
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon mobility and clustering of sodium hydroxybenzoates in aqueous solutions: a molecular dynamics simulation study
Jure
Gujt
ab,
Črtomir
Podlipnik
a,
Marija
Bešter-Rogač
a and
Eckhard
Spohr
*b
aFaculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia. E-mail: jure.gujt@fkkt.uni-lj.si
bChair of Theoretical Chemistry, Faculty of Chemistry, University of Duisburg-Essen, D-45141 Essen, Germany. E-mail: eckhard.spohr@uni-due.de
First published on 7th August 2014
Abstract
The relative position of the hydroxylic and the carboxylic group in the isomeric hydroxybenzoate (HB) anions is known to have a large impact on transport properties of this species. It also influences crucially the self-organisation of cationic surfactants. In this article a systematic investigation of aqueous solutions of the ortho, meta, and para isomers of the HB anion is presented. Molecular dynamics simulations of all three HB isomers were conducted for two different concentrations at 298.15 K and using two separate water models. From the resulting trajectories we calculated the self-diffusion coefficient of each isomer. According to the calculated self-diffusion coefficients, isomers were ranked in the order o-HB > m-HB > p-HB at both concentrations for both the used SPC and SPC/E water models, which agrees very well with the experiment. The structural analysis revealed that at lower concentration, where the tendency for dimerisation or cluster formation is low, hydrogen bonding with water determines the mobility of the HB anion. o-HB forms the least hydrogen bonds and is therefore the most mobile, and p-HB, which forms the most hydrogen bonds with water, is the least mobile isomer. At higher concentration the formation of clusters also needs to be considered. The ortho isomer predominantly forms dimers with 2 hydrogen bonds per dimer between one OH and one carboxylate group of each anion. m-HB mostly forms clusters of sizes around 5 and p-HB forms clusters of sizes even larger than 10, which can be either rings or chains.
1 Introduction
It has been well recognised that some surfactant systems form long wormlike micelles at higher concentration and/or upon the addition of electrolytes. Even though this feature has been observed in a number of systems containing nonionic and anionic surfactants, the effect is more pronounced in cationic surfactants, especially in the presence of strongly binding counterions, which usually possess an aromatic ring.1–5 Among many other systems the origin of the sphere-to-rod transition in cationic micelles with aromatic counterions has been investigated also for cetyltrimethylammonium bromide (CTAB)6–8 and dodecyltrimethylammonium bromide (DTAB).9 The experimental observations were ascribed to the specific ion hydration in the interfacial region,7 which was indicated recently also by molecular dynamics simulations.10However, it turned out that the position of the hydrophilic groups on the aromatic ring and therefore the overall hydrophobic character of the surfactant or the counterion molecules has a dramatic effect on the self-organisation of cationic surfactants, which turned out to be strongly dependent on the substitution pattern of the aromatic ring.5,11,12 This effect has been investigated recently in detail for the micellisation process of dodecyltrimethylammonium chloride (DTAC) in the presence of sodium 2-hydroxybenzoate (o-HB), 3-hydroxybenzoate (m-HB) and 4-hydroxybenzoate (p-HB),13,14 see Fig. 1. It has been found that p-HB only slightly decreases the critical micelle concentration (cmc) and is incorporated only weakly into DTA micelles, whereas the meta and ortho isomers exhibit stronger effects. In the presence of m-HB the enthalpy of micellisation is higher and changes in the diffusion coefficient of the surfactant show that the structure of the aggregate is likely to be modified. This trend is confirmed by o-HB (salicylate) for which an intermediate rod-like micelle appears due to the expulsion of water from the micelle interior and the simultaneous insertion of salicylate molecules in the vicinity of the polar head of the surfactant. The asymmetry of the salicylate molecule makes its interaction with both the hydrophilic and the hydrophobic part of the surfactant more favourable than for the two other isomers.
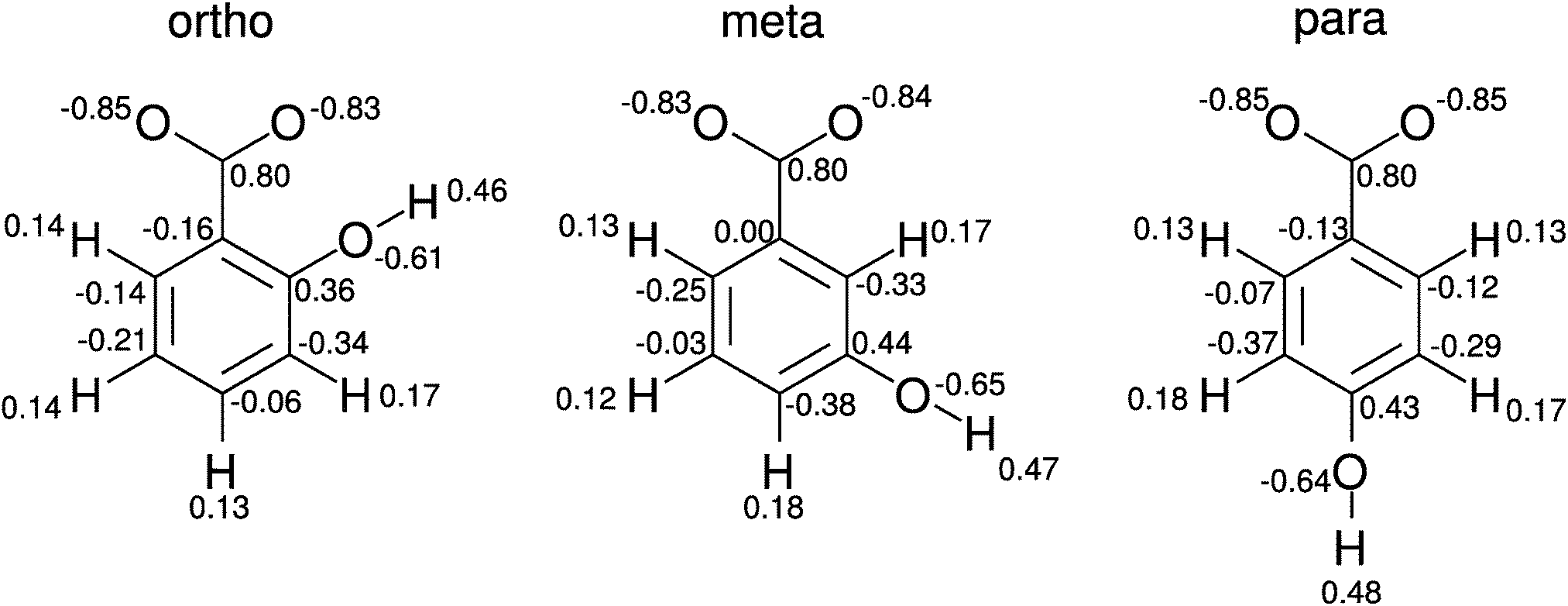 | ||
| Fig. 1 Structures and partial charges of all three hydroxybenzoate isomers. | ||
A conductivity study on diluted aqueous solutions of o-HB, m-HB and p-HB15 reveals that the mobility of the investigated anions is strongly dependent on the relative position of the carboxylic and hydroxylic groups (Fig. 1). It decreases in the order o-HB > m-HB > p-HB, which could be ascribed to the possible differences in the hydration of each isomer.
All these observations reveal that small changes in the ion structure result in quite significant differences in intermolecular interactions. To obtain more insight into the influence of the difference in the position of the –OH group on the aromatic ring in hydroxybenzoates on mobility and hydration we decided to carry out molecular dynamics simulation (MD) studies of the structure and the mobility of hydroxybenzoates in aqueous solutions. We have studied aqueous sodium salt solutions of o-HB, m-HB, and p-HB at two concentrations using two different water models, the SPC model16 and the SPC/E model.17
The next section contains a description of the details of the simulations, followed by a description of the needed lengthy equilibration process of the simulations. The main results section discusses the solvation structure on the basis of radial distribution functions and hydrogen bonding characteristics as well as ion dynamics on the basis of mean square displacements. In the final section we summarise the findings of our research.
2 Simulation details
Molecular dynamics simulations were performed with the LAMMPS simulation package (1. Dec 2012).18,19 Structures of all HB isomers were obtained from the HIC-Up database20 and GROMACS all-atom topologies were generated using the PRODRG server.21 We used VMD (1.9.1)22 with the TopoTools plug-in to solvate anions, add Na+ cations, resize systems to the desired size and write out LAMMPS data files of initial configurations. Force field parameters for non-bonded interactions (excluding partial atomic charges), bonds, angles and torsions were assigned according to the GROMOS96 45a3 force field.23 We used density functional (DFT) calculations with the B3LYP functional and a 6-311G* basis set in the effective Poisson Boltzmann Finite element method solvent as implemented in Jaguar 7.9 (Schrödinger Suite 2012)24 for the geometry minimization of HB ions and for the subsequent calculation of partial atomic charges of hydroxybenzoate's atoms that were fitted from the molecular electrostatic potential. Certainly more advanced quantum chemical methods than DFT calculations with the chosen functional and basis set to obtain partial charges of hydroxybenzoate molecules exist. Also, to describe the strong interactions of the hydroxybenzoate molecules with neighbouring water molecules using a rigid model is certainly an approximation that neglects polarisability. However, the necessity of performing very long simulations of large systems mandated the use of rigid hydroxybenzoate (and water) models. The anion models described above are thus compatible with the nature of the chosen rigid water models and suitable for the comparison of the dynamics of the three different isomers. Long-range electrostatics were dealt with by a particle–particle particle–mesh (PPPM) solver, which is available in the LAMMPS package.25 Its accuracy was set to 10−5.Here, we are interested in NaHB solutions at two different concentrations. Lower concentration systems contain one ion pair per 1000 water molecules, which corresponds to 0.055 M solutions, while systems at the higher concentration contain 120 water molecules per ion pair, which corresponds to 0.46 M solutions. The cubic simulation box with periodic boundary conditions contains 27 ion pairs at the lower and 216 ion pairs at the higher concentration. Densities of low and high concentration systems are 0.997 g cm−3 and 1.002 g cm−3 respectively. To estimate the influence of different water models on our results we have employed both the SPC16 and the SPC/E17 water model as solvents.
The initial equilibration of all systems followed the same protocol. Firstly, the starting configuration was subjected to energy minimisation. The resulting system was then gradually heated to 298.15 K in a 50 ps simulation run with a time step of dt = 0.25 fs. Temperature control was achieved by a Nosé–Hoover26 thermostat with the time constant set to 100 fs. In this simulation also the lengths of the simulation box were gradually adjusted to a value corresponding to the desired system density. This simulation was followed by a 75 ps long NVT simulation at 298.15 K with the same thermostat time constant. Finally, a 500 ps long NVT simulation at 298.15 K with an integration time step of 1 fs was performed.
Production runs for the low concentration systems were performed for 5 ns in the NVE ensemble with a time step of dt = 1 fs. The NVE ensemble was chosen since it is the native ensemble for MD, so that dynamical properties can be calculated straightforwardly. No temperature drift due to structural relaxation was observed during the simulations, so differences between NVE and NVT simulations can be regarded as negligible. For the systems at the higher concentration, an additional 15 ns of equilibration was needed; the reason for this is discussed in the next section. This was followed by 30 ns production runs, during which data for the statistical analysis were gathered. These runs were performed in the NVT ensemble at 298.15 K with 2 fs time step and a thermostat time constant of 400 fs. Since we could not rule out beforehand that further structural relaxation would occur, the NVT ensemble was chosen in order to keep the average temperature constant even if enthalpy driven association would continue. Coordinates of atoms were stored every 1 ps. To keep the water molecules rigid the SHAKE27 algorithm was employed. The cutoff distance for the Lennard-Jones and the real-space part of the Coulomb potential was set to 10 Å.
Most production runs were performed on machines with Intel Xeon E5440 and NVIDIA Tesla C2070, or AMD Phenom II X6 1090T and NVIDIA Tesla C2075 graphics processors, using the GNU compiler. The LAMMPS USER-CUDA package (double precision) was used to accelerate the simulations on the GPGPU. Approximately 1 day of run time (wallclock) was needed to compute 106 time steps on these machines.
We calculated self-diffusion coefficients of the HB anions from the mean square displacement (MSD) of the carboxylic carbon atom of an HB anion. The MSD of a particle i (a carboxylic carbon atom) was obtained as follows:28
MSDi(t) = 〈|![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) i(τ + t) − i(τ)|2〉 i(τ + t) − i(τ)|2〉 | (1) |
Here i is a position vector of the particle i and τ is the time of the time origin. Angle brackets denote averaging over all HB− ions and all time origins. The self-diffusion coefficient (D) was then calculated using Einstein's relation:28
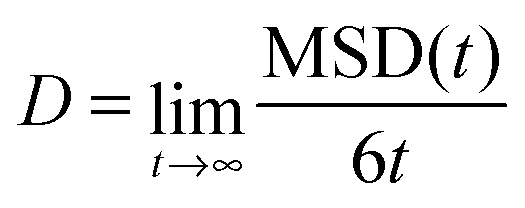 | (2) |
The self-diffusion coefficient is estimated from the slope of the MSD(t) curve. When choosing the range for which the slope will be determined, one must consider the inertial effects in the beginning of the curve, which should therefore be excluded from the calculation. On the other hand, the upper limit of the calculation range must also be determined with caution; the number of averages decreases with longer time separation, and for that reason the MSD(t) curve might deviate significantly from the straight line.29 In our case the calculation range used was between 2% and 20% of the total length of the production trajectory. When calculating the MSD(t), time origins were 1 ps apart.
Production runs at the lower concentration were performed in the NVE ensemble, i.e., without temperature control. Starting configurations for all production runs were equilibrated at 298.15 K and average temperatures for these runs ranged from 297.0 K to 299.2 K with standard deviations around 0.9 K. We neglect the small differences in average temperature in our discussion of the self-diffusion coefficient below, in spite of the fact that the SDC is a temperature-dependent quantity. In particular for the case of the lower concentration (27 HB− ions in the box) the uncertainty due to the small particle number greatly exceeds the error due to the slightly different average temperatures of systems.
3 Results
3.1 Temperature and stability
When calculating temperature dependent properties from a molecular dynamics simulation the temperature stability during the simulation run needs to be closely monitored. This is especially true for microcanonical (NVE) simulations, where there is no temperature control. The temperature, which is calculated from the kinetic energy of moving atoms, exhibits fluctuations and drift. The temperature drift is a result of approximations and deficiencies in the simulation protocol which lead to imperfect energy conservation.29Since all production runs for the lower concentration systems were NVE simulations, we have fitted the instantaneous temperature versus simulation time to a linear function. The slope, which corresponds to the temperature drift, was at most 0.069 K ns−1. In a 5 ns run this results in a total drift of approximately 0.35 K, which will have almost no influence on the calculated self-diffusion coefficients. The average temperature for any of the studied systems was in the range 298.15 K ± 1 K, and the standard deviation was approximately 0.9 K. We conclude that the system temperature is sufficiently stable during the simulation. The quotient of the standard deviation of total energy and the average kinetic energy was always less than 10−3, which indicates reasonably good energy conservation.
The high concentration systems required, due to the long equilibration time, temperature control and were simulated with a time step of 2 fs in the canonical ensemble. Under these conditions the run temperature is stable and fluctuates around the set temperature (298.15 K) with a standard deviation of approximately 1 K. The temperature drift is approximately 0.003 K ns−1. In contrast to the NVE simulations, we do not have energy conservation as kinetic energy is added to or subtracted from the system. For that reason we examine the stability of the potential energy in lieu of the total energy. In Fig. 2, we show the potential energy as a function of time for the last 10 ns of the production run. It can be seen that the potential energy is reasonably stable with a standard deviation of at most 260 kcal mol−1. It was also observed that during the course of a 35 ns production run, the potential energy increases by approximately 0.3%. This observation could be attributed to the limited precision of the computation inherent to any MD simulation protocol or more likely to the rather long timestep (2 fs) which was used in simulations of the high concentration systems.
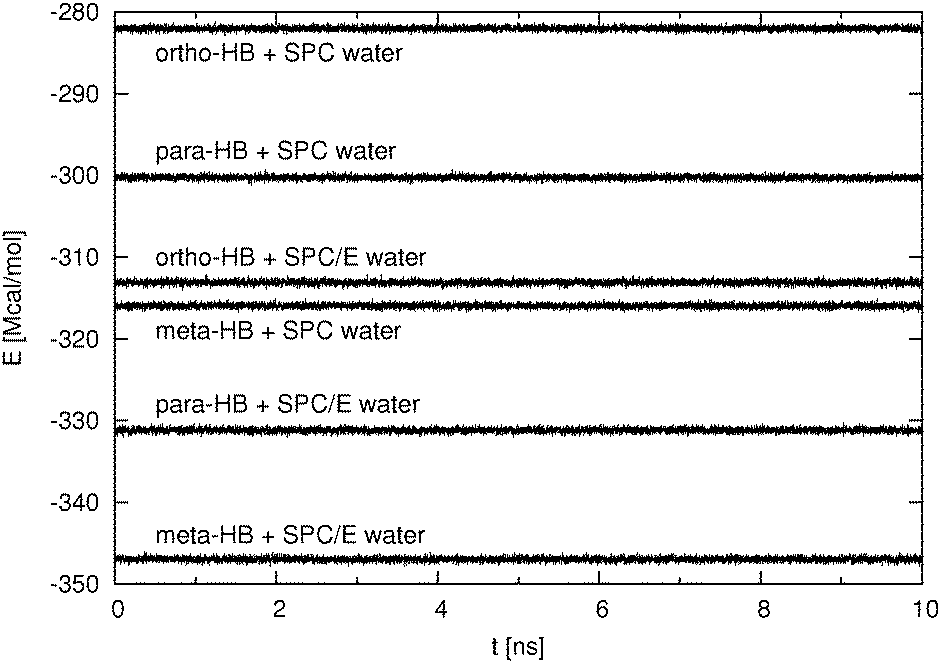 | ||
| Fig. 2 Potential energy of the high concentration systems over the last 10 ns of production runs. | ||
We performed isochoric simulations to maintain the constant experimental density, which is an important quantity when calculating transport properties. We felt that for the present study it is more important to maintain appropriate density rather than appropriate pressure. However, it is noteworthy that the average magnitude of the pressure in every simulated system was below 100 atm.
3.2 Self-diffusion coefficients
Self-diffusion coefficients were calculated from the slope of the MSD(t) curve according to eqn (2). The part from 2% to 20% of the analysed trajectory length, which corresponds to 0.9 ns of 5 ns trajectory length, was used to determine the slope. MSD(t) curves up to 2 ns are shown in Fig. 3. Results for all studied systems are presented in Table 1. The values for the lower concentration systems were obtained as an average of 3 separate 5 ns runs, each of which started from a different initial configuration, which was generated and equilibrated as outlined above. As we can see from Table 1, the MD simulations are able to correctly rank the HB isomers in the order of the experimentally determined mobilities, even at low concentration. Note that the concentrations of HB anions given in Table 1 differ between simulations and experiments, which, given that the self-diffusion coefficient depends on the concentration, might lead to difficulties when comparing these figures. Nevertheless, in the present work we are mainly interested in the correct order of the isomers' mobility based on their self-diffusion coefficient, which is compared to the experimental data.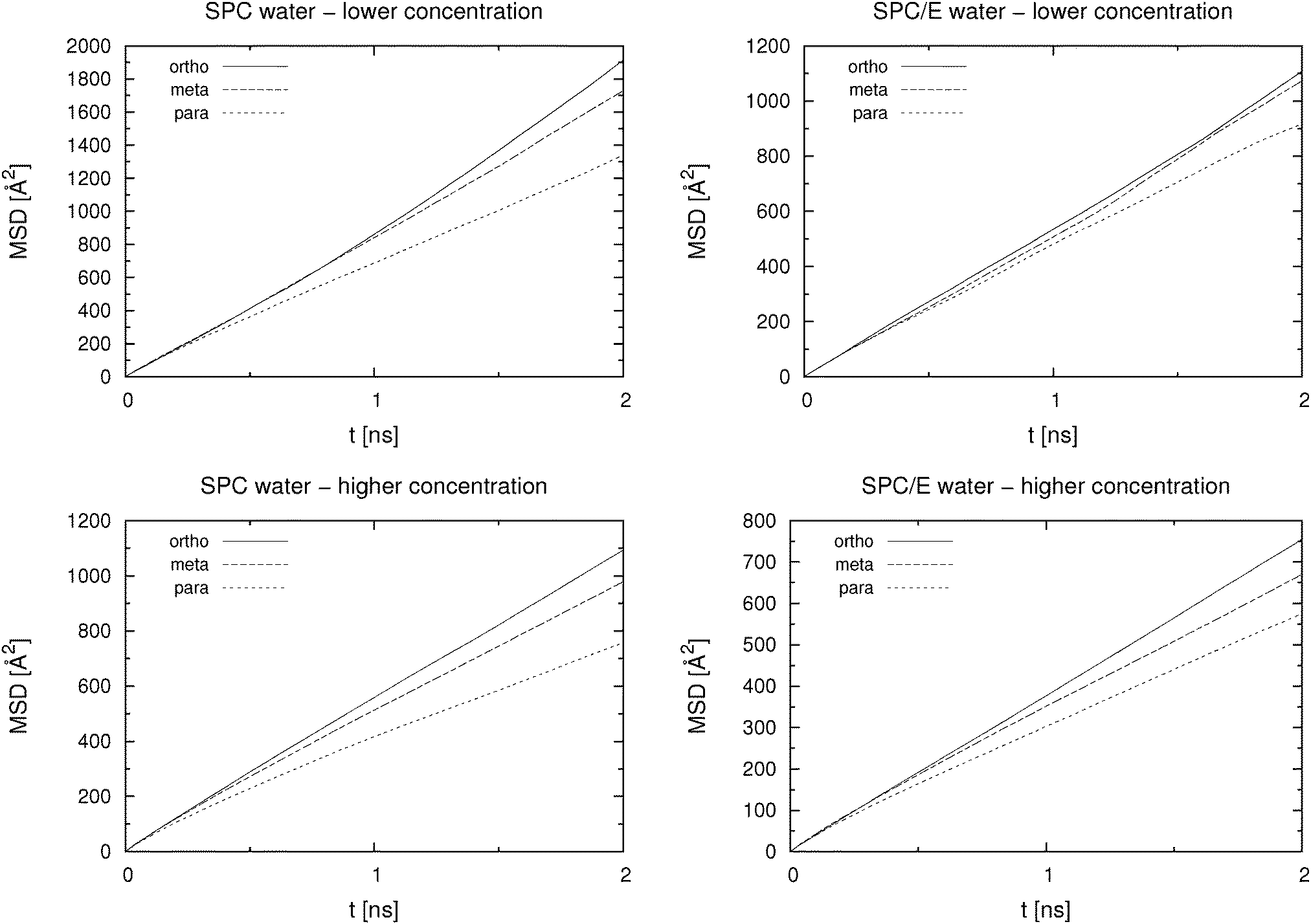 | ||
| Fig. 3 Mean square displacement of hydroxybenzoate anions in SPC (left) and SPC/E (right) water at low (c = 0.055 M; top) and at high (c = 0.46 M; bottom) concentration. Time origins, τ, are 1 ps apart. | ||
| D/10−10 m2 s−1 | Simulations | Experiment14 | ||||
|---|---|---|---|---|---|---|
| Low c (0.055 M) | High c (0.46 M) | c = 1 mM | Inf. dil. | |||
| Isomer | SPC | SPC/E | SPC | SPC/E | NMR | El. conductivity |
| ortho | 13.9 ± 0.4 | — | 9.5 ± 0.3 | 6.1 ± 0.1 | 9.3 ± 0.4 | 9.18 ± 0.01 |
| meta | 13.1 ± 0.8 | 8.7 ± 0.5 | 8.7 ± 0.5 | 5.8 ± 0.2 | 8.6 ± 0.3 | 7.90 ± 0.01 |
| para | 11.7 ± 0.5 | 7.9 ± 0.1 | 6.7 ± 0.1 | 5.0 ± 0.3 | 7.7 ± 0.4 | 7.17 ± 0.01 |
We also observe that the mobility of HB ions is smaller in SPC/E than in SPC water at both concentrations. This is not unexpected, since the SPC water has a larger self-diffusion coefficient than SPC/E water. Values are 4.2 × 10−9 m2 s−1 (298.6 K) for SPC and 2.8 × 10−9 m2 s−1 (298.2 K) for SPC/E water.29 The SPC water model has been chosen because it produces reliable results when used with the GROMOS96 force field,23 which was used for HB molecules. The decision to use also the SPC/E water model originally arose from the fact that the SPC/E model SDC is closer to the experimental value for pure water, which is 2.3 × 10−9 m2 s−1.29 The value of D for the ortho isomer in SPC/E water is not shown due to very poor statistics. While values obtained from 3 simulation runs generally agree very well for the other low concentration systems, the SDCs for the three o-HB isomer runs in SPC/E water scatter wildly, possibly due to different degrees of dimer formation (see below).
Values for the high concentration systems were obtained as averages of the three consecutive 5 ns blocks of the last 15 ns of the production run. Since there are 216 HB ions in the high concentration solution (as opposed to 27 HB ions at lower concentration) the statistics here is much better. The order of isomers according to their mobility is the same as that at lower concentration.
3.3 Hydrogen bonding, aggregate structures and possible explanations for differences in mobilities
The hydroxybenzoate anions can form, both via their carboxylic and via their hydroxylic oxygen atoms, hydrogen bonds with water molecules. Furthermore, since an HB anion can be a hydrogen bond donor and acceptor at the same time, they can also form hydrogen bonds between themselves. The differences between the hydrogen bonds formed by each isomer can shed some light on the reasons for their mobility differences. Another phenomenon that can influence the self-diffusion coefficient of HB anions is the formation of dimers or even n-mers at higher concentrations.In Table 2 we compile data for each HB isomer obtained for the number of hydrogen bonds formed per HB anion (via different donors and acceptor oxygen atoms) in both examined model solvents at both concentrations. Criteria for the hydrogen bond formation were the distance between the donor and the acceptor oxygen atoms being less than 3 Å and the angle between the O–H vector of the donor group and the vector between the hydrogen and the acceptor oxygen atoms being less than 20°. Compared to other definitions of hydrogen bonding in the literature, these conditions are rather tight and thus give rise to relatively small overall hydrogen bond numbers. As before, an average over 3 consecutive runs of 5 ns each started from different initial configurations was taken for the lower concentration systems, while at the higher concentration the three consecutive 5 ns blocks of the final 15 ns of the trajectory were analysed separately, and the average over these intervals is reported.
| Water model | Isomer | Ocar–Owat | Ohyd–Owat | Ocar–Ohyd | Total |
|---|---|---|---|---|---|
| Number of hydrogen bonds – lower concentration | |||||
| SPC | ortho | 3.17 | 1.09 | 0 | 4.26 ± 0.04 |
| meta | 3.15 | 1.38 | 0.01 | 4.54 ± 0.04 | |
| para | 3.35 | 1.34 | 0.01 | 4.70 ± 0.04 | |
| SPC/E | ortho | 3.27 | 1.17 | 0 | 4.44 ± 0.04 |
| meta | 3.26 | 1.44 | 0.01 | 4.71 ± 0.04 | |
| para | 3.48 | 1.41 | 0.02 | 4.91 ± 0.04 | |
| Number of hydrogen bonds – higher concentration | |||||
| SPC | ortho | 2.57 | 0.67 | 0.58 | 3.82 ± 0.02 |
| meta | 2.75 | 1.04 | 0.39 | 4.18 ± 0.02 | |
| para | 2.71 | 0.84 | 0.58 | 4.13 ± 0.01 | |
| SPC/E | ortho | 2.76 | 0.77 | 0.5 | 4.03 ± 0.01 |
| meta | 3.00 | 1.19 | 0.26 | 4.45 ± 0.03 | |
| para | 3.03 | 1.04 | 0.39 | 4.46 ± 0.02 | |
For the low concentration we first note that the total number of hydrogen bonds per molecule increases with increasing distance between the carboxylic and the hydroxylic group of the HB anion, i.e. in the sequence o-HB < m-HB < p-HB. This correlates well with the calculated self-diffusion coefficient: the lower the number of hydrogen bonds formed, the larger the mobility of the HB isomer. It can also be seen that there are very few hydrogen bonds between HB anions, which implies almost no dimer or cluster formation due to hydrogen bonding. However, as it can be seen from Fig. 4, which shows the pair distribution functions between the carboxylic carbon atoms, there are some dimers present even at low concentration. The pair distribution functions of hydroxylic hydrogen to carboxylic oxygen (not presented) show that hydrogen bonding is involved in the formation of such dimers. The statistical accuracy of these pair distribution functions is, due to the small number of particles (27) in the low concentration systems, relatively poor. Thus, structural properties of HB anion aggregates can only be discussed in more detail when describing higher concentration systems (see below).
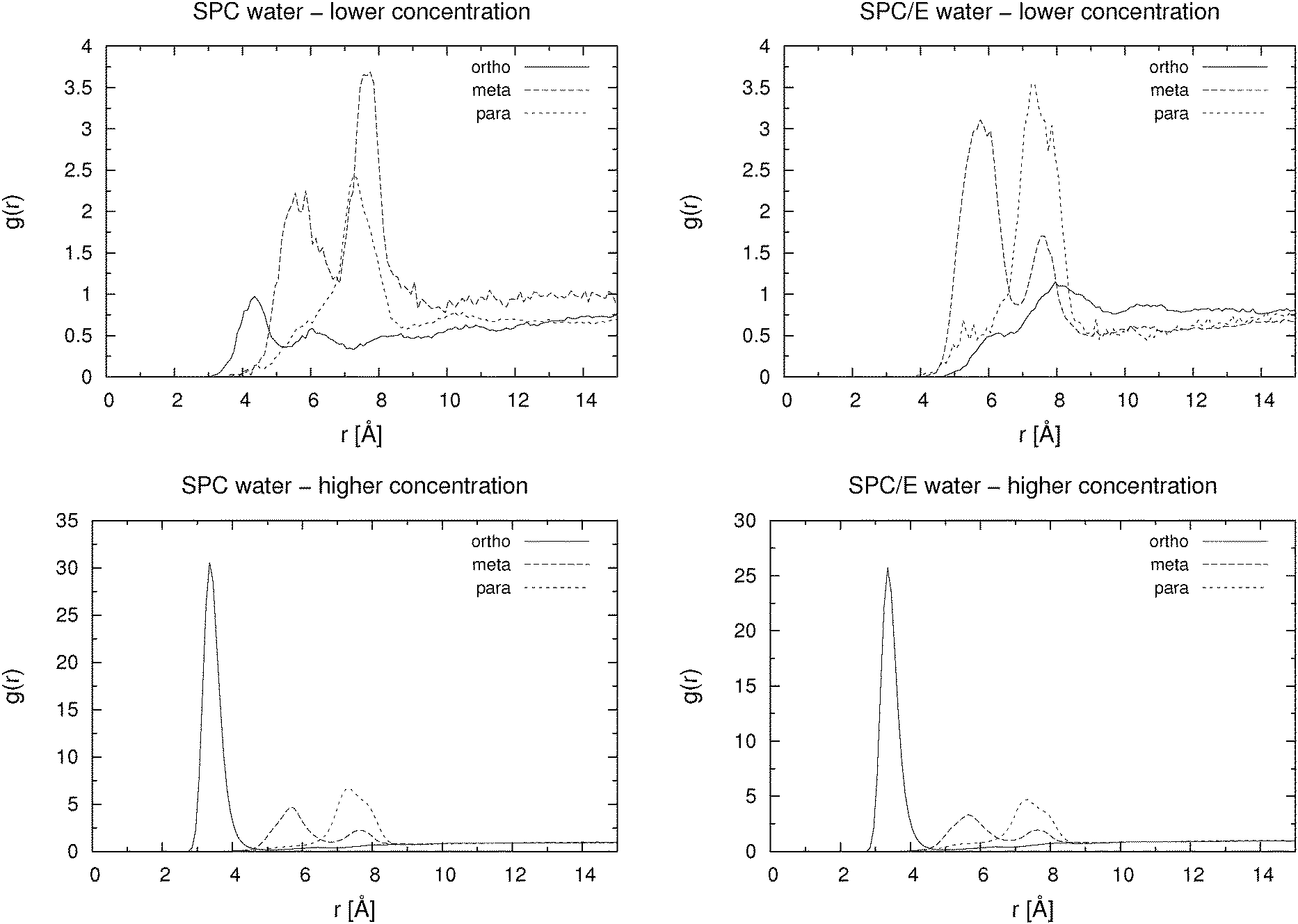 | ||
| Fig. 4 Pair distribution functions between carboxylic carbon atoms of hydroxybenzoate ions at low (0.055 M; top row) and at high (0.46 M) concentration (bottom row) for simulations using the SPC (left) and the SPC/E (right) water model. | ||
We note the same trends in the number of hydrogen bonds formed by HB anions both for the SPC and the SPC/E water model, but in the case of the SPC/E model this number is higher than in the case of the SPC model by roughly 0.2 hydrogen bonds per molecule. This could, beside the overall lower solvent mobility, explain the lower values of the calculated self-diffusion coefficients of the HB anions in SPC/E water. There are also differences in the distribution of hydrogen bonds formed by specific parts of the HB anions between the different HB isomers. The hydroxylic oxygen atoms in meta and para isomers form more hydrogen bonds with water than those of the ortho isomer. The reason for this is very likely sterical: in the case of the ortho isomer, the distance between both functional groups is too small for water molecules to be between them or close by, so that a smaller number of hydrogen bonds can be formed between hydroxylic oxygen and water. There is nevertheless no evidence for intramolecular hydrogen bond formation in the case of the ortho isomer, which is probably due to the fact that the water oxygen atom can come closer to the hydroxylic hydrogen than the carboxylic oxygen, so that hydrogen bonding with water is preferred. For the lower concentration we can conclude that hydration (formation of hydrogen bonds with water) determines how mobile the isomer is. A higher number of formed hydrogen bonds leads to lower mobility.
The behavior of the higher concentration solutions is distinctly different from the one at lower concentration. The formation of dimers or higher aggregates is very prominent. This can be seen from both Table 2 and from Fig. 4. The pair distribution functions of carboxylic carbon pairs (Ccar–Ccar) display distinct peaks which lead us to the conclusion that some cluster formation occurs. The fraction of hydrogen bonds formed between Ocar and Ohyd is almost always larger than 10%. Such an extent of ion pairing is very likely to show a large influence on the mobility of the HB anions. Because of the clustering the equilibration of the systems at high concentration took significantly longer time than for the low concentration systems. In Fig. 5 we exemplarily depict how the number of hydrogen bonds formed between different donors and acceptors for p-HB in SPC water changes over time. Similar plots can be produced for the other two isomers in either solvent. The para isomer in SPC water was chosen deliberately, because the equilibration time (with respect to the cluster formation) is the longest in this system. It is obvious that the system does not reach an equilibrium with respect to clustering until after approximately 25 ns. Note that the simulation started from an otherwise ‘equilibrated’ system, thus, all statistics is gathered starting at a time of 30 ns after system generation. In all fairness one cannot be completely certain that the system has really reached an equilibrium after 45 ns. However, in order to test for this, even much longer simulation runs would be needed, which are at the present state of computational technology not easily feasible and beyond the scope of this work.
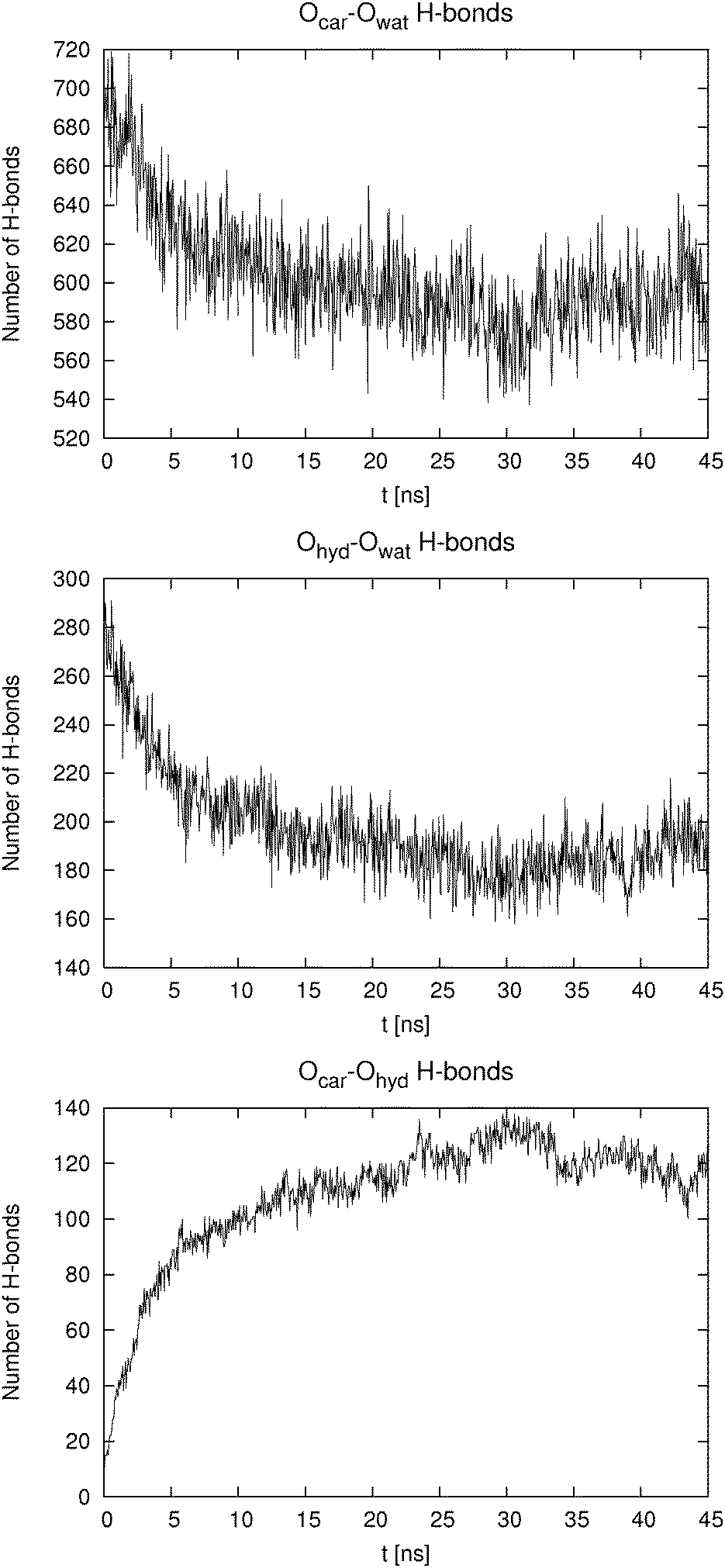 | ||
| Fig. 5 Total number of hydrogen bonds formed between Ocar and Owat (top), Ohyd and Owat (center), and Ocar and Ohyd (bottom), for p-HB in SPC water at the higher concentration as a function of time. Data points are 50 ps apart. | ||
From Table 2 one sees, for the higher concentration systems, that the total number of hydrogen bonds formed is approximately the same for the meta and para isomers, whereas it is lower for the ortho isomer. In general, numbers are smaller than for the lower concentration and higher for the SPC/E water than for the SPC water model. However, in SPC water there are more hydrogen bonds formed between HB molecules (Ocar–Ohyd), which is probably due to the lower tendency for the formation of hydrogen bonds between hydroxybenzoates and SPC water than between hydroxybenzoates and SPC/E water. The increased tendency for dimer (cluster) formation in SPC water can also be seen from Fig. 4 where the maxima for the SPC systems are higher than those for the SPC/E solutions. A possible reason for the greater affinity of SPC/E water to the HB anion is the larger value of the positive charge of the water hydrogen atoms in the SPC/E water model in comparison with the SPC water model.
The correlation of the mobility of the HB anions with the number of formed hydrogen bonds is not so simple at high concentration as it is at low concentration. The ortho isomer, which has the largest self-diffusion coefficient, forms the least hydrogen bonds, but meta and para isomers, whose self-diffusion coefficients differ, form approximately the same number of hydrogen bonds. While at lower concentration, due to the absence of significant clustering, only hydration determines the mobility of HB anions, at high concentration the formation of dimers and higher aggregates must be considered. One can see that in either solvent the para isomer forms slightly more hydrogen bonds with other HB anions (it is thus also less well hydrated), which might result in lower mobility.
The question arises, namely to what extent hydration and aggregation contribute to the overall self-diffusion coefficient. From Table 2 and Fig. 4, 6 and 7 it is clear that all three isomers at the higher concentration form some sort of aggregates. Fig. 6 and 7 show pair distribution functions for hydroxylic hydrogen to carboxylic or hydroxylic oxygen, respectively. Only the PDFs for systems with the SPC/E water model are plotted, as those for the systems with the SPC water model are very similar. When calculating these functions, we excluded pairs in which both atoms were in the same molecule. These two figures suggest that the formation of aggregates is mostly achieved through the hydrogen bond formation between hydroxylic oxygen and carboxylic oxygen. However, in Fig. 7 one also notes a very small peak around 1.5 Å, which implies that a tiny fraction of aggregates is due to hydrogen bonds in which the hydroxylic oxygen is both a donor and an acceptor at the same time.
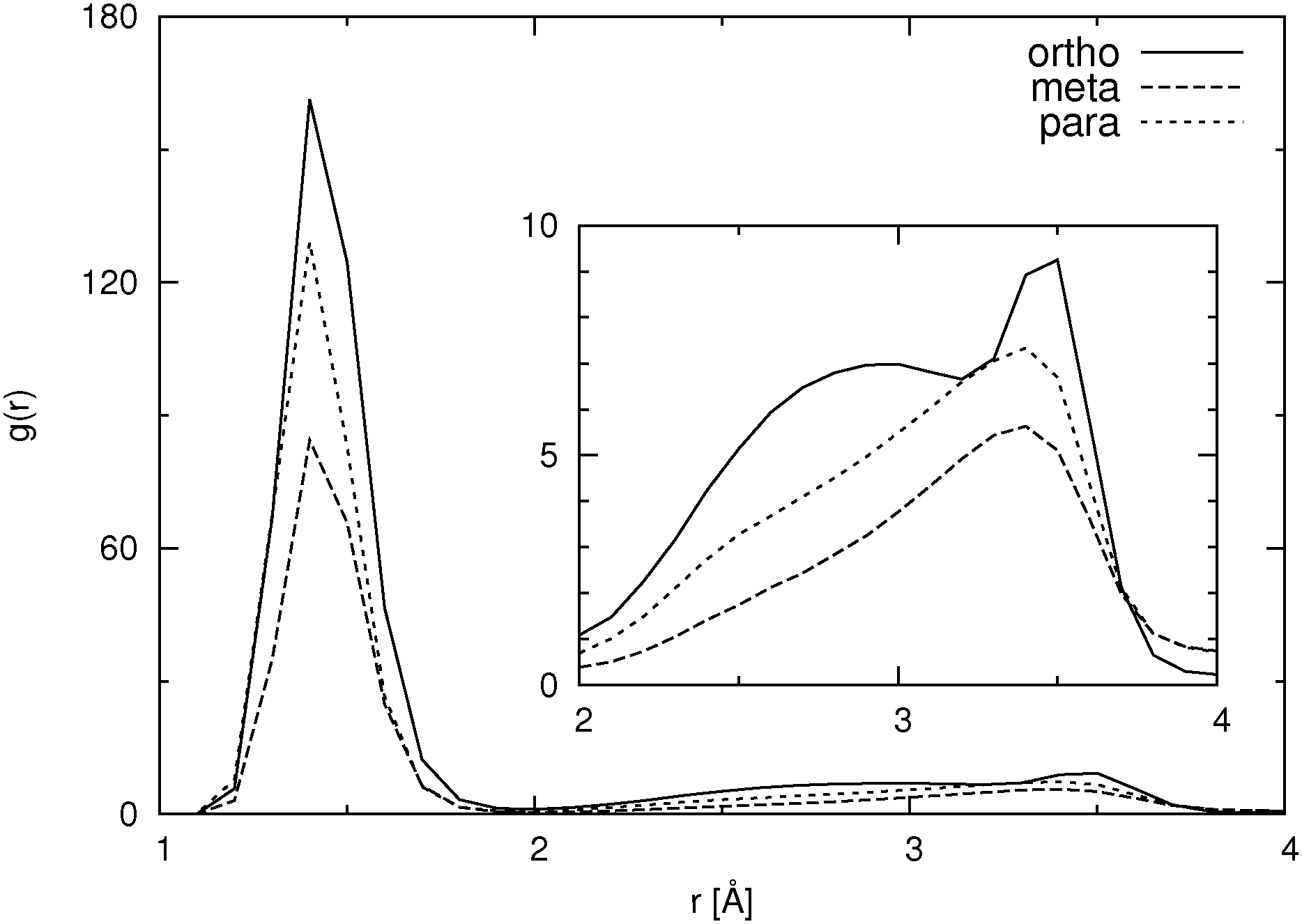 | ||
| Fig. 6 Pair distribution functions for nonaromatic (hydroxylic) hydrogen atoms to carboxylic oxygen atoms (without intramolecular pairs) for different HB isomers in SPC/E water at higher concentration. | ||
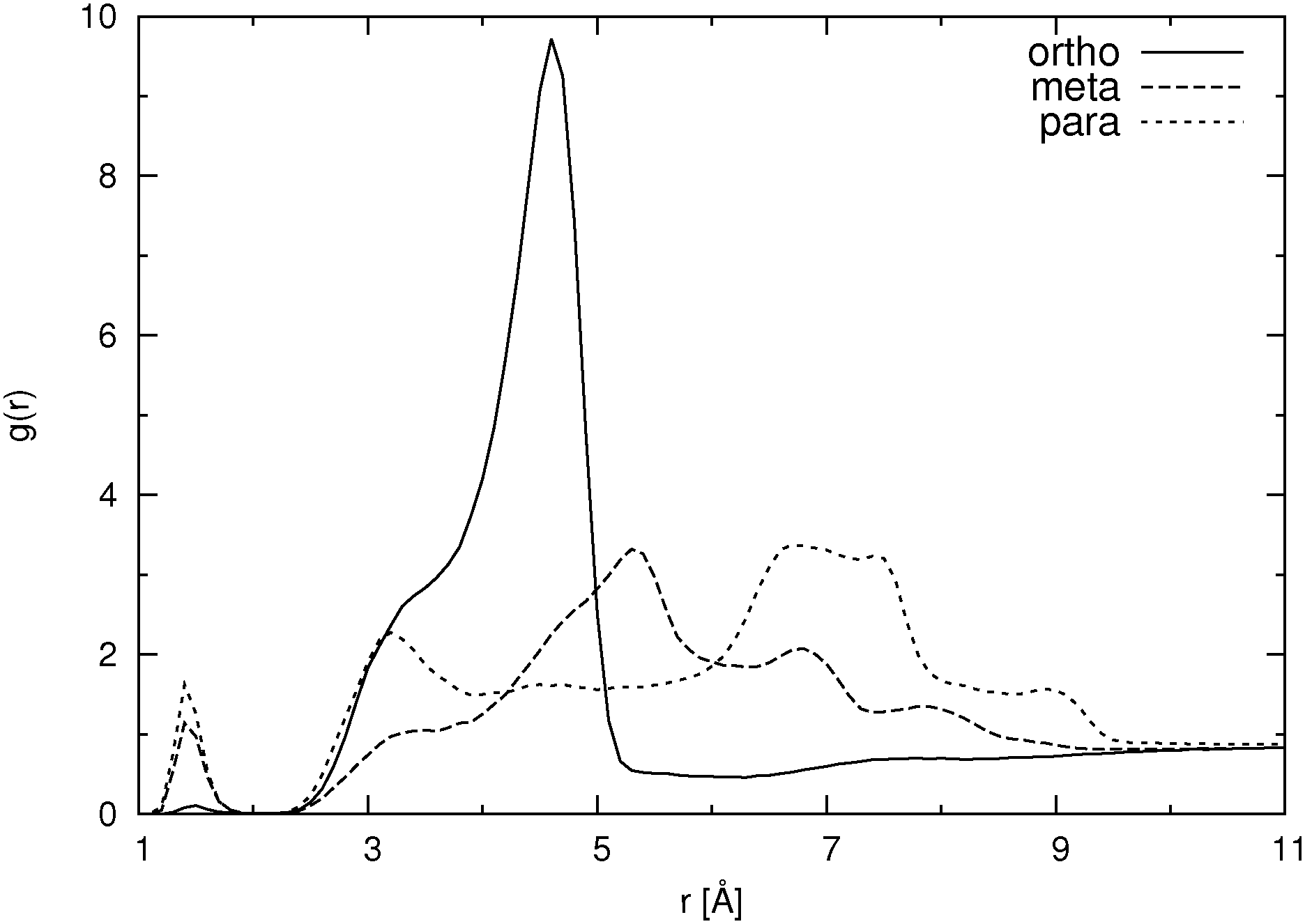 | ||
| Fig. 7 Pair distribution function for nonaromatic (hydroxylic) hydrogen atoms to hydroxylic oxygen atoms (without intramolecular pairs) for different HB isomers in SPC/E water at higher concentration. | ||
To further elucidate the aggregation in aqueous solutions of HB anions, we performed an analysis of the clusters formed by hydrogen bonding for each isomer in both solvents. The size distribution of clusters is presented in Table 3. A cluster is an aggregate of HB molecules that are connected with hydrogen bonds, where the definition of the hydrogen bond is defined geometrically as described earlier in the text. The ortho isomer tends to predominantly form clusters of size 2, i.e., dimers; almost two thirds of all ions in SPC solvent are in clusters of that size. The number of dimers is lower in SPC/E solvent, but this is expected and agrees well with the previously made statement that in this solvent there are less hydrogen bonds formed among HB anions. In both solvents we can even find trimers, but their formation is not very likely. We also notice that around 70% of clusters involving ortho isomers are cyclic. By our definition, a cluster is cyclic, when the number of HB–HB hydrogen bonds equals or exceeds the number of molecules in the cluster. While this is not a perfect classification, it should suffice for the present discussion. This means that most molecules in the clusters form two hydrogen bonds, which can also be read from Table 4. An example of a cyclic trimer is drawn in Fig. 8 for the ortho case. If we disregard the uppermost or the lowermost ion in the trimer, we see examples of the acyclic and the cyclic dimer, respectively. We note in passing that there are no cyclic clusters of size 1, which means that the rather strict definition of the hydrogen bond does not lead to intramolecular hydrogen bonds.
| SPC water model | SPC/E water model | ||||
|---|---|---|---|---|---|
| i | x i (%) | Cyclic (%) | i | x i (%) | Cyclic (%) |
| ortho | |||||
| 1 | 31.1 | 0.0 | 1 | 41.4 | 0.0 |
| 2 | 66.4 | 70.8 | 2 | 57.4 | 69.9 |
| 3 | 2.5 | 70.5 | 3 | 1.2 | 71.0 |
| meta | |||||
| 1 | 43.9 | 0.0 | 1 | 57.0 | 0.0 |
| 2 | 34.4 | 25.3 | 2 | 27.2 | 13.9 |
| 3 | 14.6 | 21.1 | 3 | 10.8 | 24.6 |
| 4 | 4.2 | 19.6 | 4 | 1.6 | 16.6 |
| 5 | 2.9 | 26.9 | 5 | 1.9 | 12.8 |
| >5 | 0.0 | — | >5 | 1.5 | — |
| para | |||||
| 1 | 23.2 | 0.0 | 1 | 41.9 | 0.0 |
| 2 | 15.6 | 0.0 | 2 | 21.9 | 0.1 |
| 3 | 15.6 | 10.9 | 3 | 12.8 | 7.3 |
| 4 | 15.1 | 36.0 | 4 | 9.3 | 22.0 |
| 5 | 13.0 | 35.0 | 5 | 6.4 | 35.9 |
| 6 | 7.4 | 47.7 | 6 | 4.1 | 31.6 |
| 7 | 4.3 | 40.7 | 7 | 1.8 | 19.0 |
| >7 | 5.8 | — | >7 | 1.8 | — |
| No. of H-bonds per molecule | Fraction of molecules (%) | ||
|---|---|---|---|
| ortho | meta | para | |
| SPC water model | |||
| 0 | 31.1 | 43.9 | 23.2 |
| 1 | 20.4 | 37.6 | 42.0 |
| 2 | 47.9 | 16.8 | 28.5 |
| 3 | 0.6 | 1.7 | 6.1 |
| 4 | 0.0 | 0.0 | 0.2 |
| SPC/E water model | |||
| 0 | 41.4 | 57.0 | 41.9 |
| 1 | 17.8 | 31.7 | 40.5 |
| 2 | 40.5 | 10.1 | 14.3 |
| 3 | 0.3 | 1.2 | 2.8 |
| 4 | 0.0 | 0.0 | 0.5 |
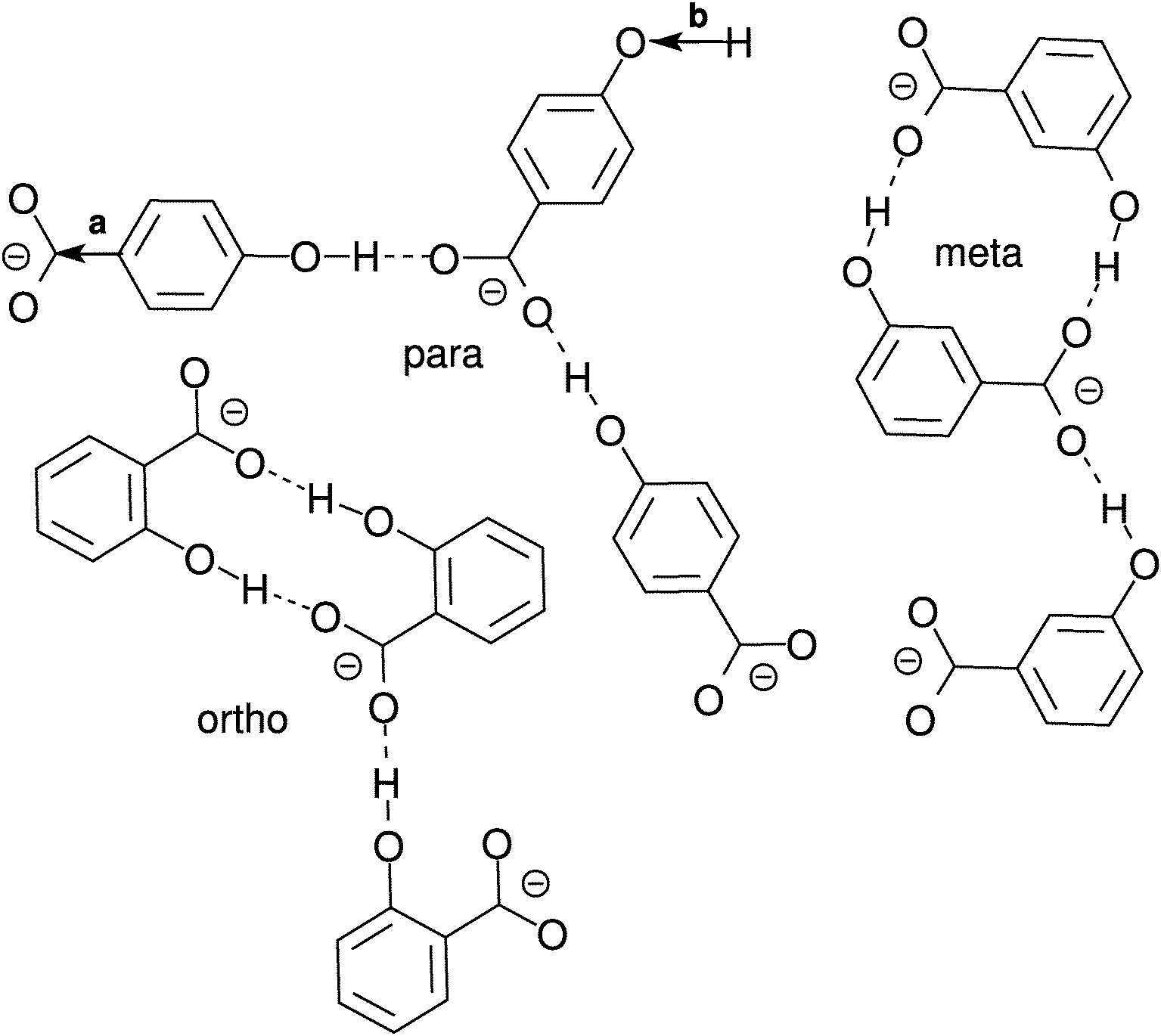 | ||
| Fig. 8 Possible hydroxybenzoate anion cluster geometries as suggested by pair distribution and angle distribution functions. Drawings are not to scale. Bonds with arrows marked with a and b denote the vectors for which the angle distributions in Fig. 10 have been calculated. | ||
The meta isomer also forms mostly clusters of size 2, but unlike the ortho isomer more than 10% of anions are members of trimers, and there is a non-negligible fraction of higher aggregates. There is yet another important difference to the ortho isomer. There are almost 43% of free (unimer) anions in SPC water and 57% in SPC/E water, which implies that the ortho isomer has a higher tendency for aggregation than the meta isomer. However, the diffusion coefficient of the ortho isomer is still higher, since the ortho isomer, on the other hand, forms less hydrogen bonds with water and, in addition, its clusters are smaller. In SPC water the largest observed cluster comprised 5 HB anions, and in SPC/E water the size of the largest observed cluster was 8. Some 25% of clusters in SPC water and roughly 20% of them in SPC/E water are cyclic. A cyclic component of a cluster is most likely very similar to the dimer consisting of the upper two m-HB ions in Fig. 8 (upper right), but it can also be a true ring with three or more HB anions. However, such behaviour is much less likely than for the para isomer. Most HB ions in the clusters form only one hydrogen bond with another HB ion (Table 4) which explains the lower fraction of cyclic clusters than in the ortho case. Cluster formation is, not surprisingly, more frequent in SPC solutions of m-HB than in SPC/E water solutions.
Para hydroxybenzoate has an even lower self-diffusion coefficient than the other two isomers, which can also be attributed to its aggregate formation. From Table 3 one can see that in SPC water only some 23% of p-HB unimers exist as free ions, which indicates an even larger tendency of the para isomer to form aggregates than of the ortho isomer. In SPC/E water this tendency is approximately the same for both isomers. It is also evident that the para isomer forms much larger clusters than the other two isomers. In SPC water the largest observed cluster contained 17 molecules (9 in SPC/E water) and some of the clusters were found to be cyclic, which indicates the formation of rings. Note that the largest clusters are found to be box-spanning (i.e. percolating), but it should also be noted that the lifetime of these large clusters is very short, since they can break and re-form at various places.
Although the analysis of hydrogen bonding shows that p-HB is less solvated than the meta isomer, the formation of clusters seems to outweigh the smaller influence of the hydration, so that the overall mobility is lower. Since there are two carboxylic oxygens, there is a possibility to form branched chains, or rings which have a chain attached to them. The formation of branched chains and rings can also be observed in solutions of the meta isomer, but because of smaller cluster sizes this phenomenon is not very pronounced. Unlike the other two isomers, the para isomer can form four hydrogen bonds per molecule (see Table 4). The observed number of such unimers is, however, very small for sterical reasons, but statistically significant. One such cluster of size 6, which is, in addition, cyclic, is shown in Fig. 11.
One can also attempt to analyse the cluster data in Table 3 within the framework of a simple statistical model analogous to the differential mass-distribution function of macromolecular assemblies.30 For the such simplest model the size distribution function fw(i) = (1 − p)2·i·pi−1 where i is the cluster size and p (denoted as 1 − a in ref. 30) is the turnover or the number of cluster bonds per monomer. This model assumes that no cyclic oligomers exist, an assumption that is not strictly fulfilled in our simulations. Nevertheless, it is instructive to use the data in Table 3 to calculate p for different cases by taking the ratio of the probability to find a cluster of size i relative to the probability to find one with a size i − 1. This ratio is given as
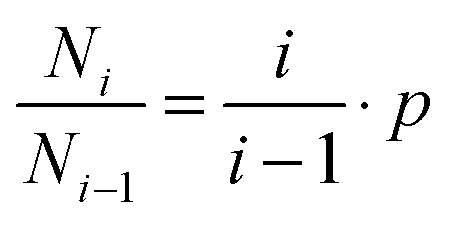 | (3) |
It is also very interesting to look at the cluster dispersity vs. time, which is plotted for the para isomer in SPC water in Fig. 9. The dispersity Đ, which is a measure of the broadness of the cluster size distribution, is calculated here as the ratio between the size average cluster size and the number average cluster size. The definition of the dispersity used here (see eqn (4)) is analogous to the definition of the dispersity (also called polydispersity) in polymer chemistry. Instead of the molar mass we use the size of the cluster.
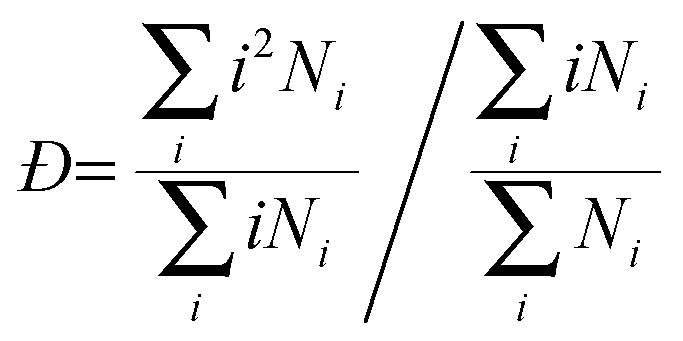 | (4) |
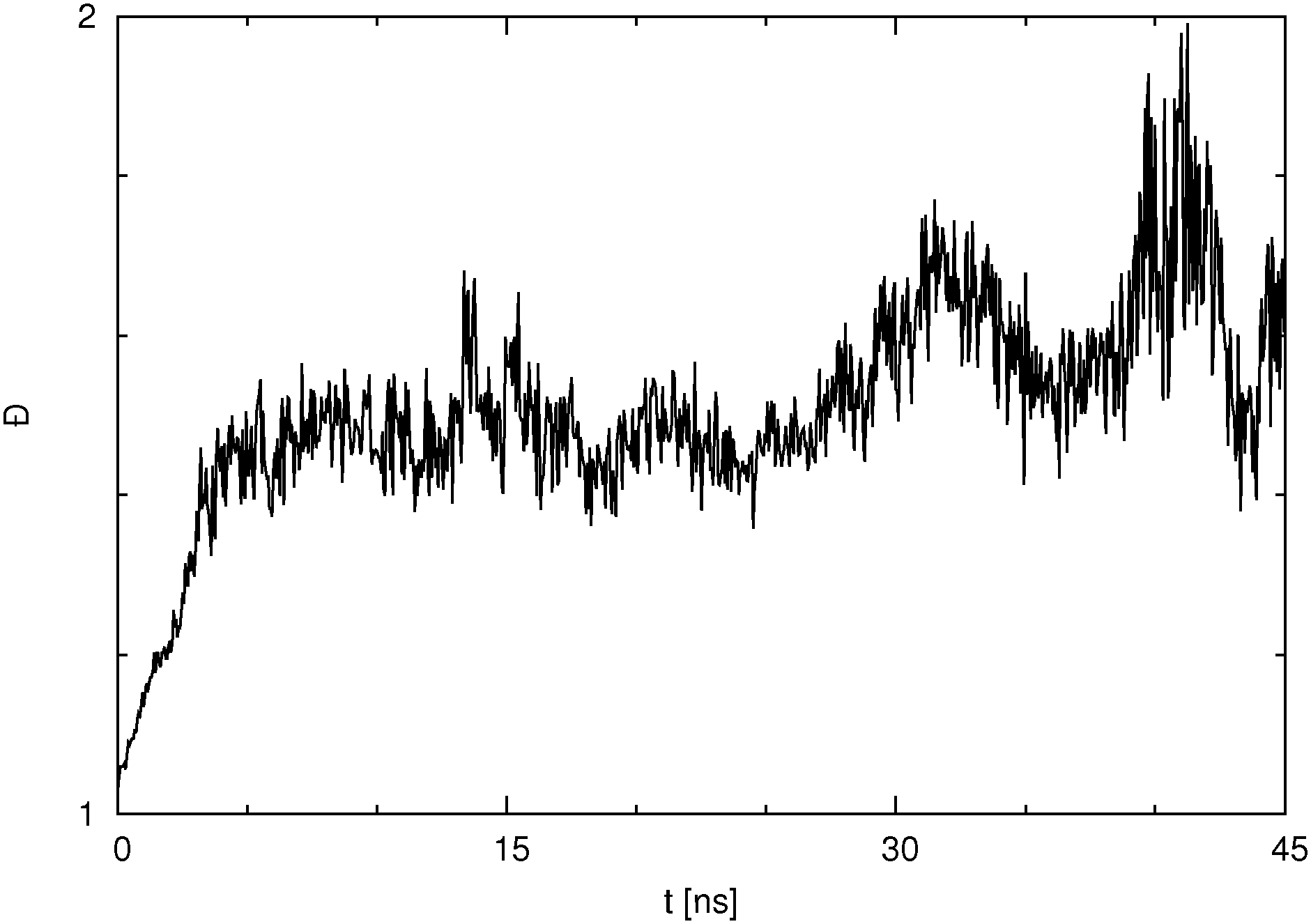 | ||
| Fig. 9 Dispersity Đ as a function of simulation time for p-HB in SPC water. Đ is taken as the ratio between the size average and the number average cluster size. | ||
In Fig. 8 a few possible aggregate structures are drawn as suggested by the pair distribution functions and by the angle distribution functions shown in Fig. 10, which show the distributions of the cosine of the angle between neighbour pairs of selected molecule fixed vectors. On the left side the molecule-fixed vector is defined from the aromatic carbon atom to which the COO− group is bound to the carboxylic carbon atom of the same molecule (vector a in Fig. 8). On the right side the vector is defined as the vector from the hydroxylic hydrogen to the hydroxylic oxygen of the same molecule (vector b in Fig. 8). The angle between pairs of defined vectors is calculated and counted if two molecules in question are less than a specified distance apart. This distance is chosen so that it corresponds to the distance of the first minimum in the pair distribution function for the carboxylic carbons (second minimum for m-HB) for the graph on the left side. For the right side a cutoff distance of 11 Å was chosen (on the basis of the RDF for the hydroxylic oxygen atoms). The distribution functions provide evidence about possible preferential orientations of the molecules that form the clusters.
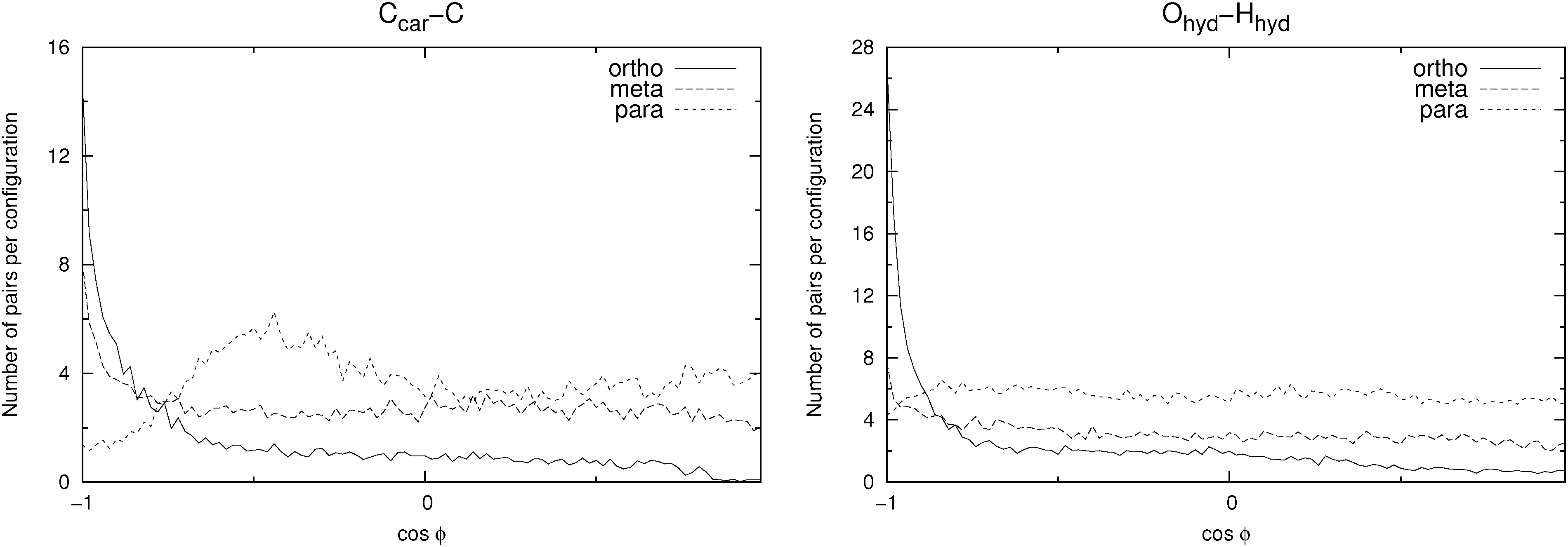 | ||
| Fig. 10 Distribution of the cosine of the angle ϕ between pairs of vectors in neighbouring molecules. Data shown are for the SPC/E solvent. Left: vector from the aromatic carbon to the carbon atom of the carboxylate group attached to it (vector a in Fig. 8). Right: vector from the hydroxylic hydrogen to the hydroxylic oxygen atom (vector b in Fig. 8). The distance between those pairs of molecules for which the angle is calculated is defined on the basis of the pair distribution functions (Ccar–Ccar for the left graph and Ohyd–Ohyd for the right graph). For details see text. | ||
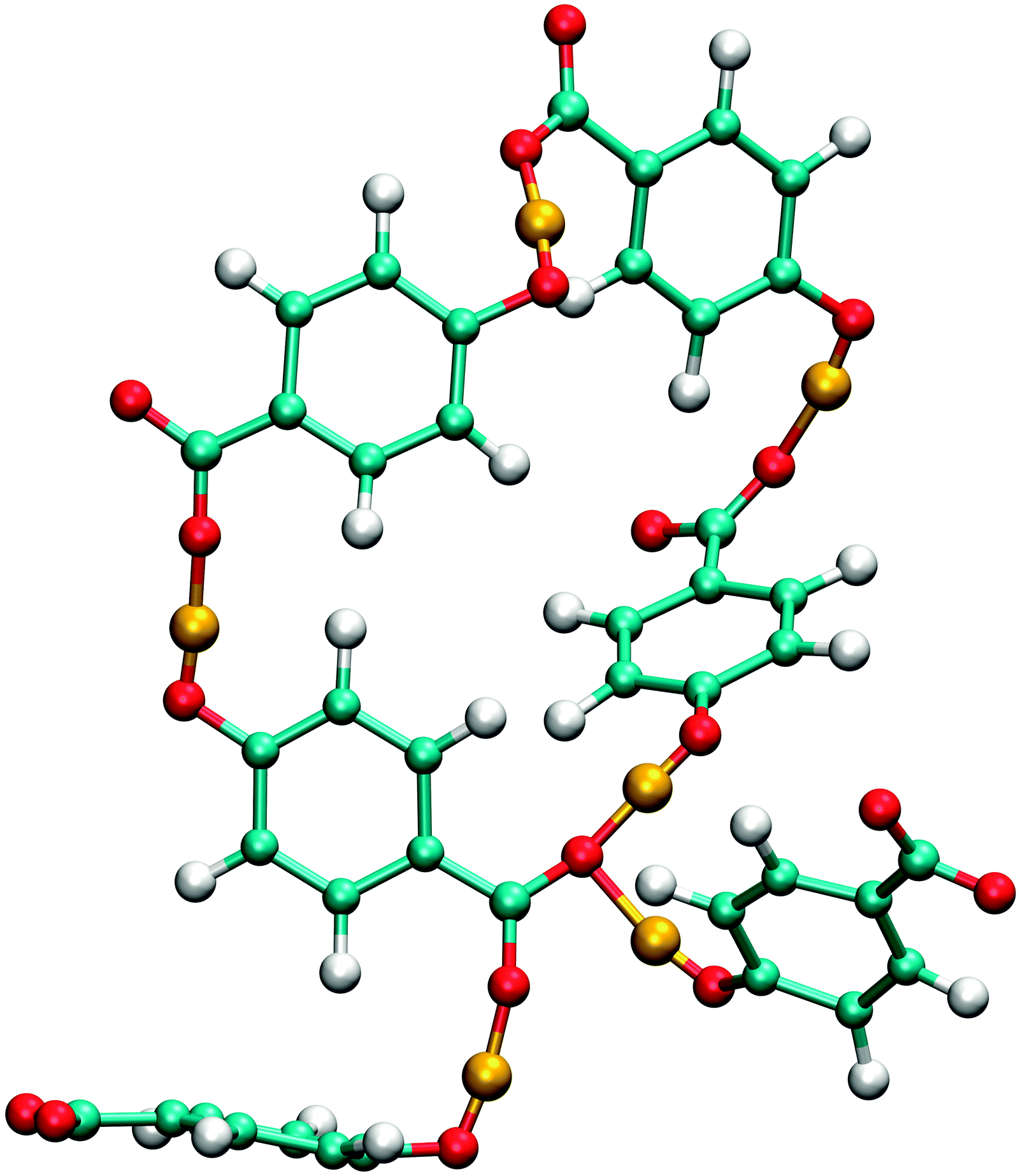 | ||
| Fig. 11 An example of a cyclic cluster of p-HB anions in SPC water. Hydroxylic hydrogens are drawn in orange colour for easier identification of hydrogen bonds. The depicted hexamer also contains a single anion that forms four hydrogen bonds. | ||
For the ortho isomer both vectors show a preferential antiparallel orientation, as can be seen from the pronounced maximum at cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ϕ = −1 (which corresponds to an angle ϕ = 180°). In Fig. 7 a relatively sharp peak at approximately 4.5 Å is visible, which corresponds to the hydroxylic hydrogen atom of the molecule that forms the hydrogen bond with a carboxylic oxygen. The proposed aggregate structure in Fig. 8 accounts for the features of these distribution functions.
ϕ = −1 (which corresponds to an angle ϕ = 180°). In Fig. 7 a relatively sharp peak at approximately 4.5 Å is visible, which corresponds to the hydroxylic hydrogen atom of the molecule that forms the hydrogen bond with a carboxylic oxygen. The proposed aggregate structure in Fig. 8 accounts for the features of these distribution functions.
In the case of the para isomer one observes a distinct peak at 7–8 Å in the RDF for carboxylic carbons and a broad peak at approximately 6–8 Å in the RDF between hydroxylic hydrogen and hydroxylic oxygen. The angle distributions show no preferential orientation for the vector corresponding to the hydroxylic group, but a very broad peak in the Ccar–C angle distribution function at cosϕ = −0.5 (ϕ = 120°), and the function increases once more slightly for the parallel orientations. The structural model of the cluster in Fig. 8 is consistent with these data; furthermore, such structural features allow the formation of chain-like aggregates of a larger number of p-HB anions.
Pair distribution functions for the carboxylic carbons of the meta isomer show two distinct peaks at about 5.5 Å and 7.5 Å respectively, suggesting two type of aggregates, or at least two types of connections between anions. In Fig. 7, which shows the RDF between hydroxylic oxygen and hydroxylic hydrogen, one detects a peak at about 5.5 Å and a smaller one at about 7 Å. Angle distributions in both cases show preferential antiparallel orientations, but this preference is not as strong as in the case of the ortho isomer. These features suggest again two type of aggregates. One is represented by the center and upper molecules in Fig. 8 (meta isomer), and the other is represented by the center and lower molecules. Of course the orientation of the lower molecule in the meta isomer can also be the opposite.
The intra-aggregate bonding exhibited by the ortho isomer does not allow for the formation of long lived higher aggregates, because it is sterically highly unlikely that the third HB anion approaches and binds to the remaining free carboxylic oxygen atom. The cyclic ortho dimer seems to be much more stable than the corresponding aggregates of meta or para hydroxybenzoates, as reflected by the fact that the number of hydrogen bonds between HB anions is the highest for o-HB. More than 47% of the ortho anions in SPC water (40% in SPC/E water) form two hydrogen bonds with another o-HB anion, which is another indication of the stability of cyclic ortho dimer. However, the ortho isomer, which is the least solvated, forms, in total, 0.3–0.4 less hydrogen bonds per molecule than the other two isomers and is therefore the most mobile. The lower hydration of the o-HB anions is the consequence of two facts: (i) the proximity of both functional groups on the benzene ring, and (ii) the stability of cyclic dimers. From the sterical point of view, the meta and para isomers are more likely to form higher aggregates. This is especially true for the para isomer where there is no sterical hindrance to extend the “chain”. Although both isomers form approximately the same number of hydrogen bonds in total, it appears that the greater fraction of hydrogen bonds with other molecules makes the para isomer less mobile than the meta isomer.
4 Conclusions
Atomistic molecular dynamics simulations have been performed to investigate transport and structural properties of hydroxybenzoate anions in aqueous solutions at 298.15 K. The self-diffusion coefficients of all three geometric isomers were calculated and the results qualitatively agree with the experimental data. MD simulations on the basis of the GROMOS96 45a3 force field and SPC or SPC/E water can correctly rank HB anions according to their mobilities (o-HB > m-HB > p-HB). The mobility of HB ions is larger in SPC than in SPC/E water. At lower concentrations only the formation of hydrogen bonds with water determines the isomer's mobility. o-HB is the least hydrated isomer and therefore the most mobile, whereas p-HB forms the most hydrogen bonds with water and has the lowest self-diffusion coefficient. At higher concentrations hydrogen bonds can also be formed between HB molecules, which results in the formation of dimers or higher aggregates. o-HB predominantly forms dimers with two hydrogen bonds per molecule, m-HB forms higher aggregates with size up to 8, and p-HB can form aggregates the size of which can exceed 10 molecules per cluster. Clusters of p-HB can be rings or chains. In SPC solvent some 30% of unimers were found to be free ions in the case of o-HB, around 50% in the case of m-HB, and about one quarter of the unimers were not clustered in the case of p-HB. In SPC/E water the tendency for formation of clusters is lower and fractions of free ions are at the studied concentration roughly 10–20 percentage points greater than in SPC water.Acknowledgements
The financial support from the Slovenian Research Agency through Grant No. P1-0201 and J1-0103-4148 is gratefully acknowledged. J.G. is grateful to Slovene Human Resources Development and Scholarship Fund for the scholarships enabling the research cooperation at University of Duisburg-Essen. E.S. acknowledges support from the Cluster of Excellence RESOLV (EXC1069) funded by the Deutsche Forschungsgemeinschaft. The work was partially supported by COST Action CM1101.References
- Y. Sakaiguchi, T. Shikata, H. Urakami, A. Tamura and H. Hirata, J. Electron Microsc., 1987, 36, 168–176 CAS.
- H. Hirata, M. Sato, Y. Sakaiguchi and Y. Katsube, Colloid Polym. Sci., 1988, 266, 862–864 CAS.
- Z. Lin, J. J. Cai, L. E. Scriven and H. T. Davis, J. Phys. Chem., 1994, 98, 5984–5993 CrossRef CAS.
- S. Kumar, D. Bansal and K. ud Din, Langmuir, 1999, 15, 4960–4965 CrossRef CAS.
- K. Bijma, E. Rank and J. B. F. N. Engberts, J. Colloid Interface Sci., 1998, 205, 245–256 CrossRef CAS.
- W. J. Kim and S. M. Yang, J. Colloid Interface Sci., 2000, 232, 225–234 CrossRef CAS PubMed.
- Y. Geng, L. S. Romsted, S. Froehner, D. Zanette, L. J. Magid, I. M. Cuccovia and H. Chaimovich, Langmuir, 2005, 21, 562–568 CrossRef CAS PubMed.
- T. Mukhim, J. Dey, S. Das and K. Ismail, J. Colloid Interface Sci., 2010, 350, 511–515 CrossRef CAS PubMed.
- N. Onoda-Yamamuro, O. Yamamuro, N. Tanaka and H. Nomura, J. Mol. Liq., 2005, 117, 139–145 CrossRef CAS PubMed.
- A. V. Sangwai and R. Sureshkumar, Langmuir, 2011, 27, 6628–6638 CrossRef CAS PubMed.
- A. L. Underwood and E. W. Anacker, J. Colloid Interface Sci., 1985, 106, 86–92 CrossRef CAS.
- K. Bijma, M. J. Blandamer and J. B. F. N. Engberts, Langmuir, 1998, 14, 79–83 CrossRef CAS.
- B. Šarac, J. Cerkovnik, B. Ancian, G. Mériguet, G. M. Roger, S. Durand-Vidal and M. Bešter-Rogač, Colloid Polym. Sci., 2011, 289, 1597–1607 Search PubMed.
- B. Šarac, G. Mériguet, B. Ancian and M. Bešter-Rogač, Langmuir, 2013, 29, 4460–4469 CrossRef PubMed.
- M. Bešter-Rogač, J. Chem. Eng. Data, 2011, 56, 4965–4971 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. van Gunsteren and J. Hermans, in Intermolecular Forces, ed. B. Pullman, D. Reidel Publishing Company, Dordrecht, 1981, p. 331 Search PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- http://lammps.sandia.gov .
- G. J. Kleywegt and T. A. Jones, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1119–1131 CrossRef CAS.
- D. M. F. van Aalten, R. Bywater, J. B. C. Findlay, M. Hendlich, R. W. W. Hooft and G. Vriend, J. Comput.-Aided Mol. Des., 1996, 10, 255–262 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- L. D. Schuler, X. Daura and W. F. van Gunsteren, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS PubMed.
- A. Bochevarov, E. Harder, T. Hughes, J. Greenwood, D. Braden, D. Philipp, D. Rinaldo, M. Halls, J. Zhang and R. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS PubMed.
- R. Hockney and J. Eastwood, Computer Simulation Using Particles, Adam Higler, New York, 1989 Search PubMed.
- W. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, New York, 1991 Search PubMed.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology, ed. A. D. McNaught and A. Wilkinson, the “Gold Book”, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
| This journal is © the Owner Societies 2014 |