
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of 1-pentyne hydrogenation on the atomic structures of size-selected AuN and PdN (N = 923 and 2057) nanoclusters†
Kuo-Juei
Hu
a,
Simon R.
Plant
a,
Peter R.
Ellis
b,
Christopher M.
Brown
b,
Peter T.
Bishop
b and
Richard E.
Palmer
*a
aNanoscale Physics Research Laboratory, School of Physics and Astronomy, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: R.E.Palmer@bham.ac.uk
bJohnson Matthey Technology Centre, Blounts Court Road, Sonning Common, Reading, RG4 9NH, UK
First published on 13th October 2014
Abstract
We report an investigation into the effects of the vapour-phase hydrogenation of 1-pentyne on the atomic structures of size-selected Au and Pd nanoclusters supported on amorphous carbon films. We use aberration-corrected high-angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) to image populations of the nanoclusters at atomic resolution, both before and after the reaction, and we assign their atomic structures by comparison with multi-slice image simulations over a full range of cluster orientations. Gold nanoclusters consisting of 923 ± 20 and 2057 ± 45 atoms are found to be robust, exhibiting high structural stability. However, a significant portion of Pd923±26 nanoclusters that appear amorphous prior to treatment are found to exhibit high symmetry structures post-reaction, which is interpreted as the reduction of oxidised Pd nanoclusters under the reaction conditions.
1 Introduction
It has long been established that the catalytic properties of supported metal particles vary as a function of size.1 The use of size-selected metal nanoclusters as model catalysts2–8 is one of the routes that has enabled catalytic activity to be accurately related to particle size. However, it is not just the size, but the full atomic structure which can regulate the performance of a catalyst, since catalytic activity can, in some cases, be correlated with specific reactive sites at the catalyst particle surface.9 Indeed, it has been demonstrated that controlling particle shape may enable improved selectivity.10 As a result, it is vital to gain an understanding of the effects upon the structure of such model catalysts arising from exposure to realistic reaction conditions, in order to achieve more robust catalyst design with improved performance. A number of techniques have been used to monitor nanocluster or nanoparticle catalysts subject to reaction conditions, including, for instance, in situ TEM,11–13 electron tomography14 and in situ STM.15 Aberration-corrected HAADF-STEM has previously been used to identify catalytically-active Au nanoclusters on oxide supports,16 and the catalytic activity of size-selected Au nanoclusters on oxides is well-established.17 For instance, amongst size-selected Au nanoclusters, Au55 has been highlighted as a high performance catalyst with pronounced oxidation resistance.18 HAADF-STEM imaging of size-selected Au nanoclusters on amorphous carbon has enabled the elucidation of their three-dimensional atomic structures.19 Nanoclusters of a specific size can exhibit a range of atomic structures that differ significantly from the bulk, and multi-slice image simulations have facilitated the identification of structures from HAADF-STEM images for size-selected Au20,20 Au55,21 Au309 (ref. 19) and Au923,22–24 regardless of cluster orientation on the substrate. Given the recent demonstration of atomic structure control during the formation of size-selected Au923,24 there is now a great potential to study the catalytic properties of such nanoclusters not only as a function of their size, but also atomic configuration, under realistic reaction conditions. Size-selected PdN nanoclusters (N = 55–400) supported on graphite have already been studied under such conditions for methane oxidation25 and 1-pentyne hydrogenation.26 The largest nanoclusters in this size range (especially Pd400) exhibited very high selectivity to the hydrogenation of 1-pentyne in the vapour phase, thus motivating the study of still larger nanocluster sizes.The selective hydrogenation of alkynes is relevant to both Au27–29 and Pd catalysts.26,30–33 The vapour-phase 1-pentyne hydrogenation reaction over Pd supported on θ-Al2O3 reveals incorporation of carbon into the catalyst to provide a Pd–C phase believed to be active for selective hydrogenation,31 and indeed, the incorporation of carbon and hydrogen into the surface has been shown to control hydrogenation events at the surface.32 However, Pd catalysts can also become deactivated and degraded by such processes.34 Aging of the catalyst as a result of thermally- or chemically-driven restructuring35–37 is important for both the catalytic activity and stability. It is therefore crucial to be able to identify changes in the structure and composition of such catalytic particles both before and after exposure to realistic reaction conditions, in order to achieve catalysts with optimised efficiency, selectivity and sustained performance.
Here, we report the atomic structures of size-selected Au and Pd nanoclusters (containing 923 and 2057 atoms) supported on amorphous carbon films, both before and after exposure to the thermal and chemical conditions for the 1-pentyne hydrogenation reaction. Observations made ex situ using aberration-corrected HAADF-STEM provide direct geometric information about these nanoclusters. Multi-slice HAADF-STEM image simulations are compared with experimental images to identify the atomic structures of the observed nanoclusters, over a full range of orientations on the substrate. Our results show that the Au nanoclusters of both sizes are very stable under the reaction conditions, remaining largely unchanged after pure thermal treatment and after full exposure to the chemical reaction conditions. However, a notable proportion of Pd923 nanoclusters, of which the vast majority (97%) appear amorphous before the reaction, are found to exhibit a face-centred cubic structure after treatment.
2 Experimental section
Gold and palladium nanoclusters were generated using a magnetron-sputtering gas-condensation cluster beam source.38,39 A lateral time-of-flight mass filter40 connected to the source permits accurate size selection prior to deposition of the nanoclusters in high vacuum conditions (10−7–10−6 mbar). Based on calibration with Ar+, the nominal mass resolutions employed were M/ΔM ≈ 18 and M/ΔM ≈ 23 for Pd and Au nanoclusters, respectively, resulting in nanoclusters that consist of 923 ± 26 and 2057 ± 57 atoms for Pd, and 923 ± 20 and 2057 ± 45 atoms for Au. The size-selected nanoclusters were deposited (at energies in the range 0.4–0.9 eV per atom) directly onto amorphous carbon films supported on molybdenum TEM grids. Following deposition, the supported Au nanoclusters were brought into air (ambient conditions) and then transferred into a vacuum desiccator for storage, both before and after imaging and treatment. Transfer of the supported Au nanoclusters from the vacuum desiccator to the microscope (in ambient conditions) was performed within ∼20 minutes. The Pd nanoclusters were stored in a desiccator at ambient temperature and pressure for 100 days prior to initial imaging, and the reaction exposure was conducted within a further 10 days. Imaging was then performed 20 days after the reaction. The samples were transferred in ambient conditions for imaging and treatment. For 1-pentyne hydrogenation treatment, each of the TEM grids was transferred into a quartz tube (length 360 mm, inner diameter 4 mm) and held at the centre of the tube by means of a quartz wool plug. A flow of pure He gas (279 ml min−1) was used to provide an inert atmosphere when investigating the effects of the thermal annealing. For the hydrogenation reaction, the carrier gas consisted of 40% H2/60% He (flow rate of 247 ml min−1). In the reaction, this carrier gas was used to vaporise a reagent solution comprising 1 M 1-pentyne plus 1 M 2-methylpentane (used in such reactions as an internal standard for gas chromatography26) dissolved in n-hexane. The temperature was increased at a rate of 2 °C min−1 from room temperature to 250 °C and then maintained at constant temperature for 2 hours. At the end of the treatment, the carrier gas was switched to pure He, and the sample was allowed to cool for 30 minutes. Atomic resolution imaging was carried out using a JEOL 2100F STEM operating at 200 keV and equipped with a spherical aberration probe corrector (CEOS GmbH) and high-angle annular dark field (HAADF) detector. The inner and outer collection angles of the HAADF detector were 62 and 164 mrad, respectively.41 Imaging was performed so as to avoid beam damage to the atomic structures of the nanoclusters (see ESI†). Following imaging, high symmetry nanocluster structures were identified by comparing experimental images with an atlas of multi-slice image simulations over a full range of orientations in three dimensions. Simulations of Au2057, Pd923 and Pd2057 were generated using the QSTEM package.42 The previously published simulation atlas was used in the case of Au923.223 Results and discussion
The high symmetry isomers of the nanoclusters observed in this study display structural motifs that are characteristic of the icosahedron (Ih), Ino-decahedron (Dh) and face-centred cubic (FCC) polyhedron, just as for size-selected Au923 nanoclusters studied previously.22–24Fig. 1 shows typical HAADF-STEM images and corresponding multi-slice image simulations of an illustrative set of high symmetry Au2057 isomers. There is always a proportion of nanoclusters within a sample population that cannot be assigned uniquely to a high symmetry structure, while some nanoclusters appear to be completely amorphous, and we designate such structures as being amorphous or unidentified (A/U). In the present study, the Dh-Au923 and Dh-Au2057 nanoclusters are found to be the most abundant; recent experimental investigations of Au923 suggest that Dh-Au923 is a low energy structure in this size regime, whereas Ih-Au923 in particular is metastable.22,24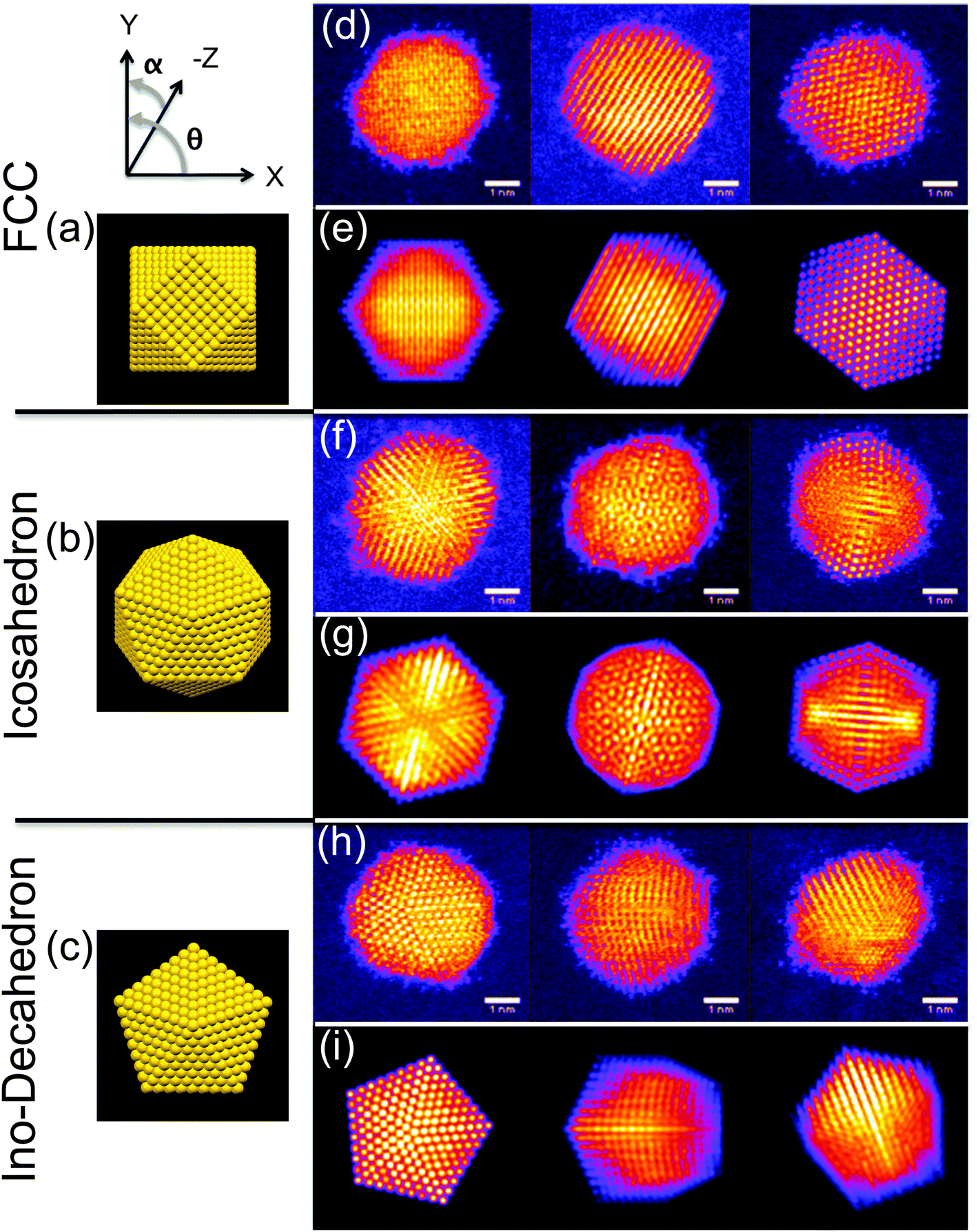 | ||
| Fig. 1 Models of the atomic structures for the (a) face-centred cubic (cuboctahedron shown), (b) icosahedral, and (c) Ino-decahedral isomers of Au2057, viewed at θ = 0°, α = 0°, as defined by the axes shown. Typical HAADF-STEM images of Au2057 nanoclusters (top) with corresponding multi-slice image simulations (below) for: (d, e) the face-centered cubic (FCC) polyhedron, (simulation orientations in row (e), from left to right, are along θ = 45°, α = 45°, and θ = 0°, α = 30°, and the 110 axis), (f, g) the icosahedron (Ih) (simulation orientations in row (g) from left to right are normal to (111) facet, along the 5-fold axis, and along the 2-fold axis), and (h, i) the Ino-decahedron (Dh) (simulation orientations in row (i) from left to right are along 5-fold axis, at θ = 0°, α = 50°, and at θ = 0°, α = 30°). | ||
Several factors might induce changes in the structure of nanoclusters in the continuous flow vapour-phase reaction. For example, thermal annealing alone can be an important driving force in triggering the structural transformation of nanoparticles.36,37,43 In order to de-couple any effects of (mere) elevated temperatures from the full reaction conditions, thermal annealing was first conducted on Au923 and Au2057 nanoclusters. Since recent work shows that the relative proportions of isomers within a sample population can be controlled by tuning the formation parameters for the generation of Au923,24 samples were produced using identical formation conditions in the cluster beam source (parameters are detailed in the figure captions). Fig. 2 shows a comparison of the proportions of Au923 isomers, before and after thermal annealing at 523 K for 2 hours in different atmospheres. (The results for Au2057 nanoclusters are very similar to those shown in Fig. 2, and are provided in the ESI.†) Fig. 2(a) presents charts showing the combined results for all 3 samples of Au923 nanoclusters prior to thermal annealing. In general, the error bars shown on the bar charts are related to the Poisson error. However, the error bars shown on the bar chart in Fig. 2(a) derive from the standard error for the 3 samples. Fig. 2(b) shows the proportions of Au923 isomers following storage in a vacuum desiccator, where the sample has been imaged within 14 days of preparation, while Fig. 2(c) and (d) show the result of heating in a pure He gas flow, and in a gas flow of 40% H2/60% He, respectively. Although some slight variations in isomer distributions can be observed, there are no significant changes in the Au923 (or Au2057) isomers as a result of subjecting them to these conditions.
 | ||
| Fig. 2 Charts showing the relative proportions of Au923 isomers (a) before thermal treatment, (b) after storage in vacuum at RT, (c) after thermal treatment under a gas flow of pure He (279 ml min−1), and (d) after thermal treatment under a gas flow of 40% H2 + 60% He (247 ml min−1). Thermal treatment was conducted at 523 K for 2 hours (ramp rate of 2 °C min−1 from RT). Related cluster formation parameters: condensation length, 250 mm; magnetron sputtering power, 10 W DC; condensation pressure, 0.60 mbar; condensation gas flows, rate 200 sccm (Ar) and 150 sccm (He); deposition energy, 0.5 eV per atom. | ||
The results may be explained by considering the temperatures required to induce melting in gold nanoparticles as a function of particle size. Thermal annealing in an inert atmosphere can be interpreted as a rapid process of bringing nanoclusters into a state of thermal equilibrium, where no reactants are involved. According to previous studies,36 the Ih-to-Dh structural transformation can occur for particles in the size range from 3 nm to 14 nm without having to reach the melting point. For instance, the gap between transition temperature and melting point is predicted to be ∼100 K for a Au particle size of 4 nm, based on the extrapolation from experimental data.36 The transformation from more stable FCC or decahedral to icosahedral structures requires temperatures above the melting point, followed by rapid cooling (freezing) of the nanoparticle.36,43 The melting point of gold nanoparticles is highly sensitive to size. Although the melting point of gold nanoparticles at ∼2.6 nm in size is close to 500 K, an increase in size to 3 (or 4) nm causes the melting point to rise to ∼750 K (or ∼900 K).37 This is consistent with the observations presented in Fig. 2. Given that the size of Au923 is ∼3 nm and Au2057 ∼ 4 nm, the temperature of 523 K employed does not approach sufficiently close to the melting point of the nanoparticles to induce an Ih-to-Dh structural transition, so even the least stable nanocluster isomer can survive the treatment.
Fig. 3 shows the proportions of size-selected Au923 and Au2057 nanocluster isomers, both before and after exposure to the full vapour-phase 1-pentyne hydrogenation reaction conditions. As shown in Fig. 3(a) and (b), with 9% Ih, 43% Dh and 35% FCC before the reaction, Au923 nanoclusters displayed similar stability to Au2057: the relative proportions of Au923 isomers after treatment are 7% Ih, 45% Dh and 35% FCC. Such variations in isomer proportions fall within the (Poissonian) error. As shown in Fig. 3(c) the proportions of as-deposited Au2057 isomers before the reaction are 9% Ih, 54% Dh and 28% FCC. The isomer proportions were found to be almost identical post reaction (see Fig. 3(d)).
 | ||
| Fig. 3 (a, b) Charts showing the relative proportions of Au923 isomers (a) before and (b) after exposure to the reaction conditions for vapour-phase 1-pentyne hydrogenation. (c, d) Charts showing the relative proportions of Au2057 isomers (c) before and (d) after exposure to the same conditions. The reaction was conducted at 523 K for 2 hours (ramp rate of 2 °C min−1 from RT) with 1 M 1-pentyne in hexane vaporised by a carrier gas of 40% H2 and 60% He (flow rate 247 ml min−1). The cluster formation parameters were: condensation length, 250 mm; magnetron sputtering power, 10 W DC; condensation pressure, 0.60 mbar (Au923) and 0.67 mbar (Au2057); condensation gas flows, 200 sccm (Ar) and 150 sccm (He); deposition energy, 0.5 eV per atom. | ||
Although there is evidence for the chemisorption of 1-pentyne at the surface of bulk gold,44 there are a limited number of investigations on the catalysis of 1-pentyne hydrogenation using gold. However, the hydrogenation of alkynes involving oxide-supported gold nanoparticles27,29 suggests that, although there is higher selectivity to semi-hydrogenation with Au, gold catalysts are considerably less active than Pd.28,29 For instance, TiO2-supported gold nanoclusters of average size 4.7 nm are reported to show poor adsorption of alkyne or alkene species, suggesting there may be a weaker interaction between gold and 1-pentyne (as compared with Pd).
Fig. 4 shows the variation in the proportions of Pd2057 and Pd923 nanocluster isomers as a result of exposure to the 1-pentyne hydrogenation reaction conditions, which were exactly the same as in the case of the Au nanoclusters. Example images of the amorphous Pd923 and Pd2057 nanoclusters observed are shown in Fig. 4(a) and (d), respectively. Fig. 4(b) reveals that the vast majority (97%) of Pd923 nanoclusters initially exhibit an amorphous appearance; the only nanoclusters with an identifiable high symmetry structure (3%) are found to be Dh. After the reaction, as shown in Fig. 4(c), a large proportion (33%) of the nanoclusters could be assigned to a high symmetry structure, dominated by FCC (32%), leaving 1% as Dh. For Pd2057, the proportion of amorphous nanoclusters pre-reaction is much lower at 37%, and almost a majority of the nanoclusters are FCC structures (see Fig. 4(e)). Both before and after reaction (Fig. 4(e) and (f)), more than three-quarters of the identified high symmetry isomers are assigned to be FCC.
 | ||
| Fig. 4 (a, d) Example HAADF-STEM images of amorphous nanoclusters for (a) Pd923 and (d) Pd2057, imaged prior to treatment. (b, c) Charts showing the relative proportions of Pd923 isomers (b) before and (c) after vapour-phase 1-pentyne hydrogenation treatment. (e, f) Charts showing the relative proportions of Pd2057 isomers both (e) before and (f) after the same treatment. The conditions for the treatments were identical to those for the Au nanoclusters shown in Fig. 3. Related cluster formation parameters: condensation length, 172 mm; magnetron sputtering power, 10 W DC; condensation pressure, 0.18 mbar (Pd923) and 0.28 mbar (Pd2057); condensation gas flows, 100 sccm (Ar) and 110 sccm (He); deposition energy, 0.9 eV per atom (Pd923) and 0.4 eV per atom (Pd2057). | ||
Pd is known to oxidise spontaneously upon contact with oxygen,31,45–47 but the oxidation depth on Pd single crystals is temperature dependent.48 On the Pd(111) surface, PdO first forms as an amorphous layer, gradually increasing in crystallinity at elevated temperatures. When the temperature reaches 500 K, amorphous PdO layers coalescence to form a PdO(101) ad-layer with a thickness of 3.5 ML at saturation.45,49 Oxygen can become incorporated into Pd nanoparticles in a number of ways, both at the surface and beneath, or through the formation of oxide shells.50,51 For (oxide-supported) Pd nanoparticles with diameters of <5 nm, both oxidation and reduction by oxygen and hydrogen, respectively, proceed readily at room temperature.52 For ZrO2-supported PdO nanoclusters with average size of 28 nm, shell-by-shell reduction in hydrogen has been observed and the clusters can be restored to Pd without fragmenting.45 In the present case, the amorphous appearance of large proportions of both Pd2057 and, in particular, Pd923 nanoclusters, prior to the reaction, may therefore be attributable to surface oxidation in air, especially given storage conditions prior to imaging (desiccator, ambient temperature and pressure, 100 days). We note, however, that only 37% of Pd2057 nanoclusters observed before exposure to the reaction conditions appear to be amorphous, compared to 97% for Pd923 stored under identical conditions, which suggests that the Pd2057 nanoclusters exhibit a marked oxidation resistance as compared to Pd923, a phenomenon that can be explored further in future work. It is possible that oxidised Pd nanoclusters can undergo reduction when exposed to the conditions of the 1-pentyne hydrogenation reaction, removing the oxide layer, and permitting the observation of (structural) motifs corresponding to high symmetry, ordered atomic structures in post-reaction HAADF-STEM imaging. As all Pd nanoclusters were stored under the same conditions both before and after the reaction, it implies that both Pd923 and Pd2057 nanoclusters are exhibiting a degree of oxidation resistance at room temperature and pressure post-reaction, at least for the high symmetry structural motifs observed. In particular, we note significant proportions of FCC structures for Pd2057 both before (49%) and after (40%) the reaction, as well as the emergence of FCC structures for Pd923 post-reaction. Indeed, the previous study of 1-pentyne hydrogenation with size-selected Pd nanoclusters suggested that there was no obvious change in the FCC-like structure of Pd400 following the reaction, although a full statistical analysis was not carried out. With regard to the present study, further investigation would be required to investigate fully the mechanisms involved and to verify whether the perceived oxidation resistance of the reduced Pd nanoclusters (post-reaction) arises due to changes to the intrinsic structure (to FCC), or results from deposits (due to surface adsorption) or carbonization (catalyst coking).
The ability to produce oxidised and oxidation-resistant Pd catalysts selectively may be advantageous,53 particularly as there is some debate as to whether Pd or PdO is the more active catalyst. An example concerns the comparative activities of alumina-supported palladium for the combustion of methane,54–56 for which early reports are conflicting. It is generally believed that methane oxidation proceeds rapidly through the chemisorption of oxygen onto PdO for alumina-supported Pd catalyst at atmospheric pressure.56 Such chemisorption would cease on the Pd catalyst surface above 650 °C, which is the decomposition temperature of PdO, and hence the catalysed methane combustion stops.55,56 Furthermore, in a more recent study conducted on a polycrystalline Pd plate in an ultrahigh vacuum system, the reaction rate of methane production drops drastically when the system goes beyond the decomposition temperature of PdO.57
4 Conclusions
We have reported an investigation into the effects of the 1-pentyne hydrogenation reaction, under realistic conditions, on the atomic structures of size-selected Au nanoclusters containing 923 ± 20 and 2057 ± 45 atoms, as well as Pd nanoclusters containing 923 ± 26 and 2057 ± 57 atoms. We find that both Au923 and Au2057 nanoclusters exhibit marked structural stability. The large majority of Pd923 nanoclusters were found to be amorphous in appearance before reaction, which may be attributable to surface oxidation. However, following exposure to the same reaction conditions, a significant proportion is found to exhibit high symmetry FCC structures, which may result from the reduction of the nanoclusters in hydrogen. Moreover, given the identical storage conditions before and after reaction, the observation of such structures implies a greater oxidation resistance post-reaction. We suggest that enhanced oxidation resistance may arise as a result of intrinsic structural changes or through protection of the surface due to carbonisation or surface absorption, but the exact origin would be the subject of further investigation. Au2057 nanoclusters, prepared under similar formation conditions to Au923, are found to consist of a large proportion of FCC-like structures, both before and after the reaction, similar to Pd400 previously studied.Acknowledgements
We acknowledge EPSRC and Johnson Matthey for funding this project. S.R.P. acknowledges support through a Science City Research Alliance Fellowship, funded by the Higher Education Funding Council for England (HEFCE).References
- M. Che and C. O. Bennett, Adv. Catal., 1989, 36, 55–172 CAS.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Angew. Chem., Int. Ed., 2009, 48, 1467–1471 CrossRef CAS PubMed.
- Y. Le, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winansl, W. Elám, R. J. Meyer, P. C. Redfern, D. Teschner, R. l. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef PubMed.
- C. Harding, V. Habibpour, S. Kunz, A. N.-S. Farnbacher, U. Heiz, B. Yoon and U. Landman, J. Am. Chem. Soc., 2009, 131, 538–548 CrossRef CAS PubMed.
- S. Kunz, K. Hartl, M. Nesselberger, F. F. Schweinberger, G. Kwon, M. Hanzlik, K. J. J. Mayrhofer, U. Heiz and M. Arenz, Phys. Chem. Chem. Phys., 2010, 12, 10288–10291 RSC.
- G. Sitja, S. L. Moal, M. Marsault, G. Hamm, F. Leroy and C. R. Henry, Nano Lett., 2013, 13, 1977–1982 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- H. Falsig, B. Hvolbæk, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Angew. Chem., 2008, 120, 4913–4917 CrossRef.
- I. Lee, F. Delbecq, R. Morales, M. A. Albiter and F. Zaera, Nat. Mater., 2009, 8, 132–138 CrossRef CAS PubMed.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Am. Chem. Soc., 2010, 132, 7968–7975 CrossRef CAS PubMed.
- S. R. Challa, A. T. Delariva, T. W. Hansen, S. Helveg, J. Sehested, P. L. Hansen, F. Garzon and A. K. Datye, J. Am. Chem. Soc., 2011, 133, 20672–20675 CrossRef CAS PubMed.
- T. Uchiyama, H. Yoshida, Y. Kuwauchi, S. Ichikawa, S. Shimada, M. Haruta and S. Takeda, Angew. Chem., Int. Ed., 2011, 50, 10157–10160 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. De Jong and P. E. De Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- F. Yang, M. S. Chen and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 254–260 CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Schneider, H. Hakkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- H.-G. Boyen, G. Kastle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmuller, C. Hartmann, M. Moller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533–1536 CrossRef CAS PubMed.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–48 CrossRef CAS PubMed.
- Z. W. Wang and R. E. Palmer, Nanoscale, 2012, 4, 4947–4949 RSC.
- Z. W. Wang and R. E. Palmer, Nano Lett., 2012, 12, 5510–5514 CrossRef CAS PubMed.
- Z. W. Wang and R. E. Palmer, Phys. Rev. Lett., 2012, 108, 245502 CrossRef CAS.
- S. R. Plant, L. Cao, F. Yin, Z. W. Wang and R. E. Palmer, Nanoscale, 2014, 6, 1258–1263 RSC.
- S. R. Plant, L. Cao and R. E. Palmer, J. Am. Chem. Soc., 2014, 136, 7559–7562 CrossRef CAS PubMed.
- F. Yin, S. Lee, A. Abdela, S. Vajda and R. E. Palmer, J. Chem. Phys., 2011, 134, 141101 CrossRef PubMed.
- V. Habibpour, M. Y. Song, Z. W. Wang, J. Cookson, C. M. Brown, P. T. Bishop and R. E. Palmer, J. Phys. Chem. C, 2012, 116, 26295–26299 CAS.
- J. A. Lopez-Sanchez and D. Lennon, Appl. Catal., A, 2005, 291, 230–237 CrossRef CAS PubMed.
- L. McEwan, M. Julius, S. Roberts and J. C. Q. Fletcher, Gold Bull., 2010, 43, 298–306 CrossRef CAS.
- Y. Segura, N. Lopez and J. Perezramirez, J. Catal., 2007, 247, 383–386 CrossRef CAS PubMed.
- M. W. Tew, M. Nachtegaal, M. Janousch, T. Huthwelker and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2012, 14, 5761–5768 RSC.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, J. Catal., 2006, 242, 26–37 CrossRef CAS PubMed.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- M. W. Tew, H. Emerich and J. A. van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465 CAS.
- P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275–286 CrossRef CAS.
- Y. Li, H. Cheng, T. Yao, Z. Sun, W. Yan, Y. Jiang, Y. Xie, Y. Sun, Y. Huang, S. Liu, J. Zhang, Y. Xie, T. Hu, L. Yang, Z. Wu and S. Wei, J. Am. Chem. Soc., 2012, 134, 17997–18003 CrossRef CAS PubMed.
- K. Koga, T. Ikeshoji and K. Sugawara, Phys. Rev. Lett., 2004, 92, 2–5 Search PubMed.
- P. Buffat and J. P. Borel, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 13, 2287 CrossRef CAS.
- I. M. Goldby, B. von Issendorff, L. Kuipers and R. E. Palmer, Rev. Sci. Instrum., 1997, 68, 3327–3334 CrossRef CAS PubMed.
- S. Pratontep, S. J. Carroll, C. Xirouchaki, M. Streun and R. E. Palmer, Rev. Sci. Instrum., 2005, 76, 045103 CrossRef PubMed.
- B. von Issendorff and R. E. Palmer, Rev. Sci. Instrum., 1999, 70, 4497–4501 CrossRef CAS PubMed.
- Z. W. Wang, O. Toikkanen, B. M. Quinn and R. E. Palmer, Small, 2011, 7, 1542–1545 CrossRef CAS PubMed.
- C. Koch, PhD thesis, Arizona Sate University, 2002.
- C. Cleveland, W. Luedtke and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 5065–5077 CrossRef CAS.
- H. Feilchenfeld and M. J. Weaver, J. Phys. Chem., 1989, 93, 4276–4282 CrossRef CAS.
- S. C. Su, J. N. Carstens and A. T. Bell, J. Catal., 1998, 176, 125–135 CrossRef CAS.
- E. Voogt, A. J. M. Mens, O. L. J. Gijzeman and J. W. Geus, Catal. Today, 1999, 47, 321–323 CrossRef CAS.
- D. Zemlyanov, B. Klötzer and H. Gabasch, Top. Catal., 2013, 56, 885–895 CrossRef CAS.
- X. Guo, A. Hoffman and J. T. Yates, J. Chem. Phys., 1989, 90, 5787 CrossRef CAS PubMed.
- H. H. Kan and J. F. Weaver, Surf. Sci., 2009, 603, 2671–2682 CrossRef CAS PubMed.
- T. Schalow, B. Brandt, D. Starr, M. Laurin, S. Shaikhutdinov, S. Schauermann, J. Libuda and H.-J. Freund, Angew. Chem., Int. Ed., 2006, 45, 3693–3697 CrossRef CAS PubMed.
- I. Meusel, J. Hoffmann, J. Hartmann, M. Heemeier, M. Bäumer, J. Libuda and H.-J. Freund, Catal. Lett., 2001, 71, 5–13 CrossRef CAS.
- A. Baylet, P. Marécot, D. Duprez, P. Castellazzi, G. Groppi and P. Forzatti, Phys. Chem. Chem. Phys., 2011, 13, 4607–4613 RSC.
- S. M. Lang, I. Fleischer, T. M. Bernhardt, R. N. Barnett and U. Landman, J. Am. Chem. Soc., 2012, 134, 20654–20659 CrossRef CAS PubMed.
- M. Lyubovsky and L. Pfefferle, Catal. Today, 1999, 47, 29–44 CrossRef CAS.
- R. Burch and F. J. Urbano, Appl. Catal., A, 1995, 124, 121–138 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227–237 CrossRef CAS.
- G. Zhu, J. Han, D. Y. Zemlyanov and F. H. Ribeiro, J. Phys. Chem. B, 2005, 109, 2331–2337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02686a |
| This journal is © the Owner Societies 2014 |