
Investigation of the bulk and surface properties of CdSe: insights from theory†
Abstract
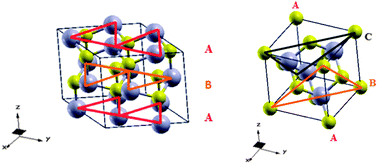
CdSe, in the form of quantum dots, is a semiconductor material extensively used for photovoltaic applications. We have performed a comprehensive density functional theory investigation of the geometrical and electronic properties of CdSe bulk crystals (for both the wurtzite and zinc blende phases) as well as their wurtzite (10−10) surface. Several combinations of Gaussian-type orbital basis sets and exchange–correlation functionals have been tested with a periodic formalism. The computational method for further studies was selected on the basis of the comparison of computed geometrical and electronic properties to available experimental data for the bulk crystals of CdSe, as well as to additional projector-augmented wave calculations. In a second step, the so-defined protocol was used to successfully simulate CdSe wurtzite nanocrystals exposing their nonpolar (10−10) surface. Most importantly, we have shown that the presented computational protocol is accurate to model not only bulk crystals but also surfaces, thus providing a powerful theoretical tool to simulate the light harvesting components of quantum dot sensitized solar cells.
 Please wait while we load your content...
Please wait while we load your content...

