 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of protein binding affinities within hydrogel-based molecularly imprinted polymers (HydroMIPs)
Hazim F.
EL-Sharif
,
Daniel M.
Hawkins
,
Derek
Stevenson
and
Subrayal M.
Reddy
*
Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: s.reddy@surrey.ac.uk; Tel: +44 (0)1483686396
First published on 16th June 2014
Abstract
Hydrogel-based molecularly imprinted polymers (HydroMIPs) were prepared for several proteins (haemoglobin, myoglobin and catalase) using a family of acrylamide-based monomers. Protein affinity towards the HydroMIPs was investigated under equilibrium conditions and over a range of concentrations using specific binding with Hill slope saturation profiles. We report HydroMIP binding affinities, in terms of equilibrium dissociation constants (Kd) within the micro-molar range (25 ± 4 μM, 44 ± 3 μM, 17 ± 2 μM for haemoglobin, myoglobin and catalase respectively within a polyacrylamide-based MIP). The extent of non-specific binding or cross-selectivity for non-target proteins has also been assessed. It is concluded that both selectivity and affinity for both cognate and non-cognate proteins towards the MIPs were dependent on the concentration and the complementarity of their structures and size. This is tentatively attributed to the formation of protein complexes during both the polymerisation and rebinding stages at high protein concentrations. We have used atomic force spectroscopy to characterize molecular interactions in the MIP cavities using protein-modified AFM tips. Attractive and repulsive force curves were obtained for the MIP and NIP (non-imprinted polymer) surfaces (under protein loaded or unloaded states). Our force data suggest that we have produced selective cavities for the template protein in the MIPs and we have been able to quantify the extent of non-specific protein binding on, for example, a non-imprinted polymer (NIP) control surface.
1. Introduction
As “smart” material polymer hydrogels have been the focus of considerable interest from both fundamental and applied perspectives, knowledge of their properties is of paramount importance for the research and development of new applications.1–3 Hydrogels are insoluble, cross-linked polymer network structures that are composed of hydrophilic homo- or hetero-co-polymers and have the ability to absorb water.4,5 The molecular imprinting community have exclusively researched the use of hydrogel-based molecularly imprinted polymers (HydroMIPs) in the past decade for their antibody-like receptor properties, and many different monomers are currently being used for different functional purposes.6,7 These monomers are generally chosen on their ability to form weak hydrogen bonds between the monomer and the template and are ideal for non-covalent molecular imprinted hydrogels.5,6 Hydrogels based on functional acrylamide monomers are known to be very inert, offer hydrogen bonding capabilities, and are biocompatible. For these reasons, functional acrylamides have been commonly used for molecular imprinting.5,6,8Molecular imprinting has been hard to adapt to aqueous conditions due to the specific polar interactions between good imprinted sites and the analyte which become weakened, and to the non-specific (hydrophobic) interactions between other small molecules and the gel which become strengthened.5 As such, common imprints have usually been low molecular weight non-biological molecules, such as drugs and pesticides.3,9–11 However, popularity for imprinting large bio-macromolecule templates such as nucleic acids, viruses and proteins has increased in the past decade, with a view to developing integrated molecularly imprinted polymer (MIP) sensors for disease markers. Furthermore, MIP selectivity is believed to depend on the orientation of the functional groups inside the cavities and the shape of the cavities. If there are two binding sites per template, several single-point bindings can occur but only one two-point binding. It is the two-point binding sites that provide high selectivity.12 The fundamental interactions between the polymer network and the imprinted template binding sites are the same attractive and repulsive interactions within the protein itself. These are van der Waals, hydrophobic, electrostatic, and hydrogen bonding. However, the challenge associated with binding in imprinted polymers is the selective template re-uptake in the cavity.
One of the principal goals of molecular imprinting is to achieve MIP binding affinities comparable to the high selectivity offered by proteins for their ligands.13
Recently, there have been reports of MIPs showing dissociation constants (Kd) of a similar magnitude to antibodies when binding proteins such as mellitin14,15 and trypsin.16Table 1 illustrates common classes of receptor–ligand interactions compared to those of previous biological MIP receptor–ligand dissociation constants. One of the most renowned interactions for having a high binding constant of 10−15 M is the biotin–avidin complex.13,17 The vitamin biotin and the egg-white protein avidin or streptavidin complex provides one of the largest measured association constants for a non-covalent interaction between a protein and small molecule.18 The strength of interaction comes from 15 amino acid residues on streptavidin. The specific positioning of the ligand in the active site allows for the formation of eight hydrogen bonds and eight sites of van der Waals interactions. The high specificity is compounded by four of these amino acids being part of a flexible loop that locks into place upon biotin binding, an “induced fit” that provides additional favourable interactions between protein and ligand.13,18 Despite the complex series of events, the process appears to come easy to such natural systems. The 15 amino acids are not all contiguous in the primary structure of streptavidin, and they are held in place by the overall fold of the protein. This is a common feature in essentially all protein–ligand interactions. The affinity of avidin for a number of biotin analogues has been determined, and small changes in structure have led to 100-fold decreases in binding affinity.13,18
| Ligand | Receptor | K d (mol L−1) |
|---|---|---|
| TDI, toluene 2,4-diisocyanate; MAA, methacrylic acid; EGDMA, ethylene glycol dimethacrylate; Ac.PABA, N-acryloyl para-aminobenzamidine; AAm, acrylamide; bis-AAm, N,N′-methylene bisacrylamide; TBAAm, N-tert-butylacrylamide; 3APM, N-(2-aminopropyl)-methacrylamide; AA, acrylic acid. Reproduced from ref. 17 with permission from Elsevier. | ||
| Classes | ||
| Ligands | Macromolecules | 10−3 to 10−15 |
| Substrate | Enzyme | 10−3 to 10−6 |
| Carbohydrate | Protein | 10−3 to 10−6 |
| Steroid hormones | Receptors at target tissue | 10−7 to 10−9 |
| Antigen | IgG antibodies | 10−8 to 10−10 |
| Specific examples | ||
| Glucose | Human red cell glucose transporter, Glut I | 1.5 × 10−2 |
| Fc portion of a mammalian IgG | Protein G | 5.2 × 10−7 |
| Tri-peptide inhibitor | Carboxypeptidase A | 10−14 |
| Pancreatic inhibitor | Trypsin | 6 × 10−14 |
| Biotin | Streptavidin | 10−15 |
| MIP examples | ||
| Cholesterol (steroid) | β-Cyclodextrin, TDI | 5.9 ± 1.2 × 10−4 |
| Leu-enkephalin (neuropeptide) | MAA, EGDMA | 1.0 ± 0.6 × 10−7 |
| Trypsin (enzyme) | Ac.PABA, AAm, bis-Aam | 3.75 × 10−8 |
| Melittin (apitoxin) | TBAAm, AAm, 3APM, AA | 25 × 10−12 |
Compared to protein–ligand complexes, protein–hydrogel complexes are not so well-studied and do not yet have the same specificities and affinities. Although protein–hydrogel complexes are believed to share the same types of interactions, the overall structural complex is the opposite to that of protein–ligand complexes, in that the receptor pocket or cavity is located within the polymer matrix and not the protein.
MIPs are typically highly cross-linked systems and by virtue of their rigid structure are therefore unable to offer many degrees of freedom to allow similar capture and locking to take place. However, HydroMIPs are able to swell and contract depending on solvent,19 ionic strength,4 buffer composition and pH,6 and the presence of other dissolved components in solution. If these parameters can be optimised to improve selective binding, compared to non-imprinted polymer controls, it could drastically improve the binding properties of such HydroMIPs.
This paper aims to investigate the rebinding affinity, selectivity and cross-selectivity of template protein molecules into hydrogel-based molecularly imprinted polymers using functional acrylamides of varying hydrophobicity.
2. Experimental
2.1. Reagents and materials
Acrylamide (AA), N-hydroxymethylacrylamide (NHMA), N-iso-propylacrylamide (NiPAm), N,N-methylenebisacrylamide (bis-AA), ammonium persulphate (APS), N,N,N,N-tetramethylethyldiamine (TEMED), sodium dodecyl-sulphate (SDS), glacial acetic acid (AcOH), bovine haemoglobin (BHb), bovine serum albumin (BSA), bovine liver catalase (BCat), and equine heart myoglobin (EMb) were all purchased from Sigma-Aldrich, Poole, Dorset, UK. Sieves (75 μm) were purchased from Inoxia Ltd., UK.2.2. Hydrogel productions
Hydrogel MIPs were synthesised by separately dissolving AA (54 mg), NHMA (77 mg), NiPAm (85.6 mg) and bis-AA as cross-linker (6 mg), (8.5 mg) and (9.5 mg) respectively along with template protein (12 mg) in 1 mL of MilliQ water. The solutions were purged with nitrogen for 5 minutes, then 20 μL of a 10% (w/v) APS solution and 20 μL of a 5% (v/v) TEMED solution were added. Polymerisation occurred at room temperature giving final crosslinking densities of 10%. For every HydroMIP created a non-imprinted ‘HydroNIP’ control was prepared in an identical manner but in the absence of protein. After polymerization, the gels were granulated separately using a 75 μm sieve. Of the resulting gels, 500 mg were conditioned by washing with five 1 mL volumes of MilliQ water followed by five 1 mL volumes of a 10% (w/v)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10% (v/v) SDS:AcOH eluent (pH 2.8). A further five 1 mL volume washes of MilliQ water followed to remove any residual SDS:AcOH eluent and equilibrated the gels. Each wash step was followed by a centrifugation, whereby the gels were centrifuged using an Eppendorf mini-spin plus centrifuge for 3 minutes at 6000 rpm (RCF: 2419 × g). All supernatants were collected for analysis by spectrophotometry.
:10% (v/v) SDS:AcOH eluent (pH 2.8). A further five 1 mL volume washes of MilliQ water followed to remove any residual SDS:AcOH eluent and equilibrated the gels. Each wash step was followed by a centrifugation, whereby the gels were centrifuged using an Eppendorf mini-spin plus centrifuge for 3 minutes at 6000 rpm (RCF: 2419 × g). All supernatants were collected for analysis by spectrophotometry.
2.3. MIP binding affinity studies
Once the gels were equilibrated, 1 mL volumes of reload protein (BHb, EMb and BCat) solutions of known concentrations (0.1–5 mg mL−1) prepared in MilliQ water were allowed to associate at room temperature with the respective imprinted gels for 20 minutes. Cross-selectivity studies were also conducted to assess the binding affinity of the original template protein. This was achieved by loading BSA and EMb on a BHb imprinted gel. Gels were then washed with four 1 ml volumes of MilliQ water solution. Each reload and wash step for all MIPs and NIP controls was followed by centrifugation at 6000 rpm (RCF: 2419 × g) for 3 minutes. All supernatants were collected for analysis by spectrophotometry.2.4. Spectrophotometric analysis
All supernatant fractions were analysed at specific peak wavelengths using a UV mini-1240 CE spectrophotometer (Shimadzu Europa, Milton Keynes, UK) to determine the protein concentrations. This was done in the appropriate wash/elution solution. Calibration curves in 10% AcOH:SDS and MilliQ water were prepared for BSA, BHb, BCat and EMb. Peak wavelengths for BHb in MilliQ water and 10% AcOH:SDS were found to be 406 nm and 395 nm respectively. Peak wavelengths for BCat in MilliQ water and 10% AcOH:SDS were found to be 404 nm and 392 nm respectively. Peak wavelengths for EMb in MilliQ water and 10% AcOH:SDS were found to be 408 nm and 396 nm respectively. Peak wavelengths for BSA in MilliQ water and 10% AcOH:SDS were found to be 288 nm and 290 nm respectively.2.5. Curve fitting
Curve fitting was carried out by non-linear least squares regression using saturation binding – one site specific binding with Hill Slope equation in GraphPad Prism 6.2.6. Atomic force spectroscopy analysis
AA MIP gels were fabricated as described in Section 2.2. Following the sieving, the MIP gels were washed with five 2 mL volumes of RO water followed by five 2 mL volumes of 10% SDS/acetic acid eluent. Each wash/elution step was performed by centrifugation. All gels were diluted 1:1 with RO water. Fifty microliters of each gel sample was pipetted into an Eppendorf tube to which 50 μL of a 5% (v/v) acrolein solution was added, and the samples were placed in a Pelco Biowave microwave (Ted Pella Inc.) and treated under vacuum at 20 °C (plate temperature) and 250 Watts for 2 min (on), 2 min (off), and 2 min (on). A 100 μL volume of RO water was added to the samples, vortex mixed, and microcentrifuged for 5 min before being treated under vacuum at 20 °C and 250 Watts for 1 min in the microwave. The supernatant was discarded. The RO water treatments were repeated in triplicate. The samples were then dehydrated using a series of 100 μL methanol washes that increased in concentration sequentially from 5% (v/v) through to 95% (v/v) (at 5% increments) in an identical manner as the RO washes. Three 100 μL volumes of 100% methanol were finally employed in an identical manner to the previous dehydration stages, which were followed by the addition of three drops of propylene oxide. The samples were treated with three 100 μL volumes of hexamethyldisilazane (HMDS), (mixed, centrifuged for 5 min, and supernatant removed after each HMDS addition) with the final treatment leaving a small dry sample at the base of the Eppendorf tube. Thermanox coverslips were dipped in 0.1% polylysine and allowed to air dry. A spatula was used to apply a small measure (ca. 0.1 g) of each HydroMIP and HydroNIP sample to a polylysine-coated Thermanox® coverslip, with the hydrogel spread homogenously across the surface of the coverslip. Each sample was then cryogenically treated as follows and stored in a dry chamber prior to analysis. A 1 μL aliquot of each gel suspension was pipetted onto 400 mesh, carbon stabilized, Formvar coated glow discharged copper grids. The grids were plunged into liquid nitrogen. Following the constant agitation of the sample in the liquid nitrogen for approximately 30 s, the grids were transferred to 100% methanol and agitated for approximately 20 s. The grids were then transferred to HMDS and again agitated for approximately 20 s.
An AFM Bioscope System (Nanoscope 3A, Digital Instruments) AFM mounted on an Axiovert 100 TV inverted microscope (Zeiss) was used in contact mode operation. The Axiovert light microscope was used to focus upon a sample region that was homogenous in appearance and devoid of any topographic features of extreme height that would impede the free movement of the cantilever across the sample surface. The probe was advanced toward the sample surface using the automated approach function. The tip was allowed to repeatedly touch and retract from the sample surface for 3 min, resulting in approximately 90 force curves. The process was repeated on the same sample in three different sample areas. For each experiment, 30 force curves were randomly selected (10 from each repeat). The binding events were quantified using a proprietary software package (NforceR) to determine the adhesion force between AFM probe and hydrogel sample and analyzed using Matlab software (Math Works). Each of the HydroMIP and HydroNIP samples, plus a polylysine-coated control coverslip, were interrogated in an identical fashion using protein (BHb) modified probes operating in the force measurement mode. From the raw values generated, a force (F) was calculated using the following formula (eqn (1)):
| F = R × Z × S × C | (1) |
3. Results and discussion
3.1. MIP binding affinity
Experimentally derived receptor–ligand binding plots of bound versus free protein concentration are not expected to yield a typical saturation profile due to linearly increasing non-specific binding.9 However, the obtained batch binding isotherms (Fig. 1) exhibited progressive saturation at higher protein concentrations for MIP. This suggests that at higher protein concentrations polymer binding occurs via a mixture of specific binding at imprinted sites and nonspecific adsorption in to the polymer matrix due to a limited number of binding sites. More strikingly with the NIP, the isotherm demonstrated a step change from near zero binding (at low protein concentration) to saturation at a higher critical protein concentration. This supports our understanding that the NIP control has no discernible features for selective protein binding. At lower protein concentrations, the non-specifically bound protein is a surface effect. However, at the higher critical protein loading, some of the surface bound protein is able to break-through the NIP surface. The immediate saturation in the isotherm suggests that the NIP is predominately impermeable to protein.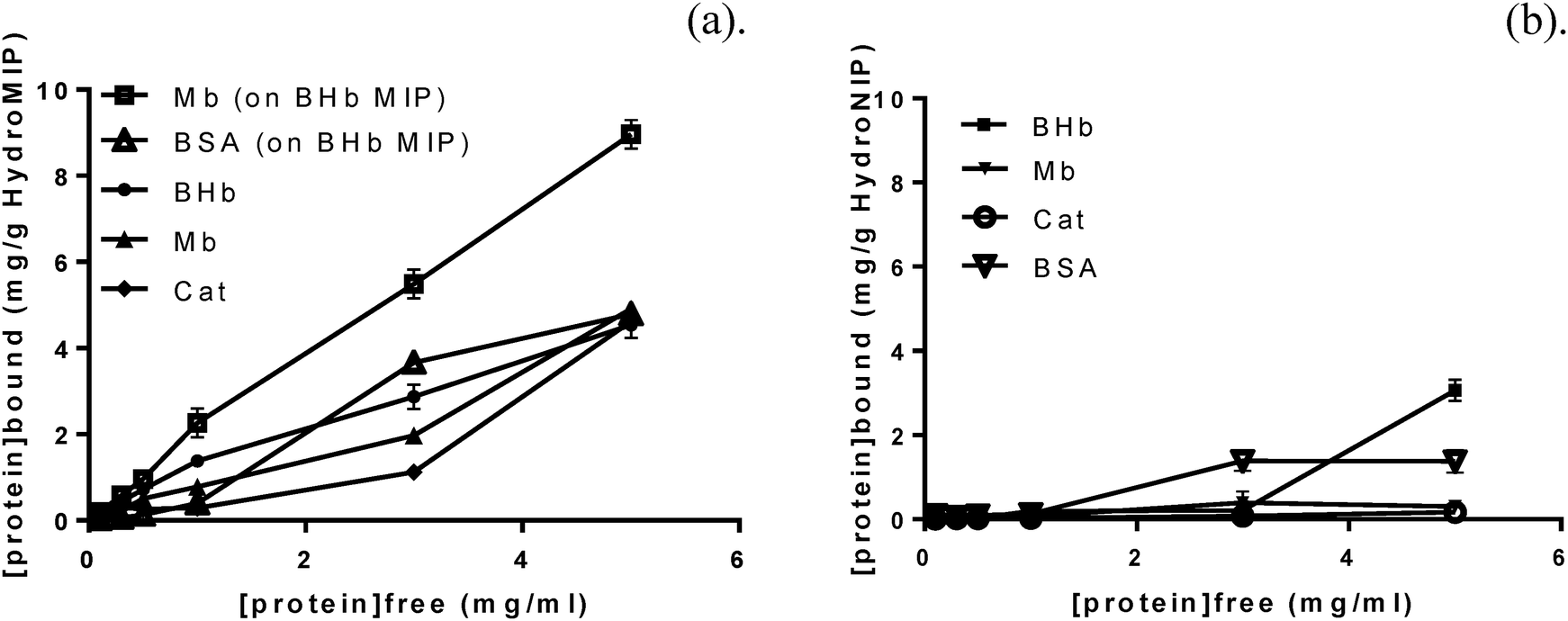 | ||
| Fig. 1 Equilibrium binding isotherms for proteins BHb, BSA, EMb and BCat for: (a) respective polyAA-MIPs, and cross-selected (BSA, Mb) on BHb–MIP; (b) NIP controls. Data represents mean ± S.E.M., n = 3. | ||
In order to determine affinity constants and binding site concentrations it is often necessary to re-plot the isotherm data in the form of a Scatchard plot using the following formula (eqn (2)).9
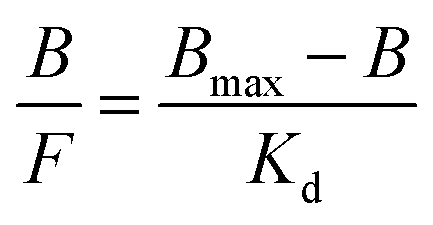 | (2) |
This is a linearized form of the Langmuir equation, of which the transformation has shown to distort experimental error, and only assumes single affinity constant binding site populations. Bmax is the apparent maximum number of binding sites, Kd the equilibrium dissociation constant, F the concentration of free protein, and B the concentration of bound protein. Moreover, due to the heterogeneous distribution of binding sites in MIP matrices, MIP–ligand binding studies for simple organic molecules, such as pesticides, herbicides and drugs, have generally reported non-linear concave curves.9 The imprinting of bio-macromolecules, such as proteins, presents a variety of challenges, i.e. proteins are relatively labile, and have changeable conformations which are sensitive to various factors, e.g. solvent environments, pH and temperature.6 Therefore, alternative approaches such as the Hill equation (eqn (3)), which is indicative of binding site cooperativity have been used for MIP–ligand binding analysis.9 In this case Y is the binding site occupancy, and nh is the Hill coefficient which relates to a linear Scatchard plot when nh is equal to 1.0, and is indicative of ligand binding with no cooperativity to one site.
 | (3) |
Variations in nh, i.e. if greater than 1.0, present a sigmoidal graph indicating receptor/ligand having multiple binding sites with positive cooperativity. Such would be expected of MIP–ligand binding due to the heterogeneous distribution of binding sites. However, if nh is less than 1.0 it can also be indicative of multiple binding sites, nonetheless with different affinities for template or negative cooperativity.9
Using the latter approach, specific binding saturation profiles were plotted (Fig. 2a), and apparent Kd (μM) and Bmax (μmol g−1 of polymer) values were determined. Proteins imprinted within polyacrylamide (polyAA), poly N-hydroxymethylacrylamide (polyNHMA) and poly N-iso-propylacrylamide (polyNiPAm) MIP gels were revealed to exhibit micromolar affinities towards their cognate proteins (Table 2). The % of theoretical total binding sites, which is a useful indication of imprinting/binding efficiency, was also determined. This was derived from the amount of the template protein used for the polymerization. Hill coefficients (nh) for all MIPs demonstrated positive cooperativity (nh > 1), implying heterogeneous binding characteristics. Positive cooperativity also implies that the first protein molecules bound to the MIP polymer with a lower affinity than did subsequent protein molecules. Our postulation is that in MIP formation the template molecules are also capable of heterogeneous populations, i.e. free and clustered proteins, when templates are imprinted at high concentrations, in this case 12 mg mL−1. The resultant population of imprinted sites would therefore contain some cavities that comprise of protein clusters. This hypothesis is supported by our force spectroscopy analysis of MIPs in Section 3.2.
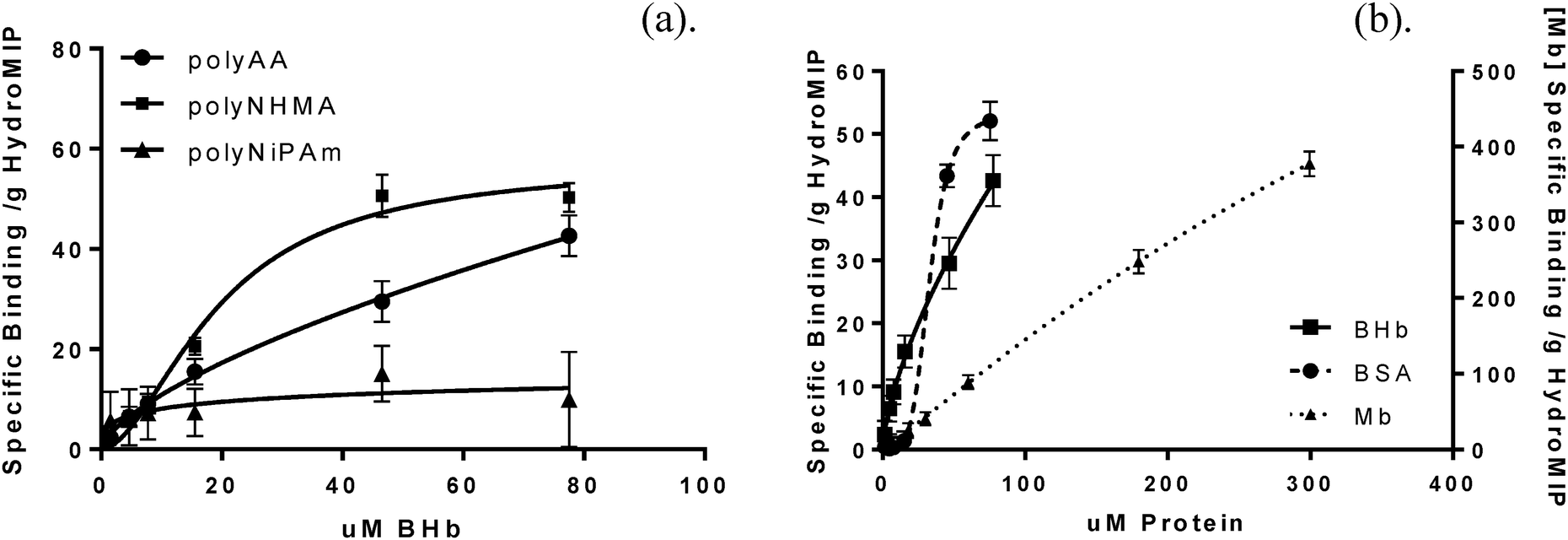 | ||
| Fig. 2 Specific binding with Hill slope saturation profiles: (a) BHb template protein recognition by cognate polyAA, polyNHMA and polyNiPAm HydroMIPs; (b) cross-selective EMb and BSA binding data in relation to template BHb on a BHb–MIPpolyAA. Specific binding was calculated by subtracting the amount of protein bound to the NIP from that bound to the MIP, based on the assumption that binding exhibited by the NIP is an estimation of non-specific, low affinity interactions. Data represents mean ± S.E.M., n = 3. | ||
| Protein | K d (μM) | B max (μmol g−1 polymer) | Hill coefficient (nh) | % of theoretical binding sites | MIP |
|---|---|---|---|---|---|
| a Denotes a 15% cross-linking density. b Denotes a 5% cross-linking density in HydroMIP synthesis. c Denotes the cross-selective EMb and BSA proteins on a BHb–MIPpolyAA. Data represents mean ± S.E.M., n = 3. | |||||
| BHb | 24.7 ± 3.8 | 53.14 | >1 | 14 | PolyAA |
| 19.4 ± 5.5 | 56 | >1 | 15 | PolyNHMA | |
| 16.1 ± 2.1 | 17.96 | >1 | 5 | PolyNiPAm | |
| Emb | 114.4 ± 3.1 | 180.1 | >1 | 13 | PolyAA |
| 315.5 ± 3.1 | 146 | >1 | 10 | PolyNHMA | |
| 345.6 ± 2.1 | 496.1 | >1 | 35 | PolyNiPAm | |
| BCat | 23.3 ± 0.6 | 17.28 | >1 | 18 | PolyAA |
| 5.5 ± 0.8 | 12.06 | >1 | 13 | PolyNHMA | |
| 20.4 ± 0.2 | 20.36 | >1 | 21 | PolyNiPAm | |
| Emb | 43.9 ± 3.1 | 479.5 | >1 | 33 | PolyAAa |
| BCat | 17.1 ± 1.8 | 12.61 | >1 | 13 | PolyAAb |
| Embc | 11.7 ± 4.1 | 194.6 | >1 | 14 | BHb–polyAAa |
| BSAc | 32.8 ± 0.6 | 53.19 | >1 | 14 | BHb–polyAA |
Interestingly, the binding affinity is highest for BHb–MIPpolyAA while both EMb and BCat exhibit the lowest affinity for a MIPpolyAA. It has previously been observed that with smaller size proteins a higher crosslinking density is necessary; the opposite is also true for larger proteins.6,13 Improved polyAA MIP affinities for EMb and BCat using optimised cross-linked densities of 15% and 5% respectively are also illustrated in Table 2. These MIPs revealed higher affinity constants for their native proteins. Therefore previous low affinities exhibited by MIPspolyAA towards BCat and EMb can be attributed to the fact that fewer cavities were imprinted due too high and too low of a crosslinking density respectively. Furthermore, HydroMIPs based on polyAA show the most promising binding affinities closely followed by polyNHMA, then polyNiPAm which is coherent with previously reported MIP selectivity trends.6 This has been attributed to the hydrophobicity of the polymers, in which the neutral polyAA is providing ideal imprinting cavities unlike the hydrophilic polyNHMA and hydrophobic polyNiPAm.
Cross-selectivity studies of the polyAA hydrogel-based MIPs were also conducted (Table 2). BSA and EMb were chosen for their similarity to BHb protein, BSA being of similar size BHb (66.5 and 64.5 kDa, respectively) and EMb (17.5 kDa) representing a single BHb sub-unit. Calculated dissociation constants for the cross-selected proteins Mb and BSA were 11.69 μM and 32.77 μM respectively. The MIPs high affinity for non-BHb target could also be justified by the previous hypothesis that protein complex formation can occur in imprinting. It is therefore possible that complementary complex formations due to the high similarities between BSA, EMb and BHb structures that further protein clustering was occurring, i.e. it would take four EMb molecules for example to aggregate or cluster to fill a single BHb recognition site or cavity. To further illustrate this theory, the equilibrium binding isotherm for cross-selective BSA and EMb binding on a BHb–MIPpolyAA (Fig. 2b) reveals that EMb increases linearly and clearly does not reach saturation at the same rate as BHb. BSA on the other hand demonstrates a curvilinear relationship and quickly reaches saturation. It has previously been postulated that when rebinding BSA to a BHb MIP the BSA due to shape and size does not bind specifically, but rather displaces the non-specific recognition sites of cavities and the nonspecific binding of BHb to BHb–MIP.20 Therefore, these results suggest that there is some degree of nonspecific cross-selectivity exhibited by the MIPs, as a saturation profile would be expected for the template BHb but not the non-cognate proteins.
Although this is a useful indication of imprinting/binding efficiency, and with the structures and populations in MIPs remaining currently unknown, it would be important to provide multipoint interacting binding sites of high selectivity in resulting MIP matrices. This would be beneficial to certain biochemical high-performance liquid chromatography (HPLC) assay screenings that use several whole blood and serum protein markers, such as liver function tests.21,22 Previous work6 shows that the application of MIPs in biocompatibility studies using human plasma and serum samples via optimised buffer conditioning strategies has major implications in improving the selectivity of MIPs in terms of rebinding efficiency. Furthermore, the micro-molar detection ranges we report are relevant with the (0.3–350 μg mL−1) range currently used in such screenings.21,22
3.2. Force spectroscopy measurements
One way in which a MIP effect can be defined is in relation to a NIP prepared in an identical manner to that of the MIP, in the absence of the template molecule. Fig. 3 displays the trends observed following the retraction force interrogation of NIP, freshly prepared BHb–MIP with protein still in cavities (MIP1), BHb–MIP with empty cavities (MIP2) and MIP2 reloaded with protein (referred to as MIP3), all interrogated with a BHb-modified AFM probe. The BHb-modified AFM tip was used to interrogate the presence of BHb-specific cavities within the MIP2 HydroMIP sample. An average force size of 23 nN was exhibited by the MIP2 sample. This force was significantly greater than the average force observed for the NIP control sample, which was 19 nN. This was an expected result, as the MIP2 sample possessed unoccupied BHb specific sites that were capable of accepting the immobilised template upon the AFM tip. Binding between these sites and the BHb molecule occurred, which in turn resulted in a greater force being required to withdraw the tip from the sample. | ||
| Fig. 3 Distribution of adhesive forces obtained between BHb functionalised AFM probe and polyAA MIP or NIP surfaces. | ||
The Gaussian distributions detail the number of adhesion events that occurred, in relation to the forces required to withdraw the AFM probe from the hydrogel surfaces. A distinctive trend is observed. The NIP control exhibited the smallest force, with a (mean) value of 18.90 nN required to withdraw the probe from the NIP surface. Similar force measurements were observed for MIP1 and MIP3. Most significantly though, a force of 23 nN was required to withdraw the template-modified AFM tip from the MIP2 sample. This occurred due to the presence of unoccupied template-specific imprinted cavities within the polymer, which accepted the template-coated probe as a result of the shape, size and charge orientation of the cavity. Typically, single antibody–antigen type molecular interactions result in force measurements ranging 100–300 pN depending on the number of intermolecular interactions (e.g. hydrogen bonds) per binding pair.23
The fact that the force values were in the nN range suggests that these larger values could be an artefact of the cryogenic preparation of the MIPs or that there are multiple protein interactions occurring between the bio-modified AFM tip and the surface. Notwithstanding this, there is a clear distinction in the force values for MIP with cavities exposed and MIP (with cavities occupied) or NIP. At best the protein-modified AFM tip would comprise of multiple protein molecules tethered to it, creating a bristle effect. Additionally, therefore, it is likely that we are seeing multi-protein interactions between AFM tip and the MIP surface. An approximate 5 nN increase in attractive force between NIP (or even protein-loaded forms of MIP) compared with MIP2 suggests that the exposed cavities in MIP2 can potentially accommodate more than one protein molecule. It is therefore plausible that during the imprinting process, cavities comprising an agglomeration of protein molecules were also being formed, rather than the generally accepted single protein cavities.
4. Conclusions
It is evident from the equilibrium binding data and supporting force spectroscopy data, that MIP cavities accommodated an agglomeration of template protein molecules rather than just a single molecule. Binding data also demonstrates micro-molar MIP affinities, and therefore our smart materials are exhibiting protein-binding affinities which are now comparable to natural receptor systems. This is an exciting and new achievement in the growing area of hydrogel imprinting. Further investigating the development of such highly selective synthetic antibody systems could provide an inexpensive, fast, sensitive and efficient diagnostic method within medical, environmental and food diagnostics in the future.Acknowledgements
The authors would like to thank the UK Engineering and Physical Sciences Research Council (EPSRC) Grants (EP/G014299/1) and NERC/ACTF (RSC) for supporting this project.References
- A. Poma, A. P. F. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637, DOI:10.1016/j.tibtech.2010.08.006.
- K. Mosbach, Anal. Chim. Acta, 2001, 435, 3–8, DOI:10.1016/S0003-2670(01)00800-5.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O'Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180, DOI:10.1002/jmr.760.
- M. E. Byrne, K. Park and N. A. Peppas, Adv. Drug Delivery Rev., 2002, 54, 149–161, DOI:10.1016/S0169-409X(01)00246-0.
- M. E. Byrne and V. Salian, Int. J. Pharm., 2008, 364, 188–212, DOI:10.1016/j.ijpharm.2008.09.002.
- H. F. El-Sharif, Q. T. Phan and S. M. Reddy, Anal. Chim. Acta, 2014, 809, 155–161 CrossRef CAS PubMed.
- D. M. Hawkins, D. Stevenson and S. M. Reddy, Anal. Chim. Acta, 2005, 542, 61–65, DOI:10.1016/j.aca.2005.01.052.
- S. M. Reddy, Q. T. Phan, H. El-Sharif, L. Govada, D. Stevenson and N. E. Chayen, Biomacromolecules, 2012, 13, 3959–3965, DOI:10.1021/bm301189f.
- N. Lavignac, K. R. Brain and C. J. Allender, Biosens. Bioelectron., 2006, 22, 138–144, DOI:10.1016/j.bios.2006.03.017.
- D. R. Kryscio and N. A. Peppas, Acta Biomater., 2012, 8, 461–473, DOI:10.1016/j.actbio.2011.11.005.
- D. Stevenson, TrAC, Trends Anal. Chem., 1999, 18, 154–158, DOI:10.1016/S0165-9936(98)00094-6.
- A. Fernández-Barbero, I. J. Suárez, B. Sierra-Martín, A. Fernández-Nieves, F. J. de las Nieves, M. Marquez, J. Rubio-Retama and E. López-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108, DOI:10.1016/j.cis.2008.12.004.
- D. E. Hansen, Biomaterials, 2007, 28, 4178–4191, DOI:10.1016/j.biomaterials.2007.06.017.
- Y. Hoshino, T. Kodama, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2008, 130, 15242–15243, DOI:10.1021/ja8062875.
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645, DOI:10.1021/ja102148f.
- A. A. Vaidya, B. S. Lele, M. G. Kulkarni and R. A. Mashelkar, J. Appl. Polym. Sci., 2001, 81, 1075–1083, DOI:10.1002/app.1529.
- J. Z. Hilt and M. E. Byrne, Adv. Drug Delivery Rev., 2004, 56, 1599–1620, DOI:10.1016/j.addr.2004.04.002.
- S. Freitag, I. Le Trong, A. Chilkoti, L. A. Klumb, P. S. Stayton and R. E. Stenkamp, J. Mol. Biol., 1998, 279, 211–221, DOI:10.1006/jmbi.1998.1735.
- S. M. Reddy, D. M. Hawkins, Q. T. Phan, D. Stevenson and K. Warriner, Sens. Actuators, B, 2013, 176, 190–197, DOI:10.1016/j.snb.2012.10.007.
- Q. Gai, F. Qu, T. Zhang and Y. Zhang, J. Chromatogr. A, 2011, 1218, 3489–3495, DOI:10.1016/j.chroma.2011.03.069.
- P. A. Lieberzeit, R. Samardzic, K. Kotova and M. Hussain, Procedia Eng., 2012, 47, 534–537, DOI:10.1016/j.proeng.2012.09.202.
- S. A. Piletsky, N. W. Turner and P. Laitenberger, Med. Eng. Phys., 2006, 28, 971–977, DOI:10.1016/j.medengphy.2006.05.004.
- F. Kienberger, G. Kada, H. Mueller and P. Hinterdorfer, J. Mol. Biol., 2005, 347, 597–606, DOI:10.1016/j.jmb.2005.01.042.
| This journal is © the Owner Societies 2014 |