
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combined IR/IR and IR/UV spectroscopy study on the proton transfer coordinate of isolated 3-hydroxychromone in the electronic ground and excited state†
A.
Stamm
,
M.
Weiler
,
A.
Brächer
,
K.
Schwing
and
M.
Gerhards
*
TU Kaiserslautern, Fachbereich Chemie & Research Center Optimas, Erwin-Schroedinger-Straße 52, D-67663 Kaiserslautern, Germany. E-mail: gerhards@chemie.uni-kl.de
First published on 19th August 2014
Abstract
In this paper the excited state proton transfer (ESPT) of isolated 3-hydroxychromone (3-HC), the prototype of the flavonols, is investigated for the first time by combined IR/UV spectroscopy in molecular beam experiments. The IR/UV investigations are performed both for the electronically excited and electronic ground state indicating a spectral overlap of transitions of the 3-HC monomer and clusters with water in the electronic ground state, whereas in the excited state only the IR frequencies of the proton-transferred monomer structure are observed. Due to the loss of isomer and species selectivity with respect to the UV excitations IR/IR techniques are applied in order to figure out the assignment of the vibrational transitions in the S0 state. In this context the quadruple resonance IR/UV/IR/UV technique (originally developed to distinguish different isomers in the electronically excited state) could be applied to identify the OH stretching vibration of the monomer in the electronic ground state. In agreement with calculations the OH stretching frequency differs significantly from the corresponding values of substituted hydroxychromones.
I Introduction
The transfer of a proton after electronic excitation (excited state proton transfer, ESPT) is an intensively investigated phenomenon due to its importance in a variety of photochemical and biological processes (cf.ref. 1–12). It e.g. plays an important role in the photoprotective mechanism of flavonoids, which are plant secondary metabolites. Here it serves to protect the plants against UV-B radiation.2,10,11Within an ESPT molecule a proton-donor and a proton-acceptor group must be present. Frequently these groups already interact in the electronic ground state via e.g. hydrogen bonds. Typical arrangements are the proton transfer of a hydroxyl to a keto group,8,13–15 between a hydroxyl group and an imine group,16,17 from an amine to a keto functionality1 and between an amine and an imine functionality.18
3-HC drawn in Fig. 1 represents the basic structure of the flavonols, a subgroup of the antioxidative and photoprotective flavonoids. After electronic excitation of the normal form (N) into a (mostly) short-lived electronically excited state (N*) the proton migration transfers the system into the tautomeric, electronically excited structure (T*). After relaxation (e.g. via fluorescence) into the tautomeric ground state T the back proton transfer regenerates the originally existing N form in the electronic ground state so that the photocycle is closed. This typical ESPT scheme is sketched in Fig. S1 of the ESI.†
 | ||
| Fig. 1 Structural isomers of 3-hydroxychromone. | ||
An intensively studied representative of the class of flavonols is 3-hydroxyflavone (3-HF) which undergoes a keto–enol tautomerism after π–π* excitation. Investigations have been performed in solution, different matrices (e.g.ref. 19–26) and supersonic expansions.27–30
Furthermore a variety of theoretical investigations have been performed.31–39 The direct structural proof for the ESPT of isolated 3-HF in the gas phase was given by our group by applying structure sensitive combined IR/UV methods (IR/R2PI for the S0 state and UV/IR/UV for the S1 state) under isolated molecular beam conditions.30 In comparison to the calculated vibrational frequencies of different (TD)-DFT optimised structures the presence of the tautomeric form in the electronically excited state could be demonstrated not only for 3-HF but also for 2-(2-naphthyl)-3-hydroxychromone (2-NHC), a derivative, differing in the naphthyl substituent on the chromone moiety.30 Beyond this, in the isolated 3-HF(H2O)2 cluster for the first time a proton wire was analysed in the electronically excited state by an IR/UV method.29
The ongoing discussion in the literature about the existence of an energy barrier for the PT (cf. e.g.ref. 21, 26, 28, 34 and 40–43), arising either from the required coplanarity of the phenyl ring with the chromone moiety (cf. e.g.ref. 21 and 40) or from the OH bending mode modulating the proton transfer distance,44 leads us to the investigation of 3-hydroxychromone (3-HC) as the basic structure. For 3-HC the PT coordinate should be easier to describe since no further aromatic substituent exists. In former studies the ESPT for 3-HC was investigated by fluorescence spectroscopy in solution40,45–47 or in a 10 K argon matrix.23,24,48 These studies mainly revealed the ESPT in 3-HC to be faster than in the case of 3-HF and the electronically excited 3-HC tautomer to be longer-lived.40
The only molecular beam investigation on 3-HC, in which the formation of water clusters was assumed to be unlikely, is a comparative fluorescence study with 3-HF and 2-NHC.28,49 The low frequency vibrations in the fluorescence excitation spectra of 3-HF and 2-NHC, which were absent for 3-HC, could be assigned to the torsional movement of the phenyl or naphthyl residue, respectively. Since the activation barrier for the ESPT depends on this movement the ESPT rate increases with a decrease in the size of the substituent on the γ-pyron ring which means that 3-HC shows the fastest proton transfer (k3-HC = 1.77 × 1012 s−1 > k3-HF = 6.55 × 1011 s−1 > k2-NHC = 1.54 × 1011 s−1).
However, new fs-UV/VIS and IR investigations in different solvents argue the converse. Here the comparison of experimental data in apolar organic and polar partially protic solvents and quantum chemical calculations suggest the involvement of a trans structure as an intermediate prior to the ESPT in the electronically excited state which is stabilised by solvent interactions. This finally effects a slower proton transfer on the picosecond time scale. Thus a substituent on the chromone unit as the phenyl group in 3-HF might even promote a faster ESPT since it hinders specific solute–solvent interactions enabling the pathway via the stabilised trans structure.50,51 Further theoretical investigations on the structure and reactivity of 3-HC are presented in ref. 32 and 52–54.
In order to achieve more information on the structure and reactivity of isolated 3-HC and defined aggregates with water we present in this paper the first molecular beam investigations of 3-HC including mass selected IR spectroscopy for the electronic ground state (IR/R2PI method) and the electronically excited state (UV/IR/UV method). Both the structurally relevant regions of the OH stretching and CO stretching/OH bending modes are analyzed. Due to the discussion of overlapping isomers/species also quadruple resonance (IR/IR) methods have to be applied,55,56 including a new variant in order to distinguish different species in the electronic ground state. The comparison with calculated vibrational frequencies of different (TD)-DFT optimised structures allows structural assignments. Those techniques/approaches represent ideal tools to investigate the intrinsic structural properties of cooled isolated molecules without the influence of an environment. These detailed investigations give comprehensive information on the hydrogen-bonding within the molecule (and its cluster with water) offering the possibility to perform direct comparisons with the results obtained for substituted 3-hydroxychromones like 3-HF and 2-NHC.
II Experimental set-up
A detailed description of the experimental set-up is given by former publications, cf. e.g.ref. 56. Thus only the main aspects for understanding the measurements should be explained here. All spectra were recorded by using a vacuum apparatus basically consisting of a differentially pumped time-of-flight mass spectrometer and a pulsed valve (General Valve Iota One, 500 μm orifice) for skimmed jet expansion. The investigated 3-HC was synthesised by our group. The two-step synthesis is described in the ESI.† The sample was heated for the jet expansion to 130 °C and helium and neon were used as carrier gases (1800 mbar). The measurements presented in this paper refer to helium expansion conditions. The results obtained for neon expansion support our assignment but they yield no new results regarding the interpretation. All aspects concerning the neon expansion experiments are given in the ESI.†To perform the quadruple resonance techniques (IR/IR/R2PI and IR/UV/IR/UV, details cf.ref. 56) four independent laser systems are required. The two UV wavelengths for excitation and ionisation were generated by two frequency-doubled dye lasers (Sirah Cobra Stretch and Sirah Precision Scan); the FWHM of the UV system is about 6 ns. The two independent IR laser systems57 have the same basic set-up. In a LiNbO3 crystal the IR light in the region of 2800–3800 cm−1 (3.57–2.63 μm) is generated by a first difference frequency mixing (DFM1) of the fundamental of a seeded Nd:YAG laser and the output of a dye laser (Sirah Precision Scan) pumped by the second harmonic of the same Nd:YAG laser. This IR radiation is further amplified in a second LiNbO3 crystal by an optical parametric amplification (OPA) of the output of the DFM1 process and the fundamental of the Nd:YAG laser. The second IR laser system offers the possibility to yield a further amplification of the IR output by introducing a second amplifier stage. Both IR laser systems have a pulse width (FWHM) of 8–10 ns. The wavelength regions of CO stretching and OH bending modes are covered by a third non-linear process (DFM II, available in the region from about 4.5 to 16 μm). Here the signal (5398–5564 cm−1) and idler (4009–3834 cm−1) of the OPA process are mixed in an AgGaSe2 crystal generating radiation from 1380 to 1730 cm−1.
The time delay of the two UV lasers was optimised to 6.8 ns since this yielded the maximum ion signal, the best two-colour effect and a sufficient delay to insert the IR laser for all measurements in the electronically excited state. To measure the IR spectra of the electronic ground state the IR laser was fired 50 ns prior to the UV excitation laser. For the UV/IR/UV spectra the IR laser was fired 4 ns after the UV excitation laser. In the case of the IR/IR/R2PI measurements the first fixed IR burn laser was fired 100 ns before the UV excitation. The second, scanned IR laser followed the IR burn laser after 50 ns. For the IR/UV/IR/UV spectra the delay of the fixed IR burn laser was set to a delay time of 50 ns prior to the UV excitation laser whereas the scanned IR laser was delayed to fire 4 ns after the UV excitation laser.
III Theory
The structures of the 3-hydroxychromone monomer (cf.Fig. 1) in the electronic ground state and the first electronically excited state were determined by geometry optimisation at the DFT and TD-DFT levels of theory using Gaussian09.58 The B3LYP functional as implemented in Gaussian09 and the TZVP basis set were chosen. The SCF convergence and geometry convergence thresholds were set to the “tight” criterion. All energies were corrected for harmonic vibrational zero-point energies. The calculated vibrational frequencies for the structures presented here were real, indicating minima on the potential energy surface. To take anharmonic effects into account the calculated vibrational frequencies were scaled with the factor of 0.99 for the CO region and 0.9613 for the OH stretching region.29,30The calculated cis structure in the electronic ground state (S0) with a stabilising intramolecular hydrogen bond between the carbonyl and hydroxyl functionalities is the most stable isomer (cf.Fig. 1a). The corresponding trans structure (S0,t, cf.Fig. 1c) and the proton-transferred tautomer form (S0′, cf.Fig. 1b) are 2915 cm−1 (35 kJ mol−1) and 5489 cm−1 (66 kJ mol−1) less stable. In the electronically excited state, the relationship changes. Here the proton-transferred tautomeric structure (S1′) represents the minimum structure whereas the non-tautomeric cis (S1) and trans structures (S1,t) are, respectively, 4050 cm−1 (49 kJ mol−1) and 4339 cm−1 (52 kJ mol−1) higher in relative energy.
The most important geometry parameters for the different 3-HC structures for both electronic states are listed in Table 1. The most important aspects regarding the calculated geometries are: (a) all structures are planar in the electronic ground and excited state and (b) the proton transfer can be followed by changing the rO2–H3 and rH3–O4 distances. The vertical electronic excitation of the cis structure S0 to the electronically excited state S1 effects an elongation of rO2–H3 and a shortening of rH3–O4 distances, which finally ends up in the proton-transferred (tautomeric) form S1′. It is just the other way around for going from the S1′ to the S0′ state structure. In the back proton transfer the hydrogen bond between O2–H3 is reconverted to a covalent bond whereas for H3–O4 it is elongated to a hydrogen bond.
| cis structure | Tautomeric structure | trans structure | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0′ | S1′ | S0,t | S1,t | |
| r C1–O2 | 1.351 | 1.319 | 1.265 | 1.335 | 1.359 | 1.370 |
| r O2–H3 | 0.976 | 1.002 | 1.812 | 2.143 | 0.965 | 0.964 |
| r H3–O4 | 2.097 | 1.871 | 1.008 | 0.975 | 3.684 | 3.649 |
| r O4–C5 | 1.232 | 1.248 | 1.312 | 1.341 | 1.220 | 1.313 |
| r C5–C1 | 1.461 | 1.510 | 1.449 | 1.414 | 1.473 | 1.408 |
| Φ C1–O2–H3 | 105.1 | 102.2 | 84.2 | 81.5 | 110.5 | 110.3 |
| Φ O2–H3–O4 | 116.4 | 123.4 | 125.2 | 114.7 | 6.8 | 6.0 |
| Φ H3–O4–C5 | 82.4 | 86.0 | 101.1 | 106.3 | 58.9 | 59.1 |
Since the IR/R2PI spectrum of the 3-HC monomer showed vibrations which were suspected to arise from a UV generated fragmentation of hydrates (details: cf. Results and discussion) the possible structures for clusters with one and two water molecules were calculated for the electronic ground state. For the most stable monohydrate the water molecule is inserted between the carbonyl and hydroxyl groups of the chromone unit (A in Fig. 2). In the second-most stable isomer, which is about 840 cm−1 (10 kJ mol−1) less stable, the water molecule is merely hydrogen-bonded to the CO group (B in Fig. 2). In the case of the Atrans structure (3107 cm−1, 37 kJ mol−1 less stable, cf.Fig. 2) water is hydrogen-bonded to the trans structure of the monomer. Additionally, further arrangements with water attached to the trans structure and the tautomeric form of the monomer were calculated (cf. Fig. S2 in the ESI†). Among these structures the tautomeric B′T arrangement (5484 cm−1, 66 kJ mol−1 less stable) will be mentioned later; it is shown in Fig. 2. Furthermore structures with water being attached to the oxygen atom of the chromone unit like structure C (2070 cm−1, 25 kJ mol−1 less stable) and to the oxygen atom of the hydroxyl group like structure B′, (1312 cm−1, 16 kJ mol−1 less stable) are illustrated in Fig. S2 (ESI†).
 | ||
| Fig. 2 Possible structures of 3-hydroxychromone with one water molecule including the two most stable arrangements A and B. For further structures cf. ESI.† | ||
The most important geometry parameters for the monohydrate isomers derived from calculations for both the electronic ground and excited state are listed in Tables S1 and S2, ESI.† Additionally structures of clusters with two water molecules and the corresponding most important geometry parameters are given in Fig. S5 and Table S3 of the ESI.†
IV Results and discussion
A mass selective two-colour R2PI spectrum of 3-HC has been recorded in the range of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210–31600 cm−1 (cf.Fig. 3). A one-colour R2PI spectrum could not be obtained since in this region of possible excitations the energy of two UV photons of the excitation laser does not exceed the ionisation potential.
210–31600 cm−1 (cf.Fig. 3). A one-colour R2PI spectrum could not be obtained since in this region of possible excitations the energy of two UV photons of the excitation laser does not exceed the ionisation potential.
 | ||
| Fig. 3 R2PI spectrum of 3-hydroxychromone. | ||
The R2PI spectrum is characterised by a broad unstructured background and an electronic origin at 30392 cm−1 which is in agreement with the origin of the laser induced fluorescence (LIF) spectrum of ref. 28. In order to get an assignment on the structure and proton transfer (PT) coordinate of 3-HC in different electronic states the R2PI spectrum is taken as a basis for further double, triple and quadruple resonance experiments (see below). An estimation of the ionization potential can be obtained by recording an ion current spectrum, for which the UV excitation laser is fixed and the ionizing laser is scanned. The recorded ion current spectrum shows no sharp increase indicating that the measured vertical ionization potential (IP) cannot directly be compared to the calculated adiabatic one.
From the ion current spectrum an IP can be estimated that is lower than 71350 cm−1, i.e. the measured vertical and the calculated adiabatic IPs differ by about 5100 cm−1. It should be mentioned that these deviations are very similar to the one obtained for 3-HF (cf.ref. 30: difference between experimental and theoretical IPs is about 5700 cm−1). In contrast to the results obtained for 3-HF (cf.ref. 29 and 30) no ion signals of clusters with water could be observed if helium is used as a carrier gas. This result was independent of the chosen excitation and ionisation wavelengths. The combined IR/UV investigations given in the next sections will show if no clusters are formed or if they fragment after electronic excitation.
The IR/R2PI spectra in the spectral region of both the OH stretching vibrations as well as the carbonyl stretching and OH bending modes obtained via the mass trace of 3-HC are depicted in Fig. 4; the spectra are identical for the different possible UV excitation wavelengths indicated in the R2PI spectrum. In the OH stretching region four transitions at 3268, 3410, 3461 and 3715 cm−1 are observed. The small shoulder at about 3300 cm−1 of the transition at 3268 cm−1 may be attributed to an overtone of the strong experimental transition at 1651 cm−1. However, in the spectral region of the OH stretching modes only one transition of the hydroxyl group of 3-HC is expected. Especially the transition at 3715 cm−1, which typically belongs to a free OH stretching mode of a water molecule with one non-hydrogen-bonded OH group, is a clear indication that not only pure 3-HC but also cluster(s) of water are indirectly investigated – although no mass signals of hydrates could be observed. The fact that IR bands of hydrates can be recorded in the IR/R2PI spectrum via the monomer ion signal is due to a UV generated fragmentation of the cluster(s) (detailed explanations for this frequently occurring behavior are given in the ESI†). These investigations already have shown the important conclusion that in contrast to previous assumptions derived from pure fluorescence spectra (cf.ref. 28 and 49) clusters with water are formed.
 | ||
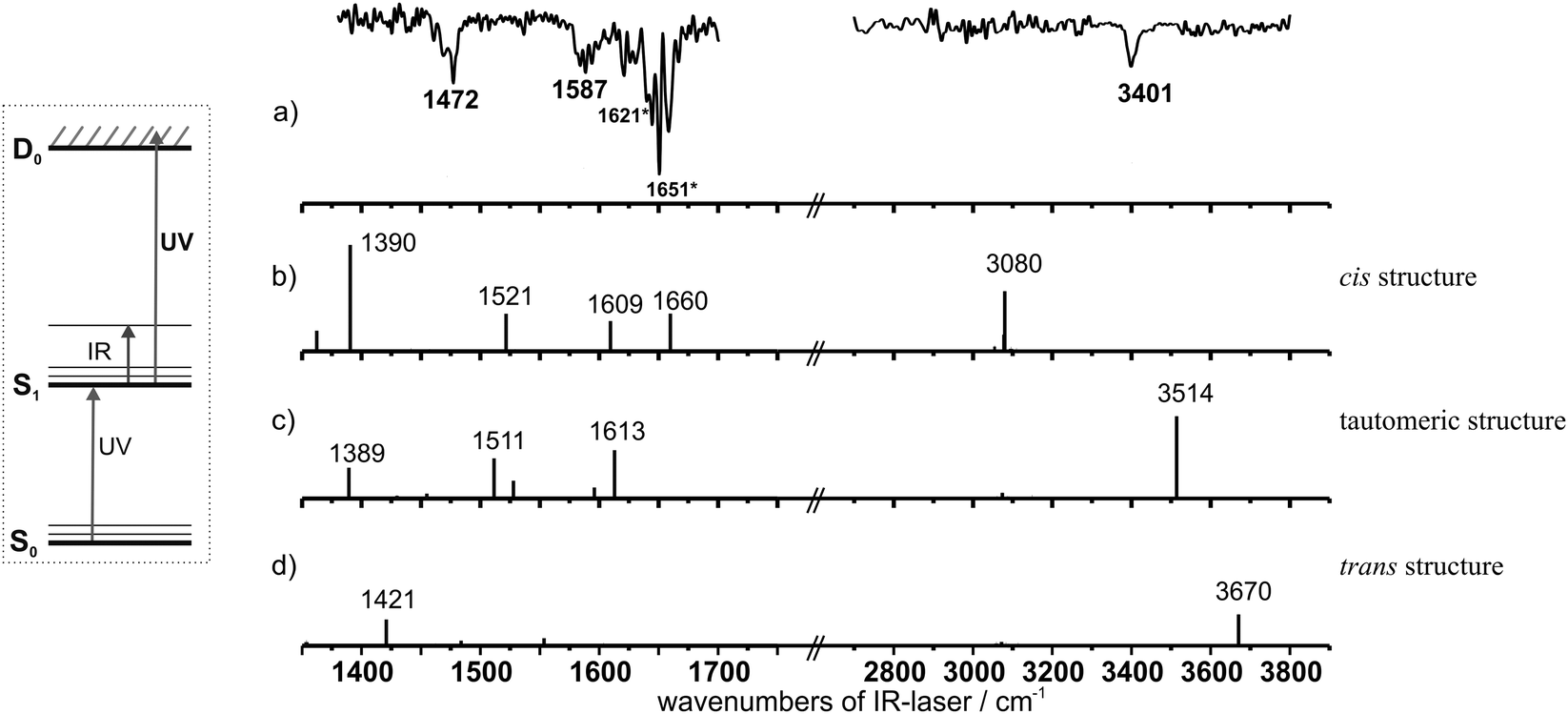
| Fig. 4 (a) Experimental IR/R2PI spectrum of 3-hydroxychromone in the electronic ground state S0 with the excitation laser at 30525 cm−1 and the ionisation laser at 45351 cm−1. (b) Calculated IR spectra (B3LYP/TZVP, scaling factor OH stretching region: 0.9613; scaling factor CO stretching/OH bending region: 0.99) of the S0 state cis structure, (c) of the tautomeric structure and (d) of the trans structure. (For a better illustration the calculated intensities in the OH stretching region were doubled.) | ||
In order to explain the presence of clusters with water in the IR spectra of 3-HC and to get a further assignment, the IR spectra are first compared with the values calculated for the different possible structures of pure 3-HC (cf.Fig. 1). In Fig. 4 the calculated IR spectra based on the harmonic approach are shown. Obviously the most stable cis structure (Fig. 1a) fits best to the experimental spectrum both in the region of OH bending and carbonyl stretching modes as well as in the OH stretching region. The OH stretching modes calculated for the trans and tautomeric structures are either too low or too high in energy with respect to the experimentally observed possible OH stretching transitions between 3268 and 3461 cm−1 (cf.Table 2).
| Experiment | cis structure | Tautomeric structure | trans structure | |||||
|---|---|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0′ | S1′ | S0,t | S1,t | |
| 3-HC | 3461 | 3401 | 3484 | 3080 | 2990 | 3514 | 3655 | 3670 |
| 3-HF | 3276 | 3350 | 3403 | 2928 | 3058 | 3436 | 3622 | 3624 |
| 2-NHC | 3280 | 3362 | 3398 | 3035 | 3091 | 3421 | 3611 | |
According to the harmonic calculations the transition at 3461 cm−1 seems to fit best but it is known from 3-HF that anharmonic effects can lead to a significant reduction of this value. In the case of 3-HF the experimentally observed OH stretching frequency is 3276 cm−1 which is closer to the transition at 3268 cm−1 of 3-HC. In order to yield an assignment, different experimental strategies have been chosen. In one attempt IR/R2PI spectra were recorded in a way that the IR laser was fixed on the different transitions of the IR spectrum and the UV laser is scanned in the region of the R2PI spectrum (cf.Fig. 5). If the IR photon excites 3-HC other R2PI transitions should be influenced (depleted) than for the case that hydrates are vibrationally excited. The spectra obtained via the transitions at 3268, 3410 and 3461 cm−1 show that all UV transitions of the original R2PI spectrum are affected in the same way, i.e. as far as pure 3-HC and clusters with water are observed all species have very similar UV spectra so that the UV excitation is not isomer and species selective in this case.
 | ||
| Fig. 5 IR/R2PI spectra of 3-hydroxychromone by using different burn laser frequencies at (a) 3268 cm−1, (b) 3410 cm−1 and (c) 3461 cm−1 for the first IR photon. The IR/R2PI spectra are given in grey and the corresponding R2PI spectra are given in black. | ||
If no isomer or species selectivity is given by the UV excitation, the application of an IR/IR/R2PI experiment55,56 may retain the isomer/species selectivity. In this case the excitation of one isomer/species with a frequency-fixed IR burn laser has an effect on all IR transitions that belong to the same isomer/species in the subsequently measured IR/R2PI spectrum. It should be mentioned that in this application the IR/IR spectra of clusters are measured on the monomer mass trace. This complication can avoid any quantitative analysis, i.e. only a qualitative correlation between peaks that belong together can be given.
For the 3-HC investigations the burn laser was set to one of the four transitions at 3268, 3410, 3461 or 3715 cm−1 (cf.Fig. 6). Unfortunately, the spectra show (like in the spectra of Fig. 5) that all transitions influence one other. Especially the excitation of the “water” characteristic band at 3715 cm−1 seems to correlate with all other transitions, i.e. all vibrations should be associated with clusters. On the other hand it cannot be excluded that one of the transitions is an overlap of a vibration belonging to pure 3-HC and a cluster with water. The IR/IR method can only be species selective if at least one IR transition exclusively belongs to the monomer. Thus, on the basis of the IR/IR/R2PI experiments a final assignment for the vibration in the S0 state cannot be given, but further information can be obtained from the investigation of the electronically excited state and on the basis of these results we can draw further conclusions for the interpretation of the electronic ground state IR spectra.
 | ||
| Fig. 6 IR/IR/R2PI spectra of 3-hydroxychromone by using different burn laser frequencies at (a) 3268 cm−1, (b) 3410 cm−1, (c) 3461 cm−1 and (d) 3715 cm−1 for the first IR photon. The IR/IR/R2PI spectra are given in grey and the corresponding IR/R2PI spectra (without using the burn laser) are given in black. | ||
The electronically excited state is analysed by applying the UV/IR/UV method both in the region of the OH stretching modes and the region of the carbonyl stretching and OH bending vibrations (cf.Fig. 7). In the region of the OH stretching vibrations only one transition at 3401 cm−1 is observed which could be a hint that only the monomer species (with one OH stretching frequency) is measured in the electronically excited state. (e.g. in the case of the monohydrated cluster three transitions in the range from 2700 to 3800 cm−1 should be observed, cf. calculated spectra in Fig. S4, ESI.†) By comparing the value of 3401 cm−1 with the experimentally obtained frequencies for the tautomeric forms of 3-HF (3350 cm−1) and 2-NHC (3362 cm−1)30 as well as their corresponding calculated values (cf.Table 2), a similar deviation between calculated and experimentally determined frequencies is observed resulting from the anharmonicity of the OH stretching vibration. The OH stretching vibrations calculated for the less stable cis and trans forms (cf. Section III) are either too low or too high compared to the experimentally observed value (cf.Table 2 and Fig. 7). This interpretation is further supported by the spectra obtained in the CO stretching/OH bending region. The two vibrational transitions observed at 1472 and 1587 cm−1 exhibit a similar distance as the two calculated frequencies at 1511 and 1613 cm−1. Compared to the OH stretching modes the absolute values of the calculated frequencies are higher due to their anharmonicities. According to the calculations performed in this spectral region for the cis and trans arrangements, both structures do not fit with respect to their peak positions and expected intensities (cf.Fig. 7). Thus an unambiguous assignment of the tautomeric form in the excited state (S1′) is obtained so that similar to 3-HF and 2-NHC an excited state proton transfer could also be proven for isolated 3-HC.
 | ||
| Fig. 7 (a) Experimental spectrum of 3-hydroxychromone in the S1 state. The asterisks indicate vibrations from the S0 state due to a partial temporal overlap of excitation (UV) and IR laser. (b)–(d) Calculated stick spectra of the S1 state (TD-DFT/B3LYP/TZVP; for scaling factors cf.Fig. 4): (b) cis structure, (c) tautomeric structure, and (d) trans structure. (For a better illustration the calculated intensities in the OH stretching region were doubled.) | ||
As mentioned before, the most striking observation is that (in contrast to the electronic ground state) no vibrations of clusters with water are observed in the IR spectrum of the electronically excited state via the 3-HC mass trace. This is a clear indication that the clusters with water fragment after UV excitation. If the hydrates fragmented during the ionisation process, additional hydrate bands should also be visible in the UV/IR/UV spectrum via the 3-HC mass trace. Since a complete fragmentation of the cluster may be less probable it can furthermore be assumed that the electronically excited state of the clusters does not live long enough for ionisation with a ns laser after electronic excitation (cf. e.g.ref. 59 and 60). It could be assumed that the broad background of the R2PI spectrum (cf.Fig. 3) with a width of several hundreds of wavenumbers may be correlated to the clusters with short lifetimes in the fs regime. Due to (a) the form of the R2PI spectrum (increasing signal), (b) the fragmentation of the hydrates and (c) the resulting overlap of 3-HC and hydrate peaks a fitting of individual peaks with Lorentzian line shapes (like in ref. 61) is not unambiguous. Nevertheless, the width of possible peaks between 20 and a few hundred wavenumbers indicate the presence of short-lived species.
As a further consequence of a short excited state lifetime the monomer and the hydrates cannot be discriminated by their ionisation potential as in the case of 3-HF.29,30 This was decisively useful to determine the 3-HF monomer band and discriminate it from the spectra of the cluster species which can also be observed on the mass traces of the clusters.
From the UV/IR/UV spectrum of 3-HC the formation of the tautomeric structure in the electronically excited state (S1′) can be derived but according to our investigations with respect to the S0 state the question remains if (a) no monomer is observed in the S0 state, i.e. all transitions arise from clusters with water which fragment in the S1 state or if (b) a monomer transition overlaps with one of the IR transitions arising from a hydrate. In the case of the dihydrated 3-HF cluster we introduced a new quadruple resonance technique (the IR/UV/IR/UV method56) in order to distinguish two different isomers within the electronically excited state if the UV excitation is not isomer selective. In the inset of Fig. 8 the method is shown schematically: The first IR laser depletes one isomer in the electronic ground state, thus (almost) only the second isomer is observed in the IR spectrum of the electronically excited state.
 | ||
| Fig. 8 IR/UV/IR/UV spectra of 3-hydroxychromone by using different burn laser frequencies at (a) 3268 cm−1, (b) 3410 cm−1 and (c) 3461 cm−1 for the first IR photon. The IR/UV/IR/UV spectra are given in grey and the corresponding UV/IR/UV spectra are given in black. | ||
In the case of 3-HC the IR/UV/IR/UV technique could be ideally suited to distinguish different species like the monomer and hydrates in the electronic ground state which have the same UV excitation energy and lead to the same species (monomer of 3-HC) in the excited state. This could work in the following way: the frequency-fixed IR burn laser (first IR laser) is set to a vibrational transition of the S0 state and a UV/IR/UV spectrum is recorded. If the IR burn laser exclusively excites a cluster with water in the electronic ground state, the monomer band(s) in the IR spectrum of the S1 state should still be conserved. If the IR burn laser excites both the cluster with water and the monomer in the electronic ground state, then (almost) no IR signal should be observed in the electronically excited state. In this case the relevant monomer species is depleted in the electronic ground state. In Fig. 8, the IR/UV/IR/UV spectra (normalised to the UV/IR/UV spectra) after excitation of the transitions at 3268, 3410 and 3461 cm−1 are shown. These spectra indicate clearly that the electronic ground state vibration at 3461 cm−1 must result from an overlap of more than one species, since – in contrast to the other IR/UV/IR/UV spectra – the S1 state vibration completely vanishes. Furthermore one of these overlapping species only has one OH transition in the spectrum of the electronic ground state at 3461 cm−1. Since this can only be the case for the monomer species the electronic ground state band at 3461 cm−1 can clearly be identified as the vibration of the monomer. By comparing this assignment with the one obtained for 3-HF (3276 cm−1) and 2-NHC (3280 cm−1) in the S0 state the vibrational frequency seems to be surprisingly high. On the other hand it has to be taken into account that in the case of 3-HC already the calculated harmonic frequency is about 100 cm−1 higher compared to 3-HF and 2-NHC. Finally one-dimensional vibrational calculations for 3-HC performed on the potential obtained from a step-wise elongation along the OH stretching normal mode yield an excellent agreement between calculated (3450 cm−1) and experimentally observed value of 3461 cm−1. The detailed theoretical analysis to describe the proton transfer coordinate performed on 1D, 2D and 3D potentials is given in another publication.62
After assigning the transition at 3461 cm−1 to the OH stretching mode of the monomer, the transitions at 3715, 3410, 3268 cm−1 and again the overlapping mode at 3461 cm−1 still belong to clusters with water. Thus we compared the spectroscopically obtained results with calculations performed for clusters with one water molecule (cf.Fig. 9). The comparison of the calculated stick spectra of 3-HC(H2O)1 with the experimentally observed IR transitions on the monomer mass show a very good agreement with transitions calculated for the two most stable forms of the monohydrated cluster (A and B). The general assignment is in agreement with the discussion on the monohydrated cluster of 3-HF for which also an overlap of the two most stable arrangements is discussed.29 Therefore all transitions in the IR/R2PI spectrum of 3-HC could be assigned, resulting from the monomer as well as from fragmenting monohydrated clusters. (For further description of the vibrations, cf. Table S4, ESI†. In the case of Ne expansion larger clusters can be formed but these do not lead to the vibrational transitions observed on the monomer trace. For further information, cf. ESI† Fig. S7, and comments).
 | ||
| Fig. 9 The upper trace schematically depicts the experimental S0 state spectrum (cf.Fig. 4) via the ion signal of 3-hydroxychromone as stick spectrum, which is compared to the calculated stick spectra of different monohydrate isomers (B3LYP/TZVP, for scaling factors, see Fig. 4.) For a better illustration the calculated intensities in the OH stretching region were doubled. | ||
Finally a further interesting feature should be addressed which can be found in the IR/IR/R2PI spectrum with a burn laser at 3268 cm−1 (cf.Fig. 6a). An additional band occurs at around 3340 cm−1 which was not recorded in the original IR/R2PI spectrum. The transition at 3268 cm−1 in the IR/R2PI spectrum is assigned to the monohydrated cluster A (cf.Fig. 9). One possibility could be that an excitation at 3268 cm−1 may (at least partially) lead to a structural rearrangement of the cluster. With a second IR photon which follows the first one in an IR/IR/R2PI spectrum the new structure would be analysed by a further decrease of the ion signal if an IR transition of this structure is excited. IR-induced structural reorganisations are known from population transfer spectroscopy introduced by Zwier and coworkers (e.g.ref. 63). In their chosen examples the population is redistributed between different isomers. In the case of 3-HC a new vibrational signature is observed which is on the blue-side of the transition at 3268 cm−1 and is therefore potentially not a hot band. Among the possible structural isomers calculated for the monohydrated cluster (cf. Fig. S3, ESI†) the B′T and Atrans structures exhibit one very intense transition at 3349 and 3439 cm−1, respectively. The value of 3349 cm−1 of the B′T structure is in perfect agreement with the experimentally observed value, but it is questionable why the UV absorption (as observed in the R2PI spectrum after excitation of the transition at 3268 cm−1) of the tautomeric form does not differ from the one of the “normal” form. In the case of the Atrans structure a one-dimensional vibrational calculation performed on the potential obtained from a step-wise elongation along the OH stretching normal mode of the 3-HC moiety yield a value of 3354 cm−1 which is also in excellent agreement with the experimentally observed value. Thus the Atrans arrangement is an appropriate candidate to explain the additional transition at around 3340 cm−1. Although this suggestion may not be taken as a final interpretation it is a very interesting effect that the formation of a new structure is induced by an IR excitation which can be probed by a successive second IR excitation in combination with a R2PI detection. These results offer the possibilities for further developments to investigate IR induced reactions which will be analysed both experimentally and theoretically in the upcoming work.
Conclusions
In this paper 3-HC a basic unit of flavonoids is investigated as an isolated species in the gas phase. In this basic system the proton transfer reaction in the electronically excited state is not influenced by side groups. Here we present the first IR/UV analysis of 3-HC in a molecular beam for both the electronic ground and excited state. In contrast to formerly investigated systems like 3-HF and 2-NHC the analysis of 3-HC was complicated by the presence of short-lived hydrates, fragmenting down onto the monomer mass trace. This leads to an overlay of the IR vibrations of 3-HC and hydrates in the IR/R2PI spectrum via the monomer mass trace. Since no mass signals of the clusters with water were observed under He expansion a direct comparison of the S0 state IR spectra for vibrational assignments was not possible. Therefore IRfixed/R2PI and IR/IR/R2PI spectroscopy were applied for the electronic ground state which did not allow concrete assignments but the conclusion that all S0 state vibrations can be related to hydrates.In contrast to the electronic ground state only the proton-transferred monomer species could be detected in the UV/IR/UV spectrum, which implied that the hydrates fragment after UV excitation. The presence of only the monomer species in the electronically excited state was used for further analysis: via IR/UV/IR/UV spectroscopy the band at 3461 cm−1 in the IR/R2PI spectrum could be assigned to the hydrogen-bonded OH stretching vibration of the 3-HC monomer, which overlaps with a hydrate vibration. A comparison with 3-HF and 2-NHC showed that the frequency of the vibration was unexpectedly high at the first sight but a simple approach with a one-dimensional potential dependent on a step-wise elongation along OH stretching normal mode allowed the calculation of the OH stretching vibration which was in good agreement with the experimentally assigned value. With respect to methodical developments it should be remarked that the quadruple IR/UV/IR/UV spectroscopy was introduced to distinguish isomers in the electronically excited state. The application of this method to 3-HC shows that this technique can also be used to distinguish species in the electronic ground state if only vibrational transitions of one species are observed in the electronically excited state.
Thus the investigations on the basic system of the flavonols, 3-HC, show (in contrast to the substituted systems) a complicated spectral behavior which had to be analysed using a variety of sophisticated, high-level spectroscopic methods. In this context the quadruple IR/UV/IR/UV method was applied to discriminate different species in the electronic ground state. The combination of experiment and theory finally allowed a successful analysis of the 3-HC system which represents a fundamental prototype for an ESPT reaction.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG; Reference No. GE 961/8-1) for financial support. This work is part of the PhD thesis of M. W. and A. S.References
- T. P. Smith, K. A. Zaklika, K. Thakur, G. C. Walker, K. Tominaga and P. F. Barbara, J. Photochem. Photobiol., A, 1992, 65, 165 CrossRef CAS.
- G. J. Smith and K. R. Markham, J. Photochem. Photobiol., A, 1998, 118, 99 CrossRef CAS.
- S. Dennison, J. Guharay and P. K. Sengupta, Spectrochim. Acta, Part A, 1999, 55, 1127 CrossRef.
- T. Schultz, Science, 2004, 306, 1765 CrossRef CAS PubMed.
- V. V. Shynkar, A. S. Klymchenko, G. Duportail, A. P. Demchenko and Y. Mély, Biochim. Biophys. Acta, Biomembr., 2005, 1712, 128 CrossRef CAS PubMed.
- Q. Chu, D. A. Medvetz and Y. Pang, Chem. Mater., 2007, 19, 6421 CrossRef CAS.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
- V. V. Shynkar, A. S. Klymchenko, E. Piémont, A. P. Demchenko and Y. Mély, J. Phys. Chem. A, 2004, 108, 8151 CrossRef CAS.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- J. B. Harborne and C. A. Williams, Phytochemistry, 2000, 55, 481 CrossRef CAS PubMed.
- R. Lois, Planta, 1994, 194, 498 CrossRef CAS.
- A. P. Demchenko, Trends Biotechnol., 2005, 23, 456 CrossRef CAS PubMed.
- P. F. Barbara, P. K. Walsh and L. E. Brus, J. Phys. Chem., 1989, 93, 29 CrossRef CAS.
- D. A. Parthenopoulos, D. P. McMorrow and M. Kasha, J. Phys. Chem., 1991, 95, 2668 CrossRef CAS.
- A. S. Klymchenko, T. Ozturk, V. G. Pivovarenko and A. P. Demchenko, Tetrahedron Lett., 2001, 42, 7967 CrossRef CAS.
- P. F. Barbara, P. M. Rentzepis and L. E. Brus, J. Am. Chem. Soc., 1980, 102, 2786 CrossRef CAS.
- M. Ikegami and T. Arai, J. Chem. Soc., Perkin Trans. 2, 2002, 1296 RSC.
- P.-T. Chou, G.-R. Wu, C.-Y. Wei, C.-C. Cheng, C.-P. Chang and F.-T. Hung, J. Phys. Chem. B, 2000, 104, 7818 CrossRef CAS.
- J. F. Ireland and P. A. H. Wyatt, Advances in Physical Organic Chemistry, 1976, p. 131 Search PubMed.
- P. K. Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382 CrossRef CAS.
- G. J. Woolfe and P. J. Thistlethwaite, J. Am. Chem. Soc., 1981, 103, 6916 CrossRef CAS.
- B. Dick and N. P. Ernsting, J. Phys. Chem., 1987, 91, 4261 CrossRef CAS.
- G. A. Brucker and D. F. Kelley, J. Phys. Chem., 1987, 91, 2856 CrossRef CAS.
- G. A. Brucker and D. F. Kelley, J. Phys. Chem., 1988, 92, 3805 CrossRef CAS.
- B. J. Schwartz, L. A. Peteanu and C. B. Harris, J. Phys. Chem., 1992, 96, 3591 CrossRef CAS.
- A. N. Bader, V. G. Pivovarenko, A. P. Demchenko, F. Ariese and C. Gooijer, J. Phys. Chem. B, 2004, 108, 10589 CrossRef CAS.
- N. P. Ernsting and B. Dick, Chem. Phys., 1989, 136, 181 CrossRef CAS.
- A. Ito, Y. Fujiwara and M. Itoh, J. Chem. Phys., 1992, 96, 7474 CrossRef CAS.
- K. Bartl, A. Funk and M. Gerhards, J. Chem. Phys., 2008, 129, 234306 CrossRef CAS PubMed.
- K. Bartl, A. Funk, K. Schwing, H. Fricke, G. Kock, H.-D. Martin and M. Gerhards, Phys. Chem. Chem. Phys., 2009, 11, 1173 RSC.
- B. Dick, J. Phys. Chem., 1990, 94, 5752 CrossRef CAS.
- G. Estiúa, J. Rama, A. Pereira, R. E. Cachau and O. N. Ventura, THEOCHEM, 1999, 221 CrossRef.
- J. C. Catalán, J. C. Del Valle, C. Díaz, J. Palomar, J. L. G. De Paz and M. Kasha, Int. J. Quantum Chem., 1999, 421 CrossRef.
- R. Casadesús, O. Vendrell, M. Moreno, J. M. Lluch and K. Morokuma, Chem. Phys., 2006, 325, 243 CrossRef.
- S. Ash, S. P. De, S. Pyne and A. Misra, J. Mol. Model., 2010, 16, 831 CrossRef CAS PubMed.
- C.-C. Hsieh, C.-M. Jiang and P.-T. Chou, Acc. Chem. Res., 2010, 43, 1364 CrossRef CAS PubMed.
- M. A. Bellucci and D. F. Coker, J. Chem. Phys., 2012, 136, 194505 CrossRef PubMed.
- S. Hayaki, Y. Kimura and H. Sato, J. Phys. Chem. B, 2013, 117, 6759 CrossRef CAS PubMed.
- S. Höfener, P. C. Kooijman, J. Groen, F. Ariese and L. Visscher, Phys. Chem. Chem. Phys., 2013, 15, 12572 RSC.
- M. Itoh, K. Tokumura, Y. Tanimoto, Y. Okada, H. Takeuchi, K. Obi and I. Tanaka, J. Am. Chem. Soc., 1982, 104, 4146 CrossRef CAS.
- D. McMorrow and M. Kasha, J. Phys. Chem., 1984, 88, 2235 CrossRef CAS.
- J. M. Petroski, C. De Sa Valente, E. P. Kelson and S. Collins, J. Phys. Chem. A, 2002, 106, 11714 CrossRef CAS.
- C. A. Kenfack, A. S. Klymchenko, G. Duportail, A. Burger and Y. Mély, Phys. Chem. Chem. Phys., 2012, 14, 8910 RSC.
- A. Peluso, C. Adamo and G. Re, J. Math. Chem., 1992, 10, 249 CrossRef CAS.
- M. Itoh, Y. Tanimoto and K. Tokumura, J. Am. Chem. Soc., 1983, 105, 3339 CrossRef CAS.
- M. Itoh and Y. Fujiwara, J. Phys. Chem., 1983, 87, 4558 CrossRef CAS.
- M. Itoh, Y. Fujiwara, M. Sumitani and K. Yoshihara, J. Phys. Chem., 1986, 90, 5672 CrossRef CAS.
- G. A. Brucker and D. F. Kelley, J. Phys. Chem., 1987, 91, 2862 CrossRef CAS.
- M. Itoh, Pure Appl. Chem., 1993, 65, 1629 CrossRef CAS.
- K. Chevalier, M. M. N. Wolf, A. Funk, M. Andres, M. Gerhards and R. Diller, Phys. Chem. Chem. Phys., 2012, 14, 15007 RSC.
- K. Chevalier, A. Grün, A. Stamm, Y. Schmitt, M. Gerhards and R. Diller, J. Phys. Chem. A, 2013, 117, 11233 CrossRef CAS PubMed.
- T. D. Bouman, M. A. Knobeloch and S. Bohan, J. Phys. Chem., 1985, 89, 4460 CrossRef CAS.
- H. M. Ishiki, C. Alemán and S. E. Galembeck, Chem. Phys. Lett., 1998, 287, 579 CrossRef CAS.
- S. Ash, S. P. De, H. Beg and A. Misra, Mol. Simul., 2011, 37, 914 CrossRef CAS.
- V. A. Shubert and T. S. Zwier, J. Phys. Chem. A, 2007, 111, 13283 CrossRef CAS PubMed.
- M. Weiler, K. Bartl and M. Gerhards, J. Chem. Phys., 2012, 136, 114202 CrossRef CAS PubMed.
- M. Gerhards, Opt. Commun., 2004, 241, 493 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2013 Search PubMed.
- Y. Nosenko, M. Kunitski, C. Riehn, R. P. Thummel, A. Kyrychenko, J. Herbich, J. Waluk and B. Brutschy, J. Phys. Chem. A, 2008, 112, 1150 CrossRef CAS PubMed.
- Y. Nosenko, M. Kunitski, C. Riehn, P. H. P. Harbach, A. Dreuw and B. Brutschy, Phys. Chem. Chem. Phys., 2010, 12, 863 RSC.
- A. Douhal, F. Lahmani, A. Zehnacker-Rentien and F. Amat-Guerri, J. Phys. Chem., 1994, 98, 12198 CrossRef CAS.
- A. Funk, T. Martin and M. Gerhards, in preparation for publication.
- B. C. Dian, A. Longarte, P. R. Winter and T. S. Zwier, J. Chem. Phys., 2004, 120, 133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02546f |
| This journal is © the Owner Societies 2014 |