
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge transfer properties of two polymorphs of luminescent (2-fluoro-3-pyridyl)(2,2′-biphenyl)borinic 8-oxyquinolinate†
Grzegorz
Wesela-Bauman
*ab,
Sergiusz
Luliński
a,
Janusz
Serwatowski
a and
Krzysztof
Woźniak
*bc
aPhysical Chemistry Department, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warszawa, Poland
bLaboratory of Crystallochemistry, Department of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warszawa, Poland. E-mail: grzegorz.wesela@chem.uw.edu.pl; kwozniak@chem.uw.edu.pl
cFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089 Warszawa, Poland
First published on 10th September 2014
Abstract
Single crystal X-ray structures of two polymorphs of (2-fluoro-3-pyridyl)(2,2′-biphenyl)borinic 8-oxyquinolinate: orthorhombic (space group Pca21), and triclinic (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) have been established and analysed. A fast rate of crystallization results in the orthorhombic polymorph, whereas slow crystallization gives the triclinic polymorph. Physicochemical and theoretical results prove that both polymorphs form similar crystals with very similar geometry of molecules. The main differences between both forms are intermolecular interactions and their impact on the charge transporting properties of both polymorphs which was evaluated through Marcus theory. The orthorhombic polymorph is a slightly more effective electron and hole transporting material than the other polymorph. In both forms the CH⋯π interactions contributed the most to the CT properties. Small changes in the molecular geometry of moieties in both polymorphs affect their molecular energies significantly.
) have been established and analysed. A fast rate of crystallization results in the orthorhombic polymorph, whereas slow crystallization gives the triclinic polymorph. Physicochemical and theoretical results prove that both polymorphs form similar crystals with very similar geometry of molecules. The main differences between both forms are intermolecular interactions and their impact on the charge transporting properties of both polymorphs which was evaluated through Marcus theory. The orthorhombic polymorph is a slightly more effective electron and hole transporting material than the other polymorph. In both forms the CH⋯π interactions contributed the most to the CT properties. Small changes in the molecular geometry of moieties in both polymorphs affect their molecular energies significantly.
Introduction
Borinic systems are of interest for many groups in the world, since they are promising materials for preparation of luminescent layers in organic light emitting diodes (OLEDs).1–7 Many of the recent papers describing applications of borinic systems in OLED devices are focused mainly on polymeric borinic derivatives.1,8–14 Such systems have excellent properties, but it is very hard to find out how the molecular or supramolecular structure of an organoboron polymer affects the properties of the diode. This is mainly due to the statistical character of the polymer but also because of lack of methods which would supply precise and accurate results of determination of their supramolecular arrangement. Coordination polymers with borinic units have not been used for application as light emitters, so far. On the other hand, studies of small molecular emitters allow for a precise determination of the structure–property relationships at the molecular level and help in the design of new interesting systems.15,16Knowledge of the influence of well-defined motifs and contacts on the properties of particular crystals should give us a better understanding of processes which take place in the diode. This is especially important because many authors were able to prove correlation between intermolecular contacts (especially via π interactions) present in their structures and optical and charge transport properties obtained for diodes. Hence, they were able to establish a structure–property relationship at the supramolecular level.16–18 The discussion in those papers was focused on charge transport properties as a subject of interest from the standpoint of optoelectronic application.19
The Marcus theory20–22 was utilised in order to establish connection between known molecular and crystal structures and observed or even predicted charge transport properties. In this theory the charge transport (CT) rate constant, kCT, can be evaluated using eqn (1):
 | (1) |
Recently, we have focused on the synthetic, physicochemical and theoretical evaluation of the properties of non-polymeric borinic 8-oxyquinolinates.23,24 In the process of preparation of our systems for creating surfaces for OLED device we have learned that one of our compounds an aromatic heteroleptic borinic system (2-fluoro-3-pyridyl)(2,2′-biphenyl)borinic 8-oxyquinolinate 1 (Scheme 1) crystallizes in two, if not more, crystallographically different polymorphs.
 | ||
| Scheme 1 Molecular structure of 1. | ||
Polymorphism has attracted much attention recently. The term “polymorphism” means that a given compound forms more than one solid-state form.25,26 Polymorphism is important in many fields e.g., drug design or crystal engineering, pharmaceutical industry, etc. As a consequence of polymorphism, the properties of a given compound in the solid state vary in different crystallographic forms.27–29 As a result we present, to the best of our knowledge, the very first example of polymorphism in the group of aromatic borinic 8-oxyquinolinate complexes with a detailed discussion of the influence of crystallographic motifs on the charge transport properties.
From the Marcus theory, one knows that, in this case, ΔGo = 0 and only the reorganization energy and electronic matrix element (charge transfer integral) do vary. The influence of the inner reorganisation energies on the CT properties were presented in a series of papers.30–34 However, in this paper we present a single molecular system in different crystallographic arrangements. Hence, the reorganization energy is a constant and for 1 it was calculated in our previous paper23 (for hole and electron transport the values of inner reorganization energies are equal to Λhole = 0.25 eV and Λelectron = 0.42 eV, respectively). These calculations indicate that our system is a hole transporting material. However, it was proven for the tris(8-oxyquinolinate)aluminium complex and for borinic 8-oxyquinolinate systems that the charge transfer integrals are more important for a proper assignment of the CT properties.23,35 The analysis of the charge transfer integrals can help tracing the influence of the intermolecular interactions on the CT properties.36–41
Experimental section
General remarks
All materials were received from Aldrich and used without further purification. Solvents used for the synthesis and analysis were distilled and, subsequently, kept under an argon atmosphere.Synthesis and analysis
The studied compound was synthesized according to the procedure published in our previous paper.23 The synthetic procedure, NMR and UV-Vis spectra for 1 are described therein.Crystal growth
Single crystals of the orthorhombic polymorph were obtained after 2–3 days of evaporation from a concentrated acetone solution, whereas crystals of the triclinic polymorph required a week to grow from a concentrated acetone solution. The volume of used solution was ca. 5 ml, and solutions were kept under ambient conditions. Crystallization was done in small vials covered with parafilm for slow evaporation. Faster crystallization was performed with perforated parafilms or without the use of parafilm cover. Crystals of both forms were also obtained after evaporation from dichloromethane, diethyl ether or THF from a concentrated solution of 1. Crystals obtained from acetone solutions were used for further studies as the quality of these crystals exceeded the quality of those obtained from other solvents. It seems that long crystallization of the triclinic polymorph makes it hard to obtain good quality single crystals, as the possibility of insertion of defects or twining increases. Low quality crystals had their diffraction pattern full of inconsistent reflexions which could not been indexed.X-ray single crystal data collection, reduction and refinement
Single crystal X-ray measurement for the triclinic polymorph was performed on a Kuma KM4CCD κ axis diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and an Oxford Cryostream cooling device. Data reduction and analysis were carried out using the CrysAlisPro program.42Diffraction data for the orthorhombic polymorph were collected on a Bruker APEX II Ultra diffractometer equipped with a TXS rotating anode and a graphite-monochromated MoKα radiation, λ = 0.71073 Å. The data were integrated using SAINT and an absorption correction was performed using the SADABS program.43 Both data sets were restricted to 0.7 Å−1 resolution in order to have a meaningful comparison of the geometries obtained from the experiments.
The structures were solved using the SUPERFLIP program implemented in CRYSTALS.44 The independent atom model (IAM) refinements based on F2 were performed using the CRYSTALS package with I > −3.0σ(I) cut-off. Reflections affected by the beam-stop were carefully removed from the refinement. In all cases the Chebychev (F2) weights were applied. Atomic scattering factors in their analytical form were taken from the International Tables for Crystallography.45 All non-hydrogen atoms were refined anisotropically and all of the hydrogen atoms were placed in idealized positions within the riding model for atomic displacement parameters (ADPs) (with UHiso = 1.2·UCeq). All hydrogen atoms were clearly visible on the difference density maps. Weighted R factors (wR2) and all goodness-of-fit (GooF) values are based on F2. Conventional R factors are based on F with F set to zero for negative F2. The Fo2 > 2σ(Fo2) criterion, adopted form Shelx, was used only for calculating R factors and is not relevant to the choice of reflections for the refinement. The R factors based on F2 are about twice as large as those based on F. Cartesian coordinates, atomic displacement parameters, bond lengths and angles are included in the ESI.†
CCDC 988296 and 988297 contain the supplementary crystallographic data (CIF files) for crystals studied in this work (for compounds I-ortho and II-tric, respectively).
X-ray powder diffraction
X-ray powder diffraction patterns were measured on a Bruker AXS WAXS D8 Discover powder diffractometer (Cu Kα radiation, λ = 1.540 Å) equipped with a VANTEC detector. Data sets were collected at room temperature within the 2θ range from 3° to 80° with a step size of 0.0061° and a counting time of 0.5 s per step. The measured patterns are included in the ESI.† Data manipulation was done using the DiffractWD program.46DSC measurements
Differential scanning calorimetric (DSC) measurements were performed on a DSC Q200 from TA Instruments. For the DSC measurements the same samples were used as for the X-ray powder diffraction data collection (mI-ortho = 1.800 mg; mII-tric = 2.2400 mg). Data were collected in two cycles of heating and cooling with the rate of heating equal to 10 °C min−1. DSC data are included in the ESI.†Theoretical calculations and visualization
Geometry optimizations for compound 1 were performed using experimental X-ray geometry (the most stable geometry) as a starting points at the RB3LYP/6-31+g(d,p) and RB97D/6-31+g(d,p) levels of theory.47–50 B97D potential was used for the correct evaluation of the energies of weak interactions. Orbital energies of the interacting molecules in the crystal structures of 1 were calculated at the same level of theory. In all cases C–H bond lengths were adjusted to standard neutron distances (1.083 Å) prior to optimization/single point calculations.51 Tight convergence criteria (opt = tight) were used along with high precision integrals (int = UltraFine) in order to obtain good quality wave functions. Normal convergence criteria and default integrals were used for the constrained θ – scan. Wave functions were calculated without use of symmetry constraints (nosymm). All calculations involving isolated molecules were performed using the Gaussian09 suits of programs.52 Cartesian coordinates for all calculations (including energy scans) are included in the ESI.†PIXEL calculations
Lattice energies were calculated using the OPiX package53–56 based on geometries taken from the X-ray experiments with C–H bonds adjusted to the standardized neutron values (1.083 Å). Electron density distributions were calculated using Gaussian09 at the recommended MP257/6-31G** level of theory. The obtained electron densities were analysed using the PIXEL module, which allows for evaluation of lattice energies. Crystal lattice energies and crystal contacts can be evaluated with total energies, and contribution to total energies (Etot) from electrostatic (Eelstat), polarization (Epol), dispersion (Edisp), and repulsion energies (Erep). A radius of 20 Å was used as a cut-off for lattice energy calculations.Results and discussion
Crystallization
The crystal structures of two polymorphic forms of 1 have been determined by using single crystal X-ray diffraction. The crystals of 1 were obtained from concentrated solutions of acetone. The rate of crystallization was the key, and the higher symmetry system (orthorhombic, I-ortho) crystallized during fast evaporation of acetone (kinetic control). The lower symmetry system (triclinic, II-tric) needed ca. a week to grow (thermodynamic control). On the basis of the Ostwald's rule26 we assumed that I-ortho, and II-tric are kinetic and thermodynamic polymorphs, respectively. However, the DSC measurements show that both forms melt at the same temperatures in two cycles of heating and cooling (219.9 °C/217.3 °C and 219.4 °C/217.2 °C for I-ortho and II-tric, respectively). It is worth mentioning that both forms were also obtained after crystallization from dichloromethane and other solvents, although the quality of those crystals was quite poor, especially the II-tric form. The rate of crystallization was determining which polymorph we were able to obtain, irrespectively of the used solvent. Crystals of both forms presented the same green colour which may suggest that that polymorphism has not affected the optical properties greatly. However, for having absolute confidence, the UV-Vis measurements for both forms should be done.Molecular structure and conformational analysis
Both forms have two molecules in the asymmetric part of the unit cells. The molecular structures along with the numbering schemes are given in Fig. 1. All molecules exhibit a typical tetrahedral geometry with 8-oxyquinolinate (Q) chelating the boron atom to form a five-membered ring. Details of data collection, data reduction and structure refinements are summarized in Table 1. Selected bond lengths and angles are listed in Table 2.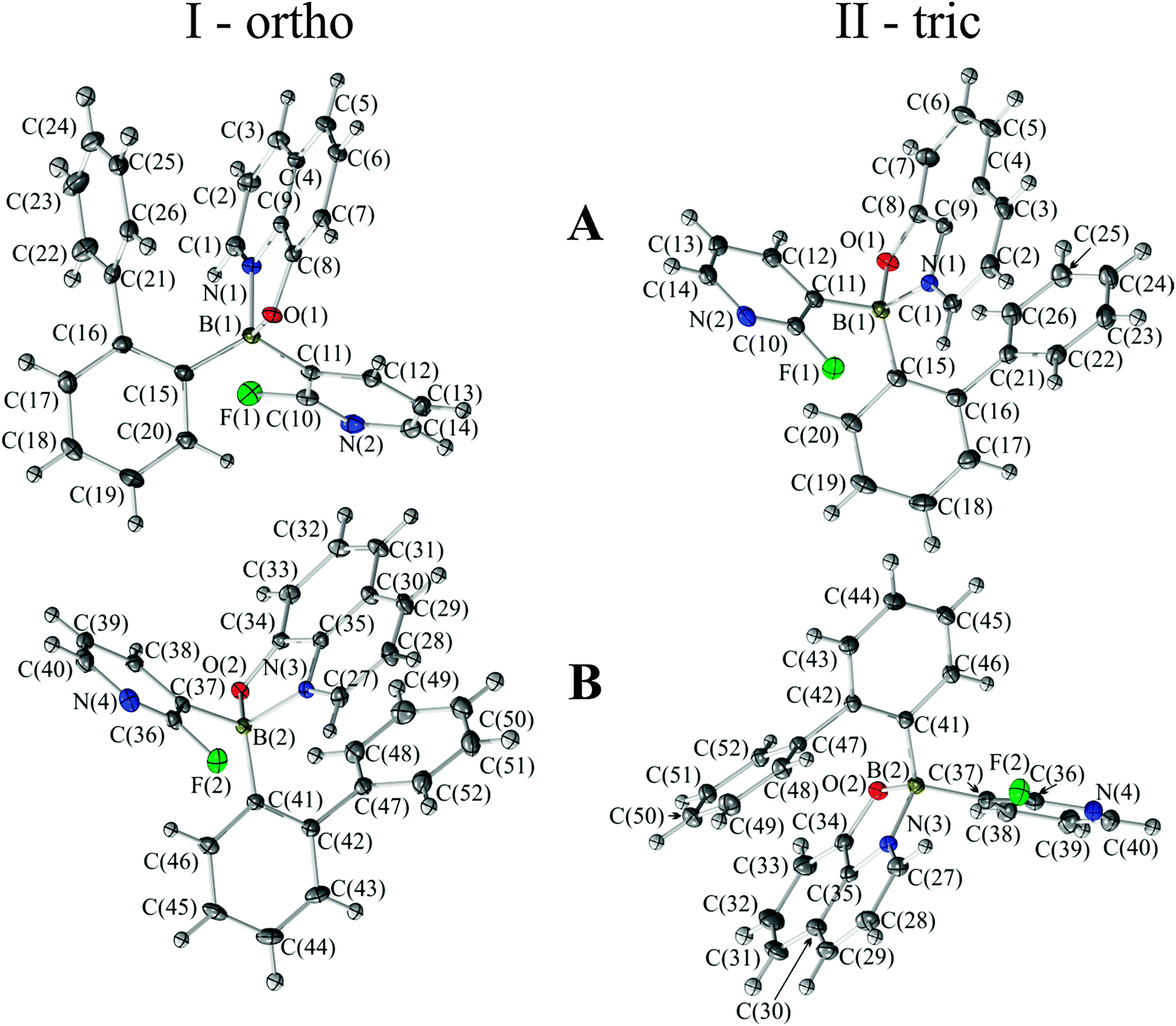 | ||
| Fig. 1 Ball-and-stick representations of the asymmetric parts of the unit cells for I-ortho and II-tric. Atomic displacements were estimated at the 50% probability level. | ||
| I-ortho | II-tric | |
|---|---|---|
| Chemical formula | C26H18BF1N2O | C26H18BF1N2O |
| Crystal system | Orthorhombic | Triclinic |
| M r | 808.50 | 808.50 |
| Space group | Pca21 |
P |
| Cell settings (Å, deg): | ||
| a | 12.3765(7) | 11.18331(13) |
| b | 14.9478(9) | 12.02706(13) |
| c | 21.9246(13) | 15.06878(16) |
| α | 90.000 | 91.5953(9) |
| β | 90.000 | 91.9937(9) |
| γ | 90.000 | 96.6478(9) |
| V (Å3) | 4056.1(4) | 2010.90(4) |
| Z/Z′ | 8/2 | 4/2 |
| d (Mg m−3) | 1.32 | 1.34 |
| Temperature (K) | 100(2) | 100(2) |
| No. of measured reflections | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 654 654 |
61584 |
| Resolution cut-off (Å−1) | 0.7 | 0.7 |
| Completeness (%) | 100 | 100 |
| R int (%) | 5.6 | 1.9 |
| θ max | 30.66 | 30.51 |
| R[F2 > 2σ(F2)] | 0.045 | 0.042 |
| wR(F2) | 0.103 | 0.076 |
| GooF | 1.00 | 1.00 |
| No. of parameters/restraints | 559/1 | 559/0 |
| Reflection used [I > −3σ(I)] | 6370 | 12226 |
| Δρmax, Δρmin (e Å−3) | 0.46, −0.25 | 0.46, −0.26 |
| X(A/B)–Y(A/B) | I-ortho | II-tric | Optimisation | |||
|---|---|---|---|---|---|---|
| A | B | A | B | B3LYPa | B97D | |
| a Data taken from ref. 23. Calculated at RB3LYP/6-31+g(d,p). | ||||||
| B(1/2)–O(1/2) | 1.511(3) | 1.508(2) | 1.512(1) | 1.523(1) | 1.526 | 1.541 |
| B(1/2)–N(1/3) | 1.631(3) | 1.642(3) | 1.641(1) | 1.630(1) | 1.651 | 1.651 |
| B(1/2)–C(11/37) | 1.620(3) | 1.616(3) | 1.625(2) | 1.616(2) | 1.632 | 1.633 |
| B(1/2)–C(15/41) | 1.605(3) | 1.612(3) | 1.613(2) | 1.615(1) | 1.621 | 1.623 |
| THC | 78(1) | 73(1) | 66(1) | 82(1) | 74 | 70 |
The geometry of the boron coordination sphere is almost the same for both crystallographic forms. Bond lengths and angles defined for the boron atom for all studied molecules are very similar (Table 2). The largest differences in the geometric parameters are found for the values of the tetrahedral parameters (THC).58 Similar values of bond lengths, close to the experimental ones, are obtained through theoretical optimization with two functionals: B3LYP and B97D. This was surprising since we were expecting that the crystal lattice will strongly affect the molecular geometry. Finally, the molecular geometries of the molecules from the asymmetric parts of the unit cells are similar for both polymorphs and they are pictured by molecular overlay shown in Fig. 2.
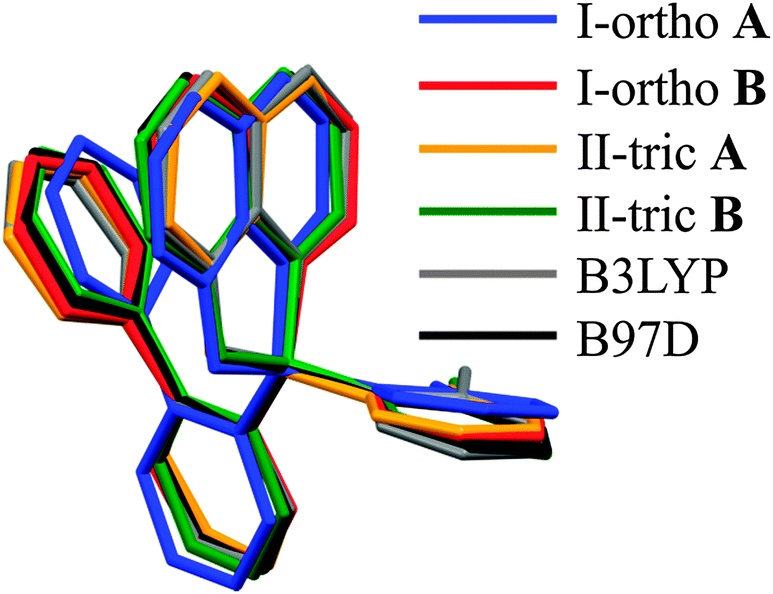 | ||
| Fig. 2 Molecular overlay of molecules from I-ortho and II-tric polymorphs and theoretical optimizations. | ||
It is intriguing that in all cases an intramolecular parallel π⋯π interaction between the biphenyl and Q rings occurs. Almost all molecules adopt conformations in which the π-interactions are face-centred. The exception is I-orthoA molecules where the aromatic ring [defined by C(21), C(22), C(23), C(24), C(25) and C(26) atoms] is in the parallel off-centred position with respect to Q, and it is closer to the pyridine part of Q. Knowing that the charge distribution around the aromatic ring creates a quadrupole moment with a partial negative charge above and below the aromatic ring plane and partial positive charge around the periphery,59 one might draw the conclusion that such a conformation is electrostatically more favourable. This is because the pyridine ring has a lower concentration of π-charge density (with respect to the phenolate ring) which minimizes the repulsion between the rings and the off-centred position enables stabilization through electrostatic interactions. This conclusion is not confirmed by the calculated energies of the isolated molecules (Table S12, ESI†). On the basis of these results, it seems that I-orthoA molecules (both A and B) exhibit the least stable conformation, whereas the molecules in the II-tric form (both A and B) seem to adopt more stable conformations. This may indicate that the II-tric form is a more stable crystallographic form of compound 1.
We wanted to rationalise these results using the electrostatic potential (ESP) mapped on the isosurface of the electron density distribution for the I-orthoA (the least stable) and II-tricB (the most stable) molecules (Fig. 3). On the basis of the generated ESPs we believe that one of the reasons for the difference in the relative stability of these geometries is the fact that the charge concentration (green/yellow area) at Q rings in I-orthoA interacts with charge concentration on the biphenyl rings (contacts indicated by red arrows). Whereas in the II-tricB molecule, the areas of charge concentrations are close to the areas where the ESP indicates charge depletion (blue areas located in the centres of the aromatic rings, contacts indicated by black arrows).
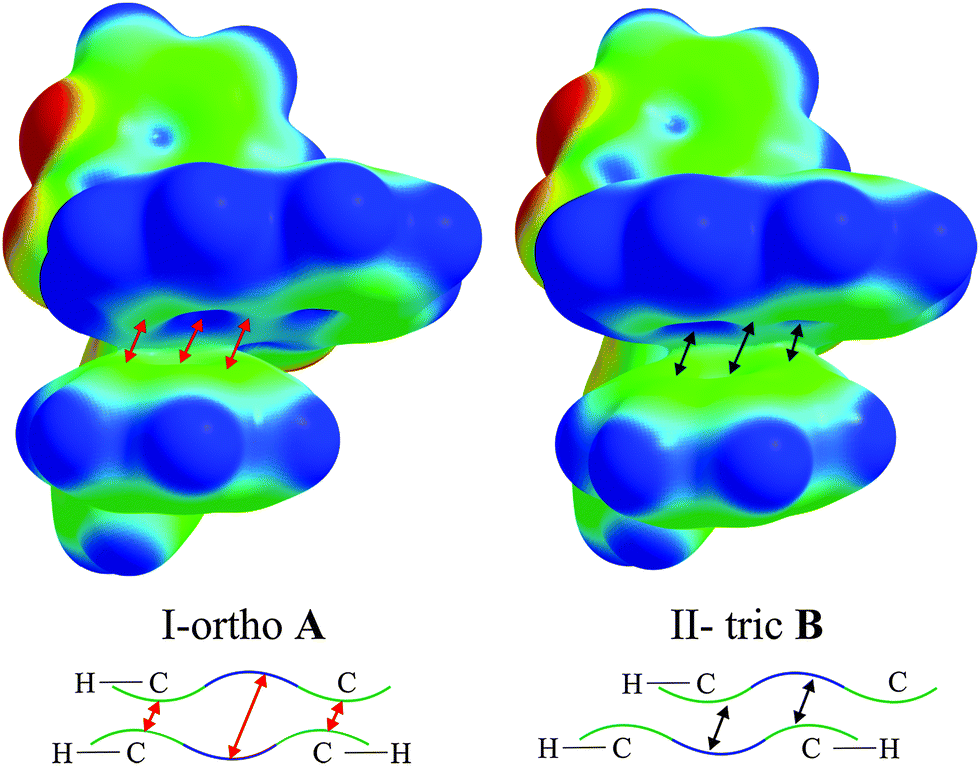 | ||
| Fig. 3 The electrostatic potential calculated for I-orthoA and II-tricB at the B97D/6-31+g(d,p) level of theory. The red arrows indicate contacts that destabilize the molecule and the black arrows the ones the stabilize the molecular geometry. | ||
If these subtle contacts/effects indeed differentiate those molecules in terms of stability then the routine analysis of π⋯π interactions based on assigning ad hoc areas of charge concentration and charge depletion to the aromatic counterparts may not be sufficient for the description of similar systems.
The above results made us wonder what is the impact of the conformation of the molecule of 1 on its energy. Hence, we performed a constrained optimization energy scan around the B–C bond (Fig. 4). The values of the Θ torsion angle for the experimental geometries are closer to the values observed for the global energy maximum rather than to the global minimum of energy. This points to the importance of the crystal field and its impact on the conformations of the molecules in the solid state. It seems that in the case of 1 we can see that even slight changes of geometry can yield in substantial changes in the energies of the molecules.
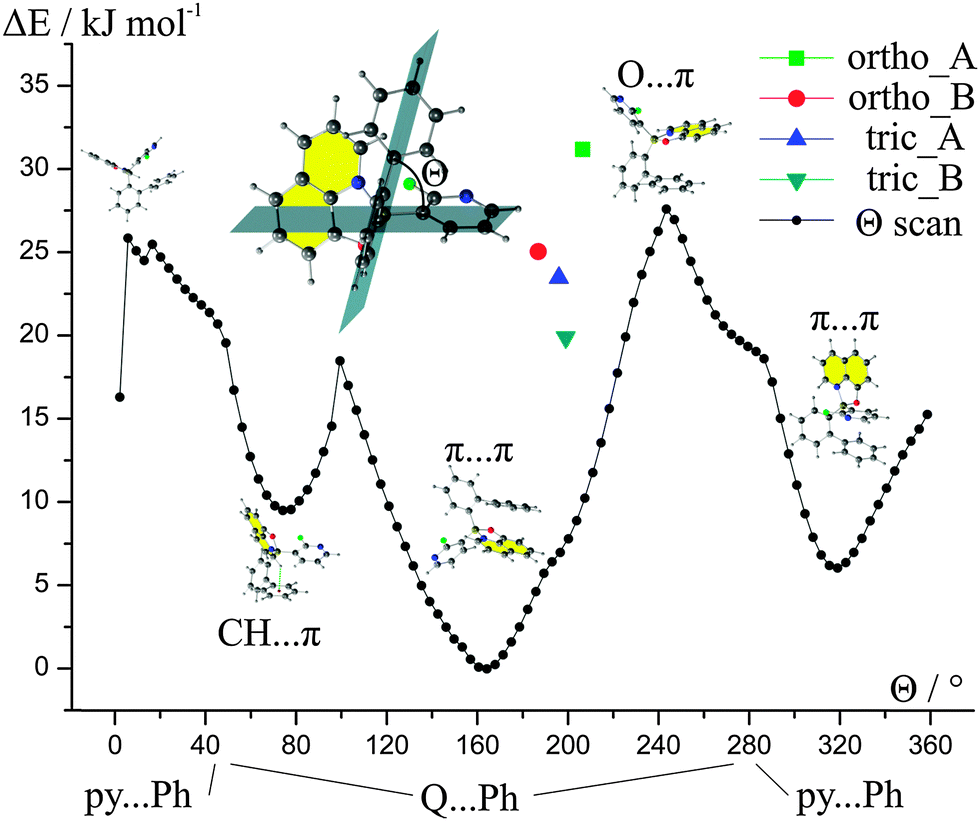 | ||
| Fig. 4 Constrained optimization of the molecular geometries around the B–C bond. Energies are presented relative to the global energy minimum. Calculations performed at the B97D/6-31+(d,p) level of theory. | ||
It may seem surprising that small changes of the Θ angle, for instance constrained by the crystal lattice, results in distinct changes in the energies of the whole molecule. A closer look at intramolecular contacts proved that such results are due to the steric hindrance. This is especially pronounced in the range of angles between 2° and 6° where H⋯H atoms repel each other or at Θ = 99.4° where two H atoms repel the F atom.
The energy scan revealed that there are three possible low-energy conformations. The most stable conformation possesses an intramolecular π⋯π interaction between Q and the phenyl ring (biphenyl ligand). The less stable conformations compromise either intramolecular π⋯π between biphenyl and 2-fluoro-3-pyridyl rings or C–H⋯π interactions between Q and biphenyl ligands.
The values of energies of rotation barriers (ca. 20–30 kJ mol−1) between the most stable geometries indicate that, theoretically, it should be possible to obtain compound 1 with a different conformation, e.g., with intramolecular π⋯π interactions between Q and 2-fluoro-3-pyridyl rings without the need for bond breaking. This means that the compound may switch between those conformations in the solution or in the gas phase (in other words, it may be the case that such rotation is present during preparation of the emitting layers in the diode). Hence, we may expect a set of conformational polymorphs for compound 1. Exact values of the energies and the Cartesian coordinates for the optimized geometries are provided in the ESI† (Table S13).
Comparison of supramolecular packing
Since the purpose of this work is the discussion of the packing and its influence on the properties of presented compounds we are presenting a detailed discussion of the nature of CH⋯O, CH⋯π, π⋯π, and CH⋯F interactions.The CH⋯O contacts
According to Jeffrey's classification,60 the CH⋯O interactions in both polymorphs (present in motifs 2, 14 and 6, 9 for I-ortho and II-tric forms, respectively) are considered as weak H-bonds on the basis of interatomic distances (Fig. 5). However, the values of the H-bond angles for I-ortho contacts: C(1)–H(1)⋯O(1) (motif 2, 163.7(1)°) and C(27)–H(27)⋯O(2) (motif 14, 163.9(1)°) exceed the values of angles assumed to be characteristic for weak interactions (90°–150°) and are in the range of medium H-bonds (130°–180°) (Table 3). Such interactions (C–H⋯O) also involve oxygen lone pair electrons. This makes possible two orientations of the hydrogen atom and oxygen lone pairs. For the discussed H-bonds, the hydrogen atom in C(1)–H(1)⋯O(1) is located in the plane created by C(8)–O(1)–B(1) atoms. This means that it occupies a position in crystal far from O(1) lone pair electrons which makes this contact a weaker one. On the other hand, the position of H(27) in the C(27)–H(27)⋯O(2) bond coincides with the position of the O(2) lone pairs deduced from the oxygen hybridization, which should strengthen this interaction.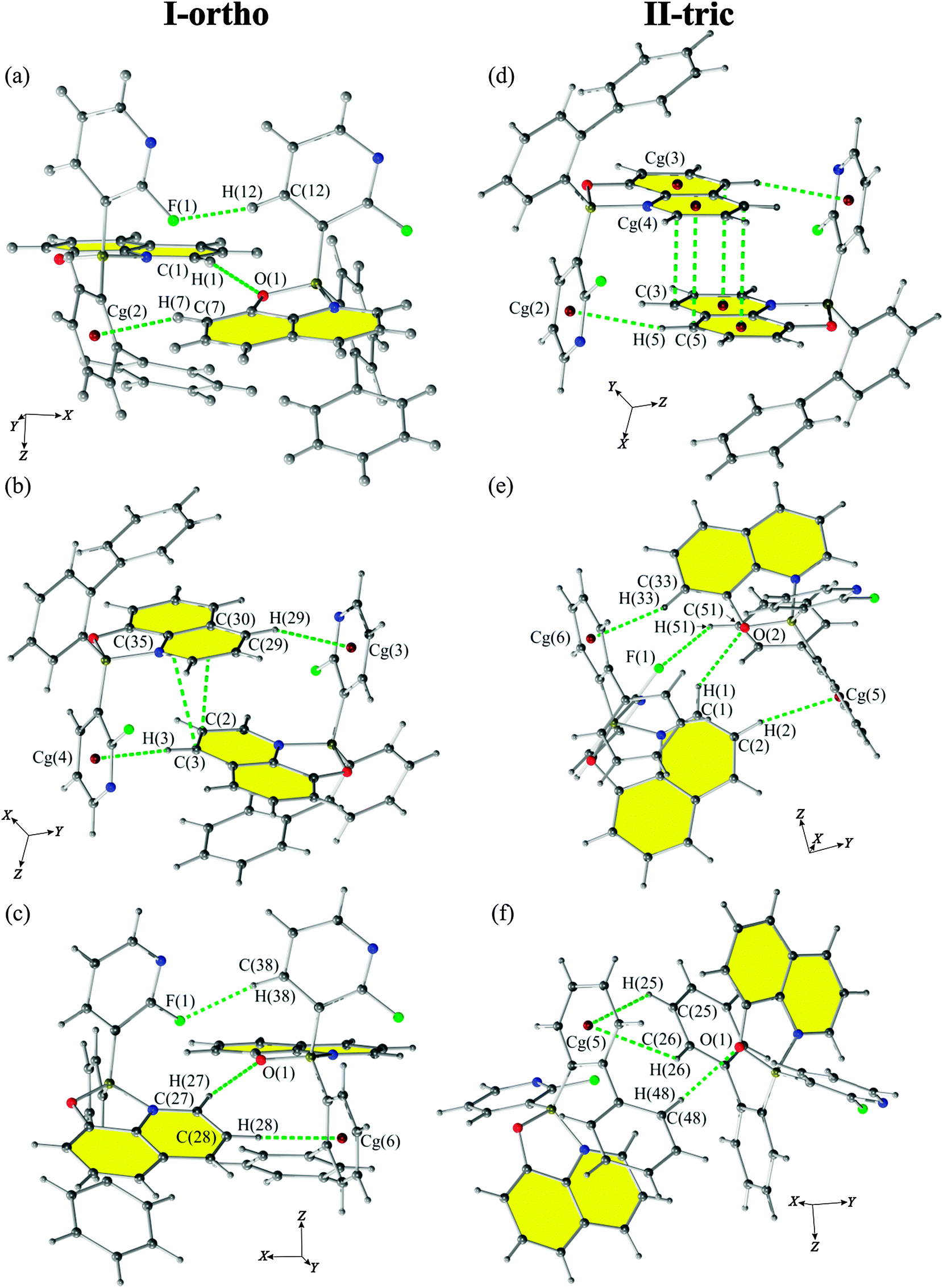 | ||
| Fig. 5 Crystal motifs 2 (a), 9 (b) and 14 (c) of the I-ortho form and motifs 2 (d), 6 (e) and 9 (f) of the II-tric form showing the strongest (according to PIXEL calculations) interactions present between two molecules of 1. | ||
| Motifs | Energy (kJ mol−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| d (Å) | E elstat | E pol | E disp | E rep | E tot | Symmetry | ||||
| Cg(1) is the centroid of the C(21)–C(26) ring; Cg(2) is the centroid of the C(15)–C(20) ring; Cg(3) is the centroid of the C(10)–C(14) ring; Cg(4) is the centroid of the C(36)–C(40) ring; Cg(5) is the centroid of the C(47)–C(52) ring; Cg(6) is the centroid of the C(41)–C(46) ring. Cg(1) is the centroid of the C(21)–C(26) ring; Cg(2) is the centroid of the C(10)–C(14) ring; Cg(3) is the centroid of the C(4)–C(9) ring; Cg(4) is the centroid of the C(1)–C(9) ring; Cg(5) is the centroid of the C(41)–C(46) ring; Cg(6) is the centroid of the C(15)–C(20) ring. | ||||||||||
| Form I-ortho | ||||||||||
| A⋯A | 1 | 10.984 | −9.5 | −5.3 | −23.0 | 22.6 | −15.1 | 0.5 − x, y, 0.5 ± z | ||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(14)–H(14)⋯Cg(1) | 3.410 | 2.630 | 139.6 | |||||||
| 2 | 6.419 | −45.4 | −19.1 | −65.0 | 52.7 | −76.8 | 0.5 ± x, 1 − y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(7)–H(7)⋯Cg(2) | 3.479 | 2.574 | 159.2 | |||||||
| C(12)–H(12)⋯F(1) | 3.179(3) | 2.263(1) | 161.7(1) | |||||||
| C(1)–H(1)⋯O(1) | 3.574(2) | 2.652(1) | 163.7(1) | |||||||
| A⋯B | 3 | 9.256 | −13.7 | −6.6 | −32.6 | 26.9 | −25.9 | x, y, z | ||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(32)–H(32)⋯Cg(3) | 3.449 | 2.555 | 156.8 | |||||||
| 4 | 11.391 | −0.4 | −0.4 | −7.1 | 1.2 | −6.7 | 0.5 − x, y, 0.5 + z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(18)–H(18)⋯H(49) | 3.567(2) | 2.834(1) | 134.7(1) | |||||||
| 5 | 9.155 | −11.9 | −6.0 | −33.7 | 25.2 | −26.3 | x, −1 + y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(6)–H(6)⋯Cg(4) | 3.539 | 2.684 | 150.0 | |||||||
| 6 | 9.426 | −12.3 | 4−4.3 | −33.1 | 20.0 | −29.7 | −x, 1 − y, 0.5 + z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(50)–H(50)⋯C(6) | 3.503(3) | 2.908(2) | 121.8(2) | |||||||
| 7 | 11.746 | −1.0 | −0.3 | −6.9 | 1.5 | −6.6 | −x, 2 − y, 0.5 + z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(18)–H(18)⋯H(43) | 4.152(3) | 2.846(1) | 119.9(1) | |||||||
| 8 | 11.309 | −1.7 | −0.6 | −7.2 | 1.7 | −7.9 | 0.5 − x, −1 + y, 0.5 + z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(32)–H(32)⋯Cg(3) | 3.449 | 2.555 | 156.8 | |||||||
| 9 | 7.306 | −27.1 | −10.9 | −55.5 | 32.4 | −61.1 | 0.5 + x, 1 − y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(3)⋯C(35) | 3.665(3) | |||||||||
| C(2)⋯C(30) | 3.618(3) | |||||||||
| C(3)–H(3)⋯Cg(4) | 3.554 | 2.613 | 171.0 | |||||||
| C(29)–H(29)⋯Cg(3) | 3.909 | 3.071 | 148.0 | |||||||
| 10 | 10.125 | −12.5 | −6.4 | −25.4 | 19.3 | −25.1 | 0.5 + x, 2 − y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(19)–H(19)⋯F(2) | 3.564(3) | 2.703(1) | 151.1(1) | |||||||
| C(45)–H(45)⋯F(1) | 3.389(3) | 2.441(2) | 174.8(1) | |||||||
| 11 | 11.892 | −3.0 | −0.3 | −1.0 | 0.0 | −4.3 | 1 + x, −1 + y, z | |||
| 12 | 11.970 | −2.8 | −0.3 | −0.8 | 0.0 | −3.9 | 1 + x, y, z | |||
| B⋯B | 13 | 11.298 | −6.8 | −2.4 | −16.1 | 10.5 | −14.8 | −x, 2 − y, 0.5 ± z | ||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(39)–H(39)⋯Cg(5) | 3.732 | 3.145 | 121.7 | |||||||
| 14 | 6.387 | −42.2 | −16.2 | −59.0 | 45.1 | −72.3 | 0.5 ± x, 2 − y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(28)–H(28)⋯Cg(6) | 3.634 | 2.699 | 168.0 | |||||||
| C(27)–H(27)⋯O(2) | 3.421(2) | 2.498(1) | 163.9(1) | |||||||
| C(38)–H(38)⋯F(2) | 3.330(2) | 2.398(1) | 167.2(1) | |||||||
| 15 | 11.655 | 1.6 | −1.8 | −9.1 | 5.1 | −4.2 | −0.5 − x, y, 0.5 ± z | |||
| Form II-tric | ||||||||||
| A⋯A | 1 | 11.183 | −7.6 | −3.3 | −20.0 | 15.0 | −15.9 | x ± 1, y, z | ||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(13)–H(13)⋯Cg(1) | 3.812 | 3.096 | 133.4 | |||||||
| 2 | 7.382 | −28.0 | −11.8 | −69.0 | 45.1 | −63.6 | 1 − x, 1 − y, 1 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(5)–H(5)⋯Cg(2) | 3.622 | 2.689 | 164.2 | |||||||
| C(4)⋯C(4) | 3.589(2) | |||||||||
| C(3)⋯Cg(3) | 3.598 | |||||||||
| C(5)⋯Cg(4) | 3.583 | |||||||||
| 3 | 10.670 | −13.0 | −6.6 | −18.1 | 18.6 | −19.1 | 1 − x, 1 − y, 2 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(19)–H(19)⋯N(2) | 3.477(1) | 2.614(1) | 151.3(1) | |||||||
| 4 | 9.506 | −13.4 | −4.3 | −34.8 | 20.3 | −32.2 | 2 − x, 1 − y, 1 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(23)–H(23)⋯Cg(1) | 4.546 | 3.834 | 134.1 | |||||||
| C(24)–H(24)⋯Cg(4) | 4.582 | 3.879 | 133.2 | |||||||
| C(23)–H(23)⋯Cg(3) | 5.090 | 4.245 | 150.0 | |||||||
| 5 | 11.747 | −1.1 | −0.2 | −6.6 | 1.1 | −6.8 | 2 − x, 1 − y, 2 − z | |||
| A⋯B | 6 | 6.335 | −42.4 | −19.1 | −64.0 | 48.7 | −76.9 | x, y, z | ||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(2)–H(2)⋯Cg(5) | 3.681 | 2.763 | 162.6 | |||||||
| C(1)–H(1)⋯O(2) | 3.256(1) | 2.528(1) | 133.5(1) | |||||||
| C(51)–H(51)⋯F(1) | 3.248(1) | 2.602(1) | 125.7(1) | |||||||
| C(33)–H(33)⋯Cg(6) | 3.624 | 2.707 | 162.4 | |||||||
| C(52)–H(52)⋯F(1) | 3.294(1) | 2.691(1) | 122.0(1) | |||||||
| 7 | 10.795 | −3.6 | −2.0 | −7.3 | 1.6 | −11.3 | x − 1, y − 1, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(12)–H(12)⋯N(4) | 3.940(1) | 3.207(1) | 135.3(1) | |||||||
| C(13)–H(13)⋯F(2) | 3.687(1) | 2.870(1) | 144.7(1) | |||||||
| 8 | 12.003 | −5.2 | −1.6 | −6.8 | 2.9 | −10.7 | x − 1, y, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(39)–H(39)⋯N(2) | 3.793(2) | 2.894(1) | 158.2(1) | |||||||
| 9 | 6.615 | −28.0 | −14.5 | −63.6 | 44.7 | −61.4 | x, y − 1, z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(48)–H(48)⋯O(1) | 3.342(1) | 2.484(1) | 150.3(1) | |||||||
| C(26)–H(26)⋯.Cg(5) | 3.962 | 3.332 | 125.6 | |||||||
| C(25)–H(25)⋯Cg(5) | 3.886 | 3.182 | 132.3 | |||||||
| 10 | 9.500 | −5.0 | −2.6 | −18.5 | 9.5 | −16.7 | 1 − x, 1 − y, 1 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(6)–H(6)⋯C(52) | 3.987(2) | 3.105(1) | 155.2(1) | |||||||
| 11 | 10.174 | −9.9 | −3.8 | −22.5 | 14.6 | −21.6 | 1 − x, 1 − y, 2 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(31)–H(31)⋯Cg(2) | 4.016 | 3.498 | 116.7 | |||||||
| C(32)–H(32)⋯Cg(2) | 3.756 | 2.944 | 144.1 | |||||||
| 12 | 10.247 | −2.4 | −2.2 | −20.2 | 9.2 | −15.6 | 2 − x, 1 − y, 1 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(40)–H(40)⋯H(6) | 3.055(1) | 2.589 | 110.6(1) | |||||||
| C(39)–H(39)⋯H(6) | 3.065(1) | 2.652 | 106.8(1) | |||||||
| 13 | 10.321 | −0.4 | −0.3 | −9.2 | 1.1 | −8.9 | 2 − x, 1 − y, 2 − z | |||
| Contacts | D⋯A (Å) | X⋯A (Å) | D–X⋯A (°) | |||||||
| C(17)–H(17)⋯C(32) | 4.204(2) | 3.441(1) | 139.0(1) | |||||||
| C(18)–H(18)⋯C(33) | 4.279(2) | 3.541(1) | 136.4(1) | |||||||
| 14 | 12.309 | −3.5 | −0.3 | −0.9 | 0.0 | −4.8 | 2 − x, 2 − y, 1 − z | |||
The CH⋯π contacts
An interesting contact was observed for the II-tric form, mainly motif 4 with a quite high value of the total energy (−32.2 kJ mol−1) and with a large contribution of dispersion energy (−34.8 kJ mol−1). Detailed analysis of this contact indicates the presence of a bifurcated CH⋯π interaction. This interaction is created by the CH group interacting with two aromatic rings (Fig. 6). Such an explanation is consistent with the recent review on the nature of CH⋯π interactions,61 where it was proven that dispersion is the major stabilizing contribution for these contacts. Probably, such an intriguing motif can exist due to the rigid structure of the discussed compound. | ||
| Fig. 6 Motif 4 of II-tric presenting bifurcated CH⋯π contacts. | ||
All of the strongest crystallographic motifs for both polymorphs have CH⋯π contacts in their structures. Almost all of them are perpendicular T-shaped type contacts except C(25)–H(25)⋯Cg(5)⋯H(26)–C(26) from motif 9 of II-tric form which has a perpendicular Y-shape. The contribution of the CH⋯π contacts to the total energy (Etot) of the motifs may be significant as interatomic distances (D–X⋯A) are in the range of 3.4–3.95 Å which is considered to be the most energetically favourable (Table 3).
The π⋯π contacts
The intermolecular π⋯π interactions for both polymorphs occur between two 8-oxyquinolinates and create parallel off-centred contacts (motif 9 for I-ortho, and motif 2 for II-tric, see Fig. 5(b) and (d), respectively). Since the Q rings are engaged in the intramolecular π⋯π interactions, it seems unexpected, due to the steric hindrance, that only intermolecular π⋯π interactions present in the crystal lattice occur between two neighbouring 8-oxyquinolinate rings, and are not engaging 2-fluoro-3-pyridyl rings or any of the biphenyl rings.The CH⋯F contacts
According to the classification proposed by A. Collas et al.,62 a fluorine atom can take part in interactions directly or it can alter the electron distribution which will activate certain atoms and engage them in new contacts. These authors designated four types of contacts: type I – occurring between perfluoroarenes (acceptors) and electron rich aromatic rings (donors); type II – between electron-poor aromatic rings (acceptors) and fluorine atom (donors); and type III – between polyfluorinated phenyl rings (acceptors) and fluorine atoms (donors). Types IVa and IVb cover the C–H⋯F and F⋯F interactions, respectively. Only contacts of type IVa are observed among contacts present in the crystal structures of the discussed polymorphs. Type IVa contacts between H-atoms and pyridyl rings are observed in motifs 2, 10 and 14 for I-ortho and motifs 6 and 7 for the II-tric form. Such interactions are known to be very weak with stabilization energies up to 2 kJ mol−1 and with the major contribution from dispersion energy. According to the review by Berger et al.,63 the C–F⋯H–C interaction is a directional bond and it is very weak when it is not collinear. The motifs compromising such interactions are stabilized by other (stronger) interactions. On the basis of deviation from linearity, one can conclude that the I-ortho polymorph features much stronger C–H⋯F interactions with angles ranging from 151.1(1)° to 174.8(1)° (motif 2, 10 and 14) than the II-tric polymorph with angles ranging from 122.0(1)° to 144.7(1)° (motifs 6 and 7). Judging from the proportion of the contacts (based on Hirshfeld surface analysis, Fig. S2, ESI†), one can see that I-ortho also has slightly more contacts of the CH⋯F type.PIXEL calculations, DSC and XRPD measurements
Calculations using PIXEL indicate that the lattice energies and contributions from electrostatic, polarization, dispersion and repulsion to total energies for both polymorphic forms are quite similar (see Table 4). This approach indicates that both forms are stabilized mainly by dispersion energy and destabilized by repulsion energy. Our results proved that the II-tric form is thermodynamically more stable than the other one. These results are consistent with the observed rates of crystallization as one can see that crystallization of the orthorhombic form is faster than that of the triclinic one. DSC measurements prove that crystals of both forms melt at the same temperature (Fig. S4–S7, ESI†) which is consistent with the fact that the presented lattice energies are very close in terms of absolute values.| Energy (kJ mol−1) | |||||
|---|---|---|---|---|---|
| E elstat | E pol | E disp | E rep | E L | |
| I-ortho | −187.2 | −72.8 | −389.2 | 264.2 | −381.9 |
| II-tric | −171.1 | −71.1 | −395.2 | 255.0 | −382.3 |
The values of lattice energies are based on the types and energies of the crystallographic motifs observed for a particular structure. It seems that the I-ortho form is dominated by interactions which occur between two different molecules from the asymmetric part of the unit cell (A⋯B, Table 3). Although there are in general only a few contacts of types A⋯A and B⋯B, among these contacts we observed one of the strongest interactions present in the structure. It is worth noticing that the II-tric form lacks B⋯B type motifs in its structure. The largest energy contributions in the discussed motifs come from dispersion and electrostatic energies which is characteristic for π interactions.59
Crystals of both polymorphs have identical melting points (219.9 °C/217.3 °C and 219.4 °C/217.2 °C for I-ortho and II-tric, respectively). These results are consistent with the calculated lattice energies which are also close in terms of absolute value (Table 4). Two separate experiments done with samples of both forms yielded the same results. The I-ortho form crystallized during the first cycle of cooling at 141.9 °C, and the II-tric form crystallized during the second cycle of heating (at 113.2 °C).
The powder X-ray diffraction patterns were collected at room temperature on the basis of samples used for single crystal X-ray determination and DSC measurements. Collected data were compared with each other and with simulated patterns. The relative shifts of some parts of simulated patterns can be shifted with respect to experimental data since simulated X-ray powder diffraction patterns were calculated on the basis of crystal structures measured at 100 K. Even though the main features of the experimental patterns were reproduced by theoretical data, both forms can be distinguished from each other on the basis of the X-ray powder diffraction patterns as we can distinguish peaks for [10−3], [211] and [202] planes observed solely for II-tric (Fig. S3, ESI†).
Hirshfeld surfaces
Subtle structural differences are revealed by the analysis of Hirshfeld surface plots (Fig. 7). Distinct features of the fingerprint plots are constructed with H⋯H, C⋯H, F⋯H and O⋯H type contacts. In the I-ortho form, the F⋯H type contacts are seen as distinct spikes, whereas O⋯H type contacts appear as spikes in the II-tric form. The proportions of intermolecular contacts are similar in both forms (Fig. S1, ESI†). However, the orthorhombic form has slightly more contacts of the CH⋯C and CH⋯H types. The latter contacts in molecules B seem to have two preferred distances as it may be concluded from their double spike representation in the corresponding fingerprint plot. Similar proportions of the contacts seem to be corresponding with the fact that lattice energies calculated using PIXEL for both forms have similar values. This also seems to be consistent with DSC measurements in which both crystals melt at the same temperatures. Hirshfeld surfaces calculated for molecules with dnorm value contacts show that the I-ortho form has a greater overall amount of intermolecular interactions (Fig. S2, ESI†). This is pronounced for the 2-fluoro-3-pyridyl ring. | ||
| Fig. 7 Fingerprint plots generated for two separate molecules in the asymmetric part of the unit cell for both polymorphs. | ||
Estimation of the charge transport properties
Evaluation of the charge transport properties through the Marcus theory indicates that both crystallographic forms can serve as electron transporting materials which is a common feature for borinic derivatives with the 8-oxyquinolinate ligand. The energy splitting of frontier orbitals is slightly more effective in the I-ortho form and so the approximate values of the charge transfer integrals are higher for this form. For both polymorphs, it seems that charge transfer is stronger between two different molecules in the asymmetric part of the unit cell (A⋯B) than transfer between symmetrically equivalent molecules (A⋯A and B⋯B). Evaluation of the charge transport properties has been ordered by contributions from crystallographic motifs and summarized in Fig. 8 (Table S5, ESI†). In the graph, the dominating transporting properties are underlined.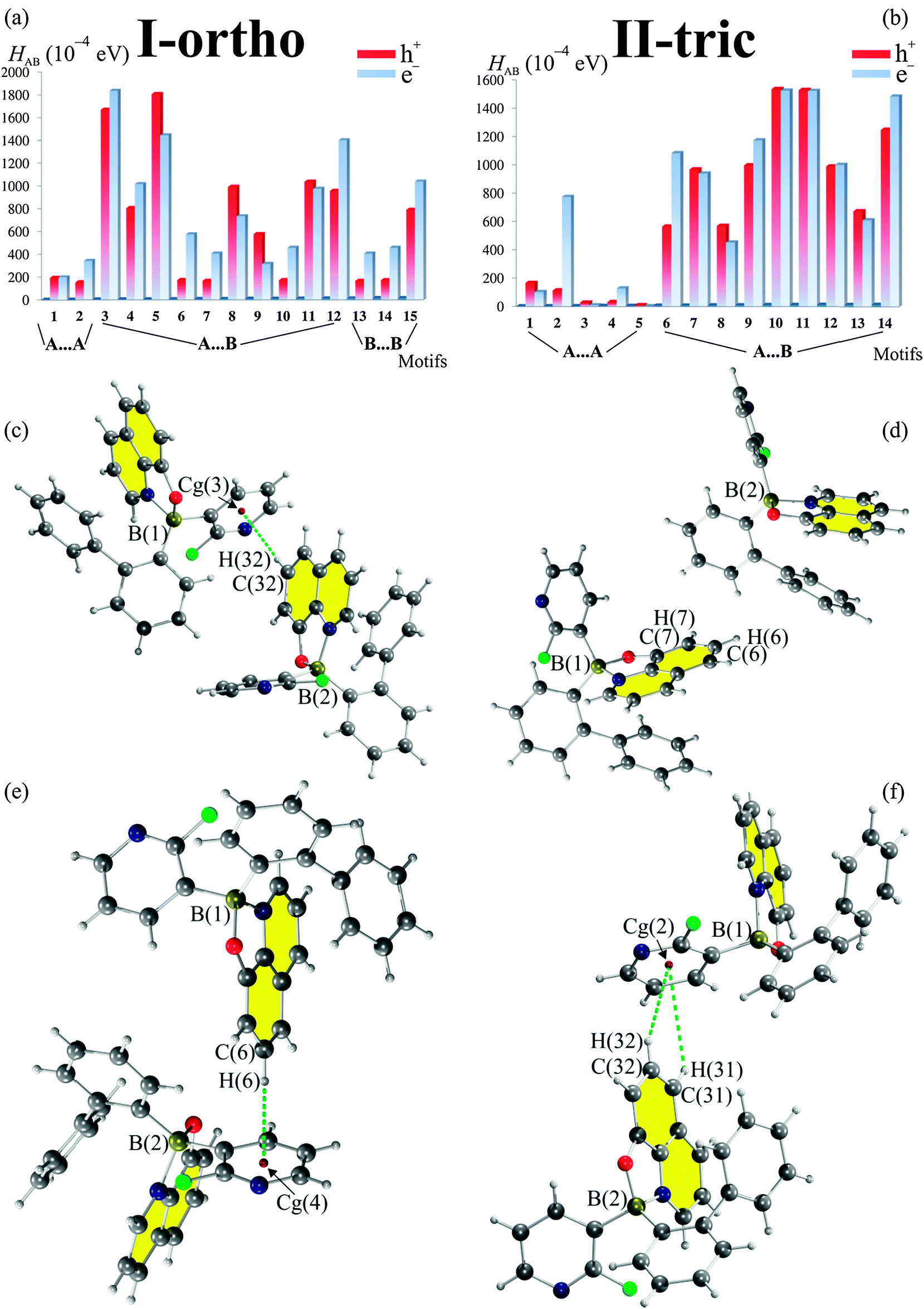 | ||
| Fig. 8 Contributions from crystallographic motifs to the charge transport properties (a and b) evaluated through Marcus theory at the B97D/6-31+g(d,p) level of theory. Most contributing motifs 3 (c), 5 (e) and 10 (d), 11 (f) for I-ortho and II-tric, respectively. | ||
These are either electron (e−, blue) or hole (h+, red) transporting properties. For the I-ortho form we see that motifs with greater stabilization energy (motifs 3 and 5) contribute more to charge transport properties. The opposite situation is observed for the II-tric form (motifs 10, 11 and 14) where less stronger contacts contribute more to the CT properties. Clearly, the strength of interactions has nothing to do with splitting of the frontier orbitals. In both structures the most contributing motifs are based on CH⋯π type interactions. Three of them involve the electron-rich phenoxyl part of the 8-oxyquinolinate and electron-poor 2-fluoro-3-pyridyl rings, which may point what sort of motifs should be introduced into newly design crystal structures.
Despite the fact that contacts with the fluorine atom are clearly visible in the fingerprint plots of the I-ortho form their presence has a minor effect on CT properties as motifs in which they appear have low values of the HAB2. Low impact of the fluorine atom is also visible for the II-tric form. However, the fluorine atom may have an indirect effect and it may activate pyridyl ring through its electron withdrawing effect. Similar conclusions concerning impact of the fluorine atom on the creation of crystallographic motifs were mentioned by Berger et al.63 However, in their review they have not combined existence of certain motifs with their CT properties.
Conclusions
The detailed analysis of the crystal structures of two polymorphs of borinic 8-oxyquinolinate supported by DFT calculations, Hirshfeld surface analysis, PIXEL calculations and Marcus theory provide insight into the impact of the polymorphism on the properties of the presented system. On the basis of the Ostwald's rule, the lattice energies of I-ortho and II-tric (−381.9 vs. −382.3 kJ mol−1, respectively) and molecular energies calculated for both polymorphs we have concluded that the orthorhombic polymorph is slightly less stable than the triclinic one. Moreover, the orthorhombic polymorph exhibits some weaker interactions than the triclinic form. However, I-ortho seems to be a more efficient electron transporting material (with charge transfer integrals higher by ca. 0.8 eV with respect to II-tric). In both systems the same type of contacts (CH⋯π) has the largest contribution to the CT properties.If it were possible to maintain the intermolecular interaction pattern characteristic for the orthorhombic polymorph during preparation of the emitting layer, then one could expect a better performance and higher output of the diode constructed with such a layer. Since the orthorhombic polymorph seems to be the kinetic product of the crystallization, the methods promoting faster production of the layers in the diode should be utilized. This means that the process used for the preparation of layers in diodes may be important for the output of organic diodes.
Acknowledgements
The MPD/2010/4 project is realized within the MPD programme of Foundation for Polish Science, cofinanced from European Union, Regional Development Fund. This work was supported by the National Science Centre (Narodowe Centrum Nauki, Grant No. DEC-2011/03/B/ST5/02755). The support from Aldrich Chemical Co., Milwaukee, WI, USA, through continuous donation of chemicals and equipment is gratefully acknowledged. G.W.B. thanks the Foundation for Polish Science for financial support within the International PhD Program. Authors gratefully acknowledge The Interdisciplinary Centre of Mathematical and Computational Modelling in Warsaw (grant no. G33-14) for providing computer facilities on which most of the calculations were done. The X-ray studies have been carried out at the Biological and Chemical Research Centre, University of Warsaw, established within the project co-financed by European Union from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013.References
- K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef PubMed.
- A. Nagai and Y. Chujo, Chem. Lett., 2010, 39, 430–435 CrossRef CAS.
- Y. Tokoro, A. Nagai and Y. Chujo, Macromolecules, 2010, 43, 6229–6233 CrossRef CAS.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- Y. Tokoro, A. Nagai and Y. Chujo, Appl. Organomet. Chem., 2010, 24, 563–568 CrossRef CAS.
- F. Cheng, E. M. Bonder and F. Jäkle, Macromolecules, 2012, 45, 3078–3085 CrossRef CAS.
- X.-Y. Wang and M. Weck, Macromolecules, 2005, 38, 7219–7224 CrossRef CAS.
- Y. Qin, C. Pagba, P. Piotrowiak and F. Jäkle, J. Am. Chem. Soc., 2004, 126, 7015–7018 CrossRef CAS PubMed.
- H. Li and F. Jäkle, Angew. Chem., Int. Ed., 2009, 48, 2313–2316 CrossRef CAS PubMed.
- F. Cheng and F. Jakle, Chem. Commun., 2010, 46, 3717–3719 RSC.
- C. Cui, E. M. Bonder and F. Jäkle, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6612–6618 CrossRef CAS.
- Y. Qin, I. Kiburu, S. Shah and F. Jäkle, Org. Lett., 2006, 8, 5227–5230 CrossRef CAS PubMed.
- L. S. Sapochak, A. Padmaperuma, N. Washton, F. Endrino, G. T. Schmett, J. Marshall, D. Fogarty, P. E. Burrows and S. R. Forrest, J. Am. Chem. Soc., 2001, 123, 6300–6307 CrossRef CAS PubMed.
- S. L. Hellstrom, J. Ugolotti, G. J. P. Britovsek, T. S. Jones and A. J. P. White, New J. Chem., 2008, 32, 1379–1387 RSC.
- M. Brinkmann, G. Gadret, M. Muccini, C. Taliani, N. Masciocchi and A. Sironi, J. Am. Chem. Soc., 2000, 122, 5147–5157 CrossRef CAS.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS.
- E. Laborda, M. C. Henstridge, C. Batchelor-McAuley and R. G. Compton, Chem. Soc. Rev., 2013, 42, 4894–4905 RSC.
- G. Wesela-Bauman, P. Ciećwierz, K. Durka, S. Luliński, J. Serwatowski and K. Woźniak, Inorg. Chem., 2013, 52, 10846–10859 CrossRef CAS PubMed.
- G. Wesela-Bauman, L. Jastrzębski, P. Kurach, S. Luliński, J. Serwatowski and K. Woźniak, J. Organomet. Chem., 2012, 711, 1–9 CrossRef CAS PubMed.
- C. Giacovazzo, Fundamentals of crystallography, Oxford Univ. Press, Oxford, 2011 Search PubMed.
- J. Bernstein, Polymorphism in Molecular Crystals, IUCr, 2002 Search PubMed.
- A. Fernández-Mato, M. D. García, C. Peinador, J. M. Quintela, M. Sánchez-Andújar, B. Pato-Doldán, M. A. Señarís-Rodríguez, D. Tordera and H. J. Bolink, Cryst. Growth Des., 2013, 13, 460–464 Search PubMed.
- S. Fanetti, M. Citroni and R. Bini, J. Phys. Chem. B, 2011, 115, 12051–12058 CrossRef CAS PubMed.
- E. A. Pozzi, L. R. Schwall, R. Jimenez and J. M. Weber, J. Phys. Chem. B, 2012, 116, 10311–10316 CrossRef CAS PubMed.
- K. Sakanoue, M. Motoda, M. Sugimoto and S. Sakaki, J. Phys. Chem. A, 1999, 103, 5551–5556 CrossRef CAS.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339–2350 CrossRef CAS PubMed.
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J.-L. Brédas, J. Am. Chem. Soc., 2002, 124, 7918–7919 CrossRef CAS PubMed.
- W. Senevirathna, C. M. Daddario and G. Sauvé, J. Phys. Chem. Lett., 2014, 5, 935–941 CrossRef CAS.
- P. Cias, C. Slugovc and G. Gescheidt, J. Phys. Chem. A, 2011, 115, 14519–14525 CrossRef CAS PubMed.
- B. C. Lin, C. P. Cheng, Z.-Q. You and C.-P. Hsu, J. Am. Chem. Soc., 2005, 127, 66–67 CrossRef CAS PubMed.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 16866–16881 CrossRef CAS PubMed.
- V. Stehr, R. F. Fink, B. Engels, J. Pflaum and C. Deibel, J. Chem. Theory Comput., 2014, 10, 1242–1255 CrossRef CAS.
- H. Mori, X. Chen, N. Chang, S. Hamao, Y. Kubozono, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 4973–4983 CrossRef CAS PubMed.
- M. Mamada, H. Katagiri, M. Mizukami, K. Honda, T. Minamiki, R. Teraoka, T. Uemura and S. Tokito, ACS Appl. Mater. Interfaces, 2013, 5, 9670–9677 CAS.
- W.-Q. Deng and W. A. Goddard, J. Phys. Chem. B, 2004, 108, 8614–8621 CrossRef CAS.
- Y.-K. Lan and C.-I. Huang, J. Phys. Chem. B, 2008, 112, 14857–14862 CrossRef CAS PubMed.
- CrysAlis Pro Software, Oxford Diffraction Ltd, 2010 Search PubMed.
- Bruker AXS Inc., APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2010 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- International Tables for Crystallography, Mathematical, Physical and Chemical Tables, ed. H. Fuess, Chester, 2006, vol. C Search PubMed.
- V. Vreshch, J. Appl. Crystallogr., 2011, 44, 219–220 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- F. H. Allen and I. J. Bruno, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 380–386 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc, Wallingford CT, 2010 Search PubMed.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Gavezzotti, J. Chem. Theory Comput., 2005, 1, 834–840 CrossRef CAS.
- A. Gavezzotti, CrystEngComm, 2003, 5, 429–438 RSC.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- H. Höpfl, J. Organomet. Chem., 1999, 581, 129–149 CrossRef.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- S. Tsuzuki, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2012, 108, 69–95 RSC.
- A. Collas, R. De Borger, T. Amanova and F. Blockhuys, CrystEngComm, 2011, 13, 702–710 RSC.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 988296 and 988297. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cp02606c |
| This journal is © the Owner Societies 2014 |