
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structure of deuterated benzene films adsorbed on the graphite (0001) basal plane: what happens below and above the monolayer coverage?
Emanuel
Bahn
*ab,
Holly
Hedgeland
c,
Andrew P.
Jardine
b,
Paul F.
Henry
d,
Thomas C.
Hansen
a and
Peter
Fouquet
*a
aInstitut Laue-Langevin, CS 20156, 38042 Grenoble Cedex 9, France. E-mail: bahn@ill.fr; fouquet@ill.fr; Tel: +33 476 20 7204
bCavendish Laboratory, J.J. Thompson Avenue, Cambridge, CB3 0HE, UK
cLondon Centre for Nanotechnology, University College London, London, WC1H 0AH, UK
dEuropean Spallation Source (ESS) AB, P.O. Box 176, 221 00 Lund, Sweden
First published on 4th September 2014
Abstract
An exact description of the interactions in aromatic carbon systems is a key condition for the design of carbon based nanomaterials. In this paper we investigate the binding and adsorbate structure of the simplest prototype system in this class – the single aromatic ring molecule benzene on graphite. We have collected neutron diffraction data of the ordered phase of deuterated benzene, C6D6, adsorbed on the graphite (0001) basal plane surface. We examined relative coverages from 0.15 up to 1.3 monolayers (ML) in a temperature range of 80 to 250 K. The results confirm the flat lying commensurate 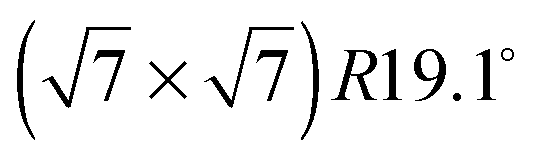 monolayer with lattice constants a = b = 6.5 Å at coverages of less than 1 ML. For this structure we observe a progressive melting well below the desorption temperature. At higher coverages we do neither observe an ordered second layer nor a densification of the structure by upright tilting of first layer molecules, as generally assumed up to now. Instead, we see the formation of clusters with a bulk crystalline structure for coverages only weakly exceeding 1 ML.
monolayer with lattice constants a = b = 6.5 Å at coverages of less than 1 ML. For this structure we observe a progressive melting well below the desorption temperature. At higher coverages we do neither observe an ordered second layer nor a densification of the structure by upright tilting of first layer molecules, as generally assumed up to now. Instead, we see the formation of clusters with a bulk crystalline structure for coverages only weakly exceeding 1 ML.
1 Introduction
The structure of adsorbed benzene has been studied for several decades and yet continues to be of active interest.1,2 From a fundamental point of view, benzene is of great importance as it represents the smallest building block of a layered hexagonal carbon system and in spite of its high symmetry it gives rise to complex structures and dynamics.3–9 A detailed understanding of benzene physisorption is also important for the development and improvement of filtering systems: benzene is a highly toxic and carcinogenic aromatic hydrocarbon molecule that occurs naturally in crude oil and coal deposits and its safe removal is thus of great concern for petrochemistry.10,11 Finally, benzene on carbon surfaces has been considered to be an important prototype for studying elementary dynamic processes and friction in view of the development of nanometer size motorization systems.8,12The structure of bulk crystalline benzene has been studied for more than fifty years.13–15 The benzene's multipole moments cause a perpendicular relative orientation of adjacent molecules (the symmetry of the molecule suppresses the net charge and dipole moment). Recently, the structure of liquid benzene has also been investigated in detail.16 The sub-monolayer structure of benzene adsorbed on graphite has been the subject of much debate. Whilst authors using nuclear magnetic resonance (NMR) measurements3,17 and quasielastic neutron scattering (QENS)4 suggested that the molecules stood upright on the graphite surface, neutron diffraction studies suggested flat lying molecules and a 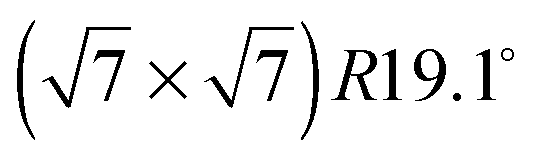 commensurate monolayer structure.5 The flat adsorption geometry was confirmed several years later by low energy electron diffraction (LEED),18,19 X-ray diffraction,20 and further NMR measurements.21,22 These heterogenous results were also reflected in the theoretical work on benzene/graphite: the first molecular dynamics (MD) simulations produced a structure that consisted mainly of flat lying molecules, but also contained a non-negligible fraction of upright molecules.23–28 A recent Grand Canonical Monte Carlo (GCMC) simulation in the liquid phase (273–373 K) produced a monolayer consisting of mostly flat lying molecules.29 It is worth noting that in the latter publication including a quadrupole interaction in the calculation was shown to be of crucial importance. However, this is not surprising in view of the alternating structure of the benzene crystal that is mainly due to dominant quadrupole interactions.13 The existence of a second surface layer has been discussed on several occasions. Whilst, to our knowledge, no experiment before has aimed to study it, several simulations on this question have been performed.23–26,28,29 The results differ, but suggest in their majority the existence of a disordered, liquid-like second layer.26,28,29 Finally, there has been a structural study using a density functional theory (DFT) approach.30 The latter does not consider, however, lateral interactions.
commensurate monolayer structure.5 The flat adsorption geometry was confirmed several years later by low energy electron diffraction (LEED),18,19 X-ray diffraction,20 and further NMR measurements.21,22 These heterogenous results were also reflected in the theoretical work on benzene/graphite: the first molecular dynamics (MD) simulations produced a structure that consisted mainly of flat lying molecules, but also contained a non-negligible fraction of upright molecules.23–28 A recent Grand Canonical Monte Carlo (GCMC) simulation in the liquid phase (273–373 K) produced a monolayer consisting of mostly flat lying molecules.29 It is worth noting that in the latter publication including a quadrupole interaction in the calculation was shown to be of crucial importance. However, this is not surprising in view of the alternating structure of the benzene crystal that is mainly due to dominant quadrupole interactions.13 The existence of a second surface layer has been discussed on several occasions. Whilst, to our knowledge, no experiment before has aimed to study it, several simulations on this question have been performed.23–26,28,29 The results differ, but suggest in their majority the existence of a disordered, liquid-like second layer.26,28,29 Finally, there has been a structural study using a density functional theory (DFT) approach.30 The latter does not consider, however, lateral interactions.
Here, we report on neutron diffraction measurements of fully deuterated benzene, C6D6, adsorbed on the basal plane surface (0001) of exfoliated graphite. Deuterium has to be used instead of hydrogen, as hydrogen scatters predominantly incoherently and, hence, contributes very little to the diffraction peaks. The measurements cover for the first time relative surface coverages between 0.15 and 1.3 monolayers (ML) in a coherent experiment. This represents a full overview of the structure from isolated molecules to the beginning of multi-layer formation. Our data indicate the formation of nano-clusters with a bulk-crystalline order at coverages of more than 1 ML. The structure was measured from low temperature across the melting transition up to 250 K.
This paper is organised as follows: after this general introduction, we describe the sample preparation procedure and the instrument parameters used for the measurements. In the subsequent section, we present the experimental results and the analysis. We finish this paper with a discussion of the results in view of past results and future research.
2 Experimental
2.1 Sample preparation
Neutron and X-ray diffraction experiments of hydrocarbon molecules adsorbed on graphite require the use of high surface density substrates due to the small surface signal. Here, we used chemically exfoliated graphite, which is a widely used high surface density material that typically has a specific surface area of about 20 m2 g−1 and retains a sufficiently low defect density.31,32 In addition, exfoliated graphite samples show a preferential orientation of the basal plane surfaces. We exploited this and oriented the basal planes parallel to the scattering plane of the neutrons. We used 25 g of Papyex exfoliated graphite of grade N998 (>99.8% C, Carbone Lorraine, Gennevilliers, France). We prepared exfoliated graphite disks of 2 cm diameter that were heated to 973 K under vacuum for 4 days and subsequently transferred to a cylindrical aluminium sample holder under a protective atmosphere. The sample holder was sealed by a lid with a steel knife edge and connected to a gas sorption system via a heated steel capillary. The surface density of the sample was measured in situ by BET (Brunauer–Emmett–Teller) nitrogen adsorption isotherms that were obtained using a high precision absolute pressure gauge (Baratron 690A, MKS Instruments). We found a specific surface area of 23 m2 g−1. Fully deuterated benzene (99.6% D, CEA Saclay, France) was used in this experiment to enhance coherent scattering. In this article we define the relative surface coverage, θ, by the ratio of the molecular density, ρ, divided by the density of the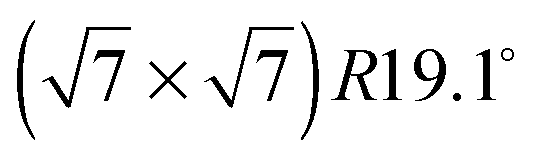 structure (ρML = 1/36.7 Å−2,19): θ = ρ/ρML. The density, ρ, was given by the change in pressure of the benzene during adsorption (exploiting the finite vapor pressure of benzene at room temperature) and by the known surface area of the sample.
structure (ρML = 1/36.7 Å−2,19): θ = ρ/ρML. The density, ρ, was given by the change in pressure of the benzene during adsorption (exploiting the finite vapor pressure of benzene at room temperature) and by the known surface area of the sample.
2.2 Diffraction measurements
Neutron diffraction measurements were carried out using the high intensity powder diffractometer D20 at Institut Laue-Langevin (ILL), Grenoble, France, using a wavelength of λ = 2.4 Å.33 Data were taken in a range of momentum transfers Q = |![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) f − i| = [0.2 to 5.1] Å−1, where i and f are the neutron wavevectors before and after scattering from the sample, respectively. The resolution in this set-up for a 2 cm diameter sample is about ΔQ/Q = 0.03.33 Measurements were performed at relative surface coverages of 0.15 ML, 0.5 ML, 0.8 ML, and 1.3 ML, respectively, and at temperatures in the range [80 to 250] K. The temperature was controlled using a standard liquid helium cryostat (‘orange’ cryostat,34). Additional diffraction measurements of the clean graphite sample were performed at all temperatures. The graphite substrate and its orientation remained the same throughout all measurements. Subsequently, the clean graphite data were subtracted from the diffraction data of the adsorbate systems at equal temperature (see Fig. 1). Five regions in our data show very strong signal from the graphite substrate that masks the signal from the benzene adsorbate and makes a meaningful interpretation of the benzene signal impossible. These regions, as well as the low Q region, are marked by grey shading in Fig. 1 and excluded in our further data analysis.
f − i| = [0.2 to 5.1] Å−1, where i and f are the neutron wavevectors before and after scattering from the sample, respectively. The resolution in this set-up for a 2 cm diameter sample is about ΔQ/Q = 0.03.33 Measurements were performed at relative surface coverages of 0.15 ML, 0.5 ML, 0.8 ML, and 1.3 ML, respectively, and at temperatures in the range [80 to 250] K. The temperature was controlled using a standard liquid helium cryostat (‘orange’ cryostat,34). Additional diffraction measurements of the clean graphite sample were performed at all temperatures. The graphite substrate and its orientation remained the same throughout all measurements. Subsequently, the clean graphite data were subtracted from the diffraction data of the adsorbate systems at equal temperature (see Fig. 1). Five regions in our data show very strong signal from the graphite substrate that masks the signal from the benzene adsorbate and makes a meaningful interpretation of the benzene signal impossible. These regions, as well as the low Q region, are marked by grey shading in Fig. 1 and excluded in our further data analysis.
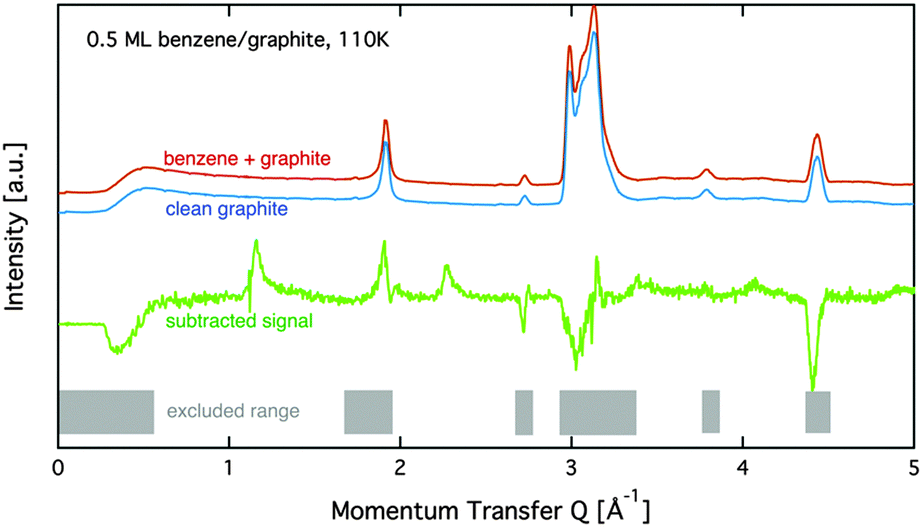 | ||
| Fig. 1 Diffraction data at 110 K for the empty graphite sample and a coverage of 0.5 ML are shown together with the subtracted data. Regions with a strong graphite signal are indicated by a grey pattern. These regions are omitted in the analysis in this paper. | ||
2.3 Neutron spin echo measurements
Supplemental neutron spin echo (NSE) measurements were performed on the NSE spectrometer IN11 at ILL using the high signal set-up IN11C, which uses a 30 degree detector bank.35 Here, we used a wavelength of λ = 5.5 Å for the maximum signal. The spectra were normalized using a standard procedure: all data were divided by a spectrum that was obtained in situ at the cryostat base temperature of 1.5 K, where the system can safely be assumed to be static in the dynamic window of the spectrometer. The experiment covered a range of momentum transfers of Q = [0.2 to 0.7] Å−1.3 Results and discussion
Fig. 2 displays an overview of the coverage dependence of the diffraction patterns of benzene/graphite at a temperature of 110 K. The graphs show significant structural differences between the sub-monolayer and the multi-layer regime as new peaks appear above 1 ML. Hence, for the sake of clarity of the description, we will describe these two regimes in separate sections.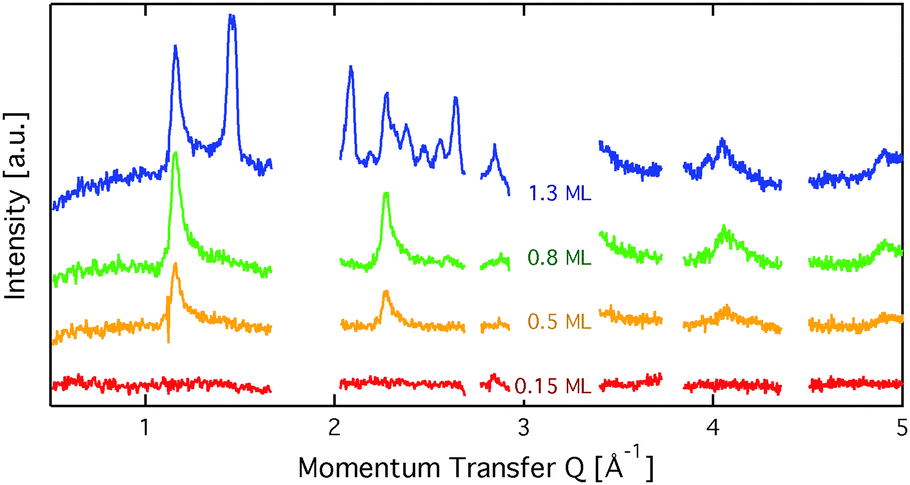 | ||
| Fig. 2 Diffraction patterns for varying coverages at a sample temperature of 110 K. Regions with strong graphite signal are omitted in the graph. At a coverage of 0.15 ML no structural peaks are seen. At a coverage of more than 1 ML additional peaks appear, which cannot be explained in the framework of a flat benzene structure. | ||
3.1 Diffraction data and structural refinement – sub-monolayer regime
The diffraction pattern of the monolayer of benzene/graphite at low temperature has been studied many times before and there exists general agreement on a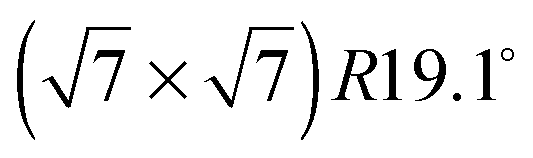 pattern with respect to the graphite (0001) surface structure both for C6H6 and C6D6.5,18–22 However, the capabilities of neutron powder diffractometers and the corresponding data treatment methods have advanced tremendously since the last data were published and we can now study the structural changes upon heating up to the monolayer desorption temperature of 150 K.36 In addition, the strong signal of the D20 diffractometer allows us to study several sub-monolayer coverages.
pattern with respect to the graphite (0001) surface structure both for C6H6 and C6D6.5,18–22 However, the capabilities of neutron powder diffractometers and the corresponding data treatment methods have advanced tremendously since the last data were published and we can now study the structural changes upon heating up to the monolayer desorption temperature of 150 K.36 In addition, the strong signal of the D20 diffractometer allows us to study several sub-monolayer coverages.
We start our analysis with the lowest measured coverage of 0.15 ML (see Fig. 2). At 0.15 ML, no diffraction peaks are observed, which signifies that no long range order arises. The graphs of the two higher sub-monolayer coverages 0.5 ML and 0.8 ML show four strong peaks outside of the regions that are dominated by graphite peaks. The benzene peaks remain at the same position up to 1 ML coverage and have an asymmetrical shape that is typical for diffraction patterns of two-dimensional poly-crystalline systems.37,38 The asymmetric shape of the diffraction peaks is caused by the random orientation of crystallographic domains, where the normal of the domain remains parallel to the surface normal.
For a more detailed analysis of the sub-monolayer and monolayer structures we have simulated neutron diffraction patterns of a flat lying monolayer using the software package nxpattern, which allowed us to adjust a number of parameters manually.39 The results of such a manual structural refinement are displayed in Fig. 3. The positions of the atoms in the deuterated benzene molecule were based on neutron diffraction data for deuterated benzene molecules.15 We have then adjusted the lattice parameters and found clear consistency with the model of a monolayer of densely packed flat lying benzene molecules. In the next step we have simulated results for out-of-plane tilting of molecules and in-plane rotations. We can exclude rotations of more than 1 degree and tilt of more than 10 degrees, due to the subsequent mismatch between the calculated and measured peak positions.
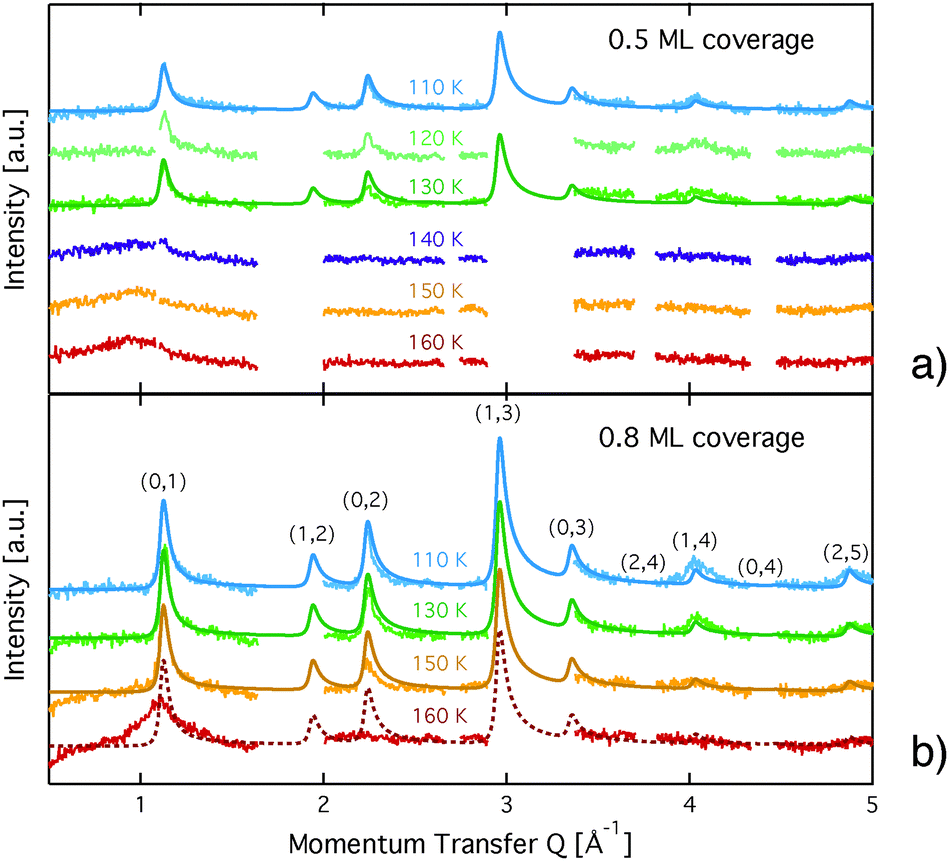 | ||
Fig. 3 Diffraction patterns for varying temperatures at a coverage of (a) 0.5 ML and (b) 0.8 ML. The respective calculated diffraction patterns for a commensurate 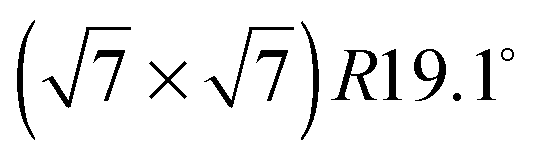 monolayer are shown by the solid lines. The pattern above 130 K at a coverage of 0.5 ML and above 150 K at 0.8 ML, respectively, cannot be approximated by a monolayer are shown by the solid lines. The pattern above 130 K at a coverage of 0.5 ML and above 150 K at 0.8 ML, respectively, cannot be approximated by a 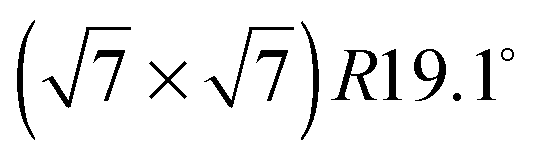 structure and the corresponding expected pattern is, therefore, indicated by a broken line. structure and the corresponding expected pattern is, therefore, indicated by a broken line. | ||
Further information could be drawn from the line-shape: in-plane neutron scattering from a flat, two dimensional layer of ordered islands that are randomly oriented produces diffraction peaks that drop sharply for lower angles and exhibit a Lorentzian decay for higher angles.37 Even better results were obtained in our case by using a squared Lorentzian peak shape that has been theoretically shown to be an indication of random layer lattices that undergo strain effects.40 The peak width of the diffraction pattern depends on the instrumental resolution ΔQ/Q and on the size of the islands. For a decreasing island size we find an increase of the width of the diffraction peak. For the 110 K data the width of the diffraction peaks was found to be equal to ΔQ/Q. Therefore, we can consider the island diameter obtained from the simulations, D = 200 ± 50 Å, a lower limit for the real island size. The graphite layers in the compressed exfoliated graphite exhibit a preferred orientation in the plane with a mosaic spread of the surface normal of 20–30 degrees full width half maximum (FWHM).41,42 This also influences the peak shape and has been taken into account in the simulation of the pattern. Finally, the Debye–Waller factor, given by DWF = exp[−Q2〈u2〉/3] (〈u2〉 is the mean square displacement of the atoms) has a significant influence on the relative amplitudes of the measured peaks (see Fig. 3): the intensity of the diffraction peaks drops with increasing momentum transfer, Q.
The diffraction patterns resulting from the structural refinement for the coverages 0.5 ML and 0.8 ML, respectively, are shown in Fig. 3. The agreement with the measured diffraction data confirms the formation of a 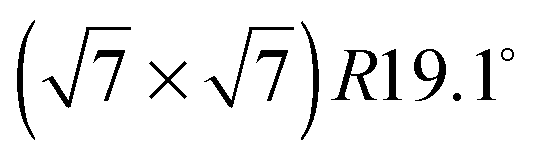 structure up to a temperature of 130 K (0.5 ML) and 150 K (0.8 ML), respectively. The measured diffraction peak positions and the simulated peak positions in the Q-range of our data are compared in Table 1. The positions of the four available peaks are reproduced well within the experimental error bar, however we find a clear deviation of the intensities of the higher order peaks from the Debye–Waller approximation at temperatures approaching 150 K (Fig. 3). This is an indication of the gradual melting of the structure. The
structure up to a temperature of 130 K (0.5 ML) and 150 K (0.8 ML), respectively. The measured diffraction peak positions and the simulated peak positions in the Q-range of our data are compared in Table 1. The positions of the four available peaks are reproduced well within the experimental error bar, however we find a clear deviation of the intensities of the higher order peaks from the Debye–Waller approximation at temperatures approaching 150 K (Fig. 3). This is an indication of the gradual melting of the structure. The 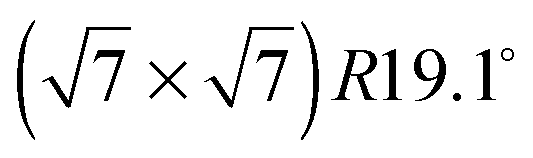 structure is depicted in Fig. 4. Here, we propose two possible variants. The structure in Fig. 4a assumes the positions of the benzene hydrogen atoms on top of the underlying carbon atoms. This structure has been preferred recently by other authors.30 In the structure of Fig. 4b the hydrogen atoms are turned to take a maximum distance from the top layer carbon atoms. Since the scattering plane is concomitant with the surface planes our diffraction data cannot distinguish between the two orientations, but orientation (b) is preferred by our MD simulations,9 where the most probable orientation of the benzene rings with respect to the carbon structure is found to be turned by about 20°. An I–V analysis of low intensity LEED measurements might resolve this issue, but it has to be kept in mind that the benzene islands remain highly dynamic even at temperatures of 60 K as shown by recent spectroscopy data.43 It would also be highly interesting to study theoretically whether many-body van der Waals interactions have an effect on the structure and phase transitions as these interactions have not yet been taken into account, to our knowledge.44
structure is depicted in Fig. 4. Here, we propose two possible variants. The structure in Fig. 4a assumes the positions of the benzene hydrogen atoms on top of the underlying carbon atoms. This structure has been preferred recently by other authors.30 In the structure of Fig. 4b the hydrogen atoms are turned to take a maximum distance from the top layer carbon atoms. Since the scattering plane is concomitant with the surface planes our diffraction data cannot distinguish between the two orientations, but orientation (b) is preferred by our MD simulations,9 where the most probable orientation of the benzene rings with respect to the carbon structure is found to be turned by about 20°. An I–V analysis of low intensity LEED measurements might resolve this issue, but it has to be kept in mind that the benzene islands remain highly dynamic even at temperatures of 60 K as shown by recent spectroscopy data.43 It would also be highly interesting to study theoretically whether many-body van der Waals interactions have an effect on the structure and phase transitions as these interactions have not yet been taken into account, to our knowledge.44
| Index | Measured position | Simulated position |
|---|---|---|
| (0,1) | 1.124 ± 0.011 | 1.116 |
| (1,2) | Hidden | 1.933 |
| (0,2) | 2.220 ± 0.022 | 2.232 |
| (1,3) | Hidden | 2.953 |
| (0,3) | Hidden | 3.486 |
| (2,4) | Hidden | 3.867 |
| (1,4) | 4.042 ± 0.040 | 4.024 |
| (0,4) | Hidden | 4.465 |
| (2,5) | 4.894 ± 0.049 | 4.865 |
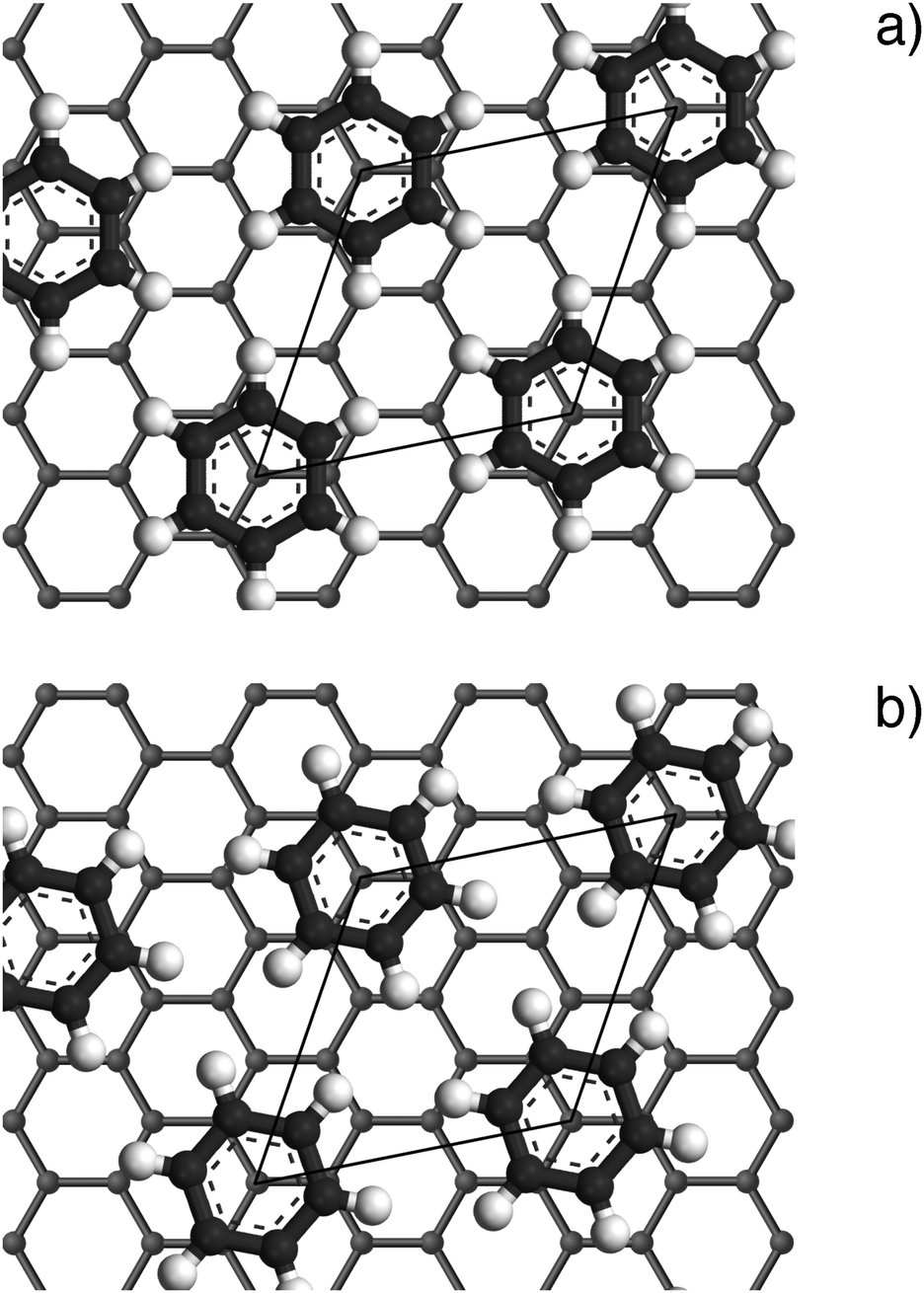 | ||
Fig. 4 Illustrations of the commensurate 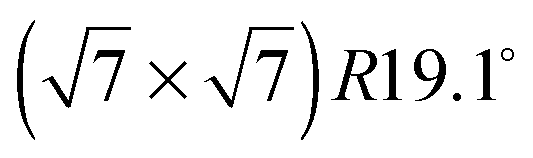 structure. The hydrogen atoms are drawn in white, the benzene carbon atoms are black and the carbon atoms of the graphite substrate are shown in grey. (a) The hydrogen atoms are positioned on top of first layer carbon atoms. (b) The hydrogen atoms are turned away from the carbon atom positions. Orientation (b) agrees with our recent MD calculations.9 structure. The hydrogen atoms are drawn in white, the benzene carbon atoms are black and the carbon atoms of the graphite substrate are shown in grey. (a) The hydrogen atoms are positioned on top of first layer carbon atoms. (b) The hydrogen atoms are turned away from the carbon atom positions. Orientation (b) agrees with our recent MD calculations.9 | ||
3.2 Comparison to neutron spin-echo data
Neutron spin-echo (NSE) spectroscopy measures the decay of structural correlation with time. NSE spectra for temperatures between 120 and 150 K are shown in Fig. 5. The NSE spectra show a decay that can be expressed by a simple exponential decay that reaches a plateau value A at large time: I(Q,t)/I(Q,0) = (1 − A) × exp[−t/τ(Q)] + A, where τ(Q) is the decay time of the structural correlation function. The level of the long time plateau A depends strongly on the sample temperature. The plateau represents the scattering from the static fraction of the sample, i.e., from the graphite substrate and from immobile islands of benzene. The change in the level of the plateau indicates a gradual melting of the layer, which corroborates our interpretation of the decay of the signal of the higher order diffraction peaks. In Fig. 6, we compare the area of the (0,2) peak of the 0.5 ML diffraction data set with the A parameter extracted from fitting the NSE data at a momentum transfer Q = 0.4 Å−1. From our diffraction and NSE data we can, thus, deduce that islands are formed at 0.5 and 0.8 ML coverages below 140 and 160 K, respectively, that are ordered in the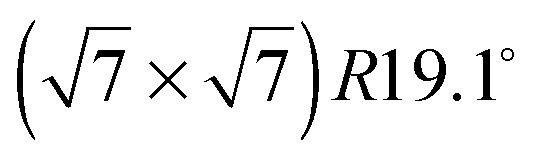 monolayer structure. As the temperature approaches 140 K and 160 K, respectively, we see a gradual melting.
monolayer structure. As the temperature approaches 140 K and 160 K, respectively, we see a gradual melting.
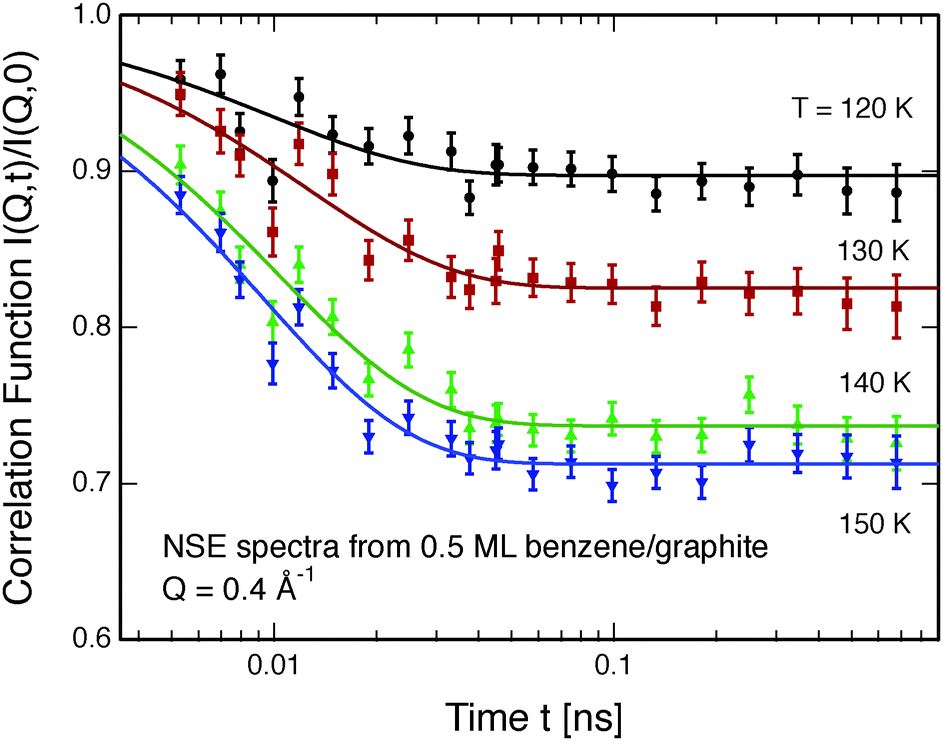 | ||
| Fig. 5 Neutron spin echo spectra of a 0.5 ML benzene layer on exfoliated graphite measured at temperatures between 120 and 150 K at a momentum transfer of Q = 0.4 Å−1. The solid lines are exponential function fits to the data. | ||
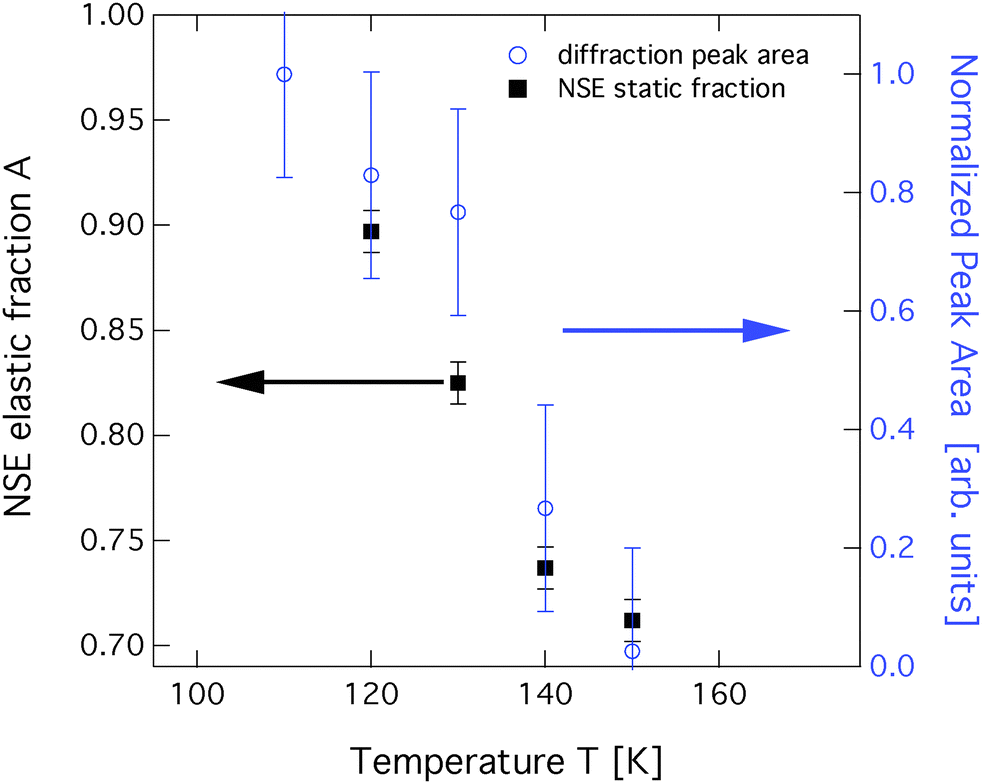 | ||
| Fig. 6 Comparison of the total area of the (0,2) diffraction peak (blue circles) to the static scattering A parameter from NSE spectroscopy at a momentum transfer Q = 0.4 Å−1 (black squares). Both data sets were obtained at a coverage of 0.5 ML. | ||
3.3 Diffraction results and structural refinement – coverage exceeding 1 ML
Diffraction data for a coverage of 1.3 ML are shown in Fig. 7. Up to a temperature of 160 K additional peaks are clearly visible and the peaks that are found for the sub-monolayer structure remain. We have performed structural refinement by assuming either flat double layers or compressed layers with alternating flat and upright molecules in various geometries. None of these models reproduce any of the observed additional lines for the 1.3 ML coverage. Furthermore, we observed that the additional peaks that appear above 1 ML coverage (see Fig. 7) disappear at around 200 K and thus at a temperature that is 50 K above the monolayer desorption temperature. We conclude, that a second layer formation is not found here. Instead, we propose the formation of bulk crystalline benzene when the first layer is complete. In Fig. 7 we also compare our data with neutron diffraction data from (deuterated) bulk benzene by Craven et al. measured at the instrument D2B at ILL.14 The similarity to the bulk data provides clear evidence that the additional structure at above monolayer coverage is bulk crystalline benzene. We have no MD calculation data for coverages of more than 1 ML, but there is one more important indication that excludes a two-dimensional structure: two-dimensional structures lead to strongly asymmetric peaks as observed for the sub-monolayer peaks,37,38 whereas the additional peaks in the 1.3 ML data are clearly symmetric and cannot be formed from randomly oriented two-dimensional ordered domains. The creation of the bulk structure at coverages with a relatively modest excess over 1 ML is at odds with the widely spread picture of structure creation in the low coverage regime.45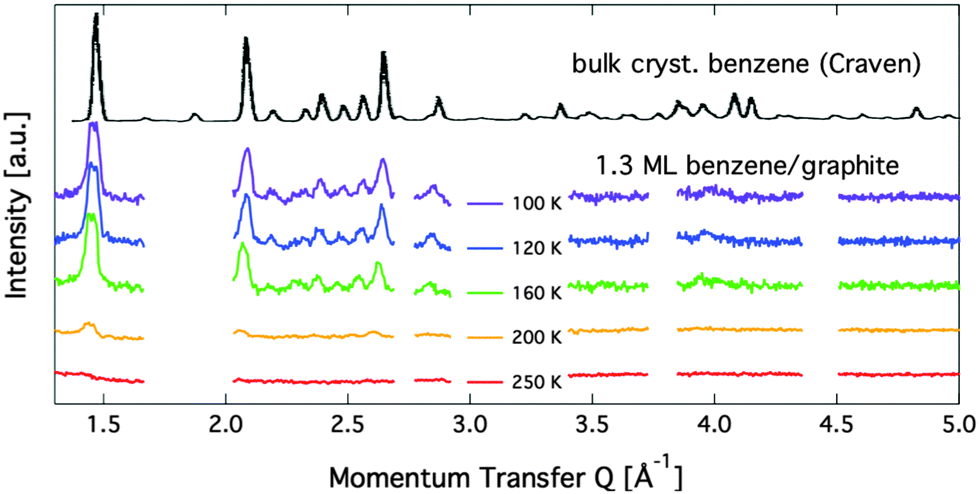 | ||
| Fig. 7 Diffraction patterns for varying temperatures at a relative benzene coverage of 1.3 ML. The black line on top shows a neutron diffraction pattern from bulk benzene at 105 K that was originally obtained by Craven et al.14 | ||
4 Conclusions
We have performed neutron diffraction studies of deuterated benzene mono- and multilayers on exfoliated graphite. For a low coverage of 0.15 ML no ordered structure was observed. For coverages between 0.5 ML and 1 ML, the existence of a flat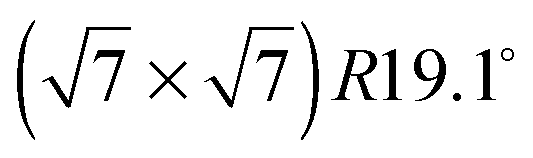 structure has been observed that can be shown to be caused by ordered islands of at least 200 Å diameter. The ordered islands gradually melt at temperatures well below the desorption temperature and we find that the melting temperature changes with coverage. The islands disappear at 140 K for a coverage of 0.5 ML and at 160 K for a coverage of 0.8 ML. The structure does not compress, within the experimental error bar, upon increasing the coverage. At a coverage of 1.3 ML, a bulk crystalline benzene structure appears rather than a second layer. This structure differs strongly from a compressed ordered structure suggested by other authors45 and indicates the formation of benzene nano-clusters in addition to an intact monolayer.
structure has been observed that can be shown to be caused by ordered islands of at least 200 Å diameter. The ordered islands gradually melt at temperatures well below the desorption temperature and we find that the melting temperature changes with coverage. The islands disappear at 140 K for a coverage of 0.5 ML and at 160 K for a coverage of 0.8 ML. The structure does not compress, within the experimental error bar, upon increasing the coverage. At a coverage of 1.3 ML, a bulk crystalline benzene structure appears rather than a second layer. This structure differs strongly from a compressed ordered structure suggested by other authors45 and indicates the formation of benzene nano-clusters in addition to an intact monolayer.
Acknowledgements
We would like to thank Dr Stuart Clarke and Adam Brewer for their elaborate help and their allocation of the structure analysis software. We would also like to thank Dr Hans Lauter for assistance with the adsorption system used in the neutron spectroscopy and diffraction studies. E.B. is grateful for financial support from the graduate school of the University of Grenoble.References
- S. Gokhale, P. Trischberger, D. Menzel, W. Widdra, H. Dröge, H.-P. Steinrück, U. Birkenheuer, U. Gutdeutsch and N. Rösch, J. Chem. Phys., 1998, 108, 5554–5564 CrossRef CAS PubMed.
- W. Liu, V. G. Ruiz, G.-X. Zhang, B. Santra, X. Ren, M. Scheffler and A. Tkatchenko, New J. Phys., 2013, 15, 053046 CrossRef.
- B. Boddenberg and J. A. Moreno, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1976, 31, 853–854 Search PubMed.
- R. Stockmeyer and H. Stortnik, Surf. Sci., 1979, 81, L315–L318 CrossRef CAS.
- P. Meehan, T. Rayment, R. K. Thomas, G. Bomchil and J. W. White, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2011–2016 RSC.
- M. Monkenbusch and R. Stockmeyer, Ber. Bunsen-Ges., 1980, 84, 808–814 CrossRef CAS PubMed.
- P. Jakob and D. Menzel, Surf. Sci., 1989, 220, 70–95 CrossRef CAS.
- H. Hedgeland, P. Fouquet, A. P. Jardine, G. Alexandrowicz, W. Allison and J. Ellis, Nat. Phys., 2009, 5, 561–564 CrossRef CAS.
- P. Fouquet, M. R. Johnson, H. Hedgeland, A. P. Jardine, J. Ellis and W. Allison, Carbon, 2009, 47, 2627–2639 CrossRef CAS PubMed.
- D. C. Glass, C. N. Gray, G. G. Adams, R. W. Manuell and J. A. Bisby, Toxicol. Ind. Health, 2001, 17, 113–127 CrossRef CAS.
- M. T. Smith, Annu. Rev. Public Health, 2010, 31, 133–148 CrossRef PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- E. G. Cox, Rev. Mod. Phys., 1958, 30, 159–162 CrossRef CAS.
- C. J. Craven, P. D. Hatton, C. J. Howard and G. S. Pawley, J. Chem. Phys., 1993, 98, 8236–8243 CrossRef CAS PubMed.
- G. A. Jeffrey, J. R. Ruble, R. K. McMullan and J. A. Pople, Proc. R. Soc. London, Ser. A, 1987, 414, 47–57 CrossRef CAS.
- T. F. Headen, C. A. Howard, N. T. Skipper, M. A. Wilkinson, D. T. Bowron and A. K. Soper, J. Am. Chem. Soc., 2010, 132, 5735–5742 CrossRef CAS PubMed.
- B. Boddenberg and J. A. Moreno, J. Phys., Colloq., 1977, 38, C4-52–C4-55 CrossRef.
- U. Bardi, S. Magnanelli and G. Rovida, Surf. Sci. Lett., 1985, 165, L7–L11 Search PubMed.
- U. Bardi, S. Magnanelli and G. Rovida, Langmuir, 1987, 3, 159–163 CrossRef CAS.
- I. Gameson and T. Rayment, Chem. Phys. Lett., 1986, 123, 150–153 CrossRef CAS.
- B. Boddenberg and R. Grosse, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1986, 41, 1361–1368 Search PubMed.
- R. Grosse and B. Boddenberg, Z. Phys. Chem., 1987, 152, 1–12 CrossRef CAS.
- C. Bondi and G. Taddei, Surf. Sci., 1988, 203, 587–593 CrossRef CAS.
- A. Vernov and W. A. Steele, Langmuir, 1991, 7, 2817–2820 CrossRef CAS.
- A. Vernov and W. A. Steele, Langmuir, 1991, 7, 3110–3117 CrossRef CAS.
- R. Hentschke and B. L. Schürmann, Surf. Sci., 1992, 262, 180–188 CrossRef CAS.
- M. A. Matties and R. Hentschke, Langmuir, 1996, 12, 2495–2500 CrossRef CAS.
- M. A. Matties and R. Hentschke, Langmuir, 1996, 12, 2501–2504 CrossRef CAS.
- N. Klomkliang, D. D. Do, D. Nicholson, C. Tangsathitkulchai and A. Wongkoblap, Chem. Eng. Sci., 2012, 69, 472–482 CrossRef CAS PubMed.
- S. D. Chakarova-Käck, E. Schröder, B. I. Lundqvist and D. C. Langreth, Phys. Rev. Lett., 2006, 96, 146107 CrossRef.
- E. P. Gilbert, P. A. Reynolds and J. W. White, J. Chem. Soc., Faraday Trans., 1998, 94, 1861–1868 RSC.
- C. Bockel, J. Coulomb and N. Dupont-Pavlovsky, Surf. Sci., 1982, 116, 369–379 CrossRef CAS.
- T. Hansen, P. Henry, H. Fischer, J. Torregrossa and P. Convert, Meas. Sci. Technol., 2008, 19, 034001 CrossRef.
- D. Brochier, ILL Technical Report 77/74, Institut Laue-Langevin technical report, 1977.
- B. Farago, Physica B, 1998, 241–243, 113–116 Search PubMed.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- B. E. Warren, Phys. Rev., 1941, 59, 693–698 CrossRef CAS.
- T. Arnold, S. Chanaa, S. M. Clarke, R. E. Cook and J. Z. Larese, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 085421 CrossRef.
- A. Y. Brewer, T. Friscic, L. M. Overvoorde, J. E. Parker, C. N. Richardson and S. M. Clarke, Mol. Phys., 2012, 111, 73–79 CrossRef.
- S. Ergun and M. Berman, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 12–17 CrossRef.
- K. W. Herwig, J. C. Newton and H. Taub, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 15287–15297 CrossRef CAS.
- T. Arnold, R. K. Thomas, M. A. Castro, S. M. Clarke, L. Messe and A. Inaba, Phys. Chem. Chem. Phys., 2002, 4, 345–351 RSC.
- I. Calvo-Almazán, E. Bahn, M. M. Koza, M. Zbiri, M. Maccarini, M. T. F. Telling, S. Miret-Artés and P. Fouquet, Carbon, 2014, 79, 183–191 CrossRef PubMed.
- V. V. Gobre and A. Tkatchenko, Nat. Commun., 2013, 4, 2341 Search PubMed.
- D. Do, D. Nicholson and H. Do, J. Colloid Interface Sci., 2008, 325, 7–22 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2014 |