 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quaternary ammonium and phosphonium based ionic liquids: a comparison of common anions†
Rebecca K.
Blundell
and
Peter
Licence
*
The GlaxoSmithKline Carbon Neutral Laboratory, School of Chemistry, The University of Nottingham, Nottingham NG7 2RD, UK. E-mail: peter.licence@nottingham.ac.uk; Tel: +44 (0)115 8466176
First published on 11th June 2014
Abstract
A series of ammonium-based ionic liquids and their phosphonium analogues have been investigated using X-ray Photoelectron Spectroscopy (XPS). A robust C 1s peak fitting model has been developed and described for the tetraalkylphosphonium and tetraalkylammonium families of ionic liquid, with comparisons made between the two series. Cation–anion interactions have been investigated to determine the impact of changing the cationic core from nitrogen to phosphorus upon the electronic environment of the anion. Comparisons between long and short chain cationic systems are also described.
Introduction
Ionic liquids (ILs) – low temperature molten salts – have received significant interest in recent years due in part to their structural diversity.1 By simply changing the ions, physico-chemical properties such as density, viscosity, melting point and polarity etc. can be tuned for specific functions. Tuning an ionic liquid to suit a specific application has attracted much interest, with this concept being taken even further by utilising mixtures of ionic liquids.2,3 By considering binary and ternary mixtures of ionic liquids the scope for potential applications considerably broadens with some examples already shown in lithium ion batteries,4 CO2 absorption,5 gas chromatography stationary phases,6 and heavy metal extraction7 to name but a few. The ability to predict and fine-tune the properties of an ionic liquid to meet a specific requirement is one of the greatest challenges in the field, and yet if fully achieved would provide an invaluable tool. To predict the behaviour for any given ionic liquid it must first be understood on a molecular level, where changes to the chemical architecture and its effect upon cation–anion interactions must be explored both theoretically and experimentally.Over the past ten years X-ray photoelectron spectroscopy (XPS) has established itself as a robust technique to characterise ionic liquid-based systems.8 To date the majority of investigations using this technique have focused upon nitrogen-based cations, particularly dialkylimidazolium systems.8,9 XPS can be used to identify different electronic environments present for each element, as well as the determination of chemical composition and purity. So far, XPS of IL-systems has provided a wealth of information in areas such as bulk and surface composition,10,11in situ reaction monitoring,12 and cation–anion interactions.13 Cation–anion interactions, in particular, have only so far been investigated for imidazolium and pyrrolidinium cations, where the effect of the anion upon the electronic environment of the cation has been explored.13,14 Correlations of these results to NMR spectroscopy, theoretical calculations and Kamlet–Taft parameters have also been achieved.13 Furthermore, XPS has also been used to monitor the electronic environment of the constituent atoms for a binary IL mixture where the mole fraction of the anionic constituents are systematically varied, thus highlighting how changes in the anionic composition of IL mixtures can be used to tune the electronic environment of their constituents.15
A particularly interesting sub-set of ionic liquids are those based upon a tetraalkylphosphonium cation; research into these compounds has often been neglected in favour of their nitrogen-based counterparts with particular emphasis on the imidazolium and pyrrolidinium cations.16 This is likely due to the high cost and air-sensitive nature of the trialkylphosphine starting materials; nevertheless, it has been observed that tetraalkylphosphonium-based ionic liquids can offer superior physico-chemical properties in comparison to their nitrogen-based relatives.16–18 Furthermore, there are only a few systematic studies that directly investigate the effect of changing the central cationic heteroatom from nitrogen to phosphorus, i.e. tetraalkylammonium versus tetraalkylphosphonium.19–23 It has been observed that changing the cationic centre from nitrogen to phosphorus has a marked effect upon the physico-chemical properties of ILs;23 however, little is known about fundamental cation–anion interactions of such systems. The work described herein utilises XPS to probe these interactions for ILs in which the cation is varied (tetraalkylammonium versus tetraalkylphosphonium) for the same anion to examine the influence of the cationic heteroatom upon the electronic environment of the anion, with an aim to determine if a degree of tunability can be achieved via the cation.
The most important output from XPS analysis are binding energies; these provide both elemental and chemical information. Small changes in the electronic environment of an element by, for example, a change in chemical bonding or oxidation state will cause a shift in the measured binding energy much like the chemical shift in NMR spectroscopy. This allows both chemical structure and inter-ion interactions to be probed on a molecular level providing vital information on the impact of structural changes upon electronic environment and communication between ions. Accordingly, it is of great importance to obtain reliable and reproducible binding energies and a number of factors must be considered. Firstly, the possibility of surface charging must be taken into account. Ionic liquids in the solid state are poor conductors and charge neutralisation must be applied. Ionic liquids in their liquid state, however, are moderately conducting and can dissipate excess charge on the sample surface, thus charge neutralisation is not necessary. Despite this, it has been observed for imidazolium ionic liquids that surface charging can occur over time especially for viscous samples, causing subtle yet measurable shifts in binding energy.24
Secondly, charge correction must be applied using a suitable internal reference post data collection to ensure binding energies obtained are accurate and allow for reliable comparison between samples, a thorough description of charge correction methods for IL systems has recently been reported.24
Finally, development of fitting models to describe and deconstruct the electronic environment for each element is particularly important. This is especially relevant to carbon, which has been described as the most important core level for XPS of ILs.8 Due to the variety and complexity in structure of ILs, C 1s fitting models must be developed on a case-by-case basis, with the aim of developing a fitting model that deconstructs these electronic environments into the minimum number of components possible.8,14
In this work we report an investigation into the XP spectra for seven tetraalkylphosphonium-based ionic liquids with comparison to their tetraalkylammonium analogues, see Table 1. The high purity of each sample is confirmed with a description of binding energies and multi-component fitting models. Furthermore, a robust C 1s peak fitting model has been developed and described for both the tetraalkylphosphonium and tetraalkylammonium families of ionic liquid. Cation–anion interactions have been probed by investigating: (1) the effect of varying the anion upon the cation for both families of ionic liquid; (2) the effect of varying the cationic core from phosphorus to nitrogen for a variety of different anions. Comparisons are also made between small and long alkyl chain systems.
| Abbreviation | Structure | Name |
|---|---|---|
| [P6,6,6,14]+ |
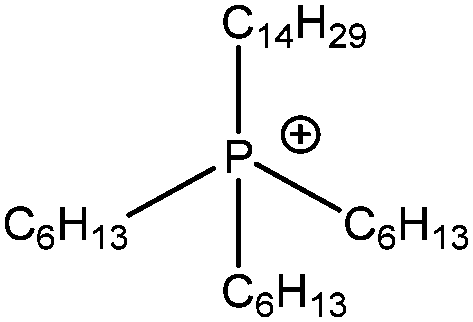
|
Trihexyl(tetradecyl)phosphonium |
| [N6,6,6,14]+ |
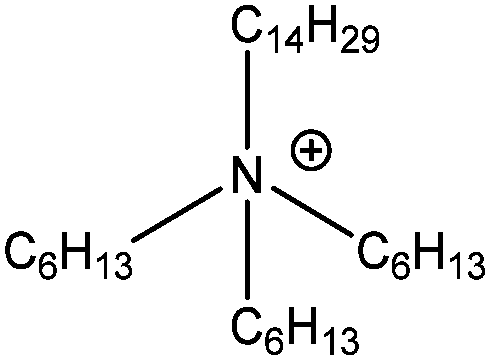
|
Trihexyl(tetradecyl)ammonium |
| [P4,4,4,1]+ |
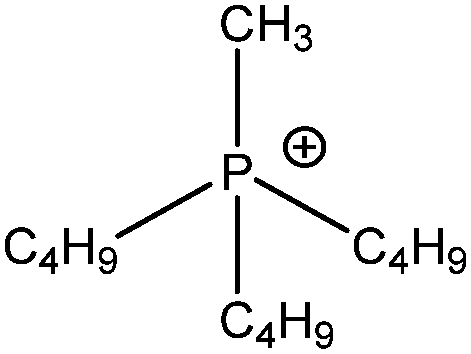
|
Tributyl(methyl)phosphonium |
| [N4,4,4,1]+ |
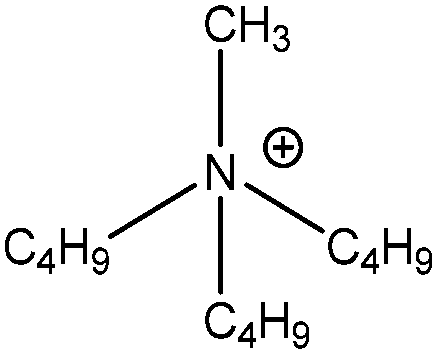
|
Tributyl(methyl)ammonium |
| [BF4]− |
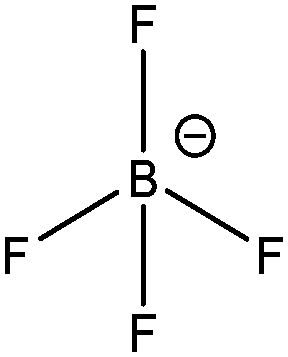
|
Tetrafluoroborate |
| [PF6]− |
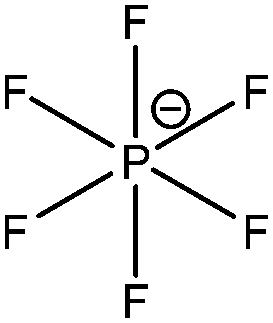
|
Hexafluorophosphate |
| [OTf]− |
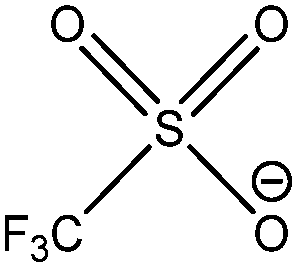
|
Trifluoromethanesulfonate |
| [NTf2]− |
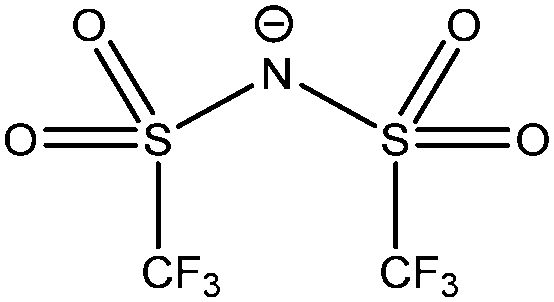
|
Bis(trifluoromethanesulfonyl)imide |
Experimental
Materials
All compounds investigated herein were prepared in our laboratory using modified literature procedures.25 A full description of each chemical synthesis is provided within the ESI.† Chemical precursors were obtained from commercial suppliers and used without further purification unless otherwise stated. Trihexyl(tetradecyl)phosphonium chloride (Cyphos IL 101) and tributyl(methyl)phosphonium methyl sulfate (Cyphos IL 108) were obtained from Cytec Industries Inc. The structures of the compounds investigated in this study are shown in Table 1. All compounds were dried in vacuo (p ≤ 10−2 mbar) at 50 °C and stored under argon before being fully characterised by 1H, 13C, 31P and 19F NMR (recorded at room temperature on a Bruker DPX-400 MHz spectrometer); IR (Perkin Elmer 1600 FT spectrometer); ESI-MS (Bruker MicroTOF 61 spectrometer) and XPS (Kratos Axis Ultra spectrometer). For syntheses which involved anion exchange chemistries, ion chromatography (Dionex ICS-3000, IonPack AS15, 4 × 250 mm analytical column) showed that residual halide was below the detection limits of the instrument (<10 ppm). Full data, including XPS spectra with peak deconstruction models, for all compounds investigated are provided within the ESI.†XPS data collection
XP spectra were recorded using a Kratos Axis Ultra spectrometer employing a focused, monochromated Al Kα source (hν = 1486.8 eV), hybrid (magnetic/electrostatic) optics, concentric hemispherical analyser, and a multi-channel plate and delay line detector (DLD) with an X-ray incident angle of 30° and a collection angle of 0° (both relative to the surface normal). X-ray source was operated at 10 mA emission current and 12 kV anode potential. All spectra were recorded using an entrance aperture of 300 × 700 mm with pass energy of 80 eV for survey scans and 20 eV for high resolution scans. The instrument sensitivity was 7.5 × 105 counts s−1 when measuring the Ag 3d5/2 photoemission peak for a clean Ag sample recorded at pass energy of 20 eV and 450 W emission power. Ag 3d5/2 full width at half maximum (FWHM) was 0.55 eV for the same instrument settings. Binding energy calibration was made using Au 4f7/2 (83.96 eV), Ag 3d5/2 (368.21 eV) and Cu 2p3/2 (932.62 eV). Charge neutralisation (used for solid samples only) was applied using a standard Kratos charge neutraliser consisting of a filament, coaxial with the electrostatic and magnetic transfer lenses, and a balance plate which creates a potential gradient between the neutraliser and sample. Charge neutralisation was applied at 1.9 A filament current and 3.3 V balance plate voltage. Sample stubs were earthed via the instrument stage using a standard BNC connector.The preparation method for each sample was dependent upon the nature of the material to be analysed. Liquid samples were prepared by placing a small drop (≈10 mg) of ionic liquid onto a stainless steel multi-sample bar; solid samples were fixed to the bar using double-sided adhesive tape. All samples were pre-pumped in a preparative chamber to pressures lower than 1 × 10−6 mbar before transfer into the main analytical chamber.
Surface contaminants such as oxygen and silicon were removed via argon ion bombardment using a Kratos Minibeam III ion gun using a sputter energy of 4 kV for three minutes. Solid samples that required argon bombardment were prepared by placing a small amount of solid in a stainless steel powder stub, then heating the sample until melted, followed by argon bombardment. Surface cleaning by argon bombardment was effective for all of the compounds investigated, where necessary, excluding [N6,6,6,14][Cl] and [N4,4,4,1][BF4] where irreparable damage to the sample occurred.
XPS data analysis
Data was analysed using the CASAXPS software. Relative sensitivity factors (RSF) were taken from the Kratos Library (RSF of F 1s = 1) and used to determine relative atomic percentages from high-resolution scans of the most intense photoelectron peak for each element. Peak areas were measured after performing a two-point linear or Shirley background subtraction. Peaks were fitted using GL(30) lineshapes (70% Gaussian, 30% Lorentzian). For compounds with a long alkyl chain (n ≥ 8) charge-referencing was achieved by setting the experimentally determined binding energy of the aliphatic carbon component (Caliphatic) equal to 285.0 eV.14,24 For compounds with a short alkyl chain (n < 8) charge-referencing was achieved by setting the measured binding energy of the cationic heteroatom photoemission peak equal to that for its long chain analogue, i.e. Ncation 1s for [N4,4,4,1][BF4] was set equal to 402.3 eV (where 402.3 eV is the measured binding energy for Ncation 1s for [N6,6,6,14][BF4]). A full description of charge-referencing for each family of ionic liquids investigated within this study is provided within the Results and discussion section.Results and discussion
Sample purity
XP survey and high resolution scans were used to establish elemental composition and purity for each ionic liquid presented in this study. Fig. 1 shows a typical survey scan for [P6,6,6,14][PF6] which displays all expected photoelectron and Auger lines. Previous XPS studies upon ionic liquids have highlighted the presence of hydrocarbon and silicone-based impurities in the near-surface region, which cannot be detected by NMR or other bulk sensitive techniques.26,28,29 A weak O 1s and Si 2p signal was observed in the case of [N6,6,6,14][Cl] and [N4,4,4,1][BF4] (see ESI†); this is likely to be a result of laboratory grease during sample preparation and is not expected to impact upon the recorded binding energies.24,26,30 Surface contaminants, such as grease, can be routinely removed through argon etching, a full description for this procedure in relation to the compounds studied herein is provided within the experimental section. There was no evidence of this contaminant in any of the other ionic liquids used in this study. Furthermore, there is no evidence of metal-based cations or halides that may be carried over from ionic liquid synthesis, particularly anion exchange chemistries. Experimental stoichiometries, calculated from high resolution spectra, are presented in Table 2. Experimental values are within the error of the nominal stoichiometry of ±10%.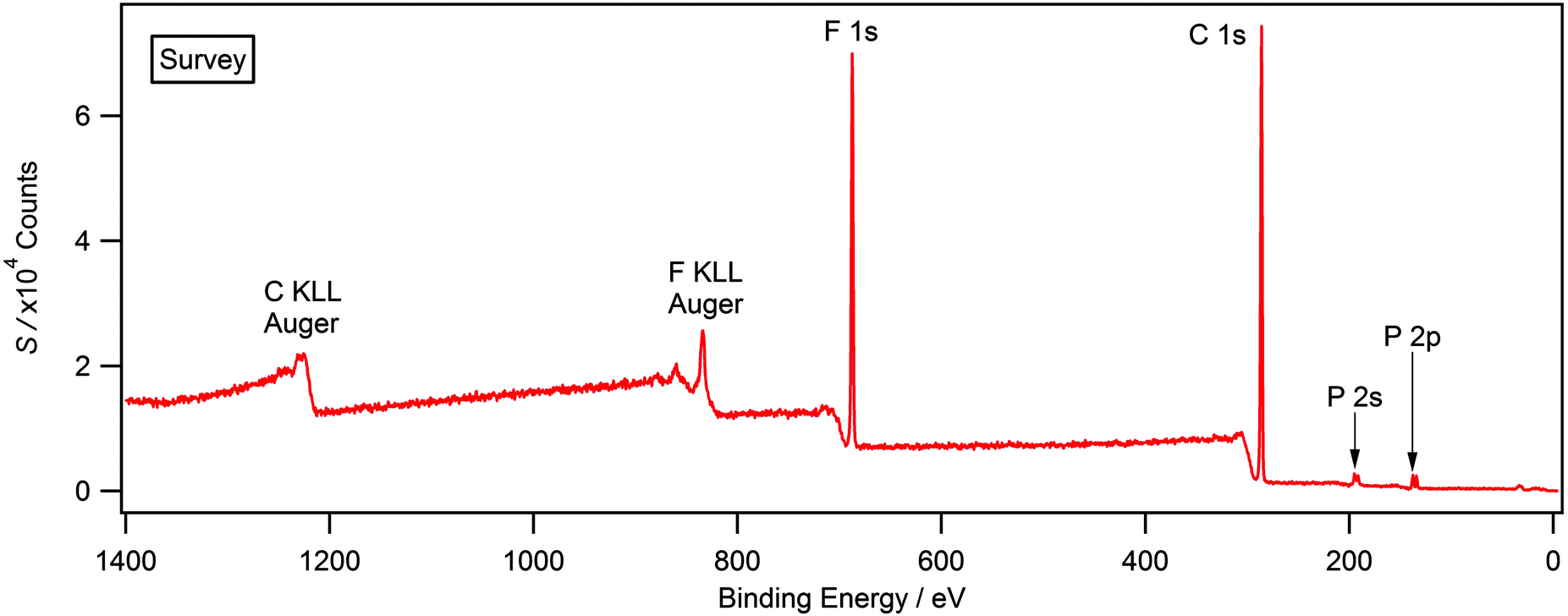 | ||
| Fig. 1 Survey XP spectrum for [P6,6,6,14][PF6]. The absence of photoelectron lines corresponding to other elements, especially oxygen and silicon, highlights the high purity of the compounds used in this study. | ||
| Compound | Compositiona (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| C | N | O | F | B | S | P | Cl | |
a Relative sensitivity factors (RSF) taken from the Kratos Library were used to derive atomic percentages from the most intense photoemission peak for each element i.e. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C 1s, NN 1s, OO 1s, FF 1s, BB 1s, SS 2p, PP 2p, ClCl 2p. C 1s, NN 1s, OO 1s, FF 1s, BB 1s, SS 2p, PP 2p, ClCl 2p.
|
||||||||
| RSF26,27 | 0.278 | 0.477 | 0.780 | 1.000 | 0.159 | 0.668 | 0.486 | 0.891 |
| [N6,6,6,14][Cl] | 95.5 (94.0) | 2.3 (2.9) | — | — | — | — | — | 2.2 (2.9) |
| [P6,6,6,14][Cl] | 95.2 (94.1) | — | — | — | — | — | 2.7 (2.9) | 2.1 (2.9) |
| [N6,6,6,14][BF4] | 84.0 (84.2) | 2.5 (2.6) | — | 10.7 (10.5) | 2.8 (2.6) | — | — | — |
| [P6,6,6,14][BF4] | 85.3 (84.2) | — | — | 10.6 (10.5) | 2.2 (2.6) | — | 2.0 (2.6) | — |
| [N6,6,6,14][PF6] | 80.5 (80.0) | 2.5 (2.5) | — | 15.0 (15.0) | — | — | 2.0 (2.5) | — |
| [P6,6,6,14][PF6] | 79.9 (80.0) | — | — | 16.3 (15.0) | — | — | 3.8 (5.0) | — |
| [N6,6,6,14][OTf] | 80.7 (80.5) | 2.4 (2.4) | 7.1 (7.3) | 7.6 (7.3) | — | 2.1 (2.4) | — | — |
| [P6,6,6,14][OTf] | 81.3 (80.5) | — | 7.4 (7.3) | 7.3 (7.3) | — | 2.1 (2.4) | 2.0 (2.4) | |
| [N6,6,6,14][NTf2] | 70.5 (70.8) | 3.9 (4.2) | 8.9 (8.3) | 12.9 (12.5) | — | 3.8 (4.2) | — | — |
| [P6,6,6,14][NTf2] | 70.8 (70.8) | 2.2 (2.1) | 8.9 (8.3) | 12.6 (12.5) | — | 3.9 (4.2) | 1.7 (2.1) | — |
| [N4,4,4,1][BF4] | 70.9 (68.4) | 4.9 (5.3) | — | 18.4 (21.1) | 5.8 (5.3) | — | — | — |
| [P4,4,4,1][BF4] | 68.6 (68.4) | — | — | 21.3 (21.1) | 5.0 (5.3) | — | 5.0 (5.3) | — |
| [N4,4,4,1][NTf2] | 52.3 (51.7) | 6.6 (6.9) | 14.2 (13.8) | 20.6 (20.7) | — | 6.4 (6.9) | — | — |
| [P4,4,4,1][NTf2] | 52.6 (51.7) | 3.5 (3.4) | 13.8 (13.8) | 20.8 (20.7) | — | 6.5 (6.9) | 2.8 (3.4) | — |
Interestingly, deviations larger than ±10% are observed for the phosphorus P 2p signal, in which the calculated value is significantly and systematically less than the theoretical value. Previous angle resolved studies upon ionic liquids have shown that long alkyl chains (n ≥ 2) point out towards the gas phase resulting in a surface enrichment of carbon.10,31 The ionic liquids in this study have a large alkyl contribution which may significantly impact the stoichiometry of other elements as it has been seen that the cationic head group is pushed deeper into the bulk with increasing alkyl chain length.32 It is feasible that differences in the conformations and mobility of the alkyl chains for tetraalkylammonium and phosphonium cations result in a nitrogen cationic core that is situated closer to the surface, but a phosphorus cationic core that is buried further within the bulk. This provides a possible explanation for the lower than expected intensity of the P 2p photoelectron peak. Earlier studies have shown a larger surface enrichment of carbon for ILs with small anions, such as chloride, whereby the anion is situated at the same level as the cation.33 This could explain the smaller than expected Cl 2p signal.
Binding energies and multi-component fitting models for [NR,R′,R′′,R′′′]X, (X = Cl−, [BF4]−, [PF6]−, [OTf]−, [NTf2]−)
The measured binding energies for the ammonium series of ionic liquids investigated are presented in Table 3. To obtain accurate and reproducible binding energies an appropriate charge-referencing method must be used. It is well established that for ionic liquids with a long alkyl chain (n ≥ 8), reliable charge-referencing can be achieved by setting the experimentally determined binding energy of the aliphatic carbon component (Caliphatic) equal to a standard reference value, in the case of this work 285.0 eV.14,24 For cations with shorter alkyl chains this procedure is not applicable. Previous studies indicate that length of the alkyl chain has negligible impact upon the binding energy of the cationic head group when the anion is kept the same.14 Charge referencing for [N4,4,4,1][BF4] was achieved by setting the measured binding energy of Ncation 1s to 402.3 eV; in this case 402.3 eV is the measured binding energy of Ncation 1s for [N6,6,6,14][BF4], which has been charge referenced to Caliphatic as discussed previously. A similar procedure is used for [N4,4,4,1][NTf2].| Compound | Binding energy/eV | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cation | Anion | |||||||||||||
| Cation | Anion | Caliphatic 1s | Cinter 1s | Chetero 1s | Ncation 1s | Pcation 2p3/2 | Canion 1s | Nanion 1s | O 1s | F 1s | B 1s | Cl 2p3/2 | S 2p3/2 | Panion 2p3/2 |
| [N6,6,6,14]+ | [Cl]− | 285.0 | 285.6 | 286.3 | 402.1 | — | — | — | — | — | — | 196.6 | — | — |
| [P6,6,6,14]+ | [Cl]− | 285.0 | — | 285.7 | — | 132.3 | — | — | — | — | — | 196.6 | — | — |
| [N6,6,6,14]+ | [BF4]− | 285.0 | 285.6 | 286.5 | 402.3 | — | — | — | — | 685.7 | 194.0 | — | — | — |
| [P6,6,6,14]+ | [BF4]− | 285.0 | — | 285.8 | — | 132.6 | — | — | — | 685.7 | 193.9 | — | — | — |
| [N6,6,6,14]+ | [PF6]− | 285.0 | 285.7 | 286.6 | 402.4 | — | — | — | — | 686.6 | — | — | — | 136.4 |
| [P6,6,6,14]+ | [PF6]− | 285.0 | — | 285.8 | — | 132.7 | — | — | — | 686.5 | — | — | — | 136.3 |
| [N6,6,6,14]+ | [OTf]− | 285.0 | 285.7 | 286.6 | 402.4 | — | 292.3 | — | 531.8 | 688.3 | — | — | 168.2 | — |
| [P6,6,6,14]+ | [OTf]− | 285.0 | — | 285.8 | — | 132.7 | 292.3 | — | 531.9 | 688.4 | — | — | 168.2 | — |
| [N6,6,6,14]+ | [NTf2]− | 285.0 | 285.7 | 286.7 | 402.5 | — | 292.8 | 399.3 | 532.5 | 688.7 | — | — | 168.9 | — |
| [P6,6,6,14]+ | [NTf2]− | 285.0 | — | 285.9 | — | 132.7 | 292.8 | 399.3 | 532.5 | 688.7 | — | — | 168.8 | — |
| [N4,4,4,1]+ | [BF4]− | 285.0 | 285.2 | 286.4 | 402.3 | — | — | — | — | 685.7 | 194.1 | — | — | — |
| [P4,4,4,1]+ | [BF4]− | 285.0 | — | 285.4 | — | 132.6 | — | — | — | 685.5 | 193.9 | — | — | — |
| [N4,4,4,1]+ | [NTf2]− | 285.1 | 285.4 | 286.6 | 402.5 | — | 292.8 | 399.3 | 532.5 | 688.7 | — | — | 168.8 | — |
| [P4,4,4,1]+ | [NTf2]− | 285.1 | — | 285.6 | — | 132.7 | 297.7 | 399.2 | 532.4 | 688.6 | — | — | 168.8 | — |
Due to the presence of several different carbon environments, peak identification and fitting of the C 1s region for ionic liquids is of particular importance. Fig. 2 shows some representative examples of the C 1s region for [N6,6,6,14]X (X = Cl−, [BF4]−, [NTf2]−). In the case of [N6,6,6,14][NTf2] there are three clear peaks, two of which are unresolved. The resolved peak at ≈293 eV is assigned to the CF3 group of the [NTf2] anion as seen previously for imidazolium and pyrrolidinium ionic liquids.14,24 The two unresolved peaks between 285–287 eV for [N6,6,6,14][Cl], [N6,6,6,14][BF4] and [N6,6,6,14][NTf2] arise from the different electronic environments of carbon present in the cation, thus it is necessary to develop a model to explain these different contributions. Previously we have developed a series of multi-component models to describe the C 1s spectra of a range of ionic liquid families including dialkyl-imidazolium, alkyl-pyridinium and pyrrolidinium based cations.14 Here we extend this to include the tetraalkylphosphonium and tetraalkylammonium based cations.
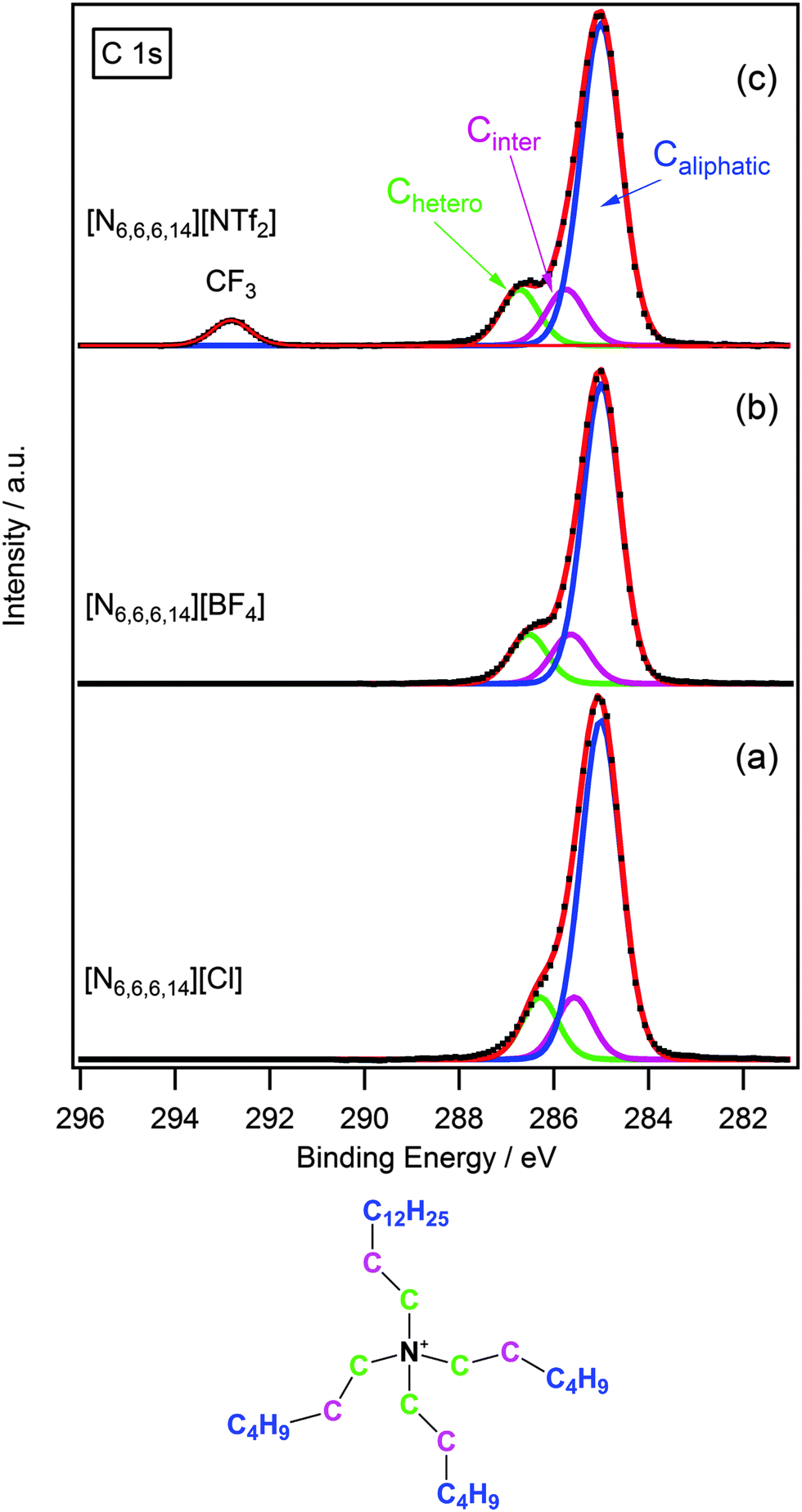 | ||
| Fig. 2 High resolution XP spectra of the C 1s region with component fittings for (a) [N6,6,6,14][Cl], (b) [N6,6,6,14][BF4] and (c) [N6,6,6,14][NTf2]. Areas are normalised to the Ncation 1s photoemission peak for [N6,6,6,14][BF4]. All XP spectra charge corrected to Caliphatic = 285.0 eV. | ||
As the structure of the ammonium cation is not dissimilar to that of the pyrrolidinium, a three-component model was adopted for the ammonium-based ionic liquids in this study. An analysis of the structure of [N6,6,6,14]+ (see structure shown in Fig. 2) highlights the three chemical environments of carbon. The carbon directly bound to the nitrogen (Chetero) is expected to have the highest binding energy as it is in closest proximity to the electropositive nitrogen, and thus feels a larger effect of the electron-withdrawing influence of the cationic nitrogen. The next carbon atom along the chain (Cinter) is designated as an “intermediate” carbon environment; here the electron-withdrawing impact of the nitrogen centre is less pronounced, resulting in a slightly lower binding energy. The remaining carbons are essentially aliphatic and give rise to the third contribution, Caliphatic, which exhibits the lowest binding energy.
To obtain a satisfactory fit that accurately describes the chemical environment it is necessary to constrain certain parameters. Component areas are constrained to their nominal stoichiometries, i.e. for [N6,6,6,14][BF4] Caliphatic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Chetero:Cinter equates to 6:1:1. The full width at half maximum (FWHM) for Chetero and Cinter are set to be equal (and constrained to be between 0.8 and 1.5 eV). The FWHM for Caliphatic is set to be 1.1 times larger than for Chetero as studies upon other similar ionic liquid systems have characteristically shown the FWHM ratio of Caliphatic:Chetero to be 1.1:1.14,24 Binding energy constraints were not applied as the binding energy shift between Chetero and Cinter is expected to be dependent upon the nature of the anion.13,14 The fitting envelopes (red curve), shown in Fig. 2a–c, demonstrate excellent agreement to the experimental spectra (black dots). C 1s fittings for [N6,6,6,14]X (X = [PF6]−, [OTf]−) and [N4,4,4,1]X (X = [BF4]−, [NTf2]−) are included in the ESI† and are also in agreement with the experimental spectra.
:Chetero:Cinter equates to 6:1:1. The full width at half maximum (FWHM) for Chetero and Cinter are set to be equal (and constrained to be between 0.8 and 1.5 eV). The FWHM for Caliphatic is set to be 1.1 times larger than for Chetero as studies upon other similar ionic liquid systems have characteristically shown the FWHM ratio of Caliphatic:Chetero to be 1.1:1.14,24 Binding energy constraints were not applied as the binding energy shift between Chetero and Cinter is expected to be dependent upon the nature of the anion.13,14 The fitting envelopes (red curve), shown in Fig. 2a–c, demonstrate excellent agreement to the experimental spectra (black dots). C 1s fittings for [N6,6,6,14]X (X = [PF6]−, [OTf]−) and [N4,4,4,1]X (X = [BF4]−, [NTf2]−) are included in the ESI† and are also in agreement with the experimental spectra.
Binding energies and multi-component fitting models for [PR,R′,R′′,R′′′]X, (X = Cl−, [BF4]−, [PF6]−, [OTf]−, [NTf2]−)
The C 1s region for phosphonium ionic liquids differs quite considerably from that of ammonium ionic liquids. Fig. 3 shows some representative examples of the C 1s region for [P6,6,6,14]X (X = Cl−, [BF4]−, [NTf2]−). In the case of [P6,6,6,14][NTf2] there are two clearly resolved peaks with the peak at higher binding energy corresponding to the CF3 group of the anion. A broad peak with slight asymmetry occurs between 284–286 eV for [P6,6,6,14]X (X = Cl−, [BF4]−, [NTf2]−); which is distinctly lacking the obvious shoulder at higher binding energy seen in the case of the ammonium analogues (Fig. 2 and 3). Consequently, this peak was fitted using a two-component model; the first contribution arising from carbons directly bound to the phosphorus atom, Chetero, and the remaining carbons considered as aliphatic, Caliphatic.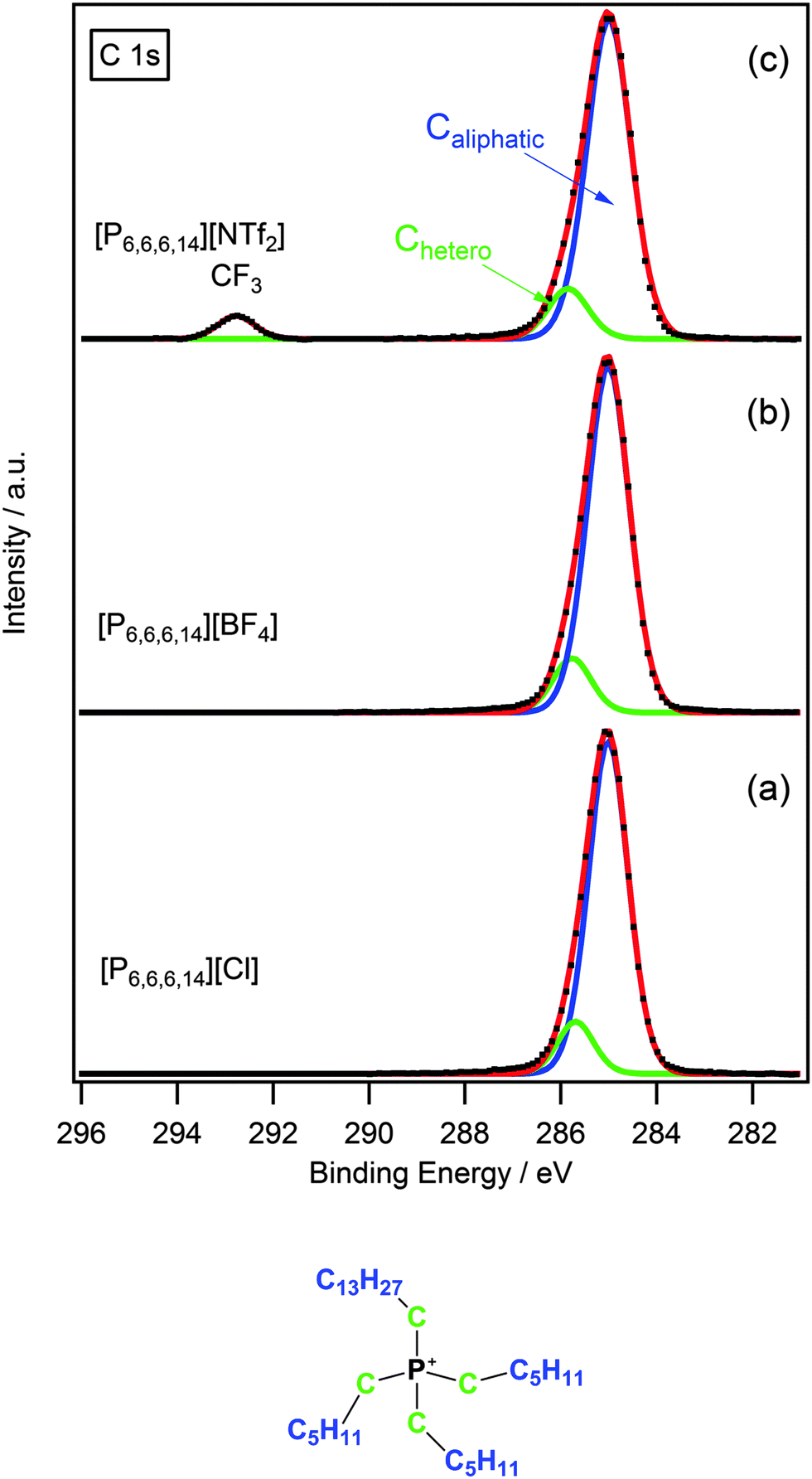 | ||
| Fig. 3 High resolution XP spectra of the C 1s region with component fittings for (a) [P6,6,6,14][Cl], (b) [P6,6,6,14][BF4] and (c) [P6,6,6,14][NTf2]. Areas are normalised to the area of Pcation 2p photoemission peak for [P6,6,6,14][BF4]. All XP spectra are charge corrected by setting the measured binding energy of Caliphatic = 285.0 eV. | ||
A comparison of binding energies (Table 3) for the Chetero component of the [N6,6,6,14]+ and [P6,6,6,14]+ cations show that Chetero for [N6,6,6,14]+ is ∼0.6–0.8 eV larger than for the [P6,6,6,14]+ analogue. This can be attributed to the difference in charge density of the nitrogen and phosphorus cationic centres. The smaller atomic radius of the nitrogen atom, in comparison to phosphorus, results in a higher charge density. Consequently, the Chetero carbons for ammonium-based ionic liquids experience a stronger electron-withdrawing effect from the positively charged nitrogen centre, which is reflected by a shift to higher binding energy.
Furthermore, from Table 3 it can be seen that Chetero binding energies for phosphonium-based ionic liquids are more similar in value to the Cinter component for the ammonium analogues. This highlights the weaker electron-withdrawing effect of the phosphorus centre, as a result of its lower charge density, and validates the use of a two-component fitting model to describe the C 1s region for phosphonium-based ionic liquids.
Cations with short alkyl chain length i.e. [P4,4,4,1]+ were fitted using the same method and charge referenced in a similar way to the short chain ammonium cations as discussed previously.
Effect of anion: cation–anion interactions
It has been previously established that XPS can be used to probe cation–anion interactions.13,14,34 Earlier work has demonstrated that binding energies of Chetero 1s and Ncation 1s for imidazolium and pyrrolidinium-based ionic liquids are dependent upon the basicity of the anion.13,14 High basicity anions such as halides result in lower binding energies, which indicate that the cation is in a more electron-rich environment. This is indicative of a significant amount of charge-transfer from anion to cation. Low basicity anions such as [NTf2]− give rise to higher binding energies signifying that the cation is more electron-poor due to the reduction in charge-transfer.The influence of the anion on the electronic environment of the cation has been investigated for three [N6,6,6,14][X] ionic liquids, where X = [NTf2]−, [BF4]− and Cl− (Fig. 4). It is seen that the binding energy of Ncation 1s follows the trend: [NTf2]− > [BF4]− > Cl−, this can be interpreted as an increase in charge transfer with increasing anion basicity, and is in accordance with earlier studies upon imidazolium13 and pyrrolidinium14 ionic liquids. Binding energies for Chetero 1s (Table 3 and Fig. 2) also exhibit an identical trend.
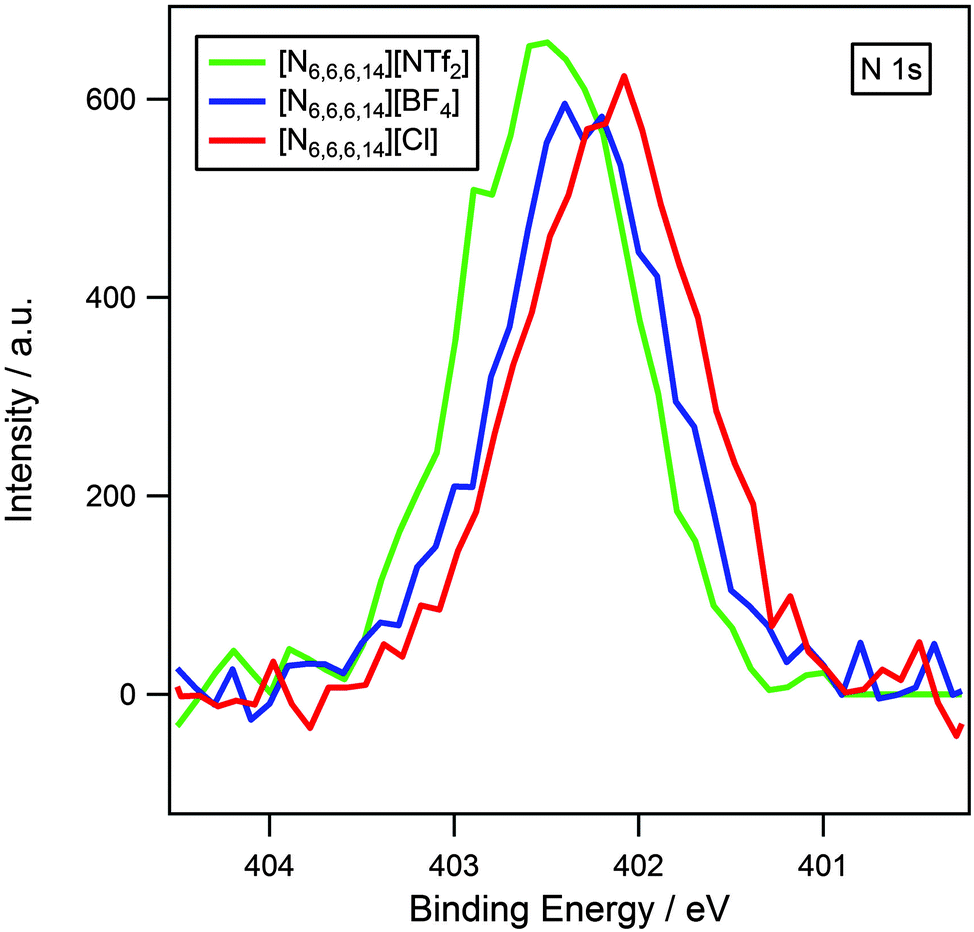 | ||
| Fig. 4 N 1s XP spectra of [N6,6,6,14][NTf2], [N6,6,6,14][BF4] and [N6,6,6,14][Cl]. Areas are normalised to the Ncation 1s peak for [N6,6,6,14][BF4]. XP spectra were charge corrected by referencing the aliphatic C 1s photoemission peak to 285.0 eV. N.B. spectra truncated to only show the cation region. | ||
The effect of varying the anion has also been investigated for the analogous phosphonium series, [P6,6,6,14][X] where X = [NTf2]−, [BF4]− and Cl− (Fig. 5). The P 2p region shows two unresolved peaks, P 2p1/2 and P 2p3/2, observed in an intensity ratio of 1:2. This is a result of spin–orbit coupling and is an inherent property of p-orbitals and does not indicate two different electronic environments of phosphorus. The same trend as for ammonium-based ionic liquids is observed in that binding energies for the Pcation 2p3/2 component are in the order: [NTf2]− > [BF4]− > Cl−, with Chetero 1s following a similar pattern.
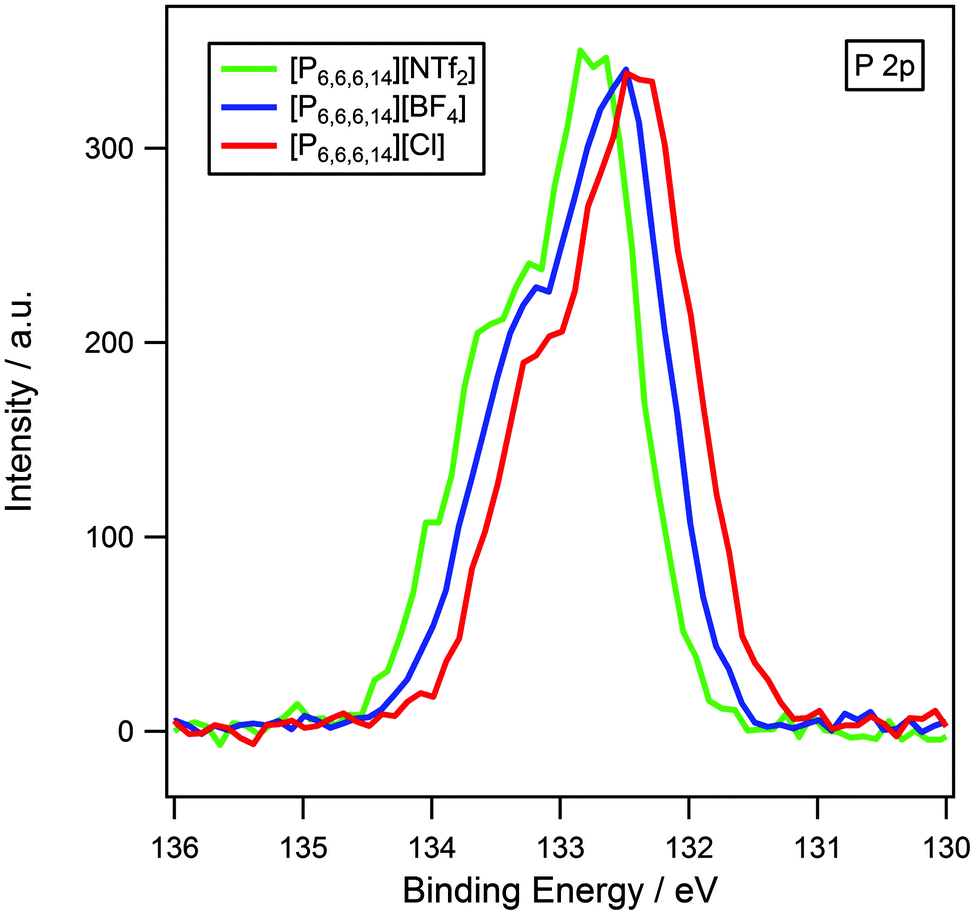 | ||
| Fig. 5 P 2p XP spectra of [P6,6,6,14][NTf2], [P6,6,6,14][BF4] and [P6,6,6,14][Cl]. Areas are normalised to the P 2p peak for [P6,6,6,14][BF4]. XP spectra were charge corrected by referencing the aliphatic C 1s photoemission peak to 285.0 eV. | ||
A linear correlation of Ncation 1s and Chetero 1s binding energies with the Kamlet–Taft β parameter (a measure of hydrogen bond acceptor ability of the anion) has been established for imidazolium-based ionic liquids.13 However, to date, β has been less extensively studied for ammonium and phosphonium ionic liquids with only a select few examples in the literature.35–39 Consequently, a comparison of β with binding energy cannot be made for the compounds investigated in this study, however it is expected that a similar trend to imidazolium-based ionic liquids would be observed.
Effect of cation: cation–anion interactions, [N6,6,6,14]+versus [P6,6,6,14]+
As discussed previously, it is expected that the difference in charge density of the ammonium and phosphonium cationic centres will affect the degree of charge-transfer, which would be reflected in the binding energy of the anionic components. We postulated that a greater degree of charge-transfer would occur for the ammonium cation, as the larger charge density would be more effective in removing excess negative charge from the anion; hence, the binding energies of the anion were expected to indicate a more electron-poor environment in contrast to the phosphonium analogue.A comparison of binding energies, however, did not support this assumption. Fig. 6 illustrates a comparison between [N6,6,6,14][BF4] and [P6,6,6,14][BF4] for all regions. All XP spectra are charge corrected by referencing Caliphatic 1s to 285.0 eV; and are normalised to the area of the F 1s photoemission peak for [N6,6,6,14][BF4] as the number of fluorine atoms are equivalent for both samples. It can be seen that the binding energies of the anionic components are identical, within the error of experiment (±0.1 eV), for example the F 1s component for [N6,6,6,14][BF4] and [P6,6,6,14][BF4] is 685.7 eV in each instance, (Fig. 6 and Table 3). This is also true for the B 1s component where binding energies are 194.0 eV and 193.9 eV for [N6,6,6,14][BF4] and [P6,6,6,14][BF4] respectively. The P 2s photoemission peak is also observed in the B 1s XP spectrum for [P6,6,6,14][BF4] at a lower binding energy of 190.4 eV.
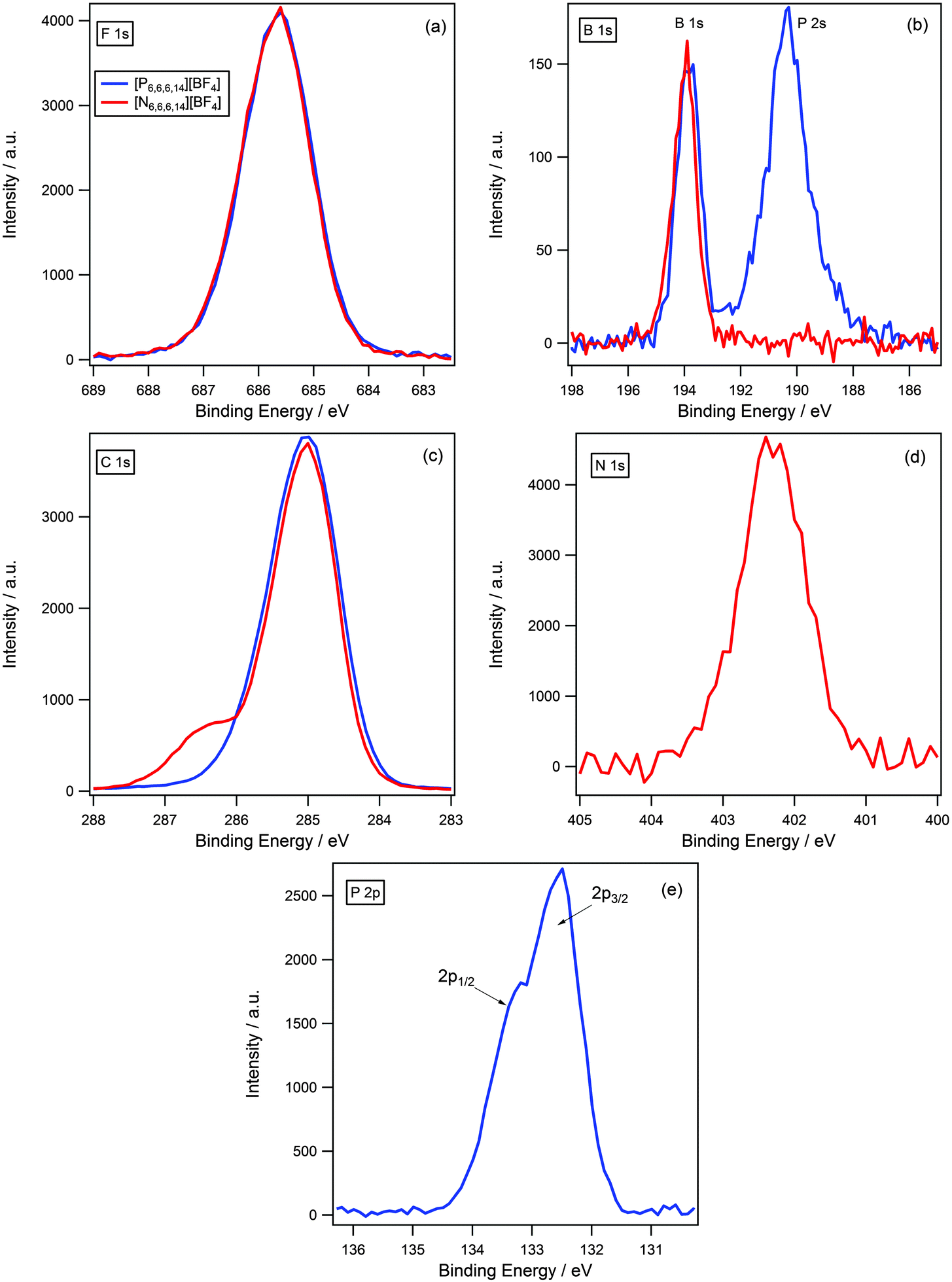 | ||
| Fig. 6 XP spectra of [N6,6,6,14][BF4] and [P6,6,6,14][BF4] for: (a) F 1s, (b) B 1s, (c) C 1s, (d) N 1s and (e) P 2p. Areas normalised to area of F 1s peak for [N6,6,6,14][BF4]. All XP spectra were charge corrected by setting Caliphatic 1s = 285.0 eV. | ||
The binding energies for a range of anions of varying size and basicity, including Cl−, [PF6]−, [OTf]− and [NTf2]−, also match within experimental error for the [N6,6,6,14]+ and [P6,6,6,14]+ cations. This suggests that the anion cannot “see” the difference in charge density of the cation, and it is proposed that the long alkyl chains are wrapped around the cationic centre effectively shielding it from the anion. This is in agreement with measurements on the enthalpy of vaporisation at 298 K, ΔvapH298, for [P6,6,6,14][BF4], which describe how the experimentally determined van der Waals contribution is significantly smaller than the theoretical value, it is suggested that parts of the alkyl chain close to the phosphorus are unavailable for van der Waals bonding with adjacent species due to shielding from the outer parts of the chain.40 In contrast, molecular dynamics simulations indicate that the alkyl chains are in a fully stretched conformation, with a small degree of bending for longer chains (n > 6) and no evidence of coiling or wrapping.41,42
It should be noted that although the anion is shielded from the difference in charge density of the two different cations, it is not shielded from “seeing” a positive charge completely. In this way, the “wrapping” of the aliphatic chains has no impact on the actual ability of the anion to transfer charge, and hence we see a shift in B.E. of the cationic core for anions of different basicity as mentioned previously in the section “Effect of the Anion: Cation–Anion Interactions”.
Effect of cation: cation–anion interactions, [N4,4,4,1]+versus [P4,4,4,1]+
In the previous section we determined that for ammonium and phosphonium cations with long alkyl chains, there is no difference in binding energy of the anionic components for a variety of different anions. To test whether the length of the alkyl chain impacts upon cation–anion interactions and the ability of the anion to “see” the difference in charge density of the cationic centres, a series of short chain analogues were investigated where the cation size has been reduced to [N4,4,4,1]+ and [P4,4,4,1]+.A visual comparison between [N4,4,4,1][BF4] and [P4,4,4,1][BF4] for all regions is shown in Fig. 7. The F 1s binding energy for [N4,4,4,1][BF4] is 0.2 eV higher than for [P4,4,4,1][BF4] (Fig. 7 and Table 3), this is slightly larger than the accepted experimental error and suggests that the shift is real. As the F 1s binding energy is higher for [N4,4,4,1]+ relative to [P4,4,4,1]+ this is indicative that the fluorine atoms are more electron-poor for [N4,4,4,1]+, which suggests a greater degree of charge-transfer is occurring for the ammonium-based ionic liquid. This supports our initial hypothesis that charge-transfer should be more significant for ammonium-based ionic liquids due to the larger charge density of the cationic centre relative to their phosphonium analogues. Additionally, it indicates that reducing the length of the alkyl chain allows for a more open structure of the cation which permits the anion to “see” the difference in charge density of the cationic centre.
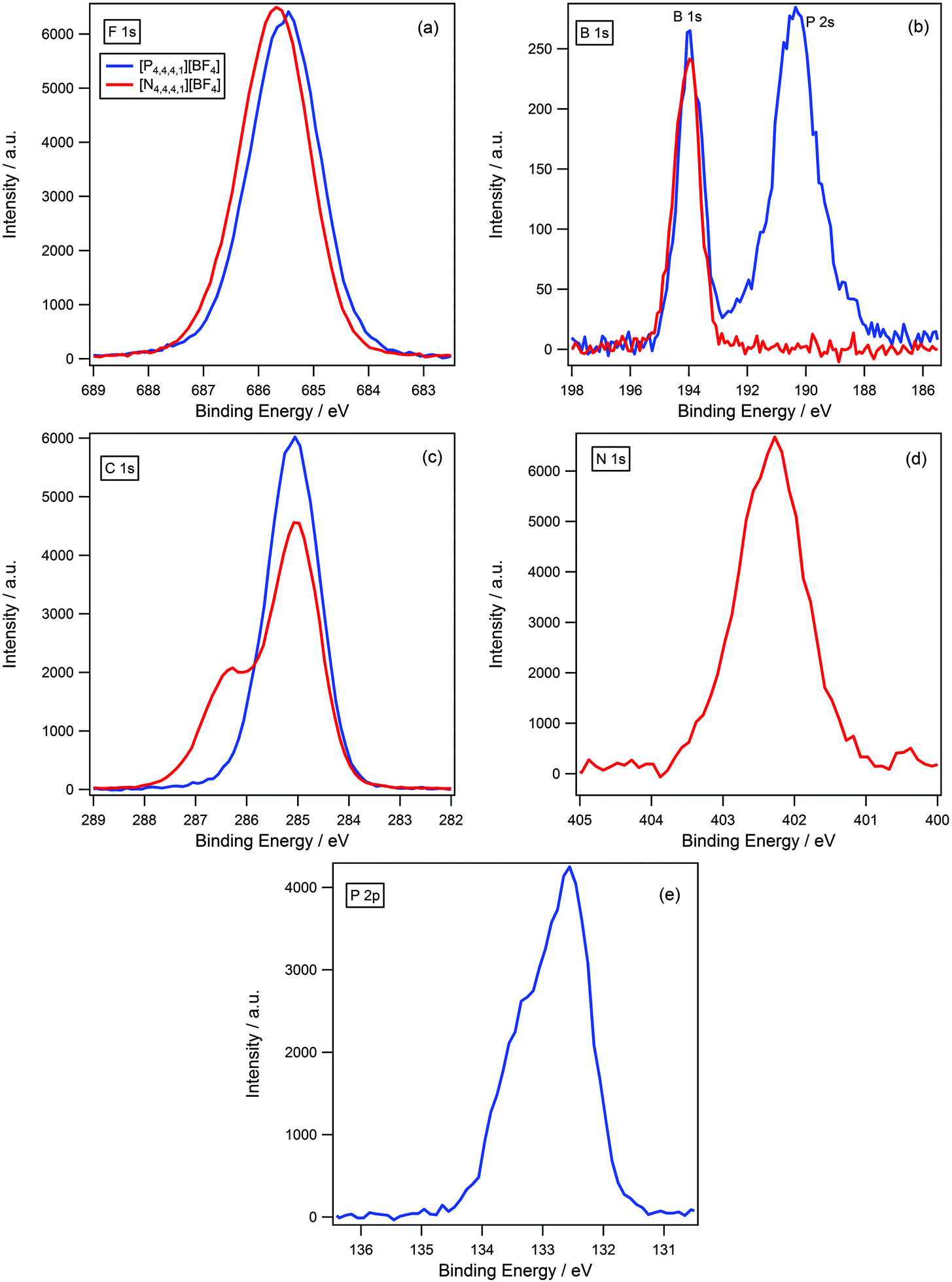 | ||
| Fig. 7 XP spectra of [N4,4,4,1][BF4] and [P4,4,4,1][BF4] for: (a) F 1s, (b) B 1s, (c) C 1s, (d) N 1s and (e) P 2p. Areas normalised to area of F 1s peak for [N4,4,4,1][BF4]. [N4,4,4,1][BF4] XP spectra are charge corrected by setting Ncation 1s to 402.3 eV. [P4,4,4,1][BF4] XP spectra are charge corrected by setting Pcation 2p3/2 to 132.6 eV. | ||
Fabris et al. found that a trioctylmethylphosphonium ([TOMP]+) cation coupled with a methylcarbonate ([MeOCOO]−) or hydrogen carbonate ([HOCOO]−) anion exhibited stronger basicity than its ammonium counterparts and other conventional basic catalysts, and as a result was a more efficient catalyst for Michael reactions.43 This could support the assertions made from the work described here, as the high basicity of [TOMP] [MeOCOO] and [TOMP] [HOCOO] may arise from a smaller degree of charge-transfer occurring from anion to cation; consequently the anion is more electron-rich thus the electrons are more available for donation. In contrast, a higher degree of charge-transfer occurs for the ammonium analogues and inorganic bases as a result of the larger charge density of the cationic centre, hence basicity is reduced as electron density is less available for donation.
It is still unclear to us at this time how the charge-transfer phenomenon is mediated. Previous studies upon 1-methyl-3-alkylimidazolium systems have shown a correlation between anion basicity and hydrogen-bonding at the C-2 position, and also with Ncation 1s and C 1s binding energy.13 However, upon methylation of the C-2 position (hence decreasing the hydrogen-bonding contribution) an anion-dependent shift of the Ncation 1s and C 1s binding energies is still observed at a similar magnitude as the non-methylated analogues.13 This indicates that charge-transfer cannot solely be mediated through hydrogen-bonding, which is especially relevant in the case of tetraalkylammonium and phosphonium ILs where the proton alpha to the heteroatom is even less available due to steric hindrance of the long alkyl chains. This suggests charge-transfer is also dependent upon the strength of the coulombic interaction of the cation and anion.
It should be noted that for [N4,4,4,1][NTf2] and [P4,4,4,1][NTf2] only a small shift (∼0.1 eV) to a higher binding energy is observed for the anionic components of [N4,4,4,1]+ relative to [P4,4,4,1]+ (Table 3), this is within the experimental error so no real conclusion can be made; however, as [NTf2]− is weakly basic it is likely that it participates in charge-transfer to a very small degree, hence little or no difference is observed when comparing the different cationic centres.
Conclusion
The impact of changing the cation core from nitrogen to phosphorus on cation–anion interactions has been investigated using XPS. For long chain systems, i.e. [N6,6,6,14]+versus [P6,6,6,14]+, it appears that changing the cation core has minimal impact on the electronic environment of the anion due to a perceived shielding effect of the long alkyl chains. In an effort to reduce the effect of hydrocarbon based shielding of the heteroatom, a second series of experiments were conducted where the alkyl components are pruned back, hence allowing a more clear investigation of the role of the heteroatom based charge carrier. A decrease in alkyl chain length to [N4,4,4,1]+ and [P4,4,4,1]+ indicates that changing the cation core from nitrogen to phosphorus does indeed influence the electronic environment of the anion; with an increase in charge-transfer shown for ammonium ILs. Further investigation into binary cationic mixtures by XPS was not attempted as any shift in the measured binding energies, with adjusted mol. fraction, would lie within the experimental error of the XPS instrument. This indicates that tunability of the electronic environment for IL systems is more strongly influenced by the anion than the cation.Interestingly, Carvalho et al.23 investigated the influence of the cationic heteroatom for ammonium- and phosphonium-based ILs on physical properties such as densities, viscosities, melting temperatures and refractive indices. A shielding effect was expected but not observed, and it was seen that the properties investigated were markedly affected by the cationic core. Taking this into consideration with the work described herein a dual tunability of IL systems could be proposed, where electronic environments, i.e. chemical properties, are tuned via the anion and physical properties are tuned via the cation. This work provides a step toward being fully able to fine-tune both the chemical and physical properties of an IL system by altering their constituent ions to suit a specific function.
Acknowledgements
We thank the EPSRC (EP/K005138/1) for financial support and provision of the Ionic Liquids XPS Facility. RKB acknowledges the University of Nottingham for support. The authors are grateful to Ms Emily F. Smith for helpful discussions and critical advice. Cytec Industries Inc. are thanked for kindly donating the samples of Cyphos 101 and Cyphos 108 used in this study.References
- Ionic Liquids in Synthesis, ed. P. Wasserscheid, T. Welton, 2nd edn, Wiley-VCH, Weinheim, 2007, ISBN 978-3-527-31239-9 Search PubMed.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- G. Chatel, J. F. B. Pereira, V. Debbeti, H. Wang and R. D. Rogers, Green Chem., 2014, 16, 2051–2083 RSC.
- M. A. Taige, D. Hilbert and T. J. S. Schubert, Z. Phys. Chem., 2012, 226, 129–139 CrossRef CAS.
- A. M. Pinto, H. Rodriguez, Y. J. Colon, A. Arce and A. Soto, Ind. Eng. Chem. Res., 2013, 52, 5975–5984 CrossRef CAS.
- Q. Q. Baltazar, S. K. Leininger and J. L. Anderson, J. Chromatogr. A, 2008, 1182, 119–127 CrossRef CAS PubMed.
- S. Katsuta, Y. Yoshimoto, M. Okai, Y. Takeda and K. Bessho, Ind. Eng. Chem. Res., 2011, 50, 12735–12740 CrossRef CAS.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinruck and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS PubMed.
- H. P. Steinruck, Phys. Chem. Chem. Phys., 2012, 14, 5010–5029 RSC.
- K. R. J. Lovelock, C. Kolbeck, T. Cremer, N. Paape, P. S. Schulz, P. Wasserscheid, F. Maier and H. P. Steinruck, J. Phys. Chem. B, 2009, 113, 2854–2864 CrossRef CAS PubMed.
- F. Maier, T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, P. S. Schulz, P. Wasserscheidc and H. P. Steinruck, Phys. Chem. Chem. Phys., 2010, 12, 1905–1915 RSC.
- I. Niedermaier, C. Kolbeck, N. Taccardi, P. S. Schulz, J. Li, T. Drewello, P. Wasserscheid, H. P. Steinruck and F. Maier, ChemPhysChem, 2012, 13, 1725–1735 CrossRef CAS PubMed.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wolfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. P. Steinruck, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244–15255 RSC.
- I. J. Villar-Garcia, K. R. J. Lovelock, S. Men and P. Licence, Chem. Sci., 2014, 5, 2573–2579 RSC.
- K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309–321 CrossRef CAS.
- R. E. Del Sesto, C. Corley, A. Robertson and J. S. Wilkes, J. Organomet. Chem., 2005, 690, 2536–2542 CrossRef CAS PubMed.
- G. Adamova, R. L. Gardas, M. Nieuwenhuyzen, A. V. Puga, L. P. N. Rebelo, A. J. Robertson and K. R. Seddon, Dalton Trans., 2012, 41, 8316–8332 RSC.
- K. Tsunashima and M. Sugiya, Electrochem. Commun., 2007, 9, 2353–2358 CrossRef CAS PubMed.
- K. Tsunashima, E. Niwa, S. Kodama, M. Sugiya and Y. Ono, J. Phys. Chem. B, 2009, 113, 15870–15874 CrossRef CAS PubMed.
- H. Shirota, H. Fukazawa, T. Fujisawa and J. F. Wishart, J. Phys. Chem. B, 2010, 114, 9400–9412 CrossRef CAS PubMed.
- H. Y. Lee, H. Shirota and E. W. Castner, J. Phys. Chem. Lett., 2013, 4, 1477–1483 CrossRef CAS.
- P. J. Carvalho, S. P. M. Ventura, M. L. S. Batista, B. Schroder, F. Goncalves, J. Esperanca, F. Mutelet and J. A. P. Coutinho, J. Chem. Phys., 2014, 140, 64505 CrossRef PubMed.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. L. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- A. Cieniecka-Roslonkiewicz, J. Pernak, J. Kubis-Feder, A. Ramani, A. J. Robertson and K. R. Seddon, Green Chem., 2005, 7, 855–862 RSC.
- E. F. Smith, F. J. M. Rutten, I. J. Villar-Garcia, D. Briggs and P. Licence, Langmuir, 2006, 22, 9386–9392 CrossRef CAS PubMed.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- E. F. Smith, I. J. Villar Garcia, D. Briggs and P. Licence, Chem. Commun., 2005, 5633–5635 RSC.
- K. R. J. Lovelock, E. F. Smith, A. Deyko, I. J. Villar-Garcia, P. Licence and R. G. Jones, Chem. Commun., 2007, 4866–4868 RSC.
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436–9445 RSC.
- V. Lockett, R. Sedev, C. Bassell and J. Ralston, Phys. Chem. Chem. Phys., 2008, 10, 1330–1335 RSC.
- C. Ridings, V. Lockett and G. Andersson, Phys. Chem. Chem. Phys., 2011, 13, 17177–17184 RSC.
- C. Kolbeck, T. Cremer, K. R. J. Lovelock, N. Paape, P. S. Schulz, P. Wasserscheid, F. Maier and H. P. Steinruck, J. Phys. Chem. B, 2009, 113, 8682–8688 CrossRef CAS PubMed.
- B. B. Hurisso, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 17737–17748 RSC.
- A. Jelicic, N. Garcia, H. G. Lohmannsroben and S. Beuermann, Macromolecules, 2009, 42, 8801–8808 CrossRef CAS.
- S. K. Poole, P. H. Shetty and C. F. Poole, Anal. Chim. Acta, 1989, 218, 241–264 CrossRef CAS.
- N. D. Khupse and A. Kumar, J. Phys. Chem. B, 2010, 114, 376–381 CrossRef CAS PubMed.
- S. Coleman, R. Byrne, S. Minkovska and D. Diamond, Phys. Chem. Chem. Phys., 2009, 11, 5608–5614 RSC.
- P. G. Jessop, D. A. Jessop, D. B. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- A. Deyko, S. G. Hessey, P. Licence, E. A. Chernikova, V. G. Krasovskiy, L. M. Kustov and R. G. Jones, Phys. Chem. Chem. Phys., 2012, 14, 3181–3193 RSC.
- L. Gontrani, O. Russina, F. Lo Celso, R. Caminiti, G. Annat and A. Triolo, J. Phys. Chem. B, 2009, 113, 9235–9240 CrossRef CAS PubMed.
- X. M. Liu, Y. L. Zhao, X. C. Zhang, G. H. Zhou and S. J. Zhang, J. Phys. Chem. B, 2012, 116, 4934–4942 CrossRef CAS PubMed.
- M. Fabris, V. Lucchini, M. Noe, A. Perosa and M. Selva, Chem. – Eur. J., 2009, 15, 12273–12282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01901f |
| This journal is © the Owner Societies 2014 |