
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium uptake in cell construction and subsequent in operando electrode behaviour of Prussian blue analogues, Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6†
James C.
Pramudita
a,
Siegbert
Schmid
*b,
Thomas
Godfrey
b,
Thomas
Whittle
b,
Moshiul
Alam
c,
Tracey
Hanley
c,
Helen E. A.
Brand
d and
Neeraj
Sharma
*a
aSchool of Chemistry, UNSW Australia, Sydney NSW 2052, Australia. E-mail: neeraj.sharma@unsw.edu.au
bSchool of Chemistry, The University of Sydney, Sydney NSW 2006, Australia. E-mail: siegbert.schmid@sydney.edu.au
cAustralian Nuclear Science and Technology Organisation, Kirrawee DC NSW 2253, Australia
dAustralian Synchrotron, 800 Blackburn Road, Clayton Victoria 3168, Australia
First published on 23rd July 2014
Abstract
The development of electrodes for ambient temperature sodium-ion batteries requires the study of new materials and the understanding of how crystal structure influences properties. In this study, we investigate where sodium locates in two Prussian blue analogues, Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6. The evolution of the sodium site occupancies, lattice and volume is shown during charge–discharge using in situ synchrotron X-ray powder diffraction data. Sodium insertion is found to occur in these electrodes during cell construction and therefore Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6 can be used as positive electrodes. NazFeFe(CN)6 electrodes feature higher reversible capacities relative to NazFeCo(CN)6 electrodes which can be associated with a combination of structural factors, for example, a major sodium-containing phase, ∼Na0.5FeFe(CN)6 with sodium locating either at the x = y = z = 0.25 or x = y = 0.25 and z = 0.227(11) sites and an electrochemically inactive sodium-free Fe[Fe(CN)6]1−x·yH2O phase. This study demonstrates that key questions about electrode performance and attributes in sodium-ion batteries can be addressed using time-resolved in situ synchrotron X-ray diffraction studies.
Introduction
Lithium-ion batteries, since their commercialisation in the 1990's,1 have dominated the market for high energy and power portable devices.2 Emerging technologies, such as energy storage from renewable energy generation, require safer, cheaper and more reliable devices with higher power and energy densities than are currently provided by lithium-ion batteries.3,4 Ambient temperature sodium-ion batteries provide a significantly cheaper (and in principle more abundant), alternative to lithium-ion batteries.4 Unfortunately, due to the inherently heavier mass of sodium compared to lithium, sodium-ion batteries are unlikely to compete in terms of energy density for mass sensitive portable or transport applications e.g. portable electronics and electric vehicles. However, for stationary energy storage, ambient temperature sodium-ion batteries are likely to provide a cheaper and more efficient alternative to lithium-ion batteries.The limiting factors for sodium-ion battery development currently are: the small number of insertion electrodes, particularly anodes; their energy densities; and the reversibility of the sodium insertion–extraction processes. Electrodes range from selected carbons, to metal oxides, phosphates, and fluorophosphates.4,5 An alternative class of materials for insertion electrodes in sodium-ion batteries are framework-based materials.6–9 Framework materials are a combination of metal nodes and organic bridging linkers or ligands, e.g. metal–organic frameworks (MOFs)/coordination polymers10,11 extended in 3-dimensional space, and these lead to an exceptionally large array of 3-dimensional materials, with seemingly infinite combinations of metals and linker units. Such frameworks in turn exhibit a vast array of properties, such as negative thermal expansion and spin-crossover, and can be used in potential applications, such as gas separation. Cyanide bridged frameworks form an important class of framework materials showing a high level of structural flexibility.12–14 Prussian blue analogues belonging to this class of framework materials, have been applied as insertion electrodes for lithium-ion batteries,15,16 and recently they have been adapted to sodium-ion batteries.6–9
The investigations of Prussian blue analogues as electrodes in sodium-ion batteries focus on the electrochemistry and require further study in order to determine what features of frameworks are ideally suited for electrode applications.8,9 Typically the positive electrode is referred to as the sodium source in a full rechargeable sodium-ion battery, providing the initial source of sodium-ions to generate battery capacity. However, sodium is also present in the electrolyte and this can provide a certain amount of capacity for the battery. The concept of where, how and by what means sodium inserts/extracts from electrode materials can shed light on electrochemical performance and further electrode/battery development. Ideally, electrodes will be able to reversibly insert/extract large quantities of sodium, i.e. high energy densities, and this process can be cycled thousands of times with only minor structural changes, i.e. will present longevity or long lifetimes. These factors can be probed using specialised techniques, in particular in situ synchrotron powder X-ray diffraction (XRD), which has the capability to provide detail on the structure and sodium site evolution while a battery is functioning.17,18 In any sodium-ion battery, sodium will carry the charge,19 and thus it is important to know what happens to sodium, and specifically how it evolves crystallographically in the electrodes. Time-resolved in situ synchrotron XRD data provide insight on the structural reaction mechanism evolution of electrodes in a battery during charge–discharge or as they function.17,18 This is distinctly different to providing a snapshot of the equilibrated structure at equilibrium conditions as ex situ (post mortem) or pseudo in situ data provide.20,21 With sufficient time-resolution, in situ data show what happens under non-equilibrium real battery operation conditions.
The capability of time-resolved determination of sodium atomic parameters was only recently demonstrated for Na3V2O2x(PO4)2F3−2x electrodes with in situ synchrotron XRD detailing reaction mechanism, lattice and sodium site occupancy evolution as a function of charge–discharge.17,22 Considering the larger quantity of lithium-ion battery literature, only a few in situ studies describe the lithium site occupancy and evolution as a function of charge–discharge.21,23,24 Note, for lithium-ion batteries, neutron powder diffraction (NPD) is required due to the weak signal lithium generates in XRD data. Thus XRD has a distinct advantage in the study of sodium-ion batteries; it is sensitive to sodium atomic parameters, due to the larger atomic number of sodium relative to lithium. Additionally, smaller samples, e.g., simple coin cells can be probed with XRD, as compared to the larger, more challenging batteries required for in situ NPD. Limited research has been conducted utilising in situ XRD at both laboratory and synchrotron based sources on sodium batteries.17,18,25–37 There is clearly the ability to track in detail the evolution of sodium in coin cells as a function of time during charge–discharge using in situ synchrotron XRD.
In this study, we determine the sodium uptake, insertion and extraction in Prussian blue analogues, Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6, using time-resolved in situ synchrotron XRD data. We detail how cell preparation can turn the sodium-free versions of these compounds into sodium-containing compounds and thus act as a positive electrode in sodium-ion batteries. Further insight is detailed on storage time of the electrode and the influence of charging versus discharging the electrode as a first step.
Experimental
Fe[Fe(CN)6]1−x·yH2O and Fe[Co(CN)6] were synthesised according to the previously reported method.38 50 mL of a 0.1 M aqueous solution of K3[Fe(CN)6] or K3[Co(CN)6] was added dropwise to 100 mL of 0.1 M aqueous FeCl3. For Fe[Fe(CN)6]1−x·yH2O, the reagents were heated to 53 °C, while for Fe[Co(CN)6] they were kept at room temperature. The resulting mixtures were stirred at room temperature overnight to allow the precipitates to mature. The mixtures were centrifuged and the precipitates repeatedly washed with deionised water, followed by methanol, until the supernatant was colourless. Fe[Fe(CN)6]1−x·yH2O was obtained as a dark green solid and Fe[Co(CN)6] as a yellow solid.The positive electrodes were manufactured by mixing 80 wt% of the active material, 10 wt% conductive carbon (Super C65, Timcal) and 10 wt% polyvinylidenefluoride binder (PVDF, MTI Corporation). A few mL N-methylpyrrolidone (NMP, MTI Corporation) were added and the resulting slurry was stirred overnight. This slurry was then coated on aluminium foil using a notch bar. The electrode film was dried at 100 °C in a vacuum oven overnight. The electrode sheets were pressed to 100 kN using a flat plate press (MTI corporation) and dried overnight at 100 °C before transfer to the Ar-filled glovebox. Coin cells with 3 mm diameter holes in the casing and 5 mm diameter holes in the stainless spacer were used for the construction of the coin cells for the in situ measurements. The coin cells contained Na metal (∼1 mm thickness), glass fibre separators with 1 M NaPF6 in dimethyl carbonate and diethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt%) electrolyte solution. Further details regarding coin cell construction and beamline setup can be found in ref. 17, 18, 39 and 40.
:1 wt%) electrolyte solution. Further details regarding coin cell construction and beamline setup can be found in ref. 17, 18, 39 and 40.
Laboratory XRD experiments were conducted on a Panalytical X'pert MPD employing Cu Kα radiation. In situ synchrotron X-ray diffraction experiments were performed within 3–4 days after cell construction. The first Fe[Fe(CN)6]1−x·yH2O cell was initially discharged to 1 V and then charged at 0.1 mA, while the second cell was initially charged to 3.8 V at 0.2 mA held at 3.8 V for 10 minutes and then discharged to 0.1 V at 0.2 mA. The FeCo(CN)6 cell was cycled twice, by discharging to 1 V at 0.05 mA and charging to 4 V at 0.2 mA. These procedures were used to ensure sufficient information could be extracted from the limited beamtime available.
In situ synchrotron XRD data were collected on the powder diffraction beamline41 at the Australian Synchrotron with a wavelength (λ) of 0.73716(2) Å, determined using the NIST 660b LaB6 standard reference material. Data were collected continuously in 4.36 minute acquisitions on the coin cell in transmission geometry throughout the charge–discharge cycles described above. Further details about this experimental setup can be found in ref. 39 and 42. The powder diffraction beamline employs two detector positions, and each position was exposed for 2 minutes and the changeover time between positions was 0.36 minutes, resulting in an overall collection time of 4.36 minutes. These two detector positions are merged to provide a diffraction pattern. Rietveld refinements were carried out using the GSAS43 software suite with the EXPGUI44 software interface. For the in situ data, in the first dataset the lattice parameters and atomic displacement parameters (ADPs) for all atoms were refined. The ADPs were then fixed and the sodium occupancies refined. For the sequential refinements, the ADPs were kept fixed and the lattice and sodium occupancies refined.
Results and discussion
Initial structure of Fe[Fe(CN)6]1−x·yH2O in a sodium-ion cell
The Fe[Fe(CN)6]1−x·yH2O electrode in the as-constructed sodium-ion cell shows the presence of two phases that have distinctly different lattice parameters but adopt the same space-group symmetry, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and structure type. The major phase has a refined lattice parameter of a = 10.36543(27) Å and comprises 78.5(1)% of the electrode while the minor phase is a = 10.2222(3) Å and comprises 21.5(5)% of the electrode (Fig. 1 and Table 1).
m, and structure type. The major phase has a refined lattice parameter of a = 10.36543(27) Å and comprises 78.5(1)% of the electrode while the minor phase is a = 10.2222(3) Å and comprises 21.5(5)% of the electrode (Fig. 1 and Table 1).
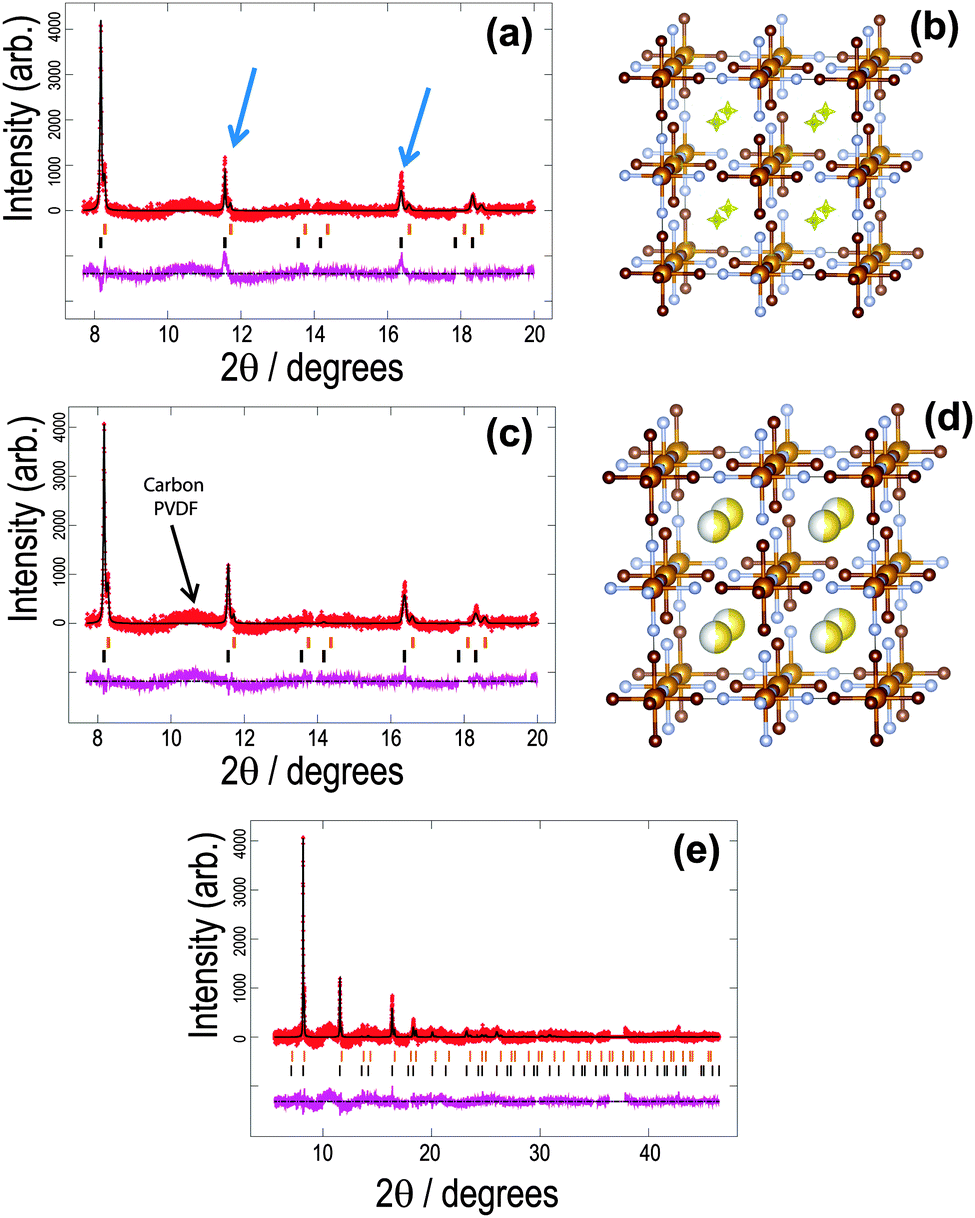 | ||
| Fig. 1 The structure of the initial cathode material in the sodium-ion battery using the vacancy-free model. Rietveld refined fit of the (a) FeFe(CN)6 and (c), (e) Na0.516(12)FeFe(CN)6 models to the synchrotron XRD data in the (a), (b) 8 ≤ 2θ ≤ 20° and (e) 6 ≤ 2θ ≤ 48° regions. Data are shown as crosses, calculated data resulting from the Rietveld refinement of the model are shown as a line through the crosses, and the difference between the observed and calculated data as the line below. Arrows indicate data and model mismatches. The vertical reflection markers are for the two FeFe(CN)6 phases present in the electrode. The crystal structure and remaining Fourier electron density (yellow) of the major (b) FeFe(CN)6 and (d) Na0.516(12)FeFe(CN)6 phase with iron in light brown, carbon in dark brown, nitrogen in blue and sodium in yellow with the shading indicating occupancy. Note in (a), (c) and (e) there are excluded regions for the sodium metal and aluminium reflections, in addition the features in the background (indicated in (c)) arise in part due to carbon-containing components in the electrode, e.g. PVDF and carbon black.47 | ||
| Atom | x | y | z | SOF | Isotropic ADPa (×100)/Å2 |
|---|---|---|---|---|---|
|
a Refined for the sample-only pattern and subsequently fixed. Space group = Fmm, a = 10.36543(27) Å, 26 refinement parameters of which 20 are background parameters, χ2 = 1.35, Rp = 2.61%, wRp = 3.33%, atomic displacement parameter (ADP), site occupancy factor (SOF). The sodium content in the formula unit would be 0.498(24) or Na0.498(24)FeFe(CN)6.
|
|||||
| Fe(1) | 0 | 0 | 0 | 1 | 2.91 |
| Fe(2) | 0.5 | 0 | 0 | 1 | 2.89 |
| C | 0.196 | 0 | 0 | 1 | 3.89 |
| N | 0.303 | 0 | 0 | 1 | 0.92 |
| Na | 0.25 | 0.25 | 0.227(11) | 0.083(4) | 6.50 |
The as-synthesized dry Fe[Fe(CN)6]1−x·yH2O powder was found to be single-phase with a refined lattice parameter of a = 10.2260(6) Å (see ESI,† Fig. S1) which corresponds to the minor phase of the electrode inside a sodium-ion cell. Before proceeding further it is important to note that there are structural features that may not be wholly captured by structural models38 in the literature for FeFe(CN)6. The simple model (termed the vacancy-free model in this paper) assumes Fe3+ is present at both metal centres and any water remaining in the pores is removed by drying and/or electrode manufacturing conditions resulting in the composition Fe[Fe(CN)6]. If these assumptions are made the major phase in the electrode shows the structural features presented in Table 2. The location of the sodium sites for the vacancy-free model is determined by investigating the Fourier difference maps, locating the site at x = y = z = 0.25 which results in a significant improvement of the fit (Fig. 1 and detailed below). The more complex model (vacancy-containing model) involves considering the potential for mixed valent Fe sites, e.g. some of Fe3+ reduced to Fe2+. In order for this to occur, vacancies are required for Fe(1), C and N sites with precedents existing in the literature,45,46 and which also show OH groups replacing CN. A further complicating factor is the potential for water to remain in the pores of the structure as Prussian blue analogues are known to absorb water. Laboratory powder XRD data fitted to the water-free vacancy-free model show mismatched observed and calculated reflection intensities (ESI,† Fig. S1). Using Fourier analysis, this leads to a model where Fe, C and N vacancies are modelled and are found to be ∼12% and O is located at the x = y = z = 0.25 site, leading to a = 10.2260(5) Å and a composition of Fe[Fe0.85(7)(C0.88(4)N0.88(4))6]·0.473(24)H2O or x ∼ 0.15, y ∼ 0.47 in Fe[Fe(CN)6]1−x·yH2O. Note, OH groups are not considered to substitute on the CN sites. Crystallographic data for the refined model and fits to the XRD data are shown in ESI,† Fig. S2 and Table S1. In this analysis of the in situ synchrotron XRD data we have utilised both models (vacancy-free and vacancy-containing) to gauge whether any differences are found.
| Atom | x | y | z | SOF | Isotropic ADPa (×100)/Å2 |
|---|---|---|---|---|---|
|
a Refined and subsequently fixed. Space group = Fmm, a = 10.36571(27) Å, 24 refinement parameters of which 20 are background parameters, χ2 = 1.34, Rp = 2.59%, wRp = 3.30%, atomic displacement parameter (ADP), site occupancy factor (SOF), Na0.516(12)FeFe(CN)6.
|
|||||
| Fe(1) | 0 | 0 | 0 | 1 | 0.57 |
| Fe(2) | 0.5 | 0 | 0 | 1 | 1.35 |
| C | 0.19 | 0 | 0 | 1 | 0.96 |
| N | 0.31 | 0 | 0 | 1 | 0.96 |
| Na | 0.25 | 0.25 | 0.25 | 0.516(12) | 0.24 |
For the first in situ synchrotron XRD dataset the structural model of the major phase is detailed in Tables 1 and 2 for each option. In the case of the vacancy-free model, the major phase intensities matched well with the 200 and 331 Fe[Fe(CN)6]1−x·yH2O reflections but were poorly fitted (under-calculated) to the 220 and 400 Fe[Fe(CN)6]1−x·yH2O reflections (indicated by the arrows in Fig. 1a). Investigating the Fourier difference maps a positive scattering intensity is located at the x = y = z = 0.25 site (Fig. 1b), placing sodium at this site (Fig. 1d) improves the fit (Fig. 1c) resulting in the refined formula of Na0.516(12)FeFe(CN)6. Interestingly for the vacancy containing model the minor phase still shows evidence of Fe(1), C and N vacancies and O on the x = y = z = 0.25 site, but for the major phase refined occupancies of Fe(1), C and N converge to 1, full occupation. Furthermore, removing the O x = y = z = 0.25 site from the calculated pattern and probing the difference Fourier densities reveals Fourier intensity in an octahedral arrangement around the x = y = z = 0.25 site (Fig. 2a). This indicates a distribution of ions/molecules along these pores in a disordered manner. Placing oxygen on these sites, refining their positions and occupancies, illustrates a large under-calculation of the electron density. The Fourier difference map shown in Fig. 2b, shows that the electron density contributed by oxygen at this site is not sufficient to capture the intensity observed. The replacement of O with Na (Fig. 2c), where Na contains more electrons or a larger electron density in Fourier maps, results in minimal remaining Fourier density and the fit to the synchrotron XRD data is shown in Fig. 2d. Clearly Na captures the electron density of the species in the pores of the framework more precisely than O in the in situ electrochemical cell. There still exists the possibility of both O and Na occupancy on this site, a mixed site however, this is unlikely as the Na+ is likely to coordinate to H2O in these pores leading to significant local strain on the framework. The refined atomic coordinates of Na are x = y = 0.25 and z = 0.227(11) and a composition of Na0.498(24)FeFe(CN)6. Notably both models provide virtually the same scenario a composition close to Na0.5FeFe(CN)6 with Na located near the centre of the pores. Where the models differ is the location of Na, either at the special position at the centre of the pore or in an octahedral arrangement surrounding this special position. In any case, both models indicate that Na is likely to be in the pores, and if this is the case then it should be possible to remove Na, i.e., use this material as a positive electrode.
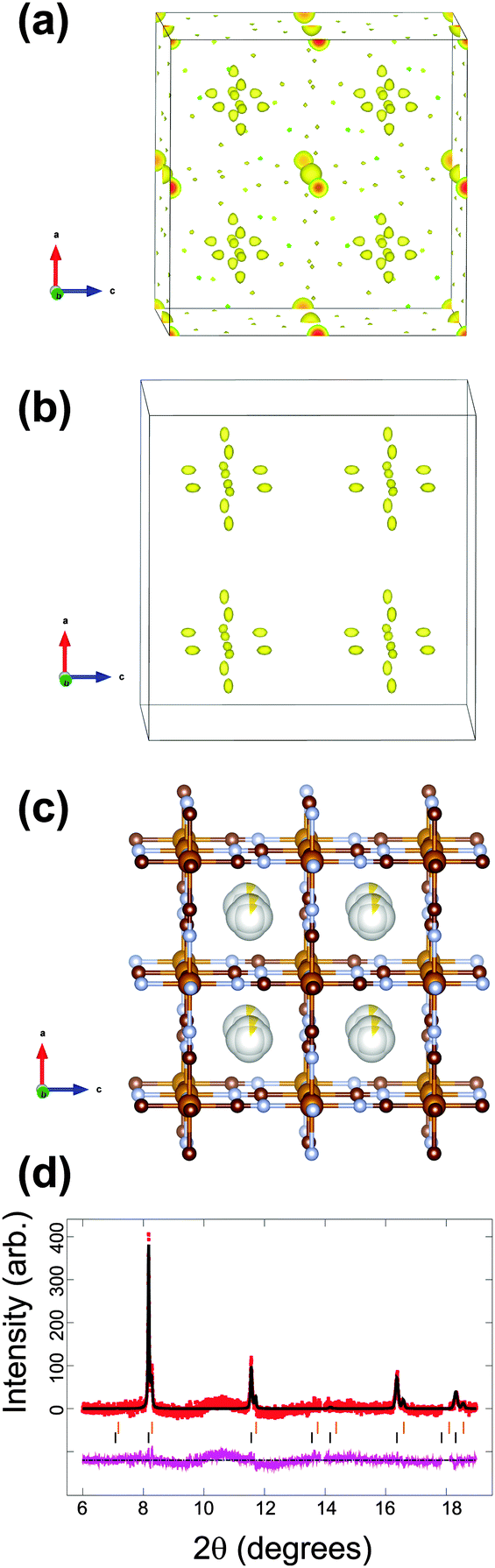 | ||
| Fig. 2 The structure of the initial cathode material in the sodium-ion battery using the vacancy-containing model. Fourier electron density (yellow) difference maps of the major phase using the (a) FeFe(CN)6 and (b) FeFe(CN)6·0.473(24)H2O models (atoms omitted for clarity). (c) The structural model best capturing the electron density with iron in light brown, carbon in dark brown, nitrogen in blue and sodium is yellow with the shading indicating occupancy. Rietveld refined fit of the (d) Na0.498(24)FeFe(CN)6 model to the synchrotron XRD data in the 8 ≤ 2θ ≤ 20° region. Data are shown as crosses, calculated data resulting from the Rietveld refinement of the model are shown as a line through the crosses, and the difference between the observed and calculated data as the line below. Note there are excluded regions for the sodium metal and aluminium reflections, in addition the features in the background arise in part due to carbon-containing components in the electrode, e.g. PVDF and carbon black.47 | ||
Therefore, the sodium-free Fe[Fe(CN)6]1−x·yH2O electrode has chemically inserted a proportion of sodium presumably from the electrolyte into its pores prior to electrochemical cycling. By placing the Fe[Fe(CN)6]1−x·yH2O electrode in a battery in contact with the electrolyte there appears to be a spontaneous reaction that allows sodium insertion into the Fe[Fe(CN)6]1−x·yH2O transforming it to a sodium-free FeFe(CN)6 (with or without water) minor phase and a sodium-containing ∼Na0.5FeFe(CN)6 (water free) major phase. The observed insertion of sodium maybe the result of either only the presence of electrolyte solution (i.e., chemical insertion) or the presence of the electrode in an electrochemical cell with an open circuit potential. It is likely that the rate of sodium uptake is dependent on time. To investigate chemical insertion, dried electrodes were soaked in electrolyte solution inside an Ar-filled glovebox for 1 and 24 h. The extracted electrodes were sealed and analysis of XRD data revealed a larger time-dependent decrease in lattice parameters of Fe[Fe(CN)6]1−x·yH2O (1 h a = 10.144(2) Å and 24 h a = 10.109(1) Å) compared to FeCo(CN)6 (1 and 24 h a = 10.192(2) Å). The slight change in lattice parameter indicates that the electrolyte solution influences the structure but other factors are required to account for the observed changes in the first in situ dataset, e.g. the electrochemical cell. The reduction in lattice parameters may suggest dehydration and further experiments are proposed where vacancy content and time dependence of sodium content changes in the electrode are probed.
Furthermore, the transformation of the electrode to the sodium-containing and sodium-free phases appears to be two-phase in character. The ∼Na0.5FeFe(CN)6 phase forms at the expense of the Fe[Fe(CN)6]1−x·yH2O phase in both models. It is interesting to note that a range of NazFeFe(CN)6 phases where say z = 0.2, 0.4 and so on were not formed. This may indicate an energetically favourable composition of z ∼ 0.5 and it may also suggest that this is the maximum amount of sodium that can be reversibly inserted/extracted. Additionally, in the vacancy containing model, the Fe, C and N vacancies and water-containing pores are segregated to the minor phase, while the major phase is vacancy and water-free. The question remains whether the minor phase is electrochemically active or acts to ‘trap’ water and vacancies allowing the majority phase to be electrochemically active.
In situ evolution of the Fe[Fe(CN)6]1−x·yH2O electrode
This study examined two Fe[Fe(CN)6]1−x·yH2O-containing batteries in situ. For the first battery, the hypothesis of whether further sodium could be electrochemically inserted into the major ∼Na0.5FeFe(CN)6 or minor FeFe(CN)6 phases were tested by discharging the cell to 1 V. No changes are observed in the FeFe(CN)6 or FeFe(CN)6·0.473(24)H2O minority phase. For ∼Na0.5FeFe(CN)6 small changes are observed in reflection positions (Fig. 3), the refined lattice parameters (volume) and sodium content (Fig. 4). A small lattice and volume expansion of 0.23(1)% and 0.68(1)% respectively with minimal change in sodium content is noted. This indicates that any further sodium insertion into this electrode is likely via surface reactions or in a disordered manner, i.e., the sodium ions do not locate on a crystallographic site in a long-range ordered manner and therefore are not detectable using XRD. This could also suggest that the current rate used for discharge does not allow sufficient time for the inserted sodium ions to locate onto crystallographic sites, i.e., in non-equilibrium conditions. On charge the volume contracts gradually and the sodium content marginally changes until the later stages of charge (from 110 to 140 minutes), where the sodium content rapidly decreases. | ||
| Fig. 3 Selected 2θ region of in situ synchrotron XRD data highlighting the evolution of the 200 reflections of the two FeFe(CN)6 phases by a colour scale and the potential profile. | ||
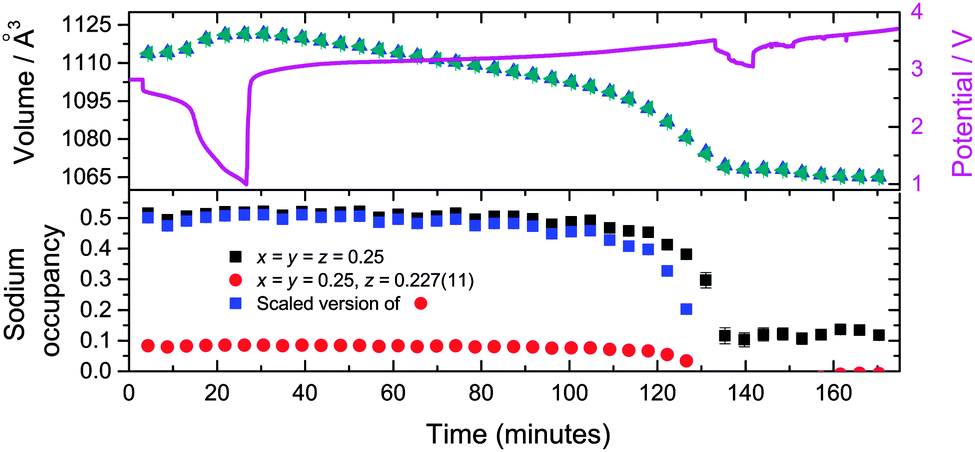 | ||
| Fig. 4 The time evolution of the sodium site occupancy of the major Na0.516(12)FeFe(CN)6 (black squares) and major Na0.498(24)FeFe(CN)6 (red circles) phases in the two models. The sodium evolution of the major Na0.498(24)FeFe(CN)6 phase (blue squares) scaled (to the corresponding formula unit) to the equivalent Na0.516(12)FeFe(CN)6 phase. The unit cell volume as a function of time for both models (triangles), with the potential profile included. | ||
In terms of the two models used, the vacancy-containing and vacancy-free models both follow the same trend in lattice and volume expansion/contraction. The only discrepancy observed between these models is between 110 and 140 minutes where the equivalent sodium content of the vacancy-containing model appears to decrease more rapidly than the vacancy-free model. The vacancy-containing model subsequently shows no sodium occupation while the vacancy free model shows on average 0.12(2) sodium occupancy. It is worthwhile noting that the vacancy-free and vacancy-containing models place sodium on different sites, with the former at the x = y = z = 0.25 site. Investigating the Fourier difference map of the vacancy-containing model, at 140 minutes there is no evidence of electron density near this position.
A second Fe[Fe(CN)6]1−x·yH2O containing battery was examined to determine whether the Na0.516(12)FeFe(CN)6 or majority phase can be cycled as a positive electrode or sodium source, extracting the 0.516(12) sodium ions by initially charging this electrode. Note this battery was stored for a longer period of time before the in situ experiment was undertaken (>1 day relative to the battery discussed above). Thus, first, the initial structure of the electrodes for the two batteries will be compared. Fig. 5 shows the Fourier difference map of the majority phase in the electrode without sodium for both models (vacancy-containing and vacancy-free). In both cases positive electron density is found at the centre of the pores at the x = y = z = 0.25 site. This differs from the 1st battery where the vacancy-containing model shows an octahedral arrangement around the x = y = z = 0.25 site. This may indicate that extended storage helps to locate sodium at the centre of the pores.
 | ||
| Fig. 5 The crystal structure and remaining Fourier electron density (yellow) of the major FeFe(CN)6 phase in the 2nd battery, with iron in light brown, carbon in dark brown and nitrogen in blue. (a) and (b) show the vacancy-free and vacancy-containing models respectively. | ||
For the initial electrode, the overall sodium content and lattice are within error in both batteries using the vacancy-containing model. The vacancy-free model shows subtle differences, slightly larger refined sodium content by 7(3)% (0.550(11) Table 3) and a 0.0020(4) Å difference in the lattice parameter. However, the major difference between the batteries appears to be the phase fractions of the constituent phases where both models show a larger phase fraction of the major phase compared in the 2nd battery relative to the 1st battery, 89.9(7)% which is 5.2(7)% larger for the vacancy-free model and 84.6(1)% which is 6.1(1)% larger for the vacancy-containing model. This evidence indicates that extended storage may generate a more sodium rich electrode or a larger proportion of the electrode that contains sodium prior to first use.
| Atom | x | y | z | SOF | Isotropic ADPa (×100)/Å2 |
|---|---|---|---|---|---|
|
a Kept consistent with first electrode. Space group = Fmm, a = 10.36367(25) Å, 24 refinement parameters of which 20 are background parameters, χ2 = 1.3, Rp = 2.57%, wRp = 3.32%, atomic displacement parameter (ADP), site occupancy factor (SOF).
|
|||||
| Fe(1) | 0 | 0 | 0 | 1 | 0.57 |
| Fe(2) | 0.5 | 0 | 0 | 1 | 1.35 |
| C | 0.19 | 0 | 0 | 1 | 0.96 |
| N | 0.31 | 0 | 0 | 1 | 0.96 |
| Na | 0.25 | 0.25 | 0.25 | 0.550(11) | 0.24 |
On charging the ∼Na0.5FeFe(CN)6 electrode, a reduction in the lattice and volume is observed (Fig. 6). Therefore, the sodium in the electrode from the construction process can be used as a sodium source – it behaves as a positive electrode. Notably, similar to the battery above, the sodium content is fairly stable using both the vacancy-free and vacancy-containing models, decreasing marginally, until the later stages of charge, 36 to 52 minutes where the sodium content drops rapidly. This may suggest that the actual sodium content in the electrode is higher than 0.550(11) or 0.520(12) per formula unit, but the reminder is in the form of disordered sodium that is not detected by diffraction techniques which are sensitive to the long-range ordered occupancy of crystallographic sites.
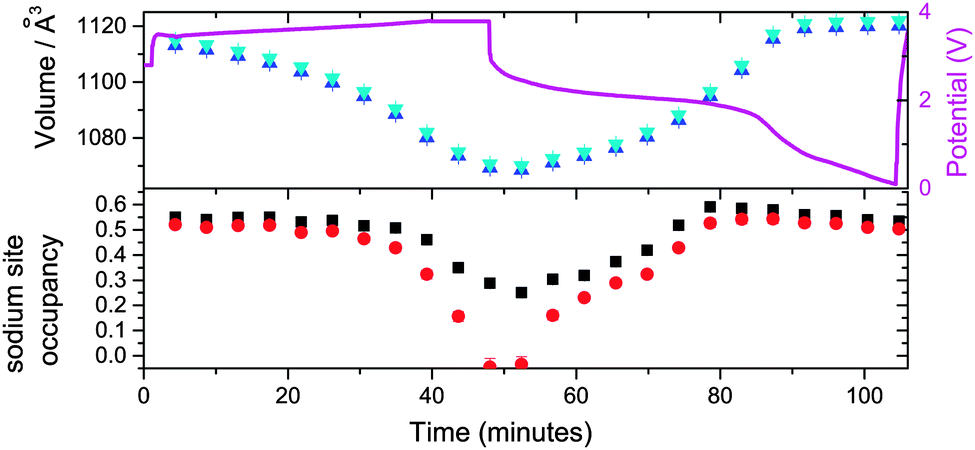 | ||
| Fig. 6 The evolution of the sodium site occupancy for the vacancy-free (black) and vacancy-containing models (red), and unit cell volume (blue and cyan for the vacancy-free and vacancy-containing models respectively) of the major ∼Na0.5FeFe(CN)6 phase as a function of time, with the potential profile included. | ||
On discharge, there appears to be a pseudo-plateau region around 2 V that leads to most of the ordered sodium on the x = y = z = 0.25 site re-inserting and the lattice parameter and volume reaching their respective maximum values. These values are close to the fresh electrode. For example, comparing 0 and 87 minutes or 4.33 and 1.31 V, sodium contents are 0.550(11) and 0.578(15), lattice parameters are 10.3637(3) and 10.3707(4) Å, and volumes are 1113.12(8) and 1115.38(12) Å3 respectively for the vacancy-free model. Below 1.3 V the sodium content, lattice parameter and volume show minimal change in both models. This may indicate that this positive electrode should be cycled to ∼1.2 V in order to re-insert all the crystallographically ordered sodium that was extracted during charge. Additionally, in both Fe[Fe(CN)6]1−x·yH2O batteries using both models, the minor sodium-free FeFe(CN)6 phase is inactive during charge–discharge cycling.
Kinetics of sodium insertion/extraction in Fe[Fe(CN)6]1−x·yH2O
With time-resolved data, the rates and evolution of various processes can be compared. The volume evolution in Fig. 6 of the NazFeFe(CN)6 electrode appears linear during charge, but closer inspection indicates a more complex relationship. This is correlated to the non-uniform removal of sodium from the crystallographic site. Linear fits of the two distinct sodium regions during charge, minimal sodium removal from the crystallographic site (between 0 and 31 minutes) and dramatic removal from this crystallographic site (between 36 and 52 minutes) show rate constants of −0.0010(4) SOF units per minute (R2 = 0.53) and −0.016(2) SOF units per minute (R2 = 0.96) for the vacancy-free model. There is at least an order of magnitude faster removal of sodium from the crystallographically ordered site during the last part of charge starting at 3.73 V than the initial charge process. Linear fitting of the volume in these regions show similar trends, with the rate constants derived from the linear fits, increasing by more than a factor of two from −0.68(5) to −1.5(1) Å3 per minute (R2 = 0.96). Similar trends in the volume rate constant are noted on discharge, with a component corresponding to the filling of the x = y = z = 0.25 crystallographic site and another presumably to disordered sodium in the structure. These trends are mirrored in the vacancy-containing model, with a more dramatic change in the last part of charge and first part of discharge. A rate constant of −0.034(4) SOF units per minute (R2 = 0.97) is noted during the last part of charge which is double that of the vacancy-free model.Initial structure of FeCo(CN)6 in a sodium-ion cell
For FeCo(CN)6, a dramatically different initial phase composition is found with phase 1 showing the larger lattice parameter of a = 10.36663(19) Å and comprising 55.7(2)% of the electrode and phase 2 a smaller lattice parameter of a = 10.28105(22) Å and comprising 44.3(2)% of the electrode. Thus one phase does not dominate the composition of the electrode. Notably, the single-phase raw material has a smaller lattice parameter then either of the phases in the coin cell, with a = 10.2127(3) Å and there is no indication of vacancies in this structure (see ESI,† Fig. S1). This initially indicates that both phases of the electrode in the coin cell contain sodium and intuitively phase 1 with the larger lattice parameter is expected to contain more sodium. Using a sodium-free model clearly presents mismatches between the refined model and the data, indicated by the arrows in Fig. 7a. The Fourier difference maps of each phase illustrate regions of positive electron density (Fig. 7b and c). Phase 1 is similar to the majority phase in Fe[Fe(CN)6]1−x·yH2O, with the electron density situated at the x = y = z = 0.25 site, while phase 2 differs in electron density with the site corresponding to x = y = 0.25, z = 0.2. The site in phase 2 produces an octahedral distribution around the x = y = z = 0.25 site similar to the arrangement found in the vacancy-containing model of the first Fe[Fe(CN)6]1−x·yH2O battery. The corresponding formula for phase 1 is Na0.224(12)FeCo(CN)6, while phase 2 is Na0.108(12)FeCo(CN)6 and the fit of these models to the synchrotron XRD data is shown in Fig. 7d and the crystallographic details of the two phases presented in Table 4. The total long-range ordered sodium content in both phases is 0.332(17) which is lower than either of the NazFeFe(CN)6 electrodes within a coin cell prior to electrochemical cycling. This indicates that the capacity obtained from the long-range ordered sodium content prior to cycling is smaller in the FeCo(CN)6 electrodes relative to Fe[Fe(CN)6]1−x·yH2O.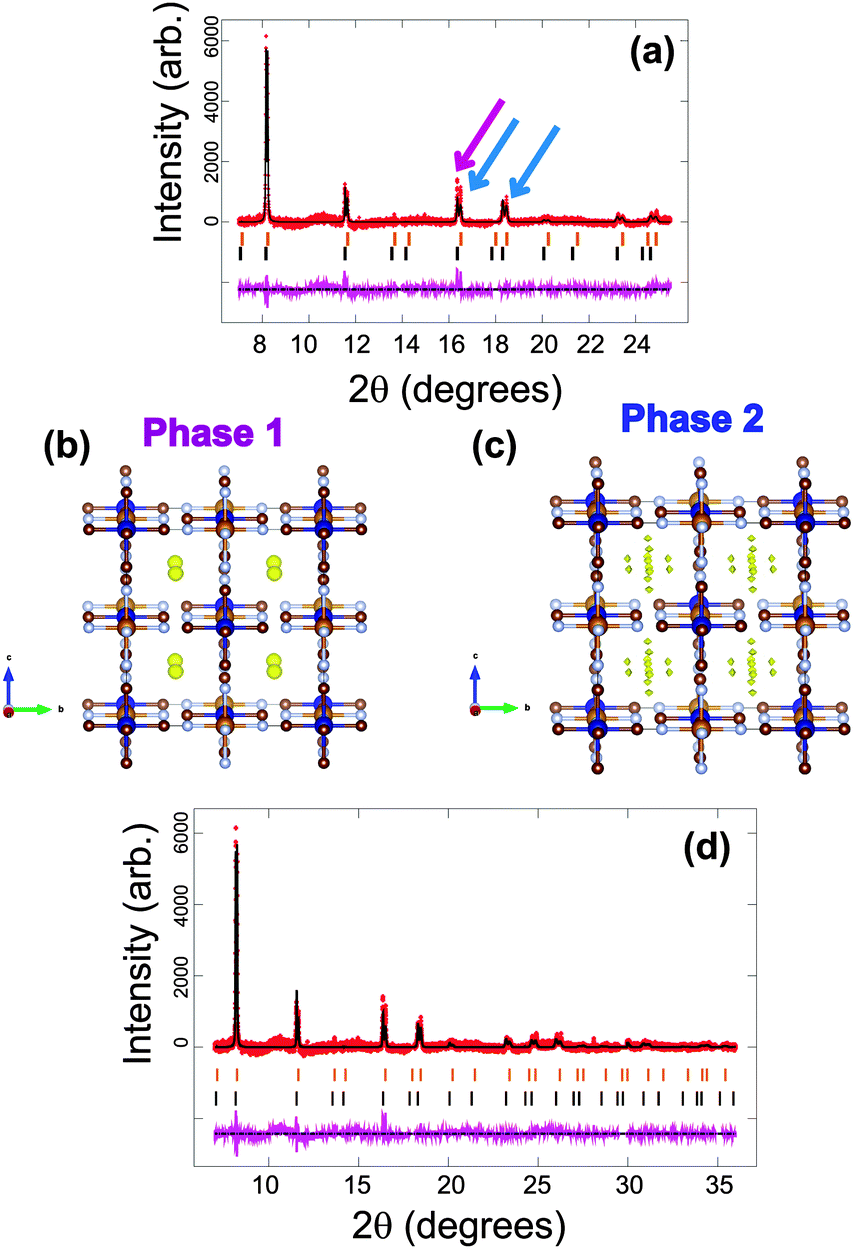 | ||
| Fig. 7 Rietveld refined fit of the (a) FeCo(CN)6 and (d) Na0.332(17)FeCo(CN)6 models to the synchrotron XRD data in the (a) 8 ≤ 2θ ≤ 25° and (d) 6 ≤ 2θ ≤ 36° regions. Data are shown as crosses, calculated data resulting from the Rietveld refinement of the model are shown as a line through the crosses, and the difference between the observed and calculated data as the line below. Arrows indicate data and model mismatches. The vertical reflection markers are for the two FeCo(CN)6 phases present in the electrode. The crystal structure and remaining Fourier electron density (yellow) of the two (b), (c) FeCo(CN)6 phases with iron in light brown, cobalt in dark blue, carbon in dark brown and nitrogen in light blue. | ||
| Atom | x | y | z | SOF | Isotropic ADPa (×100)/Å2 |
|---|---|---|---|---|---|
|
a Refined and subsequently fixed. Space group = Fmm, a = 10.36663(19), 10.28105(22) Å, 26 refinement parameters of which 20 are background parameters, χ2 = 1.43, Rp = 2.70%, wRp = 3.54%, atomic displacement parameter (ADP), site occupancy factor (SOF). The sodium sites for phase 2 equates to 0.108(12) in terms of the phase 1 SOF.
|
|||||
| Co | 0 | 0 | 0 | 1 | 0.44 |
| 0.24 | |||||
| Fe | 0.5 | 0 | 0 | 1 | 0.05 |
| 0.26 | |||||
| C | 0.19 | 0 | 0 | 1 | 0.28 |
| 0.16 | |||||
| N | 0.31 | 0 | 0 | 1 | 0.28 |
| 0.16 | |||||
| Na | 0.25 | 0.25 | 0.25 | 0.224(12) | 0.25 |
| 0.20 | 0.027(3) | 0.10 | |||
In situ evolution of the FeCo(CN)6 electrode
Considering the evolution of the NazFeCo(CN)6 phases during discharge–charge, there is no apparent change with initial discharge, e.g. no change in the structure with further insertion of sodium. This is evidenced by the 200 reflections of NazFeCo(CN)6 in Fig. 8 and the refined structural parameters in Fig. 9 prior to 65 minutes showing virtually no change. Similar to NazFeFe(CN)6 this indicates that any charge storage or further sodium insertion occurs either in a disordered manner or on the surface of the electrode/particles which is not resolved with synchrotron XRD. Upon charge phase 1, Na0.224(12)FeCo(CN)6 shows the largest change in the lattice parameter and volume, both of which decrease by Δa = 0.0530(3) Å and Δvolume = 17.0(1) Å3. A decrease in the lattice and volume is also noted for phase 2 but to a smaller extent by Δa = 0.0179(3) Å and Δvolume = 5.7(1) Å3. The charge regions are shaded blue in Fig. 9. Phase 1 appears to converge with phase 2, see Fig. 8, but closer inspection of the XRD patterns clearly shows two distinct reflections corresponding to each phase throughout the electrochemical cycling performed in this experiment. The 2nd discharge shows an increase in lattice and volume for both phases of similar extents (phase 1 increases by Δa = 0.0122(3) Å and Δvolume = 3.9(1) Å3 while phase 2 increases by Δa = 0.0189(4) Å and Δvolume = 6.0(1) Å3), potentially suggesting similar levels of sodium insertion in both phases. Additionally, the changes in phase 2 seem to be of the same extent suggesting a maximum reversible amount of the structural change within this voltage window. However, the lattice and volume of phase 1 remains overall higher than phase 2. The 2nd charge repeats the behaviour for the 1st charge noted above. Overall, phase 1 contains more sodium initially, but over the course of the experiment the lattice and volume parameters appear come closer to the phase 2 values although not completely overlapping. Both phases are active in the electrochemical process, with phase 1 showing slightly larger changes in the lattice and volume during charge–discharge.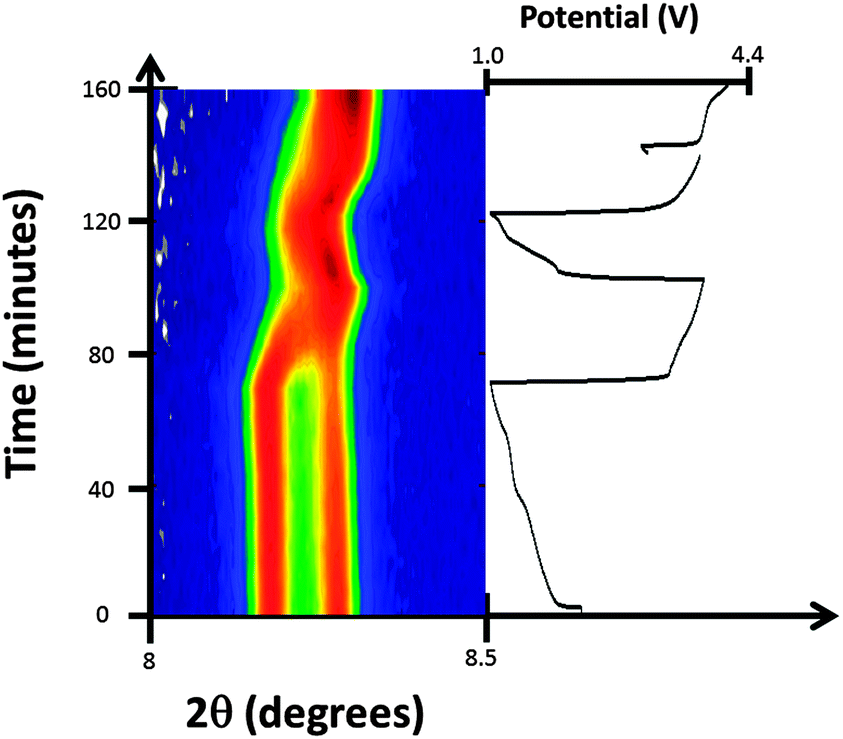 | ||
| Fig. 8 Selected 2θ region of in situ synchrotron XRD data highlighting the evolution of the 200 reflections of the two FeCo(CN)6 phases by a colour scale and the potential profile. | ||
 | ||
| Fig. 9 The evolution of the unit cell volume of the NazFeCo(CN)6 phases as a function of time, with the potential profile included, where the black and red symbols represent phase 1 and phase 2, respectively. | ||
Comparison of Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6
In both Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6, sodium insertion occurs during the construction of the coin cell batteries. The fact that Fe[Fe(CN)6]1−x·yH2O can be charged initially suggests the electrode is indeed sodium-inserted FeFe(CN)6 as opposed to water-inserted FeFe(CN)6. In addition, the presence of the second phase in Fe[Fe(CN)6]1−x·yH2O corresponding to dry (or vacancy-containing) Fe[Fe(CN)6]1−x·yH2O confirms that any water and vacancy that is present in the pores of the electrodes is limited to this inactive phase. The NazFeFe(CN)6 electrode contains more sodium than the NazFeCo(CN)6 electrode and thus shows higher capacity, both initially and over a number of cycles. Although, both electrodes contain two phases, these phases are different, with the NazFeFe(CN)6 electrode showing a sodium-containing and an inactive sodium-free phase, while the NazFeCo(CN)6 electrode shows a sodium-rich and sodium-poor phase both of which are electrochemically active. This phase distribution, the resulting phase evolution during charge–discharge and the amount of sodium each electrode can reversibly insert/extract are directly related to the battery capacity and performance parameters.It is interesting to note the shrinking of the NazFeCo(CN)6 lattice/volume (Fig. 9) relative to the NazFeFe(CN)6 lattice/volume (Fig. 6). Although this is not a direct comparison due to the different number of cycles and procedure undertaken first between the batteries, a result of limited beamtime, it may shed light on the differences between the electrodes. Typically the NazFeCo(CN)6 electrode shows lower capacities relative to the NazFeFe(CN)6 electrode (see ESI,† Fig. S4–S7), thus the reduction in the lattice/volume is likely to correspond to lower insertion of sodium into the electrode as shown in Fig. 9. Other factors that may contribute to this observation include irreversible processes at the electrode and CN vibrations similar to that found during negative thermal expansion of this material.12–14
Initial studies were performed to explore the applicable voltage ranges and the longevity of the electrode, i.e. does cycling to 1.2 V enhance the lifetime of the electrode or is cycling to 0.1 or 1 V more optimal? This was undertaken for both Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6. The key findings were the same for both chemistries, the longevity, or maintenance of battery capacity and cyclability of the battery, is optimal if the batteries containing these electrodes are discharged first to 1 V and then charged. Discharging to 0.1 V renders them inoperative after a few cycles and charging first also limits battery lifetime (see ESI,† Fig. S3–S7). In situ synchrotron XRD data of the 2nd cycle of the NazFeFe(CN)6 battery shown in Fig. 6, which was discharged to 0.1 V in the 1st cycle, shows minimal changes that correspond to minimal capacity (ESI,† Fig. S3c). Therefore, a discharge to 1 V as the first step appears to be beneficial for battery performance, which presumably acts to activate the electrode, although further work is required to determine precisely why this is so.
Conclusions
Both Fe[Fe(CN)6]1−x·yH2O and FeCo(CN)6 electrodes insert sodium during the construction of sodium-ion batteries forming phases that contain long-range ordered sodium and from which sodium extraction can be performed. Both electrodes phase separate, with Fe[Fe(CN)6]1−x·yH2O forming a major sodium-containing phase and a minor sodium-free phase which is equivalent to the as-prepared powder, while FeCo(CN)6 forms a sodium-rich and sodium-poor phase in approximately equal quantities. The Fe[Fe(CN)6]1−x·yH2O electrode features a larger overall long-range ordered sodium content which is situated in the major phase at the x = y = z = 0.25 or x = y = 0.25 and z = 0.227(11) sites depending on whether a vacancy-free or vacancy-containing model is used to for the initial structure. Storage of the battery appears to converge the sodium ions to the x = y = z = 0.25 site. In the FeCo(CN)6 electrode, sodium is located on the x = y = z = 0.25 site for the sodium-rich phase and on the x = y = 0.25, z = 0.2 site for the sodium-poor phase. Initial discharge or insertion of further sodium into the NazFeM(CN)6 electrode does not result in the insertion of further long-range ordered sodium. In Fe[Fe(CN)6]1−x·yH2O only the sodium-containing major phase is electrochemically active while in FeCo(CN)6 both phases are electrochemically active. It appears that a minor concentration of an inactive sodium-free phase in these Prussian blue analogues is beneficial for battery performance, presumably assisting in the electronic conductivity of the electrode. Following sodium in such frameworks opens up the possibility of understanding why certain frameworks work better as electrodes in sodium-ion batteries relative to others and what structural and phase composition features may be necessary to maximise performance of such frameworks.Acknowledgements
Dr Sharma would like to thank AINSE Ltd for providing support through the research fellowship scheme. A part of this research was undertaken on the powder diffraction beamline at the Australian Synchrotron, Victoria, Australia.Notes and references
- T. Nagaura and K. Tozawa, Prog. Batteries Sol. Cells, 1990, 9, 209 CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, J. Power Sources, 2011, 196, 6688–6694 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342–3345 CAS.
- C. Combelles, M. Ben Yahia, L. Pedesseau and M.-L. Doublet, J. Phys. Chem. C, 2010, 114, 9518–9527 CAS.
- H. Lee, Y. I. Kim, J. K. Park and J. W. Choi, Chem. Commun., 2012, 48, 8416–8418 RSC.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- X. Wu, W. Deng, J. Qian, Y. Cao, X. Ai and H. Yang, J. Mater. Chem. A, 2013, 1, 10130–10134 CAS.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734–777 CrossRef PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- K. W. Chapman, P. J. Chupas and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 15630–15636 CrossRef CAS PubMed.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- A. E. Phillips, A. L. Goodwin, G. J. Halder, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 1396–1399 CrossRef CAS PubMed.
- A. Eftekhari, J. Power Sources, 2004, 126, 221–228 CrossRef CAS PubMed.
- P. J. Kulesza and Z. Galus, Electrochim. Acta, 1997, 42, 867–872 CrossRef CAS.
- P. Serras, V. Palomares, T. Rojo, H. E. A. Brand and N. Sharma, J. Mater. Chem. A, 2014, 2, 7766–7779 CAS.
- N. Sharma, P. Serras, V. Palomares, H. E. A. Brand, J. Alonso, P. Kubiak, M. L. Fdez-Gubieda and J. M. Rojo, Chem. Mater., 2014, 25, 4917–4925 Search PubMed.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 CAS.
- N. Sharma, X. Guo, G. Du, Z. Guo, J. Wang, Z. Wang and V. K. Peterson, J. Am. Chem. Soc., 2012, 134, 7867–7873 CrossRef CAS PubMed.
- N. Sharma, D. Yu, Y. Zhu, Y. Wu and V. K. Peterson, Chem. Mater., 2013, 25, 754–760 CrossRef CAS.
- P. Serras, V. Palomares, J. Alonso, N. Sharma, J. M. Lopez del Amo, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2013, 25, 4917–4925 CrossRef CAS.
- W. K. Pang, N. Sharma, V. K. Peterson, J.-J. Shiu and S. H. Wu, J. Power Sources, 2014, 246, 464–472 CrossRef CAS PubMed.
- W. K. Pang, V. K. Peterson, N. Sharma, J.-J. Shiu and S. H. Wu, Chem. Mater., 2014, 26, 2318–2326 CrossRef CAS.
- F. Sauvage, L. Laffont, J. M. Tarascon and E. Baudrin, Inorg. Chem., 2007, 46, 3289–3294 CrossRef CAS PubMed.
- X. Yu, H. Pan, W. Wan, C. Ma, J. Bai, Q. Meng, S. N. Ehrlich, Y.-S. Hu and X. Q. Yang, Nano Lett., 2013, 13, 4721–4727 CrossRef CAS PubMed.
- H. Chen, Q. Hao, O. Zivkovic, G. Hautier, L.-S. Du, Y. Tang, Y.-Y. Hu, X. Ma, C. P. Grey and G. Ceder, Chem. Mater., 2013, 25, 2777–2786 CrossRef CAS.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- M. Guignard, C. Didier, J. Darriet, P. Bordet, E. Elkaim and C. Delmas, Nat. Mater., 2013, 12, 74–80 CrossRef CAS PubMed.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- C. Didier, M. Guignard, J. Darriet and C. Delmas, Inorg. Chem., 2012, 51, 11007–11016 CrossRef CAS PubMed.
- M. Sathiya, K. Hemalatha, K. Ramesha, J.-M. Tarascon and A. S. Prakash, Chem. Mater., 2012, 24, 1846–1853 CrossRef CAS.
- C. Vidal-Abarca, J. M. Ateba Mba, C. Masquelier, J. L. Tirado and P. Lavela, J. Electrochem. Soc., 2012, 159, A1716–A1721 CrossRef CAS PubMed.
- F. Tournadre, L. Croguennec, P. Willmann and C. Delmas, J. Solid State Chem., 2004, 177, 2803–2809 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS PubMed.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef CAS PubMed.
- S. Patoux, G. Rousse, J.-B. Leriche and C. Masquelier, Chem. Mater., 2003, 15, 2084–2093 CrossRef CAS.
- A. Kumar, S. M. Yusuf and L. Keller, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 054414 CrossRef.
- W. R. Brant, S. Schmid, G. Du, Q. Gu and N. Sharma, J. Power Sources, 2013, 244, 109–114 CrossRef CAS PubMed.
- R. J. Gummow, N. Sharma, R. Feng, G. Han and Y. He, J. Electrochem. Soc., 2013, 160, A1856–A1862 CrossRef CAS PubMed.
- K. S. Wallwork, B. J. Kennedy and D. Wang, AIP Conf. Proc., 2007, 879 CrossRef CAS PubMed.
- R. A. Leising, M. J. Palazzo, E. S. Takeuchi and K. J. Takeuchi, J. Electrochem. Soc., 2001, 148, A838–A844 CrossRef CAS PubMed.
- A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory Report LAUR 86-748, 1994.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- T. Godfrey and S. Schmid, manuscript in preparation.
- J. Agrisuelas, J. J. García-Jareno and F. Vicente, J. Phys. Chem. C, 2012, 116, 1935–1947 CAS.
- N. Sharma, G. Du, A. J. Studer, Z. Guo and V. K. Peterson, Solid State Ionics, 2011, 199–200, 37–43 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld-refined fits of structural models to XRD data, Fourier difference maps, crystallographic table for the vacancy-containing model, electrochemical cycling curves under various conditions (e.g. cutoff potentials, charging first, discharging first). See DOI: 10.1039/c4cp02676d |
| This journal is © the Owner Societies 2014 |