
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited electronic states of thiophene: high resolution photoabsorption Fourier transform spectroscopy and ab initio calculations†‡
D. M. P.
Holland
*a,
A. B.
Trofimov
bc,
E. A.
Seddon
a,
E. V.
Gromov
bd,
T.
Korona
e,
N.
de Oliveira
f,
L. E.
Archer
f,
D.
Joyeux
f and
L.
Nahon
f
aDaresbury Laboratory, Daresbury, Warrington, Cheshire WA4 4AD, UK. E-mail: david.holland@sftc.ac.uk
bLaboratory of Quantum Chemistry, Irkutsk State University, 664003 Irkutsk, Russia
cFavorsky Institute of Chemistry, SB RAS, 664033 Irkutsk, Russia
dTheoretische Chemie, Physikalisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 229, D-69120 Heidelberg, Germany
eFaculty of Chemistry, University of Warsaw, ul. Pasteura 1, PL-02-093 Warsaw, Poland
fSynchrotron Soleil, Orme des Merisiers, St Aubin BP 48, 91192 GIF sur Yvette Cedex, France
First published on 8th September 2014
Abstract
The recently introduced synchrotron radiation-based Fourier transform spectroscopy has been employed to study the excited electronic states of thiophene. A highly resolved photoabsorption spectrum has been measured between ∼5 and 12.5 eV, providing a wealth of new data. High-level ab initio computations have been performed using the second-order algebraic-diagrammatic construction (ADC(2)) polarization propagator approach, and the equation-of-motion coupled-cluster (EOM-CC) method at the CCSD and CC3 levels, to guide the assignment of the spectrum. The adiabatic energy corrections have been evaluated, thereby extending the theoretical study beyond the vertical excitation picture and leading to a significantly improved understanding of the spectrum. The low-lying π → π* and π → σ* transitions result in prominent broad absorption bands. Two strong Rydberg series converging onto the ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2A2 state limit have been assigned to the 1a2 → npb11B2 and the 1a2 → nda21A1 transitions. A second, and much weaker, d-type series has been assigned to the 1a2 → ndb11B2 transitions. Excitation into some of the Rydberg states belonging to the two strong series gives rise to vibrational structure, most of which has been interpreted in terms of excitations of the totally symmetric ν4 and ν8 modes. One Rydberg series, assigned to the 3b1 → nsa11B1 transitions, has been identified converging onto the à 2B1 state limit, and at higher energies Rydberg states converging onto the
2A2 state limit have been assigned to the 1a2 → npb11B2 and the 1a2 → nda21A1 transitions. A second, and much weaker, d-type series has been assigned to the 1a2 → ndb11B2 transitions. Excitation into some of the Rydberg states belonging to the two strong series gives rise to vibrational structure, most of which has been interpreted in terms of excitations of the totally symmetric ν4 and ν8 modes. One Rydberg series, assigned to the 3b1 → nsa11B1 transitions, has been identified converging onto the à 2B1 state limit, and at higher energies Rydberg states converging onto the ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 2A1 state limit could be identified. The present spectra reveal highly irregular vibrational structure in certain low energy absorption bands, and thus provide a new source of information for the rapidly developing studies of excited state non-adiabatic dynamics and photochemistry.
2A1 state limit could be identified. The present spectra reveal highly irregular vibrational structure in certain low energy absorption bands, and thus provide a new source of information for the rapidly developing studies of excited state non-adiabatic dynamics and photochemistry.
1. Introduction
Thiophene (Fig. 1) is one of the most studied heterocyclic aromatic compounds,1–3 yet it remains a focal point of much on-going research due to the central role it continues to play in the challenges facing modern science and technology. Materials that include thiophene units, such as thiophene polymers and oligomers, possess various important properties,4–6 which make them promising as photochromic molecular switches,7–9 organic semiconductors,10,11 solar cells,12,13 light-emitting diodes and field-effect transistors.14–16 For many of these applications, understanding the fundamental electronic structure, spectroscopy, and photophysics of thiophene is of primary importance, and considerable effort is constantly being made to gain further knowledge in these fields. A prominent example is the remarkable increase in recent years of the interest in the ultrafast decay of the lowest excited states of thiophene.17–22 Despite the unprecedented activity of several theoretical research groups, some basic photochemical aspects related to the evolution of the lowest excited states of thiophene have yet to be clarified. The main factor hindering this progress is the absence of high resolution photoabsorption data which would allow the peak profiles associated with these short lived states to be observed. | ||
| Fig. 1 Schematic representation of thiophene showing the atomic labelling and axis orientation. | ||
The excited electronic states of thiophene have been studied extensively in the past. The gas phase UV photoabsorption spectrum of thiophene23–32 exhibits several prominent bands associated with valence states. In addition, some much sharper but less intense structure has been assigned to Rydberg series converging onto the ground state ( 2A2) ionization threshold. More recently, Rennie et al.33 have measured the absolute photoabsorption cross section up to ∼36 eV, and have observed Rydberg series converging onto the 2A1 or the ![[G with combining tilde]](https://www.rsc.org/images/entities/char_0047_0303.gif) 2A1 state ionization limits. Complementary theoretical investigations, using various computational approaches, have been undertaken to obtain transition energies and oscillator strengths for excitation into low-lying valence or Rydberg states.17–22,32,34–39
2A1 state ionization limits. Complementary theoretical investigations, using various computational approaches, have been undertaken to obtain transition energies and oscillator strengths for excitation into low-lying valence or Rydberg states.17–22,32,34–39
Of relevance to our Rydberg state studies are previous measurements of the photoelectron spectrum of thiophene using H (121.5 nm),40 NeI,41 HeI,42–45 HeII,42–44 and synchrotron46,47 radiation. The valence shell Penning ionization spectrum has also been recorded.41,45 The high resolution photoelectron spectra40,42 show vibrational progressions, all of which were attributed to totally symmetric modes, in the 2A2, 2A1 and 2A1 state bands. More recently,48 the vibrational structure in the 2A2 state has been examined in greater detail using the ZEKE technique. The vibrational structure in the 2A2 and the à 2B1 state photoelectron bands has been investigated theoretically.49,50 Various theoretical approaches have been used to investigate the complete valence shell photoelectron spectrum of thiophene.51–54
Finally, the low-lying neutral excited states have been studied using electron impact55–58 and magnetic circular dichroism (MCD).59–61 The polarized crystal spectrum of thiophene has also been recorded.34
Despite numerous experimental and theoretical investigations of the excited electronic states of thiophene, a consistent set of assignments for the complex and irregular bands observed in the UV photoabsorption spectrum has yet to emerge. Amongst the difficulties contributing to these inconsistencies are: (1) insufficient experimental resolution, as already mentioned, to observe rotational band contours or to separate peaks due to vibrational modes of similar energies; (2) the possible appearance of hot-bands in the experimental spectrum; (3) the excitation of non-totally symmetric vibrational modes due to vibronic coupling; and (4) configuration interaction resulting in final states having mixed (Rydberg–valence, Rydberg–Rydberg or valence–valence) character. It should be emphasized that condition (1) is especially critical for theoretical studies which include nuclear dynamics modelling. In such work it is essential to know whether the absence of resolved structure is due to experimental limitations or whether it is a manifestation of genuine molecular effects, such as vibronic coupling, non-adiabatic nuclear dynamics or some photochemical process.
The recent advent of Fourier transform (FT) spectroscopy62 using synchrotron radiation63 enables, for the first time, high resolution photoabsorption spectra to be recorded at energies above 8.8 eV. Thus, this new experimental technique not only allows significantly improved absorption data of thiophene to be obtained but it also allows access to high-lying excited states which are predominantly due to transitions from orbitals other than the outermost. Since the theoretical excited state studies rely heavily on the experimental results, our FT data facilitate major advances in the modelling of the non-adiabatic phenomena.
The aim of the present work is to extend our knowledge of the electronic states of thiophene in the energy range between 5 and 12.5 eV, with particular emphasis on the Rydberg states belonging to series converging onto the 2A2 state. The interpretation of our high resolution FT photoabsorption spectrum is guided by vertical transition energies and oscillator strengths computed using high-level ab initio methods, namely the second-order algebraic-diagrammatic construction (ADC(2)) polarization propagator approach64–68 and the equation-of-motion coupled-cluster (EOM-CC) method at the level of the CCSD and CC3 models.69–72 The large basis sets, which include polarization and diffuse functions, used in the computations ensure that the valence states and the many Rydberg states converging onto each of the ionization limits are reliably reproduced. In addition, the adiabatic corrections to the energies evaluated at the ADC(2) level, within the linear vibronic coupling model, extend the theoretical study beyond the vertical excitation picture.
2. Experimental apparatus and procedure
The photoabsorption spectrum of thiophene was recorded using the Fourier transform spectrometer (FTS)62 attached to the DESIRS beamline63 at the Soleil synchrotron radiation source. Detailed descriptions of the beamline and spectrometer have already been reported so only a brief summary is given here.The undulator-based beamline provides highly coherent radiation, with a bell-shaped intensity distribution, spanning the energy range ∼5–40 eV. The central energy of this distribution is tunable and can be selected by adjusting the magnetic field in the undulator.73 A typical bandwidth corresponds to ∼7% of the central energy. This continuum radiation forms the source for the FTS.
The spectrometer, which is a modified version of a Fresnel bimirror interferometer,74 employs only plane mirrors to perform the wavefront division. In this modified configuration, the Fresnel bimirrors are replaced by two roof-shaped reflecting mirrors and the spatial division of the coherent photon beam occurs at the interface of the two reflectors. One of these reflectors is fixed and the other can be translated, thereby providing a means to vary the optical path difference. The fixed and the mobile reflectors are tilted so as to make the two beams recombine and form an interference pattern. This interferogram is detected with a photodiode.
Prior to entering the FTS, the photon beam passes through a 100 mm long windowless absorption cell with 150 mm long entrance and exit capillaries. The cell could be filled with thiophene vapour obtained from a liquid sample, situated outside the vacuum chamber, held at room temperature. This arrangement, whilst providing a quasi-static column of gas in the absorption cell, due to the low conductance through the capillaries, also ensured that the gaseous sample was continuously refreshed. Before use, the liquid sample was subjected to several freeze–pump–thaw cycles.
The photon energy scale, which is intrinsically linear, obtained from the Fourier transform of the interferogram was calibrated by using the absorption line due to the Ar 3p6 1S0 → 3p5(2P1/2)4s transition at 11.8282 eV. A small peak due to this state appears in the spectrum and originates from the gas filter used to attenuate the high harmonics produced in the undulator.
The composite photoabsorption spectrum plotted in Fig. 2 has been assembled from seventeen individual overlapping spectral windows (undulator settings) and was measured with a resolution of 2.1 meV (FWHM). Although the FTS is capable of achieving a much higher resolving power (∼106), spectra recorded with an improved resolution did not reveal any significant additional structure. This suggests that the absorption band profiles in our spectrum of thiophene are determined mainly by the rapid decay of the short lived excited states. Rotational broadening from the room temperature sample may also contribute to the observed widths.
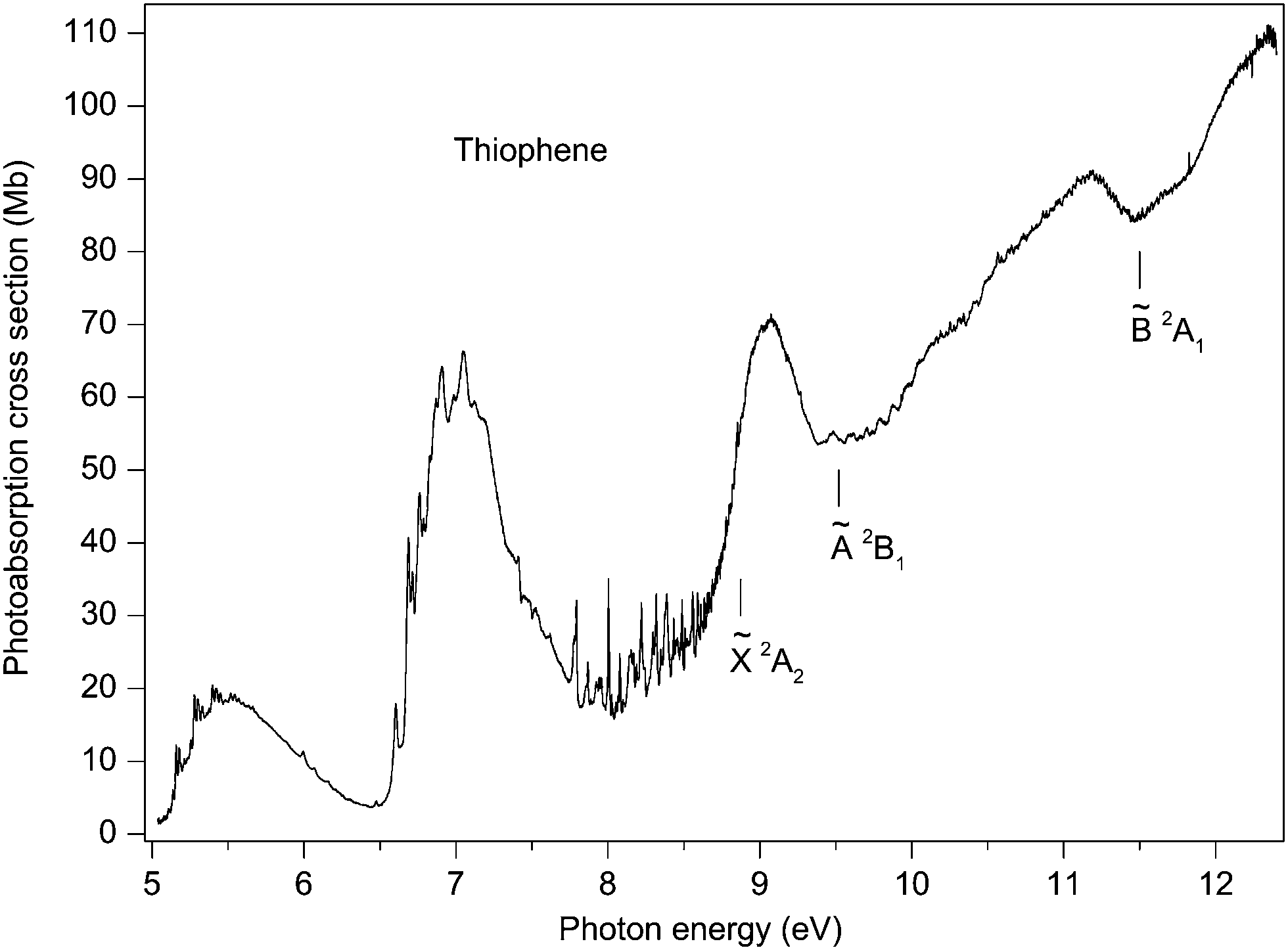 | ||
| Fig. 2 The photoabsorption spectrum of thiophene. The 2A2, Ã 2B1 and 2A1 state ionization thresholds are marked. | ||
The relative photoabsorption spectrum obtained from the FTS has been normalised, at an energy of 9.1 eV, to the absolute photoabsorption cross section measured by Rennie et al.33 At this energy the spectrum is devoid of sharp features.
3. Computational details
The EOM-CCSD approach,69–72 as implemented in MOLPRO,75,76 was employed to calculate the vertical absorption spectrum, the final state dipole moments and the second electronic moments of thiophene. The equilibrium ground state geometrical parameters were taken from the experimental work of Bak et al.77 The basis set was constructed from the aug-cc-pVTZ set78,79 on atoms and from s, p, d and f type Rydberg functions,80 each taken with effective quantum numbers n = 3, 3.5, 4, 4.5, 5, placed in the molecular centre of gravity. The total number of molecular orbitals in this basis (henceforth referred to as basis A) was 406. In the EOM-CCSD calculations, a molecular symmetry of C2v was adopted, with 25 roots being evaluated in each symmetry. For selected excited states, coupled-cluster calculations employing the next higher level CC3 (basis set A) model were performed, using the CFOUR program.81Additional calculations were carried out using the approach introduced in our previous theoretical studies of pyrrole82 and furan,83 thereby allowing the results for all three molecules (thiophene, pyrrole and furan) to be compared directly. In these calculations, the electronic excitations were treated using the ADC(2) approximation for the polarization propagator64–68 in the so-called ‘strict’ version.65 In this scheme, the vertical excitation energies and dipole transition moments are computed directly, and are accurate through to second and zero order of many-body perturbation theory with respect to singly and doubly excited configurations, respectively. The strict ADC(2) scheme can be compared to the CC2 model of the equation-of-motion (or linear response) coupled-cluster hierarchy, but the ADC(2) method has the advantage of having a Hermitian secular matrix and also it does not suffer from instabilities near conical intersections.84 The ADC(2) scheme is therefore often preferred in studies that go beyond the vertical excitation picture.82,83,85–88
As in our previous work,82,83 the information about the vibrational structure associated with the electronic transitions was obtained from the linear vibronic coupling (LVC) model.89,90 This model allows the adiabatic (Ω0–0) transition energies to be determined and the Franck–Condon spectrum for a given excited state to be constructed, once the linear vibrational coupling constants have been evaluated. These coupling constants are defined as the excitation energy derivatives with respect to the ground state normal coordinates of the totally symmetric vibrations. The LVC model works well if the excited state frequencies can be approximated with the ground state harmonic frequencies and if the adiabatic approximation is valid.89,90 This limitation means that the LVC model may have difficulties, and hence produce less reliable results, if the excited state frequencies differ significantly from those of the ground state, as, for example, can often be the case with excitations into anti-bonding orbitals. Another limitation of the present model is that it does not take into account the interaction between different states (all interstate couplings are assumed to be zero). As a result, the LVC model cannot explain the minima on the excited state potential energy surfaces which do not correspond to the symmetric C2v structures of the thiophene molecule (Fig. 1). Moreover, excitations of non-totally symmetric modes are not taken into account. Thus, the present treatment of the excited state vibrational structure should be considered as the first step beyond the vertical excitation picture. It does, nevertheless, provide additional information which is useful in the assignment of the experimental spectra.
The ADC(2) calculations for the various geometrical parameters required for determining the coupling constants were performed with a smaller basis set (henceforth referred to as basis B). This basis was constructed in a manner similar to that used for pyrrole and furan,82,83 and comprised the aug-cc-pVDZ set78,79 without the diffuse polarization functions on the sulphur and carbon atoms, and the cc-pVDZ set78,79 on the hydrogen atoms. Also, s, p and d Rydberg functions,80 each with effective quantum numbers n = 3, 3.5, 4, were placed in the molecular centre of gravity. Six Cartesian components of the d functions were used in the calculations. The total number of molecular orbitals for basis B was 149. In the ADC(2) computations, the carbon K-shell atomic orbitals, as well as the sulphur K- and L-shell atomic orbitals, were kept frozen. The original polarization propagator ADC software91 linked to the GAMESS-US package92 was used in these calculations.
In order to enable strict comparisons with the EOM-CCSD results, the ADC(2) calculations were performed also for basis A. Since these computations were beyond the capabilities of our ADC software,91 they were carried out using the ADC(2) implementation in the MOLPRO program.93
The evaluation of the vibrational coupling constants was accomplished by means of a numerical differentiation of the ADC(2) vertical excitation energies with respect to the dimensionless normal coordinates of the totally symmetric vibrations. The equilibrium geometry, the normal modes, and the harmonic frequencies of the totally symmetric vibrations were computed using second order Møller–Plesset perturbation theory (MP2), as implemented in the GAUSSIAN-94 program.94 The results of the geometry optimization are shown in Table S1 in the ESI,‡ and compared with the experimental data of Bak et al.77 The calculated ground state harmonic frequencies are compared to the experimental results95 in Table S2 in the ESI.‡ These tables show that the calculated ground state characteristics are in reasonable agreement with the experimental data. The calculated vibrational coupling constants, used within the LVC model for evaluation of the adiabatic excitation energies, can be found in Table S7 in the ESI.‡
4. Preliminary considerations and review of the theoretical results
According to our calculations, the ground state Hartree–Fock outer valence shell electronic configuration of thiophene may be written as| (9a1)2 (6b2)2 (10a1)2 (7b2)2 (2b1(π1))2 (11a1(S3p))2 (3b1(π2))2 (1a2(π3))2 1A1 |
Electron promotion from either the π2 or the π3 orbital, into various vacant valence or Rydberg orbitals, gives rise to the majority of the structure observed in the UV absorption spectrum. Four valence excited singlet states are expected from the π → π* transitions. Two of these states, of B2 symmetry, arise from the 1a2(π3) → 4b1(π4*) and the 3b1(π2) → 2a2(π5*) transitions. The other two states, due to the 3b1(π2) → 4b1(π4*) A1 and the 1a2(π3) → 2a2(π5*) A1 transitions, mix to form an antisymmetric (A1−) and a symmetric (A1+) pair of states. The A1− state has the lower excitation energy and spectral intensity. A distinctive feature of thiophene, compared to furan and pyrrole, is the existence of a low-lying σCS*(8b2) orbital with S–C antibonding properties.18,21,22,38,39 The promotion of an electron into this orbital appears as low-lying π2 → σ* and π3 → σ* valence-type transitions in the photoabsorption spectrum. As already discussed in relation to the two low-lying π* orbitals, the numbering for the low-lying vacant orbital of b2 symmetry should also be regarded as symbolic. At higher energies, valence states due to n → π* and n → σ* transitions (where n denotes the sulphur lone-pair orbital (S3p)) may occur.
The present EOM-CCSD and ADC(2) vertical excitation energies and oscillator strengths for the subset of the lowest π3 → Rydberg, π2 → Rydberg and valence-type transitions are given in Table 1. A more complete list of our EOM-CCSD results, for all transitions, obtained with basis set A can be found in Tables S3–S5 (ESI‡). These tables include the quantum defects (δ) for the vertical transitions, the dipole moments and the second electronic moments for the final states.
| Excitations | EOM-CCSD | ADC(2)a | ADC(2)b | SAC-CIc | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ω | f | Ω | f | Ω (S = 0) | Ω (S = 1) | f | Ω | f | ||
| a Present results, basis set A, experimental geometry (see text for details). b Present results, basis set B, optimized geometry (see text for details). Singlet (S = 0) and triplet (S = 1) excitation energies are shown. c Wan et al.37 | ||||||||||
| A1 | 1a2 → 3da2 | 7.61 | 0.056 | 7.46 | 0.032 | 7.20 | 7.17 | 0.006 | 7.08 | 0.018 |
| 1a2 → 2a2(π5*) | 7.74 | 0.232 | 7.61 | 0.265 | 7.62 | 6.23 | 0.211 | 7.32 | 0.361 | |
| 1a2 → 4da2 | 8.24 | 0.011 | 8.01 | 0.009 | 7.78 | 7.75 | 0.012 | 7.70 | 0.001 | |
| 3b1 → 4b1(π4*) | 5.78 | 0.081 | 5.72 | 0.094 | 5.73 | 4.87 | 0.099 | 5.41 | 0.091 | |
| 3b1 → 3pb1 | 7.23 | 0.080 | 7.17 | 0.078 | 7.02 | 6.99 | 0.040 | 6.73 | 0.066 | |
| 3b1 → 3db1 | 7.97 | 0.013 | 7.86 | 0.007 | 7.67 | 7.62 | 0.109 | 7.47 | 0.034 | |
| 3b1 → 4pb1 | 8.31 | <0.001 | 8.14 | <0.001 | 7.95 | 7.93 | 0.002 | 7.79 | <0.001 | |
| 3b1 → 4db1 | 8.52 | 0.001 | 8.35 | <0.001 | 8.23 | 8.20 | 0.001 | 8.06 | 0.001 | |
| B2 | 1a2→ 4b1(π4*) | 6.13 | 0.084 | 6.08 | 0.104 | 6.09 | 3.96 | 0.129 | 5.72 | 0.113 |
| 1a2 → 3pb1 | 6.93 | 0.044 | 6.84 | 0.043 | 6.60 | 6.57 | 0.024 | 6.41 | 0.038 | |
| 1a2 → 3db1 | 7.65 | 0.016 | 7.49 | 0.019 | 7.23 | 7.23 | 0.001 | 7.12 | 0.003 | |
| 1a2 → 4pb1 | 7.99 | 0.004 | 7.78 | 0.003 | 7.49 | 7.49 | 0.012 | 7.46 | 0.024 | |
| 1a2 → 4db1 | 8.21 | 0.002 | 7.98 | 0.001 | 7.81 | 7.79 | 0.041 | 7.69 | 0.002 | |
| 3b1→ 2a2(π5*) | 7.78 | 0.111 | 7.61 | 0.134 | 7.74 | 6.78 | 0.106 | 7.40 | 0.120 | |
| 3b1→ 3da2 | 7.95 | 0.002 | 7.84 | <0.001 | 7.65 | 7.65 | <0.001 | 7.43 | 0.004 | |
| 3b1→ 4da2 | 8.54 | 0.001 | 8.22 | 8.22 | <0.001 | 8.02 | <0.001 | |||
| A2 | 1a2 → 3sa1 | 6.19 | 6.12 | 5.88 | 5.83 | 5.70 | ||||
| 1a2 → 3pa1 | 6.89 | 6.77 | 6.51 | 6.49 | 6.41 | |||||
| 1a2 → 3da1 | 7.20 | 7.13 | 6.89 | 6.82 | 6.73 | |||||
1a2 → 3![[d with combining tilde]](https://www.rsc.org/images/entities/char_0064_0303.gif) a1 a1 |
7.24 | 7.15 | 6.98 | 6.92 | 6.75 | |||||
| 1a2 → 4sa1 | 7.76 | 7.57 | 7.28 | 7.27 | 7.23 | |||||
| 1a2 → 4pa1 | 7.98 | 7.76 | 7.48 | 7.47 | 7.46 | |||||
| 1a2 → 4da1 | 8.06 | 7.86 | 7.61 | 7.58 | 7.54 | |||||
| 1a2 → 4a1 |
8.11 | 7.89 | 7.63 | 7.60 | 7.59 | |||||
| 3b1 → 8b2(σ*) | 6.37 | 6.39 | 6.42 | 6.21 | 6.03 | |||||
| 3b1 → 3pb2 | 7.32 | 7.23 | 7.08 | 7.03 | 6.89 | |||||
| 3b1 → 3db2 | 7.91 | 7.81 | 7.66 | 7.60 | ||||||
| 3b1 → 4pb2 | 8.32 | 8.15 | 7.95 | 7.95 | ||||||
| B1 | 1a2 → 8b2(σ*) | 6.33 | 0.013 | 6.26 | 0.015 | 6.14 | 6.01 | 0.021 | 5.87 | 0.011 |
| 1a2 → 3pb2 | 6.94 | 0.024 | 6.85 | 0.019 | 6.66 | 6.58 | 0.011 | 6.47 | 0.017 | |
| 1a2 → 3db2 | 7.63 | <0.001 | 7.49 | <0.001 | 7.27 | 7.23 | <0.001 | 7.21 | <0.001 | |
| 1a2 → 4pb2 | 7.98 | 0.006 | 7.77 | 0.005 | 7.49 | 7.47 | 0.005 | 7.45 | 0.005 | |
| 1a2 → 4db2 | 8.15 | <0.001 | 7.94 | <0.001 | 7.76 | 7.74 | <0.001 | 7.68 | <0.001 | |
| 3b1 → 3sa1 | 6.53 | <0.001 | 6.47 | <0.001 | 6.32 | 6.26 | <0.001 | 6.12 | <0.001 | |
| 3b1 → 3pa1 | 7.15 | 0.025 | 7.08 | 0.027 | 6.89 | 6.85 | 0.027 | 6.73 | 0.019 | |
| 3b1 → 3da1 | 7.52 | <0.001 | 7.47 | <0.001 | 7.33 | 7.19 | 0.001 | 7.14 | <0.001 | |
| 3b1 → 3a1 |
7.61 | <0.001 | 7.53 | <0.001 | 7.41 | 7.36 | <0.001 | 7.15 | <0.001 | |
| 3b1 → 4sa1 | 8.09 | 0.003 | 7.93 | <0.001 | 7.72 | 7.71 | <0.001 | 7.61 | <0.001 | |
| 3b1 → 4pa1 | 8.29 | 0.007 | 8.12 | 0.008 | 7.91 | 7.88 | 0.009 | 7.80 | 0.007 | |
| 3b1 → 4da1 | 8.40 | <0.001 | 8.02 | 7.98 | 0.002 | 7.95 | <0.001 | |||
| 3b1 → 4a1 |
8.43 | 0.002 | 8.07 | 8.06 | <0.001 | 8.33 | 0.001 | |||
The proposed assignments of the calculated transitions, which take into account symmetry considerations as well as the computed quantum defects, oscillator strengths and second electronic moments, look reasonably consistent. The calculated quantum defects are within the expected range and the members of the same orbital multiplet lie fairly close in energy. As expected, the valence-type excited states, unlike the Rydberg states, are characterised by more compact electron density distributions. The corresponding transitions have considerable oscillator strengths and their excitation energies do not fit into Rydberg series. Valence states can perturb Rydberg states of the same symmetry. Thus, Rydberg-valence mixing, in certain cases, cannot be excluded.
Table 1 also contains the ADC(2) excitation energies of the triplet transitions (basis set B). As expected, the triplet excitation energies are systematically lower than the singlet counterparts. Whereas for Rydberg transitions the triplet and singlet excitation energies are almost degenerate, for valence-type transitions they can be quite different. This separation is especially pronounced for π → π* transitions but is smaller for π → σ* transitions. The present ADC(2) singlet–triplet splittings support our assignments of transitions to being of either Rydberg or valence type.
The present theoretical results provide, to the best of our knowledge, the most complete and accurate description of the thiophene absorption spectrum. Moreover, the predictions obtained from our various calculations are quite consistent (Table 1). In particular, the ADC(2) excitation energies seem to be in satisfactory agreement with those given by the EOM-CCSD method (using the same basis set), with the latter results being 0.1–0.2 eV higher. Also, a comparison between the ADC(2) results obtained with basis set A and the corresponding values obtained with basis set B shows that the improvement in the basis set systematically increases the excitation energies of the Rydberg transitions by ∼0.1 to 0.2 eV but has little effect on those of valence-type transitions.
The previous SAC-CI calculations of Wan et al.37 yield distinctly lower vertical excitation energies (Table 1). Moreover, the SAC-CI oscillator strengths do not match our EOM-CCSD values particularly well. The observed discrepancies are rather unexpected, since, from the theoretical standpoint, the EOM-CCSD and the SAC-CI schemes are essentially the same methods, and the basis set used by Wan et al.37 is only a moderately truncated version of our basis set A. In contrast, the agreement between the other CCSD results found in the literature22,39 and our EOM-CCSD data is much better (Table S6, ESI‡).
In order to evaluate the quality of our EOM-CCSD data we have performed more sophisticated CC3 calculations for the singlet excited states of thiophene which are the lowest in energy in each spatial symmetry (Table S6, ESI‡). The corresponding CC3 vertical excitation energies show only minor energy corrections (ranging from 0.06 to 0.16 eV) compared to the CCSD results. Similar observations can be made from a comparison between our data and the results of Stenrup22 and Pastore et al.,39 which were obtained using the CCSDR(3) scheme where the electronic excitations are treated beyond the CCSD level (Table S6, ESI‡).
In Table 2 we present the adiabatic excitation energies (Ω0–0) which were evaluated by combining the EOM-CCSD vertical excitation energies with the adiabatic lowerings (Table S7, ESI‡) obtained using the ADC(2) data and the LVC model. Table 2 also lists our best theoretical estimates for the vertical and the adiabatic excitation energies. These have been obtained by correcting our original EOM-CCSD results for expected inaccuracies related to the method and basis set used.
| Excitation | Theory | Best estimate | |||
|---|---|---|---|---|---|
| Ω v | Ω 0–0 | Ω v | Ω 0–0 | ||
| a EOM-CCSD vertical excitation energy. b Adiabatic transition energy (obtained from EOM-CCSD vertical excitation energy and ADC(2) adiabatic energy lowering). c Corrected EOM-CCSD vertical excitation energy (see text). d Corrected adiabatic transition energy (see text). e Repulsive state.22 f No data available. | |||||
| 1A1 | 3b1 → 4b1(π4*) | 5.78 | 5.57 | 5.64 | 4.99 |
| 1B2 | 1a2 → 4b1(π4*) | 6.13 | 5.75 | 5.97 | —e |
| 1B1 | 1a2 → 8b2(σ*) | 6.33 | 6.06 | 6.17 | 5.96 |
| 1A2 | 1a2 → 3sa1 | 6.19 | 6.03 | 6.23 | 6.07 |
| 1A2 | 3b1 → 8b2(σ*) | 6.37 | 6.15 | 6.33 | 6.11 |
| 1B1 | 3b1 → 3sa1 | 6.53 | 6.46 | 6.68 | 6.61 |
| 1B1 | 1a2 → 3pb2 | 6.94 | 6.74 | 6.98 | 6.78 |
| 1B2 | 1a2 → 3pb1 | 6.93 | 6.76 | 6.97 | 6.80 |
| 1A2 | 1a2 → 3pa1 | 6.89 | 6.77 | 6.93 | 6.81 |
| 1A2 | 1a2 → 3da1 | 7.20 | 7.03 | 7.24 | 7.07 |
| 1A2 | 1a2 → 3a1 |
7.24 | 7.11 | 7.28 | 7.15 |
| 1A1 | 1a2 → 2a2(π5*) | 7.74 | 7.66 | —f | —f |
| 1B2 | 3b1 → 2a2(π5*) | 7.78 | 7.67 | —f | —f |
| 1B1 | 3b1 → 3pa1 | 7.15 | 7.07 | 7.30 | 7.22 |
| 1A1 | 3b1 → 3pb1 | 7.23 | 7.15 | 7.38 | 7.30 |
| 1A2 | 3b1 → 3pb2 | 7.32 | 7.27 | 7.47 | 7.42 |
| 1A1 | 1a2 → 3da2 | 7.61 | 7.45 | 7.65 | 7.49 |
| 1B2 | 1a2 → 3db1 | 7.65 | 7.46 | 7.69 | 7.50 |
| 1B1 | 1a2 → 3db2 | 7.63 | 7.50 | 7.67 | 7.54 |
| 1B1 | 3b1 → 3da1 | 7.52 | 7.43 | 7.67 | 7.58 |
| 1A2 | 1a2 → 4sa1 | 7.76 | 7.59 | 7.80 | 7.63 |
| 1B1 | 3b1 → 3a1 |
7.61 | 7.54 | 7.76 | 7.69 |
| 1A2 | 1a2 → 4pa1 | 7.98 | 7.81 | 8.02 | 7.85 |
| 1B2 | 1a2 → 4pb1 | 7.99 | 7.83 | 8.03 | 7.87 |
| 1B1 | 1a2 → 4pb2 | 7.98 | 7.86 | 8.02 | 7.90 |
| 1B2 | 3b1 → 3da2 | 7.95 | 7.85 | 8.10 | 8.00 |
| 1A2 | 1a2 → 4da1 | 8.06 | 7.97 | 8.10 | 8.01 |
| 1A2 | 1a2 → 4a1 |
8.11 | 7.98 | 8.15 | 8.02 |
| 1A2 | 3b1 → 3db2 | 7.91 | 7.89 | 8.06 | 8.04 |
| 1A1 | 3b1 → 3db1 | 7.97 | 7.93 | 8.12 | 8.08 |
| 1B1 | 1a2 → 4db2 | 8.15 | 8.10 | 8.19 | 8.14 |
| 1A1 | 1a2 → 4da2 | 8.24 | 8.10 | 8.28 | 8.14 |
| 1B2 | 1a2 → 4db1 | 8.21 | 8.13 | 8.25 | 8.17 |
| 1B1 | 3b1 → 4sa1 | 8.09 | 8.06 | 8.24 | 8.21 |
| 1A1 | 3b1 → 4pb1 | 8.31 | 8.23 | 8.46 | 8.38 |
| 1B1 | 3b1 → 4pa1 | 8.29 | 8.23 | 8.44 | 8.38 |
| 1A2 | 3b1 → 4pb2 | 8.32 | 8.29 | 8.47 | 8.44 |
| 1B1 | 3b1 → 4da1 | 8.40 | 8.31 | 8.55 | 8.46 |
| 1B1 | 3b1 → 4a1 |
8.43 | 8.33 | 8.58 | 8.48 |
| 1A1 | 3b1 → 4db1 | 8.52 | 8.43 | 8.67 | 8.58 |
| 1B2 | 3b1 → 4da2 | 8.54 | 8.45 | 8.69 | 8.60 |
For the Rydberg states, the corrections were defined as the differences between the vertical ionization energies, 9.06 and 9.37 eV for the (1a2)−1 2A2 and the (3b1)−1 Ã 2B1 states, respectively, as computed using the IP-EOM-CCSD method, and the corresponding values of 9.10 and 9.52 eV, which we consider to represent the experimental energies. The value of 9.10 eV for the vertical ionization energy of the 2A2 state was derived from the experimental adiabatic ionization energy of 8.874 eV48 in the course of our vibronic modelling96 and reflects that fact that the centre of gravity of the 2A2 state vibrational progression does not coincide with its origin. The difference (0.23 eV) between the two quantities is referred to as the adiabatic lowering for the (1a2)−1 2A2 transition. By contrast, the experimental value of 9.52 eV corresponds to the (3b1)−1 Ã 2B1 state band maximum42 and therefore needs no correction.96 The corrections entail systematic shifts of +0.04 and +0.15 eV for the 1a2 and the 3b1 Rydberg series, respectively.
Since the results for valence-type transitions cannot be corrected in a systematic manner, the vertical excitation energies obtained from the present CC3 calculations were used for the 3b1(π2) → 4b1(π4*) 1A1, 1a2(π3) → 4b1(π4*) 1B2, 1a2(π3) → 8b2(σ*) 1B1 transitions, and the CCSDR(3) results22 were used for the 3b1(π2) → 8b2(σ*) 1A2 transition. For the higher-lying valence-type transitions, 1a2(π3) → 2a2(π5*) 1A1 and 3b1(π2) → 2a2(π5*) 1B2, to the best of our knowledge, no results of calculations beyond the CCSD level are available.
The adiabatic excitation energies were corrected in the same way as were the vertical energies, with the exceptions being the values for the lowest 3b1(π2) → 4b1(π4*) 1A1 and 1a2(π3) → 4b1(π4*) 1B2 transitions. As follows from the MS-CASPT2 excited state geometry optimisation,22 the actual adiabatic energy lowering for the 1A1 (π2–π4*) state is ∼0.7 eV, which is much larger than our present value of 0.2 eV found from the LVC model. Since the predictions of the explicit geometry optimisation are more reliable, we have adopted the value given by Stenrup.22 This yields the best theoretical estimate of 4.99 eV for the adiabatic excitation energy of the 3b1(π2) → 4b1(π4*) 1A1 transition. For the 1B2 (π3–π4*) state, the calculations performed by Stenrup22 do not predict a minimum on the potential energy surface within the spectroscopically significant domain of geometries. Therefore, we refrain from giving an estimate for the adiabatic energy of this transition.
5. Results and discussion
5.1 Overview
At photon energies below 10 eV, the photoabsorption spectrum (Fig. 2) is dominated by three prominent bands, centred at ∼5.5, 7.0 and 9.1 eV. The high intensity and broadness of these bands indicate that they are associated with valence states. The present calculations show, in accord with earlier theoretical results,22,32,35–39 that the band at 5.5 eV is due to the 3b1(π2) → 4b1(π4*) 1A1 and the 1a2(π3) → 4b1(π4*) 1B2 transitions. An additional contribution on the high energy side of this peak arises from the 1a2(π3) → 8b2(σ*) 1B1 transition. The prominent band around 7 eV is due to the closely spaced 1a2(π3) → 2a2(π5*) 1A1 and 3b1(π2) → 2a2(π5*) 1B2 transitions. Thus, there is general agreement that each of the two low-lying prominent broad bands contains major contributions from two π → π* transitions.At higher energies, in addition to the π2, π3 → σ* and the π2, π3 → π* valence transitions, excitations involving the sulphur lone-pair orbital start to appear. The lowest energy n → π* transition is predicted around 8 eV.32,35,37,39
Between ∼7.7 and 8.9 eV the experimental spectrum exhibits many sharp peaks, most of which can be attributed to two Rydberg series (one p-type and one d-type) converging onto the 2A2 state limit. In the present work, an additional d-type series, also converging onto this limit, is tentatively proposed. Two members of an s-type Rydberg series converging onto the à 2B1 state limit are associated with two broad absorption bands. Finally, numerous weak features are observed at excitation energies above 9.4 eV, presumably due to one or more Rydberg series converging onto the 2A1 state limit.
5.2 Excited states in the energy range 5.0 to 6.4 eV
Although the principal absorption band occurring in the energy range ∼5.0 to 6.4 eV (Fig. 3) has been studied extensively, the assignments of the vibrational structure have undergone several revisions. It is well established that most of the oscillator strength associated with this band arises from two overlapping valence transitions. The existence of two valence states in this energy region was confirmed long ago by MCD measurements59–61 which showed two bands, having different polarizations, with absorption maxima at 5.33 and 5.70 eV.61 However, the symmetry assignments proposed for these two states, based upon a comparison between the measured and the predicted Faraday B values, appear inconsistent.59–61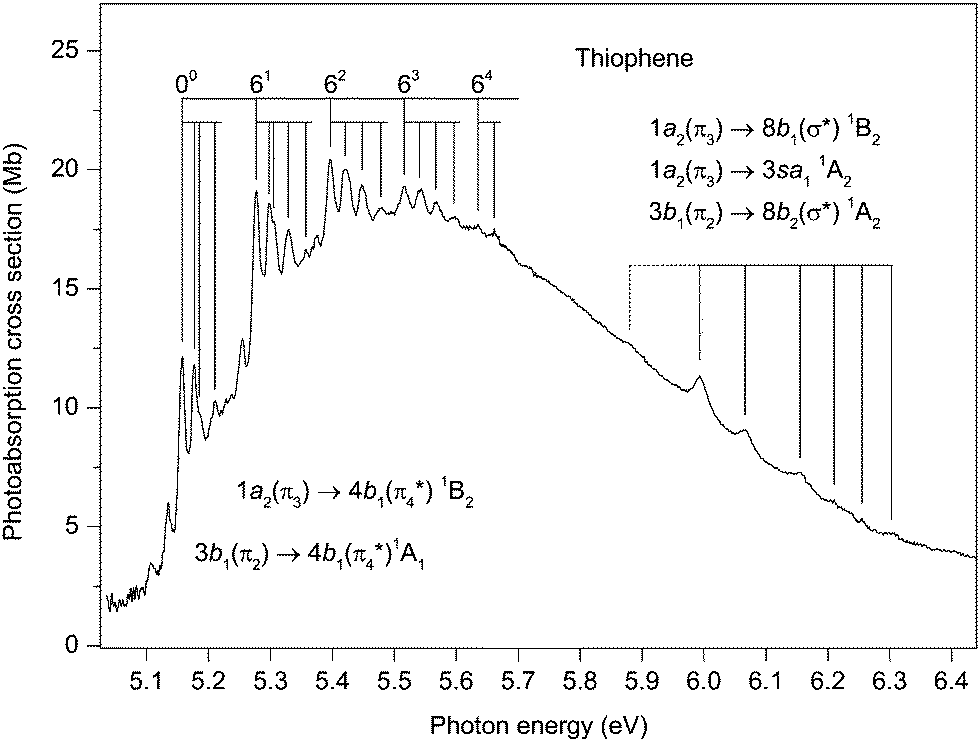 | ||
| Fig. 3 The photoabsorption spectrum of thiophene showing structure due to the 1a2(π3) → 4b1(π4*) 1B2 and the 3b1(π2) → 4b1(π4*) valence transitions. | ||
The vibrational structure in the near-threshold region has been studied recently by Weinkauf et al.19 In addition to performing calculations which showed that the ordering of the electronic states changes when the excited states are allowed to relax their geometry, the one-colour resonant, two-photon ionization spectrum from a beam of cooled thiophene molecules was measured and compared to the room temperature spectrum. Based upon this comparison, an absorption band at 5.156 eV was attributed to the origin of the 3b1 → 4b11A1 transition. Furthermore, vibrational structure observed at lower excitation energies was tentatively associated with the 1a2 → 4b11B2 transition, whose adiabatic ionization energy was calculated to lie below the origin of the 3b1 → 4b11A1 transition. Their calculations also showed that some of the vibrational energies were considerably reduced if the excited states possessed a non-planar geometry.
Our calculations show, in accord with recent theoretical studies,18,19,22,35–39 that the vertical excitation energy for the 3b1 → 4b11A1 transition lies below that for the 1a2 → 4b11B2 transition. The potential energy surface for the 1A1 (π2–π4*) state develops a double-well minimum,22 indicative of vibronic coupling with the upper 1B2 (π3–π4*) state, thus supporting the conclusions reached by Köppel et al.17 in their study of multi-mode−multi-state vibronic interactions on the low-lying excited electronic states of thiophene. However, in contrast to the results obtained with the earlier model,17 the calculations of Stenrup22 show that the 1B2 (π3–π4*) state may be unbound and therefore that the associated vibronic structure may not be discernible in the absorption spectrum.
The present theoretical results, in combination with the previous theoretical data of Stenrup,22 allow one to speculate that the structure observed between 5.1 and 5.6 eV is most likely due to excitations of the totally symmetric vibrational modes of the 1A1 (π2–π4*) state accompanied by excitations of the non-totally symmetric modes which gain their intensity from the 1a2(π3) → 4b1(π4*) 1B2 transition. A large portion of the oscillator strength associated with the latter transition goes into the excitation of these modes. According to our calculations, no other state can contribute to the absorption in this energy region.
Previous interpretations of the vibrational structure observed in the ∼5.1–5.7 eV range have included hot-bands, the excitation of non-totally symmetric vibrational modes, and the excitation of totally symmetric modes whose energies have changed significantly from those in the neutral ground state. The present absorption spectrum (Fig. 3) has been recorded at a resolution higher than used in earlier studies and therefore reveals additional details. The main structure seems to involve the excitation of a vibrational mode with an energy of 119 meV. Inspection of the spectrum readily allows a series of repeating vibrational progressions to be recognised, built upon the excitation of 0,1,2,3 or 4 quanta of this principal vibrational mode. The first members of these five progressions occur at 5.157, 5.277, 5.397, 5.516 and 5.635 eV (Table 3) and we ascribe the peak at 5.157 eV to the origin of the 3b1 → 4b11A1 transition, in agreement with previous assignments. The first peak in each of the progressions (especially apparent in those progressions at lower energy) is sharp, which suggests that it is due to the excitation of a single vibrational mode. However, the next peak in each progression is a doublet (for example, the two overlapping bands with maxima at 5.298 and 5.305 eV), indicative of the excitation of two vibrational modes with slightly different energies. The progressions whose first members are observed at 5.157 and 5.277 eV have noticeably sharper structure than those whose first members occur at 5.397, 5.516 and 5.635 eV.
The identification of the vibrational mode having an energy of 119 meV is not straightforward as this energy does not readily match any of the known energies of the totally symmetric modes in the molecular ground state. Since the absorption structure associated with this mode is relatively intense, we assume that it involves the excitation of a totally symmetric mode. Taking into account the reduction in the calculated vibrational energies if the geometry of the excited state becomes non-planar,19 we tentatively attribute the mode having an energy of 119 meV to ν6, which has energies of 128.5 meV95 and 130.2 meV48 in the neutral and ionic ground states, respectively. Assignments for the other structure, involving modes having very low vibrational energies, are even more difficult to propose. Excitation of modes having b2 symmetry might be expected, due to vibronic coupling.
As is evident from the above discussion, some of assignments are of a speculative nature and a proper understanding of the absorption structure observed between ∼5.0 and 6.4 eV requires additional calculations, specifically including vibronic coupling and nuclear dynamics. Such calculations are now in progress and will be reported in a future article.
Our EOM-CCSD calculations predict vertical excitation energies of 6.19, 6.33 and 6.37 eV for the 1a2 → 3sa11A2, 1a2 → 8b2(σ*) 1B1 and 3b1 → 8b2(σ*) 1A2 transitions, respectively (Tables S3 and S5, ESI‡), and, according to our best estimates for these excitation energies (Table 2), the three transitions almost coincide. Strictly, only the 1a2 → 8b21B1 transition is optically allowed (and has an appreciable oscillator strength of 0.013 (Table S5, ESI‡)). However, the electric dipole forbidden 1a2 → 3sa11A2 and 3b1 → 8b21A2 transitions can acquire intensity through vibronic coupling with the allowed 1a2 → 8b21B1 transition. Some intensity can also be borrowed through coupling with the 3b1(π2) → 4b1(π4*) 1A1 transition. Thus, theoretically, vibrational progressions associated with these three closely spaced excited states can be anticipated between ∼6.0 and 6.2 eV in the absorption spectrum.
| Assignment | Energy (eV) | Assignment | Energy (eV) | ||
|---|---|---|---|---|---|
| 3b1 → 4b11A1 (1a2 → 4b11B2) | 1a2 → npb11B2 | ||||
| 00 | 5.157 | ||||
| 5.177 | n = 6 | 00 | 8.433 (δ = 0.446) | ||
| 5.185 | 81 | 8.507 | |||
| 5.210 | 82 | 8.583 | |||
| 61 | 5.277 | 41 | 8.595 | ||
| 5.298 | 4181 | 8.672 | |||
| 5.305 | |||||
| 5.329 | n = 7 | 00 | 8.558 (δ = 0.438) | ||
| 5.357 | 81 | 8.635 | |||
| 62 | 5.397 | ||||
| 5.421 | n = 8 | 00 | 8.635 (δ = 0.455) | ||
| 5.448 | |||||
| 5.478 | n = 9 | 00 | 8.687 (δ = 0.470) | ||
| 63 | 5.516 | ||||
| 5.541 | n = 10 | 00 | 8.723 (δ = 0.508) | ||
| 5.567 | |||||
| 5.596 | n = 11 | 00 | 8.753 (δ = 0.396) | ||
| 64 | 5.635 | ||||
| 5.661 | 1a2 → nda21A1 | ||||
| Unidentified | n = 4 | 1411 | 7.992 | ||
| 5.880 | 00 | 8.003 (δ = 0.048) | |||
| 5.993 | 211 | 8.067 | |||
| 6.066 | 81 | 8.078 | |||
| 6.155 | 201 | 8.107 | |||
| 6.210 | 82 | 8.153 | |||
| 6.255 | 41 | 8.169 | |||
| 6.303 | 4181 | 8.242 | |||
| 3b1 → 3sa11B1 | |||||
| 00 | 6.474 (δ = 0.887) | n = 5 | 00 | 8.318 (δ = 0.053) | |
| 81 | 8.392 | ||||
| 1a2 → 3pb11B2 | 82 | 8.467 | |||
| 00 | 6.603 (δ = 0.552) | 41 | 8.480 | ||
| 81 | 6.686 | ||||
| 82 | 6.759 | n = 6 | 00 | 8.487 (δ = 0.071) | |
| 83 | 6.830 | 81 | 8.561 | ||
| 82 | 8.635 | ||||
| 1a2 → 3pb21B1 | n = 7 | 00 | 8.591 (δ = 0.066) | ||
| 00 | 6.713 (δ = 0.491) | 81 | 8.666 | ||
| 81 | 6.785 | 41 | 8.757 | ||
| 1a2 → npb11B2 | n = 8 | 00 | 8.657 (δ = 0.082) | ||
| n = 4 | 1422 | 7.774 | n = 9 | 00 | 8.704 (δ = 0.054) |
| 1411 | 7.782 | ||||
| 00 | 7.792 (δ = 0.454) | n = 10 | 00 | 8.733 (δ = 0.177) | |
| 211 | 7.855 | ||||
| 81 | 7.868 | n = 11 | 00 | 8.761 (δ = 0.027) | |
| 201 | 7.887 | ||||
| 81211 | 7.924 | n = 12 | 00 | 8.778 (δ = 0.095) | |
| 82 | 7.943 | ||||
| 41 | 7.956 | 1a2 → ndb11B2 | |||
| 4181 | 8.029 | ||||
| 82212 | 8.053 | n = 4 | 00 | 8.025 (δ = −0.003) | |
| 81 | 8.099 | ||||
| n = 5 | 00 | 8.220 (δ = 0.439) | 41 | 8.189 | |
| 81 | 8.295 | ||||
| 82 | 8.371 | n = 5 | 00 | 8.330 (δ = −0.001) | |
| 41 | 8.383 | ||||
| 4181 | 8.457 | n = 6 | 00 | 8.498 (δ = −0.016) | |
| n = 7 | 00 | 8.595 (δ = 0.017) | |||
In the present spectrum (Fig. 3), irregular and rather broad absorption structure can be observed between ∼5.9 and 6.3 eV. The first significant peak occurs at 5.993 eV, but an additional, rather weak, feature is discernible at 5.88 eV and may be part of the progression. None of this structure appears in the corresponding condensed phase spectrum28,31,34 where Rydberg states are considered to play a minor role. Thus we attribute the vibrational progression, which does not resemble that in the 2A2 state photoelectron band,40 observed in the gas phase absorption spectrum, to the vibronically induced 1a2 → 3sa11A2. The peak at 5.993 eV probably corresponds to the lowest energy, vibronically allowed transition into this Rydberg state, resulting in a quantum defect of ∼0.83. Such a value lies on the low side of the accepted range for a 3s Rydberg state. However, it appears reasonable to assume that the weak peak at 5.88 eV might be due to the forbidden adiabatic transition, leading to a quantum defect of ∼0.87.
It should be noted that the 1a2 → 8b2(σ*) 1B1 and 3b1 → 8b2(σ*) 1A2 valence transitions may, as seems likely by promotion of electrons into σ* orbitals, have dissociative potential energy surfaces in the spectroscopically relevant domain of nuclear coordinates. These states have been shown to be involved in the photochemical ring opening reaction in thiophene,17–22 and, more generally, such ring opening mechanisms are known to be an important ultrafast radiationless decay channel for the lowest excited states of the five membered heterocyclic molecules.97–100 The influence of ring opening reactions on the photoabsorption spectrum of thiophene is, however, not yet fully understood and requires additional theoretical investigations. At this stage, it seems likely that the dissociative character of the potential energy surfaces, as well as the vibronic coupling between the excited states, contribute to the observed line broadening in the spectral region under discussion.
Another factor which may influence the appearance of the absorption spectrum is Rydberg–valence mixing between the 1A2 (π3–3s) and the 1A2 (π2–σ*) states. According to the present EOM-CCSD calculations, the 1A2 (π3–3s) state is characterised by a rather compact electronic density distribution (Table S3, ESI‡), which is comparable to that of the 1A2 (π2–σ*) state (Table S5, ESI‡), and is typical of those for valence states.
5.3 Excited states in the energy range 6.4 to 7.7 eV
The first absorption band in this region (Fig. 4) occurs at 6.474 eV and is ascribed to the 3b1 → 3sa11B1 transition. Although the calculated oscillator strength (<0.001 (Table S4, ESI‡)) of this transition is rather low, the isolated nature of the state allows a firm identification. Use of the experimental transition energy and the vertical ionization energy of 9.52 eV for the à 2B1 state,42 leads to a quantum defect of 0.89 (Table 3).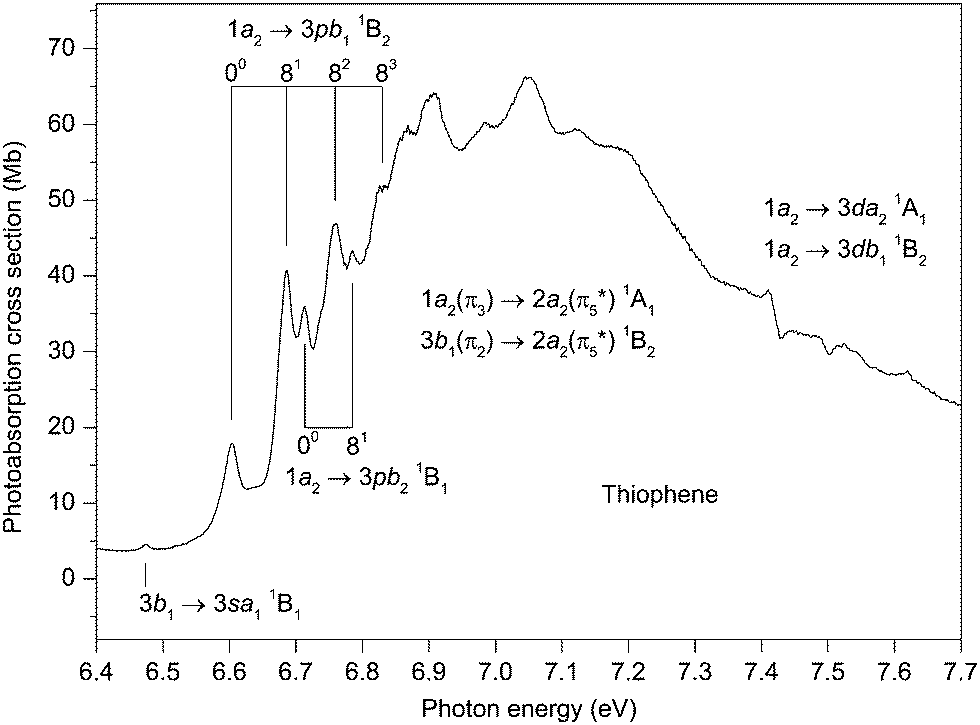 | ||
| Fig. 4 The photoabsorption spectrum of thiophene showing structure due to the 1a2(π3) → 2a2(π5*) 1A1 and the 3b1(π2) → 2a2(π5*) 1B2 valence transitions and to the 3b1 → 3sa11B1, 1a2 → 3pb11B2 and 1a2 → 3pb21B1 Rydberg transitions. | ||
At slightly higher energy, an intense band, which dominates the absorption spectrum below the ionization threshold, has an onset at about 6.6 eV and extends to at least 8 eV. The band is asymmetric, with a maximum at ∼7 eV, but with its centre of gravity, which should correspond to the vertical excitation energy of a very strong transition (or transitions), estimated to occur at ∼7.2–7.3 eV. The only plausible transitions are the valence-type 1a2(π3) → 2a2(π5*) 1A1 and the 3b1(π2) → 2a2(π5*) 1B2 transitions, with EOM-CCSD vertical excitation energies of 7.74 and 7.78 eV, respectively (Table S5, ESI‡). According to our results, these transitions, with oscillator strengths of 0.232 and 0.111 for the 1A1 and 1B2 states, respectively (Table S5, ESI‡), are the two most intense transitions occurring in the energy range encompassed by our theoretical investigation. For reasons which are not yet fully understood, it seems that the EOM-CCSD calculations overestimate, by about 0.5 eV, the vertical excitation energies of these two transitions. By analogy with furan and pyrrole, where results of the next higher scheme, CC3, of the coupled-cluster hierarchy are available,101,102 an energy lowering of 0.2–0.3 eV can be expected for the 1a2 → 2a21A1 transition at the CC3 level of theory compared to the present CCSD excitation energy. This lowering, however, is insufficient to reach a good agreement with the experimental energy, so a further improvement in the basis set and/or in the treatment of electron correlation is required to describe these transitions properly.
Interestingly, NEVPT calculations give vertical excitation energies of 7.94 and 8.13 eV39 for the 1a2 → 2a21A1 and 3b1 → 2a21B2 transitions, respectively, whereas the corresponding excitation energies obtained from CCSDR(3) calculations are 7.71 and 7.85 eV.39 The discrepancy between these predictions and the experimental values was attributed to the known difficulty in obtaining accurate theoretical results for ionic high energy π → π* states.39 In contrast, the SAC-CI calculations gave significantly lower energies, of 7.32 and 7.40 eV for the 1a2 → 2a21A1 and the 3b1 → 2a21B2 transitions, respectively.37
At present it is not clear whether the 1A1 (π3–π5*) and the 1B2 (π2–π5*) states are bound. At least one of these states should be bound otherwise an absorption maximum would not be observed in the spectrum. The coupling constants (Table S7, ESI‡) for both of these states are relatively small for all the totally symmetric vibrational modes. Thus, only rather moderate adiabatic lowerings of 0.08 and 0.1 eV occur for the 1A1 (π3–π5*) and the 1B2 (π2–π5*) states, respectively. This suggests that both states are bound. However, a more detailed study of the potential energy surfaces is required, preferably using explicit geometry optimisation procedures, to confirm this interpretation and also to characterise the adiabatic transitions more accurately.
Vibrational structure, which is always rather broad and becomes progressively so towards higher energy, appears in the absorption spectrum between ∼6.6 and 7.1 eV (Fig. 4). In addition to the two valence transitions already discussed, two relatively intense Rydberg transitions are predicted on the low energy side of the main absorption band. Thus, the vibrational structure on the rising edge might be associated with either valence states or Rydberg states, or with a combination of both. Our proposed assignments, to Rydberg transitions, are based upon the similarity between the quantum defects derived for the suggested 3p Rydberg states, and those obtained for the well resolved higher members (Table 3). Also taken into account is the resemblance between the vibrational structure in the absorption bands and that in the 2A2 state photoelectron band40,42 where excitation of the ν4 and ν8 modes plays a major role.
The EOM-CCSD calculations predict vertical excitation energies (and oscillator strengths) of 6.93 eV (f = 0.044) and 6.94 eV (f = 0.024) for the 1a2 → 3pb11B2 and the 1a2 → 3pb21B1 transitions, respectively (Table S3, ESI‡). Our best estimates for the adiabatic excitation energies into the 3pb11B2 and the 3pb21B1 states are 6.80 and 6.78, respectively, (Table 2). One vibrational progression (Table 3) involving the excitation of a mode having an energy of ∼76 meV is evident in the experimental spectrum, with the band at 6.603 eV being the origin. This vibrational spacing probably corresponds to excitation of the ν8 mode, which has measured energies of 75.5 meV95 and 74.6 meV48 in the neutral and ionic ground states, respectively. A second progression, consisting of two or possibly three members, involving excitation of the same mode, can also be observed in the experimental spectrum (Fig. 4) with the first peak in this progression occurring at 6.713 eV. The separation of 110 meV, between the peaks ascribed to the origins of these two progressions, does not correspond to the energy of a totally symmetrical vibrational mode. Therefore, we associate the absorption bands to two progressions, belonging to two separate Rydberg states, rather than to one more complex progression belonging to a single Rydberg state. The vibrational progression with the adiabatic transition occurring at 6.603 eV is stronger than that at 6.713 eV. Thus, taking into account that the calculated oscillator strength for the 1a2 → 3pb11B2 transition is significantly greater than that calculated for the 1a2 → 3pb21B1 transition, we tentatively assign the progression with the lower energy adiabatic peak to the 1a2 → 3pb11B2 transition and the second progression to the 1a2 → 3pb21B1 transition.
There are several broad peaks, with maxima at 6.868, 6.904, 6.984, 7.049 and 7.123 eV, superimposed upon the main band around 7 eV. In this energy region, the calculations predict, in addition to the two strong valence transitions, the 3b1 → 3pa11B1 and the 3b1 → 3pb11A1 Rydberg transitions, both of which have a reasonably high oscillator strength (Table S4, ESI‡). Our best estimates for the vertical excitation energies of the 3pa11B1 and the 3pb11A1 states are 7.30 and 7.38 eV, respectively (Table 2). Note, however, that due to an expectedly strong overlap of the valence and Rydberg progressions it remains uncertain whether the cluster of peaks observed around 7 eV should be associated with valence or with Rydberg states, or with a combination of both.
Between 7.4 and 7.7 eV some highly irregular features appear in the experimental spectrum (Fig. 4). Theoretically, the 1a2 → 3da21A1 (f = 0.056) and the 1a2 → 3db11B2 (f = 0.016) transitions (Table S3, ESI‡) are predicted to occur in the relevant energy range, together with several other transitions of negligible oscillator strength. However, as the absorption structure bears no resemblance to that in the 2A2 state photoelectron band, assignments are difficult to propose.
Since the energy range between 6.6 and 7.7 eV encompasses contributions from two intense valence transitions and from several Rydberg transitions with significant calculated oscillator strength, the question arises as to the validity of describing a particular absorption feature in terms of a transition into a specific excited (Rydberg or valence) state. Although an assignment to a single configuration may be a reasonable approximation for some of the structure observed between ∼6.6 and 6.8 eV, at higher energies it appears that configuration mixing probably invalidates such simple interpretations.
5.4 Excited states in the energy range 7.7 to 8.9 eV
The photoabsorption spectrum covering the energy range 7.7 to 8.9 eV (Fig. 5 and 6) displays extensive well resolved structure, the majority of which can be attributed to two Rydberg series converging onto the 2A2 state limit. Some of this structure has been observed previously and assigned to one p-type and one d-type Rydberg series. The present higher resolution spectrum allows both of these series to be observed to higher (n ∼ 11) members (Table 3). Many of the Rydberg states have accompanying vibrational structure which resembles that in the 2A2 state photoelectron band.40
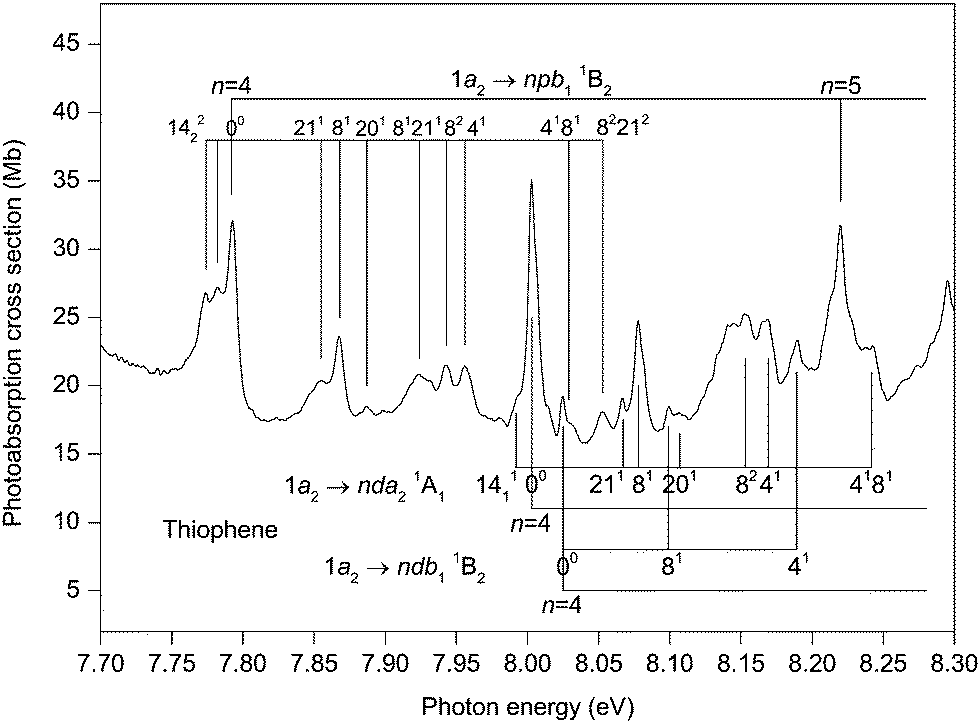 | ||
| Fig. 5 The photoabsorption spectrum of thiophene showing structure due to the 1a2 → npb11B2, 1a2 → nda21A1 and 1a2 → ndb11B2 Rydberg transitions. | ||
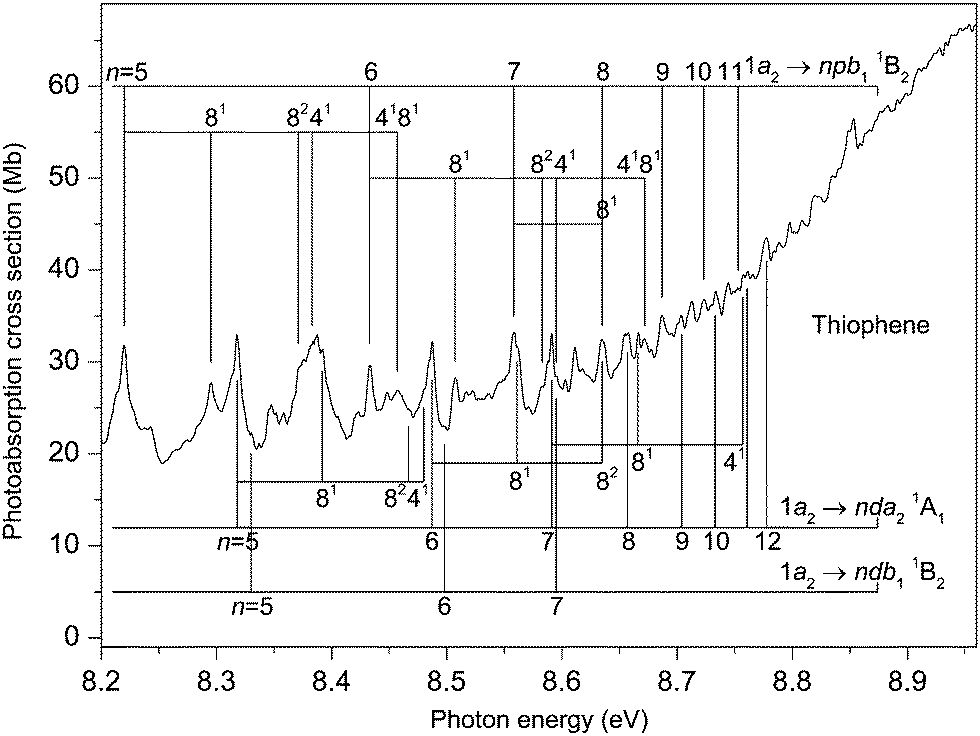 | ||
| Fig. 6 The photoabsorption spectrum of thiophene showing structure due to the 1a2 → npb11B2. 1a2 → nda21A1 and 1a2 → ndb11B2 Rydberg transitions. | ||
Before discussing assignments for the absorption bands occurring in this region due to Rydberg states, it should be mentioned that several of the more intense peaks exhibit accompanying hot-band structure. This is particularly evident in the structure associated with the 1a2 → 4pb11B2 transition where peaks located 10 and 18 meV to the low energy side of the strong band due to the adiabatic transition might be attributed to the 1411 and the 1422 hot-band transitions. The ν14 (b1) vibrational mode has energies of 56.195 and 46.148 meV in the neutral and ionic ground states, respectively. A peak due to the 1411 transition was observed in the ZEKE spectrum of the 2A2 state.48
The absorption band occurring at 7.792 eV in the experimental spectrum (Fig. 5) undoubtedly corresponds to the adiabatic transition into a Rydberg state. Assuming n = 4, the resulting quantum defect of δ = 0.45 suggests that the peak is due to a 1a2 → 4p transition. In this energy region, the calculations predict the 1a2 → 4pb21B1 transition with a vertical excitation energy of 7.98 and an oscillator strength of 0.006, and the 1a2 → 4pb11B2 transition with a vertical excitation energy of 7.99 eV and an oscillator strength of 0.004 (Table S3, ESI‡). Our best theoretical estimates of the adiabatic excitation energies for the 1a2 → 4pb21B1 and the 1a2 → 4pb11B2 transitions are 7.90 and 7.87 eV, respectively (Table 2). For the 1a2 → 5pb11B2 and the 1a2 → 6pb11B2 transitions with vertical excitation energies of 8.41 and 8.65 eV, respectively (Table S3, ESI‡), the calculated oscillator strengths are 0.004 and 0.003, respectively, which are similar to that (f = 0.004) for the 1a2 → 4pb11B2 transition. On the other hand, the calculated oscillator strengths for the 1a2 → 5pb21B1 and the 1a2 → 6pb21B1 transitions are negligible. Experimentally, the p-type Rydberg series is observed to high principal quantum number. Therefore, if this series is to be associated with a well-defined (single) symmetry, then the theoretical results suggest an assignment of 1a2 → npb11B2. Alternatively, the peak at 7.792 eV might arise through both the 1a2 → 4pb11B2 and the 1a2 → 4pb21B1 transitions, with the separate contributions not being resolved experimentally. In this interpretation, the calculations indicate that the contribution to the absorption bands from the npb11B2 transitions would become progressively larger, relative to that from the npb21B1 transitions, as n increases.
Another Rydberg series, with the absorption band observed at 8.003 eV ascribed to the adiabatic transition into the n = 4 member, appears prominently in the experimental spectrum (Fig. 5). The quantum defect of 0.048 derived for this peak is indicative of a d-type series, as suggested previously.32,37,42 According to our calculations, the most likely candidates are the 1a2 → 4db11B2 (f = 0.002) transition with a vertical excitation energy of 8.21 eV and the 1a2 → 4da21A1 (f = 0.011) transition with a vertical excitation energy of 8.24 eV (Table S3, ESI‡). Our best theoretical estimates of the adiabatic excitation energies for the 1a2 → 4db11B2 and the 1a2 → 4da21A1 transitions are 8.17 and 8.14 eV, respectively (Table 2). Thus, we assign the strong d-type Rydberg series appearing in the experimental spectrum to the 1a2 → nda21A1 transitions, in agreement with Derrick et al.,42 with the band at 8.003 eV corresponding to the origin of the n = 4 member.
Many of the Rydberg states belonging to the 1a2 → npb11B2 or the 1a2 → nda21A1 series exhibit vibrational structure (Fig. 5 and 6) consisting, mainly, of progressions involving excitation of the ν8 mode, either alone or in combination with a single quantum of the ν4 mode. In these Rydberg states, the vibrational energies of the ν4 and ν8 modes are ∼164 and 75 meV, respectively. The original assignment of the vibrational structure observed in the 2A2 state photoelectron band involved only the totally symmetric modes.40 A subsequent reinterpretation50 resulted in the identification of two additional peaks which Yang et al.48 have ascribed to the ν20 (b2) and ν21 (b2) modes. The excitation of a single quantum of these modes is normally forbidden but may occur through vibronic coupling. In the 2A2 state, vibrational energies of 104.3 and 59.9 meV have been determined for the ν20 and ν21 modes, respectively.48 Absorption bands due to the excitation of these non-totally symmetric modes are observed in the structure associated with the 1a2 → 4pb11B2 transition and, to a lesser extent, in that attributed to the 1a2 → 4da21A1 transition (Fig. 5). For the first of these transitions, peaks occur due to the excitation of the ν20 or the ν21 mode either alone or together with the totally symmetric ν8 mode.
There appears to be some experimental evidence of a second, and weaker, d-type Rydberg series whose members lie at excitation energies slightly higher than those of the 1a2 → nda21A1 series. The peak observed at 8.025 eV is tentatively ascribed to the adiabatic transition into the n = 4 member of this second d-type series, with the peaks at 8.099 and 8.189 eV corresponding to excitation of the ν8 and the ν4 modes, respectively, in this Rydberg state. Such an assignment results in a quantum defect of −0.003. Some weak absorption bands at higher energies are tentatively attributed to the n = 5, 6, 7 members (Table 3). Our theoretical predictions suggest that this weaker d-type series should be assigned to the 1a2 → ndb11B2 transitions.
Absorption bands due to excitation from the 3b1 orbital may also be expected in the energy region under discussion. The 3b1 → 3sa11B1 transition has been assigned to the peak at 6.474 eV, resulting in δ = 0.89. Use of this quantum defect in the Rydberg formula leads to an excitation energy of 8.12 eV for the n = 4 member, compared to 8.24 eV for our best theoretical estimate of the vertical excitation energy (Table 2). The experimental spectrum (Fig. 5) contains a relatively intense peak, centred at ∼8.15 eV, with complex structure. Only some of this structure can be attributed, with confidence, to the 1a2 → 4da21A1 transition. Therefore, it appears reasonable to conclude that this band contains a contribution from the 3b1 → 4sa11B1 transition, in accord with the assignment proposed by Palmer et al.32
Another intense structured peak is observed around 8.38 eV (Fig. 6) and, again, only part of the structure can be ascribed to either the 1a2 → 5pb11B2 or the 1a2 → 5da21A1 transitions. Under the assumption that this peak contains a contribution from a state belonging to a series converging onto the à 2B1 limit, then the most likely candidate appears to be that arising from the 3b1 → 4pa11B1 transition (f = 0.007 (Table S4, ESI‡)). Our best theoretical estimate of the vertical excitation energy for the 3b1 → 4pa11B1 transition is 8.44 eV (Table 2). Finally, the peak observed at 8.611 eV might correspond to the 3b1 → 4fa11B1 transition whose vertical excitation energy and oscillator strength are 8.51 eV and f = 0.010 (Table S4, ESI‡).
5.5 Excited states in the energy range 8.8 to 12.5 eV
Above the 2A2 state threshold, the photoabsorption cross section of thiophene gradually increases (Fig. 2) with prominent broad bands appearing at ∼9.1 and 11.2 eV. Such rises in cross section in this energy range are commonly observed in five- or six-membered ring-type molecules, and are generally due to transitions into many, closely spaced, valence states. For example, the photoabsorption cross sections of furan,103 pyrrole104 and the diazines105,106 show a similar energy dependence. According to Wan et al.,37 the most intense valence states in thiophene in this region arise through π → σ* or π → π* transitions. Their calculations predict several transitions, of high oscillator strength, into mixed valence–Rydberg states in the excitation range 8.9–9.3 eV, which might account for the broad peak observed at 9.1 eV. Excitations involving the 11a1 orbital may also occur and the lowest n → π* transition, of B1 symmetry, is predicted at 8.83,32 7.77,35 7.8637 and 8.26 eV.39
Our absorption spectrum exhibits extensive, highly irregular, vibrational structure throughout the excitation range ∼9.4 to 11.8 eV (Fig. 7). This structure is similar to that observed previously by Rennie et al.33 but is better developed. The structure is most probably associated with Rydberg states belonging to series converging onto the 2A1 state which has adiabatic and vertical ionization energies of 11.5 and 12.1 eV, respectively.42 An analysis of the vibrational structure appearing in the 2A1 state photoelectron band indicated that the progressions involved excitation of the ν4, ν7 and ν8 modes.42
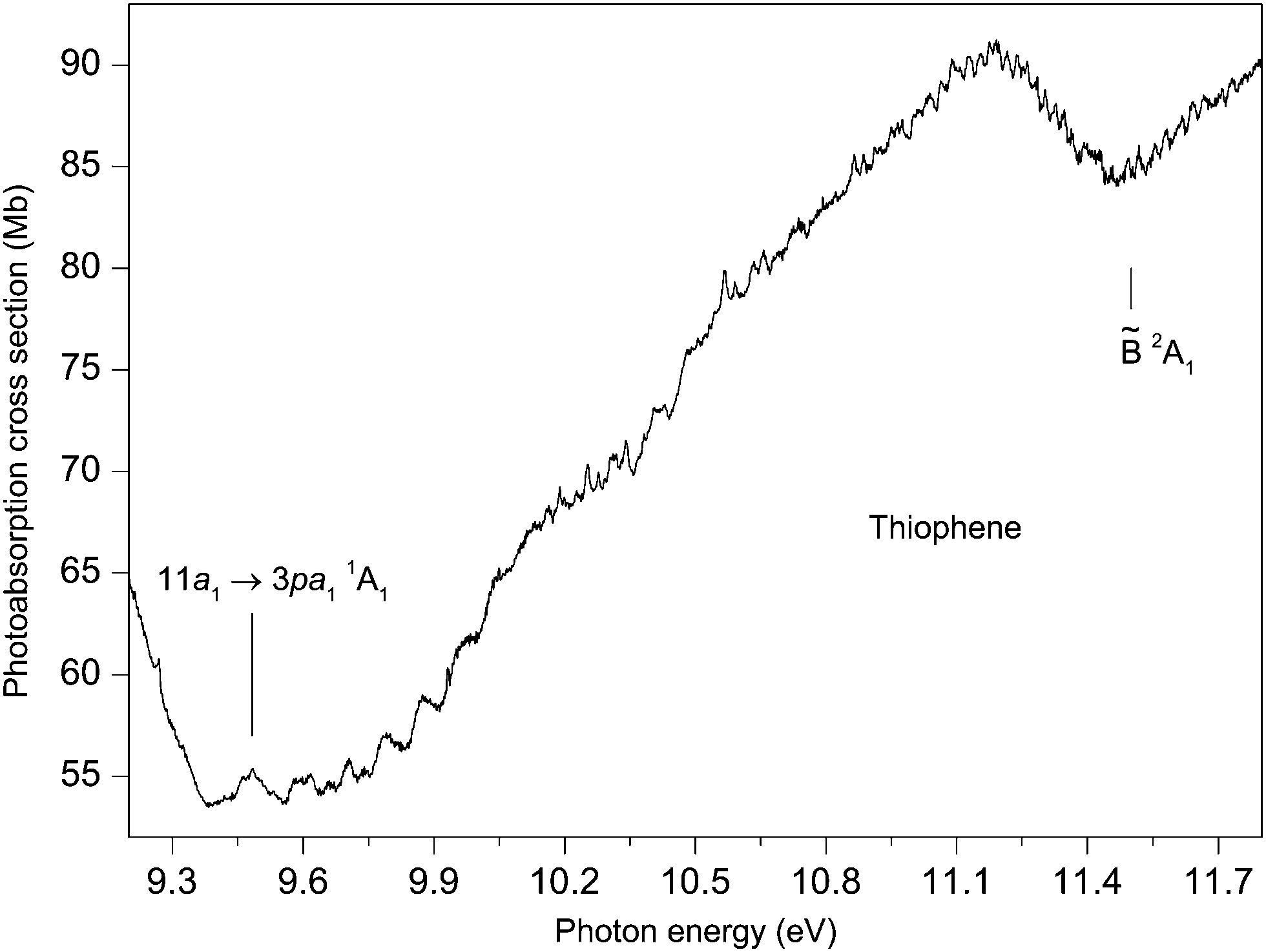 | ||
| Fig. 7 The photoabsorption spectrum of thiophene showing structure probably due to Rydberg states belonging to series converging onto the 2A1 state limit. | ||
Rydberg states arising from transitions originating from the 11a1 orbital have been studied theoretically by Serrano-Andrés et al.35 and by Wan et al.37 Serrano-Andrés et al. give an excitation energy of 9.96 eV for the 11a1 → 3sa11A1 transition, and Wan et al. give energies of 8.77, 9.32, 9.68 and 9.85 eV for the 11a1 → 3sa11A1, 11a1 → 3pa11A1, 11a1 → 3pb21B2 and the 11a1 → 3da11A1 transitions, respectively.
If we assume that the lowest lying absorption peak, centred at 9.483 eV in the experimental spectrum (Fig. 7), corresponds to the adiabatic transition into a Rydberg state, then for n = 3, a quantum defect of 0.40 is obtained. This suggests that the peak should be associated with an 11a1 → 3p transition and, based upon the theoretical predictions of Wan et al., the 11a1 → 3pa11A1 transition has the lower of the two calculated 11a1 → 3p excitation energies. As the predicted excitation energies of the 11a1 → 3pa11A1 and the 11a1 → 3pb21B2 transitions differ by only 0.36 eV, and as the width of the vibrational envelope associated with the 2A1 state photoelectron band is at least 0.5 eV,42 the vibrational structure corresponding to the 11a1 → 3pa11A1 transition probably overlaps that associated with the 11a1 → 3pb21B2 transition. The interpretation of the absorption structure is further complicated by the close proximity of the 11a1 → 3da11A1 transition which is calculated to lie only slightly higher in energy and to possess a high oscillator strength.37
Derrick et al.42 assigned the vibrational structure in the 2A1 state photoelectron bands purely in terms of excitations involving the totally symmetric modes, of which ν8 has the smallest energy of ∼75 meV. The structure in the absorption spectrum shows substantially smaller separations, with many occurring in the 20–40 meV range. One plausible interpretation is that the absorption bands are due to overlapping vibrational envelopes, involving totally symmetric modes, associated with closely spaced Rydberg states. An alternative explanation is that low energy, non-totally symmetric vibrational modes are being excited. The clarification of these assignments requires additional investigation.
6. Summary
The highly resolved photoabsorption spectrum of thiophene has been measured between 5 and 12.5 eV using a new Fourier transform spectrometer. The spectrum displays several prominent bands due to transitions into valence states and some much narrower structure ascribed to Rydberg series. The overall spectral profile is distinctly more detailed than any hitherto published and provides much new information that has yet to be analysed. Vertical excitation energies and oscillator strengths have been calculated with the ADC(2), EOM-CCSD and EOM-CC3 approaches, in conjunction with large basis sets, yielding a very consistent description of the valence and many Rydberg states. The vibrational coupling constants for the electronic transitions have also been evaluated and these have enabled adiabatic transition energies to be estimated. The results of the calculations are in good qualitative, and even semi-quantitative, agreement with the measurements and proved useful in assigning the experimental spectra.Guided by the theoretical results, a reasonable interpretation has been obtained for the low-lying valence bands due to π → π* or π → σ* excitations as well as for the principal Rydberg series converging onto the 2A2 state limit. For this lowest ionization threshold, one p-type and one d-type Rydberg series have definitely been identified and assigned, and there is some experimental evidence of a second d-type series. One s-type series converging onto the à 2B1 state limit has been identified. The analysis and assignment of the observed absorption bands is complicated by irregular intensity distributions and by shifts in the transition energies from those estimated through application of the Rydberg formula. This suggests that in many cases the assignment of an absorption band to a single excited configuration is probably invalid and that Rydberg–valence, Rydberg–Rydberg or valence–valence state mixing needs to be taken into account to obtain a proper description.
Many of the absorption bands associated with Rydberg states display vibrational structure which resembles that in the corresponding photoelectron band. However, the assignment of vibrational structure associated with excited valence states has proved difficult due to the disparity between the vibrational spacings observed in the absorption bands and the established vibrational energies in the neutral and ionic ground states. The low vibrational energies measured for some of the valence state structure suggest that non-totally symmetric modes may be excited.
The highly irregular vibrational structure observed in the high resolution FT absorption spectra represent a new source of information for the rapidly developing studies of thiophene excited state non-adiabatic dynamics and photochemistry. The assignment of this structure cannot be accomplished within the normally adopted Born–Oppenheimer and Franck–Condon approximations. Indeed, it appears likely that a successful interpretation will require calculations which specifically include interactions amongst the closely spaced valence and Rydberg states.
Further theoretical studies are now underway to investigate the vibronic coupling which seems particularly important in some of the neutral excited states.
Acknowledgements
The authors are grateful to the technical staff of the Soleil synchrotron radiation facility for their efficient operation of the storage ring. Alexander Trofimov is grateful to Jochen for the chance once given to him to enter the fascinating world of quantum chemistry, for the education in the field of many-body Green's functions, and for many years of support and interesting collaborations. The authors are indebted to Jochen for developing excellent theoretical tools, particularly the ADC methods, for interpreting molecular ionization and excitation data collected with synchrotron radiation.References
- T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structures, Reactions, Syntheses, and Applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
- H. D. Hartough, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1952, vol. 3 Search PubMed.
- S. Gronowitz, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1992, vol. 44 Search PubMed.
- C. Taliani and W. Gebauer, in Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley-VCH, Weinheim, 1998, p. 361 Search PubMed.
- J. L. Brédas, J. Cornil, F. Meyers and D. Beljonne, in Handbook of conducting polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker, New York, 1998, p. 1 Search PubMed.
- G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581 CrossRef CAS PubMed.
- G. M. Tsivgoulis and J. M. Lehn, Adv. Mater., 1997, 9, 39 CrossRef CAS PubMed.
- M. Irie, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001, p. 37 Search PubMed.
- A. Staykov, J. Areephong, W. R. Browne, B. L. Feringa and K. Yoshizawa, ACS Nano, 2011, 5, 1165 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS PubMed.
- Y. Y. Liang and L. P. Yu, Polym. Rev., 2010, 50, 454 CrossRef CAS.
- A. R. Murphy and J. M. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS PubMed.
- T. Noda, H. Ogawa, N. Noma and Y. Shirota, Adv. Mater., 1997, 9, 720 CrossRef CAS PubMed.
- M. G. Harrison and R. H. Friend, in Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998, p. 516 Search PubMed.
- H. Köppel, E. V. Gromov and A. B. Trofimov, Chem. Phys., 2004, 304, 35 CrossRef PubMed.
- S. Salzmann, M. Kleinschmidt, J. Tatchen, R. Weinkauf and C. M. Marian, Phys. Chem. Chem. Phys., 2008, 10, 380 RSC.
- R. Weinkauf, L. Lehr, E. W. Schlag, S. Salzmann and C. M. Marian, Phys. Chem. Chem. Phys., 2008, 10, 393 RSC.
- X.-F. Wu, X. Zheng, H. G. Wang, Y.-Y. Zhao, X. Guan, D. L. Phillips, X. Chen and W. Fang, J. Chem. Phys., 2010, 133, 134507 CrossRef PubMed.
- G. Cui and W. Fang, J. Phys. Chem. A, 2011, 115, 11544 CrossRef CAS PubMed.
- M. Stenrup, Chem. Phys., 2012, 397, 18 CrossRef CAS PubMed.
- W. C. Price and A. D. Walsh, Proc. R. Soc. London, Ser. A, 1941, 179, 201 CrossRef CAS.
- G. Horváth and Á. I. Kiss, Spectrochim. Acta, Part A, 1967, 23, 921 CrossRef.
- G. Di Lonardo, G. Galloni, A. Trombetti and C. Zauli, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 2009 RSC.
- G. Varsányi, L. Nyulászi, T. Veszprémi and T. Narisawa, J. Chem. Soc., Perkin Trans. 2, 1982, 761 RSC.
- E. J. Beiting, K. J. Zeringue and R. E. Stickel, Spectrochim. Acta, Part A, 1985, 41, 1413 CrossRef.
- L. Nyulászi and T. Veszprémi, J. Mol. Struct., 1986, 140, 253 CrossRef.
- L. Nyulászi and T. Veszprémi, J. Mol. Struct., 1986, 140, 353 CrossRef.
- L. Nyulászi and T. Veszprémi, Chem. Scr., 1988, 28, 331 Search PubMed.
- L. Nyulászi and T. Veszprémi, Acta Chim. Hung., 1993, 130, 811 Search PubMed.
- M. H. Palmer, I. C. Walker and M. F. Guest, Chem. Phys., 1999, 241, 275 CrossRef CAS.
- E. E. Rennie, D. M. P. Holland, D. A. Shaw, C. A. F. Johnson and J. E. Parker, Chem. Phys., 2004, 306, 295 CrossRef CAS PubMed.
- G. L. Bendazzoli, F. Bertinelli, P. Palmieri, A. Brillante and C. Taliani, J. Chem. Phys., 1978, 69, 5077 CrossRef CAS PubMed.
- L. Serrano-Andrés, M. Merchán, M. Fülscher and B. O. Roos, Chem. Phys. Lett., 1993, 211, 125 CrossRef.
- F. Negri and M. Z. Zgierski, J. Chem. Phys., 1994, 100, 2571 CrossRef CAS PubMed.
- J. Wan, M. Hada, M. Ehara and H. Nakatsuji, J. Chem. Phys., 2001, 114, 842 CrossRef CAS PubMed.
- M. Kleinschmidt, J. Tatchen and C. M. Marian, J. Comput. Chem., 2002, 23, 824 CrossRef CAS PubMed.
- M. Pastore, C. Angeli and R. Cimiraglia, Theor. Chem. Acc., 2007, 118, 35 CrossRef CAS PubMed.
- P. J. Derrick, L. Åsbrink, O. Edqvist and E. Lindholm, Spectrochim. Acta, Part A, 1971, 27, 2525 CrossRef CAS.
- T. Munakata, K. Kuchitsu and Y. Harada, J. Electron Spectrosc. Relat. Phenom., 1980, 20, 235 CrossRef CAS.
- P. J. Derrick, L. Åsbrink, O. Edqvist, B. Ö. Jonsson and E. Lindholm, Int. J. Mass Spectrom. Ion Phys., 1971, 6, 177 CrossRef CAS.
- J. W. Rabalais, L. O. Werme, T. Bergmark, L. Karlsson and K. Siegbahn, Int. J. Mass Spectrom. Ion Phys., 1972, 9, 185 CrossRef CAS.
- L. Klasinc, A. Sabljić, G. Kluge, J. Rieger and M. Scholz, J. Chem. Soc., Perkin Trans. 2, 1982, 539 RSC.
- N. Kishimoto, H. Yamakado and K. Ohno, J. Phys. Chem., 1996, 100, 8204 CrossRef CAS.
- A. D. O. Bawagan, B. J. Olsson, K. H. Tan, J. M. Chen and B. X. Yang, Chem. Phys., 1992, 164, 283 CrossRef CAS.
- D. M. P. Holland, L. Karlsson and W. von Niessen, J. Electron Spectrosc. Relat. Phenom., 2001, 113, 221 CrossRef CAS.
- J. Yang, J. Li and Y. Mo, J. Chem. Phys., 2006, 125, 174313 CrossRef PubMed.
- K. Takeshita and Y. Yamamoto, Chem. Phys., 1994, 189, 489 CrossRef CAS.
- A. B. Trofimov, H. Köppel and J. Schirmer, J. Chem. Phys., 1998, 109, 1025 CrossRef CAS PubMed.
- W. von Niessen, W. P. Kraemer and L. S. Cederbaum, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 179 CrossRef CAS.
- A. B. Trofimov, J. Schirmer, D. M. P. Holland, A. W. Potts, L. Karlsson, R. Maripuu and K. Siegbahn, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 5051 CrossRef CAS.
- M. Ehara, Y. Ohtsuka, H. Nakatsuji, M. Takahashi and Y. Udagawa, J. Chem. Phys., 2005, 122, 234319 CrossRef PubMed.
- A. B. Trofimov, I. L. Zaitseva, T. E. Moskovskaya and N. M. Vitkovskaya, Chem. Heterocycl. Comp., 2008, 44, 1101 CrossRef CAS PubMed.
- W. M. Flicker, O. A. Mosher and A. Kuppermann, Chem. Phys. Lett., 1976, 38, 489 CrossRef CAS.
- E. H. van Veen, Chem. Phys. Lett., 1976, 41, 535 CrossRef CAS.
- W. M. Flicker, O. A. Mosher and A. Kuppermann, J. Chem. Phys., 1976, 64, 1315 CrossRef CAS PubMed.
- H. Haberkern, K. R. Asmis, M. Allan and P. Swiderek, Phys. Chem. Chem. Phys., 2003, 5, 827 RSC.
- R. Håkansson, B. Nordén and E. W. Thulstrup, Chem. Phys. Lett., 1977, 50, 305 Search PubMed.
- B. Nordén, R. Håkansson, P. B. Petersen and E. W. Thulstrup, Chem. Phys., 1978, 33, 355 CrossRef.
- N. Igarashi, A. Tajiri and M. Hatano, Bull. Chem. Soc. Jpn., 1981, 54, 1511 CrossRef CAS.
- N. de Oliveira, D. Joyeux, D. Phalippou, J. C. Rodier, F. Polack, M. Vervloet and L. Nahon, Rev. Sci. Instrum., 2009, 80, 043101 CrossRef CAS PubMed.
- L. Nahon, N. de Oliveira, G. A. Garcia, J.-F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508 CrossRef CAS PubMed.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299 CrossRef CAS.
- A. B. Trofimov, G. Stelter and J. Schirmer, J. Chem. Phys., 1999, 111, 9982 CrossRef CAS PubMed.
- A. B. Trofimov, G. Stelter and J. Schirmer, J. Chem. Phys., 2002, 117, 6402 CrossRef CAS PubMed.
- J. H. Starcke, M. Wormit and A. Dreuw, J. Chem. Phys., 2009, 130, 024104 CrossRef PubMed.
- H. Sekino and R. J. Bartlett, Int. J. Quantum Chem., Symp., 1984, 18, 255 CrossRef CAS PubMed.
- J. Geertsen, M. Rittby and R. J. Bartlett, Chem. Phys. Lett., 1989, 164, 57 CrossRef CAS.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029 CrossRef CAS PubMed.
- T. Korona and H. J. Werner, J. Chem. Phys., 2003, 118, 3006 CrossRef CAS PubMed.
- O. Marcouille, P. Brunelle, O. Chubar, F. Marteau, M. Massal, L. Nahon, K. Tavakoli, J. Veteran and J.-M. Filhol, Am. Inst. Phys., Conf. Proc., 2007, 879, 311 CAS.
- F. Polack, D. Joyeux, J. Svatoš and D. Phalippou, Rev. Sci. Instrum., 1995, 66, 2180 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, F. R. Manby, M. Schütz, P. Celani, G. Knizia, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2010.1, a package of ab initio programs, 2010, see http://www.molpro.net Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242 CrossRef CAS PubMed.
- B. Bak, D. Christensen, L. Hansen-Nygaard and J. Rastrup-Andersen, J. Mol. Spectrosc., 1961, 7, 58 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS PubMed.
- K. Kaufmann, W. Baumeister and M. Jungen, J. Phys. B: At., Mol. Opt. Phys., 1989, 22, 2223 CrossRef CAS.
- CFOUR, a quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding and P. G. Szalay, with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, O. Christiansen, M. Heckert, O. Heun, C. Huber, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. Matthews, T. Metzroth, D. P. O’Neill, D. R. Price, E. Prochnow, K. Ruud, F. Schimann, S. Stopkowicz, A. Tajti, M. E. Varner, J. Vázquez, F. Wang and J. D. Watts, and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen and J. Olsen), and ecp routines by A. V. Mitin and C. van Wüllen. For the current version, see http://www.cfour.de.
- A. B. Trofimov and J. Schirmer, Chem. Phys., 1997, 214, 153 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, Chem. Phys., 1997, 224, 175 CrossRef CAS.
- C. Hättig, in Response Theory and Molecular Properties, Adv. Quantum Chemistry, Academic Press, New York, 2005, vol. 50, p. 37 Search PubMed.
- S. Yamazaki, A. L. Sobelewski and W. Domcke, Phys. Chem. Chem. Phys., 2009, 11, 10165 RSC.
- P. G. Szalay, A. J. A. Aquino, M. Barbatti and H. Lischka, Chem. Phys., 2011, 380, 9 CrossRef CAS PubMed.
- J. H. Starcke, M. Wormit, J. Schirmer and A. Dreuw, Chem. Phys., 2006, 329, 39 CrossRef CAS PubMed.
- J. H. Starcke, M. Wormit and A. Dreuw, J. Chem. Phys., 2009, 131, 144311 CrossRef PubMed.
- L. S. Cederbaum and W. Domcke, Adv. Chem. Phys., 1977, 36, 205 CrossRef CAS PubMed.
- H. Köppel, W. Domcke and L. S. Cederbaum, Adv. Chem. Phys., 1984, 57, 59 CrossRef PubMed.
- A. B. Trofimov, I. L. Rusakova (Krivdina), G. Stelter and J. Schirmer, Polarization propagator ADC/ISR code, Irkutsk State University, Irkutsk Institute of Chemistry, SB RAS, Heidelberg University, 1991–2011 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS PubMed.
- G. Wälz, D. Kats, D. Usvyat, T. Korona and M. Schütz, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 86, 052519 CrossRef.
- M. J. Frisch, et al., Gaussian 94, Revision C.2, Gaussian Inc., Pittsburgh, PA, 1995 Search PubMed.
- T. D. Klots, R. D. Chirico and W. V. Steele, Spectrochim. Acta, Part A, 1994, 50, 765 CrossRef.
- A. B. Trofimov, H. Köppel and J. Schirmer, J. Chem. Phys., 1998, 109, 1025 CrossRef CAS PubMed.
- N. Gavrilov, S. Salzmann and C. M. Marian, Chem. Phys., 2008, 349, 269 CrossRef CAS PubMed.
- B. Sellner, M. Barbatti and H. Lischka, J. Chem. Phys., 2009, 131, 024312 CrossRef PubMed.
- E. V. Gromov, A. B. Trofimov, F. Gatti and H. Köppel, J. Chem. Phys., 2010, 133, 164309 CrossRef CAS PubMed.
- M. Stenrup and Å. Larson, Chem. Phys., 2011, 379, 6 CrossRef CAS PubMed.
- O. Christiansen, J. Gauss, J. F. Stanton and P. Jørgensen, J. Chem. Phys., 1999, 111, 525 CrossRef PubMed.
- M. Schreiber, M. R. Silva-Junior, S. P. A. Sauer and W. Thiel, J. Chem. Phys., 2008, 128, 134110 CrossRef PubMed.
- E. E. Rennie, C. A. F. Johnson, J. E. Parker, D. M. P. Holland, D. A. Shaw, M. A. MacDonald, M. A. Hayes and L. G. Shpinkova, Chem. Phys., 1998, 236, 365 CrossRef CAS.
- E. E. Rennie, C. A. F. Johnson, J. E. Parker, R. Ferguson, D. M. P. Holland and D. A. Shaw, Chem. Phys., 1999, 250, 217 CrossRef CAS.
- M. Stener, P. Decleva, D. M. P. Holland and D. A. Shaw, J. Phys. B: At., Mol. Opt. Phys., 2011, 44, 075203 CrossRef.
- D. M. P. Holland, D. A. Shaw, S. Coriani, M. Stener and P. Decleva, J. Phys. B: At., Mol. Opt. Phys., 2013, 46, 175103 CrossRef.
Footnotes |
| † This paper is dedicated to Professor Jochen Schirmer on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Calculated and experimental geometry of thiophene; calculated and measured frequencies; vibrational coupling constants, vibrational widths and adiabatic energy lowering of Rydberg and valence-type excitations of thiophene derived from the ADC(2) calculations. See DOI: 10.1039/c4cp02420f |
| This journal is © the Owner Societies 2014 |