
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical imaging of live fibroblasts by SERS effective nanofilm†
D.
Radziuk
*a,
R.
Schuetz
b,
A.
Masic
b and
H.
Moehwald
a
aDepartment of Interfaces, Max-Planck Institute of Colloids and Interfaces, D14476 Potsdam, Germany. E-mail: darya.radziuk@mpikg.mpg.de; Fax: +49-331-567-9202; Tel: +49-331-567-9235
bDepartment of Biomaterials, Max-Planck Institute of Colloids and Interfaces, D14476 Potsdam, Germany
First published on 7th October 2014
Abstract
Reliable and strong surface enhanced Raman scattering (SERS) signatures of intracellular compartments in live NIH3T3 fibroblasts are collected in real time by means of SERS active thin nanofilm (30 nm) on colloidal silica (1.5 μm). Nanofilm is composed of preformed silver nanoparticles in the matrix of polyacrylic acid, protecting against heating (37 °C) in water, or culture medium or phosphate buffered saline aqueous solution. The SERS enhancement factors (EFs) of the order 108 allow single biomolecule detection in the native environment of a single live cell. Primary and secondary SERS hot spots of nanofilm are responsible for such high EFs. A slow SERS EF intensity decay occurs over a broader distance of micron silica with nanofilm, not achievable in a common core–shell model (silver nanoparticle coated with a thin silica layer). Extensive local field EFs and SERS EFs are mainly delivered by prolate silver nanoparticles (“rugby-like” shape). This is achieved if an incident field is polarized along the z-axis and the direction of incident polarization and main axis (z) are perpendicular to each other, not observable in water or on gold.
Introduction
Investigating proteins and other biomolecules in a living cell environment is a major goal of molecular biology, which has been achieved to a very limited extent so far. Traditional methods for intracellular studies are indirect and require millions of cells to extract specific biomolecules or organelles.1 However, the behavior of a single cell varies even in a small population and the true cell metabolism might be obscured.2 Moreover, the extraction can cause significant artifacts in the final data.3 Therefore there is a need in single-cell techniques to characterize the organelles preserved in their native environment.A palette of spectroscopy and imaging techniques to study single cells, presumably developed for the early detection of diseases and drug delivery promotion, relies mostly on fluorescence-based methods.4,5 Nondestructive analysis with high lateral image resolution enables the morphological study of individual organelles in their native state. However, the need of a fluorescent dye is still a problem for time-dependent studies because of toxicity.6 In addition, nonspecific binding can cause a strong background signal lowering the detection sensitivity. Moreover, fluorescence methods are sensitive to quenching processes distorting quantification.
Distinct from fluorescence, Raman imaging is a vibrational technique that is based on scattering excluding the necessity of dyes, the problem of toxicity and photobleaching.7 Raman vibrational spectroscopy is a powerful diagnostic tool that provides characteristic molecular signatures non-invasively and in a straightforward manner.8 Its sensitivity allows single biomolecule detection and diagnostics of structural conformation in native biological environment. However, the intrinsically low cross section results in weak Raman signals (10−30 and 10−25 cm2 compared to fluorescence with 10−17–10−16 cm2), and the reproducibility is low. Since long it is known that Raman scattering can be strikingly increased at a metallic surface (surface enhanced Raman scattering, SERS) or in the vicinity of plasmonic (e.g. Au or Ag) nanostructures that enable a local electromagnetic field increase.9–11 As a great advantage a molecule can perform more SERS than fluorescence cycles per time interval due to the shorter vibrational relaxation times and the molecular prints that can be collected from all molecules in a probed sample even at a minute volume (advantage over IR spectroscopy). On a nano-structured surface single molecule SERS (SM-SERS) arises in the gaps of metallic nanoparticles with the surface plasmon resonance for the local electromagnetic field increase. It usually depends on the nanostructure geometry with the highest values of electromagnetic field at a distance <10 nm (SERS hot spots).12,13
In a single cell study to make advantage of the SERS hot spots colloidal particles can be adsorbed on a glass nanopipette to increase the Raman scattering cross section of cellular compounds (e.g. DNA, RNA, proteins, and phospholipids), but the orientation of analyte molecules with respect to the SERS substrate must be taken into account.14 Another strategy towards reliable SERS signals includes the fabrication of core–shell nanostructures bearing a plasmonic-core material and a protective dielectric shell.15 However, the narrow choice of composition and breakdown at high or low pH sufficiently limits its application.
One conceptually simple approach is through chemical binding of the colloids using appropriate surface chemistry. This is however challenging and has not yet been fully explored. As a recent successful example, the nanoshell16 approach which is based on gold or silver nanostructures (nanoshell) adsorbed on larger dielectric pearls (silica, polystyrene, etc.) yields more reliable SERS signals.17 The term “nanoshell” means a designed shell of metallic nanoparticles bearing plasmonic properties with thickness at nm scale. Such thin plasmonic films can be formed on micrometer substrates of different curvatures which greatly increase the SERS effective area over the entire surface. The recently introduced silver nanofilm can be formed in a matrix of polyethylene glycol (PEG), and the interparticle junctions (SERS hot spots) can be controlled by the polymeric chain length.18 Chain-like silver nanostructures with stable and defined interparticle gaps can be prepared in PEG with 8000 Da yielding strong reliable SERS spectra of live fibroblasts in real time. As another advantage, such thin nanofilm can overcome a common problem of dimers: it will become less and less exponentially probable to add an additional particle to the cluster because of the increased barrier height for dimer-sphere interactions and beyond.
Distinct from the core–shell approach,19 nanofilms are more flexible and versatile in chemical functionalization and can promote the chemical and physical adsorption of an analyte over a larger SERS effective surface area. As a supporting substrate, silica microparticle, can increase the SERS contact area and nano-roughness of the plasmonic nanofilm allowing SERS detection of proteins, lipids and, due to the broad aromatic ring vibrations of DNA/RNA, of live fibroblasts in real time.20 By this approach the problem of uncontrolled silver nanoparticle aggregation can be solved by the protecting polymeric chain groups yielding a SERS effective substrate with geometrically confined stable hot spots. Analytes can be bound to suitable polymers via electrostatic, biospecific or other noncovalent interactions, as the polymers can be coupled in most cases by electrostatic attraction. Design flexibility in the coating density, conformation and porosity of the bound polymer can make advantage at a SERS hot spot with the enhancement values sufficient for single molecule detection. To note, a single polymeric layer can add an additional thickness of less than 1.5 nm to trap analytes and benefit high electromagnetic field increase.21 As another advantage the surface charge density oscillations associated with surface plasmons at the interface between a metal and a dielectric can give rise to strongly enhanced optical near-fields which are spatially confined to the interface, contributing to the SERS enhancement factors.22 Moreover, the self-assembly of silica colloids bearing plasmonic nanofilm can provide secondary hot spots in the silica–silica junctions (primary hot spots are formed in the gaps of plasmonic nanoparticles in the nanofilm).
As we have shown above, in SERS a thin plasmonic nanofilm has a number of advantages to chemically image analyte(s) in real time with a relatively low uncertainty. However, the reproducibility of SERS signals (especially of single molecules) is still a problem: probes should have uniformly distributed nanoparticles (uncontrolled aggregation must be avoided), the concentration of plasmonic nanostructures should correlate with the number of analyte(s) and the enhanced SERS signals should be reproducibly strong. As another challenge, most of the cells imaging techniques have to be combined with confocal fluorescence measurements. Based on our previous successful results18 we advance a nanofilm approach for SERS studies of single live fibroblasts in real time. We show strong and reproducible SERS signals of single biomolecules in their native environment followed by the label free chemical imaging of the entire cell.
In details the nanoshell approach is advanced by the formation of primary and secondary SERS hot spots consisting of preformed silver nanoparticles in the polymeric matrix of polyacrylic acid (PAA) on colloidal silica, SiO2@Ag–PAA particles (Scheme 1). Primary hot spots are formed in the gaps of preformed silver nanoparticles (<10 nm), which are protected by the biocompatible PAA matrix, Ag–PAA (Scheme 1A). Secondary hot spots appear in the silica–silica junctions of the Ag–PAA nanofilm (Scheme 1B). Our approach addresses the problem of low laser intensity for SERS spectra collection and opens the way to a coupled SERS spectral knowledge and a Raman imaging analysis. We also explain how the SERS signals can be intensified by theoretical calculation of the local field intensity enhancement factors (LFI EFs) and SERS EFs of Ag–PAA nanofilm on silica. These findings are thoroughly compared with Ag–PAA in water or with gold (Au) instead of silver. Moreover, we demonstrate how Ag–PAA nanofilm on colloidal silica exhibit the superior advantage of SERS EFs decays versus distance and molecular coverage over a common SERS active “core–shell” model (plasmonic nanoparticles coated with a thin outer layer of silica).
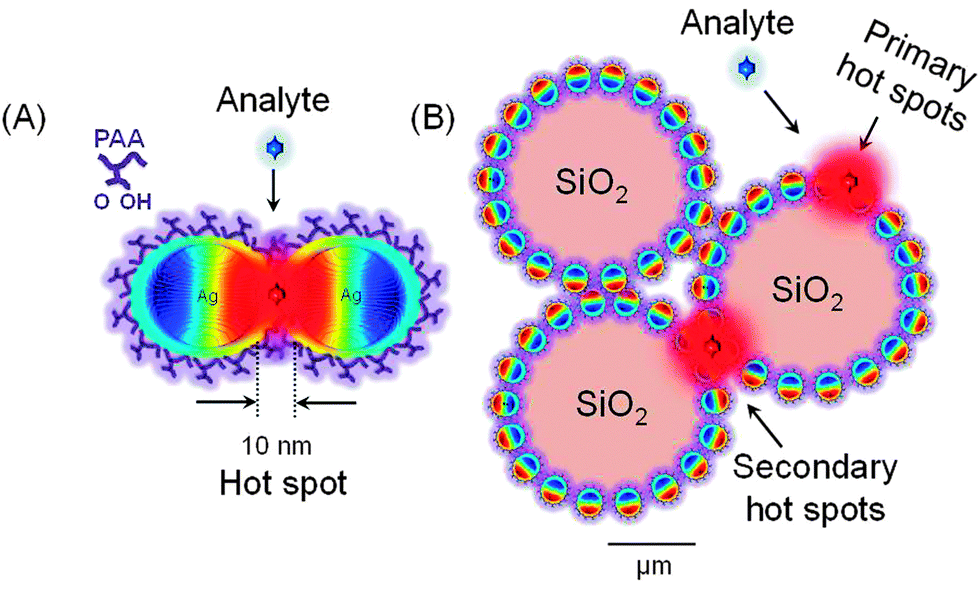 | ||
| Scheme 1 Schematic illustration of primary and secondary SERS hot spot formation by Ag–PAA nanofilms on colloidal silica. (A) Pre-formed silver nanoparticles are embedded in the matrix of polyacrylic acid (Ag–PAA nanofilms) with fixed gaps (∼10 nm) that can serve as primary SERS hot spots. (B) Ag–PAA nanofilms are deposited on silica microparticles (∼1.5 μm) that can form secondary SERS hot spots at the interparticle junctions during self-assembly. | ||
Results and discussion
Formation of Ag–PAA nanofilm on silica microparticles
To prepare monodisperse spherical silver nanoparticles (NPs) (25 ± 5 nm) we modified the well-known procedure of reduction of silver nitrate with trisodium citrate dihydrate23 (Fig. 1A). Silver nanoparticles have a spherical and oval shape with either prolate or oblate spheroid surface (indicated by white arrows in Fig. 1A). The ζ-potential of freshly formed silver NPs was negative −35 ± 5 mV and no sediment was observed within a month. As the next step these silver NPs were used for a triple stepwise adsorption on hydrophilic silica microparticles (MPs) (1.55 μm ± 0.04 μm) with terminal Si–OH bonds. Prior to silver NPs deposition the initially negative ζ-potential of silica MPs (−22.4 ± 5 mV) was reversed to a positive +44 ± 5 mV value by functionalizing the hydroxyl groups with aminosilanes24,25 (Fig. 1B). After silver NP adsorption the ζ-potential of the silica MPs was reversed to −45 ± 5 mV (pH = 8.5). Silica MPs were non-uniformly coated with aggregated silver NPs as isolated islands (average diameter 89 ± 3 nm) (Fig. 1C and Fig. S1A, ESI†).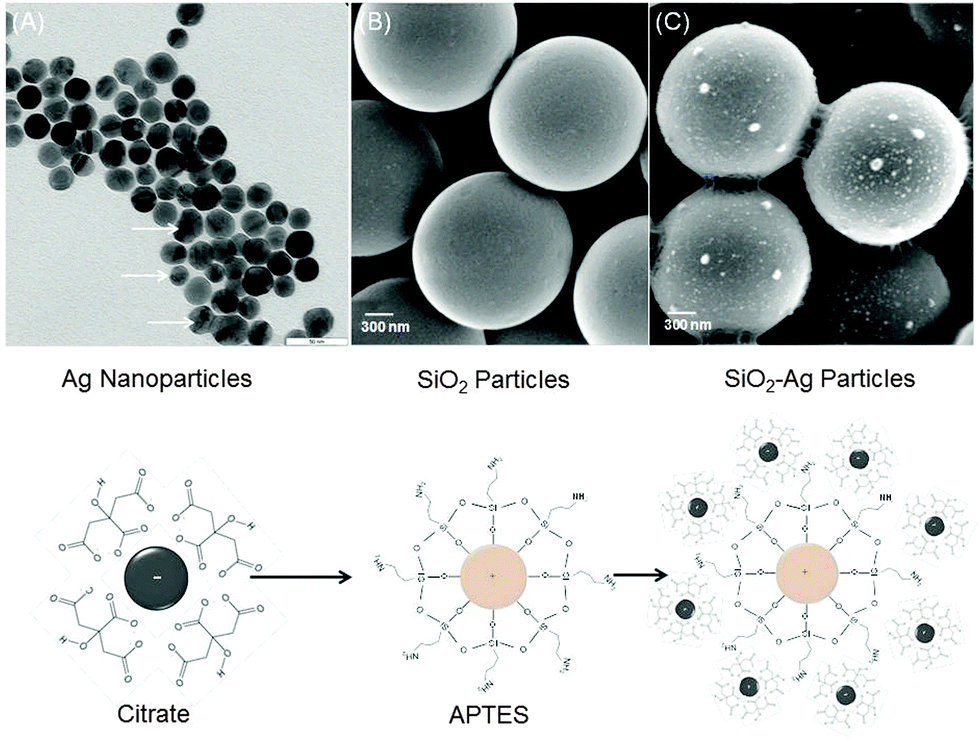 | ||
| Fig. 1 (A) TEM image of silver–citrate nanoparticles prepared after reduction of silver nitrate with trisodium citrate dihydrate in water (scale bar is 50 nm). (B) and (C) SEM images of colloidal silica before and after adsorption of silver–citrate nanoparticles (scale bar is 300 nm). The sketch below shows the chemical structure of particles in A, B and C. | ||
To avoid aggregation during deposition on silica MPs, silver NPs were incubated in the matrix of polyacrylic acid (Ag–PAA NPs). The stability of nanoparticles was tested during heating at 37 °C in water (Fig. S2, ESI†). After heat treatment the negative ζ-potential value of Ag–PAA NPs did not change and no sediment appeared on the bottom within several months which is distinct from bare silver–citrate NPs formed by a Meisel's procedure (Fig. S3, ESI†). The UV-Vis absorbance spectrum of Ag–PAA NPs on silica MPs shows a strong band with a slightly red-shifted peak (405 nm) (Fig. 2A). For comparison bare Ag–citrate NPs that are formed by a Meisel's procedure23 or modified Xia's method26 exhibit absorbance as broad bands with twice or lower intensity (Fig. 2A). Distinct from bare Ag–citrate NPs, silica MPs acquired more uniform coverage by Ag–PAA NPs (Fig. 2B and Fig. S1B, ESI†).
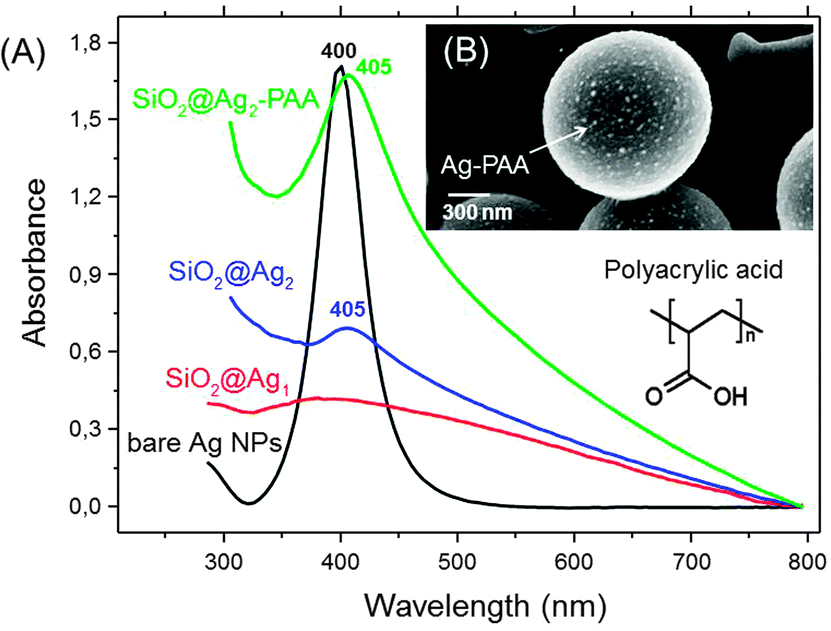 | ||
| Fig. 2 (A) UV-Vis absorbance spectra of silver nanoparticles (Ag NPs) before and after adsorption on colloidal silica (silica MPs) in water. SiO2@Ag1 (red) and SiO2@Ag2 (blue) indicate that Ag NPs are formed according to the Meisel's (red) and modified Xia's (blue) procedures followed by their adsorption on silica MPs. SiO2@Ag2–PAA (green) indicates Ag nanofilms in the polyacrylic acid matrix (Ag–PAA) after deposition on silica MPs. (B) SEM image of SiO2@Ag2–PAA (green) with the scale bar 300 nm. | ||
Before adsorption of Ag–PAA NPs, the positive surface of silica MPs appeared smooth (Fig. 3A and B). After deposition of Ag–PAA NPs the contact surface area between neighbouring silica MPs was increased fourfold with the formation of sharp plasmonic edges in between (Fig. 3C). Fig. 3D shows a TEM image of an individual silica MP with a non-uniform thin layer of Ag–PAA nanofilm, no agglomerates were observed.
 | ||
| Fig. 3 TEM images of positively charged hydrophilic silica microparticles (silica MPs) (average diameter = 1.5 μm) (A and B) before and (C and D) after adsorption of Ag nanofilms. The scale bars are 500 nm (A), 100 nm (B), 250 nm (C) and 100 nm (D). | ||
As the next step described below, SiO2@Ag–PAA particles were examined for the SERS efficiency of rhodamine 6G (Rh6G) molecules in aqueous solution.
SERS efficiency of rhodamine 6G on Ag–PAA nanofilm
To test the SERS efficiency, SiO2@Ag–PAA particles were incubated in the presence of Rh6G with 10−4 mol L−1 NaCl. From our previous SERS measurements of Rh6G on silver NPs:18 it takes less than seconds for SERS spectra detection. However, an incubation time of at least tens of minutes is necessary to achieve a thermodynamic equilibrium between a dye molecule and a silver nanofilm on silica MPs. To note, an incubation period of aqueous solution with nanoparticles can be longer (up to several hours). However, in medical diagnostics in a flow rate detection system, where a SERS effective substrate (not a bulk colloidal solution) can be fixed, no incubation time is required.The presence of Cl− anions is necessary in order to assist specific chemisorption of dye molecules on Ag NPs via a N–Ag covalent bond.27 Rh6G aqueous solution at 10−8 mol L−1 concentration was sufficient for low threshold surface enhancement (106), which is accompanied by suppression of fluorescence28 (Fig. S4, ESI†). The effective number of Rh6G on the SiO2@Ag–PAA surface is about 14 dye molecules per silver NP, assuming the surface areas of silica and silver as 1.89 μm2 and 3.14 × 10−4 μm2 (more details can be found in the Experimental section).
The SERS spectra of Rh6G were collected on single SiO2@Ag–PAA particles, their dimers or closely packed self-assembly consisting of four-five particles (Fig. 4A), that are designated as A, B or C, respectively (Fig. 4B). The SERS spectra are reproducible and cumulatively averaged over at least ten particles of each type (Fig. S5, ESI†). Overall the intensity of SERS bands was increased by two orders of magnitude during the self-assembly of particles due to the increased number of interparticle junctions. The estimated SERS EFs of Rh6G on the closely packed SiO2@Ag–PAA particles are of the order of 108 being sufficient for single molecule detection (details can be found in the Experimental section).
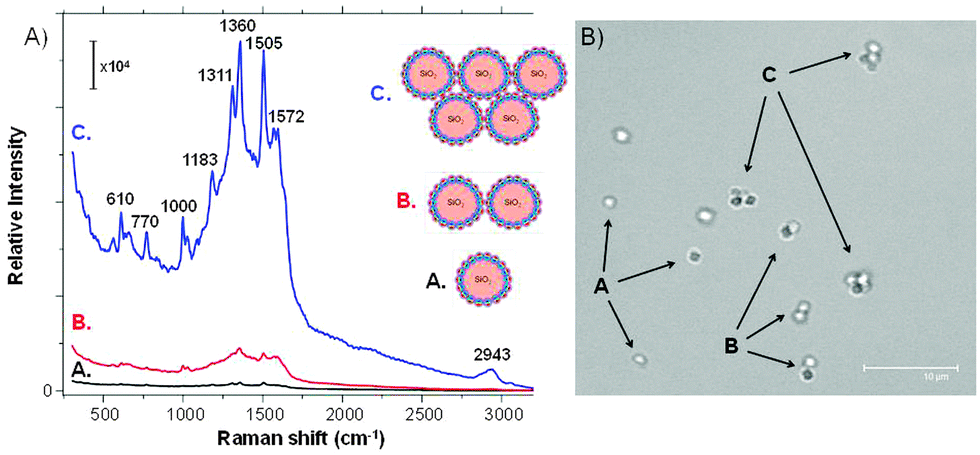 | ||
| Fig. 4 (A) Surface enhanced Raman scattering (SERS) spectra of Rh6G (10−8 mol L−1) in aqueous solution of SiO2@Ag–PAA particles as single (A), dimers (B) or closely-packed self-assembly (C). The excitation wavelength was 532 nm with the grating 600 g mm−1 (BLZ = 500 nm) and a spectral resolution of 3 cm−1. The SERS spectra are cumulative and averaged over at least ten particles of each type. (B) Confocal laser scanning microscopy (CLSM) image of these particles in a bulk aqueous solution (scale bar is 10 μm). | ||
The density of Ag NPs is higher on C than on A or B as silver–halide vibration (246 cm−1) appears with increased broadening and smoothing (Cl− anions are forced to desorb) followed by a shift to a higher frequency30 (Fig. 4A).
The conformational distortion of the hydrogen atoms of the xanthene skeleton (at 610 cm−1 and 770 cm−1)29 is the strongest on C due to a weaker chemisorption of the Rh6G dye. To note, the full width at half maximum (FWHM) of this band does not change, as only one vibration contributes to this signal. Strong bands of pyridine (992 cm−1 and 1030 cm−1) are intensified on C, but at shifted frequencies (1000 cm−1, 1026 cm−1 and 1086 cm−1), not observable in free Ag NPs.18 The absolute intensity of the C–H in-plane bending coupled to the C–C stretching (1181 cm−1)28 is fivefold stronger on C (at 1183 cm−1) as well as the C–H stretching at 1311 cm−1 and 1360 cm−1.
Another C–C band (1509 cm−1) appears at 1505 cm−1 as a strong band on C, while the aromatic ring of this vibration is split into a doublet (1572 cm−1 and 1595 cm−1) and the band at 1650 cm−1 gradually disappears. It is known, that this carbon skeleton band is extremely sensitive to molecular distortion, i.e. coupling between the Cl− and silver–Rh6G complexes. This sensitivity is enhanced by two orders of magnitude on C and is not observable in free Ag NPs.18
However, SiO2@Ag–PAA particles do not enhance the in-plane N–H deformations at 1240–1250 cm−1 and the pyridinium halide C5H5NH+Cl− vibration (2380 cm−1), similar to free Ag NPs.18 Although the characteristic ring vibrations with considerable N–O stretching are not observed at 1252 cm−1 this vibration appeared as small peaks with lower frequency shifts (543 cm−1 and 835 cm−1), distinct from free Ag NPs. To note, the O–H vibrations of water (2943 cm−1) that are initiated by the electromagnetic field enhancement are also strongly enhanced on C.
Overall, Ag–PAA nanofilm efficiently enhance Raman signals of Rh6G with the EF (108), more than sufficient for single molecule detection. The SERS increase is higher from several self-assembled silica MPs due to the formation of interparticle junctions between Ag–PAA nanofilms, which can act as secondary SERS hot spots. However, it is unclear how such high SERS EFs can be achieved and whether the nanoparticle geometry in the nanofilm can be responsible for the local field intensity EFs. Moreover, it is unknown if there is a dominance in the SERS EF increase of silica as a micron-substrate or as a thin outer layer (typically several nm only). Can silver be substituted by gold in the introduced nanofilm in order to reach such high SERS EFs and what are the possible conditions for this? To start with, we decided to calculate EFs of the local field intensity as well as maximum and average SERS EFs on Ag–PAA nanofilm on silica, in water and compare them with gold and a common core–shell model (silver or gold nanoparticle coated with a thin silica layer).
Theoretical calculation of SERS-EF and LFI EF (local field intensity) enhancement factors of Ag–PAA nanofilm
In the frame of the generally accepted distance dependence in the electrostatic approximation (ESA) we calculated the cumulated SERS EFs in a SiO2@Ag–PAA particle (Fig. 5A) and compared it with a core–shell model consisting of Ag NP as a core coated with a thin silica-shell (Fig. 5B) over the distance from the SERS effective surface. In our calculation we restrict ourselves to situations of reasonably large enhancements, where the scattered field dominates over the incident field.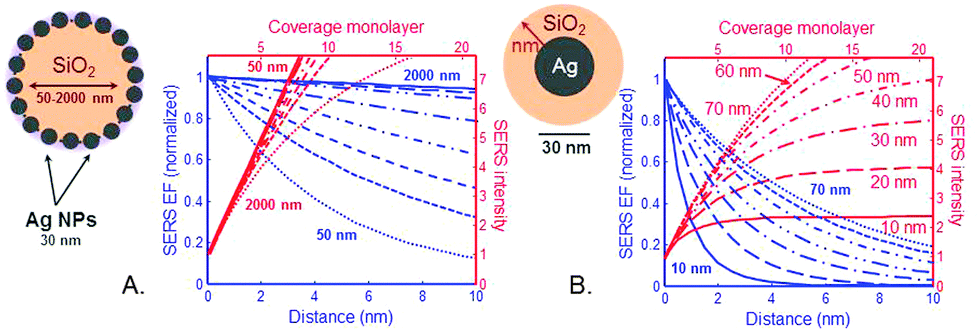 | ||
| Fig. 5 Dependence of electromagnetic SERS enhancement factors (EFs) with the distance d and coverage. (A) Ag–PAA nanofilm (radius 30 nm) on silica MP designated as SiO2@Ag–PAA particle with a radius of silica MP that is varied from 50 nm to 2000 nm. (B) Ag NP (radius 30 nm) as a core coated by a thin silica outer layer with the thickness from 10 nm to 70 nm. The cumulated average SERS intensity (on the right axis) is plotted as a function of analyte coverage assuming a monolayer thickness of about 0.5 nm. | ||
The electric field outside the particle in the ESA is created by an electrostatic dipole at the center of a particle. For a molecule at a distance d from the particle surface, the electric field amplitude should decay as 1/(a + d)3, where “a” is a radius of a particle and “d” is a distance from its surface. Punctual and average SERS EFs should decay as 1/(a + d).12 Despite a dramatic decay due to an exponent as large as 12 a SERS signal can be dominated by more than the first layer of adsorbed molecules because a ≫ d in both examples of particles.
Although the SERS EF is maximum at d = 0 (first layer), the EM enhancements are long-range effects and can extend to at least 10 nm away from the metallic surface. In the frame of the ESA one can directly predict the coverage dependence of the SERS signal (i.e. the number of molecular monolayers on the surface) from the distance dependence of the SERS EF. Because the SERS EF decreases with distance, the cumulated SERS signal varies sub-linearly with coverage and saturates.
In our calculation the SERS-EFs are plotted versus coverage at Ag–PAA nanofilm on silica MPs with their radius varied from 50 nm to 2000 nm (Fig. 5A). A monolayer thickness is assumed to be 0.5 nm. Sublinear decay of the SERS EF is slower on SiO2 of micron size (e.g. 1 μm or 2 μm) with saturation approaching at far above 10 nm followed by a larger coverage of analytes experiencing high SERS EF. This means more and different analytes can be potentially detected and examined at a single molecule level in this configuration. This is in contrast with smaller SiO2 particles (i.e. <several hundred nm) where the SERS EF saturates faster in the proximity of 10 nm. This means that the challenge of analyte positioning directly on a hot spot (typically 10 nm distance from the metal surface) can be overcome by using SiO2 particles rather of micron than nm size. However, for practical purposes many additional effects should be also considered at multilayer coverages. For example, the molecules in the first layer may be chemically influenced by the metallic surface and the adsorption geometry may be different in subsequent layers. To note, the average and maximum SERS EF is rather a widespread property of the enhancement on the entire surface of our particle(s). This is in agreement with the tendency of higher electromagnetic field increase with the increase surface area of plasmonic nanoparticles.31
To remind, the local surface plasmon (LSP) resonances are mostly radiative (especially dipolar one), resulting in a larger radiative decay rate when the dipole couples to them efficiently. These radiative resonances and the main dipolar LSP resonance are strongly size-dependent for spheres and more generally are geometry-dependent, being largest for smaller spheres (nm range). For comparison we calculated the cumulated SERS EFs versus distance and coverage for a common SERS system: core–shell particle, consisting of Ag NP (30 nm) as a core being coated by a thin outer silica layer. The thickness of the silica coating is varied from 10 nm to 70 nm (Fig. 5B). From our calculation the SERS EF intensity saturates fivefold faster at a distance <2 nm with a silica shell thickness of 10 nm and at ∼5 nm with a silica shell thickness of 70 nm. At this distance range the coupling of an analyte to these resonances is not strongly distance-dependent. However, it is sensitive to the dipole orientation: the perpendicular dipole couples more efficiently to the LSP resonances. To note, in this SERS system an analyte should be closely brought to the metallic surface (d = 0–2 nm) or positioned at the interparticle junction in order to benefit higher SERS EFs, which is problematic for single molecule studies. As another fact, the coverage thickness of analytes is several nm less in this SERS system at a minimum detection distance of about 8 nm, which further lowers the potential ability of this system to overcome problems in single molecule studies.
In the frame of the ESA we also calculated the local field intensity enhancement factors (LFI EFs) of Ag–PAA nanofilm by applying an analytical solution (details in the Experimental section) (Fig. 6). As the shape of Ag NPs in the PAA matrix strongly changes from spherical to ellipsoidal at 37 °C (conditions for living cell studies) (Fig. S1, ESI†) the LFI EFs were estimated for prolate or oblate silver spheroids (30 nm) (Fig. 6).
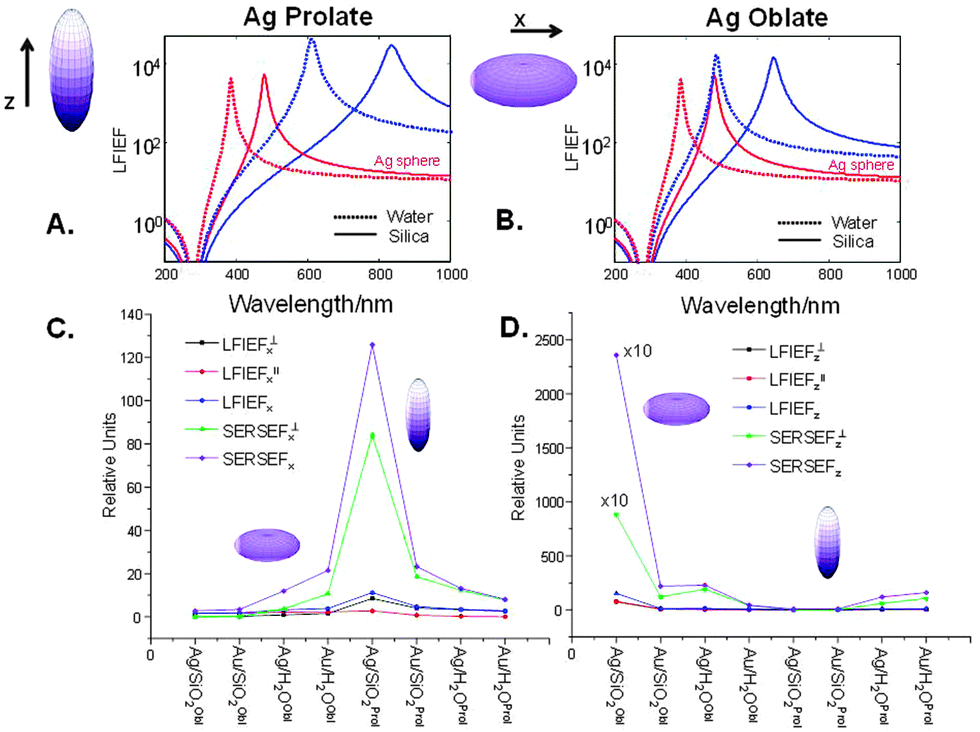 | ||
| Fig. 6 (A) and (B) Wavelength dependence of the maximum (Aperp) and minimum (Apara) of the local field intensity enhancement factor (LFI EF) for the prolate and oblate Ag spheroid with aspect ratio 3 (dashed lines indicate those in water and solid lines – in silica). “Ag sphere” is the corresponding quantity for a sphere given for comparison. (C) and (D) LFI EFs and SERS EFs on Ag oblate or prolate spheroids in nanofilm on silica or in water in comparison with gold at the incident polarization along x or z axes. | ||
The calculated LFI EFs of SiO2@Ag–PAA particles were compared with Ag nano-spheres (30 nm). As for the sphere we can consider the LFI EF at selected positions on the ellipsoid surface (at the corners along the main axes) at an incident field polarized along one of the axes of the spheroid (z for prolate or x for oblate). To note, the LFI EFs at all points on the surface can be expressed in terms of the non-dimensional polarizability βi(ω) along the incident polarization axis (z or x), distinguishing between local field perpendicular and parallel to the surface. The maximum (Aperp) and minimum (Apara) values characterize the maximum and minimum LFI EFs for incident polarization along ei. The maximum enhancement, Aperp, is obtained at the “tips” of the axis, and the local field is perpendicular to its surface. The minimum enhancement, Apara, is obtained at the four points on the two other axes and the local field is tangential to the surface.
In the ESA for LFI EFs we have to distinguish between excitations along a “pointy” or “flat” axis:
– For excitation along axes with “flat” curvatures (Li > 1/3), the factor (3Li)−2 contributes to a decrease of the LFI EF at a resonance compared to the sphere case. This decrease is even larger for Aperp because of the dependence of |ε(λi)|2 on Li.
– For excitation along axes with “pointy” curvatures (Li < 1/3) the factor (3Li)−2 contributes to an increase of the LFI EF at a resonance compared to the sphere case. This increase is further magnified for Aperp because |ε(λi)|2 increases as the resonance wavelength red-shifts (which is the case for decreasing Li).
Overall, Fig. 6 illustrates the wavelength dependence of the maximum (Aperp) and minimum (Apara) LFI EFs of prolate or oblate spheroids with aspect ratio 3 (blue curves) in comparison with a sphere (Agsphere, red curves) in silica (solid lines) or water (dashed lines). The LFI EFs are higher for Ag prolate spheroids with a strong red-shift near 850 nm (Fig. 6A), but being close to 650 nm for Ag oblate spheroids (Fig. 6B). This effect is more pronounced on silica than in water due to the higher value of the dielectric constant. It is important to emphasize, that LFI EFs are of the order between 104 and 105 (corresponding to the SERS EF between 108 and 1010), in agreement with the experimental data and potential ability for meaningful single molecule studies. In contrast, LFI EFs of bulk Agsphere are slightly below 104 on silica or in water (Table S1, ESI†).
High enhancement values are observed in the Ag–silica system when the directions of incident polarization and main axis of a spheroid are perpendicular to each other, being several times larger than in the Ag–water or Au–water systems. To note, the LFI EFs in Au spheroids are much smaller with a resonance that is strongly damped, because it occurs at wavelengths where the optical absorption is large. However, for prolate Au spheroids these values are slightly larger as the red-shifted resonance is no longer affected by this problem (Table S1, ESI†). This resonance should have comparable properties as that of silver ellipsoid. This means that SERS studies of live cells can be potentially improved by minimizing drawbacks of particle toxicity, increasing biocompatibility and broadening the surface functionality of a potential SERS effective platform based on the nanofilm approach.
Moreover, prolate spheroids (Ag or Au) with a “rugby-like” shape exhibit larger maximum EFs than oblate spheroids (“pumpkin-like” shape) of both metals and can reach the SERS EF of 1010 (LFI EF ∼ 105) more than sufficient for single molecule studies. In this way, these results illustrate an important point, often the source of confusion: in SERS gold substrates can be as efficient as those of silver when resonances are sufficiently red-shifted (i.e. Au prolate spheroids with the aspect ratio ≥3 and those of Ag being sufficient with ≥2). In this way, a nanofilm approach, based on the stability of preformed Ag or Au nano-geometries in the polymeric matrix of a specific interest, can offer a great variety of functional hot spots, hence reliable LFI EFs to perform single molecule studies.
From the above arguments that large additional enhancements can be achieved at specific points of prolate spheroids, let us consider also the average SERS EFs on these particles when analytes are randomly adsorbed over the surface (Fig. 6C and D). General expressions are described in details in the Experimental section. To note, the average SERS-EFs can be higher by two orders of magnitude for Ag–silica oblate spheroids, when the incident polarization is along z direction. However, it can be about ten times larger for Ag–silica prolate spheroids, when the incident polarization is along x direction, in agreement with the theoretical predictions elsewhere.32 This is true, bearing in mind the same aspect ratio, that minimum average SERS-EFs can be observed for both Ag–silica oblate or prolate spheroids, when the incident polarization coincides with their main axes. In the latter case, minimum enhancement values for silver are comparable with those for gold. However, as a result of a “localization” effect the maximum SERS EF can be also increased, but applied to a smaller proportion of the surface area (oblate shape). Upon surface averaging the average SERS EF can be increased, but partly compensated by localization, hence the importance of hot spots (interparticle junctions) is critical.
From calculation above we found out that the proposed Ag–PAA nanofilm approach offers a number of benefits for not only higher SERS EFs, but also LFI EFs. These values are several fold higher on silica as a micron substrate than as a coating shell of nm thickness. Considering the lower aspect ratio, Ag–PAA NPs with prolate shape exhibit stronger EFs (up to 1010) on silica MPs than a similar system in water or with gold. However, at the equivalent higher aspect ratio such high EFs can be achieved also with gold instead of silver, which are important for live cellular studies. Lower EFs (106–108), however sufficient for single molecule detection, can be provided by oblate silver or gold nanoshells in a comparable fashion. As we have demonstrated above (experimentally and theoretically) Ag–PAA nanofilm can be used as SERS efficient platforms to intensify weak Raman signals of biomolecules from a live cell in real time. Let us further demonstrate how Ag–PAA nanofilm can effectively increase molecular prints of biomolecules of a single live fibroblast in real time.
Chemical imaging of live fibroblasts by Ag–PAA nanofilm
SiO2@Ag–PAA particles were embedded into the interior of live NIH/3T3 fibroblasts by electro-permeabilization33 of the cell membrane. The optical phase contrast images of live fibroblasts before (Fig. S6A, ESI†) and after (Fig. S6B, ESI†) electroporation show no visible changes of the cell damage or apoptosis. In general, in all experiments the cell viability after electroporation was optically monitored and compared with control cells free of particles. For comparison SERS effective nanofilm was prepared also with gold nanoparticles instead of silver.After electroporation we observed fibroblasts, which preserved their native morphology (designated as “normal”), those with the acquired round shape due to the reduced adhesion affinity (designated as “round”) and those that acquired an increased volume of a vacuole (designated as “vacuole”). To remind, a vacuole changes its structure to the needs of the cell, e.g. when it isolates materials that might be harmful or a threat to the cell, among other functions.
Fig. S7A (ESI†) shows the statistics of live fibroblasts with normal or round morphology followed by those with increased number of visible vacuoles after electroporation. Overall, the majority of cells were observed as normal (about 75–95%) with almost no difference between silver or gold nanofilms. Less than 20% of the cells acquired round shape and vacuoles with increased volume. The number of cells with embedded silver nanoshells is about the same in both types, but it is different with gold nanofilms: the number of round cells is slightly higher and fewer those with vacuoles. This means that only one fifths of all the cells experience reasonable changes of the physiological conditions in order to maintain the main cellular functions. To note, similar changes after electroporation were observed with the control cells free of particles. It is important to emphasize, that for a development of a non-destructive label-free chemical imaging electroporation should be replaced by alternative non-damaging methods.
In addition, the applied voltage and the duration of electroporation were varied in order to find out the appropriate conditions at which the number of normal cells can be higher. The statistics of live cells after different types of experiment is plotted in Fig. S7B (ESI†). At applied voltage below a certain minimum threshold value (i.e. ±200 V) during 750 ms the effect is comparable to incubation/aging. However, above ±500 V in 350 ms most of the cells were damaged (independently on the number of SiO2@Ag–PAA particles). The majority of normal cells (about 95%) were observed without any visible morphological changes comparable to the control cells before electroporation (types of experiment # 9 and 11). As shown later these conditions were used as default set of parameters for electroporation of SiO2@Ag–PAA particles (Fig. 7 and 8).
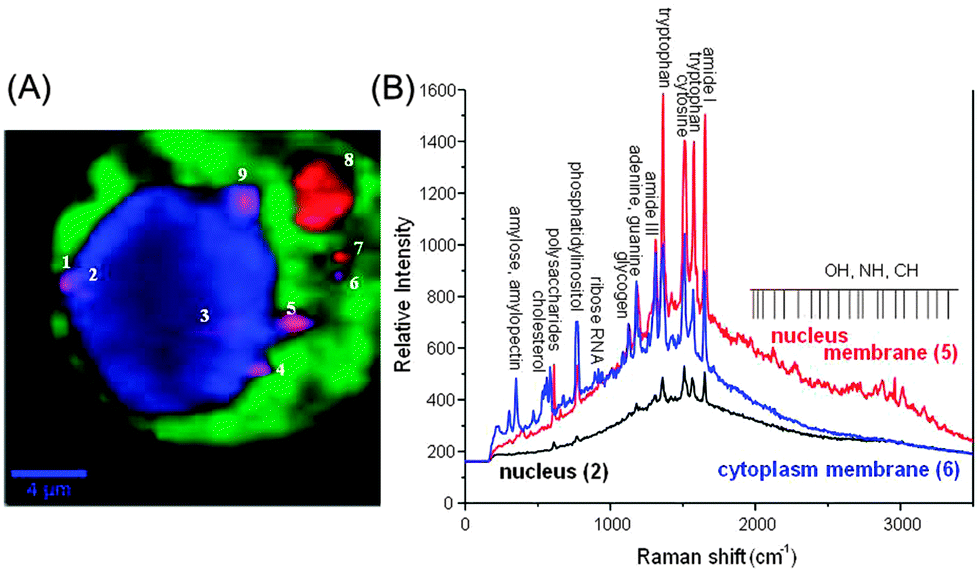 | ||
| Fig. 7 (A) Raman spectroscopic imaging of a live NIH/3T3 fibroblast with embedded SiO2@Ag–PAA particle in a coloured spectral map (scale bar is 4 μm). The map reflects the differences found in the Raman data and SERS active Ag–PAA nanofilms. This map is a linear combination of the averaged single spectra and characteristic for cell compartments (green cytoplasm and blue – nucleus). Intense green and blue colors of spots separated by a darker background contrast indicate the presence of SERS effective SiO2@Ag–PAA particles inside the cellular medium. Spots with violet and red colors show SiO2@Ag–PAA particles that are located at the membrane interface or at its surface (highlighted with numbers). A confocal Raman image is generated by integration of the intensity of the strongest bands in the three spectral ranges: (i) <1000 cm−1, (ii) 1000–2000 cm−1 and (iii) 2500–3500 cm−1 after local baseline subtraction using the hypercluster analysis (HCA) as one of the efficient label-free methods for the visualization of intracellular components and processes. (B) Selected SERS spectra that are collected from nucleus (2 in A) and periphery membranes of the nucleus (5 in A) or cytoplasm (6 in A). The laser excitation wavelength was 532 nm and the grating was 600 g mm−1 (BLZ = 500 nm) and spectral resolution of 3 cm−1. | ||
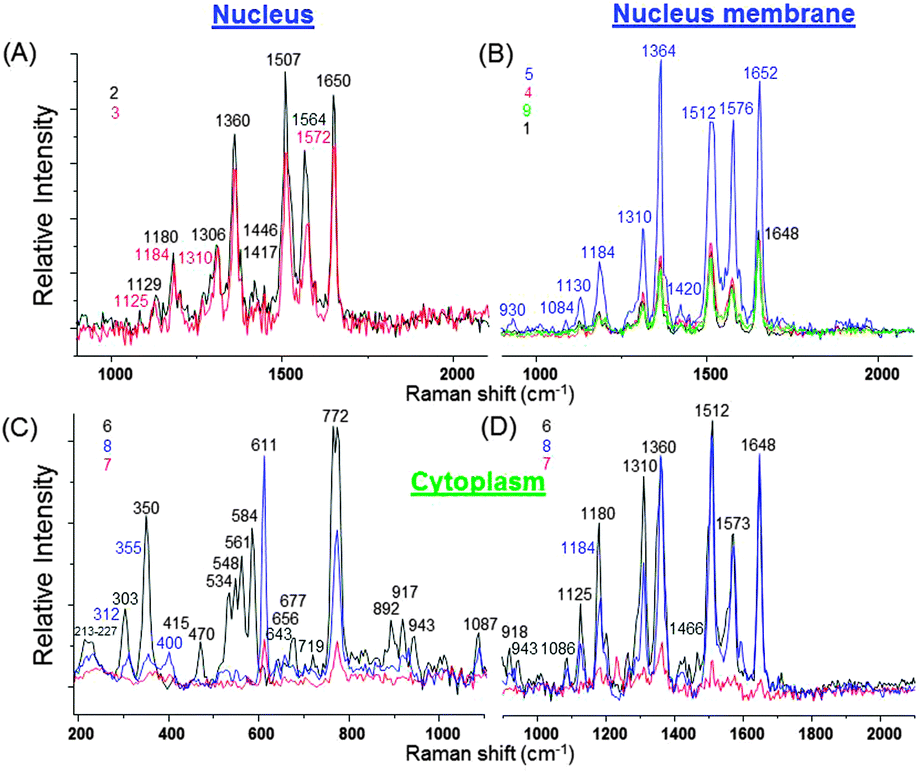 | ||
| Fig. 8 Cumulated average SERS spectra collected at different locations inside live NIH/3T3 fibroblasts with the numbers indicating position of SiO2@Ag–PAA particles from the HCA of the SERS image in Fig. 7A. The laser excitation wavelength was 532 nm and the grating was 600 g mm−1 (BLZ = 500 nm) with a spectral resolution of 3 cm−1. | ||
For SERS measurements the culture medium was removed and the petri dish with incubated live fibroblasts was triply rinsed with phosphate buffered saline (PBS) solution. As one of the examples, most of the SERS active particles are closely-packed in the area close to or surrounding the nucleus of live fibroblasts (Fig. S8, ESI†). The SERS effective Ag–PAA nanofilm appear as bright scattering spots of relatively high intensity as indicated by arrows (Fig. S8A and B, ESI†).
Fig. 7A shows a Raman spectroscopic image of a live NIH/3T3 fibroblast with embedded SiO2@Ag–PAA particles as a colourful spectral map. This image was reconstructed by using the hypercluster analysis (HCA) from the strongest bands that are cumulative and averaged from at least ten fibroblasts. To note, the HCA can provide label-free methods for the visualization of intracellular components and processes based on strong Raman/SERS signals (Fig. S9, ESI†). The confocal laser scanning microscopy image shows the same cell with embedded SERS effective SiO2@Ag–PAA particles that are closely-packed throughout the intracellular interior, being in the nucleus and cytoplasm (Fig. S9A, ESI†). Colourful regions of the nucleus and cytoplasm are clearly distinguishable by the contrast differences (brighter regions are due to strong SERS activity) (Fig. 7A). The numbered spots with a violet or red color are those closely-packed SERS effective SiO2@Ag–PAA particles that appear at the membrane interface or at the outer surface of the cell, respectively.
It is well-known that, the cellular nuclei can be distinguished from the cytoplasm, but Raman signals due to DNA are barely visible. This suggests that HCA identifies the nucleus mostly by protein composition (the nuclear proteins differ from cytoplasmic proteins), rather than the actual DNA signal. However, during certain stages of mitosis, the DNA can be so condensed that it exhibits such a strong signal to be relatively easily observable, which is not the case here. In the HCA image analysis the intensity at each pixel is used to threshold the image, which is segmented into pseudocolors. In this way, the intensity is necessary to display those regions of the image corresponding to chemical information and for the computer to correlate or discriminate regions based on spectroscopic data. In our spectral data the intensity is stronger than the necessary threshold value in order to perform the HCA analysis and clearly distinguish the intracellular compartments. To note, the HCA method can be also supported by a reliable technique of principle component analysis (PCA) based on the group discrimination in SERS imaging of organelles.34
For analysis we select and number the following locations inside the cell where the SERS intensity is high, i.e. (i) inside the nucleus (2 and 3), on its periphery membrane (1, 4, 5, and 9) and inside the cytoplasm (6, 7 and 8) (Fig. 7A). Strong SERS spectra exhibit multiple peaks over the entire spectral region (Fig. 7B) with different shape profiles from the nucleus and periphery membrane of the nucleus or cytoplasm.
Strong SERS signals arise from amide (–CONH2, neutral), tryptophan (negatively charged at 6 < pH < 8.5), cytosine (negatively charged), adenine (interact with the Ag surface due to the highly negative charge density of the N1 nitrogen36), phosphatidylinositol (COO−), polysaccharides (NH3−, COO−), cholesterol (negative, OH−, CH3−) or amylose (negative, CH2OH, H and OH), among others. To note, the SERS spectra of SiO2@Ag–PAA particles in culture medium or PBS solution without fibroblasts exhibit small peaks at 788 cm−1 and 965 cm−1 (in CM) or 1321 cm−1 and 1593 cm−1 (in PBS) (Fig. S10, ESI†) not observable with live fibroblasts (Fig. 7B).
As some fibroblast cell lines prefer slightly more basic environment (pH 7.4–7.7), an anionic PAA corona over Ag NPs does not have free carboxylic acid groups, contributing to the electrostatic stabilization of Ag NPs. In this way, aggregation of Ag NPs is unlikely as most citrate ions are not displaced. The COO− groups of the detected molecules can displace original citrate ions of the PAA and come closer to the metallic silver surface. Hence the SERS signals of molecular prints are strong and repeatable, and any analyte effect on the SERS effective Ag–PAA nanofilm is avoided. Moreover, from a SERS point of view the ideal system would be that of a stable solution of small nanoparticle clusters over the microscale surface, perfectly matching stable SiO2@Ag–PAA particles. As no aggregates of silver nanoparticles appeared either in the bulk solution of CM or PBS (Fig. S11, ESI†) or inside live fibroblasts (SiO2@Ag–PAA are only closely packed to each other) undesirable time-dependent and irreproducible results can be avoided.
In Fig. 8 multiple strong SERS spectra from intracellular compartments are extensively informative and reproducible. Because of this we decided to group these SERS spectra in three spectral regions in order to perform the intracellular study: (I) 200–1000 cm−1; (II) 1000–2000 cm−1 and (III) 2000–3800 cm−1 (Fig. 8 and Tables S2–S4, ESI†).
Inside the nucleus the SERS spectra exhibit slight changes in its center or close to the periphery membrane (Fig. 8A). The most prominent SERS peaks appear from tryptophan (strong at 1360 cm−1 and 1564–1572 cm−1), cytosine (very strong at 1507 cm−1) and amide I (very strong at 1650 cm−1), that is important for the dehydration synthesis reaction (condensation reaction) and usually occurs between aminoacids (Table S3, ESI†). However, at the nucleus membrane these peaks are much stronger (nine fold higher intensity), especially at a hot spot 5, (Fig. 8B) and appear at higher frequency shifts due to the decreased concentration (Table S3, ESI†). In addition, a strong SERS peak from cytosine, adenine or guanine (1184 cm−1) is observed at seven fold higher intensity, while it appears only as a weak signal inside the nucleus (Fig. 8A and B; Table S3, ESI†).
It is important to note, that adenine is an interesting molecule in the field of SERS, which interacts with the silver surface at the N1 site due to the highly negative charge density of this site.36 It is the first small organic molecule used in single-molecule SERS measurements.35–37 This means that SERS effective SiO2@Ag–PAA particles can be capable to perform single molecule SERS studies of single live cells.
Other important biomolecules such as cholesterol and phosphatidylinositol exhibit stronger SERS prints at the nucleus membrane than inside the nucleus or at its periphery (Table S2, ESI†). And the extensive OH, NH and CH stretching groups appear as stronger SERS signals from the nucleus membrane rather than from its center (Table S4, ESI†).
It is remarkable, that in the cytoplasm the overall SERS spectra appear with a far larger number of strong signals and from much more biomolecules in all three selected spectral regions (Fig. 8C and D; Tables S2–S4, ESI†). In particular, very strong SERS signatures from cholesterol (at 534 cm−1, 548 cm−1 and 611 cm−1), phosphatidylinositol (at 772 cm−1) and new moderate signals from polysaccharides, amylose, amylopectin (470 cm−1), ring breathing in the DNA bases (677 cm−1), saccharide band, C–C skeletal backbone (892 cm−1), ν(C–C) gauche (1086 cm−1) and antisymmetric phosphate stretching vibration (1230 cm−1) are not observable in the nucleus or at its membrane (Tables S2 and S3, ESI†). To remind, cholesterol is important for the permeability and fluidity of the membrane. In this way, we may assume that its strong SERS signature arises from the periphery membranes of organelles inside the cytosol. Distinct from the nucleus or its membrane, numerous resolved SERS peaks appear from the phospholipids in the region below 800 cm−1 which can be assigned to different residues at the phosphate-ester head-groups.38–40
Extensive SERS prints appear with multiple less strong new peaks of symmetric stretching of the choline group, phospholipids (719 cm−1)41 ribose vibration, that is one of the distinct RNA modes (918 cm−1 and 975 cm−1), PO2− stretching (DNA/RNA) (1060 cm−1 and 1069 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching in a quinoid ring (1594 cm−1) (Tables S2 and S3, ESI†). It is possible to monitor the changes of other components such as glycogen (1047 cm−1), triglycerides in fatty acids with δ(C–H) (phospholipids) (1264–1268 cm−1) and lipids (1466 cm−1) due to the high quality of SERS signals (Tables S2 and S3, ESI†). Furthermore, multiple arrays of the SERS signatures of OH, NH and CH stretching groups exhibit peaks that can be found either in the nucleus or at its membrane (Table S4, ESI†).
N and CC stretching in a quinoid ring (1594 cm−1) (Tables S2 and S3, ESI†). It is possible to monitor the changes of other components such as glycogen (1047 cm−1), triglycerides in fatty acids with δ(C–H) (phospholipids) (1264–1268 cm−1) and lipids (1466 cm−1) due to the high quality of SERS signals (Tables S2 and S3, ESI†). Furthermore, multiple arrays of the SERS signatures of OH, NH and CH stretching groups exhibit peaks that can be found either in the nucleus or at its membrane (Table S4, ESI†).
To remind, the spectral shift indicates changes in the concentration of detected molecules.42,43 In our SERS spectra the largest shifts were observed in the ν(C–C) stretching in amino acids and skeletal modes of polysaccharides and amylose (930 cm−1 and 943 cm−1), indicating the concentration decrease of the last ones. Less pronounced shifts were examined in the ν(C–C) skeletal of the acyl backbone in lipids (1125 cm−1 and 1130 cm−1), that correspond to the trans conformation. Fewer molecules can contribute to this vibrational signal inside the nucleus and its corresponding periphery membrane than in the cytosol. A very strong SERS signature from tryptophan (1360 cm−1) appears at higher frequency (1364 cm−1) at the nucleus membrane due to the higher concentration. This is an interesting finding as tryptophan is an amino acid being essential in the human diet, as it was demonstrated by its growth effects on rats.22 Overall it is observed that the cytoplasm region shows relatively high concentrations of lipids compared to the nucleus region. Notable nucleic acid related vibrations in the cytoplasm regions are attributed to the ribose contribution, one of the distinct RNA modes.
Conclusions
In this work the SERS effective nanofilm approach has been advanced by a Ag–PAA matrix which exhibits enhancement factors, EFs ∼ 108 more than sufficient for single molecule studies. High EFs can be achieved by the coupling of primary and secondary SERS hot spots of Ag–PAA nanofilm on silica MPs (SiO2@Ag–PAA particles). From theoretical calculation in the electrostatic approximation such high local field intensity (LFI) and SERS EFs can arise from prolate Ag–PAA (low aspect ratio) on silica, not in water or with gold (instead of silver). However, at higher aspect ratio the EFs on silver or gold can be comparable. Oblate nanoparticles can exhibit EFs of two orders of magnitude lower than those with prolate shape.SiO2@Ag–PAA particles can have a number of superior advantages to achieve a LFI EFs (SERS EFs) increase than a common core–shell model (Ag NPs which is coated by a thin outer silica layer):
– SERS EFs decay very slowly with the distance from the nanofilm surface and still have high values at distances above 10 nm;
– Larger coverage by analytes (up to 10 nm) experiencing higher EFs;
– The cumulated SERS intensity saturates slower by at least two fold with the coverage increase;
– The average SERS EFs are higher by two orders of magnitude Ag oblate nanoparticle, when the incident polarization is along z direction and about ten times larger for Ag prolate nanoparticles, when the incident polarization is along x direction.
Because of the strong SERS intensity from Ag–PAA nanofilms the hypercluster analysis (HCA) of live single fibroblasts can be carried out in real time. This is important as HCA is one of the most efficient label-free methods for the visualization of intracellular components and processes. SERS bands can be reproducible with multiple strong peaks throughout the whole spectral region with clearly distinguishable shapes from nucleus, periphery membrane and cytosol. As a proof of single molecule detection – adenine can be detected from live fibroblast by a Ag–PAA nanofilm.
Our results demonstrate how live single cells can be chemically imaged with single molecule precision by a SERS effective Ag–PAA nanofilm on silica MPs as reliable substrates. These findings can be of potential practical importance to conduct systematic analyte analysis of a live single cell in real time. Moreover, cell biological aspects such as the intracellular metabolism, drug uptake, apoptosis, and other effects could, in principle, be also investigated.
Experimental part
Materials
Silver nitrate (AgNO3, analytical grade, 99.8%) was purchased from Serva (Germany). Trisodium citrate dihydrate (99%) was obtained from Acros Organics Thermo Fischer scientific (Belgium). Polyacrylic acid sodium salt (PAA, MW ≈ 5100 Da), polyvinyl pyrrolidone (PVP K 30, MW ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), rhodamine 6G (99%) and 3-aminopropyltriethoxysilane (APTES) were purchased from Aldrich. Silica particles (SiO2, 1.55 ± 0.04 μm, 50 mg mL−1) were obtained from Microparticles GmbH (Germany). Methanol was purchased from Merck KGaA (Germany) and toluene was obtained from AppliChem (Germany). Sodium chloride (NaCl, ≥99.5%, biograd, suitable for the cell culture medium), DMEM, gentamicin, glucose and phosphate buffered saline (PBS, bioperformance certified, suitable for cell culture) were purchased from Sigma-Aldrich Co. (Germany). Calf serum was purchased from PAA Laboratories GmbH (Austria).
000 Da), rhodamine 6G (99%) and 3-aminopropyltriethoxysilane (APTES) were purchased from Aldrich. Silica particles (SiO2, 1.55 ± 0.04 μm, 50 mg mL−1) were obtained from Microparticles GmbH (Germany). Methanol was purchased from Merck KGaA (Germany) and toluene was obtained from AppliChem (Germany). Sodium chloride (NaCl, ≥99.5%, biograd, suitable for the cell culture medium), DMEM, gentamicin, glucose and phosphate buffered saline (PBS, bioperformance certified, suitable for cell culture) were purchased from Sigma-Aldrich Co. (Germany). Calf serum was purchased from PAA Laboratories GmbH (Austria).
The water used in all the experiments was prepared in a three-stage Millipore Milli-Q Plus 185 purification system and had a resistivity higher than 18.2 MΩ cm−1. For the experiments with living cells Milli-Q water was autoclaved.
Preparation of silver nanoparticles (Ag NPs)
:4 at room temperature in darkness. The Ag–PAA colloidal mixture was incubated at mild shaking (300 rpm) in an Eppendorf Thermomixer compact (Hamburg, Germany) at 37 °C for 20 min, then triply washed by centrifugation (14000 rpm for 30 min). The Ag–PAA surface potential was −45 ± 5 mV.
:9 and incubated at room temperature for 15 min at mild shaking 300 rpm. The silica–silver mixture was triply washed with Milli Q autoclaved water by centrifugation (8000 rpm for 5 min). The adsorption procedure was repeated three times. The average surface potential of the SiO2@Ag–PAA particles was −45 ± 5 mV.
:5 of salt to sol followed by the addition of fresh 1 μL Rh6G solution. The fluorescence of SiO2@Ag–PAA particles with adsorbed dye molecules was quenched due to electron transfer (Fig. S4, ESI†).
SiO2@Ag–PAA particles were delivered into the cellular interior by the electro-permeabilization of the cell membranes in a BIO-RAD gene-pulser cuvette with 0.4 cm electrode gap inserted inside a home-made electric cell connected to the GHT-Bi500 electroporator (βtech, France). 200 × 10−6 L (5.34 × 104 cells) in a culture medium were mixed with 400 × 10−6 L of silver colloidal solution at a sequence of pulse voltages of 250 V for the positive and negative parts with a pulse length 500 ms within the sequence. After electroporation the colloidal mixture with the cell suspension was mixed with 1 × 10−3 L of culture medium and deposited on to the glass bottom of a petri dish in the incubator. The inspection of the incubated cells was checked by optical microscopy. Before Raman measurements the cells were washed at least three times with PBS aqueous solution.
Characterization
The second confocal Raman microscope (alpha 300, WITec, Ulm, Germany) was employed with a frequency doubled Nd:YAG (532 nm) laser excitation and piezoscanner (P-500, Physik Instrumente, Karlsruhe, Germany). The spectra were acquired with a thermoelectrically cooled CCD detector (DU401A-BV, Andor, UK), with an integration time 1 s of fifty accumulations with a grating 600 g mm−1 (BLZ = 500 nm) and spectral resolution of 3 cm−1. The excitation laser intensity was in the range from 7.9 kW cm−2 to 23.8 kW cm−2 (i.e. laser power from 1 mW to 3 mW) with a spectral resolution of 3 cm−1.
The signal to noise ratio was high enough to ensure the low value of the root mean square fluctuations averaged in time (typically below 5%). Basic alignment was carried out by a Raman spectrum of a silicon wafer with a characteristic Si line at 520 cm−1 during integration times from 0.3 to 1 s. The SERS measurements of live cells were conducted in real time in a μ-dish (35 mm, Ibidi, Munich, Germany) equipped with a heating stage and external temperature probe (Bioscience Tools, USA) to maintain 37 °C of the cells. The acquired Raman and SERS spectra were corrected for the baseline, background of the μ-dish substrate and the SiO2 (485 cm−1 band). For imaging integration time of 0.1 s per pixel was used. The Scan Ctrl Spectroscopy Plus software (version 1.38, WITec) was used for measurement, WITec Project Plus (version 2.02, WITec) for spectra processing. Raman images were generated with the WITec software using a sum filter, which integrates the intensity of the signal for a defined wavenumber range of interest and subtracts the background as a linear baseline from the upper to lower border. Hypercluster analysis (HCA) of Raman images was performed with the OPUS 6.5 software. Briefly, HCA was used to sort objectives according to a defined similarity based on Raman/SERS spectra. HCA builds the cluster by combining the objectives, which are treated and linked into larger groups by calculating criterion (spectral bands and regions) distance step by step.
where EF is the average enhancement factor (averaged over all possible positions on the metallic surface and also from molecules randomly adsorbed on the surface as compared to the same number of non-adsorbed molecules), Cnorm and CSERS are concentrations of Rh6G aqueous solutions for SERS and normal Raman measurement (i.e. number of molecules on the surface of a single nanoparticle and in bulk solution effectively excited by the laser beam); ISERS and Inorm are corresponding intensities of these vibrational modes. The Raman peak of Rh6G at 1365 cm−1 was taken for the EF estimation. To estimate the molecular density of Rh6G adsorbed on Ag NP (average radius 15 × 10−9 m) surface we consider the aqueous solution of Ag NPs at 0.58 × 10−3 mL−1 concentration in a volume 0.6 mL incubated with 10−8 mol L−1 Rh6G aqueous solution. The mole number of silver is νAg = 0.3 × 10−6 mol
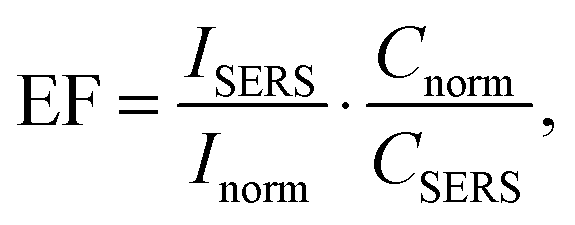
where nAg is the molecular density of silver, NAg is the number of silver atoms in a volume, NA is Avogadro's number (6.02 × 1023 mol−1). The total average density of silver is 3.48 × 1020 L−1 and of Rh6G molecules is 6.02 × 1015 L−1. As the silver crystals with the atoms are arranged in the face-centred cubic stacking cell with about four atoms, N, inside the unit cell for each of the packing arrangements, the 74% packing efficiency, h, as follows
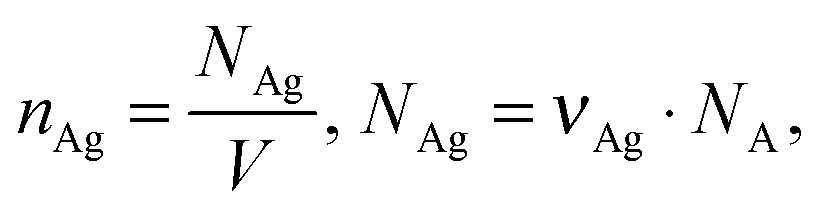
where Vsphere and Vtotal are the volume occupied by a sphere and a total volume, r is a radius of an atom. The covalent radius of a silver atom is 145 ± 5 × 10−12 m and its volume is 1.28 × 10−29 m3 (spherical geometry). The radius of a particle is 15 × 10−9 m and the volume of a spherical particle is 1.41 × 10−23 m. In this way, taking the packing efficiency into account,
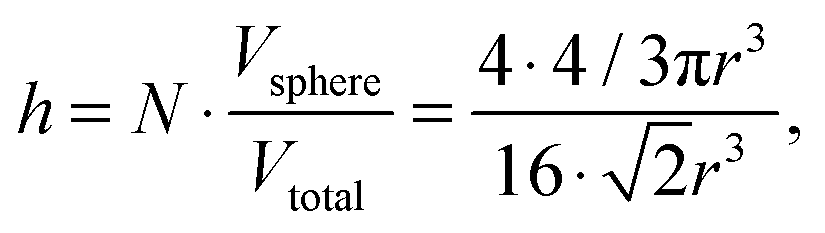
one single Ag NP consists of 8.15 × 105 silver atoms. The average silver density is 4.3 × 1014 L−1 and when it is divided by the dye density, we have about 14 Rh6G molecules adsorbed on one single Ag NP.
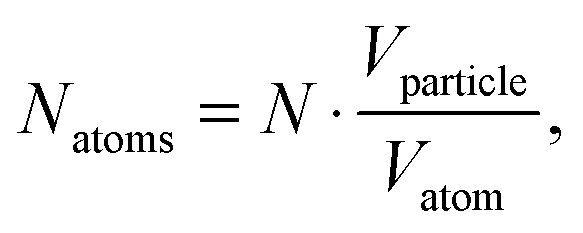
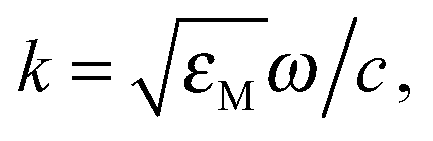
As for a single sphere, the electric field solution is expanded for each spherical symmetry in every volume. The extension of Mie theory (generalized Mie theory, GMT) is applied considering two spheres and each of them contributes to a scattering wave that is seen by the other one. The electromagnetic response of the spheres interferes with each other through the respective scattered fields. The scattered field has a contribution from spherical waves (VSHs) emanating from two individual spheres. For a system of two spheres the scattered field is expanded as
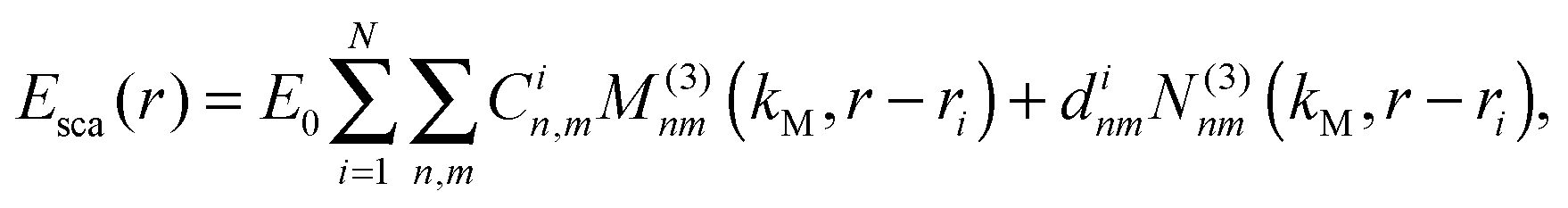
The scattered fields are a superposition of VSHs originating from the centers of two different spheres with the interaction of the respective VSHs aligned along z due to the symmetry. For a system of two spheres the internal field is expanded as
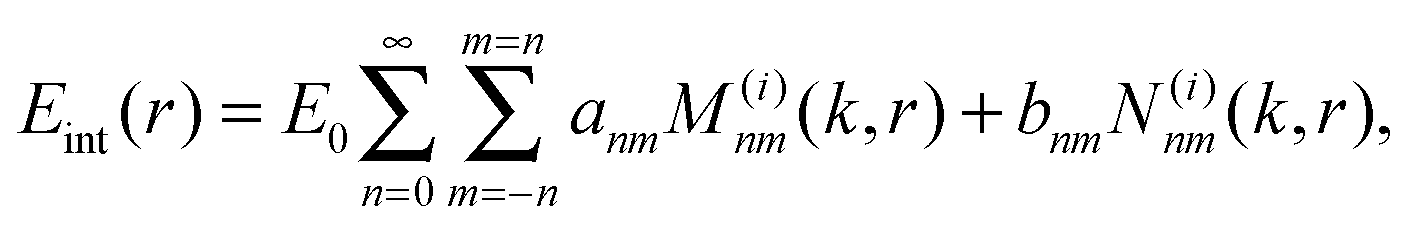
This interpretation is a direct consequence of the spherical symmetry and is completely analogous to the same classification done in quantum mechanics for potentials with spherical symmetry. Vectors M(i)nm(k,r) and N(i)nm(k,r) are called vector spherical harmonics (VSHs) or multipole fields and have the following physical meaning: magnetic and electric field that is created by a given multipolar distribution of sources at the origin. M(i)nm(k,r) describes a magnetic field of an electric dipole and N(i)nm(k,r) determines an electric field of that dipole. For n > 1 they correspond to the electric fields of higher order electric and magnetic multipoles (quadrupole, etc.). The angular dependence of the VSHs is described in terms of spherical harmonics, while their r-dependence involves spherical Bessel functions.
| Mnm(k,r) = Δ × (ςnm(k,r),r) |
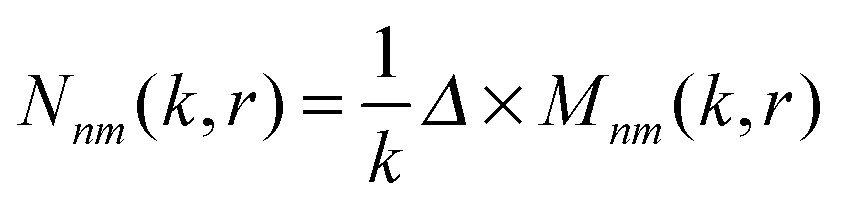
where Zn – spherical Bessel functions, Ynm(θ,φ) – spherical harmonics with the following standard convention:
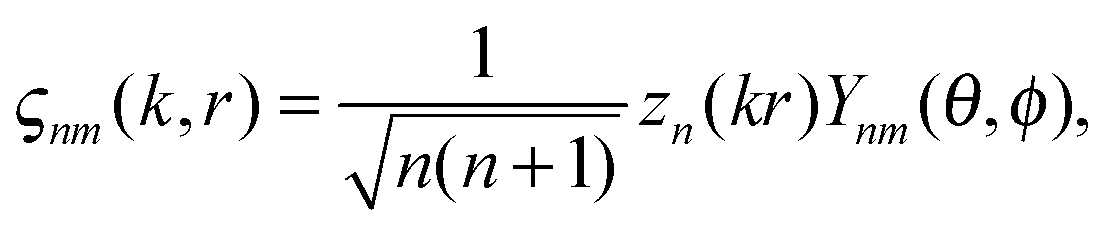
where Pnm(cos(θ)) are the associated Legendre functions, defined using the Condon–Shortley phase.
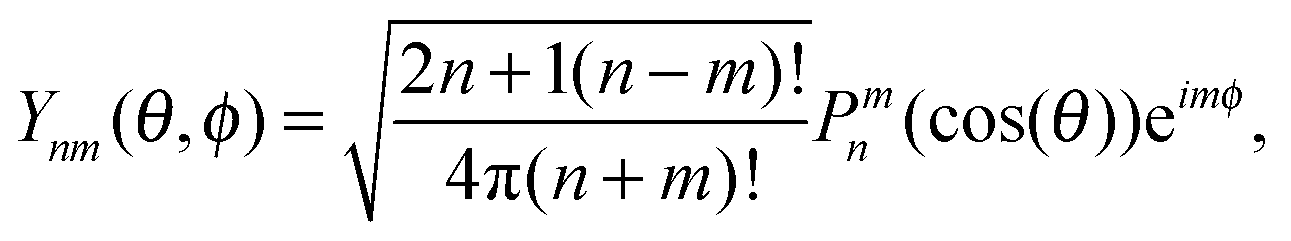
Unknown complex coefficients must be determined by imposing two independent EM boundary conditions at the sphere surface (r = a). For the scattered field
| Cnm = Γnanm and dnm = Δnbnm, while for the internal field the coefficients are αnm = Ananm and βnm = Bnbnm. |
For small spheres
with
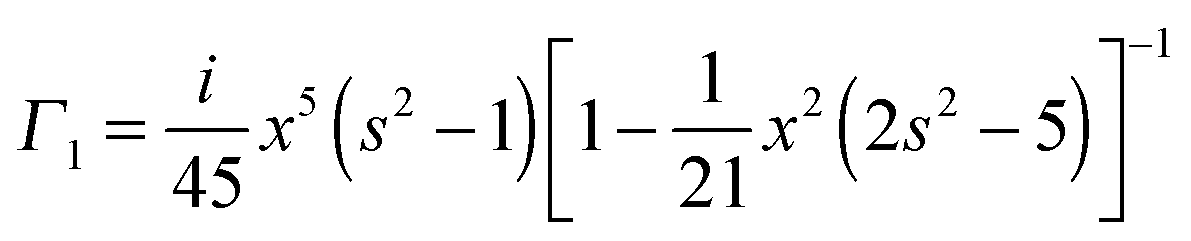
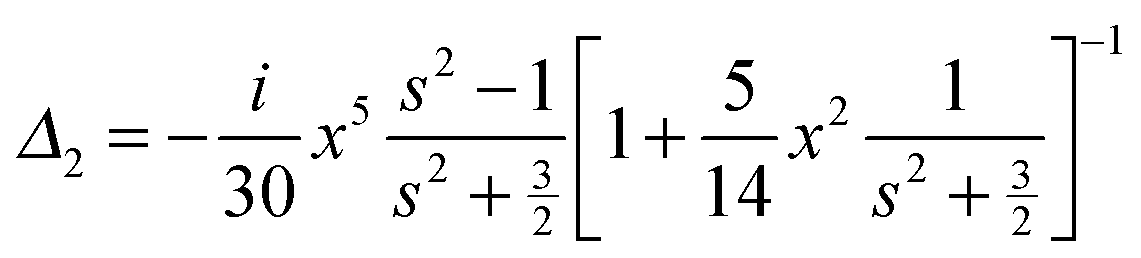
We use analytical formulas of the electrostatic approximation (ESA) for a qualitative understanding of scattering field enhancements at the silica/silver interface. We assume that the electric field outside the sphere is created by an electrostatic dipole at the sphere center.
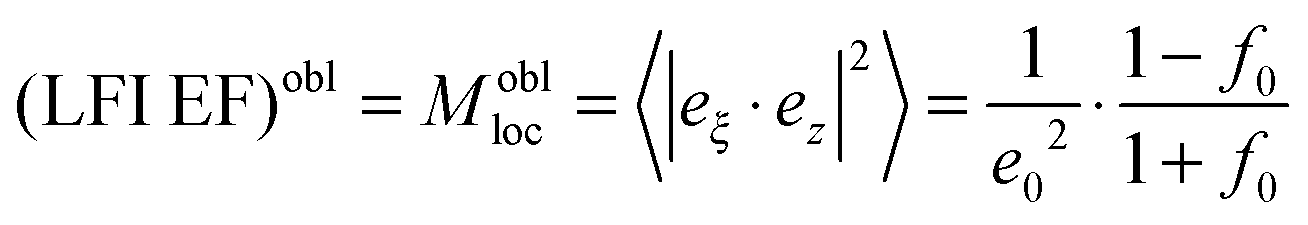 with the surface average
with the surface average 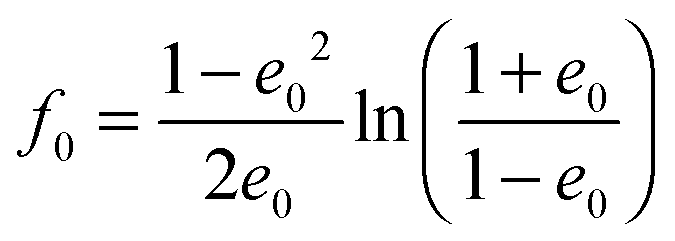 and surface area S = 2πa2[1 + f0] and
and surface area S = 2πa2[1 + f0] and 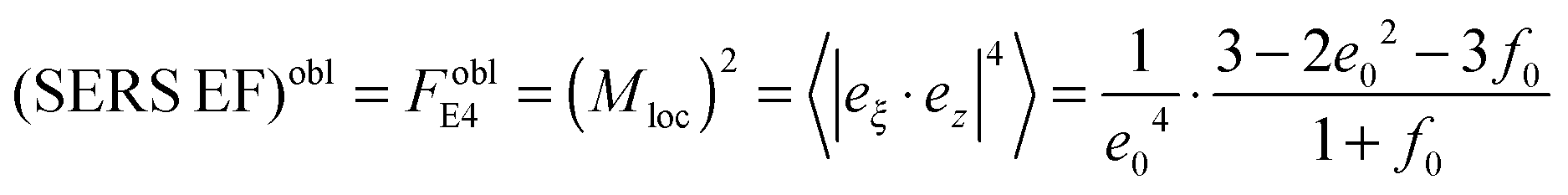 for oblate spheroid
for oblate spheroid 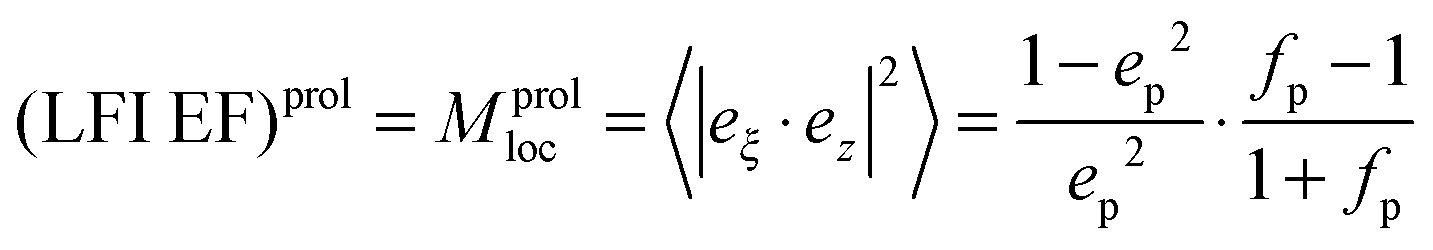 with the surface average
with the surface average 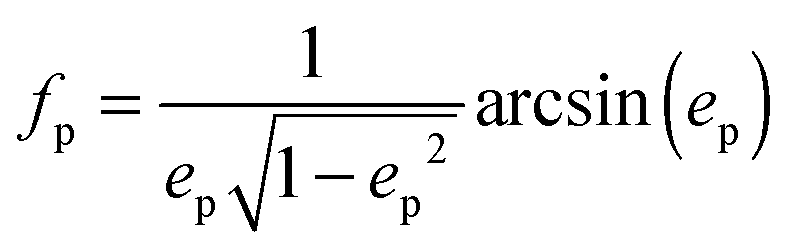 and surface area S = 2πb2[1 + fp] and
and surface area S = 2πb2[1 + fp] and 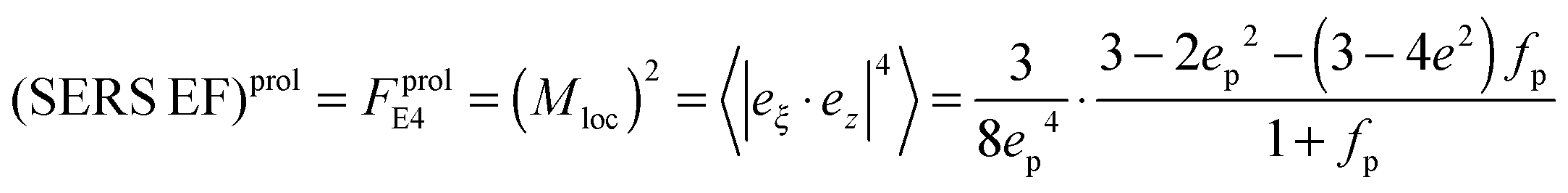 for prolate spheroid.
for prolate spheroid.
We assume for axes with “flat” curvature (Li > 1/3) the resonant wavelength is blue shifted compared to the sphere, but remains bound by the bulk plasmon resonance. For axes with “pointy” curvature (Li < 1/3) the resonant wavelength is red shifted compared to the sphere and the amount of the red-shift depends on the magnitude of the coefficient is 1/Li − 1, i.e. the smaller the L value (more pronounced corners of the prolate spheroid) the larger the red-shift.
| Mperploc(r,ω) = Aperp(ω)|eξ·ez|2, |
| Aperp(ω) = |1 + (3 − 3L)β(ω)|2, |
| Mparaloc(r,ω) = Apara(ω)[1 − |eξ·ez|2], |
| Apara(ω) = |1 − 3Lβ(w)|2, |
where ε and is εM are dielectric constants in the metal and the medium.
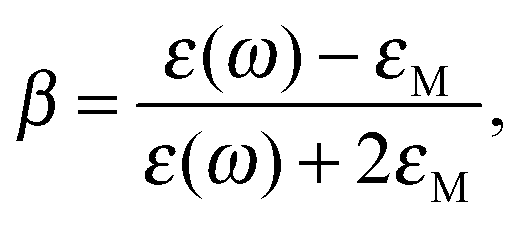
The geometrical or depolarization factors Li (i = 1, 3 correspond to x and z) for oblate spheroids are L1 = L2 < 1/3 < L3 and prolate spheroids are L1 < 1/3 < L2 = L3. They are defined as following for an oblate spheroid:
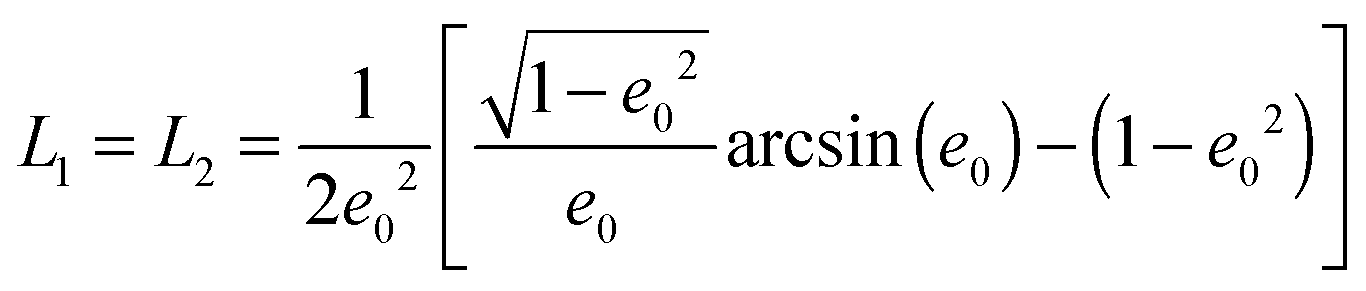
and prolate spheroid:
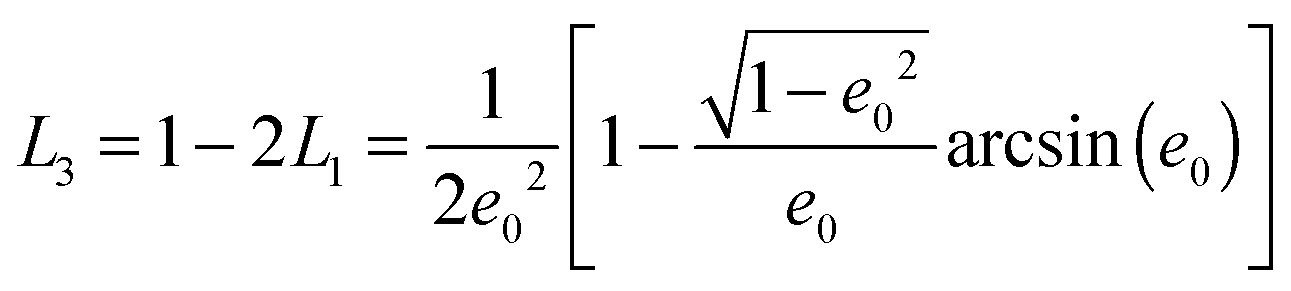
and
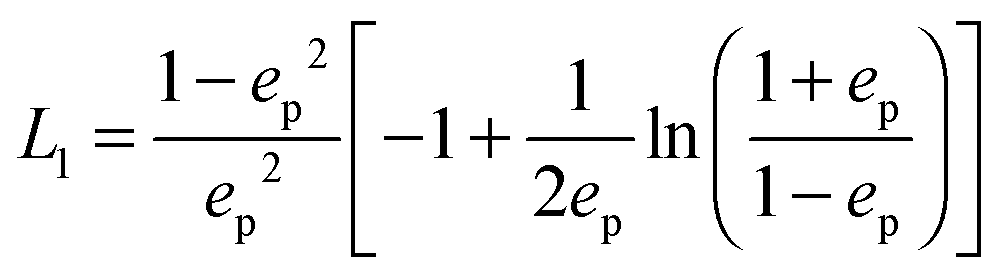
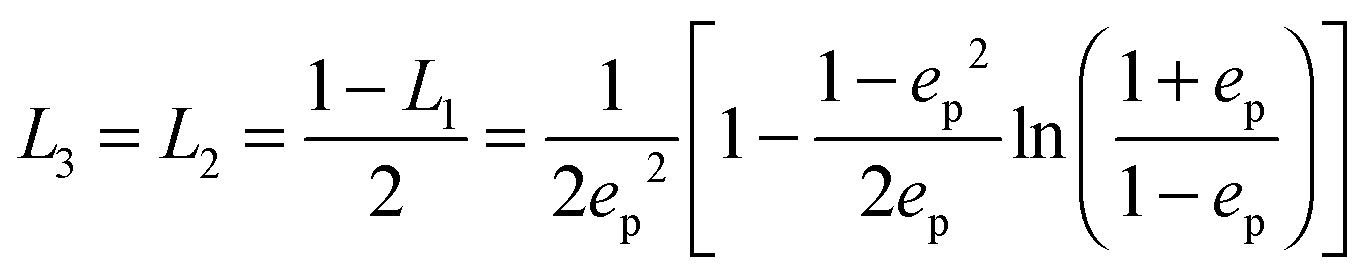 .
.| Mloc(r,ω) = Apara(ω) + (Aperp(ω) − Apara(ω))|eξ − ez|2 |
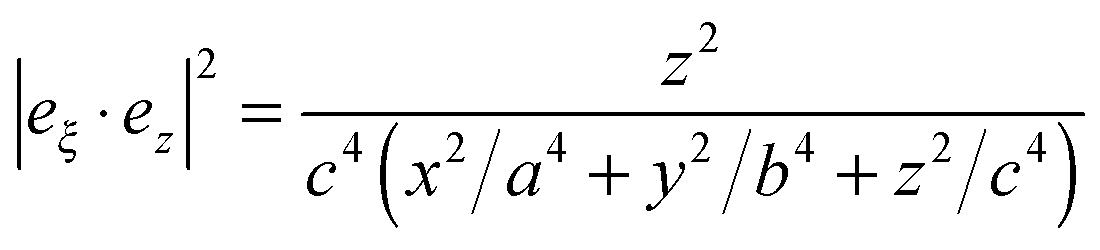 .
.
A perp(ω) and Apara(ω) are maximum and minimum LFI EF for incident polarization along the electric field ei. The maximum enhancement is obtained at two points on this axis (the tips) and the local field is normal to the surface at these points. The minimum enhancement is obtained at four points on two other axes (perpendicular to the incident polarization) and is then tangential to the surface. For other points the wavelength dependence of the LFI EF is determined by Aperp(ω) and Apara(ω), while the position dependence is governed by the factor |eξ·ei|2, where i = x or z. For SERS red-shifted resonances are the most important and they can be excited with polarization along the most “pointy” axes.
Acknowledgements
We thank Rona Pitschke and Heike Runge (Departments of Colloids at MPI) for electron microscopy analysis. We appreciate the allowance to use equipment of the high voltage electroporator from Dr Ronald Knorr and Prof. Rumiana Dimova (Theory Department at MPI). The help with the cell culture assistance by Christine Pilz (Department of Biomaterials at MPI) is greatly appreciated. Research grants from the Max-Planck society and Alexander von Humboldt foundation are greatly acknowledged.Notes and references
- D. Willis, K. W. Li, J. Q. Zheng, J. H. Chang, A. Smit, T. Kelly, T. T. Merianda, J. Sylvester, J. van Minnen and J. L. Twiss, J. Neurosci., 2005, 25, 778 CrossRef CAS PubMed.
- S. L. Spencer, S. Gaudet, J. G. Albeck, J. M. Burke and P. K. Sorger, Nature, 2009, 459, 428 CrossRef CAS PubMed.
- N. Lafuente, V. Azcutia, N. Matesanz, E. Cercas, L. Rodriguez-Manas, C. F. Sanchez-Ferrer and C. Peiro, J. Cardiovasc. Pharmacol., 2005, 45, 193 CrossRef CAS PubMed.
- M. Mir, A. Bergamaschi, B. S. Katzenellenbogen and G. Popescu, PLoS One, 2014, 9, 1 Search PubMed.
- W. K. Huh, J. V. Falvo and L. C. Gerke, Nature, 2003, 425, 686 CrossRef CAS PubMed.
- A. Jean-Jacques and S. Trajkovska, Fluoresc. Spectrosc., 2006, 7, 1067 Search PubMed.
- M. Hedegaard, C. Matthaus and S. Hassing, Theor. Chem. Acc., 2011, 130, 1249 CrossRef CAS.
- J. Ando, K. Fujita, N. I. Smith and S. Kawata, Nano Lett., 2011, 11, 5344 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. Mcquillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem., 1977, 84, 1 CrossRef CAS.
- S. Emory and S. M. Nie, Anal. Chem., 1997, 69, 2631 CrossRef CAS.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616 CrossRef CAS PubMed.
- E. A. Vitol, Z. Orynbayeva, M. J. Bouchard, J. Azizkhan-Clifford, G. Friedman and Y. Gogotsi, ACS Nano, 2009, 3, 3529 CrossRef CAS PubMed.
- J. Feng Li, X. D. Tian, S. B. Li, J. R. Anema, Z. L. Yang, Y. Ding, Y. F. Wu, Y. M. Zeng, Q. Z. Chen, B. Ren, Z. L. Wang and Z. Q. Tian, Nat. Protoc., 2013, 8, 52 Search PubMed.
- F. Tam, A. L. Chen, J. Kundu, H. Wang and N. J. Halas, J. Chem. Phys., 2007, 127, 204703 CrossRef PubMed.
- K. N. Yu, S. M. Lee, J. Y. Han, H. Park, M. A. Woo, M. S. Noh, S. K. Hwang, J. T. Kwon, H. Jin, Y. K. Kim, P. J. Hergenrother, D. H. Jeong, Y. S. Lee and M. H. Cho, Bioconjugate Chem., 2007, 18, 1155 CrossRef CAS PubMed.
- D. Radziuk and H. Moehwald, Nanoscale, 2014, 6, 6115 RSC.
- A. F. McCabe, C. Eliasson, R. A. Prasath, A. Hernandez-Santana, L. Stevenson, I. Apple, P. A. G. Cormack, D. Graham, W. E. Smith, P. Corish, S. J. Lipscomb, E. R. Holland and P. D. Prince, Faraday Discuss., 2006, 132, 303 RSC.
- A. Yashchenok, A. Masic, D. Gorin, B. S. Shim, N. A. Kotov, P. Fratzl, H. Moehwald and A. Skirtach, Small, 2013, 9, 351 CrossRef CAS PubMed.
- B. M. DeVetter, S. T. Sivapalan, D. D. Patel, M. V. Schulmerich, C. J. Murphy and R. Bhargava, Langmuir, 2014, 30, 8931–8937 CrossRef CAS PubMed.
- L. Novotny and B. Hecht, Principles of Nano-optics, Cambridge University Press, 2006, ch. 12, pp. 378–418 Search PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391 CrossRef CAS.
- N. E. Cant, K. Critchley, H. L. Zhang and S. D. Evans, Thin Solid Films, 2003, 426, 31 CrossRef CAS.
- R. D. Badley, W. T. Ford, F. J. McEnroe and R. A. Assink, Langmuir, 1990, 6, 792 CrossRef CAS.
- Y. Sun, B. Mayers and Y. Xia, Nano Lett., 2003, 3, 675 CrossRef CAS.
- P. Hildebrandt and M. Stockburger, J. Phys. Chem., 1984, 88, 5935 CrossRef CAS.
- B. Pettinger, K. Krischer and G. Ertl, Chem. Phys. Lett., 1988, 151, 151 CrossRef CAS.
- H. Wetzel, H. Gerischer and B. Pettinger, Chem. Phys. Lett., 1981, 78, 392 CrossRef CAS.
- F. R. Dollish, W. G. Fateley and F. F. Bentley, Characteristic Raman frequencies of organic compounds, Wiley, New York, USA, 1974 Search PubMed.
- V. Amendola and M. Meneghetti, Adv. Funct. Mater., 2012, 22, 353–360 CrossRef CAS.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493 CrossRef CAS.
- J. C. Weaver, J. Cell. Biochem., 1993, 51, 426 CrossRef CAS PubMed.
- A. Huefner, W.-L. Kuan, R. A. Barker and S. Mahajan, Nano Lett., 2013, 13, 2463–2470 CrossRef CAS PubMed.
- C. Krafft, L. Neudert, T. Simat and R. Salzer, Spectrochim. Acta, Part A, 2005, 61, 1529 CrossRef PubMed.
- S. K. Kim, T. H. Joo, S. W. Suh and M. S. Kim, J. Raman Spectrosc., 1986, 17, 381 CrossRef CAS.
- K. Kneipp, H. Kneipp, V. B. Kartha, R. Manoharan, G. Deinum, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 6281 CrossRef.
- Y. Maruyama, M. Ishikawa and M. Futamata, Chem. Lett., 2001, 834 CrossRef CAS.
- A. Barhoumi, D. Zhang, F. Tam and N. J. Halas, J. Am. Chem. Soc., 2008, 130, 5523 CrossRef CAS PubMed.
- Y. S. Huang, T. Karashima, M. Yamamoto and H. Hamaguchi, Biochemistry, 2005, 44, 10009 CrossRef CAS PubMed.
- C. Matthaus, T. Chernenko, J. A. Newmark, C. M. Warner and M. Diem, J. Biophys., 2007, 93, 668 CrossRef CAS PubMed.
- E. A. Vitol, Z. Orynbayeva, G. Friedman and Y. Gogotsi, J. Raman Spectrosc., 2012, 43, 817 CrossRef CAS.
- A. Barhoumi, D. M. Zhang and N. J. Halas, J. Am. Chem. Soc., 2008, 130, 14040 CrossRef CAS PubMed.
- E. Le Ru and P. Etchegoin, Principles of Surface Enhanced Raman Spectroscopy and related plasmonic effects, Elsevier, Amsterdam, Netherlands, 1st edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04034a |
| This journal is © the Owner Societies 2014 |