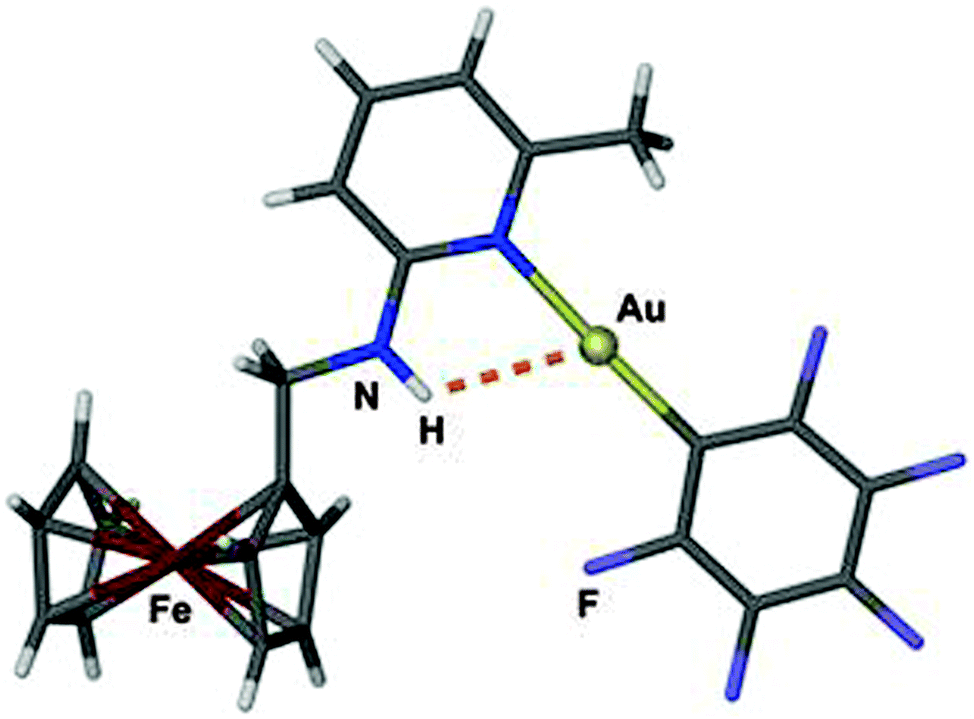 | ||
| Fig. 6 Structure of the monomer in a crystal of {2-methyl-6-(ferrocenylmethylamino)pyridine}AuC6F5; the Au⋯H–N contact is shown as a dashed orange line.129 | ||
A similar case was found in the product of the insertion of benzonitrile into a [{(NHC)Au}2OH]+ complex cation (30, Fig. 7). The conformation of the cation in the crystal allows the N–H group of the imide unit to maintain a close contact to one of the two gold atoms. The angle Au⋯H–N of 143° may indicate that the H atom is drawn to the gold atom. However, the chemical shift of the NH proton concerned is not unusual (δ 5.13 ppm) and no IR anomalies have been reported.130 It is to be noted also that any rotation of structural units about the C–N bond of the molecule away from OCNH coplanarity would be detrimental to the amide resonance system.

 | ||
| Fig. 7 Structure of the cation [{(IPr)Au}2(PhCONH)]+ of the BF4− salt; irrelevant H atoms are omitted for clarity; the Au⋯H–N contact is shown as a dashed orange line.130 | ||
The largest part of the remaining examples with Au⋯H–N contacts has the common structural principles shown in formulae 31 and 32. Therein, one or two ligands are attached to the gold atom through a thione function bearing an N–H group in the γ-position relative to the gold atom. In the simplest case, the ligands are thiourea molecules or their selenium analogs. It should be noted that rotation of the thiourea unit about the Au–S bond does not change the Au⋯H distance and leaves the π-delocalization of the ligand unaffected. However, any rotations about the S–C and C–N bonds make the Au⋯H distance longer and would be detrimental to the π-system. Therefore, the conformation with a planar urea core structure and with the Au atom in the thiourea plane is certainly the preferred array even in the absence of any Au⋯H attraction.
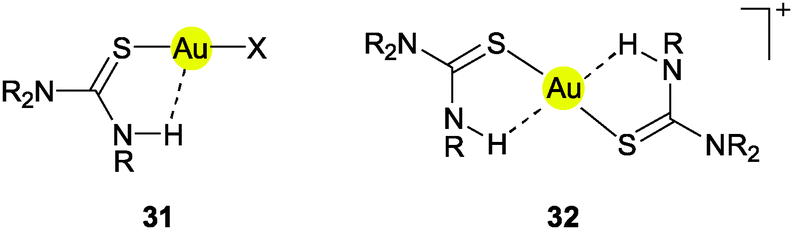
The crystal structures of bis(thiourea)gold(I) complexes [{(H2N)2CS}2Au]X (32X, R = H) have been determined with X = Cl, Br or ½SO4.131,132 In all cases the ligand molecules adapt small torsional angles Au–S–C–N (between 4 and 26°) which bring the N-bound H atom closest to the gold atom. The relevant distances Au⋯H and Au⋯N are in the ranges of 2.75–2.95 and 3.30–3.40 Å, respectively, with angles Au⋯H–N close to 120°. As explained above, the observed conformation is probably largely determined by the influence of the π-system within the ligand and not by Au⋯H–N hydrogen bonding. Complexes of the type 32 of substituted thioureas (RHN)2CS with R = Me, Et show the same standard structures.133
A closer investigation of possible Au⋯H–N interactions in crystalline gold(I) complexes of thioureas shows that in addition to the intra-cationic contacts there are also comparable inter-cationic contacts. Two out of four crystallographically independent cations present in the crystal structure of 32X (R = H, X = Cl− and ½SO42−) are organized in chains through two types of Au⋯H–N contacts with Au⋯H and Au⋯N distances of 2.812/2.687 and 3.636/3.521 Å and large angles of 161.1 and 163.8°. The intercationic contacts are in an interplay with contacts to the anions, and several conformations with different dihedral angles between the two SCN2 planes can be present in one crystal. The dihedral angles between the two urea planes may assume values between 0 and 180°, and the axis S–Au–S may be bent. The Au⋯H–N contacts have not been considered by the authors.131
In partially alkyl-substituted derivatives (32 with in part R ≠ H), the intermolecular contacts are absent owing to steric hindrance.132,133
In the crystal structure of the AuCl complex of N,N-diethyl-N′-camphanoylthiourea (33, Cam = camphanoyl) two crystallographically independent molecules are present in the asymmetric unit. It is an interesting observation that the C3N plane of the Et2N–C fragments again has the expected coplanarity with the thiourea reference plane N2CS, but the NH–C(O)– plane is rotated out of this reference plane. Obviously, the resonance of the amide group reduces the energy barrier for (S)C–N rotation in the latter. In the resulting conformation, the N–H groups are coming closer to the gold atoms of adjacent molecules with Au⋯N distances of 3.6 and 3.7 Å and angles 125 and 127° respectively. However, based purely on these values, it is hard to estimate the contribution of such Au⋯H–N contacts to the stability of the complex.134

Selenourea compounds of the type [(L)AuSeC(NH2)2]+ with L = PMe3, PPh3, and ½(dppm) have the expected related characteristics,135,136 known also for the thiourea complex [(Cy3P)AuSC(NH2)2]+.137
A group of related structures has been found for the gold(I) thiocarbazone complexes in which the thiourea unit is extended as shown in formulae 34 and 35. Hydrogen atoms of NH(R) groups are again positioned near the gold atom with Au⋯H distances of ca. 2.80 Å and with angles Au⋯H–N near 115°. The ν(NH) vibrations are found ca. 100 cm−1 lower than for the free ligand (3100 vs. 3210 cm−1). The atoms S2AuH2 are in a common plane. Another typical example is the cationic 2:1 complex of the related 2-pyridylformaldehyde derivative (36, R = 2-pyridyl)138 where the geometry of the Au⋯H–N contacts (Au⋯H 2.70–2.80 Å) is very similar to that in the parent thiourea complexes. The structure of a trinuclear complex with a thiosemicarbazone derived from 2-diphenylphosphino-phenylformaldehyde (36, R = (C6H4-2-PPh2)AuCl) shows the same characteristics.139 The authors of the report concerned have pointed out that there may be an Au⋯H–N contact and addressed it as a “weak agostic interaction”. The Au⋯H and Au⋯N distances are 2.89 and 3.421 Å with an angle Au⋯H–N of 120.4°, and in the related dinuclear complex 37 the corresponding data are 2.84 and 3.327 Å, and 119.4°. As indicated for 37, the molecule also features a short Au⋯H–C contact (Au⋯H 2.86, Au⋯C 3.389 Å, Au⋯H–C 115.1°). The hydrogen atoms concerned are further involved in stronger intermolecular N/C–H⋯Cl contacts with the corresponding angles 158.0 and 136.6° (not shown in formula 37).139

In crystals of gold(I) complexes with 3-nitrobenzaldehyde thiocarbazone ligands (R = 3-NO2-C6H4 in 36), the cations form dimers through Au⋯Au and S⋯S contacts integrating the chloride anions through N–H⋯Cl hydrogen bonding. As expected for the fundamental geometry, there are also Au⋯H–N contacts of the types indicated in 35 and 36, but the authors have not referred to this aspect.140
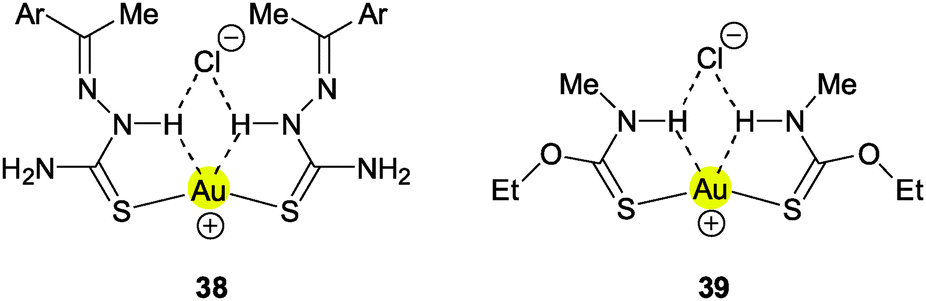
Simple ion pair formation is observed with the corresponding 4-bromo- and 3,4-dichlorobenzaldehyde thiosemicarbazone ligands (38, Ar = C6H4Br, C6H3Cl2). In both cases the S–Au–S axes are bent significantly, away from linearity, which brings the Au atom closer to the N–H hydrogen atoms, but the distances are still quite long (Au⋯H 2.859 and 2.885; Au⋯N 3.378 and 3.393 Å), and therefore a bonding contribution from Au⋯H–N remains uncertain because of the presence of competing N–H⋯Cl hydrogen bonds and an attraction of the AuI center to the Cl− anion.141 This is also true for the (triethylphosphine)gold(I) complex of the menadione bisulfite thiosemicarbazone ligand.142
Gold(I) thiocarbamate complexes are closely related to thiourea complexes. The cationic 2:1 complex with N-methyl-O-ethyl-thiocarbamate therefore has a similar structure forming also a cyclic ion pair with the chloride anion through N–H⋯Cl hydrogen bonding (39). In this association, the S–Au–S angle is again bent (155.3°), probably owing to AuI⋯Cl− attraction. The long Au⋯H distances of 2.987 and 2.993 Å show that these contacts are unlikely to contribute significantly to the overall stability.143
Another closely related group of gold(I) complexes with potential for Au⋯H–N hydrogen bonding is formed with imidazolidine-2-thione ligands which also contain the thiourea framework.144 Structures of 1:1 complexes have been determined for examples with imidazolidine-2-thione and its 1-methyl derivative (40, R = H, Me),144,145 and with 1-ethyl-imidazolidine-2-thione (40, R = Et).146 The intramolecular contacts Au⋯H–N have very long Au⋯H distances of 2.963 and 2.868 Å (two crystallographically independent molecules in the asymmetric unit). Therefore, the authors have drawn more particular attention to some intermolecular Au⋯H–C contacts.144 For the same reason, Au⋯H–N interactions have not been considered by the authors for (imidazolidine-2-thione)AuCN (type 40) and [(dimethylthiourea)2Au]+ [Au(CN)2]− (type 32), where again Au⋯Au contacts appear to dominate.145 The 2:1 complex with imidazolidine-2-thione and the counterion Y− = AuI2− has a centrosymmetrical cation with an Au⋯H distance of 2.871 Å, but the major emphasis in the structural discussion has been directed to intermolecular N–H⋯I contacts (N⋯I = 3.818 Å, angle N–H⋯I = 146.23°) not considering the possibility of intermolecular Au⋯H–N contacts (N⋯Au = 3.430 Å, angle N–H⋯Au = 146.35°).145,147 Using 1-propylimidazolidine-2-thione 41b (R = Pr) with Cl− as the counterion, the Au⋯H distance is slightly reduced to 2.777/2.805 Å, but the cations are associated with the Cl− anions via hydrogen bonds N–H⋯Cl.148 Without substituents R, the form 41a is preferred. In the related compound with 3-methylimidazolidine-2-thione and a bis(arylsulfonyl)amide anion the Au⋯H distances are increased to 2.971 Å probably owing to the interference of interionic N–H⋯O hydrogen bonds.149 A Ci-symmetrical structure has been found for the related 2:1 gold(I) complexes with 4-amino-3-methyl-1,2,4-Δ2-triazoline-5-thione (42), where the Au⋯H contact is long at 2.967 Å, probably also due to an interference of concomitant hydrogen bonding N–H⋯Cl to the anion.150
An intramolecular Au⋯H–N contact appears in the crystal structure of the 1:1 complex of ω-thiocaprolactam-κS with AuBr (43). The alignment of the N–H and Au⋯S vectors arising from a small Au–S–C–N torsional angle leads to intramolecular Au⋯H distances of 2.814 and 2.819 Å which are at the lower end of the range observed for most of the related compounds. Two independent molecules present in the asymmetric unit are associated via aurophilic contacts. The authors did not comment on this structural detail, the focus being mainly on the aurophilic interactions in a chain-like organization of the molecules. There is little doubt that the approximate coplanarity of the Au–S and N–H atoms is largely determined by the thioamide resonance.151

Close Au⋯H distances of 2.651 Å can be found in a cation of the 2:1 complex with pyridine-2-thione (44) with perchlorate as the counterion. The crystals contain a pentanuclear unit (aggregated via aurophilic interactions) and a mononuclear unit, the individual cations having different conformations regarding the positioning of the NH groups. In these orientations they become involved in hydrogen bonding with the anions. The authors did not draw attention to the contacts of N–H or C–H units with the metal centers.152 The corresponding system with chloride cations was studied later in the context of Au⋯H–C contacts (65, below). The pyridine-2-thione in the 1:1 complex with AuCl has a similar conformation with a small Au–S–C–N dihedral angle, and the intramolecular distance Au⋯H is found at 2.820 Å, not considered by the authors (45). The overall coplanarity of the relevant atoms in the monomer is mainly due to the extended resonance system. The monomers are aggregated through aurophilic bonding into chains where the N–H groups also engage in intermolecular hydrogen bonding, N–H⋯Cl. Both these interactions are held as the major structure-determining forces.153

In a complex of AuCl with a tertiary phosphine bearing an enamine substituent, the amino group is clearly orientated towards the gold atom, but the conformation detected in the crystals has an Au⋯H distance of 2.912 Å which does not indicate any major interaction (46).154 A conformational preference could be inferred in gold(I) complexes of 2-aminophenylphosphines (47) with a gold atom in a rare trigonal planar configuration. However, the authors have pointed out that the Au⋯H distances below and above the trigonal plane are in the range of 2.91–3.13 Å and the Au⋯H–N angles are quite small (99–115°), casting doubt on the significance of these contacts.155 The Au⋯H–N contacts in a two-coordinate complex with (2-aminophenyl)diphenylphosphine ligands are much shorter, at 2.767 Å. The neighbouring diphosphinimide counterions Ph2P(O)NPPh2S form N–H⋯O hydrogen bonds to the amino group concerned linking the monomers into chains. The possible influence of an Au⋯H–N interaction has not been discussed.156 In a very special example, crystals of a complex of AuCl with 2-aminopyridine contain both the molecular 1:1 and the cationic 2:1 complex (48), with Au⋯H distances as short as 2.607 Å and angles Au⋯H–N at 118.3°.157 A pyridine ligand with an amide group in the 2-position also becomes attached to a gold(I) center in a conformation bringing the amide N–H group close to the metal atom (Au⋯H 2.851 Å) (49, R = ferrocenyl). This is the expected arrangement considering the pyridine–amide conjugation.158

In the polymer of gold(I) 2-amino-benzimidazolide (50), each of the hydrogen atoms of the amino groups are placed near a gold atom with a distance Au⋯H of 2.849 Å. On the one hand, in this particular case the overall arrangement is of course determined by the rigid framework and cannot be attributed only to a significantly attractive Au⋯H interaction (not considered by the authors), but on the other hand such an interaction cannot be excluded.159 In a cationic complex [(Ph3P)AuL]+, with L being an N-bound 3,5-diarylpyrazole ligand (51, Ar = 4-BuO-C6H4, X = tosylate) the N–H group is in closer proximity (Au⋯H 2.772 Å) to gold, but the hydrogen atom is also engaged in an N–H⋯O hydrogen bond with a sulfonate counterion. The same observation has been made for the corresponding [AuL2]+ cation. The short distance and the small angle Au⋯H–N of only 94.9° are of course determined solely by the standard configuration of the heterocycle, and the authors have refrained from a discussion of this structural detail.160 The 1:1 complex of AuCl with a “calixpyrrole” that contains two NH, one PhP and one S units at the rim of the ligand was shown to appear in two different configurations (out and in). There are contacts Au⋯H–N (Au⋯N = 3.738 and 3.797 Å) in the latter which are much longer in the former, and this may be the reason for a clear preference of the in isomer (ratio 3:97) in equilibrated solutions (Fig. 8).161
 | ||
| Fig. 8 Schematic representation of the orientation of the Au–Cl unit in the two conformers of the complex with a “calixpyrrole”; Au⋯H–N contacts are shown as dashed orange lines; H(C) atoms are omitted for clarity.161 | ||
In the discussion of the interplay of hydrogen bonding with aurophilic interactions in crystals of the dimeric Schiff base complex C6F5Au(HNCPh2), the authors have focused mainly on intermolecular N–H⋯F interactions ignoring the possibility of Au⋯H–N contacts (Fig. 9). Although the corresponding Au⋯H/Au⋯N distances are long (2.943/3.536 Å; Au⋯H–N 127.7°), the energy of the contacts may be competitive with regard to the proposed interaction across distances H⋯F/F⋯N of 2.75/3.224 Å and the angle F⋯H–N = 116.1°. It is noteworthy that the structure of the analogous AgI complex is very different (parallel vs. antiparallel orientation of the ligands at neighboring metal atoms), but this change of the relative orientation of the individual units may have several reasons.162
 | ||
| Fig. 9 Molecular structure of the dimer [(HN = CPh2)AuC6F5]2. Dashed lines indicate aurophilic interactions (golden), N–H⋯F hydrogen bonds (bluegreen) and Au⋯N–H contacts (orange).162 | ||
Intermolecular Au⋯H–N associations may be present in crystals of a (triarylphosphine)gold(I) alkynyl complex bearing an urea function designed for anion sensing (52).163 The molecules can be described as tetramers associated through Au⋯H–N contacts with standard dimensions of 2.944 and 3.658 Å and 141.8°, seconded by Au⋯H–C contacts (2.954, 3.474 Å, 116.8°).
Finally, Au⋯H–N contacts have also been observed in a variety of dicyanoaurates(I) that are combined with divalent transition metal cations (Cu, Zn, Ni) carrying amine ligands like ethylenediamine (en), tris(2-aminoethyl)amine, or propanolamine. The distances Au⋯H and Au⋯N are all large (3.084, 2.957, 2.928, 2.917; 3.841, 3.848, 3.813, 3.683 Å) and associated with large angles (162.0, 161.1, 161.5, 144.04°). The similarity of the values for the three structures originates from their isostructurality.164–167 Formula 53 shows the interionic contacts in [Cu(en)2]+ [Au(CN)2]−.164 However, in no case there is an obvious and unequivocal preference for an orientation of N–H functions towards the Au atoms.
In crystals of alkynylgold(I) complexes where the auxiliary ligand features a terminal amino group, as in (4-amino-pyridine)AuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6F5 and (m-Tol)3PAu(4-aminophenylethynyl), intermolecular Au⋯H–N contacts are present. The Au⋯H and Au⋯N distances are quite long (2.902 and 3.114; 3.752 and 3.899 Å), but the angles (169.5 and 152.9°) show consistent directionality (Fig. 10). The authors have pointed out that these Au⋯H–N contacts are a rare phenomenon which should not be ignored but rather be considered as weak hydrogen bonds.168,169
CC6F5 and (m-Tol)3PAu(4-aminophenylethynyl), intermolecular Au⋯H–N contacts are present. The Au⋯H and Au⋯N distances are quite long (2.902 and 3.114; 3.752 and 3.899 Å), but the angles (169.5 and 152.9°) show consistent directionality (Fig. 10). The authors have pointed out that these Au⋯H–N contacts are a rare phenomenon which should not be ignored but rather be considered as weak hydrogen bonds.168,169

 | ||
| Fig. 10 1D zig-zag motif in a crystal of {(3-MeC6H4)3P}Au–CCC6H4-4-NH2 assembled via intermolecular Au⋯H–N contacts, shown as dashed orange lines.169 | ||
To close this section which illustrates the uncertainties about the significance of the Au⋯H–N contacts considered, a particularly interesting case is to be mentioned. In 1995 the crystal structure of chloro(dicyclohexylphosphinoyl-N-methyl-thioformamide)gold(I) was published. In the search for any short Au⋯H–N distances in this molecule it has now been discovered that in its monomeric units (several independent molecules in the unit cell), conspicuously short Au⋯H contacts are indeed established, the shortest being 2.248 Å, but it involves a methyl hydrogen atom (Au–C 3.191 Å; Au⋯H–C angle 160.2°) (54a).170 Upon inspection of the formula of the monomers one may find it surprising that the NHMe group is in a conformation with the methyl group close to the gold atom, because a rotation by 180° about the N–C(S) bond (syn–anti) would give the methyl group more space and place the N–H group near the gold atom. However, when taking into account that the monomers are in fact associated to dimers via two strong N–H⋯O hydrogen bonds (54b) it becomes obvious that there is no reason to believe that the orientation of the N–Me unit is determined by Au⋯H–C interactions. Even one of the shortest Au⋯H contacts so far found in the literature is thus not an example of significant Au⋯H–X hydrogen bonding interactions.

Results compiled from crystallographic data for Au⋯H–N contacts are summarized in the histograms shown in Fig. 11. Within the applied cut-off the Au⋯H distances do not show a really pronounced maximum up to the 3.0 Å limit which indicates that there is no real optimum distance for the underlying contacts. This is in contrast to standard hydrogen bonding like O–H⋯O, where such a maximum is immediately obvious. The Au–N distances go through a maximum near 3.3 Å mainly because the associated Au⋯H–N angles also go through an early maximum at ca. 113°. This result suggests that the energy profile of the underlying interactions for the approach of the H–N group to the Au center must be very flat with no pronounced preference for a certain distance. It appears that in almost all cases examined, the geometry is predominantly influenced by other effects which overrule the very minor contributions from the Au⋯H–N contact. Again, for Au⋯H–N contacts, as for their Au⋯H–O counterparts, with very few exceptions,138 there is no evidence as yet for the solution state. No spectroscopic investigations have been reported.
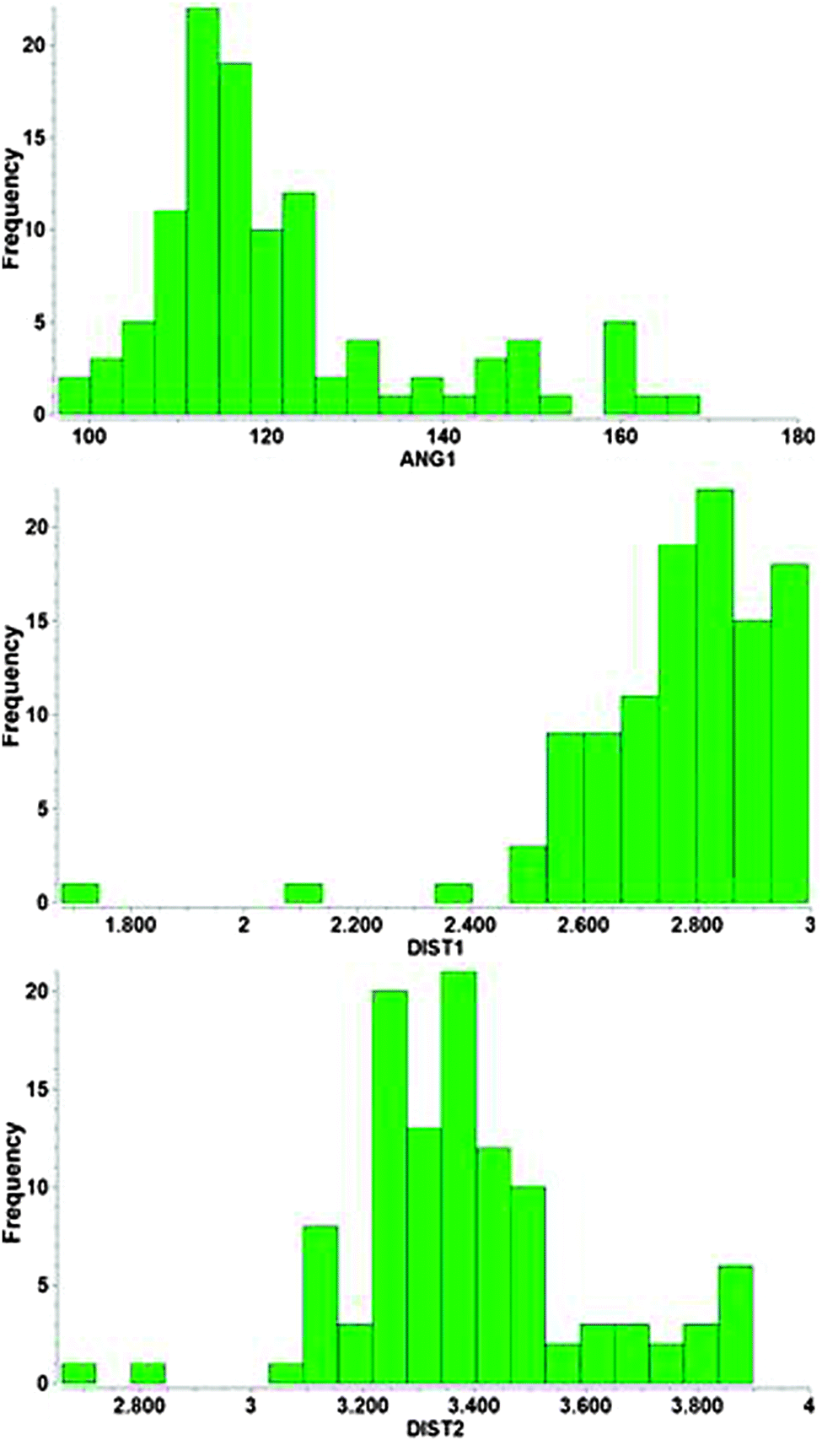 | ||
| Fig. 11 Histograms representing the parameters for Au(I)⋯H–N contacts of 66 hits from a CSD search with cut-offs applied for angles Au⋯H–N (ANG1, in °) and distances Au⋯H (DIST1, in Å) and Au⋯N (DIST2, in Å). | ||
(Triphenylphosphine)gold(I) halides, (Ph3P)AuX, have been the most widely used precursors in the coordination chemistry of gold, and hundreds of crystal structures of compounds with (Ph3P)Au units have been documented. From this work there is a plethora of structural data which provide an excellent basis for a screening of their conformational details.
In early statistical analyses174–176 of the conformation of adducts Ph3PE (where E stands for acceptor elements like the chalcogens, but also for metal atoms with their set of ligands) it has been shown that the three phenyl groups may be arranged not only in the two intuitively expected enantiomeric rotor or propeller type conformations (C3 symmetry), but also with one phenyl ring either parallel or orthogonal to the relevant C–P–E plane and the other two at angles as in the rotor type (Fig. 12). The energy differences between these conformations are obviously surprisingly small and many minor variations are tolerated which are best described by their three torsional angles. These angles may deviate from the three standards within large margins. An evaluation of more than 8000 structures known in the year 2000 (and there are many more examples now) has shown for the solid state an almost equal distribution over the three alternatives for E taken from various parts of the periodic table. In the crystals (not in solution) specific intermolecular interactions between Ph3P ligands of molecules or ions are established, which are described as various forms of “phenyl embraces” (PEs). For a participation of all six phenyl groups (6PE) in a fully symmetrical “embrace”, the energy of the interaction established upon pairing may reach as much as 12.5 kcal per pair. The pairing mode depends on the influence of E and its ligands.
 | ||
| Fig. 12 Space-filling models of the three most abundant conformations of the Ph3P ligand in a Ph3P–M moiety (from left to right: rotor-type, parallel – with one of the phenyl rings ca. parallel to the P–M vector, orthogonal – with one of the phenyl rings ca. orthogonal to the P–M vector).174 | ||
Surprisingly, a careful consideration of the examples with E = Au has shown that for linearly coordinated gold(I) there is a significant anomaly in that the PEs are formed more frequently from units with parallel P–C vectors than for complexes with E ≠ Au.175 An examination of many examples shows, however, that the conformations may be mainly due to the fact that the absence of ligands at the gold atom other than the one trans to the phosphine ligand allows particularly close contacts which may also include aurophilic interactions. The conformation of the PPh3 phenyl groups thus does not reflect the preferred orientation of one phenyl group which would optimize an Au⋯H–C interaction which is the focus of the present screening.
For the specific conformation concerned, a simple calculation can give relevant benchmark values: the conformation with the parallel orientation of one phenyl ring to the C–P–E plane concerned brings an ortho-hydrogen atom closest to the attached atom in E (Au) (55). Based on standard bond lengths and angles which appear with only small variations in all (Ph3P)AuX complexes [Au–P 2.260, P–C(aryl) 1.828, C–C(aryl) 1.395, H–C(aryl) 1.10 Å; Au–P–C 115°, P–C–C 120°, C–C–H 120°] the distance Au⋯H under relaxed conditions appears to be 2.933 Å. In this conformation, the angle Au⋯H–C is 117°. Both values may vary considerably (±0.15 Å, ±3°) upon minor alterations of e.g. the Au–P–C angle which is given as an average over many experimental data. All other conformations have the hydrogen atoms further apart from the gold atom. Therefore, other influences excluded, a high abundance of this parallel conformation which has the shortest possible Au⋯H distance in strain-free (Ph3P)Au units could have been indicative of an energetic preference originating from Au⋯H–C interactions. An inspection of the structures has shown, however, that the particularly open coordination sphere of two-coordinate gold favours specific forms of the ubiquitous intermolecular phenyl embraces in the crystals. An example of a common mode is shown in Fig. 13, taken from the crystal structure of (Ph3P)AuSC(S)OEt. The association may be complemented in some cases by aurophilic bonding.177,178 The results of the extensive statistical investigations therefore gave no obvious reason to assume that Au⋯H–C bonding has a significant influence on the molecular structures of (R3P)AuE compounds.174,175

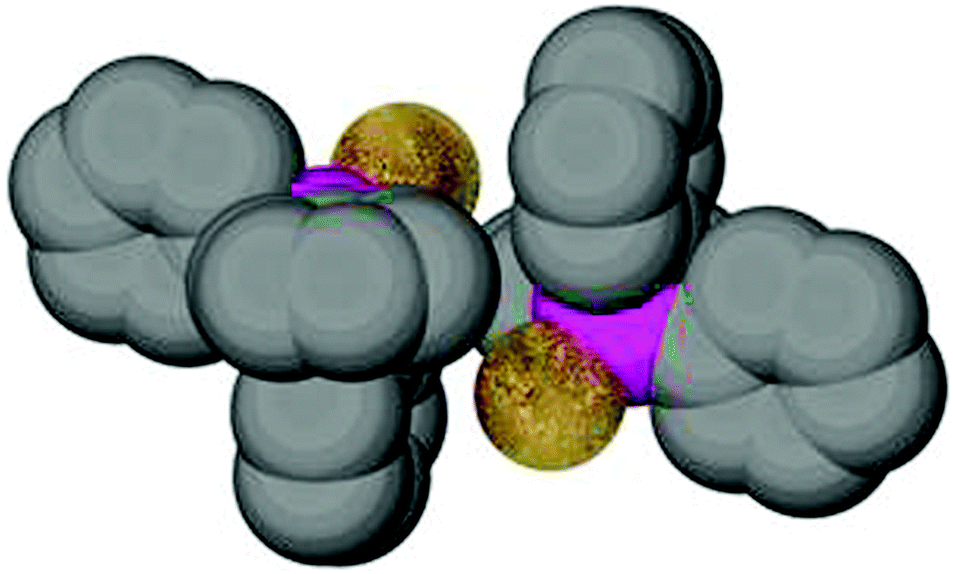 | ||
| Fig. 13 Arrangement of two (Ph3P)AuS units of the dimers present in a crystal of (Ph3P)AuSC(S)OEt. The dimers are further involved in multiple phenyl embraces with neighboring molecules.175 | ||
In solutions of (Ph3P)AuX complexes, there are generally monomers with virtually free rotation of the phenyl groups about the P–C bonds as demonstrated by NMR spectroscopy indicating that there is no significant extra barrier other than common steric interactions between phenyl groups. The low barrier to rotation of a phenyl group shows that if there is any Au⋯H–C interaction, it must be only very weakly attractive. The mechanism and pathways of interconversions of propeller rotamers have been studied in an early work by Mislow.179
In gold(I) complexes with triarylphosphines bearing substituents at the phenyl rings, the rotation about the P–C bonds is sterically hindered as the number and size of these substituents are increased and as they are placed close to the ipso carbon atoms (i.e. in ortho-positions). Trimesityl- and tri-o-tolylphosphine are examples in case, and their AuCl complexes have the rotor conformation with a high barrier to rotation as demonstrated by solution NMR.180–182 There is no evidence that Au⋯H–C interactions of ortho- or methyl hydrogen atoms would co-determine the structures and the limitations of the internal mobility. A comparison with the tri-m-tolylphosphine or diphenyl(2-pyridyl)phosphine complexes shows that there is no difference in the general conformational performance.183,184 Even in cases where the aryl rotation is less hindered owing to reduced crowding, such as in complexes with secondary or primary arylphosphines, the conformation observed in crystal structures is not indicative of any Au⋯H–C attraction. The examples investigated were (o-tolylphosphine)gold bromide and {bis(o-tolyl)phosphine}gold chloride.185,186
The validity of these general considerations can be tested by an inspection of representative compounds with Ph3P or other tertiary phenylphosphine ligands. In cases where for instance one or two (Ph3P)Au units are present at the sulfur donor atom of the quinoline-8-thiolate anion (56a,b), the orientation of the phenyl groups is indeed almost random and gives no hint of a conspicuous preference for parallel phenyl orientations.187 The same is true for examples with alkynyl substituents at (Ph3P)Au (57), even though therein the linearity of the –CC– group leaves the space at the gold atom wide open.188
In crystals of dinuclear gold(I) complexes with bis(diphenylphoshino)methane (dppm) or -ethane (dppe) and 1-methyl-tetrazole-5-thiolato ligands (58, n = 1, 2; Fig. 14 for n = 1) the Au⋯H–C contacts involving phenyl groups have Au⋯H distances of 2.82 – 3.10 Å and the Au⋯H–C angles fall in the range of 115–123°. These data reflect the random orientation of the aryl groups. The authors of this work have discussed the structural data in terms of “weak agostic interactions”, but there is no evidence for Au⋯H–C bonding from spectroscopic data, and the distances and angles are not unusual as shown above.189

 | ||
| Fig. 14 Molecular structure of the 1:2 complex of bis(diphenylphoshino)methane (dppm) and gold(I) 1-methyl-tetrazole-5-thiolate, with an aurophilic interaction (golden) and Au⋯H–C contacts (green), shown for one of the independent molecules.189 | ||
In the literature of the 1980s, several cases of surprisingly short Au⋯H–C contacts were presented and discussed in terms of attractive interactions. For example, an intramolecular Au⋯H–C contact involving a methylene group of the tripodal ligand MeC(CH2PPh2)3 in its trinuclear complex with AuCl was observed to show a distance Au⋯H of only 2.485 Å (59). For two legs of the tripod ligand, the conformation appears to be determined by aurophilic interactions, but for the third leg concerned there is no obvious conformational pressure that would direct the gold atom to methylene hydrogen atoms. Therefore, this example is one of the puzzling cases in the present context. It is very likely that the pressure of molecular packing in the crystal causes the backbending of all parts of the molecule. Unfortunately, no such crystallographic details are available.190
A similar situation was detected in the dinuclear complex of 2,2′-bis(triphenylphosphinegold)-diphenylmethane (60). Each of the two hydrogen atoms in the methylene bridge shows a contact with a different gold atom, but the Au⋯H distances are very long (in the range of 2.92–3.06 Å). The IR bands for ν(CH2) of the complex are shifted by ca. 100 cm−1 as compared to the bands of free diphenylmethane. Moreover, the 1H NMR spectrum is temperature-dependent and shows diastereotopic CH2 protons at −95 °C, which become virtually equivalent at room temperature. Because of the absence of aurophilic bonding and no obvious other causes for restricted molecular dynamics, the authors have attributed their observations to significant Au⋯H–C bonding (60). Unfortunately again, no crystallographic details are available.191 In the light of the above considerations, the Au⋯H distances of ca. 3.00 Å do not suggest significant interactions, and the IR and NMR characteristics are probably due to the different structural restraints in the free and complexed ligands, the influence of the metal coordination, and tentative assignments.
In the cyclic dinuclear complex obtained upon substitution of the two Ph3P ligands of 60 by 1,2-bis(diphenylphosphino)ethane (dppe) only one short Au⋯H–C contact is established with d(Au⋯H) at 2.62 Å while the other one is much longer at 3.20 Å (61). The representation in the scheme in the publication is not very clear and may be misleading.192 In the related 1,2-diphenyl-ethane compound (62) both of the bridging methylene groups are close to a gold atom. The two types of Au⋯H distances (there is a center of inversion in the molecule) are 2.75 and 2.99 Å and the Au⋯H–C angles are 107 and 120°. The ν(C–H) bands are shifted to lower wavelengths as observed for the diphenylmethane analog, but the 1H NMR signal of the CH2 groups is not resolved at low temperature (–90 °C in CH2Cl2). The tetranuclear dication 63 (Ci symmetry) has four intramolecular Au⋯H contacts involving all four gold atoms and all four methylene hydrogen atoms with Au⋯H distances of 2.6 and 3.0 Å (Fig. 15).192 In a later communication, correct data for the same compound, d(Au⋯H) 2.71 and 2.88 Å, are reported.193

 | ||
| Fig. 15 Structure of the tetranuclear dication with pairs of Ph3Au units attached geminally to the 2,2′-positions of 1,2-diphenylethane in the BF4− salt; irrelevant H atoms are omitted for clarity; aurophilic interactions and Au⋯H–C contacts are shown as dashed golden and green lines, respectively.192 | ||
According to the proposed assignments, the ν(C–H) bands in the IR spectrum of 63 are shifted to a surprisingly low value of ca. 2690 cm−1 and the 1H NMR signal of the CH2 protons becomes broadened at low temperature, but the origin of these phenomena is not clear. Notably, all four P atoms give only one sharp 31P NMR signal even at −95 °C (in CD2Cl2).192 The many conformational variations observed for all four examples 60–63 with concomitant consequences for intramolecular distances make it unlikely that the alleged Au⋯H–C interactions have a major influence on the individual structures. Some of the changes in the spectroscopic features occurring for the ligands upon complexation to AuI can be ascribed to the enforced alterations of their structures in the complexed form, but for the IR-assignments (above) there is as yet no further confirmation.
To rationalize the conformation found in crystals of (triphenylphosphine)dibenzoylmethylgold(I) a conspicuous Au⋯H–C contact has been discussed, the closest approach (Au⋯H 2.607, Au⋯C 3.413 Å; Au⋯H–C 129.02°) being found for an aryl group of the dibenzoylmethyl unit (64). However, other factors cannot be excluded to explain the arrangement of this molecule.194
Interionic Au⋯H–C (aryl) contacts between cations and anions have been considered very recently for the solid state structure of [(Ph3P)2Ag]+ [Au(CN)2]− where three long Au⋯C distances of 3.668, 3.662 and 3.645 Å were measured. The authors do not mention the angles which are 163.31, 81.60 and 81.73°, respectively. These geometrical parameters do not indicate any contacts with bonding significance.195
Turning from mixed complexes with phosphine ligands to coordination compounds with heterocyclic ligands, the structural situation regarding intramolecular Au⋯H–C contacts is much more diverse. A detailed study of complexes formed between chlorogold(I) and 2- and 4-pyridine thione ligands has led to remarkable results.196 In the bis(pyridinethione) gold complexes obtained, the two ligands were found to be coplanar or twisted symmetrically in both isomers to form centrosymmetric conformers which have an o-C–H group of each pyridine ring close to the metal atom (65a, 65d). DFT calculations have shown that the energies of these and the other two possible rotamers of 65 (Ci and Cs symmetry for 65b and 65c, all in the gas phase) are very close. The experimental data for the structures have been well reproduced (Au⋯H found 2.83, 2.88, calcd 2.884 Å in 65a; found 2.86, calcd 2.863 Å in 65d). The overall results could mean that the Au⋯H–C contacts in 65a are clearly preferred over the equally possible Au⋯H–N contacts (65c). However, since in both types of crystals that contain 65a and 65d, respectively, there are hydrogen bonds N–H⋯Cl between cations and anions, in the condensed phase these interactions may overrule the Au⋯H–N alternative. The calculations for the isolated cation 65c have indeed shown that the Au⋯H–N contacts would be much shorter (at 2.659 Å) than the calculated (and experimentally observed) Au⋯H–C distances even though there may be Coulomb repulsion (N+ ↔ Au+) in this conformer. The IR or NMR spectroscopic results for compounds 65a, 65d have not been evaluated regarding the effects of the Au⋯H–C contacts. In mass spectrometric studies a mixed ligand cation with thione-bonded bis(pyridine-4-thione)sulfide and 4-pyridinethionato groups was observed, for which DFT calculations also showed two similar Au⋯H–C contacts (Au⋯H 2.891 and 2.900 Å). The authors of this important contribution196 have discussed their results in the context of those available on other structures for which Au⋯H–C interactions had been considered (and which are also discussed in the present review). It has been noted that the Au⋯H distances cover a broad range from 2.50 to 3.20 Å and that it is as yet unclear if significant energy contributions arise from these contacts, which were addressed as “γ-agostic interactions”. The centrosymmerical structures of the cations 65a and 65d probably represent the energy minimum in the chloride salts largely owing to the minimization of Coulomb repulsions compared to effective molecular stacking in the crystal and to cation–anion hydrogen bonding. It is important to remember the report on the corresponding perchlorate salt, [(2-pyridinethione)2Au]ClO4,152 where, amongst others, cation conformations of type 65c have been found which appear to be directed by N–H⋯O hydrogen bonding with the counterions (compare 44, above).

It has been shown above that 2:1 complexes of thiocarbazone ligands with AuI feature intra-cationic contacts involving Au⋯H–N units and Cl− counterions (35, 36).138 If the substitution pattern is modified by introducing a hexamethyleneimine group, the conformation of the cation is changed to a rotamer in which Au⋯H–C contacts are established instead of Au⋯H–N contacts (66).138 The two Au⋯H distances in the centrosymmetrical cation are short at 2.793 Å and the two Au⋯H–C angles are relatively large at 141.9°. In the absence of other experimental evidence, it remains unclear if this conformation is directed by this Au⋯H–C contact.
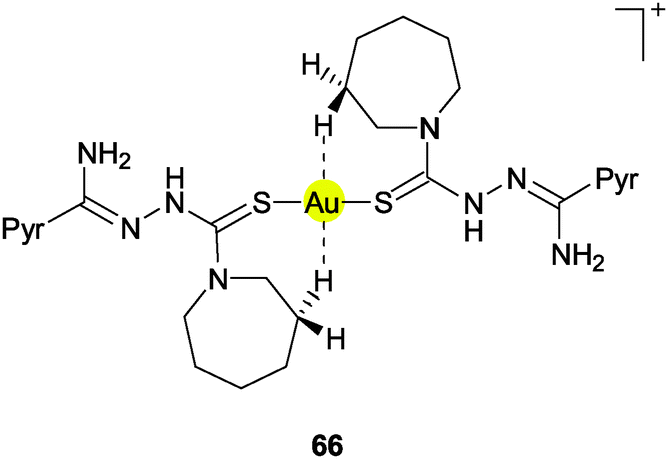
In crystals of neutral carbene complexes of the type (IPr)AuX (67), and of salts of the cationic complexes [(IPr)2Au]+ (68) and [{(IPr)Au}2OH]+ with various anions, some of the iPr groups are always held in close intramolecular contact with a gold atom.129

The situation is similar for alkyl-substituted heterocyclic carbenes. Obviously determined by the steric interactions of the substituents, the planes of the heterocycles RImR′ are either coplanar (R = R′ = Me; R = Me, R′ = Et) or twisted (R = R′ = tBu, 69). Invariably, this leads to longer and shorter Au⋯H–C contacts, but these show no conformational preference.197 The situation is comparable in the dinuclear complexes with N-methyl-N′-2-pyridylmethyl-imidazolylidene (70). Both homonuclear Au/Au and heteronuclear Au/Ag complexes have either methyl or methylene hydrogen atoms near the metal atoms. The contacts can be intra- and intermolecular and appear to arise primarily from the optimized packing of the components accounting for the structural requirements of the bidentate ligands.198

Considerable structural work has also been carried out on gold(I) complexes of nitrogen acyclic carbenes (NACs).199 The authors have not drawn attention to the surprisingly close Au⋯H–C contacts that occur in several of the investigated examples: in the doubly tbutyl-substituted compound 71, the shortest intramolecular Au⋯H distance is 2.204 Å with an angle Au⋯H–C of 141.15°. A close inspection of the syn,syn-conformation in the crystal shows that two methyl groups of each tbutyl substituent are staggered relative to the gold atom and that the overall structure is mainly determined by steric interactions. In crystals of analogs with smaller substituents and different substitution patterns, the syn,syn-conformation also prevails indicating that there is no specific additional interaction present in the tbutyl-substituted cases. NMR studies have shown that in solution other conformers (syn, anti) are also present which can be thermally interconverted.
In a much earlier study the carbene complex {Ph(Me2N)C}AuCl has been structurally characterized (72). It shows a close contact Au⋯H of only 2.204 Å, but the position of the methyl group is determined by the planarity of the substituents at the carbene carbon and at the nitrogen atom. The angles at the former do not suggest that the Au atom is drawn towards the methyl hydrogen atoms. Note also that the phenyl group is rotated out of the plane of the core unit Ccarbene(Au, N, Cphenyl) avoiding a close contact of the o-hydrogen atom at the gold atom.200
Short Au⋯H–C contacts have been discovered in crystals of chloro[(methoxy)(1-phenyl-thiazol-5-yl)carbene]gold(I). The molecules are arranged in tetranuclear assemblies held together mainly by aurophilic interactions (Fig. 16). The hydrogen atoms of the thiazole rings as well as of the methoxy groups are involved in intramolecular contacts with Au⋯H distances of 2.87 and 2.97 Å and 2.77 and 2.91 Å, respectively (two independent molecules in the asymmetric unit, 73a). The former interaction appears to also have consequences for the signal of this proton in the 1H NMR spectrum of solutions in CD2Cl2. The resonance is very broad at 10 °C, but sharpens to a narrow line at −55 °C, and the chemical shift value goes through a minimum at −10 °C, while there are no anomalies for other resonances. The reason for this phenomenon is not fully clear, but an Au⋯H–C bonding effect, assisting or assisted by the aurophilic bonding between various oligomers that may be present in solution, is most plausible. Interestingly, another rotamer with potential for C–H⋯O contact (73b) is not observed.201

 | ||
| Fig. 16 Tetramers of chloro{methoxy(2-phenylthiazol-5-yl)methylidene}gold(I) linked through aurophilic interactions; aurophilic interactions (golden) and Au⋯H–C contacts (green) are shown as dashed lines.201 | ||
Pending iPr groups in a crowded ditertiary phosphine are positioned close to gold centers in the dinuclear complex 74, however, again with no discernible orientational preference.202 There is a conspicuous folding of the alkylidene chains of Ph2P–(CH2)3–PPh2 ligands in catena complex 75, which brings the central CH2 group closer to the metal atoms of the penetrating metallacycle, with angles Au⋯H–C as large as 153°. Yet again, this conformation could also be largely determined by the folding required for a best fit of the two macrocyclic rings.203
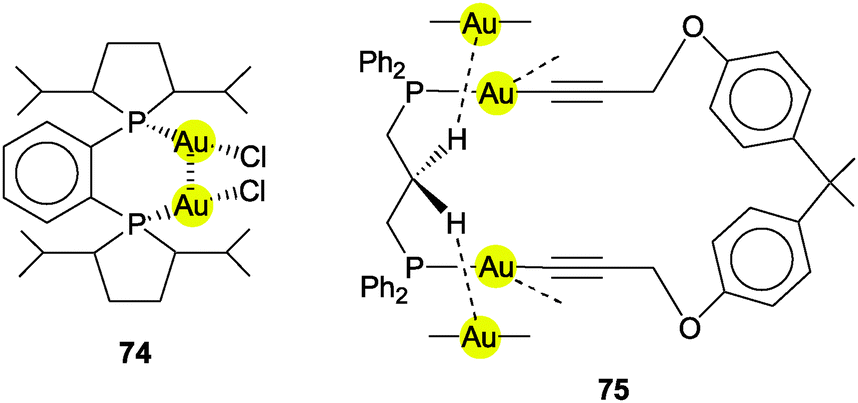
Excessive crowding of substituents in multinuclear gold(I) phosphide oligomers leads to some particularly short enforced Au⋯H contacts. Au⋯H and Au⋯C distances as small as 2.277/2.299 and 3.223/3.258 Å, respectively, were found in the mesityl-substituted 1,3-diphophido-propane complex {MesP(CH2)3PMes}4Au8 and in tetrameric gold(I) dimesitylphosphide, respectively. In both cases the authors have seen no reason to refer to any Au⋯H bonding.204,205
In addition to all the representative examples of intramolecular Au⋯H–C contacts discussed in foregoing sections, there are many examples other than 75 in which intermolecular, mostly (charge-assisted) interionic Au⋯H approaches have been observed and discussed by the authors of the reports. The trinuclear mixed metal complex anions shown in 76 are stacked in the crystal of the PPN salt {PPN = (Ph3P)2N+} establishing short Au⋯H–C contacts between the gold atoms and methyl groups of Me3P ligands of neighbouring anions. The Au⋯H distances and the angles Au⋯H–C are in the narrow ranges of 2.89–2.97 Å and 158–161°, respectively. These relatively short distances and large angles may suggest hydrogen bonding, but no pertinent data obtained by other analytical methods have been provided. It should be noted that in these large triangular anions the particularly open coordination sphere of the gold atom facilitates the interionic approach, and that the delocalization of the negative charge reduces any major Coulomb repulsions.206

Au⋯H–C contacts have also been observed between bis(1-methylimidazolidine-2-thione)gold(I) (as contained e.g. in 41) and bis(1,3-thiazolidine-2-thione)gold cations,149 but they appear to be rather non-specific. For anions with complex hydrogen bonding multifunctionalities such as camphorsulfonates, the situation is particularly complicated.207 The results were also extended by employing smaller di(methanesulfonyl)amide counterions, which led again to a different organizational pattern with both ring methylene and methyl hydrogen atoms showing Au⋯H contacts.208 In all of these systems, the Au⋯H–C contacts are just one component in the manifold of interactions in extended networks including strong hydrogen bonds like N–H⋯N and N–H⋯O, but also weaker bonding as suggested for C–H⋯O and C–H⋯S. There is no way to estimate the individual contributions to the overall stability of the end products. This is true in particular for cases with multiple intermolecular Au⋯H–C contacts.
Cation–anion Au⋯H–C contacts have been described in crystals of a compound composed of cations with a gold(III) center N,N,N,N-coordinated by a macrocyclic ligand and dibromoaurate(I) anions (77). Both CH and CH3 groups could be seen as orientated towards the linear Br–Au–Br unit, but the distances Au⋯H of 3.16, 3.23 and 3.30 Å are very long (with angles 173, 171 and 163° respectively). The existing Br⋯H distances are much shorter (2.95, 2.95 and 3.00 Å, with angles 160, 168 and 158°, respectively) which indicates that the Au⋯H–C contacts should at best be considered as very weak.209 In the same crystals there are also Au⋯H–C contacts between gold(III) centers and methyl groups of neighboring cations (Au⋯H 3.28 Å, Au⋯C–H 138°). It is obvious that structurally there is not much of a difference between these approaches (AuI⋯H–C and AuIII⋯H–C) which both may not be significant contributors to the organization and stability of the crystal.209 However, there are a few cases where the structural evidence for AuIII⋯H–X bonding must also be considered (see Section 3.3).

Fig. 17 shows the distribution of relevant structural parameters for Au⋯H–C contacts in the compounds considered. Just as found in Fig. 11 for the Au⋯H–N contacts, there is no clear evidence for a distinct structural preference.
 | ||
| Fig. 17 Histograms representing the parameters for Au(I)⋯H–C contacts of 2579 hits from a CSD search with cut-offs applied for angles Au⋯H–C (ANG1, in °) and distances Au⋯H (DIST1, in Å) and Au⋯C (DIST2, in Å). | ||
3.3 Experimental evidence for Au⋯H–X hydrogen bonding in AuIII complexes
The gold atoms in gold(III) complexes are generally in a square planar environment of four donor atoms. Potential hydrogen bond donors will thus approach the metal center from an apical position to establish an Au⋯H contact which is in a vertical orientation relative to the molecular plane on one side or the other. Interactions of this type have been discussed for only a few examples with both N–H and C–H donors, mainly in an intramolecular mode (78). Intermolecular AuIII⋯H–N/C contacts may be abundant in crystals of gold(III) compounds, but there are only very few cases for which significant bonding contributions to the supramolecular arrays could be invoked.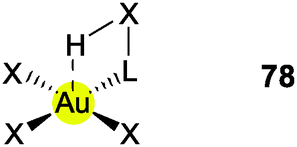
AuIII⋯H–N interactions were first considered for the complex [(NC3H5S2)Au(C6H4-2-CH2NHMe2)Cl]Cl2 (79). However, the authors noticed that there was no clear orientation of the three N–H functions (one at the dimethylammonium group, two at the thiazolidine rings) towards the gold atom. In fact the former is pointing away from the metal center, while the other two prefer contacts to the chloride counterions. Similar observations were made with pyridine-2-thione instead of thiazolidine-2-thione ligands. The results have shown that for this type of compounds Au(III)⋯H–N contacts are not discernible as significant interactions. These results are at variance with observations in Pt(II) chemistry, where Pt⋯HNMe2–CH2– contacts have been identified.210 The work with thiazolidine-thiones was extended to include an anionic dithizonate ligand PhNHNC(S−)NNPh in 80 and a dianionic dithizonate ligand PhNNC(S−)N(N−)Ph, combined with coordinated 2-methylmercaptotetrazolate, in 81. In the latter of these two complexes with a freely dangling 2-(dimethylammoniomethyl)phenyl group, the N–H function approaches gold more closely (2.817, 3.518 Å, 132.50°) whereas in the former it has an adverse orientation.211
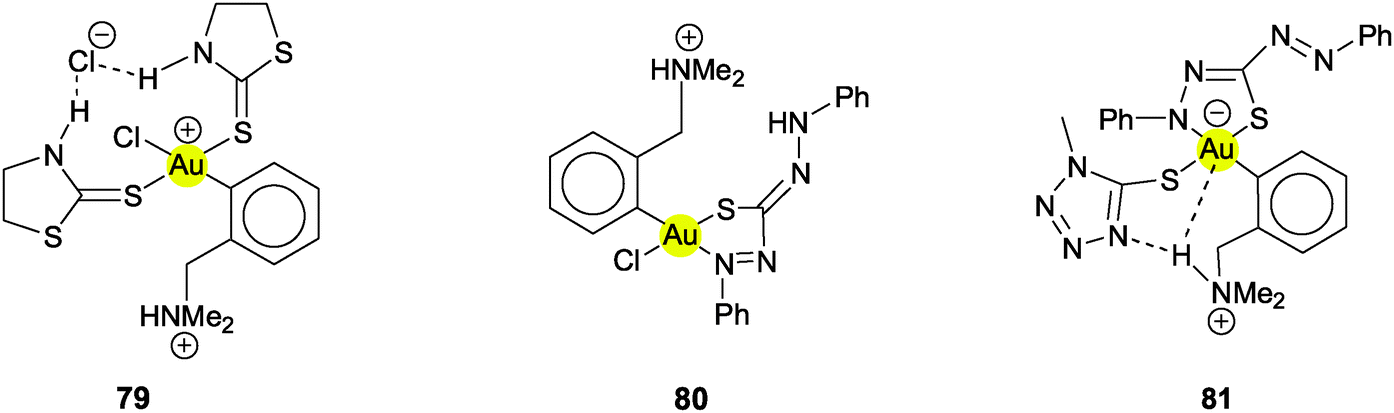
Another candidate for intramolecular Au⋯H–N bonding was seen in the 1:1 complex of (C6F5)3Au with 2-amino-thiazoline (82). The contact of one proton of its amino group with the metal center has a distance Au⋯H of 2.91 Å with an angle Au⋯H–N of 110°. However, the same hydrogen atom also entertains a contact with one of the ortho F atoms of a neighboring pentafluorophenyl group which may at least codetermine the orientation of the neutral ligand.212 The 1:1 complexes of (C6F5)3Au or (C6F5)2AuCl with 6-ferrocenylmethylamino-2-picoline also have the amino group close to the metal center affording Au⋯H distances of 2.62 and 2.67 Å and Au⋯H–N angles of 117.7 and 115.2°, respectively (83 shows the latter example), but again there are also N–H⋯F/Cl contacts which may have a major influence on the molecular conformation. The authors have considered the Au⋯H–N contacts as “the weaker parts of three-coordinate interactions”.128 A similar situation has been encountered in the structure of the 1:1 adduct of (C6F5)2AuCl with 2-aminophenyl(diphenyl)phosphine (84), where the amino group is also associated with one of the ortho-F atoms of a pentafluorophenyl substituent. Owing to these structural details, the authors did not draw attention to a hydrogen bonding alternative involving the gold atom.213

The 2:1 complex of AuCl3 with 1,4,7-triazacyclononane (tacn) has an ionic structure, [(tacn)AuCl2]+ [AuCl4]−. In the cation, the tacn ligand is in a crown-like conformation chelating the metal atom through two of its nitrogen atoms (Fig. 18). The third NH unit is orientated towards the metal atom establishing a contact Au⋯H as short as 1.91 Å. The authors have pointed out that this type of potential interaction is a rare phenomenon. According to NMR studies, in solution the structure is fluxional.214
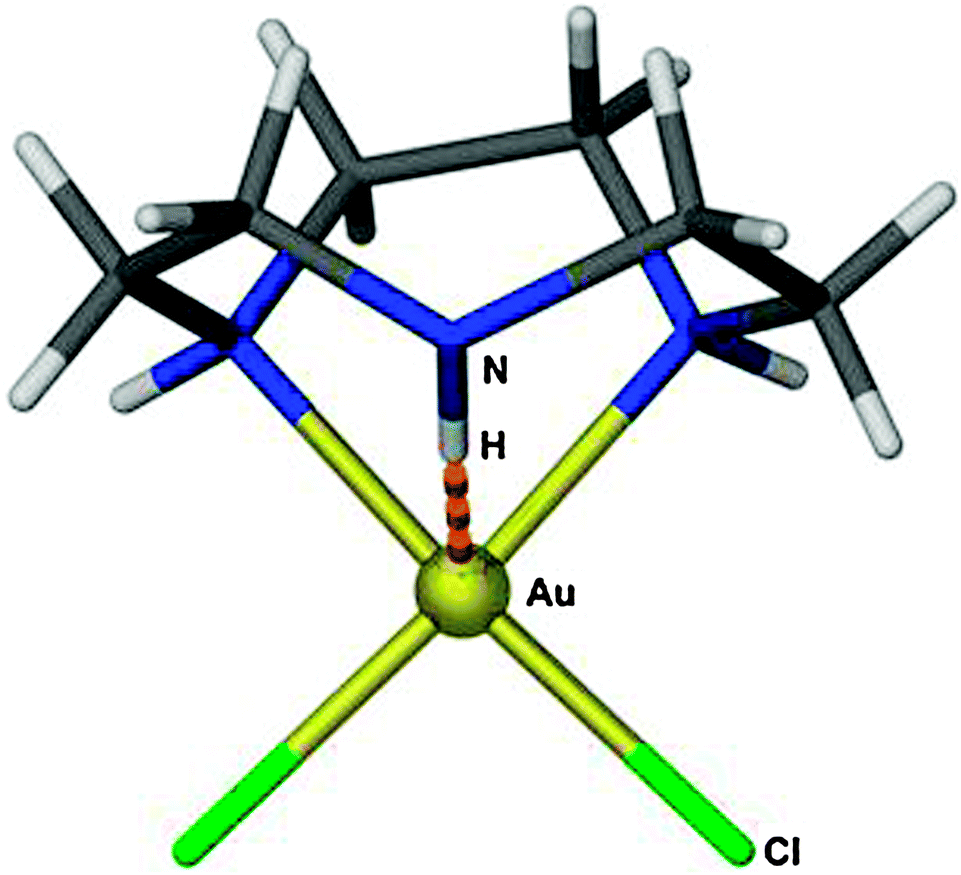 | ||
| Fig. 18 Structure of the cationic complex [(tacn)AuCl2]+ in the [AuCl]4− salt; the Au⋯H–N contact is shown as a dashed orange line.214 | ||
A short contact AuIII⋯H–C was first observed in one example of three dichloro{2-(2-pyridylalkyl)phenyl}gold complexes (85, (a) R, R′ = H; (b) R = H, R′ = Me; (c) R, R′ = Me).215 According to NMR studies in CD2Cl2 solution, the six-membered metallacycles appear to be all folded into a conformer with at least the bridging methylene carbon atom out of the best plane. This conformation is fixed on the NMR time scale rendering the methylene H atoms or Me groups in 85a and 85c inequivalent, respectively, and gives rise to two isomers for 85b. The crystal structure of 85c has been determined (Fig. 19). The metallacycle is in a boat conformation with folding angles of 44.6(3) and 38.2(2)°. The two aromatic rings form a dihedral angle of 136.8(2)° with each other. Any inversion of the boat thus requires extensive wing movements. In this conformation exclusively present in the crystal, one of the two methyl groups shows a transannular Au⋯H distance to the metal atom of only 2.56(6) Å (85c′, R′ = Me). This contact was addressed by the authors as even shorter than a “long range metal hydrogen interaction” suggested for related Pt(II) complexes.49,216
 | ||
| Fig. 19 Molecular structure of dichloro[2-(2-pyridyl)-2-(o-phenylene)propane]gold(III); the Au⋯H–C contact is shown as a dashed green line.215 | ||
However, it is unlikely that the structure and the rigidity of the conformation of the metallacycle 85c with the two methyl groups are due predominantly to this supporting interaction because the rigidity in solution is also observed for the CH2-bridged homolog 85a where an Au⋯H contact is ruled out by the long transannular distance. Moreover it should be considered that the parent hydrocarbon 9,10-dihydroanthracene is also folded to assume a boat conformation with similar dihedral angles. There is a further structural analogy with the corresponding Pd(II) and Pt(II) complexes.217,218 Recent quantum chemical calculations have shown that the influence of the transannular Au⋯H–C contact on the structure and its dynamics is indeed very limited.219

A similar structure has subsequently been discovered for a case where the chelating ligand in 85 is extended by an additional pyridyl group (86).220 In crystals of the AuCl4− salt of the cation with its N,N,C-bonded tridentate ligand, one of the two methyl groups of the bridging unit again approaches the gold atom quite closely to a distance Au⋯H of 2.62 Å. The metallacycle is in a boat conformation with the Au atom and the bridging carbon CMe2 on the same side of the folded ring displaced from the bottom by 0.550(1) and 0.657(6) Å, respectively. The two reference wings (above) form a dihedral angle of 124.6(2)°. And yet, in acetone-d6 solution, the two methyl groups are NMR-equivalent at room temperature which indicates that the conformation of the metallacycle cation is more flexible, probably owing to the influence of the bipyridyl rigidity which lowers the barrier to inversion. It therefore again appears that the transannular Au⋯H–C contact – even though it is rather short – does not decisively determine the conformation of the molecules 85 and 86.

It is interesting to note that the metallacycle in the cation shown in formula 87 with a hydroxyl group in the δ-position relative to the gold atom also has a distorted boat conformation, but the folding of the ring is rather flat and there is no evidence for any transannular Au⋯H–O interaction. The crystals of a methanol solvate of the corresponding ketone complex 88 have the CH3OH molecule in close proximity of the metal atom, but it remains unclear if the arrangement is well defined.221
Finally it should be noted that many rather short intramolecular AuIII⋯H–C contacts can be found in several complexes of gold trihalides with tertiary phosphines bearing bulky alkyl groups. While such short distances are not established with triarylphosphines, the presence of tricyclohexylphosphine, for example, leads to a crowding of substituents reducing also the intramolecular non-bonding distances between the metal atom and the hydrogen atoms of its ligands. Most authors of papers describing prototypical structures of this kind have wisely refrained from ascribing any significant bonding effects to these steric observations.222
Apart from the well-documented cases of intramolecular AuIII⋯H–X contacts (X = N, C) described above, there are several reports of such intermolecular or interionic contacts. It has for example been pointed out that in crystals of tetrabutylammonium bis(quinoxaline-2,3-dithiolato)gold(III) (89) the gold atom of the anionic complex is not solely in a common square planar coordination of sulfur atoms, but also near to at least two hydrogen atoms of CH2 or CH3 groups of neighboring cations.223 The Au⋯H distances are 3.038(9) and 3.17(1) Å and the Au⋯H–C angles are 142(1) and 142(1)°. The authors propose that these “C–H⋯M interactions [are] acting as a supramolecular motif” and should be “best described as 3c–4e hydrogen bonds”. They also draw attention to the fact that the interactions may benefit from the ionic nature of the compounds which may induce a charge effect that is well known to be reinforcing weak H-interactions between anions and cations.223 Most interestingly, crystals of the corresponding copper complex are isomorphous and show very similar dimensions, ruling out any selective intermolecular interactions for the gold compound.

An investigation of crystals of bis(trimethylenetetrathiafulvalenedithionato)gold at high pressure (up to 10.7 GPa) has shown that the pressure affects mainly the intermolecular S⋯S contacts. Any Au⋯H–C contacts change as well, but have not been considered.224
Contacts Au⋯H–C have also been observed and discussed for gold(III) complexes with tetradentate macrocyclic ligands bearing methyl groups in their periphery (77). It appears that these contacts are clearly non-directional and unspecific, with Au⋯H distances near 3.28 Å and angles near 138°. The contacts between these cations and the [AuBr2]− anions have already been considered above.209
3.4 Agostic hydrogen bonding in alkylgold(I) and alkylgold(III) complexes?
It is worthwhile to ask the very general question if there is any structural or other evidence for intramolecular Au⋯H contacts in simple alkylgold(I) or alkylgold(III) complexes that would resemble typical agostic interactions known in particular for the early transition metals. Very recent work has made an important pertinent contribution by investigating inter alia the simple carbene complex (IPr)AuEt.69 This ethylgold(I) complex can be viewed as a source for the corresponding hydride (IPr)AuH4 through β-elimination of ethene (Scheme 1).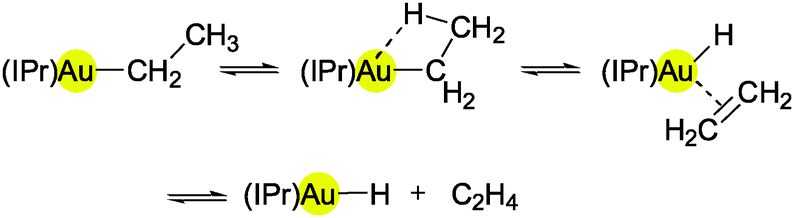 | ||
| Scheme 1 Idealized β-elimination of ethene from an ethylgold(I) complex showing a mechanism involving an agostic interaction. | ||
The β-elimination may proceed through agostic interactions bringing one of the methyl hydrogen atoms in a transition state, or even an intermediate, close to the metal atom. In order to detect any preorganization of the molecule for this approach, the crystal structure of (IPr)AuEt has been determined. The unit cell contains three independent molecules, and all three structures exhibit no sign of any back-bending of the ethyl group. The geometry of the Au–CH2–CH3 units shows no anomalies, with Au–C–C angles in the range of 110.6–116.2°, and inconspicuous distances Au–C (2.119–2.133 Å) and C–C (1.488–1.509 Å). The Au⋯H(Cβ) distance is 3.128 Å (av.). A very similar value has also been found for the gas phase ground state in quantum chemical calculations (3.245 Å). The IR and NMR characteristics correspond to those of many compounds with unconstrained M–Et units (M = late transition metal). The compound is thermally very stable, well beyond 120 °C, and shows no tendency to ethene elimination. This is in line with the exceedingly high calculated activation barriers for the β-elimination of ethene (57.1 kcal mol−1).69 In this context it should be remembered that the corresponding phosphine complexes such as (Ph3P)AuEt (m.p. 135 °C)225,226 are also known to be thermally stable compounds. The kinetic study of their decomposition did not provide any evidence for ethene β-elimination.225
Ethylgold(III) compounds have been known for more than a century. Ethylation of gold(III) halides (AuX3 or NaAuX4) using Grignard reagents EtMgX or ethyllithium EtLi affords stable compounds of the formulae (L)AuEt2X or (L)AuEt3. The thermal stability of these compounds is not very different from that of the corresponding methylgold(III) compounds and their decomposition does not produce ethene.171 The crystal structure of (Et2AuBr)2 has been determined as early as 1937 and showed no anomalies regarding the conformation of the ethyl groups (90).227 Several later studies have confirmed these findings.171

In summary, therefore, no evidence exists for ethylgold(I) and ethylgold(III) compounds to feature agostic interactions. In both of its two major oxidation states the gold atom has no attraction for hydrogen atoms in the β-position of its alkyl substituents and does not induce relevant thermal decomposition pathways including β-elimination of alkene. This observation confirms that gold(I) and gold(III) centers are generally very poor acceptors for Au⋯H–C interactions.
3.5 Gold–hydrogen interactions in catalytic processes
Enantioselective hydrogenation of olefins and imines is selectively catalyzed by the gold compound [(AuCl)2{R,R-Me-DuPhos}] {Me-DuPhos = cis-1,2-bis(2,5-dimethylphospholanyl)benzene}.228 By studying simplified model complexes and finally the chiral dinuclear complex itself in DFT calculations at the B3LYP level of theory, Comas-Vives and Ujaque49 obtained results consistent with an ionic mechanism for such hydrogenation processes.A simplified version of the results obtained employing a model diphosphine gold complex and cyclohexene as the olefin is given in Scheme 2. The heterolytic hydrogen activation involves both catalyst and solvent and leads to a gold hydride complex, 91, and a protonated solvent molecule, 92. As discussed earlier, hydride complexes of gold are still rare although some have been invoked in catalytic cycles.53 The cyclic hydrocarbon in the transition state 93 for the hydride transfer has already been protonated by 92 and participates in trapping a hydride ion between two cationic centres. Although this dispersion-effect-stabilized transition state (hydride hydrogen associated with gold and carbon atoms, complemented by gold–carbon association) would be an ideal starting point for an agostic or pseudo-agostic interaction by Au–H bond lengthening to ca. 2 Å31 and H–C bond shortening to approach ca. 1.1 Å, no such intermediate or transition state bonding situations have been identified. Many other gold(I)-catalyzed reactions may follow this mechanistic pathway.
 | ||
| Scheme 2 Schematic representation of the proposed mechanistic pathway for the hydrogenation of cyclohexene using a dinuclear gold complex in ethanol as precatalyst (dmpey = cis-1,2-bis(dimethylphosphino)ethene).49 | ||
3.6 Theoretical approaches to Au⋯H–X hydrogen bonding
Attempts to identify and characterize hydrogen bonding to gold atoms and ions by quantum chemical calculations published in recent years have not only considered simple and small model systems, but have even included aggregates of gold clusters and cluster anions with glucose, amino acids, peptides and the bases or base pairs of RNA and DNA. The latter developments have their origin in the rapidly growing application of gold nanoparticles in medicine and biotechnology.DFT calculations (at the B3LYP or MP2 level, usually with BSSE or ZPVE corrections) carried out since 2005 have indicated strong hydrogen bonding interactions between Au− anions and NH3, H2O and HF molecules. The increase in the binding energies of the three 1:1 complexes 17–19 formed, i.e. 6 kcal mol−1 for [Au⋯NH3]−,110 12.43,111 11.93,114 11.36,28 and 12.18 (ref. 91) kcal mol−1 for [Au⋯H2O]−, and 20.3 kcal mol−1 for [Au⋯HF]−,114 corresponds with decreases in the proton affinities of the anions NH2− (1689), OH− (1635) and F− (1554 kcal mol−1)229 and in the calculated Au⋯H separations 2.690,113 2.242 – 2.45,28,91,111,114 and 2.156 Å114 within the respective three complexes. The calculated binding energies for [Au⋯H2O]− can be compared with an approximated experimental value of 10.38 kcal mol−1.112 The calculations for the same adduct are also supported by the satisfactory correspondence between experimental and theoretical VDEs and νAu⋯HOH vibrational frequencies (Table 1).
Outstanding features of all the results are the large Au⋯H–XHn−1 (X = N, O, F; n = 3, 2, 1) angles found which vary between 156.3 and 169.9° for OH2 as the donor28,114 to 157.4° for NH3,110 and approach almost linearity (178.2°) in [Au⋯H–F]−.113 Calculations not only show a significant red shift in the H–O stretching frequency upon hydrogen bonding of water to gold but also an enhancement of the Raman intensities. The changes are less marked for Au− as compared to the same stretching mode for Ag− and Cu−, corresponding to an increasing polarizability in the series. It has been proposed to use the strong Raman line of the H–O–H bonding mode as a fingerprint suggesting significant hydrogen bonding of a water molecule.28
With more than one proton donor molecule attached to the auride ion, different bonding schemes have been calculated, depending on the manner in which the proton donors arrange themselves by means of conventional hydrogen bonding. Binding energies are not always directly comparable because the chosen reference dissociation products may differ. The structures of the lowest energy adducts between Au− and two ligand molecules are shown as 20–22.114 Whereas the associated ligand pairs in [Au⋯(NH3)2]− and [Au⋯(H2O)2]− appear as bidentately bonded, the two HF molecules act as monodentate proton donors. The variation in the strength of the Au⋯H hydrogen bonds in the series NH3, H2O, and HF, as manifested in the shortest Au⋯H separation in each complex, remains unchanged from those in the 1:1 adducts. In the cyclic products, deviations from linearity in the Au⋯H–X angles can be quite significant.
Interactions between monoanionic gold clusters and NH3 molecules have also been investigated theoretically.110 With only one NH3 molecule attached to anionic Au3 or Au4 clusters, the resultant hydrogen bonding is weaker than in the 1:1 complexes. With more than one ligand linked by hydrogen bonds, a variety of bonding motifs become available; however, the binding energy remains low. The connecting Au⋯H distances are approximately 3 Å – too long to be considered as proper H-bonds. Two examples (94 and 96) are shown in Scheme 3. Note that in 94 only one Au⋯H interaction occurs whereas in 96 three are present. In 95, uncharacteristically, the two mutually H-bonded NH3 molecules are participating in both anchoring interactions and hydrogen bonding to only one gold atom of the kite-like tetranuclear cluster. Here the Au⋯H distance is just shorter than the sum of the van der Waals radii (∼2.8 Å). The bonding between neutral (and cationic) gold atoms or clusters and NH3 or H2O is dominated by classical Lewis-adduct formation, and hydrogen bonding, if present at all, is extremely weak.
 | ||
| Scheme 3 Lowest energy complexes between anionic gold clusters and H-bonded NH3 molecules; internuclear distances (Å) and angle (°) from ref. 110. | ||
In one of the earlier theoretical publications by the Kryachko and Remacle group,113 the hydrogen bonding between HF molecules and neutral gold clusters has been described. Surprisingly, in the complexes formed between neutral clusters and one or more HF molecules, apart from one example, hydrogen bonding apparently plays the most important role in the stabilisation of the ground state structures. In 97 (Scheme 4) this is the only mode of attachment. Only with HF bonded to triangular Au3 is the anchor bond strong enough to alone support a structure with nearly the same binding energy as 98 in Scheme 4, which, however, exhibits both anchoring interaction and hydrogen bonding. In the series [(Au)3⋯(HF)2], [(Au)3⋯(HF)3], and [(Au)4⋯(HF)2] (Scheme 4, 99–101), the Au–F- and Au⋯H-bonding work in tandem in stabilising low energy complex formation. The anchor bond strengthens all hydrogen bonds in the structures wherein they occur. Notably, the Au⋯H (and Au–F) distances decrease from 2.560 to 2.167 Å (2.781–2.354 Å) in the triangular complexes when the number of mutually H-bonded HF molecules increases. Complex 101, in which the synergy between anchor and hydrogen bonds is particularly effective, has by far the largest binding energy (17.6 kcal mol−1) and strongest electron pair and proton donor interactions in the entire series of complexes that have been studied in this investigation.
 | ||
| Scheme 4 Examples of calculated linkages in the ground state conformations of complexes between neutral gold clusters and HF molecules; distances (Å) and angles (°) from ref. 113. | ||
 | ||
| Scheme 5 Association of formamide and formic acid with neutral gold clusters (a);230 simplified resonance structures explaining co-operative coordination interactions (b). | ||
It was later established theoretically that when organic molecules such as ketones or aldehydes that lack an N–H or O–H functionality serve as ligands, C–H units could play the role of proton donors.120 Using DFT calculations and concentrating mainly on the interaction between acetone and a series of odd-numbered neutral gold clusters, it has been found, as expected, that Au–O anchor bonding dominates the adduct formation, but that significant Au⋯H–C interactions (2.1–2.7 Å) are also present. The strongest bond formation of the latter type occurs with Au5, Au7 and Au13 clusters.
Two research groups have reported results describing the interactions between gold clusters and glycine or cysteine (Scheme 6).231,232 Complexes formed from anionic systems exhibit the highest bonding energies but no hydrogen bonding.232 Cationic systems associate only weakly. Unfortunately, the two groups do not fully agree on the relative stability of neutral adducts and by which of them hydrogen bonding is exhibited. Nonetheless, it seems clear that such bonding may occur, albeit not necessarily in the most stable structure. An example (104) of a neutral low energy, bidentately coordinated isomer involving glycine (Gly–Au3) for which relevant distances have been reported is shown in Scheme 6. The coordination is very similar to that illustrated for formamide and formic acid in Scheme 5. Singh and co-workers investigated interactions between proline and neutral gold clusters233 and reached conclusions similar to those made independently by Pakieri and Jamshidi231 and Xie et al.,232i.e., (i) the strongest anchoring interaction is found at the amine center, but then H-bonding with gold is negligible; (ii) other higher energy isomers do exhibit such bonding when anchoring occurs through the carbonyl oxygen atom.
 | ||
| Scheme 6 Structures of the amino acids glycine and cysteine, and the peptide glutathione (GSH); hydrogen bonding between glycine and a neutral Au3 cluster with distances from ref. 231. | ||
Fattahi and co-workers234 investigated the interaction of neutral and deprotonated forms of the tripeptide glutathione (Scheme 6) with a trinuclear gold cluster. Complexation within the neutral system is extremely weak – binding energies ≤3.0 kcal mol−1. Unfortunately, the numbering and information given in their tables and their Fig. 3 are confusing and it is not adequately clear whether the lowest energy anionic systems – although certainly the most stable – are involved in meaningful hydrogen bonding at all. Very little resemblance, if any, is seen between the interactions of the multifunctional glutathione and those of its constituent amino acids, cysteine and glycine, with Au3 clusters.
The anionic clusters bind exclusively through Au⋯H–O bonds, which may have bonding energies >20 kcal mol−1 in bidentate coordinations. An example, 105, in which hydroxyl hydrogens at C1 and the hydroxymethyl group attached to C5 are involved, is shown in Scheme 7. Utilising the same latter proton donor group, but the oxygen atom of the hydroxymethyl group as the electron pair donor in 106, neutral [Au3] forms relatively strong bonds with the glucose molecule. Being a multifunctional hydroxyl compound, glucose offers many other opportunities for such bidentate O–H∧O chelate formation. The cationic gold cluster, though very strongly bonded to the sugar molecule, forms no hydrogen bonds.
 | ||
| Scheme 7 Coordination interactions in the lowest energy complexes formed between α-D-glucose (Glu) and neutral or anionic Au3 clusters; bond distances from ref. 235. | ||
In all the calculated relevant low energy complexes between glucose and gold clusters with hydrogen bonds, calculated stretching frequencies of the O–H bond become red-shifted upon complex formation by as much as 308 cm−1. NBO calculations again confirm electron transfer from gold to the H–O unit during hydrogen bond formation. In the final analysis, by means of Bader's quantum theory of atoms in molecules, very little electron density is found along the bond path, which, according to the authors, might indicate an electrostatic attraction mechanism. [Ideal conditions for H-bonding at the bond critical point (BCP) have been defined by Popelier et al.102,236 Recent calculations have indicated that a “BCP for M⋯H interactions is an unreliable criterion which usually fails to reveal the interaction, even in established agostic systems.”]237
 | ||
| Scheme 8 The DNA (A, G, T, C) and RNA (A, G, C, U) bases. | ||
 | ||
| Scheme 9 Hydrogen bonds with anionic gold atoms in the thymine and cytosine complexes.239 | ||
 | ||
| Scheme 10 The lowest energy calculated complex isomers of guanine and adenine [A–Au]−, [G–Au]− and the complexes identified in the mass spectrometer by PES [A′–Au]−.238–241 | ||
In all the calculated ground-state structures, at least one Au⋯N–H interaction is present: one in the isomer [A–Au]− of adenine and two in the isomer [G–Au]− of guanine. The Au⋯H–O and Au⋯H–N separations in [G–Au]− are identical. The Au⋯H–C interactions that also assist in the gold bonding are weak, with distances of 3.0 Å or longer. On the basis of their approximated bonding energies (0.63–1.54 eV) and bond distances (av. 2.3 Å), the heteroatom-assisted hydrogen attraction to anionic gold atoms can be classified as medium or strong hydrogen bonding.78 Natural bond orbital (NBO) analysis indicates that the stronger hydrogen bonds are formed by electron donation from Au− into the σ* orbitals of the N–H or O–H bonds.
Using different Watson–Crick tautomers of the nitrogen bases and even different A–U bonding schemes, while positioning the Au− unit at all the different atoms of the A–U base pair, it was found that in the most stable structure gold preferably associates with N9–H and C8–H of adenine in almost exactly the same manner as depicted in the structure 111 of [A′–Au]− (Scheme 10).242 Two other low energy structures have gold H-bonded to either N1–H and C6–H of uracil as in the structure 107 of [T–Au]−, or to one N–H proton donor from each base (N9–H of A and N1–H of U) while disrupting the hydrogen bonds in the conventional base pair.
Vargas and Martínez243 recently considered three pterins as bases in their calculations. These compounds prefer to form hydrogen bonds with gold atoms in anionic systems via their N1–H and N2–H units, as illustrated by 113 for pterin (Ptr) and by 114 for isoxanthopterin (7-Xap) in Scheme 11. The combined binding energies of respectively 28.6 and 31.5 kcal mol−1 (1.24 and 1.37 eV) in the two examples can be compared with a binding energy of 1.45 eV calculated for the structurally related [G–Au]−.238 Notably, useful direct correlations between calculated VDEs and hydrogen bonding energies were established and, furthermore, it was concluded that
 | ||
| Scheme 11 Preferred bonding of gold atoms to two pterins in anionic structures.243 | ||
(i) bond lengths and angles of the Au⋯H–X hydrogen bonds are not always reliable indicators of the strength of the interaction;
(ii) standard hydrogen bonds (N–H⋯N, etc.) in pterin dimers and tetramers are not affected by interaction with gold anions;
(iii) gaseous copper and silver anions also form hydrogen bonds, and although the total bond energies are of the same order and bonding occurs generally in the same positions as with gold anions, the hydrogen bonding interactions are weaker for copper and silver;
(iv) linear trinuclear gold clusters [Au3]− (see the following section) coordinate in the same position as monoanions Au−, but the interactions are weaker because the negative charge is distributed over more than one atom.
 | ||
| Scheme 12 The most stable isomers formed between adenine and uracil and two anionic gold clusters.24 | ||
In all the stable isomers of the [A–Aun]− and [U–Aun]− systems studied, exo-bidentate coordination occurs with the involvement of N9–H and C8–H in adenine, and of N1–H and C6–H in uracil. Although now more than one gold atom is involved per base, the protonated sites remain the same as in [A–Au]− and [U–Au]− (or [T–Au]−) (compare 111 and 107 in Schemes 9 and 10). The dinuclear gold complexes in which the negative charge is delocalised over only two atoms have higher dissociation energies than the higher nuclearity cluster products.
In another publication not specifically dealing with hydrogen bonding interactions to gold, Suhai and co-workers244 also studied the influence of anionic gold clusters [Aun]− (n = 4, 8) on A–T and G–C base pairs, using quantum mechanical calculations. They split the [Au4]− cluster into two dinuclear units, or placed two tetranuclear clusters near the electron-rich sites of the base pairs. Only small changes in the hydrogen bonding distances of the A–T base pair were induced by hydrogen bonding to gold.
 | ||
| Scheme 13 Most important binding sites of adenine (N3, N9) and thymine (O2, N3) to a neutral triangular gold cluster; bond distances from ref. 245; compare also ref. 247. | ||
Using NBO analysis, Singh and co-workers248,249 confirmed the important role of Au⋯H–N and even Au⋯H–C interactions in stabilising complexes formed between small gold clusters and extended DNA (xDNA) bases or base pairs. Referring to such hydrogen bonding for extended guanine, they state that the Au6⋯H–N association “is among the major interactions present in the xG–Au6 complex”.249
Although Kryachko and Remacle established N3 to be the preferred site for anchoring a neutral Au3 cluster to guanine,245 other evidence250,251 indicated N7 to be the major groove site for gold aggregates inside cells. Leszczynski and co-workers,252 therefore, chose N7 for the major interaction with various gold clusters Aun (n = 2, 4, 6, 8) in their calculations. They have only reported reinforcing hydrogen bonds in two of their complexes; both are weak Au⋯H–C interactions (>3.4 Å).
The coordinate anchor bonds as well as the structure of the gold cluster predetermine the formation and location of Au⋯H–N and Au⋯H–C hydrogen bonds during the interaction of DNA base pairs and neutral gold clusters. In certain calculations, one or both of the main attachment positions and the structure of the cluster have been fixed,246,252 whereas in others neither was pre-established, and more than one cluster unit were simultaneously employed.244 Hence, different calculations of base pair interactions with gold clusters are not always comparable. The results of DNA single base and base pair coordination investigations are also not necessarily comparable, even within the same research group, because certain electron pair and proton donors, and thus anchor or hydrogen bond positions, are excluded by the base pairing. Nevertheless, the anchoring and supporting hydrogen bonds in the base pairs [A–Au3 (N3, N9)]T and [G–Au3(N3, N9)]C are practically identical to similar bonds in the single base complexes [A–Au3] (117, Scheme 13) and [G–Au3].246 Note, however, that coordination at the most favourable anchoring and co-operative proton donor sites of cytosine (N3, N4–H) is prevented by the Watson–Crick base pairing. In DNA or RNA, the attachment of sugar residues to the bases further excludes, in particular, N1–H (of pyrimidines) or N9–H (of purines) from participating in hydrogen bonding.
Here it should be mentioned that, thus far, theoretical approaches do not directly explain the binding adsorption affinities of nucleobases on polycrystalline gold films,253 although the exceptionally weak single-site bonding of thymine with neutral gold clusters is in accordance with the experimental results.254
4. Summary
After several decades of unsuccessful endeavors into the chemistry of gold hydrides, recent preparative advances have provided several examples of well-defined compounds with the metal atom in both of its most important oxidation states, AuI and AuIII. These compounds will open up new synthetic routes into other areas of gold chemistry. By contrast, a screening of the literature on gold compounds in which the gold atoms may be involved in intra- and intermolecular hydrogen bonding has not produced consistent evidence for any major influences of interactions of the type Au⋯H–X on the molecular structure and dynamics of the systems concerned. There are very few cases, if any, where e.g. details of crystal and molecular structures complemented by vibrational or NMR spectral data could be traced consistently to anomalies originating from discrete Au⋯H–X interactions. Strong hydrogen bonding in the sense of the latest IUPAC definition may only be assumed for compounds of gold in its oxidation state Au−. Towards typical hydrogen bond donor functions H–X (X = N, C), these auride anions can act as hydrogen bonding acceptors just like halide anions X−. Owing to the large anionic radius of Au−, the distances Au⋯H are large and the associated energies very small. The experimental data are still very limited, but extensive quantum chemical calculations on model systems, including not only Au− but also anionic clusters [Aun]m−, have provided a large set of parameters for further orientation. This work has also considered model complexes with ligands taken from DNA bases to shed light on bonding to negatively charged gold nanoparticles.For AuI or AuIII as hydrogen bond acceptors, several groups of authors have postulated significant Au⋯H–X interactions based mainly on the observation of short Au⋯H distances (2.3–3.0 Å), medium to large Au⋯H–X angles (120–180°) and associated conformational anomalies. A screening of the literature for other systems with a geometry fitting into these ranges provided many more examples, but in almost all cases other effects may be found responsible for the conspicuous structural details. It is particularly interesting that there are virtually no examples for a specific approach of the H atom of H–O functions to AuI or AuIII centers, while there are many more for H–N and H–C functions, in particular owing to the greater abundance of the latter. However, the latter show no clear directionality and appear to contribute very little to the energy of a molecule in a given structure. This is also true for intermolecular contacts of this type, which have not been found to overrule significantly conventional van der Waals interactions. This does not mean that this type of contacts can be ignored completely. In the same way as many other “weak” and “very weak” hydrogen bonds, the Au⋯H–X contacts may complement other contributions and thus co-determine structural nuances of molecules and intermolecular contacts, or have an influence on molecular dynamics. This may become obvious only in favorite cases where multihapto interactions can be established leading to what has been called the “Gulliver effect”. There is currently no evidence for “agostic” interactions in organogold compounds as defined for standard cases of such interactions, and this term therefore should not be used when referring to Au⋯H–C contacts. Other designations like “non-conventional” or “unconventional” hydrogen bonding, “anagostic”, “pseudo-agostic”, “pre-agostic” or “electron-rich agostic” interactions are possibilities, but will not be very helpful in characterizing the abundant Au⋯H–X contacts. Future experimental and theoretical work will finally provide more detailed data of the energetics and of the optimum geometry of this type of interactions and their specific consequences for the properties of the systems concerned. From presently available information, no major anomalies are to be expected for gold compounds as compared to complexes of other late transition metals. This result would correspond to the development of the understanding of “aurophilic” bonding which is now accepted as just a prominent variant of “metallophilic” bonding. However, much lower bond energies, in the range of up to a few kcal mol−1, are to be expected for Au⋯H–X contacts, while aurophilic bonding can contribute energies of 6–12 kcal mol−1, truly comparable to standard hydrogen bonding.
Acknowledgements
This work was supported by Fonds der Chemischen Industrie, Frankfurt/Main, Germany (H.S.), by the Alexander von Humboldt Stiftung, Bonn, Germany, the Oppenheimer Memorial Trust, Johannesburg, South Africa, and the National Research Foundation of South Africa (all H.G.R.), and by the Deanship of Scientific Research, KAU, Jeddah, Saudi Arabia (grant 22-3-1432/HiCi) (H.S.). The authors thank Professor I. Dance, Univ. New South Wales, Australia, for a very helpful discussion.References
- Gmelin Handbook of Inorganic and Organometallic Chemistry, Au, Suppl. Vol. B1, Springer, Berlin, 1992, pp. 28 ff Search PubMed.
- R. J. Puddephatt, The Chemistry of Gold, Elsevier, Amsterdam, 1978 Search PubMed.
- Gold Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, J. Wiley, Chichester, 1999 Search PubMed.
- E. Y. Tsui, P. Müller and J. P. Sadighi, Angew. Chem., Int. Ed., 2008, 47, 8937 CrossRef CAS PubMed.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 CrossRef PubMed.
- H. Lv, J.-H. Zhan, Y.-B. Cai, Y. Yu, B. Wang and J.-L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216 CrossRef CAS PubMed.
- H. G. Raubenheimer and H. Schmidbaur, Organometallics, 2011, 31, 2507 CrossRef.
- A. Kant and K. A. Moon, High Temp. Sci., 1979, 11, 55 CAS.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967 RSC.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412 CrossRef PubMed.
- P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113 CrossRef PubMed.
- P. Schwerdtfeger, P. D. W. Boyd, A. K. Burrell, W. T. Robinson and M. J. Taylor, Inorg. Chem., 1990, 29, 3593 CrossRef CAS.
- P. Schwerdtfeger, M. Dolg, W. H. E. Schwerm, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys., 1989, 91, 1762 CrossRef CAS.
- M. B. Cortie and A. McDonagh, in Gold Chemistry, ed. F. Mohr, Wiley-VCH, Weinheim, 2009, p. 321 Search PubMed.
- B. M. Trapnell, Proc. R. Soc. A, 1953, 218, 566 CrossRef CAS.
- G. C. Bond, Gold Bull., 1972, 5, 11 CrossRef CAS.
- N. Bartlett, Gold Bull., 1998, 31, 22 CrossRef CAS.
- J. Pritchard and F. C. Tompkins, Trans. Faraday Soc., 1960, 56, 540 RSC.
- W. Lisowski, L. Stobinski and R. Dus, Surf. Sci., 1987, 188, L735 CrossRef.
- L. Stobinski, L. Zommer and R. Dus, Surf. Sci., 1993, 298, 101 CrossRef CAS.
- R. B. McLellan, J. Phys. Chem. Solids, 1973, 34, 1173 CrossRef.
- E. S. Kryachko, J. Mol. Struct., 2008, 880(23), 114 Search PubMed.
- A. Martínez, J. Phys. Chem. A, 2009, 113, 1134 CrossRef PubMed.
- H. Nuss and M. Jansen, Angew. Chem., Int. Ed., 2006, 45, 4369 CrossRef CAS PubMed.
- H. Nuss and M. Jansen, Z. Naturforsch., 2006, 61b, 1205 Search PubMed.
- P. D. C. Dietzel and M. Jansen, Chem. Commun., 2001, 2208 RSC.
- D.-Y. Wu, S. Duan, X.-M. Liu, Y.-C. Xu, Y.-X. Jiang, B. Ren, X. Xu, S. H. Lin and Z.-Q. Tian, J. Phys. Chem. A, 2008, 112, 1313 CrossRef CAS PubMed.
- Y. X. Chen, S. Z. Zou, K. Q. Huang and Z. Q. Tian, J. Raman Spectrosc., 1998, 29, 749 CrossRef CAS.
- L. Brammer, Dalton Trans., 2003, 3145 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, PNAS, 2007, 104, 6908 CrossRef CAS PubMed.
- W. I. Sundquist, D. P. Bancroft and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 1590 CrossRef CAS.
- M. Bortolin, U. E. Bucher, H. Rüegger, L. M. Venanzi, A. Albinati, F. Lianza and S. Trofimenko, Organometallics, 1992, 11, 2514 CrossRef CAS.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782 CrossRef CAS PubMed.
- S. J. La Placa and J. A. Ibers, Inorg. Chem., 1965, 4, 778 CrossRef CAS.
- N. A. Bailey, J. M. Jenkins, R. Mason and B. L. Shaw, Chem. Commun., 1965, 237 RSC.
- J. F. Van Baar, K. Vrieze and D. J. Stufkens, J. Organomet. Chem., 1974, 81, 247 CrossRef CAS.
- D. M Roe, P. M. Bailey, K. Moseley and P. M. Maitlis, J. Chem. Soc., Chem. Commun., 1972, 1273 RSC.
- S. Trofimenko, Inorg. Chem., 1970, 9, 2493 CrossRef CAS.
- H. M. Echols, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1627 CrossRef.
- F. A. Cotton, T. LaCour and A. G. Stanislowski, J. Am. Chem. Soc., 1974, 96, 754 CrossRef CAS.
- F. A. Cotton and V. W. Day, J. Chem. Soc., Chem. Commun., 1974, 415 RSC.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS.
- F. A. Cotton and R. L. Luck, Inorg. Chem., 1989, 28, 3210 CrossRef CAS.
- A. Albinati, C. G. Anklin, F. Ganazzoli, H. Rüegg and P. S. Pregosin, Inorg. Chem., 1987, 26, 503 CrossRef CAS.
- A. Albinati, C. Arz and P. S. Pregosin, J. Organomet. Chem., 1988, 356, 367 CrossRef CAS.
- A. Albinati, P. S. Pregosin and F. Wombacher, Inorg. Chem., 1990, 29, 1812 CrossRef CAS.
- P. S. Pregosin and F. Wombacher, Magn. Reson. Chem., 1991, 29, 106 CrossRef.
- A. Comas-Vives and G. Ujaque, J. Am. Chem. Soc., 2013, 135, 1295 CrossRef CAS PubMed.
- H. G. Raubenheimer and S. Cronje, in ref. 3, p. 584 ff.
- G. N. Khairallah, R. A. J. O'Hair and M. I. Bruce, Dalton Trans., 2006, 3677 Search PubMed.
- A. Escalle, G. Mora, F. Gagosz, N. Mézailles, X. F. Le Goff, Y. Jean and P. Le Floch, Inorg. Chem., 2009, 48, 8415 CrossRef CAS PubMed.
- H. Ito, T. Saito, T. Miyahara, C. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829 CrossRef CAS.
- E. Wiberg and H. Neumaier, Inorg. Nucl. Chem. Lett., 1965, 1, 35 CrossRef CAS.
- U. Ringström, Ark. Fys., 1964, 27, 227 Search PubMed.
- U. Ringström, Nature, 1963, 198, 981 CrossRef.
- D. D. Wagman, W. H. Evans, V. B. Parker, I. Halow, S. M. Bailey and R. H. Schuman, NBS Technical Note Nr. 270-4, US Dept. of Comm., Washington D.C., 1969 Search PubMed.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules, Van Nostrand-Reinhold, New York, 1979 Search PubMed.
- D. A. Weil and C. L. Wilkins, J. Am. Chem. Soc., 1985, 107, 7316 CrossRef CAS.
- X. Wang, L. Andrews, L. Manceron and C. Marsden, J. Phys. Chem. A, 2003, 107, 8492 CrossRef CAS.
- X. Wang and L. Andrews, J. Phys. Chem. A, 2002, 106, 3744 CrossRef CAS.
- P. Schwerdtfeger, P. D. W. Boyd, S. Brienne and A. K. Burrell, Inorg. Chem., 1992, 31, 3411 CrossRef CAS.
- L. Andrews, Chem. Soc. Rev., 2004, 33, 123 RSC.
- C. A. Bayse, J. Chem. Phys. A, 2001, 105, 5902 CrossRef CAS.
- N. B. Balabanov and J. E. Boggs, J. Chem. Phys. A, 2001, 105, 5906 CrossRef CAS.
- L. Andrews and X. Wang, J. Am. Chem. Soc., 2003, 125, 11751 CrossRef CAS PubMed.
- H.-T. Liu, Y.-L. Wang, X.-G. Xiong, P. D. Dau, Z. A. Piazza, C.-L. Huang, C.-Q. Xu, J. Li and L.-S. Wang, Chem. Sci., 2012, 3, 3286 RSC.
- M. J. Crawford and T. M. Klapötke, Angew. Chem., Int. Ed., 2002, 114, 2373 CrossRef.
- G. Klatt, R. Xu, M. Pernpointner, L. Molinari, T. Q. Hung, F. Rominger, A. S. K. Hashmi and H. Köppel, Chem.–Eur. J., 2013, 19, 3954 CrossRef CAS PubMed.
- R. J. F. Berger, Z. Naturforsch., 2009, 64b, 388 Search PubMed.
- T. J. Robilotto, J. Basca, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2012, 51, 12077 CrossRef CAS PubMed.
- G. N. Khairallah, R. A. J. O'Hair and M. I. Bruce, Dalton Trans., 2006, 3699 RSC.
- A. Escalle, G. Mora, F. Gagosz, N. Mezailles, X. F. Le Goff, Y. Jean and P. le Floch, Inorg. Chem., 2009, 48, 8415 CrossRef CAS PubMed.
- H. Ito, T. Yajima, J. Tateiwa and A. Hosomi, Chem. Commun., 2000, 981 RSC.
- H. Ito, K. Takagi, T. Miyahara and M. Sawamura, Org. Lett., 2005, 7, 3001 CrossRef CAS PubMed.
- L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, T. F. Koetzle and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1987, 443 RSC.
- M. Lusi and L. J. Barbour, Cryst. Growth Des., 2011, 11, 5515 CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford Univ. Press, Oxford, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford Univ. Press, Oxford, 1999, p. 246 Search PubMed.
- M. J. Calhorda, Chem. Commun., 2000, 801 RSC.
- G. R. Desiraju, J. Chem. Soc., Dalton Trans., 2000, 3745 RSC.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Oxford Univ. Press, Oxford, 2009, pp. 49 ff Search PubMed.
- M. Etienne, J. E. McGrady and F. Maseras, Coord. Chem. Rev., 2009, 253, 635 CrossRef CAS PubMed.
- A. Martin, J. Chem. Educ., 1999, 76, 578 CrossRef CAS.
- I. Alkorta and J. Elguero, Chem. Soc. Rev., 1998, 27, 163 RSC.
- W. Yao, O. Eisenstein and R. H. Crabtree, Inorg. Chim. Acta, 1997, 254, 105 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52 CrossRef CAS PubMed.
- W. Aquilanti, E. Cornicchi, M. M. Teixidor, N. Saendig, F. Pirani and D. Cappelletti, Angew. Chem., Int. Ed., 2005, 44, 2356 CrossRef PubMed.
- Y. Gao, W. Huang, J. Woodford, L.-S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2009, 131, 9484 CrossRef CAS PubMed.
- T. S. Thakur and G. R. Desiraju, THEOCHEM, 2007, 810, 143 CrossRef CAS PubMed.
- D. Braga, F. Grepioni, E. Tedesco, K. Birasha and G. R. Desiraju, Organometallics, 1997, 16, 1846 CrossRef CAS.
- D. Braga, F. Grepioni and G. T. Desiraju, Chem. Rev., 1998, 98, 1375 CrossRef CAS PubMed.
- M. E. Olmos, in Modern Supramolecular Gold Chemistry, ed. A. Laguna, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M. Brookhart, M. L. H. Green and L. L. Wong, Prog. Inorg. Chem., 1988, 36, 1 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS.
- R. H. Crabtree, Angew. Chem., Int. Ed., 1993, 32, 789 CrossRef.
- L. Brammer, D. Zhao, F. T. Ladipo and J. Braddock-Wilking, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 632 CrossRef.
- O. Eisenstein and Y. Jean, J. Am. Chem. Soc., 1985, 107, 1177 CrossRef CAS.
- E. Clot and O. Eisenstein, Struct. Bonding, 2004, 113, 1 CrossRef CAS.
- P. L. A. Popelier and G. J. Logothetis, J. Organomet. Chem., 1998, 555, 101 CrossRef CAS.
- S. Pillet, G. Wu, V. Kulsomphob, B. G. Harvey, R. D. Ernst and P. Coppens, J. Am. Chem. Soc., 2003, 125, 1937 CrossRef CAS PubMed.
- I. Vidal, S. Melchor, I. Alkorta, J. Elguero, M. R. Sundberg and J. A. Dobado, Organometallics, 2006, 25, 5638 CrossRef CAS.
- A. H. Sommer, Nature, 1943, 152, 215 CrossRef CAS.
- A. Zachwieja, 1999, in ref. 3, p. 495.
- M. Jansen and A. V. Mudring, in ref. 3, p. 747.
- F. Hensel, Z. Phys. Chem., 1987, 154, 201 CrossRef CAS.
- W. Biltz, F. Weibke, H. J. Ehrhorn and R. Z. Wedemeyer, Z. Anorg. Allg. Chem., 1938, 236, 12 CrossRef CAS.
- E. S. Kryachko and F. Remacle, J. Chem. Phys., 2007, 127, 194305 CrossRef CAS PubMed.
- H. Schneider, A. D. Boese and J. Weber, J. Chem. Phys., 2005, 123, 084307 CrossRef PubMed.
- W. Zheng, X. Li, S. Eustis, A. Grubisic, O. Thomas, H. de Clercq and K. Bowen, Chem. Phys. Lett., 2007, 444, 232 CrossRef CAS PubMed.
- E. S. Kryachko, A. Karpfen and F. Remacle, J. Phys. Chem. A, 2005, 109, 7309 CrossRef CAS PubMed.
- E. S. Kryachko and F. Remacle, in Recent Advances in Theory of Chemical and Physical Systems, ed. J.-P. Julien, Springer, Dordrecht, 2006, vol. 15, p. 433 Search PubMed.
- J. Strähle, in ref. 3, p. 311.
- J. Hrušák, D. Schröder and H. Schwarz, Chem. Phys. Lett., 1994, 225, 416 CrossRef.
- R. H. Hertwig, J. Hrušák, D. Schröder and H. Schwarz, Chem. Phys. Lett., 1995, 236, 194 CrossRef CAS.
- Q. Li, H. Li, R. Li, B. Jing, Z. Liu, W. Li, F. Luan, J. Cheng, B. Gong and J. Sun, J. Phys. Chem. A, 2011, 115, 2853 CrossRef CAS PubMed.
- D. Feller, E. D. Glendening and W. A. de Jong, J. Chem. Phys., 1999, 110, 1475 CrossRef CAS.
- G. S. Shafai, S. Shetty, S. Krishnamurty, V. Shah and D. G. Kanhere, J. Chem. Phys., 2007, 126, 014704 CrossRef PubMed.
- P. Kasák, V. B. Arion and M. Widhalm, Tetrahedron Lett., 2007, 48, 5665 CrossRef PubMed.
- D. E. Berning, K. V. Katti, C. L. Barnes and W. A. Volkert, Chem. Ber./Recl., 1997, 130, 907 CrossRef CAS.
- M. B. Smith, S. H. Dale, S. J. Coles, T. Gelbrich, M. B. Hursthouse and M. E. Light, CrystEngComm, 2006, 8, 140 RSC.
- L. Ray, V. Katiyar, M. J. Raihan, H. Nanvati, M. M. Shaik and P. Ghosh, Eur. J. Inorg. Chem., 2006, 3724 CrossRef CAS.
- L. Ray, V. Katiyar, S. Barman, M. J. Raihan, H. Nanavati, M. M. Shaik and P. Ghosh, J. Organomet. Chem., 2007, 692, 4259 CrossRef CAS PubMed.
- M. H. Mir, J. X. Ong, G. K. Kole, G. K. Tan, M. J. McGlinchey, Y. Wu and J. J. Vittal, Chem. Commun., 2011, 47, 11633 RSC.
- J. M. López-de-Luzuriaga, A. Sladek and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1996, 4511 RSC.
- L. M. Scherf, S. A. Baer, F. Kraus, S. W. Bawaked and H. Schmidbaur, Inorg. Chem., 2013, 52, 2157 CrossRef CAS PubMed.
- E. M. Barranco, O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Eur. J. Inorg. Chem., 2004, 4820 CrossRef CAS.
- R. S. Ramón, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2011, 17, 1238 CrossRef PubMed.
- O. E. Piro, E. E. Castellano, R. C. V. Piatti, A. E. Bolzan and A. J. Arvia, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m252 Search PubMed.
- L. C. Porter, J. P. Fackler, J. Costamagna and R. Schmidt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1751 CrossRef.
- R. J. Staples, J. P. Fackler and J. Costamagna, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1555 Search PubMed.
- R. C. Luckay, F. Mebrahtu, C. Esterhuysen and K. R. Koch, Inorg. Chem. Commun., 2010, 13, 468 CrossRef CAS PubMed.
- M. Fettouhi, M. I. M. Wazeer, S. Ahmad and A. A. Isab, Polyhedron, 2004, 23, 1 CrossRef CAS PubMed.
- P. G. Jones and C. Thöne, Chem. Ber., 1991, 124, 2725 CrossRef CAS.
- A. A. Isab, M. Fettouhi, S. Ahmad and L. Ouahab, Polyhedron, 2003, 22, 1349 CrossRef CAS.
- A. Castiñeiras, N. Fernández-Hermida, R. Fernández-Rodríguez and I. García-Santos, Cryst. Growth Des., 2012, 12, 1432 Search PubMed.
- A. Castiñeiras and R. Pedrido, Dalton Trans., 2012, 41, 1363 RSC.
- T. S. Lobana, S. Khanna and R. J. Butcher, Inorg. Chem. Commun., 2008, 11, 1433 CrossRef CAS PubMed.
- S. D. Khanye, N. B. Bathori, G. S. Smith and K. Chibale, Dalton Trans., 2010, 39, 2697 RSC.
- J. S. Casas, E. E. Castellano, M. D. Couce, J. Ellena, A. Sanchez, J. Sordo and C. Taboada, J. Inorg. Biochem., 2006, 100, 1858 CrossRef CAS PubMed.
- U. Casellato, G. Fracasso, R. Graziani, L. Sindellari, A. S. Gónzalez and M. Nicolini, Inorg. Chim. Acta, 1990, 167, 21 CrossRef CAS.
- S. Friedrichs and P. G. Jones, Z. Naturforsch., 2004, 59b, 49 Search PubMed.
- F. B. Stocker and D. Britton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 798 Search PubMed.
- M. S. Hussain and A. A. Isab, Transition Met. Chem., 1984, 9, 398 CrossRef CAS.
- S. Friedrichs and P. G. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1625 Search PubMed.
- M. S. Hussain and A. A. Isab, Transition Met. Chem., 1985, 10, 178 CrossRef CAS.
- P. G. Jones and S. Friedrichs, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m623 CAS.
- M. B. Cingi, F. Bigoli, M. Lanfranchi, E. Leporati, M. A. Pellinghelli and C. Foglia, Inorg. Chim. Acta, 1995, 235, 37 CrossRef.
- M.-E. N. Gaytan, S. Bernes, E. Rodrigez de San Miguel and J. de Gyves, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m414 Search PubMed.
- R. Usón, A. Laguna, M. Laguna, J. Jiménez, M. P. Gómez and A. Sainz, J. Chem. Soc., Dalton Trans., 1990, 3457 RSC.
- G. Jia, R. J. Puddephatt and J. J. Vittal, Polyhedron, 1992, 11, 2009 CrossRef CAS.
- R. J. Bowen, M. A. Fernandes, P. W. Gitari, M. Layh and R. M. Moutloali, Eur. J. Inorg. Chem., 2005, 1955 CrossRef CAS.
- O. Crespo, E. J. Fernández, M. Gil, M. C. Gimeno, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga and M. E. Olmos, J. Chem. Soc., Dalton Trans., 2002, 1319 RSC.
- A. M. Z. Slawin and M. B. Smith, New J. Chem., 1999, 23, 777 RSC.
- J. H. K. Yip, R. Feng and J. J. Vittal, Inorg. Chem., 1999, 38, 3586 CrossRef CAS.
- J. E. Aguado, O. Crespo, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, Dalton Trans., 2010, 39, 4321 RSC.
- G. A. Ardizzoia, S. Brenna, F. Castelli, S. Galli, N. Masciocchi and A. Maspero, Inorg. Chem. Commun., 2008, 11, 502 CrossRef CAS PubMed.
- M. L. Gallego, P. Ovejero, M. Cano, J. V. Heras, J. A. Campo, E. Pinilla and M. R. Torres, Eur. J. Inorg. Chem., 2004, 3089 CrossRef CAS.
- T. Nakabuchi, Y. Matano and H. Imahori, Organometallics, 2008, 27, 3142 CrossRef CAS.
- A. Codina, E. J. Fernández, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, J. Pérez and M. A. Rodriguez, J. Am. Chem. Soc., 2002, 124, 6781 CrossRef CAS PubMed.
- Y.-P. Zhou, M. Zhang, Y.-H. Li, Q.-R. Guan, F. Wang, Z.-J. Lin, C.-K. Lam, X.-L. Feng and H.-Y. Chao, Inorg. Chem., 2012, 51, 5099 CrossRef CAS PubMed.
- D. B. Leznoff, B.-Y. Xue, R. J. Batchelor, F. W. B. Einstein and B. O. Patrick, Inorg. Chem., 2001, 40, 6026 CrossRef CAS PubMed.
- F. Baril-Robert, X. Li, M. J. Katz, A. R. Geisheimer, D. B. Leznoff and H. Patterson, Inorg. Chem., 2011, 50, 231 CrossRef CAS PubMed.
- E. Calacio, F. Lloret, R. Kivekäs, J. Suarez-Varela, M. R. Sundberg and R. Uggla, Inorg. Chem., 2003, 42, 560 CrossRef.
- C. Paraschiv, M. Andruh, S. Ferlay, M. W. Hosseini, N. Kyritsakas, J.-M. Planeix and M. Stanica, Dalton Trans., 2005, 1195 RSC.
- K. J. Kilpin, R. Horvath, G. B. Jameson, S. G. Telfer, K. C. Gordon and J. D. Crowley, Organometallics, 2010, 29, 6186 CrossRef CAS.
- G. Hogarth and M. M. Alvarez-Falcon, Inorg. Chim. Acta, 2005, 358, 1386 CrossRef CAS PubMed.
- G. Siasios and E. R. T. Tiekink, Aust. J. Chem., 1995, 48, 757 CrossRef CAS.
- H. Schmidbaur, Gmelin Handbuch der Anorganischen Chemie: Organogold Compounds, Springer-Verlag, Berlin, 1980 Search PubMed.
- A. Laguna, in ref. 3, p. 349.
- J. P. Fackler, in ref. 3, p. 795.
- I. Dance and M. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1579 RSC.
- I. Dance and M. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1587 RSC.
- E. Bys, W. B. Schweizer and J. D. Dunitz, J. Am. Chem. Soc., 1982, 104, 5893 CrossRef.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- K. Mislow, Acc. Chem. Res., 1976, 9, 26 CrossRef CAS.
- A. Bayler, G. A. Bowmaker and H. Schmidbaur, Inorg. Chem., 1996, 35, 5959 CrossRef CAS.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006 CrossRef CAS.
- C. S. W. Harker and E. R. T. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1546 CrossRef.
- C. S. W. Harker and E. R. T. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 878 CrossRef.
- C. J. L. Lock and M. A. Turner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2096 CrossRef.
- H. Schmidbaur, G. Weidenhiller, A. A. M. Aly, O. Steigelmann and G. Müller, Z. Naturforsch., 1989, 44b, 1503 Search PubMed.
- H. Schmidbaur, G. Weidenhiller, O. Steigelmann and G. Müller, Z. Naturforsch., 1990, 45b, 747 Search PubMed.
- B.-C. Tzeng, C.-K. Chan, K.-K. Cheung, C.-M. Che and S.-M. Peng, Chem. Commun., 1997, 135 RSC.
- P. Li, B. Ahrens, L. Feeder, P. R. Raithby, S. J. Teat and M. S. Khan, Dalton Trans., 2005, 874 RSC.
- A. Ilie, C. I. Rat, S. Scheutzow, C. Kinske, K. Lux, T. M. Klapötke, C. Silvestru and K. Karaghiosoff, Inorg. Chem., 2011, 50, 2675 CrossRef CAS PubMed.
- M. K. Cooper, K. Henrick, M. McPartlin and J. L. Latten, Inorg. Chim. Acta, 1982, 65, L185 CrossRef CAS.
- T. V. Baukova, V. P. Dyadchenko, N. A. Oleinikova, D. A. Lemenovskii and L. G. Kuz'mina, Russ. Chem. Bull., 1994, 43, 1063 CrossRef.
- T. V. Baukova, L. G. Kuz'mina, N. A. Oleinikova, D. A. Lemenovskii and A. L. Blumenfel'd, J. Organomet. Chem., 1997, 530, 27 CrossRef CAS.
- L. G. Kuz'mina, A. V. Churakov and J. A. K. Howard, Russ. J. Coord. Chem., 1998, 24, 282 CAS.
- L. G. Kuz'mina, Koord. Khim., 1994, 20, 547 CAS.
- M. Ghazzali, M. H. Jaafar, K. Al-Farhan, S. Akerboom and J. Reedijk, Inorg. Chem. Commun., 2012, 20, 188 CrossRef CAS PubMed.
- M. T. Räisänen, N. Runeberg, M. Klinga, M. Nieger, M. Bolte, P. Pyykkö, M. Leskelä and T. Repo, Inorg. Chem., 2007, 46, 9954 CrossRef PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Huckey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708 RSC.
- V. J. Catalano and A. L. Moore, Inorg. Chem., 2005, 44, 6558 CrossRef CAS PubMed.
- A. S. K. Hashmi, T. Hengst, C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315 CrossRef CAS.
- U. Schubert, K. Ackermann and R. Aumann, Cryst. Struct. Commun., 1982, 11, 591 CAS.
- C. E. Strasser, S. Cronje and H. G. Raubenheimer, New J. Chem., 2010, 34, 458 RSC.
- M.-Z. Wang, C.-Y. Zhou, Z. Guo, E. L.-M. Wong, M.-K. Wong and C.-M. Che, Chem.–Asian J., 2011, 6, 812 CrossRef CAS PubMed.
- C. P. McArdle, M. J. Irwin, M. C. Jennings and R. J. Puddephatt, Angew. Chem., Int. Ed., 1999, 38, 3376 CrossRef CAS.
- E. M. Lane, T. W. Chapp, R. P. Hughes, D. S. Glueck, B. C. Feland, G. M. Bernard, R. E. Wasylishen and A. L. Rheingold, Inorg. Chem., 2010, 49, 3950 CrossRef CAS PubMed.
- D. M. Stefanescu, H. F. Yuen, D. S. Glueck, J. A. Golen and A. L. Rheingold, Angew. Chem., Int. Ed., 2003, 42, 1046 CrossRef CAS PubMed.
- J. Vicente, M.-T. Chicote, M. M. Alvarez-Falćon and P. G. Jones, Chem. Commun., 2004, 2658 RSC.
- S. Friedrichs and P. G. Jones, Z. Naturforsch., 2004, 59b, 793 Search PubMed.
- S. Friedrichs and P. G. Jones, Z. Naturforsch., 2004, 59b, 1429 Search PubMed.
- V. A. Afanas'eva, L. A. Glinskaya, R. F. Klevtsova and I. V. Mironov, Russ. J. Coord. Chem., 2011, 37, 325 CrossRef CAS.
- U. Abram, J. Mack, K. Ortner and M. Müller, J. Chem. Soc., Dalton Trans., 1998, 1011 RSC.
- K. Ortner and U. Abram, Polyhedron, 1999, 18, 749 CrossRef CAS.
- M. Bardaji, A. Laguna and M. Reyez Perez, Organometallics, 2002, 21, 1877 CrossRef CAS.
- E. J. Fernandez, M. Gill, M. E. Olmos, O. Crespo, A. Laguna and P. G. Jones, Inorg. Chem., 2001, 40, 3018 CrossRef CAS PubMed.
- P. Shi, Q. Jiang, J. Lin, Y. Zhao, L. Lin and Z. Guo, J. Inorg. Biochem., 2006, 100, 939 CrossRef CAS PubMed.
- M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1995, 2865 RSC.
- A. Albinati, C. Arz and P. S. Pregosin, Inorg. Chem., 1987, 26, 508 CrossRef CAS.
- G. Minghetti, M. A. Cinellu, S. Stoccoro and A. Zucca, Gazz. Chim. Ital., 1992, 122, 455 CAS.
- G. Minghetti, A. Zucca, S. Stoccoro, M. A. Cinellu, M. Manassero and M. Sansoni, J. Organomet. Chem., 1994, 481, 195 CrossRef CAS.
- F. Kraus, H. Schmidbaur and S. S. Al-juaid, Inorg. Chem., 2013, 52, 9669 CrossRef CAS PubMed.
- M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1996, 4217 RSC.
- E. Bulak, O. Sarper, A. Dogan, F. Lissner, T. Schleid and W. Kaim, Polyhedron, 2006, 25, 2577 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7411 CrossRef CAS PubMed.
- S. Lo Schiavo, F. Nicoló, R. Scopelliti, G. Tresoldi and P. Piraino, Inorg. Chim. Acta, 2000, 304, 108 CrossRef CAS.
- Y. Okano, B. Zhou, H. Tanaka, T. Adachi, Y. Ohishi, M. Takata, S. Aoyagi, E. Nishibori, M. Sakata, A. Kobayashi and H. Kobayashi, J. Am. Chem. Soc., 2009, 131, 7169 CrossRef CAS PubMed.
- A. Tamaki and J. K. Kochi, J. Organomet. Chem., 61, 441 CrossRef CAS.
- A. Tamaki, S. A. Magennis and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 6140 CrossRef CAS.
- A. Burawoy, C. S. Gibson, G. C. Hampson and H. M. Powell, J. Chem. Soc., 1937, 1690 RSC , and references therein.
- C. Conzález-Arellano, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2005, 3451 RSC.
- J. E. Bartmess and R. T. McIver Jr, in Gas-Phase Ion Chemistry, ed. M. T. Bowers, Academic Press, New York, ch. 11, vol. 2, 1979 Search PubMed.
- E. S. Kryachko and F. Remacle, Chem. Phys. Lett., 2005, 404, 142 CrossRef CAS PubMed.
- A. H. Pakieri and Z. Jamshidi, J. Phys. Chem. A, 2007, 111, 4391 CrossRef PubMed.
- H.-J. Xie, Q.-F. Lei and W. J. Fang, J. Mol. Model., 2012, 18, 645 CrossRef CAS PubMed.
- S. Rai, N. V. S. Kumar and H. Singh, Bull. Mater. Sci., 2012, 35, 291 CrossRef CAS PubMed.
- Z. A. Tehrani, Z. Jamshidi, M. J. Javan and A. Fattahi, J. Phys. Chem. A, 2012, 116, 4338 CrossRef PubMed.
- Z. Jamshidi, H. Farhangian and Z. A. Tehrani, Int. J. Quantum Chem., 2013, 113, 1062 CrossRef CAS.
- U. Koch and P. Popelier, J. Phys. Chem., 1995, 99, 9747 CrossRef CAS.
- M. Lein, Coord. Chem. Rev., 2009, 253, 625 CrossRef CAS PubMed.
- G.-J. Cao, H.-G. Xu, R.-Z. Li and W. Zheng, J. Chem. Phys., 2012, 136, 14305 CrossRef PubMed.
- E. S. Kryachko, Pol. J. Chem., 2009, 83, 917 CAS.
- E. S. Kryachko, in Quantum Biochemistry, ed. C. F. Matta, Wiley-VCH,Weinheim, 2009, vol. 1, p. 245 Search PubMed.
- J. Valdespino-Saenz and A. Martínez, THEOCHEM, 2010, 939, 34 CrossRef CAS PubMed.
- J. Valdespino-Saenz and A. Martínez, J. Phys. Chem. A, 2008, 112, 2408 CrossRef CAS PubMed.
- R. Vargas and A. Martínez, Phys. Chem. Chem. Phys., 2011, 13, 12775 RSC.
- A. Kumar, P. C. Mishra and S. Suhai, J. Phys. Chem. A, 2006, 110, 7719 CrossRef CAS PubMed.
- E. S. Kryachko and F. Remacle, Nano Lett., 2005, 5, 735 CrossRef CAS PubMed.
- E. S. Kryachko and F. Remacle, J. Phys. Chem. B, 2005, 109, 22746 CrossRef CAS PubMed.
- G. Lv, F. Wei, H. Jiang, Y. Zhou and X. Wang, THEOCHEM, 2009, 915, 98 CrossRef CAS PubMed.
- P. Sharma, S. Sharma, A. Mitra and H. Singh, J. Biomol. Struct. Dyn., 2009, 27, 65 CAS.
- P. Sharma, H. Singh and S. Sharma, J. Chem. Theory Comput., 2007, 3, 2301 CrossRef CAS.
- P. A. Beal and P. B. Dervan, J. Am. Chem. Soc., 1992, 114, 4976 CrossRef CAS.
- K. N. Ganesh, V. A. Kumar and D. A. Barakwar, in Supramolecular Control of Structure and Reactivity, ed. A. D. Hamilton, J. Wiley, Chichester, 1996, p. 263 Search PubMed.
- M. S. Shukla, M. Dubey, E. Zakar and J. Leszczynski, J. Phys. Chem. C, 2009, 113, 3960 CAS.
- H. Kimura-Suda, D. Y. Petrovykh, M. J. Tarlov and L. J. Whitman, J. Am. Chem. Soc., 2003, 125, 9014 CrossRef CAS PubMed.
- A. Gourishankar, S. Shukla, K. N. Ganesh and M. Sastry, J. Am. Chem. Soc., 2004, 126, 3186 CrossRef PubMed.
Footnotes |
| † It should be noted already at this stage that to date there is no case where neutron diffraction has been used for the determination of a crystal structure of a compound with an Au···H–X contact. This point is important because structural data from single crystal X-ray and neutron diffraction show significant differences, which need to be taken into account in evaluating experimental results.77 Moreover, X-ray diffraction data generally did not provide accurate geometrical parameters for hydrogen atoms positioned in the vicinity of gold atoms. |
| ‡ In “hydrogen bonding terminology” the donor is the electron-poor hydrogen atom H in X–H and the acceptor is the electron-rich atom Y in Y–Z; in “Lewis acid base terminology” it would be the opposite. |
| This journal is © The Royal Society of Chemistry 2014 |