
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixtures of protic ionic liquids and propylene carbonate as advanced electrolytes for lithium-ion batteries
T.
Vogl
,
S.
Menne
and
A.
Balducci
*
Institute of Physical Chemistry & MEET Battery Research Center University of Muenster Corrensstr. 28/30, 48149 Muenster, Germany. E-mail: andrea.balducci@uni-muenster.de
First published on 6th October 2014
Abstract
In this study we investigated the chemical–physical properties of mixtures containing the protic ionic liquid (PIL) N-butyl-pyrrolidinium bis(trifluoromethanesulfonyl)imide (PYRH4TFSI), propylene carbonate (PC) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in view of their use as electrolytes for lithium-ion batteries (LIBs). We showed that these electrolytic solutions might display conductivity and viscosity comparable to those of conventional electrolytes. Depending on the amount of PIL present inside the mixtures, such mixtures might also display the ability to suppress the anodic dissolution of Al. Furthermore, we showed that the coordination of lithium ions by TFSI in PIL–PC mixtures appears to be different than the one observed for mixtures of PC and aprotic ionic liquids (AILs). When used in combination with a battery electrode, e.g. lithium iron phosphate (LFP), these mixtures allow the achievement of high performance also at a very high C-rate.
1. Introduction
Lithium-ion batteries (LIBs) are nowadays considered one of the most important electrochemical storage devices.1–5 LIBs are used in portable electronics and they have been proposed for electric vehicles, aerospace applications or delocalized energy storage.1–5 In commercially available LIBs usually an electrolyte based on organic carbonates (e.g. ethylene carbonate (EC), diethyl carbonate (DEC)) with a lithium salt is used. This class of electrolytes features good conductivities and thus the capability to be used in high power devices. However, as organic solvents are volatile and highly flammable, their use is limiting the safety of LIBs.2 Therefore, in the last few years many efforts have been dedicated to the development of alternative electrolytes.Ionic liquids (ILs) are presently considered as one of the most interesting class of alternative electrolytes for the realization of safer LIBs.6–8 ILs typically display large electrochemical stability, high thermal stability and, moreover, they offer the advantage of non-volatility and thereby non-flammability.6,7,9–12 In the past years, a large number of studies have been dedicated to IL-based LIBs, demonstrating that the use of these electrolytes might increase the safety of LIBs.7,12 Nevertheless, IL-based LIBs are still not able to match the performance of LIBs containing conventional electrolytes, especially during tests in which high current densities are applied. As a matter of fact, under these conditions the rather high viscosity and the relatively low mobility of the lithium ions of IL-based electrolytes, compared to those of conventional electrolytes, limit the performance of LIBs.10,13,14 Furthermore, ILs are rather expensive. For these reasons, to date no IL-based LIBs have been commercialized.
In the past years almost all studies dedicated to IL-based electrolytes considered aprotic ionic liquids (AILs). However, we recently showed that protic ionic liquids (PILs) can also be successfully introduced as electrolytes for LIBs.15–19 One very interesting feature of PIL-based electrolytes is that the lithium ion coordination in these electrolytic solutions is typically lower than that of AIL-based electrolytes.20 As a consequence of this different coordination, the environment of the lithium ions in AILs and PILs is very different.20 We showed that the use of PIL-based electrolytes produces a much higher performance, particularly during test at high current densities, compared to the “traditional” AILs. Taking into account that the limited performance at a high rate of IL-based LIBs is presently considered as one of the main limitations of these devices, the use of PIL-based electrolytes can be therefore regarded as a new and promising strategy to overcome this drawback. Additionally, since PILs are typically cheaper than AILs, the introduction of these innovative electrolytes could also be of importance for the development of safer and cheaper IL-based LIBs.
The use of mixtures of AILs and organic solvents, e.g. propylene carbonate (PC), is becoming a popular strategy for the realization of advanced IL-based electrolytes.7,21–27 The main advantage related to the use of these mixtures is the possibility to tune their properties by changing the ratio of IL/organic solvent28–30 and it has been shown that their use might allow the realization of high performance LIBs.23,24,26,30 So far, almost all studies dedicated to these mixtures have been made using AILs. To the best of our knowledge, only few reports described the physicochemical properties of mixtures of PIL and PC16,31 and the use of these mixtures as electrolytes for LIBs has been only marginally considered. Nevertheless, considering the promising results obtained with pure PIL-based electrolytes, the investigation of such mixtures could be of high interest for the development of advanced IL-based electrolytes suitable for high power applications.
In this study we considered several mixtures containing the PIL N-butyl-pyrrolidinium bis(trifluoromethanesulfonyl)imide (PYRH4TFSI), PC and lithium bis(trifluoromethanesulfonyl) imide (LiTFSI). Initially, several chemical–physical properties, including conductivity, viscosity and thermal stability of the prepared mixtures, were investigated. As the anodic dissolution of Al in PIL-based electrolytes has not been investigated so far, this aspect has also been considered. Afterward, the lithium coordination of these electrolytes was studied using Raman spectroscopy with the aim to understand the impact of the solvent on the lithium environment of the PIL-based electrolyte. Finally, the electrochemical performance of lithium iron phosphate (LFP) electrodes in the considered mixtures was investigated with particular regard to the performance at high current density.
2. Experimental section
2.1. Electrolyte preparation
The pyrrolidinium-based PIL N-butyl-pyrrolidinium bis(trifluoro-methanesulfonyl)imide (PYRH4TFSI) was synthesized following a procedure similar to that described elsewhere.32 As there is no data available in the literature, this new compound has been characterized via NMR: 1H-NMR (CDCl3): δ = 0.93–0.97 (t, 3H, CH3); 1.34–1.43 (m, 2H, CH2); 1.66–1.74 (m, 2H, CH2); 2.05–2.18 (m, 4H, CH2); 2.96–3.05 (m, 2H, CH2); 3.09–3.15 (m, 2H, CH2); 3.68–3.75 (m, 2H, CH2); 7.48 (s, 1H, NH) ppm. 13C-NMR (CDCl3): δ = 13.39; 19.72; 22.89; 27.81; 55.00; 56.16; 115.01; 118.20; 121.39; 124.58 ppm. 19F-NMR (CDCl3): δ = −78.88 ppm. 1-Butylpyrrolidine (Aldrich, >98%) was distilled directly before use. HCl (30%, Merck Suprapur®) and LiTFSI (3 M) were used as received. At the end of the synthesis, the obtained PIL was dried under vacuum at 60 °C. Propylene carbonate (PC, UBE) was used without further purification. The water contents of the PIL and PC were measured using coulometric Karl-Fischer titration, and were found to be lower than 10 ppm. Three different mixtures of PYRH4TFSI, PC and LiTFSI were considered. Additionally, also electrolytes containing only PC and LiTFSI and only PYRH4TFSI and LiTFSI were investigated. The compositions, in terms of weight fraction as well as molar fraction, of all 5 investigated electrolytes are reported in Table 1. In the same table are indicated also the acronyms used for each electrolyte in this work. All electrolytes were prepared in an argon-filled glove box with oxygen and water contents lower than 1 ppm.| Weight fraction/wt% | Molar fraction/mol% | |||||
|---|---|---|---|---|---|---|
| PYRH4TFSI | PC | LiTFSI | PYRH4TFSI | PC | LiTFSI | |
| H4_100 | 89.76 | 0 | 10.24 | 86.04 | 0 | 13.96 |
| H4_80 | 71.59 | 17.90 | 10.51 | 45.27 | 45.18 | 9.45 |
| H4_50 | 44.49 | 44.49 | 11.02 | 18.68 | 74.74 | 6.58 |
| H4_20 | 17.70 | 70.82 | 11.48 | 5.58 | 89.27 | 5.51 |
| H4_0 | 0 | 88.27 | 11.73 | 0 | 95.49 | 4.51 |
2.2. Ionic conductivity, viscosity and thermal stability
The conductivity and the viscosity of the prepared mixtures were determined as reported in ref. 29. VTF parameters have been calculated using the following two equations. Eqn (1) shows the VTF-equation for the conductivity, eqn (2) for the viscosity. The estimated maximum error for the reported viscosities and conductivities values is lower than 5%.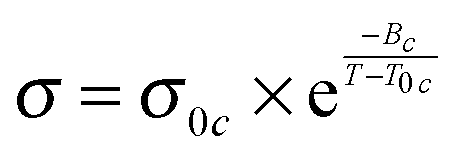 | (1) |
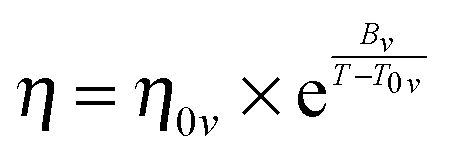 | (2) |
2.3. Raman spectroscopy
Raman spectra of all mixtures were recorded and analyzed as indicated in ref. 20.2.4. Electrochemical characterization
All the electrochemical tests were carried out with 3-electrode Swagelok® type cells. The cells were assembled in an argon-filled glove box with oxygen and water contents lower than 1 ppm. Electrochemical stability windows (ESW) were determined by linear sweep voltammetry (LSV) as reported in the literature.34 All measurements were carried out using a silver wire as reference. The anodic dissolution of Al was investigated using a VMP multichannel potentiostatic–galvanostatic system (Biologic Science Instrument, France). As a working electrode, discs of 12 mm diameter cut out of aluminum foil (30 μm, rinsed with purified water and ethanol, then vacuum dried) were used. The tests were carried out at 30 °C and the temperature was controlled using a Binder MK 53 climatic chamber. The potential at the working electrode was swept linearly from OCP to 2.0 V vs. Ag at a scan rate of 5 mV s−1. Then a CV was carried out using the cut-off potentials of 0.3 V vs. Ag and 2.0 V vs. Ag with the same scan rate for 100 cycles. Lithium iron phosphate (LFP) composite electrodes were prepared as in ref. 34. The composition of the dry electrodes was 85 wt% of the active material LFP, 10 wt% of conductive agent Super C65® and 5 wt% of sodium carboxymethyl cellulose (CMC) as the binder. The electrode mass loading was 1.5 mg cm−2; the electrode area was 1.13 cm2. LFP-based electrodes were used as working electrodes, a silver wire was used as a pseudo-reference electrode and an oversized activated carbon-based electrode was used as a counter electrode. As a separator, a Whatman GF/D glass microfiber filter (675 μm thickness) drenched with 200 μL of electrolyte was used. All the electrochemical measurements were carried out using a MACCOR Series 4000 battery tester. Constant current cycling (CC) was carried out at 30 °C using current densities ranging from 1 C to 20 C taking into account the theoretical capacity of LFP of 170 mA h g−1 when cycled between 2.8 and 4.2 V vs. Li/Li+.3. Results and discussion
3.1. Conductivity and viscosity
Fig. 1a compares the conductivities of all investigated electrolytes in the temperature range between −40 °C and 80 °C. For the electrolytes containing high amounts of ionic liquid the conductivity is just reported for temperatures above 20 °C (H4_100) and −10 °C (H4_80) as below these temperatures the electrolytes become solid. As shown in the figure, the conductivity of the pure PC-based electrolyte is higher than that of the electrolyte containing only PYRH4TFSI over the whole range of investigated temperatures. Nevertheless, with increasing temperature the conductivity of this latter electrolyte rises faster than the one of the former, and at 80 °C their conductivities become rather similar (8.29 vs. 8.56 mS cm−1 for H4_100 and H4_0, respectively). Among all investigated electrolytes, H4_50 and H4_20 display the highest conductivity over the considered range of temperatures. Fig. 1b shows an Arrhenius plot of the electrolyte conductivities. As visible, none of the electrolytes follow an Arrhenius-like behavior. However, the temperature dependence of the conductivity of all electrolytes could be well described by the Vogel–Tammann–Fulcher (VTF) model. Such behavior has been already reported in the literature for various IL-based electrolytes and therefore is not surprising. Table 2 reports the VTF fitting parameters for the conductivity. The viscosity of the considered electrolytes is shown in Fig. 1c. As expected, the electrolyte H4_100 is the most viscous while the H4_0 has the lowest viscosity. All the electrolytes containing mixtures of PYRH4TFSI and PC display a viscosity dependent on the amount of PC and in the range between the two non-mixed electrolytes. The viscosity of the considered electrolytes is shown in Fig. 1c. As expected, the electrolyte H4_100 is the most viscous while the H4_0 has the lowest viscosity. All the electrolytes containing mixtures of PYRH4TFSI and PC display a viscosity dependent on the amount of PC and in the range between the two non-mixed electrolytes. PC was chosen as the solvent because of its low melting point of −48.8 °C,35 but due to the relatively high melting point of the pure PIL, it was not possible to measure some of the electrolytes down to a temperature of −40 °C. As visible in the figure, at higher temperature the viscosity of the investigated electrolytes became more similar. As for the conductivity, the temperature dependence of the viscosity also does not follow an Arrhenius-like behavior but it could be well described by the VTF model (Fig. 1d). The VTF fitting parameters for the viscosities are reported in Table 2. | ||
| Fig. 1 Conductivity (a) and viscosity (c) with the corresponding Arrhenius plots (b, d) of the investigated mixtures. The solid lines represent the VTF fittings. | ||
| Conductivity | Viscosity | |||||||
|---|---|---|---|---|---|---|---|---|
| T 0c/K | σ 0/mS cm−1 | B c /K |
R
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
T 0v/K | η 0/mPa s | B v /K |
R
2
|
|
| a Correlation coefficient. | ||||||||
| H4_100 | 230.5 | 76.72 | 268.9 | 0.9988 | 190.0 | 0.186 | 710.3 | 1 |
| H4_80 | 235.0 | 53.21 | 179.7 | 0.9908 | 170.5 | 0.174 | 683.7 | 0.9993 |
| H4_50 | 193.0 | 59.53 | 245.2 | 0.9946 | 181.0 | 0.241 | 436.2 | 0.9994 |
| H4_20 | 190.0 | 41.74 | 206.4 | 0.9967 | 157.5 | 0.156 | 513.9 | 0.9999 |
| H4_0 | 175.0 | 45.89 | 286.4 | 0.9986 | 149.5 | 0.125 | 525.1 | 0.9999 |
Fig. 2 shows a comparison of the conductivity and viscosity of all investigated electrolytes at 30 °C. At this temperature the conductivities of pure PC-based and the pure PIL-based electrolytes are 5.15 and 1.83 mS cm−1, respectively. The maximum conductivity (7.23 mS cm−1) is displayed by the electrolyte H4_20, which contains 20 wt% of PYRH4TFSI. Nevertheless, it is interesting to notice that the conductivity of this electrolytes is only slightly higher than the conductivity of the electrolyte H4_50, (7.16 mS cm−1), which contains 50 wt% of PYRH4TFSI. As displayed in the figure, an inverse correlation between conductivity and viscosity cannot be observed. As indicated before, the viscosity simply decreases with increasing PC content while the conductivity reaches a maximum. Such behavior has been already reported for mixtures of AILs, e.g. containing N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYR14TFSI), and PC.18,19,29,30 Taking into account these results it appears that the behavior of dry PIL (as those used for this investigation) in combination with an organic solvent is rather comparable to that of AILs.
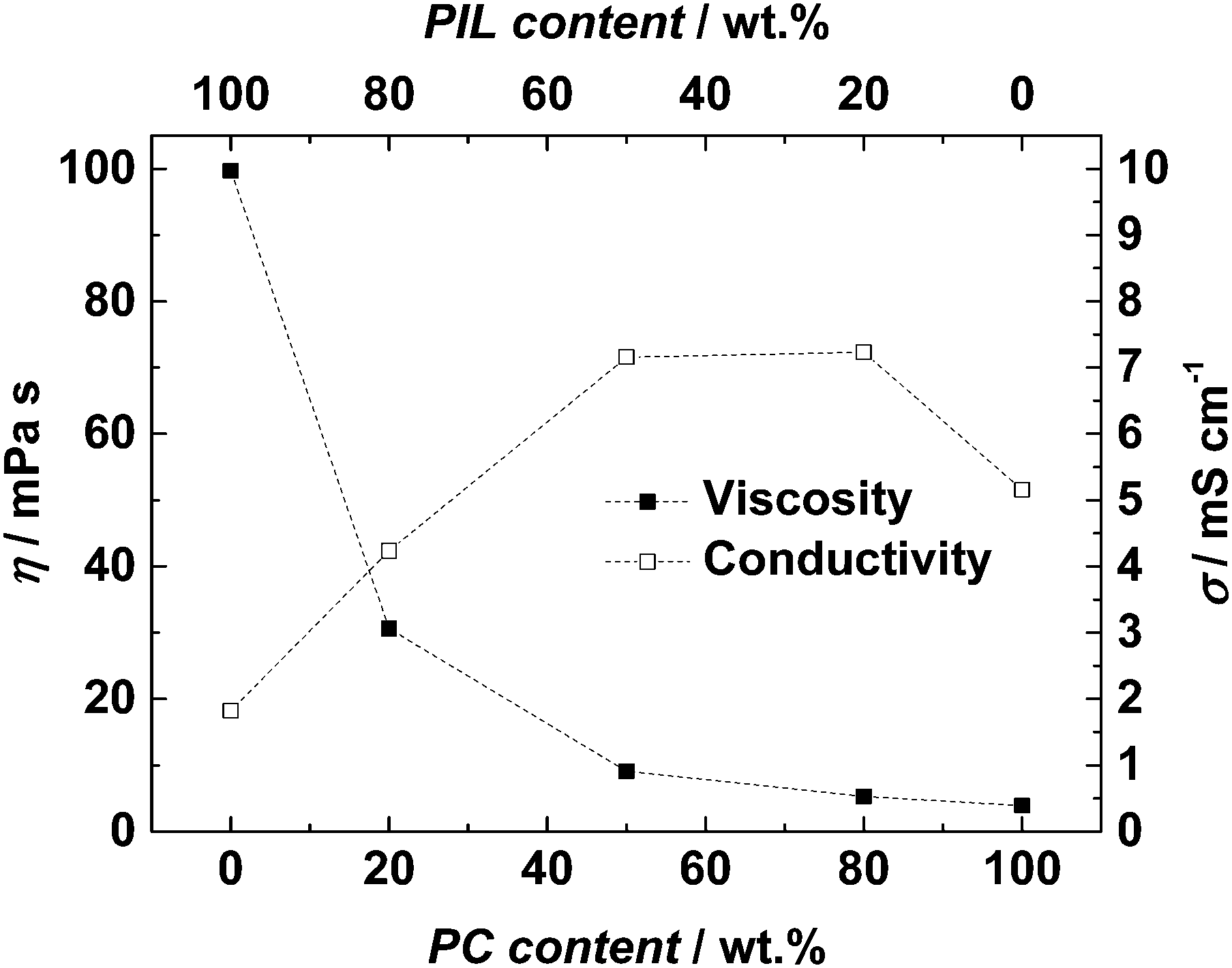 | ||
| Fig. 2 Conductivity and viscosity of the investigated mixtures at 30 °C. | ||
3.2. Thermal stability
Fig. 3a compares the thermal stability of all investigated electrolytes. As expected, the pure PIL-based electrolyte displays the highest thermal stability and it starts to decompose at about 300 °C.19 Upon PC addition, the stability of the electrolytes decreases with increasing PC content. The reason for this behavior is obviously related to the evaporation of the PC, which has a boiling point of 242 °C.35 As shown, the electrolyte H4_0 displays a residual weight above 250 °C. This residual mass originates due to the presence of LiTFSI inside the electrolytic solution, which has been shown to decompose above 250 °C.34 From the figure, it appears that the PC evaporation temperatures shift toward higher values in electrolytes with high PIL content. Such shift has been already reported in the literature for mixtures of PC and AILs.30 Nevertheless, it is important to notice that several studies showed that in the case of these mixtures such a shift is not originated by an interaction between the solvent and the AILs, but it rather indicates a reduction of the kinetic of the PC evaporation process.30,33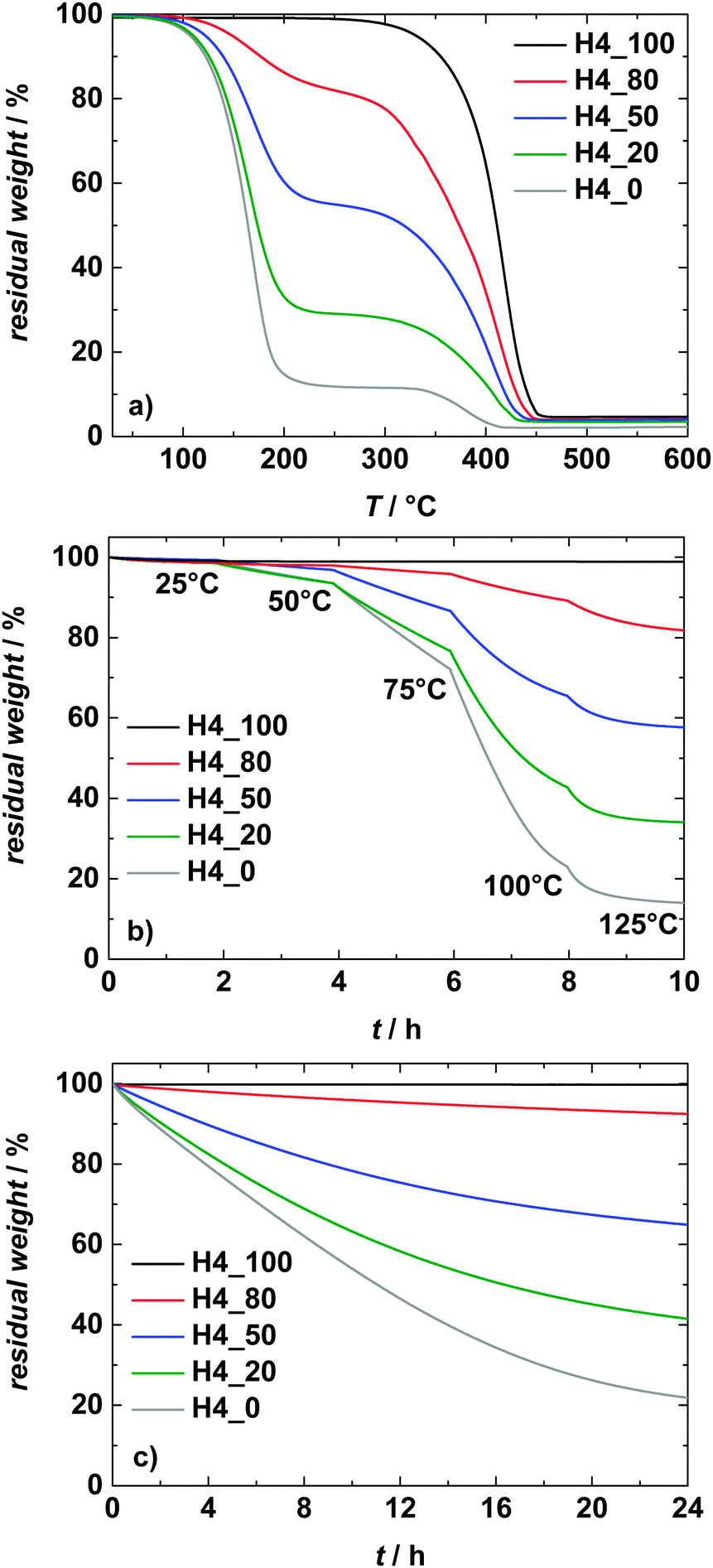 | ||
| Fig. 3 Thermogravimetric analysis of the investigated electrolytes. (a) Temperature ramp: 10 °C min−1, (b) isothermal at temperatures between 25 °C and 125 °C and (c) isothermal at 60 °C for 24 hours. | ||
In order to investigate this latter point more in detail, isothermal TGA experiments have also been carried out. In the first experiment (Fig. 3b), the temperature was increased stepwise from 25 °C to 150 °C and at each step the temperature was kept constant for 2 hours. As shown, during this test substantial weight losses have been recorded at temperature well below those observed during the first TGA experiment.
During each isothermal step a weight loss was observed, but the biggest occurred during the step at 100 °C. A similar trend was also observed in mixtures containing the AIL PYR14TFSI and PC.30 Nevertheless, it is interesting to notice that in the case of these mixtures the biggest weight loss was observed at a temperature of 75 °C.30 In the second isothermal TGA experiment (Fig. 3c), the investigated electrolytes were kept at a temperature of 60 °C for 24 hours. As shown, during this experiment in all electrolytes containing PC this solvent volatilizes well before its boiling point. In contrast, the PIL does not evaporate. Taking into account these results, it is reasonable to suppose that in mixtures of PYRH4TFSI and PC a weak interaction between ions and solvents is occurring and the volatility of PC is not altered by the presence of the PIL. Nevertheless, a reduction of the kinetic of the PC evaporation process is clearly observed and the higher is the content of PIL inside the electrolytes, the more significant appears such a reduction. The same type of behavior has been observed for several mixtures of PC and AILs.33 As already mentioned in the literature, even if no strong interaction between ILs and PC takes place in these mixtures, the reduction of the kinetic of the solvent evaporation might represent an advantage in the case of cell overheating.30,33
3.3. Electrochemical stability windows and anodic dissolution of aluminum
Fig. 4 compares the electrochemical stability windows (ESWs) of the investigated electrolytes at 30 °C. All electrolytes contain the TFSI− anion and their anodic stability, which is limited by the decomposition of this ion, is therefore very comparable.36 In contrast, the cathodic stability of the electrolyte appears very different. As a matter of fact, while the stability of the electrolyte containing only PC is limited by the lithium plating process,30,34 those of the electrolytes containing the PIL are limited by the deprotonation of the cation PYRH4+.32 As a consequence of these different cathodic stabilities, the electrolytes containing only PC display an overall ESW of ca. 2 V larger than those of the mixtures. Although the ESWs of electrolytes containing the PIL appear significantly lower than that of the PC-based electrolytes, their use in combination with cathodic materials, e.g. LFP, appears possible.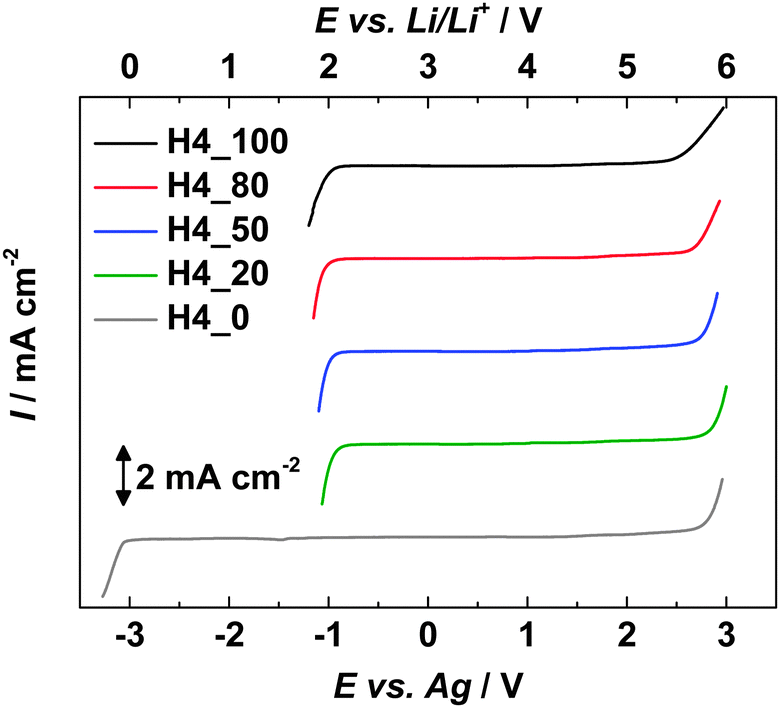 | ||
| Fig. 4 Electrochemical stability windows of the considered electrolytes. | ||
All electrolytes investigated in this study contain only TFSI− as the anion. In the past, it was widely reported that electrolytic solutions containing this anion dissolved in organic solvents, e.g. PC, do not display the ability to prevent the anodic dissolution of Al, which is used as a current collector for battery electrodes.37,38 Therefore, when these electrolytes are used in LIBs corrosion processes might occur, reducing the life of the devices.39,40 Nevertheless, it has been shown that TFSI-based AILs display the ability to suppress the anodic dissolution of Al.41–43 Such ability is originated by the low solubility of the Al–TFSI complexes formed on the Al surface, which makes possible the formation of a stable protective passivation layer and prevent the aluminium dissolution process.44 Furthermore, in recent studies we also showed that mixtures of TFSI-based AILs and PC might display the ability to suppress the anodic dissolution of Al.34 Such ability, however, strongly depends on the amount of IL present inside the mixtures and the higher is the AIL content, the stronger is the ability to prevent the anodic dissolution process.
To the best of our knowledge, the anodic dissolution of Al in TFSI-based PIL has not been considered in the past. Taking into account the influence that such a process might have on the cycle life of LIBs, we therefore investigated this aspect. Fig. 5 compares the SEM images of Al foil as obtained after CV experiments (the test conditions are reported in the experimental section) carried out on the 5 electrolytes investigated in this work. As shown in the figure, in the electrolyte containing only PC a massive dissolution process occurred (as expected). However, upon addition of PYRH4TFSI into the electrolyte such a process becomes less intense and in the case of the electrolytes containing only PIL no signs of anodic dissolution are visible in the Al foil. These results clearly indicate that also in the case of TFSI-based PILs, as for AILs, the anodic dissolution is suppressed. Moreover, they also indicate that a mixture of PIL and PC might display this ability. As shown, in the electrolyte containing 50% of PYRH4TFSI no holes on the surface of the Al foil are visible and only superficial modification of the Al surface seems to take place. In the electrolyte containing 80% of PYRH4TFSI the surface appears almost clean. Table 3 reports the charge densities recorded at the end of the CV experiments for each electrolyte. The values in the table are also graphically reported in Fig. 5. These values clearly indicate that the addition of PYRH4TFSI on PC based electrolytes has the ability to reduce the entity of the dissolution process orders of magnitudes and that in the electrolyte containing only the PIL such dissolution is practically completely suppressed. Taking these results into account, it is therefore possible to affirm that mixtures of TFSI-based PILs and PC might also display the ability to suppress the anodic dissolution of Al. As for the mixtures containing AILs, such ability strongly relates to the amount of ILs present inside the mixture.
 | ||
| Fig. 5 Comparison of SEM images and of the charge densities recorded at the end of the corrosion experiment for each electrolyte investigated in this study. From the left to the right, the PC content is increasing. | ||
| PC content/wt% | Charge density/C cm−2 |
|---|---|
| H4_0 | 0.018 |
| H4_20 | 0.035 |
| H4_50 | 11.43 |
| H4_80 | 27.24 |
| H4_100 | 277.5 |
3.4. Lithium-ion coordination
In the past, several studies showed that in electrolytes containing TFSI-based AILs, e.g. PYR14TFSI, and LiTFSI, strong interactions between the anion TFSI− and the lithium ions are taking place. At the same time, due to the sterical shielding of the IL cations, the interactions between the cations of the ILs and TFSI− anions are very weak.45–47 In a recent work we showed that in the case of electrolytes containing TFSI-based PILs the ion–ion interactions are considerably different compared to those of AILs.20 As a matter of fact, due to the occurrence of interactions between the cation of the PIL and the anion, the TFSI− ions are coordinated by both, the lithium ions and the cation of the IL. As a result of this sort of “competition” for the coordination of the TFSI− anion, in the case of equal lithium ion concentration, the lithium coordination number in PIL-based electrolytes is always lower than that of AIL-based electrolytes.As mentioned in the introduction, the use of mixtures of AILs and PC is becoming a popular strategy for the realization of advanced IL-based electrolytes. Recently, we investigated the lithium coordination in mixtures of PC and PYR14TFSI having a fixed amount of LiTFSI. We showed that in these mixtures the lithium ions are coordinated with both, the PC and the TFSI− anions. Nevertheless, the result of this investigation indicated that the lithium ions are preferentially coordinated by the PC solvent and in diluted electrolytes, meaning electrolytes with low concentration of PYR14TFSI, most of the lithium ions are coordinated by PC.29
To the best of our knowledge, no data are available about the lithium coordination in mixtures of PILs and PC. Nevertheless, taking into account the findings reported above, such investigation appears certainly of interest. Therefore, we carried out a preliminary investigation of the Li+ solvation in the considered electrolytes taking advantage of Raman spectroscopy. As reported in the literature, Li+ coordination by TFSI− anions can be investigated by analysing the strong band at ca. 742 cm−1.45–47 This band, which corresponds to the expansion and contraction of the whole TFSI− anion, is known to shift to higher wavenumbers upon coordination of TFSI− to Li+.45–47 Due to the relatively small energy difference of this band between the non-coordinated and coordinated TFSI−, the band representing coordinated TFSI− is often only visible as a shoulder.45–47
Fig. 6 compares the Raman spectra in the region of this TFSI− band of all electrolytes investigated in this study. The contributions of coordinated and non-coordinated TFSI− were calculated via the deconvolution of the peak and are also shown in the graphs. As clearly visible, the contribution of TFSI− to the coordination of Li+ decreased upon substitution of PYRH4TFSI by PC. The same trend was also observed for mixtures of PC and the AILs.29 To quantify this change in coordination, coordination numbers have been calculated for all the electrolytes using the equation (eqn (3)).29
 | (3) |
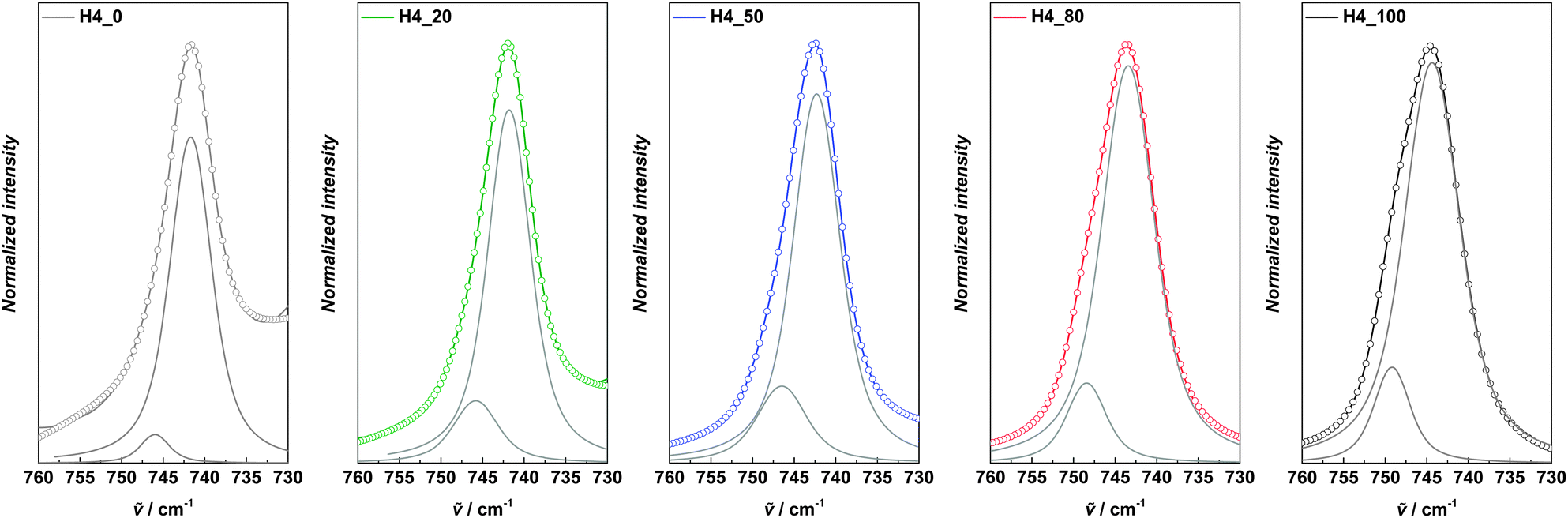 | ||
| Fig. 6 Raman spectra of the investigated electrolytes in the wavenumber range between 760 and 730 cm−1. The colored lines represent the measured data, the circles represent the fitted curve and the grey lines the two individual fitted peaks for coordinated and non-coordinated TFSI− anions. | ||
Fig. 7 shows the average numbers of TFSI− anions coordinating one Li+ cation in the different electrolytes dependent on the amount of PC. It is important to mention that the number of TFSI− anions (n) per Li+ cation derived from the Raman measurements should only be considered as average number. The complexity of Li+ solvation in liquid electrolytes is high and most likely different complexes are formed inside the solvent.29 Nevertheless, the trend indicated in Fig. 7 can be seen as a useful indication of the variation of the Li+ coordination upon PC addition into the electrolyte. As shown, the value of n in the solvent-free electrolyte was ca. 1, in agreement with the value already reported for PYRH4TFSI.20 It is important to remark that such a value is significantly lower than those observed for the PYR14TFSI (which is in the order of 2). As mentioned above, the “competition” between the lithium ions and the cation for the coordination of the TFSI− ions is responsible for such a low (compared to AIL) coordination number. In the solution without PIL, no contribution of TFSI− to the coordination of Li+ was found. Also this value is in agreement with the results available in the literature.29,48 The mixtures of PC–PYRH4TFSI showed decreasing n with increasing PC weight fraction of the electrolyte. When the PC weight fraction was 0.2 and 0.8 the value of n was 0.8, and 0.3, respectively. As shown in Fig. 6, together with the decrease in n, the peak representing TFSI− coordination to Li+ shifted to smaller wavenumbers. This indicates that not only fewer TFSI− anions are present in the Li+ solvation shell of the mixtures, but also that a weakened interaction of the remaining TFSI− anions and Li+ is taking place. This behaviour was also reported for mixtures of PC and PYR14TFSI. Nevertheless, it is very interesting to notice that the decrease of n upon addition of PC seems to follow a different trend in PIL and AIL. In our previous investigation we showed that in mixtures of PC and PYR14TFSI containing a PC weight fraction of 0.2, n was reduced already 50% compared to the solvent free electrolyte. When a PC weight fraction of 0.8 was considered, the reduction was above 90%.29 As shown in Fig. 7, in the investigated mixtures such reduction does not appear so dramatic. As a matter of fact, in mixtures containing a PC weight fraction of 0.2, n was reduced of ca. 20% compared to the solvent free electrolyte. When a PC weight fraction of 0.8 was considered, the reduction was ca. 70%.29
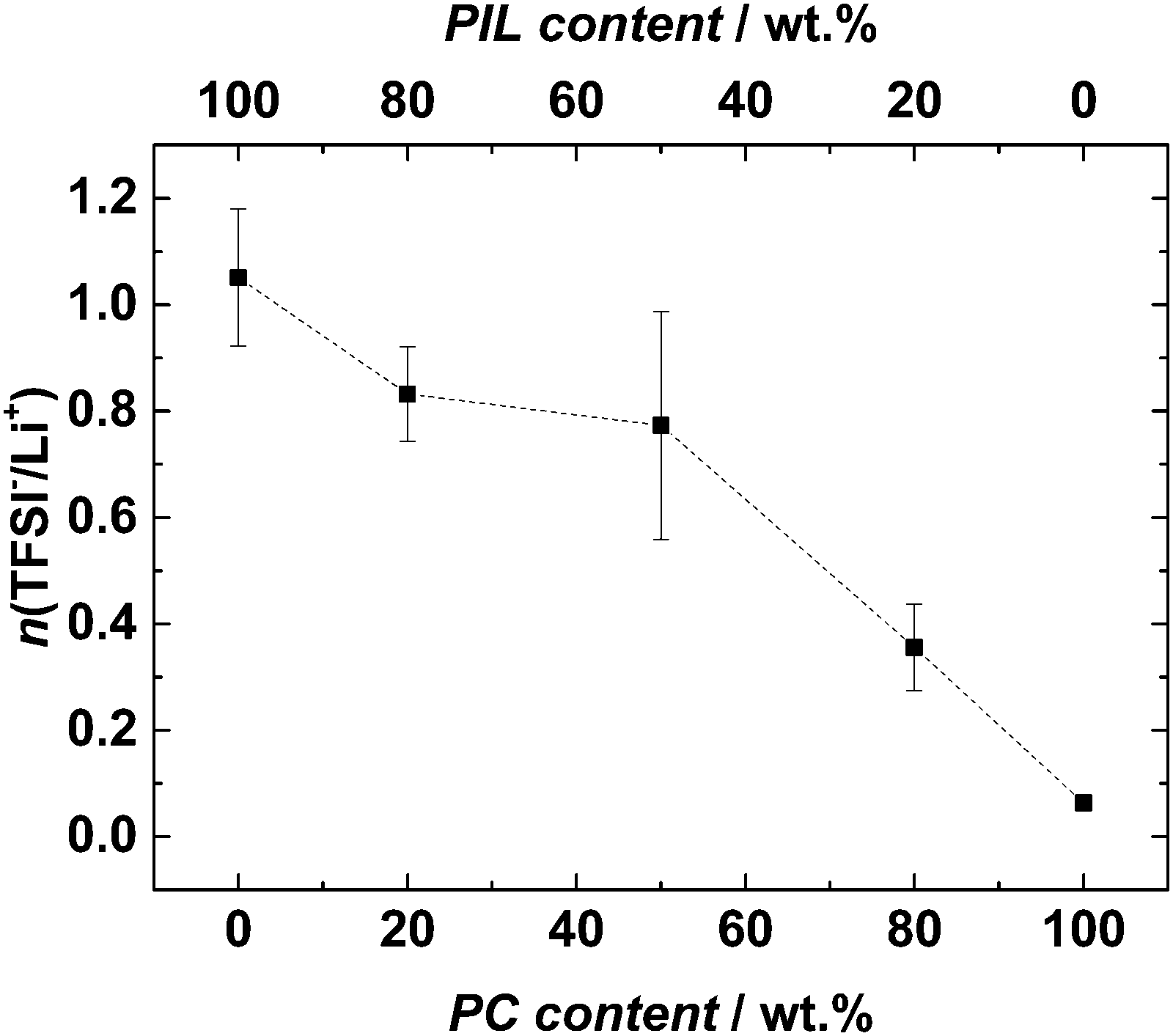 | ||
| Fig. 7 Average number of TFSI−anions (n) coordinating each Li+ ion for the investigated electrolytes, derived from the Raman intensities of the characteristic TFSI− band at ca. 740 cm−1. | ||
As mentioned above, the trend indicated in Fig. 7 can only be seen as an indication of the variation of the Li+ coordination upon PC addition into the electrolyte. Nevertheless, these results seem to indicate that the interactions between the cation and anion in PILs not only have an effect on Li+ coordination in solvent-free electrolytes, but they also affect the coordination between the PC and the Li+. It is reasonable to suppose that the proton of the pyrrolidinium-based cation might interact with the PC molecule and that such interaction might in turn affect the coordination between PC and Li+. Further investigation about PIL and PC is certainly necessary, but as they are beyond the aim of this work they will not be considered further. Nevertheless, taking into account the results of the investigation, it appears that the coordination of Li+ by TFSI in mixtures of AIL–PC and PIL–PC might be different due to the different ion–ion interaction taking place in these ILs.
3.5. Electrochemical characterization
As mentioned in the introduction, in a recent work we showed that the use of PIL-based electrolytes (solvent-free) results in a much higher performance, particularly during test at high current densities, compared to the “traditional” AILs.19 At the same time, many reports showed that also the use of mixtures of organic carbonates-AIL represents a promising strategy for the realization of advanced IL-based electrolytes.24,30,33,34 Taking these points into account, it is reasonable to suppose that mixtures containing PC and PIL could also represent a very interesting class of electrolytes for the realization of high performance LIBs.In order to verify this point, we tested the behavior of LFP electrodes in combination with mixtures of PC–PYRH4TFSI at different current densities, ranging from 1 C to 50 C, at 30 °C. For comparison, the electrolyte containing only PC and the solvent-free electrolyte have also been tested under the same conditions.
As shown in Fig. 8, the electrodes cycled in the solvent-free electrolyte display the lowest performance at all investigated C-rates. This behavior has been already reported in the literature and it is, therefore, not unexpected. Nevertheless, it is important to notice that the capacity delivered by the electrode in the PIL-based electrolyte is significantly higher than that observed for the same type of electrodes in AIL-based electrolytes, e.g. containing PYR14TFSI (ca. 40 mA h g−1 at 1 C).30 This result confirms the advantage related to the use of PIL-based electrolyte during test at a high C-rate. As shown, the addition of 20 wt% of PC leads to a significant enhancement of the electrode capacity.
 | ||
| Fig. 8 Variation of the specific capacity of LFP electrodes used in combination with the electrolytes investigated in this work. The tests were carried out at 30 °C using C-rates ranging from 1 C to 50 C. | ||
As a matter of fact, at 1 C the electrode used in combination with the solvent-free electrolyte delivers a specific capacity of ca. 95 mA h g−1, while the one cycled in H4_80 shows a capacity of 135 mA h g−1. At 5 C, the difference between the two electrodes becomes even more marked as the delivered capacity was 100 mA h g−1 and 50 mA h g−1 in H4_80 and H4_100, respectively. Very interestingly, the use of mixtures containing 50 and 20 wt% of PYRH4TFSI allowed the achievement of performance practically identical of those possible with conventional electrolytes. As shown in the figure, when these electrolytes were used, the capacities of the LFP electrodes at 1 C were in the order of 140 mA h g−1. At a current corresponding to a rate of 10 C, all electrodes delivered a specific discharge capacity of more than 100 mA h g−1. At the highest considered current (corresponding to 50 C) the electrodes cycled in these three electrolytes still deliver nearly 50 mA h g−1. Taking this performance into account, the use of mixtures of PIL and PC can certainly be considered as a very promising strategy for the realization of high performance LIBs. To the best of our knowledge, this is the first example of IL-based electrolytes, with a content of 50 wt% of IL, able to match the performance achievable with conventional electrolytes up to 50 C. As shown in the previous section, the mixture H4_50 displays conductivity and viscosity comparable with those of conventional electrolytes. However, because of its relatively high IL content, it displays a reduced kinetic of the PC evaporation and an improved behavior in terms of suppression of anodic dissolution of Al compared to the conventional electrolyte. Thus it can be considered as a very promising electrolytic solution.
An aspect of particular interest on the use of mixtures of PC and PIL is related to the amount of IL inside the electrolyte. In the past several reports indicated that in the case of mixtures of PC–AILs a large amount of PC is needed to reach the performance of a conventional electrolyte, thus reducing the safety of these electrolytes. The results reported above indicate that in the case of mixtures of PIL and PC it is possible to achieve very high performance also when a large amount of ILs are present inside the solution. Taking this point into account, the use of mixtures of PIL and PC appears to be of great interest not only because it results in high performance, but also because it might improve the safety of these types of electrolyte mixtures.
Anode materials have so far not been tested with these mixtures, but in previous work we showed, that also typical anode materials like graphite or LTO can be cycled successfully with pure PILs.15,18
Finally, the cycling stability of the investigated electrodes was also investigated using a current density corresponding to 1 C. In this case, we selected 4 electrolytes: H4_0, H4_20, H4_80 and H4_100. As shown in Fig. 9, all electrodes displayed an initial capacity comparable with those observed during the C-rate test, which was practically maintained over 1000 cycles. For all electrodes, the coulombic efficiency of the charge–discharge process was close to 100% for all tests. Considering these results, it is evident that the use of mixtures of PC–PYRH4TFSI allows the realization of electrodes with both, high performance and high stability during test at relatively high current density.
 | ||
| Fig. 9 Long term cycling stability of LFP electrodes in combination with the electrolytes H4_20, H4_80 and H4_100. The test has been carried out at 30 °C using a charge–discharge rate corresponding to 1 C. | ||
4. Conclusions
Mixtures of PC and PILs are interesting electrolyte candidates for LIBs. In this work we investigated mixtures of PC and PYRH4TFSI and we showed that these electrolytic solutions might display conductivity and viscosity comparable to those of conventional electrolytes. Furthermore, depending on the amount of PIL present inside the mixtures, such mixtures might also display the ability to suppress the anodic dissolution of Al.We showed that the interactions between cations and anions in PILs have not only an effect on Li+ coordination on solvent-free electrolytes, but they also affect the coordination between the PC and the Li+ in these mixtures. As a consequence of these interactions, the coordination of Li+ by TFSI in PIL–PC mixtures appears to be different than that of mixtures of AILs–PC.
When used in combination with a battery electrode, e.g. LFP, these mixtures allow the achievement of high performance. Using mixtures of PC and PYRH4TFSI containing 50 wt% of IL it is possible to match the performance achievable with conventional electrolytes up to 50 C. Furthermore, the use of these innovative electrolytes also provides an excellent cycling stability.
In the past several reports indicated that in the case of mixtures of PC–AILs a large amount of PC is needed to reach the performance of a conventional electrolyte, thus reducing the safety of these electrolytes. The results of this study show that using mixtures of PIL and PC it is possible to achieve very high performance also when a large amount of ILs are present inside the solution. Taking this point into account, mixtures of PIL and PC can be considered as very promising electrolytes for the realization of safe and high performance LIBs.
Acknowledgements
The authors wish to thank the University of Münster and the Ministry of Innovation, Science and Research of North Rhine-Westphalia (MIWF) within the project “Superkondensatoren und Lithium-Ionen-Hybrid-Superkondensatoren auf der Basis ionischer Flüssigkeiten” and the Bundesministerium für Bildung and Forschung (BMBF) within the project IES (contract number 03EK3010) for the financial support.Notes and references
- R. A. Marsh, S. Vukson, S. Surampudi, B. V. Ratnakumar, M. C. Smart, M. Manzo and P. J. Dalton, J. Power Sources, 2001, 97–98, 25–27 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS PubMed.
- T. Takamura, Solid State Ionics, 2002, 152–153, 19–34 CrossRef CAS.
- T. Tanaka, K. Ohta and N. Arai, J. Power Sources, 2001, 97–98, 2–6 CrossRef CAS.
- K. Zaghib, P. Charest, A. Guerfi, J. Shim, M. Perrier and K. Striebel, J. Power Sources, 2004, 134, 124–129 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. Balducci, S. S. Jeong, G. T. Kim, S. Passerini, M. Winter, M. Schmuck, G. B. Appetecchi, R. Marcilla, D. Mecerreyes, V. Barsukov, V. Khomenko, I. Cantero, M. I. De, M. Holzapfel and N. Tran, J. Power Sources, 2011, 196, 9719–9730 CrossRef CAS PubMed.
- J. F. Wishart, Energy Environ. Sci., 2009, 2, 956–961 CAS.
- A.-O. Diallo, A. B. Morgan, C. Len and G. Marlair, Energy Environ. Sci., 2013, 6, 699–710 CAS.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS PubMed.
- X.-W. Gao, C.-Q. Feng, S.-L. Chou, J.-Z. Wang, J.-Z. Sun, M. Forsyth, D. R. MacFarlane and H.-K. Liu, Electrochim. Acta, 2013, 101, 151–157 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- A. Guerfi, M. Dontigny, Y. Kobayashi, A. Vijh and K. Zaghib, J. Solid State Electrochem., 2009, 13, 1003–1014 CrossRef CAS.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- S. Menne, M. Schroeder, T. Vogl and A. Balducci, J. Power Sources, 2014, 266, 208–212 CrossRef CAS PubMed.
- N. Böckenfeld, M. Willeke, J. Pires, M. Anouti and A. Balducci, J. Electrochem. Soc., 2013, 160, A559–A563 CrossRef PubMed.
- A. Brandt, J. Pires, M. Anouti and A. Balducci, Electrochim. Acta, 2013, 108, 226–231 CrossRef CAS PubMed.
- S. Menne, J. Pires, M. Anouti and A. Balducci, Electrochem. Commun., 2013, 31, 39–41 CrossRef CAS PubMed.
- T. Vogl, S. Menne, R.-S. Kuhnel and A. Balducci, J. Mater. Chem. A, 2014, 2, 8258–8265 CAS.
- S. Menne, T. Vogl and A. Balducci, Phys. Chem. Chem. Phys., 2014, 16, 5485–5489 RSC.
- C. Arbizzani, G. Gabrielli and M. Mastragostino, J. Power Sources, 2011, 196, 4801–4805 CrossRef CAS PubMed.
- P. M. Bayley, G. H. Lane, N. M. Rocher, B. R. Clare, A. S. Best, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2009, 11, 7202–7208 RSC.
- A. Chagnes, M. Diaw, B. Carré, P. Willmann and D. Lemordant, J. Power Sources, 2005, 145, 82–88 CrossRef CAS PubMed.
- A. Guerfi, M. Dontigny, P. Charest, M. Petitclerc, M. Lagace, A. Vijh and K. Zaghib, J. Power Sources, 2010, 195, 845–852 CrossRef CAS PubMed.
- K. Kim, Y.-H. Cho and H.-C. Shin, J. Power Sources, 2013, 225, 113–118 CrossRef CAS PubMed.
- B. S. Lalia, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2010, 195, 7426–7431 CrossRef CAS PubMed.
- A. Lewandowski and A. Olejniczak, J. Power Sources, 2007, 172, 487–492 CrossRef CAS PubMed.
- E. T. Fox, E. Paillard, O. Borodin and W. A. Henderson, J. Phys. Chem. C, 2013, 117, 78–84 CAS.
- R.-S. Kuehnel and A. Balducci, J. Phys. Chem. C, 2014, 118, 5742–5748 CAS.
- R.-S. Kühnel, N. Böckenfeld, S. Passerini, M. Winter and A. Balducci, Electrochim. Acta, 2011, 56, 4092–4099 CrossRef PubMed.
- J. Pires, L. Timperman, J. Jacquemin, A. Balducci and M. Anouti, J. Chem. Thermodyn., 2013, 59, 10–19 CrossRef CAS PubMed.
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Beguin and M. Anouti, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207 RSC.
- A. Brandt, C. Ramirez-Castro, M. Anouti and A. Balducci, J. Mater. Chem. A, 2013, 1, 12669–12678 CAS.
- S. Menne, R. S. Kühnel and A. Balducci, Electrochim. Acta, 2013, 90, 641–648 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320–325 CrossRef CAS.
- M. Morita, T. Shibata, N. Yoshimoto and M. Ishikawa, Electrochim. Acta, 2002, 47, 2787–2793 CrossRef CAS.
- L. Peter and J. Arai, J. Appl. Electrochem., 1999, 29, 1053–1061 CrossRef CAS.
- C. Peng, L. Yang, Z. Zhang, K. Tachibana, Y. Yang and S. Zhao, Electrochim. Acta, 2008, 53, 4764–4772 CrossRef CAS PubMed.
- S.-W. Song, T. J. Richardson, G. V. Zhuang, T. M. Devine and J. W. Evans, Electrochim. Acta, 2004, 49, 1483–1490 CrossRef CAS PubMed.
- B. Garcia, S. Lavallée, G. Perron, C. Michot and M. Armand, Electrochim. Acta, 2004, 49, 4583–4588 CrossRef CAS PubMed.
- D. Moosbauer, S. Zugmann, M. Amereller and H. J. Gores, J. Chem. Eng. Data, 2010, 55, 1794–1798 CrossRef CAS.
- C. Peng, L. Yang, Z. Zhang, K. Tachibana and Y. Yang, J. Power Sources, 2007, 173, 510–517 CrossRef CAS PubMed.
- B. Garcia and M. Armand, J. Power Sources, 2004, 132, 206–208 CrossRef CAS PubMed.
- D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski and J. E. Desnoyers, Phys. Chem. Chem. Phys., 2002, 4, 6063–6071 RSC.
- M. Castriota, T. Caruso, R. G. Agostino, E. Cazzanelli, W. A. Henderson and S. Passerini, J. Phys. Chem. A, 2004, 109, 92–96 CrossRef PubMed.
- M. Herstedt, M. Smirnov, P. Johansson, M. Chami, J. Grondin, L. Servant and J. C. Lassègues, J. Raman Spectrosc., 2005, 36, 762–770 CrossRef CAS.
- Z. Wang, W. Gao, X. Huang, Y. Mo and L. Chen, J. Raman Spectrosc., 2001, 32, 900–905 CrossRef CAS.
| This journal is © the Owner Societies 2014 |