
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electrochemical in situ study of freezing and thawing of ionic liquids in carbon nanopores†
Daniel
Weingarth
a,
Robert
Drumm
a,
Annette
Foelske-Schmitz
bc,
Rüdiger
Kötz
b and
Volker
Presser
*ad
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@inm-gmbh.de
bPaul Scherrer Institute, 5232 Villigen PSI, Switzerland
cVienna University of Technology, 1060 Vienna, Austria
dSaarland University, Campus D2 2, 66123 Saarbrücken, Germany
First published on 26th August 2014
Abstract
Room temperature ionic liquids (RTILs) are an emerging class of electrolytes enabling high cell voltages and, in return, high energy density of advanced supercapacitors. Yet, the low temperature behavior, including freezing and thawing, is little understood when ions are confined in the narrow space of nanopores. This study shows that RTILs may show a tremendously different thermal behavior when comparing bulk with nanoconfined properties as a result of the increased surface energy of carbon pore walls. In particular, a continuous increase in viscosity is accompanied by slowed-down charge–discharge kinetics as seen with in situ electrochemical characterization. Freezing reversibly collapses the energy storage ability and thawing fully restores the initial energy density of the material. For the first time, a different thermal behavior in positively and negatively polarized electrodes is demonstrated. This leads to different freezing and melting points in the two electrodes. Compared to bulk, RTILs in the confinement of electrically charged nanopores show a high affinity for supercooling; that is, the electrode may freeze during heating.
Introduction
Room-temperature ionic liquids (RTILs)1 are an emerging class of advanced electrolytes for electrochemical energy storage systems, such as lithium ion batteries2,3 and carbon supercapacitors.4,5 The unique feature of supercapacitors is that electrical charge is stored at an electrified fluid/solid interface via the formation of an electrical double-layer as a result of ion electrosorption in porous carbon electrodes.6,7 This feature makes this technology excel in cycling stability, longevity, and power handling compared to battery technologies.8 RTILs are unique solvent-free electrolytes enabling high operating voltages (at or above 3.5 V) and superior safety because of the noninflammable and non-volatile character with an excellent high temperature stability compared to organic electrolytes. While the electrochemical behavior of RTIL supercapacitors is fairly well studied at room temperature or above,9 only limited investigations have been carried out at low temperatures let alone in nanoconfinement.10,11 As a result, the thermophysical behavior of RTILs in carbon nanopores and the in situ electrochemical performance remain highly speculative. In our study, we apply differential scanning calorimetry (DSC) and electrochemical measurements at low temperatures to study the freezing and thawing of RTILs (with less than 10 ppm H2O) in the confinement of carbon nanopores. Step by step, we are going to establish that the thermal properties of RTILs may be affected by nanoconfinement but are vastly different from carbon electrodes before and after charging and that the electrochemical performance can be fully restored even after freezing.It is well-known from the literature that fluids, including ionic liquids, exhibit different freezing and melting points when confined in nanoporous materials (e.g., silica).12–18 This different behavior is commonly attributed to the additional interaction with the pore walls along the large contact (surface) area in porous media. The presence of an electrical charge at the fluid/solid interface can also have an effect on the thermal behavior.19 Yet, for RTIL supercapacitors, we have to consider that not only nanoconfinement, but also local changes in the anion/cation ratio have to be considered.20 This is related to the ejection of co-ions (ions with the same charge as the electrode) and the insertion of counter-ions (ions with opposite charge) from carbon nanopores during the charging of a supercapacitor electrode. While there are some thermal data for neat ionic liquids available, the effect of nanoconfinement on the thermophysical properties of RTILs remains poorly investigated.21,22 From the limited literature, it seems that a drastic difference for RTILs is expected to emerge once the confined geometry is smaller than circa 10 ion radii (i.e., below 10 nm).23 In the work of Kanakubo et al.24 the smallest pores of silica (7.5 nm) yielded the lowest melting point for 1-butyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide [BMIM][TFSI] (Δ = −30 °C) which is in line with the Gibb–Thomson effect.15 In contrast, Chen et al.25 reported a severely increased melting point (Δ = +194 °C) of 1-butyl-3-methyl-imidazolium hexafluorophosphate [BMIM][PF6] inside multi-wall carbon nanotubes (5–10 nm diameter) which possibly indicates the importance of surface charge and the intricacies of RTIL phase transitions as shown by Jeon et al.26 From an application point of view, it is essential to understand if, once frozen, an RTIL supercapacitor can regain its original electrochemical performance. In addition, it is essential to understand how anion/cation separation during charging affects the thermal behavior and may lead to different freezing points at the anode and the cathode. To address these questions, we carried out experiments using PTFE-bound electrodes with nanoporous carbon black (BP2000, Cabot Corporation; see ESI†) with a large specific area of 1374 m2 g−1 as a model material representative for many activated carbons. This material was chosen since the small particle size (<50 nm) and the additional presence of mesopores are highly suitable factors to severely mitigate diffusional limitations within a pore network similar to many activated carbons.27
Experimental description
DSC thermograms were recorded using a DSC 1 system from Mettler Toledo ANALYTICAL in combination with the STAR software using standard aluminum crucibles (40 µL). The scan rate for all thermograms was 10 °C min−1.The main purpose of the DSC measurements in this work is to show qualitative differences in the freezing and melting behavior of bulk RTIL and IL confined in a porous medium. The DSC thermograms are normalized by the mass of the RTIL. The DSC pans were filled in an Ar filled glove box with O2 and H2O below 1 ppm.
Polytetrafluoroethylene (PTFE) bound carbon electrodes were prepared by mixing the carbon powder (BP2000, Cabot Corp.) with ethanol forming a slurry which was sonicated for 15 minutes. The PTFE (60 wt% in H2O, Sigma-Aldrich) was added under constant stirring until a final amount of 10 wt% PTFE with respect to the carbon mass was reached. The slurry was stirred and heated until a dough-like mass was achieved. This mass was rolled into self-standing electrode sheets with 200 µm thickness using a hot rolling machine (MTI Corp.). Afterwards, the electrodes were dried for at least 24 h at 120 °C and 20 mbar.
The electrochemical measurements were carried out in custom-built electrochemical cells. Two electrodes (diameter 12 mm) were punched out from the electrode sheets and contacted using two spring loaded Ti pistons. The electrodes were separated using a cellulose based separator (TF 4425, Nippon Corp.) and carbon coated aluminum foil (type Zflo 2653, Exopack technologies) was used for better contact. Activated carbon (AC, YP50, Kuraray Chemical) was used as a reference electrode28 in order to monitor the potential of the positive and the negative electrode separately in the symmetric cell arrangement.
The temperature dependent cyclic voltammetry measurements were carried out in an environmental simulation chamber (Binder MK53). The temperature of the cell was not additionally monitored. The final cell temperature was determined from the display of the oven. Cyclic voltammetry was carried out using a VMP3 potentiostat (Bio-Logic Instruments).
For the DSC investigation of the charged electrodes a symmetric cell containing a reference electrode was used. The cell was charged up to a cell voltage of 2.5 V (see electrode potentials in ESI†) and afterwards carefully disassembled inside the glove box. Finally, a piece of each electrode (positive and negative) was taken, put into a DSC pan, and DSC measurements were conducted.
The ionic liquids 1-ethyl-3-methyl imidazolium tetrafluoroborate [EMIM][BF4], 1-butyl-3-methyl imidazolium tetrafluoroborate [BMIM][BF4], and N-methyl-N-propyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide [PYR13][TFSI] were purchased from Iolitec and dried at 100 °C and a vacuum better than 10−3 mbar for at least 6 h, leading to a water content of less than 10 ppm as measured by Karl Fisher titration.
Results and discussion
First, we investigated the influence of nanoconfinement on the thermophysical properties in an uncharged state. We see that the thermal behavior is strongly dependent on the RTIL ions and their interaction with each other and the carbon pore walls. For example, bulk N-methyl-N-propylpyrrolidinium bis(trifluoromethylsulfonyl) imide [PYR13][TFSI] shows in Fig. 1A a melting point depression of only Δ 1 °C from +13 °C to +12 °C compared to the bulk liquid. Yet the non-equilibrium nucleation process of freezing is drastically affected and shifted to +2 °C from originally −24 °C. This increase is related to the strong interactions with pore walls at the large liquid–solid interface in the presence of surface defects of partially graphitic carbon. This finding aligns well with the enhanced ion mobility and ability to rearrange in nanoconfined RTILs compared to the bulk.29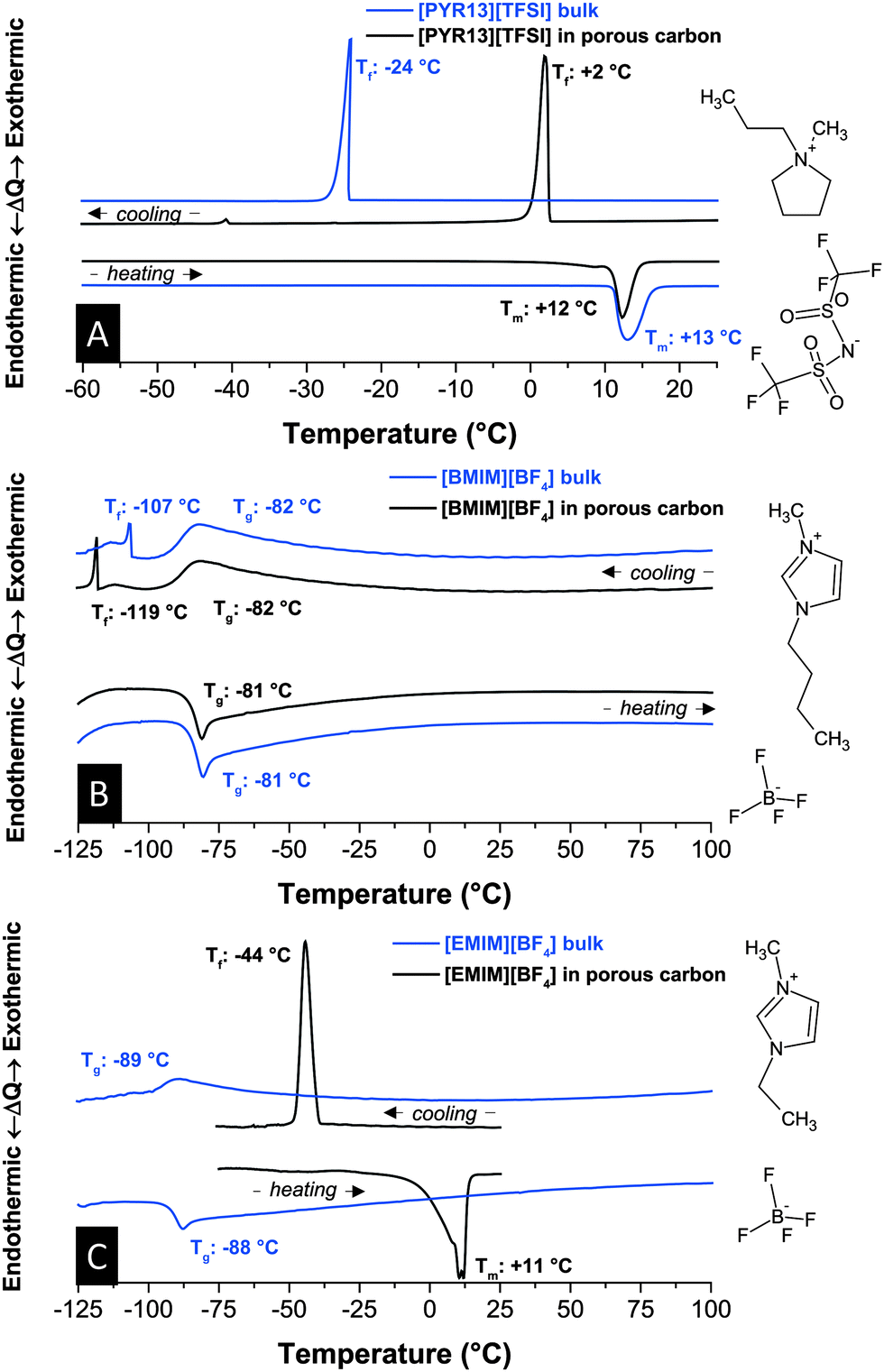 | ||
| Fig. 1 Differential thermograms of [PYR13][TFSI] (A), [BMIM][BF4] (B), and [EMIM][BF4] (C) in bulk form and in confinement of nanoporous carbon. Scan rate: 10 °C min−1. | ||
When looking at a typical imidazolium-based RTIL (Fig. 1B), such as 1-butyl-3-methyl-imidazolium tetrafluoroborate [BMIM][BF4], we now have a very different situation since this ionic liquid does not undergo crystallization but only one glass transition and a minor phase transition within a large temperature range (−125 °C to +100 °C). Nanoconfinement does not change this behavior significantly and only the ultralow-temperature phase transition at −107 °C is shifted to −119 °C when the RTIL is soaked in nanoporous carbon. Such a decrease aligns well with the downshift of phase transition temperatures in glass-like materials following the Ehrenfest equation30 although during cooling also interactions between the pore walls and the ions15 need to be considered. The minor phase transitions might be attributed to an incomplete glass formation and probably small crystallite formation. Upon heating, only one endothermal glass transition at −81 °C occurs in either case which is close to what has been reported by Nishida et al., namely at −85 °C.31 No specific peak for melting can be identified.
Staying in the imidazolium system but using a cation with a much shorter chain length, namely 1-ethyl-3-methyl-imidazolium [EMIM]+, we see for the bulk one glass transition at −89 °C and −88 °C during cooling and heating, respectively (Fig. 1C). Compared to [BMIM][BF4], this downshift of Tg can be explained by less entwining of the shorter cation chains.22,31,32 The highly reproducible absence of crystallization and subsequent melting of the bulk RTIL is in contrast to the literature where a melting point at 11 °C (13 °C) and a freezing point at −63 °C (−58 °C) have been reported in ref. 33 (ref. 31). It is possible that earlier reports of bulk [EMIM][BF4] crystallization were influenced by impurities, such as water which is well known to severely influence physical properties of RTILs.34 For this reason, the content of H2O of all RTILs studied in this work was well below 10 ppm as determined by Karl Fischer titration. In nanoconfinement, the RTIL shows pronounced exo- and endothermal signals related to crystallization (−44 °C) and melting (+11 °C). We also have to note the broadening of the onset temperature of endothermal signals in carbon nanopores. This feature can be explained by the presence of RTILs in small nanopores (esp. around 1 nm) and larger mesopores (see pore size distribution in the ESI†) leading to a “smearing” effect of overlying thermal signatures. In addition the crystallization of [EMIM][BF4] in nanopores was highly dependent on the history of the RTIL (see Fig. S2, ESI†). A lower limit for the upper temperature yields a higher temperature for the exothermic peak during cooling. Thus, all comparisons between bulk and nanoconfined RTILs were carried out within the same temperature window.
Charging and discharging of a carbon supercapacitor locally change the ion concentration inside the pores of the positively and negatively polarized electrode.35–37 Theoretical work36 on 1-ethyl-3-methylimidazoliumbis(trifluoromethylsulfonyl)imide [EMIM][TFSI] in a 1.2 nm pore shows that the overall number of ions per unit volume only slightly increases (<20%) when using an electrode potential of up to ±1.5 V; yet, the ratio of cations and anions greatly varies by a factor of ∼300%. Thus, we expect the strong change in local composition to have also a significant influence on the thermophysical behavior since a recent theoretical work revealed an accelerating charging dynamics in subnanometer pores due to the diffusion variation of inside ions under polarization.38
In our experiments, we charged the electrodes of a symmetrical carbon supercapacitor cell, disassembled the device in an inert gas glove box, and analyzed, separately, the negatively and positively polarized electrodes. By doing so, some of the electrical charge may be lost, but the anion/cation ratio remains unchanged. For each experiment, a symmetric cell was fully charged to a voltage of 2.5 V (charging potential of each electrode see Fig. S4, ESI†). In the case of [PYR13][TFSI], the melting point was severely suppressed compared to the measurements at 0 V (Fig. 2A). The small endothermic peak indicative of melting during heating is located at significantly lower temperatures for positive polarization (8 °C) compared to negative polarization (10 °C) or not-polarized carbon (12 °C). We attribute the downshift of the melting point to the higher concentration of anions or cations resulting in non-stoichiometric local ion arrangement impeding the formation of extended RTIL crystal arrangements. The onset for the crystallization is in a narrower range and seems to be minor affected by polarization.
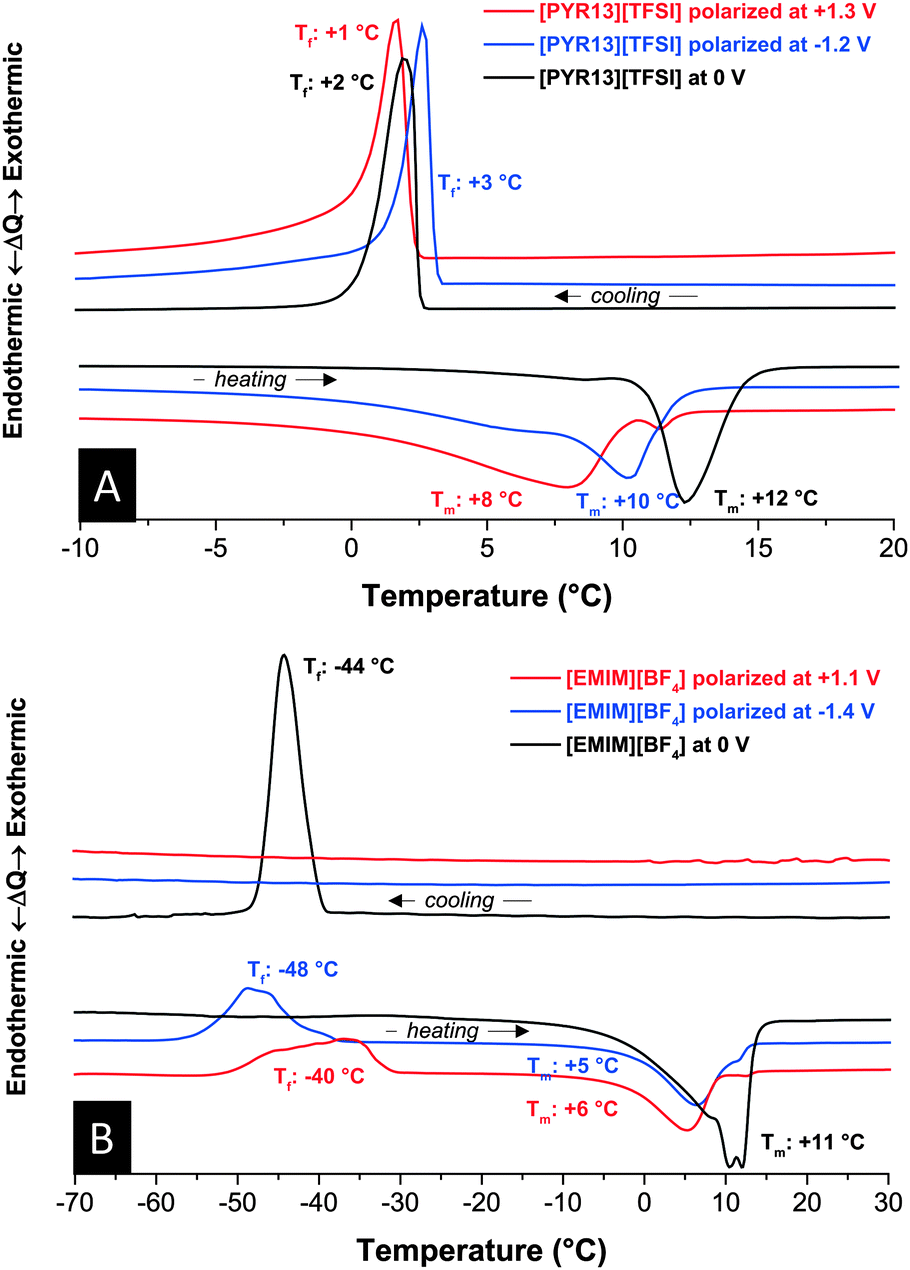 | ||
| Fig. 2 Differential thermograms of [PYR13][TFSI] (A) and [EMIM][BF4] (B) with and without polarization at +1.3 V or −1.2 V vs. AC. Scan rate: 10 °C min−1. | ||
A similar trend of melting point depression is found for [EMIM][BF4] (Fig. 2B). Here, the anion is much smaller and much more mobile than the cation. Ion separation, thus, is expected to have an even larger influence on the thermophysical response. Interestingly, when decreasing the temperature, [EMIM][BF4] continuously supercools in polarized carbon electrodes without any phase or glass transition down to −70 °C. Instead, [EMIM][BF4] undergoes crystallization between −40 °C and −45 °C during heating. This feature of some ionic liquids, especially imidazolium-based ones, is related to overall high viscosity and steric effects of the ions. A similar trend could be seen in the vertex temperature dependence, presented in the (Fig. S2, ESI†). The freezing behavior of [EMIM][BF4] is highly dependent on the history of the sample and, hence, leads to either supercooling or a crystallization during the cooling cycle. This effect has never been reported for [EMIM][BF4] as it is absent in the bulk form or nanoconfined without a modified anion/cation ratio.
Finally, we investigated the influence of freezing and thawing on the electrochemical performance. The equilibrium capacitance of RTIL supercapacitors should not be affected by temperature.39 However, since capacitance is usually determined via dynamic methods (such as galvanostatic charge–discharge or cyclic voltammetry), the measured capacitance is susceptible to temperature variations and different degrees of ion mobility. Thus, a severe decrease of the capacitance at low temperatures is commonly found in RTIL systems that show neither crystallization nor glass transition within the studied range.10
To investigate two extreme cases, we used [BMIM][BF4] (no crystallization) and [EMIM][BF4] (crystallization) and cycled constantly at 10 mV s−1 while cooling down to −20 °C. The voltage range was kept small (±0.25 V) to be more sensitive to abrupt changes in the capacitive current. In the case of [BMIM][BF4], we see a continuous decrease of the capacitive current; yet we note that while the overall current decreases with temperature (Fig. 3A) the system still behaves as a capacitor even at −20 °C. This remnant capacitive behavior is more evident when normalizing to the maximum current (Fig. 3B) and the narrowing of the cyclic voltammogram curve is a result of the decrease in ion mobility and increase of cell resistance at lower temperatures.10 Noticeably, [EMIM][BF4] exhibits a complete loss of capacitance when cooling the device to −20 °C (Fig. 3C) indicating a structural change already at this temperature initiated by ion movement during electrochemical charging. Thermal analysis of porous carbon soaked with [EMIM][BF4] had shown freezing at much lower temperatures (−44 °C), but the RTIL appears fully frozen near −20 °C as evidenced by the electrochemical response. Upon heating, both RTILs (Fig. 3D: [EMIM][BF4]) fully regain their initial capacitance. Thus, no significant deterioration of the carbon had been encountered and thawing restored the initial energy storage capacity. As discussed in ref. 31 [BMIM][BF4] most probably does not undergo crystallization due to steric hindrance because of the length of the alkyl chain.
 | ||
| Fig. 3 Cyclic voltammograms using [BMIM][BF4] (A) and (B) and [EMIM][BF4] (C) during continuous cooling (A–C) and comparison before and after freezing for [EMIM][BF4] (D). Scan rate: 10 mV s−1. | ||
Lastly, we studied the behavior of an ionic liquid mixture of 10 wt% [BMIM][BF4] in the [EMIM][BF4] electrolyte upon freezing and re-heating. The idea was to investigate if the residual capacitive performance of [BMIM][BF4] can be partially transferred to a [EMIM][BF4]-based mixed ionic liquid electrolyte or if we could even completely hinder the crystallization of [EMIM][BF4]. The system was operated in three electrode mode, which enabled the monitoring of the working electrode potential and the counter electrode potential separately. Cooling the cell from room temperature to −20 °C we observed reproducibly that the cell exhibited capacitive response much longer (ca. 3-times) at −20 °C compared to a pure [EMIM]+. However, ultimately also for the mixture the cell capacitance dropped to zero comparable to Fig. 3C. The DSC thermogram of the respective mixture in a nanoporous environment can be found in the Fig. S3, ESI.† The behaviour is similar to [EMIM][BF4] except for the presence of a much broader peak and mixed freezing during cooling and heating.
We also observed at −20 °C an asymmetric current signal which is highly unusual for a supercapacitor setup. Such behavior can only be explained by vast differences in the ion mobility of anions and cations. For positive polarization the system behaves at −20 °C as if the ions were immobile while, for negative polarization, ion mobility is still observed (Fig. 4A). Upon heating, the normal behavior of the cell was restored (Fig. 4B). This in situ electrochemical probing of cation and anion dynamics at the anode and cathode complements the results shown in Fig. 2 where only the global thermal response of positively or negatively polarized electrodes is depicted. Yet, we see from these results that even under nominally “frozen” conditions (in the sense of Fig. 2) some ions, which do not seem to undergo crystallization (such as [BMIM]+), may retain some residual ion mobility. We explain this behavior by the residual mobility of excess [BMIM]+ cations within a partially solidified [EMIM][BF4] network.
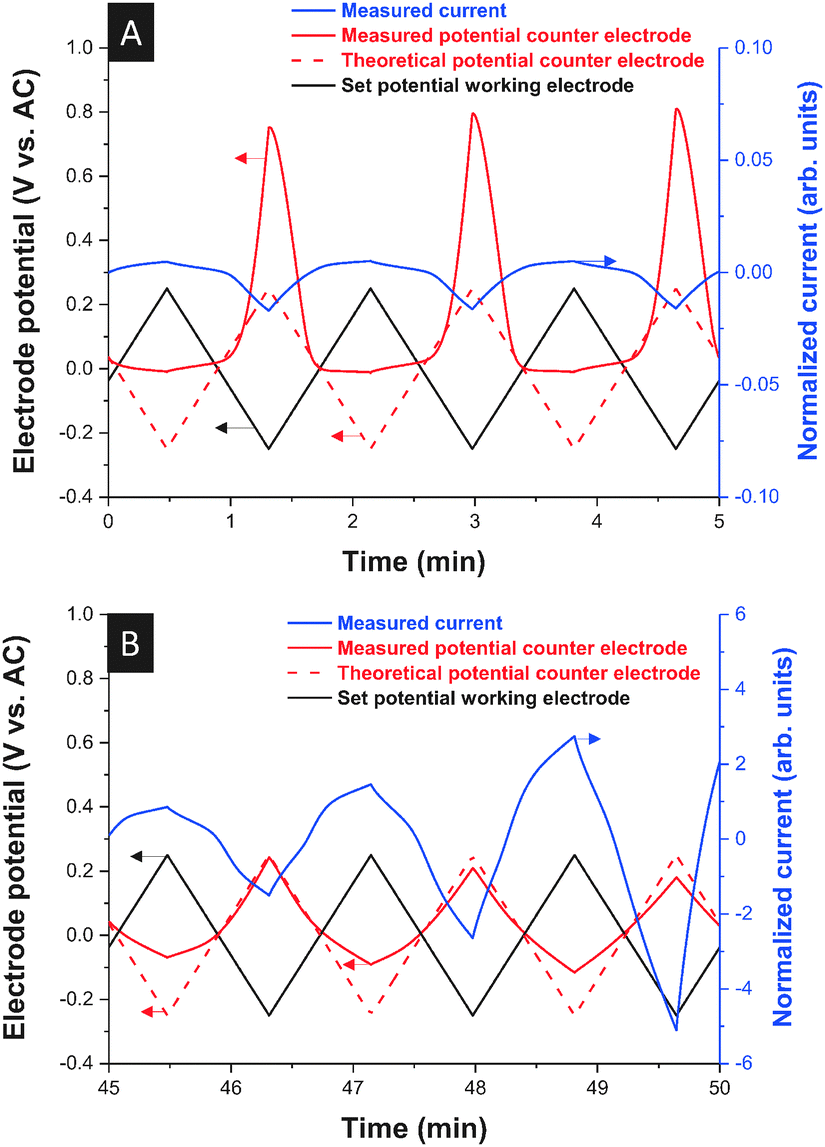 | ||
| Fig. 4 Voltage and current response of a 10 wt% [BMIM][BF4] in [EMIM][BF4] mixture at −20 °C (A) and during re-heating (B). Scan rate: 10 mV s−1. | ||
Conclusions
In summary, we have demonstrated that the thermophysical behavior of RTIL carbon supercapacitors can be very different from the bulk properties of the electrolyte. A main mechanism of this difference may be the molecular interaction between ionic liquid and carbon pore walls in nanoconfinement. The freezing behavior is a non-equilibrium process that may be fully suppressed in bulk and only occur in nanoconfinement ([EMIM][BF4]) or it may occur at much higher temperatures (e.g., Δ +26 °C; [PYR13][TFSI]). Also, supercooling is an important mechanism in understanding the crystallization properties of RTIL supercapacitors. Yet, even if no crystallization occurs at low temperatures for a charged supercapacitor, the device may nevertheless freeze either during re-heating or when discharging the system. Carbon-based RTIL supercapacitors exhibit fully reversible electrochemical behavior and the initial capacitance is fully recovered once the device is re-heated. Thus, low temperatures provide much less of a concern to the device safety compared to high temperatures at which (electro-)chemical decomposition may be encountered.Acknowledgements
The INM (http://www.inm-gmbh.de) is part of the Leibniz Research Alliance Energy Transition (LVE). DW and VP acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework and thank Prof. Eduard Arzt (INM) for his continuing support. Benjamin Krüner (INM) is thanked for fruitful discussions.Notes and references
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- A. Lewandowski and A. Świderska-Mocek, J. Power Sources, 2009, 194, 601–609 CrossRef CAS PubMed.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS PubMed.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS PubMed.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS PubMed.
- R. Lin, P.-L. Taberna, S. Fantini, V. Presser, C. R. Perez, F. Malbosc, N. L. Rupesinghe, K. B. K. Teo, Y. Gogotsi and P. Simon, J. Phys. Chem. Lett., 2011, 2, 2396–2401 CrossRef CAS.
- W.-Y. Tsai, R. Lin, S. Murali, L. Li Zhang, J. K. McDonough, R. S. Ruoff, P.-L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS PubMed.
- C. Alba-Simionesco, B. Coasne, G. Dosseh, G. Dudziak, K. E. Gubbins, R. Radhakrishnan and M. Sliwinska-Bartkowiak, J. Phys.: Condens. Matter, 2006, 18, R15 CrossRef CAS PubMed.
- S. Jahnert, F. Vaca Chavez, G. E. Schaumann, A. Schreiber, M. Schonhoff and G. H. Findenegg, Phys. Chem. Chem. Phys., 2008, 10, 6039–6051 RSC.
- A. K. Gupta, Y. L. Verma, R. K. Singh and S. Chandra, J. Phys. Chem. C, 2014, 118, 1530–1539 CAS.
- M. P. Singh, R. K. Singh and S. Chandra, J. Phys. Chem. B, 2011, 115, 7505–7514 CrossRef CAS PubMed.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, J. Am. Chem. Soc., 2005, 127, 16792–16793 CrossRef CAS PubMed.
- Y. Liu, G. Wu, H. Fu, Z. Jiang, S. Chen and M. Sha, Dalton Trans., 2010, 39, 3190–3194 RSC.
- J. Zhang, Q. Zhang, X. Li, S. Liu, Y. Ma, F. Shi and Y. Deng, Phys. Chem. Chem. Phys., 2010, 12, 1971–1981 RSC.
- D. Ehre, E. Lavert, M. Lahav and I. Lubomirsky, Science, 2010, 327, 672–675 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- W. M. Reichert, J. D. Holbrey, K. B. Vigour, T. D. Morgan, G. A. Broker and R. D. Rogers, Chem. Commun., 2006, 4767–4779 RSC.
- G. Raabe and J. Köhler, J. Chem. Phys., 2008, 128, 154509 CrossRef PubMed.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- M. Kanakubo, Y. Hiejima, K. Minami, T. Aizawa and H. Nanjo, Chem. Commun., 2006, 1828–1830 RSC.
- S. Chen, G. Wu, M. Sha and S. Huang, J. Am. Chem. Soc., 2007, 129, 2416–2417 CrossRef CAS PubMed.
- Y. Jeon, D. Vaknin, W. Bu, J. Sung, Y. Ouchi, W. Sung and D. Kim, Phys. Rev. Lett., 2012, 108, 055502 CrossRef.
- M. Rose, Y. Korenblit, E. Kockrick, L. Borchardt, M. Oschatz, S. Kaskel and G. Yushin, Small, 2011, 7, 1108–1117 CrossRef CAS PubMed.
- D. Weingarth, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochem. Commun., 2012, 18, 116–118 CrossRef CAS PubMed.
- S. M. Chathoth, E. Mamontov, S. Dai, X. Wang, P. F. Fulvio and D. J. Wesolowski, EPL, 2012, 97, 66004 CrossRef.
- R. O. Davies and G. O. Jones, Adv. Phys., 1953, 2, 370–410 CrossRef.
- T. Nishida, Y. Tashiro and M. Yamamoto, J. Fluorine Chem., 2003, 120, 135–141 CrossRef CAS.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- A. B. McEwen, H. L. Ngo, K. LeCompte and J. L. Goldman, J. Electrochem. Soc., 1999, 146, 1687–1695 CrossRef CAS PubMed.
- G. Yu, D. Zhao, L. Wen, S. Yang and X. Chen, AIChE J., 2012, 58, 2885–2899 CrossRef CAS PubMed.
- F. W. Richey, B. Dyatkin, Y. Gogotsi and Y. A. Elabd, J. Am. Chem. Soc., 2013, 135, 12818–12826 CrossRef CAS PubMed.
- J. M. Black, G. Feng, P. F. Fulvio, P. C. Hillesheim, S. Dai, Y. Gogotsi, P. T. Cummings, S. V. Kalinin and N. Balke, Adv. Energy Mater., 2014, 4, 1300683 Search PubMed.
- M. Deschamps, E. Gilbert, P. Azais, E. Raymundo-Piñero, M. R. Ammar, P. Simon, D. Massiot and F. Béguin, Nat. Mater., 2013, 12, 351–358 CrossRef CAS PubMed.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Nat. Mater., 2014, 13, 387–393 CrossRef CAS PubMed.
- G. Feng, S. Li, J. S. Atchison, V. Presser and P. T. Cummings, J. Phys. Chem. C, 2013, 117, 9178–9186 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02727b |
| This journal is © the Owner Societies 2014 |