
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced desorption of CO from rutile TiO2: elucidation of a new desorption mechanism using first principles
Hendrik
Spieker
and
Thorsten
Klüner
*
Institut für Chemie, Universität Oldenburg, 26129 Oldenburg, Germany. E-mail: hendrik.spieker@uni-oldenburg.de; thorsten.kluener@uni-oldenburg.de
First published on 23rd July 2014
Abstract
Due to its high photocatalytic activity, TiO2 is of eminent interest for a manifold of applications ranging from surface catalysis up to solar energy conversion. However, the fundamental mechanisms of the underlying surface photochemistry are by no means understood. Focussing on this drawback, laser-induced photodesorption of CO on an ideal rutile TiO2(110) surface is studied using first principles in terms of a model system. This paper presents both quantum chemical as well as quantum dynamical results, taking into account the desorption coordinate Z, the polar angle θ, and the azimuth angle ϕ of the adsorbate. Detailed insight into the fundamental mechanisms of the photodesorption process in this model system is obtained, and a new desorption scenario is elucidated.
1 Introduction
As photocatalysis, solar energy conversion and quantum control of chemical reactions represent an emerging field of contemporary surface science, a detailed mechanistic understanding of the underlying surface photochemistry is crucial. The complexity of this effort may be reduced by studying the most fundamental photochemical reaction on surfaces which is photodesorption of small molecules. It has been shown that very accurate quantum chemical and quantum dynamical studies concerning surface photochemistry are consistent with experimental data1 and may therefore serve as a prognosis for future experiments. Using an embedded cluster approach and stochastic wave packet calculations diatomic adsorbates like CO or NO on oxide surfaces such as NiO(100), Cr2O3(0001), and TiO2(110) have been studied.2–6 The underlying theoretical approach is addressed in an extensive review.7In this paper, we simulate the photodesorption of CO on an ideal rutile(110)-surface using first principles. While it is experimentally known that surface defects are the favoured adsorption sites for small molecules such as carbon monoxide, a detailed understanding of the reactivity of the ideal surface is indispensably important. Although first experiments concerning the adsorption and desorption of CO on titanium dioxide using reflection-absorption-spectroscopy8,9 were performed, no state-resolved experimental data concerning photodesorption in the CO–TiO2(110)-system are available so far.
At first, we present three-dimensional potential energy surfaces for the electronic ground state and the first excited state of the adsorbate–substrate-system, which serve as potential candidates for subsequent quantum dynamical studies. Accordingly, the motion of the adsorbate after vertical excitation is simulated using a stochastic jumping wave packet approach. For the first time, and as a consistent further development,10,11 this approach allows for the investigation of the complete rotational properties of a CO molecule desorbing from a rutile TiO2(110)-surface. So far, only two four-dimensional studies of the photodesorption of small molecules from surfaces using first principles exist.1,4 Nonetheless, this three-dimensional study of the CO–TiO2(110)-system is considered state-of-the-art, as the simulation of the rutile surface is much more complicated and incorporates a bigger model system than in previous high-dimensional studies.
2 Methods
Consistent with previous studies, a Ti9O18Mg714+ cluster (see Fig. 1) embedded in a point charge field (PCF) of 4421 charges with values qTi = +2 and qO = −1, respectively, served as a model for the ideal rutile TiO2(110)-surface.10,11 The charge values are obtained from a Mulliken population analysis of the cluster and are consistent with the literature.12 Surface relaxation due to adsorption of one CO molecule has been taken into account. The displacements of the surface atoms are taken from slab calculations obtained within density functional theory (DFT).13,14 Throughout all calculations, the relaxed positions of the cluster atoms and the point charges were kept frozen. Moreover, the lattice constants a = c = 4.649 Å and b = 2.966 Å of the ideal rutile structure have been used.14 Lateral relaxations have been neglected in this study due to their small amount and the possibility of a symmetry breaking in the cluster. Since the latter is finite, it needs to be saturated to avoid an artificial polarisation of the anionic charge density towards bare positive point charges. In our study, Mg2+-ions have been used in consistency with previous studies.10,11 A customized basis set of approximately triple zeta quality and a total number of 628 basis functions has been used.10,11 All quantum chemical calculations regarding the potential energy surfaces have been performed using the Molcas program package.15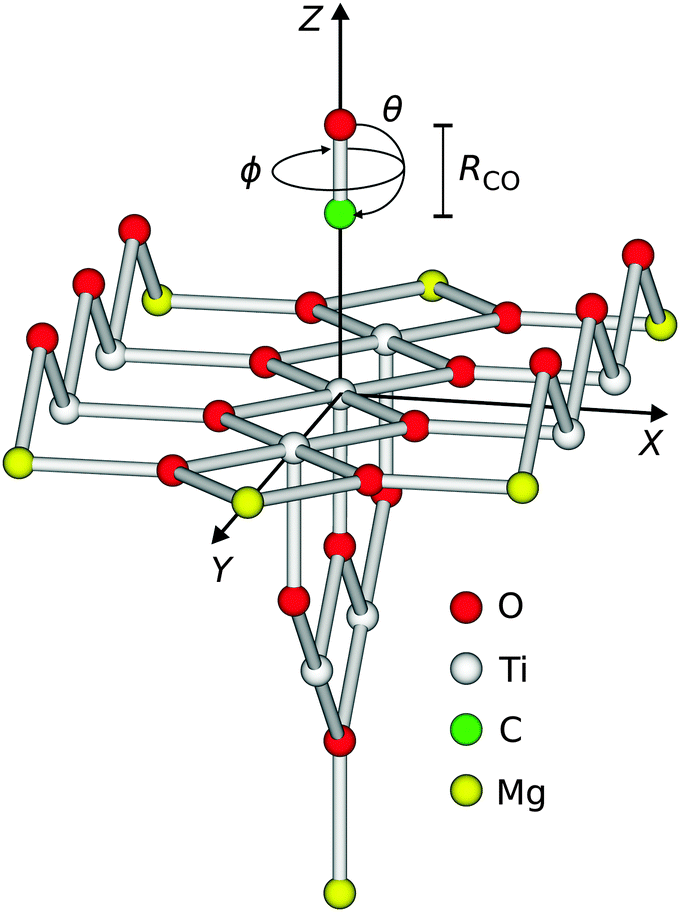 | ||
| Fig. 1 Illustration of the degrees of freedom of the diatomic adsorbate on the surface. The point charge field is not shown. | ||
The diatomic CO adsorbate on top of the central titanium atom of the cluster exhibits six degrees of freedom with respect to its center of mass, as shown in Fig. 1.
In detail, these are the three translational coordinates X, Y, and Z, the polar and the azimuth angle θ, and ϕ, and eventually the bond length R of the CO molecule. Here, the polar angle defines the angle between the CO-bond and the surface normal, with the result that the adsorption geometry is linear with the carbon atom pointing towards the surface for θ = 0°. The azimuth angle constitutes the angle between the projection of the CO-bond axis on the XY-plane and the X-axis, thus with a varying angle θ at a constant ϕ = 0° the CO molecule turns within the XZ-plane. For the first time, three-dimensional calculations of the CO–TiO2(110)-system taking into account Z, ϕ and θ are presented in this paper. The CO bond length has been kept fixed at 1.14 Å10 throughout the study as distortion effects of the CO molecule upon electronic excitation have been found to be negligible in a similar case.4 Since finite basis sets are being used, the calculated interaction energy is affected by a basis set superposition error (BSSE), and its correction is crucial. As the error depends on the geometry of the system, the counterpoise correction16 has been applied to all ab initio data points of the electronic ground state and the excited state potential energy surfaces (PES), respectively.
The quantum dynamical calculations were performed using our highly efficient dyn5d program package.5,17 Although the lifetime of the excited state is experimentally unknown, Gadzuk's jumping wave packet method can be applied.18 It has been shown that this approach, where the total propagation time is subdivided into the propagation on the excited state PES and the ground state PES (wave packet jumping) to simulate the desorption process induced by an electronic excitation, yields results which can be equivalent to a numerical solution of the Liouville–von Neumann equation.19 For the sake of completeness it should be mentioned that the resonance lifetime can be estimated using a surrogate Hamiltonian approach.20,21 Both, the potentials as well as the wave functions are represented on a grid whose size and resolution need to be chosen carefully. Initially, the primary wave packet of the simulation is calculated as the rovibronic ground state of the system. Subsequently, the latter is transferred to the PES of the electronically excited state and then propagated for specific residence lifetimes tn before all of these trajectories are relaxed to the ground state PES, and are propagated until convergence is achieved with respect to the asymptotic observables of interest. Here, a significantly large total propagation time is needed to achieve convergence concerning observables like the desorption probability. Those (partial) wave packets which reach the interaction free asymptotic area of the ground state PES are considered desorbed, and the corresponding observables are calculated. To reduce the computational costs, the split propagator is used to approximate the exact time evolution operator throughout our quantum dynamical simulations. The split propagator is defined as:
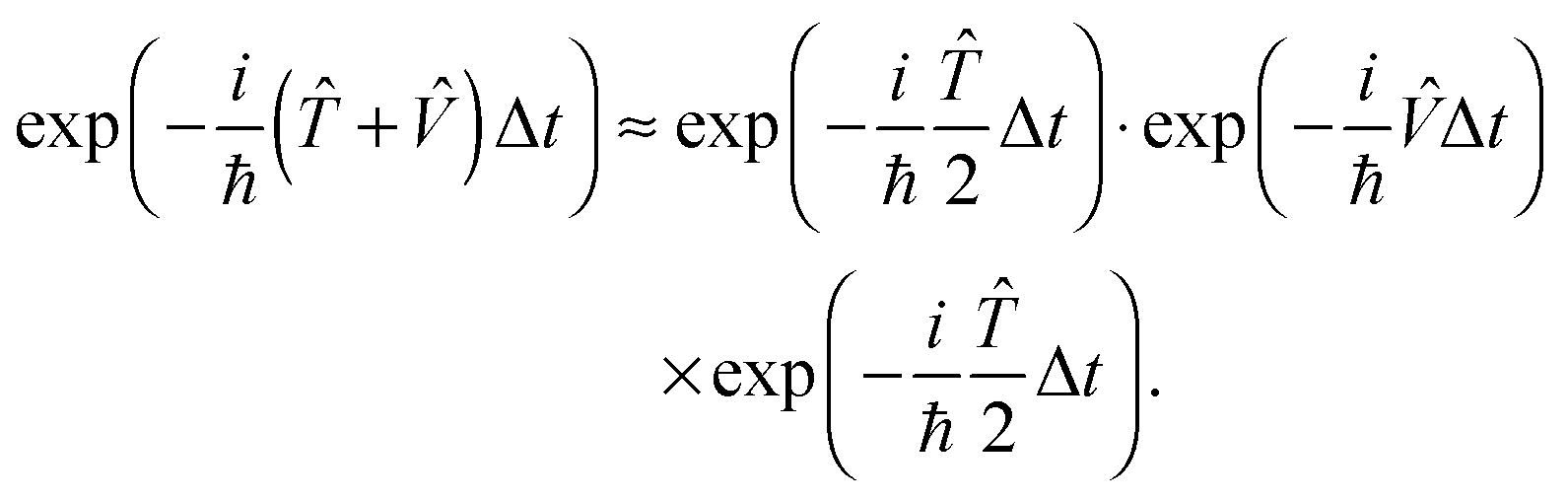 | (1) |
As the accompanying error is of the order ![[scr O, script letter O]](https://www.rsc.org/images/entities/char_e52e.gif) (Δt)3, the time step Δt must be chosen small. The jumping wave packet approach comprises as few as one empirical parameter which is the resonance lifetime τ of the system. Its value may be interpreted as the spectroscopic lifetime of the excited state and therefore is usually adjusted to match experimental data. On this basis, the observables of the quantum dynamical simulations are calculated by averaging over the total number of generated quantum trajectories N. For example, the averaged desorption probability P(t; τ) is given by
(Δt)3, the time step Δt must be chosen small. The jumping wave packet approach comprises as few as one empirical parameter which is the resonance lifetime τ of the system. Its value may be interpreted as the spectroscopic lifetime of the excited state and therefore is usually adjusted to match experimental data. On this basis, the observables of the quantum dynamical simulations are calculated by averaging over the total number of generated quantum trajectories N. For example, the averaged desorption probability P(t; τ) is given by
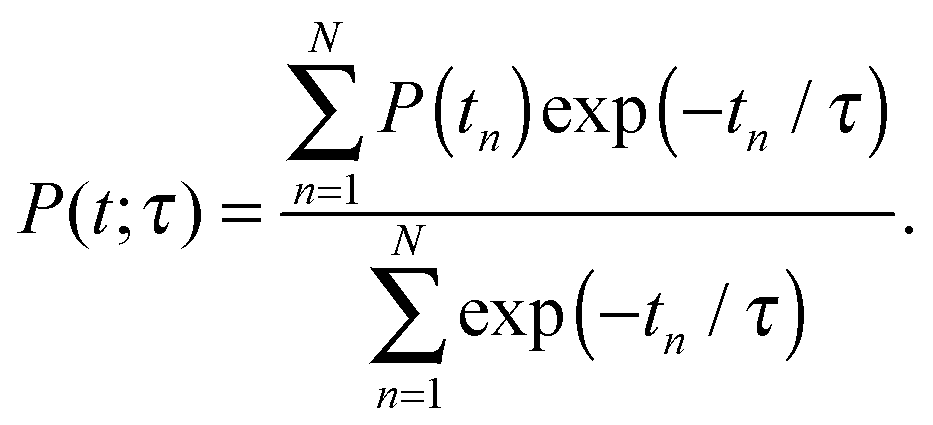 | (2) |
3 Quantum chemical results
Using a total number of 4180 ab initio data points calculated on the MP2 level of theory, an analytical expression for the ground state PES was found by adapting a three-dimensional polynomial function. In the minimum energy configuration, the CO molecule is oriented parallel to the surface normal with an adsorption energy of −0.73 eV and a C–Ti-distance of 2.32 Å, while the carbon atom is pointing towards the surface (which corresponds to θ = 0°). A second but local minimum is found for the reverse orientation of the adsorbate (corresponding to θ = 180°) at almost the same distance to the surface and an interaction energy of −0.22 eV. As shown on the upper panel of Fig. 2, a variation of ϕ lowers the energy barrier between these two minima by approximately 0.1 eV.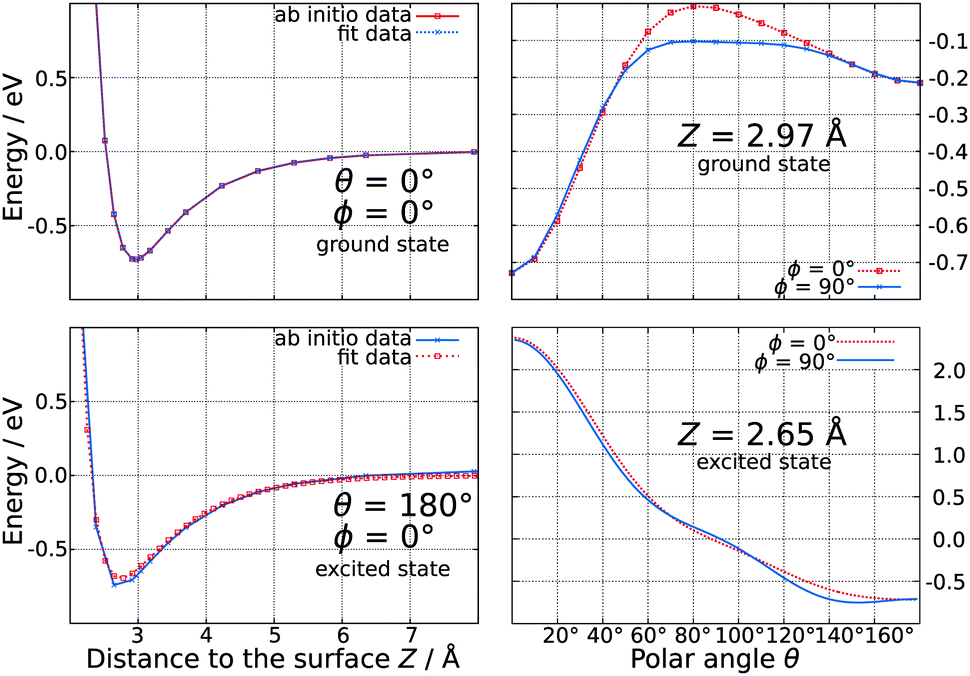 | ||
| Fig. 2 Representative one dimensional cuts of the three-dimensional ground state (upper panel) and excited state (lower panel) potential energy surfaces. | ||
As demonstrated earlier for other substrates such as NiO(100)2 and Cr2O3(0001),17 a 5σ → 2π* excitation of the adsorbate is assumed to be relevant for desorption also in the CO–TiO2-system. We assume, that this excitation is a consequence of a rather complex nonadiabatic mechanism, where a laser pulse initially generates electron–hole pairs in the surface. Accordingly, the latter induce an internal excitation of the CO molecule rather than its ionization via charge transfer processes. The calculations have been performed at the CASSCF level of theory with a CAS(2,3) including the 5σ and the two antibonding 2π* orbitals of the CO molecule, yielding an excitation energy of 6.08 eV in the gas phase on the CASPT2 level of theory. This value is in good accordance with the experimental results revealing an excitation energy of 6.32 eV.23 As our desired excited state is not the first one of our model system, CAS-techniques have to be used to restrict the number of possible configurations and to generate the qualitatively correct reference function. Convergence checks showed that the vertical excitation energy of CO in the gas phase does not substantially depend on the size of the active space on the CASPT2 level. The topology of the PES of the electronically excited state consisting of a total number of 1600 ab initio data points is dominated by a global minimum of −0.74 eV for a linear adsorption geometry, where the oxygen atom points towards the surface. Furthermore, the adsorbate is located much closer to the surface compared to the electronic ground state as the O–Ti-distance is only 2.16 Å. Interestingly, the PES is purely repulsive in the Franck–Condon-area exhibiting no local minimum. A variation of ϕ only marginally affects the interaction energy as illustrated in the lower panel of Fig. 2.
4 Quantum dynamical results
Applying Gadzuk's stochastic jumping wave packet method, the photodesorption of CO from an ideal rutile TiO2(110) surface with respect to three degrees of freedom Z, ϕ, and θ has been simulated on the basis of the three dimensional quantum chemical calculations presented above. The corresponding potentials as well as the nuclear wave functions are represented on a grid of NZ × Nϕ × Nθ = 640 × 255 × 128 points. The convergence of the asymptotic observables with respect to the grid resolution was carefully checked. First, an initial parametric Gaussian wave packet has been propagated in imaginary time24 on the electronic ground state PES using a time step of 1.21 fs and a total propagation time of 2.42 ps, yielding the rovibronic ground state Ψ0 of the system. Its expectation values are in accordance with the minimum energy configuration of the PES as shown in Table 1. Additionally, the imaginary time propagation also yields the zero-point energy (ZPE) of the system.| 〈Z〉 (Å) | 〈ϕ〉 (°) | 〈θ〉 (°) | 〈EZPE〉 (eV) |
|---|---|---|---|
| 2.97 | 180.00 | 5.92 | 0.69 |
In the second step, this wave function is transferred vertically to the excited state PES (Franck–Condon transition). Subsequently, it is propagated in real time for predefined residence lifetimes tn with a time step of Δtn = 0.24 fs generating a total number of 200 quantum trajectories as the maximum residence lifetime is chosen to be 480 fs. Each single trajectory is subsequently transferred vertically to the ground state PES and propagated for 2.42 ps with a time step of 1.21 fs. Choosing τ according to a physical relevant value of P(τ) ≈ 15% yields τ = 12.0 fs as illustrated in Fig. 3. Additionally, attaching weight to later trajectories (τ = 96.0 fs) yields rotationally highly excited wave packets, as shown in the right handside of Fig. 3. It needs to be clearly stated that although this large value of τ should be regarded as unphysical it is used for analysis and interpretation within the scope of our study.
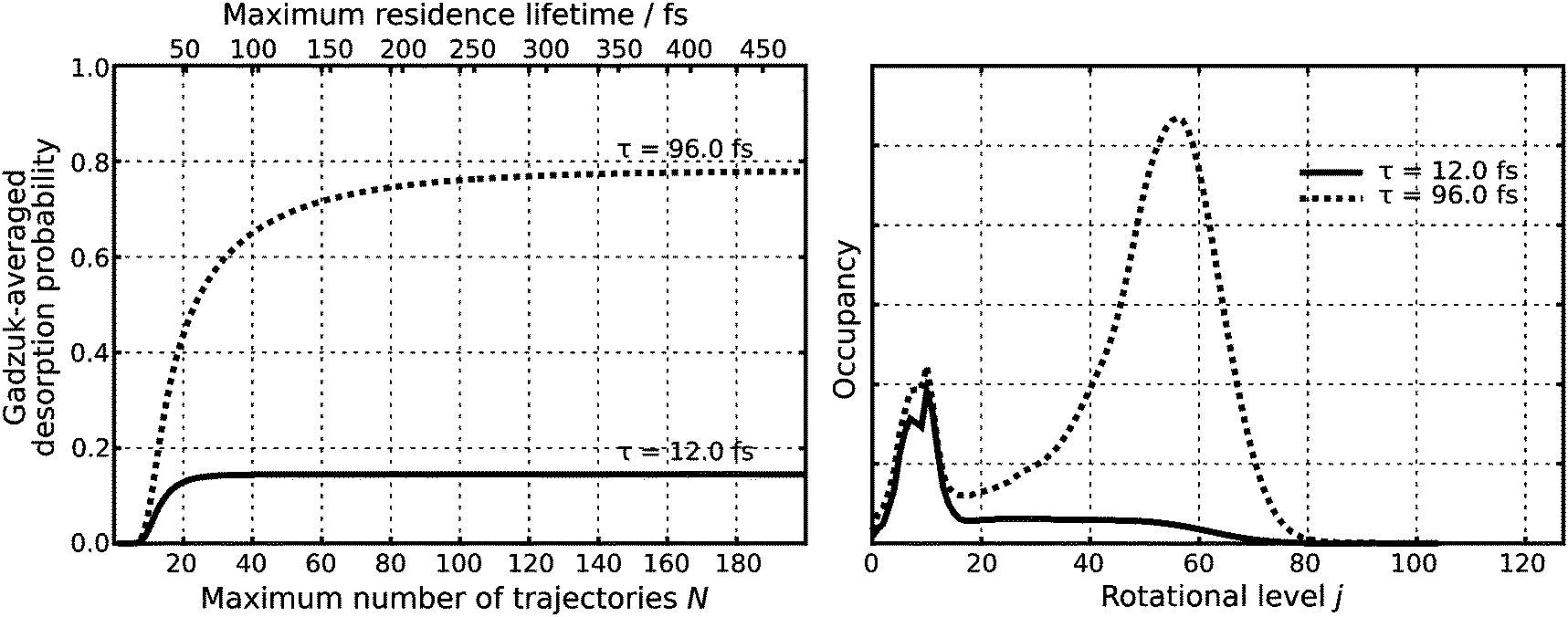 | ||
| Fig. 3 Lifetime averaged desorption probability for specific resonance lifetimes τ in dependence of the maximum residence lifetime on the excited state PES (left panel) and the corresponding final asymptotic occupancy of the rotational levels (right panel). | ||
The rotational excitation is rooted in the topology of the excited state PES with the large gradient both in θ and Z. As depicted in Fig. 4, the wave packet is accelerated, moving fast in the positive direction of Z and rotating vigorously in the polar angle θ. In comparison, its motion in the azimuth angle is rather minor as the CO molecule is merely rotated around ϕ ≈ 61°, which is also shown in Fig. 4. This corresponds to a movement of the adsorbate away from the bridging oxygen atoms. Note that the expectation value 〈ϕ〉 is obtained in the interval 0° < 90° due to the symmetry of the system.
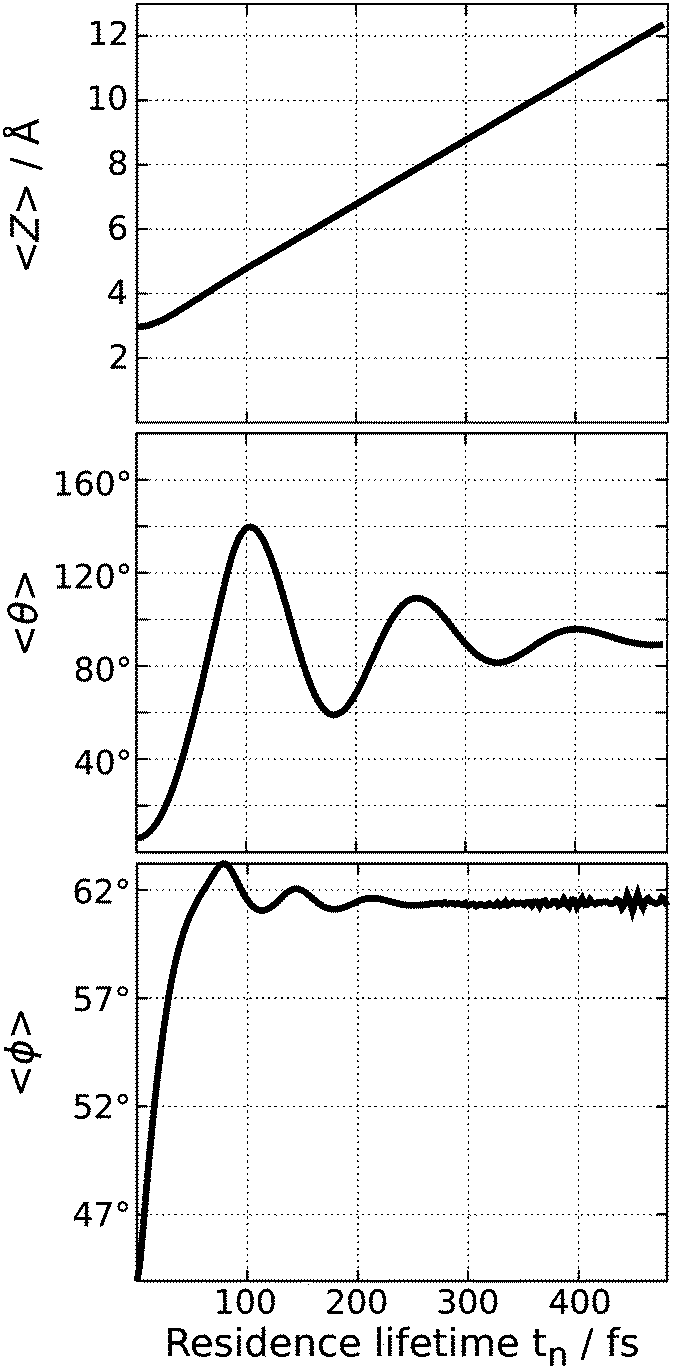 | ||
| Fig. 4 Expectation values of the three examined geometrical degrees of freedom of the wave function during the propagation on the excited state PES. | ||
Both chosen resonance lifetimes τ = 12.0 fs or τ = 96.0 fs are exemplary for relatively extreme desorption scenarios (as shown in Fig. 5), which allows for a detailed comparison of different desorption scenarios due to non-available experimental data. However, in most other studies, the resonance lifetime was chosen to be in accordance with a desorption probability of a few percent.3 Along these lines a resonance lifetime of similar to τ = 12.0 fs seems more likely according to Fig. 5. In the scope of our analysis of the time evolution of the wavefunction, we will not overemphasize those extreme and empirical resonance lifetimes. Instead, we will investigate the terms of the sum in the numerator P(tn)exp(−tn/τ) of eqn (2) and will identify the most relevant quantum trajectory according to its weight choosing either τ = 12.0 fs or τ = 96.0 fs. This analysis revealed the importance of the residence lifetimes tn = 28.8 fs (τ = 12.0 fs) and tn = 40.8 fs (τ = 96.0 fs), respectively.
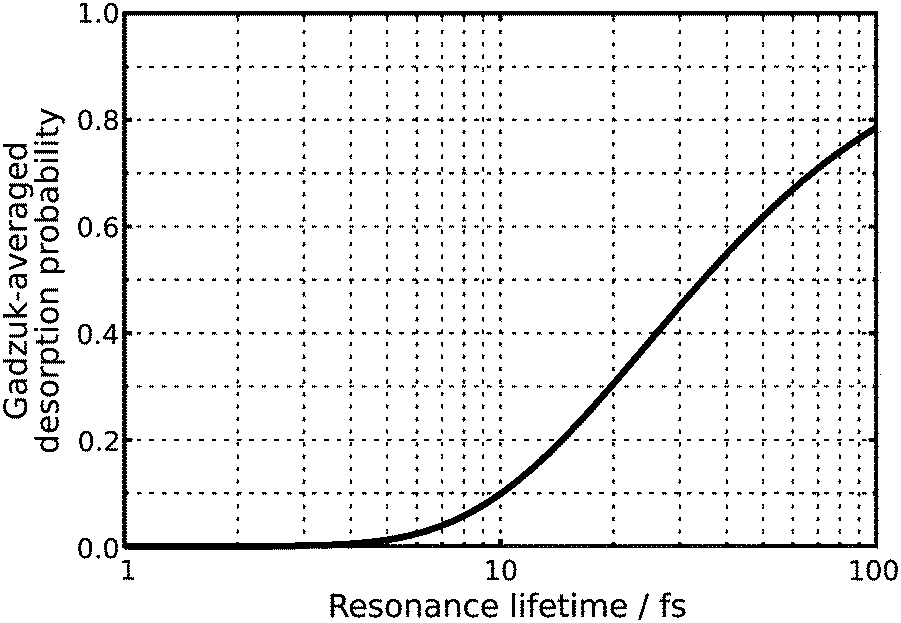 | ||
| Fig. 5 Gadzuk-averaged desorption probability in dependence of the resonance lifetime τ. | ||
Upon investigating the probability distribution of the wave function during propagation on the electronically excited state PES for those residence lifetimes, a negative correlation of translation and rotation of the CO molecule is found in both cases. Those partial wave packets at higher polar angles θ move comparably slower and are located closer to the surface than those at lower angles, as shown in the upper half of Fig. 6. As indicated by the black dotted guide lines, a negative correlation of rotational and translational motion of the CO molecule after the laser excitation is clearly visible. Following the gradient at θ = 0° the motion of the wave packet in positive direction of Z can be characterized as MGR-like (Menzel–Gomer–Redhead, repulsive excited state),25,26 while it is translated closer to the surface for higher polar angles θ, which is related to an Antoniewicz-like (attractive excited state)27 mechanism. The latter is caused by the attractive gradient of the electronic potential at θ = 180°.
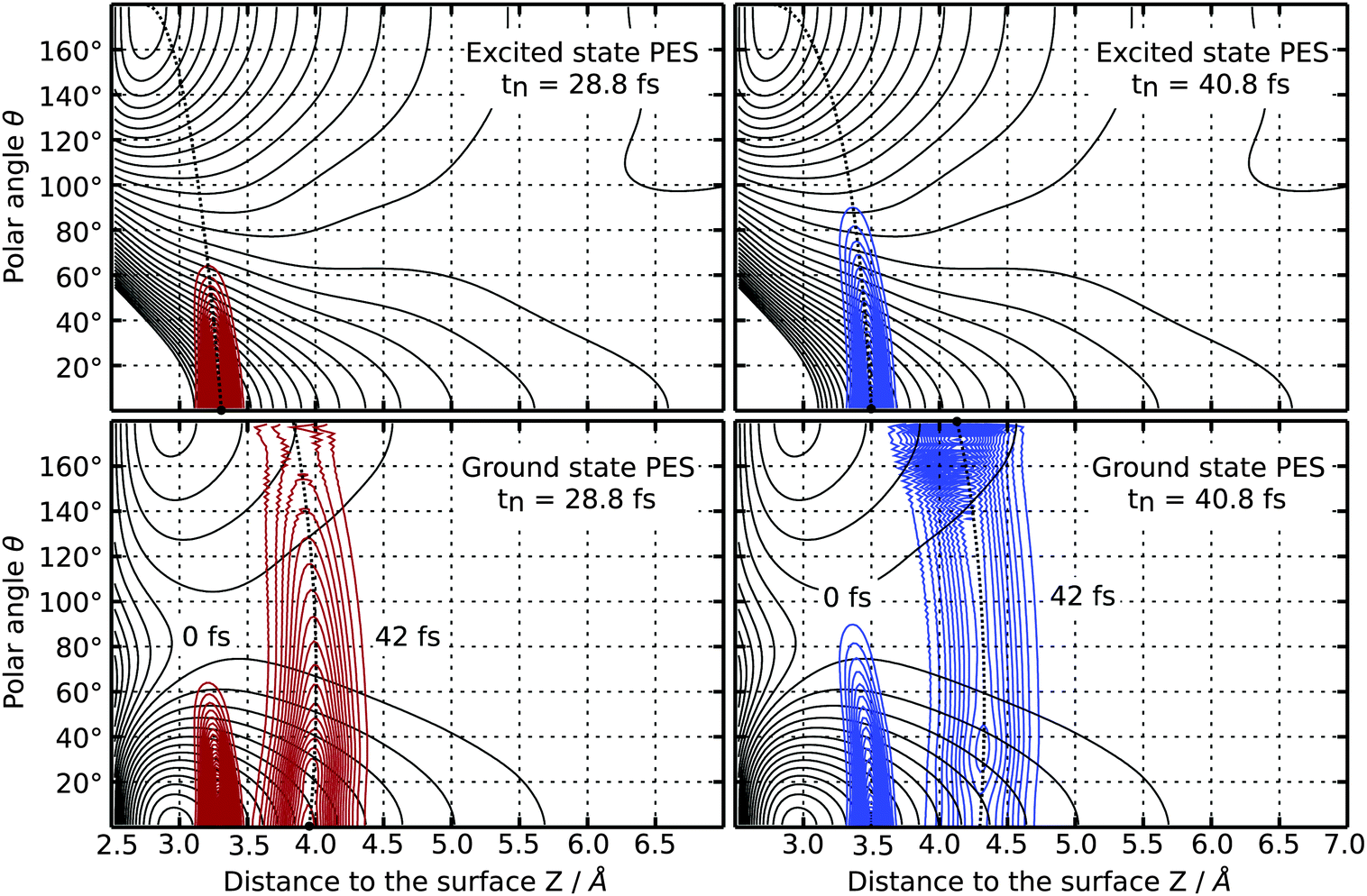 | ||
| Fig. 6 Two-dimensional contour plots of the probability distribution of the wave function after a residence lifetime of tn = 28.8 fs (red lines) and tn = 40.8 fs (blue lines), respectively. In each case a black dot indicates the maximum of the probability distribution. The PES of both electronic states are also shown (black lines). The dotted black lines are guide lines of the motion of the wave packet at the respective simulation time. | ||
After relaxation to the ground state the afar rotated partial wave packet is located closer to the local minimum of the ground state PES at θ = 180° and encounters a smaller gradient than the other part of the wave packet which relaxes closer to the global minimum energy configuration at θ = 0°, as illustrated in the lower half of Fig. 6. To show the effect of the relaxation back to the electronic ground state, the wavefunction has been propagated for an additional time of 42.0 fs on the ground state PES. The resulting motion gives rise to an inversion of the correlation of rotation and translation of the CO molecule which is now positive for high polar angles. In fact, this alteration opens up different desorption channels for the CO molecule which are dependent on the level of rotational excitation in the electronically excited state. Both of them can be traced back by calculating the final averaged velocity distributions for both selected resonance lifetimes which are presented in Fig. 7. Attaching weight to higher residence lifetimes by applying τ = 96.0 fs to the simulation results in rotationally excited desorbates with a high velocity of 2050 ms−1. The latter arises from the positive correlation of rotation and translation on the ground state PES. Supportingly, the velocity distribution is monomodal as depicted in the right panel of Fig. 7. Applying τ = 12.0 fs instead, two more desorption channels at 700 ms−1 and 450 ms−1 are identifiable along with the fastest at 1550 ms−1, as displayed in the left handside of Fig. 7. Those channels originate from trajectories in the repulsive Franck–Condon area, where translation dominates over rotational excitation. This motion reveals a new desorption scenario, which could be interpreted as a mixture between an Antoniewicz-like27 and an MGR-like25,26 mechanism of desorption. To provide further evidence for these results, rotational alignment during desorption needs to be studied for different desorption channels. The results of these investigations will be presented in a future publication.
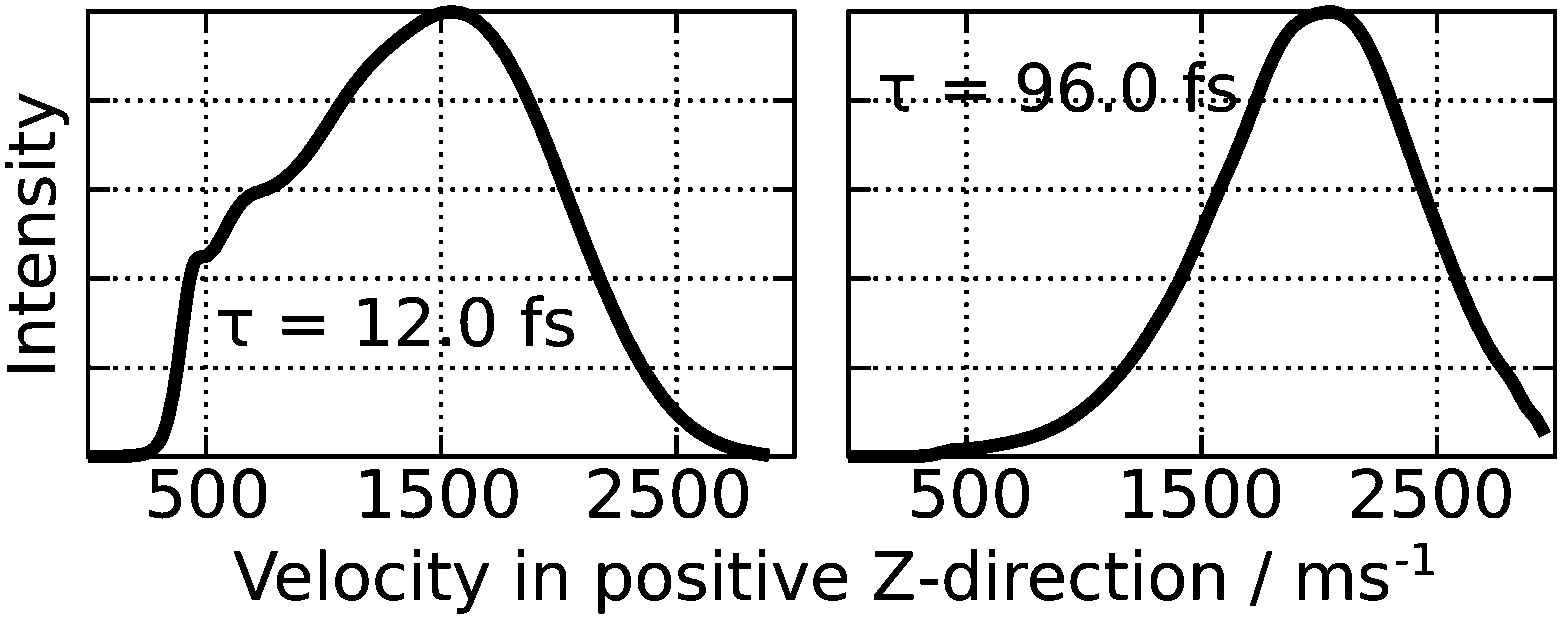 | ||
| Fig. 7 Final averaged velocity distributions of the desorbed CO molecule for τ = 12.0 fs and τ = 96.0 fs, respectively. | ||
5 Conclusion
In conclusion, the laser-induced photodesorption of CO on an ideal rutile TiO2(110)-surface has been studied using first principles. During the desorption process, the wave packet experiences a delicate interplay of gradients in Z, and θ, respectively. This results in a completely new multi-dimensional desorption mechanism which can be classified as an angle-dependent superposition of the conventional one-dimensional MGR- (where the electronically excited state is repulsive) and Antoniewicz (with an attractive electronically excited state) mechanisms. As a consequence, a strong correlation between translation and rotation of the adsorbate was observed although this effect was found in the opposite direction for the ground state and excited state PES, respectively. Due to the interesting topology of the excited state PES, surface temperature might affect the desorption dynamics significantly. In particular, the population of higher rotational states might influence the superposition of the MGR- and Antoniewicz mechanism, which opens the perspective of temperature control of this photochemical reaction. The underlying quantum dynamical calculations will also allow an enhanced insight into the rotational alignment of the CO molecule during desorption. Additionally, we might also add further geometrical degrees of freedom of the adsorbate in our simulation in the future. In summary, our results not only shine new light on the mechanisms of surface photochemistry in general, but also pave the way for a faithful interpretation of future experiments possibly including even defect sites on the surface or dopants within the rutile bulk structure.Acknowledgements
The authors thank F. Habecker for support concerning the analytical fits of the PESs. This work was funded by the Deutsche Forschungsgemeinschaft through the priority program 1613 (KL 1175/12-1). The simulations were performed at the HPC Cluster HERO, located at the University of Oldenburg (Germany) and funded by the DFG through its Major Research Instrumentation Programme (INST 184/108-1 FUGG) and the Ministry of Science and Culture (MWK) of the Lower Saxony State.References
- I. Mehdaoui, D. Kröner, M. Pykavy, H.-J. Freund and T. Klüner, Phys. Chem. Chem. Phys., 2006, 8, 1584–1592 RSC.
- T. Klüner, H.-J. Freund, J. Freitag and V. Staemmler, J. Chem. Phys., 1996, 104, 10030–10040 CrossRef PubMed.
- T. Klüner, H.-J. Freund, V. Staemmler and R. Kosloff, Phys. Rev. Lett., 1998, 80, 5208–5211 CrossRef.
- M. Pykavy, S. Thiel and T. Klüner, J. Phys. Chem. B, 2002, 106, 12556–12562 CrossRef CAS.
- S. Borowski, T. Klüner and H.-J. Freund, J. Chem. Phys., 2003, 119, 10367–10375 CrossRef CAS PubMed.
- M. P. de Lara-Castells and J. L. Krause, J. Chem. Phys., 2003, 118, 5098–5105 CrossRef CAS PubMed.
- T. Klüner, Prog. Surf. Sci., 2010, 85, 279–345 CrossRef PubMed.
- C. Rohmann, Y. Wang, M. Muhler, J. Metson, H. Idriss and C. Wöll, Chem. Phys. Lett., 2008, 460, 10–12 CrossRef CAS PubMed.
- N. G. Petrik and G. A. Kimmel, J. Phys. Chem. Lett., 2013, 4, 344–349 CrossRef CAS.
- M. Mehring and T. Klüner, Chem. Phys. Lett., 2011, 513, 212–217 CrossRef CAS PubMed.
- M. Mehring and T. Klüner, Mol. Phys., 2013, 111, 1612–1621 CrossRef CAS.
- C. Sousa and F. Illas, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 13974–13980 CrossRef CAS.
- B. Meyer, private communication, 2009.
- M. Kunat, F. Traeger, D. Silber, H. Qiu, Y. Wang, A. C. van Veen, C. Wöll, P. M. Kowalski, B. Meyer, C. Hättig and D. Marx, J. Chem. Phys., 2009, 130, 144703 CrossRef CAS PubMed.
- G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrady and L. Seijo, Comput. Mater. Sci., 2003, 28, 222–239 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Borowski, T. Klüner, H.-J. Freund, I. Klinkmann, K. Al-Shamery, M. Pykavy and V. Staemmler, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 223–230 CrossRef CAS PubMed.
- J. Gadzuk, L. Richter, S. Buntin, D. King and R. Cavanagh, Surf. Sci., 1990, 235, 317–333 CrossRef CAS.
- P. Saalfrank, Chem. Phys., 1996, 211, 265–276 CrossRef CAS.
- S. Dittrich and T. Klüner, Chem. Phys. Lett., 2006, 430, 443–447 CrossRef CAS PubMed.
- E. Asplund and T. Klüner, Mol. Phys., 2013, 111, 2377–2386 CrossRef CAS.
- M. Menges, B. Baumeister, K. Al-Shamery, H.-J. Freund, C. Finkscher and P. Andresen, J. Chem. Phys., 1994, 101, 3318–3325 CrossRef CAS PubMed.
- E. S. Nielsen, P. Jørgensen and J. Oddershede, J. Chem. Phys., 1980, 73, 6238–6246 CrossRef CAS PubMed.
- R. Kosloff and H. Tal-Ezer, Chem. Phys. Lett., 1986, 127, 223–230 CrossRef CAS.
- D. Menzel and R. Gomer, J. Chem. Phys., 1964, 41, 3311–3328 CrossRef CAS PubMed.
- P. A. Redhead, Can. J. Phys., 1964, 42, 886–905 CrossRef CAS.
- P. R. Antoniewicz, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 21, 3811–3815 CrossRef CAS.
| This journal is © the Owner Societies 2014 |