
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shining new light on the multifaceted dissociative photoionisation dynamics of CCl4
Jonelle
Harvey†
a,
Richard P.
Tuckett
*a and
Andras
Bodi
*b
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.p.tuckett@bham.ac.uk
bMolecular Dynamics Group, Swiss Light Source, Paul Scherrer Institut, Villigen 5232, Switzerland. E-mail: andras.boedi@psi.ch
First published on 12th August 2014
Abstract
Internal energy selected carbon tetrachloride cations have been prepared by imaging photoelectron photoion coincidence (iPEPICO) spectroscopy using synchrotron vacuum ultraviolet radiation. The threshold photoelectron spectrum shows a newly observed vibrational progression corresponding to the ν2(e) scissors mode of CCl4+ in the third, ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 2E band. Ab initio results on the first four doublet and lowest-lying quartet electronic states along the Cl3C+–Cl dissociation coordinate show the state to be strongly bound, and support its relative longevity. The
2E band. Ab initio results on the first four doublet and lowest-lying quartet electronic states along the Cl3C+–Cl dissociation coordinate show the state to be strongly bound, and support its relative longevity. The ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2T1 and à 2T2 cationic states, on the other hand, are barely bound and dissociate promptly. The
2T1 and à 2T2 cationic states, on the other hand, are barely bound and dissociate promptly. The ![[C with combining tilde]](https://www.rsc.org/images/entities/char_0043_0303.gif) 2T2 state may intersystem cross to the quartet ã state, which dissociates to a triplet state of the CCl3+ fragment ion. This path is unique among analogous MX4+ (M = C, Si, Ge; X = F, Cl, Br) systems, among which several have been shown to have long-lived states, which decay by fluorescence. The breakdown diagram, recorded here for the first time for the complete valence photoionisation energy range of CCl4, is interpreted in the context of literature based and CBS-QB3, G4, and W1U computed dissociative photoionisation energies. No Cl2-loss channel is observed in association with the CCl2+ or CCl+ fragments below the 2 or 3 Cl-loss reaction energies, and Cl2 loss is unlikely to be a major channel above them. The breakdown diagram is modelled based on the calculated dissociative photoionisation onsets and assuming a statistical redistribution of the excess energy. The model indicates that dissociation is not impulsive at higher energies, and confirms that the 2T2 state of CCl4+ forms triplet-state CCl3+ fragments with some of the excess energy trapped as electronic excitation energy in CCl3+.
2T2 state may intersystem cross to the quartet ã state, which dissociates to a triplet state of the CCl3+ fragment ion. This path is unique among analogous MX4+ (M = C, Si, Ge; X = F, Cl, Br) systems, among which several have been shown to have long-lived states, which decay by fluorescence. The breakdown diagram, recorded here for the first time for the complete valence photoionisation energy range of CCl4, is interpreted in the context of literature based and CBS-QB3, G4, and W1U computed dissociative photoionisation energies. No Cl2-loss channel is observed in association with the CCl2+ or CCl+ fragments below the 2 or 3 Cl-loss reaction energies, and Cl2 loss is unlikely to be a major channel above them. The breakdown diagram is modelled based on the calculated dissociative photoionisation onsets and assuming a statistical redistribution of the excess energy. The model indicates that dissociation is not impulsive at higher energies, and confirms that the 2T2 state of CCl4+ forms triplet-state CCl3+ fragments with some of the excess energy trapped as electronic excitation energy in CCl3+.
Introduction
A chemical reaction can take place if there is more kinetic energy in one degree of freedom, the reaction coordinate, than the activation energy. Therefore, unveiling internal energy flows and their effect on the reactive flux are central to understanding chemical dynamics. Among different electronically excited states, the reactive flux is guided by internal conversion and intersystem crossings. The non-ergodic nature of the former has been the subject of a recent minireview by Sølling et al.1 Among other phenomena, they discuss the localization of the internal energy in one degree of freedom, e.g., a reactive mode, as dictated by internal conversion as well as the induction of internal conversion by a certain molecular motion. In the neutral, such processes can be followed directly by femtosecond pump–probe spectroscopy.2,3 In the ionic manifold, internal energy selected parent ions can be prepared using threshold photoionisation and tunable vacuum ultraviolet (VUV) light.4 Their unimolecular dissociation dynamics can then be studied by imaging photoelectron photoion coincidence (iPEPICO) spectroscopy at the VUV beamline of the Swiss Light Source.5,6 Dissociative photoionisation thresholds and measured dissociation rates in the 103–107 s−1 range can routinely be interpreted by statistical theory,7 to derive thermochemical data with accuracy sometimes below 1 kJ mol−1.8,9 Statistical theory assumes that the complete phase space is explored by the system, and the dissociation rate essentially corresponds to the probability that sufficient kinetic energy is present in the reaction coordinate.Statistical processes pre-suppose the presence of a bound state of the parent cation. As, at any internal energy, the phase space volume of the ground state is much larger than that of the electronically excited states, dissociation normally takes place on this potential energy surface, following internal conversion from higher-lying states. Thus, the assumption of statistical behaviour relies on strong coupling between different degrees of freedom. Anharmonicity is the driving force of intramolecular vibrational energy redistribution (IVR), which explains why non-statistical behaviour is observed for low-barrier isomerization reactions of large molecules in which the internal energy per oscillator, and thus, the anharmonicity, are small at the activation energy.10,11 Electronic degrees of freedom are coupled by conical intersections. The large density of states in cations allows for the observation of Franck–Condon prohibited non-resonant transitions in threshold photoionisation, and ensures that the electronic excitation energy is also available for dissociation.12 In recent years, we made use of the tunability and energy resolution of the iPEPICO endstation and revisited several halogenated systems to study their dissociative photoionisation properties. In agreement with previous observations, we have found a number of non-statistical processes. As best shown in the case of the fluoroethene cations, C2H4−nFn+ (n = 1–4), new experimental and theoretical approaches have yielded a deeper understanding of the underlying reaction mechanism than was possible before.13,14 These cations dissociate statistically in the low-energy region along several parallel dissociation channels. In the mid- to high-valence ionisation energy region, they lose F atoms in a non-statistical process. In mono-, di-, and trifluoroethene, it was shown that -state cations can undergo internal conversion to the state, either on a bound or a repulsive domain along the C–F bond stretch coordinate. Should the latter take place, the product state cation suffers non-statistical F-loss promptly. Long-lived -state ions, on the other hand, can redistribute their internal energy, and dissociate statistically. In C2F4+, the electronically excited à state is disconnected from the ground state and only correlates with ground state C2F3+ + F products. Hence, the à state establishes a second dissociation regime, which is disconnected from the low-energy regime solely because of an absence of electronic coupling.
In this paper, we report on the dissociative photoionisation dynamics of carbon tetrachloride. We show that, while the dissociation is impulsive close to the onset of ionisation, it takes on a statistical character as coupling to the repulsive ground state of the parent cation gets weaker with higher electronic excitation.
Another non-statistical decay process should now be mentioned, namely fluorescence.15 The cations of analogous halogenated compounds are known to fluoresce,16 but it is often difficult to identify fluorescence competing with fragmentation processes.17 If dissociation is energetically prohibited, the only decay process for excited electronic states in the gas phase is fluorescence, and such ions will indeed fluoresce. Radiative decay decreases the available energy for fragmentation processes. As will be discussed later, unlike in analogous MX4+ ions, fluorescence processes have not been observed in CCl4+, and fluorescence cannot compete effectively with dissociative photoionisation whenever the latter is energetically allowed.
Several of the carbon group tetrahalogenides of MX4 (M = C, Si, Ge; X = F, Cl, Br, I) are unstable with respect to photoionisation, i.e. the ground electronic state of MX4+ in the Franck–Condon window is unbound, but the cations also possess bound excited electronic states. Carbon tetrachloride, CCl4 dissociatively photoionises into daughter ions CCl3+, CCl2+, CCl+ in the 11–30 eV photon energy range, and a very weak CCl4+ peak can only be observed under special circumstances.18,19 Similar to CCl4+, the lower electronic states of CF4+ were also proposed to be repulsive, resulting in a significant force towards C–F bond length increase in the Franck–Condon allowed photoionisation energy range, and giving rise to an impulsive dissociation to form CF3+ + F + e−.20 Kinugawa et al. published photoion and photoelectron angular distribution data on VUV ionisation of CF4 and CCl4, and, based on the observed correlation, suggested that electron and nuclear motion may take place on similar time-scales.21 As seen from our work on fluoroethenes and from the amount of new thermochemical information derived from the breakdown diagrams of halogenated methanes,22 the higher internal energy resolution of the iPEPICO experiment can offer further insights into dissociative photoionisation.
In threshold photoion photoelectron coincidence (TPEPICO) spectroscopy, virtually zero kinetic energy electrons are detected and used as the start signal for ion time-of-flight (TOF) analysis. Method developments in the last decade included the application of velocity map imaging for high collection efficiency,23 slow extraction fields for high residence times to measure dissociation rates,4 the use of synchrotron radiation6,24,25 together with fast position-sensitive detectors and triggerless data acquisition26 setups, and, most recently, double imaging experiments.27,28 In addition to yielding more accurate onsets and broadening the spectrum of possible samples, these improvements have led to a refined understanding of threshold photoionisation,12 as well as detailed models for the non-statistical aspects of the dissociative photoionisation of fluoroethenes,13,14 halogenated tin compounds29 and methanol.30 Consequently, it seemed fitting to re-visit the impulsive halogen loss from CCl4+, a system known to exhibit non-statistical behaviour, in more detail using the iPEPICO experiment.31
Experimental methods
Experiments were performed using the iPEPICO endstation at the VUV beamline of the Swiss Light Source (SLS) at the Paul Scherrer Institut in Villigen, Switzerland. The setup has been described in detail in several publications and only an overview is given here.5,6,26 Carbon tetrachloride vapour was introduced effusively into the ionisation chamber of the iPEPICO endstation at room temperature, where it was ionised by the incident synchrotron radiation. Typical operating pressures within the chamber were 7–9 × 10−7 mbar. The synchrotron radiation is dispersed by a grazing-incidence monochromator with laminar 600 and 1200 mm−1 gratings for photon energies below and above 15 eV, respectively. The photon energy at a resolving power, E/ΔE, of ca. 5000 was calibrated against the 11s′ to 14s′ argon autoionisation lines in both first and second order. In addition to the laminar gratings, higher harmonic radiation is further suppressed by a compact differentially pumped rare-gas filter operating at 10 mbar. Pure neon was used in the gas filter, applicable to the 11–21 eV photon energy range used herein.Photoelectrons are extracted with a continuous field and are velocity map imaged onto a DLD40 Roentdek position sensitive delay-line detector. Threshold electrons are focussed into a small centre spot on the detector with a kinetic energy resolution better than 1 meV. Photoions are extracted in the opposite direction by the same, constant 120 V cm−1 field in the first 5 cm long acceleration region. Afterwards, they are accelerated further to achieve space focussing, pass through a 50 cm long field-free drift region, then are detected by a Jordan TOF C-726 microchannel plate detector. Photoelectrons are position and time stamped, and serve as the start signal for the ion time-of-flight analysis.
Some energetic (or hot) electrons are produced with negligible off-axis momentum, and are also focussed into the centre spot, contaminating the true threshold signal. The hot electron contamination is accounted for by a subtraction process,32,33 whereby a small ring area around the centre is assumed to represent the hot electron background in the centre, and the time-of-flight mass spectrum in coincidence with ring electrons is multiplied with a factor corresponding to the centre-to-ring area ratio and subtracted from the mass spectrum in coincidence with the central electrons. Photoion mass selected threshold photoelectron spectra (ms-TPES) indicate the threshold photoionisation yields of different ions as a function of photon energy. The fractional ion abundances can be plotted as a function of photon energy in the breakdown diagram, whereas all electrons can be used to plot the threshold photoelectron spectrum of the sample.
Computational methods
Calculations were carried out using the Gaussian 0934 as well as the QChem 4.0.1 programs.35 The former was used to determine the reaction energies of different dissociative photoionisation channels of CCl4 using the CBS-QB3, G4 and W1U composite methods. DFT geometry optimizations at the B3LYP/6-311+G(d) level mapped the potential energy surface. In these geometry optimizations, two bond lengths were scanned in C3v symmetry, namely a single C–Cl bond length for the leaving chlorine atom, and a C–Cl bond length in the remaining CCl3+ fragment. The reaction coordinate was then defined as a minimum energy path on this two-dimensional surface.EOM-IP-CCSD (equation-of-motion coupled-cluster singles and doubles for ionisation potentials) calculations36 were carried out with the cc-pVTZ basis set to obtain the energies of the doublet 2, 2Ã, 2, and 2 cation states along the C–Cl dissociation coordinate in CCl4+. Second order Møller–Plesset perturbation theory (MP2) was also used along the same path to calculate the energy difference between the doublet 2 and the quartet 4ã states, which was then added to the ground state reaction energy curve to obtain a path to Cl-loss on the lowest-lying quartet surface.
Results and discussion
Threshold photoelectron spectrum
The photoelectron spectrum of CCl4 was previously measured by von Niessen et al.,37 and our threshold photoelectron spectrum is shown in Fig. 1. The photoelectron peak for the 2T1 state is broad and structureless, as is expected for a vertical transition into an unbound domain of the cation potential energy surface. The spin–orbit splitting of the next, à 2T2 peak, ca. 0.22 eV, is, however, readily observable. The higher degenerate and hence more intense G3/2 component has lower energy than the lower degenerate E5/2 component, with an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively, confirming that the sign of the spin–orbit splitting parameter is negative.38 No vibrational structure is observed in the à peak. This either indicates a short lifetime and/or large geometry change upon ionisation. To the best of our knowledge, no one has yet reported the vibrational progression clearly seen in the third photoelectron band belonging to the 2E state (inset in Fig. 1). The 0.025 eV (200 cm−1) splitting suggests that the ν2(e) scissors mode is active, as the corresponding harmonic frequency in neutral CCl4 is 214 cm−1 (B3LYP/cc-pVTZ+d result). This means that the geometry change when ionising to the state is only moderate, and the Franck–Condon factors for ionisation into the potential energy well are large. Excitation of non-totally-symmetric vibrations such as ν2 of e symmetry are formally forbidden in photoelectron spectroscopy unless the point group of the molecule changes. Therefore, this mode can only gain intensity through Jahn–Teller distortion of the doubly-degenerate 2E electronic state. Herzberg39 has shown that the symmetry of the vibration which causes the distortion must be contained in the direct product of the symmetry of the degenerate electronic state with itself. In Td geometry, E ⊗ E = A1 + A2 + E, therefore the ν2(e) mode possesses the correct symmetry to cause Jahn–Teller distortion. At the same time, internal conversion of to the shorter-lived à and states, surely driven by its Jahn–Teller distorted structure, must be slow enough that the -state potential energy surface can support bound nuclear vibrational wave functions.
:1, respectively, confirming that the sign of the spin–orbit splitting parameter is negative.38 No vibrational structure is observed in the à peak. This either indicates a short lifetime and/or large geometry change upon ionisation. To the best of our knowledge, no one has yet reported the vibrational progression clearly seen in the third photoelectron band belonging to the 2E state (inset in Fig. 1). The 0.025 eV (200 cm−1) splitting suggests that the ν2(e) scissors mode is active, as the corresponding harmonic frequency in neutral CCl4 is 214 cm−1 (B3LYP/cc-pVTZ+d result). This means that the geometry change when ionising to the state is only moderate, and the Franck–Condon factors for ionisation into the potential energy well are large. Excitation of non-totally-symmetric vibrations such as ν2 of e symmetry are formally forbidden in photoelectron spectroscopy unless the point group of the molecule changes. Therefore, this mode can only gain intensity through Jahn–Teller distortion of the doubly-degenerate 2E electronic state. Herzberg39 has shown that the symmetry of the vibration which causes the distortion must be contained in the direct product of the symmetry of the degenerate electronic state with itself. In Td geometry, E ⊗ E = A1 + A2 + E, therefore the ν2(e) mode possesses the correct symmetry to cause Jahn–Teller distortion. At the same time, internal conversion of to the shorter-lived à and states, surely driven by its Jahn–Teller distorted structure, must be slow enough that the -state potential energy surface can support bound nuclear vibrational wave functions.
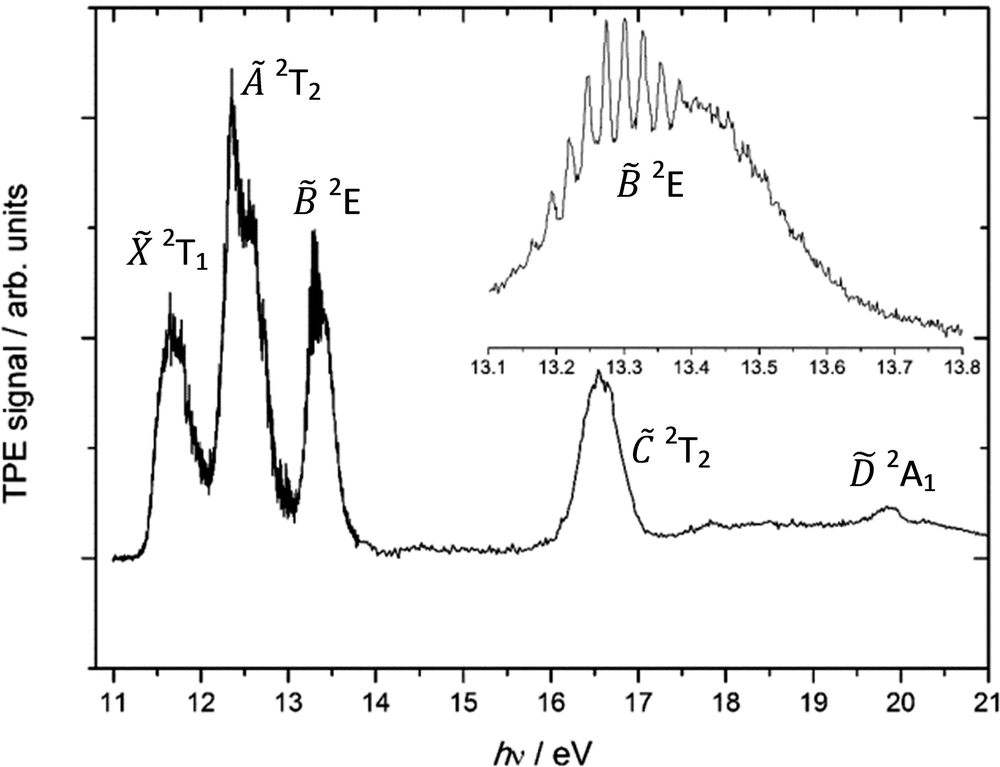 | ||
| Fig. 1 Threshold photoelectron spectrum of CCl4. The inset shows an enlarged scale TPES of the third band belonging to the state from 13.1 to 13.8 eV with 0.002 eV resolution. | ||
In the Koopmans picture, these first three states correspond to ionisation from the chlorine 3pπ lone pair orbitals with symmetry e + t1 + t2. The next band in the TPES of CCl4, the 2T2 peak, corresponds to ionisation from a C–Cl bonding orbital and is centred at 16.58 eV, followed by the band corresponding to ionisation to the highest-lying valence electronic state of the parent cation, ![[D with combining tilde]](https://www.rsc.org/images/entities/char_0044_0303.gif) 2A1, centred at hν = 19.86 eV.
2A1, centred at hν = 19.86 eV.
Breakdown diagram and calculated energetics
Based on the photoion mass selected threshold photoelectron signal, fractional fragment ion abundances have been determined and are plotted as a function of photon energy in the 14–21 eV range in Fig. 2. As with CF4, it has long been accepted that CCl4 dissociatively photoionises and the intact parent ion can scarcely be detected.21 Thus, in agreement with previous reports,31 only CCl3+ with 100% fractional abundance was observed in the, Ã, and peaks of the spectrum below 14 eV. Between 15 and 16 eV, the CCl3+ signal disappears and signal due to CCl2+ rises, only for the heavier CCl3+ fragment ion to make a brief and weak return between 17 and 18 eV. This observation is strong evidence that dissociation cannot be impulsive in the Franck–Condon gap just below the 2T2 peak. If most of the excess energy could be released as kinetic energy in the dissociation, there would always be low internal energy CCl3+ intermediates with insufficient energy to dissociate further; this contradicts the negligible CCl3+ signal between 16 and 17 eV. For the same reason, the parent ion cannot lose internal energy by fluorescent decay. At higher energies, the CCl2+ signal decays and the 50% crossover energy with CCl+ lies at 19.2 eV. The breakdown diagram remains unaffected by the presence of the 2A1 peak in the TPES at 19.9 eV, and the CCl+ fractional abundance reaches almost 100% at 20.5 eV. C+, Cl+ and Cl2+ have not been observed in the energy range studied, which is also well below the 27.99 eV double photoionisation threshold (CBS-QB3 calculation to form triplet ground-state CCl42+).
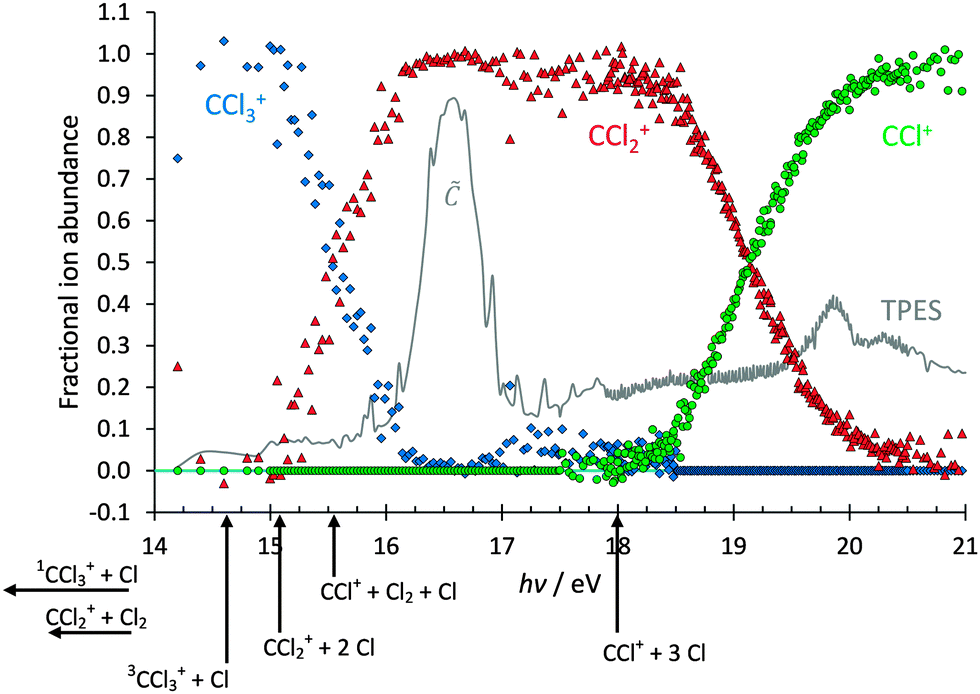 | ||
| Fig. 2 Breakdown diagram of CCl4 in the 14–21 eV photon energy range. Below 14 eV, only CCl3+ was observed in the mass spectrum. The W1U calculated 0 K dissociative photoionisation energies are also shown. | ||
The breakdown diagram can be interpreted in the context of energetically allowed dissociation reactions. Based on the 0 K heats of formation of CCl4, CCl3+ and Cl determined from a global fit to a halomethane thermochemical network,22 we derive the CCl4 → CCl3+ + Cl + e− threshold at 0 K to be 11.021 ± 0.047 eV. Furthermore, Rademann et al.41 measured ΔfHo298K(CCl2+) = 1107.9 ± 7.5 kJ mol−1, which can be converted to 1106.9 ± 7.5 kJ mol−1 at 0 K using the W1U calculated thermal enthalpy of CCl2+, 11.18 kJ mol−1, and the published thermal enthalpies of C and Cl2 of 1.05 and 9.18 kJ mol−1,40 respectively. Thus, the 0 K dissociative photoionisation threshold for CCl4 → CCl2+ + 2Cl + e− lies at 14.926 ± 0.085 eV. In order to confirm these values, we have calculated the thresholds for dissociative photoionisation of CCl4 to form CCl3+, CCl2+ and CCl+, including the formation of the lowest triplet (excited) state of CCl3+, using the CBS-QB3, G4 and W1U composite methods. The results are summarized in Table 1.
| CCl4 − e− → | E 0/eV | |||
|---|---|---|---|---|
| CBS-QB3 | G4 | W1U | Literaturea | |
| a See text for derivation. b The Cl2 energy was calculated, and the Cl energy was obtained as half the Cl2 energy plus the 0 K 1/2Cl2 → Cl reaction energy of 119.6 kJ mol−1.40 | ||||
| 1CCl3+ + Clb | 11.154 | 11.057 | 11.074 | 11.021 ± 0.047 |
| 3CCl3+ + Cl | 14.658 | 14.693 | 14.711 | |
| CCl2+ + 2Cl | 15.199 | 15.055 | 15.087 | 14.926 ± 0.085 |
| CCl2+ + Cl2 | 12.719 | 12.576 | 12.607 | |
| CCl+ + 3Cl | 18.126 | 18.006 | 18.015 | |
| CCl+ + Cl2 + Cl | 15.647 | 15.526 | 15.535 | |
The agreement between computational methods and literature data is reassuringly good. Furthermore, the rise of the CCl2+ and CCl+ signals (at ca. 15.0 and 18.1 eV, respectively) in the breakdown diagram corresponds to the calculated thermochemical onsets of 2Cl and 3Cl losses. Because of the uncertainty in some of the energetics and the fact that the CCl2+ appearance energy is in a Franck–Condon gap, inaccessible with non-threshold ionisation, previously the possibility of Cl2 formation could not be ruled out completely.21,42 Based on these results, we can now say that no Cl2 loss takes place below the CCl2+ + 2Cl + e− or CCl+ + 3Cl + e− thresholds. Nonetheless, preliminary reaction path calculations, similar to the Cl-loss channels discussed in the next section, indicate that there may be unbound electronic states of CCl4+ leading to production of CCl2+ + Cl2. While the smaller-than-expected kinetic energy release (see breakdown diagram fits below) indicates that these electronic states do not play a significant role in the dissociative photoionisation mechanism, they may play a minor role above the CCl2+ + 2Cl + e− and CCl+ + 3Cl + e− thresholds. However, based on the computed energetics and the appearance of the breakdown diagram, we can assume that little if any Cl2 is produced in the valence dissociative photoionisation of CCl4.
Cl-loss potential energy curves, CCl4+ → CCl3+ + Cl
Notwithstanding symmetry, the electronic ground state potential energy surface of CCl4+ is a function of nine internal coordinates. To address fully the Cl-loss reaction on the ground and electronic excited states of CCl4+, the electronic degree of freedom and spin–orbit effects would have to be included. Evidently, the computational problem needs to be simplified to become tractable. First, we have restricted the symmetry of the geometries along the reaction coordinate to C3v to correspond to that of the products. There are then only three internal coordinates: the C–Cl bond length in the CCl3+ fragment ion, the C–Cl bond length in the breaking bond, and the angle between the C–Cl bonds in CCl3+ with the rotational symmetry axis which converges to 90° as the Cl leaves and planar ground-state singlet 1CCl3+ is formed. The two bond lengths were scanned and the bond angle optimized to obtain the ground state potential energy surface of CCl4+ as shown in Fig. 3. The Cl-loss reaction coordinate was then defined as the minimum energy path connecting the shallow tetrahedral minimum in C3v symmetry with the 1CCl3+ + Cl products. Thereafter, the ground and excited doublet state energies were calculated along this path using EOM-IP-CCSD, and the lowest quartet state curve was obtained by calculating the doublet–quartet splitting using MP2 and adding it to the doublet ground-state energy. The results are shown in Fig. 4.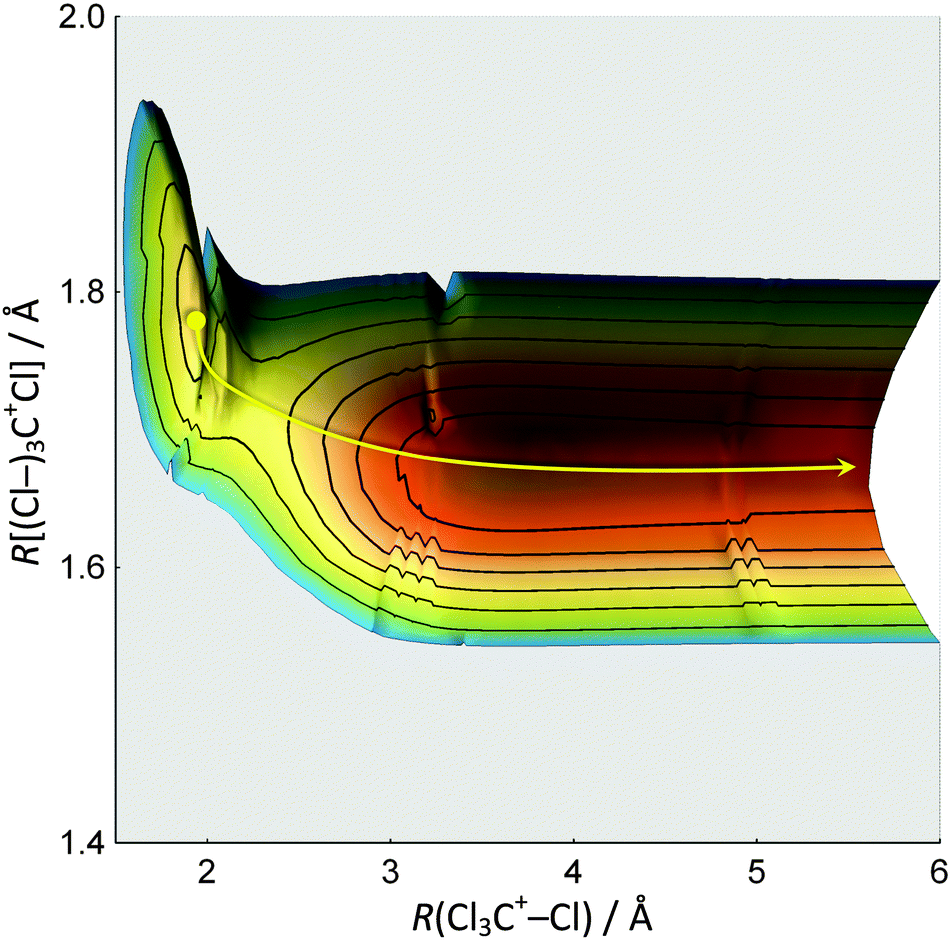 | ||
| Fig. 3 Ground electronic state potential energy surface of Cl-loss from CCl4+ in C3v symmetry, shown from the energy minimum up to 1 eV. The Td structure at a bond length of about 1.8 Å corresponds to a local minimum in C3v symmetry. The loss of a Cl is simultaneous with the contraction of the other C–Cl bonds, and proceeds over a small barrier to yield the minimum energy CCl3+ + Cl products. The minimum energy path on this surface is also shown. | ||
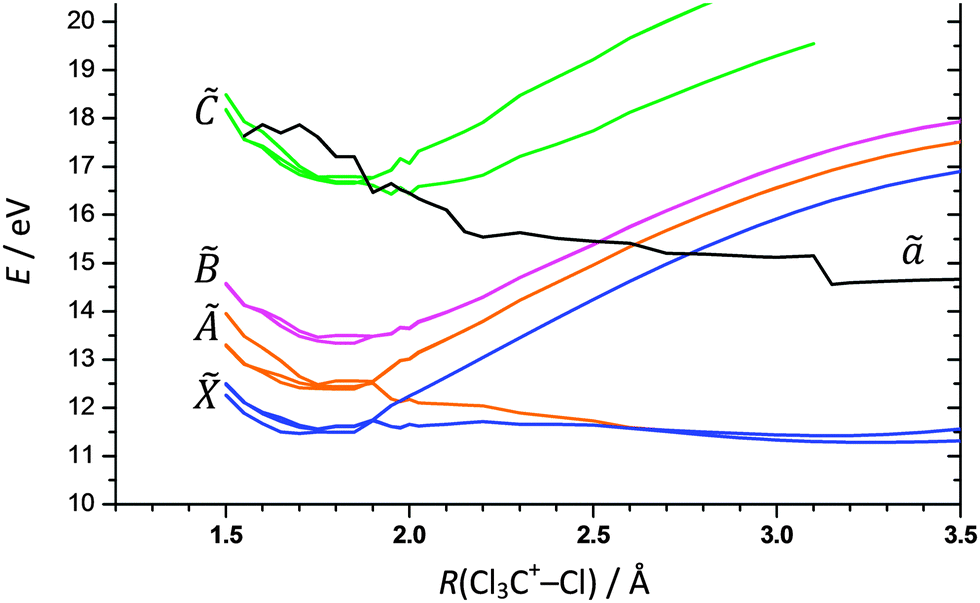 | ||
| Fig. 4 Calculated potential energy curves for the ground and first three excited doublet and the lowest quartet states along the Cl3C+–Cl dissociation coordinate. | ||
The triply-degenerate 2T1 state of CCl4+ splits into a doubly-degenerate unbound and a singly-degenerate bound state along the reaction coordinate. On the other hand, the triply degenerate à 2T2 state splits into a doubly-degenerate bound and a singly-degenerate repulsive state; the latter state is degenerate with the repulsive component of the state. Strictly speaking, these states are not purely repulsive. The lowest lying ion curve appears to be bound at tetrahedral geometry but the potential well is probably insufficient to support a vibrational state. Further out, the electronic energy plateaus and leads to a second shallow minimum at ca. 3.25 Å. However, in the Franck–Condon envelope, the internal energy of the ion is more than sufficient to lead to prompt dissociation. At higher energies, the 2E and 2T2 doublet states of CCl4+ appear to be bound and do not correlate with low-energy CCl3+ + Cl products. These dissociative potential energy surfaces are consistent with the absence of vibrational fine structure in the first two bands of the TPES of CCl4 and the observation of the ν2(e) scissors mode in the 2E state. The calculated potential energy curve is not as smooth for the lowest quartet state of CCl4+, but it shows an intersection with the 2T2 state around 16 eV. Furthermore, the quartet state leads to the production of triplet 3CCl3+ + Cl. Keeping in mind that the 2E state, but not the 2T2 state, of CCl4+ supports bound vibrational wave functions, there is some evidence from the TPES that, among the first four doublet CCl4+ states, 2E is the longest lived. Smith et al. proposed that CCl4+ behaves analogously to CF4+, suggesting that dissociation from the 2E state occurs via internal conversion into the à 2T2 state.42 This conclusion is supported by the experimental and computational results reported here.
Statistical modelling and dissociation mechanism
In a statistical dissociation, the excess energy, i.e. the energy available to the system above the dissociation threshold, is partitioned among the internal degrees of freedom of the fragments and their translational kinetic energies according to the respective phase space volumes.7,43 Aside from spin–orbit and electronically excited states, the chlorine atom cannot accommodate internal energy, therefore most of the excess energy is expected to stay in the fragment ion produced from CCl4+. In terms of which vibrational modes of CCl3+ might be excited, one would only expect the umbrella motion and the symmetric stretch to be active, as the reaction coordinate is a combination of C–Cl bond contraction, Cl3C+–Cl bond elongation, and increasing planarity of CCl3+ (see also previous section).44 During impulsive dissociations a significant amount of the excess energy can be released as kinetic energy of the fragments,45 as the force and the resulting acceleration along the reaction coordinate is such that the internal energy cannot be redistributed quickly enough before fragmentation is complete. High kinetic energy release leads to low internal energy fragments, which may not have enough energy to dissociate further, and are stable well above their dissociative photoionisation energies.As mentioned earlier, the breakdown diagram of CCl4 in the 14–21 eV photon energy range (Fig. 2), observed for the first time with better than 10 meV internal energy resolution and good signal-to-noise ratio, appears to tell a different story from that of an impulsive dissociation. The CCl3+/CCl2+ and CCl2+/CCl+ crossovers are quite sharp, and do not suggest suprastatistical kinetic energy release. The weak but reproducible recurrence of the CCl3+ signal between 17 and 18 eV indicates a decrease in the CCl3+ internal energy as the photon energy increases, and suggests that there are two dissociation mechanisms at play.
Unlike the equivalent 2T2 state of CF4+, SiCl4+ and GeCl4+ where fluorescence is a major decay channel with lifetimes of 9, 38 and 65 ns, respectively,46,47 the 2T2 state of CCl4+ does not appear to decay radiatively. Its apparently short, sub-ns lifetime, together with an available intersystem crossing path onto the lowest (ã) quartet state which dissociates to triplet 3CCl3+, offers an alternative dissociation path to fluorescence, direct dissociation, or internal conversion to lower doublet states. Because of the different level spacing and the required closeness of the and ã states in energy near the minimum of the state, such a fast dissociative decay route as opposed to radiative decay may be unique to CCl4+ within this family of compounds. Furthermore, such fast intersystem crossing paths have been detected in water,9,48 and the larger spin–orbit coupling in CCl4+ makes them just as plausible here. If the 2T2 state of CCl4+ decays in this way, the energy of the singlet–triplet gap in CCl3+ of ca. 3.6 eV (Table 1), will be ‘trapped’ in the fragment ion and be unavailable for kinetic energy release. If singlet CCl3+ production is allowed again at higher energies, the effective internal energy of the fragment ion may indeed decrease with increasing photon energy, as appears to be observed in our experiment.
We have modelled the breakdown diagram (Fig. 2) assuming the W1U-determined onset energies (Table 1), two dimensional kinetic energy release,49 and, consistent with symmetric daughter ion peak shapes, fast sequential dissociations.7 The calculated breakdown diagram is shown in Fig. 5(a) for singlet, and in Fig. 5(b) triplet CCl3+ being the intermediate ion. The CCl2+ signal is reproduced well in Fig. 5(a) at its onset in the Franck–Condon gap at 15 eV, albeit at a slightly lower appearance energy than observed. The calculated curve disagrees with the measured one more as we approach the 2T2 peak in the photoelectron spectrum. We propose that the reason for this disagreement is 2T2 state Rydberg series involvement below ca. 16 eV. As observed for the dissociation mechanism change in F-atom loss in the 1,1-difluoroethene cation,14 Rydberg states may behave similarly to their convergent ionic state also regarding their dissociative decay. In CCl4+, this means that state Rydberg series may autoionise to the quartet ã state at geometries where the two are close to degenerate. Cl loss will then yield triplet 3CCl3+. As the photon energy reaches 16 eV, which corresponds to the onset of the 2T2 ion state, the fractional abundance of CCl2+ rises sharply at the cost of that of CCl3+. This indicates that the photoionisation mechanism is completely dominated by the state, which leads to the production of triplet-state 3CCl3+ fragments. The singlet–triplet excitation energy stays trapped in the fragment ion, which limits the excess energy release in the first Cl-loss channel. With less kinetic energy being available for release and more excess energy trapped in the system, it is more likely to be sufficiently energetic to lose a further Cl to form CCl2+. The limited regeneration of the CCl3+ signal at ca. 17 eV, past the Franck–Condon zone of the 2T2 state, is again well reproduced by the assumption that the intermediate is singlet 1CCl3+, again suggesting autoionisation of these states to the 2E state prior to singlet 1CCl3+ formation on the à or state potential energy surface. In summary, the triplet state involvement is inferred chiefly from the disappearance of the CCl3+ signal in the energy range of the state and its faint and fleeting reapparition at slightly higher energies.
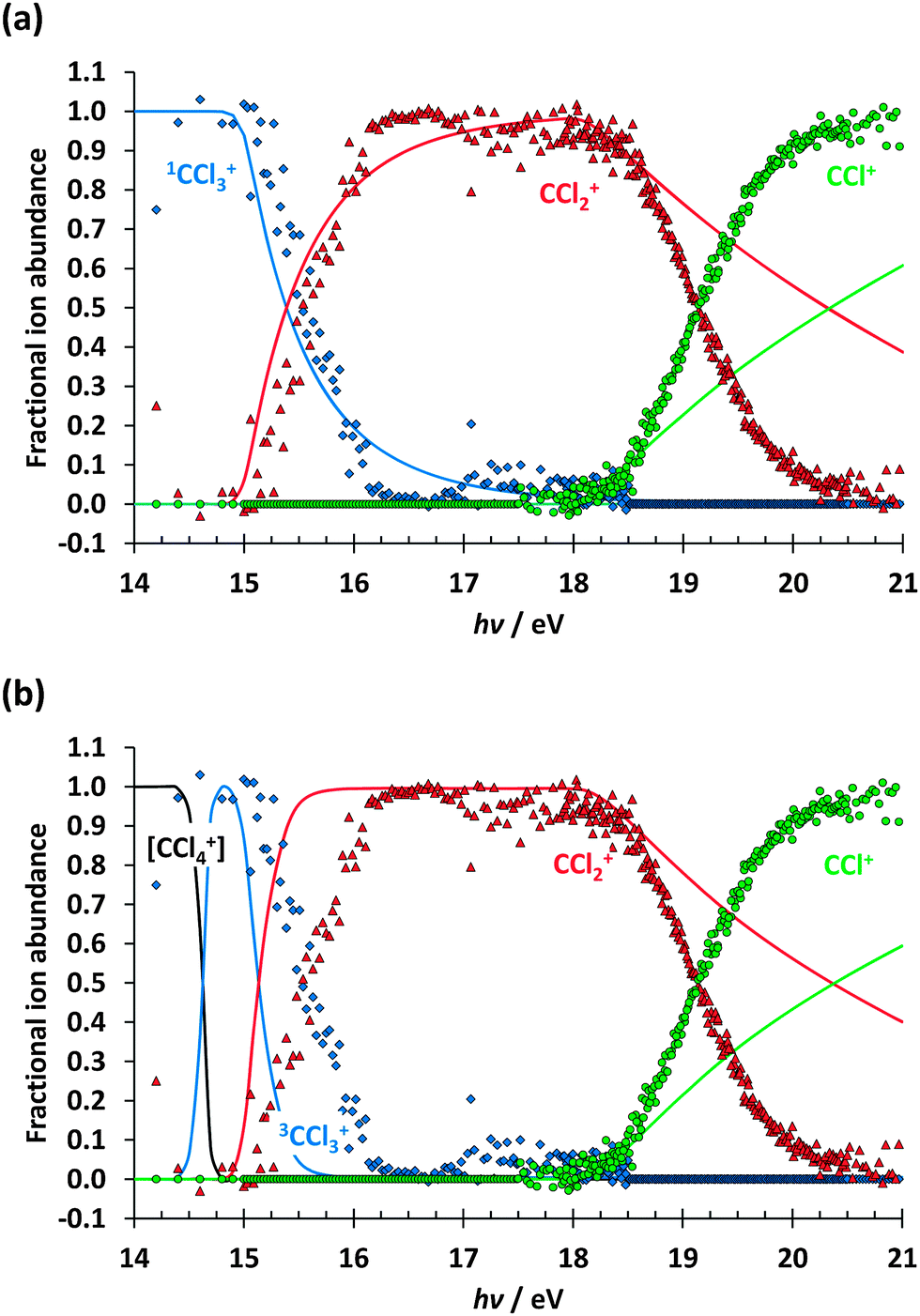 | ||
| Fig. 5 Calculated CCl4 breakdown diagram assuming statistical and fast, k(E0) > 107 s−1, dissociation, using ab initio onsets for (a) singlet and (b) triplet CCl3+ formation. The dots are the experimentally measured fractional ion abundances, continuous lines correspond to the model calculation. | ||
The onset of the CCl+ signal is also relatively well reproduced by the statistical model, but since the overall excess energy is independent of the intermediate energy, the shape of the breakdown curve is hardly affected by the choice of CCl3+ spin state. Above 18.5 eV, the excess energy released is actually less than predicted by statistical theory, and the CCl+ abundance rises more quickly with increasing photon energy. Such discrepancies are not unheard of at high excess energies,50 but its magnitude here suggests possible electronically excited CCl2+ participation, similarly to the triplet CCl3+ intermediates. At the same time, the W1U onset energy of CCl+ with three Cl atoms is confirmed by the well-reproduced rising edge of the CCl+ breakdown curve.
Conclusions
We have revisited the dissociative photoionisation of CCl4 between its ionisation energy and 21 eV, covering the five valence ion states. Composite calculations were carried out to confirm the dissociative photoionisation onset energies. The minimum energy Cl-loss path in CCl4+ to singlet 1CCl3+ + Cl was found by a 2-dimensional potential energy scan, and electronic excited state energies in the parent cation were calculated using EOM-IP-CCSD. We have found a direct path from the à 2T2 state of CCl4+ to the promptly-dissociating 2T1 state, which explains the lack of vibrational structure in the TPES of both bands. The 2E state, on the other hand, supports bound vibrational wave functions, shown by a progression being observed in the ν2(e) scissors mode in this band of the TPES. This mode can only show activity in a photoelectron spectrum of a tetrahedral molecule if the symmetry is broken. The 2E electronic state of the ion is doubly degenerate. Since E ⊂ E ⊗ E in Td, the vibration has the correct e symmetry to induce Jahn–Teller distortion from Td symmetry; the vibration then becomes totally symmetric in the lower-symmetry D2d point group, making the vibrational progression observable.
A statistical model reproduces the CCl2+ fractional abundance in the breakdown curve at the CCl2+ + 2Cl + e− threshold well, which is further evidence for the presence of a long-lived CCl4+ intermediate undergoing IVR at higher photon energies. However, the CCl3+ signal then drops rapidly in the vicinity of the 2T2 band of CCl4+. An intersystem crossing pathway via the lowest quartet state yielding triplet 3CCl3+ + Cl is proposed to be the reason. By decreasing the excess energy available for kinetic energy release, this allows for quantitative CCl2+ production over the energy range of the 2T2 band. CCl3+ makes a weak return in the breakdown diagram to high energy of the peak, which is reproduced by the statistical ‘singlet-CCl3+’ model. At a higher energy of ca. 18 eV, CCl+ is produced from the CCl2+ cation. The onset of this process is predicted by the statistical model well, but the CCl+ signal rises more steeply than predicted shortly after its onset, indicating suppressed kinetic energy release.
In conclusion, there are non-statistical aspects to the dissociative photoionisation of CCl4+ in the 2T2 band, which increase the internal energy available for sequential Cl losses, but the breakdown curves in the onset region for production of the CCl2+ and CCl+ daughter ions are both described well by a statistical model. When the initial ion state is the state or higher, the dissociative ground and first excited electronic states of CCl4+ are only accessed by internal conversion after IVR has taken place. As triplet-state 3CCl3+ production recedes to higher energy of the 2T2 band in the TPES, the internal energy available for dissociation actually decreases with increasing photon energy, resulting in a brief re-appearance of the CCl3+ signal.
This behaviour is in contrast with that of some of the fluoroethene cations, in which the dissociative photoionisation starts out as a statistical process and then exhibits non-statistical F-loss, or, in the case of C2F4+, a second, de-coupled statistical dissociation regime.13,14 The parent cation of tetrachloromethane dissociates impulsively at low energies, but then behaves almost statistically at higher energies. The decay of the 2T2 state of CCl4+ ions also appears to be distinctively different from that of analogous state of other MX4+ ions, which have previously been reported to be quite long-lived and decay by fluorescence. The opportune quasi-degeneracy of the doublet and quartet ã states in CCl4+ therefore seems unlikely to occur in the other members of this family of compounds.
Acknowledgements
The iPEPICO experiments were carried out at the VUV beamline of the Swiss Light Source of the Paul Scherrer Institut. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 226716. JH thanks the University of Birmingham for a Research Studentship.Notes and references
- T. I. Sølling, T. S. Kuhlman, A. B. Stephansen, L. B. Klein and K. B. Møller, ChemPhysChem, 2014, 15, 249–259 CrossRef PubMed.
- S. Ullrich, T. Schultz, M. Z. Zgierski and A. Stolow, Phys. Chem. Chem. Phys., 2004, 6, 2796 Search PubMed.
- Y. Liu, G. Knopp, P. Hemberger, Y. Sych, P. Radi, A. Bodi and T. Gerber, Phys. Chem. Chem. Phys., 2013, 15, 18101–18107 Search PubMed.
- T. Baer, B. Sztaray, J. P. Kercher, A. F. Lago, A. Bodi, C. Skull and D. Palathinkal, Phys. Chem. Chem. Phys., 2005, 7, 1507–1513 CAS.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 Search PubMed.
- A. Bodi, M. Johnson, T. Gerber, Z. Gengeliczki, B. Sztaray and T. Baer, Rev. Sci. Instrum., 2009, 80, 34101 Search PubMed.
- B. Sztáray, A. Bodi and T. Baer, J. Mass Spectrom., 2010, 45, 1233–1245 Search PubMed.
- A. Bodi, P. Hemberger and T. Gerber, J. Chem. Thermodyn., 2013, 58, 292–299 CrossRef CAS PubMed.
- A. Bodi, J. Csontos, M. Kállay, S. Borkar and B. Sztáray, Chem. Sci., 2014, 5, 3057–3063 RSC.
- U. Lourderaj and W. L. Hase, J. Phys. Chem. A, 2009, 113, 2236–2253 CrossRef CAS PubMed.
- T. Baer and A. R. Potts, J. Phys. Chem. A, 2000, 104, 9397–9402 CrossRef CAS.
- A. Bodi, N. S. Shuman and T. Baer, Phys. Chem. Chem. Phys., 2009, 11, 11013–11021 RSC.
- J. Harvey, A. Bodi, R. P. Tuckett and B. Sztaray, Phys. Chem. Chem. Phys., 2012, 14, 3935–3948 RSC.
- J. Harvey, P. Hemberger, A. Bodi and R. P. Tuckett, J. Chem. Phys., 2013, 138, 124301 CrossRef PubMed.
- J. P. Maier, Acc. Chem. Res., 1982, 15, 18–23 CrossRef CAS.
- D. M. Smith, R. P. Tuckett, K. R. Yoxall, K. Codling, P. A. Hatherly, J. F. M. Aarts and M. Stankiewicz, J. Chem. Phys., 1994, 101, 10559–10575 CrossRef CAS PubMed.
- P. M. Guyon, T. Baer and I. Nenner, J. Chem. Phys., 1983, 78, 3665–3672 CrossRef CAS PubMed.
- L. A. Shadoff, L. Prókai, G. B. Anderson, R. G. Gills, Q. N. Porter, L. Bencze, G. W. Dillow, I. K. Gregor, P. R. Ashton and M. E. Rose, Org. Mass Spectrom., 1986, 21, 381–390 CrossRef CAS PubMed.
- C. E. C. A. Hop, J. L. Holmes, F. P. Lossing and J. K. Terlouw, Int. J. Mass Spectrom. Ion Processes, 1988, 83, 285–294 CrossRef CAS.
- J. C. Creasey, H. M. Jones, D. M. Smith, R. P. Tuckett, P. A. Hatherly, K. Codling and I. Powis, Chem. Phys., 1993, 174, 441–452 CrossRef CAS.
- T. Kinugawa, Y. Hikosaka, A. M. Hodgekins and J. H. D. Eland, J. Mass Spectrom., 2002, 37, 854–857 CrossRef CAS PubMed.
- J. Harvey, R. P. Tuckett and A. Bodi, J. Phys. Chem. A, 2012, 116, 9696–9705 CrossRef CAS PubMed.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477 CrossRef CAS PubMed.
- G. A. Garcia, H. Soldi-Lose and L. Nahon, Rev. Sci. Instrum., 2009, 80, 23102 CrossRef PubMed.
- X. F. Tang, X. G. Zhou, M. L. Niu, S. L. Liu, J. D. Sun, X. B. Shan, F. Y. Liu and L. S. Sheng, Rev. Sci. Instrum., 2009, 80, 113101 CrossRef PubMed.
- A. Bodi, B. Sztaray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 84102 CrossRef PubMed.
- G. A. Garcia, B. K. Cunha de Miranda, M. Tia, S. Daly and L. Nahon, Rev. Sci. Instrum., 2013, 84, 053112 CrossRef CAS PubMed.
- A. Bodi, P. Hemberger, T. Gerber and B. Sztáray, Rev. Sci. Instrum., 2012, 83, 083105 CrossRef PubMed.
- T. Baer, A. Guerrero, J. Z. Davalos and A. Bodi, Phys. Chem. Chem. Phys., 2011, 13, 17791–17801 RSC.
- S. Borkar, B. Sztáray and A. Bodi, Phys. Chem. Chem. Phys., 2011, 13, 13009–13020 RSC.
- J. C. Creasy, I. R. Lambert, R. P. Tuckett, K. Codling, L. J. Frasinski, P. A. Hatherly, M. Stankiewicz and D. M. P. Holland, J. Chem. Phys., 1990, 93, 3295–3306 CrossRef PubMed.
- B. Sztaray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef CAS PubMed.
- A. Bodi and P. Hemberger, Phys. Chem. Chem. Phys., 2014, 16, 505–515 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, S. Nakatsuji, M. Caricato, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V Ortiz, J. Cioslowski, D. J. Fox, H. Nakatsuji, X. Li and J. N. Millam, Gaussian 09, Revision C.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- J. F. Stanton and J. Gauss, J. Chem. Phys., 1994, 101, 8938 CrossRef CAS PubMed.
- W. Von Niessen and L. Asbrink, J. Electron Spectrosc. Relat. Phenom., 1982, 26, 173 CrossRef CAS.
- R. N. Dixon and R. P. Tuckett, Chem. Phys. Lett., 1987, 140, 553–557 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecular Structure III. Electronic Spectra and Electronic Structure of Polyatomic Molecules, Van Nostrand, Princeton, 1966 Search PubMed.
- M. W. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, New York, 1998 Search PubMed.
- K. Rademann, H. W. Jochims and H. Baumgaertel, J. Phys. Chem., 1985, 89, 3459–3464 CrossRef CAS.
- D. M. Smith, R. P. Tuckett, K. R. Yoxall, K. Codling and P. A. Hatherly, Chem. Phys. Lett., 1993, 216, 493–502 CrossRef CAS.
- B. Sztáray and T. Baer, J. Phys. Chem. A, 2002, 106, 8046–8053 CrossRef.
- I. Powis, Mol. Phys., 1980, 39, 311–327 CrossRef CAS.
- P. A. Hatherly, D. M. Smith and R. P. Tuckett, Z. Phys. Chem., 1996, 195, 97–136 CrossRef CAS.
- I. R. Lambert, S. M. Mason, R. P. Tuckett and A. Hopkirk, J. Chem. Phys., 1988, 89, 2675 CrossRef CAS PubMed.
- I. R. Lambert, S. M. Mason, R. P. Tuckett and A. Hopkirk, J. Chem. Phys., 1988, 89, 2683 Search PubMed.
- A. G. Sage, T. A. A. Oliver, R. N. Dixon and M. N. R. Ashfold, Mol. Phys., 2010, 108, 945–955 CrossRef CAS.
- T. Baer, S. H. Walker, N. S. Shuman and A. Bodi, J. Phys. Chem. A, 2012, 116, 2833–2844 CrossRef CAS PubMed.
- E. M. Russell, E. Cudjoe, M. E. Mastromatteo, J. P. Kercher, B. Sztáray and A. Bodi, J. Phys. Chem. A, 2013, 117, 4556–4563 CrossRef CAS PubMed.
Footnote |
| † Current address: The Royal Society of Chemistry, Thomas Graham House, Science Park, Milton Road, Cambridge CB4 0WF, UK. |
| This journal is © the Owner Societies 2014 |