
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimised photocatalytic hydrogen production using core–shell AuPd promoters with controlled shell thickness†
Wilm
Jones
ab,
Ren
Su
*c,
Peter P.
Wells
ad,
Yanbin
Shen
ce,
Nikolaos
Dimitratos
ab,
Michael
Bowker
ab,
David
Morgan
 b,
Bo B.
Iversen
ce,
Arunabhiram
Chutia
ad,
Flemming
Besenbacher
c and
Graham
Hutchings
*ab
b,
Bo B.
Iversen
ce,
Arunabhiram
Chutia
ad,
Flemming
Besenbacher
c and
Graham
Hutchings
*ab
aThe UK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Oxfordshire OX11 0FA, UK
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: Hutch@cardiff.ac.uk
cInterdisciplinary Nanoscience Center (iNANO) and Department of Physics and Astronomy, Aarhus University, Gustav Wieds Vej 14, DK-8000 Aarhus C, Denmark. E-mail: rensu@inano.au.dk
dUniversity College London, Kathleen Lonsdale Materials, Department of Chemistry, Gordon Street, London, WC1H 0AJ, UK
eDepartment of Chemistry, Aarhus University, Langelandsgade 140, DK-8000 Aarhus C, Denmark
First published on 27th October 2014
Abstract
The development of efficient photocatalytic routines for producing hydrogen is of great importance as society moves away from energy sources derived from fossil fuels. Recent studies have identified that the addition of metal nanoparticles to TiO2 greatly enhances the photocatalytic performance of these materials towards the reforming of alcohols for hydrogen production. The core–shell structured Au–Pd bimetallic nanoparticle supported on TiO2 has being of interest as it exhibited extremely high quantum efficiencies for hydrogen production. However, the effect of shell composition and thickness on photocatalytic performance remains unclear. Here we report the synthesis of core–shell structured AuPd NPs with the controlled deposition of one and two monolayers (ML) equivalent of Pd onto Au NPs by colloidal and photodeposition methods. We have determined the shell composition and thickness of the nanoparticles by a combination of X-ray absorption fine structure and X-ray photoelectron spectroscopy. Photocatalytic ethanol reforming showed that the core–shell structured Au–Pd promoters supported on TiO2 exhibit enhanced activity compared to that of monometallic Au and Pd as promoters, whilst the core–shell Au–Pd promoters containing one ML equivalent Pd provide the optimum reactivity.
Introduction
Photocatalytic hydrogen (H2) production is an attractive method of converting solar energy into chemical energy,1,2 with H2 showing great promise as a future fuel. Indeed, the move towards a H2 economy is driven by the impact of CO2 emissions and ever diminishing reserves of fossil fuels. Currently, more than 90% of H2 is produced via steam reforming of hydrocarbons, an energy intensive process which requires high temperature (700–1100 °C).3 However, as this process is still fundamentally driven by the consumption of fossil fuels, there has been a desire to harness the energy from the sun to produce H2. The photocatalytic reforming of alcohols (i.e., methanol, ethanol, and glycerol) using semiconductor based materials offers an alternative route for the production of H2 at ambient conditions.4Titanium dioxide (TiO2) has received significant attention as an efficient semiconductor for photocatalysis applications,4–9 which is largely as a consequence of its relative low recombination rate, non-toxicity, stability, and abundance.10 Photocatalytic H2 production by TiO2 based materials relies on the generation of electron–hole (e− and h+) pairs via absorption of photons with energy equal to or greater than the band gap (3.2 eV for anatase).11 The pairs either recombine or react with surface adsorbed species; therefore an effective photocatalyst should be able to inhibit recombination whilst utilising the electron–hole pairs to perform redox reactions efficiently. However, previous studies have shown that pristine TiO2 based materials are poor photocatalyst materials for reforming reactions,12 as the charge transfer from TiO2 to surface adsorbed alcohols is significant slower than that of the recombination kinetics.
The addition of precious metal (i.e., Pt, Pd, Au, and Ag) nanoparticles (NPs) on the surface of TiO2 can greatly enhance the H2 production efficiency.13–16 The promotional effect of the metal NPs can be understood in terms of the energy levels of the semiconductor–metal system, where the photo-excited conduction band (CB) electrons on the surface of TiO2 can be rapidly transferred and trapped at the surface of NPs.17 This prolongs the lifetime of the trapped electrons thus improving the photoreactivity.18,19 To further tune the charge separation efficiency, modification of TiO2 with bimetallic NPs has also been studied (i.e., AuPt, AgPt, and AuPd).2,20,21 Recent work by Su et al. on AuPd NPs as promoters has shown the structure of the NPs to be important to the activity of the photocatalyst. Their study highlighted Pdshell–Aucore structured NPs demonstrated the highest photoreactivity with excellent reusability in comparison to other possible structures.18,22 This increase in activity was attributed to the structured NPs increased ability to inhibit the reverse transfer of trapped electrons back to TiO2, whilst simultaneously providing a fast reduction rate of the surface adsorbed reagents. The enhanced photocatalytic performance caused by the interaction between AuPd alloy and TiO2 was also reported by Mizukoshi et al.23 Moreover, recent research on photocatalytic benzene oxidation further suggested that the interplay of AuPd alloy and TiO2 led to a significantly improvement of selective conversion of phenol.24 These investigations also indicate the thickness and composition of the shell layer may influence the electronic properties of the promoters. Chemical vapour impregnation (CVI) approach have been also reported as a promising technique for the decoration of semiconductor photocatalyst with surface clean metal promoter NPs.22 However, synthesis of metal NPs with controlled shell thickness and composition has been seldom performed due to the challenges in preparation. As a consequence, although the physical properties of the shell layer of the promoter was considered to be an essential parameter, how it influence the photocatalytic performance was still unclear. Moreover, the properties of the shell layer have been normally determined by microscopic methods, which left the overall features of the shell layer remaining uncertain.
This study reports the synthesis of core–shell structured Au–Pd metal NPs with controlled shell thickness using both photodeposition and sequential reduction routes. Moreover, by depositing only thin layers of Pd it allows bulk characterisation techniques, such as X-ray absorption fine structure (XAFS), to provide some degree of surface sensitive information.25 Indeed, the thin layers of Pd deposited here allow us to ascertain the core–shell nature of these materials in more detail. Herein, this study we demonstrate how a change in the microstructure of the metal promoter influences the photocatalytic H2 evolution performance.
Experimental
Catalyst preparation
For the synthesis of core–shell structured metal NPs supported on TiO2, we first prepared 1 wt% of Au supported on TiO2 (Au/TiO2) by standard sol immobilisation.26 Then thin layers of Pd were deposited on Au using either by photodeposition or sequential reduction method, as demonstrated in below:Catalyst characterisation
| Sad = 1/(∑nd3/∑nd2) | (1) |
| D = 1/Sad = NS/NT | (2) |
Photocatalytic performance evaluation
Photocatalytic evolution of H2 was carried out in a leak-tight reactor and products were analysed using a quadrupole mass analyser (MS, Hiden HPR-20). For all measurements, 25 mg of the photocatalyst was dispersed into 18.75 mL of DI water in the reactor and subsequently UV irradiated using a UV-A LED light (WL: 365 nm, FWHM: 10 nm, Tritan™ 365, Spectroline) for 2.5 h to remove the protecting ligands. 6.25 mL of absolute ethanol (99%) was then added in and the suspension was kept in the dark for 30 min to establish adsorption equilibrium prior to experiment. The reactor was then carefully evacuated for 30 min using a bypass rotary pump that was attached to the reactor. The dissolved O2 in the solution was calculated to be ∼5 μM after evacuation for all tests. A two-hour irradiation was employed for the H2 evolution process under continuous stirring and the partial pressures of m/z− = 2(H2), 18(H2O), 28(N2), 32(O2), and 44(CO2) were monitored. The light source was a UV LED (365 nm, Optimax 365) with a photon flux of 4 × 1017 photons s−1 for all H2 evolution experiments. Note that all incidents UV light can be sufficiently absorbed by the photocatalyst suspension (see assessment in ESI†). All samples were characterised by similar optical properties, as shown in diffuse reflectance spectra (see Fig. S2 in ESI†). Details of the photo-reactivity measurement and related calculations have been reported elsewhere.30 The produced H2 [n(H2)gas] and the apparent quantum efficiency (AQE) can be therefore derived using the following equations:18| n(H2)gas = p(H2)rea × Vrea/RT | (3) |
| p(H2)rea = RSF(H2) × p(H2)det × p(Air)rea/p(Air)det | (4) |
| AQE = 2n(H2)gas/n(photons) × 100% | (5) |
n(photons) is the number of incident photons within two hours, which is estimated to be 4.78 mmol by using the photon flux of the light source (4 × 1017 photons s−1).
Results and discussion
Structure characterisation
TEM characterisation was performed on all materials to determine the metal particle size distribution. Representative TEM images and the derived histograms are shown in Fig. 1, and the calculated average particle sizes along with standard deviations are shown in Table 1. It is clear from the TEM images that all metal NPs were deposited on the TiO2 homogeneously, and there is no significant difference in particle size within all samples. The values of the standard deviation are roughly one third of the average particle size. As such, any particle size increase as a result of the Pd modification is not observable. As the particle size has remained roughly constant, it is thought possible to rule out two potential scenarios, (i) Pd was deposited in small particles away from the Au only and (ii) Pd was deposited in large particles away from the Au only. In both cases the average particle size would be shifted to either smaller or larger values. This can be seen more clearly from TEM images and the particle size distribution histograms (Fig. 1). In fact, the Pd deposition is thought to proceed through a seed growth mechanism on the surface of the Au NPs in both preparation approaches. During the photo excitation of Au/TiO2 it has been established that electrons will migrate to the metal surface,19 therefore the reduction and deposition of Pd should be predominately onto the surface of the Au. | ||
| Fig. 1 Representative TEM images of (a) 1 ML (PD), (b) 1 ML (SR), (c) 2 ML (PD), (d) 2 ML (SR), and (e) Au nanoparticles supported on TiO2, respectively. The metal loading was 1 wt% for all samples (f) particle size distributions derived from corresponding TEM images. | ||
| Catalyst | Average particle size (nm) | Standard deviation (nm) |
|---|---|---|
| Au/TiO2 | 3.0 | 1.2 |
| Pd/TiO2 | 3.2 | 1.0 |
| 1 ML (SR) | 3.1 | 1.0 |
| 2 ML (SR) | 2.8 | 1.0 |
| 1 ML (PD) | 3.2 | 1.1 |
| 2 ML (PD) | 3.3 | 1.1 |
It is also possible that the Pd has been deposited separately on the surface of TiO2 with a particle size similar to that of initial Au NPs, which would mask their detection from this analysis. To support our claim that Pd has predominately deposited on Au, XPS analysis was performed for all samples, as shown in Fig. 2(a)–(d) and summarised in Table 2 (O 1s spectra were shown in Fig. S4 in ESI†). The survey scans (Fig. 2a) suggest all samples are mainly TiO2 with surface adventitious carbon and minor amount of metals (Au and Pd). Ti 2p spectra clearly show that neither deposition methods alter the oxidation states of Ti, which remained to be Ti4+ in all cases. Meanwhile, both Pd 3d and Au 4f spectra indicate Pd and Au to be in their metallic form (Fig. 2c, d and Table 2).31,32 It is worth noting that when Pd NPs were deposited on TiO2 by conventional impregnation methods without the presence of capping ligands (i.e., PVA), the dominant species was PdO.33 Similar result was observed for the chemical vapour impregnated Pd NPs supported on TiO2 in our recent work, where the molar ratio of Pd0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd2+ was ∼1:1.22 This is not the case for the sol immobilisation method prepared Pd/TiO2, as the NPs are encapsulated by the stabiliser.34 Interestingly, the deposition of metallic Pd in this study was performed in the absence of any stabiliser, which could be beneficial to photocatalysis applications due to the relative clean surface for interfacial charge transfer.
:Pd2+ was ∼1:1.22 This is not the case for the sol immobilisation method prepared Pd/TiO2, as the NPs are encapsulated by the stabiliser.34 Interestingly, the deposition of metallic Pd in this study was performed in the absence of any stabiliser, which could be beneficial to photocatalysis applications due to the relative clean surface for interfacial charge transfer.
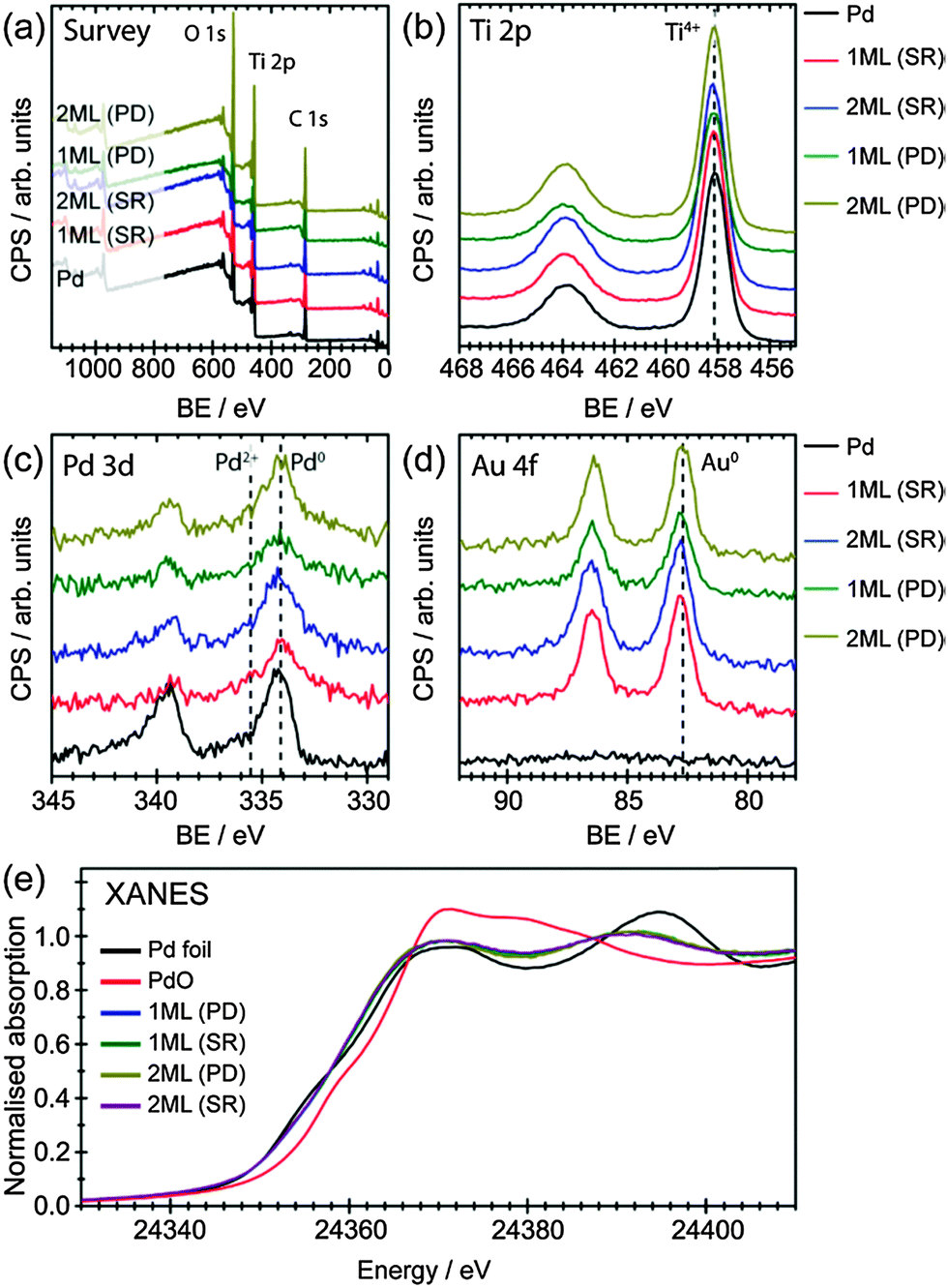 | ||
| Fig. 2 (a) XPS survey spectra of the as-prepared metal NPs supported on TiO2. (b)–(d) High resolution XPS spectra of Ti 2p, Pd 3d, and Au 4f, respectively. (e) XANES spectra of Pd foil, PdO, 1 ML (PD), 1 ML (SR), 2 ML (PD) and 2 ML (SR). | ||
| Catalyst | Peak name | BE (eV) | Pd/Au ratio |
|---|---|---|---|
| 1 ML (SR) | Pd 3d | 334.1 | 0.38 |
| Au 4f | 82.8 | ||
| 2 ML (SR) | Pd 3d | 334.2 | 0.92 |
| Au 4f | 82.7 | ||
| 1 ML (PD) | Pd 3d | 334.2 | 0.42 |
| Au 4f | 82.8 | ||
| 2 ML (PD) | Pd 3d | 334.3 | 0.63 |
| Au 4f | 82.7 | ||
The oxidation state of Pd, determined by XPS, confirmed that only metallic Pd was found in all samples, indicating an interaction between Au and Pd that prevents the oxidation of Pd. Surface atomic ratios of Pd/Au were also calculated from the XPS spectra to give information on the structure on the NPs (Table 2). As expected increasing the thickness of the shell layer results in an increase of the Pd/Au atomic ratio. Moreover, the surface atom ratio shows that the dominant component at the surface is Au in all cases. This suggests that the Pd may not be present at the surface as a segregated overlayer, with the EXAFS analysis providing more detail on this.
X-ray absorption near edge structure (XANES) analysis have also been used to understand the oxidation states of these materials, as shown in Fig. 2(e). The position of the main edge in the XANES spectrum was affected by the oxidation state of the element of interest, with the initial maxima in the 1st derivative of the normalised XANES of Pd foil and PdO separated by 4 eV. It was clear that the main edge positions of all core–shell structured AuPd promoters were in agreement with what would be expected for metallic Pd, with no edge shifted to higher energy. The XANES spectra of a Pd foil exhibits two peaks in the 1st derivative spectrum, due to 1s to 4d (these are hybridised p and d orbitals with a high degree of d character) and 1s to 5d transitions.35 For the PdAu/TiO2 samples reported here the position of these peaks were shifted upon alloying,36 however, the centroid position of both features was the same for all samples (Fig. S5 in ESI†). The differences in the XANES spectra beyond the main edge position between Pd foil and the core–shell structured AuPd spectra can be accounted for by the reduction of metal particle size as well as the alloying with Au.37 Unlike XPS, XANES provides a per atom average, therefore probing all environments – both surface and bulk – equally. By combining both XPS and XANES, it can be inferred that the majority of Pd, in all samples, was presented in its metallic form, confirming the successful deposition of Pd onto the Au core.
The determined 1st shell EXAFS fitting parameters are shown in Table 3, as well as the k2 weighted magnitude of Fourier transform data and corresponding fit shown in Fig. 3a (Au L3) and b (Pd K).
| Sample | Abs Sc | N | R/Å | 2σ2/Å2 | E f/eV | R factor | |
|---|---|---|---|---|---|---|---|
| 1 ML (PD) | Au L3 | Au–Au | 8.7 (5) | 2.84 (1) | 0.008 (1) | 4 (1) | 0.02 |
| Au–Pd | 1.5 (4) | 2.82 (3) | 0.009 (3) | ||||
| Pd K | Pd–O | 0.3 (1) | 1.95 (1) | 0.001 (1) | 1 (1) | 0.003 | |
| Pd–Pd | 0.8 (3) | 2.78 (2) | 0.009 (2) | ||||
| Pd–Au | 8.3 (3) | 2.78 (1) | 0.011 (1) | ||||
| 2 ML (PD) | Au L3 | Au–Au | 8.3 (6) | 2.81 (1) | 0.008 (1) | 4 (1) | 0.02 |
| Au–Pd | 2.2 (4) | 2.80 (2) | 0.009 (2) | ||||
| Pd K | Pd–O | 0.3 (1) | 1.96 (1) | 0.001 (1) | 1 (1) | 0.007 | |
| Pd–Pd | 3.1 (5) | 2.76 (1) | 0.011 (1) | ||||
| Pd–Au | 6.9 (8) | 2.78 (1) | 0.011 (1) | ||||
| 1 ML (SR) | Au L3 | Au–Au | 8.4 (5) | 2.84 (1) | 0.008 (1) | 4 (1) | 0.02 |
| Au–Pd | 1.7 (4) | 2.84 (2) | 0.010 (2) | ||||
| Pd K | Pd–Pd | 0.6 (2) | 2.75 (4) | 0.010 (4) | 1 (1) | 0.006 | |
| Pd–Au | 8.9 (3) | 2.78 (1) | 0.011 (1) | ||||
| 2 ML (SR) | Au L3 | Au–Au | 8.5 (6) | 2.81 (2) | 0.009 (2) | 4 (1) | 0.02 |
| Au–Pd | 2.1 (4) | 2.84 (2) | 0.008 (1) | ||||
| Pd K | Pd–O | 0.6 (1) | 1.97 (2) | 0.002 (1) | 1 (1) | 0.004 | |
| Pd–Pd | 1.5 (2) | 2.76 (1) | 0.011 (2) | ||||
| Pd–Au | 7.4 (3) | 2.78 (2) | 0.011 (1) | ||||
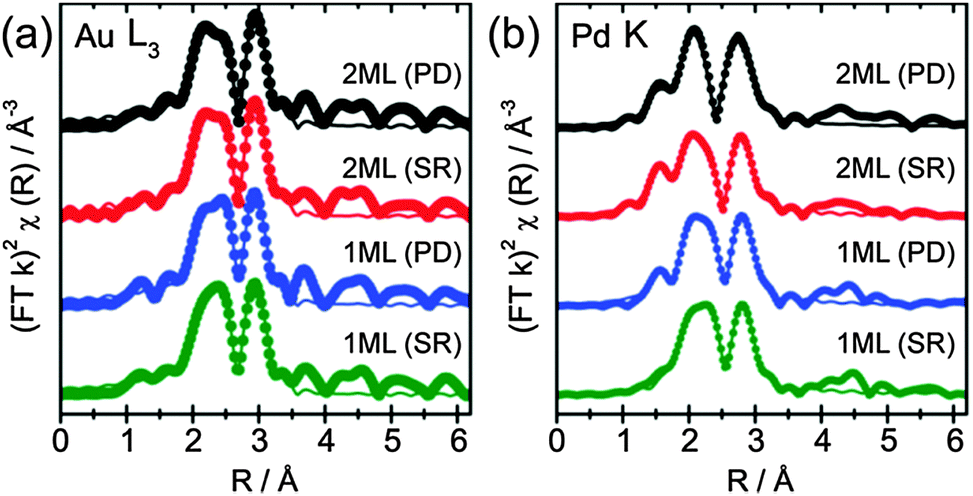 | ||
| Fig. 3 (a) Au L3 edge and (b) Pd K edge for the EXAFS of the core–shell structured promoters with different thickness prepared by PD and SR methods, respectively. Dots and lines represent the magnitude of the k2 weighted Fourier transform and associated 1st shell fittings, respectively. | ||
The EXAFS data show that for all catalysts prepared, both the Au and Pd environments were dominated by a primary Au coordination shell, with similar values observed for both preparation methods. The Au–Au and Pd–Au coordination numbers ranged between 8.3–8.7 and 6.9–8.9, respectively, and was the most significant coordination shell in both systems. The large Pd–Au contribution confirms that Pd was presented within an Au rich alloy, and not segregated as Pd overlayers. However, further addition of Pd, from the 1 ML to 2 ML systems, showed little influence on the Au EXAFS (e.g., the Au–Au coordination was not significantly altered), confirming that the majority of Au within the core of the particle remained unchanged. Moreover, although the Au–Pd coordination number increases with Pd loading, a much larger change would be expected if Pd was evenly distributed throughout the entirety of the particle. This can be evidenced by assessing the surface composition from XPS, which reports Au:Pd ratios of between 1:0.9 and 1:0.4. If one assumes these ratios broadly represent compositions of bulk structures of AuPd and Au3Pd random alloy, they would expect to yield 1st shell Au–Pd coordination numbers of 6 and 3, respectively. As the Au–Pd coordination number was around 2, in all cases we can confirm that the exterior of the particle was enriched in Pd compared to the bulk. Indeed, previous work by Dimitratos et al. has demonstrated that the sequential reduction method yields particles where the Pd content remains on the exterior of the particle.34,38 Crucially, by depositing only small amounts of Pd, in conjunction with EXAFS measurements, it can be demonstrated that the outer Pd was presented within an Au rich alloy environment. The presence of significant quantities of Pd not associated with Au can be ruled out by the relatively low Pd–Pd coordination number, which ranges from 0.6 to 3.1. Any significant amount of Pd NPs not associated with Au would result in a much larger Pd–Pd coordination number. Moreover, a larger component of oxidic Pd would be expected for individual Pd NPs, which the XPS and XANES have shown not to be the case. The EXAFS identified a small component of Pd–O interactions, which, with the relatively low coordination numbers (0.3 to 0.6), can easily be ascribed to surface oxides as opposed to bulk PdO. An example where discrete Pd over-layers have been deposited has been demonstrated by Russell et al.39 They used controlled surface modification routes with organometallic precursors to prepare single monolayer PdshellPtcore nanoparticles on carbon. Here, cyclic voltammetry (supported by TEM-EDX line scans) was able to show surface characteristics consistent with a Pd surface. EXAFS analysis of these materials showed that the relative Pd–Pd and Pd–Pt coordination neighbours were comparable to each other, as one would expect for Pd exclusively segregated at the surface. Comparison to studies such as these provides further evidence that the Pd in these samples has been incorporated further into the NP to make an Au rich, AuPd alloy, at the surface.
As a result of the characterisation preformed on the 1 ML and 2 ML NPs, the model of an Au core and AuPd shell structure is supported. These NPs are of the order ∼3 nm with predominant metal oxidation states of Au(0) and Pd(0). Fig. 4 represents illustrative diagrams showing the supported model of the AuPd NPs after deposition of Pd.
 | ||
| Fig. 4 Schematic diagram representing the supported model of the AuPd NPs, containing a gold core and an AuPd alloyed shell, where Au atoms are yellow and Pd atoms are blue. The scheme dose not quantitatively presents the composition information of the NPs. | ||
Photocatalytic performance
Fig. 5(a) depicts the time-resolved photocatalytic H2 evolution from 25 vol% EtOH solution. For comparison, monometallic Au and Pd as promoters supported on TiO2 with a loading of 1 wt% have been prepared via sol immobilisation method and evaluated. Metal NPs decorated on TiO2 photocatalysts have shown to generate H2 under UV irradiation, however, their performance varies depending on the identity and microstructure of the promoters. Pure Au as the promoter showed comparable but slightly enhanced H2 production performance compared to that of pure Pd. It was noticed that the Pd/TiO2 sample showed a slight drop in photocatalytic performance compared to that of previous observations. This may arise from the difference in oxidation states of the supported Pd NPs. Whilst mainly Pd0 was observed in the current study (Fig. 2c), a mixture of Pd0 and Pd2+ exists in our previous investigations.18 Since it has been reported that the presence of noble metal oxides (PtO) supported on TiO2 can inhibit the oxidation of generated H2,40 the drop in photocatalytic performance may be associated to the absence of PdO in this study. Interestingly, the core–shell structured PdshellAucore promoters showed further improvement in photocatalytic performance, where the 1 ML AuPd samples yield the slightly improved performance synthesised by both photodeposition and sequential reduction methods. Evidence from XAFS characterisation indicates that the Pd is associated with the Au NPs surface, therefore the increase in activity can be attributed to the presence of a surface AuPd alloy at the Au NPs. Previous studies have reported that core–shell structured Au–Pd NPs demonstrate a higher activity compared to that of the random Au–Pd alloys NPs.18 It is therefore proposed that the improved activity reported here results from the interplay between the Au-core and the AuPd alloy shell of the promoter NPs. It has been reported that while Pd and Pt normally provide an ohmic contact with the semiconductor, Au and Ag normally show capacitive properties.17 Thus the AuPd alloy shell is beneficial for the rapid interfacial charge transfer of the accumulated electrons on the Au core that originated from the CB of TiO2. Furthermore, it was observed that the activity decreased slightly for the 2 ML promoters compared to the 1 ML promoters, regardless of synthesis methods, indicating the electronic properties of the promoter can be tuned precisely by engineering the thickness of surface layer. Although the total metal loading has increased after the deposition of Pd, this is not considered to be the primary reason for the increased activity. The Pd is primarily associated with the Au and not present as isolated Pd NPs, therefore the number of NPs would not have increased significantly. This leads to the conclusion that there has been no significant rise in the number of active sites and the increase in activity can be attributed to the structure of the NPs.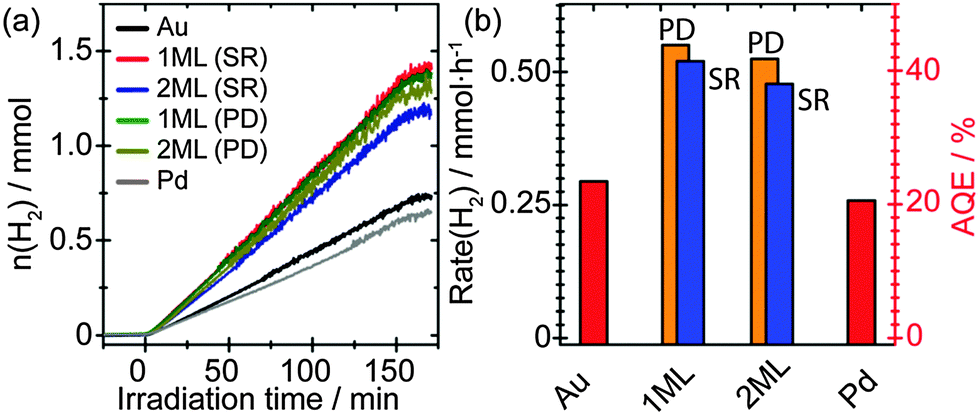 | ||
| Fig. 5 (a) Photocatalytic H2 evolution from a 25 vol% of ethanol solution using the metal/TiO2 photocatalyst materials. (b) The derived H2 production rates and apparent quantum efficiencies (AQE) of the metal/TiO2 photocatalyst materials. All catalysts have a metal loading of 1 wt%. The orange and blue bars indicate the core–shell NPs prepared by photodeposition and sequential reduction methods, respectively. | ||
Fig. 5(b) depicts the derived H2 evolution rate and apparent quantum efficiencies (AQE) of all samples. Specifically, the 1 ML Pd promoter on TiO2 prepared by photodeposition approach showed an impressive H2 production rate of ∼0.55 mmol h−1 that corresponding to an AQE of ∼44%, which presented a two-time enhancement than that of monometallic Au promoter.
Conclusions
We demonstrate two promising approaches for the deposition of thin layers of Pd on to the surface of Au NPs using both a traditional colloidal approach and a novel photodeposition method. XPS and XAFS characterisations confirmed that the microstructures of these promoters are in the form of PdAushell–Aucore, with no evidence of isolated Pd or Au NPs. The particle size of all core–shell NPs was determined to be ∼3 nm regardless of the shell thickness and preparation method. The Au and Pd were found to be predominately in their metallic form. Photocatalytic performance evaluations suggested that the PdAushell–Aucore promoters exhibited improved H2 evolution compared to the monometallic (Au and Pd) references. The optimum reactivity was observed when 1 ML equivalent of Pd was deposited on Au, suggesting that a thin layer of AuPd covering an Au core can maximise the alteration in electronic properties of surface atoms therefore boost the photocatalytic hydrogen production rate. We expect by fine tuning the surface Pd composition of the Au NPs the hydrogen production performance can be further enhanced.Acknowledgements
We acknowledge EPSRC for funding (EP/I019693/1, EP/K014714/1, and EP/I038748/1) and Diamond Light Source for provision of beam time (SP8071-5) and support of beamline scientists Giannantonio Cibin, Diego Gianolio, Stephen Parry and Andy Dent. The Research Complex at Harwell is also acknowledged for use of facilities and support of their staff. We also thank the financial support from the iNANO Centre through the Danish Strategic Research Council and the Carlsberg Foundation and the Centre for Materials Crystallography.Notes and references
- C. G. Silva, R. Juarez, T. Marino, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef PubMed.
- M. Bowker, C. Morton, J. Kennedy, H. Bahruji, J. Greves, W. Jones, P. R. Davies, C. Brookes, P. P. Wells and N. Dimitratos, J. Catal., 2014, 310, 10–15 CrossRef CAS PubMed.
- J. M. Ogden, Annu. Rev. Energy Environ., 1999, 24, 227–279 CrossRef.
- L. Saeed Al-Mazroai, M. Bowker, P. Davies, A. Dickinson, J. Greaves, D. James and L. Millard, Catal. Today, 2007, 122, 46–50 CrossRef PubMed.
- H. Bahruji, M. Bowker, P. R. Davies, L. S. Al-Mazroai, A. Dickinson, J. Greaves, D. James, L. Millard and F. Pedrono, J. Photochem. Photobiol., A, 2010, 216, 115–118 CrossRef CAS PubMed.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269–8285 CrossRef CAS.
- A. Mattsson and L. Osterlund, J. Phys. Chem. C, 2010, 114, 14121–14132 CAS.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS PubMed.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 CAS.
- K. M. Reddy, S. V. Manorama and A. R. Reddy, Mater. Chem. Phys., 2003, 78, 239–245 CrossRef.
- J. Zhang, S. Yan, S. L. Zhao, Q. Xu and C. Li, Appl. Surf. Sci., 2013, 280, 304–311 CrossRef CAS PubMed.
- J. Greaves, L. Al-Mazroai, A. Nuhu, P. Davies and M. Bowker, Gold Bull., 2006, 39, 216–219 CrossRef CAS.
- K. Mogyorosi, A. Kmetyko, N. Czirbus, G. Vereb, P. Sipos and A. Dombi, React. Kinet. Catal. Lett., 2009, 98, 215–225 CrossRef CAS.
- Y. Z. Yang, C. H. Chang and H. Idriss, Appl. Catal., B, 2006, 67, 217–222 CrossRef CAS PubMed.
- M. S. Park and M. Kang, Mater. Lett., 2008, 62, 183–187 CrossRef CAS PubMed.
- A. Takai and P. V. Kamat, ACS Nano, 2011, 5, 7369–7376 CrossRef CAS PubMed.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- Z. F. Jiang, J. J. Zhu, D. Liu, W. Wei, J. M. Xie and M. Chen, CrystEngComm, 2014, 16, 2384–2394 RSC.
- A. Gallo, M. Marelli, R. Psaro, V. Gombac, T. Montini, P. Fornasiero, R. Pievo and V. Dal Santo, Green Chem., 2012, 14, 330–333 RSC.
- R. Su, M. M. Forde, Q. He, Y. Shen, X. Wang, N. Dimitratos, S. Wendt, Y. Huang, B. B. Iversen, C. J. Kiely, F. Besenbacher and G. J. Hutchings, Dalton Trans., 2014, 43, 14976–14982 RSC.
- Y. Mizukoshi, K. Sato, T. J. Konno and N. Masahashi, Appl. Catal., B, 2010, 94, 248–253 CrossRef CAS PubMed.
- R. Su, L. Kesavan, M. M. Jensen, R. Tiruvalam, Q. He, N. Dimitratos, S. Wendt, M. Glasius, C. J. Kiely, G. J. Hutchings and F. Besenbacher, Chem. Commun., 2014, 50, 12612–12614 RSC.
- C. Brookes, P. P. Wells, G. Cibin, N. Dimitratos, W. Jones, D. J. Morgan and M. Bowker, ACS Catal., 2014, 4, 243–250 CrossRef CAS.
- N. Dimitratos, A. Villa, C. L. Bianchi, L. Prati and M. Makkee, Appl. Catal., A, 2006, 311, 185–192 CrossRef CAS PubMed.
- W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 264–273 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2012, 6, 6284–6292 CrossRef CAS PubMed.
- A. Zwijnenburg, A. Goossens, W. G. Sloof, M. W. J. Craje, A. M. van der Kraan, L. J. de Jongh, M. Makkee and J. A. Moulijn, J. Phys. Chem. B, 2002, 106, 9853–9862 CrossRef CAS.
- G. B. Hoflund, H. A. E. Hagelin, J. F. Weaver and G. N. Salaita, Appl. Surf. Sci., 2003, 205, 102–112 CrossRef CAS.
- Z. B. Wu, Z. Y. Sheng, H. Q. Wang and Y. Liu, Chemosphere, 2009, 77, 264–268 CrossRef CAS PubMed.
- N. Dimitratos, J. A. Lopez-Sanchez, J. M. Anthonykutty, G. Brett, A. F. Carley, R. C. Tiruvalam, A. A. Herzing, C. J. Kiely, D. W. Knight and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 4952–4961 RSC.
- M. Tromp, J. A. van Bokhoven, G. P. F. van Strijdonck, P. van Leeuwen, D. C. Koningsberger and D. E. Ramaker, J. Am. Chem. Soc., 2005, 127, 777–789 CrossRef CAS PubMed.
- A. F. Lee, C. J. Baddeley, C. Hardacre, R. M. Ormerod, R. M. Lambert, G. Schmid and H. West, J. Phys. Chem., 1995, 99, 6096–6102 CrossRef CAS.
- J. W. J. Hamilton, J. A. Byrne, P. S. M. Dunlop, D. D. Dionysiou, M. Pelaez, K. O'Shea, D. Synnott and S. C. Pillai, J. Phys. Chem. C, 2014, 118, 12206–12215 CAS.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63–86 RSC.
- P. P. Wells, E. M. Crabb, C. R. King, R. Wiltshire, B. Billsborrow, D. Thompsett and A. E. Russell, Phys. Chem. Chem. Phys., 2009, 11, 5773–5781 RSC.
- Y. H. Li, J. Xing, Z. J. Chen, Z. Li, F. Tian, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 2500–2505 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04693e |
| This journal is © the Owner Societies 2014 |