
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast photoinduced charge transport in Pt(II) donor–acceptor assembly bearing naphthalimide electron acceptor and phenothiazine electron donor†
Igor V.
Sazanovich
*ab,
Jonathan
Best
a,
Paul A.
Scattergood
a,
Michael
Towrie
b,
Sergei A.
Tikhomirov
c,
Oleg V.
Bouganov
c,
Anthony J. H. M.
Meijer
 *a and
Julia A.
Weinstein
*a
*a and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: Julia.Weinstein@sheffield.ac.uk; A.Meijer@sheffield.ac.uk
bCentral Laser Facility, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Chilton, Oxfordshire OX11 0QX, UK. E-mail: igor.sazanovich@stfc.ac.uk
cB. I. Stepanov Institute of Physics, Prosp. Nezavisimosti, 68, Minsk, 220072, Belarus
First published on 24th October 2014
Abstract
Visible light-induced charge transfer dynamics were investigated in a novel transition metal triad acceptor–chromophore–donor, (NDI–phen)Pt(II)(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph–CH2–PTZ)2 (1), designed for photoinduced charge separation using a combination of time-resolved infrared (TRIR) and femtosecond electronic transient absorption (TA) spectroscopy. In 1, the electron acceptor is 1,4,5,8-naphthalene diimide (NDI), and the electron donor is phenothiazine (PTZ), and [(phen)Pt(–CC–Ph–)], where phen is 1,10-phenanthroline, represents the chromophoric core. The first excited state observed in 1 is a 3MLCT/LL′CT, with {Pt(II)–acetylide}-to-phen character. Following that, charge transfer from the phen-anion onto the NDI subunit to form NDI−–phen–[Pt–(CC)2]+–PTZ2 occurs with a time constant of 2.3 ps. This transition is characterised by appearance of the prominent NDI-anion features in both TRIR and TA spectra. The final step of the charge separation in 1 proceeds with a time constant of ∼15 ps during which the hole migrates from the [Pt–(CC)2] subunit to one of the PTZ groups. Charge recombination in 1 then occurs with two distinct time constants of 36 ns and 107 ns, corresponding to the back electron transfer to each of the two donor groups; a rather rare occurrence which manifests that the hole in the final charge-separated state is localised on one of the two donor PTZ groups. The assignment of the nature of the excited states and dynamics in 1 was assisted by TRIR investigations of the analogous previously reported ((COOEt)2bpy)Pt(CC–Ph–CH2–PTZ)2 (2), (J. E. McGarrah and R. Eisenberg, Inorg. Chem., 2003, 42, 4355; J. E. McGarrah, J. T. Hupp and S. N. Smirnov, J. Phys. Chem. A, 2009, 113, 6430) as well as (bpy)Pt(CC–Ph–C7H15)2, which represent the acceptor-free dyad, and the chromophoric core, respectively. Thus, the step-wise formation of the full charge-separated state on the picosecond time scale and charge recombination via tunnelling have been established; and the presence of two distinct charge recombination pathways has been observed.
C–Ph–CH2–PTZ)2 (1), designed for photoinduced charge separation using a combination of time-resolved infrared (TRIR) and femtosecond electronic transient absorption (TA) spectroscopy. In 1, the electron acceptor is 1,4,5,8-naphthalene diimide (NDI), and the electron donor is phenothiazine (PTZ), and [(phen)Pt(–CC–Ph–)], where phen is 1,10-phenanthroline, represents the chromophoric core. The first excited state observed in 1 is a 3MLCT/LL′CT, with {Pt(II)–acetylide}-to-phen character. Following that, charge transfer from the phen-anion onto the NDI subunit to form NDI−–phen–[Pt–(CC)2]+–PTZ2 occurs with a time constant of 2.3 ps. This transition is characterised by appearance of the prominent NDI-anion features in both TRIR and TA spectra. The final step of the charge separation in 1 proceeds with a time constant of ∼15 ps during which the hole migrates from the [Pt–(CC)2] subunit to one of the PTZ groups. Charge recombination in 1 then occurs with two distinct time constants of 36 ns and 107 ns, corresponding to the back electron transfer to each of the two donor groups; a rather rare occurrence which manifests that the hole in the final charge-separated state is localised on one of the two donor PTZ groups. The assignment of the nature of the excited states and dynamics in 1 was assisted by TRIR investigations of the analogous previously reported ((COOEt)2bpy)Pt(CC–Ph–CH2–PTZ)2 (2), (J. E. McGarrah and R. Eisenberg, Inorg. Chem., 2003, 42, 4355; J. E. McGarrah, J. T. Hupp and S. N. Smirnov, J. Phys. Chem. A, 2009, 113, 6430) as well as (bpy)Pt(CC–Ph–C7H15)2, which represent the acceptor-free dyad, and the chromophoric core, respectively. Thus, the step-wise formation of the full charge-separated state on the picosecond time scale and charge recombination via tunnelling have been established; and the presence of two distinct charge recombination pathways has been observed.
Introduction
The dynamics of photoinduced charge transport in molecular systems underpins a significant proportion of the research in artificial photosynthesis and photocatalysis,1 and accordingly is a subject of much fundamental studies. The first step in initiating charge transport is usually the formation of a charge-separated excited state within a light-absorbing “chromophoric” unit. This state subsequently engages in dark electron–hole transfer events to propagate the charges to the periphery of the molecule and create an independently reactive electron–hole pair. Accordingly, much research has focused on understanding the rules governing forward and back electron transfer in such systems.Transition metal complexes have been at the forefront of developments of artificial light-harvesting systems. There, judicious modification of the metal chromophoric core in the donor–chromophore–acceptor assembly with various electron donors and acceptors in a “Lego”-type fashion allows one to vary the driving force of charge recombination and the distance between the ultimately separated charges without altering its light-absorbing properties. In such systems, absorption of light will lead to the formation of an initial donor–[chromophore]*–acceptor state, with the D+˙–[chromophore]–A˙− being an ultimate target state. The ultrafast nature of the forward charge separation requires accordingly fast methods to elucidate each step in the cascade of the charge-transfer events.2–5
Amongst transition metal chromophores, much attention has been recently directed towards square-planar, Pt(II)-based systems, which are characterised by intense absorption of light in the visible region as well as by the directionality of the electron transfer. Pt(II) diimine (bis)acetylide chromophores in particular have been a subject of intense research in this regard, owing to the presence of a charge-transfer, ML/LLCT, excited state that usually possesses a relatively long lifetime due to the strong-field acetylide ligand,6–27 which is generally sufficient to initiate an electron transfer cascade.
In this paper we present an acceptor–chromophore–donor molecular triad (NDI–phen)Pt(II)(–CC–Ph–CH2–PTZ)2 (1), based on a Pt(II) diimine chromophoric core, [(phen)Pt(–CC–Ph–)2], where phen is 1,10-phenathroline. Triad 1 combines a strong electron acceptor, 1,4,5,8-naphthalene diimide (NDI, E1/2(NDI/NDI˙−) of ca. −0.94 V vs. Fc+/Fc),28 and a strong electron donor, phenothiazine (PTZ, E1/2(PTZ˙+/PTZ) of ca. 0.32 V vs. Fc+/Fc),7 which can potentially engage in a cascade electron transfer process following initial excitation of the core.
The choice of the PTZ donor, and the NDI acceptor, is also determined by the drastic differences between absorption spectra of their neutral and ionic forms which are expected to be formed in the charge-separated state. Both PTZ and NDI do not absorb in the visible region, whilst PTZ˙+ possesses a characteristic absorption band at ca. 510–530 nm,7 and NDI˙− has strong absorbences at ca. 470 nm and in the 600 nm region,29–34 enabling the use of electronic transient absorption spectroscopy to monitor transient changes. Finally, the presence of the strong infrared reporters, ν(CO) on the NDI, and acetylide stretching vibrations on the donor ligand, will enable the use of time-resolved infrared spectroscopy to monitor light-induced processes in 1.9,11,24,27
Previously, the donor part of this design motif, –CC–Ph–CH2–PTZ, was successfully incorporated into [chromophore–donor] molecular dyads,7,10 using bipyridine and phenanthroline derivatives as diimine ligands, where the formation of the charge-separated state on the picosecond time scale has been demonstrated. Likewise, the dyad using only the acceptor part, NDI–phen–PtCl2, has been studied.35 However, no charge separation was observed due to the extremely short, ∼2 ps, lifetime of the initially formed MLCT excited state, which made the forward electron transfer process to NDI not competitive.
Here, a combination of time-resolved infrared (TRIR) and femtosecond electronic transient absorption (TA) spectroscopy, supported by DFT calculations and electrochemical methods, is applied to resolve the light-induced dynamics for 1 on the femto-to-microsecond time scale. The interpretation of the results is assisted by their comparison with the behavior of [donor–chromophore] (2) and [chromophore–acceptor] (3) dyads, and compound (4) as an analog of the chromophoric core.
Experimental
Synthetic procedures
Pt(phen–NDI)Cl2 was prepared as described previously.35 Synthesis of the Phenothiazine containing ligand N-(4-ethynylbenzyl)-phenothiazine36 and compound 4, ((COOEt)2bpy)Pt(CC–C7H15)2, is detailed in the ESI.†
Electronic transient absorption spectroscopy
Femtosecond transient absorption experiments were performed at B.I. Stepanov Institute of Physics, Minsk, on a pump–probe spectrometer based on a home-made original fs Ti:sapphire pulsed oscillator and regenerative amplifier system both operated at a 10 Hz repetition rate.37 Nanosecond flash photolysis data were obtained in Sheffield. The third harmonic (355 nm) of a picosecond Nd:YAG laser (PY61-10, Continuum) was used as the excitation source, with the excitation pulse energy at the sample attenuated down to 1 mJ to avoid non-linear sample response. The excitation beam was focused into 3 mm spot on the sample. The changes in absorbance of the photoexcited sample were probed using a pulsed 150 W Xe arc lamp (Applied Photophysics). A Si PIN photodiode detector (DET10A/M, Thorlabs) coupled into a digital oscilloscope (Tektronix TDS 3032B) was used to monitor the changes in the probe light intensity. The spectral selectivity of the detection system was achieved by narrow band-pass filters (CVI Technical Optics or Thorlabs, spectral bandwidth ca. 10–12 nm). The home-built delay-generator/trigger-suppressor electronic unit was used to synchronise the slow pulse rate of the Xe lamp (0.3 Hz) with the repetition rate of the picosecond laser (10 Hz). For fine timing adjustment of the Xe lamp pulses, a digital delay generator was employed (Princeton Applied Research, Model 4144). An electro-mechanical beam shutter (SH05, Thorlabs) was placed in the excitation beam to avoid unnecessary sample irradiation by unused laser pulses. All the measurements were performed in quartz cells with 1 mm path, the samples were freeze–pump–thaw deoxygenated. The overall time resolution of the detection system is estimated to be ca. 1.8 ns FWHM.Femtosecond time-resolved IR spectroscopy
The studies of compound 2 were performed in the laser for science facility (LSF), Rutherford Appleton Laboratory, on the PIRATE setup,38 based on 1 kHz, 800 nm, 150 fs, 1 mJ Ti:sapphire oscillator/regenerative amplifier; tuneable mid-infrared pulses (ca. 150 cm−1 FWHM, 0.1 µJ) were used as a probe, the estimated time resolution is ca. 0.4 ps. The studies of compound 1 were performed in the LSF on the PIRATE and ULTRA39 setups. ULTRA is based on a 65 MHz Ti:sapphire oscillator synchronously seeding a pair of regenerative amplifiers (Thales) operating at a 10 kHz repetition rate. One amplifier was configured to produce 40 fs duration pulses with broad bandwidth. Second harmonic generation of part of the 800 nm output from the femtosecond amplifier produced pulses of ∼50 fs at 400 nm for sample excitation. Approximately 0.4 mJ of the 50 fs, broadband output pulse train pumped a TOPAS OPA, and difference frequency mixing of the signal and idler components generated mid-IR pulses with ∼400 cm−1 bandwidth and ∼50 fs pulse duration at the output of the OPA to be used as a probe beam.Time-resolved data analysis
The analysis of time-resolved data to obtain decay lifetimes was performed using Igor Pro software (WaveMetrics, Inc.). The decay kinetics were fitted to the sum of exponentials (with an addition of a long-lived component when required) using the least-squares algorithm built into Igor Pro. In the case of picosecond transient absorption data, the deconvolution of the instrument response function (approximated as the Gaussian profile) from experimental decay kinetics was employed. The determination of actual zero time delay for each spectral position was incorporated in the data analysis algorithm, which automatically provided the chirp correction. For picosecond transient absorption and TRIR data, global fitting was applied to analyse simultaneously decay kinetics obtained for a number of spectral points, which considerably increased reliability of the fitted lifetimes and also provided the individual spectra of decaying species in the case of transient absorption data.DFT calculations
All calculations were performed using Gaussian 09, version C.01,40 compiled using Portland compiler v 8.0-2 with the Gaussian-supplied versions of ATLAS and BLAS,41,42 using the B3LYP functional of DFT.43 In all cases an extensive basis set was used, consisting of 6-311G**44,45 on all elements apart from platinum, which was described using a Stuttgart–Dresden pseudo-potential.46,47 In the previous work it was found that this results in a reasonably accurate description of transition-metal complexes and their properties,48–52 allowing for semi-quantitative comparison with experiment. In all calculations the bulk solvent was described using PCM,53,54 whereby the standard parameters for dichloromethane as supplied in Gaussian were used.Upon convergence frequencies within the harmonic approximation were calculated to check whether a local minimum was found and for use in comparison with the IR spectra. For all converged structures the UV-Vis spectrum was calculated using TD-DFT for 100 states.
The initial analysis on our calculations was done using the GaussSum programme v. 2.2.5.55
Calculated IR spectra were generated using in-house developed software. We scaled the frequencies by 0.98 for the fingerprint region and by 0.97 for the acetylide region to account for anharmonicity.56 Structures were created using Jmol57 and Povray.58 The overlay in Scheme 2 was created using ROCS.59,60 The images of the orbitals in Fig. S7, S8 and S10 (ESI†) as well as the mapped electrostatic potentials were created using GaussView v. 5.61
Results and discussion
Synthesis and characterisation
The synthesis of the target compound 1, (NDI–phen)Pt(CC–Ph–CH2–PTZ)2 (Scheme 1), was achieved as follows: N-(4-ethynylbenzyl)-phenothiazine (420 mg, 1.34 mmol), CuI (30 mg, 0.16 mmol), (NDI–phen)PtCl2 (3) (145 mg, 0.18 mmol) and diisopropylamine (5 ml, 3.61 g, 35.74 mmol) were sonicated in degassed CH2Cl2 (30 ml) for 3 h. The reaction was monitored by TLC (Al2O3, CH2Cl2). Once the reaction was complete, the solvent was reduced in volume and the product purified by column chromatography (Al2O3, CH2Cl2). A yellow band was collected, reduced in volume, and the product precipitated by the addition of hexane, being subsequently collected by filtration and dried in vacuo. Yield 82 mg, 0.06 mmol (33%). Detailed characterisation and assignment of the NMR spectra using COSY are given in the ESI.†
 | ||
| Scheme 1
Donor–acceptor Pt(II) compounds investigated: top left: (NDI–phen)Pt(CC–Ph–CH2–PTZ)2 (1); top right: ((COOEt)2bpy)Pt(CC–Ph–CH2–PTZ)2 (2); bottom left: (NDI–phen)PtCl2 (3); bottom right: ((COOEt)2bpy)Pt(CC–Ph–C7H15)2 (4).
| ||
Compound 2, ((COOEt)2bpy)Pt(CC–Ph–CH2–PTZ)2 was synthesized using a slightly different synthetic approach than that reported previously.7 Here, N-(4-ethynylbenzyl)-phenothiazine and Pt((COOEt)2bpy)Cl2 were reacted in the presence of CuI and diisopropylamine in degassed CH2Cl2. Full details are given in the ESI.†
Cyclic voltammetry
Cyclic voltammograms (Fig. S3, ESI†) were recorded for the model complex (phen)Pt(CC–Ph–CH3)2 across the potential window −1.95 to +0.90 V (vs. Fc+/Fc). A single irreversible oxidation was observed at +0.73 V which is likely to be localised on the central Pt–acetylide fragments. Similar electrochemical behaviour has been observed previously in related Pt bisacetylide complexes.7,12,19 One fully reversible reduction process observed at −1.75 V is assigned to the reduction of the coordinated phen unit, with the electrochemical reversibility being characteristic of Pt(II) diimine species.12,62
Electronic absorption spectra
The electronic absorption spectra of 1 along with the parent compound 3 and the related complex 2 are shown in Fig. 1. A set of well-structured intense absorption bands observed in the range 330–400 nm for 1 and 3 corresponds to intra-NDI transitions.35 A broad shoulder is also observed for both 1 and 3 at the long-wavelength edge of NDI bands, being positioned at ca. 400 nm for 3. In the case of 1, this shoulder is composed of at least two overlapping broad bands, with the lowest-energy one positioned at ca. 480 nm. The magnitude of the extinction coefficient of this shoulder ε ≈ 3 × 103 M−1 cm−1 is typical for MLL′CT (mixed MLCT/LL′CT) transitions7,9,12,35 and was assigned accordingly. Such an assignment fully corresponds to the calculated absorption spectrum (see below) which predicts that the lowest-energy absorption band is composed of HOMO − 3 to LUMO + 1 and HOMO − 2 to LUMO + 2 transitions. Here LUMO + 1/LUMO + 2 are predominantly localized on phenanthroline entity, while HOMO − 3/HOMO − 2 are localized on Pt–acetylide. In 3, the shoulder at 400 nm is assigned to the MLCT transition.35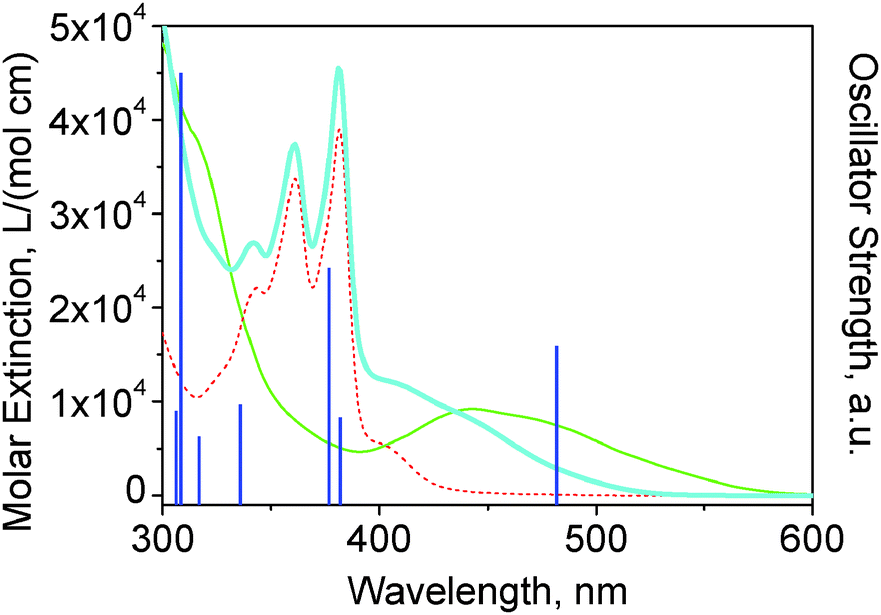 | ||
| Fig. 1 Ground state electronic absorption spectra of 1 (thick solid blue line), 2 (thin solid green line), and 3 (dashed red line) in CH2Cl2 at r.t. Blue vertical bars represent main transitions of the calculated absorption spectrum for 1. | ||
The well-structured intra-NDI absorption bands in the range 330–400 nm for 1 and 3 corresponding to intra-NDI transitions show virtually identical maxima positions as those for the free NDI,35 which indicates very little electronic interaction between the NDI and the diimine–Pt fragments of those molecules. This finding agrees well with the theoretical predictions that terminal nitrogen atoms (through which NDI is linked to the rest of the molecule in 1 and 3) of naphthalimides are not involved into either HOMO or LUMO.63
The absorption spectrum of 2 (Fig. 1) agrees fully with that published previously.7
FTIR spectra
The Fourier transform infra-red absorption spectra of 1 and 3 in the range 1550–1775 cm−1 (Fig. 2) exhibit absorption bands characteristic of NDI symmetric–asymmetric combination of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes.35 When NDI is modified asymmetrically as in 1 and 3, those bands split into two groups, with a shift to higher energy. In the range 1640–1750 cm−1 compound 1 shows four bands at 1670, 1690, 1709 and 1723 cm−1, compound 3 shows NDI ν(CO) bands at 1670, 1688, 1709 and 1723 cm−1. The ν(CO) mode of the bipyridyl-ester ligand in 2 appears at 1732 cm−1. The strong band observed for 1 and 3 at 1582 and 1583 cm−1, respectively, is assigned primarily to ν(CC) of the aromatic rings of NDI.35
O stretching modes.35 When NDI is modified asymmetrically as in 1 and 3, those bands split into two groups, with a shift to higher energy. In the range 1640–1750 cm−1 compound 1 shows four bands at 1670, 1690, 1709 and 1723 cm−1, compound 3 shows NDI ν(CO) bands at 1670, 1688, 1709 and 1723 cm−1. The ν(CO) mode of the bipyridyl-ester ligand in 2 appears at 1732 cm−1. The strong band observed for 1 and 3 at 1582 and 1583 cm−1, respectively, is assigned primarily to ν(CC) of the aromatic rings of NDI.35
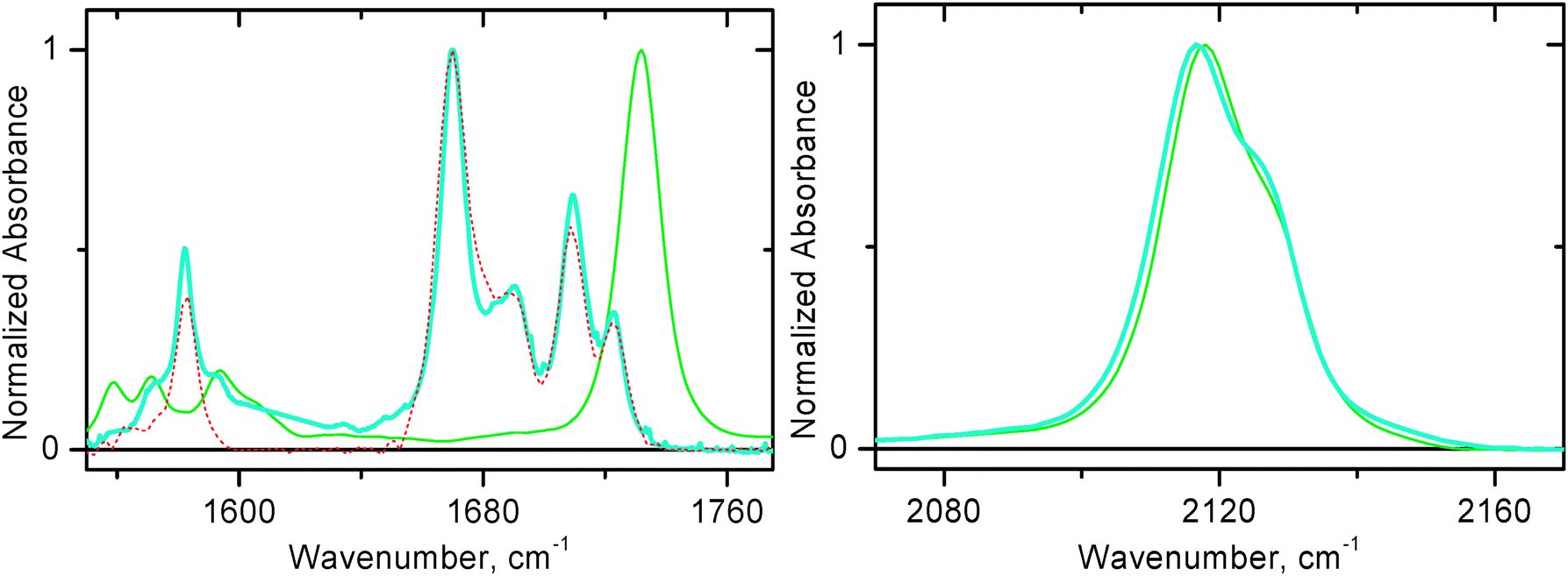 | ||
| Fig. 2 FTIR spectra in CH2Cl2 at r.t. Left panel: 1550–1775 cm−1 region for 1 (thick solid blue line), 2 (thin solid green line), and 3 (dashed red line). Right panel: 2070–2170 cm−1 region for 1 (thick solid blue line) and 2 (thin solid green line). | ||
The high-frequency (2000–2200 cm−1) region of the infra-red spectrum for both compounds 1 and 2 is rather similar (Fig. 2, right panel) and demonstrates symmetric–asymmetric combination of ν(CC) vibration. A maximum is observed at approximately 2117/8 cm−1 with a shoulder shifted 10 cm−1 higher in energy for 1 and 2.
Femtosecond time-resolved IR (TRIR) data
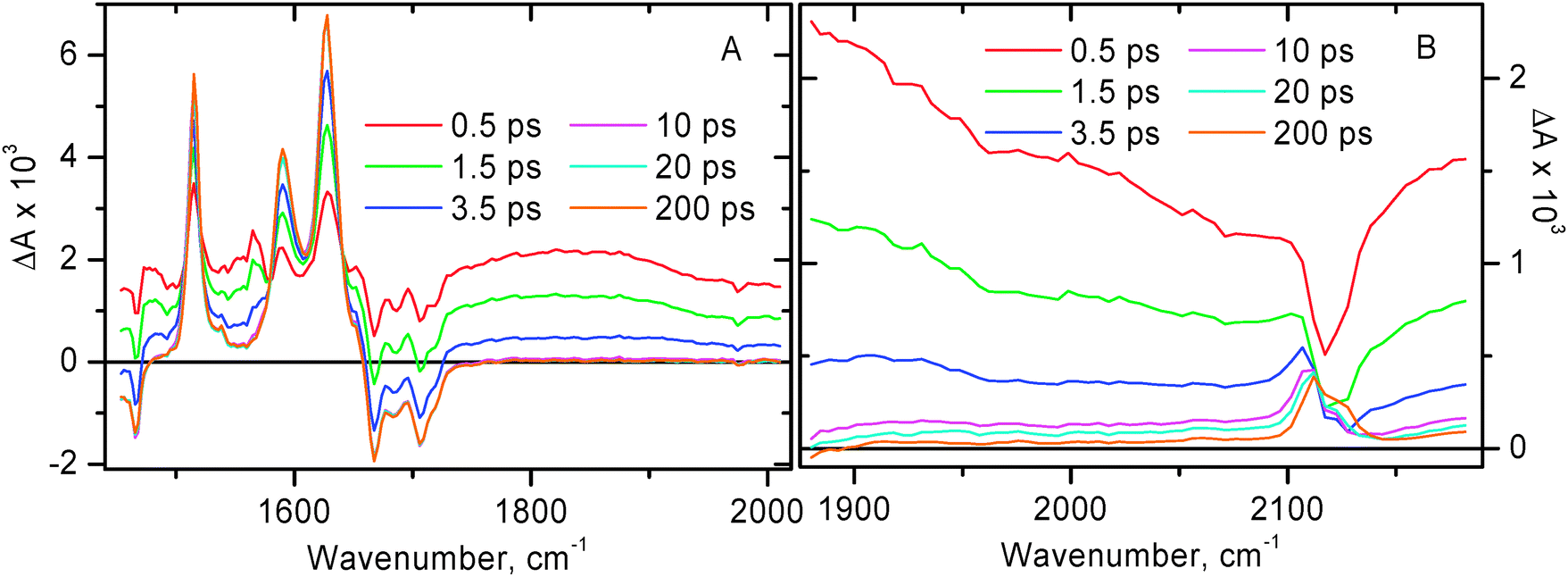
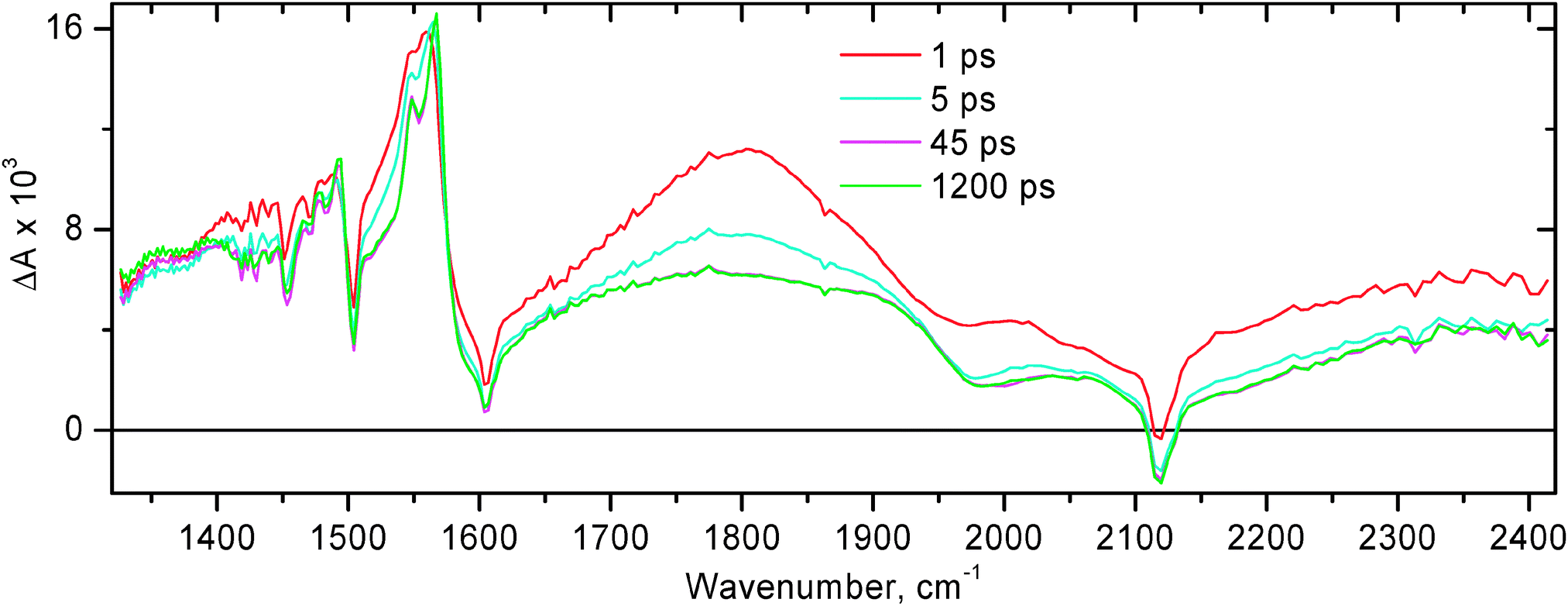
The time-resolved infrared data obtained for compound 1 following excitation with 50 fs, 400 nm laser pulse are shown in Fig. 3 and 4. The extinction coefficient of 1 at 400 nm is ca. 1.2 × 104 M−1 cm−1, whilst the extinction coefficient of free NDI at 400 nm is less than 50 M−1 cm−1 (Fig. 1).35 It follows that upon pulsed laser excitation at 400 nm the initial excited state produced is almost entirely of MLL′CT nature. It has been reported previously that the rate of intersystem crossing (ISC) is less than (1 ps)−1 for the related Pt(II) diimine complex ((tBu)2bpy)Pt(CC–Ph)2.64 Since the MLL′CT excited state in 1 is formed in the immediate Pt(II) coordination environment, we assume a similar rate of ISC in 1. Therefore, the early time transient spectra of 1 are assigned to 3MLL′CT. At 0.5 ps after photoexcitation, the TRIR spectrum is dominated by a broad transient IR absorption signal covering the entire region probed, complemented with ground state bleaches at ca. 1580, 1668, 1687, 1707, 1719, and 2117 cm−1. Sharp transient bands at 1515, 1589, and 1628 cm−1 can already be seen at early times. We believe that the broad absorption in the mid-IR is mainly caused by two reasons: (i) an extremely broad and intense electronic transient absorption band extending into the IR range which is inherent to many diimine–Pt(II)–acetylide systems in the 3MLCT excited state; and (ii) a contribution from a transient ν(CC) band in the 3MLL′CT excited state which is expected to be very intense and be positioned at ca. 1800 cm−1 as suggested by the DFT calculations for (Mes-BIAN)Pt(CC–Ph–CH3)2.24 TD-DFT calculations on the 3MLL′CT state of the chromophoric core of 1 show a strong IR absorption band precisely at this position (see Fig. S12(b), ESI†), confirming our assignment. This hypothesis is supported by the flash photolysis experiments on the related complex ((tBu)2bpy)Pt(CC–Ph)2 which showed intense extremely broad structureless absorption extending far into the IR range beyond the spectral window of the setup (1400 nm).18 The lifetime of this broad structureless absorption in the NIR range matches the 3MLCT excited state lifetime. Similar extremely broad absorption in the mid-IR range was also observed at early times in the TRIR experiments with compound 2 (see below), and in many other diimine–Pt(II)–acetylide systems that we have studied, such as (bpy)Pt(CC–Ph–C7H15)2 and 4 (Fig. 5 and 8, respectively), and in (MesBIAN)Pt(CC–Ph–CH3)2.24
 | ||
| Fig. 3
Time resolved infra-red spectra at selected time delays for 1 in CH2Cl2 after excitation with a 400 nm, 50 fs laser pulse. (A) Spectral data in the region of CO stretch. (B) Spectral data in the region of CC stretch. Note the different intensity scales of panels A & B.
| ||
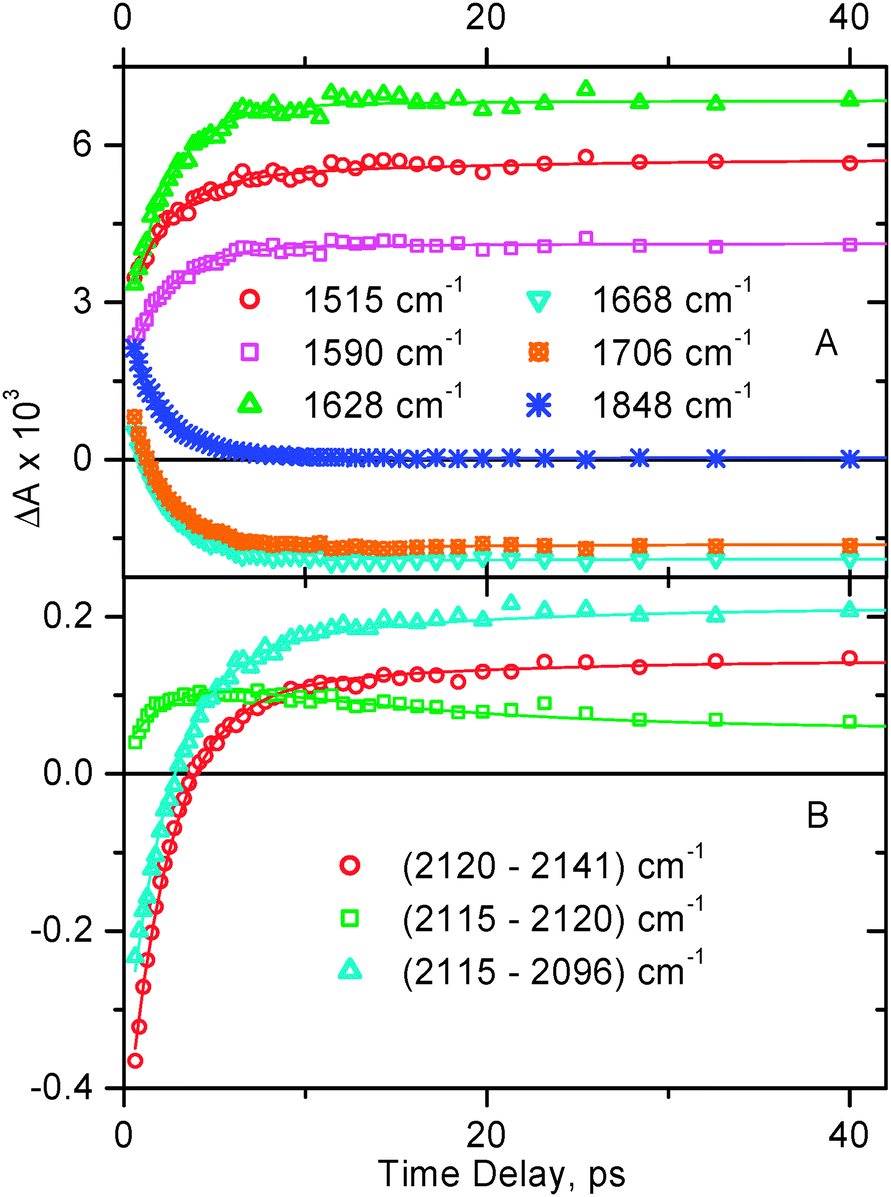 | ||
| Fig. 4
Representative decay traces obtained in time-resolved infrared experiments for 1 in CH2Cl2. Markers show the experimental data, the corresponding fitting curves are shown by solid lines. Panel A: in the spectral window 1450–2000 cm−1. The data were globally fitted with a bi-exponential function (plus a long-lived signal) with lifetimes τ1 = 2.0 ± 0.5 ps and τ2 = 15 ± 3 ps. Panel B: spectral data in the region of ν(CC). The decay traces shown are plotted as a difference between the indicated spectral points to emphasize the charge separation step. The data were globally fitted with a bi-exponential function (plus a long-lived signal) with lifetimes τ1 = 2.3 ± 0.5 ps and τ2 = 14 ± 2 ps.
| ||
 | ||
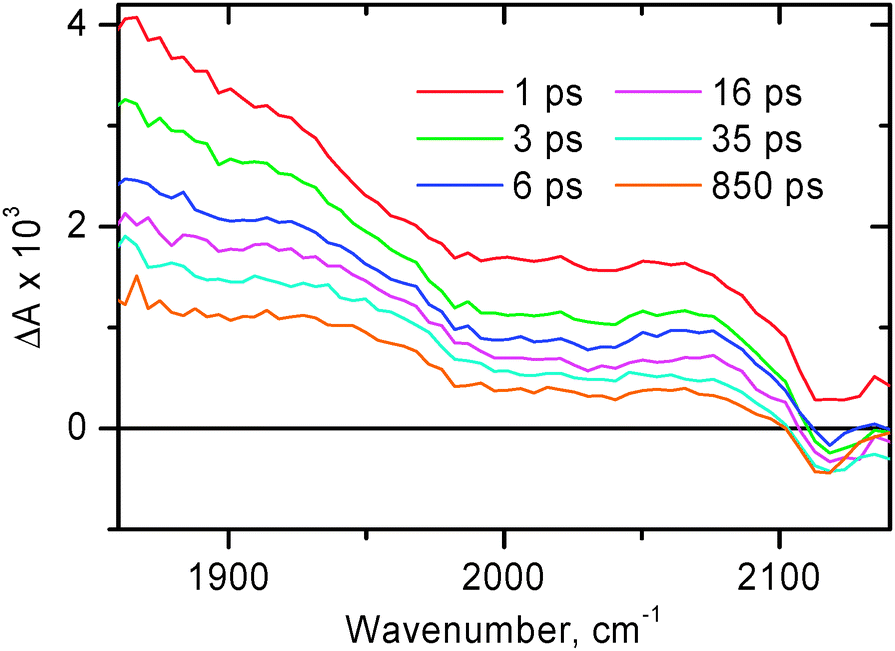
| Fig. 5
Time resolved infra-red spectra at selected time delays for (bpy)Pt(CC–C7H15)2 in CH2Cl2 after excitation with a 400 nm, 50 fs laser pulse. Presented spectra were obtained in separate spectral windows over the range 1300–2450 cm−1 and combined afterwards.
| ||
The narrow transient bands detected for 1 at 1515, 1589, and 1628 cm−1 correspond to the radical anion of naphthalene diimide observed spectroelectrochemically.35 As time elapses, the broad structureless transient absorption decays uniformly across the entire spectral range with a time constant ca. 2 ps, and the narrow transient bands of NDI anions along with NDI ground state bleaches grow in with the same rate (Fig. 4A). The key lifetimes obtained throughout the study for compounds 1 & 2 are summarised in Table 1.
| Exc. state | Compound | |
|---|---|---|
| 1 | 2 | |
| a Averaged over picosecond TA and TRIR data. b From TRIR experiments. c From nanosecond TA experiments; the amplitudes of the two exponentials are equal in value within experimental uncertainty. d Averaged over picosecond TA and TRIR data in the high-frequency region. e From ref. 65. | ||
| 3MLL′CT | 2.3 ± 0.5 psa | 12 ± 2 psd |
| [NDI−–Pt+–PTZ] | 15 ± 3 psb | N.A. |
| [{(COOEt)2bpyPt}−–PTZ+] | N.A. | 2.7 nse |
| [NDI−–Pt–PTZ+] | 36 ± 4 nsc | N.A. |
| 107 ± 11 nsc | ||
The fact that the transient bands inherent to NDI anions are resolved at the very early time after photoexcitation reflects the fast rate of the first step of spectral dynamics, i.e. decay of the 3MLL′CT state and formation of the subsequent charge-separated state involving NDI, i.e. [NDI−–Pt+–PTZ]. It shall be noted that in our TRIR experiments, the earliest accessible time delay is 0.5 ps, the signal at earlier times is severely affected by coherence artefacts.
The rise time of NDI anion bands of ca. 2 ps is interpreted as the time constant for the first step of the forward electron transfer. The first stage of spectral dynamics is complete by 10 ps. By that time, in addition to NDI anion transient features in the 1450–1650 cm−1 range, a clearly resolved transient band is also formed in the high-frequency region of the spectrum at 2110 cm−1. This is assigned to ν(CC) in the initial charge-separated excited state [NDI−–Pt+–PTZ] in which the negative charge resides on the NDI.
Only minor subsequent dynamics is observed in the 1450–1800 cm−1 range on the time scale up to a 2 ns time limit of the setup (Fig. 3A and 4A). However in the high-frequency region, one can see small but noticeable shift of ν(CC) to 2113 cm−1, with a shoulder appearing to the other side of the ground state bleach, i.e. at 2124 cm−1 (Fig. 3B). This is interpreted as the next step in the charge separation process with a positive charge moving from acetylide groups towards stronger electron donating phenothiazines, to form the final charge-separated state [NDI−–Pt–PTZ+]. This step occurs with a time constant of ca. 15 ps. The small magnitude of the shift in ν(CC) indicates that there is a very small difference in electron density on CC between the ground state and the final charge-separated state [NDI−–Pt–PTZ+], as could be anticipated. The fact that the excited state transient absorption and the ground state bleaching signals do not cancel out completely is ascribed to the slightly higher extinction coefficient in the excited state, in agreement with the theoretical calculations (vide infra).
Vibrational cooling is expected to occur on a similar time scale (10–20 ps time constant) as was evidenced in the earlier study on 3,35 and on (MesBIAN)Pt(–CC–Ph–CH3)2.24 Therefore the spectral dynamics on the time scale longer than 10 ps could involve a contribution from the cooling of vibrationally hot state, indistinguishable from the second step of charge separation.
At time scales longer than 40 ps, no spectral dynamics has been observed for 1 in the TRIR data. Thus, upon photoexcitation of 1, an initial MLL′CT state [NDI–phen−–Pt–acetylide+–PTZ] is formed within our instrument resolution, which is followed by two steps of electron transfer to form [NDI−–Pt+–PTZ] (the rate constant is ca. 0.5 × 1012 s−1) and finally [NDI−–Pt–PTZ+] (the rate constant is ca. 6.7 × 1010 s−1).
The TRIR data for compound 2 are summarised in Fig. 6, the corresponding decay traces are shown in Fig. 7. Immediately after photoexcitation the transient signal is dominated by a broad absorption spanning across the whole range, complemented with two ground state bleaches of ν(CO) at 1732 cm−1 and ν(CC) at 2118 cm−1, and a new transient band at 1703 cm−1 due to ν(CO) in the excited state.
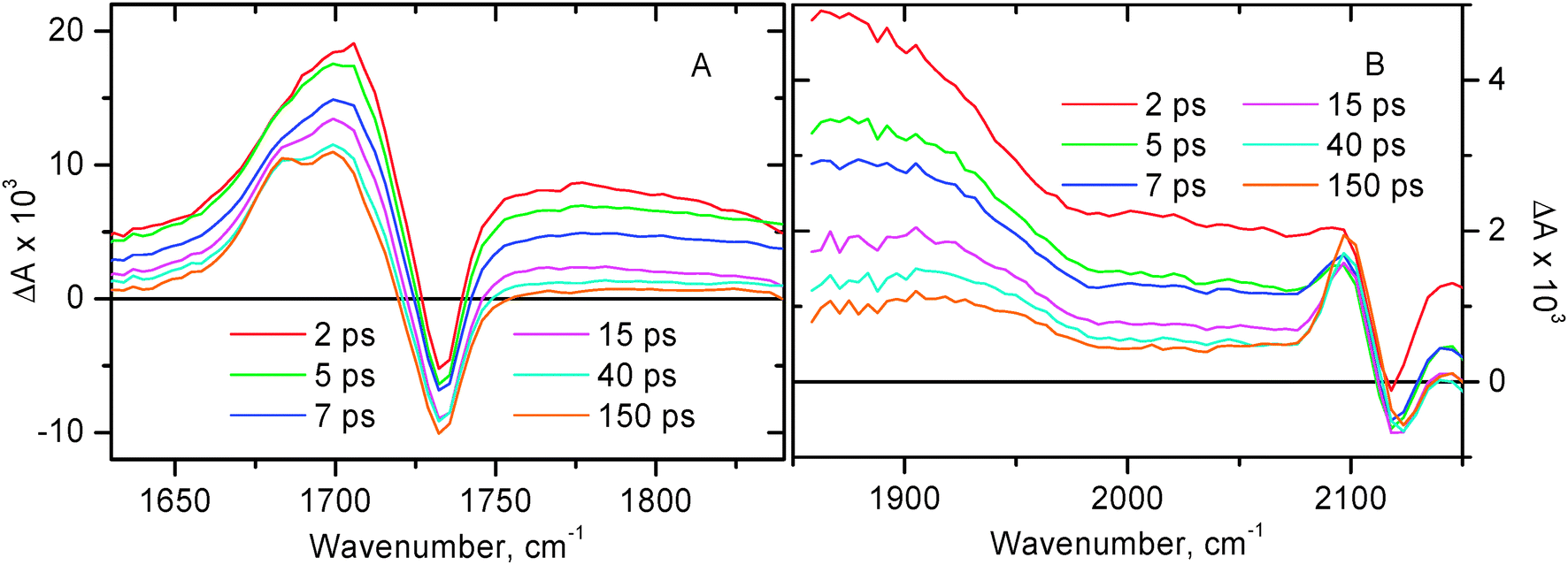 | ||
| Fig. 6
Time-resolved infrared spectra at selected time delays for 2 in CH2Cl2 following excitation with a 400 nm, 150 fs laser pulse. Panel A: spectral data in the region of ν(CO). Panel B: spectral data in the region of ν(CC). Note the different intensity scales of A & B.
| ||
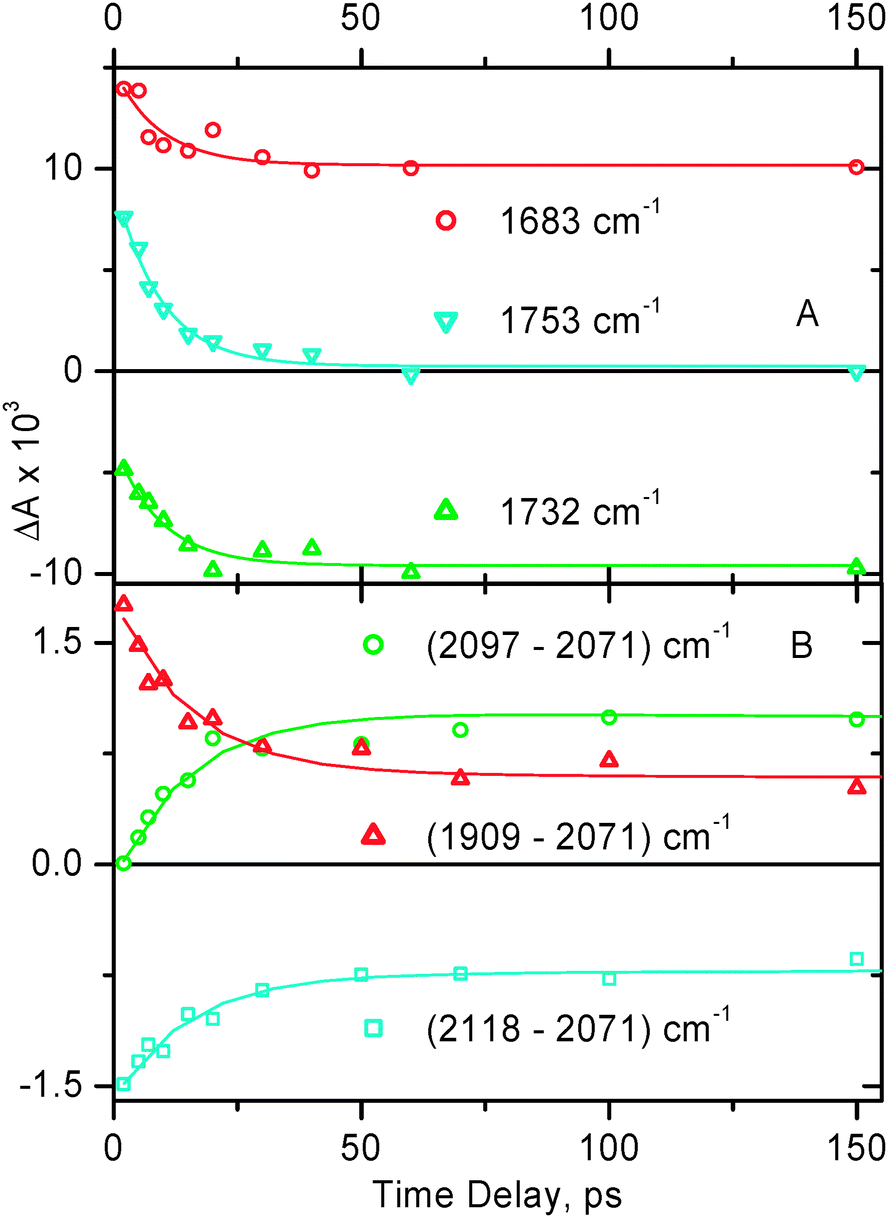 | ||
| Fig. 7
Representative decay traces obtained in time-resolved infrared experiments for 2 in CH2Cl2. Markers show the experimental data, the corresponding fitting curves are shown by solid lines. Panel A: spectral data in the region of CO stretch. The data were fitted with a single-exponential function (plus a long-lived signal) with a lifetime of 9.3 ± 1.0 ps. Panel B: spectral data in the region of ν(CC). The decay traces shown are plotted as a difference between the indicated spectral points to emphasize the charge separation step. The data were fitted with a dual-exponential function with lifetimes τ1 = 15 ± 2 ps and τ2 = 4.8 ± 2.0 ns.
| ||
 | ||
| Fig. 8
Time-resolved infrared spectra at selected time delays for 4 in CH2Cl2 following excitation with a 400 nm, 150 fs laser pulse in the region of CC stretch.
| ||
The broad structureless transient absorption spanning across the whole spectral range at early times resembles those seen for 1 and some other model compounds in the 3MLCT excited state (see above), and accordingly is assigned to the 3MLCT excited state of diimine–Pt(II)–acetylide nature. The broad absorption band decays considerably by 50 ps, after that the remaining fraction of this broad signal persists up to the time limit of the setup (2 ns).
The transient band at 1703 cm−1 observed for 2 (Fig. 6A) corresponds to ν(CO) in the vibrationally hot MLCT excited state. By 50 ps time delay narrowing and splitting of this band into two maxima positioned at ca. 1685 and 1699 cm−1 are observed. There are no further significant spectral changes in the ν(CO) region up to 2 ns. Overall, the spectral dynamics in the ν(CO) region can be well described by concomitant electronic evolution and vibrational cooling. The average time constant obtained by the mono-exponential fit in the ν(CO) region is 9.3 ± 1.0 ps.
To adequately fit the spectral dynamics in the region 1850–2150 cm−1, two decay times were required, τ1 = 15 ± 2 ps and τ2 = 4.8 ± 2.0 ns. The faster decay time describes both the decay of a broad initial transient absorption and structuring of the new transient band at 2097 cm−1 which corresponds to ν(CC) of the excited molecule in the charge-separated state [((COOEt)2bpy)−–Pt–PTZ+], when the positive charge is transferred from acetylide groups towards stronger electron donating phenothiazines. The slower decay component corresponds to the charge recombination with return to the ground state. Given the limited time range accessible in the TRIR setup (up to 2 ns), the value of 4.8 ± 2.0 ns cannot be considered as accurate, however it is in reasonable agreement with 1.4 ns or 2.7 ns obtained in ref. 7 and in ref. 65, respectively, in electronic transient absorption experiments.
As is evidenced by the new ν(CC) transient band at 2097 cm−1 which is formed by 50 ps, the nature of the excited state has changed considerably by that time and is different from the original diimine–Pt(II)–acetylide 3MLL′CT state. The reference compound 4 was investigated in order to evaluate the origin of the broad transient (Fig. 8). For compound 4 no formation of a narrow transient ν(CC) band near 2100 cm−1 has been observed, the broad transient absorption dominates the acetylide region up to the latest accessible time delays. With the only difference between compounds 2 and 4 being the presence of two phenothiazine groups attached to the acetylides in 2, we consider the described behaviour to be an indication that in compound 2 there is an additional step of charge separation reaction, i.e. hole transfer from acetylide groups onto stronger electron-donating phenothiazines, forming a full CS state.
Electronic transient absorption (TA)
Transient absorption spectra for compound 1 in the visible range, following excitation with a 395 nm, 150 fs laser pulse, are shown in Fig. 9A. The representative transient absorption decay traces are shown in Fig. 9C. The laser excitation at 395 nm promotes the sample exclusively into the MLL′CT excited state as the ratio of extinction coefficients for 1vs. that for parent free NDI is >50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.35 Compound 1 shows rather complex spectral evolution at early times, the observed spectral dynamics being convolved with the chirp of a probe pulse. To obtain a chirp-free shape of an early-time spectrum, and to obtain the true transient spectra of the excited states involved, global fitting was performed throughout the entire spectral range. This enabled us to obtain more reliable lifetimes and to reconstruct the transient spectra assuming a consecutive reaction scheme (Fig. 9B). The initial excited state transforms into the product state with a time constant of 2.5 ± 0.3 ps. After the product excited state is formed, there is only negligible decay if any on the set-up limited time scale of 1.6 ns. The spectrum of the initial excited 3MLL′CT state is rather flat in the range 475–625 nm. The transient spectrum of a product excited state (Fig. 9A at 300 ps time delay, and Fig. 9B, circles) shows a main peak at 478 nm, and a minor peak at 606 nm, the features characteristic of the NDI anion.33–35 Thus, the picosecond TA data confirm population of a charge separated excited state in which the negative charge resides on the NDI fragment, with the formation time constant of 2.5 ± 0.3 ps (very similar to 2.0–2.3 ps obtained in TRIR). Notably, the PTZ radical-cation is not pronounced in the transient absorption spectra due to its extinction coefficient being significantly smaller than that of the radical anion of NDI.66
:1.35 Compound 1 shows rather complex spectral evolution at early times, the observed spectral dynamics being convolved with the chirp of a probe pulse. To obtain a chirp-free shape of an early-time spectrum, and to obtain the true transient spectra of the excited states involved, global fitting was performed throughout the entire spectral range. This enabled us to obtain more reliable lifetimes and to reconstruct the transient spectra assuming a consecutive reaction scheme (Fig. 9B). The initial excited state transforms into the product state with a time constant of 2.5 ± 0.3 ps. After the product excited state is formed, there is only negligible decay if any on the set-up limited time scale of 1.6 ns. The spectrum of the initial excited 3MLL′CT state is rather flat in the range 475–625 nm. The transient spectrum of a product excited state (Fig. 9A at 300 ps time delay, and Fig. 9B, circles) shows a main peak at 478 nm, and a minor peak at 606 nm, the features characteristic of the NDI anion.33–35 Thus, the picosecond TA data confirm population of a charge separated excited state in which the negative charge resides on the NDI fragment, with the formation time constant of 2.5 ± 0.3 ps (very similar to 2.0–2.3 ps obtained in TRIR). Notably, the PTZ radical-cation is not pronounced in the transient absorption spectra due to its extinction coefficient being significantly smaller than that of the radical anion of NDI.66
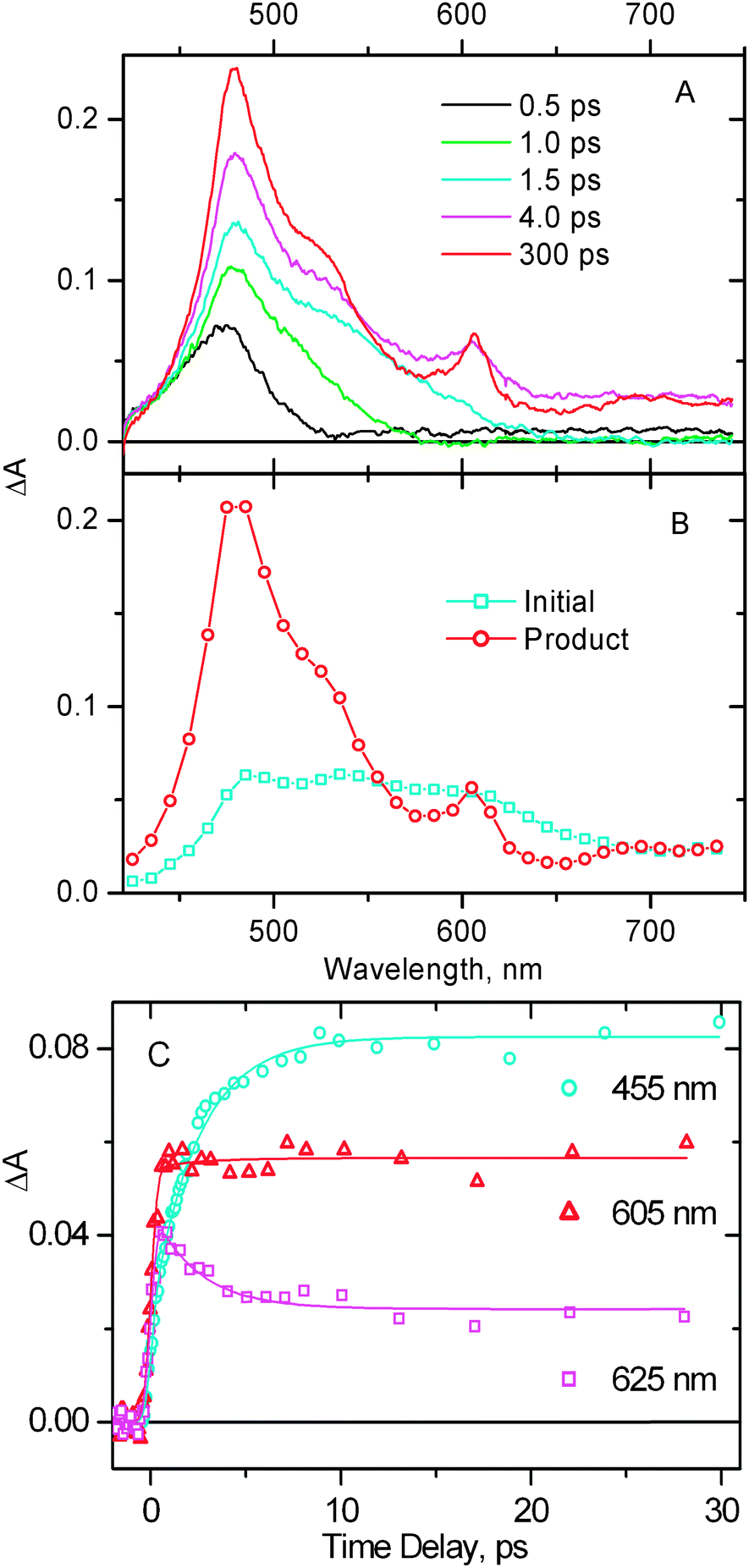 | ||
Fig. 9
(A) Representative transient absorption spectra at selected time delays obtained for 1 in CH2Cl2, following excitation with a 395 nm, 150 fs laser pulse. The observed spectral dynamics is convolved with the chirp of a probe pulse, which was corrected by the data analysis with the results shown on panel B. (B) Reconstructed transient spectra of the two excited states involved into the spectral evolution, assuming a consecutive reaction scheme “A → B”. ( ) The initial excited state populated immediately after the photoexcitation, ( ) The initial excited state populated immediately after the photoexcitation, ( ) the successor excited state populated from the former one. (C) Representative transient absorption decay traces. Markers show the experimental data, the corresponding fitting curves are shown by solid lines. The lifetime obtained in single-exponential (plus long-lived component) global fit is 2.5 ± 0.3 ps. ) the successor excited state populated from the former one. (C) Representative transient absorption decay traces. Markers show the experimental data, the corresponding fitting curves are shown by solid lines. The lifetime obtained in single-exponential (plus long-lived component) global fit is 2.5 ± 0.3 ps.
| ||
As was mentioned above in the TRIR section, the time constant for the final charge separation in compound 2 is ca. 12 ps, which is the mean value over the two spectral windows. This number matches well the rate of the corresponding process in compound 1, however considerably differs from the value of 80 ± 20 ps obtained for the same compound 2 in tetrahydrofuran by electronic transient absorption.65 A plausible explanation for such a difference would be the solvent effect. One would expect the free energy for charge separation to be greater in more polar CH2Cl2vs. that in tetrahydrofuran, accounting for faster charge separation. To test this idea, we performed electronic transient absorption experiments with compound 2 in CH2Cl2. The representative transient absorption spectra along with the decay traces obtained for 2 on the time scale up to 60 ps are shown in Fig. 10.
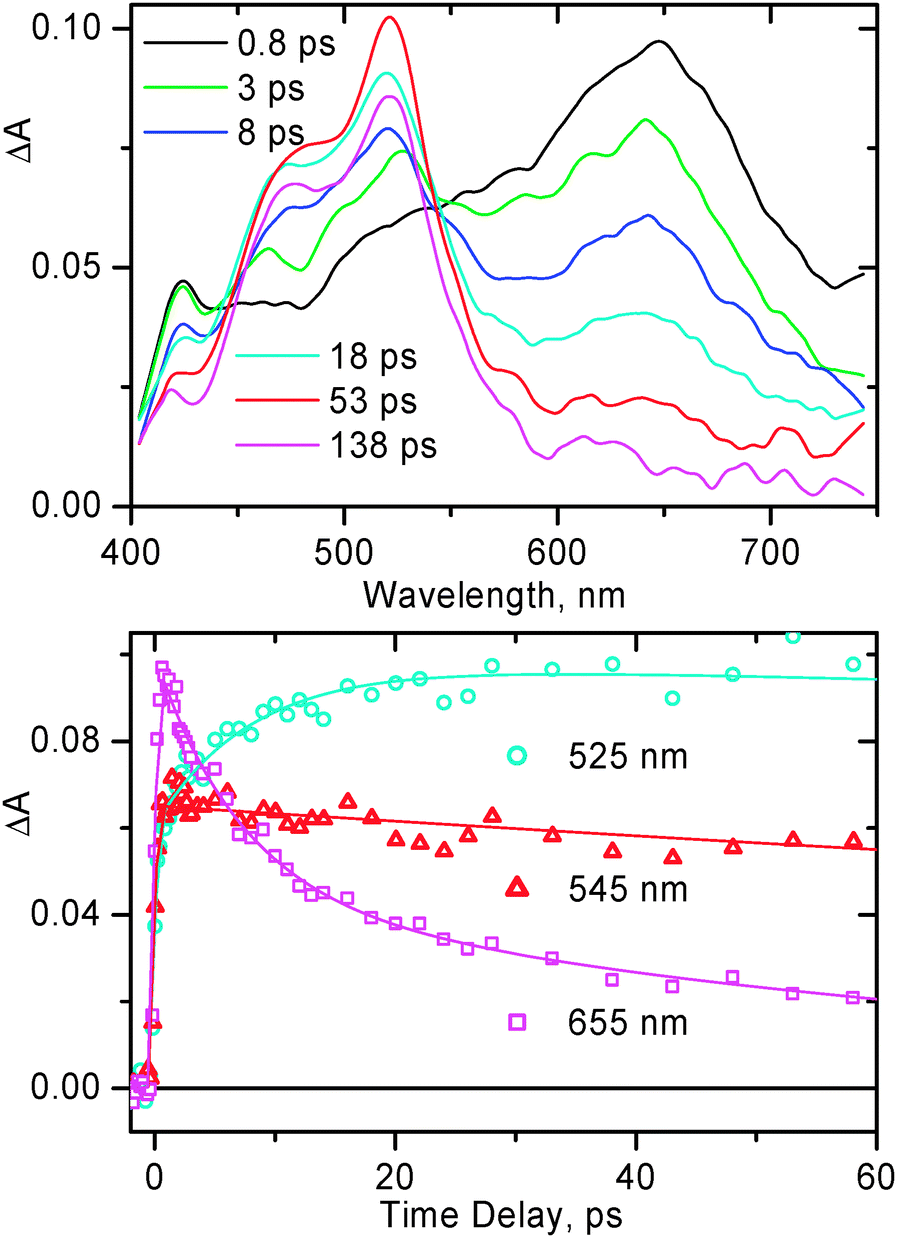 | ||
| Fig. 10 Representative transient absorption spectra (top) and selected decay traces (bottom) obtained for 2 in CH2Cl2, following excitation with a 395 nm, 150 fs laser pulse. For kinetic traces, markers show the experimental data, the corresponding fitting curves are shown by solid lines. The rise of the signal of PTZ anions at 525 nm and the main decay component of the 3MLL′CT band at 655 nm occur with an identical lifetime of 8 ± 2 ps. | ||
Immediately after photoexcitation, the transient spectrum of 2 features the broad absorption band with a maximum at ca. 650 nm, similar to that observed previously65 and is typical for bpy–Pt(II)–acetylide in the 3MLCT excited state. This band decays on the picosecond time scale giving rise to another band at ca. 520 nm, characteristic of the PTZ radical cation.67–71 The spectral evolution observed in CH2Cl2 is similar to what was observed in tetrahydrofuran.65 As obtained in the TA experiments of current study for 2 in CH2Cl2, the grow-in of the PTZ anion band at 520 nm and main decay of the 3MLCT band at 650 nm occur with an identical lifetime of 8 ± 2 ps (Fig. 10). This value is significantly smaller than that obtained in tetrahydrofuran65 but matches reasonably well the average 12 ± 2 ps observed in the TRIR experiments. This finding emphasizes the important role of the solvent in the charge separation process in compound 2.
To obtain the decay time of the final charge separated state in 1, the nanosecond flash photolysis setup with ca. 2 ns time resolution has been used (see Fig. S6 (ESI†) for decay kinetics). The decay of the final charge separated state for 1 occurs on the sub-microsecond time scale, and is not mono-exponential, however can be approximated as double-exponential with the two lifetimes being 36 ± 4 ns and 107 ± 11 ns. The same values were obtained when the experiments were performed at different concentrations and laser excitation energies, confirming that the two lifetimes are intrinsic for the decay of the final charge separated state in 1.
DFT calculations
DFT calculations were performed on 1 using the procedure described in the Experimental section, with the resultant structure given in Part S3.1 in the ESI.† We note that the NDI unit is orthogonal to the phenanthroline unit. Moreover, the PTZ units are orthogonal to the phenyl groups. Since it is appreciated that torsional freedom may lead to a number of different local minima, the NDI orientation was investigated by starting optimizations of the full complex in a number of different orientations. All these relaxed back to the structure given in Part S3.1 (ESI†).The PTZ units lie far apart and therefore do not interact at all (N–N distance 15.5 Å and 15.6 Å for the singlet and triplet states, respectively). However, here also other conformations may be energetically accessible, given the torsional degrees-of-freedom. This was investigated in two ways. First, a torsional scan was performed around the phenyl–CH2 bond, focusing only on the acetylide–phenyl–PTZ unit for reasons of computational efficiency. It is clear from those calculations that the barrier to rotation around this bond (approximately 17 kJ mol−1) means that rotation at room temperature is not feasible. However, rotation around the Pt–acetylide bond is most likely facile, leading to a number of alternative conformations, of which representative examples (for both singlet and triplet states) are shown in the ESI.†
This alternative conformation (Fig. S4, ESI†) has approximately the same singlet and triplet energies as the conformation in Part S3.1 (ESI†) (differences <500 J mol−1), although the relative orientation of the PTZ units is different. However, the IR spectra calculated for both singlet and triplet states are indistinguishable from the ones calculated for the first conformation. Therefore, the remainder of the discussion will be based on the conformation of Part S3.1 (ESI†). It needs to be noted that the conformation with both PTZ units pointing towards each other was not investigated. However, even in that case the closest approach between the two units is around 10 Å, and therefore again this conformation is expected to have similar singlet and triplet energies and spectra to the conformations already discussed.
The calculated electronic absorption spectra for both singlet and triplet states (Fig. S5 and S9, ESI†) match the experimental data well. It should be noted that the vibrational progression in the electronic absorption spectrum of the ground state cannot be reproduced in our calculations.
For the singlet ground state, the calculations (see ESI† and Fig. 1) show that there are three major groups of transitions, at similar wavelengths as the experimental spectra in Fig. 1.
The orbital breakdown for the strongest transitions in the singlet manifold is given in Table 2 (for more complete breakdown, see ESI†). The corresponding frontier orbitals given in Fig. S7 and S8 (ESI†) support the assignment of the lowest-energy transition, experimentally observed near 480 nm, as an MLL′CT transition. The calculated transition around 380 nm is largely based on NDI, however there is a component to this peak (namely from HOMO − 16 to LUMO), which is a ligand-to-ligand charge transfer transition mainly between an orbital delocalized over phenanthroline–Pt–acetylide, and NDI, with some noticeable contribution from a PTZ-to-NDI transition. Finally, the band at ca. 300 nm is a transition largely from the acetylide to the PTZ ligands.
| Energy (cm−1) | Wavelength (nm) | f | Major contributions | Origin of transition |
|---|---|---|---|---|
| a Calculated oscillator strength. | ||||
| 20754 |
481.83 | 0.23 | H − 3 → L + 1 (47%), H − 2 → L + 2 (52%) | Pt–acetylide → phenanthroline |
| 26184 |
381.91 | 0.12 | H − 16 → LUMO (13%), H − 15 → LUMO (12%), H − 14 → LUMO (14%), H − 13 → LUMO (12%), H − 9 → LUMO (25%) | Phenanthroline–Pt–acetylide–PTZ → NDI; (π,π*) NDI; acetylide–PTZ → NDI |
| 26552 |
376.62 | 0.35 | H − 21 → LUMO (14%), H − 16 → LUMO (19%), H − 15 → LUMO (55%) | Pt–acetylide–PTZ → NDI; (π,π*) NDI |
| 29778 |
335.82 | 0.14 | H − 2 → L + 5 (95%) | Pt–acetylide → phenanthroline |
| 31563 |
316.83 | 0.09 | H − 16 → L + 1 (28%), H − 3 → L + 5 (21%) | Phenanthroline–Pt–acetylide–PTZ → phenanthroline; Pt–acetylide → phenanthroline |
| 32441 |
308.25 | 0.65 | H − 2 → L + 7 (84%) | Pt–acetylide → phenanthroline–Pt–acetylide |
| 32681 |
305.99 | 0.13 | HOMO → L + 14 (31%), HOMO → L + 15 (47%) | (π,π*) PTZ |
| 34121 |
293.08 | 0.11 | H − 16 → L + 1 (10%), H − 15 → L + 2 (16%), H − 14 → L + 2 (17%), H − 11 →L + 2 (17%) | Phenanthroline–Pt–acetylide–PTZ → phenanthroline; NDI → phenanthroline |
| 34956 |
286.08 | 0.15 | H − 11 → L + 2 (12%), H − 2 → L + 8 (31%), H − 2 → L + 18 (16%) | Pt–acetylide → Pt–acetylide–PTZ; Pt–acetylide → phenanthroline–Pt–acetylide |
The agreement between the calculated electronic absorption spectrum for the lowest triplet state (see ESI†) and the experimental spectrum (Fig. 9A, 300 ps trace) is also very good. The spectrum is dominated by a single peak at around 450 nm, which is dominated by a combination of intra-ligand CT transitions on both the α- and β-manifolds (see ESI†).
The simulated IR spectra for the singlet and triplet states as well as the TRIR spectra are given in Fig. 11. The agreement with the FTIR spectrum in Fig. 2 above is excellent with all major features accounted for, both in the acetylide region and in the fingerprint region. The two regions are treated with a slightly different scaling factor, which is appropriate, given that the two regions cover different types of vibration. If the calculated TRIR spectrum (green line in Fig. 11) is compared against the longest time-delay TRIR spectrum in Fig. 3, then again the excellent agreement is noted. It is clear from Fig. 11 that the intensity of the IR spectrum increases upon excitation to the lowest triplet state, in accordance with experimental data. When the electrostatic potential is mapped on top of a density isosurface (density value = 0.004) it is clear that this triplet state is a charge-separated state. Comparison of Fig. 12(a) (S0) with Fig. 12(b) and (c) (T1 and T2, respectively) clearly shows the charge redistribution upon excitation into the lowest triplet states. Our results predict the existence of two sub-sets of molecule 1 in the final charge-separated state (with the hole residing either on one or the other PTZ group) differing by 1.118 kJ mol−1 in energy. The lowest energy is obtained for the variety shown in Fig. 12(b), with the smaller donor–acceptor separation.
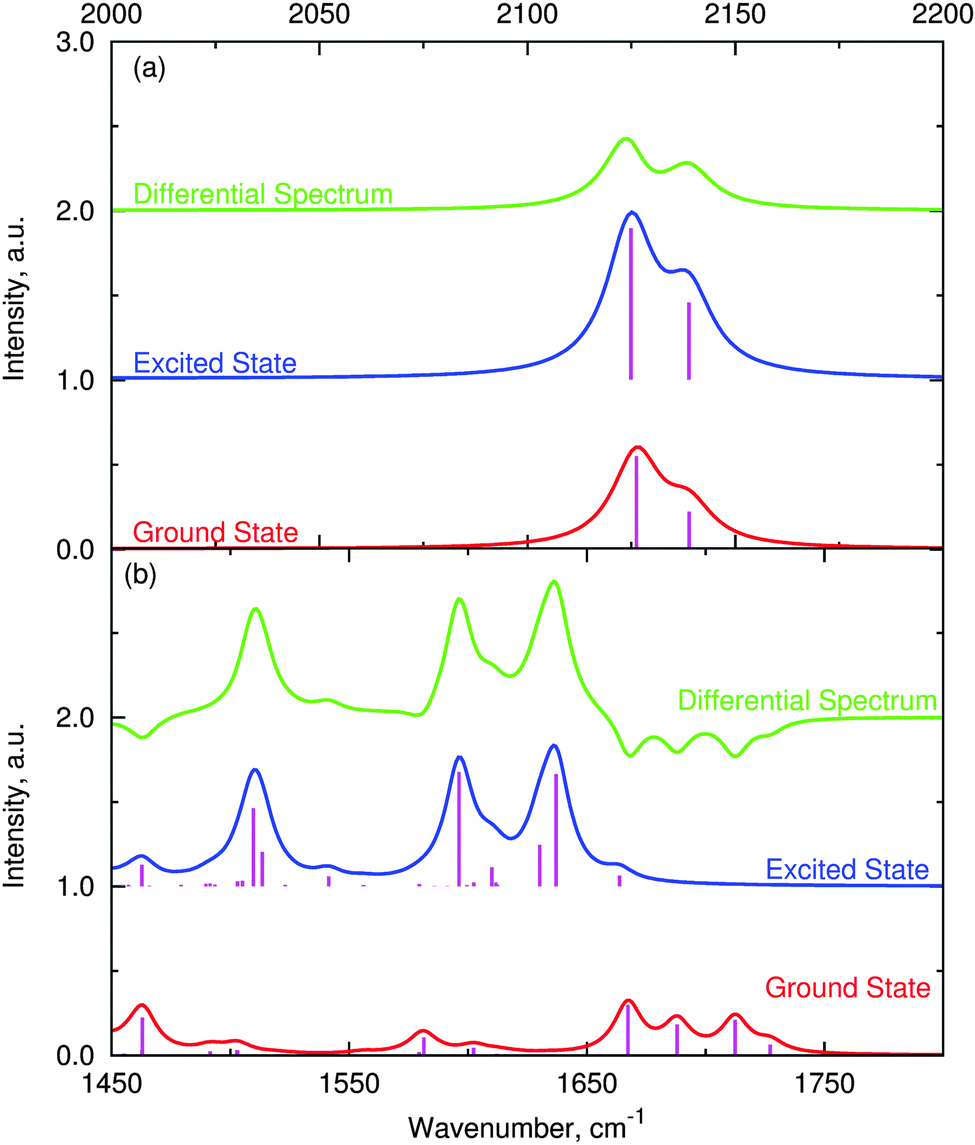 | ||
| Fig. 11 Calculated IR spectra of ground singlet and lowest triplet states of 1, including the TRIR spectrum. Panel (a) shows the acetylide stretch region (scaling 0.97). Intensity scaled by 500 for the ease of presentation. Panel (b): fingerprint region between 1450 and 1800 cm−1 (scaling 0.98). Intensity scaled by 3000 for the ease of presentation. | ||
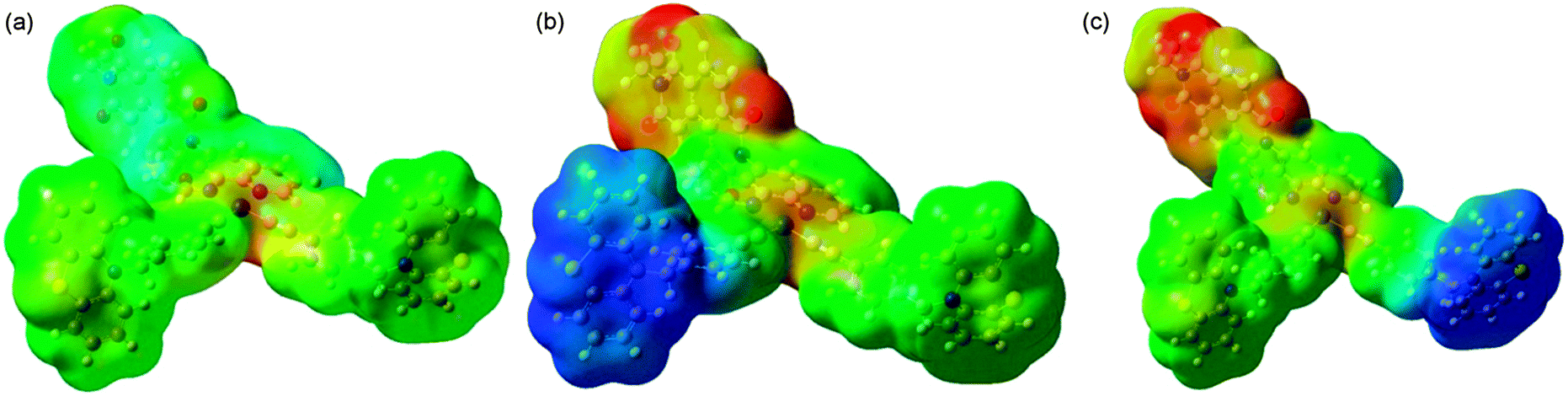 | ||
| Fig. 12 Electrostatic potential map for the ground singlet [panel (a)] and lowest triplet state with the hole localised either on one PTZ group [panel (b)] or the other PTZ group [panel (c)]. The configuration shown in (b) is 1.118 kJ mol−1 lower in energy than the one in (c). Colours correspond to charges with red being −0.1 and blue being +0.1. | ||
Analysis of the two lowest triplet states (Scheme 2, right) shows that the structural differences are very minor and localized on the PTZ units; the formation of a hole on a PTZ unit causes a flattening of the central ring of the PTZ unit.
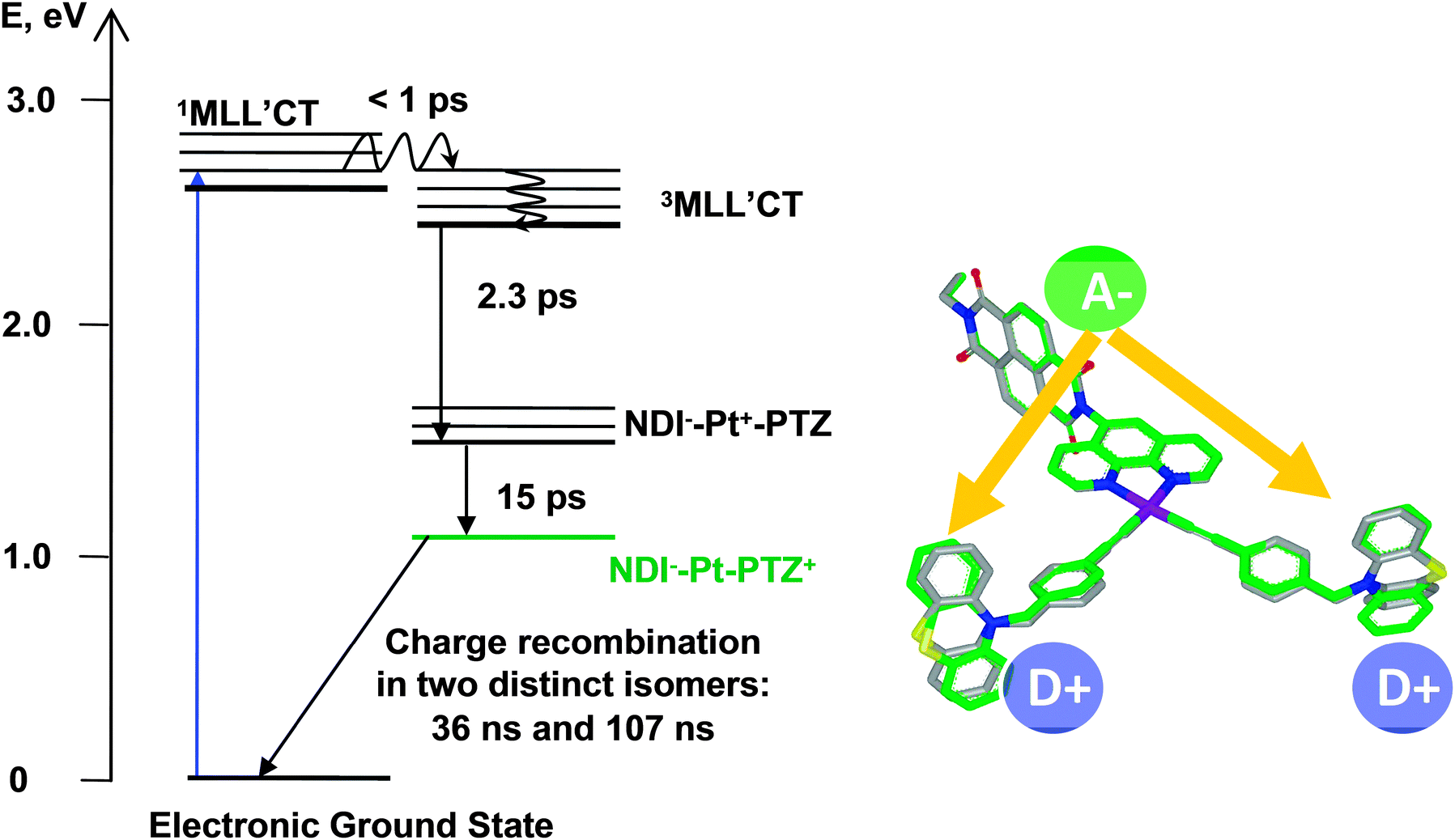 | ||
| Scheme 2 Photophysical diagram of 1 in CH2Cl2, following excitation with ca. 400 nm, 50–150 fs laser pulse. Shown on the right-hand side is the overlay of the T1 (grey bonds) and T2 (green bonds) triplet states in 1. | ||
Discussion
The diagram summarising the excited state dynamics of compound 1 is shown in Scheme 2. The energy of 1MLL′CT state was estimated to be 2.6 eV from the electronic absorption spectrum of 1 (Fig. 1) using the 480 nm position of the lowest-energy component of the broad MLL′CT absorption band. The upper limit for 3MLL′CT energy for 1 was assumed to be ca. 2.5 eV by analogy to the 3MLL′CT energy level in the closest analogue of 1, i.e. (phen)Pt(CC–C6H5)2; the value was obtained from the emission peak position at 77 K.12
For the charge-separated states involving NDI as an electron acceptor and phen–Pt–acetylide or PTZ as electron donors, energy levels of those with respect to the ground state were estimated using a simplified Rehm–Weller equation:
| ECS = E+/01/2 − E0/−1/2 − (Δe)2/(4πε0εr) | (1) |
The first reduction potential for 1 is assumed to be very similar to that of 3, for which reduction is localised on NDI with the value of −0.94 V (NDI/NDI−˙) in CH2Cl2vs. Fc+/Fc.35 The oxidation of phen–Pt–acetylide fragment in 1 is assumed to be similar to that of related (phen)Pt(CC–Ph–CH3)2 for which the value of +0.73 V vs. Fc+/Fc in CH2Cl2 is obtained here. The separation between reduced NDI and oxidised phen–Pt–acetylide is considered to be 12 Å, an average center-to-center distance between NDI and CC obtained in DFT calculations. Hence, the energy of the charge-separated state composed of reduced NDI and oxidised phen–Pt–acetylide in CH2Cl2 can be estimated to be ca. 1.5 eV.
The energy of the final charge-separated state, [NDI−–Pt–PTZ+], could be also estimated from eqn (1) from the electrochemical reduction potential of NDI and the oxidation potential of the PTZ, respectively. The former value has been quoted above; while the latter one was reported to be +0.32 V for PTZ in CH2Cl2vs. Fc+/Fc.7,73 The donor–acceptor separation for 1 is 17.1 Å and 20.1 Å (center-to-center) for each of the two PTZ groups as suggested by the optimised molecular structure in the ground state, the average value over the two PTZ groups being ca. 18.6 Å. Therefore, considering the full transfer of one electron, one obtains from eqn (1) the estimated energy value of ca. 1.2 eV for the final charge separated state in 1.
For the charge recombination process, i.e. the decay of the final charge-separated state back to the ground state, the free energy change equals the energy of that charge-separated state, i.e. −ΔG0 = ECS = 1.2 eV. The superexchange route for the back electron transfer in 1 should be constrained by the presence of the saturated linker between the PTZ group and phenyl–acetylide. Lack of spatial overlap between the orbitals of the donor and the acceptor directly with the Pt(II) atom may diminish the heavy atom effect, and therefore T–S conversion in the course of charge recombination will be considerably slower than that observed in the MLCT states when participating orbitals directly involve a heavy atom.74,75
The bi-exponential decay law observed for the final charge separated state in 1 provides an unusual insight into the mechanism of back electron transfer in this molecule which contains two electron-donating PTZ groups. The through-bond distance between the NDI subunit and each of the two PTZ groups is virtually identical. Therefore, if the charge recombination was dictated exclusively by the superexchange mechanism, this would result in the same rate of back electron transfer to both PTZ groups, which is certainly not the case. However, if the through-space distance of charge recombination is considered, the difference in the distance between the electron-accepting NDI subunit and each of the phenothiazine groups may be of importance. The results of DFT calculations suggest that the through-space separation from NDI to the two PTZ groups in 1 is 14.7 Å and 15.5 Å (edge-to-edge, as seen from the optimized molecular structure in the lowest triplet state).
The rate of through-space electron tunnelling kET depends on the direct overlap of electronic wavefunctions of both donor and acceptor, and is expected to decrease exponentially with distance (eqn (2)):
| kET = k0·exp[−β(r − r0)] | (2) |
Therefore, the difference in donor–acceptor separation for the two PTZ groups would lead to two distinct charge recombination rates. That is exactly what was observed in the nanosecond transient absorption experiments for 1, which yielded τ1 = 36 ± 4 ns and τ2 = 107 ± 11 ns. The donor–acceptor separation in 1 is somewhat too large for an efficient overlap, however there are some examples in the literature demonstrating electron tunnelling on the scales up to 25 Å from excited triplet states in rigid media, the system studied possessed comparable driving force (1–2 eV).77 The values of β obtained for a number of different charge-transfer systems generally fall in the range 0.8–2 Å−1.76,78 Given the two time constants of 36 ns and 107 ns for the charge recombination in 1, and the edge-to-edge through space separations of 14.7 Å and 15.5 Å between NDI and either of the two PTZ groups in the final charge-separated state, eqn (2) gives an estimate of β = 1.36 Å−1, which fits well with the results of other studies. Therefore we conclude that the charge recombination in 1 occurs mainly through the electron tunnelling mechanism.
The DFT calculations support the hole localization on either of the two PTZ groups, but only on a single group at a time. The population ratio between those was experimentally estimated to be ca. 1:1 as suggested by the relative amplitudes of the two decay components for the back electron transfer. As the DFT calculations also predict, the two isomers for 1 in the final charge-separated state are different in energy by 1.118 kJ mol−1, which means a 61:39 population ratio at room temperature, agreeing well with the 1:1 ratio observed experimentally.
Conclusions
The present paper reports a time-resolved spectroscopic study of excited state dynamics in transition metal complexes 1, 2 and 4 developed for photoinduced charge separation. Complex 1 involves a 1,4,5,8-naphthalene diimide (NDI) subunit as an ultimate electron acceptor and two phenothiazine (PTZ) groups as electron donors bridged by a [phenanthroline–Pt(II)–(CC)2] chromophore. Ground state electronic absorption, time-resolved infrared (TRIR) and electronic transient absorption (TA) spectroscopy provide evidence that the initial excited state of 1 observed after photoexcitation is of 3MLL′CT diimine–Pt(II)–acetylide character. Following that, the subsequent step of charge separation occurs with a time constant of 2.3 ps in which the negative charge migrates from the [phen–Pt] subunit onto the NDI subunit to form NDI−–phen–[Pt–(CC)2]+–PTZ2. This transition is characterised by appearance of the prominent NDI-features in both TRIR and TA spectra. The final step of charge separation in 1 proceeds with a time constant of 15 ps during which the hole migrates from the [Pt–(CC)2] subunit to one of the PTZ groups. This step occurs with the rate similar to that for NDI-free analogue 2, as measured by TA and TRIR methods. This transition completes the formation of a long-lived charge separated state in 1 with positive and negative charges residing on the termini of the molecule. There are two isomers of 1 in the final charge-separated state with distinct localization of the positive charge on one of the two PTZ groups. This behaviour is manifested by the bi-exponential character of the charge recombination with the lifetimes of 36 ns and 107 ns. It is also supported by the DFT calculations, which predict the two isomers separated by ca. 1 kJ mol−1 in energy, implying approx. a 60:40 population ratio between the two isomers at room temperature. The collected evidence indicates that the main mechanism of charge recombination in 1 is electron tunnelling. The fact that the two isomers in the final charge-separated state of 1 are distinguished in the experiment is quite rare.
Acknowledgements
We thank the EPSRC and the University of Sheffield for support, and STFC for TRIR beam time at RAL (programme access). S.A.T. and O.V.B. thank the Belarusian Republican Foundation for Fundamental Research for the funding (Project F 13V-011). All calculations were performed on the ‘Jupiter’ cluster of the Theoretical Chemistry Group as well as on the central ‘Iceberg’ cluster of the University of Sheffield.Notes and references
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS PubMed.
- A. Vlček, Coord. Chem. Rev., 2000, 200–202, 933 CrossRef.
- M. Chergui, Dalton Trans., 2012, 41, 13022 RSC.
- A. C. Bhasikuttan, M. Suzuki, S. Nakashima and T. Okada, J. Am. Chem. Soc., 2002, 124, 8398 CrossRef CAS PubMed.
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275(5296), 54 CrossRef CAS PubMed.
- C.-W. Chan, L.-K. Cheng and C.-M. Che, Coord. Chem. Rev., 1994, 132, 87 CrossRef CAS.
- J. E. McGarrah and R. Eisenberg, Inorg. Chem., 2003, 42, 4355 CrossRef CAS PubMed.
- Y. Zhang, K. D. Ley and K. S. Schanze, Inorg. Chem., 1996, 35, 7102 CrossRef CAS PubMed.
- C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053 CrossRef CAS PubMed.
- J. E. McGarrah, Y.-G. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510 CrossRef CAS PubMed.
- E. A. Glik, S. Kinayyigit, K. L. Ronayne, M. Towrie, I. V. Sazanovich, J. A. Weinstein and F. N. Castellano, Inorg. Chem., 2008, 47, 6974 CrossRef CAS PubMed.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS PubMed.
- S.-C. Chan, M. C. W. Chan, Y. Wang, C.-M. Che, K.-K. Cheung and N. Zhu, Chem. – Eur. J., 2001, 7, 4180 CrossRef CAS.
- S. Chakraborty, T. J. Wadas, H. Hester, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6865 CrossRef CAS PubMed.
- T. J. Wadas, S. Chakraborty, R. J. Lachicotte, Q.-M. Wang and R. Eisenberg, Inorg. Chem., 2005, 44, 2628 CrossRef CAS PubMed.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802 CrossRef CAS PubMed.
- M. L. Muro, S. Diring, X. Wang, R. Ziessel and F. N. Castellano, Inorg. Chem., 2009, 48, 11533 CrossRef CAS PubMed.
- A. A. Rachford, S. Goeb, R. Ziessel and F. N. Castellano, Inorg. Chem., 2008, 47, 4348 CrossRef CAS PubMed.
- F. Hua, S. Kinayyigit, J. R. Cable and F. N. Castellano, Inorg. Chem., 2006, 45, 4304 CrossRef CAS PubMed.
- I. E. Pomestchenko and F. N. Castellano, J. Phys. Chem. A, 2004, 108, 3485 CrossRef CAS.
- I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394 CrossRef CAS PubMed.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS.
- C. J. Adams, N. Fey and J. A. Weinstein, Inorg. Chem., 2006, 45, 6105 CrossRef CAS PubMed.
- C. J. Adams, N. Fey, Z. A. Harrison, I. V. Sazanovich, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 8242 CrossRef CAS PubMed.
- M. L. Muro, A. A. Rachford, X. Wang and F. N. Castellano, Top. Organomet. Chem., 2009, 29 Search PubMed.
- M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115 CrossRef CAS.
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 2530 CrossRef CAS.
- I. V. Sazanovich, M. A. H. Alamiry, A. J. H. M. Meijer, M. Towrie, E. S. Davies, R. D. Bennett and J. A. Weinstein, Pure Appl. Chem., 2013, 85, 1331 CrossRef CAS.
- D. W. Dixon, N. B. Thornton, V. Steullet and T. Netzel, Inorg. Chem., 1999, 38, 5526 CrossRef CAS PubMed.
- D. S. Tyson, C. R. Luman, X. Zhou and F. N. Castellano, Inorg. Chem., 2001, 40, 4063 CrossRef CAS PubMed.
- O. Johansson, H. Wolpher, M. Borgström, L. Hammarström, J. Bergquist, L. Sun and B. Akermark, Chem. Commun., 2004, 194 RSC.
- L. Flamigni, E. Baranoff, J.-P. Collin and J.-P. Sauvage, Chem. – Eur. J., 2006, 12, 6592 CrossRef CAS PubMed.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545 CrossRef CAS.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011 CrossRef CAS.
- I. V. Sazanovich, M. A. H. Alamiry, J. Best, R. D. Bennett, O. V. Bouganov, E. S. Davies, V. P. Grivin, A. J. H. M. Meijer, V. F. Plyusnin, K. L. Ronayne, S. A. Tikhomirov, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 10432 CrossRef CAS PubMed.
- J. E. McGarrah, Y.-G. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510 CrossRef CAS PubMed.
- V. I. Stsiapura, A. A. Maskevich, S. A. Tikhomirov and O. V. Buganov, J. Phys. Chem. A, 2010, 114, 8345 CrossRef CAS PubMed.
- M. Towrie, D. C. Grills, J. Dyer, J. A. Weinstein, P. Matousek, R. Barton, P. D. Bailey, N. Subramaniam, W. M. Kwok, C. Ma, D. Phillips, A. W. Parker and M. W. George, Appl. Spectrosc., 2003, 57, 367 CrossRef CAS PubMed.
- G. Greetham, P. Burgos, Q. Cao, I. Clark, P. Codd, R. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. Pollard, D. Robinson, Z.-J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.2), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- R. C. Whaley and A. Petitet, Softw.: Pract. Exp., 2005, 35, 10 CrossRef.
- R. C. Whaley, A. Petitet and J. J. Dongarra, Parallel Comput., 2001, 27, 3 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- X. Y. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348 CrossRef CAS.
- A. Nicklass, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1995, 102, 8942 CrossRef CAS.
- J. Best, I. V. Sazanovich, H. Adams, R. D. Bennett, E. S. Davies, A. J. H. M. Meijer, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, M. D. Ward and J. A. Weinstein, Inorg. Chem., 2010, 49, 10041 CrossRef CAS PubMed.
- C. S. Grange, A. J. H. M. Meijer and M. D. Ward, Dalton Trans., 2010, 39, 200 RSC.
- S. P. Foxon, C. Green, M. Walker, A. Wragg, H. Adams, J. A. Weinstein, S. C. Parker, A. J. H. M. Meijer and J. A. Thomas, Inorg. Chem., 2012, 51, 463 CrossRef CAS PubMed.
- J. A. Thomas, H. Ahmad and A. J. H. M. Meijer, Chem. – Asian J., 2011, 6, 2339 CrossRef PubMed.
- A. B. Wragg, S. Derossi, T. L. Easun, M. W. George, X.-Z. Sun, F. Hartl, A. H. Shelton, A. J. H. M. Meijer and M. D. Ward, Dalton Trans., 2012, 41, 10354 RSC.
- B. Mennucci and J. Tomassi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS.
- M. Cossi, V. Barone, B. Menucci and J. Tomassi, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
- Jmol: an open-source Java viewer for chemical structures in 3D, http://www.jmol.org/, accessed: 11 March 2014 .
- Povray: Persistence of Vision Pty. Ltd (2004). Persistence of Vision Raytracer (Version 3.6) [Computer software], http://www.povray.org/download/, accessed 11 March 2014 .
- K. K. Irikura, R. D. Johnson and R. N. Kacker, J. Phys. Chem. A, 2005, 109, 8430 CrossRef CAS PubMed.
- J. A. Grant, M. A. Gallardo and B. T. Pickup, J. Comput. Chem., 1996, 17(1653), 166 Search PubMed.
- ROCS. OpenEye Scientific Software, Inc., Santa Fe, NM, USA, 2008. Available at http://www.eyesopen.com, accessed: 11 March 2014 .
- R. Dennington, T. Keith and J. Millam, GaussView (Version 5), Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- E. J. L. McInnes, R. D. Farley, S. A. Macgregor, K. J. Taylor, L. J. Yellowlees and C. C. Rowlands, J. Chem. Soc., Faraday Trans., 1998, 94, 2985 RSC.
- D. Jacquemin, E. A. Perpète, G. Scalmani, M. J. Frisch, I. Ciofini and C. Adamo, Chem. Phys. Lett., 2007, 448, 3 CrossRef CAS.
- E. O. Danilov, I. E. Pomestchenko, S. Kinayyigit, P. L. Gentili, M. Hissler, R. Ziessel and F. N. Castellano, J. Phys. Chem. A, 2005, 109, 2465 CrossRef CAS PubMed.
- J. E. McGarrah, J. T. Hupp and S. N. Smirnov, J. Phys. Chem. A, 2009, 113, 6430 CrossRef CAS PubMed.
- R. Sugimura, S. Suzuki, M. Kozaki, K. Keyaki, K. Nozaki, H. Mastsushita, N. Ikeda and K. Okada, Res. Chem. Intermed., 2013, 39, 185 CrossRef CAS.
- H. J. Shine and E. E. Mach, J. Org. Chem., 1965, 30, 2130 CrossRef CAS.
- J. Daub, R. Engl, J. Kurzawa, S. E. Miller, S. Schneider, A. Stockmann and M. R. Wasielewski, J. Phys. Chem. A, 2001, 105, 5655 CrossRef CAS.
- S. A. Alkaitis, G. Beck and M. Graetzel, J. Am. Chem. Soc., 1975, 97, 5723 CrossRef CAS.
- E. Danielson, C. M. Elliott, J. W. Merkert and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 2519 CrossRef CAS.
- W. D. Bates, P. Chen, D. M. Dattelbaum, W. E. Jones, Jr. and T. J. Meyer, J. Phys. Chem. A, 1999, 103, 5227 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2009, ch. 7, pp. 421–424 Search PubMed.
- R. H. Goldsmith, L. E. Sinks, R. F. Kelley, L. J. Betzen, W. Liu, E. A. Weiss, M. A. Ratner and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3540 CrossRef CAS PubMed.
- K. N. Solovyov and E. A. Borisevich, Phys.-Usp., 2005, 48, 231 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS PubMed.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148 CrossRef CAS.
- J. R. Miller, W. Hartman and S. J. Abrash, J. Am. Chem. Soc., 1982, 104, 4296 CrossRef CAS.
- M. R. Wasielewski, in Photoinduced Electron Transfer, ed. M. A. Fox and M. Chanon, Elsevier, Amsterdam, 1988, part A, p. 161 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S12, further synthetic details, description of the equipment used for spectroscopic characterisation, details of experimental procedures, and computational details. See DOI: 10.1039/c4cp03995e |
| This journal is © the Owner Societies 2014 |