
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic domain magnetic nanoalloys: interplay between molecular structure and temperature dependent magnetic and dielectric properties in manganese doped tin clusters†
Urban
Rohrmann
*a,
Peter
Schwerdtfeger
b and
Rolf
Schäfer
a
aEduard-Zintl-Institut, Technische Universität Darmstadt, Darmstadt, Germany. E-mail: rohrmann@cluster.pc.chemie.tu-darmstadt.de
bCentre for Theoretical Chemistry and Physics, Massey University, Albany, New Zealand
First published on 3rd September 2014
Abstract
We present extensive temperature dependent (16–70 K) magnetic and electric molecular beam deflection measurements on neutral manganese doped tin clusters Mn/SnN (N = 9–18). Cluster geometries are identified by comparison of electric deflection profiles and quantum chemical data obtained from DFT calculations. Most clusters adopt endohedral cage structures and all clusters exhibit non-vanishing magnetic dipole moments. In the high temperature regime all species show exclusively high field seeking magnetic response and the magnetic dipole moments are extracted from the shift of the molecular beam. At low nozzle temperatures, some of the clusters show considerably broadened beam profiles due to non-uniform deflection in the magnetic field. The results reflect the influence of the chemical environment on the magnetic properties of the transition metal in atomic domain magnetic nanoalloys. Different ground state spin multiplicities and coupling of rotational and vibrational degrees of freedom with the spin angular momentum of isolated clusters of different size apparently cause these variations of spin orientation. This is discussed by taking electronic and molecular structure data into account.
1 Introduction
Transition metal doped clusters are a promising class of nanoalloys with interesting composition and size dependent properties.1,2 Affecting electronic and molecular structure, physical and chemical properties can be tuned by the choice of the transition metal and host element and their atomic composition.2,3 Clusters with unpaired electrons are promising candidates for the development of nanostructured magnetic materials.4,5 While our interest is focused on the properties of the isolated clusters and especially the influence of the molecular topology on the magnetic properties, some of these clusters are highly stable and can possibly serve as building blocks for the synthesis of advanced materials.6,7 In order to understand and to systematically design materials based on the integrity of nanostructures that form a super lattice, a better understanding of the cluster building blocks is highly desirable. An improved fundamental knowledge of the intrinsic properties of clusters will allow us to get a better view of the interactions in nanostructured materials. To explore the third dimension of the periodic table,7–9 isolated clusters in the gas phase are an ideal starting point.7 Beam deflection experiments have been adapted from Stern and Gerlach's original work on silver atoms10,11 to investigate the magnetic properties of isolated clusters. Early work was reported by Knight et al.12 and the method was considerably refined by de Heer and coworkers,13–15 Cox et al.16 and Bucher et al.17 to name just a few pioneering approaches. Subsequently, Hihara, Pokrant and Becker extended the method to the investigation of heterogeneous systems18,19 and in the meantime a vast of gas phase species has been examined by this technique. An outline of the topic is given by Knickelbein20 and Kresin and de Heer.21 The interpretation of these experiments is, however, still a challenging task. Therefore, we have started to analyse the deflection behaviour of clusters doped with a single paramagnetic atom, to examine the impact of the host structures on their magnetic response.In recent publications22,23 we investigated the dependence of spin angular momentum orientation on the size and temperature of Mn@SnN clusters (N = 12, 13, 15). The specific molecular structure of Mn@Sn12 (Ih) gives rise to quasi atomic magnetic response in the vibrational ground state, whereas excited normal modes that break the spin microstate degeneracy induce spin orientation. This purely internal vibrationally induced spin orientation process has to our knowledge not been considered in related work. It is an example of how the uncommon chemistry of semimetal/metal clusters can contribute to a more complete fundamental understanding of molecular systems in general. In the present work we focus on the effect of the individual molecular cage structure depending on the number of Sn atoms. We show results from electric beam deflection experiments on the Mn/SnN clusters (N = 9–18) together with an extended investigation of the magnetic response. In addition the clusters are studied extensively in terms of density functional theory (DFT) to obtain the ground state electronic spin multiplicity, geometry, dielectric properties and vibrational normal modes. The molecular topologies, vibrational spectra and spin multiplicities of the clusters are taken into account in order to rationalize the influence of the tin cage on the magnetic response of the nanoalloys.
2 Dielectric and magnetic properties from molecular beam deflection
The properties of small clusters do not only depend on size and composition of the objects, but also on their environment. Experiments on isolated clusters allow us to study size selected neutral clusters in a vacuum, without obscuring the intrinsic properties. In molecular beam deflection experiments, the spatial distribution (beam profile) is measured size selectively without and with an applied inhomogeneous magnetic or electric field in order to investigate the response of the clusters (Stern–Gerlach and Stark experiment, respectively).Electric beam deflection experiments are established as a tool to probe the dielectric properties of neutral clusters and molecules.24–32 The influence of the electric dipole moment and polarizability of a cluster, the role of its molecular structure and thermal effects have been studied extensively. Experimental observations on homo-30,33–36 and hetero-atomic31,37 clusters and molecules27,28 can be reproduced by models taking these properties into account, extracted from electronic structure calculations. If magnitude and orientation of the electric dipole moment allow to discriminate possible isomers, matching the simulations with the experimental results allows to identify the topology of the cluster.30–32 However, thermal effects can considerably affect the results.30,38 While this is an opportunity to study interesting dynamical effects like isomerisation30 and thermally induced symmetry breaking,39,40 for structure determination the clusters have to be vibrationally cold (i.e. rigid) to exclude such effects.
In magnetic beam deflection experiments the response of the cluster ensemble to an inhomogeneous magnetic field is measured. Like in Stern and Gerlach's original experiment on silver atoms,10,11 the component of the electronic angular momentum in the direction of the field (defined by the z-axis) causes this effect. The electronic angular momentum states are eigenstates of the Hamiltonian in isolated atoms and the magnitude of the angular momentum and the component along the field axis are constants of motion (in the case of small perturbation). Hence, the quantum numbers representing total electronic angular momentum J and its z-component MJ are good quantum numbers at commonly available magnetic fields.41 A beam of atoms is therefore split up into components with MJ = −J, −J + 1,…, + J (or with zero electronic orbital angular momentum this reduces to MJ = MS = −S, −S + 1,…, + S, with the principal electronic spin quantum number S and its z-component MS), because each atom experiences a constant force proportional to MJ (MS). Recently we demonstrated the same effect experimentally on the cluster Mn@Sn12 in its vibrational ground state.23 In general, however, clusters in molecular beams show net deflection due to orientation of the magnetic dipole moment in the magnetic field. Hence, in the case of clusters (and other molecular systems) the component of the electronic angular momentum is commonly observed not to be a constant of motion.
The magnetic response of tin clusters doped with a single Mn atom depends critically on the size and temperature of the clusters.22,23 In order to rationalize these observations, we study the Mn/SnN clusters by quantum chemistry (QC) methods combined with an evolutionary inspired optimization of the tin cage. Stark experiments are analyzed in order to identify the molecular structure of the clusters from simulations of the dielectric response. The experimentally verified topologies are then used to elucidate the magnetic response of the clusters in Stern–Gerlach experiments at cryogenic temperatures.
3 Quantum chemical and experimental procedure
3.1 Quantum chemistry
In order to obtain possible molecular structures of the manganese doped tin clusters and the ground state spin-configurations, the Mn/SnN clusters (N = 9–18) were studied by means of DFT in combination with global optimization of the tin cages.The hybrid functional B3P8642,43 is used throughout the computational studies. It was found to reproduce the Sn2 bond distance and electronic polarizability in very good agreement with experimental data and coupled cluster calculations.30,44 The agreement of dielectric properties of the tin clusters with experimental observations is appealing,30,44 and in addition the functional is known to give good accuracy for transition metal compound geometries.45,46 It performs superior in reproducing the molecular vibrational spectrum in far-infrared multi photon dissociation experiments on manganese doped silicon cluster cations among a choice of functionals.47 Additionally, test calculations for Mn@Sn12 in the framework of long range corrected (LC) Tamm–Dancoff time dependent density functional theory48 (TDDFT) reproduce the energy gap between highest occupied and lowest unoccupied molecular orbitals (HOMO–LUMO gap) obtained in our DFT studies,‡ reflecting the reliability of the chosen functional for electronic structure calculations of transition metal doped tetrel clusters. However, one has to keep in mind that common hybrid functionals tend to over stabilize high-spin configurations in transition metal compounds, while pure generalized gradient approximation (GGA) favours low spin configurations.51 In analogy to transition metal compounds, the tin cages can be considered as ligand spheres complexing a transition metal centre. Complexes of weak-field ligands require significant contribution of exact exchange to provide agreement with experimental observations, while there is increasing evidence that with strong-field ligands the relative spin-state energy is in better agreement with reduced Hartree–Fock exchange.51 To investigate the influence of the choice of functional on the relative energy of the spin state, single point calculations are performed also with the pure GGA functional BP8643,52 at the optimized geometries.
Tin tends to form polyanions and the difference in electronegativity among Sn and Mn (0.41 on the Pauling scale) induces the idea that the clusters Mn/SnN might be understood as consisting of a Zintl polyanion SnN2− and a Mn2+ ion. In analogy to Mg@PbN,53 BiMSnN31,37 and NaMAlN,54 the Mn doped SnN clusters are referred to as gas phase Zintl-compounds. According to Wade's rules, in the simplified picture of the charge transfer complexes Mn2+/SnN2− particularly stable tin cages are formed with 2N + 2 skeletal bonding electrons and another two electrons localized at each Sn atom.22 In a first approximation we expect diamagnetic closo tin cages,§ hosting a paramagnetic transition metal ion with localized spin (according to large HOMO–LUMO gaps spin polarization on the tin cages is neglected).
Therefore, only the bare doubly charged SnN2− anions were optimized using a genetic algorithm (GA) coupled with the Gaussian09 quantum chemistry program.55,56 Starting geometries for the GA were taken from a large pool of known metal, semimetal and rare gas clusters. The population of structures considered during global optimization had a maximum size of 100 individuals. After running for at least 250 generations the lowest energy structures were constant and convergence was assumed. During global optimization of the tin cages, the computationally efficient Los Alamos National Laboratory (LANL2) effective core potential (ECP) and corresponding double zeta basis sets (DZ) were employed.57 We considered singlet (closed shell) and triplet (open shell) electronic configurations of the dianions in spin unrestricted self-consistent field (SCF) calculations. As discussed briefly in the ESI,† with N = 13 and 14 the global minima of the polyanions do not correspond to hollow cage structures and we did not proceed with this method for N > 14. To obtain (nearly) spherical cage structures for the range N = 13–18 and in order to check the GA approach of the smaller cages, a simple electrostatic model is used to obtain start geometries for local optimization. The electrostatic energy of N equal charges on a unit sphere is minimized with respect to the position of the charges. The resulting geometries are scaled in order to obtain reasonable distances among neighbouring tin atoms.
We further investigated the lowest lying isomers of SnN2− within a range of up to 1 eV (depending on the number of low lying isomers) by formally introducing a Mn2+ ion at the centre of mass of the host structures. To verify this simple, chemically inspired approach for the complex problem of optimizing binary cluster structures, we also added Mn2+ exohedrally to the low lying dianionic tin structures as well as by taking the lowest energy isomer of the next larger SnN+1 cluster30,44 and substituting one of the Sn atoms by Mn.
The doped structures were relaxed within unrestricted open shell DFT calculations (B3P86/LANL2-DZ), taking total electronic spin angular momentum quantum numbers S = 1/2, 3/2, 5/2 and 7/2 into account. To finally obtain proper energies and dielectric properties we switched to the more accurate small core Stuttgart–Dresden–Bonn ECP (SDB)58,59 and corresponding triple zeta basis sets (TZ). The lowest energy isomer and structures within 0.5–1 eV above were relaxed and harmonic frequency analysis of the vibrational normal modes was performed to verify the structures found to be true minima on the potential energy surface (PES).
To calculate the dielectric properties and vibrational normal modes in the harmonic approximation, tight convergence is afforded in all SCF calculations and geometry optimizations.
3.2 Beam deflection experiments
A cluster with electric (magnetic) dipole moment![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) el (mag) travelling through an inhomogeneous field
el (mag) travelling through an inhomogeneous field ![[capital Gamma, Greek, vector]](https://www.rsc.org/images/entities/i_char_e1d1.gif) =
= ![[e with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0065_20d1.gif) zΓz with gradient
zΓz with gradient 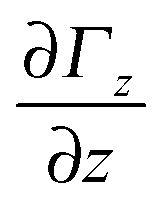 is deflected by a force
is deflected by a force | (1) |
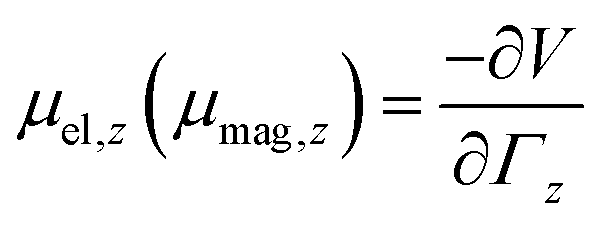 the z-component of the corresponding dipole moment.
the z-component of the corresponding dipole moment.
The cluster enters the electric (magnetic) field of length l1,Γ with velocity ![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) = xvx and the deflection measured at the detector is
= xvx and the deflection measured at the detector is
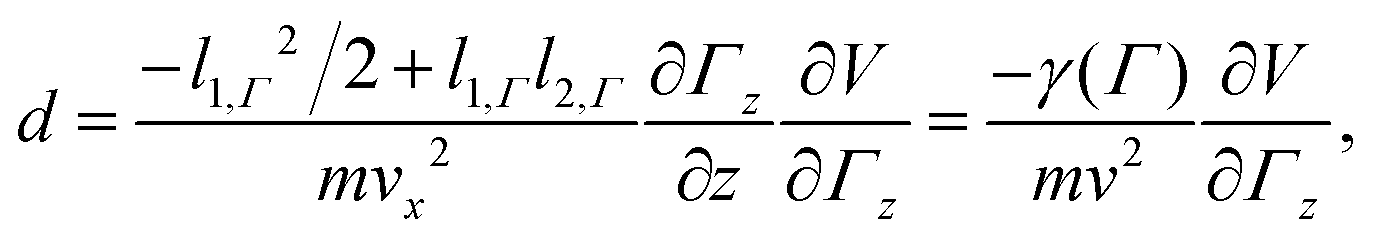 | (2) |
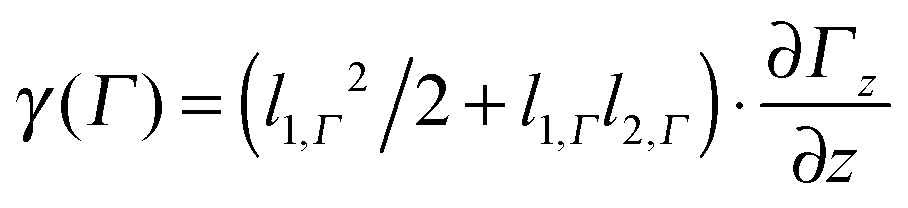 .
.
In molecular beam deflection experiments, however, not the response of single clusters is measured, but of an ensemble of clusters of each size. If Φ0 is the beam profile at zero field, the beam profile with an applied electric (magnetic) field is the convolution of Φ0 with the deflections according to eqn (2) and taking the distribution function ρ(μel,z) (ρ(μmag,z)) of the corresponding dipole moment into account. The principle of electric and magnetic beam deflection hence is the same; however, the distribution function of the dipole moments and their temporal evolution while passing the field depends on the kind of interaction (electric/magnetic).
The experimental setup for electric and magnetic beam deflection studies has been reported before22,23,33,60 and hence we focus only on experimental modifications made in the meantime. Manganese doped tin clusters are produced in a laser ablation source with a temperature controlled nozzle at a repetition rate of 10 Hz. Targets are prepared from an alloy of 5 at% Mn in tin, fused in a furnace at 800 °C under vacuum. After a period of 2 h the sample is removed from the furnace and rapidly cooled down to room temperature to avoid segregation.
Before a part of the Mn–Sn mixture is evaporated by a short pulse of 1064 nm wavelength from a Nd:YAG laser, a small amount of helium (7 bar background pressure) is introduced into the source chamber. The clusters grow in the helium atmosphere and subsequently the gas–cluster mixture passes a nozzle of 2 mm diameter and 60 mm length. Before reaching the cold copper part of the nozzle (25 mm), the mixture passes a leading section of the nozzle made from Teflon to isolate the chamber (which is at approximately room temperature) from the nozzle that can be cooled to 15 K while operating the source. Collisions of the carrier gas rapidly transfer heat from the clusters to the nozzle and in general we assume the clusters to reach thermal equilibrium in the nozzle, before the carrier gas is expanded into high vacuum, forming a molecular beam of doped and pure tin clusters.
We worked with rather short valve opening durations (ca. 250 μs vs. 400–600 μs in our earlier work), in order to achieve slow clusters in the beam with sufficient dwell time (≈1.5 ms) for the helium–cluster mixture. An important difference to earlier work is the extended delay of the laser vaporization pulse (now ca. 600 μs vs. 200–400 μs in previous work), relative to the signal for the helium valve. In previous studies the laser was triggered within the opening time of the valve, which effectively generated clusters in the early stage of rising pressure in the chamber. This leads to sharper supersonic expansions and generated clusters with rotational temperatures well below Tnozzle. The supersonic expansion into high vacuum is slightly softer with late evaporation. As discussed in Section 4.2.1, this causes rotational temperatures Trot of the clusters of approximately 10 K, compared to 3–8 K commonly considered in our earlier investigations.30,31 The influence of the adiabatic expansion on the vibrational temperature of clusters in seeded supersonic molecular beams depends on the carrier gas. It has been found that He carrier gas hardly reduces the vibrational temperature, even with sharp supersonic expansion.61,62 Therefore, if equilibrium of clusters and nozzle is reached, Tvib ≈ Tnozzle. To elucidate this assumption Tvib is extracted from the magnetic deflection experiments of Mn@Sn12 (see ESI†) and while Tvib ≈ Tnozzle in the range of 30–70 K has been found, at lower nozzle temperatures, equilibration is insufficient and Tvib > Tnozzle.¶
After expansion into high vacuum, the molecular beam passes a double skimmer with 2 mm diameter and subsequently a fast shutter which is employed to measure the velocity of the clusters in the molecular beam. Further downstream the molecular beam is collimated by two slits (400 μm × 2 mm), forming a molecular beam with rectangular shape which passes the deflection units (the electrodes and pole faces have the well known shape to produce two-wire field analogues, l1,E = 150 mm, l1,B = 80 mm).63,64 The clusters then pass a drift region (l2,E = 159.0 cm, l2,B = 123.3 cm) before entering the time-of-flight (TOF) mass spectrometer through a scanning slit (400 μm × 1.5 cm) which is moved by a step motor to measure the spatial distribution of the molecular beam. The clusters are ionized by a He/F2 excimer laser (157 nm) approximately 20 cm before the ions are extracted orthogonally by an acceleration unit working in Wiley–McLaren time focus mode. At each position of the scanning slit, mass spectra are measured without and with an electric/magnetic field applied. To reach acceptable signal to noise ratios 100–150 spectra are average. The positions are chosen randomly from a set of equally spaced coordinates and every position is measured three times in total. In the ESI† we present an exemplary mass spectrum and explain briefly a method employed in this work to fit Gaussian functions to the mass spectra. The molecular beam profiles are obtained from the integrated signals of the mass spectra as a function of the slit position. The experimental uncertainties in Stark deflection experiments are Δμel ≈ ±0.2 D, depicted by the shaded grey area in Fig. 3 to reflect the experimental resolving capabilities. In the magnetic deflection studies we assume uncertainties of the magnetic dipole moments of Δμmag ≈ ±1 μB. The estimated measurement errors are mean values. Especially for clusters with very low intensity (N = 9, 10, 18) augmented errors should be considered.
4 Results and discussion
This section is organized as follows. The QC studies are presented first, focusing on the dielectric properties and relative energies of the doped clusters with respect to the electronic spin configuration and molecular structure. These results in combination with electric deflection experiments are then used to identify the structural isomers present in the molecular beam. The magnetic beam deflection experiments at Tnozzle = 30, 50, 70 K allow us to extract the magnitude of the magnetic dipole moment from the shift of the molecular beam. At Tnozzle = 16 K an appreciable amount of each cluster species is expected to populate the vibrational ground state, distinctively affecting the magnetic response. A microscopic model is employed, taking the Zeeman energy levels into account to qualitatively investigate the influence of the rotational energy spectrum and spin multiplicity of the isomers on the magnetic response of the ensemble of rigid clusters.4.1 Quantum chemistry – manganese doped clusters
The dianionic tin cage structures are briefly discussed in the ESI.† For all cluster sizes the exohedrally and substitutionally doped clusters have considerably higher energies (2–4 eV at the LANL2-DZ level of theory), regardless of the spin state. To exemplify this, the exohedrally doped isomer of Mn@Sn12 (12.2.s) is included at the B3P86/SDB-TZ level of theory. In the range of N = 10–16 the symmetry of the lowest energy hollow tin cages is retained on most doped clusters with sextet electronic spin multiplicity and in several cases the geometries of different cages converge at the same optimized structure after doping. Only with N = 11 and S = 5/2 (11.1.s), the Sn atom sitting on the C2 axis of the C2v symmetric cage is slightly tilted, resulting in a Cs symmetric structure. On the other hand, the clusters with quartet states are distorted due to the Jahn–Teller (JT) effect, and consequently greater numbers of low lying isomers are found. Up to N = 12 the minimum energy structure corresponds to the global minimum molecular cage identified by the GA. However, the Mn/Sn9 cluster 9.0.s with S = 5/2 does not retain the D3h symmetry of the empty host structure. The cage opens on one of the trigonal sites of the prism and distorts into a bowl-shaped structure. The remaining clusters have closo cages, apart from one exception; the Mn@Sn11 cluster 11.0.q (S = 3/2), for which the geometry optimization after doping of the C2v cage resulted in a C5v symmetric structure, an icosahedron with one missing vertex. The clusters Mn@Sn17 and Mn@Sn18 are unstable to distortion of the Mn atom from the centre of mass, reflecting the large size of the host structures.The resulting optimized cluster geometries are shown in Fig. 1 together with their relative energies (B3P86 bold, BP86 italic) and electric dipole moments, electronic polarizabilities and spin multiplicities. Additional data required for the molecular dynamics (MD) simulations of the dielectric response (components of the electric dipole moments and moments of inertia in the molecular fixed frame) are presented in Table S1 in the ESI.† The isomers are designated within the following discussion, in Fig. 1, 2 and 5 and in Table S1 (ESI†) by labels according to: [number of Sn atoms].[energetic order (B3P86)].[letter for spin multiplicity (2 = d, 4 = q, 6 = s)].
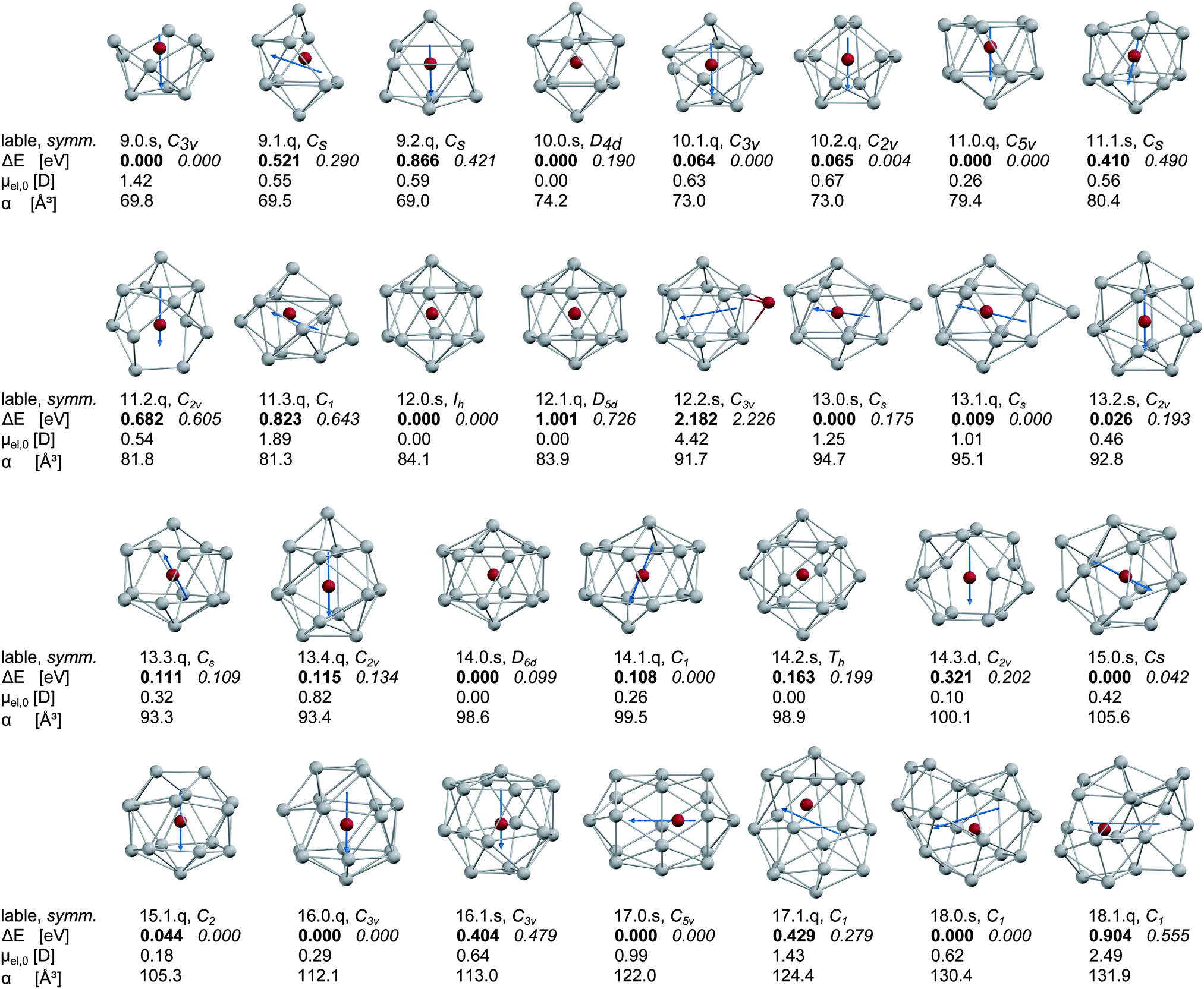 | ||
| Fig. 1 Lowest energy isomers of the Mn/SnN clusters with N = 9–18 as obtained by DFT (Mn red, Sn gray). The arrows represent the orientation of the electric dipole moment in the cluster. The first line of tabulated data of each structure is the label of the isomer as used in the text with [number of Sn atoms].[energetic order (B3P86)].[letter for spin multiplicity (2 = d, 4 = q, 6 = s)] and the point group symmetry. The second line shows the relative energy ΔE compared to the global minimum obtained with B3P86 hybrid functional (bold) and the relative energy obtained in single point calculations with BP86 pure GGA functional (italic) at the same geometry. The last two lines represent the magnitude of the electric dipole moment μel,0 and the isotropic static electronic polarizability α. For more details (components of the electric dipole moment and the components of the moment of inertia) refer to the ESI.† | ||
In the case of Mn/Sn12 we also include the exohedrally doped species at the same level of theory to point out the sensitivity of the electric dipole moment on the position of the manganese atom. According to the difference in electronegativity of the elements involved, exohedrally doped binary clusters are expected to have considerably larger dipole moments than the corresponding pure tin cluster. This can be observed for the 12.2.s structure, which has the largest magnitude of the electric dipole moment μel,0 of all structures in Fig. 1 and approximately twice that of the global minimum structure of Sn12 as obtained in ref. 30. The lowest energies of the doped clusters are obtained for those geometries with Mn close to the centre of mass. Although the electric dipole moment depends on the geometrical isomer and the spin state, all clusters investigated here except Mn/Sn9 have considerably reduced electric dipole moments in the ground state compared to the corresponding pure tin clusters as presented in ref. 30. While in Mn/Sn9 the large dipole moment results from the small size of the host structure, the clusters Mn@Sn17 and Mn@Sn18 have cages too large to retain nearly spherical symmetry. The Mn atom is distorted from its central position, causing enhanced electric dipole moments for the larger endohedrally doped clusters, compared to the range N = 10–16. The relative energies of the clusters obtained at various electronic spin multiplicities suggest quartet electronic ground states for Mn@Sn11 and Mn@Sn16 at the B3P86/SDB-TZ level of theory. The remaining clusters all have lowest energy with S = 5/2. However, the difference among spin isomers is in some cases below 0.1–0.2 eV, and the energies obtained in single point calculations with the BP86 GGA functional are lower for the quartet spin states. For clusters with near degenerate spin isomers (N = 10, 13, 14, 15, 18) it is not possible to clearly identify the ground state electronic spin configuration from the DFT calculations. However, Mn/Sn9, Mn@Sn12 and Mn@Sn17 have high-spin electronic ground states using the hybrid or GGA functionals. Neglecting electronic orbital contributions we expect the corresponding clusters to have spin only magnetic dipole moments of approximately 5.9 μB (taking into account the Landé factor of the free electron g = 2). The remaining clusters should have magnetic dipole moments of 3.8 μB or 5.9 μB (corresponding to S = 3/2 and S = 5/2), or intermediate values if quasi-degenerate spin states exist and become populated with significant probability.
4.2 Beam deflection experiments
Experimentally measured beam profiles of the Mn doped clusters with N = 9–18 are displayed in Fig. 2. For each cluster size the beam profiles measured with an applied magnetic field of 1.5 T at Tnozzle = 16 K (upper panel, red squares, Mn/Sn9: 30 K) and with an applied electric field of 15 kV (lower panel, red squares) at Tnozzle = 30 K are presented together with the beam profiles at 0 kV (T) (blue dots). The measured data points at zero field and with an applied field are fitted by a Gaussian function (blue solid line and red dashed line), with the constraint of a constant integral value. In addition we show, as described in the corresponding subsections, beam profiles obtained by MD simulation of the response to the electric field according to the structural and dielectric properties obtained from the DFT calculations (solid red line, solid grey line, and solid green line for S = 5/2, 3/2 and 1/2 isomers, respectively). In the case of Mn@Sn11, Mn@Sn12 and Mn@Sn14 the beam profiles according to quasi-atomic response with S = 3/2 (dotted grey line), 5/2 (dotted red line) and 1/2 (dotted green line) with an applied magnetic field are also displayed (upper panels). We show only one set of beam profiles for each cluster species. But the electric and magnetic dipole moments presented in Fig. 3 and 4 and also the vibrational temperatures extracted in the ESI† are the mean value of two measurements at each temperature, with identical settings of the experimental parameters.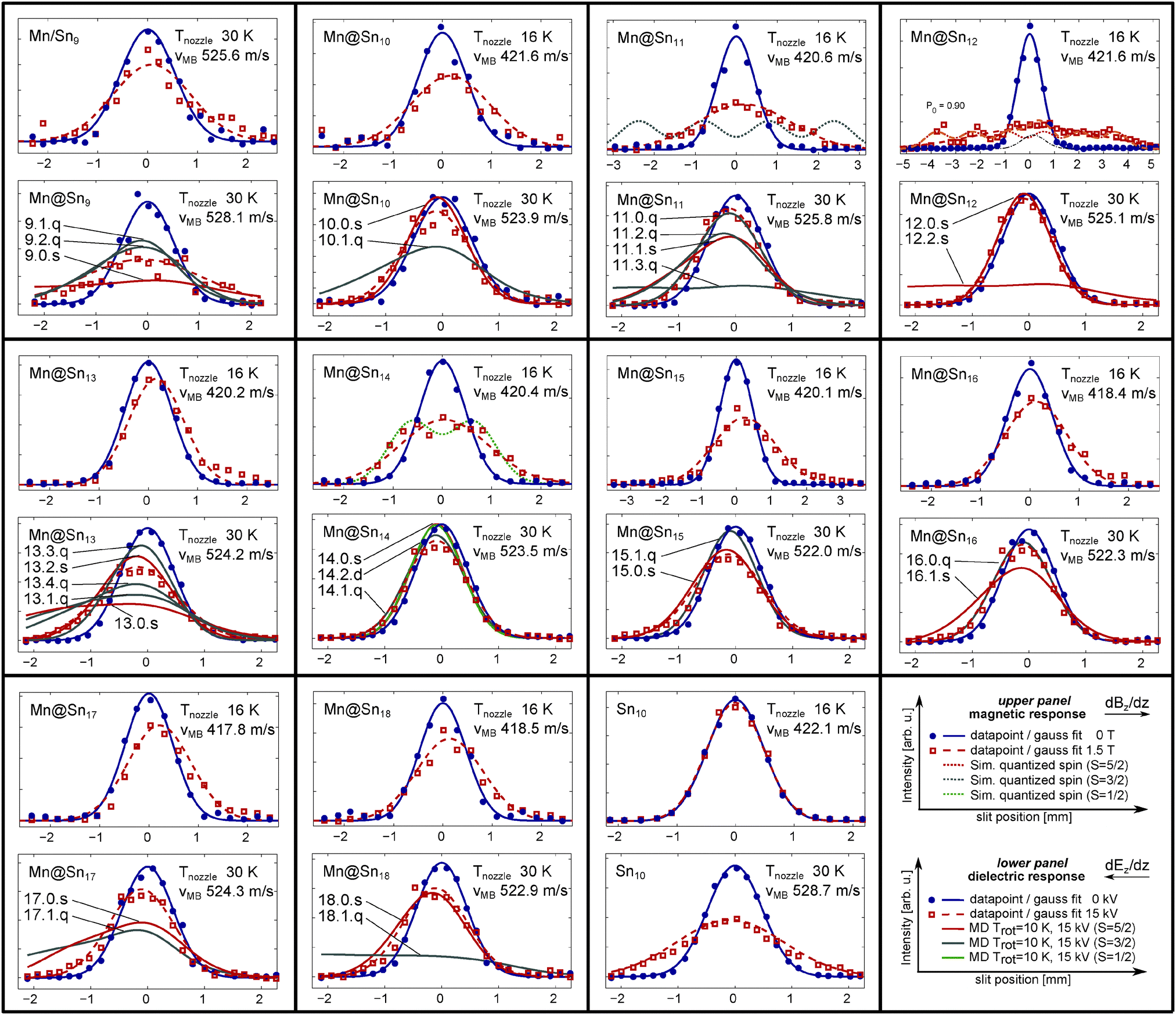 | ||
| Fig. 2 Beam profiles of the clusters Mn/SnN with N = 9–18 and Sn10 measured with an inhomogeneous magnetic field (upper panel, Bz = 1.5 T) and electric field (lower panel, deflection voltage Udefl = 15 kV). In order to analyze by first order perturbation theory the measured beam profiles (blue dots without and red square with an applied field), Gaussian functions are fitted to the data points (solid blue line and dashed red line). Upper panels: for Mn@Sn11, Mn@Sn12 and Mn@Sn14 we show the beam profiles according to quasi-atomic paramagnetic response for S = 3/2 (grey dotted line), S = 5/2 (red dotted line) and S = 1/2 (green dotted line), respectively. For Mn@Sn12 a two-component fit23 is shown (dashed orange line), corresponding to Tvib = 28 K (see ESI†). Lower panels: the dielectric response of the isomers shown in Fig. 1 obtained by MD simulation is displayed in the lower panels, with solid green, grey and red lines for S = 1/2, S = 3/2 and S = 5/2 spin-isomers. Additionally the labels corresponding to Fig. 1 are included. | ||
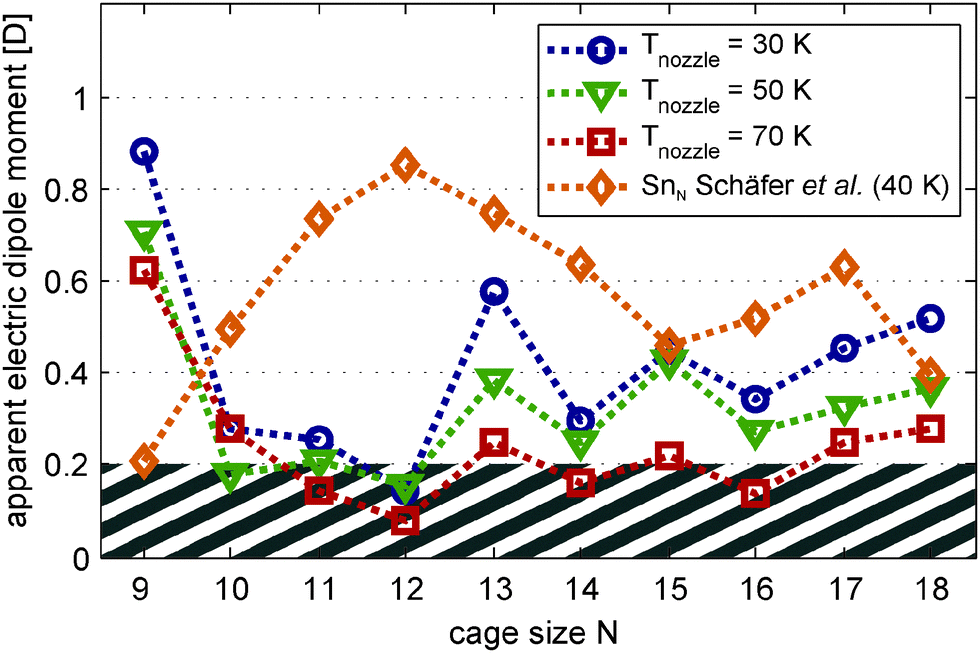 | ||
| Fig. 3 Apparent magnitude μel,0 of the electric dipole moments of Mn/SnN clusters produced at nozzle temperatures Tnozzle = 30 K (blue circles), 50 K (green triangles) and 70 K (red squares). The orange diamond shape is the measured electric dipole moment of the pure clusters SnN as presented in ref. 30. Data points are connected by a dotted line with corresponding colour as a guide to the eye. The electric dipole moments are extracted from the variance of Gaussian functions fitted to the molecular beam profiles without and with an electric field applied (deflection voltage Udefl = 15 kV). | ||
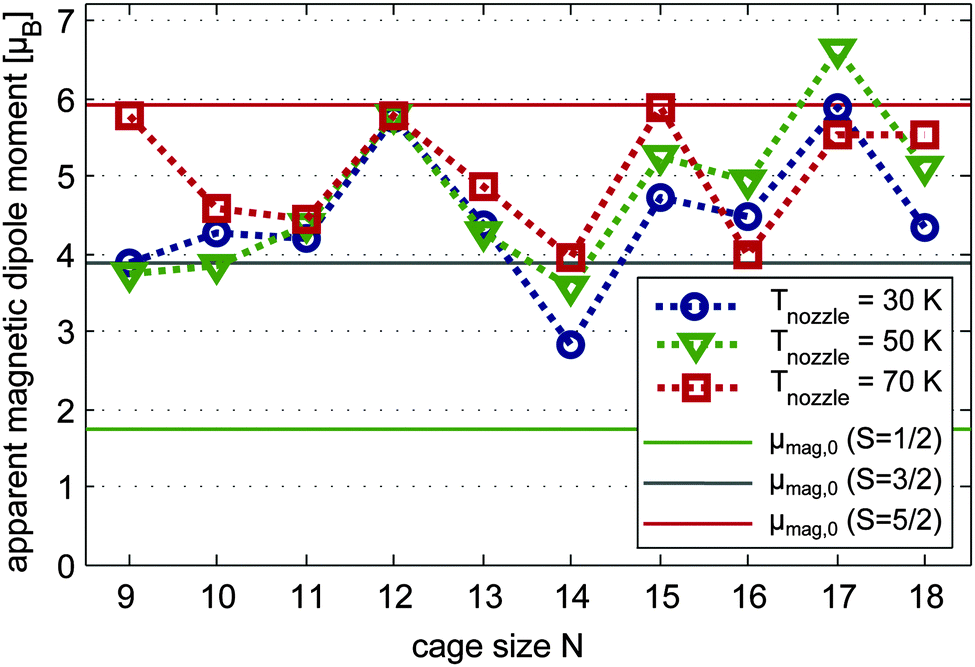 | ||
| Fig. 4 Apparent magnitude μmag,0 of the magnetic dipole moments of Mn/SnN clusters produced at nozzle temperatures Tnozzle = 30 K (blue circles), 50 K (green triangles) and 70 K (red squares). As a guide to the eye the data points are connected by a dotted line with corresponding colour. The magnetic dipole moments are extracted from the shift of gauss functions fitted to the molecular beam profiles without and with a magnetic field (Bz = 1.53 T, ∂B/∂z = 335 T m−1) applied. The straight solid lines represent the magnitude of a magnetic dipole moment resulting from S = 1/2 (green), S = 3/2 (grey) and S = 5/2 (red) with a spin-only Landé factor g = 2. | ||
(a) Extraction of the electric dipole moments. Fig. 3 shows the apparent electric dipole moment of the doped clusters, obtained by first-order perturbation theory (FOPT) from the broadening of the molecular beam recorded at Tnozzle = 30, 50 and 70 K. The electric dipole moment is extracted from the variance of the Gauss fits with (σon2) and without (σoff2) applied field.30,32 We do not show the effective polarizabilities of the clusters obtained from the shift of the maxima, but taking into account the static electric polarizabilities and polarization of the dipole distribution32 allows us to quantitatively reproduce the trend observed for the doped clusters with Trot = 10 K. The cluster Mn@Sn12, which has no permanent electric dipole moment according to its molecular structure, shows negligible broadening of the molecular beam. The static electronic polarizability extracted from the shift of the beam profile is 89.3 Å3 at 30 K and 86.6 Å3 averaged over 30–70 K, in good agreement to the isotropic polarizability of 84.1 Å3 obtained from DFT (relative error: 6.2%).
To demonstrate the effect of the dopant atom on the electric dipole moment of the host structure, we have included the measured electric dipole moments of the pure tin clusters from ref. 30. In Fig. 2 we show in addition to the doped clusters the beam profiles of Sn10 obtained from our new datasets. The apparent electric dipole moment obtained by FOPT is 0.63 D in excellent agreement to the value of 0.63 D predicted by DFT.30 We want to point out that in ref. 30 the dipole moment obtained by FOPT is only 0.49 D but MD simulations of the response taking into account a rotational temperature of 3.5 K allow to reproduce the experimental data. In the experiments presented here the rotational temperature of the clusters is expected to be higher due to late evaporation (refer to Section 3.2). The assumption of lower interaction energy with the external field compared to the rotational energy of the cluster hence is a more reasonable approximation and the result indeed provides evidence for higher rotational temperatures.
With increasing nozzle temperature, as shown in Fig. 3, the magnitude of the apparent electric dipole moment is reduced, reflecting the sensitivity to thermal excitation of vibrational modes. Comparison of the electric dipole moments obtained at Tnozzle = 30 K and 50 K shows considerable quenching of the apparent electric dipole moment for clusters with N = 9, 13, 17 and 18. Accordingly, even at 30 K not all clusters can be expected to be strictly rigid and the electric dipole moments reported here for Tnozzle = 30 K are considered as lower limits. However, except for Mn@Sn17 the values obtained experimentally agree very well with the trend apparent in the DFT studies. Mn/Sn9 has a large dipole moment compared to the remaining clusters, reflecting the effect of the bowl shaped host structure. The doped clusters with N = 10–16 have considerably smaller electric dipole moments, compliant to the endohedrally doped structures predicted. Except for Mn@Sn15 and Mn@Sn18, even the more asymmetric cluster species have small electric dipole moments compared to the pure tin clusters with the same number of Sn atoms.
(b) Molecular dynamics simulations. In Fig. 2 (lower panels) we show the beam profiles of the doped clusters obtained by MD simulation of the dielectric response as reported in ref. 30–32 and 65. The experimental parameters (deflection voltage, velocity of the clusters and rotational temperature), dielectric properties and moments of inertia obtained from the QC studies are used to simulate the response of the clusters, taking into account the data from Table S1 (ESI†) of the structures depicted in Fig. 1.
The large drop of intensity of Mn/Sn9 in the central region of the experimental beam profile (z0) allows us to identify this cluster as a non-closed host structure, with the manganese atom close to the centre of mass. The beam profile measured with an applied field (red squares) shows slightly better agreement with the simulation of the higher lying quartet isomers. However, the precision of the relative intensities with/without field is doubtful due to the very low intensities, and it is therefore not possible to uniquely identify the spin isomer of this cluster by its dielectric response. But the electric dipole moment of a tin cluster with a Mn atom attached exohedrally can be expected to be much larger, which would cause the molecular beam to be almost depleted, ruling out such topologies. The electric dipole moment of exohedrally doped Mn/Sn9 obtained at the B3P86/LANL2-DZ level of theory reflects this. With the D3h (minimum energy) cage found by the GA and after relaxation of the Mn doped cluster we obtain for the structure corresponding to the 9.0.s (S = 5/2) cluster 0.89 D, and for the exohedrally doped (S = 5/2) cluster 3.93 D.
The experimental data of Mn@Sn10 correspond much better to the simulated beam profile of 10.0.s than to 10.1.q, but the vanishing dipole moment of 10.0.s does not reproduce the small drop of intensity observed at z0. The isomer 10.2.q is only a slightly distorted geometry of 10.1.q (both closely related to the tetra-capped trigonal prismatic geometry of Sn10) with similar dielectric properties and is not included in the figure. The augmented uncertainty due to low intensity possibly causes the observed drop of the beam profile with an applied field. Hence, the extracted dipole moment in Fig. 3 as well as the discrepancies in the experimental data and the simulated response of 10.0.s are considered primarily as experimental error. But it might also be related to presence of multiple isomers in the molecular beam. The relative energy of the two isomers is only 64 meV (−190 meV) with B3P86 (BP86) functional, and regarding typical errors of total energies in DFT, the spin isomers might both be present in the molecular beam. If the beam does contain both isomers, the isomer 10.0.s contributes stronger and hence can be expected as lower in energy as predicted by the hybrid functional.
The next larger cluster, Mn@Sn11, shows dielectric response in excellent agreement to the lowest energy, quartet electronic ground state isomer 11.0.q. As expected, the next higher sextet spin isomer 11.1.s is further destabilized with the BP86 functional, but the relative energy of 410 meV obtained with B3P86 is large enough to expect only the ground state isomer to contribute significantly to the ensemble in the molecular beam.
The beam profile of the icosahedral cluster Mn@Sn12 is only shifted in the direction of the gradient of the electric field, as mentioned in ref. 23. This reflects a vanishing permanent electric dipole moment as dictated by the inversion symmetry of the molecular cage structure of 12.0.s, as expected from earlier studies in the context of quantum chemical investigations and photoelectron spectroscopy on similar species.1,2 The small drop of intensity is regarded as measurement error, which is likely to result from intensity fluctuations and small errors in beam alignment. The observed shift is well explained by the isotropic electronic polarizability of the cluster obtained from DFT as mentioned above.
Mn@Sn13 has the largest apparent electric dipole moment of the ground state geometries of doped clusters with N > 9. All isomers investigated by DFT are close in energy and exhibit non-vanishing electric dipole moments, but the simulated beam profile of each isomer deviates considerably from the measured data. The two lowest energy quasi-degenerate isomers 13.0.s and 13.1.q produce simulated beam profiles with considerable intensity on the left edge of the figure. This is not observed at all, instead the beam profile intensity follows closely on both sides the simulation according to 13.2.s, and in the central region lies between 13.2.s and 13.4.q. Harmonic analysis of the vibrational normal modes shows that all Mn@Sn13 isomers considered have low vibrational frequencies. Hence, even at Tnozzle = 30 K the majority of clusters are vibrationally excited, affecting the apparent electric dipole moment. The dielectric response does not allow us to assign a molecular structure in this case, but the magnitude of the electric dipole moment points towards an endohedrally doped molecular cage cluster.
The beam profile of Mn@Sn14 measured with applied electric field is reproduced rather well by the simulations of the spin-isomer 14.1.q with S = 3/2. The lowest energy isomer 14.0.s obtained with B3P86/SDB-TZ and 14.2.s (the simulated beam profile is not shown) have vanishing permanent electric dipole moment due to the symmetries of the tin cages and accordingly cannot reproduce the drop of intensity at z0. The electric dipole moment of 14.3.d is also too low to reproduce the measured data. The energetic order is however different with BP86, suggesting 14.1.q as the ground state isomer. Here B3P86 seems to over stabilize the sextet spin isomer.
The dielectric response of the clusters Mn@Sn15 and Mn@Sn16 measured experimentally is reproduced very well by the simulations of the response according to the sextet (15.0.s) and quartet (16.0.q) isomers, respectively. The relative energies of the isomers obtained by DFT (B3P86) indeed suggest the corresponding isomers as the ground state, although in the case of Mn@Sn15 the relative energy between S = 3/2 and S = 5/2 isomer is again very small and inverted with BP86. The much better agreement of the experimentally obtained beam profile with the simulation according to the lowest energy configuration with B3P86 again provides evidence for the reliability of the chosen functional.
The experimentally obtained beam profile of the Mn@Sn17 clusters is not well reproduced by the MD simulations. Although distinct broadening of the molecular beam is observed, indicating a nonvanishing electric dipole moment in the experiment (see Fig. 3), the large electric dipole moments of the quartet and sextet isomer obtained by DFT causes considerably stronger broadening of the simulated beam profiles. Similar to Mn@Sn13, the reason for these discrepancies can possibly be attributed to soft molecular vibrations. Qualitatively, the enhanced flexibility with rising nozzle temperature causes reduced effective electric dipole moments (see Fig. 3), resulting in lower experimentally observed broadening of the molecular beam.30
The beam profile of Mn@Sn18 is reproduced by the simulations of the isomer 18.0.s (S = 5/2), while with S = 3/2 (18.1.q) the electric dipole moment is much too large. Similar to Mn@SnN with N = 9 and 10, the intensities in the MS are small, causing enhanced scattering of the measured data points.
The possibility of weakly quenched electric dipole moments with N = 9, 13, 17 and 18 is noted earlier and the magnitude of the apparent electric dipole moment is possibly enhanced in the limit of the rigid clusters. Keeping this in mind, the ground state isomers of all species identified at our level of theory are in principle in accordance with the dielectric response.
(a) High temperature regime (30–70 K). As discussed in detail in ref. 23 and in agreement with the literature,66–68 the magnetic response of a cluster ensemble with thermally excited vibrational normal modes that break the spin microstate degeneracy, i.e. Jahn Teller (JT) active normal modes,69 can be described by Brillouin's function.70 In this case the ratio of magnetic interaction energy to internal thermal energy x = μmag,0Bz/(kBTint) depends on the vibrational temperature of the clusters (i.e. Tint = Tvib). For the temperature range and the maximum value of the magnitude of the magnetic dipole moment
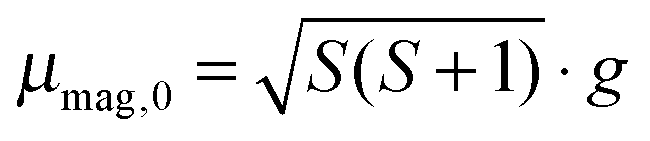 μB of 5.9 μB at 1.5 T the ratio x is smaller than 0.25 in our experiment. Accordingly the low field approximation of Brillouin's function is used here. From the deflection of the molecular beam 〈d〉 the magnitude of the magnetic dipole moment μmag,0 is extracted by using
μB of 5.9 μB at 1.5 T the ratio x is smaller than 0.25 in our experiment. Accordingly the low field approximation of Brillouin's function is used here. From the deflection of the molecular beam 〈d〉 the magnitude of the magnetic dipole moment μmag,0 is extracted by using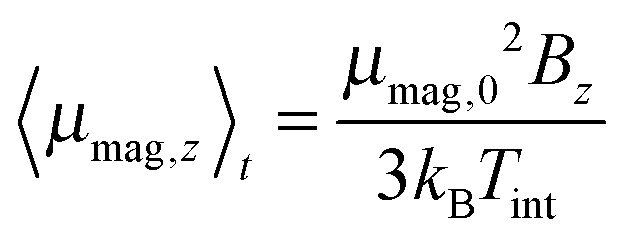 | (3) |
In Fig. 4 the apparent magnitude of the magnetic dipole moment of the doped clusters is reported, obtained from the shift of the beam profile (Gauss fit) at Tnozzle = 70 K (red squares), 50 K (green triangles) and 30 K (blue circles). At 70 K the magnitude of the magnetic dipole moment μmag,0 corresponds in most cases well with the spin only magnetic dipole moments of the isomers as identified by the Stark experiments in conjunction with DFT. Again, augmented errors are expected especially for Mn/Sn9 and also Mn@Sn10. With N = 9 the high temperature magnetic response corresponds well to the spin magnetic dipole moment of the ground state isomer. In the case of Mn@Sn10 the dielectric response pointed at the ground state isomer 10.0.s, possibly with minor contribution of the quartet isomer 10.1.q. If the assumption of spin only magnetism is reasonable, the magnetic response on the other hand corresponds better to primary contribution of the quartet isomer. As the electric dipole moment obtained experimentally is a lower limit (refer to Section 4.2.1), the combined experimental observations in fact can be rationalized by the isomer 10.1.q. Mn@Sn11 was identified by QC and electric beam deflection as 11.0.q with S = 3/2 and the magnitude of the magnetic dipole moment is in very good agreement, corroborating the former results. The Mn@Sn12 cluster reflects a magnetic dipole moment in excellent correlation with the electronic spin of the icosahedral ground state isomer 12.0.s. In the case of Mn@Sn13 the dielectric response cannot be allocated to a certain isomer. But quasi-degeneracy of the two lowest energy isomers 13.0.s and 13.1.q gives a reasonable explanation of the intermediate spin magnetic dipole moment of the ensemble obtained by magnetic beam deflection due to similar population of both S = 3/2 and S = 5/2 isomers (or possibly a spin mixed state of a single isomer as discussed for ferric compounds71). The cluster Mn@Sn14 was identified by its dielectric response as 14.1.q, which is obtained only with BP86 as the ground state isomer. The magnetic response at 70 K is in very good agreement with these observations. Also for Mn@Sn15 and Mn@Sn16, the magnetic deflection experiments correlate well with the S = 5/2 and S = 3/2 ground state isomers identified by the dielectric properties. The magnetic dipole moment measured at 70 K for Mn@Sn17 and Mn@Sn18 both correspond well to the ground state isomers as obtained in the DFT studies, although in the case of Mn@Sn17 it is not possible to verify the ground state geometry of the cluster via electric beam deflection.
With reduced nozzle temperatures of 50 K and 30 K the average magnetic dipole moment of the clusters Mn/Sn9 and Mn@Sn14 is reduced by 1.5–2 μB. The clusters with N = 13, 15 and 18 also show slightly reduced response. But taking into account the estimated mean uncertainty of the experimentally obtained magnetic dipole moments of ±1 μB, the magnitudes of the apparent dipole moment are not very sensitive to the temperature in this range.
In order to investigate the influence of the vibrational temperature also on the variance of the beam profiles with applied magnetic field, clusters are selected for which the dielectric response and beam profile shift in the magnetic field can be related to a specific isomer. In Fig. 5 the relative field induced molecular beam broadening ((σon2 − σoff2)·mv2, normalized to the maximum value) of Mn@SnN with N = 11, 12, 14, 15, 16, 18 at Tnozzle = 16, 30, 50 and 70 K is presented as a function of the fraction P0 of clusters populating the vibrational ground state. The populations P0 are determined by harmonic analysis, taking Tvib (obtained by the procedure described in the ESI†) and a Boltzmann distribution into account. Similar to ref. 23 only JT active normal modes are considered in the case of Mn@Sn12 (due to Ih symmetry only 14 out of 33 in total). The reduced symmetries of the other clusters compared to Mn@Sn12 determine a larger ratio of JT active normal modes. For simplicity in Fig. 5 with N ≠ 12 we include all vibrational normal modes for the determination of P0 to demonstrate qualitatively the correlation of beam broadening and vibrational excitation. To calculate P0 we used the vibrational spectra obtained from the DFT data of those isomers which can be identified by their dielectric response.
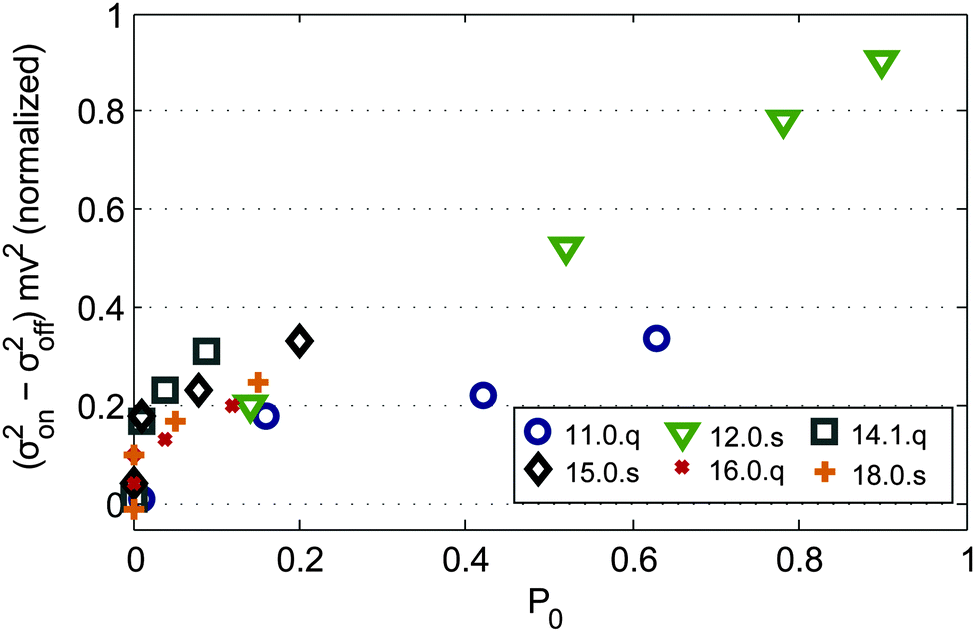 | ||
| Fig. 5 Difference of the variance of the molecular beam profile induced by the magnetic field as a function of the population of the vibrational ground state P0, corrected for mass and velocity of the clusters and normalized to the maximum value. Only those cluster species are considered for which experimental dielectric response and the properties according to our DFT studies allow us to allocate isomers. In the range of negligible population of the vibrational ground state the field induced broadening vanishes for all cluster sizes. With reduced vibrational temperatures the increasing fractions of rigid clusters in the ensemble cause nonuniform deflection of the cluster beam. | ||
For Mn@Sn12 and Mn@Sn11 the populations P0 cover a large range for the temperature interval we investigated. In both cases, but also for the less rigid clusters, the field induced broadening correlates well with P0. While small or vanishing P0 results in uniform deflection of the beam of clusters, with increasing population of the vibrational ground state all clusters show more pronounced broadening of the beam profile. Individual topologies of the rigid clusters then have a distinct influence on the magnetic response, causing non-uniform deflection. While the microscopic mechanism induced by the vibrational degrees of freedom remains still unclear, this correlation provides evidence that in Stern–Gerlach experiments excited (JT active) vibrational normal modes couple electronic spin microstates with the rotational motion of the clusters, inducing net orientation of the magnetic dipole moment.
(b) Low temperature regime (16 K). In the following, the beam profiles measured by magnetic beam deflection experiment at Tnozzle = 16 K are discussed (upper panels in Fig. 2). According to Fig. 5 at least a fraction of the clusters of each species is expected to be rigid. The beam profiles of all clusters in Fig. 2 show broadening, although with N = 13 the effect is small. Besides the tremendous beam broadening of Mn@Sn12, distinct broadening is also observed for clusters with N = 11, 14 and 15. The clusters with N = 11, 12 and 15 are selected as exemplary systems in order to explore the influence of the rigid molecular cages on the magnetic response of the clusters.
The superatomic response of Mn@Sn12 has been discussed in detail in ref. 23. The experimental data in Fig. 2 are fitted by a least square procedure with the two-component model (orange dashed line) presented there. The two components, i.e. the rigid superatom (dotted red line) and vibrationally excited fraction (dash-dotted black line), correspond to the response of a S = 5/2 superatom and vibrationally mediated spin orientation in the latter fraction. As described in the ESI† we use this procedure to obtain experimental estimates of Tvib. The data in Fig. 2 correspond to Tvib = 28 K.
Mn@Sn11 on the other hand is expected in any regard (ground state in the QC studies, dielectric response according to the ground state isomer, magnetic response at Tnozzle 30–70 K) as a S = 3/2 cluster. Mn@Sn11 like Mn@Sn12 is expected to substantially populate the vibrational ground state (refer to Fig. 5) and if the rigid, diamagnetic environment was inert to the magnetic centre, a fraction would show response according to the grey dotted line (Fig. 2). The measured beam profile is considerably broadened by the inhomogeneous magnetic field, reflecting non-uniform deflection. But although a fraction is deflected in the direction of decreasing field, the fraction of rigid clusters is not split up into components of the spin microstates but instead effects asymmetric broadening of the beam.
The molecular beam of Mn@Sn15 still shows field induced broadening, but is exclusively shifted in the direction of the field gradient, with hardly any increase in beam intensity observed on the low field side of the beam profile. Tailing of the beam profile in the direction of the field gradient on the other hand indicates a small fraction of rigid clusters in the beam.
An adiabatic magnetization model has been proposed by de Heer and co-workers to explain the magnetization of rigid clusters, isolated in molecular beams.72,73 In this model, orientation of the (spin) magnetic dipole moment results from the coupling of spin and rotational degrees of freedom. This interaction is rather small and the quantum state of a cluster is represented to a good approximation by the product of the uncoupled basis states, if there is no other state close in energy. In the vicinity of level crossings among states with equal total angular momentum, however, the perturbation of spin-rotation coupling causes the states to repel and the crossing is avoided. The z-components of the spin angular momentum of the interacting states mutually transform while passing the avoided crossing (where Sz is in fact not well defined) and apparently the spin flips. Simultaneously the rotational state undergoes a similar transformation and while no energy is redistributed (the cluster remains in the same total energy eigenstate), total angular momentum (z-component) is also conserved.
In order to establish the relation of molecular topology and the response of the cluster to the inhomogeneous magnetic field, we want to point out the influence of the density of (avoided) crossings. A cluster entering the deflection magnet experiences a relatively fast changing magnetic field, on the order of 105 T s−1. But it is important to remember that in the experiment the clusters also experience a change of magnetic flux as they pass the field region of length l1,B. Due to experimental imperfections (molecular beam alignment, shape of pole faces, homogeneity of the pole face material, nonzero divergence of the molecular beam) and the small but nonzero deflection already present while travelling through the magnet, each cluster is situated in a slowly varying magnetic field. The response measured after the cluster has passed the magnet and the drift region therefore corresponds to the time averaged mean z-component of the magnetic dipole moment 〈μmag,z〉t. In this model the magnetic response of a rigid rotor results from adiabatic state propagation. As discussed in ref. 72 and 73, and in close analogy to the electric beam deflection MD simulations of the former section, the rotational states are thermally populated at zero field. The rotational temperature Trot of the clusters is accordingly required to understand and possibly simulate the response of rigid clusters in the picture of the adiabatic avoided crossing model.
It has been shown that in the limit of a very large number of avoided crossings the z-component of the average magnetic dipole moment corresponds to Brillouin's function also for rigid clusters, with Tint = Trot then determining the ratio of magnetic to internal energy. Rapid oscillations of 〈Sz〉 cause the average z-component of the magnetic dipole moment of all clusters to be equal, resulting in a shifted beam profile, with vanishing broadening.72,73 But adiabatic magnetization can also lead to broadening of the molecular beam, due to non-uniform deflection of the clusters, if the density of avoided crossings is small or vanishes. The beam of clusters then is broadened or split up according to the electronic spin multiplicity, as with Mn@Sn12 in its vibrational ground state. If the clusters are assumed as rigid, it is the manifold of rotational levels and the multiplicity of the spin state that determine the density of crossings. On the other hand, spin-rotation coupling in the first place results from zero field splitting (ZFS) of the spin microstates, caused by the molecular environment of the cluster. In the case of the vibrationally frozen superatom Mn@Sn12 it has been discussed that ZFS vanishes due to its Ih symmetry, and hence the crossings are not avoided.23 If the topology of a cluster on the other hand gives rise to non-vanishing permanent ZFS, crossings among states of equal total angular momentum are in principle avoided. Opposed to homoatomic magnetic clusters, only a single magnetic atom is located in the nanoalloys investigated here and spin–spin coupling effects can be neglected as a source of ZFS. The atomic domain magnetic nanoalloys studied here have small total spin angular momenta, and the number of level crossings is accordingly small and depends considerably on the density of rotational levels.
In Fig. 6 the sum of rotational and magnetic (Zeeman) energies of Mn@Sn12, Mn@Sn11 and Mn@Sn15 are shown as a function of the flux density Bz. In the case of the spherical top cluster Mn@Sn12 the deflection of the cluster populating states with MS = ±5/2 is 3.8 mm. Assuming a constant force acting on the clusters within the deflection unit, the maximum shift within the deflection unit amounts to about 250 μm. With ∂Bz/∂z = 335 T m−1 the range of ΔBz the clusters sweeps through then corresponds to about 0.1 T. Rotational states of a spherical rotor with principal rotational quantum number R are (2R + 1)2-fold degenerate. Even with S = 5/2, i.e. spin multiplicity M = 6, the density of crossings is so small, that within this range of Bz only a small number of crossings is observed, compared to a symmetric or an asymmetric rotor (Fig. 6b and c, respectively). Note that superatomic magnetic response requires vanishing ZFS, i.e. no crossings are avoided. Otherwise, even a single avoided crossing encountered while passing the magnet would suppress the equidistant splitting of the molecular beam components.
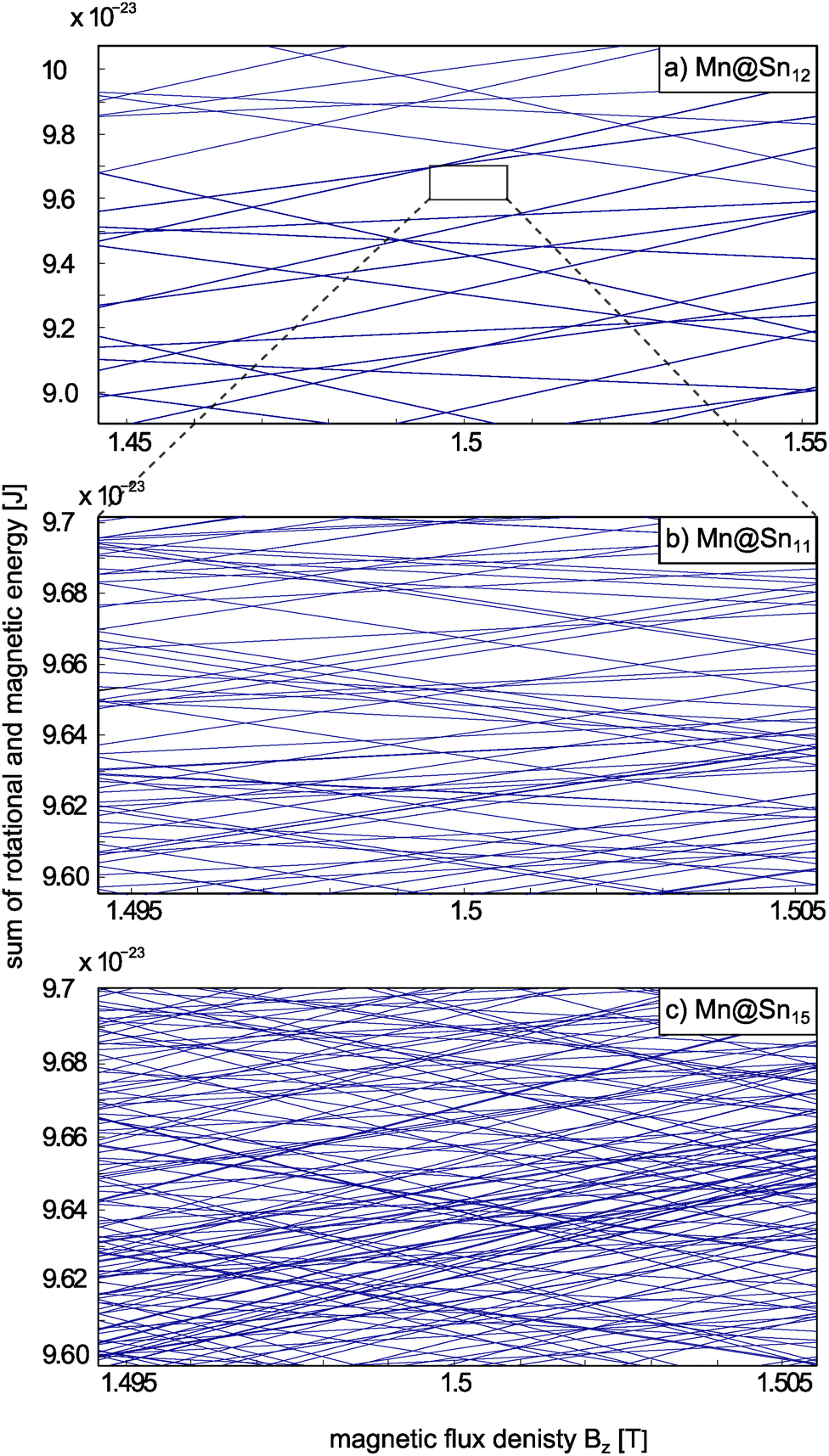 | ||
| Fig. 6 Sum of rotational and magnetic energies (rigid rotor + Zeeman energy) of (a) Mn@Sn12 (S = 5/2), (b) Mn@Sn11 (S = 3/2) and (c) Mn@Sn15 (S = 5/2), i.e. spherical, symmetrical and asymmetric rotors, respectively. In (a) and (b) the rotational energies are the analytic eigenvalues of the rotational Hamiltonian. In (c) the rotational energies are obtained by diagonalization of the rotational Hamilton matrix obtained on the basis of the symmetric rotor states.74 Please note the different range of energy and Bz in (a) compared to (b) and (c). Even with this extended range the number of intersections among states in (a) is much smaller than in (b) and (c), due to the low density of rotational states of a spherical top cluster. The density of states in (c) is again much larger than for (b), because degeneracy of the states corresponding to different projections of rotational angular momentum on the body-fixed axis is split by the lower symmetry, but also because of sextet instead of quartet spin multiplicity. | ||
With typical total deflections of the other clusters in the range of 0.1 mm the shift within the deflection unit amounts to roughly 10 μm. The range of ΔBz the clusters sweep through then corresponds to a few mT and therefore as an estimate of the experimentally relevant range 0.01 T is arbitrarily chosen for (b) and (c). The rotational spectrum of a symmetric top cluster like Mn@Sn11 (Fig. 6b) already shows a considerably richer structure, even with the much smaller range of Bz depicted by the box in Fig. 6a. The reduced rotational symmetry partially breaks the degeneracy of the rotational states and the number of intersections is considerably larger, even with smaller spin multiplicity compared to Mn@Sn12. With a finite range of Bz being sampled, in the experiment in general an arbitrary state will encounter some crossings while passing the deflection magnet. However, the number of crossings is still limited and apparently not sufficient to effectively generate the same average slope, i.e. the same magnetic dipole moment for all clusters. This confirms the idea that in the lower symmetry rigid cluster spin and rotational degrees of freedom are coupled. However, the medium spin multiplicity and the symmetric rotor environment of 11.0.q cause only a limited density of avoided crossings. A cluster then might pass some avoided crossings, but the number is not large enough to statistically enforce similar average values of 〈μmag,z〉t for all clusters. Instead, for some clusters the average orientation might cancel while others have more contribution of high- or low-field seeking states and hence are deflected accordingly.
However, in the experiment all clusters pass at least one avoided crossing, because otherwise some contribution according to the quasi-atomic response (grey dotted line in Fig. 2) would be observed. Finally, in the case of asymmetric top clusters with high spin configuration like Mn@Sn15 the number of avoided level crossings is even larger. In the course of traversing the magnet the spin of each cluster flips many times. The number of avoided crossings traversed by each cluster is apparently sufficient to cause net orientation of the magnetic dipole moment. However, for Mn@Sn15 the fraction of rigid clusters is expected to be rather small (refer to Fig. 5) and it is not possible to conclude whether the field induced broadening of the molecular beam merely results from the superposition of flexible and rigid clusters (with vanishing broadening of the individual fractions), or if the rigid fraction itself is deflected non-uniformly.
The same applies for Mn@Sn10, Mn@Sn13, Mn@Sn15, Mn@Sn16, Mn@Sn17 and Mn@Sn18 at Tnozzle = 16 K. Although the contributions of vibrating and rigid clusters vary significantly with the size of the cluster (refer to Fig. 5), a fraction of rigid clusters of each size should be present and clearly an increase in the broadening of the molecular beams is observed. However, no or only very small increase of intensity on the low field side is observed, in accord with the idea of adiabatic magnetization of the rigid clusters induced by spin-rotation coupling.
Interestingly, the beam profile of Mn@Sn14 at low temperature can be reproduced with a superatomic model by taking a doublet state into account. Although it is the only cluster for which a low lying doublet states is found, even with BP86 the S = 1/2 isomer is higher in energy compared to the S = 3/2 and S = 5/2 spin isomers. On the other hand, the dielectric response in Section 4.2.1 and the high temperature magnetic response both agree well to the properties of 14.1.q. Harmonic analysis in this case reveals very soft vibrational modes (average wave number of the five lowest modes 27 cm−1 with 9 cm−1 of the first mode, while the average of the five lowest modes (ground state isomers, B3P86/SDB-TZ) of the other clusters is 30–65 cm−1). While for most species studied in this work the observed response is rationalized in the full range of temperatures by determining the same spin isomer as rigid or vibrating, this model possibly breaks down as a result of strong vibronic interactions. At the current level of theory we therefore cannot explain the temperature dependent magnetic response of the Mn@Sn14 cluster. It is an interesting candidate to explore the possibility of temperature dependent ground state spin configurations by further beam deflection studies and extended quantum chemical investigations.
5 Conclusions
Electric beam deflection experiments allow to verify the structural motif of the neutral manganese doped clusters Mn/SnN, N = 9–18. The Mn atom is located close to the centre of mass in all investigated clusters. Very small intensities of Mn/Sn9 are observed in the mass spectrum, reflecting the low stability/high reactivity of the cluster as discussed before.22,75 The electric dipole moment is larger in the doped cluster, compared to Sn9, while the remaining doped clusters Mn@SnN have reduced electric dipole moments compared to the homo-atomic clusters SnN. This confirms the tendency of Mn/Sn to form nanoalloy structures with maximum coordination of the Mn centre by Sn. However, the host structure Sn9 is too small to fully encapsulate the dopant atom. Simulations of the dielectric response allow us to confirm the structure of most of the investigated clusters, but also the electronic spin configuration of the clusters.Quantum chemical studies reveal the total spin angular momentum to depend on the size and geometry of the clusters. In terms of transition metal complexes this can be understood phenomenologically as a nephelauxetic effect, or in a more sophisticated picture, it is the result of electron exchange and correlation effects. Considering Sn atoms as rather weak field ligands explains the large abundance of high- or medium-spin clusters found experimentally, in contrast for example to the magnetic properties of endohedrally doped Mn@SiN+ ions probed by X-ray magnetic circular dichroism.76 The model chemistry we employed to investigate the clusters allows us to reproduce these observations. In general we obtain with B3P86 not only very good agreement of the dielectric properties, but also the energetic order of spin isomers is in line with the observed magnetic response in the high temperature regime, except for Mn@Sn10 and Mn@Sn14. In the case of Mn@Sn14 and possibly Mn@Sn10 the magnetic and dielectric response agrees better to the second lowest isomer found with B3P86, which is however the ground state with the pure GGA functional. Accordingly, it might be necessary to employ a hybrid functional with reduced exact Hartree–Fock exchange, as proposed by others,51 to obtain the correct energetic order in the full range of compositions. The better option would be to perform multi-reference configuration interaction calculations, but such calculations are currently prohibitively expensive in computer time.
We investigated the effect of thermal excitation of the clusters on the response to inhomogeneous electric and magnetic fields. Clusters with excited vibrational normal modes are deflected uniformly in the magnetic field and the field induced broadening of the beam vanishes. The magnetic dipole moments of most thermally excited clusters obtained from the deflection of the molecular beam compare well to the predicted spin-only magnetic dipole moments of the clusters.
At low temperature, depending on the magnitude of the spin angular momentum, the molecular structure of the cluster and the vibrational spectrum, a considerable effect of the magnetic field on the beam profile width is noticed. In some cases significant fractions of low field seeking clusters are observed. This is explained by taking adiabatic magnetization of the clusters into account, induced by spin-rotation coupling. Nonvanishing permanent ZFS causes net orientation of the magnetic dipole moment of rigid clusters in the direction of the magnetic field. For asymmetric rigid clusters with sufficiently large densities of avoided crossings this results in single sided deflection. In cases where low spin multiplicity and high molecular symmetry cause a very limited number of crossings, the beam profile with an applied field can show large intensity also on the low field side, but the response of all clusters except Mn@Sn12 and possibly Mn@Sn14 is affected by avoided crossings, resulting in net magnetization of the rigid rotating clusters.
The observations presented in this work reflect the relation of topology and magnetic response of isolated rigid magnetic nanoalloy clusters. Taking into account the molecular parameters related to spin orientation induced by spin-rotation coupling allows us to rationalize these observations. The quantitative treatment of the terms related to ZFS and hence spin-rotation coupling (spin–orbit interaction and in the case of multicenter magnetic clusters spin–spin interaction) is an intricate task and beyond the scope of the present work. But the observations give hope that simulations of the magnetic response of rigid clusters are possible by qualitatively taking symmetry related laws of angular momentum conservation into account to identify avoided crossings and to simply follow the uncoupled states between avoided crossings.
Acknowledgements
We thank the Deutsche Forschungs Gemeinschaft for financial support through grant SCHA 885/10-2. U.R. gratefully thanks the Deutscher Akademischer Austausch Dienst (DAAD) for a travelling stipend, and sincere thanks are given to Dr. Behnam Assadollahzadeh for the introduction to the GA code and helpful discussions regarding the quantum chemical procedure.Notes and references
- V. Kumar and Y. Kawazoe, Appl. Phys. Lett., 2003, 83, 2677 CrossRef CAS PubMed.
- L.-F. Cui, X. Huang, L.-M. Wang, J. Li and L.-S. Wang, Angew. Chem., 2007, 119, 756 CrossRef.
- M. Sargolzaei and N. Lotfizadeh, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155404 CrossRef.
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770 CrossRef CAS.
- J. Bansmann, S. H. Baker, C. Binns, J. A. Blackman, J.-P. Bucher, J. Dorantes-Dávila, V. Dupuis, L. Favre, D. Kechrakos, A. Kleibert, K.-H. Meiwes-Broer, G. M. Pastor, A. Perez, O. Toulemonde, K. N. Trohidou, J. Tuaillon and Y. Xie, Surf. Sci. Rep., 2005, 56, 189 CrossRef CAS PubMed.
- M. Qian, A. C. Reber, A. Ugrinov, N. K. Chaki, S. Mandal, H. M. Saavedra, S. N. Khanna, A. Sen and P. S. Weiss, ACS Nano, 2010, 4, 235 CrossRef CAS PubMed.
- P. Jena, J. Phys. Chem. Lett., 2013, 4, 1432 CrossRef CAS.
- A. C. Reber, S. N. Khanna and A. W. Castleman, J. Am. Chem. Soc., 2007, 129, 10189 CrossRef CAS PubMed.
- A. W. Castleman, J. Phys. Chem. Lett., 2011, 2, 1062 CrossRef CAS.
- W. Gerlach and O. Stern, Z. Phys., 1922, 9, 349 CrossRef CAS.
- W. Gerlach and O. Stern, Z. Phys., 1922, 9, 353 CrossRef CAS.
- W. D. Knight, R. Monot, E. R. Dietz and A. R. George, Phys. Rev. Lett., 1978, 40, 1324 CrossRef CAS.
- W. A. de Heer, PhD thesis, University of California, Berkeley, 1985.
- P. Ballone, P. Milani and W. A. de Heer, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 350 CrossRef.
- W. A. de Heer, P. Milani and A. Chatelain, Z. Phys. D: At., Mol. Clusters, 1991, 19, 241 CrossRef CAS.
- D. M. Cox, D. J. Trevor, R. L. Whetten, E. A. Rohlfing and A. Kaldor, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 7290 CrossRef CAS.
- J. Bucher, D. Douglass and L. Bloomfield, Phys. Rev. Lett., 1991, 66, 3052–3055 CrossRef CAS.
- T. Hihara, S. Pokrant and J. A. Becker, Chem. Phys. Lett., 1998, 294, 357 CrossRef CAS.
- S. Pokrant and J. A. Becker, J. Magn. Magn. Mater., 2001, 230, 1921 CrossRef.
- M. B. Knickelbein, in Nanoclusters - A Bridg. across Discip., ed. P. Jena and A. W. Castleman Jr., Elsevier, Amsterdam, 2010, ch. 11, vol. 1, p. 415 Search PubMed.
- W. A. de Heer and V. V. Kresin, in Handb. Nanophysics Clust. Fullerenes, ed. K. D. Sattler, CRC Press, Taylor & Francis Group, Boca Raton, 2011, ch. 10, pp. 10–11 Search PubMed.
- U. Rohrmann, S. Schäfer and R. Schäfer, J. Phys. Chem. A, 2009, 113, 12115 CrossRef CAS PubMed.
- U. Rohrmann and R. Schäfer, Phys. Rev. Lett., 2013, 111, 133401 CrossRef.
- R. Schäfer, S. Schlecht, J. Woenckhaus and J. A. Becker, Phys. Rev. Lett., 1996, 76, 471 CrossRef.
- R. Antoine, P. Dugourd, D. Rayane, E. Benichou, M. Broyer, F. Chandezon and C. Guet, J. Chem. Phys., 1999, 110, 9771 CrossRef CAS PubMed.
- G. Tikhonov, V. Kasperovich, K. Wong and V. Kresin, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 64, 063202 CrossRef.
- M. Abd El Rahim, R. Antoine, M. Broyer, D. Rayane and P. Dugourd, J. Phys. Chem. A, 2005, 109, 8507 CrossRef CAS PubMed.
- R. Antoine, M. A. El Rahim, M. Broyer, D. Rayane and P. Dugourd, J. Phys. Chem. A, 2006, 110, 10006 CrossRef CAS PubMed.
- R. Moro, X. Xu, S. Yin and W. A. de Heer, Science, 2003, 300, 1265 CrossRef CAS PubMed.
- S. Schäfer, B. Assadollahzadeh, M. Mehring, P. Schwerdtfeger and R. Schäfer, J. Phys. Chem. A, 2008, 112, 12312 CrossRef PubMed.
- S. Heiles, R. L. Johnston and R. Schäfer, J. Phys. Chem. A, 2012, 116, 7756 CrossRef CAS PubMed.
- S. Heiles and R. Schäfer, Dielectric Properties of Isolated Clusters, Springer, Dordrecht, 2014 Search PubMed.
- S. Schäfer and R. Schäfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 205211 CrossRef.
- S. Heiles, S. Schäfer and R. Schäfer, J. Chem. Phys., 2011, 135, 034303 CrossRef CAS PubMed.
- D. A. Götz, S. Heiles, R. L. Johnston and R. Schäfer, J. Chem. Phys., 2012, 136, 186101 CrossRef PubMed.
- D. A. Götz, A. Shayeghi, R. L. Johnston, P. Schwerdtfeger and R. Schäfer, J. Chem. Phys., 2014, 140, 164313 CrossRef PubMed.
- S. Heiles, K. Hofmann, R. L. Johnston and R. Schäfer, ChemPlusChem, 2012, 77, 532 CrossRef CAS.
- S. M. Kast, S. Schäfer and R. Schäfer, J. Chem. Phys., 2012, 136, 134320 CrossRef PubMed.
- I. Compagnon, R. Antoine, D. Rayane, M. Broyer and P. Dugourd, Phys. Rev. Lett., 2002, 89, 253001 CrossRef CAS.
- F. Rabilloud, R. Antoine, M. Broyer, I. Compagnon, P. Dugourd, D. Rayane, F. Calvo and F. Spiegelman, J. Phys. Chem. C, 2007, 111, 17795 CAS.
- M. A. Morrison, T. L. Estle and N. F. Lane, Understanding more quantum physics, Prentice Hall Inc., Englewood Cliffs, 1991, p. 170 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- B. Assadollahzadeh, S. Schäfer and P. Schwerdtfeger, J. Comput. Chem., 2010, 31, 929 CAS.
- M. Bühl and H. Kabrede, J. Chem. Theory Comput., 2006, 2, 1282 CrossRef.
- M. Bu, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, J. Chem. Theory Comput., 2008, 4, 1449 CrossRef.
- V. T. Ngan, E. Janssens, P. Claes, J. T. Lyon, A. Fielicke, M. T. Nguyen and P. Lievens, Chemistry, 2012, 18, 15788 CrossRef CAS PubMed.
- S. Hirata and M. Head-gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- A. Shayeghi, R. L. Johnston and R. Schäfer, Phys. Chem. Chem. Phys., 2013, 15, 19715 RSC.
- A. Shayeghi, C. J. Heard, R. L. Johnston and R. Schäfer, J. Chem. Phys., 2014, 140, 054312 CrossRef CAS PubMed.
- J. N. Harvey, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 203 RSC.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- S. Schäfer and R. Schäfer, ChemPhysChem, 2008, 9, 1925 CrossRef PubMed.
- H. Wang, X. Zhang, Y. J. Ko, A. Grubisic, X. Li, G. Ganteför, H. Schnöckel, B. W. Eichhorn, M.-S. Lee, P. Jena, A. K. Kandalam, B. Kiran and K. H. Bowen, J. Chem. Phys., 2014, 140, 054301 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, 2009 Search PubMed.
- B. Assadollahzadeh, P. R. Bunker and P. Schwerdtfeger, Chem. Phys. Lett., 2008, 451, 262 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS PubMed.
- B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563 CrossRef CAS PubMed.
- S. Schäfer, M. Mehring, R. Schäfer and P. Schwerdtfeger, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 052515 CrossRef.
- J. B. Hopkins, P. R. R. Langridge-Smith, M. D. Morse and R. E. Smalley, J. Chem. Phys., 1983, 78, 1627 CrossRef CAS PubMed.
- B. A. Collings, A. H. Amrein, D. M. Rayner and P. A. Hackett, J. Chem. Phys., 1993, 99, 4174 CrossRef CAS PubMed.
- N. F. Ramsey, Molecular Beams, Oxford University Press, Oxford, 1956 Search PubMed.
- G. Tikhonov, K. Wong, V. Kasperovich and V. Kresin, Rev. Sci. Instrum., 2002, 73, 1204 CrossRef CAS PubMed.
- P. Dugourd, R. Antoine, M. Abd El Rahim, D. Rayane, M. Broyer and F. Calvo, Chem. Phys. Lett., 2006, 423, 13 CrossRef CAS PubMed.
- M. Knickelbein, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 014424 CrossRef.
- D. C. Douglass, A. J. Cox, J. P. Bucher and L. A. Bloomfield, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 12874 CrossRef CAS.
- I. Billas, A. Chatelain and W. de Heer, Science, 1994, 265, 1682 CAS.
- M. C. M. O'Brien and C. C. Chancey, Am. J. Phys., 1993, 61, 688 CrossRef.
- C. Kittel, Introduction to Solid State Physics, John Wiley and Sons, Inc., Hoboken, 8th edn, 2005 Search PubMed.
- G. Harris, Theor. Chim. Acta, 1968, 10, 119 CrossRef CAS.
- X. Xu, S. Yin, R. Moro and W. A. de Heer, Phys. Rev. Lett., 2005, 95, 237209 CrossRef.
- X. Xu, S. Yin, R. Moro and W. A. de Heer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 054430 CrossRef.
- H. W. Kroto, Molecular Rotation Spectra, Dover Publications, 2003 Search PubMed.
- S. Neukermans, X. Wang, N. Veldeman, E. Janssens, R. Silverans and P. Lievens, Int. J. Mass Spectrom., 2006, 252, 145 CrossRef CAS PubMed.
- V. Zamudio-Bayer, L. Leppert, K. Hirsch, A. Langenberg, J. Rittmann, M. Kossick, M. Vogel, R. Richter, A. Terasaki, T. Möller, B. von Issendorff, S. Kümmel and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115425 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: The structures of the dianionic tin cages, an exemplary mass spectrum of the cluster species including a brief description of a procedure implemented to fit the mass spectra by Gaussian functions, detailed geometrical parameters of the doped clusters required for the molecular dynamics simulations and a method to obtain experimental estimates of the vibrational temperature of the clusters in the molecular beam. See DOI: 10.1039/c4cp02994a |
| ‡ Private communication, A. Shayeghi. The TDDFT calculations were performed employing the same basis set and ECP used here, but LC ω-PBEh functional. For more details on the TDDFT calculations refer to ref. 49 and 50. |
| § The singlet electronic configurations of SnN2− are indeed lower in energy compared to the putative global minimum triplet structures according to our GA-DFT investigations. |
| ¶ The limited thermalization at very low Tnozzle is also reflected by the velocity of the clusters.23 |
| This journal is © the Owner Societies 2014 |