
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the phase space of time of flight mass selected PtxY nanoparticles
Federico
Masini
a,
Patricia
Hernández-Fernández†
a,
Davide
Deiana
b,
Christian Ejersbo
Strebel
c,
David Norman
McCarthy
c,
Anders
Bodin
a,
Paolo
Malacrida
 a,
Ifan
Stephens
a and
Ib
Chorkendorff
*a
a,
Ifan
Stephens
a and
Ib
Chorkendorff
*a
aCenter for Individual Particle Functionality (CINF), Department of Physics, DTU, DK-2800 Kgs. Lyngby, Denmark. E-mail: ibchork@fysik.dtu.dk
bCenter for Electron Nanoscopy (CEN), Department of Physics, DTU, DK-2800 Kgs. Lyngby, Denmark
cHaldor Topsøe A/S, Nymøllevej 55, DK-2800 Kgs Lyngby, Denmark
First published on 17th July 2014
Abstract
Mass-selected nanoparticles can be conveniently produced using magnetron sputtering and aggregation techniques. However, numerous pitfalls can compromise the quality of the samples, e.g. double or triple mass production, dendritic structure formation or unpredicted particle composition. We stress the importance of transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and ion scattering spectroscopy (ISS) for verifying the morphology, size distribution and chemical composition of the nanoparticles. Furthermore, we correlate the morphology and the composition of the PtxY nanoparticles with their catalytic properties for the oxygen reduction reaction. Finally, we propose a completely general diagnostic method, which allows us to minimize the occurrence of undesired masses.
Introduction
Heterogeneous catalysis of nanoparticles has been the subject of numerous investigations in recent years.1–15Industrial catalysts are almost always in the nanoparticulate form due to their high surface area to volume ratio. Traditionally, the fundamental properties of catalysts were investigated on extended surfaces, e.g. single crystals. However, in recent years, an increasing number of model studies have been devoted to the direct investigation of nanoparticles. In some cases it turns out that the surface chemistry of nanoparticles is quite distinct from that of extended surfaces.
Size dependent effects have been observed in numerous catalytic systems such as those for Co nanoparticles in the Fischer–Tropsch reaction, where activity drops at particle sizes greater than 6–8 nm.16 Furthermore, Au nanoparticles showed a remarkably high activity for CO oxidation below 5 nm particle diameter, reaching a maximum in activity at 2–3 nm size.17 Ruthenium has interesting catalytic properties for CO oxidation if prepared in the form of nanoparticles of diameter between 2 nm and 6 nm.18
The reason for size dependence is still not entirely clear and has been debated for a long time. However, there are cases in which a combination of theoretical and experimental work has elucidated the role of different types of sites in the catalytic activity, whose abundance is related to particle size and structure.
On the Ru(0001) surface, the so called B5 step sites were found to yield a much higher contribution to N2 dissociation (about nine orders of magnitude) than the facets.19 Recently, the same sites (B5) were shown to have analogous properties in Ru nanoparticles,10 and the concentration of these sites has been correlated with particle size.
The relevance of surface steps and defects for catalytic purposes has been elucidated by the work of Vendelbo et al.,20,21 who investigated the reactivity of the Ru(0154) surface (Ru(0001) with 4% steps) for methanation. Not only the steps proved to be of paramount importance for successful methane formation, but defects induced by sputtering readily improved the catalytic activity. Importantly, sulfur poisoning of the step sites reduced the catalytic activity, providing further evidence that all the catalytic properties are concentrated in the few defect sites.
A particularly illustrative example of where single crystal surfaces do not model the catalytic properties of nanoparticles can be found in the case of oxygen electroreduction. This reaction limits the efficiency of low temperature fuel cells, and prohibitively high loadings of Pt-based nanoparticles are required to catalyse the reaction. Most, albeit not all, theoretical and experimental studies seem to suggest that the surface specific activity for oxygen reduction should decrease with a decrease in particle size.12,22–28 However, this notion is in conflict with the experimental observation that extended single crystal stepped Pt surfaces sometimes exhibit higher oxygen reduction activity than unstepped surfaces.29–31
One strategy to improve the activity of Pt is to alloy it with other metals. Most research has been devoted to the study of alloys of Pt and late transition metals such as Ni, Co, Cu and Fe.32–38 However, in the harsh acidic and oxidising environment of a fuel cell, these alloys degrade by dealloying.39–41 In our laboratory we have taken a different approach, namely to study alloys of Pt and rare earths such as Y, Gd, Ce and La.33,42–48 These alloys have a particularly negative heat of formation, which should provide them with long term-kinetic stability against dealloying at the cathode of a fuel cell. Extended surfaces of Pt3Y and Pt5Gd show particularly high activity for oxygen reduction.42,43,47 On the other hand, upon exposure to reaction conditions, alloys such as Pt2Y or PtY corroded extensively, due to Y dissolution; this suggests that an excessive amount of Y is detrimental to catalyst stability.44 Most recently, we demonstrated that PtxY is also highly active for the oxygen reduction reaction in nanoparticulate form. The activity increases monotonically with particle size; we attributed this to an increased degree of compressive strain exerted by the alloy core onto the pure Pt shell.48
Importantly, size effects are not the only significant characteristics of nanoparticles. Improvements can be made to catalysts by forming non-equilibrium morphologies.14,38,49–53 Very recent results by Chen et al.13 have shown unprecedented capabilities of PtxNi “nanoframes” to catalyze oxygen reduction.
It is of utmost importance to conduct studies on monodisperse, size selected nanoparticles, to be able to correlate size, shape and surface structure with the catalytic activity of the nanoparticles. Common problems which could arise are (i) too large particle size distribution, (ii) contaminations, (iii) polydispersity, (iv) undesired morphology e.g. dendritic shapes and (v) inadequate particle composition.
To circumvent the first and the second issues, we produce nanoparticles using a cluster source,54 interfaced with a von Issendorff (lateral time-of-flight) mass selector.55 The source produces particles with narrow size distribution by Ar+ plasma sputtering followed by gas phase aggregation under ultra-clean conditions. Nonetheless, it is still necessary to avoid the last three problems to be able to study the catalytic properties of the deposited nanoparticles. To do so, greater attention must be paid to undesired higher mass production. Additionally, particular attention must be paid to the particle morphology and chemical composition.
These issues stress the necessity of checking the quality of the deposited samples, e.g. via transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and ion scattering spectroscopy (ISS).
In this paper, we show how different PtxY chemical composition affects the particle activity for ORR.
Furthermore, we discuss the conditions under which dendritic PtxY nanostructures are produced; we show that their electrochemical activity for ORR varies as a function of the morphology of the PtxY nanostructure.
Finally, we qualitatively correlate the quality of the deposited nanoparticle sample with the evolution in time of the deposition current versus mass curves, measured using a time of flight analyzer (TOF). We present a method to minimize the problem of multiple masses production, thereby greatly reducing the risk of compromising an experimental sample. Although the study is conducted on PtxY particles, the method is completely general.
Experimental
The PtxY nanoparticles were prepared using a magnetron sputter gas aggregation source (Birmingham Instruments Inc.), combined with a von Issendorff time-of-flight mass filter, and deposited onto Lacey Carbon-coated Au or Si3N4 (for the Ru reference) TEM grids mounted in a multichamber ultrahigh vacuum (UHV) system (Omicron, Multiscan Lab) with a base pressure in the low 10−10 mbar region. The gas aggregation technique involves Ar+ sputtering of a target (in this study Ru and Pt 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Y, Pt 3:1 Y alloy targets from Kurt J. Lesker Inc.), to produce an atomic vapor that is condensed into nanoparticles through collisions with cooled Ar and He gas. The gas aggregation method is particularly useful in the context of formation of metallic nanoparticles from materials such as yttrium that have high affinities for oxygen, as it is an ultrahigh vacuum compatible method where oxygen is present at extremely low levels. The second chief advantage arises from the fact that a high fraction of the nanoparticles produced via Ar+ sputtering are ionized,54 thus the particles can be filtered based on their mass-to-charge ratio, which in turn allows the deposition of particles with narrow size distributions. In our experiments the Pt–Y and the Ru nanoparticles are filtered using a time-of-flight mass filter.55
:1 Y, Pt 3:1 Y alloy targets from Kurt J. Lesker Inc.), to produce an atomic vapor that is condensed into nanoparticles through collisions with cooled Ar and He gas. The gas aggregation method is particularly useful in the context of formation of metallic nanoparticles from materials such as yttrium that have high affinities for oxygen, as it is an ultrahigh vacuum compatible method where oxygen is present at extremely low levels. The second chief advantage arises from the fact that a high fraction of the nanoparticles produced via Ar+ sputtering are ionized,54 thus the particles can be filtered based on their mass-to-charge ratio, which in turn allows the deposition of particles with narrow size distributions. In our experiments the Pt–Y and the Ru nanoparticles are filtered using a time-of-flight mass filter.55
Nanoparticle size distributions were obtained from TEM and high angle annular dark field (HAADF) STEM micrographs were acquired using a probe Cs-corrected FEI Titan Analytical 80–300 electron microscope operated at 300 kV accelerating voltage.
The electrochemical experiments were performed using a Bio-Logic Instruments' VMP2 potentiostat, controlled by a computer. The rotating ring disk electrode (RRDE) assemblies and the glassy carbon substrates were provided by Pine Instruments Corporations. All glassware was cleaned in 96% H2SO4 (Merck, Emsure) and 30% H2O2 (Analar, Normpur) (3:1 v/v). Those were subsequently rinsed several times in hot Millipore water (85 C, >18.2 MΩ cm−1, TOC < 5 ppb). A standard three-compartment glass cell was used for all the experiments, equipped with an external jacket attached to a water bath with temperature control. The electrolyte, 0.1 M HClO4 (Merck, Suprapur), was prepared using Millipore water. The counter electrode was a platinum wire and the reference was the Hg/Hg2SO4 electrode separated from the working electrode compartment using a ceramic frit. The measurements were conducted at 23 ± 1 °C. All potentials in the manuscript are quoted with respect to the RHE and corrected for Ohmic losses. The catalyst prepared under UHV conditions was inserted into the arbor of a RRDE and was immersed into the electrochemical cell under potential control at 0.05 V in a N2 saturated 0.1 M HClO4 electrolyte. The potential was then constantly cycled between 0.05 V and 1.0 V until a stable cyclic voltammogram was recorded. The ORR activity was measured by cycling the potential between 0 V and 1 V at 50 mV s−1 and 1600 rpm, after saturating the solution in O2 (N55, AGA), whereas the stability tests were carried out by cycling the potential between 0.6 V and 1 V at 100 mV s−1 and 0 rpm for 9000 potential cycles. The ORR specific activity, mA cmPt−2, was calculated from the anodic sweep of the polarization curve. To that purpose, the kinetic current density, jk, for the ORR was calculated by using the following equation: 1/j = 1/jk + 1/jd, where j is the measured current density and jd is the diffusion limiting current density. The active surface area of each PtxY sample, cmPt2, was measured by CO stripping analyses, which were carried out in a CO-free Ar electrolyte at 50 mV s−1 after CO adsorption. The active surface area was estimated using the area under the CO-stripping peak assuming a charge of 420 μC cmPt−2.56
Low-energy ion scattering spectroscopy (ISS) was performed after deposition of the nanoparticles. A pressure of 5 × 10−8 mbar of He was used for the analysis and a primary energy of 1000 eV was used.
XPS measurements of the samples after electrochemical testing were taken on a Theta Probe instrument (Thermo Scientific). This instrument is equipped with a monochromatized Al Kα (1486.7 eV) source, and XPS spectra were obtained with a pass energy of 100 eV. The analyzer has an acceptance angle of 60°, between 20° and 80° to the surface normal. An X-ray beam size of 400 μm was used for all measurements. The atomic concentrations were quantified by integration of the Pt 4f, Y 3d peaks after background removal. A Shirley-type background was chosen for this purpose. The intensities were corrected for the transmission function of the analyzer, relative sensitivity factors and the electron mean free path. The relative sensitivity factors of Pt and Y were experimentally determined, as described earlier,57 from the Pt 4f and Y 3d spectra acquired after sputter-cleaning on a Pt(111) single crystal and Y foil, respectively.
Results
As mentioned in the Introduction, the electrochemical stability of polycrystalline Pt2Y and PtY was rather poor. Therefore, it was decided to decrease the yttrium content of the polycrystalline samples. Further electrocatalytic tests on the polycrystalline Pt3Y showed that this alloy is a stable catalyst for ORR.PtxY nanoparticles were obtained by sputtering targets with decreasing yttrium content i.e. Pt3Y and Pt9Y targets.
The nanoparticles produced by sputtering the Pt3Y target were tested for ORR under the same conditions used for the nanoparticles obtained using the Pt9Y target. The ORR kinetic current densities at 0.9 V as a function of the particle size for the samples prepared with the Pt3Y target and the Pt9Y target are displayed in Fig. 1. The nanoparticles prepared from the Pt9Y target display a higher activity than the ones produced using the Pt3Y target. For instance, for the 5 nm case, the specific activity varies from 4.02 mA cmPt−2 to 2.25 mA cmPt−2. This great difference in activity is due to the difference in nanoparticle surface composition (Pt/Y ratio), as demonstrated in Fig. 1b, where the ion scattering spectroscopy (ISS), taken on the as deposited particles, are shown for both samples. The ISS spectrum of the Pt9Y target has an atomic Pt/Y ratio of 2.5, substantially higher than 0.11 for the Pt3Y target. TEM (Fig. 1d) shows that the particles obtained from the Pt3Y target are spherical as for the nanoparticles (NPs) obtained sputtering the Pt9Y target (shown in Fig. 3). This is not surprising: the sputtering conditions for the Pt3Y target were similar to the sputtering conditions for the Pt9Y target, and the two targets have similar densities.
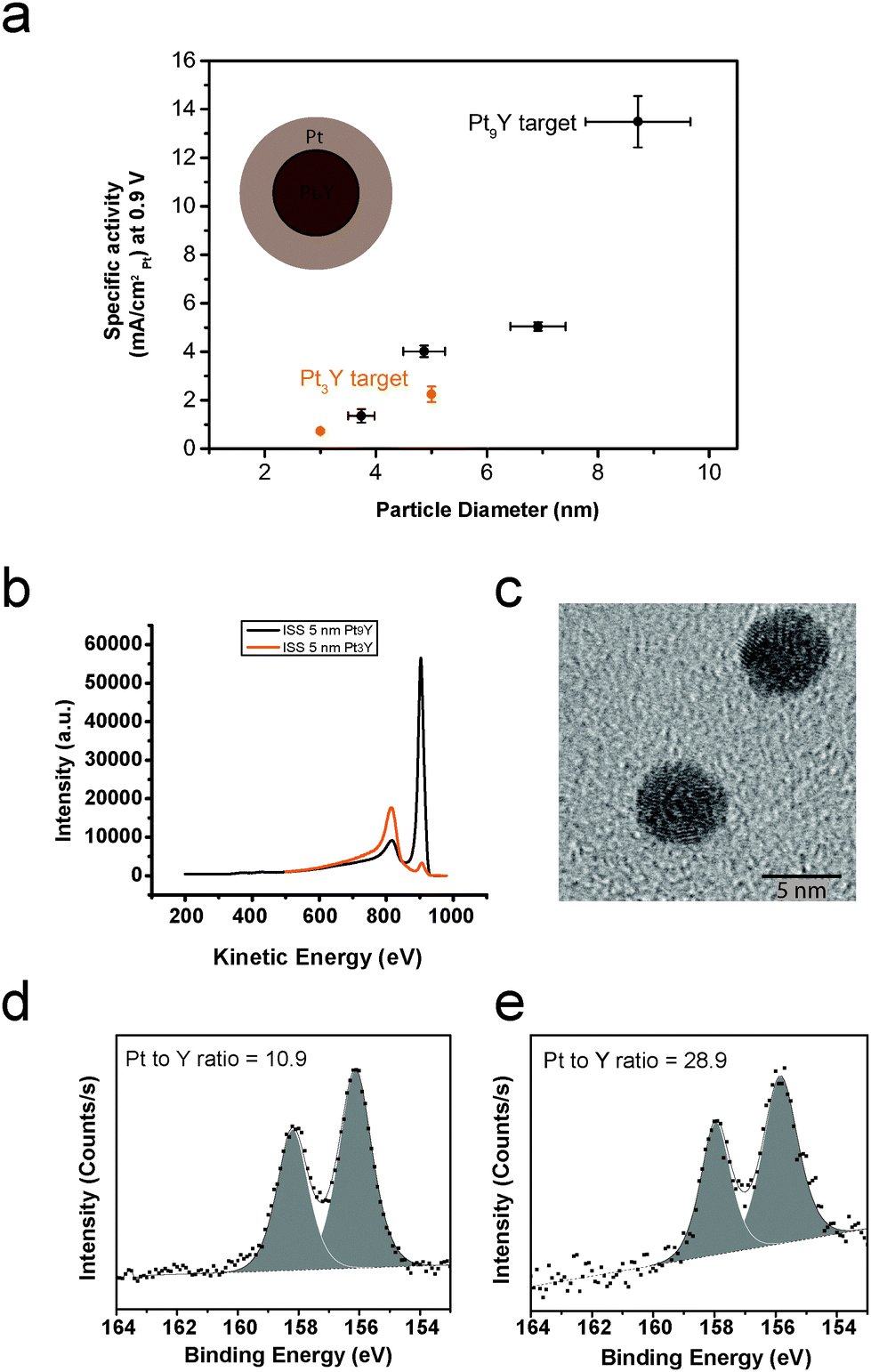 | ||
| Fig. 1 (a) Specific activity at 0.9 V versus particle size for PtxY nanoparticles generated by sputtering two targets, Pt9Y (black) and Pt3Y (orange). The cartoon nanoparticle shows the formation of an overlayer of platinum on a PtxY core, as a consequence of Y leach-out during ORR. The electrochemical measurements were recorded at 50 mV s−1, 1600 rpm and 23 ± 1 °C in O2-saturated 0.1 M HClO4 electrolyte. The current densities have been corrected for mass transport limitations. Each data point is the result of at least three independent measurements. The error bars show the standard deviation. (b) ISS spectra on 5 nm as-deposited particles produced from Pt3Y target (orange) and Pt9Y (black). The peak at ∼920 eV is attributed to Pt, whereas the peak at ∼800 eV is attributed to Y. (c) Representative TEM image of the nanoparticles obtained by sputtering the Pt3Y target. XPS spectra of the Y3d core level region for 10 nm NPs after ORR (the peaks are attributed to metallic yttrium), obtained from (d) Pt3Y target and (e) Pt9Y target. The Pt/Y ratio is reported for each spectrum. | ||
Fig. 1d and e show the XPS spectra of the Y3d core level region after ORR for 10 nm nanoparticles. The nanoparticles were fabricated from Pt3Y and Pt9Y targets, respectively. The Pt/Y ratios, displayed in the corresponding spectra, are 10.9 and 28.9, respectively.
The chemical composition of the sample is not the only issue which deserves attention when preparing a sample. Morphology also matters.
Fig. 2 displays representative TEM images of two 9 nm PtxY nanoparticulate samples displaying different morphologies. In one image, Fig. 2a, the particles have dendritic morphology, whereas in the other image, Fig. 2b, the particles are spherical. The conditions under which the particles were prepared were very different. In the case of the dendritic particles, 70 W and a flow of Ar of 120 ml min−1 and He of 20 ml min−1 were used. The selected particles were positively charged. In the case of the spherical particles, 9 W and an Ar flow of 30 ml min−1 were used. The particles were negatively charged.
 | ||
| Fig. 2 Representative TEM images of dendritic (a) and spherical (b) nanoparticles obtained by sputtering a Pt9Y target at high and low rates, respectively. (c) ORR Tafel plots for the PtxY dendritic and spherical nanoparticles after activity test (black triangles and circles, respectively). The Tafel plots after 9000 potential cycles are also shown (white triangles and circles, respectively). The electrochemical measurements were recorded at 50 mV s−1, 1600 rpm and 23 ± 1 °C in O2-saturated 0.1 M HClO4 electrolyte. The current densities have been corrected for mass transport limitations. | ||
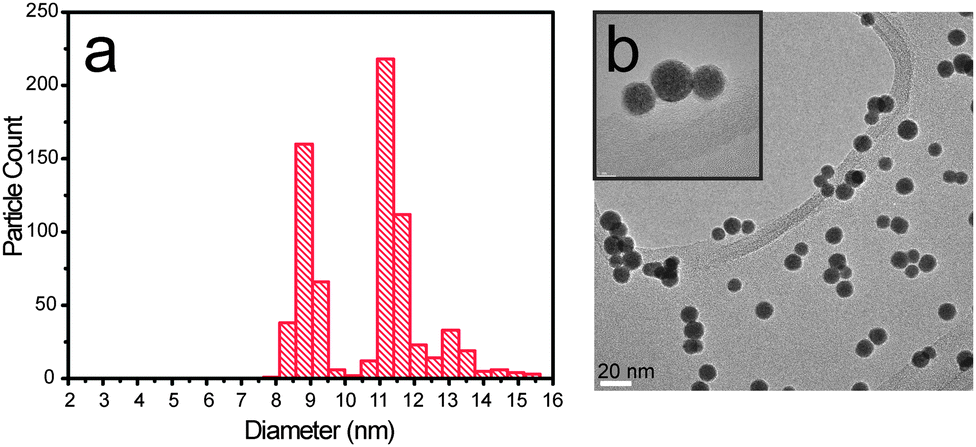 | ||
| Fig. 3 (a) Particle size distribution for a 9 nm PtxY deposition and (b) bright field TEM image. In the inset (top left corner) three particles are magnified to show the presence of undesired double and triple mass particles. | ||
The specific activity after ORR and stability studies as a function of the applied potential for both dendritic and spherical PtxY particles are shown in Fig. 2c. The two morphologies present dramatically different activities and corrosion resistance. The initial ORR specific activity at 0.9 V is ca. 13.5 mA cmPt−2 for the spherical nanoparticles, while for the dendritic ones it decreases to 8.7 mA cmPt−2. Not only is the activity, but also the stability influenced by the shape of the particles. Spherical particles only lose around 35 ± 5% of their activity after 9000 potential cycles. Dendritic particles lose around 60 ± 10% of their initial activity.
The first requirement for correct size-selection is the monodispersion of the nanoparticles. Using magnetron sputtering techniques, one typically scans a large range of masses to determine the distribution of masses that exit the sputtering chamber. Fig. 3 shows the particle size distribution (PSD) and a TEM image of a PtxY sample, where the peak intensity was intended to be 9 nm. It appears obvious that the deposition of monodispersed nanoparticles failed, since the PSD is trimodal and peaked at 8.75 nm, 11.2 nm and 13 nm.
Fig. 4a shows two deposition curves for PtxY 7 nm. In this case, a shift in the deposition curve minima was detected (from 2 million amu to 1.6 million amu) and some change in the shape of the deposition curves is noticeable.
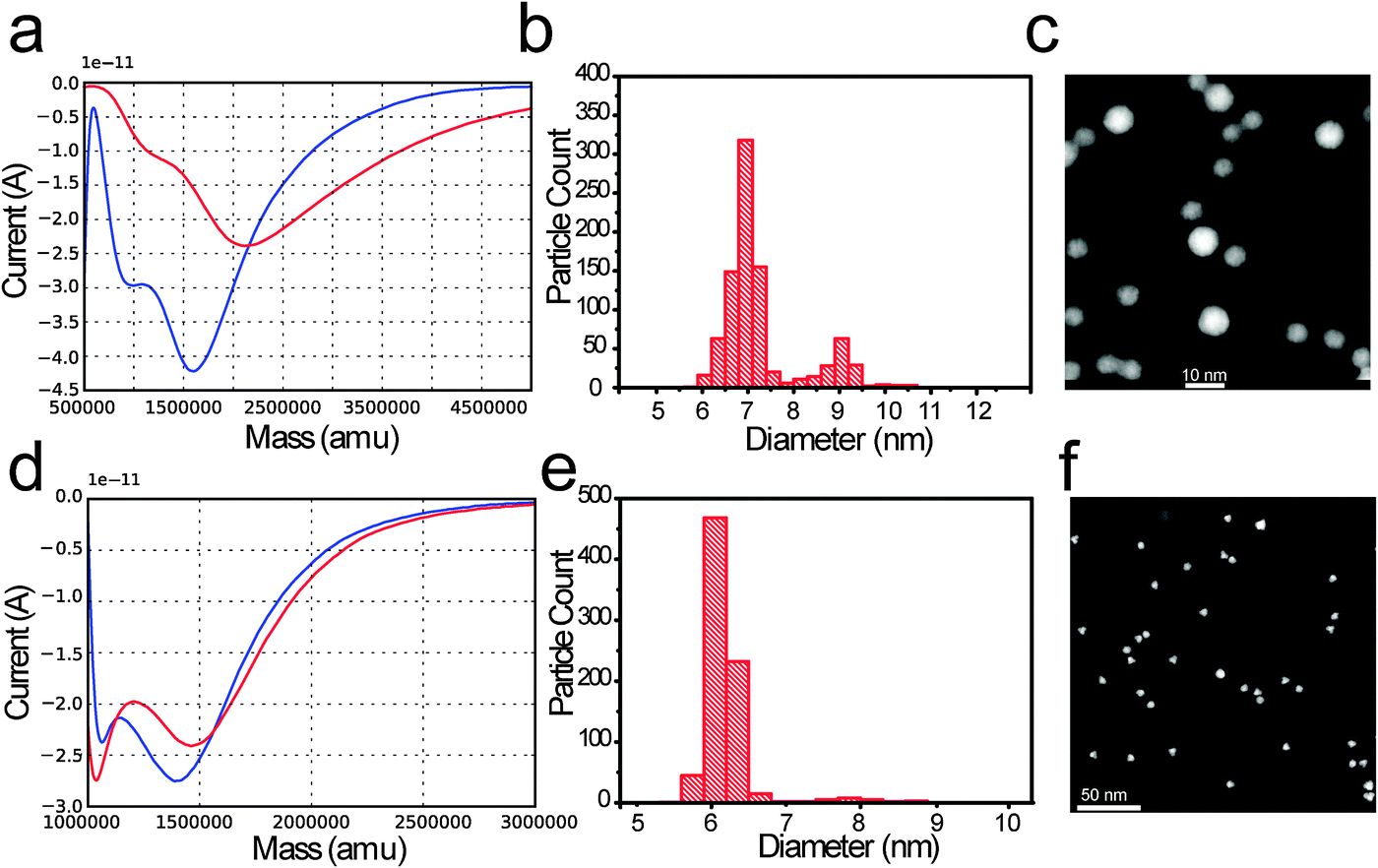 | ||
| Fig. 4 (a) Current versus mass curve (measured with TOF mass selector) before (red curve) and after (blue curve) PtxY NPs deposition. (b) Corresponding particle size distribution and (c) representative dark field STEM image relative to the deposited sample. The intended size was 7 nm. As an ideal deposition reference: (d) current versus mass curve (measured with TOF mass selector) before (red curve) and after (blue curve) Ru NPs deposition. (e) Corresponding particle size distribution and (f) representative dark field STEM image relative to the deposited sample. The intended size was 7 nm. | ||
A deposition curve is a mass distribution which is recorded before and after the deposition; before the deposition we select a target mass, M, which is kept fixed throughout the deposition.
As the deposition is completed, the features of the profile appear less smeared out, compared to the starting deposition curve. Nonetheless, the qualitative aspect of the curve is preserved. Furthermore, after deposition, an increase in the M/2M ratio was observed; the M/2M ratio shifts from 0.76 to 4.75.
A fraction of undesired particles, ca. 20%, is observed in both the particle size distribution and TEM micrograph displayed in Fig. 2c for the PtxY 7 nm sample. The intended size was ∼7 nm.
As a reference for an ideal deposition we show, in Fig. 4d, two current versus mass curves for the Ru nanoparticle. In red, the current versus mass curve taken before the deposition is shown, and in blue the current versus mass curve collected after the deposition is shown. The curves do not vary significantly. The starting value of the M/2M ratio is 24, where M is the selected mass, and the M/2M ratio slightly increases throughout the deposition. The corresponding PSD and TEM shown in Fig. 4e and f indicate a very little amount of undesired mass (about 4%) at ∼7.8 nm.
Discussion
In the first part of the “Results” section, we presented a case which highlights the dependence of the nanoparticle composition on the composition of the sputtering target. The ISS spectra confirm that, at least on the surface, the nanoparticles produced from the Pt3Y are richer in Y than the ones prepared from the Pt9Y target. Furthermore, under the assumption that the composition of the PtxY alloy is uniform throughout the particle,48 XPS after ORR show that the NPs produced from the Pt3Y target are richer in Y than the NPs produced from the Pt9Y. This is similar to what was observed by Gan et al. in an earlier study on PtxNi nanoparticles.58As we proposed recently,48 the presence of Y in the Pt lattice induces a compressive surface strain in the Pt overlayer of the NP (Fig. 1a), destabilising OH intermediates of the ORR. However, a Pt/Y ratio different from the optimum one might change the amount of the binding energy shift, resulting in a less efficient catalyst.
Besides particle chemical composition, other parameters must be kept under control. In Fig. 2 we displayed two entirely different morphologies of PtxY nanoparticles obtained by sputtering a Pt9Y target. The conditions of operation were totally different, though. To produce dendritic nanoparticles both the Ar flux and the power employed were around five times to one order of magnitude higher than those required to produce spherical nanoparticles. This provided a much higher rate of sputtering for the dendritic case than for the spherical case, which resulted in a higher rate of aggregation, as discussed by Alayan et al.59
Importantly, these high sputtering rate conditions are imposed by the choice of positive particle deposition for the dendritic case. Negative particle deposition requires substantially lower sputtering rate, and therefore results in spherical particle production, rather than dendrite formation.
Different morphologies showed different reactivity in the electro-reduction of oxygen and also different corrosion resistance, with the spherical particles losing about a half in activity compared to the dendritic particles. Tentatively, the reduced corrosion resistance for the dendritic nanoparticles might be ascribed to easier diffusion of yttrium out of the dendritic nanostructures. Furthermore, the dendritic structure might modify the extent of the Pt lattice strain, thereby affecting the activity.
Besides morphology and chemical composition of the particle, one should also concentrate on monodispersion. The peaks in the trimodal distribution showed in Fig. 3 are located at particle diameters which have ratios of (11.2/8.75) = 1.28 and (13/8.75) = 1.48, respectively. These values are remarkably close, within the error bar, to the cube root of 2 (∼1.26), and to the cube root of 3 (∼1.45). This suggests that particles with mass double or triple of the intended mass were also deposited. TEM images shown in Fig. 3 and 4 confirm that double or triple mass particles were deposited.
This is a serious problem which can occur while depositing clusters and nanoparticles synthesized using sputtering and aggregation techniques. The presence of significant amounts of nanoparticles with double or triple masses may completely compromise the result of a catalytic experiment.
Masses are effectively filtered through a Von Issendorff type Time of Flight (TOF) mass filter, however, the chance for the deposition of undesired masses is not zero. Masses which are multiples according to an integer n of the intended mass M can also be filtered; all that is needed is that their charge e is a multiple according to the same integer n, so that the mass to charge ratio is equal to the desired one. The relevant quantity here is the mass to charge ratio: 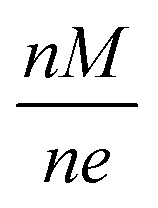 .
.
The presence of multiple negative charges (particularly with n < 4) is not unlikely for nanoparticles with diameter in the range 1–10 nm. This is due to the presence of a Coulomb barrier inhibiting the loss of electrons, as discussed by other authors.60,61 However, it seems that charges higher than 3e do not represent a serious threat for the deposition. This is corroborated by the fact that, in Fig. 3, the quadruple mass peak at 9 × 1.59 = 14.3 nm is really negligible compared to the other peaks.
Nanoparticles with a charge higher than 3e would probably rapidly discharge through the electron tunnelling mechanism. However, double (n = 2) or even triple negative charges (n = 3) can be retained by the nanoparticle for time intervals long enough to represent a serious problem in the particle mass selection process.
The case presented in Fig. 4a–c represents an acceptable case of deposition.
The deposition starts at 0.76 M/2M. This would be a disastrous ratio to start with, and a majority of 2M should be deposited. However, the ratio changes during the deposition, and the final ratio is 4.75. This prevents a large quantity of 2M from passing through the mass filter.
The reference case presented in Fig. 4d–f represents the ideal case of deposition. The M/2M ratio is very high (>20) compared to Fig. 4a, and remains high throughout the deposition. As a consequence, very few 2M are filtered through the time of flight analyzer. The particles filtered are of a different element (Ru) compared to the ones shown in Fig. 4a–c, to stress the generality of the concept.
In summary, great care should be taken in choosing deposition profiles (deposition current versus deposited mass curves) which present an M/2M ratio as high as possible. The outcome of the deposition should be checked at the end of the deposition, to ensure that such a ratio has not changed significantly, compared to the starting curve.
Deposition curves offer a fast and reliable diagnostic method to ensure that the deposition was performed correctly.
If the ratio of M/2M stays at high values (>20 at least) throughout the deposition, one can be confident the amount of multiple masses will be minimized. This threshold value is expected to vary with particle size, resulting in larger particles (diameter > 5 nm) being the most exposed to this kind of problem.
To achieve a high M/2M ratio, two phenomena should be avoided: (i) the shift of deposition minima and (ii) the appearance of new minima, compared to the pre-deposition curves. These two phenomena can mar the deposition outcome by changing the M/2M ratio beyond the allowed threshold.
Interestingly, we have noticed that in the cluster source we use, a potentially dangerous shift in the deposition minimum can occur as a result of a variation in pressure in the aggregation chamber. In Fig. 5b, three deposition curves taken at 15 minutes time from each other are shown. In this case, the pressure at the “Exit Aperture” (Fig. 5a) was kept constant by regulating the flow of He/Ar sputter gas with a feedback loop. The deposition curves show no significant change over a time that is comparable with a typical deposition time. Therefore, we can conclude that by keeping the pressure constant, depositions which are more reliable can be achieved.
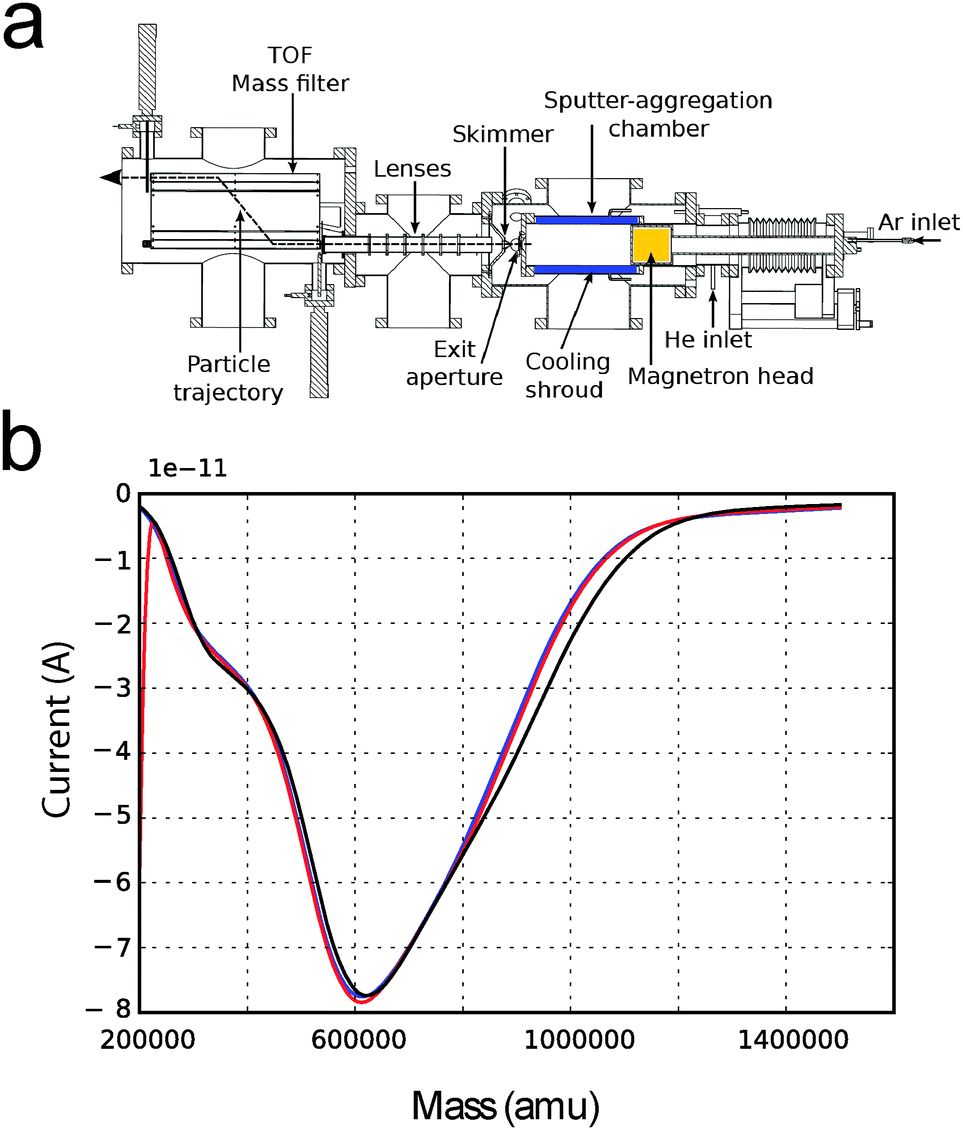 | ||
| Fig. 5 (a) Scheme of the Birmingham cluster source used to deposit size selected nanoparticles. (b) Current versus mass deposition curves taken keeping the pressure in the sputter-aggregation chamber constant. The three deposition curves were collected at time intervals of 15 minutes. | ||
We note that the double or triple mass problem presented here might be, in rare cases, caused by other factors such as sudden changes in the plasma state or abrupt change in the temperature of the sputter-aggregation chamber.
Conclusions
Using the magnetron sputtering and gas aggregation technique, we have shown how composition and morphology influence the catalytic activity of PtxY nanoparticles. The interplay between microscopic, spectroscopic techniques and nanoparticle production is of paramount importance, if the catalytic properties of nanoparticles are to be correctly understood and predicted.We have analysed the deposition curves (deposition current versus deposited mass) for size-selected nanoparticles produced using magnetron sputtering techniques. We proposed a simple protocol to gain feedback regarding whether the depositions contain negligible or intolerable amounts of double/triple mass. Although here we have focussed on PtxY nanoparticles, our approach is general and could be applied to any particle produced using this method.
Acknowledgements
The authors gratefully acknowledge financial support from the Danish Ministry of Science's UNIK initiative, Catalysis for Sustainable Energy. The Center for Individual Nanoparticle Functionality is supported by the Danish National Research Foundation (DNRF54). The A. P. Møller and Chastine Mc-Kinney Møller Foundation is gratefully acknowledged for its contribution towards the establishment of the Centre for Electron Nanoscopy in the Technical University of Denmark. The Interdisciplinary Centre for Electron Microscopy (CIME) at EPFL is gratefully acknowledged for the use of the FEI Tecnai Osiris TEM. Support for this work was received from the Danish Council for Strategic Research's project NACORR (12-133817) and MEDLYS (10-093906). I.E.L.S. was supported by the ForskEL programme's project CATBOOSTER (2001-1-10669). D.N.M. is the recipient of a HC Ørsted postdoctoral fellowship.References
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651–4654 CrossRef CAS PubMed.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222–234 CrossRef CAS PubMed.
- A. Tuxen, S. Carenco, M. Chintapalli, C.-H. Chuang, C. Escudero, E. Pach, P. Jiang, F. Borondics, B. Beberwyck, A. P. Alivisatos, G. Thornton, W.-F. Pong, J. Guo, R. Perez, F. Besenbacher and M. Salmeron, J. Am. Chem. Soc., 2013, 135, 2273–2278 CrossRef CAS PubMed.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2013, 303, 31–40 CAS.
- M. Nesselberger, M. Roefzaad, R. Faysal Hamou, P. Ulrich Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 3712, 1–6 Search PubMed.
- S. Kunz, K. Hartl, M. Nesselberger, F. F. Schweinberger, G. Kwon, M. Hanzlik, K. J. J. Mayrhofer, U. Heiz and M. Arenz, Phys. Chem. Chem. Phys., 2010, 12, 10288–10291 RSC.
- J. Gavnholt and J. Schiøtz, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035404 CrossRef.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282–290 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed.
- F. Behafarid, L. K. Ono, S. Mostafa, J. R. Croy, G. Shafai, S. Hong, T. S. Rahman, S. R. Bareb and B. Roldan Cuenya, Phys. Chem. Chem. Phys., 2012, 14, 11766–11779 RSC.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS PubMed.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef CAS PubMed.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- S. B. Vendelbo, M. Johansson, J. H. Nielsen and I. Chorkendorff, Phys. Chem. Chem. Phys., 2011, 13, 4486–4493 RSC.
- S. B. Vendelbo, M. Johansson, D. J. Mowbray, M. P. Andersson, F. Abild-Pedersen, J. H. Nielsen, J. K. Nørskov and I. Chorkendorff, Top. Catal., 2010, 53, 357–364 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS PubMed.
- S. W. Lee, S. Chen, W. Sheng, N. Yabuuchi, Y.-T. Kim, T. Mitani, E. Vescovo and Y. Shao-Horn, J. Am. Chem. Soc., 2009, 131, 15669–15677 CrossRef CAS PubMed.
- J. P. Greeley, J. Rossmeisl, A. Hellman and J. K. Nørskov, Z. Phys. Chem., 2007, 221, 1209–1220 CrossRef CAS.
- G. A. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141, 909–913 CrossRef CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- W. Sheng, S. Chen, E. Vescovo and Y. Shao-Horn, J. Electrochem. Soc., 2012, 159, B96–B103 CrossRef CAS PubMed.
- A. Kuzume, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2007, 599, 333–343 CrossRef CAS PubMed.
- A. Hitotsuyanagi, M. Nakamura and N. Hoshi, Electrochim. Acta, 2012, 82, 512–516 CrossRef CAS PubMed.
- A. S. Bandarenka, H. A. Hansen, J. Rossmeisl and I. E. L. Stephens, Phys. Chem. Chem. Phys., 2014, 16, 13625–13629 RSC.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 CAS.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750–3756 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- C. H. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- H. L. Xin, J. A. Mundy, Z. Liu, R. Cabezas, R. Hovden, L. F. Kourkoutis, J. Zhang, N. P. Subramanian, R. Makharia, F. T. Wagner and D. A. Muller, Nano Lett., 2011, 12, 490–497 CrossRef PubMed.
- S. Chen, H. A. Gasteiger, K. Hayakawa, T. Tada and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A82–A97 CrossRef CAS PubMed.
- L. Dubau, F. Maillard, M. Chatenet, J. André and E. Rossinot, Electrochim. Acta, 2010, 56, 776–783 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Grønbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, L. Bech and I. Chorkendorff, ChemCatChem, 2012, 4, 341–349 CrossRef CAS.
- T. P. Johansson, E. T. Ulrikkeholm, P. Hernandez-Fernandez, P. Malacrida, H. A. Hansen, A. S. Bandarenka, J. K. Nørskov, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Top. Catal., 2014, 57, 245–254 CrossRef CAS.
- P. Malacrida, M. Escudero-Escribano, A. Verdaguer-Casadevall, I. E. L. Stephens and I. Chorkendorff, J. Mater. Chem. A, 2014, 2, 4234–4243 CAS.
- T. P. Johansson, E. T. Ulrikkeholm, P. Hernandez-Fernandez, M. Escudero-Escribano, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, Phys. Chem. Chem. Phys., 2014, 16, 13718–13725 RSC.
- P. Hernandez-Fernandez, F. Masini, D. N. McCarthy, C. E. Strebel, D. Friebel, D. Deiana, P. Malacrida, A. Nierhoff, A. Bodin, A. M. Wise, J. H. Nielsen, T. W. Hansen, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, Nat. Chem., 2014 DOI:10.1038/nchem.2001.
- V. Komanicky, H. Iddir, K.-C. Chang, A. Menzel, G. Karapetrov, D. Hennessy, P. Zapol and H. You, J. Am. Chem. Soc., 2009, 131, 5732–5733 CrossRef CAS PubMed.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 3588–3591 CrossRef CAS PubMed.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- J. X. Wang, C. Ma, Y. Choi, D. Su, Y. Zhu, P. Liu, R. Si, M. B. Vukmirovic, Y. Zhang and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 13551–13557 CrossRef CAS PubMed.
- L. Dubau, M. Lopez-Haro, L. Castanheira, J. Durst, M. Chatenet, P. Bayle-Guillemaud, L. Guétaz, N. Caqué, E. Rossinot and F. Maillard, Appl. Catal., B, 2013, 142–143, 801–808 CrossRef CAS PubMed.
- H. Haberland, M. Karrais, M. Mall and Y. Thurner, J. Vac. Sci. Technol., A, 1992, 10, 3266–3271 CAS.
- B. von Issendorff and R. E. Palmer, Rev. Sci. Instrum., 1999, 70, 4497–4501 CrossRef CAS PubMed.
- F. Maillard, E. R. Savinova and U. Stimming, J. Electroanal. Chem., 2007, 599, 221–232 CrossRef CAS PubMed.
- M. P. S. D. Briggs, Practical Surface Analysis, Auger and X-ray Photoelectron Spectroscopy, 1996 Search PubMed.
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423–5430 CrossRef CAS PubMed.
- R. Alayan, L. Arnaud, M. Broyer, E. Cottancin, J. Lermé, J. L. Vialle and M. Pellarin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 125444 CrossRef.
- R. N. Compton, A. A. Tuinman, C. E. Klots, M. R. Pederson and D. C. Patton, Phys. Rev. Lett., 1997, 78, 4367–4370 CrossRef CAS.
- C. Yannouleas and U. Landman, Chem. Phys. Lett., 1994, 217, 175–185 CrossRef CAS.
Footnote |
| † Present address: DanishTechnological Institute (DTI), Gregersensvej 1, DK-2630 Taastrup, Denmark. |
| This journal is © the Owner Societies 2014 |