The calculation of 29Si NMR chemical shifts of tetracoordinated silicon compounds in the gas phase and in solution†
Received
22nd April 2014
, Accepted 25th June 2014
First published on 27th June 2014
Abstract
Aiming at the identification of an efficient computational protocol for the accurate NMR assessment of organosilanes in low-polarity organic solvents, 29Si NMR chemical shifts of a selected set of such species relevant in organic synthesis have been calculated relative to tetramethylsilane (TMS, 1) using selected density functional and perturbation theory methods. Satisfactory results are obtained when using triple zeta quality basis sets such as IGLO-III. Solvent effects impact the calculated results through both, changes in substrate geometry as well as changes in the actual shieldings. Spin–orbit (SO) corrections are required for systems carrying more than one chlorine atom directly bonded to silicon. Best overall results are obtained using gas phase geometries optimized at MPW1K/6-31+G(d) level in combination with shielding calculations performed at MPW1K/IGLO-III level in the presence of the PCM continuum solvation model.
Introduction
The measurement of 29Si NMR chemical shifts is exceedingly helpful in elucidating the identity of silicon-containing molecular systems.1–13 This is not only true for stable reactants and products of well-defined transformations, but also for silicon-based species generated as transient intermediates in the course of a reaction.14 In this latter case the combination of theoretically predicted and experimentally measured 29Si NMR chemical shifts is particularly helpful, as was amply demonstrated in detailed studies of, for example, silylenes15 and disilenes.16 When attempting to follow the course of base-catalyzed silylation reactions of alcohols, which constitute an important protecting group strategy in organic synthesis,17,18 we were confronted with the appearance of a number of silicon-based species with rather similar chemical shifts in the solution-phase 29Si NMR spectra.19 With the goal of identifying a computational protocol for the accurate theoretical prediction of the respective chemical shifts in low-polarity organic solvents, we analyze here the performance of strategies based on DFT- and MP2-level calculations.20–27 Our particular emphasis will be on the effects of geometry optimization, methods for actual shift calculations, the modeling of solvent effects, and the evaluation of relativistic effects.
Results and discussion
The effects of geometry optimization
We first investigated the influence of molecular structures optimized using several computationally efficient methods on the quality of 29Si chemical shifts evaluated on these structures at MP2(FULL)/IGLO-III level. These calculations were performed using SiMe3Cl (2) as a model system for silyl chloride reagents used in organic synthesis. Experimentally measured chemical shifts for this system are available in selected solvents at room temperature such as benzene with δ(29Si, 2) = +30.21 ppm,28 benzene-d6 with δ(29Si, 2) = +30.64 ppm,29 toluene-d8 with δ(29Si, 2) = +30 ppm,30 and CDCl3 with δ(29Si, 2) = +30.7 ppm.31 It thus appears that organic solvents of intermediate polarity have only a very limited influence on the 29Si chemical shift in 2. Using microwave spectroscopy the gas phase Si–Cl bond distance in 2 has been determined in two separate studies as r(Si–Cl, 2) = 202.2 ± 5 pm32 and 203 pm.33 In the solid state the Si–Cl bond distance has been determined to be of comparable length at r(Si–Cl, 2) = 209 pm,34 201.0 pm (0.23 GPa and 296 K),35 and 208.6 pm (0.1 MPa and 157 K).36
The marked dependence of computed chemical shifts on the molecular structure is illustrated in Fig. 1, which displays 29Si chemical shifts calculated for SiMe3Cl (2) at MP2(FULL)/IGLO-III level employing structures optimized at various levels of theory in combination with the 6-31+G(d) basis set (the results obtained from MP2(FULL)/IGLO-III geometry optimizations are included as a reference). 29Si chemical shifts evaluated for various points along a relaxed scan (MPW1K/6-31+G(d) structures) of the Si–Cl distance in 2 from 200 to 300 pm (solid line in Fig. 1) show an almost linear dependence with larger Si–Cl distances leading to systematically higher chemical shifts. This trend is also visible in the δ(29Si) data evaluated on molecular structures fully optimized with different methods in the gas phase (Fig. 1, solid circles): the shortest Si–Cl bond distances (about 208.5 pm) are obtained in MPW1K and MP2(FC) optimizations, while the other density functionals tested yield longer Si–Cl bonds reaching 211 pm obtained with the B3LYP hybrid functional. The shift values calculated for these gas phase geometries at MP2(FULL)/IGLO-III level vary by approx. 2 ppm from +29.7 to +32.0 ppm. As detailed further below, use of the PCM solvent model for structure optimization (Fig. 1, empty squares) leads to systematically longer Si–Cl bonds and, correspondingly, to systematically higher δ(29Si) values, with larger deviations from experiment for the cases tested.
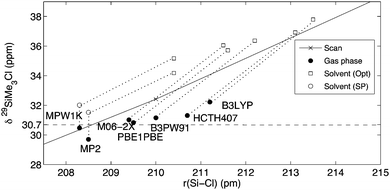 |
| | Fig. 1 Calculated δ(29Si) values for SiMe3Cl (2) (MP2: solid circles and PCM/MP2: empty symbols; IGLO-III basis set) using molecular structures optimized at various levels of theory using the 6-31+G(d) basis set. Dashed line: experimental value in CDCl3; solid line: relaxed scan along the Si–Cl bond (gas phase, MPW1K structures). Empty circles: PCM/MP2 shifts on gas phase structures; empty squares: PCM/MP2 shifts on solution-phase geometries (all solvent calculations: CHCl3, UAHF radii). | |
We further explored whether chemical shift predictions can be improved through the use of more sophisticated basis sets in gas phase geometry optimizations for selected Pople-style and correlation consistent (cc) basis sets (Table 1). Use of the systematically developed cc basis sets results in a systematic decrease of Si–Cl bond distances in the order cc-pVDZ, cc-pVTZ, and cc-pVQZ, irrespective of the theoretical method used. This directly impacts the 29Si chemical shift calculations performed at MP2(FULL)/IGLO-III level, where shorter Si–Cl bonds are again observed to yield lower shift values. A comparable trend is not observed for Pople-style basis sets of double- and triple-zeta quality. Among all quantum mechanical methods the MP2(FC) level is most strongly affected by these variations. Taking the MP2(FC)/cc-pVQZ structure as reference, the best agreement is observed at the MPW1K/6-31+G(d) level, which was therefore chosen for all subsequent geometry optimizations.
Table 1 Influence of basis set choice on the gas phase geometry optimized with various methods and the gas phase 29Si chemical shift of 2 (evaluated at MP2(FULL)/IGLO-III level)
| Basis set |
PBE1PBE |
MPW1K |
M06-2X |
MP2(FC) |
|
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
| 6-31+G(d) |
209.5 |
+30.84 |
208.3 |
+30.32 |
209.4 |
+31.02 |
208.5 |
+29.70 |
| 6-31+G(2d,p) |
209.6 |
+31.81 |
208.5 |
+31.37 |
209.5 |
+31.90 |
210.2 |
+32.60 |
| 6-31++G(2d,p) |
209.6 |
+31.82 |
208.5 |
+31.38 |
209.5 |
+31.92 |
210.3 |
+32.63 |
| 6-31++G(2df,p) |
208.9 |
+30.90 |
207.8 |
+30.43 |
211.4 |
+34.14 |
208.5 |
+30.44 |
| 6-311++G(2d,p) |
209.4 |
+31.82 |
208.3 |
+31.31 |
209.3 |
+31.90 |
209.9 |
+32.30 |
| cc-pVDZ |
211.2 |
+32.98 |
210.2 |
+32.63 |
211.1 |
+33.16 |
210.7 |
+31.83 |
| cc-pVTZ |
209.1 |
+31.23 |
208.1 |
+30.81 |
209.1 |
+31.39 |
208.6 |
+30.79 |
| cc-pVQZ |
208.5 |
+30.77 |
207.6 |
+30.36 |
208.5 |
+30.92 |
207.8 |
+30.24 |
Theoretical methods for chemical shift calculations
Chemical shift calculations at MP2(FULL)/IGLO-III level are quite challenging for larger molecular systems and an effort has therefore been made to identify more economical approaches. Table 2 displays 29Si shift values for SiMe3Cl (2) computed with selected combinations of methods and basis sets based on the MPW1K/6-31+G(d)-optimized geometry. Compared to the experimental value of δ(29Si, 2) = +30.7 ppm31 largest deviations are found for the 6-311++G(2d,2p)26 basis set, while the def2-TZVP,37,38 cc-pVTZ,39 and IGLO-III40 triple zeta basis sets yield generally better results. The quadruple zeta pcS-3 basis set specifically developed for NMR calculations41 performs as well as IGLO-III, but its use is computationally significantly more demanding. We note that second-order perturbation theory in its canonical (MP2) or local (LMP2) variants perform best, closely followed by the HCTH40742 and MPW1K20 hybrid functionals. Interestingly, the DF-LMP2/IGLO-III43 calculations closely reproduce the much more costly MP2(FULL)/IGLO-III calculations, which we chose as reference method for 29Si chemical shift calculations. The worst performer found here is clearly the M06-2X44 functional, which responds more sensitively to the individual basis set design than any other method listed in Table 2. The deviations of MPW1K and HCTH407 are comparable to those of the MP2 methods and the performance of these two methods is better than that of the other DFT methods tested here.
Table 2 Calculated δ(29Si) chemical shifts for 2 using different theoretical methods and basis setsa
| Methodb |
6-311++G(2d,2p) |
Def2-TZVP |
cc-pVDZ |
cc-pVTZ |
cc-pVQZ |
pcS-3 |
IGLO-III |
|
Geometries optimized with MPW1K/6-31+G(d).
Method used for NMR chemical shift calculations.
|
| HCTH407 |
+35.00 |
+32.37 |
+32.63 |
+32.91 |
+26.37 |
+31.94 |
+31.40 |
| B3LYP |
+36.37 |
+34.29 |
+34.13 |
+34.14 |
+27.60 |
+33.44 |
+32.94 |
| B3PW91 |
+36.84 |
+34.92 |
+35.31 |
+34.64 |
+28.78 |
+34.15 |
+33.66 |
| PBE1PBE |
+36.56 |
+34.68 |
+35.35 |
+34.83 |
+29.37 |
+33.93 |
+33.31 |
| MPW1K |
+34.83 |
+33.06 |
+33.89 |
+33.59 |
+28.09 |
+32.44 |
+31.88 |
| M06-2X |
+42.67 |
+38.10 |
+38.85 |
+31.00 |
+36.82 |
+35.64 |
+37.24 |
| DF-LMP2 |
+33.05 |
+31.47 |
+31.36 |
+33.09 |
+29.55 |
+30.25 |
+28.40 |
| MP2(FULL) |
+33.65 |
+31.92 |
+30.71 |
+32.54 |
+28.59 |
+31.31 |
+30.32 |
Solvent effects
The experimental reference shift data for SiMe3Cl (2) has been determined in apolar organic solvents such as CDCl3 or benzene. As a first straightforward approach to account for solvent effects we performed shift calculations in combination with the polarizable continuum model (PCM) for chloroform at PCM/MP2(FULL)/IGLO-III level based on the MPW1K/6-31+G(d) gas-phase geometry, which leads to downfield shifts of the 29Si signal in SiMe3Cl (2) by 1.5–2.0 ppm relative to the gas phase value (+30.32 ppm), depending on the particular PCM variant employed (Table 3). An additional downfield shift of the same magnitude is obtained upon inclusion of the polarizable continuum also for geometry optimization at PCM/MPW1K/6-31+G(d) level owing to the elongated Si–Cl bond present in the optimized structure (Table 3). From a glance at the data compiled in Table 3 it is apparent that the particular choice of the atomic radii used to construct the solute cavity in the PCM calculations has only a minute influence on structures and chemical shifts.
Table 3 Influence of continuum solvation models on 29Si chemical shift calculations (PCM/MP2(FULL)/IGLO-III) for SiMe3Cl (2)
| |
Gas-phase structurea |
Solution-phase structureb |
|
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
r(Si–Cl) (pm) |
δ(29Si) (ppm) |
|
Optimized at MPW1K/6-31+G(d) level.
Optimized in CDCl3 with different solvation models at MPW1K/6-31+G(d) level.
|
| PCM(UFF) |
208.4 |
+31.80 |
210.19 |
+34.34 |
| PCM(UAHF) |
208.4 |
+32.15 |
210.40 |
+35.00 |
| PCM(UAKS) |
208.4 |
+32.15 |
210.40 |
+35.00 |
| SMD |
208.4 |
+32.28 |
210.48 |
+35.22 |
A more detailed picture is presented in Fig. 1 above, where the PCM/MP2(FULL)/IGLO-III shift data using the gas-phase geometries (empty circles) and the solution-phase geometries (empty squares) for selected hybrid DFT methods are shown. Taking the MPW1K/6-31+G(d) gas phase geometries as an example, we can see that the PCM(UAHF)/MP2(FULL)/IGLO-III shifts are 1.7 ppm larger as compared to the gas phase MP2(FULL)/IGLO-III shifts. This is also found for all other methods used for geometry optimization. Reoptimizing the geometry of SiMe3Cl (2) in the presence of the PCM reaction field leads to elongation of the Si–Cl bond, which at PCM/MPW1K/6-31+G(d) level reaches up to 210.4 pm. This geometrical change is accompanied by a downfield shift in the 29Si resonance to +35.0 ppm. This is found in a very similar manner also for all other methods used for geometry optimization and we may thus conclude that the PCM continuum solvation model impacts 29Si shift calculations in a significant way through changes in the molecular structure.
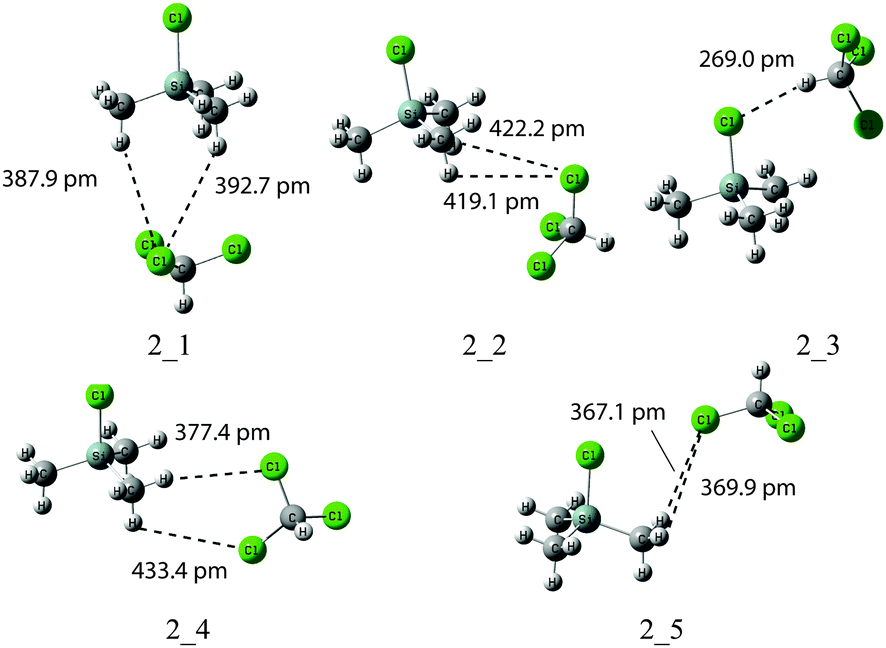
Effects of explicit solvation were subsequently explored for complexes of SiMe3Cl (2) with one molecule of CHCl3. A number of minima were identified in MPW1K/6-31+G(d) geometry optimizations (Fig. 2). The longest Si–Cl bond with 209.5 pm (and thus the largest downfield shifted 29Si signal) is found for solute–solvent complex 2_3, in which the chlorine atom of SiMe3Cl forms a hydrogen bond to chloroform (Table 4). In terms of gas phase free energies ΔG298 structure 2_3 is not the most stable conformer, but is 7 kJ mol−1 less stable than the best structure 2_1. Due to only weak interactions between the methyl groups of 2 and chlorine atoms of the solvent molecule, the Si–Cl distance in structure 2_1 is hardly different from that of 2 alone. After Boltzmann averaging, δ(29Si) is only +0.18 ppm higher than the experimental result. In solution phase, the most stable structure is 2_3. The continuum solvation model increases the Si–Cl distance and also reduces the energy gap between different conformers. With the PCM solvent model δ(29Si) values are 5 ppm higher than the experimental value. We thus have to conclude that, at least for model system 2, the gas phase predictions are significantly closer to the solution phase experiment as compared to solution-phase calculations based on a combined implicit–explicit solvent model.
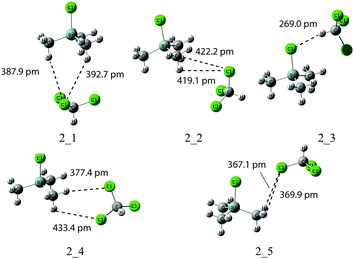 |
| | Fig. 2 Structures of SiMe3Cl (2) complexed to CHCl3 obtained at MPW1K/6-31+G(d) level in the gas phase (dashed lines show shortest contacts between solvent and solute molecules). | |
Table 4
29Si chemical shifts for complexes 2_1–2_5 of SiMe3Cl (2) with one chloroform molecule at MP2(FULL)/IGLO-III//MPW1K/6-31+G(d) level (bond distances are in pm, free energies ΔG298 are in kJ mol−1 and chemical shifts are in ppm)
| |
Gas phase |
Solution phasea |
|
r(Si–Cl) |
ΔG298 |
δ(29Si) |
r(Si–Cl) |
ΔG298 |
δ(29Si) |
|
Solvent effects calculated at PCM(UAHF)/MP2(FULL)/IGLO-III//MPW1K/6-31+G(d) level.
δ(29Si) shift calculated as Boltzmann average, for detail see ESI.
|
|
2_1
|
208.6 |
0.00 |
+30.70 |
210.50 |
1.02 |
+35.09 |
|
2_2
|
208.6 |
0.99 |
+30.89 |
210.50 |
3.02 |
+35.43 |
|
2_3
|
209.5 |
7.01 |
+33.81 |
210.70 |
0.00 |
+36.24 |
|
2_4
|
208.5 |
7.44 |
+30.69 |
210.40 |
1.29 |
+35.16 |
|
2_5
|
208.7 |
7.91 |
+31.28 |
210.40 |
2.06 |
+35.06 |
| Aveb |
|
|
+30.88 |
|
|
+35.52 |
Spin–orbit corrections for chlorosilanes
29Si Chemical shifts calculated at MPW1K/IGLO-III//MPW1K/6-31+G(d) level for the SiMe4−xClx compounds show a systematic deviation between theoretically calculated and experimentally measured shift data (Fig. 3). That the deviations are systematic in nature can readily be seen from the good linear correlation between the actual deviation and the number of chlorine atoms (Fig. 3).15 The importance of heavy-atom-induced spin–orbit (SO) effects for nuclear magnetic shifts of group 14 element halides have been highlighted by several groups.45–50 According to these earlier reports, the experimentally observed normal halogen dependence (NHD), i.e. the characteristic high-field shift of the nucleus bound directly to the halogen substituents with increasing atomic number of the halogen, is mainly a result of spin–orbit effects. In these studies it was found that shifts calculated with the inclusion of SO effects agree significantly better with the experiment than their non-relativistic counterparts.
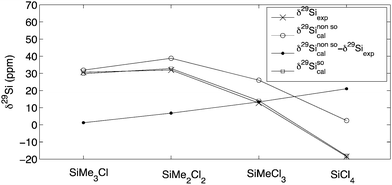 |
| | Fig. 3 Comparison between experimentally measured and theoretically calculated shift values at MPW1K/IGLO-III//MPW1K/6-31+G(d) level for alkylchlorosilanes SiMe4−xClx with x = 1–4. SO corrections are obtained from ZORA-SO-PBE0/TZ2P single-point calculations. Geometries are obtained at MPW1K/6-31+G(d) level in gas phase. | |
We evaluated contributions of relativistic spin–orbit effects to the nuclear magnetic shielding constants for the series of SiMe4−xClx compounds employing the two-component zeroth-order regular approximation (ZORA)51,52 formalism, as implemented in ADF. The SO correction per chloro-substituent increases monotonously from approximately −2 ppm (SiMe3Cl) to −3 ppm for SiMe2Cl2, −4 ppm for SiMeCl3, and −5 ppm for SiCl4. In line with the discussion of Kaupp et al. on SO-induced heavy-atom effects on NMR chemical shifts,47 this trend can readily be explained based on the analogy to the Fermi-contact mechanism of spin–spin coupling. The increasing involvement of valence silicon s-type orbitals in the bonding orbitals to the chloro-substituents results in larger SO contributions in the series from SiMe3Cl to SiCl4.53 Aside from the chlorine count there is only a rather limited influence of other silicon substituents on the magnitude of the SO corrections. A SO correction of around −2 ± 0.5 ppm per chlorine substituent will thus be typical for different chloromonosilanes (see also Table 5). For systems with more than one conformer, Boltzmann-weighted SO corrections are close to the SO-correction for the most stable conformer.54
Table 5 The 29Si NMR chemical shifts of selected chemical species
| |
System |
δ
exp
(ppm) |
δ
gascal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (ppm) b (ppm) |
δ
gas+socalc (ppm) |
δ
sp+socald (ppm) |
δ
opt+socale (ppm) |
|
Experimentally measured δexp(29Si) values for all systems. The solvent is given in parenthesis.
δ
gascal(29Si) chemical shift in gas phase using gas phase structure at MPW1K/IGLO-III//MPW1K/6-31+G(d) level.
δ
gas+socal(29Si) gas phase chemical shift with added Boltzmann-weighted SO corrections. SO corrections are obtained at ZORA-SO-PBE0/TZ2P level in gas phase.
δ
sp+socal(29Si) chemical shifts are calculated with PCM(UAHF)/MPW1K/IGLO-III(+ZORA-SO)//MPW1K/6-31+G(d). Boltzmann-averaged SO corrections are calculated at ZORA-SO-PBE0/TZ2P in gas phase based on gas phase structures.
δ
opt+socal(29Si) chemical shifts are calculated with PCM(UAHF)/MPW1K/IGLO-III//PCM/UAHF-MPW1K/6-31+G(d) level and corrected with Boltzmann-weighted SO corrections at ZORA-SO-PBE0/TZ2P level based on gas phase geometries. For more detailed information on the SO corrections see ESI.
|
| 2 |
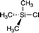
|
+30.70 (CDCl3)31 |
+31.92 |
+29.82 |
+31.90 |
+34.90 |
| 3 |
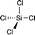
|
−18.50 (CDCl3)15 |
+2.49 |
−18.11 |
−18.05 |
−18.13 |
| 4 |
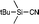
|
−1.57 (CDCl3) |
−5.39 |
−5.19 |
−3.01 |
−1.15 |
| 5 |
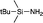
|
+8.40 (C6D6)55 |
+8.97 |
+8.57 |
+8.18 |
+10.41 |
| 6 |
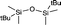
|
+9.91 (CDCl3) |
+11.59 |
+10.79 |
+11.21 |
+12.78 |
| 7 |
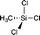
|
+12.70 (CDCl3) |
+26.04 |
+14.04 |
+15.30 |
+16.53 |
| 8 |
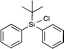
|
+12.86 (CDCl3) |
+16.12 |
+14.42 |
+14.73 |
+15.22 |
| 9 |
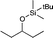
|
+15.54 (CDCl3) |
+17.07 |
+16.37 |
+16.88 |
+18.16 |
| 10 |
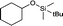
|
+15.80 (CDCl3) |
+17.40 |
+16.60 |
+17.28 |
+18.51 |
| 11 |
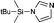
|
+17.08 (CDCl3) |
+16.41 |
+16.01 |
+17.13 |
+19.06 |
| 12 |
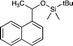
|
+18.42 (CDCl3) |
+21.10 |
+20.40 |
+20.68 |
+21.01 |
| 13 |
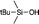
|
+20.34 (CDCl3) |
+19.48 |
+19.08 |
+19.09 |
+20.10 |
| 14 |
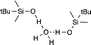
|
+20.39 (CDCl3) |
+17.69 |
+17.09 |
+17.84 |
+18.70 |
| 15 |
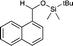
|
+20.58 (CDCl3) |
+21.46 |
+20.66 |
+20.95 |
+18.51 |
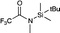
| 16 |
|
+26.69 (CDCl3) |
+22.95 |
+22.55 |
+23.36 |
+24.16 |
| 17 |
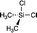
|
+32.00 (CDCl3)3 |
+38.81 |
+32.81 |
+35.13 |
+37.66 |
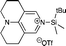
| 18 |
|
+32.16 (CDCl3) |
+35.70 |
+35.30 |
+36.18 |
+34.47 |
| 19 |
|
+33.25 (CDCl3) |
+38.44 |
+37.94 |
+36.40 |
+37.00 |
| 20 |
|
+35.89 (CDCl3) |
+36.81 |
+35.31 |
+37.13 |
+39.80 |
| 21 |
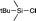
|
+36.09 (CDCl3) |
+37.78 |
+35.78 |
+37.62 |
+39.98 |
| 22 |
|
+43.71 (CDCl3) |
+44.50 |
+44.00 |
+46.55 |
+51.65 |
| 23 |
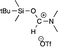
|
+45.21 (CDCl3) |
+51.41 |
+50.81 |
+49.83 |
+49.12 |
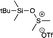
| 24 |
|
+45.96 (CDCl3) |
+43.30 |
+42.70 |
+43.68 |
+48.94 |
29Si chemical shifts for larger molecular systems
29Si chemical shifts for a larger group of “typical” systems are collected in Table 5 and graphically shown in Fig. 4. Already the calculation of gas phase 29Si chemical shifts at MPW1K/IGLO-III//MPW1K/6-31+G(d) level provides a rather accurate agreement with experimental results obtained in low polarity organic solvents with R2 = 0.872 and MD = 2.54 ppm (panel (a) in Fig. 4). The largest deviations occur for systems containing more than one chlorine atom directly attached to silicon, and inclusion of SO-corrections evaluated at ZORA-SO-PBE0/TZ2P single point level improves correlation with experiment significantly for the entire dataset to R2 = 0.978 and MD = 0.18 ppm (compare panel (a) and (b) in Fig. 4). Best theoretical predictions are then obtained through shielding calculations in the presence of the PCM continuum solvation model with gas phase structures and including the SO corrections. It is remarkable to see that the correlation for this approach based on gas phase structures (R2 = 0.984, MD = 0.97 ppm) is even slightly better as compared to the approach based on solution-phase structures with R2 = 0.983 and MD = 2.34 ppm (compare panel (c) and (d) in Fig. 4). Performing the actual shielding calculations with different functionals such as HCTH407 or with the local DF-LMP2 method lead to no further systematic improvement (see ESI† for details).
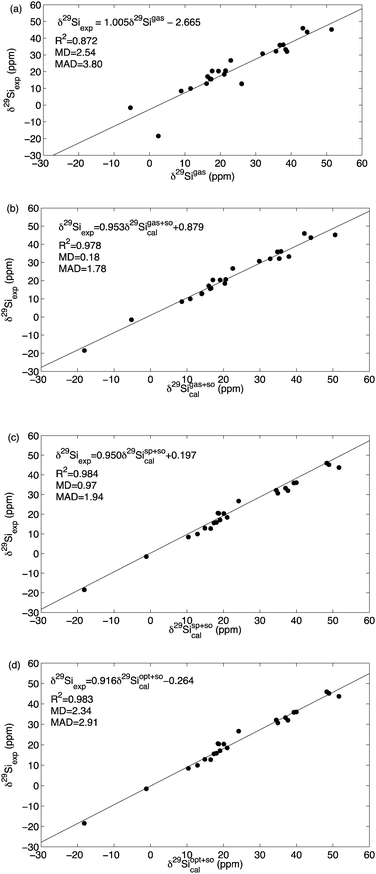 |
| | Fig. 4 Theoretically calculated vs. experimentally measured 29Si chemical shifts for the data set collected in Table 5. (a) δ(29Si) calculated at MPW1K/IGLO-III//MPW1K/6-31+G(d) level. (b) δ(29Si) calculated at MPW1K/IGLO-III(+ZORA-SO)//MPW1K/6-31+G(d). (c) δ(29Si) calculated at PCM(UAHF)/MPW1K/IGLO-III(+ZORA-SO)//MPW1K/6-31+G(d) level using gas phase structures. (d) δ(29Si) calculated at PCM(UAHF)/MPW1K/IGLO-III(+ZORA-SO)//PCM(UAHF)MPW1K/6-31+G(d) level using solution phase structures. | |
Chemical shifts for ion pairs
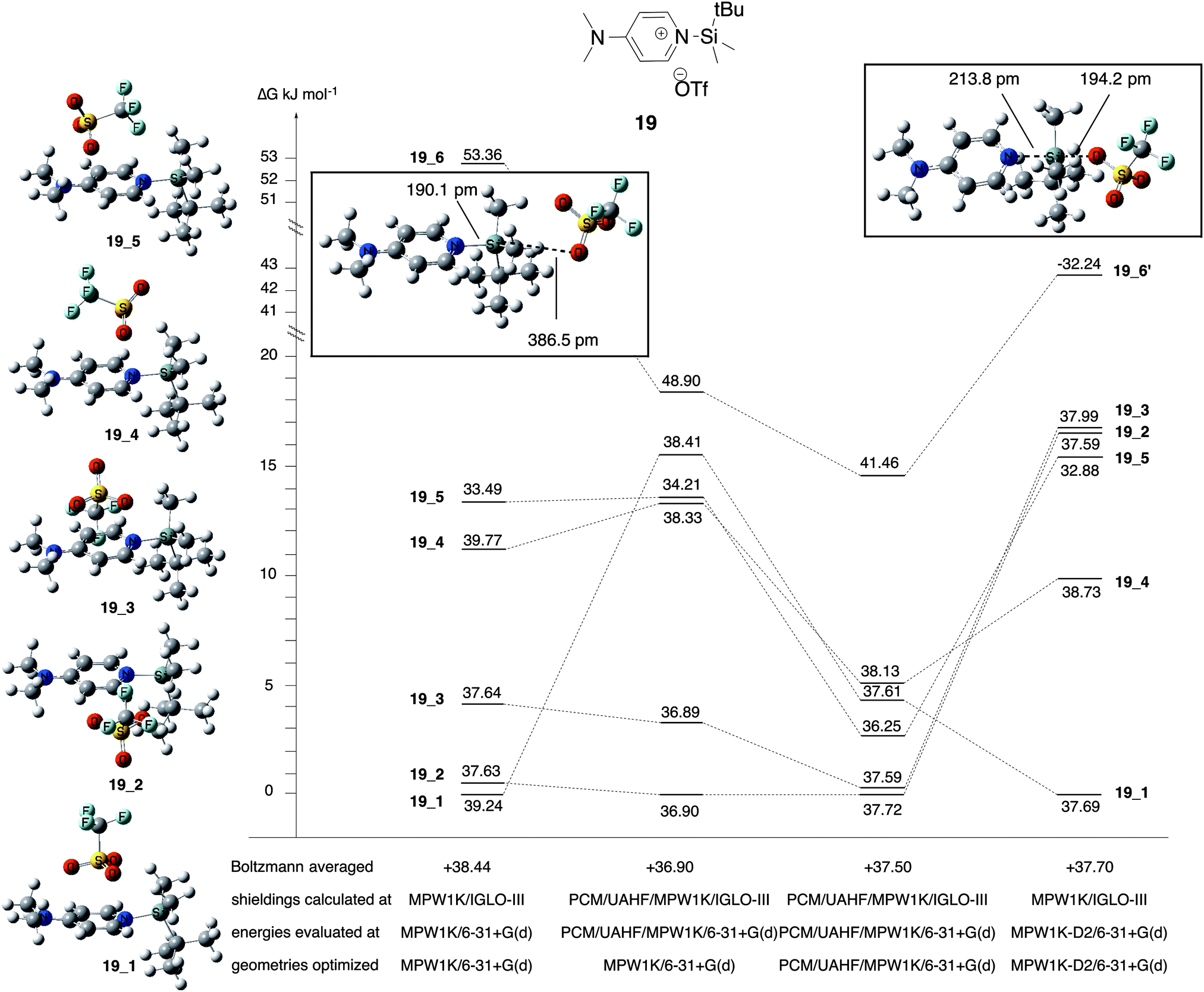
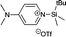
Chemical shift calculations for the ion pair intermediates 18, 19, 23, and 24 included in Table 5 are somewhat less accurate as compared to all other systems at the MPW1K/IGLO-III(+ZORA-SO)//MPW1K/6-31+G(d) level. This may, in part, be due to significantly larger solvent effects for these polar species as well as their rather large conformational flexibility. How the latter actually impacts chemical shift calculations is illustrated in detail in Fig. 5 for silylpyridinium ion pair 19, whose experimentally measured 29Si chemical shift amounts to +33.25 ppm in chloroform. For ion pairs, which have more than one conformer, chemical shifts are evaluated by Boltzmann averaging over individual conformers based on their relative free energies. When using gas-phase free energies the structures with the largest molecular dipole moment make the smallest contribution to the Boltzmann average due to their high relative energies (Table 6). In solution, however, these structures lead to the largest solvation energies: the dipole moment of conformer 19_1 is comparatively low at 10.30 D (MPW1K/6-31+G(d) level) and thus has the smallest solvation energy of −81.50 kJ mol−1 (gas-phase geometry). Conformers 19_4 and 19_5 have slightly larger dipole moments and also slightly larger solvation energies around −96 kJ mol−1. Conformer 19_6 has the largest dipole moment at 23.93 D, leading to a rather large solvation energy of −131.50 kJ mol−1 (gas phase geometry) or −162.72 kJ mol−1 (solution phase geometry). As a consequence of these variations in solvation energies, relative free energies in solution (ΔGopt) are significantly smaller as compared to the gas phase. Despite these large changes in relative energies, the actual ion pair structure shows rather little change in gas-phase vs. solution-phase geometry optimization as exemplified for the Si–N bond distance (Table 6). This can also be stated for the electronic character of these ion pairs as characterized by the triflate ion charge, which assumes values between −0.94 and −0.96 for the best conformers 19_1 and 19_2 in gas-phase as well as in solution (Table 6).
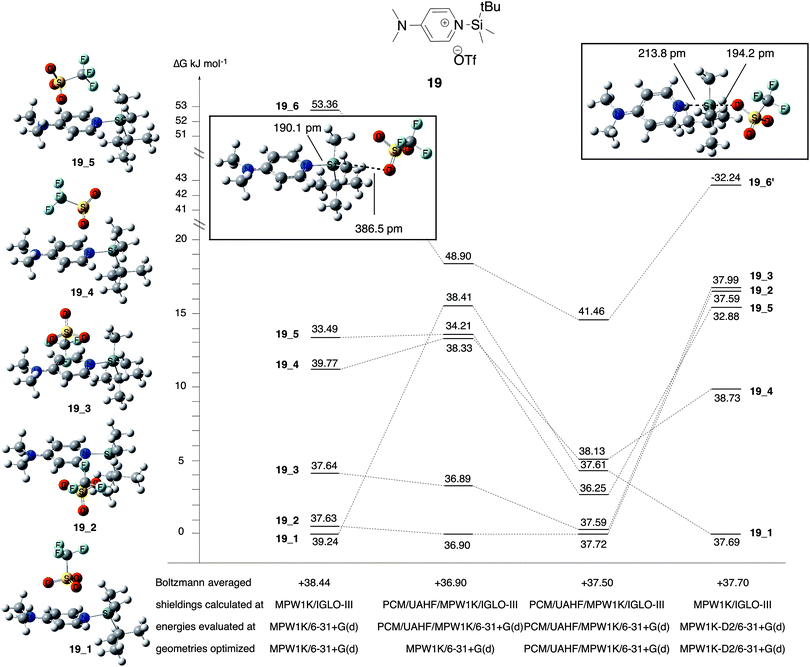 |
| | Fig. 5 Relative free energies and chemical shifts of individual conformers of ion pair system 19 at different theory levels. | |
Table 6 Relative free energies, structural and charge parameters, and 29Si chemical shifts for individual conformational isomers of ion pair 19. Charge parameters have been obtained from the NBO analysis56
| Level for free energy calculation |
|
19_1
|
19_2
|
19_3
|
19_4
|
19_5
|
19_6
|
| MPW1K/6-31+G(d)//MPW1K/6-31+G(d) |
μ (Debye) |
10.30 |
17.27 |
17.36 |
13.16 |
14.39 |
23.93 |
| ΔG (kJ mol−1) |
0.00 |
0.67 |
4.01 |
11.81 |
13.35 |
52.89 |
|
r(Si–N) (pm) |
184.3 |
184.9 |
184.9 |
184.7 |
182.9 |
190.1 |
|
q(OTf) |
−0.96 |
−0.94 |
−0.94 |
−0.95 |
−0.96 |
−0.95 |
|
δ(29Si) (ppm) |
+39.24 |
+37.63 |
+37.64 |
+39.77 |
+33.49 |
+53.36 |
|
|
| PCM/UAHF/MPW1K/6-31+G(d)//MPW1K/6-31+G(d) |
ΔGspsolv (kJ mol−1) |
−81.5 |
−97.65 |
−97.86 |
−95.60 |
−96.86 |
−131.50 |
| ΔG (kJ mol−1) |
15.54 |
0.00 |
3.13 |
13.20 |
13.47 |
18.23 |
|
r(Si–N) (pm) |
184.3 |
184.9 |
184.9 |
184.7 |
182.9 |
190.1 |
|
q(OTf) |
−0.96 |
−0.95 |
−0.95 |
−0.96 |
−0.97 |
−0.96 |
|
δ(29Si) (ppm) |
+38.41 |
+36.90 |
+36.89 |
+38.33 |
+34.21 |
+48.90 |
|
|
| PCM/UAHF/MPW1K-6-31+G(d)//PCM/UAHF/MPW1K-6-31+G(d) |
ΔGoptsolv (kJ mol−1) |
−125.81 |
−119.41 |
−119.79 |
−133.05 |
−141.04 |
−162.72 |
| ΔG (kJ mol−1) |
4.31 |
0.00 |
0.29 |
5.10 |
2.69 |
14.51 |
|
r(Si–N) (pm) |
184.4 |
184.5 |
184.4 |
184.5 |
184.2 |
186.0 |
|
q(OTf) |
−0.98 |
−0.97 |
−0.97 |
−0.99 |
−0.99 |
−0.98 |
|
δ(29Si) (ppm) |
+37.61 |
+37.72 |
+37.59 |
+38.13 |
+36.25 |
+41.64 |
|
|
| MPW1K-D2/6-31+G(d)//MPW1K-D2/6-31+G(d) |
ΔG (kJ mol−1) |
0.00 |
16.54 |
16.76 |
9.91 |
15.39 |
42.78 |
|
r(Si–N) (pm) |
183.0 |
183.5 |
183.2 |
183.2 |
181.6 |
213.8 |
|
q(OTf) |
−0.95 |
−0.94 |
−0.94 |
−0.95 |
−0.95 |
−0.78 |
|
δ(29Si) (ppm) |
+37.69 |
+37.59 |
+37.99 |
+38.73 |
+32.88 |
−32.24 |
The largest effects are observed for ion pair 19_6, whose structure also depends significantly on the chosen level of theory in the gas phase: geometry optimization at MPW1K/6-31+G(d) level leads to the structure shown as “19_6” in Fig. 5, which can be characterized as a true ion pair with a comparatively long (387 pm) distance between silicon atom and triflate counter ion. Reoptimization with the same functional and added dispersion corrections (MPW1K-D2/6-31+G(d)) leads to a structure best described as pentacoordinated silicon intermediate (termed 19_6′) with a much shorter Si–O(triflate) distance of 194 pm and a largely changed 29Si chemical shift of −32.24 ppm.57 More importantly, the very different chemical shift for pentacoordinate structure 19_6′ clearly supports the assignment of the experimentally detected ion pair species as systems with tetracoordinate silicon atoms. The “collapse” of the ion pair structure to a pentacoordinated intermediate upon geometry optimization at MPW1K-D2/6-31+G(d) level has only been observed for conformer 19_6 and not for any of the other (low-energy) conformers.
Conclusions
Calculated 29Si chemical shifts of (chloro)organosilanes depend strongly on structural details of the respective systems. MPW1K/6-31+(d) gas phase optimizations give structures close enough to experiment for reliable NMR shielding calculations. Relativistic spin–orbit effects are large for chlorosilane systems and reliable shielding calculations thus require SO corrections for these systems. For non-halosilane systems the SO effects are rather small and 29Si shift predictions are thus possible without any relativistic corrections. Solvent effects on 29Si NMR chemical shifts are quite systematic, but only of moderate size, and reliable shift predictions can thus be made using gas phase geometries and, in many cases, also using gas phase shielding calculations. For the (chloro)organosilane compound family studied here MPW1K/IGLO-III(+ZORA-SO)//MPW1K/6-31+G(d) calculations are thus recommended for 29Si NMR shift calculation.
Computational details
Chemical shifts of 29Si containing species (δ(29Si)) are calculated using eqn (1), where σ(29SiTMS) is the chemical shielding for the reference compound tetramethylsilane (Si(CH3)4, TMS, 1) and σ(29Si) is the shielding of the 29Si nucleus in the compound under investigation:| | | δ(29Si) = σ(29SiTMS) − σ(29Si) | (1) |
Shieldings are calculated using the Gauge-Independent Atomic Orbital (GIAO) method.58 Shielding values of chemicals are calculated with Gaussian 09, revision C.0159 and MOLPRO, Version 2012.1.60,61
The relativistic spin–orbit corrections to the nuclear magnetic shielding constants for 29Si were calculated with the two-component zeroth-order regular approximation (ZORA)51,52 using the Amsterdam Density Functional (ADF 2013.01) code.62 The hybrid functional PBE063–65 in combination with the Slater-type basis set TZ2P66 optimized for relativistic ZORA calculations is used for these single point calculations at MPW1K/6-31+G(d) gas phase structures. The spin–orbit correction is calculated as δ(29Si)ZORA–δ(29Si)non-relativistic and then Boltzmann-averaged for compounds with more than one conformer. For more details on different functionals and basis sets see the ESI.†
Experimental details
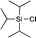
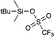
All commercial chemicals were of reagent grade and were used as received unless otherwise noted. The substances tert-butyl-dimethylsilyl cyanide (4), tert-butyldiphenylsilyl chloride (8), 1-(tert-butyldimethylsilyl)imidazole (11), N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (16), and triisopropylsilyl chloride (20), were purchased in the highest purity from Sigma Aldrich and measured in freshly distilled CDCl3 over CaH2. 1H and 13C NMR spectra were recorded on Varian 300 or Varian INOVA 400 machines at room temperature. All 1H chemical shifts are reported in ppm (δ) relative to CDCl3 (+7.26); 13C chemical shifts are reported in ppm (δ) relative to CDCl3 (+77.16). All other chemicals were purchased from commercial suppliers at the highest available grade, store over orange gel in an desiccator and used without any further purification.
General procedure 1
For all purchased substances this procedure was performed. In a pre-dried NMR tube 30 mg of the substance were dissolved in 0.6 mL of freshly distilled CDCl3. The tube was closed with a cap and sealed with parafilm.
General procedure 2
For the ion pairs (18, 19, 23, 24) two stock solutions have been prepared in the concentration of 0.5 M in freshly distilled CDCl3. Both solutions (Lewis base and TBSOTf) are mixed in a ratio 1:1 and therefore the concentration of the intermediates can be 0.25 M max. The tube was closed with a cap and sealed with parafilm.
General procedure 3 (silylation of alcohols)
One equivalent of alcohol and 1.2 equivalents of used silyl chloride were dissolved in 20 mL DCM and 1.2 equivalents triethylamine was added. After adding up to 30 mmol% of a catalyst DMAP the reaction mixture stirred at RT up to 7 d. The reaction mixture was washed with sat. aq. NH4Cl solution, extracted three times with DCM (10 mL) and the combined organic phases dried over MgSO4. The solvent was removed under reduced pressure and column chromatography was used to purify the product.
References
- T. M. Alam and M. Henry, Phys. Chem. Chem. Phys., 2000, 2, 23–28 RSC.
- C. van Wüllen, Phys. Chem. Chem. Phys., 2000, 2, 2137–2144 RSC.
- U. Herzog, J. Prakt. Chem., 2000, 342, 379–388 CrossRef CAS.
- X. Xue and M. Kanzaki, Phys. Chem. Miner., 1998, 26, 14–30 CrossRef CAS.
- J. A. Tossell and P. Lazzeretti, J. Chem. Phys., 1986, 84, 369–374 CrossRef CAS PubMed.
- D. H. Brouwer, J. Magn. Reson., 2008, 194, 136–146 CrossRef CAS PubMed.
- D. Auer, M. Kaupp and C. Strohmann, Organometallics, 2004, 23, 3647–3655 CrossRef CAS.
- C. Corminboeuf, T. Heine and J. Weber, Chem. Phys. Lett., 2002, 357, 1–7 CrossRef CAS.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed.
- J. Casanovas, F. Illas and G. Pacchioni, Chem. Phys. Lett., 2000, 326, 523–529 CrossRef CAS.
- R. Herges and F. Starck, J. Am. Chem. Soc., 1996, 118, 12752–12757 CrossRef CAS.
- J. Ambati and S. E. Rankin, J. Phys. Chem. A, 2010, 114, 5279–5286 CrossRef CAS PubMed.
- T. Heine, A. Goursot, G. Seifert and J. Webert, J. Phys. Chem. A, 2001, 105, 620–626 CrossRef CAS.
- A. I. Poblador-Bahamonde, R. Poteau, C. Raynaud and O. Eisenstein, Dalton Trans., 2011, 40, 11321–11326 RSC.
- F. Meyer-Wegner, A. Nadj, M. Bolte, N. Auner, M. Wagner, M. C. Holthausen and H. W. Lerner, Chem. – Eur. J., 2011, 17, 4715–4719 CrossRef CAS PubMed.
- M. Karni, Y. Apeloig, N. Takagi and S. Nagase, Organometallics, 2005, 24, 6319–6330 CrossRef CAS.
- M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed.
- R. P. Singh and J. M. Shreeve, Tetrahedron, 2000, 56, 7613–7632 CrossRef CAS.
-
P. Patschinski, C. Zhang and H. Zipse, unpublished results.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- A. F. Wallace, G. V. Gibbs and P. M. Dove, J. Phys. Chem. A, 2010, 114, 2534–2542 CrossRef CAS PubMed.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2001, 114, 5497 CrossRef CAS PubMed.
- V. A. Du, G. N. Stipicic and U. Schubert, Eur. J. Inorg. Chem., 2011, 3365–3373 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- B. Maryasin and H. Zipse, Phys. Chem. Chem. Phys., 2011, 13, 5150–5158 RSC.
- S. Sklenak, J. Dědeček, C. Li, B. Wichterlová, V. Gábová, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1237–1247 RSC.
- E. V. van den Berghe and G. P. van der Kelen, J. Organomet. Chem., 1973, 59, 175–187 CrossRef CAS.
- M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982–15989 CrossRef CAS PubMed.
- K. Ohmatsu, Y. Hamajima and T. Ooi, J. Am. Chem. Soc., 2012, 134, 8794–8797 CrossRef CAS PubMed.
- B. J. Albert and H. Yamamoto, Angew. Chem., Int. Ed., 2010, 49, 2747–2749 CrossRef CAS PubMed.
- J. R. Durig, Y. S. Li and R. O. Carter, J. Mol. Spectrosc., 1972, 44, 18–31 CrossRef.
- R. C. Mockler, J. H. Bailey and W. Gordy, J. Chem. Phys., 1953, 21, 1710–1713 CrossRef CAS PubMed.
- P. W. Allen and L. E. Sutton, Acta Crystallogr., 1950, 3, 46–72 CrossRef CAS.
- R. Gajda, K. Dziubek and A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 86–93 Search PubMed.
- J. Buschmann, D. Lentz, P. Luger and M. Rottger, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56(Pt 1), 121–122 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS PubMed.
-
W. Kutzelnigg, U. Fleischer and M. Schindler, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck, H. Günther, R. Kosfeld and J. Seelig, Springer, Berlin/Heidelberg, 1991, vol. 213, pp. 165–262 Search PubMed.
- F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2001, 114, 5497–5503 CrossRef CAS PubMed.
- H.-J. Werner, F. R. Manby and P. J. Knowles, J. Chem. Phys., 2003, 118, 8149 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- H. Nakatsuji, T. Nakajima, M. Hada, H. Takashima and S. Tanaka, Chem. Phys. Lett., 1995, 247, 418–424 CAS.
- T. Helgaker, S. Coriani, P. Jørgensen, K. Kristensen, J. Olsen and K. Ruud, Chem. Rev., 2012, 112, 543–631 CrossRef CAS PubMed.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem. – Eur. J., 1998, 4, 118–126 CrossRef CAS.
- A. Bagno, M. Bonchio and J. Autschbach, Chem. – Eur. J., 2006, 12, 8460–8471 CrossRef CAS PubMed.
- L. A. Truflandier, E. Brendler, J. Wagler and J. Autschbach, Angew. Chem., Int. Ed., 2011, 50, 255–259 CrossRef CAS PubMed.
- J. Autschbach, K. Sutter, L. A. Truflandier, E. Brendler and J. Wagler, Chem. – Eur. J., 2012, 18, 12803–12813 CrossRef CAS PubMed.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef PubMed.
- S. K. Wolff, T. Ziegler, E. van Lenthe and E. J. Baerends, J. Chem. Phys., 1999, 110, 7689 CrossRef CAS PubMed.
- V. G. Malkin, O. L. Malkina and D. R. Salahub, Chem. Phys. Lett., 1996, 261, 335–345 CrossRef CAS.
- Taking ion pair system 19 as an example, the SO correction for the six different conformers shown in Fig. 5 vary from −0.3 to −0.5 ppm at ZORA-SO-PBE0/TZ2P level. After Boltzmann averaging, the SO correction amounts to −0.5 ppm, which is identical to the value of the most stable conformer 19_1 (see ESI† for details).
- B. Wrackmeyer, C. Stader and H. Zhou, Spectrochim. Acta, Part A, 1989, 45, 1101–1111 CrossRef.
- NBO calculations were performed using the NBO 3.1 program as implemented in the Gaussian 09 C.01 package.
- This type of geometry is also obtained upon geometry optimization at MP2(FC)/6-31+G(d) level.
- R. Ditchfied, Mol. Phys., 1974, 27, 789–807 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2010.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
-
H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz and others, see also http://www.molpro.net.
- ADF2013, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, Netherlands; see also: http://www.scm.com.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01736f |
|
| This journal is © the Owner Societies 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence