Preparation of carbon nanomaterials from molecular precursors
Simon
Rondeau-Gagné
and
Jean-François
Morin
*
Department of Chemistry and Centre de Recherche sur les Matériaux Avancés (CERMA), 1045 Ave de la Médecine, Pavillon A.-Vachon, Université Laval, Québec (QC), Canada G1V 0A6. E-mail: jean-francois.morin@chm.ulaval.ca; Fax: +1 418-656-7916; Tel: +1 418-656-2812
First published on 5th September 2013
Abstract
The preparation of carbon nanomaterials has been the focus of many research groups over the last twenty years. Traditionally, the synthesis of these materials has been accomplished using physical methods involving harsh conditions, limiting their applicability in numerous technological areas. Thus, researchers have been constantly working on developing new methods involving organic molecules possessing highly reactive functional groups that can be graphitized under mild conditions. In this tutorial review, we highlight recent approaches for the preparation of various carbon nanomaterials using physical stimuli (heat and/or light) to trigger graphitization of both non-organized and self-assembled molecular precursors. Special attention is devoted to the preparation of carbon nanomaterials from alkyne-containing molecules.
 Simon Rondeau-Gagné | Simon Rondeau-Gagné received his BSc in chemistry from the Université du Québec à Chicoutimi in 2008. In 2010, he completed his master's degree at Université Laval under the supervision of Prof. Jean-François Morin and Prof. Mario Leclerc. His master's project focused on the synthesis and characterization of new fullerene derivatives as electron acceptors for organic photovoltaics. Since 2010, he has been working as a PhD candidate in the group of Prof. Jean-François Morin. His main interests include supramolecular self-assembly of rigid macrocycles and the cross-linking of organic architectures using the topochemical photopolymerization of diynes. |
 Jean-François Morin | Jean-François Morin was awarded a PhD in chemistry from Université Laval, Quebec City, Canada, in 2004 under the guidance of Prof. Mario Leclerc. The same year, he joined The Richard E. Smalley Institute for Nanoscale Science and Technology at Rice University, Houston, Texas, USA as a postdoctoral research fellow where he worked on the development of self-propelled nanomachines under the supervision of James M. Tour. In 2006, he moved back to Université Laval as an Assistant Professor where his current research activities include the synthesis and characterization of carbon-rich materials, organic nanotubes, two-dimensional polymers, organic semiconductors and biocompatible dendrimers. He is now Associate Professor (since 2011) and Head of NanoULaval research facility. |
Key learning points• Elements of design for self-assembly of alkyne-containing molecules.• Structural requirements for topochemical polymerization. • Dependence of supramolecular architectures on the sizes and shapes of carbon nanomaterials. • Effects of reaction conditions on the final carbon nanomaterial properties. |
1. Introduction
Since the discovery of fullerenes, carbon nanotubes (CNTs) and, more recently, graphene, carbon nanomaterials have emerged amongst the most promising materials for various types of applications owing to their outstanding electronic and optical properties.1 The high number of scientific articles published annually in this area (more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 articles overall having either graphene, carbon nanotubes or fullerenes in the article title according to SciFinder®) is a direct measure of the popularity of these materials. The interest for these materials comes in major part from their unique network of sp2 carbon atoms that allow electron delocalization all over the molecules, either in one or two dimensions. Depending on different structural parameters, these nanomaterials can be semiconducting or metallic, which make them potentially useful in a wide range of electronic applications such as transistors, ultracapacitors, batteries, transparent electrodes, sensors, solar cells, and so on. As most materials, the properties and functions of carbon nanomaterials can be tuned by changing their size, shape and dimensionality. For example, graphene, which is a two-dimensional zero-gap material, could be transformed into a one-dimensional, nonzero gap semiconducting CNT just by folding the graphene sheet on itself following a precise vector.2 Thus, the control of these structural parameters is of utmost importance when specific applications are targeted. The best way to obtain a high level of preciseness for these parameters remains the modulation of the reaction conditions used to prepare carbon nanomaterials. For instance, synthetic methods that use carbon feedstock (gas, graphite, amorphous carbon, solvents, etc.) to prepare carbon nanomaterials such as laser ablation, arc discharge and hydrocarbon combustion are all methods in which reaction conditions can be adjusted to give preferentially one specific carbon nanomaterial.3 Although very valuable, these synthetic methods, called “physical methods”, possess serious disadvantages regarding materials purity, uniformity, batch-to-batch consistency and industrial applicability.4 Because physical methods often involved elevated temperature processes, the control over the structural parameters is possible, but rather limited in large-scale production of well-defined carbon nanomaterials.
000 articles overall having either graphene, carbon nanotubes or fullerenes in the article title according to SciFinder®) is a direct measure of the popularity of these materials. The interest for these materials comes in major part from their unique network of sp2 carbon atoms that allow electron delocalization all over the molecules, either in one or two dimensions. Depending on different structural parameters, these nanomaterials can be semiconducting or metallic, which make them potentially useful in a wide range of electronic applications such as transistors, ultracapacitors, batteries, transparent electrodes, sensors, solar cells, and so on. As most materials, the properties and functions of carbon nanomaterials can be tuned by changing their size, shape and dimensionality. For example, graphene, which is a two-dimensional zero-gap material, could be transformed into a one-dimensional, nonzero gap semiconducting CNT just by folding the graphene sheet on itself following a precise vector.2 Thus, the control of these structural parameters is of utmost importance when specific applications are targeted. The best way to obtain a high level of preciseness for these parameters remains the modulation of the reaction conditions used to prepare carbon nanomaterials. For instance, synthetic methods that use carbon feedstock (gas, graphite, amorphous carbon, solvents, etc.) to prepare carbon nanomaterials such as laser ablation, arc discharge and hydrocarbon combustion are all methods in which reaction conditions can be adjusted to give preferentially one specific carbon nanomaterial.3 Although very valuable, these synthetic methods, called “physical methods”, possess serious disadvantages regarding materials purity, uniformity, batch-to-batch consistency and industrial applicability.4 Because physical methods often involved elevated temperature processes, the control over the structural parameters is possible, but rather limited in large-scale production of well-defined carbon nanomaterials.
To circumvent the lack of control inherent to the physical methods, chemists and materials scientists proposed fancier alternatives that rely on synthetic organic chemistry strategies. With the help of modern and classical synthetic tools, total synthesis of carbon nanomaterials can be achieved, although these methods are far from being efficient to prepare large quantities of materials. Beautiful examples of synthesis of fullerenes, CNT fragments, nanographene and graphene nanoribbons have been published in the last twenty years and works toward more efficient synthetic paths are still underway in many research groups.5 A mid-point strategy between the physical and all-organic methods, called the “hybrid method”, is to use a mix of both in which reactive, carbon-rich precursors are synthesized in a few synthetic steps and exposed to physical stimuli such as heat or light. Depending on their organization in the solution, solid or thin-film state, these precursors can be transformed into graphitic carbon materials of various sizes, shapes and functions. Although this method is not perfect and effective for routine production of materials yet, it could provide the best long-term alternative to overcome some major disadvantages of both physical and all-organic methods. One of the major benefits of this method is the possibility of preparing carbon nanomaterials under very mild conditions (with temperature usually below 250 °C) without any complex experimental setup. Mild conditions are particularly important for the growth or preparation of carbon nanomaterials directly on sensitive substrates such as silicon and semiconducting electrodes used by the electronics industry. Nevertheless, more research must be conducted to evaluate the real potential of this increasingly popular alternative in large-scale materials production.
In this review, the strategies developed in the past twenty years or so to prepare carbon nanomaterials using reactive, carbon-rich precursors are surveyed. A particular emphasis will be put on reactive precursors that contain alkyne moieties, especially oligoynes, although other carbon-rich molecules will also be covered.
2. Solid-state polymerization of oligoynes
In order to prepare thermodynamically stable carbon nanomaterials under mild conditions and low temperatures, carbon-rich precursors with reactive functional groups that can be transformed without the use of excessive energy input have to be used. Unfortunately, very few functional groups fit that category and the one that has been studied the most is undoubtedly the alkyne. In fact, an alkyne, or a carbon–carbon triple bond, is a reactive functional group owing to its two π-bonds that provide intrinsic thermodynamic instability. On the one hand, this reactivity enables, among others, hydrogenation, electrophilic additions, hydroboration and oxidation reactions that are very precious to the organic chemists' toolbox. On the other hand, alkynes are sufficiently stable to be prepared and handled without significant decomposition. Moreover, alkynes can be installed on organic substrates using different, very versatile set of reactions6 and coupled together under various conditions to give oligoynes and polyynes.7Considering their relative thermodynamic instability and ease of preparation using common organic transformations, alkynes are definitely the ideal functional groups for the preparation of carbon nanomaterials under mild conditions, especially in the solid state. One of the most exploited reactions for the transformation of alkynes in the solid state is undoubtedly the 1,4-addition of 1,3-butadiynes, called topochemical polymerization, to give polydiacetylenes (PDAs, Fig. 1). Gerhard Wegner, whose goal was to prepare highly crystalline polymers in the solid state, discovered this reaction in 19698 and since then, thousands of articles have been published on this topic, mostly because of the excellent chromic properties of PDAs that could potentially be used in sensor devices.
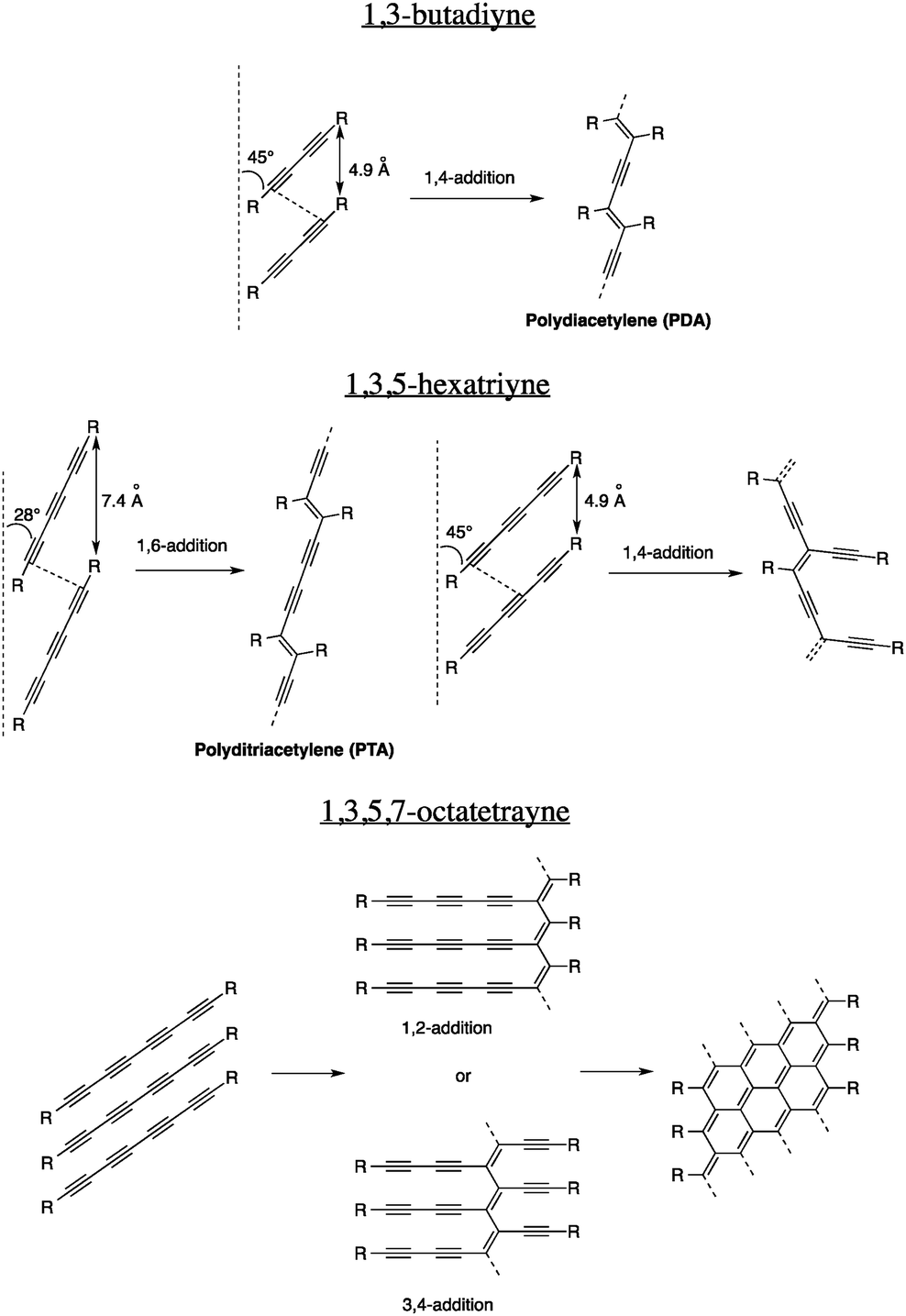 | ||
| Fig. 1 Possible addition pathways in topochemical polymerizations of oligoynes. | ||
However, not all oligoyne-embedded compounds can react to give PDAs and eventually carbon nanomaterials. For the reaction to occur, rigorous structural parameters including the intermolecular distance and the tilt angle have to be respected. For the 1,3-butadiyne derivatives, an intermolecular distance of 4.9 Å and a tilt angle of 45° are required in the solid state. These strict prerequisites mean that proper elements of design that will orient the molecules properly in the solid state must be embedded within their scaffolds. Usually, H-bonding, π–π and, to a lesser extent, van der Waals interactions can be very efficient to organize molecules in a particular orientation to allow topochemical polymerization. Over the years, the scope of this reaction has been extended to longer oligoynes,9 giving new sets of conjugated polymers. The topochemical polymerizations of different oligoynes and their structural requirements are summarized in Fig. 1.
3. Carbon-rich materials from topochemical polymerization
3.1 Carbon-rich materials from non-organized oligoynes
Besides the preparation of chromic materials, the topochemical polymerization of oligoynes can also be used to prepare carbon-rich materials with amorphous graphitic structure. The first example of preparation of graphitic materials from alkyne precursors involved the pyrolysis of poly(diethynylbenzene). In 1992, Economy and co-workers showed that poly(1,3-diethynylbenzene) underwent crosslinking reactions to form condensed aromatic structures, or amorphous graphitic carbon, that could be transformed into carbon-fiber–carbon-matrix composites.10 The authors associated the formation of amorphous graphitic carbon with multiple Diels–Alder reactions between two 1,3-butadiynes in adjacent polymer chains, although this hypothesis was not demonstrated experimentally. It is worth mentioning that this particular Diels–Alder reaction would be very difficult to drive because of the high rigidity of the alkynes. To the best of our knowledge, no such reaction has been ever reported in the literature.A few years after the Economy's report, Kijima and co-workers reported the carbonization reaction of poly(phenylenebutadiynylene)s (PPBs) at 900 °C to give amorphous graphitic carbon in very high yields.11 Interestingly, differential scanning calorimetry (DSC) analysis on the powder sample of PPBs suggested that an exothermic reaction occurred at 200 °C with enthalpy change (ΔH) values of approximately −100 kJ mol−1, which is in perfect agreement with the enthalpy change observed during the 1,4-additions of 1,3-butadiyne units to form PDAs.12 This result indicates that graphitization reaction at higher temperature is attributable to the formation of a pre-carbon material (PDA) that undergoes a cycloaromatization reaction upon further heating.
In 2006, Ozaki and co-workers undertook the detailed study on pyrolysis of PPBs by thermoanalysis.13 Using a combination of spectroscopic tools, they showed that PPBs do not actually graphitize upon heat treatment, even when temperature as high as 3000 °C was used. Instead, amorphous carbon containing sp3-hybridized carbon atoms is produced. The plausible mechanism of carbonization involved the cross-linking of polymer chains through a Diels–Alder mechanism involving both butadiyne or terminal alkyne and a phenyl group, as shown in Fig. 2. These results clearly indicate that PPBs can serve as carbon-rich reactive precursors for the preparation of amorphous graphitic carbon but that high quality, defect-free graphite or graphene would be difficult to prepare using this strategy.
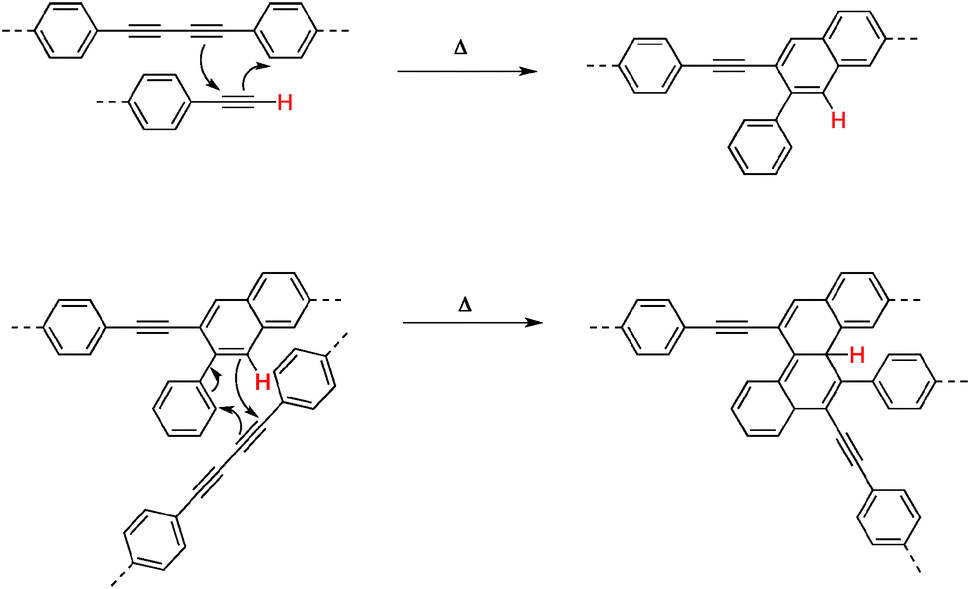 | ||
| Fig. 2 Preparation of graphitic materials from diphenylacetylene under elevated temperature and high pressure.13 | ||
Another way of preparing amorphous graphitic carbon is to use low molecular-weight molecules containing butadiyne units rather than polymers such as PPBs. Kojima and co-workers studied the transformation of 1,4-diphenyl-1,3-butadiyne by thermal annealing under elevated pressure (0.1–500 MPa).14 Interestingly, the reaction proceeds with a gas evolution, indicating that hydrogen was lost during the annealing process. This result, along with the absence of alkyne signals in the infrared spectrum, suggests that the reaction did not proceed through a 1,4-addition mechanism to form PDAs. In fact, the Raman spectrum of the insoluble black material confirms the presence of graphitized carbon materials, mostly CNTs, that are likely to be the product of dehydrogenation of polyacene-like precursors as shown in Fig. 3.
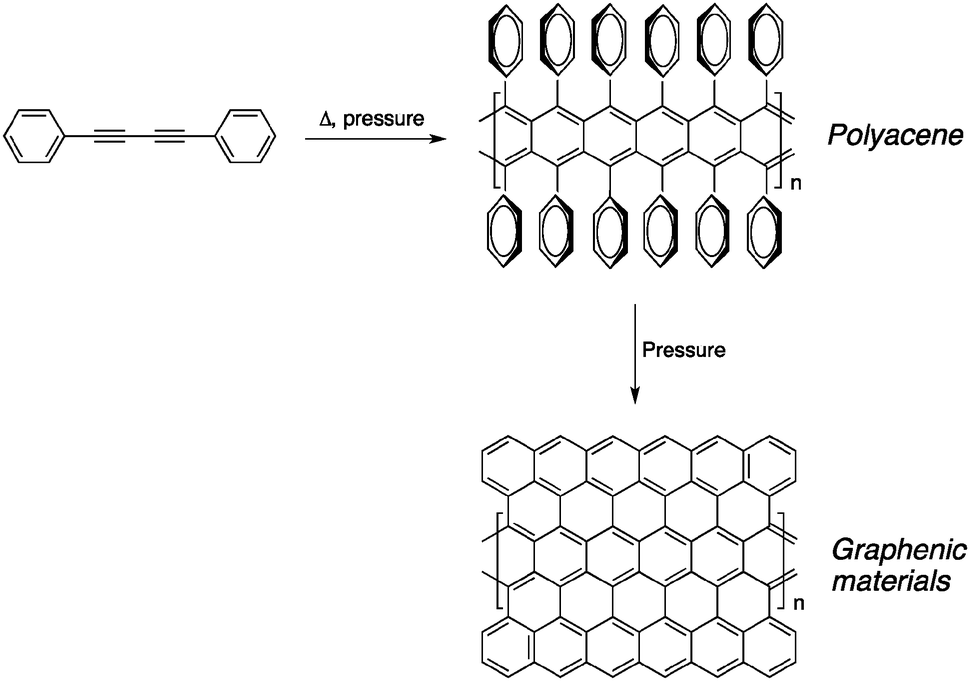 | ||
| Fig. 3 Possible pathway proposed by Kojima and co-workers for the formation of amorphous cross-linked carbon from PPBs upon heat treatment.14 | ||
In the early 2000s, the group of Hlavaty explored the reaction of very simple oligoyne derivatives for the preparation of carbon nanomaterials. In their first report in this area, Hlavaty and co-workers discovered that multi-walled CNTs and graphene-like materials could be prepared through low temperature processes using 1-iodohexa-1,3,5-triyne and hexa-1,3,5-triyne.15 Because of their instability, both derivatives undergo carbonization in solution at room temperature when stored under argon for one to two days, the iodo derivative being a little less reactive than the free hexa-1,3,5-triyne. Interestingly, two reaction mechanisms can be predominant depending on the concentration of the starting materials during the carbonization: the so-called “two-dimensional” polymerization took place when the solution was diluted to form thermodynamically stable graphene-like structures, while increasing the concentration yield the “one-dimensional” polymerization favourably (see Fig. 4). Transmission electron microscopy (TEM) analysis revealed that the graphitic materials thus produced are made of multi-walled CNTs with diameter ranging from 10 to 20 nanometers. Similar results were obtained when α,ω-dialkalihexatriynides were used as starting materials. This was among the first examples of low-temperature (≤20 °C) chemical synthesis of CNTs from carbon-rich, reactive molecules.
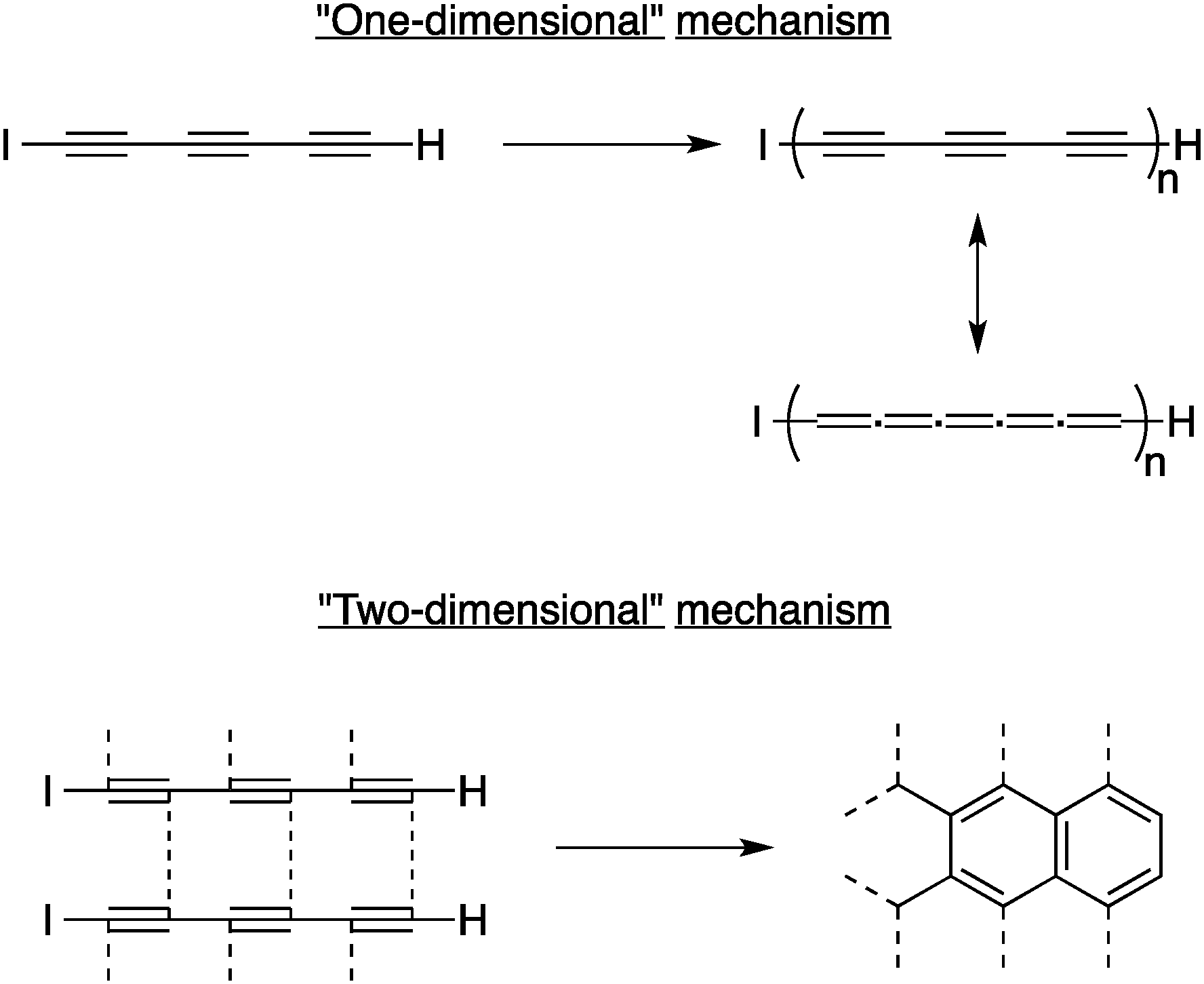 | ||
| Fig. 4 The “one-dimensional” and “two-dimensional” mechanisms for the solid-state polymerization of 1-iodohexa-1,3,5-triyne at room temperature.15 | ||
Solid-state polymerization of oligoynes can also be used to prepare nanostructured surfaces made of carbon-rich nanomaterials. In 2005, Zhao and co-workers exploited the high reactivity of tetraynes to prepare C60-embedded carbon nanospheres on a mica surface.16 After spin-coating of a C60-terminated tetrayne derivative, called the “molecular dumbbell” (Fig. 5), the surface was heated up at 160 °C for one hour, allowing intermolecular reactions between the tetrayne moieties to produce monodisperse, carbon-rich nanospheres over a relatively large scale. Because the starting material cannot stack following the proper orientation for the topochemical polymerization owing to the presence of bulky C60, the polymerization mechanism is considered to be rather random, as observed by the large exothermic peak in DSC measurements. The high steric hindrance also leads to a quite low degree of polymerization, which is believed to be beneficial to obtain homogeneous nanospheres across the surface.
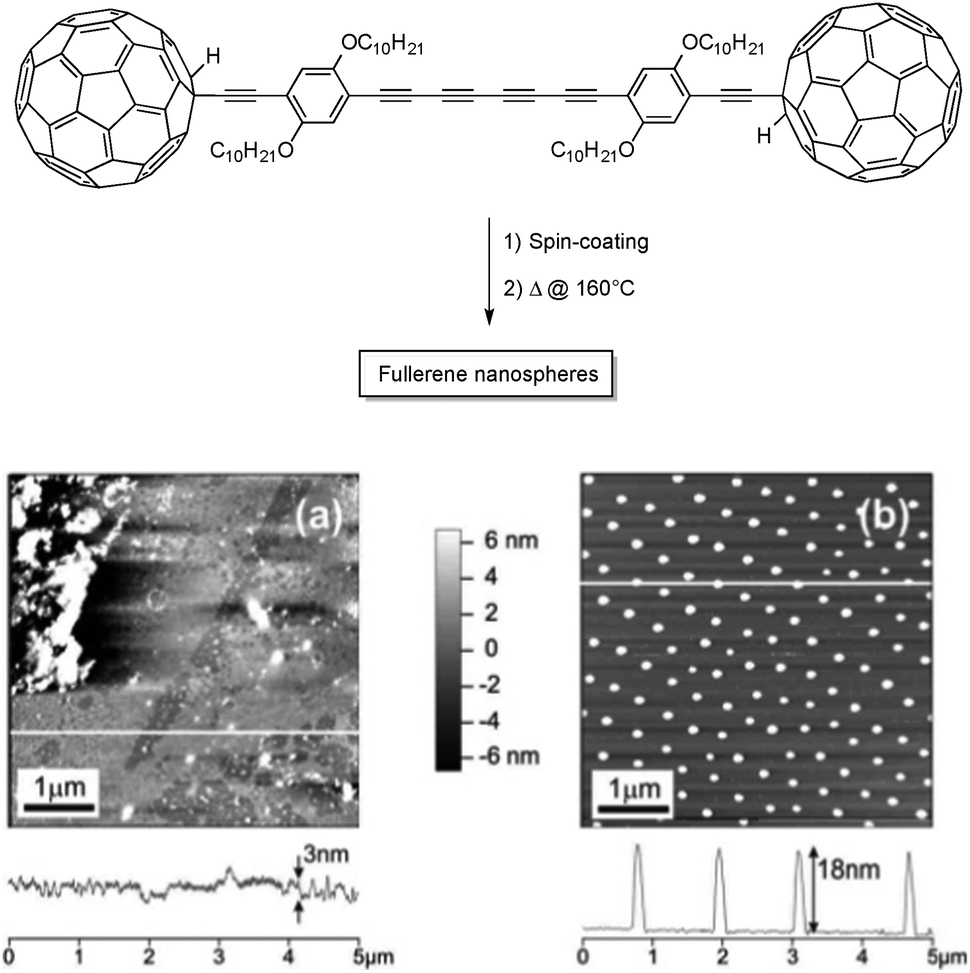 | ||
| Fig. 5 The “molecular dumbbell” used as starting materials for the preparation of carbon nanospheres (above) and AFM images of the “molecular dumbbell” before (a) and after (b) thermal annealing at 160 °C for 1 h.16 Copyright 2005 American Chemical Society. | ||
All the examples above are strong indication of the relatively high reactivity of alkynes upon heating. However, these high-temperature processes are conceptually not very different to what can be achieved with simpler carbon molecule feedstock to form amorphous graphitic carbon. In this regard, the strategy based on the use of alkyne-containing molecules is not that advantageous, especially if it does not lead to materials with unique properties. Therefore, efforts have been devoted recently to prepare carbon nanomaterials from oligoyne derivatives using milder conditions. These strategies exploit the high reactivity of oligoyne moieties toward topochemical polymerization in the solid state to prepare reactive intermediates, mostly PDAs, which can be transformed into graphitic materials using mild conditions. The most representative example of this strategy was reported by Goroff and co-workers who used co-crystals of 1,4-diiodo-1,3-butadiyne and a nitrogen-containing host to prepare poly(diiododiacetylene) (PIDA)17 that further undergo a graphitization reaction upon treatment with Lewis bases at room temperature as shown in Fig. 6.18 The Lewis base-driven transformation of PIDA into graphitic material is likely to be the result of the formation of the polyynes intermediate, accompanied by the loss of iodine (I2), which rapidly decomposes to form a network of sp2 carbon atoms. This hypothesis has been verified by treating low molecular weight 1,2-diiodoalkene with pyrrolidine that induced the dehalogenation of the alkene to give the corresponding alkyne in high yield. The final black material appeared as long fibers that exhibited higher electrical conductivity than the starting PIDA and resembles in many points the nanofibers obtained from pyrolysis of PIDA at 900 °C.19
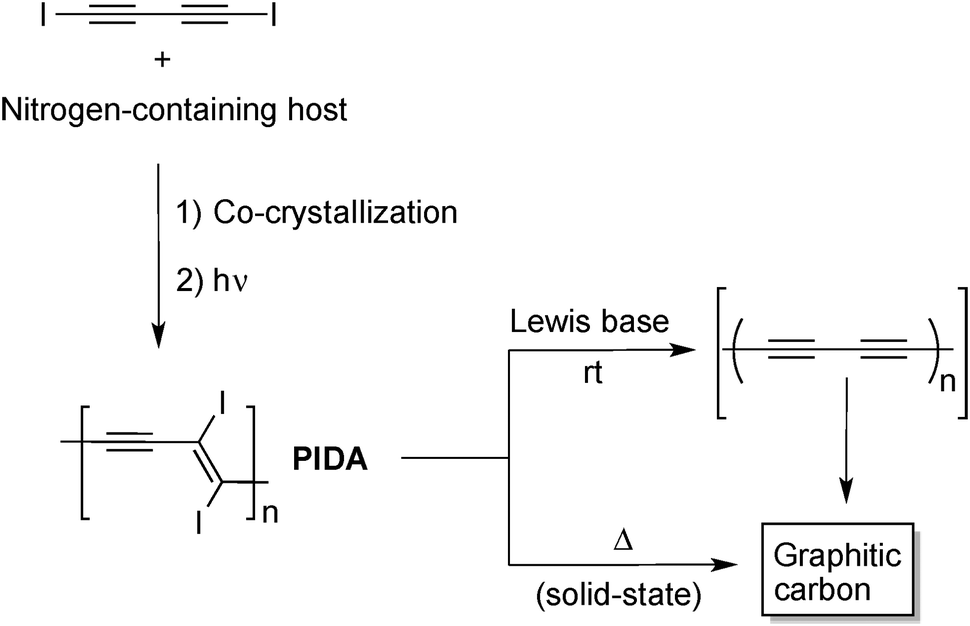 | ||
| Fig. 6 Preparation of graphitic carbon from readily accessible poly(diiododiacetylene) (PIDA).18,19 | ||
3.2 Carbon-rich materials from self-assembled oligoynes
Using the knowledge acquired on the topochemical polymerization of oligoynes, researchers recently made a step further by preparing nanomaterials with precise sizes and shapes. Using supramolecular chemistry, oligoyne-containing building blocks possessing specific functional groups that allow intermolecular interactions can be assembled to form ordered arrays of molecules capable of reacting through topochemical polymerizations to stabilize the assembly.One of the first attempts of the covalent fixation of a supramolecular assembly through topochemical polymerization was achieved on dehydrobenzo[n]annulenes (DBAs) that can assemble into tubular structures in the solid state depending on their functional group substitution patterns. Moreover, a DBA offers significant ring strain that can be exploited to make the butadiyne more reactive, thus driving the topochemical polymerization, although it was proved that solid-state packing is even more important than ring strain to drive this reaction.20 The first attempt to cross-link butadiyne-containing macrocycles through topochemical polymerization was accomplished in 1974 by Baughman and co-workers and later on by Bloor on a macrocycle (CMA) with only one butadiyne unit as shown in Fig. 7.21
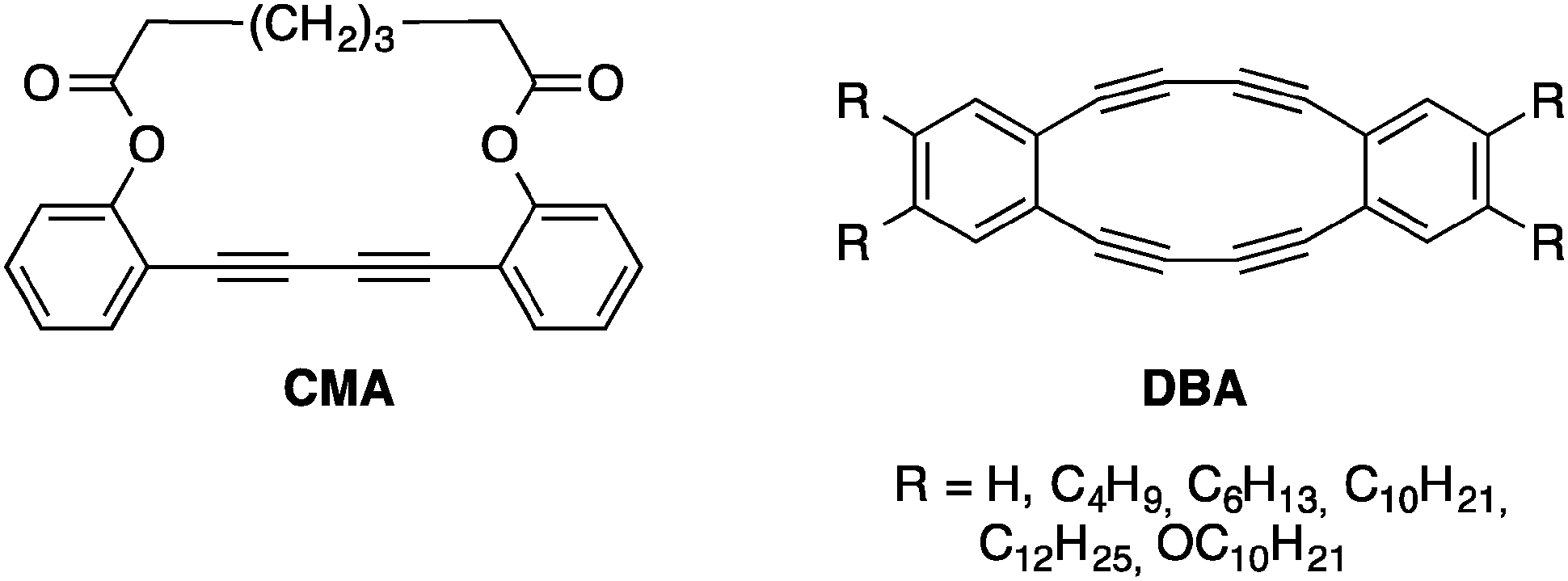 | ||
| Fig. 7 CMA and DBAs prepared by Baughman and Swager, respectively.21,22 | ||
Upon thermal annealing or exposure to UV or γ-ray irradiation, colorless crystals of CMA became deeply colored and the product formed was insoluble in organic solvents. The infrared and Raman spectroscopy and X-ray diffraction pattern clearly indicated that a PDA was formed from this macrocycle. However, there was no evidence of the formation of a tubular structure.
It was not until twenty years later that another attempt to obtain controlled polymerization of butadiyne-containing macrocycles to make nanotubes was reported. In 1994, Swager and co-workers prepared soluble macrocycles of the DBA family (Fig. 7) and tested their reactivity upon heating using DSC analyses.22 Interestingly, the apparent stability of the different DBAs depends on the substitution pattern on the phenyl groups. While electron-donating groups prevent the macrocycles from decomposing under ambient conditions, their hydrogen analogues decompose very rapidly at room temperature and exploded under gentle heating.23 DSC analysis reveals a very intense, exothermic sharp peak between 100 and 120 °C when DBA bearing alkyl chains are heated and the ΔH values of these processes are in agreement with the values reported for the topochemical polymerization of butadiyne units.12 However, infrared spectroscopy showed no evidence of PDA after the thermal process, suggesting that the starting DBAs did not undergo a typical 1,4-addition reaction. In fact, the polymerization reaction is rather random and an intramolecular reaction to form a 6-5-6-5-6 fused ring system seemed to occur as well. This reaction has been tested in solution with molecular iodine as the electrophile to initiate the reaction and yielded a tetraiodo-fused system that can be oxidized to a fused fluorenone upon exposure to oxygen.
A year after Swager's report, Vollhardt and co-workers showed that a three-membered, one butadiyne-containing DBA underwent a clean topochemical polymerization to give a PDA.24 The final PDA seemed to appear as nanotubes based on the packing mode of the DBAs as observed by X-ray diffraction. However, no experimental evidence proved the presence of such a tubular structure. Two years after their initial report, the same group reported the preparation of onion- and tube-like closed-shell carbon particles from the explosive decomposition of four-membered DBAs, as shown in Fig. 8.25 While this DBA did not undergo a topochemical polymerization under UV irradiation, it literally exploded violently when heated under vacuum at 245 °C with a flash of orange light, resulting in gas evolution (CH4 and H2) and the formation of a black powder. As shown by the TEM image in Fig. 8, the carbon materials thus produced are made of graphitic onions and capped multi-walled CNTs. Although this method leads to useful carbon materials, it does not allow control over their sizes, shapes and properties. Haley26 and Komatsu27 have prepared other DBAs in order to test their reactivity upon heating. However, the materials obtained after the thermal process were not characterized in detail.
 | ||
| Fig. 8 Vollhardt's DBAs and TEM image of the carbon materials obtained after their thermal decomposition.25 Copyright 1997 American Chemical Society. | ||
In order to prepare more reactive macrocycles, Rubin and co-workers prepared non-benzoannulated dehydroannulene macrocycles (Fig. 9).28 Benzoannulation is believed to have a stabilizing effect on the dehydroannulene scaffold in addition to hindering the reaction site. While the non-functionalized (R = H) and methyl acetate-decorated macrocycles did not stack properly to undergo topochemical polymerization, the methyl alcohol derivatives stacked properly in the crystal to create nanochannels of macrocycles in which the butadiyne moieties face each other due to the macrocycle's planar conformation. One could expect that upon polymerization, covalently linked organic nanotubes could be created. Despite the almost ideal conformation for the topochemical polymerization, the crystals of methyl alcohol-decorated macrocycles are stable under ambient conditions and start turning brown at 110 °C. It is noteworthy that crystals of the non-functionalized macrocycles exploded very rapidly under ambient conditions due to the close C–C contact and the lack of solvent inclusion within the crystals. However, the materials produced from this explosion were not further characterized.
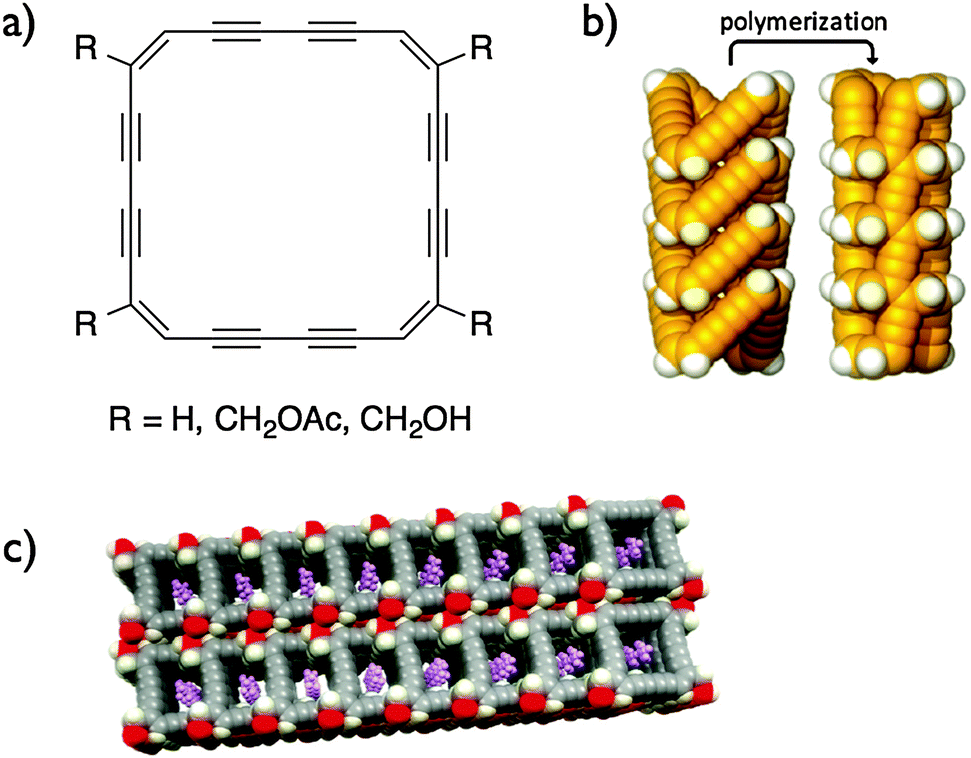 | ||
| Fig. 9 Structures of non-benzoannulated dehydroannulenes (a), schematic of a stack of the same macrocycle undergoing a topochemical polymerization (b) and the packing diagram of macrocycles where R = CH2OH (c).28 Copyright 2010 American Chemical Society. | ||
The group of Shimizu was the first to report the preparation of nanotubes using topochemical polymerization of butadiyne-embedded macrocycles.29 Starting from very simple butadiyne-containing macrocycles obtained in two steps, they were able to grow crystals in which the inter-macrocycles distance and the tilt angle are close to the ideal values to allow topochemical polymerization, as shown in Fig. 10. The choice of amide functions to drive the self-assembly into columns through H-bonding was motivated by their ability to provide structural parameters that are close to that required for the topochemical polymerization. As expected, the polymerization can be initiated upon thermal annealing (180 °C for 12 h) or UV irradiation to afford dark purple/brown crystals that were insoluble in organic solvent suggesting the formation of PDAs. Besides the ideal structural parameters, the enhanced reactivity of this macrocycle can be attributed to the presence of a more flexible bonding environment nearby the butadiyne moieties, thus enabling conformational change within the crystal lattice. Although it leads to insoluble carbon-rich materials, this strategy proved that it was possible to obtain nanostructured materials from controlled topochemical polymerization.
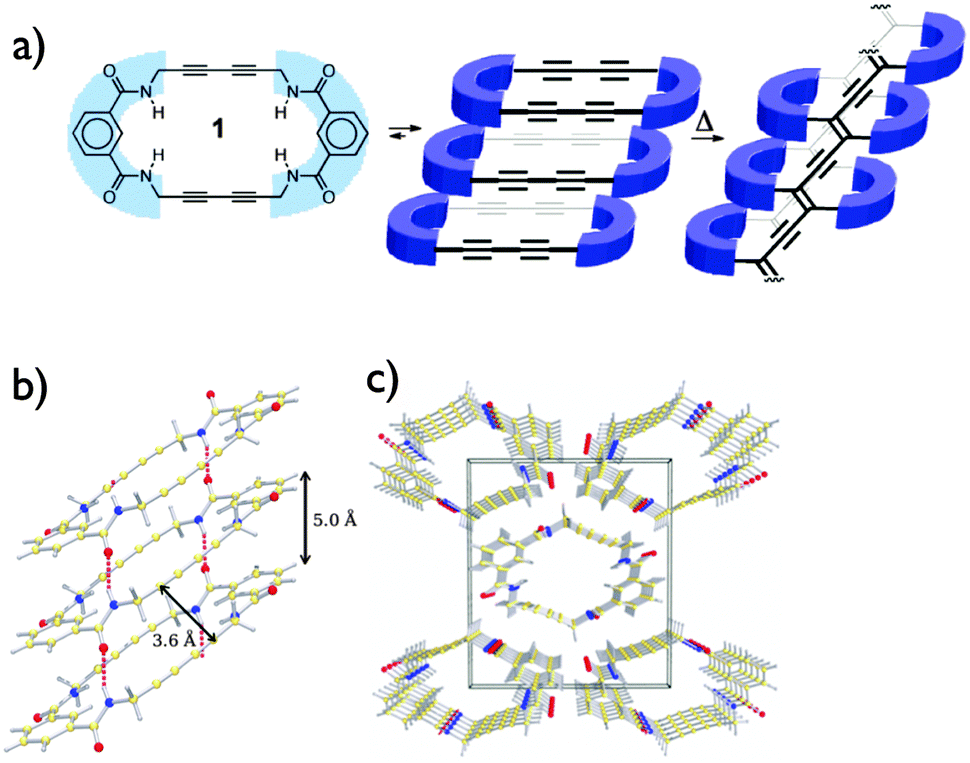 | ||
| Fig. 10 Structures (a) and X-ray crystal structure (b and c) of the butadiyne-containing macrocycle prepared by Shimizu and co-workers.29 Copyright 2010 American Chemical Society. | ||
Two years after Shimizu's report, Fowler and co-workers also reported the preparation of nanotubes from similar macrocycles but again the nanotubes formed were not soluble enough to be further characterized.30
Inspired by the strategy used by Rubin and Shimizu to create nanochannels from stacks of macrocycles within crystals, Morin and co-workers designed six-membered phenylacetylene macrocycles, or PAMs, with the aim of preparing soluble organic nanotubes with unique properties. Because the formation of crystals is tedious and very time consuming, they decided to use the formation of organogels to assemble the molecules. In fact, organogels can be prepared within minutes by simply dissolving the molecule to assemble, PAMs in this case, in a solvent near its boiling point before the solution is slowly cooled down to room temperature. The organogels thus formed are usually made of one-dimensional arrays of molecules that assembled into larger fibers or ribbons. Organogels proved to be efficient to organize molecules for topochemical polymerization of butadiyne units and many examples have been published over the past ten years. Moreover, organogels are more suitable for topochemical polymerization when bulky substituents are present at both ends of the butadiyne unit. In this regard, Morin and co-workers recently showed that aryl-appended PDAs can be obtained in relatively good yield by irradiating xerogels of 1,4-diaryl-1,3-butadiyne derivatives under UV light.31 In order to have good gelation properties, molecules have to possess specific elements of design that enhance intermolecular interactions. Usually, organogel formation relies on H-bonding, van der Waals and π–π interactions. Thus, macrocycles were designed in order to exploit these three different interactions: amide functions were introduced into the top and bottom part of the PAMs (H-bonding), long alkyl chains were attached to the four remaining phenyl groups of the PAMs' scaffold (van der Waals) and the presence of six phenyl groups ensured π–π interactions.32 PAMs of three different generations are shown in Fig. 11.
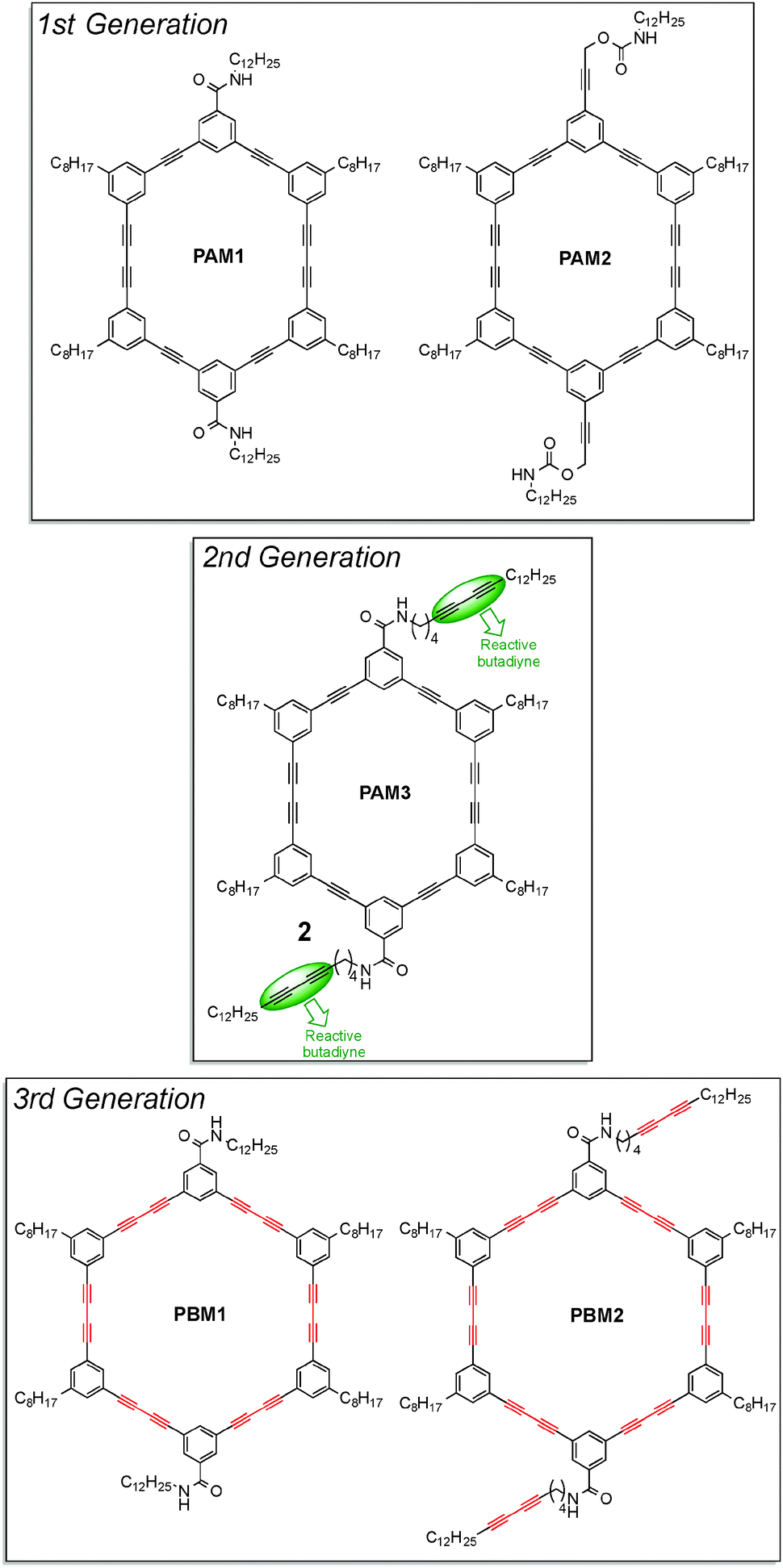 | ||
| Fig. 11 Structures of alkyne-containing macrocycles used as building blocks to prepare soluble nanorods and nanotubes.32–34 | ||
The first generation of macrocycles allowed to study in greater detail the intermolecular interactions involved within the organogels. While PAM2 did not form organogels in all the solvents tested, PAM1 formed organogels in many solvents, especially in aromatic ones. Among other features, it was shown using scanning electron microscopy (SEM) and powder X-ray diffraction (PXRD) that organogels are made of micrometer-long nanofibers, indicating that PAMs are stacking on top of each other in a columnar fashion. Also, it was demonstrated by temperature-dependent infrared spectroscopy that the amide functions of PAM1 participate in H-bonding.32 However, xerogels of PAM1 experienced only little colour change upon UV light irradiation, indicating that topochemical polymerization surely begins but is limited to very few macrocycles.
Thus, a second generation of macrocycles, with additional butadiyne units on the outer part of the macrocycles that should provide a more flexible environment enabling the topochemical polymerization in the xerogel state, was prepared.33 As expected, the xerogel of PAM3 cross-linked very rapidly under UV light to form a deep purple material. Surprisingly, Raman spectroscopy on the size-exclusion chromatography (SEC) purified material reveals that all the butadiyne moieties of PAM3, including those embedded within the macrocycle skeleton, reacted to form PDAs. The resulting soluble nanostructures appeared as nanorods rather than nanotubes by TEM since an internal void was not observed.
Encouraged by this success, they decided to extend the scope of this strategy by preparing larger macrocycles possessing six butadiyne units embedded within the macrocycle scaffold. This new generation of macrocycles, called phenylenebutadiynylene macrocycles, or PBMs, is also depicted in Fig. 11 (3rd generation).34 Unlike the PAM series, PBM2 with the external butadiyne units did not form organogels in all the solvents tested. PBM1 forms organogels in many solvents, cyclohexane being the best of all. Interestingly, the xerogel of PBM1 experienced colour change from off-white to dark purple under UV light. After purification by SEC, a soluble blue material was recovered. Again, Raman spectroscopy revealed that all the butadiynes have reacted, indicating that PBM1 underwent a six-fold polymerization to form very rigid, long nanotubes, which possess a formal internal void as determined using HRTEM. This nanostructure is a rare example of soluble, non-graphitic semiconducting nanotubes.
The topochemical polymerization of butadiyne can also be used to prepare semi-conducting, one-dimensional nanowires very efficiently. In this regard, Morin and co-workers recently demonstrated that a star-shaped molecule containing three butadiyne moieties could be used to prepare tens of nanometers-long nanowires.35 The covalent interconnection between the PDA chains within a nanowire could be potentially used to extend the conjugation of PDA in the second dimension, thus decreasing its band gap. The absence of chromic properties for these nanowires proved that the PDA adopted a rigid and planar configuration.
In addition to nanotubes and nanowires, butadiyne-containing molecules can be used to prepare various types of nanoarchitectures and some of the most common and useful in nanoscience are nanoparticles. The first to apply the topochemical polymerization of oligoynes to the preparation of nanoparticles was Olesik's group.36 In order to obtain spherical particles in the micrometers regime, they employed an emulsion polymerization using dibutyloctatetrayne as the carbon-rich source dissolved in poly(vinyl alcohol), using water as the continuous phase. After heating the suspension for 48 h at 80 °C, the particles were isolated and heated at 800 °C in a furnace to yield the final graphitized particles. The resulting particles can still be easily dispersed in various organic solvents, meaning that the pyrolysis step did not lead to aggregation between particles. Also, depending on the polymerization conditions, particles from a few micrometers to a few hundreds of micrometers were prepared. Interestingly, some of these spherical particles exhibit rather high porosity (SABET = 611 m2 g−1), making these particles potential candidates for gas storage and catalyst applications. Almost at the same time, the same group reported the preparation of much smaller carbon nanospheres by self-assembling an amphiphilic octatetrayne derivative in water that undergo a topochemical polymerization at 65 °C.37 Surprisingly, the same suspension yielded very thin films of carbon rather than nanospheres when it is kept at room temperature, meaning that various shapes of carbon materials can be obtained by carefully adjusting the kinetic parameters.
More recently, Morin and co-workers reported that an organogel of a tetrayne derivative could be polymerized to yield green fluorescent graphitic nanoparticles that are soluble in common organic solvents.38 After purification using SEC, TEM images showed that the polydispersity index of these nanoparticles was rather high with diameters ranging from 130 to 350 nm. In this case, the production of nanoparticles is quite surprising considering that the starting tetrayne organogel was made of long one-dimensional nanofibers. One would have expected a morphological retention upon polymerization as Olesik showed in the case of emulsion polymerization. This nanofibers-to-nanoparticles transformation is not understood yet and did not occur when the xerogel rather than the organogel was irradiated. To the best of our knowledge, this was the example of the preparation of graphitic nanoparticles at room temperature.
Hollow carbon nanospheres can also be obtained using the topochemical polymerization of oligoynes. Frauenrath and co-workers recently reported the preparation of such nanoarchitectures by using an amphiphilic hexayne derivative that spontaneously self-assembled into water to give vesicles whose walls can be stabilized covalently through polymerization of the hexayne moieties (Fig. 12).39 The polymerization is conducted under UV irradiation at 1 °C in water, proving the high reactivity of the hexayne moieties upon irradiation. In order to narrow the size distribution of these nanospheres, extrusion through a polycarbonate membrane can be performed, giving nanospheres with diameter of ca. 100 or 60 nm on average, depending on the pore sizes of the membranes. The main advantage of the low temperature process is the possibility of introducing biologically active molecules at the surface of the vesicles, opening the way to the use of this material in biochemical or sensing applications.
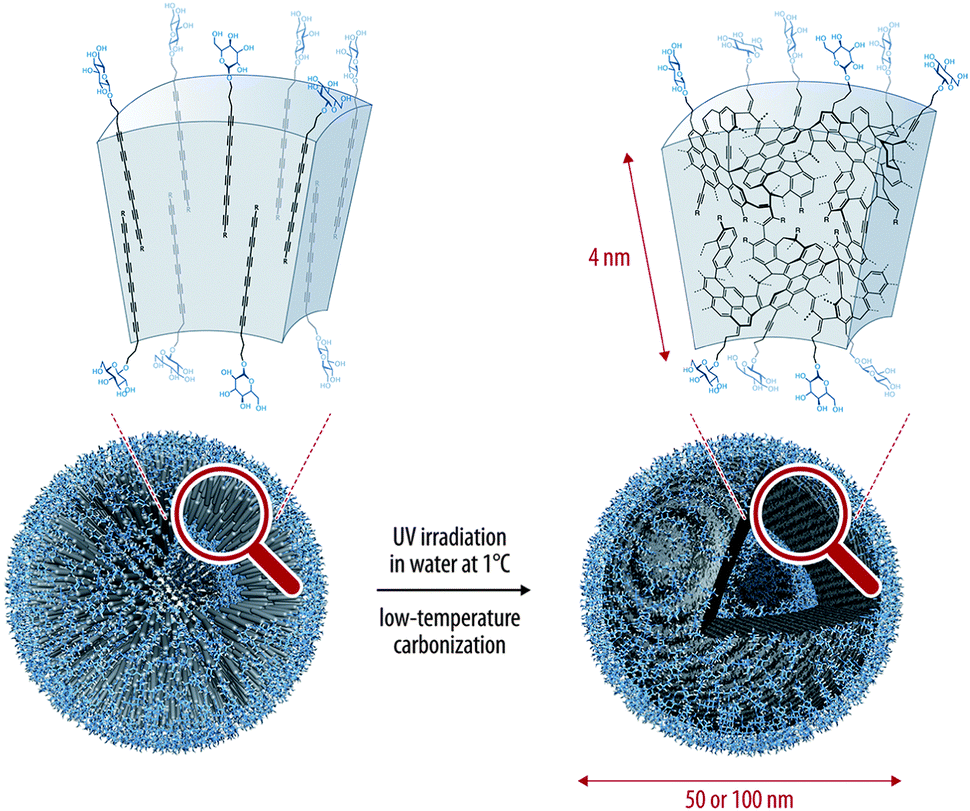 | ||
| Fig. 12 Preparation of hollow carbon nanospheres with controlled morphology through topochemical polymerization of an amphiphilic hexayne derivative.39 Copyright 2012 American Chemical Society. | ||
4. Carbon-rich nanomaterials from organometallic precursors
Metal complexation with precursors possessing specific functionality has proved to be an interesting avenue toward nanostructured materials with new properties and their large-scale preparation. A particularly interesting method was developed using the affinity between cobalt complexes and alkynes leading to highly reactive organometallic complexes upon heating. In some cases, supramolecular chemistry can be used to guide the assembly of these complexes, leading to more interesting architectures.4.1 Carbon-rich nanomaterials from cobalt-encapsulated dehydroannulene
In the late 90s, Vollhardt and co-workers revisited their work (vide supra) on DBAs by increasing their reactivity through complexation of their alkyne moieties with an organocobalt compound. This was simply achieved by mixing a DBA and Co2(CO)8.40 Upon heating of the resulting complex, it was observed that the carbonization occurred at much lower temperature (200 °C) than that required for the carbonization of cobalt-free DBA. TEM analysis on the powder obtained after the pyrolysis showed that even though carbonization already took place at low temperature, thermal treatment (800 °C for 6 h in optimal cases) produced a much better yield of onions and multi-walled CNTs, graphitic and some amorphous carbon (Fig. 13). In addition, dark spots can be noticed on carbon nanoarchitectures due to cobalt deposition on the sidewalls of the formed materials. | ||
| Fig. 13 TEM images of CNTs containing crystalline cobalt from dehydroannulene cobalt complexes.40 Copyright 1999 American Chemical Society. | ||
The observation of the high reactivity of Co-containing dehydroannulene when heated to undergo carbonization led to the synthesis of a complete family of new cobalt complexes.41 With the synthesis of new macrocycles, it was possible to vary the sizes and properties of precursors in order to study the influence of these parameters on the materials resulting from the thermal decomposition. For example, it was noted that, by introducing a ferrocenyl group into the dehydroannulene, molecular arrangement was modified and more ordered. Interestingly, the properties of explosive decomposition depended on supramolecular order and not on the intrinsic strain contained within the molecular structure.
The results obtained with dehydroannulene also bring research a step further by bringing the use of pyrolysis of cobalt–alkyne complexes in the synthesis of CNTs and nano-onions on the gram scale. Vollhardt and co-workers showed that by using small molecules such as ethynylbenzene and diphenylbutadiyne as sources of alkynes for the complexation of organocobalt compounds, it was possible to obtain a good amount of carbon-rich materials with a relatively good control over their structures and properties.42 Moreover, addition of another carbon source such as fullerene and anthracene or catalysts such as [Ni(cod)2]/tolane (1:1; cod = cycloocta-1,5-diene) directly influenced the width, the length and the amount of nanotubes formed during the pyrolysis.
4.2 Carbon-rich materials from organometallic complexes of hexabenzocoronene and related structures
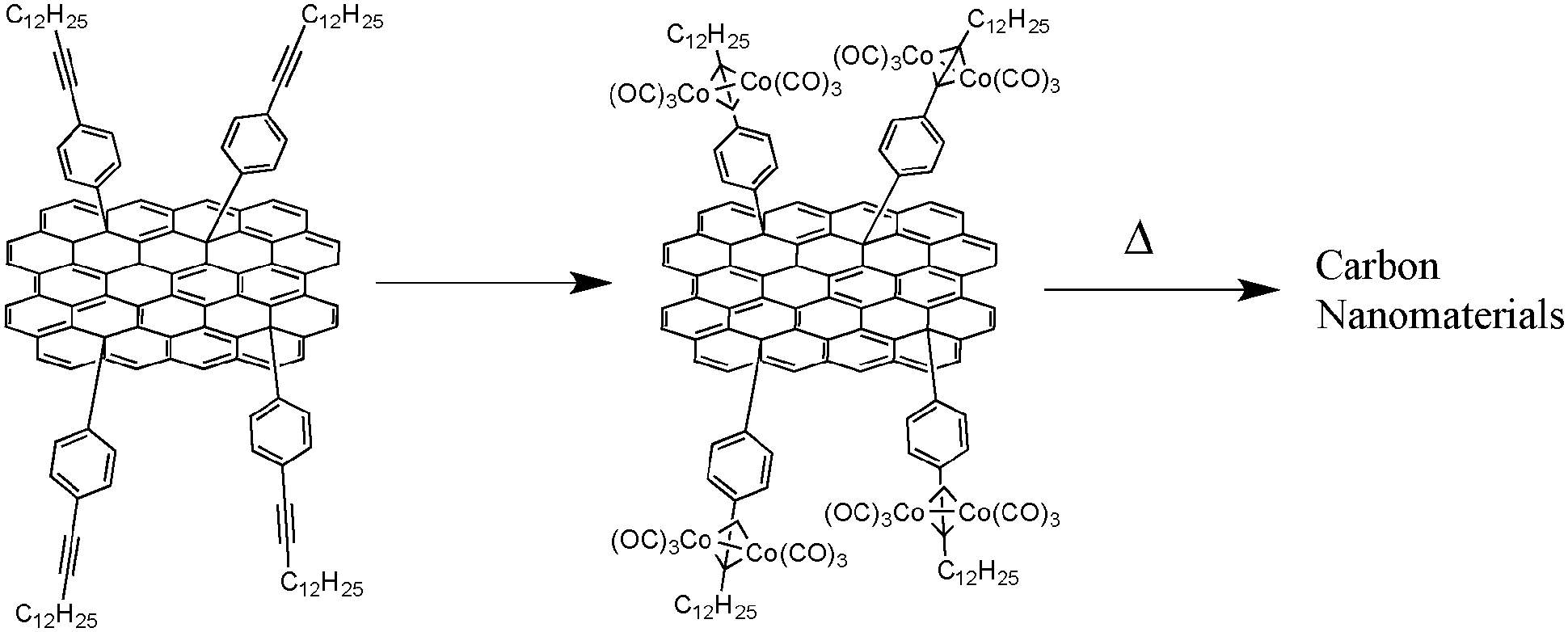
As previously demonstrated, the chemical nature of the carbon source is a crucial parameter for the control of the structural features of the resulting carbon nanomaterials. Thus, researchers investigated pyrolysis of molecules with different sizes and shapes to study the structure–property relationship. Among the different families of reactive precursors used, hexa-peri-hexabenzocoronenes (HBCs) are particularly interesting since they contain a high carbon/hydrogen ratio. In 2005, Müllen and co-workers reported the synthesis and pyrolysis of novel HBCs containing alkyne functionalities complexed with cobalt (see Fig. 14).43 By performing the pyrolysis of these materials (250 °C for 3 h and 600 to 1000 °C for 8 h), a black powder was recovered. SEM and TEM analysis revealed that the black residue was made of CNTs and bamboo-like structures with different aspect ratios. Moreover, these well-defined structures have been obtained in near-quantitative yield, meaning that this material was obtained without any production of amorphous carbons or other graphitic carbon nanostructures such as onions. The nanotubes and bamboo-like structures have showed diameters of about 33 ± 2 nm and inner diameters of about 16 ± 1 nm. TEM analysis also revealed that higher pyrolysis temperatures lead to more ordered nanotube walls. It is also important to mention that the cobalt content in the precursors is mainly found at the tips of the nanotubes, as it is the case for CNTs produced through the CVD process.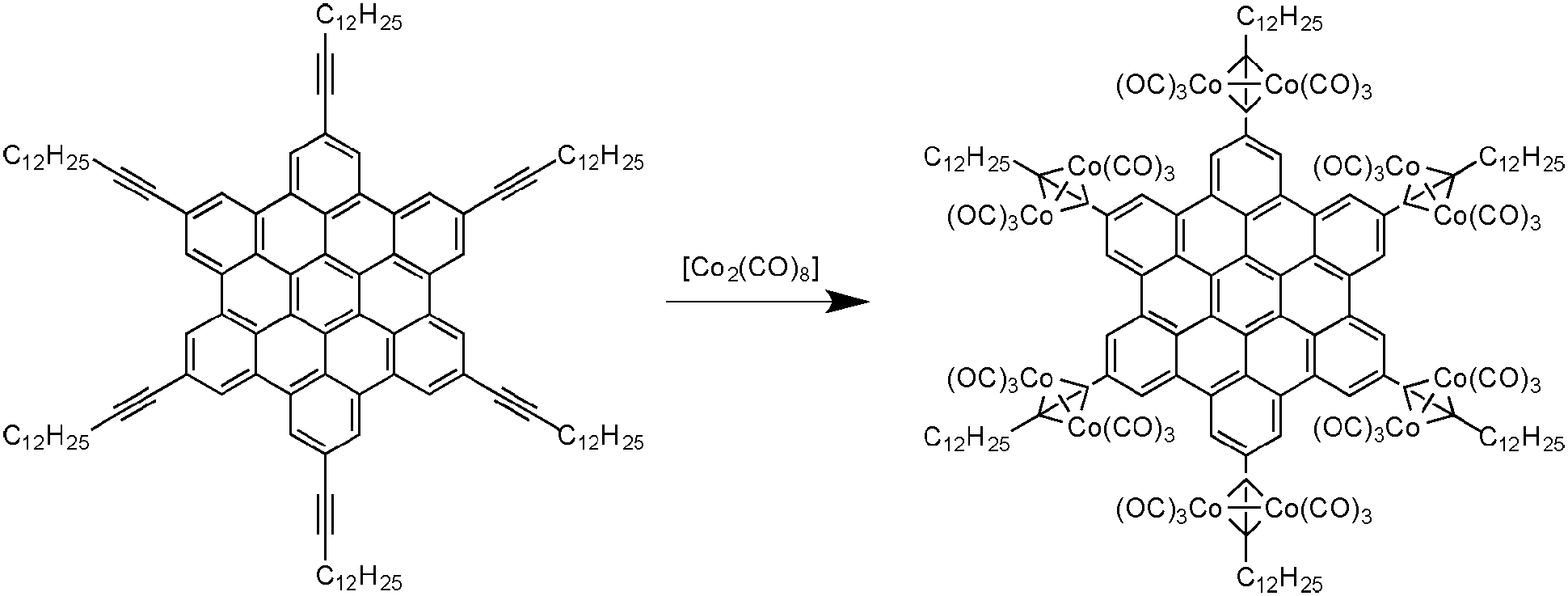 | ||
| Fig. 14 HBC–Co complex leading to carbon nanomaterials upon pyrolysis.43 | ||
In order to demonstrate the versatility of the solid-state pyrolysis using disk-like molecules and to demonstrate the importance of the precursor's structure on the final nanoarchitecture, Müllen and co-workers carried out the pyrolysis of several different compounds.44 Generally, CNTs have been obtained by pyrolysis of molecules with graphenic structure in high yields. In addition, by reducing the cobalt/carbon ratio in the precursor, a larger amount of CNTs was obtained. Alternatively, by performing the pyrolysis of materials with a non-graphenic, linear structure, carbon nanorods were obtained. Instead of cobalt, a ruthenium complex could also be used. The major difference with these complexes is the attachment point onto the molecules. With ruthenium, the metal is positioned over a π–electrons system (face-on) rather than on a specific functional group. With these complexes, larger nanorods and a significant amount of graphitic materials were obtained, demonstrating the importance of the position and the nature of the metal in the pyrolysis process.
Given that solid-state pyrolysis can lead to well-defined nanoarchitectures, synthesis of CNTs doped with nitrogen was carried out in order to obtain nanoarchitectures with improved electronic properties. To do this, the pyrolysis of metallo phthalocyanine derivatives, easily accessible, was realized.45 Pyrolysis of cobalt and iron phthalocyanines at different temperatures led to metal–organic hybrid nanowires and nanotubes with well-defined parameters. TEM analysis showed the presence of multi-walled CNTs with diameters of 20 to 60 nm and a wall thickness of 5 to 10 nm. In addition, the presence of some notable shaded area indicated twists in the structure, suggesting the introduction of five and seven membered cycles into the graphitic structures. Also, the presence of cobalt and iron crystals as well as 2 to 4% pyridine-like molecules within the final carbon-rich materials had also been observed.
4.3 Carbon-rich materials from polymeric organometallic precursors
Carbon-rich polymers are also interesting precursors for the preparation of carbon nanomaterials, although there are only a few examples reported in the literature. Reported in 2005 by Müllen and co-workers, the pyrolysis of polyphenylene dendrimers, a particularly rigid class of macromolecules, proved to be a very efficient method to obtain CNTs, nanospheres and nanorods.46 Given the monodispersity of the dendrimer unit and ease of complexation between peripheral functions and organocobalt compounds, pyrolysis yields are generally excellent. By heating polyphenylene dendrimer–cobalt complexes at 160 °C, large nanorods (size from 200 to 500 nm) can be obtained. Using this same organometallic complex but by subjecting it to a fast pyrolysis, it is possible to make hybrid carbon–cobalt nanoparticles rather than nanorods. When the heating rate is slow, CNTs can be obtained. This significant difference in the nanostructures obtained from the same carbon-rich precursor illustrates the importance of pyrolysis conditions on the final structure.In a similar trend, Bunz and co-workers reported the synthesis of carbon/cobalt nanospheres through pyrolysis of a specific cobalt-containing poly(p-phenyleneethynylene).47 By heating the hybrid polymer, large microspheres, with sizes up to 0.03 mm, were obtained. However, smaller microspheres with size averaging 1.8 microns with a narrow size distribution can be prepared.
Ferrocene derivatives are particularly interesting organometallic compounds to induce carbonization of carbon-rich precursors since it allows the preparation of hybrid carbon–iron nanoarchitectures. Generally used as catalysts and/or as feedstock in the synthesis of CNTs by CVD, π-extended ferrocenyl complexes could directly lead to well-defined carbon nanomaterials upon pyrolysis.48 The major part of the collected material appeared as nanocapsules, sometimes filled with Fe and P from starting precursors, having an average diameter of 100 nm, an average wall thickness of 16 nm and a length varying from 220 to 700 nm with no other amorphous carbon by-products. This result suggested that a near-quantitative yield of nanocapsules was obtained.
Synthesis of multi-walled CNTs containing iron nanoparticles was also achieved by pyrolysis of butadiynyl-ferrocene-containing compounds.49 The pyrolysis reaction was followed by TGA and the formation of nanotubes occurs at around 700 °C. TEM analysis shows the formation of carbonaceous materials as a complex assortment of randomly oriented multi-walled CNTs with metal nanoparticles and a small amount of amorphous carbon scattered throughout the sample. It has been shown that the mechanism of formation of multi-walled CNTs occurs from the thermal decomposition of the ferrocene units, which initially produces Fe atoms that act as a seed for the multi-walled CNT formation. In fact, the aromatic units in the precursor interact with the Fe atoms to form carbon-containing nanoparticles such as Fe3C, which appear to be a precursor to the multi-walled CNTs. Pyrolysis of such compound therefore leads to production of multi-walled CNTs at much lower temperatures than that used in the CVD method.
The formation of carbon-rich nanostructured materials allows the production of new materials with very interesting properties. In the field of molecular electronics, hybrid materials are particularly interesting for the preparation of new types of electrodes. More precisely, theoretical calculation showed that nanostructured materials containing tin could present interesting properties as electrode materials. Müllen and co-workers reported the encapsulation of tin in a carbon matrix for the first time in 2008.50 Their strategy relied on the pyrolysis of carbon–organotin complexes, more precisely allyltriphenyltin (ATP-Sn), a particularly reactive reagent in polymerization reactions. When heated at 700 °C, a black powder is produced and XRD and TEM analyses revealed that tin nanoparticles encapsulated in a carbon matrix with sizes ranging from 2 to 10 nm were produced. These new materials have also shown interesting properties for lithium storage batteries.
A very recent approach reported by Müllen and co-workers uses the formation of an organometallic complex on the surface of graphene with the aim of further growing CNTs (Fig. 15).51 To perform the in situ synthesis of such carbon nanoarchitectures, the authors had the idea of using the post-functionalization of graphene followed by insertion of alkyne-containing molecules capable of forming cobalt complexes. These cobalt complexes can then serve as seeds for growing CNTs. To verify the presence of an alkyne on the graphene sheet prior to pyrolysis, IR spectroscopy was used and revealed the presence of strong absorption bands associated with alkyne functionalities. Complexation of cobalt was made by mixing the functionalized graphene with Co2(CO)8. After being extensively rinsed with THF, the resulting powder was used as a precursor in CVD fed by a mixture of CH2–H2–Argon. TEM analysis of the resulting material clearly demonstrated the presence of CNTs on the graphene sheet, which is due to carbon growing on the complexed cobalt onto the graphene layer. Interestingly, the CNTs are randomly and horizontally stacked between the graphene layers. Such graphene–CNT composite materials are expected to be promising electrode materials in various energy storage devices.
 | ||
| Fig. 15 Cobalt-containing graphene layer to graphene–CNTs composite film.51 | ||
5. Carbon-rich materials from hexabenzocoronene (HBC) and polymer precursors
Polymers organizing in interconnected 3D networks can also be used as reactive precursors, with the potential advantage of leading to porous carbon networks once pyrolyzed. The 3D structure already present before the formation of carbon nanomaterials can also be modified or controlled by the polymerization method used, choice of monomers and chemical modifications. In addition, pyrolysis of these molecules generally gives a high rate of carbonization and pyrolysis yields. Despite the need to conduct pyrolysis, unlike some approaches previously discussed with oligoynes, temperatures required to obtain such nanomaterials are relatively low, which makes this method more accessible than what is generally used for the synthesis of such materials.In 2002, Müllen and co-workers reported the use of hexa-peri-hexabenzocoronenes (HBCs) as high molecular weight precursors without metal complexation for the preparation of carbon nanomaterials.52 These molecules, because of their disc shape, have very interesting liquid crystalline properties, which provide a very organized 3D structure. This arrangement was used to pre-orient the molecules before performing a pyrolysis at relatively low temperature. After heating to the mesophase transition temperature in order to form a highly ordered and stable columnar mesophase, the material was heated to 400 °C. MALDI-TOF mass spectrometric analysis showed partial oligomerization and revealed that it occurs in two steps, which are (1) preferential cleavage of the alkyl bonds and formation of free radicals followed by (2) an intermolecular cross-linking. It is important to note here that the aromatic nature of the HBCs and most of the alkyl chains remain intact during the oligomerization at 400 °C. Following this first stage of pyrolysis, a final pyrolysis at 800 °C was achieved. Carbonization occurred at this temperature and led to a mixture of nanostructured carbon nanomaterials, including carbon nanospheres (with diameters ranging from a few hundred nanometers to 2 μm), carbon microfibers (up to 10 μm in cross section, with a peculiar bamboo-like shape) and bundles of carbon nanowires (Fig. 16). This study also highlighted the importance of the heating steps, choice of substrate on which pyrolysis takes place and on the alkyl substitution pattern on HBC precursors over the resulting structure of resulting carbon nanomaterials.
 | ||
| Fig. 16 SEM images of (a) carbon nanospheres (scale bar 5 μm) (b) carbon microfibers (scale bar 100 μm) and (c) bundles of carbon nanowires (scale bar 200 nm) formed after pyrolysis at 800 °C of HBCs precursors in an ordered mesophase.52 Copyright 2002 American Chemical Society. | ||
To increase the control over nanoarchitectures obtained after pyrolysis of HBCs, Müllen and co-workers also performed pyrolysis of the same precursors in a porous alumina membrane.53 The trapping and confinement of these large molecules inside mesopores promotes π-stacking and allows the formation of a rigid three-dimensional structure, as in the formation of a liquid crystalline mesophase. After pyrolysis (up to 900 °C), the formation of graphitized nanotubes of exactly the same diameter as the original mesopores was observed. After full removal of the template, single CNTs with lengths of several to tens of microns were obtained. The average outer diameter is about 100 nm and the wall thickness is around 20 nm. HRTEM measurements on the resulting CNTs showed structured graphitic walls formed of highly ordered graphene layers with a layer-to-layer distance of about 0.34 nm.
More recently, the same group used a polymer network made of 1,3,5-tris-2′-biphenylbenzene and different cross-linker co-monomers (1,4-diethynylbenzene or 4,4′-diethynylbiphenyl) to obtain highly cross-linked, high molecular weight polymers.54 In addition, because of its rigidity and twisted 3D structure, 1,3,5-tris-2′-biphenylbenzene prevents the formation of tightly packed polymer chains and allows the formation of well-defined 3D structures. When these polymers are precipitated from solution, long tubular assemblies with open ends and diameters ranging from 250 to 400 nm and wall thickness between 50 to 100 nm were formed. Pyrolysis at 600 °C was achieved to graphitize the structures with full retention of the structure morphology. The porosities of the materials obtained were measured and very high specific surface areas (up to 900 m2 g−1) were obtained, opening the way for their use as electrode materials in electrochemical double layered capacitors (EDLCs).
Control of chirality can also be obtained through proper templating of carbon-rich precursors. In this regard, the group of Che and co-workers reported the synthesis of nanotubes with a partially graphitized enantiopure structure (Fig. 17).55 Using a self-assembled polypyrrole derivative forming helical nanotubes, those were heated to form carbonaceous structured architectures. Interestingly, conservation of helical tubular structure was observed, despite the use of high temperature (up to 950 °C). The supramolecular self-assembly step was directed by the electrostatic interaction between polypyrrole and an amphiphilic chiral enantiopure N-acylamino acid molecule. Then, a subsequent polymerization of the pyrrole units was achieved. The nanotubes present unique optical properties and enantiomeric purity. These properties are also observed after pyrolysis, leading the way for the synthesis of enantiopure nanotubes, which are very difficult to prepare using traditional CVD methods. These materials also present high reversible storage capacity of lithium ions.
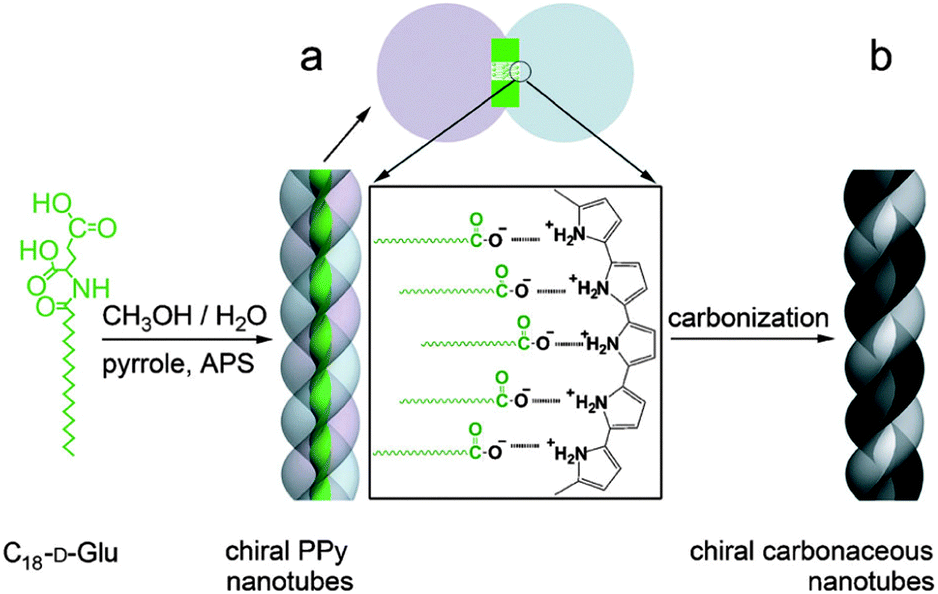 | ||
| Fig. 17 Synthesis of chiral polypyrrole nanotubes and CNTs. (a) Polypyrrole nanotubes prepared by self-assembly of C18-D-Glu (template) with ammonium persulfate (APS) as an initiator and (b) carbonaceous CNTs prepared by carbonization of polymeric nanotubes under an Ar atmosphere.55 Copyright 2013 Wiley. | ||
6. Conclusions
Although research on carbon nanomaterials has been a hot topic for a while, research on hybrid methods between all-organic and physical strategies to prepare them is still in its infancy. Alkyne-containing molecules, because of their high reactivity toward physical stimuli, proved to be promising building blocks when self-assembled to prepare carbon nanomaterials of various sizes and shapes. Using oligoyne derivatives, nanotubes, nanoparticles, nanovesicles, nanowires and other nanostructures have been prepared. As demonstrated in this review, the advantage of these precursors (Sections 2 and 3) compared to other non-alkynylated derivatives (Sections 4 and 5) is the possibility to conduct chemical transformations towards carbon nanomaterials at relatively low temperatures, often below 300 °C. This is particularly important when carbon nanomaterials have to be grown on temperature-sensitive surfaces used in the electronics industry (silicon for example). Moreover, low temperature processes are expected to provide a better control over the size and shape of the final nanomaterials through morphological retention of the initial assemblies of reactive precursors. Low-temperature processes are also compatible with the introduction of functional groups to provide the newly formed carbon nanomaterials with unique properties.Nonetheless, a significant amount of work still needs to be done in order to extend even more the scope of this strategy and to make it a real useful method that materials scientists will use on a regular basis to prepare carbon nanomaterials. As of now, the hybrid strategy showed great promise towards the preparation of well-defined carbon nanomaterials. The fact that the sizes and shapes of carbon nanomaterials can be controlled by the organization mode of the reactive precursors is probably the most important intrinsic advantage of the hybrid methods over the physical ones and many examples of that have been presented in this review. However, the hybrid methods do not offer an alternative to traditional physical methods for the large-scale production of materials yet. Before hybrid methods become the benchmark of carbon nanomaterial production methods, several requirements need to be fulfilled:
(1) low-cost, expeditious synthesis of reactive precursors;
(2) efficient and reproducible self-assembly of these precursors;
(3) morphological retention of the assemblies during the carbonization process to prepare on-demand, specific nanostructures;
(4) clean conversion to well-defined carbon materials with minimal side reactions;
(5) modulation of the properties of the carbon nanomaterials through variation in the chemical composition of the precursors.
Although these challenges will certainly take time to overcome, one can expect that the increasing number of research team working to tackle these problems will accelerate the pace of discoveries in this area and that large-scale production of carbon nanomaterials using hybrid methods will arrive sooner than later.
Notes and references
- A. Hirsch, Nat. Mater., 2010, 9, 868 CrossRef CAS PubMed; E. H. L. Falcao and F. Wudl, J. Chem. Technol. Biotechnol., 2007, 82, 524 CrossRef.
- M. S. Dresselhaus, G. Dresselhaus and P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996 Search PubMed.
- O. Zhou, H. Shimoda, B. Gao, S. Oh, L. Fleming and G. Yue, Acc. Chem. Res., 2002, 35, 1045 CrossRef CAS PubMed.
- T. N. Hoheisel, S. Schrettl, R. Szilluweit and H. Frauenrath, Angew. Chem., Int. Ed., 2010, 49, 6496 CrossRef CAS PubMed; E. T. Chernick and R. R. Tykwinski, J. Phys. Org. Chem., 2013, 26, 742, DOI:10.1002/poc.3160.
- M. M. Boorum, Y. V. Vasilev, T. Drewello and L. T. Scott, Science, 2001, 294, 828 CrossRef CAS PubMed; L. T. Scott, M. M. Boorum, B. J. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner and A. de Meijere, Science, 2002, 295, 1500 CrossRef PubMed; C. D. Simpson, G. Mattersteig, K. Martin, L. Gherghel, R. E. Bauer, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2004, 126, 3139 CrossRef PubMed; E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006 CrossRef PubMed; L. Dössel, L. Gherghel, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2540 CrossRef PubMed; K. Matsui, Y. Segawa, T. Namikawa, K. Kamada and K. Itami, Chem. Sci., 2013, 4, 84 RSC.
- P. Fritsch, Liebigs Ann. Chem., 1894, 279, 319 CrossRef CAS; W. P. Buttenberg, Liebigs Ann. Chem., 1894, 279, 324 CrossRef; H. Wiechell, Liebigs Ann. Chem., 1894, 279, 337 CrossRef; D. Seyferth, R. S. Marmor and P. Hilbert, J. Org. Chem., 1971, 36, 1379 CrossRef; E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769 CrossRef; J. C. Gilbert and U. Weerasooriya, J. Org. Chem., 1982, 47, 1837 CrossRef; K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef.
- P. Cadiot and W. Chodkiewicz, in Chemistry of Acetylenes, ed. H. G. Viehe, Marcel Dekker, New York, ch. 9, 1969 CrossRef; P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2633 CrossRef; E. Métay, Q. Hu and E. Negishi, Org. Lett., 2006, 8, 5773 CrossRef PubMed.
- G. Wegner, Natureforsch., B: Chem. Sci., 1969, 24, 824 Search PubMed; G. Wegner, Makromol. Chem., 1972, 154, 35 CrossRef CAS.
- J. Xiao, M. Yang, J. W. Lauher and F. W. Fowler, Angew. Chem., Int. Ed., 2000, 39, 2132 CrossRef CAS; A. Sarkar, S. Okada, H. Matsuzawa, H. Matsuda and H. Nakanishi, J. Mater. Chem., 2000, 10, 819 RSC; J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215 CrossRef PubMed; R. H. Baughman and K. C. Yee, J. Polym. Sci., Macromol. Rev., 1978, 13, 219 CrossRef; Y. Zhao, T. Lu, G. M. Bernard, T. Taerum, R. McDonald, R. E. Wasylishen and R. R. Tykwinski, Can. J. Chem., 2012, 90, 994 CrossRef PubMed.
- J. Economy, H. Jung and T. Gogeva, Carbon, 1992, 30, 81 CrossRef CAS.
- M. Kijima, H. Tanimoto, H. Shirakawa, A. Oya and T.-T. Liang, Carbon, 2001, 39, 287 CrossRef.
- A. Sarkar and S. S. Talwar, Bull. Chem. Soc. Jpn., 1999, 72, 859 CrossRef CAS.
- J.-I. Ozaki, M. Ito, H. Fukazawa, T. Yamanobe, M. Hanaya and A. Oya, J. Anal. Appl. Pyrol., 2006, 77, 56 CrossRef CAS PubMed.
- Y. Kojima, M. Tsuji, T. Matsuoka and H. Takahashi, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1371 CrossRef CAS.
- J. Hlavaty, L. Kavan, N. Kasahara and A. Oya, Chem. Commun., 2000, 737 RSC; J. Hlavaty, L. Kavan and J. Kubista, Carbon, 2002, 40, 345 CrossRef CAS; J. Hlavaty, L. Kavan, K. Okabe and A. Oya, Carbon, 2002, 40, 1147 CrossRef.
- N. Zhou, S. E. F. Merschrod and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 14154 CrossRef CAS PubMed.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030 CrossRef CAS PubMed.
- L. Luo, D. Resch, C. Wilhelm, C. N. Young, G. P. Halada, R. J. Gambino, C. P. Grey and N. S. Goroff, J. Am. Chem. Soc., 2011, 133, 19274 CrossRef CAS PubMed.
- L. Luo, C. Wilhelm, C. N. Young, C. P. Grey, G. P. Halada, K. Xiao, I. N. Ivanov, J. Y. Howe, D. B. Geohegan and N. S. Goroff, Macromolecules, 2011, 44, 2626 CrossRef CAS.
- V. Enkelmann and H. J. Graf, Acta Crystallogr., 1978, B34, 3715 CAS.
- R. H. Baughman and K. C. Yee, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 2467 CrossRef CAS; D. Bloor, Chem. Phys. Lett., 1976, 42, 174 CrossRef.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294 CrossRef CAS.
- O. M. Behr, G. Eglinton, A. R. Galbraith and R. A. Raphael, J. Chem. Soc., 1960, 3614 RSC.
- K. P. Baldwin, A. J. Matzger, D. A. Scheiman, C. A. Tessier, K. P. C. Vollhardt and W. J. Youngs, Synlett, 1995, 1215 CrossRef CAS PubMed.
- R. Boese, A. J. Matzger and K. P. C. Vollhardt, J. Am. Chem. Soc., 1997, 119, 2052 CrossRef CAS.
- M. M. Haley, M. L. Bell, J. J. English, C. A. Johnson and T. J. R. Weakly, J. Am. Chem. Soc., 1997, 119, 2956 CrossRef CAS.
- T. Nishinaga, N. Nodera, Y. Miyata and K. Komatsu, J. Org. Chem., 2002, 67, 6091 CrossRef CAS PubMed.
- M. Suzuki, A. Comito, S. I. Khan and Y. Rubin, Org. Lett., 2010, 12, 2346 CrossRef CAS PubMed.
- Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334 CrossRef CAS PubMed.
- T.-J. Hsu, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2012, 134, 142 CrossRef CAS PubMed.
- J. R. Néabo, K. I. S. Tohoundjona and J.-F. Morin, Org. Lett., 2011, 13, 1358 CrossRef PubMed.
- K. Cantin, S. Rondeau-Gagné, J. R. Néabo, M. Daigle and J.-F. Morin, Org. Biomol. Chem., 2011, 9, 4440 CAS.
- S. Rondeau-Gagné, J. R. Néabo, M. Desroches, J. Larouche, J. Brisson and J.-F. Morin, J. Am. Chem. Soc., 2013, 135, 110 CrossRef PubMed; S. Rondeau-Gagné, J. R. Néabo, M. Desroches, K. Cantin, A. Soldera and J.-F. Morin, J. Mater. Chem. C, 2013, 1, 2680 RSC.
- S. Rondeau-Gagné, J. R. Néabo, M. Desroches, I. Levesque, M. Daigle, K. Cantin and J.-F. Morin, Chem. Commun. 10.1039/c3cc43177k , Advance Article.
- J. R. Néabo, S. Rondeau-Gagné, C. Vigier-Carrière and J.-F. Morin, Langmuir, 2013, 29, 3446 CrossRef PubMed.
- L. Ding and S. V. Olesik, Chem. Mater., 2005, 17, 2353 CrossRef CAS.
- L. Ding and S. V. Olesik, Nano Lett., 2004, 4, 2271 CrossRef CAS.
- J. R. Néabo, C. Vigier-Carrière, S. Rondeau-Gagné and J.-F. Morin, Chem. Commun., 2012, 48, 10144 RSC.
- R. Szilluweit, T. N. Hoheisel, M. Fritzsche, B. Ketterer, A. F. Morral, D. Demurtas, V. Laporte, R. Verel, S. Bolisetty, R. Mezzenga and H. Frauenrath, Nano Lett., 2012, 12, 2573 CrossRef CAS PubMed.
- P. I. Dosa, C. Erben, V. S. Iyer, K. P. C. Vollhardt and I. M. Wasser, J. Am. Chem. Soc., 1999, 121, 10430 CrossRef CAS.
- M. Laskoski, W. Steffen, J. G. Morton, M. D. Smith and U. H. Bunz, J. Am. Chem. Soc., 2002, 124, 13814 CrossRef CAS PubMed.
- V. S. Iyer, K. P. C. Vollhardt and R. Wilhelm, Angew. Chem., 2003, 115, 4515 CrossRef.
- J. Wu, B. El Hamaoui, J. Li, L. Zhi, U. Kolb and K. Müllen, Small, 2005, 1, 210 CrossRef CAS PubMed.
- B. El Hamaoui, L. Zhi, J. Wu, J. Li, N. T. Lucas, Z. Tomović, U. Kolb and K. Müllen, Adv. Funct. Mater., 2007, 17, 1179 CrossRef CAS; L. Zhi, Y. S. Hu, B. E. Hamaoui, X. Wang, I. Lieberwirth, U. Kolb, J. Maier and K. Müllen, Adv. Mater., 2008, 20, 1727 CrossRef.
- L. Zhi, T. Gorelik, R. Friedlein, J. Wu, U. Kolb, W. R. Salaneck and K. Müllen, Small, 2005, 1, 798 CrossRef CAS PubMed; L. Zhi, T. Gorelik, J. Wu, U. Kolb and K. Müllen, J. Am. Chem. Soc., 2005, 127, 12792 CrossRef PubMed.
- B. El Hamaoui, L. Zhi, J. Wu, U. Kolb and K. Müllen, Adv. Mater., 2005, 17, 2957 CrossRef CAS.
- S. Scholz, P. J. Leech, B. C. Englert, W. Sommer, M. Weck and U. H. F. Bunz, Adv. Mater., 2005, 17, 1052 CrossRef CAS.
- D. Jain, A. Winkel and R. Wilhelm, Small, 2006, 2, 752 CrossRef CAS PubMed.
- M. Laskoski, T. M. Keller and S. B. Qadri, Carbon, 2007, 45, 443 CrossRef CAS PubMed.
- G. L. Cui, Y. S. Hu, L. J. Zhi, D. Q. Wu, I. Lieberwirth, J. Maier and K. Müllen, Small, 2007, 3, 2066 CrossRef CAS PubMed.
- Q. Su, Y. Liang, X. Feng and K. Müllen, Chem. Commun., 2010, 46, 8279 RSC.
- L. Herghel, C. Kübel, G. Lieser, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2002, 124, 13130 CrossRef PubMed.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 2120 CrossRef CAS PubMed.
- F. Xinliang, Y. Liang, L. Zhi, A. Thomas, D. Wu, I. Lieberwirth, U. Kolb and K. Müllen, Adv. Funct. Mater., 2009, 19, 2125 CrossRef.
- S. Liu, Y. Duan, X. Feng, J. Yang and S. Che, Angew. Chem., Int. Ed., 2013, 52, 6858 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |