
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradation of Fe/N/C catalysts upon high polarization in acid medium†
Vincent
Goellner
a,
Claudio
Baldizzone
b,
Anna
Schuppert
b,
Moulay Tahar
Sougrati
a,
Karl
Mayrhofer
b and
Frédéric
Jaouen
*a
aInstitut Charles Gerhardt de Montpellier – UMR 5253, 2 Place Eugène Bataillon, 34095 Montpellier Cedex 5, France. E-mail: frederic.jaouen@univ-montp2.fr; Tel: +33 467143211
bMax Planck Institut für Eisenforschung, Department of Interface Chemistry and Surface Engineering, Max Planck Strasse 1, 40237 Düsseldorf, Germany
First published on 17th July 2014
Abstract
A comprehensive study of the degradation of a highly active Fe/N/C catalyst in acid medium is reported. An accelerated aging protocol was applied in the temperature range of 20 to 80 °C. From fundamental rotating-disc electrode studies and polymer electrolyte membrane fuel cell investigations combined with identical-location electron microscopy and Mößbauer spectroscopy at various stages of degradation, important insights into the structural and chemical changes of the catalyst were obtained. Most importantly, the degradation is strongly enhanced at elevated temperature, which is correlated to (i) increased carbon-corrosion rate and (ii) parallel non-preferential dissolution of the FeNx-based active sites. The degradation not only leads to a decreased ORR kinetics over time but also induces significant charge- and mass-transport resistances due to the collapse of the electrode structure.
Introduction
Electrochemical energy conversion is expected to play a key role in the future energy landscape. Whether as a means of efficiently storing off-peak grid power, producing hydrogen or propulsing electric vehicles, the high efficiency, scalability and possible reversibility of these systems offer key advantages of enabling the integration of a higher load of renewable energy in the power grid as well as of reducing the dependence on fossil fuels. Among today's electrochemical devices, the polymer electrolyte membrane fuel cell (PEMFC) is perceived as the best contender to replace internal combustion engines in the near future. While PEMFC-powered automobile fleets have been demonstrated, remaining challenges are increased durability and reduced cost, while a longer-term challenge is the replacement of the rare-element platinum used in its catalysts.1,2 The development of catalysts free of platinum-group metals is crucial, in particular for the cathode, which typically accounts for 80% of the Pt content in a PEMFC.While Pt particles dispersed on carbon are still the state-of-the-art catalysts for the oxygen reduction reaction (ORR), non-precious metal catalysts involving Fe or Co, nitrogen and carbon have shown fast progress since 2008.3–9 One aspect common to Fe(Co)/N/C and Pt/C catalysts is the presence of a carbon support and/or anchoring matrix for the active sites. Carbon corrosion is a major source of performance loss in Pt/C-catalyzed PEMFC. Rapid carbon corrosion occurs during transient events that polarize either the anode or the cathode to high electric potentials of 1.5 V against the reversible hydrogen electrode (RHE). These transients are caused by either fuel starvation or H2/air bleed front at the anode, strongly polarizing the anode or cathode, respectively.10–13 While complete fuel starvation is a rare incident, build-up of a H2/air front at the anode systematically occurs during start up and shutdown, unless specific procedures implying additional cost or increased system size are applied.14,15 Ever since these transient phenomena were identified,10,16,17 carbon corrosion of Pt/C catalysts has been a major research topic.17–20
While a comprehensive body of work exists on the degradation mechanisms of Pt particles supported on carbon or non-carbon supports, few such investigations have focused on Fe(Co)/N/C catalysts.21–26 Two facts account for this: (i) research on Fe(Co)/N/C catalysts has focused on increasing their initial activity and power performance in PEMFC and (ii) recording changes in the active sites of Fe(Co)/N/C catalysts during their degradation remains a major scientific challenge. In particular, the effect of high-voltage transients on Fe(Co)/N/C catalysts has rarely been studied.24,27 Important questions about the effect of high-voltage transients on Fe(Co)/N/C catalysts include whether the degradation of the FeNxCy active sites precedes or follows the carbon corrosion process and whether the degradation rate of Fe(Co)/N/C catalysts is faster than that of Pt/C catalysts subjected to identical start/stop protocols. High-voltage cycling offers the possibility to degrade a catalyst without performing the ORR, which avoids a number of complex parameters such as the potential- and catalyst loading-dependent formation of H2O2. In addition, start/stop cycling protocols may be used as an interesting means of identifying the active sites in Fe(Co)/N/C catalysts through a reverse strategy. By degrading a catalyst to different extents, a wide range of ORR activities may be obtained and compared to post mortem spectroscopic characterisation.
This reduces the number of variables compared to the classical approach where a set of catalysts are synthesized and their initial ORR activities are correlated to spectroscopic features of the pristine catalysts.28–31 X-ray absorption and 57Fe Mössbauer spectroscopy are the most suited techniques to investigate the electronic environment and coordination of the metal atoms in Fe(Co)/N/C-catalysts.8,28–35 A common drawback is however their inability to differentiate the surface signal from the bulk signal, which complicates tentative correlations to the ORR activity. This drawback can be partially overcome by investigating the post mortem spectroscopic signal of a pristine catalyst degraded to different extents, thereby highlighting changes that necessarily occurred at the electrochemical interface.
The present study investigates the changes induced by high-voltage transients in the ORR activity, morphology and spectroscopic response of a single pristine Fe/N/C catalyst. For the first time, identical-location electron microscopy and post mortem57Fe Mössbauer spectroscopy were applied to investigate the degradation of an Fe/N/C-catalyst.
Experimental
Catalyst synthesis
One Fe-based catalyst, labelled Fe/N/C, was mostly investigated in this study, while a second catalyst, labelled Fe/N/C-dry and having a smaller content of ORR-inactive crystalline Fe species, was additionally investigated by post mortem Mössbauer spectroscopy. The catalyst precursor for the Fe/N/C catalyst was prepared from optimized amounts of FeII acetate (FeIIAc), 1,10-phenanthroline (phen) and a commercially available ZnII imidazolate metal–organic framework labelled ZIF-8. These compounds were added in sequence to an ethanol:water solution where the Fe(phen)3 complex formed. After evaporation of the solvents overnight, the catalyst precursor was collected. This powder was then ballmilled to homogenize the Fe(phen)3 distribution on ZIF-8 and pyrolyzed in Ar at 1050 °C. During this process, the microporous ZIF-8 compound was transformed into N-doped microporous carbon with embedded FeNxCy active sites. Finally, the resulting catalyst powder was acid-washed with a pH 1 sulfuric-acid solution, yielding the Fe/N/C catalyst. The second catalyst, labelled Fe/N/C-dry, was synthesized identically except for the omission of (i) the initial wet impregnation step and (ii) the final acid-washing step. In practice, weighed amounts of dry powders of FeIIAc, phen and ZIF-8 were directly ballmilled and the resulting powder was pyrolyzed at 1050 °C in Ar.Rotating disk electrode (RDE) measurements
RDE measurements were performed in the range 20–80 °C, which required the following adjustments: (i) a glassy-carbon tip shrouded in polyether ether ketone was used, (ii) the electrolyte was heated via a temperature-controlled oil bath and (iii) a reversible H2 electrode was used as a reference electrode in order to be free from the temperature drift of classical reference electrodes based on a salt-precipitation reaction. The catalyst loading was always 800 μg cm−2. Unless otherwise mentioned, measurements were performed in 0.1 M H2SO4 electrolyte.PEMFC measurements
Pure H2 and O2 were fed at 50–70 sccm into the humidifiers held at 80 °C; the cell temperature was 80 °C and the gas inlet pressure was 1 bar gauge. The anode and cathode loadings were 0.5 mgPt and 4.0 mg cm−2, and the membrane was Nafion™ NRE-211.Results
Deterioration of ORR kinetics due to high-potential cycling
We investigated the effect of high-voltage transients on the ORR activity measured first with the RDE technique (Fig. 1). The applied square-wave signal of electric potential is schematized in the inset of Fig. 1A, featuring one cycle with a 3 second hold at 0.9 V vs. RHE and a 3 second hold at 1.4 V vs. RHE. Following the measurement of the initial ORR activity for the Fe/N/C catalyst (solid black curves), 300 such cycles were applied in a N2-saturated pH 1 electrolyte and the ORR activity was re-measured (solid red curves). The same experiment was performed at room temperature, 40, 60 and 80 °C (Fig. 1A–D). The results show a clear effect of temperature on the activity decay, especially above 40 °C. The temperature of 80 °C is used for most PEMFC tests and is therefore particularly relevant. | ||
| Fig. 1 ORR activity of the Fe/N/C catalyst at 20 (A), 40 (B), 60 (C) and 80 °C (D) after 0, 300 or 900 square wave signals of 0.9 to 1.4 V vs. RHE (inset). In (D), the dashed curves correspond to the N/C catalyst. | ||
Interestingly, Fig. 1D reports the initial activity at 80 °C of the iron-free N/C catalyst (dashed black curve) obtained after pyrolyzing phenanthroline and ZIF-8 at 1050 °C in argon. Its half-wave potential is 650–670 mV vs. RHE, which is fairly high by all accounts for a metal-free catalyst but still 110–150 mV below the initial half-wave potential of the Fe/N/C catalyst. The activity of the Fe/N/C catalyst after 300 cycles at 80 °C becomes lower than the initial activity of the N/C catalyst (Fig. 1D, solid red curve and dashed black curve), suggesting that most FeNxCy sites did degrade during the cycles. The N/C catalyst also degraded during cycling (dashed curves in Fig. 1D), with a seemingly smaller decay than Fe/N/C. This impression is largely due to the modified Tafel slope for Fe/N/C (see later), but unchanged Tafel slope for N/C. The removal or inactivation of the FeNxCy sites during cycling implies that they were mostly located on the surface of the pristine Fe/N/C catalyst. The higher activity of Fe/N/C after 300 cycles relative to N/C after 300 cycles (red curves in Fig. 1D) may come from a few intact FeNxCy sites, or from the intrinsically more resistant carbon support of Fe/N/C. Iron is known to catalyze graphitization at high temperature.
During high-potential cycling, the Fe/N/C-catalyst may have degraded through either (i) oxidation of the iron cations coordinated in the FeNxCy active sites, leading to demetallation or (ii) destruction of the FeNxCy active sites resulting from the corrosion of the surrounding carbon support. Mechanism (i) is apparented to the deactivation of unpyrolyzed FeII-phthalocyanine (FeIIPc) in acid medium. Oxidation of FeIIPc first leads to a coordinated FeIII cation, whose smaller ionic radius favours the demetallated H2-Pc form.36 This hypothetical deactivation mechanism for Fe/N/C would not require degradation of the carbon support itself. In contrast, if mechanism (ii) is operational, the ORR activity decay should be positively correlated with the carbon corrosion rate. The latter was measured at various temperatures for the Fe/N/C catalyst. Fig. 2A shows its cyclic voltammograms measured up to 1.4 V vs. RHE in the N2-saturated electrolyte, while Fig. 2B shows the corresponding Tafel plots. The assignment of the current in Fig. 2A to carbon corrosion is supported by the near superimposition of the curves for the Fe/N/C and N/C samples at 80 °C (not shown). An increase in the carbon-corrosion rate with temperature is observed and the Arrhenius law was verified, yielding an activation energy of 38 kJ mol−1. The faster rate of carbon corrosion at high temperature explains the increased ORR activity decay during cycling at high temperature (Fig. 1). Hence, mechanism (ii) seems to be the main degradation route for the Fe/N/C-catalyst during high potential cycling.
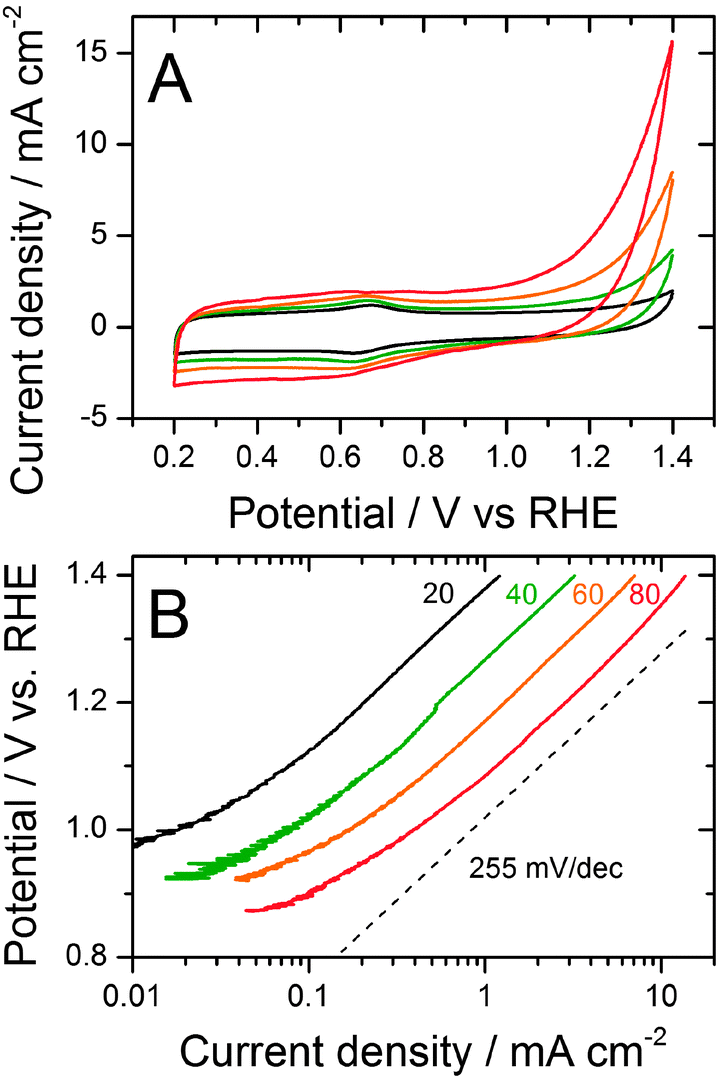 | ||
| Fig. 2 Carbon corrosion rates of the Fe/N/C catalyst as a function of temperature. (A) Cyclic voltammograms measured at 10 mV s−1 in a N2-saturated pH 1 electrolyte at 20, 40, 60, and 80 °C. (B) Tafel plots for the Faradaic currents of carbon corrosion, as measured from the positive-going scans shown in (A) and corrected for the capacitive current. | ||
Surface changes following high-potential excursion were also investigated with cyclic voltammetry (Fig. S1, ESI†). The increased capacitive current after cycling is due to new oxygen groups,24 and so is the redox peak observed at 620/660 mV vs. RHE.37,38
Fig. 3 reports the progressive decay of the ORR activity measured in RDE at 80 °C as a function of cycle number. In the Tafel plot (Fig. 3B), an increase in the apparent Tafel slope below 0.7 V vs. RHE takes place. This increase might come from a change in the ORR mechanism in this potential region or might be the outcome of the decreased density of active sites on the catalyst surface. The decrease in mass activity at 0.8 V vs. RHE as a function of cycle number is shown in Fig. 3C for the RDE measurements performed at 20 and 80 °C (filled circles and squares, respectively) and also for the PEMFC measurements at 80 °C (open squares). Fig. 3C demonstrates the excellent agreement between RDE and PEMFC regarding the ORR activity decay resulting from the same high-voltage cycling protocol if, and only if, they are compared at the same temperature of 80 °C. This is an important conclusion for the application of the RDE technique.
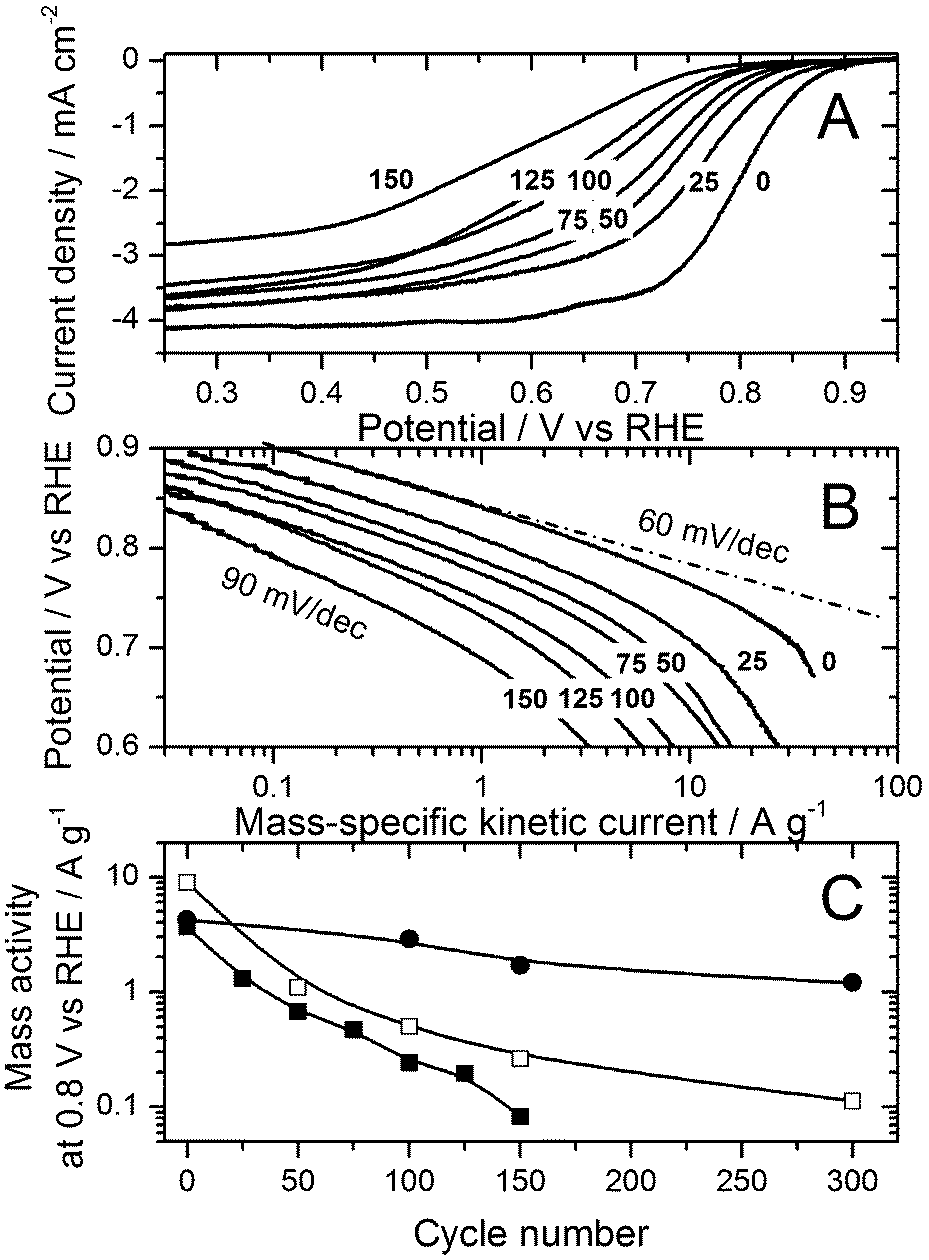 | ||
| Fig. 3 Activity decay of the Fe/N/C catalyst as a function of cycle number. (A) RDE polarisation curves measured at 80 °C from 0 to 150 cycles, by steps of 25 cycles. (B) Tafel plots corresponding to panel A. (C) ORR mass activity at 0.8 V vs. RHE as a function of cycle number as measured in RDE at 20 °C (filled circles) and 80 °C (filled squares) and in PEMFC at 80 °C (open squares). | ||
After 150 cycles, the ORR mass activity measured by the RDE technique was reduced by factors of 2.5 and 44 at 20 °C and 80 °C, respectively (Fig. 3C). The activity decay is therefore 18 times larger at 80 °C than at 20 °C, which correlates well with the corresponding 14-fold increase in the carbon corrosion rate calculated from its activation energy of 38 kJ mol−1. This positive correlation further points to carbon corrosion as the main degradation mechanism for the FeNxCy active sites.
Carbon corrosion effects on mass and charge transport
While ORR kinetics is an essential aspect, electrodes operating at high current density in PEMFC also necessitate the study of other characteristics, which however cannot be directly assessed with the RDE technique. In addition to the obvious ORR activity decay with cycling (Fig. 3C and 4B), high-voltage transients resulted in additional negative changes in the cathode layer, leading to a far more pronounced decay of the fuel cell performance than could be expected solely on the basis of the decrease of the cathode ORR kinetics. This is evidenced by comparing the PEMFC polarisation curve after 50 and 300 cycles (Fig. 4B). The corresponding differential decrease in cell voltage at 1 mA cm−2 is only 80 mV while it is 340 mV at 100 mA cm−2. As previously mentioned, decreased density of active sites upon high-voltage cycling may have resulted in increased mass-transport resistance locally around the remaining active sites. In addition, the high-voltage cycling and concomitant carbon corrosion may have led to (i) the collapse of the electrode structure leading to a reduced electrode porosity,13,39 and (ii) reduced electron conductivity throughout the cathode active layer. | ||
| Fig. 4 Degradation in PEMFC of the Fe/N/C-based cathode as a function of cycle number. (A) Polarisation curves as a function of cycle number indicated below each curve, (B) corresponding Tafel plots, (C) impedance spectroscopy measured at 100 mA cm−2, (D) high-frequency resistance. | ||
The latter is one possible explanation for the increase in high-frequency resistance (HFR) with increasing cycle number (Fig. 4C and D), the second explanation being increased membrane resistance. Indeed, the membrane may have been polluted with iron cations leached out during degradation. However, this second hypothesis did not withstand the following test experiment: we cycled an MEA for which only the anode had been hot-pressed onto the membrane, while the cathode was simply positioned against the membrane during cell assembly.
The initial polarisation curve of this MEA was similar to the one shown in Fig. 4A. After an extensive degradation and measurement of the final polarisation curve, the cell was opened and the cycled anode-membrane part was replaced by a new one, while the cycled cathode was re-utilized. As can be seen in Fig. S2A (ESI†), replacement of the anode-membrane part did not lead to any improvement, implying that only the cathode layer was damaged during cycling. While some iron was detected in the membrane (Fig. S2B and C, ESI†), its amount was too small (8 μg Fe cm−2) to significantly impact the proton conductivity of the membrane. It can thus be concluded that the increase in the HFR of the MEA during cycling (Fig. 4D) comes from the cathode layer. From an analytical expression for the HFR of the entire MEA and including a component arising from the thick cathode layer, effective cathode electronic conductivities of 100 and 5 S m−1 were estimated initially and after 300 cycles, respectively (see ESI†). This huge decrease of the electronic conductivity is a consequence of carbon corrosion during cycling.
An important question is whether the reduced electronic conductivity in the cathode fully accounts for the much higher PEMFC voltage drop observed than expected from a decrease in the ORR kinetics alone(Fig. 4B). This question was investigated with a mathematical model for porous electrodes, involving Tafel kinetics, electron and proton conduction within the active layer, and local O2 diffusion within catalytic agglomerates. The model is similar to that described in ref. 40 but additionally takes into account limitation by electron conduction.41
The model predicted a current density of ca. 600 mA cm−2 at 0.3 V after 300 cycles (assuming a final electronic conductivity of 5 S m−1 and a drop in ORR activity by a factor of 40, Fig. S3, ESI†), much higher than the experimental current density of ca. 100 mA cm−2 at 0.3 V after 300 cycles (Fig. 4A). Thus, while reduced electronic conductivity slightly increases the charge-transport resistance of the cycled cathode, at least another factor plays a key role in its huge mass-transport resistance. The calculation also predicted that the performance should not be much improved if the electronic conductivity could be restored. To test this, we opened a cell that had undergone 300 cycles of degradation, and re-hot-pressed the cycled MEA. The hot-pressing step restored to some extent the electric connection between the catalyst particles. However, the ensuing improvement of the polarisation curve was minor (Fig. S2D, ESI†).
In summary, carbon corrosion leads to deep morphological changes witnessed by the increase in the HFR value of the cathode. Decreased electron conductivity and ORR activity of the cathode cannot fully explain the experimental data. Other factors seem to play a key role. The decreased density of active sites after cycling may have led to additional mass-transport resistances.
In order to estimate the weight loss of carbon during high-voltage cycling in PEMFC, we switched to a potentiostatic degradation mode applying 1.3 V vs. RHE for a longer time in order to integrate the current vs. time curves without interference of the capacitive current that is predominant in short cycles. From these curves, we estimated the weight loss of carbon on the assumptions that (i) the oxidation current related to O2 evolution reaction is negligible and (ii) carbon corrosion proceeded through the 4e mechanism to produce CO2. The first hypothesis is valid for Fe/N/C catalysts that do not catalyze the O2 evolution. The second hypothesis introduces only a minor error since the ratio CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 from carbon corrosion is usually 10%.18 Based on these assumptions, the relative losses in the current density at 0.7 and 0.5 V could be plotted on the polarisation curve against the relative weight loss of carbon that occurred during the potential holds (Fig. 5). After only 15 wt% carbon loss, the ORR activity (current at 0.7 V) dropped by 90%, indicating that most of the FeNxCy active sites are surface-located.
:CO2 from carbon corrosion is usually 10%.18 Based on these assumptions, the relative losses in the current density at 0.7 and 0.5 V could be plotted on the polarisation curve against the relative weight loss of carbon that occurred during the potential holds (Fig. 5). After only 15 wt% carbon loss, the ORR activity (current at 0.7 V) dropped by 90%, indicating that most of the FeNxCy active sites are surface-located.
 | ||
| Fig. 5 Relative decrease of the current density for the Fe/N/C cathode in PEMFC against carbon weight loss. Potential holds were performed at 1.3 V vs. RHE and cumulative durations at 1.3 V are indicated as labels. | ||
The morphology of the Fe/N/C-catalyst may oversimplistically be represented as a 3 nm-thick carbon slab, resulting in a theoretical specific surface area of 370 m2 g−1, in line with its experimental value of 370–420 m2 g−1. A 15% volume decrease of such a carbon slab corresponds to the removal of 2.25 Å on each side. This corresponds to the removal of either (i) less than one monolayer of graphene (the graphene interlayer distance in graphite is 3.35 Å) or (ii) almost one full row of carbon atoms situated at the zigzag or armchair edges of a graphene sheet (the width of a C6 ring in graphite is 2.46 Å). Carbon atoms situated on the edges are more reactive than those found in the basal plane,42 making the latter analogy more meaningful. The fastest decrease in ORR activity occurs at ca. 3 wt% loss (upper curve in Fig. 5), corresponding to the removal of ca. 1/5th of the C or N atoms found at the edges, and to a 30% decrease in ORR activity. The remaining low ORR activity measured beyond 15 wt% loss of carbon comes either from N-groups on the carbon support or from the few FeNxCy sites initially situated within a graphene layer. While a strong performance decrease was also observed with Pt/C, the Fe/N/C catalyst degraded faster. For example, in order to reach a 60% relative decay of the current density at 0.7 V, a 1.4 V potential hold of only 85 s was necessary for Fe/N/C, but was 800 s for Pt/C (Fig. S4, ESI†).
In order to visualize the effect of high-voltage cycling on the morphology of the Fe/N/C catalyst, we employed the so-called identical-location electron-microscopy approach (IL-STEM and IL-EDS, see ESI†).43 This technique allowed us to follow the morphological evolution of specific areas of the catalyst throughout the degradation. The experiment involved the transmission electron microscopy (TEM) characterisation of several isolated agglomerates on a TEM-grid in their initial state as well as after 300 and 900 cycles at 60 °C. Fig. 6 provides a selection of STEM and EDS spectrum images of selected locations representative of the average degradation of the catalyst. The corresponding electrochemical results are shown in Fig. 1C. As shown in Fig. 6 (upper row), the catalyst agglomerate appears to rapidly decrease in size, thickness and density with the number of cycles. Analysis of the images reveals that the cross-section area of the agglomerate decreased by 17 and 40% after 300 and 900 cycles, respectively. This visual size reduction due to carbon corrosion results from both a direct loss by oxidation and CO2 evolution and physical disconnection of agglomerate parts having initially narrow connections to the remainder of the agglomerate. While the IL-STEM experiments were performed directly on a TEM finder grid with low catalyst loading, the observed shrinkage of the agglomerates due to the imposed degradation protocol also occurs in PEMFCs. This inevitably leads to the collapse of the electrode structure, which plays a key role in the increased charge- and mass transfer resistances, and thus in the performance decrease.
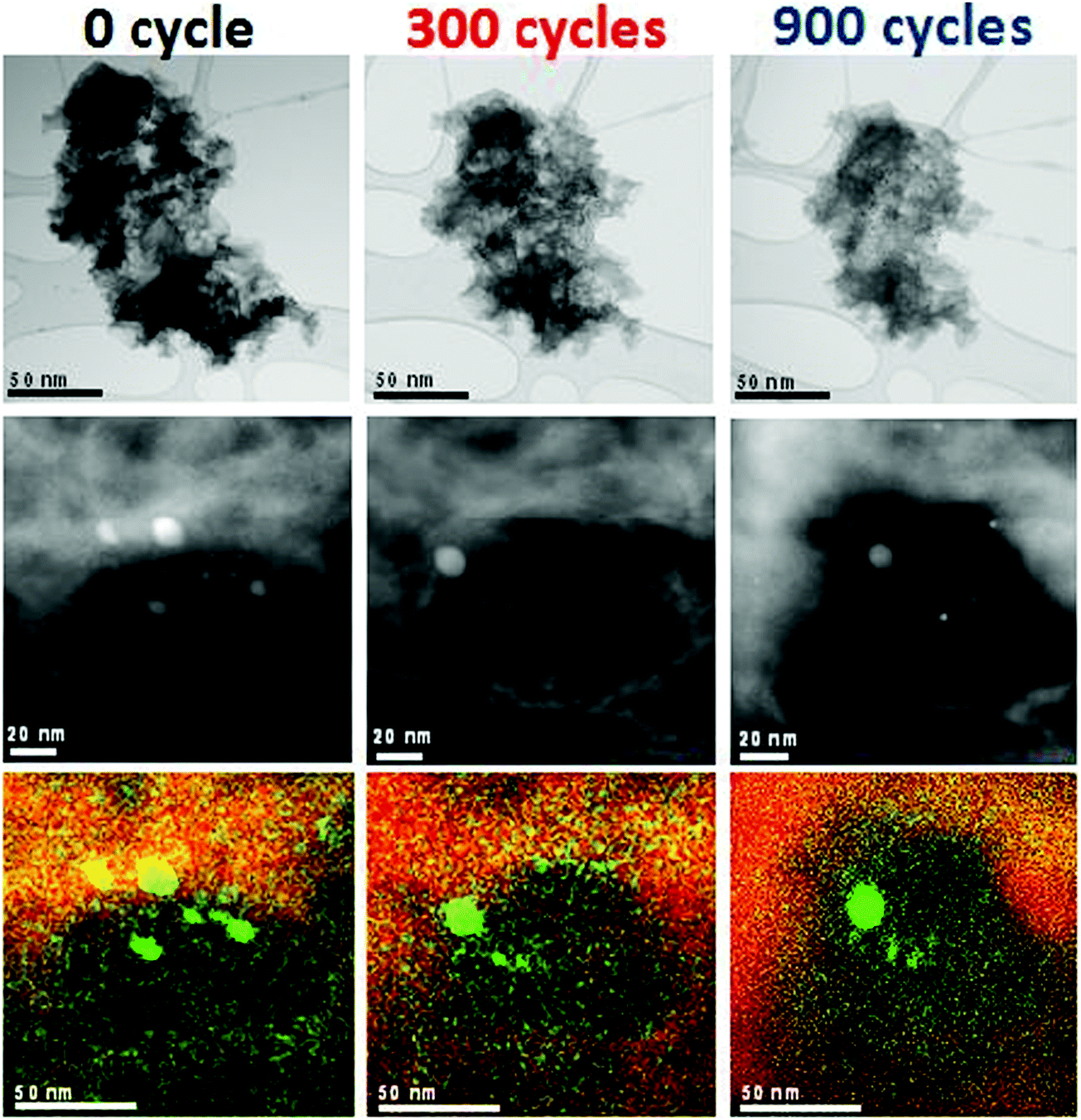 | ||
| Fig. 6 Identical-location STEM and EDS of two agglomerates of the Fe/N/C catalyst after 0, 300 and 900 electrochemical cycles performed at 60 °C. First row: bright-field STEM images of a single agglomerate; 2nd and 3rd rows: dark-field STEM images and the corresponding EDS spectrum images of a single agglomerate region (iron atoms are green and carbon is red). The high voltage cycling was performed in 0.1 M HClO4. | ||
Carbon corrosion effects on Fe distribution and speciation
The fate of iron during cycling was then investigated in detail, which is important to better understand changes in the ORR activity. At the magnification used for Fig. 6 (upper row), the density of iron nanoparticles remained unchanged after 900 cycles at 60 °C. This correlates with the Mössbauer intensities of the signals assigned to Fe0 (Fig. 7, yellow and brown sextets and orange singlet) which only slightly decreased upon cycling. Higher magnification images of the upper row of Fig. 6 however show better defined Fe nanoparticles after cycling, probably as an optical consequence of the thinning of the carbon matrix. The second row in Fig. 6 focuses on another agglomerate initially showing iron nanoparticles. While some Fe particles were no longer visible after only 300 cycles at 60 °C, others and including small ones were still present after the extensive 900-cycle degradation. The elemental mapping (3rd row in Fig. 6) shows a localized Fe-signal assigned to particles and a delocalized signal assigned to either 1–2 nm Fe nanoparticles or FeNxCy active sites. These two types of signals are also identified by Mössbauer spectroscopy (Fig. 7, upper row, filled and unfilled peaks for Fe0 and FeNxCy sites, respectively). Fig. 6 shows no obvious increase nor decrease of the Fe-signal relative to the carbon signal, suggesting that the high-voltage cycling did not preferentially attack the Fe particles or the Fe-based active sites. | ||
| Fig. 7 Mössbauer transmission spectra of PEMFC cathodes vs. number of cycles (0.9/1.4 V–3 s/3 s, 80 °C). Left: Fe/N/C, right: Fe/N/C-dry. | ||
We then investigated the Fe-speciation in pristine and cycled cathodes using Mössbauer spectroscopy. For Fe/N/C, the Mössbauer spectra of the pristine cathode and of the cathode after 150 and 600 cycles at 1.4 V are shown on the left-hand side of Fig. 7. The corresponding polarisation curves were very similar to those seen in Fig. 4A and B after 0, 150 and 300 cycles. The Mössbauer spectrum of the pristine Fe/N/C cathode shows two sextets assigned to α-Fe and iron carbide, and one singlet assigned to superparamagnetic-Fe or γ-Fe. Doublets D1 and D2 are assigned to FeIIN4/C in low and medium spin, respectively.28,30,35,44 Doublet D1 has Mössbauer parameters similar to those of FeII-phthalocyanine (FeII-Pc) adsorbed on carbon.45 D2 has parameters similar to those of crystalline FeIIPc in which stacked FeIIPcs interact with each other. In pyrolyzed Fe/N/C-catalysts, the interactions leading to D2 likely arise from a 2nd graphene plane parallel to the FeN4 site.
This is supported by the similar plane-to-plane distances in crystalline FeIIPc and graphite (3.35–3.40 Å). Among Fe-species present in the pristine Fe/N/C-cathode, only D1 or D2 may account for the ORR activity.
Major changes are observed in the spectrum of the Fe/N/C-cathode after 150 cycles at 80 °C (Fig. 7, left-hand side). The amplitudes of D1 and D2 were divided by 6.6 and 1.6, respectively. In contrast, the amplitudes related to α-Fe and Fe-carbide decreased by only 14–22%. These phases are usually surrounded by a graphitic shell, explaining their better resistance to corrosion. Last, a new doublet labelled D* appeared (Fig. 7, red doublet), with a quadrupole splitting significantly different from that of D1 and D2 (Table 1). After 600 cycles of degradation, the amplitude of D* increased much while the D1 and D2 doublets nearly disappeared. Last, the signal of α-Fe significantly decreased by 35% relative to the initial spectrum. The simultaneous large increase in the amplitude of D* is understandable if it is a degradation product of α-Fe. The Mössbauer parameters of D* match those of amorphous FeIII hydroxide particles of 2–3 nm size.46 The latter may have precipitated from FeIII cations produced when initially carbon-embedded α-Fe particles became unprotected, due to carbon corrosion. The Pourbaix diagram of Fe shows that FeIII ions may precipitate as Fe(OH)3 at high electric potential, even at low pH.
| Component | IS (mm s−1) | QS (mm s−1) | LW (mm s−1) | HF (Tesla) | Assignment |
|---|---|---|---|---|---|
| D1 | 0.4 | 1.1 | 0.8 | — | FeIIN4/C, LS |
| D2 | 0.4–0.5 | 2.1–2.8 | 1.3–1.9 | — | FeIIN4/C, MS |
| D* | 0.4 | 0.7 | 0.5 | — | FeIII oxide, HS |
| Sextet 1 | 0.0 | — | 0.3–0.4 | 33–34 | α-Fe |
| Sextet 2 | 0.2 | — | 0.4 | 20 | FeXC |
| Singlet | −0.1 | — | 0.4 | — | Param. Fe or γ-Fe |
Quantification may be derived from Mössbauer spectroscopy if the Lamb–Mössbauer factors for each Fe-species are known. Such factors have previously been assumed to be identical for all Fe-species.28,29,35 However, from the present data, it is demonstrated that this assumption was not valid. Since the pristine Fe/N/C catalyst contains a significant amount of Fe0, we modified the synthesis procedure to minimize this. A second catalyst, labelled Fe/N/C-dry, was synthesized (experimental). Its electrochemical behaviour is similar to that of Fe/N/C. However, its Mössbauer spectrum shows a tiny signal for α-Fe (Fig. 7, upper row, right). Moreover, while the Fe/N/C-dry and Fe/N/C catalysts have similar Fe contents (3 wt%), the integral below the spectrum of the latter is twice larger than that of the former. This is possible only if the Lamb–Mössbauer factors of the Fe0-species are 5–10 times larger than those for D1 and D2. With this assumption, the 60% relative area assigned to Fe0-species in the spectrum of the Fe/N/C catalyst translates into a relative Fe0 content of 13–23% (i.e. 77–87% for D1 + D2). In the Fe/N/C-dry catalyst, the Fe atoms mostly formed the D1 and D2 species (92% for D1 + D2). This explains why the Fe/N/C-dry catalyst shows only a moderate 10% increase in the absolute signals of D1 and D2 (i.e. an increase from 77–87% to 92% relative to the total Fe content of 3 wt% in both catalysts). Determination of the Lamb–Mössbauer factors will be necessary for accurate quantification.
The Fe/N/C-dry catalyst allows us to better identify changes in the active sites with cycling. Decreased absolute amplitude of D1 and D2 after 150 and 600 cycles is obvious (right-hand side of Fig. 7). The amplitude of D1 had decreased by a factor of 5.2 after 150 cycles. This correlates well with the decrease in ORR activity at 0.8 V (6.7 and 1.1 A g−1 after 150 cycles, factor 6). After 600 cycles, the ORR activity is so low that the contribution from the NCz active sites is non-negligible, which prevents a direct correlation between D1 and the ORR activity. Next, the smaller decrease of the D2 signal from 0 to 150 cycles (divided by 2.8) and its unmodified amplitude between 150 and 600 cycles suggest that the site structure related to D1 is more active than that of D2. This conclusion is supported by the trends in the amplitudes of D1 and D2 in the Fe/N/C-catalyst (left-hand side of Fig. 7) and in line with conclusions drawn from Mössbauer spectra of various pristine catalysts.28,29,35 In addition, the degradation of D1 and D2 mostly results in solvated FeII or FeIII being removed from the cathode with water, since the combined decreased amplitude of D1 + D2 with cycling is not compensated by the new doublet D*. The higher solubility of FeII than FeIII points to the former as the Fe species existing in the pristine FeNxCy sites. Last, the much smaller amplitude for D* after 600 cycles in Fe/N/C-dry than in Fe/N/C supports the hypothesis that D*, assigned to FeIII hydroxides, arises from the Fe0 species, in particular from α-Fe.
From these observations, a scheme of catalyst degradation at the atomic scale can be proposed (Fig. 8). The Fe/N/C-catalyst free of crystalline Fe species initially contains two types of FeNx sites assigned to micropore-hosted and graphene-defect sites (D1–D2 doublets). Since the carbon oxidation rate is faster on zigzag and armchair edges than on the basal plane,47 micropore-hosted FeNxCy sites are eliminated faster than graphene-defect FeNxCy sites (Fig. 7, right-hand side). Loss of the ligand environment of the Fe ions leads to their solvation, most likely as FeII (Fig. 8), and removal from the cathode with PEMFC exhaust water.
 | ||
| Fig. 8 Scheme depicting carbon corrosion and the ensuing loss of micropore-hosted and graphene-defect FeNxCy sites. N: blue, Fe: red. Oxygen atoms are omitted for clarity. | ||
Conclusions
A comprehensive study of the degradation of a particular, highly active Fe/N/C catalyst in acidic medium was performed by applying potential cycles beyond the reversible ORR potential. These accelerated-aging tests simulating start–stop conditions in fuel cells have shown that RDE and PEMFC investigations do correlate well when performed at the same elevated temperature of 80 °C. By combining both approaches and complementary structural and chemical analysis of the catalyst at various stages throughout the degradation, the following insights were gained:• Degradation of the FeNx-based active sites and of the supporting N-doped carbon occurs in parallel at high potential
• No preferential destruction of the FeN4 active sites occurs
• The degradation is strongly enhanced at high temperature, which is correlated with an increased carbon-corrosion rate
• Destruction of the FeNx-based active sites mostly leads to the removal of Fe cations by water, most probably as FeII.
Degradation of the catalyst at high temperature and potential leads to decayed ORR kinetics due to the loss of active Fe sites. Moreover, huge charge- and mass-transport resistances are also induced by the collapse of the electrode structure. Future research efforts being directed towards understanding the degradation at the atomic level and stabilization of the carbon matrix in non-precious metal catalysts are required.
Acknowledgements
We acknowledge funding from ANR, contract 2011 CHEX 004 01.Notes and references
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864 CrossRef CAS.
- M. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- T. E. Wood, Z. Tan, A. K. Schmoekel, D. O'Neill and R. Atanasoski, J. Power Sources, 2008, 178, 510 CrossRef CAS PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef PubMed.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- J. Y. Cheon, T. Kim, Y. Choi, H. Y. Jeong, M. G. Kim, Y. J. Sa, J. Kim, Z. Lee, T.-H. Yang, K. Kwon, O. Terasaki, G.-G. Park, R. R. Adzic and S. H. Joo, Sci. Rep., 2013, 3, 2715 Search PubMed.
- S.-T. Chang, C.-H. Wang, H.-Y. Du, H.-C. Hsu, C.-M. Kang, C.-C. Chen, C.-S. Wu, S.-C. Yen, W.-F. Huang, L.-C. Chen, M.-C. Lin and K.-H. Chen, Energy Environ. Sci., 2012, 5, 5305 CAS.
- D. Zhao, J.-L. Shui, L. R. Grabstanowicz, C. Chen, S. M. Commet, T. Xu, J. Lu and D.-J. Liu, Adv. Mater., 2014, 26, 1093 CrossRef CAS PubMed.
- Z. Siroma, N. Fujiwara, T. Ioroi, S.-I. Yamazaki, H. Senoh, K. Yasuda and K. Tanimoto, J. Power Sources, 2007, 172, 155 CrossRef CAS PubMed.
- J. P. Meyers and R. M. Darling, J. Electrochem. Soc., 2006, 153, A1432–A1442 CrossRef CAS PubMed.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273 CrossRef CAS PubMed.
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306 CrossRef CAS PubMed.
- Y. Yua, H. Li, H. Wang, X.-Z. Yuan, G. Wang and M. Pan, J. Power Sources, 2012, 205, 10 CrossRef PubMed.
- A. Oyarce, E. Zakrisson, M. Ivity, C. Lagergren, A. Baumann Ofstad, A. Bodén and G. Lindbergh, J. Power Sources, 2014, 254, 232 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, J. C. Meier, S. J. Ashton, G. K. H. Wiberg, F. Krausc, M. Hanzlik and M. Arenz, Electrochem. Commun., 2008, 10, 1144 CrossRef CAS PubMed.
- N. Linse, L. Gubler, G. G. Scherer and A. Wokaun, Electrochim. Acta, 2011, 56, 7541 CrossRef CAS PubMed.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, J. Power Sources, 2008, 176, 444 CrossRef CAS PubMed.
- L. M. Roen, C. H. Paik and T. D. Jarvi, Electrochem. Solid-State Lett., 2004, 7, A19 CrossRef CAS PubMed.
- K. H. Kangasniemi, D. A. Condit and T. D. Jarvi, J. Electrochem. Soc., 2004, 151, E125 CrossRef CAS PubMed.
- N. Larouche, R. Chenitz, M. Lefèvre, E. Proietti and J. P. Dodelet, Electrochim. Acta, 2014, 115, 170 CrossRef CAS PubMed.
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J. P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-wurmbach, P. Bertrand, T. M. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087 CAS.
- M. Ferrandon, X. Wang, J. Kropf, D. Myers, G. Wu, C. M. Johnston and P. Zelenay, Electrochim. Acta, 2013, 110, 282 CrossRef CAS PubMed.
- T. Han, N. Dale, K. Adjemian, V. Nallathambi and S. C. Barton, ECS Trans., 2011, 41, 2289 CAS.
- G. Liu, X. Li and B. N. Popov, ECS Trans., 2009, 25, 1251 CAS.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Appl. Catal., B, 2009, 93, 156 CrossRef CAS PubMed.
- S. Mukerjee, Development of Novel Non Pt Group Metal Electrocatalysts for Proton Exchange Membrane Fuel Cell Applications, 2012. http://www.hydrogen.energy.gov/pdfs/review12/fc086_mukerjee_2012_o.pdf.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J. P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673 RSC.
- U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2013, 136, 978 CrossRef PubMed.
- U. I. Kramm, I. abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69 CrossRef CAS PubMed.
- M. Ferrandon, J. A. Kropf, D. J. Myers, K. Artyushkova, U. I. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001 CAS.
- G. Wu, K. Artyushkova, M. Ferrandon, J. Kropf, D. Myers and P. Zelenay, ECS Trans., 2009, 25, 1299 CAS.
- J. M. Ziegelbauer, T. S. Olson, S. Pylypenko, F. Alamgir, C. Jaye, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2008, 112, 8839 CAS.
- M. Bron, J. Radnik, M. Fieber-Erdmann, P. Bogdanoff and S. Fiechter, J. Electroanal. Chem., 2002, 535, 113 CrossRef CAS.
- A. Morozan, M. T. Sougrati, V. Goellner, D. Jones, L. Stievano and F. Jaouen, Electrochim. Acta, 2014, 119, 192 CrossRef CAS PubMed.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J. M. Léger, J. Electroanal. Chem., 2005, 577, 223 CrossRef CAS PubMed.
- K. Kinoshita, Carbon, electrochemical and physicochemical properties, John Wiley & Sons, 1988 Search PubMed.
- J. Maruyama and I. Abe, Electrochim. Acta, 2001, 46, 3381 CrossRef CAS.
- Z. Y. Liu, B. K. Brady, R. N. Carter, B. Litteer, M. Budinski, J. K. Hyun and D. A. Muller, J. Electrochem. Soc., 2008, 155, B979 CrossRef CAS PubMed.
- F. Jaouen, G. Sundholm and G. Lindbergh, J. Electrochem. Soc., 2002, 149, A437 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 CAS.
- L. R. Radovic, J. Am. Chem. Soc., 2009, 131, 17166 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, S. J. Ashton, J. C. Meier, G. K. H. Wiberg, M. Hanzlik and M. Arrenz, J. Power Sources, 2008, 185, 734 CrossRef CAS PubMed.
- U. I. Koslowski, I. Abs-wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356 CAS.
- C. A. Melendres, J. Phys. Chem., 1980, 84, 1936 CrossRef CAS.
- L. Machala, R. Zboril and A. Gedanken, J. Phys. Chem. B, 2007, 111, 4003 CrossRef CAS PubMed.
- M. Matsumoto, T. Manako and H. Imai, J. Electrochem. Soc., 2009, 156, B1208 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, test experiments on membrane replacement after cycling, model-predicted polarisation curves, effect of high potential cycling on Pt/C. See DOI: 10.1039/c4cp02882a |
| This journal is © the Owner Societies 2014 |