
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-color delay dependent IR probing of torsional isomerization in a [AgL1L2]+ complex†
Johannes
Lang
,
Maximilian
Gaffga
,
Fabian
Menges‡
and
Gereon
Niedner-Schatteburg
Fachbereich Chemie and Forschungszentrum OPTIMAS, Technische Universität Kaiserslautern, 67663 Kaiserslautern, Germany
First published on 11th July 2014
Abstract
Two-color infrared multiple photon dissociation (2c-IR-MPD) spectroscopy with delayed pulses indicates a torsional isomerization in a “ligand–metal–chelate” complex [AgL1L2]+. Ab initio calculations reveal the torsional barrier as well as the change in vibrational frequencies and IR intensities along the isomerization pathway. The current approach bears prospects for further elucidation of competing interactions within naked or microsolvated complexes in gas phase coordination chemistry.
Metal ions stabilize through reversible solvation or persistent coordination. In appropriate mixtures, ligand exchange drives equilibria towards the formation of complexes with as many strongly coordinating ligands as possible within steric and electronic constraints.
Mass produced chelating agents like ethylenediaminetetraacetic acid (EDTA)1 serve in the extraction of heavy metal ions from waste waters and in alkaline earth metal ion scavenging for water softening on a large scale.2,3 Environmental issues4 call for biodegradability (as e.g. fulfilled by Trilon M®5,6) and for further optimization of the chelation processes.7 The finely tuned interplay between ligand-to-ligand and ligand-to-solvent interactions (including hydrogen bonding) plays a crucial role in the reversible stabilization of metal ions within such chelated complexes.8,9 It is paramount to acquire insight into their structure and vibrational dynamics in order to advance our understanding of the dominating processes and the prevailing equilibria.
The combination of mass spectrometry with infrared (IR) laser spectroscopy is suitable for the characterization of stoichiometrically defined coordination complexes, and recent two-color double resonance studies proved to be instrumental in obtaining isomer sensitivity of various gas phase complexes and clusters.10–14 Experimental spectra recorded in such solvent-free environments correspond to ab initio calculations of the isolated species, thus yielding insight into the structure and dynamics of likely binding motifs. We have recently applied a two-color IR-MPD detection scheme to study an isolated di-nuclear silver complex which spontaneously forms in solution by coordination with nucleobase mimicking ligands.15
Here, we investigate a flexible arrangement of a single Ag(I) center with two coordinating ligands, α-cyano-4-hydroxycinnamic acid (HCCA = L1, monodentate ligand) and 2-(5-methyl-1H-pyrazol-3-yl)pyridine16 (MPP = L2, bidentate chelate), one each. The resulting [AgL1L2]+ complex serves as a model for a ligand-to-chelate hydrogen bonding within a “ligand–metal–chelate” complex. A possible torsional isomerization (cf.Scheme 1) is the subject of verification and characterization by this study.
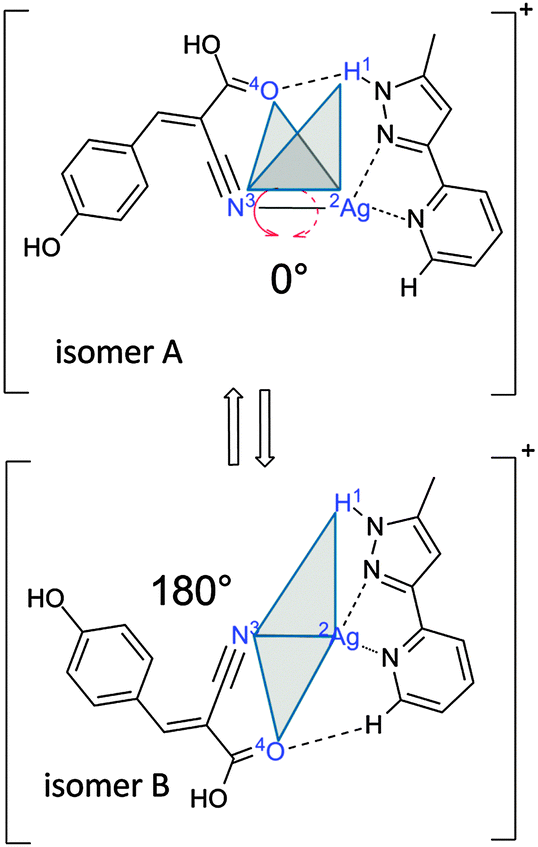 | ||
| Scheme 1 Definition of the dihedral angle that describes the torsional isomerization in the [AgL1L2]+ complex. Note, that isomer A stabilizes through a NH–O hydrogen bond and isomer B through a CH–O hydrogen bond. | ||
A sample solution of AgNO3, HCCA and MPP17 in acetonitrile at a concentration of 1 × 10−5 M was used without further purification and continuously infused into an electrospray ion trap instrument (Bruker amaZon SL, cf. Supplement for further experimental details). We deduced the formation of the self-assembled [AgL1L2]+ complex from the signal of a cation recorded at m/z = 455 and 457 (due to the 107Ag and 109Ag isotopes). The isotopic pattern clearly proves a complexation of a Ag(I) ion (cf. Fig. S1, middle inset, ESI†) by neutral L1 and L2 ligands leading to a singly charged ligand–metal–chelate complex. Collision induced dissociation inside the ion trap exhibits the exclusive loss of the monodentate L1 ligand (cf. Fig. S1, top inset, ESI†).
We employed IR-MPD spectroscopy in the 1200–3800 cm−1 range on the [AgL1L2]+ complex (cf. top of Fig. 1) using a tunable high power OPO/OPA IR laser (IRscan, LaserVision, cf. SI. 2 and SI. 3 for further experimental details, ESI†). The observed bands can be assigned by comparison with DFT derived vibrational spectra: OH stretching vibration of the phenolic OH group (ṽPh![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) = 3639 cm−1), OH stretching vibration of the carboxyl group (ṽCO =3581 cm−1), free NH stretching vibration of the L2 chelate (ṽNH = 3490 cm−1) and hydrogen bonded NH stretching vibration of the L2 chelate (ṽH-boundNH at around 3400 cm−1), aliphatic CH stretching vibrations (ṽaliph.CH at around 2960 cm−1), C
= 3639 cm−1), OH stretching vibration of the carboxyl group (ṽCO =3581 cm−1), free NH stretching vibration of the L2 chelate (ṽNH = 3490 cm−1) and hydrogen bonded NH stretching vibration of the L2 chelate (ṽH-boundNH at around 3400 cm−1), aliphatic CH stretching vibrations (ṽaliph.CH at around 2960 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations (ṽCC = 1600 cm−1) as well as the carboxylic CO stretching band in L1 (ṽ
C vibrations (ṽCC = 1600 cm−1) as well as the carboxylic CO stretching band in L1 (ṽ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OH at around 1780 cm−1). A spurious dip in the ṽPh band at 3630 cm−1 is an artefact of known fluctuations in the IRscan laser power. The simultaneous occurrence of a free and a hydrogen bonded NH stretching vibration cannot result from a single isomer, since there is only one NH group in the [AgL1L2]+ complex. This indicates the coexistence of at least two isomers in the gas phase. The ṽCO band at 3581 cm−1 is red shifted with respect to the calculation which is explained by a higher anharmonicity of the carboxylic ṽCO relative to the phenolic ṽPh.18–20
OH at around 1780 cm−1). A spurious dip in the ṽPh band at 3630 cm−1 is an artefact of known fluctuations in the IRscan laser power. The simultaneous occurrence of a free and a hydrogen bonded NH stretching vibration cannot result from a single isomer, since there is only one NH group in the [AgL1L2]+ complex. This indicates the coexistence of at least two isomers in the gas phase. The ṽCO band at 3581 cm−1 is red shifted with respect to the calculation which is explained by a higher anharmonicity of the carboxylic ṽCO relative to the phenolic ṽPh.18–20
 | ||
| Fig. 1 IR-MPD spectrum of the [AgL1L2]+ complex (green, L1 loss recorded) in comparison with the DFT calculations at the B3LYP/cc-pVTZ (H, C, N, and O) and Stuttgart (1997) ECP (Ag) level of theory (black). Frequencies are scaled with the factor 0.96 (1.0) above (below) 2000 cm−1. Intensities are multiplied by the photon energy (in cm−1) for normalization. | ||
Minimum energy structures, relative energies and linear IR spectra were calculated at the B3LYP21–24/cc-pVTZ25 level of theory as implemented in the Gaussian 09 program package.26 The Stuttgart–Dresden effective core potential basis set was used to represent the Ag atom.27 Harmonic vibrational frequencies were scaled by the factor 0.96 (1.0) for the stretching modes above (below) 2000 cm−1 to match the most intense experimental bands (e.g. ṽPh at 3639 cm−1). Calculated intensities were multiplied by the photon energy (in cm−1) for normalization. We found at least four energetically favored coordination motifs in [AgL1L2]+ (cf.Fig. 1, structures A–D). Structure A is the energetically most favorable. Energies of structures B–D are given relative to the energy of structure A. Within the structures A and B, the silver ion is coordinated by the nitrile group of the L1 ligand and two nitrogen atoms of the L2 chelate. Structure A allows for the formation of a NH–O hydrogen bond (d(![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) H–) = 3.011 Å, d(N–) = 2.035 Å) between the NH group of L2 and the carboxyl group of L1. Structures A and B are rotational conformers, where L1 and L2 twist against each other by about 180°. This rotation allows for the formation of a new CH–O hydrogen bond (d(H–) = 4.058 Å, d(C–) = 2.989 Å) between a pyridine CH in L2 and the carboxyl group of L1. The computed bond distances are only slightly above typical values of similar bands in the solid state.28 Nevertheless, the CH–O hydrogen bonded structure B is less stable by 8 kJ mol−1 than the NH–O hydrogen bonded structure A. In isomers C and D, the silver ion is coordinated by the same two nitrogen atoms of L2 as in A and B, whereas – from the L1 point of view – the terminal oxygen atom of the carboxyl group serves as an electron donor in C and D, instead of the nitrile group in isomers A and B. A mere rotation by 180° transforms structure C (21 kJ mol−1) into structure D (33 kJ mol−1), which makes them mutual rotamers.
H–) = 3.011 Å, d(N–) = 2.035 Å) between the NH group of L2 and the carboxyl group of L1. Structures A and B are rotational conformers, where L1 and L2 twist against each other by about 180°. This rotation allows for the formation of a new CH–O hydrogen bond (d(H–) = 4.058 Å, d(C–) = 2.989 Å) between a pyridine CH in L2 and the carboxyl group of L1. The computed bond distances are only slightly above typical values of similar bands in the solid state.28 Nevertheless, the CH–O hydrogen bonded structure B is less stable by 8 kJ mol−1 than the NH–O hydrogen bonded structure A. In isomers C and D, the silver ion is coordinated by the same two nitrogen atoms of L2 as in A and B, whereas – from the L1 point of view – the terminal oxygen atom of the carboxyl group serves as an electron donor in C and D, instead of the nitrile group in isomers A and B. A mere rotation by 180° transforms structure C (21 kJ mol−1) into structure D (33 kJ mol−1), which makes them mutual rotamers.
The experimental one-color IR-MPD spectrum can be explained by a mixture of isomers A and B. There is no evidence of isomers C and D, which would exhibit a red-shifted CO stretching vibration due to the coordination of the Ag(I) ion to the terminal carboxyl oxygen atom. The ṽPh and ṽCO bands, corresponding to free OH and COOH stretching motions, result from both isomers A and B. The hydrogen bonded NH stretching vibration ṽH-boundNH exclusively results from isomer A, while the NH stretching band ṽNH is indicative of isomer B. The predicted intensity of ṽH-boundNH is much higher than observed, which is a known finding in hydrogen bonding that broadens and smears out the affected vibrations. Observed bands in the CO stretching region at around 1800 cm−1 correlate well with those of isomers A and B. The recorded spectra do not reveal the predicted strong bands (at around 1640–1650 cm−1) of the high energy isomers C and D. It is a pending task to record two-color IR-MPD spectra in the fingerprint region below 2000 cm−1 and to perform an in-detail interpretation. However, it is already warranted at this stage that the current findings on [AgL1L2]+ conclude in the coexistence of isomers A and B most likely in the absence of C and D.
We investigated by further DFT calculations the torsional conversion of isomer A to B through parametrical variation of an appropriate dihedral angle (between H1Ag2N3O4, cf.Scheme 1), which can be thought of as the intersection angle of the two planes spanned by H1Ag2N3 and Ag2N3O4 atoms. Our calculations performed full structural relaxation at fixed dihedral angles in steps of 10° with closer intervals of 4° around the minima at 0° and 180° in order to extract pointwise values of relative energies, vibrational frequencies and IR intensities (cf.Fig. 2, from bottom to top).
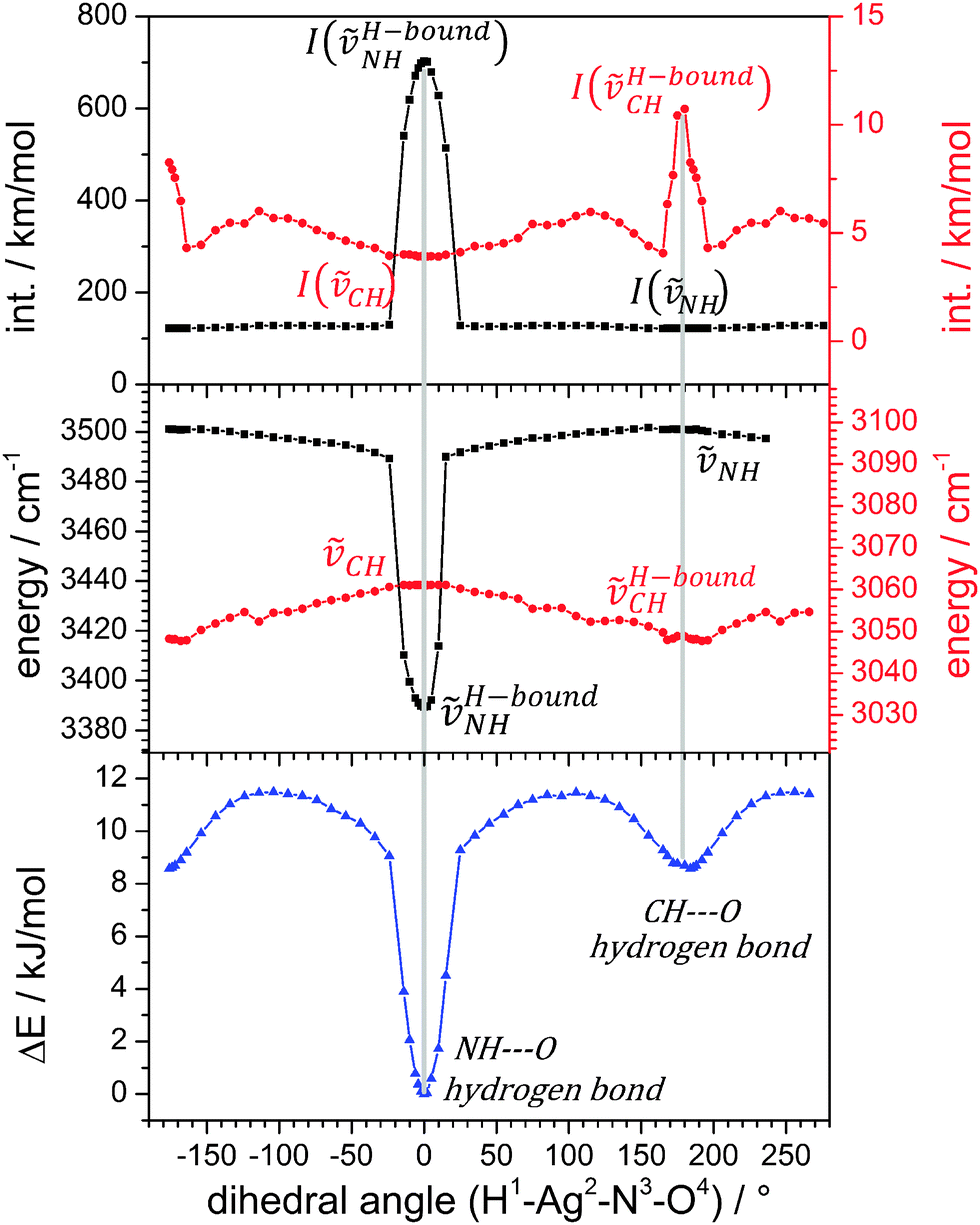 | ||
| Fig. 2 Total energy of torsional twisting in the [AgL1L2]+ complex (bottom), frequencies of hydrogen bonding vibrations (middle) and their IR intensities (top) derived by DFT calculations at the B3LYP/cc-pVTZ (H, C, N, O) and Stuttgart (1997) RSC ECP (Ag) level of theory. Formation and breaking of the hydrogen bonds by torsional isomerization leaves marks in all three diagrams. Note that the angular width of the strong NH–O bond is narrow while that of the weaker CH–O bond is wider. | ||
Isomers A and B are well confirmed by the energetic minima at 0° and 180° (cf.Fig. 2, bottom), and we find a higher (lower) torsional stiffness in the OH (CH) hydrogen bonded more (less) stable isomer A (B) with a torsional barrier of 12 kJ mol−1. The majority of vibrational modes are decoupled from the torsional isomerization and their frequencies persist without a significant change in the geometries of isomers A and B and – most notably – at all dihedral angles in between. However, there are considerable red shifts of those vibrational bands that are involved in hydrogen bonding, and concomitant variations of their IR intensities. At a dihedral angle of 0°, isomer A is the favorable structure and ṽH-boundNH is red shifted due to the hydrogen bond and its IR intensity is strongly enhanced. At 180°, isomer B, the pyridinic CH group is involved in a CH–O hydrogen bond. There are at least four IR active normal modes of the pyridine ring that involve this CH group. We chose to plot the mode with the strongest change in IR intensities (cf. Fig. S2 for its definition and Fig. S3 for torsional plots of IR intensity changes in other CH stretching modes, ESI†). Its frequency shifts slightly to the red upon hydrogen bonding while its IR intensity increases considerably. All findings of the parametric torsional variation nicely confirm strong NH–O hydrogen bonding in isomer A and weak CH–O hydrogen bonding in isomer B.
In order to investigate the predicted interconversion of isomers A and B experimentally, we applied the two-color IR-MPD technique to the [AgL1L2]+ complex. An attenuated IR laser (IRfix) is set resonant to ṽPh at 3639 cm−1, and a high power scanning IR laser (IRscan) was used to measure delay dependent IR-MPD spectra (cf.Fig. 3a). The time delay, Δt, between the two laser pulses (6 ns each) is defined as:
| Δt = t(IRfix) − t(IRscan) | (1) |
It depends on the time delay Δt between the IRscan and IRfix pulses whether the hydrogen bound NH stretching band at around 3400 cm−1 is observable or not. Note, that the application of a second laser pulse causes some increase of the baselines in the two-color IR-MPD spectra as compared to the one-color spectrum.
 | ||
| Fig. 3 (a) Details from the infrared spectra of stretching vibrations in the [AgL1L2]+ complex. The delay dependent two-color IR-MPD spectra with selected time delay reveal heating effects and/or isomerization. (b) Fragmentation efficiency at the peak of the ṽNH band (filled squares) and the ṽH-boundNH band (empty squares) in dependence of the time delay Δt = t(IRfix) − t(IRscan). A step-like switch amongst both vibrations around Δt = 0 ns is clearly visible. | ||
When the IRscan pulse excites the [AgL1L2]+ complex ahead of the IRfix pulse (Δt positive, red line spectrum in Fig. 3a) then there is some increase in the IR-MPD fragmentation yield with respect to the one-color spectrum (black curve) on all bands. In this case, the IRfix pulse serves to enhance the absolute fragmentation yield. It does not, however, alter the isomer population as recorded by the one-color experiment. The two-color spectrum thus probes “cold” (here: room temperature) complexes. In particular, the hydrogen bonded NH vibration (ṽH-boundNH) becomes strongly enhanced and clearly observable.
However, when the time delay reverses (Δt negative, blue curve in Fig. 3a), such that the IRfix pulse excites first, the IR-MPD yield of ṽH-boundNH diminishes and the intensity of ṽNH, the free NH vibration, increases with respect to the two-color IR-MPD spectrum with positive Δt. A pointwise scan of Δt reveals a fast switch amongst both vibrations (cf.Fig. 3b). We interpret this finding as follows: firstly, the IRfix laser heats the complexes and induces an isomerization such that the population of isomer A (evidenced by the hydrogen bonded ṽH-boundNH vibration) is diminished and the population of isomer B (free ṽNH vibration) is enhanced. The possibility of some isomers at skewed dihedral angles, which are void of any L1 to L2 bonding, is acknowledged; however, present results do not find any evidence. Subsequently, the changed isomer population is detected by the IRscan laser. This interpretation matches the observation of a significant broadening and red shifting of the three recorded stretching vibrations, as known to occur from a rovibrational preheating of part of the [AgL1L2]+ ensemble by the IRfix pulse – prior to the scanning IRscan pulse.
Our study revealed that strong and weak hydrogen bonds29–31 within a given complex may break and form by torsional rearrangements that take place perpendicular to the hydrogen bond directions (here, through torsional twisting of the two ligands against each other). It confirms that the forming and breaking of hydrogen bonds is of a multidimensional character – much beyond an intuitive pulling apart of a hydrogen bond along its bond direction. It further elucidates how weak CH–O hydrogen bonds may come into play to stabilize conformers other than the global minimum structure. The importance of torsional rearrangements becomes clear once more by the gained picture, as was similarly unravelled in a previous study on intraligand torsion in a Ru complex that precedes and enables activation of the catalytic center in a somewhat surprising way.16 Our studies benefit from two-color IR pulse probing as a valuable tool for such investigations (two-color IR-MPD). Variable time delays between the two laser pulses allow us to probe ion ensembles with variable internal energies. The preheating effect of the additional IRfix pulse could be demonstrated in terms of laser induced torsional isomerization as well as a red shifting and broadening of the IR-MPD bands. The torsional isomerization path was examined by concomitant DFT calculations that served to elucidate the torsional barrier, the relative stabilities of participating isomers, and the change of vibrational frequencies and IR intensities in the course of formation and breaking of strong and weak hydrogen bonds amongst chelates and ligands in a semiflexible ligand–metal–chelate complex.
The present study presents the work in progress. Δt tuning two-color IR-MPD spectra of the ṽaliph.CH range (at around 3000 cm−1) and in the fingerprint region (below 2000 cm−1) as well as temperature dependent measurements will help to provide more insight into the details of population dynamics in the [AgL1L2]+ model system. Further complexes are to follow in order to learn more about the competing interactions of strong coordination and weak auxiliary bonds in multiple coordinated metal complexes.
Acknowledgements
This work was supported by the German research foundation DFG within the transregional collaborative research center SFB/TRR 88 “Cooperative effects in homo and heterometallic complexes” (3MET) and by the state research center OPTIMAS. We acknowledge valuable suggestions by the referees and conceptual discussions by Otto Dopfer.References
- M. S. Vohra, Int. J. Environ. Sci. Technol., 2010, 7, 687–696 CrossRef.
- L. Suarez, M. A. Diez, R. Garcia and F. A. Riera, Sep. Purif. Technol., 2013, 118, 144–150 CrossRef CAS PubMed.
- D. Kołodyńska, Desalination, 2011, 267, 175–183 CrossRef PubMed.
- P. Römkens, L. Bouwman, J. Japenga and C. Draaisma, Environ. Pollut., 2002, 116, 109–121 CrossRef.
- J. Jachuła, D. Kołodyńska and Z. Hubicki, Chem. Eng. Res. Des., 2012, 90, 1671–1679 CrossRef PubMed.
- J. Jachuła, D. Kołodyńska and Z. Hubicki, Cent. Eur. J. Chem., 2011, 9, 52–65 CrossRef.
- P. Muller, Pure Appl. Chem., 1994, 66, 1077 CrossRef.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed.
- K. Biradha, G. R. Desiraju, D. Braga and F. Grepioni, Organometallics, 1996, 15, 1284–1295 CrossRef CAS.
- C. M. Leavitt, A. B. Wolk, J. A. Fournier, M. Z. Kamrath, E. Garand, M. J. Van Stipdonk and M. A. Johnson, J. Phys. Chem. Lett., 2012, 3, 1099–1105 CrossRef CAS.
- T. R. Rizzo, J. A. Stearns and O. V. Boyarkin, Int. Rev. Phys. Chem., 2009, 28, 481–515 CrossRef CAS.
- A. Fujii and K. Mizuse, Int. Rev. Phys. Chem., 2013, 32, 266–307 CrossRef CAS.
- N. Heine, M. R. Fagiani, M. Rossi, T. Wende, G. Berden, V. Blum and K. R. Asmis, J. Am. Chem. Soc., 2013, 135, 8266–8273 CrossRef CAS PubMed.
- K. Tanabe, M. Miyazaki, M. Schmies, A. Patzer, M. Schutz, H. Sekiya, M. Sakai, O. Dopfer and M. Fujii, Angew. Chem., Int. Ed., 2012, 51, 6604–6607 CrossRef PubMed.
- Y. Nosenko, F. Menges, C. Riehn and G. Niedner-Schatteburg, Phys. Chem. Chem. Phys., 2013, 15, 8171–8178 RSC.
- L. Taghizadeh Ghoochany, C. Kerner, S. Farsadpour, F. Menges, Y. Sun, G. Niedner-Schatteburg and W. R. Thiel, Eur. J. Inorg. Chem., 2013, 4305–4317 CrossRef CAS.
- K. Muller, Y. Sun, A. Heimermann, F. Menges, G. Niedner-Schatteburg, C. van Wüllen and W. R. Thiel, Chem. – Eur. J., 2013, 19, 7825–7834 CrossRef CAS PubMed.
- G. M. Florio, T. S. Zwier, E. M. Myshakin, K. D. Jordan and E. L. Sibert, J. Chem. Phys., 2003, 118, 1735–1746 CrossRef CAS PubMed.
- J. L. Leviel and Y. Marechal, J. Chem. Phys., 1971, 54, 1104–1107 CrossRef CAS PubMed.
- J. Antony, G. von Helden, G. Meijer and B. Schmidt, J. Chem. Phys., 2005, 123, 014305 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Wallingford, CT, 2009.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS.
- I. Rozas, Phys. Chem. Chem. Phys., 2007, 9, 2782–2790 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02045f |
| ‡ Current address: Dept. of Chemistry, Yale University, New Haven, CT 06520, USA. |
| This journal is © the Owner Societies 2014 |