DOI:
10.1039/C4CP02442G
(Perspective)
Phys. Chem. Chem. Phys., 2014,
16, 21172-21182
Dihydroazulene: from controlling photochromism to molecular electronics devices
Received
3rd June 2014
, Accepted 19th August 2014
First published on 20th August 2014
Abstract
Recent synthetic advances allowing large-scale preparation and systematic functionalization of the dihydroazulene (DHA)–vinylheptafulvene (VHF) photo-/thermoswitch have enabled detailed studies on how to tune optical and switching properties and have paved the way for using this system as a functional unit in molecular electronics and materials chemistry. Since discovery of its photochromism in the 1980'ies, numerous examples of DHA–VHF systems have been developed, allowing multimode switching, fluorescence-control and fine tuning of absorbance and VHF half-lives, giving insights into the mechanism of the switching event. Here, we present an overview of the properties of the DHA–VHF system, together with some selected synthetic procedures which have paved the way for its development.
Introduction
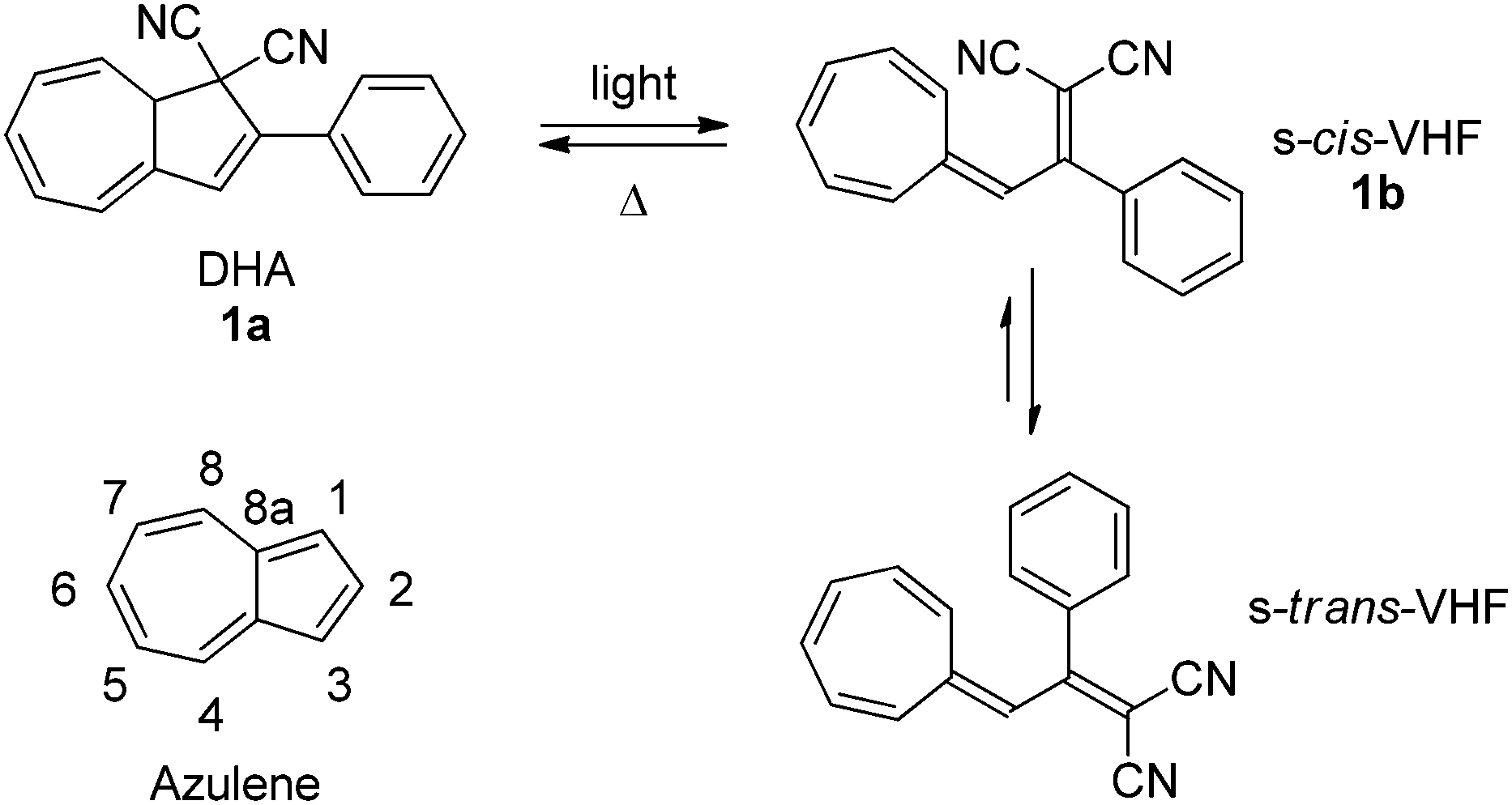
The 1,8a-dihydroazulene-1,1-dicarbonitrile (DHA) molecule is photochromic.1 Thus, by irradiation with light DHA can undergo a ring-opening to vinylheptafulvene (VHF) which in turn can return to DHA by a thermally induced ring-closure (VHF → DHA). The initial product of the ring-opening is the VHF in its s-cis-conformation but as shown by X-ray crystallography2 and calculations3 this will convert into its more stable s-trans-conformer as shown for DHA 1a–VHF 1b in Scheme 1.
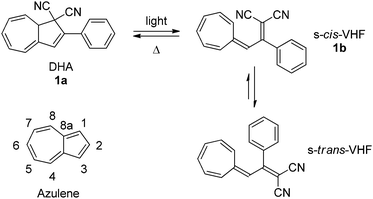 |
| | Scheme 1 Dihydroazulene (DHA)–vinylheptafulvene (VHF) photo-/thermoswitch.1 Inset: numbering of the azulene core. | |
The ring-opening reaction (DHA → VHF) occurs with a high quantum yield4 and is accompanied with significant changes in the physical properties of the molecule, such as dipole-moment,5 emission-4 and absorption spectra4 and even its single molecule conductance.6 The back-reaction is relatively slow, but very solvent dependent4,7 and can easily be monitored either by UV-Vis or NMR spectroscopies,7 often without the need for special precautions such as air or water free conditions. The fact that only the ring-opening reaction is light-induced and the VHF-isomer is relatively long-lived (t1/2 = 21 to 1545 min) makes this system convenient to study and an interesting candidate for the development of advanced photochromic devices.
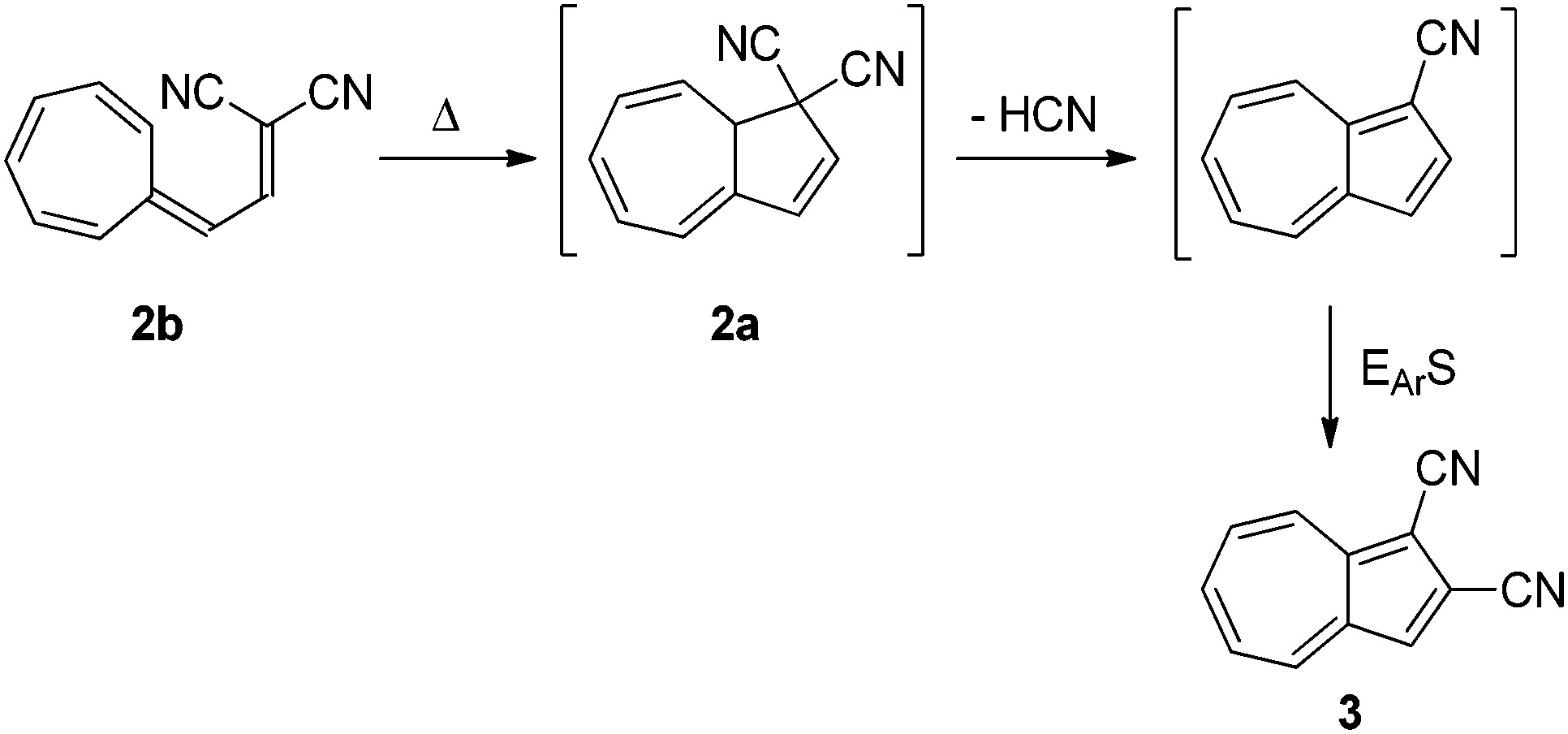
Though the first VHF was prepared in 1964 by Jutz,8 no ring-closure was reported until Tsuruta et al.9 suggested the thermally induced ring-closure of VHF 2b to DHA 2a as a route to an azulene derivative 3 as shown in Scheme 2.
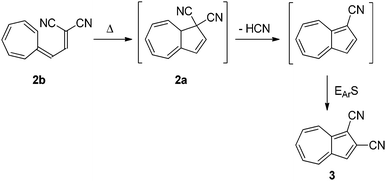 |
| | Scheme 2 Proposed thermally induced ring-closure of VHF to form azulene. EArS = electrophilic aromatic substitution.9 | |
Later, in 1984, Daub and co-workers reported the light-induced ring-opening and the reversible thermally induced back-reaction of the system.1 Several non-photochromic derivatives have also been reported, thus it seems that strongly electron withdrawing groups in positions 1 and/or 3 are necessary for the photochromism though several papers report the cyclization of VHF-like structures.10
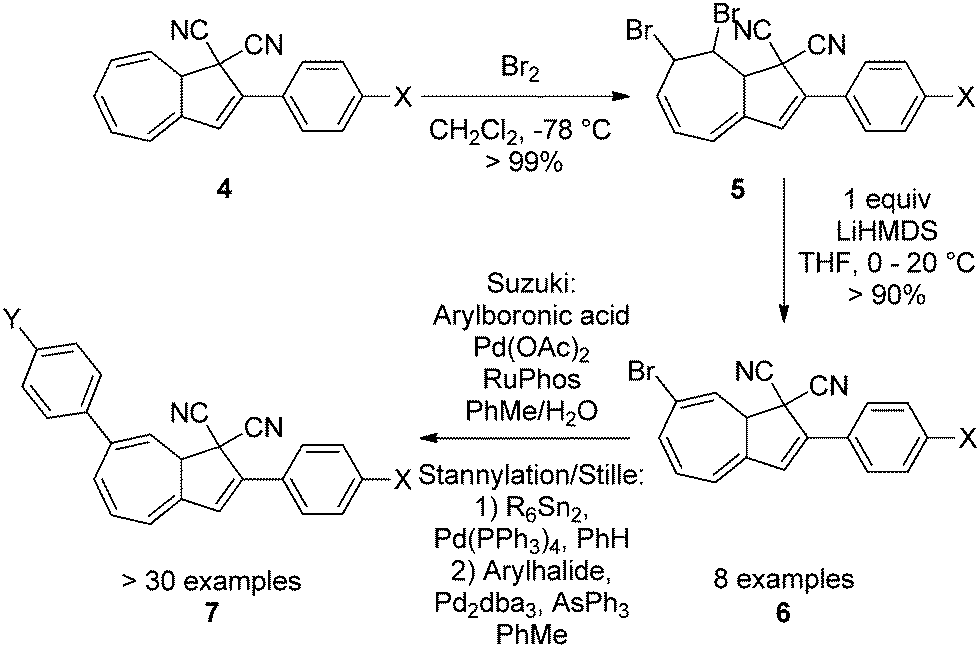
Up until 2007, the focus had been on the functionalization of the five-membered ring of the DHA. Numerous synthetic approaches to large-scale preparation of systems with both electron withdrawing and electron donating groups have been developed (4).4,11 But in 2009, Nielsen and co-workers developed the first regioselective bromination–elimination procedure for incorporation of a bromo-substituent in position 7 of the seven-membered ring (numbering according to azulene, see Scheme 1).12 This substituent has been used as a handle for further functionalizations by either Sonogashira12,13 or Suzuki cross-coupling reactions11c,14 or by first a stannylation followed by a Stille coupling15 (4–7, Scheme 3) thereby giving the opportunity to study, for the first time, the influence of placing a substituent in the seven-membered ring. A review on synthetic protocols has recently been published and shall not be covered in detail here.16
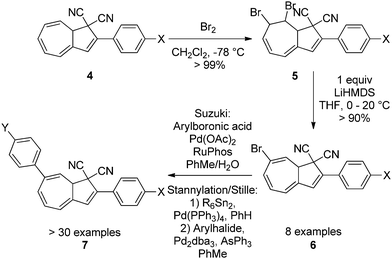 |
| | Scheme 3 Functionalization of the seven-membered ring via bromination–elimination-cross-coupling procedure. Arylation can be done by Suzuki cross-coupling or stannylation followed by a Stille coupling.11c,12,14,15 | |
Physical properties
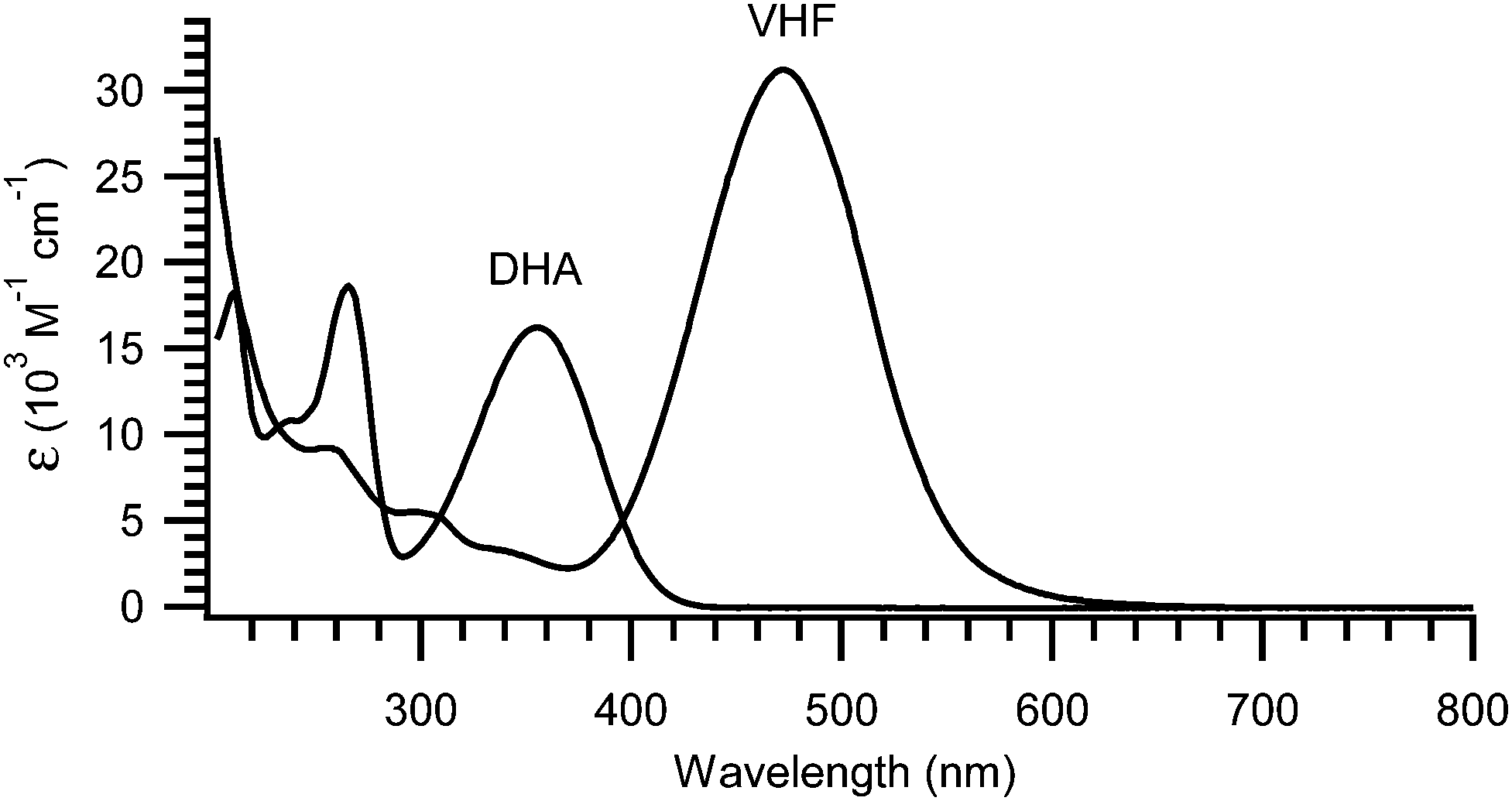
The light-induced ring-opening reaction is easily monitored by the changes in the absorption spectrum or even by the naked eye. The DHA exhibits a characteristic absorption around 350 nm (yellow compound) and by irradiation with light (300–400 nm), a significant redshift of the absorption spectrum is observed due to the formation of VHF. Thus, the VHF has a more intense absorption band around 470 nm (red compound).4,7,11–15,17 An example of absorption spectra of DHA 1a and VHF 1b is shown in Fig. 1.7
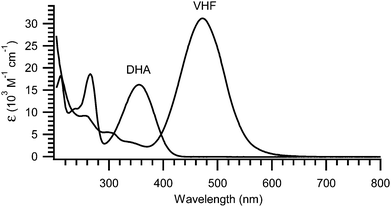 |
| | Fig. 1 UV-Vis absorption spectra of 1a (DHA) and 1b (VHF) in MeCN. The longest wavelength absorption of VHF is redshifted by 120 nm and is approximately twice as intense as that of DHA.7 | |
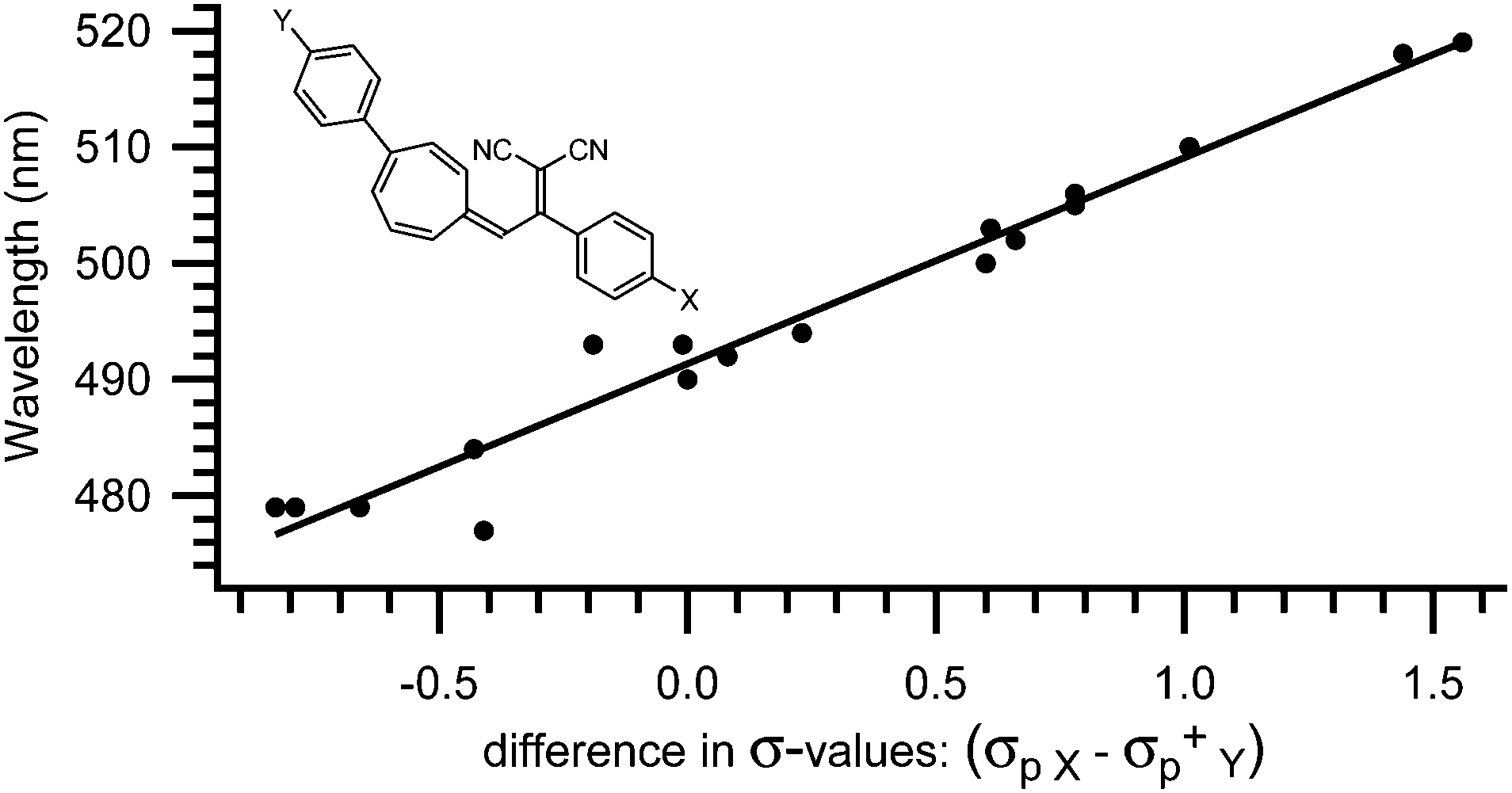
The absorption spectrum of the VHF is systematically influenced by both solvent polarity4,7 and substituents positioned in either the five- or seven-membered ring.4,11c,12–15,17 Thus, the absorption maximum is redshifted in polar solvents, such as ethanol (EtOH) or acetonitrile (MeCN) and blueshifted in non-polar solvents, such as methylcyclohexane (MCH) or toluene (PhMe). Also, the absorption maximum of the VHF is redshifted by electron withdrawing groups and blueshifted by electron donating groups positioned in the five-membered ring.4,17 Thus, the absorption of a VHF with a p-NO2 phenyl substituent in what corresponds to position 2 in the DHA (see Scheme 1) is redshifted by 14 nm (λmax = 484 nm) and blueshifted by 15 nm (λmax = 455 nm) with a p-NH2 phenyl substituent relatively to phenyl VHF (λmax = 470 nm).17 This systematic behaviour is also observed for substituents positioned in the seven-membered ring though the influence has the exact opposite effect. Here, p-NO2 phenyl in what corresponds to position 7 in the DHA caused a blueshift of 11 nm (λmax = 479 nm) relative to parent VHF (λmax = 490 nm) and p-OMe phenyl caused a redshift of 15 nm (λmax = 505 nm).14 Even more interestingly, these two effects can be combined when a molecule contains a substituent in both the five- and the seven-membered rings.17 So, if a VHF is substituted with both a nitrophenyl-group in the five-membered ring and a methoxyphenyl-group in the seven-membered ring (positions 2 and 7, respectively), both a redshift of 14 nm and 15 nm adds up to an absorption maximum of 519 nm, which is exactly what is observed experimentally.17 For a series of more than 30 VHFs with two substituents, an empirical relationship between λmax and Hammett σ-values was established as a straight line was observed when plotting the absorption maximum against the difference in Hammett σ-values (σpX − σpY+) between the two substituents (Fig. 2). In this way the absorption maxima can be fine-tuned between 478 and 563 nm.17
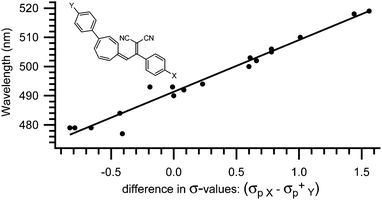 |
| | Fig. 2 Empirical relationship between λmax of substituted VHFs and the difference in σ-values (σpX − σpY+).17 | |
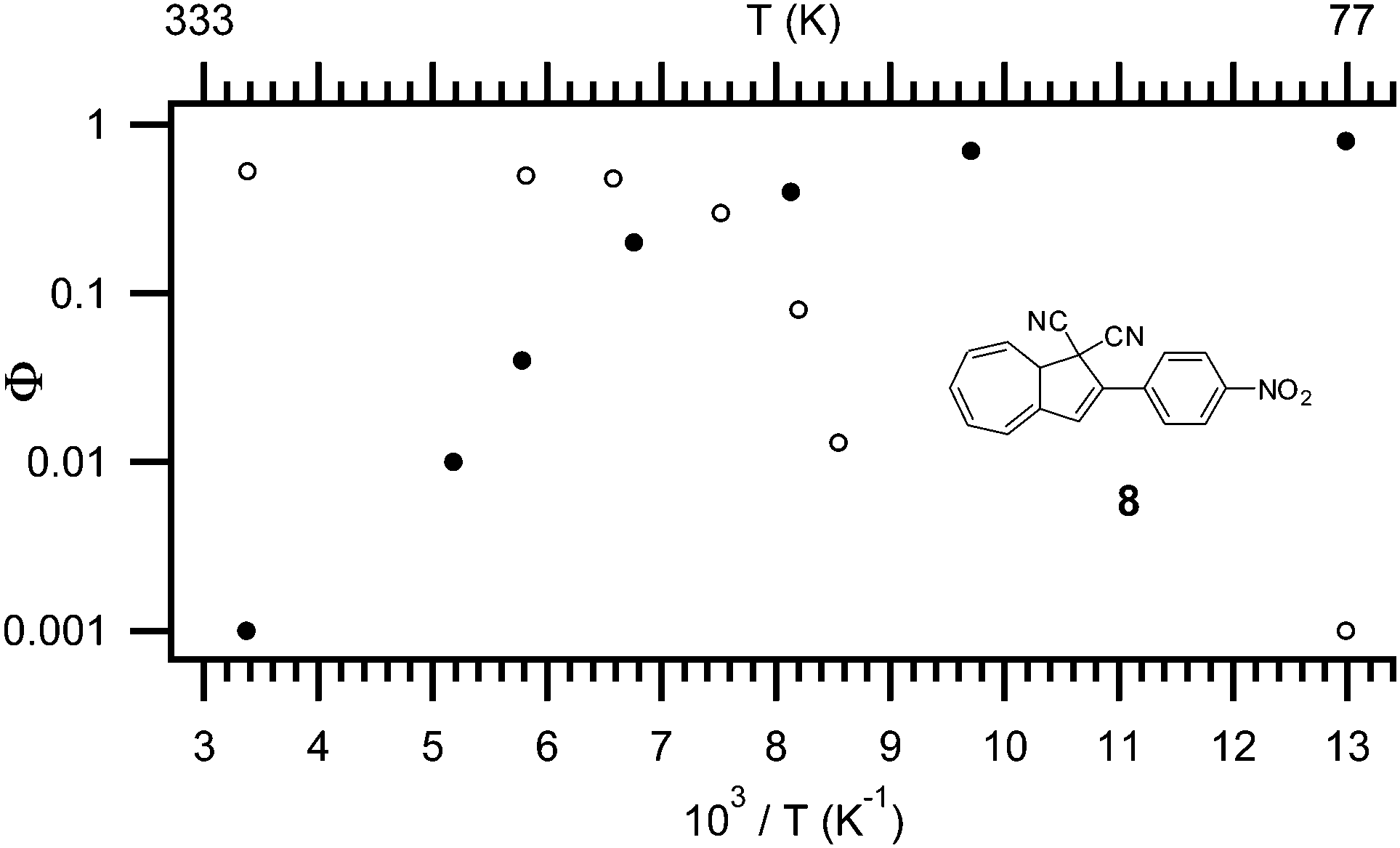
The quantum yield of the ring-opening is highly affected by both temperature,4 solvent polarity,4 and the substituent positioned in the five-4 or seven-membered ring.14 As shown in Fig. 3, at higher temperatures the main mechanism of relaxation upon light-excitation is rearrangement of the orbitals in a 10-π electron retro-electrocyclization; the ring-opening reaction from DHA to VHF. While at low temperature (−196 °C), fluorescence is the main mechanism following excitation. As depicted for DHA 8 (Fig. 3) the quantum yield is 0.53 for ring-opening and as low as 0.001 for fluorescence at room temperature. At −196 °C, the quantum yield for ring-opening is <0.001 but the quantum yield of fluorescence is 0.8 (in EtOH). The change in mechanism is explained by a change in viscosity of the solvent, since the ring-opening of DHA to VHF requires a change in molecular structure and geometry.4b
 |
| | Fig. 3 Temperature dependence of quantum yield for both light-induced ring opening reaction (open symbols) and fluorescence (filled symbols), model compound: 8, solvent: EtOH.4b | |
Interestingly, in polar solvents the quantum yield of the ring-opening reaction is very high (NO2 and CN; Φ = 0.6), but there is a clear tendency that the stronger the electron donation of an attached substituent, the lower the quantum yield at room temperature (NH2; Φ = 0.15).4a It appears that a substituent dependency is not present in non-polar solvents. Note that the quantum yield of the fluorescence is not influenced by the substituents at room temperature, but it decreases systematically with stronger electron donation at −196 °C. Also, a significant change in the quantum yield of ring-opening is observed in different solvents, though no systematic behavior is reported.1b,4
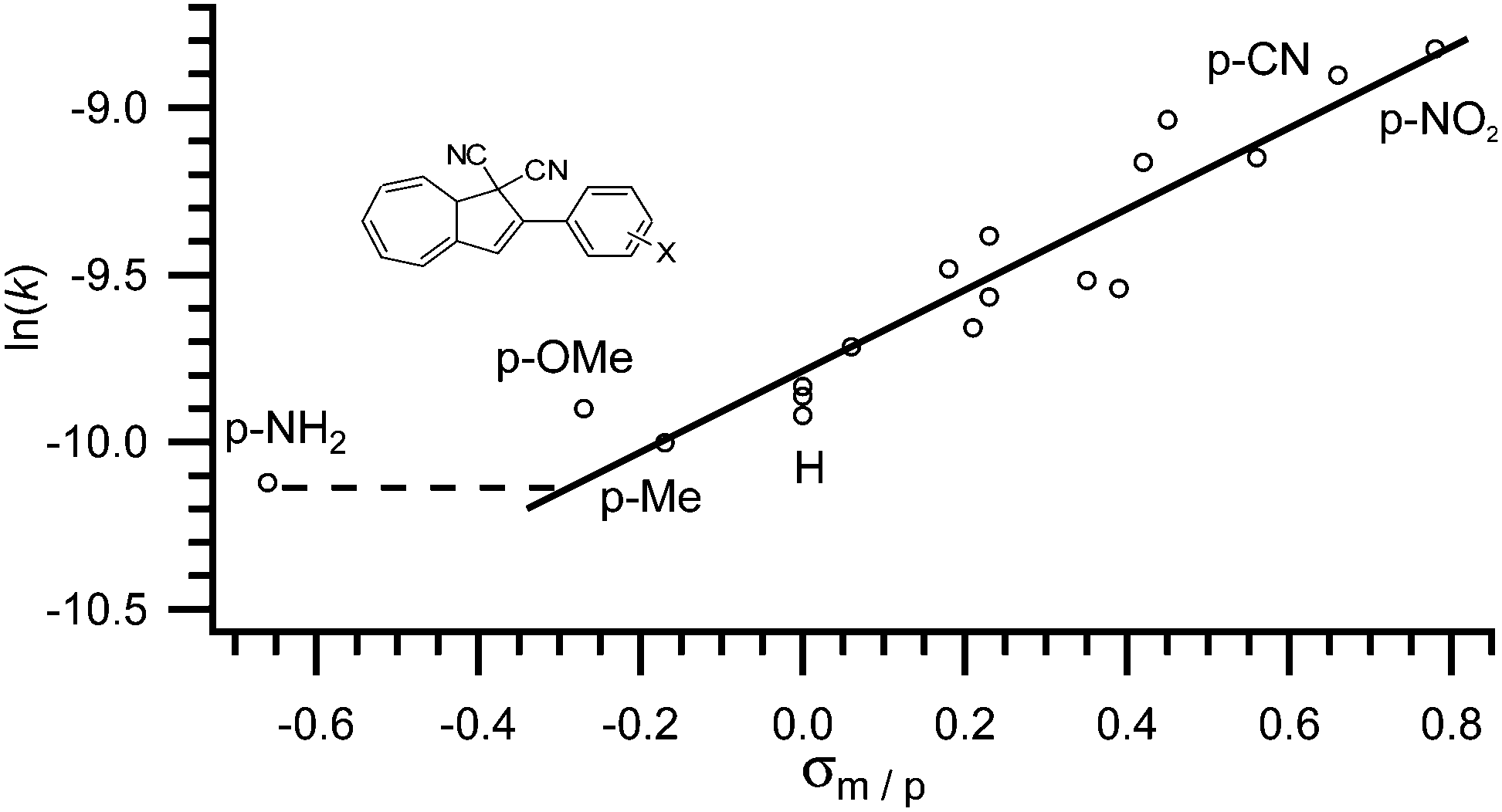
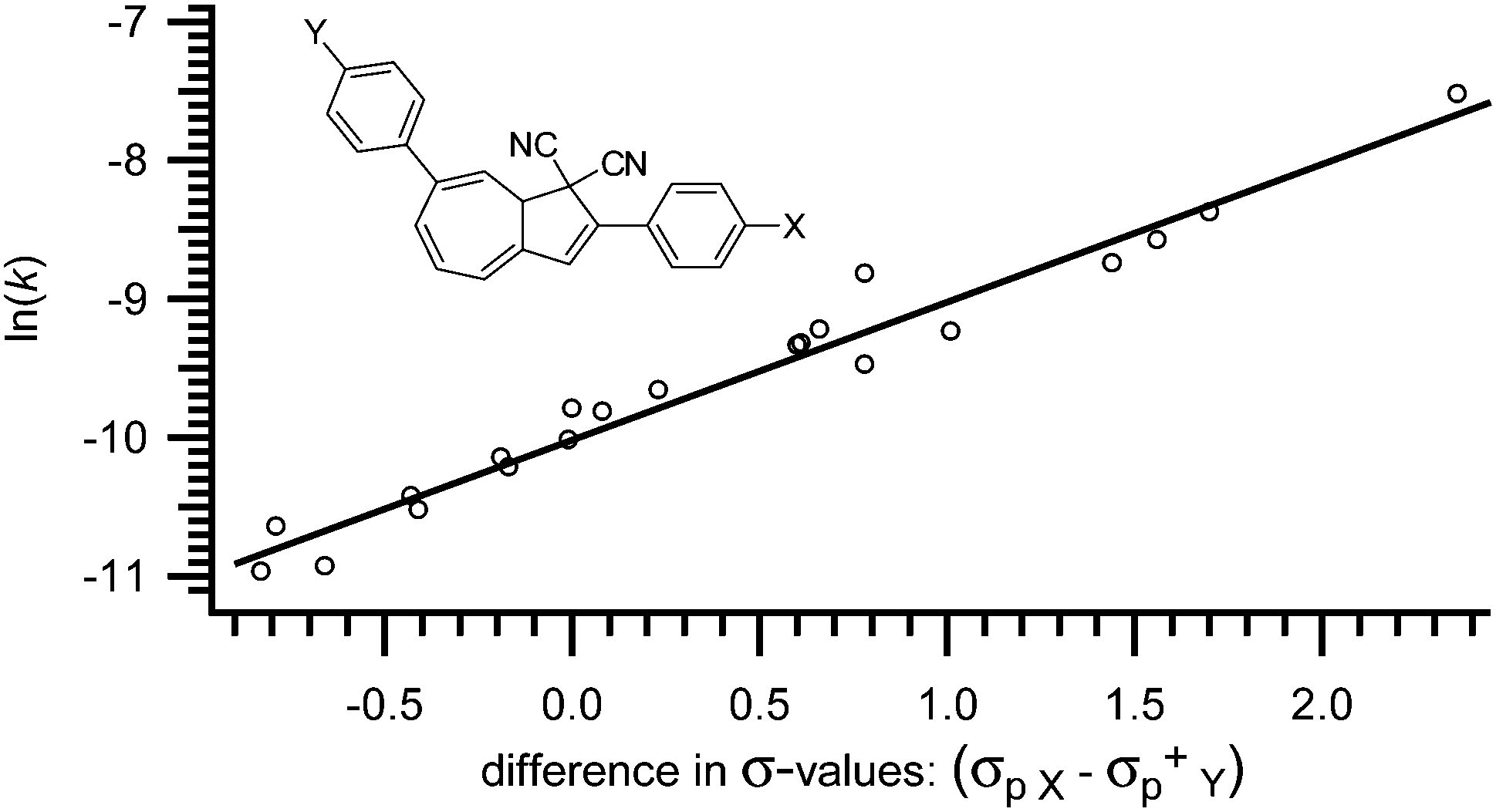
As observed by Daub and co-workers in 1993,4a some correlation between the substituent's ability to withdraw or donate electrons and the rate of back-reaction can be made. Clearly, when placed in the five-membered ring, an electron withdrawing group increases the rate of ring-closure and an electron donating group decreases the rate of ring-closure. The half-life of the VHF in MeCN is reported to increase from 79 to 230 minutes, when the electron withdrawing nitro-group is exchanged for an electron donating methoxy-group (X = NO2 to X = OMe in Fig. 4).17 In fact, as shown by Nielsen and co-workers,17 the rate of back-reaction (VHF → DHA) follows a Hammett correlation, thus, when plotting ln(k) against the appropriate σ-value a straight line is observed as shown in Fig. 4. Yet, some change in mechanism is causing the rate to be faster than predicted for an electron donating p-NH2 group.17
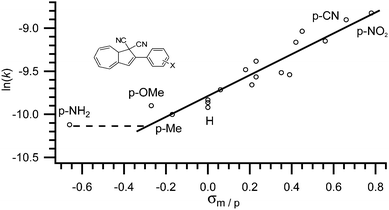 |
| | Fig. 4 Hammett correlation for thermally induced ring-closure (VHF → DHA); ln(k) against σp. [k] = s−1.17 | |
This systematic behaviour has enabled the estimation of σ-values where the value has not already been determined by the classical method. For example some triazole, thiophene and furan substituents, alkynes, butadienes, sulfones and sulfoxides were studied and their σ-values estimated from linear interpolation.17,18
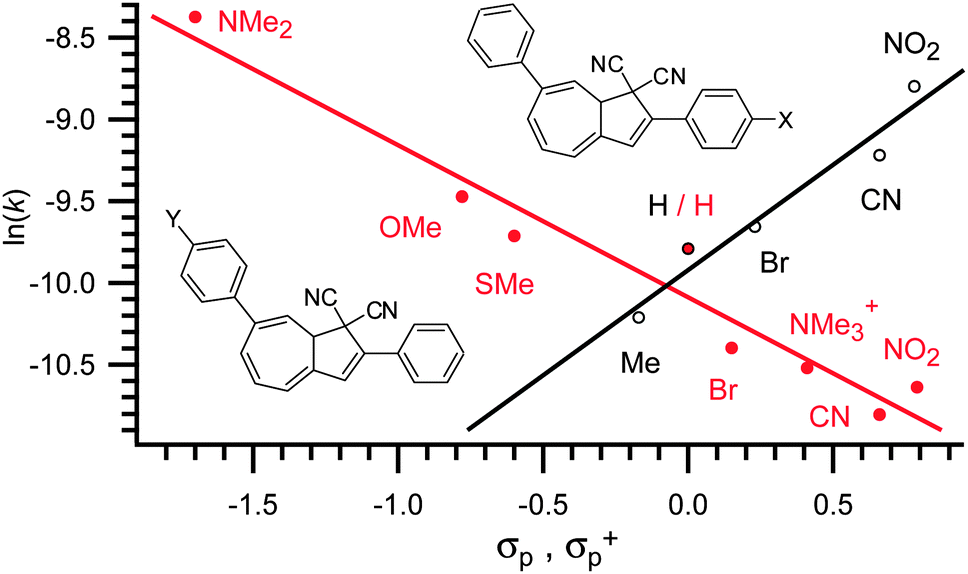
As for the absorption maxima, the complete opposite effect is observed for substituents placed in the seven-membered ring. Thus, when placed in position 7, electron donating groups tend to increase the rate of ring-closure whereas electron withdrawing groups tend to retard it. Exchanging the electron withdrawing nitro-group with the strongly electron donating dimethylamino-group caused the half-life of the back reaction to go from 490 minutes to only 50 minutes. In seven model systems, the effect of electron withdrawal and donation on the rate of ring-closure was established.13,14,17 It was shown that the rate of ring-closure obeys a Hammett correlation, thus a straight line can be fitted when plotting ln(k) against σp+.13,14,17 The Hammett correlations of two closely related systems are shown in Fig. 5 from which it is seen that a positive slope is observed for substituents-exchange in the five-membered ring (thus (de)stabilization of a negative charge in the TS) and a negative slope is observed for substituents exchange in the seven-membered ring (thus (de)stabilization of a positive charge in the TS).
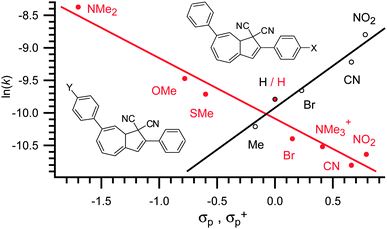 |
| | Fig. 5 Hammett correlation for thermally induced ring-closure (VHF → DHA).17 | |
Even more interesting, the two effects can be added together. Thus, if a VHF contains a substituent in both the five- and the seven-membered rings, the two effects are opposite of each other and can be simply subtracted and a straight line is again observed. Thus, when plotting ln(k) against the difference in σ-values for the two substituents, one gets a straight line (Fig. 6).
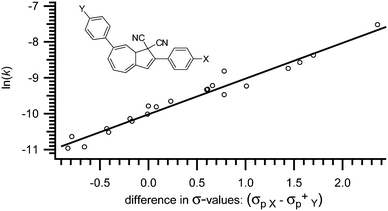 |
| | Fig. 6 Merged Hammett correlation for thermally induced ring-closure (VHF → DHA) for substituted VHFs; ln(k) against difference in σ-values (σp X − σp+Y). [k] = s−1.17 | |
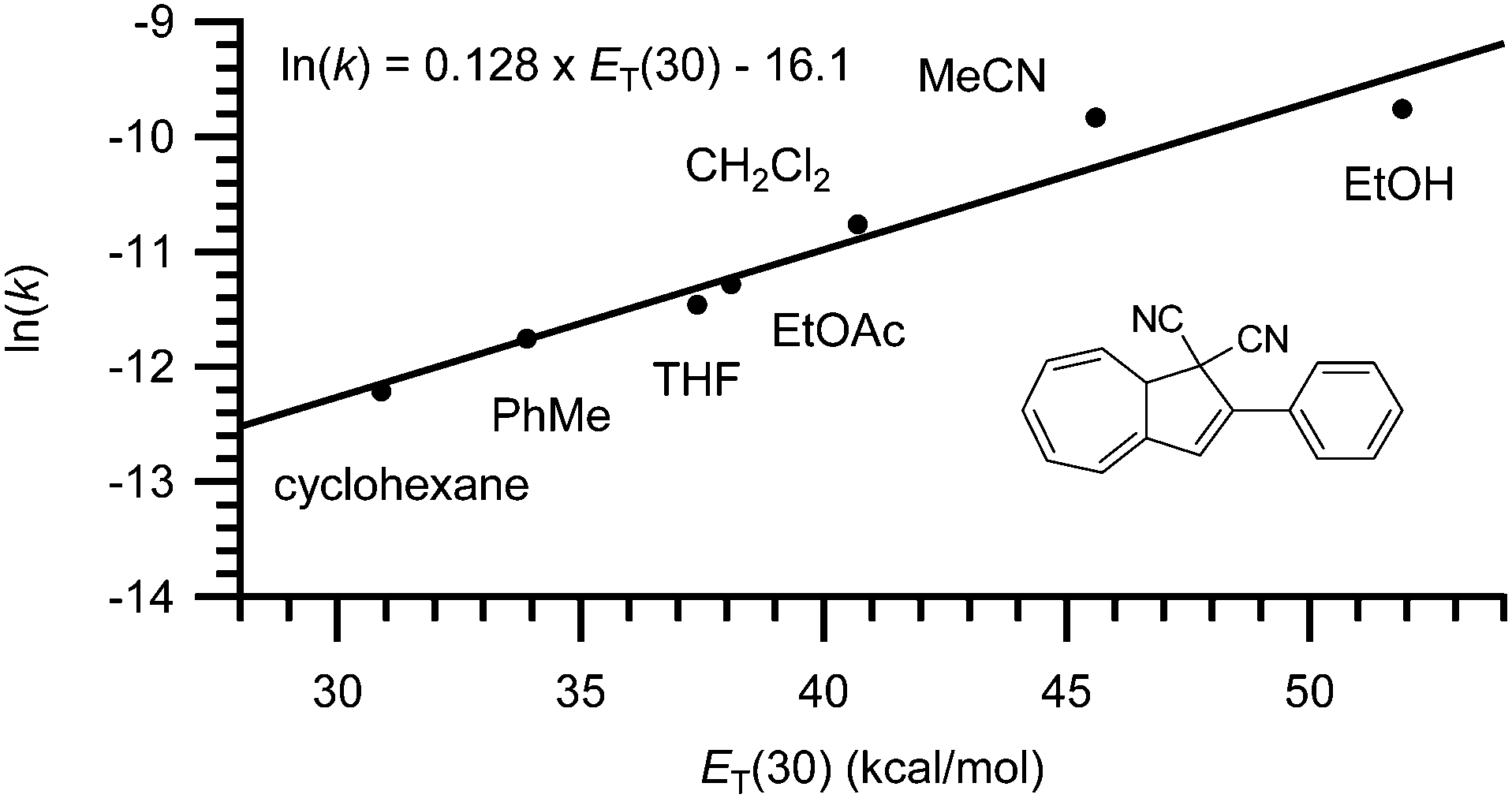
Finally, the rate of back-reaction (VHF → DHA) is also solvent dependent.4,7 Thus, in polar solvents (such as EtOH or MeCN) the rate of ring-closure is faster whereas in nonpolar solvents (such as PhMe or cyclohexane) the rate is slowed down. As shown for the conversion of VHF 1b to DHA 1a in Fig. 7, a linear trend is observed when plotting ln(k25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C) against the ET(30) value of the solvent.7
°C) against the ET(30) value of the solvent.7
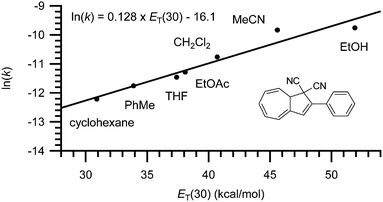 |
| | Fig. 7 Rate constant dependency on solvent polarity; ln(k) against ET(30).7 | |
In Table 1, the absorption maxima, quantum yields for the ring-opening reaction and rates of back-reaction for some representative DHA and VHFs are listed.
Table 1 Absorption maxima of DHA and VHF (for structures; see Fig. 4), quantum yield of ring-opening (DHA → VHF) and fluorescence, and rate constant for the ring closure (VHF → DHA) at 25 °Ca
| X |
Solvent |
λ
max DHA [nm] |
λ
max VHF [nm] |
Φ
DHA→VHFb (rt) |
Φ
fluorescenceb (−196 °C) |
k
25°C VHF→DHA [s−1 × 10−5] |
| EtOH = ethanol. MeCN = acetonitrile. THF = tetrahydrofuran. CH = cyclohexane. If not otherwise stated, data is taken from ref. 17. Ref. 4a. Ref. 4b. Ref. 7. |
|
p-NO2 |
EtOH |
350c |
474c |
0.53 |
0.8 |
— |
| MeCN |
386 |
484 |
0.59 |
0.8 |
14.7 |
|
p-CN |
EtOH |
362 |
474 |
0.35 |
0.9 |
20 |
| MeCN |
368 |
482 |
0.6 |
— |
13.6 |
|
p-I |
MeCN |
361 |
476 |
— |
— |
7.62 |
|
p-Br |
EtOH |
— |
— |
— |
0.8 |
— |
| MeCN |
360 |
474 |
0.5 |
— |
7.01 |
| H |
EtOH |
354d |
471d |
0.5 |
0.9 |
5.72d |
| MeCN |
353d |
470d |
0.55 |
— |
5.36d |
| CH2Cl2 |
357d |
475d |
— |
— |
2.12d |
| EtOAc |
355d |
463d |
— |
— |
1.28d |
| THF |
357d |
467d |
— |
— |
0.98d |
| PhMe |
360d |
464d |
— |
— |
0.78d |
| CH |
354d |
446d |
— |
— |
0.50d |
|
p-Me |
EtOH |
— |
— |
— |
0.8 |
— |
| MeCN |
358 |
470 |
0.4 |
— |
4.53 |
|
p-OMe |
EtOH |
— |
— |
— |
0.4 |
— |
| MeCN |
365 |
471 |
0.4 |
— |
5.02 |
|
p-NH2 |
EtOH |
— |
— |
— |
0.15 |
— |
| MeCN |
389 |
455 |
0.4 |
— |
4.02 |
|
p-NH3+ |
MeCN |
358 |
479 |
— |
— |
9.22 |
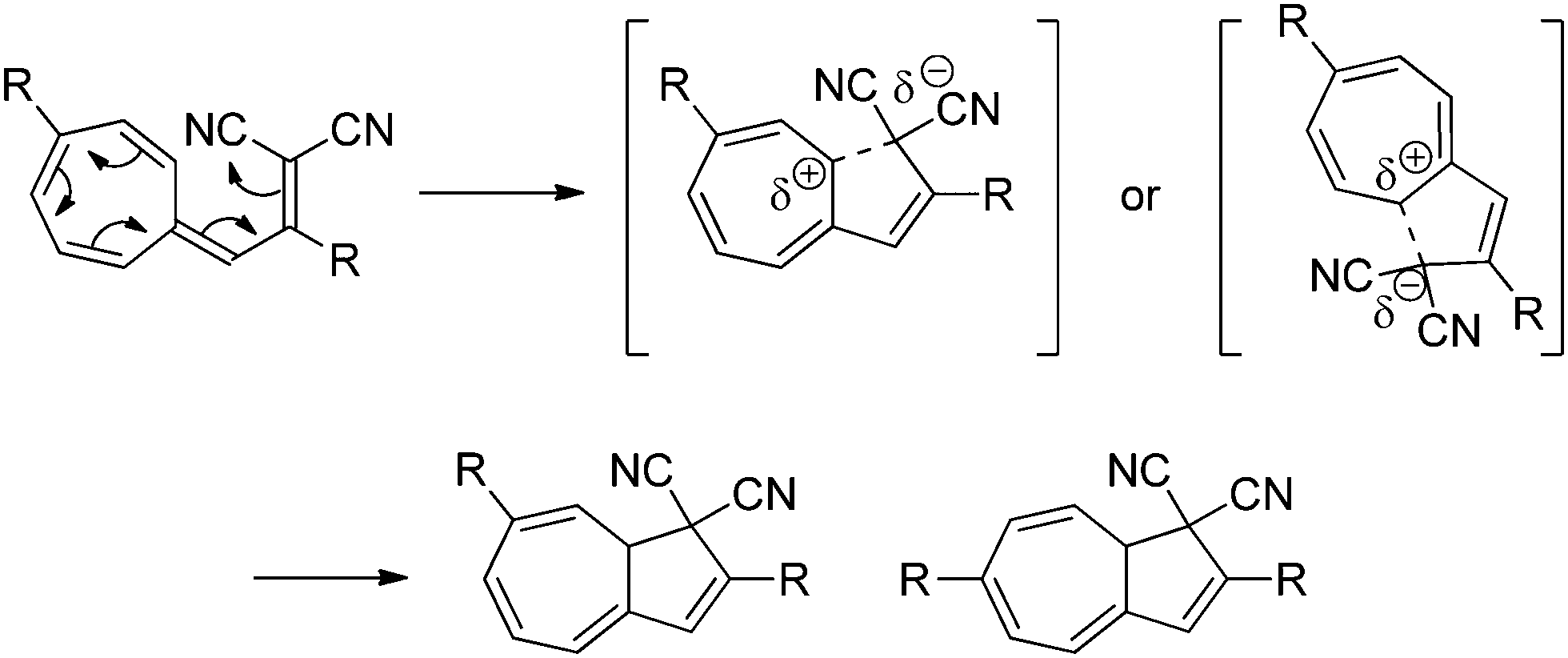
The substituent and solvent polarity dependency of the ring-closure (VHF → DHA) can be explained by the presence of a highly polarized or even charge-separated intermediate or transition state (TS), with a positively charged seven-membered ring and a negatively charged malononitrile moiety. This TS is then stabilized by polar solvents or destabilized by non-polar solvents. In the seven-membered ring, the positive charge building up in the TS is destabilized by electron withdrawing groups, thus decreasing the rate of ring-closure, and stabilized by electron donating groups, thereby increasing the rate of ring-closure. The suggested mechanism for the thermally induced ring-closure is depicted in Scheme 4.
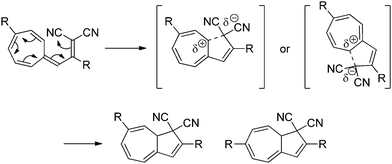 |
| | Scheme 4 Proposed mechanism of the thermally induced ring-closure reaction. Two different conformations of the TS results in two different DHAs. | |
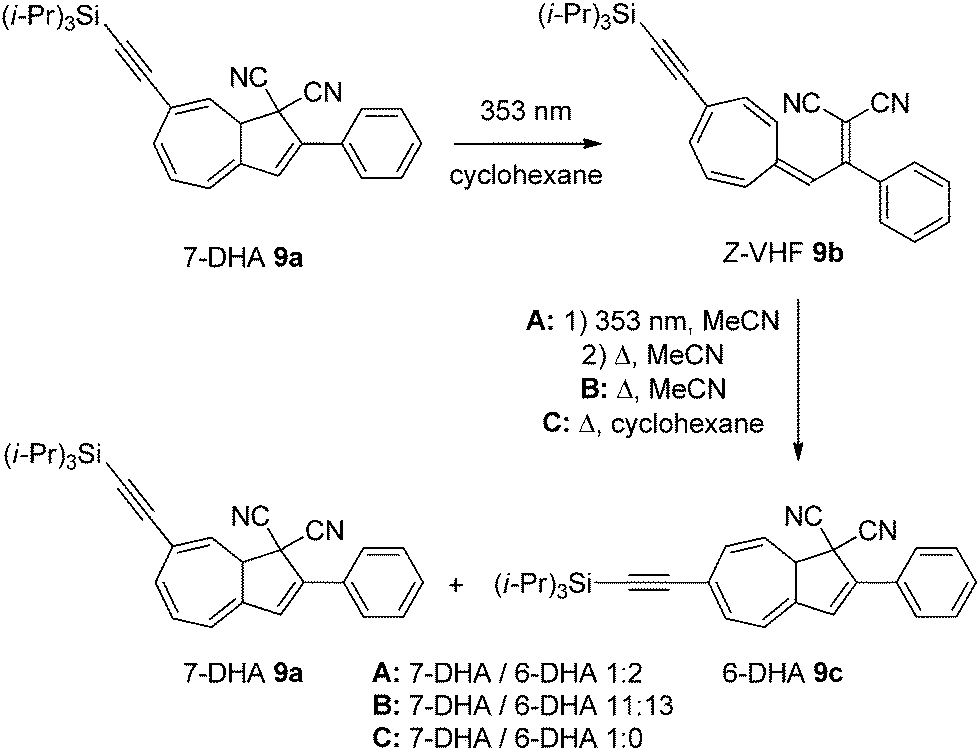
After one light–heat cycle (DHA → VHF → DHA), the pure 7-substituted DHA 9a had partly isomerized to the corresponding 6-isomer 9c,12,13 indicating that the VHF 9b can isomerize via a zwitterionic intermediate that allows the rotation around the 3aC–3C single bond to give a mixture of the two DHA isomers (numbering according to azulene, see Scheme 1). Light-induced cis–trans isomerization of the exocyclic double bond in the VHF 9b also takes place (situation A), but it has been shown that a pure Z-VHF (prepared by irradiation in cyclohexane) reverts to a mixture of DHAs when heated in MeCN (situation B) but to pure 7-DHA 9a when heated in cyclohexane (situation C). This indicates that while cis–trans isomerization may take place both during the ring-opening reaction and by light-irradiation of the VHF, it also happens during the ring-closure (in polar solvents).3 The outcomes of a single light–heat cycle under three different conditions are depicted in Scheme 5.
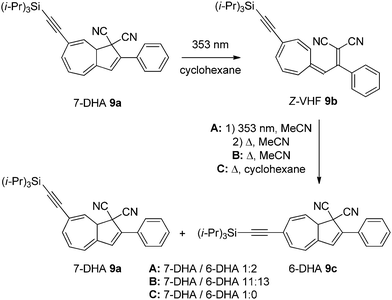 |
| | Scheme 5 Solvent dependent ratios between isomers of DHAs after one light–heat cycle under three different sets of conditions. | |
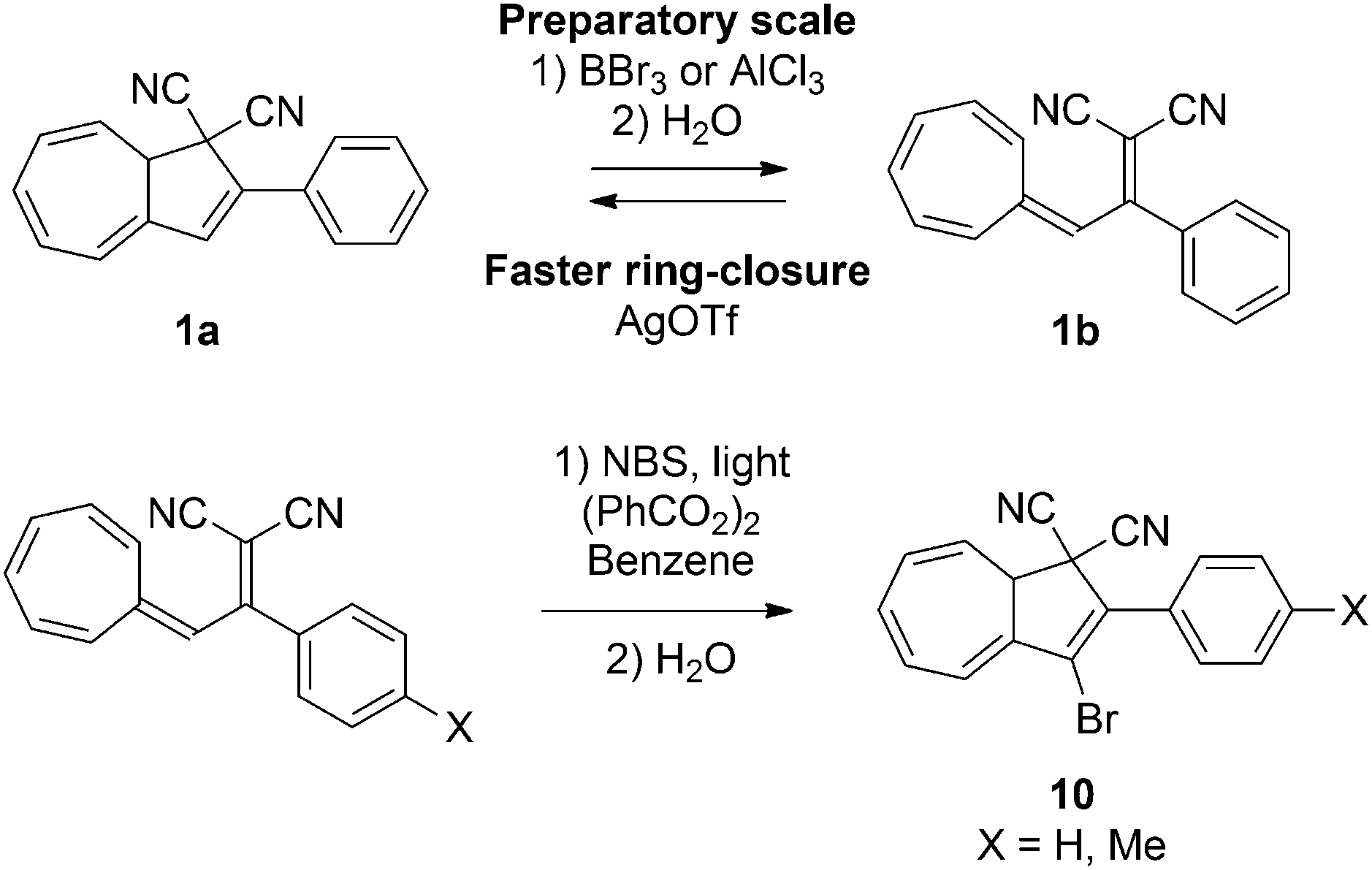
Lewis acid controlled switching
By the means of a relatively weak light source such as a standard laboratory UV-lamp, up to 10 mg of DHA can easily and quickly be converted to its corresponding VHF, but on a preparatory scale the rate of back-reaction becomes overwhelmingly fast. In the quest of being able to prepare VHF on a larger scale it was found that coordination between Lewis acids like AlCl3 or BBr3 to the nitriles is so strong that it in fact can induce the ring-opening reaction without the need for light (Scheme 6).19 This chemically-induced ring-opening has been used to incorporate a bromo-substituent at position 3 (10) by treatment of the VHF with N-bromosuccinimide (NBS) in the presence of benzoyl peroxide and light (Scheme 6).2b Only using this procedure has the VHF so far been prepared on gram-scale. While strong Lewis acids induce the ring-opening, weak Lewis acids (AgOTf or ZnCl2) increase the rate of the thermally induced back-reaction (half-life can be reduced from 597 min to 9 min in 1,2-dichloroethane).
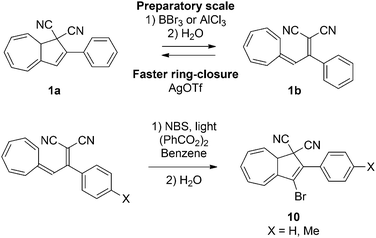 |
| | Scheme 6 Strong Lewis acid induced ring-opening and its application for post-functionalization of the DHA. NBS = N-bromosuccinimide. X = H, Me. | |
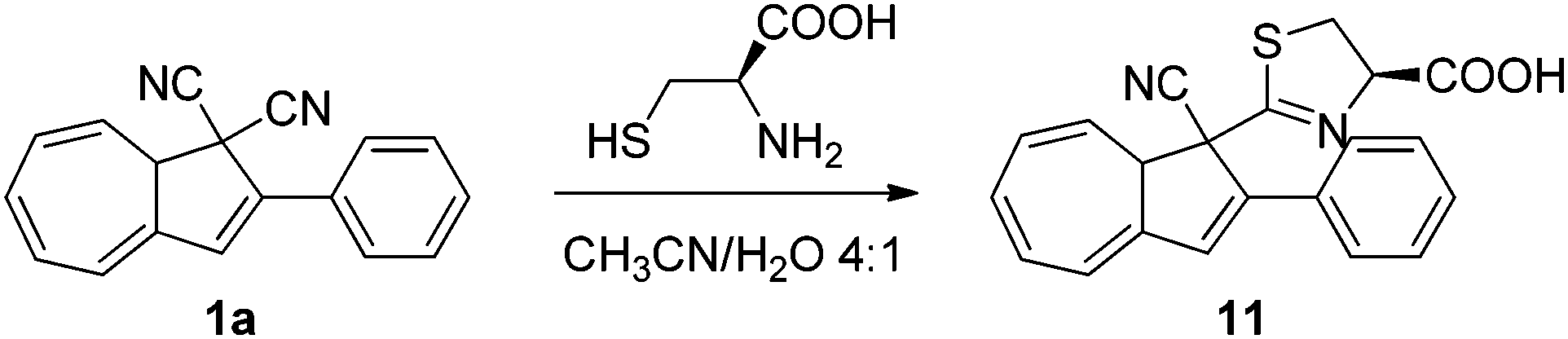
Colorimetric probing of cysteine
The colour change associated with the DHA–VHF conversions was recently used in a colorimetric probe for the detection of thiols.20 While a solution of DHA–VHF 1a–1b in MeCN–H2O turns more and more red upon exposure to light owing to the gradual conversion of DHA to VHF, a solution containing the amino acid cysteine first turns red but then in time changes colour to yellow. It seems that cysteine reacts with one of the cyano groups on DHA, forming a 4,5-dihydro-1,3-thiazole ring, corresponding to the DHA product 11 (Scheme 7). Ring-opening of this DHA generates a VHF with a blueshifted absorption maximum relative to that of the VHF 1b. This VHF very slowly returns to the DHA 11. Detection of the antioxidant agent dithiothreitol and ethanethiol was also accomplished based on visible colour changes, but the products formed between 1a and these thiols has so far not been elucidated in detail.
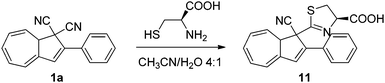 |
| | Scheme 7 Reaction between DHA and cysteine. | |
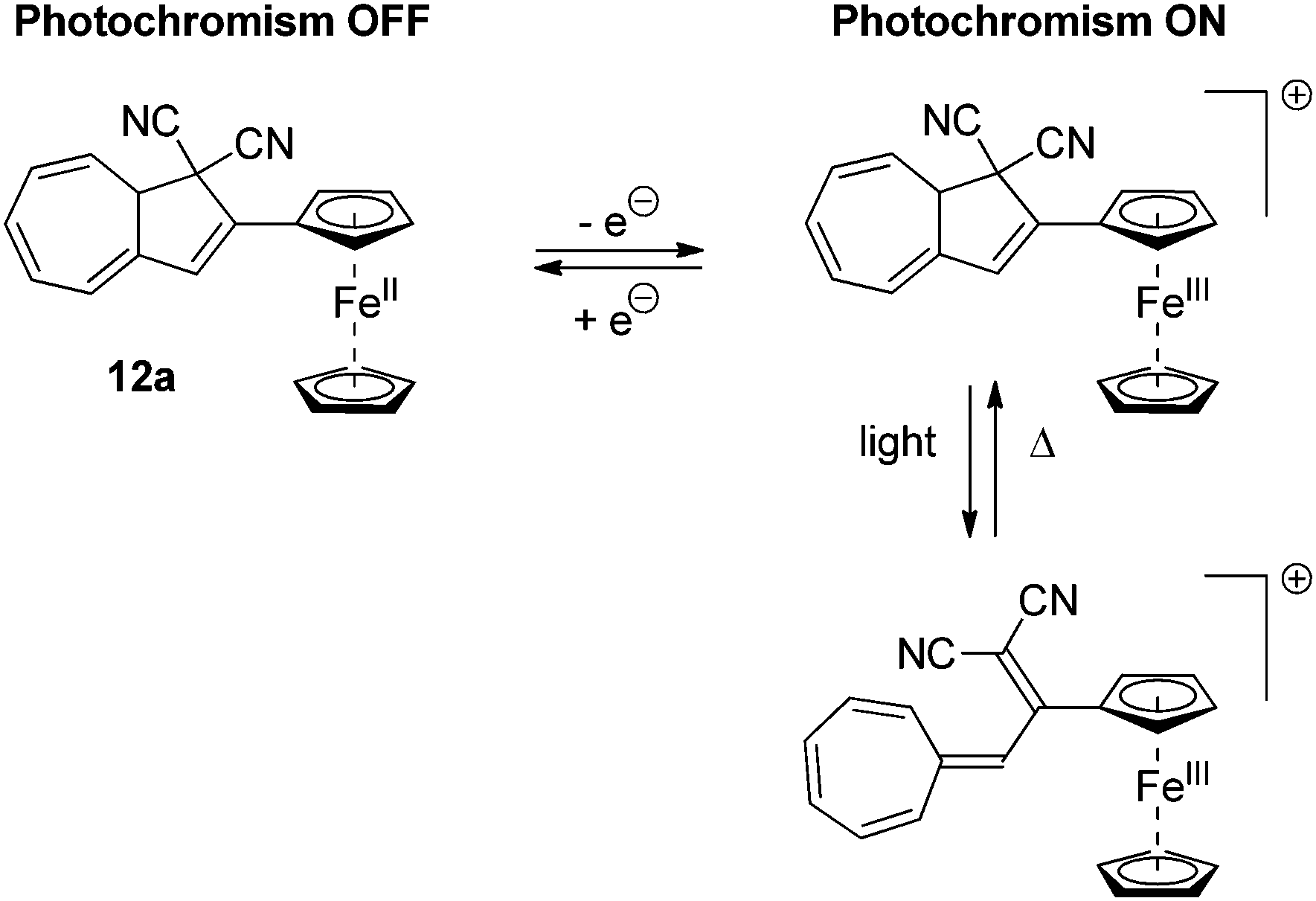
Redox-controlled and multimode photoswitching
Several examples of DHAs, which can be controlled by a redox-active substituent, have been reported. The ferrocene DHA 12a, shown in Scheme 8, can be reversibly electrochemically oxidized to the radical cation, which, in contrast to the neutral state, is photochromic.21 By light-irradiation of the neutral state of the DHA, no change in colour was observed. On the other hand, by electrochemical oxidation or addition of [Ph3N˙+/SbCl6˙−] and subsequent irradiation with light caused the disappearance of a band in the UV-Vis absorption spectrum at 362 nm and the formation of a band at 470 nm corresponding to ring-opening.
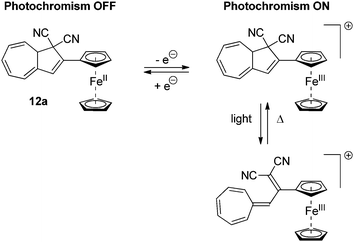 |
| | Scheme 8 Ferrocene substituted redox-controllable ON/OFF switch of the DHA–VHF-system. By reversible oxidation of the ferrocene unit, the photochromism is activated. | |
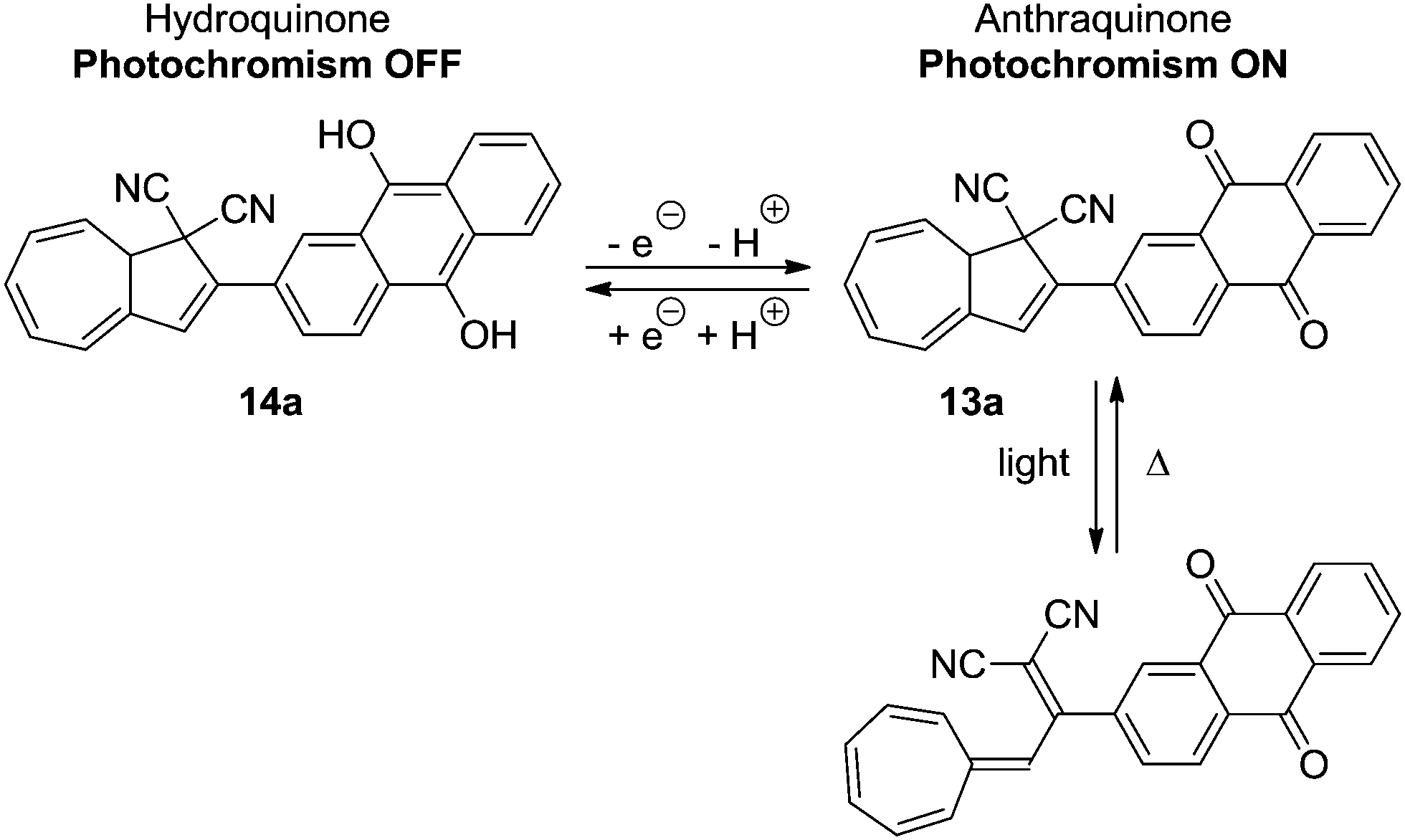
In another system, 13a, by the oxidation state of a covalently bound anthraquinone, the quantum yield of the ring-opening reaction is also affected (Scheme 9).22 Reversible reduction of the anthraquinone unit to the hydroquinone 14a caused the quantum yield of the light-induced reaction to be significantly lowered.
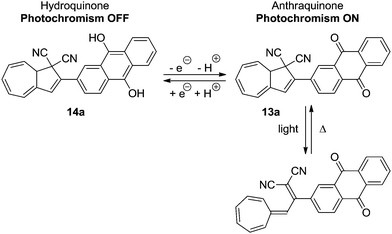 |
| | Scheme 9 Anthraquinone incorporated DHA–VHF. By reduction of the anthraquinone unit, the quantum yield of the DHA is significantly reduced. | |
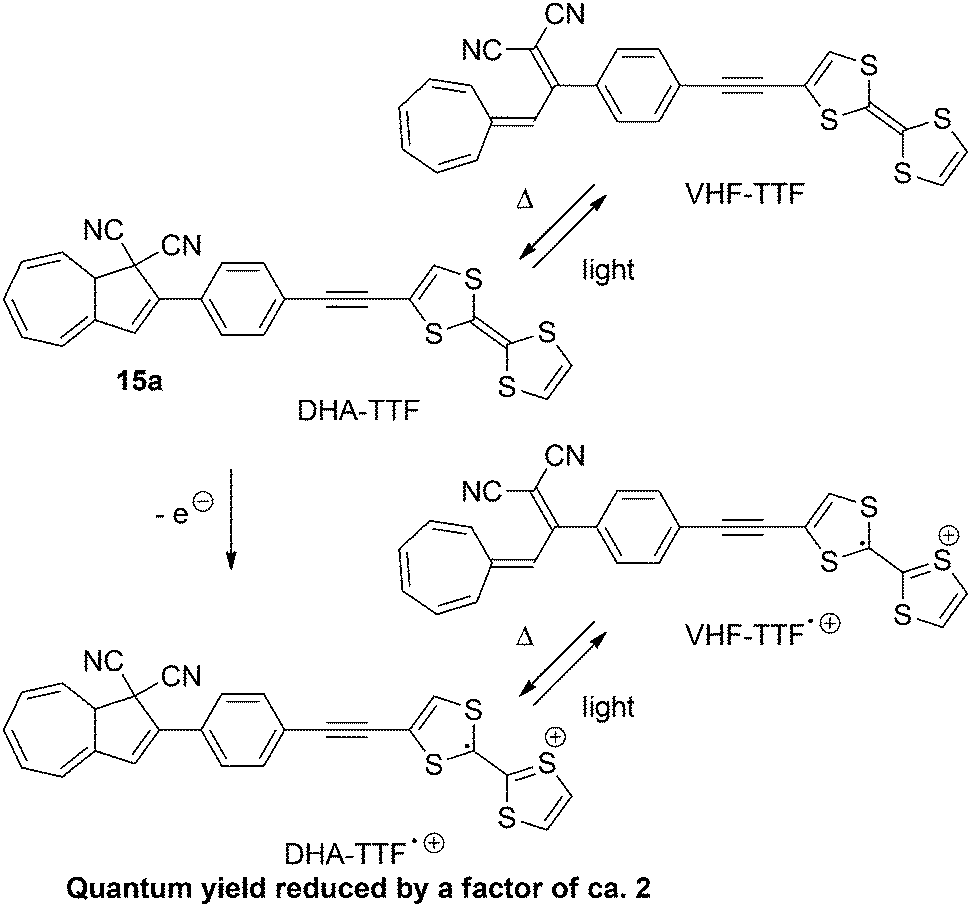
Similar results were found by Nielsen and co-workers23 with DHA–VHF-derivatives of dithiafulvene (DTF) or tetrathiafulvalene (TTF). The low quantum yields of the two DHAs can be explained by the presence of the electron donor. By (electro)chemical oxidation of the TTF-unit of 15a the quantum yield is further decreased, presumably because of an electron transfer from the excited state of DHA to radical cation of TTF. The switching between states of the TTF-substituted DHA 15a are shown in Scheme 10.23b
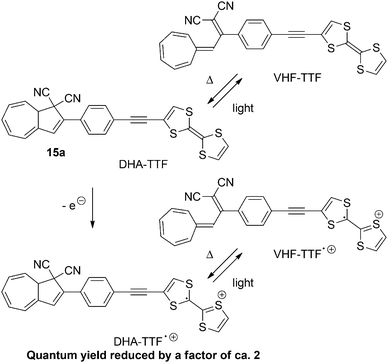 |
| | Scheme 10 Switching of the TTF derivative of the DHA–VHF-system. By oxidation of the TTF-unit to the radical cation, the quantum yield of the ring-opening (DHA → VHF) is further reduced. | |
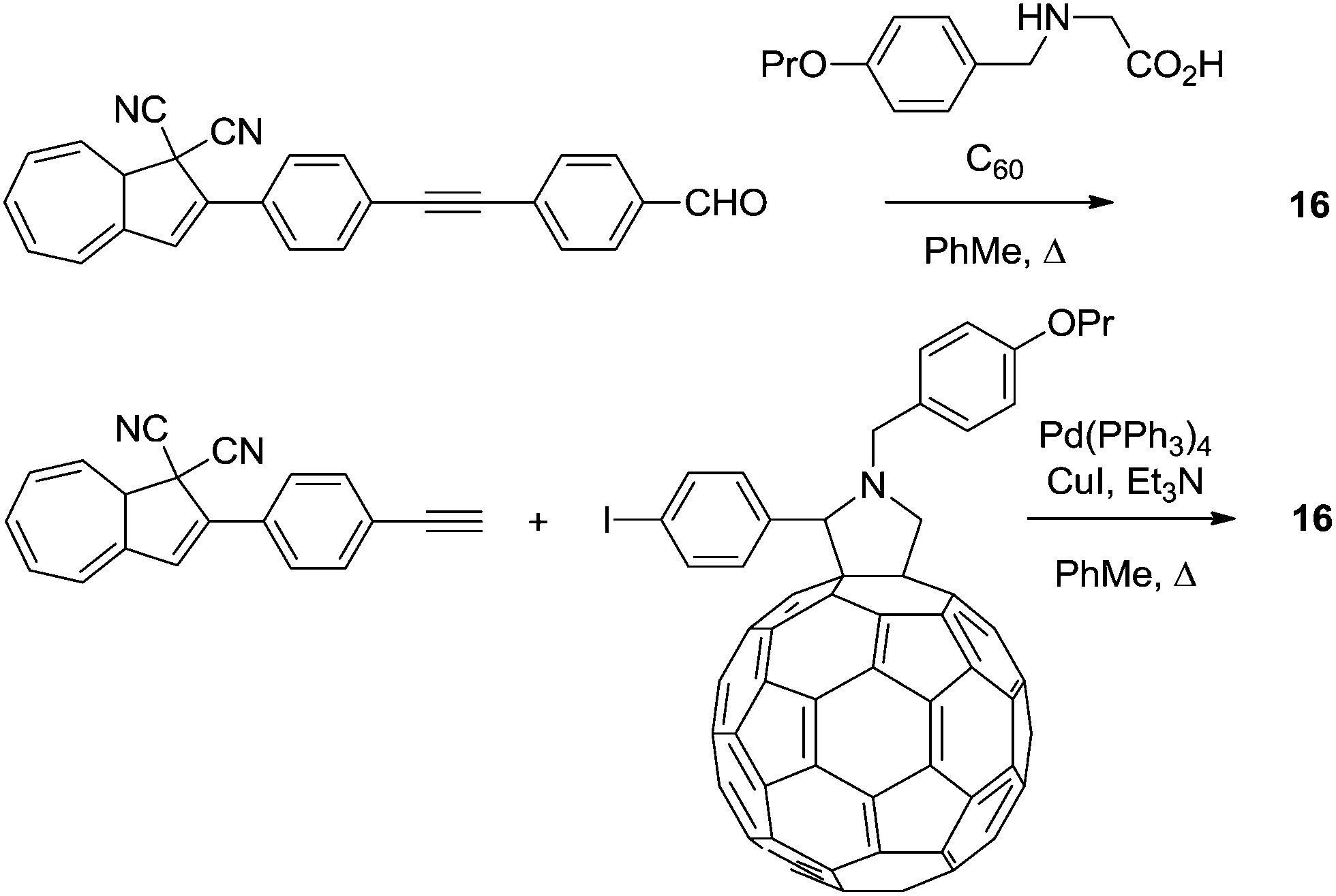
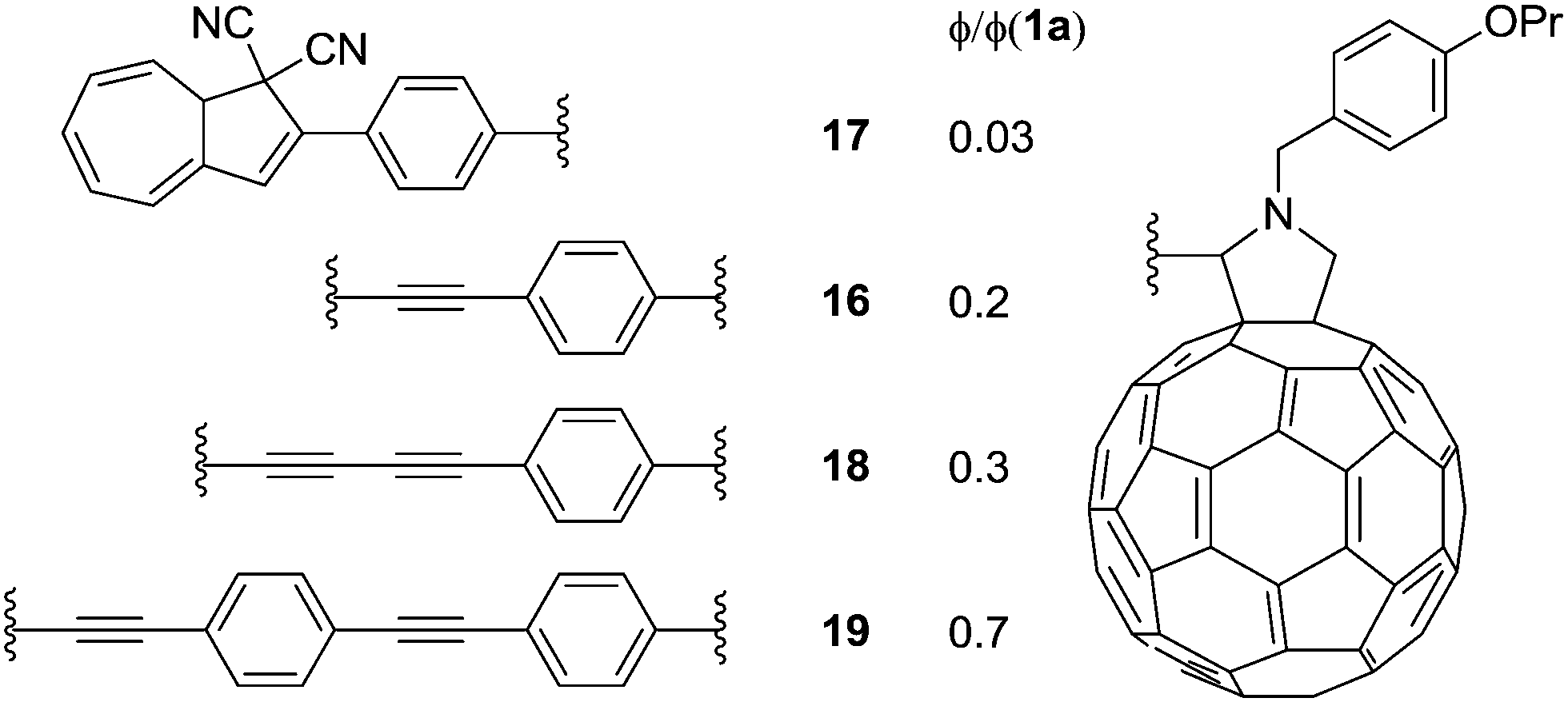
Later, in 2012,24 a series of DHAs connected to buckminsterfullerene (C60) by a π-spacer with varying length was prepared (16–19), by either Prato or Sonogashira reactions. Representative syntheses of 16 are shown Scheme 11. Here, it was shown that the ring-opening of the DHA to VHF was significantly quenched and the closer the fullerene was placed to the photoswitch, the lower the quantum yield, presumably because of a light-induced electron-transfer from the excited state of the DHA to the C60 electron acceptor (Fig. 8).
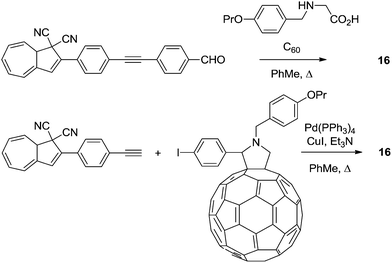 |
| | Scheme 11 Two approaches to the synthesis of DHA–fullerene conjugates via Prato or Sonogashira reactions. | |
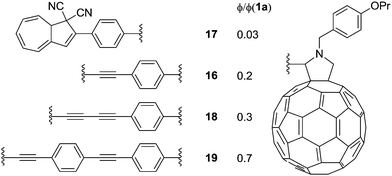 |
| | Fig. 8 Structures of DHA–C60 conjugates and estimated quantum yields (ϕ) for the DHA → VHF conversion relative to that of DHA 1a, i.e., quantum yields ratios. | |
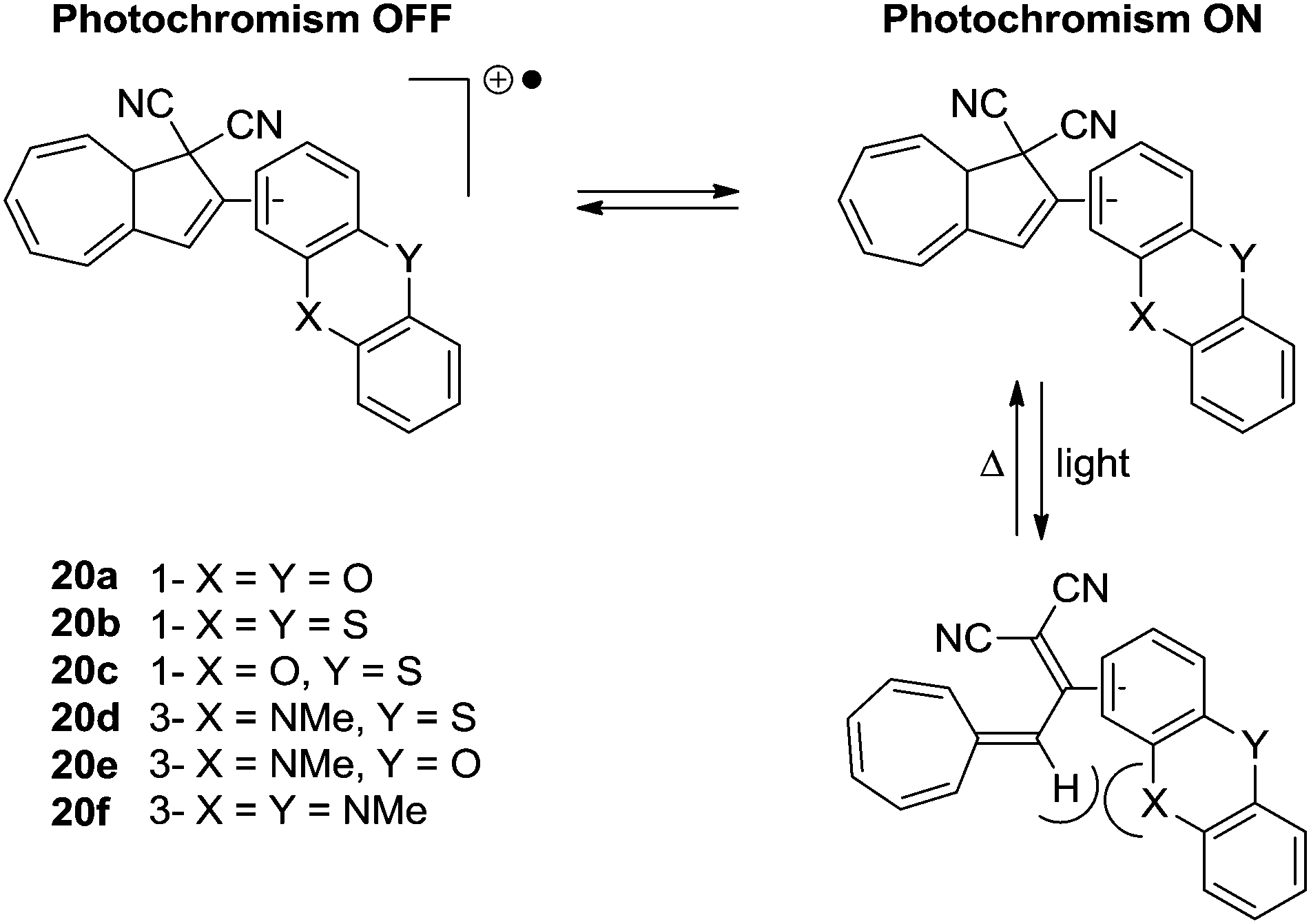
The first approach to a multimode switch partly based on the DHA–VHF-system is that of 20a–f and is shown in Scheme 12.25 By connecting the DHA–VHF to an electron-rich heteroaromatic subunit the photochromic behavior was attempted altered by the oxidation state. By the reversible oxidation of the subunit, the photochromism could successfully be suppressed. Interestingly, the rate of the ring-closure for VHF 20b was exceptionally low which is explained by the peri interaction between the sulfur atom and the hydrogen as indicated in the s-cis conformer of the VHF, thus, inhibiting the s-trans-VHF to s-cis-VHF conversion.
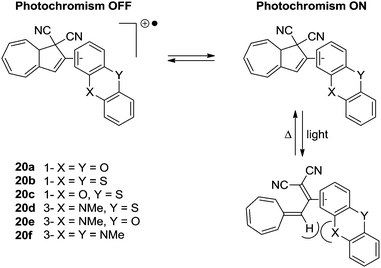 |
| | Scheme 12 Multimode redox-controlled DHA–VHF switch. The quantum yields for the ring-opening of the DHAs are reduced by oxidation of the subunit. | |
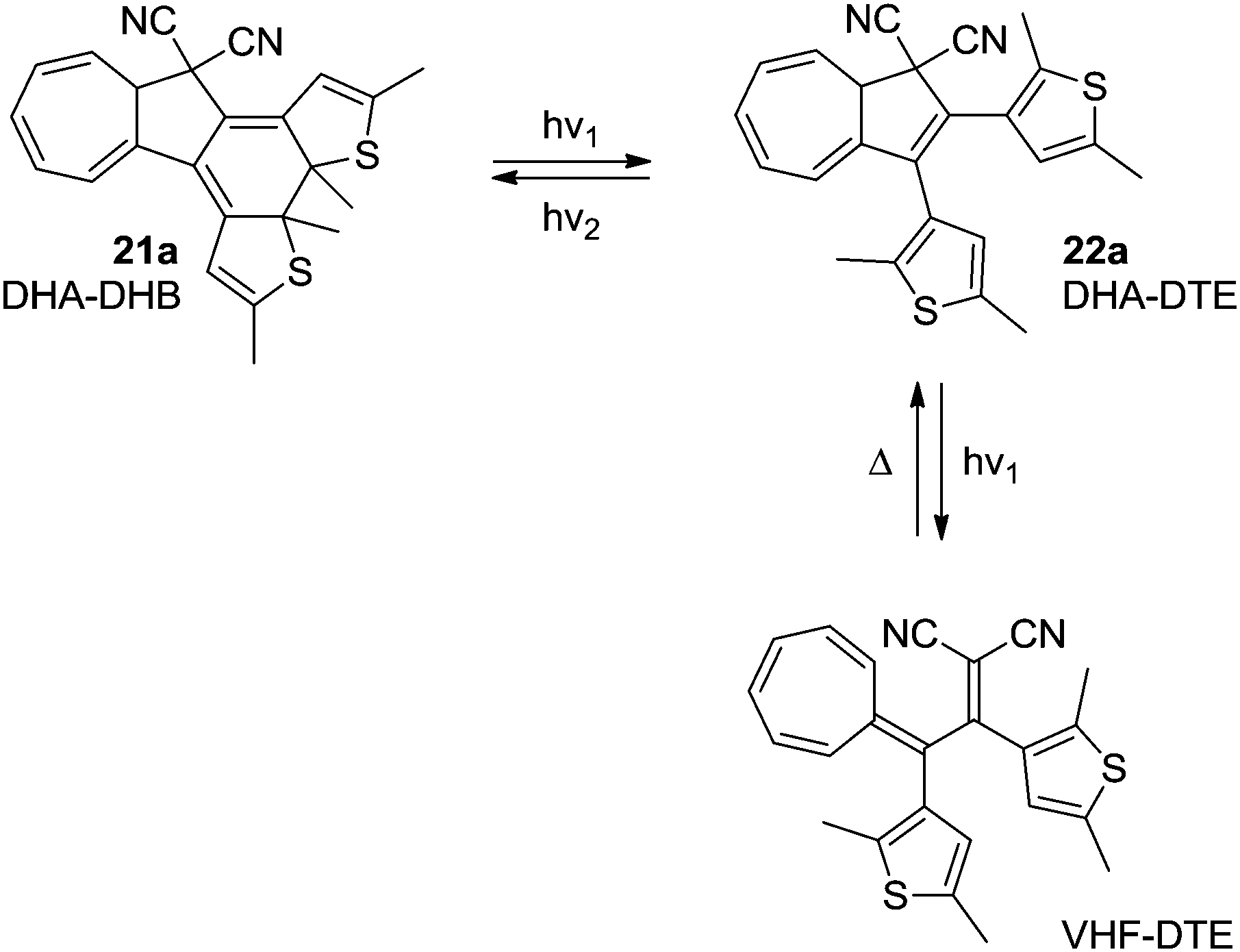
In the system 21a–22a, the combination of the photochromic dithienylethene (DTE)–dihydrothienobenzothiophene (DHB) unit, with the DHA–VHF, theoretically gives the potential to switch between four different states using light and heat as stimuli; (1) DHA–DHB (2) DHA–DTE (3) VHF–DTE (4) “VHF–DHB” (zwitterionic).26 Unfortunately, the excitation wavelengths of the DHA and DTE units are very similar, which means that a photostationary state between VHF–DTE (DHA → VHF) and DHA–DHB (DTE → DHB) is observed. Even though this is solvent dependent, no wavelength of excitation was found that exclusively excites only one of the units. The DHA–DHB state can be reversed to DHA–DTE photochemically (not thermally). The observed conversions are shown in Scheme 13.
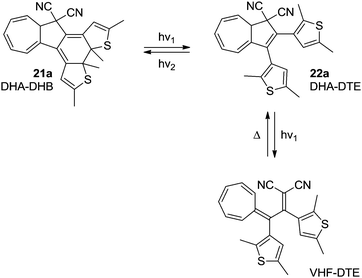 |
| | Scheme 13 Multimode-switching of DHA–DTE between states of the units; DHA–VHF and DTE–DHB. By irradiation with light with different wavelengths, switching of either the DHB–DTE or the DHA–VHF can be triggered. | |
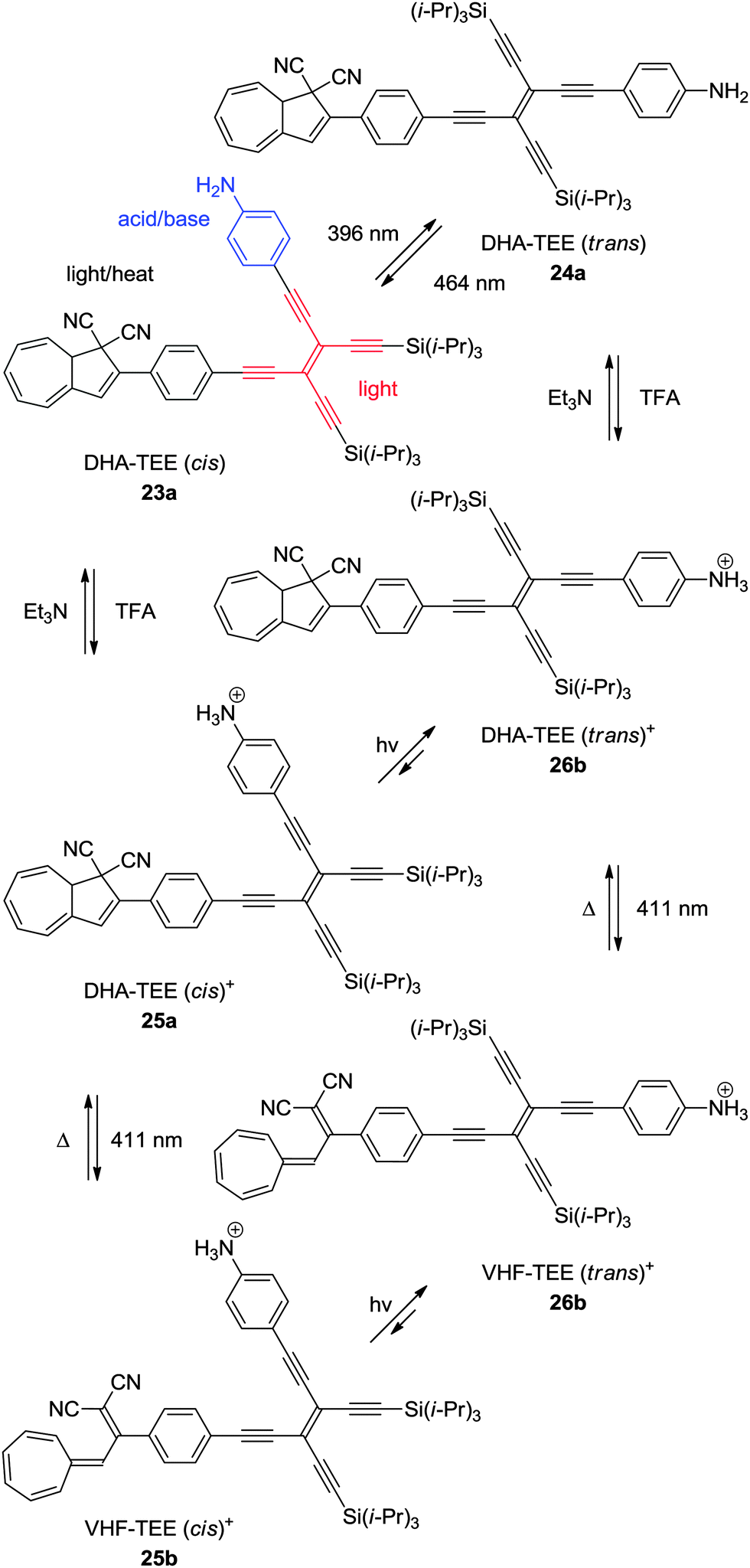
An elegant example of a multimode switch, including the DHA–VHF-system is that of 23a.11b Here, the photochemically induced cis–trans isomerization of tetraethynylethene (TEE) is combined with the photochemically and thermally induced switching of the DHA–VHF-system, together with the acid–base control of an aniline. Of the eight theoretical states, six of them have been observed as shown in Scheme 14. The quantum yield of ring-opening reaction (DHA → VHF) is quite low for the unprotonated DHA–TEE (trans) 24a, but can be reversibly interchanged between the states of the TEE (trans → cis) by changing the wavelength of light irradiation (396 nm or 464 nm). By protonation of DHA–TEE (cis) 23a or DHA–TEE (trans) 24a into 25a and 26a, respectively, the DHAs can then be converted into the VHFs 25b–26b by irradiation with light at 411 nm to obtain the VHF–TEE (cis)+25b and VHF–TEE (trans)+26b, respectively. By manipulation using the five stimuli of light at three different wavelengths (396 nm, 411 nm and 464 nm), heat, or with addition of acid or base, the six different states of the multimode system can be obtained.
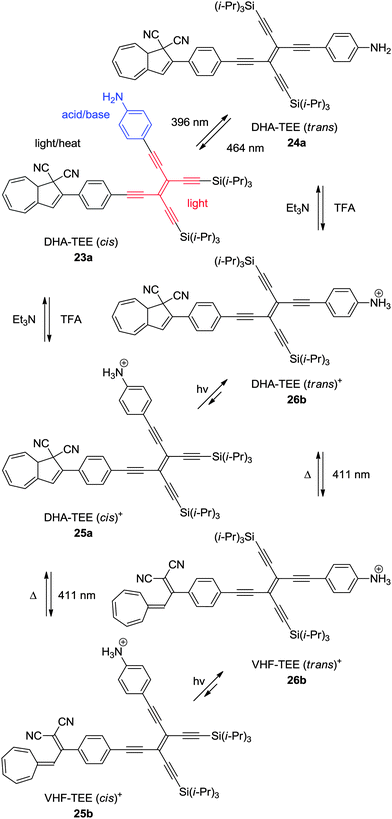 |
| | Scheme 14 Multimode switch based on an acid–base (aniline), light (TEE), and light–heat (DHA–VHF) controlled DHA–TEE derivative.11b | |
Acid–base control
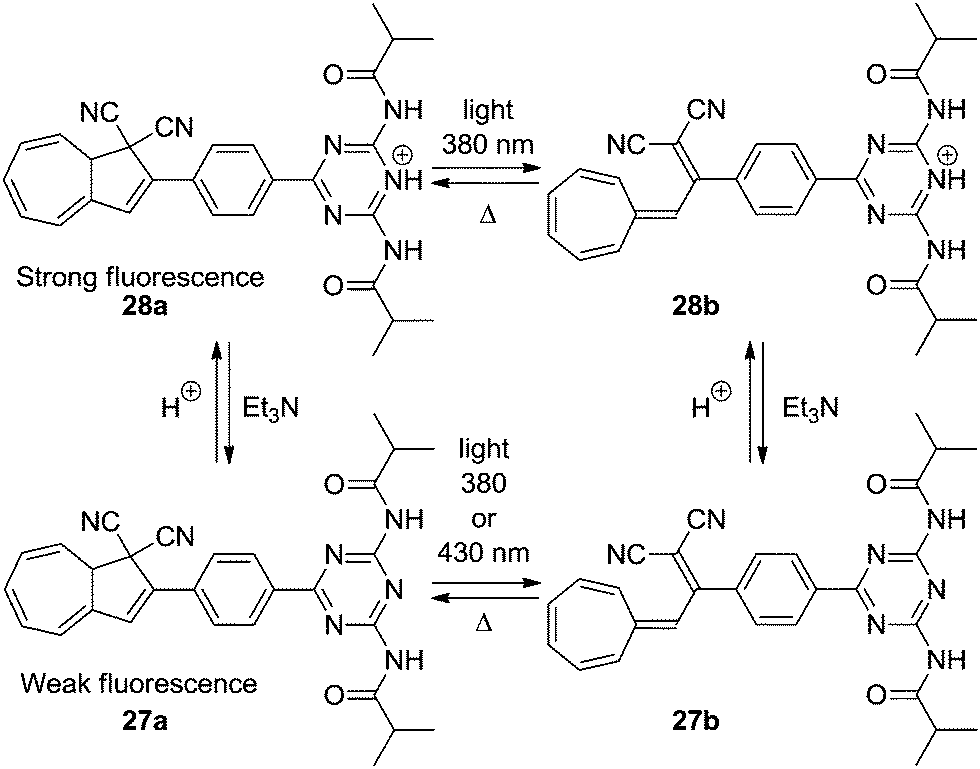
Compound 23a presents an example of an acid–base controlled switch, but also other acid–base sensitive systems have been developed. By connecting a diamidotriazine (DT) to the DHA/VHF the emission of the photoswitch could be controlled.27 The neutral DHA–DT 27a has only a weak emission at room temperature, but upon protonation (trifluoroacetic acid, TFA), the system 28a becomes highly fluorescent (Scheme 15). Since the VHFs 27b and 28b are not fluorescent, the emission of the system can be controlled by light/heat.
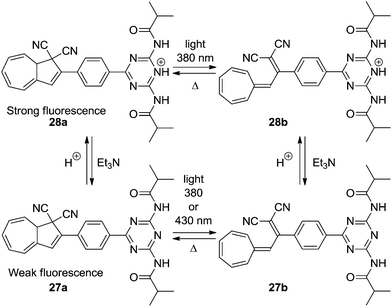 |
| | Scheme 15 Acid–base controlled emission of the DHA–VHF system by protonation of diamidotriazine using TFA. | |
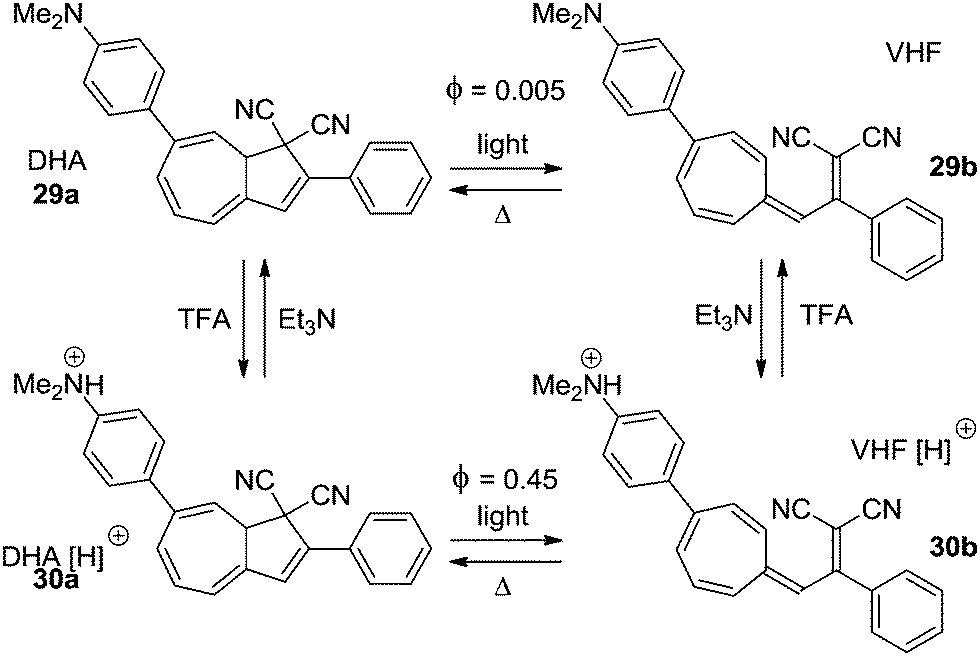
Electron donors affect the quantum yield of the ring-opening reaction (DHA → VHF) when positioned in the seven-membered ring13,14 as well as in the five-membered ring.4a The strongly electron donating amino- or dimethylamino-group can be turned into an electron withdrawing group upon protonation. This has been exploited in two examples to control the switching ability of the DHA–VHF system. The most significant change is seen in the DHA 29a (Scheme 16).14 While the neutral form 29a has a quantum yield for the ring-opening reaction of less than 0.005, the protonated form 30a has a 90 times higher quantum yield (Φ = 0.45). By irradiation with light the neutral DHA 29a could not be fully converted into VHF 29b, but upon protonation a quantitative conversion to VHF 30b was observed within 60 seconds. Then, by addition of Et3N, deprotonation gave the neutral VHF 29b.
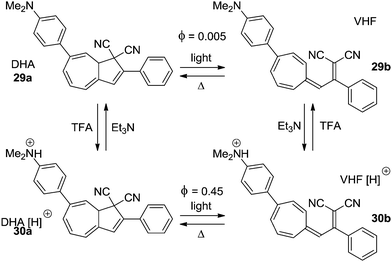 |
| | Scheme 16 Acid–base controlled ON/OFF switching of photochromism of the DHA–VHF-system by changing a strong electron donor into an electron acceptor.14 | |
Light–heat controlled fluorescence
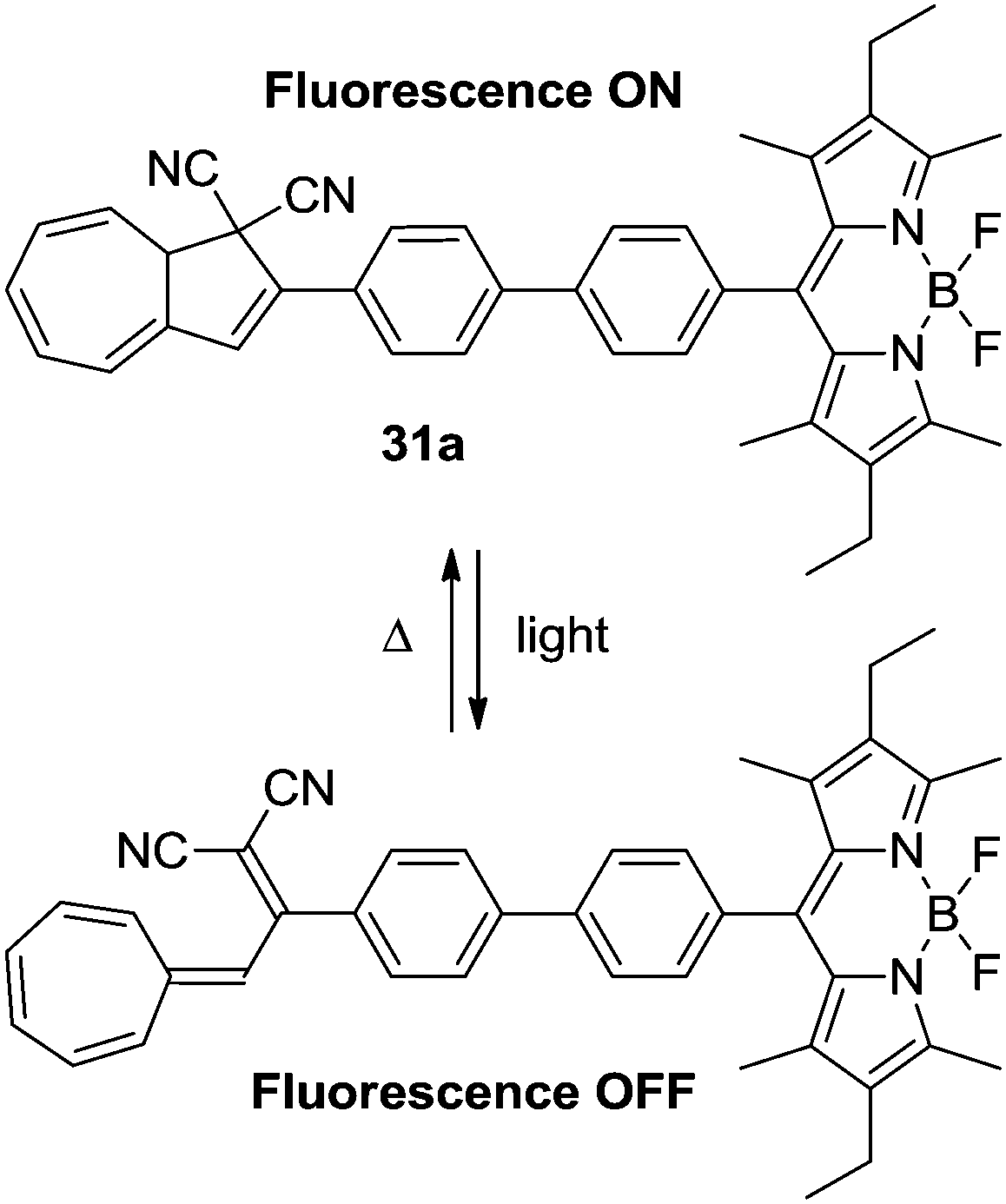
The DHA–VHF-system can be used to control the fluorescence of the boron dipyrromethene (BDP) dyad. Since the VHF is generally non-fluorescent because of a radiation-less deactivation of the excited state, this could be used to disable the fluorescence of the BDP unit (Scheme 17). Thus, the DHA 31a is strongly fluorescent with a high quantum yield (Φ = 0.62–0.67) and upon irradiation with light (366 nm) the fluorescence is turned almost completely off (Φ = 0.01).28
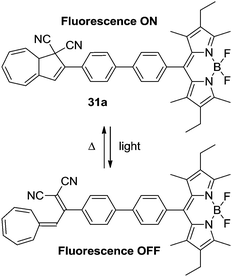 |
| | Scheme 17 DHA–VHF-controlled boron dipyrromethene fluorescence. The fluorescence of the BDP dyad is effectively quenched upon ring-opening.28 | |
Quenching of electrochemically generated luminescence was also reported, of relevance for the development of light-emitting diodes.29
Ultrafast switching
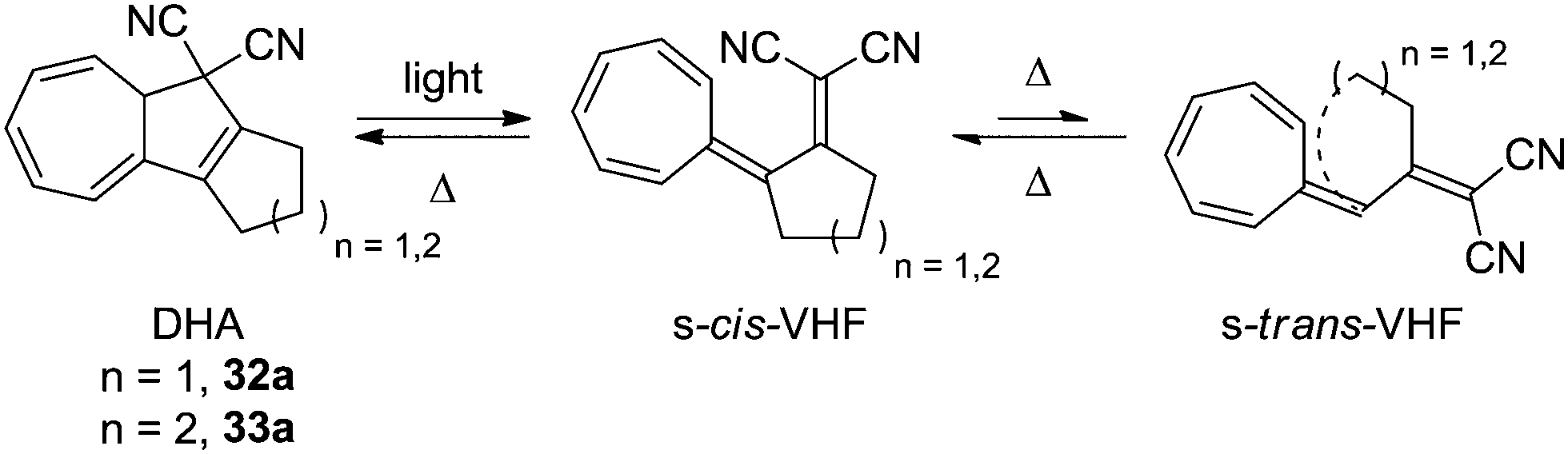
To the naked eye, the light induced ring-opening seems to happen instantaneously while the thermally induced ring-closure varies from derivative to derivative, but is generally quite slow. This is because of the large structural changes which the VHF has to undergo in order to get a good overlap between the two orbitals; first, an s-trans to s-cis conformational change, after which the dicyanoethylene unit has to approach the cycloheptatriene. By annulation of a cyclopentyl- or cyclohexyl ring, however, the rate of back-reaction is significantly increased, since s-cis/s-trans isomerization around the C2–C3 single bond is strained because of the cycloalkylidene ring, which puts the dicyanoethylene unit right in front of the cycloheptatriene and pushes the equilibrium far towards the s-cis conformer. The structures of the cyclopentanyl and cyclohexanyl annulated derivatives 32a and 33a are shown in Scheme 18.30
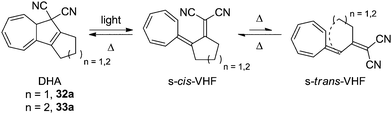 |
| | Scheme 18 Ultrafast switching between cycloalkylidene-interlocked DHAs and VHFs. The combining orbitals are brought into close proximity by the steric constraints of the cycloalkylidene ring. | |
A time resolved investigation of the ring-opening dynamics has revealed that the light-induced ring-opening reaction of 34a takes place within 1.2 ps on the DHA–VHF S1 potential energy surface, which is followed by the internal conversion from VHF-S1 to VHF-S0 in 13 ps.31 More interestingly, while the thermally more stable s-trans VHF is not photochromic, the s-cis can undergo a photochemically induced back-reaction upon a laser-pulse at 530 nm delayed 25 ps from a 360 nm laser-pulse (Scheme 19).32
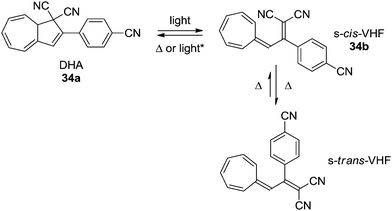 |
| | Scheme 19 Light-induced ultrafast switching between states of the DHA–VHF. * Light pulse delayed by 25 ps from ring-opening pulse. | |
Molecular electronics
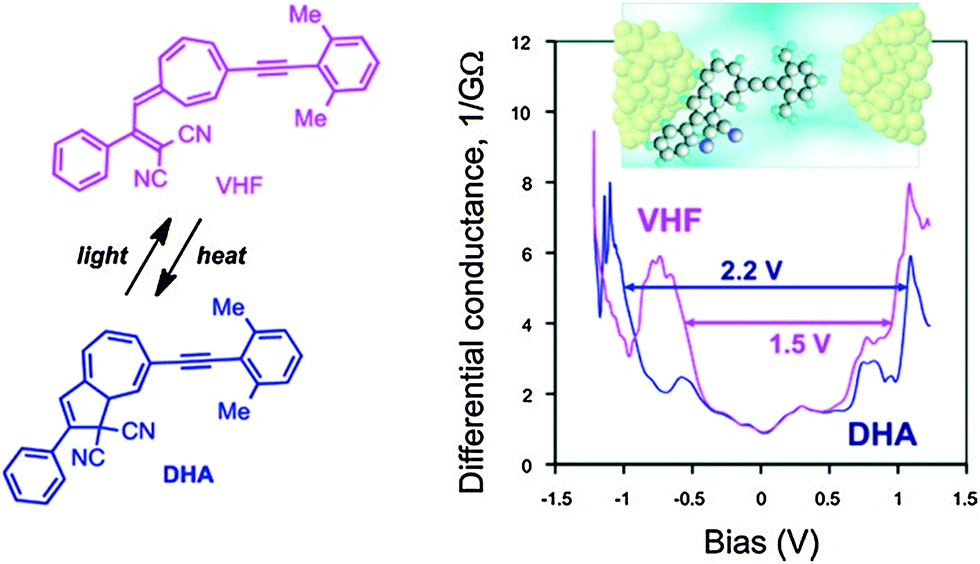
Daub and co-workers described in 1989 gating of the current flux in an electric circuit by DHA–VHF photochromism.33 Later steps towards incorporating the DHA–VHF-system into single-molecule molecular electronic devices were taken in 2011 by the groups of Nielsen and Kubatkin.6a By sublimation, the DHA could be trapped in between silver electrodes in a pre-fabricated nano-gap and light-induced switching between two conduction states was observed. Four independent samples demonstrated reproducible tunneling spectra with a transport gap being in the range of 2.1–2.4 V before light-irradiation and this low conducting (high resistivity) state of the molecule could be assigned to the DHA-state. Since none of the samples revealed gate-induced conductance modulation or Coulomb blockade effects, the molecule was believed to be strongly coupled to at least one of the electrodes. But since the resistivity was determined to be between 20 MΩ and 20 GΩ, the molecule was, in all samples, separated from one electrode with a tunneling gap as schematically depicted in Fig. 9 (inset).
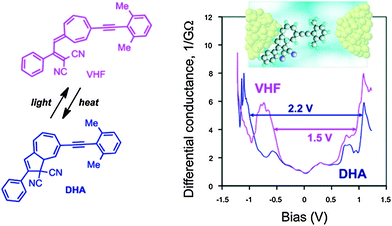 |
| | Fig. 9 Arylethynyl-functionalized DHA–VHF incorporated in a single-molecule silver electrode nanojunction (schematic illustration of a possible sample geometry shown). The differential conductance as a function of the bias potential is plotted for each isomer. Reprinted with permission from S. Lara-Avila, A. V. Danilov, S. E. Kubatkin, S. L. Broman, C. R. Parker and M. B. Nielsen, J. Phys. Chem. C, 2011, 115, 18372. Copyright 2014 American Chemical Society. | |
By irradiation with light (10–20 min), a significant change in resistivity was observed to a higher conducting state (lower resistivity) which could be assigned to VHF. This is in accordance with an observed reduction in tunneling gap; transport gap 1.5–1.75 V. In one sample it was possible to reset the state of conductance back to the original state as such; DHA (A) → VHF (B) → DHA (A′) → VHF (B′) → DHA (A′′) → VHF (B′′) → DHA (A′′′) and three reversible switching events could be observed. The estimated difference in HOMO–LUMO gap between the DHA and VHF was determined to be 0.7 eV (2.2–1.5 V) when positioned in between the silver nano-gap, which was almost the same value determined by cyclic voltammetry and UV-Vis spectroscopy in solution.
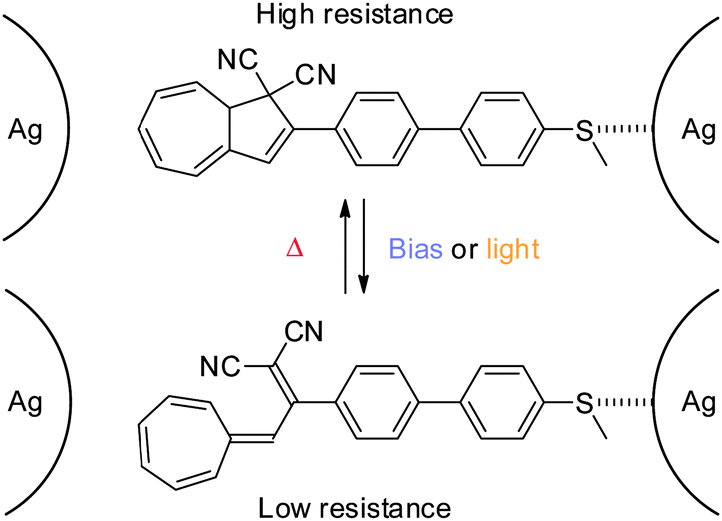
As the lack of reversible switching between states of the system might be due to a strong coupling to (at least) one of the electrodes, a DHA–VHF with a weaker coupling to the electrode was developed in 2012 (Scheme 20).6b
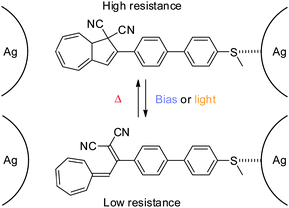 |
| | Scheme 20 Reversible switching between states of conductance of a thiomethyl-anchored DHA–VHF positioned in a silver nanogap. A possible anchoring configuration shown.6b | |
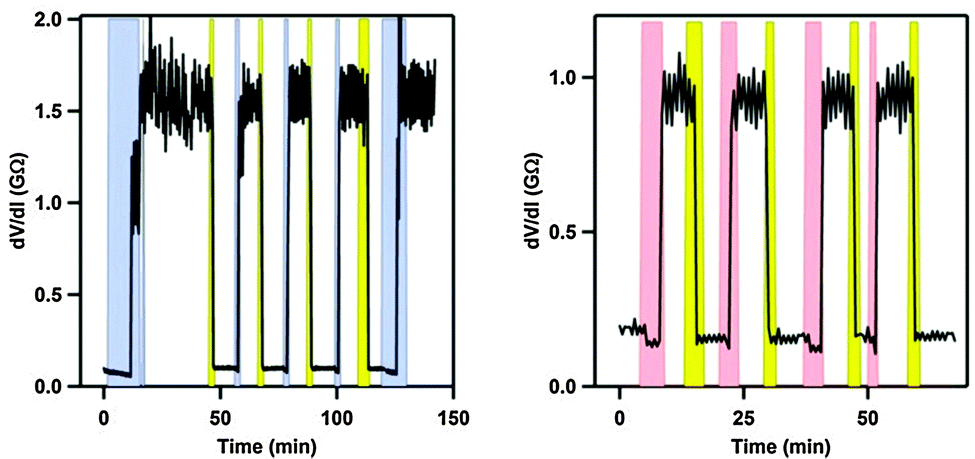
By anchoring the photoswitch to the silver-electrode, via a thiomethyl end-group, gate-dependence and reversible switching between states of conductance was obtained (Coulomb Blockage regime). Thus, an anchored molecule could be switched between states of conductance at a fixed gate voltage (VG = 2.67 V) by illumination with light (DHA → VHF) or changing the bias voltage from 25 mV to 80 mV as shown in Fig. 10 (left). Also, at a fixed gate voltage (VG = 2.7 V), reversible switching between the DHA and VHF states could be obtained by using heat and light as stimuli as shown in Fig. 10 (right).6b
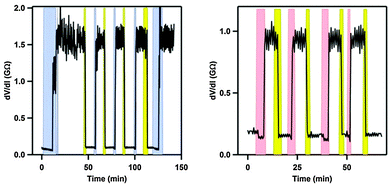 |
| | Fig. 10 On–off switchings (differential resistance, taken at bias voltage 25 mV, plotted against time) of the molecule shown in Scheme 20 in a silver electrode nanojunction at a fixed gate potential of 2.7 V and at 4 K. Left: switchings induced by increased bias voltage (blue; 80 mV) and light (yellow). Right: switchings induced by increased temperature (rose; from 4 to 25 K) and light (yellow). Copyright © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Conclusions
In conclusion, the DHA–VHF system shows great potential in multiple exciting fields with the many possible ways to tune the properties of the system; like acid–base, redox-state and Lewis acids. While the first steps towards the exploitation of the DHA–VHF in molecular electronics devices have been taken, new more controllable systems are to be targeted with a more well-defined contact to the electrode(s). Another interesting exploitation of photochromism, which has attracted significant attention recently, is to employ photoswitches for light-harvesting and solar energy storage (“solar heat batteries”).34 For the specific DHA–VHF system, the challenge within this field is to store a sufficient amount of energy in the metastable VHF isomer and at the same time retard its back-reaction so that the energy is first released as heat when needed, preferably triggered by an external stimulus. As hopefully apparent from this review, significant steps have already been taken in regard to controlling the VHF to DHA conversion by for example Brønsted acids–bases and Lewis acids.
Acknowledgements
We acknowledge the Danish Research Council and the University of Copenhagen for support.
Notes and references
-
(a) J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., Int. Ed. Engl., 1984, 23, 960 CrossRef PubMed;
(b)
T. Mrozek, A. Ajayaghosh and J. Daub, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2001, pp. 63–106 Search PubMed.
-
(a) J. Daub, S. Gierisch, U. Klement, T. Knöchel, G. Maas and U. Seitz, Chem. Ber., 1986, 119, 2631 CrossRef CAS PubMed;
(b) V. Mazzanti, M. Cacciarini, S. L. Broman, C. R. Parker, M. Schau-Magnussen, A. D. Bond and M. B. Nielsen, Beilstein J. Org. Chem., 2012, 8, 958 CrossRef CAS PubMed.
- O. Schalk, S. L. Broman, M. Å. Petersen, D. V. Khakhulin, R. Y. Brogaard, M. B. Nielsen, A. E. Boguslavskiy, A. Stolow and T. I. Sølling, J. Phys. Chem. A, 2013, 117, 3340 CrossRef CAS PubMed.
-
(a) H. Görner, C. Fischer, S. Gierisch and J. Daub, J. Phys. Chem., 1993, 97, 4110 CrossRef;
(b) H. Görner, C. Fischer and J. Daub, J. Photochem. Photobiol., A, 1995, 85, 217 CrossRef.
- A. Plaguet, B. Champagne, F. Castet, L. Ducasse, E. Bogdan, V. Rodriguez and J.-L. Pozzo, New J. Chem., 2009, 33, 1349 RSC.
-
(a) S. Lara-Avila, A. V. Danilov, S. E. Kubatkin, S. L. Broman, C. R. Parker and M. B. Nielsen, J. Phys. Chem. C, 2011, 115, 18372 CrossRef CAS;
(b) S. L. Broman, S. Lara-Avila, C. L. Thisted, A. D. Bond, A. V. Danilov, S. E. Kubatkin and M. B. Nielsen, Adv. Funct. Mater., 2012, 22, 4249 CrossRef CAS PubMed.
- S. L. Broman, S. L. Brand, C. R. Parker, M. Å. Petersen, C. G. Tortzen, A. Kadziola, K. Kilså and M. B. Nielsen, ARKIVOC, 2011, ix, 51 CrossRef.
- C. Jutz, Chem. Ber., 1964, 97, 2050 CrossRef CAS PubMed.
- H. Tsuruta, T. Suguyama and T. Mukai, Chem. Lett., 1972, 185 CrossRef CAS.
-
(a) T. Toda, K. Saito and T. Mukai, Tetrahedron Lett., 1972, 1981 CrossRef CAS;
(b) M. A. O'Leary and D. Wege, Tetrahedron Lett., 1978, 2811 CrossRef;
(c) M. A. O'Leary, G. W. Richardson and D. Wege, Tetrahedron, 1981, 37, 813 CrossRef;
(d) N. Harada, J. Kohori, K. Uda and R. Takada, J. Am. Chem. Soc., 1985, 107, 423 CrossRef CAS;
(e) R. Gleiter, H. Zimmermann and W. Sander, Angew. Chem., 1986, 98, 893 CrossRef CAS PubMed;
(f) N. Harada, J. Kohori, H. Uda and K. Toriumi, J. Org. Chem., 1989, 54, 1820 CrossRef CAS;
(g) A. Frimer, J. Weiss and Z. Rosental, J. Org. Chem., 1994, 59, 2516 CrossRef CAS.
-
(a) J. Daub, S. Gierisch, T. Knöchel and E. Salbeck, Z. Naturforsch., 1986, 41b, 1151 CAS;
(b) L. Gobbi, P. Seiler and F. Diederich, Helv. Chim. Acta, 2001, 84, 743 CrossRef CAS;
(c) S. L. Broman, M. Jevric, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2014, 79, 41 CrossRef CAS PubMed.
- M. Å. Petersen, S. L. Broman, A. Kadziola, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2009, 2733 CrossRef CAS PubMed.
- S. L. Broman, M. Å. Petersen, C. G. Tortzen, K. Kilså, A. Kadziola and M. B. Nielsen, J. Am. Chem. Soc., 2010, 132, 9165 CrossRef CAS PubMed.
- M. Å. Petersen, S. L. Broman, A. Kadziola, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2011, 1033 CrossRef CAS PubMed.
- M. Jevric, S. L. Broman, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2013, 78, 4348 CrossRef CAS PubMed.
- M. Cacciarini, S. L. Broman and M. B. Nielsen, ARKIVOC, 2014, i, 249 CrossRef.
- S. L. Broman, M. Jevric and M. B. Nielsen, Chem. – Eur. J., 2013, 19, 9542 CrossRef CAS PubMed.
- H. Lissau, S. L. Broman, M. Jevric, A. Ø. Madsen and M. B. Nielsen, Aust. J. Chem., 2014, 67, 531 CrossRef CAS.
- C. R. Parker, C. G. Tortzen, S. L. Broman, M. Schau-Magnussen, K. Kilså and M. B. Nielsen, Chem. Commun., 2011, 47, 6102 RSC.
- M. Cacciarini, E. A. Della Pia and M. B. Nielsen, Eur. J. Org. Chem., 2012, 6064 CrossRef CAS PubMed.
- J. Daub, S. Gierisch and J. Salbeck, Tetrahedron Lett., 1990, 31, 3113 CrossRef CAS.
- J. Achatz, C. Fischer, J. Salbeck and J. Daub, J. Chem. Soc., Chem. Commun., 1991, 504 RSC.
-
(a) M. Å. Petersen, L. Zhu, S. H. Jensen, A. S. Andersson, A. Kadziola, K. Kilså and M. B. Nielsen, Adv. Funct. Mater., 2007, 17, 797 CrossRef CAS PubMed;
(b) M. Å. Petersen, A. S. Andersson, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2009, 1855 CrossRef CAS PubMed.
- M. Santella, V. Mazzanti, M. Jevric, C. R. Parker, S. L. Broman, A. D. Bond, A. Kadziola and M. B. Nielsen, J. Org. Chem., 2012, 77, 8922 CrossRef CAS PubMed.
- H. Spreitzer and J. Daub, Chem. – Eur. J., 1996, 2, 1150 CrossRef CAS PubMed.
-
(a) T. Mrozek, H. Görner and J. Daub, Chem. Commun., 1999, 1487 RSC;
(b)
T. Mrozek, Neue photochrome Systeme basierend auf Zehn- und Sechselektronenumlagerungen, Doctoral dissertation, University of Regensburg, 2000 Search PubMed;
(c) T. Mrozek, H. Görner and J. Daub, Chem. – Eur. J., 2001, 7, 1028 CrossRef CAS.
-
(a) J. Daub, C. Trieflinger, O. Kushnir and R. Procházka, Mol. Cryst. Liq. Cryst., 2005, 430, 115 CrossRef CAS;
(b)
C. Trieflinger, Mehrfach adressierbare molekulare Schalter auf Basis von funktionellen Farbstoffen, Doctoral dissertation, University of Regensburg, 2004 Search PubMed.
- C. Trieflinger, H. Röhr, K. Rurack and J. Daub, Angew. Chem., Int. Ed., 2005, 44, 6943 CrossRef CAS PubMed.
- H. Spreitzer and J. Daub, Liebigs Ann. Chem., 1995, 1637 CrossRef CAS PubMed.
-
(a) J. Ern, M. Petermann, T. Mrozek, J. Daub, K. Kuldová and C. Kryschi, Chem. Phys., 2000, 259, 331 CrossRef CAS;
(b) S. Gierisch and J. Daub, Chem. Ber., 1989, 122, 69 CrossRef CAS PubMed.
- V. De Waele, M. Beutter, U. Schmidhammer, E. Riedle and J. Daub, Chem. Phys. Lett., 2004, 390, 328 CrossRef CAS PubMed.
- V. De Waele, U. Schmidhammer, T. Mrozek, J. Daub and E. Riedle, J. Am. Chem. Soc., 2002, 124, 2438 CrossRef CAS PubMed.
- J. Daub, J. Salbeck, T. Knöchel, C. Fischer, H. Kunkely and K. M. Rapp, Angew. Chem., Int. Ed. Engl., 1989, 28, 1494 CrossRef PubMed.
-
(a) T. J. Kurcharski, Y. Tian, S. Akbulatov and R. Boulatov, Energy Environ. Sci., 2011, 4, 4449 RSC;
(b) K. Moth-Poulsen, D. Coso, K. Börjesson, N. Vinokurov, S. K. Meier, A. Majumdar, K. P. C. Vollhardt and R. A. Segalman, Energy Environ. Sci., 2012, 5, 8534 RSC;
(c) T. J. Kurcharski, N. Ferralis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441 CrossRef PubMed.
|
| This journal is © the Owner Societies 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a