 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereomeric preference of a triply axial chiral binaphthyl based molecule: a concentration dependent study by chiroptical spectroscopies†
Zahra
Dezhahang
a,
Mohammad Reza
Poopari
a,
Florencio Eloy
Hernández
b,
Carlos
Diaz
b and
Yunjie
Xu
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2. E-mail: Yunjie.xu@ualberta.ca; Fax: +1-780-492-8231; Tel: +1-780-492-1244
bDepartment of Chemistry and CREOL/the College of Optics and Photonics, University of Central Florida, P.O. Box 162366, Orlando, Florida 32816-2366, USA. E-mail: Florencio.Hernandez@ucf.edu; Fax: +1-407-823-5222; Tel: +1-407-823-0843
First published on 8th May 2014
Abstract
We have examined the effects of environmental perturbations, specifically solvents and concentrations, on axial chirality of a recently synthesized axially chiral binaphthyl fluorene based salen ligand, named AFX-155, {[2,2′-(1E,1′E)-(R)-1,1′-binaphthyl-2,2′-diylbis(azan-1-yl-1-ylidene)bis(methan-1-yl-1-ylidene)bis(4-((7-(diphenylamino)-9,9-dihexyl-9H-fluoren-2-l)ethynyl)-phenol)]}. Chirality and dominant conformations of AFX-155 in CDCl3 solvent have been characterized using vibrational absorption (VA) and vibrational circular dichroism (VCD) spectroscopy in combination with DFT calculations. AFX-155 exhibits triple axial chirality: one is at the binaphthyl ring and the other two are related to the axes of chirality along the –C–N bonds where Cs are part of the binaphthyl group. To evaluate solvent and concentration dependence, complementary VA and VCD experiments in both THF-d8 and CDCl3 have been performed, as well as the optical rotatory dispersion (ORD) and electronic CD (ECD) measurements in CDCl3 under much diluted conditions. While the binaphthyl chirality is determined by the synthetic route, the results show that the latter two axial chirality labels of the dominant diastereomers are concentration dependent. Under much diluted conditions, R-binaphthyl, R_intra_HB//R_extra_HB (R-RR) is favoured, whereas R-binaphthyl, S_intra_HB//S_extra_HB (R-SS) is the dominant species in a concentrated solution. This diastereomeric interconversion is found to be independent of the two solvents used. To provide insights into this interesting finding, conformational searches and the related spectral simulations have been carried out at the DFT/B3LYP/6-31G(d) level.
1 Introduction
It is well known that solvent can exert a deciding influence on the preferred conformation and even chirality of a chiral solute in solution.1,2 Such effects are often manifested in severe and sometimes non-intuitive effects on chiroptical spectral signatures. Both solvent polarity and hydrogen (H)-bonding formation between solute and solvent molecules can influence the chiroptical response.3,4 For example, the experimental vibrational circular dichroism (VCD) spectra of methyl mandelate look quite different in chloroform, methanol, and dimethyl sulfoxide (DMSO). While the spectrum in chloroform can be well reproduced by the gas phase simulation, it is necessary to apply the explicit solvention model to account for the spectra in the other two solvents.5 In another example, chiral poly(ureidophthalimide) foldamers were reported to adopt two opposite helical handedness in water and in tetrahydrofuran (THF) where mirror imaged electronic CD (ECD) bands were detected.6 Such solvent effects allow one to control over the helical architecture and direct the supramolecular synthesis. Similarly, solvent-mediated chirality switching was reported for dendrimer folding in aqueous solution.7 Generally speaking, polar protic solvents, such as water can disrupt intra- and intermolecular electrostatic and H-bonding interactions of the example systems discussed above thereby changing the relative stabilities of various conformational assemblies.8Much less reported is the concentration induced conformational change in common organic solvents used for chiroptical spectroscopic studies, such as DMSO, deuterated chloroform (CDCl3) or THF. This is an interesting and important topic since several commonly used chiroptical spectroscopic tools for absolute configuration and conformational determination of chiral compounds, such as VCD and ECD, Raman optical activity (ROA), and optical rotatory dispersion (ORD) spectroscopy, operate under very different concentration regimes and each has its own advantages and disadvantages. For example, VCD spectroscopy generally provides very rich band structures which often contain detailed structural information. Furthermore, VCD simulations are considerably more reliable than ECD and ORD since only ground electronic state calculations are needed.9 On the other hand, an ECD or ORD experiment requires much less sample than VCD. While a typical concentration used for an ECD or ORD experiment is in the range of 10−5 to 10−3 M, the concentrations needed for VCD experiments are normally 10−1 to a few M and often saturated solution is used. In addition, each of these spectroscopy techniques probes only a certain molecular property. Consequently, a number of researchers have advocated using more than one of the spectroscopic monitoring tools in order to reach a reliable absolute configuration determination, providing there are no major structural changes with concentration.10–13
The susceptibility of axial chirality to the environmental perturbations such as the choice of different solvents and concentrations of the sample is the subject of significant practical and fundamental importance in supramolecular assembly.14 We chose AFX-155, a triply axial chiral binaphthyl fluorene based salen ligand, i.e. [2,2′-(1E,1′E)-(R)-1,1′-binaphthyl-2,2′-diylbis(azan-1-yl-1-ylidene)bis(methan-1-yl-1-ylidene)bis(4-((7(diphenylamino)-9,9-dihexyl-9H-fluoren-2-l)ethynyl)-phenol)], as our targeted molecular system. AFX-155 is a model axial chiral system, with a good number of possible diastereomers and a few potential H-bonding sites. This molecule was recently synthesized for its potential applications in homogeneous catalysis, biophotonics, and biosensing.15,16 It was also characterized using ECD and two-photon CD (TPCD) spectroscopy by Hernández and co-workers.15,16
One of our goals is to utilize VCD spectroscopy and DFT calculations to clearly establish the axial chirality of AFX-155 in solution. A second goal of our study is to examine the effects of environmental perturbations, i.e. solvent and concentrations, on axial chirality of AFX-155. To achieve that, we carried out VA and VCD experiments in THF and CDCl3, as well as ORD and ECD measurements in CDCl3 under much diluted conditions. From the combined experimental and theoretical results, we reached a surprising and interesting conclusion that the preferred axial chirality at the –C–N bonds is switched under concentrated and much diluted conditions. Further theoretical modeling was also performed to help to rationalize the observed phenomena.
2 Experimental and theoretical details
AFX-155 was synthesized using the reported Scheme 1 according to ref. 15 and 16. VA and VCD spectra were measured using a Bruker FTIR (Vertex 70) spectrometer equipped with a PMA 50 module for VCD measurements.17 The data were collected at 4 cm−1 resolution in the wavenumber region of 1700–1100 cm−1 using a liquid nitrogen cooled MCT detector for a period of 4 hours (∼4300 scans per an hour). 0.046 M solutions of AFX-155 in CDCl3 and THF-d8 were used for the VA and VCD measurements. The solution samples were placed between a pair of BaF2 windows with a path-length of 0.125 mm.For ORD measurements, a solution of 6.5 × 10−4 M AFX-155 in CDCl3 was prepared using a 10 cm cell. The ORD data were collected at a series of wavelengths, namely at 589 nm of a sodium D line, and at 578 and 546 nm of a mercury lamp by means of a Perkin-Elmer 240 polarimeter. The solution with the same concentration is used to carry out the ECD data acquisitions. To meet spectroscopic criteria for CD measurements, the magnitude of the absorption is adjusted using a UV spectrometer and a 1 mm cell was used. An Olis DSM 17 CD spectrometer and a Hewlett Packard 8453 UV-VIS Spectrophotometer were used for ECD and UV measurements, respectively.
The Gaussian 0918 program was used for all geometrical searches and optimizations, harmonic frequency calculations, and VA and VCD intensity predictions using density functional theory (DFT).19 The well-known Becke three parameter, Lee–Yang–Parr, functional (B3LYP)20 was used for the calculations. AFX-155 contains 224 atoms. This makes the usage of a high level of quantum chemical calculations prohibitively expensive. The 6-31G(d) basis set21 was chosen since it offers a good combination of accuracy and computational efficiency. In an effort to better capture the relative stability of the potential conformers, single point energy calculations were also performed with the cc-pVTZ basis set.22 In addition, we also carried out geometry optimizations for the targeted conformers with the D3 version of Grimme's dispersion method23 using B3LYP/6-31G(d).
To account for the bulk solvent environment, the integral equation formalism (IEF) version of the polarizable continuum model (PCM) using the universal force field radii24,25 was used. In a few previous studies, it was shown that the strength of H-bonding interaction between the chiral solute and CDCl3 is not strong enough to demand an explicit treatment.26,27 For this purpose, the dielectric constant of chloroform, ε = 4.7113, was used. A Lorentzian line shape with a half-width at half-height of 4 cm−1 was used for the simulations of VA and VCD spectra.
The time dependent density functional theory (TD-DFT) method with B3LYP/6-31G(d) was employed for all excited state energies, oscillator strength and optical rotation dispersion calculations. The first 80 electronic states were taken into account for theoretical ECD spectral simulations. To account for bulk of the solvent environment, the PCM model of chloroform was applied, as it was used for VA and VCD spectral simulations. For comparison to the experimental measurements, the half-width at half height (HWHH) of 0.2 eV with a Gaussian line-shaped function was used to simulate ECD spectra. We noted that in the previous work,15 a broader Gaussian line-shaped function with 0.35 eV HWHH was used on the truncated systems where four –C6H13 groups were replaced by four methyl groups.15
3 Results and discussion
3.1 Possible diastereomers of AFX-155
Fig. 1 shows the chemical structure of this relatively large and flexible triply axial chiral molecule. AFX-155 possesses three chiral axes: one chiral axis for the binaphthyl ring and two others along the –C–N bonds where the carbon atoms are part of the binaphthyl group. The three chiral axes are highlighted in Fig. 1. In addition to the chirality labels, there are cis- and trans-conformations about the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, intra and extra relative orientations of the salicylaldehyde moiety with respect to the other binaphthyl half, finally HB and NHB labels to indicate the existence or the non-existence of the –OH⋯NC– intramolecular HB interaction, respectively. Based on the synthesis procedure of AFX-155, the chirality of the binaphthyl ring is determined to be R. For easy comparison with the previous publications on AFX-155, we have adopted the same axial chirality labels along the–C–N bonds as used in ref. 15. The details about the axial chirality and intra and extra labels are provided in Fig. S1, ESI.†
N bond, intra and extra relative orientations of the salicylaldehyde moiety with respect to the other binaphthyl half, finally HB and NHB labels to indicate the existence or the non-existence of the –OH⋯NC– intramolecular HB interaction, respectively. Based on the synthesis procedure of AFX-155, the chirality of the binaphthyl ring is determined to be R. For easy comparison with the previous publications on AFX-155, we have adopted the same axial chirality labels along the–C–N bonds as used in ref. 15. The details about the axial chirality and intra and extra labels are provided in Fig. S1, ESI.†
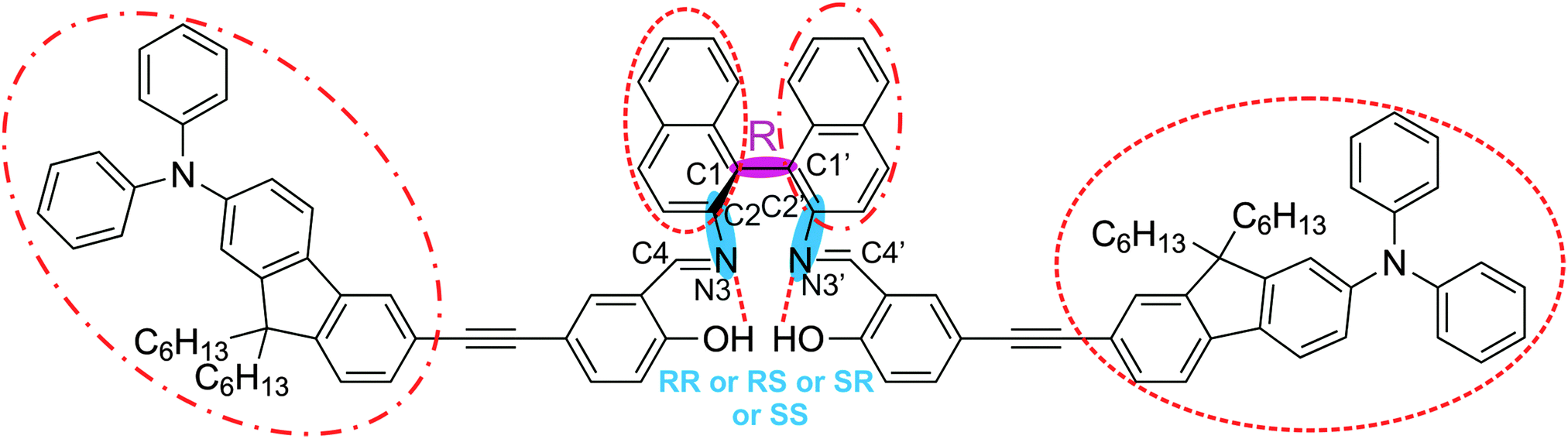 | ||
| Fig. 1 Chemical structure of AFX-155 studied in this paper. There are three chiral axes: one at the binaphthyl ring and two along the C–N bonds. All of the chiral axes are highlighted with shades. Chirality of the binaphthyl ring is R, while the associated chirality at the –C–N bonds can be RR, RS, SR or SS. The two large substitutes about the –CN bond take on the trans-arrangement shown here. The pair of the bulky group and the opposite binaphthyl half marked with dashed circles (or with dotted–dashed circles) can be in either extra or intra orientation (see Fig. S1 for further details, ESI†). In addition, the –OH⋯NC intramolecular H-bonding interactions are indicated with dashed lines. See the text for further details. | ||
3.2 Experimental VCD spectra of AFX-155
VA and VCD spectra of the AFX-155 molecule in weakly polar deuterated chloroform, i.e. CDCl3, are presented in Fig. 2. CDCl3 was chosen as it offers sufficient solubility and does not interfere in the IR window from 1700–1100 cm−1. Since the previous ECD study15,16 was carried out in deuterated THF solvent, i.e. THF-d8, we also perform further VA and VCD measurements in THF-d8 for comparison. The resulting VA and VCD spectra in THF are also included in Fig. 2 for comparison. Generally, the raw VA spectra of AFX-155 in these two solvents, i.e. without solvent subtraction, look somewhat different because of different solvent bands. In the 1700–1200 cm−1 region, the VCD band features, on the other hand, look very much the same in both solvent, suggesting that the conformational landscape and the associated axial chirality of AFX-155 remain unchanged in these two solvents. Since CDCl3 provides a wider VCD spectral window in the current study, for simplicity, we will use VA and VCD spectra in CDCl3 for the spectral analyses in the remainder of this paper, unless indicated otherwise.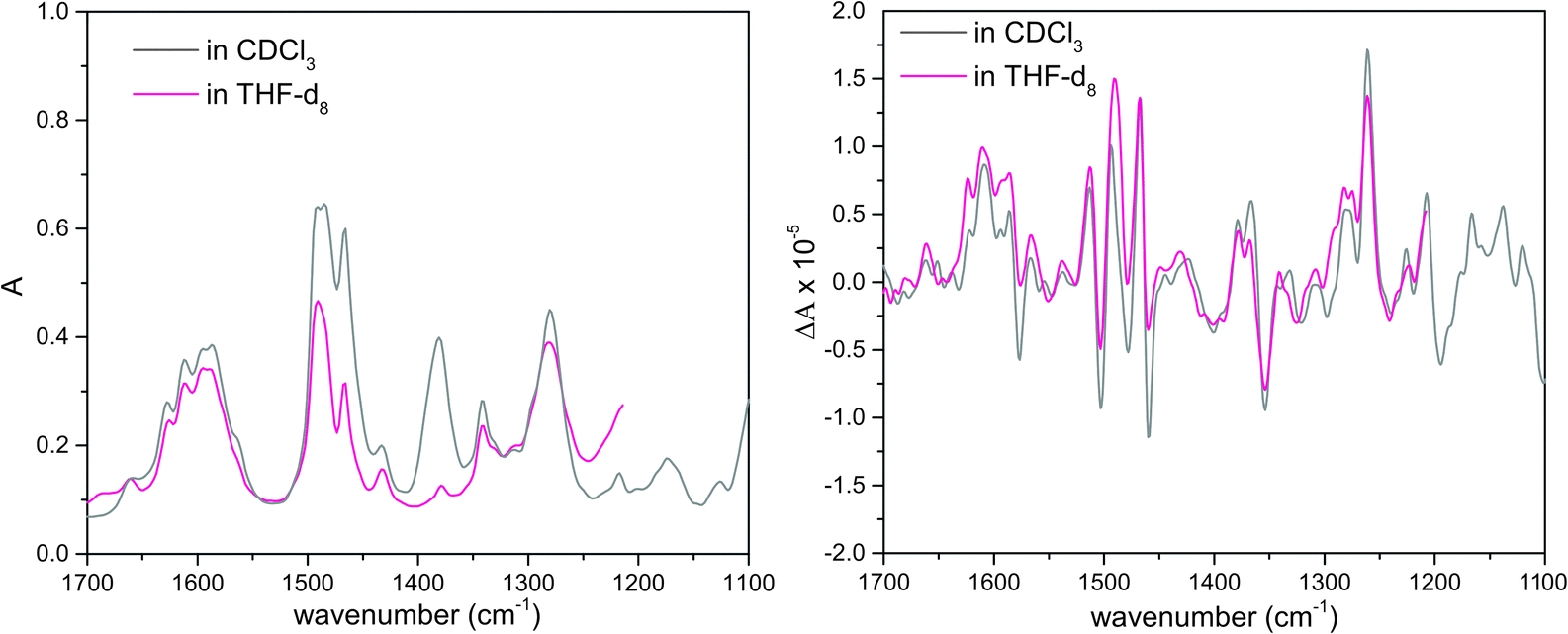 | ||
| Fig. 2 Raw experimental VA (left) and VCD (right) spectra of AFX-155 in deuterated chloroform and THF-d8 solvents. The measurements below 1200 cm−1 in THF are removed due to strong THF solvent absorption. | ||
3.3 Comparison of experimental and simulated VCD spectra of AFX-155
A systematic conformational search for AFX-155 was reported in ref. 15. All cis-conformers were found to be substantially less stable than trans-conformers due to severe steric effects in the former case. Therefore only trans-conformers are considered here. For simplicity, we drop the prefix trans for all conformers in the current study. We would also like to point out that in the previous study, the four alkyl chains (–C6H13) were replaced by four –CH3 subunits to reduce computational cost because it was rationalized that these chains do not have any significant contributions to the electronic transitions. Since VCD spectral features are much more sensitive to subtle conformational changes, we decide to include these alkyl chains in our geometry optimizations and VA and VCD spectral simulations.One noticeable outcome with the inclusion of the four alkyl chains is that the number of possible conformers is reduced because of the additional steric hindrance introduced by these bulky subunits. In total nine HB conformers are re-optimized and are confirmed to be real minima. Their geometries are presented in Fig. 3 where we separate these structures obtained into three groups, based on their associated axial chirality at the –C–N bonds, i.e. RR, RS, or SS. It is interesting to note that the long bulky side chains form cavities of different sizes in different conformers. As a result, one may anticipate that accessibility to solvent molecules can vary quite differently among various conformers. For example, the R_intra_HB//S_extra_HB conformer is structurally fully extended whereas R_extra_HB//R_extra_HB is considerably more compact, with a much smaller cavity size (see Fig. S2, ESI†).
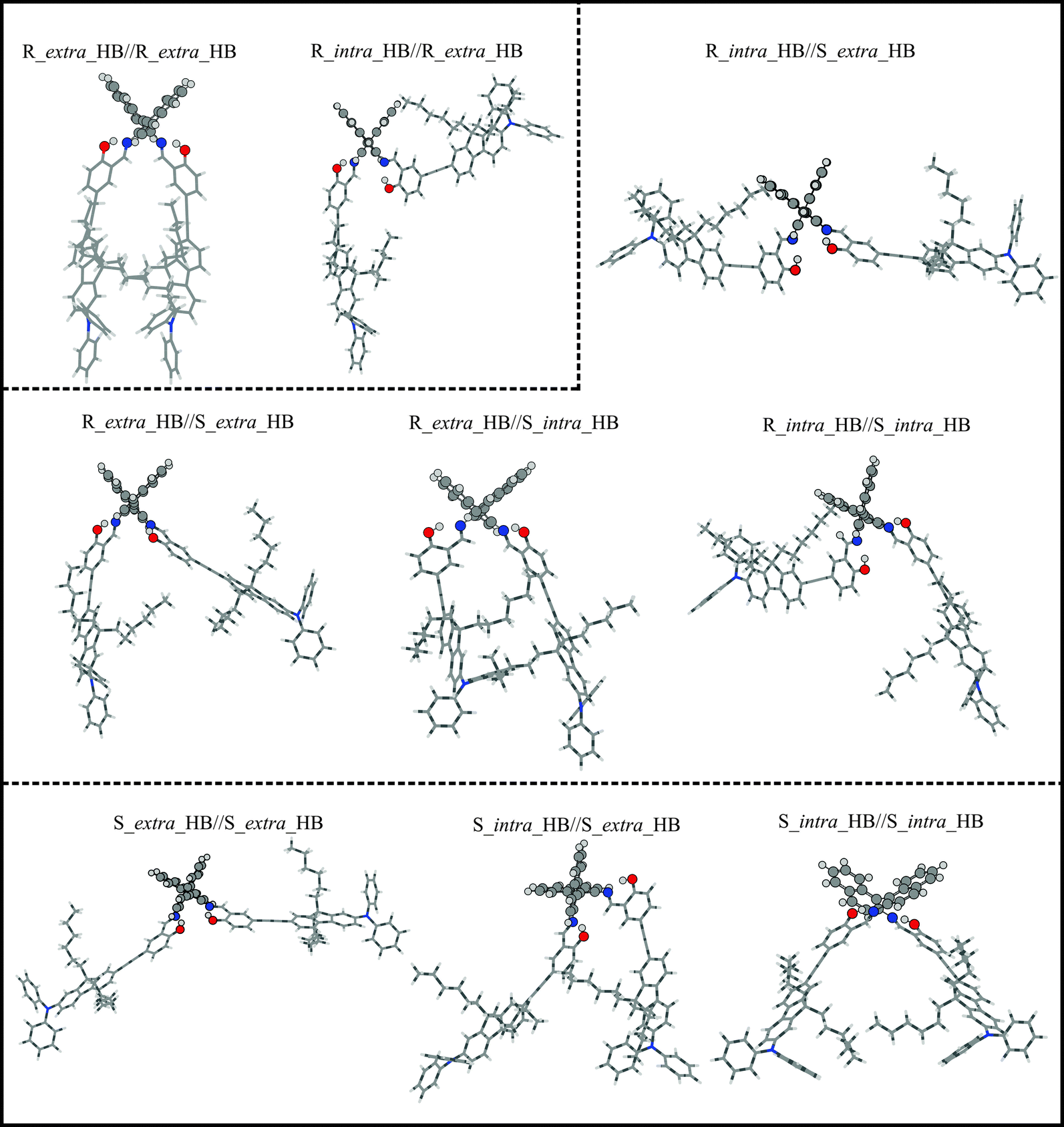 | ||
| Fig. 3 Optimized geometries of the HB conformers of AFX-155 in CDCl3 obtained at the PCM/B3LYP/6-31G(d) level of theory. The geometries are separated into three groups based on their associated axial chirality at the –C–N bonds, i.e. RR, RS, or SS. | ||
The relative energies and free energies of these three groups of conformers using B3LYP/6-31G(d), single point energy at B3LYP/cc-pVTZ//6-31G(d) and using B3LYP/6-31G(d) with the D3 Grimme's dispersion correction are summarized in Table S1, ESI.† We note that the inclusion of the dispersion interactions can change the relative energies significantly. For example, S_extra_HB//S_extra_HB is more stable than S_intra_HB//S_extra_HB and S_intra_HB//S_intra_HB by a few kcal mol−1 without the inclusion of dispersion interactions. With the inclusion of dispersion interactions, S_intra_HB//S_extra_HB becomes the most stable one by more than 15 kcal mol−1 compared to the other two. It is no surprise that dispersion energy becomes important for systems like AFX-155, but we also stress the considerable current challenges in obtaining reliable relative energies for a large system like AFX-155 in general.
In addition to the HB conformers, the relevant HB//NHB and NHB conformers are also considered and their geometries re-optimized. We note that those conformers with two intramolecular HB bonds are considerably more stable than those with one and even more so than those without in each of the three groups indicated above. This is consistent with other previous studies that suggest that species with the intramolecular H-bonding interactions become dominant over those without in the presence of non-polar or weakly polar solvent such as CH2Cl2 and CHCl3.28
In Fig. 4, we compare the calculated VA and VCD spectra of all HB conformers of AFX-155. Since AFX-155 is fairly large and has a significant number of vibrational modes in the finger print region, many of these VA bands overlap with each other. This often makes direct and detailed comparison between experimental and theoretical VA spectra ambiguous and prevents one to draw decisive structural information. On the other hand, VCD spectra tend to show a considerable amount of unique spectral signatures related to stereogenic and conformational structural information. For comparison between theory and experiment, we divide the finger print region into three sections from high to low wavenumbers and refer to Fig. 1 for the functional groups indicated below. Region I from 1700 to 1550 cm−1 is composed of several strong vibration bands due to the CN stretching, O–H bending, and CC stretching of the binaphthyl rings. Region II from 1550 to 1400 cm−1 contains mostly vibrational motions associated with the C–H and O–H bending of the two phenyl groups which are in close proximity to the binaphthyl rings, the C–H bending of the aromatic rings of the two bulky end substitutes, and the CH2 scissoring of the four C6H13 alkyl chains. Region III from 1400–1100 cm−1 contains mainly C–N stretching at the two bulky terminal groups and CH2 bending of the long alkyl chain. Overall, the simulated IR spectra of all nine HB conformers presented in Fig. 4 look all alike. Furthermore, they all agree well with the experimental VA data. Therefore, one cannot differentiate these conformers on the basis of their VA spectral signatures.
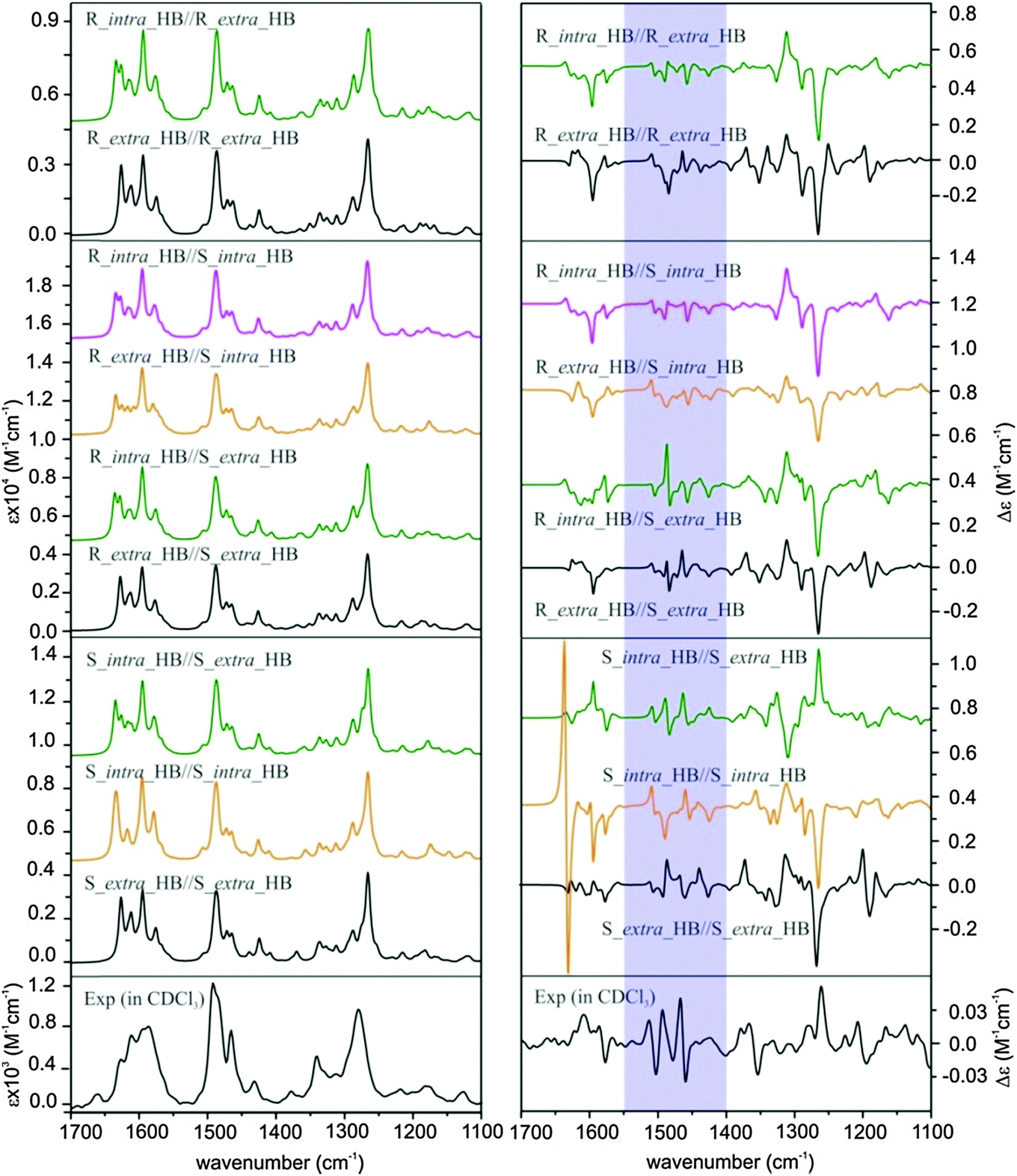 | ||
| Fig. 4 Calculated VA (left) and VCD (right) spectra of all nine HB conformers of AFX-155 obtained at the PCM/B3LYP/6-31G(d) level compared to the experimental data at the bottom. For easy comparison, these conformers are separated into three groups based on the associated axial chirality at the –C–N bonds, i.e. RR, RS, or SS. | ||
The theoretical VCD spectral features, on the other hand, look substantially different from one conformer to the next, enabling potentially clear identification of the dominant conformational species in solution. This highlights the significant advantages of VCD spectroscopy in providing structural information of chiral systems in solution. Below, we compare the simulated VCD features of each HB conformers among themselves and with the major experimental VCD spectral features in all three finger print regions. In region I, the prominent experimental positive and negative VCD band at 1610 cm−1 and 1577 cm−1, respectively, can only be matched by conformers R_extra_HB//S_extra_HB and S_intra_HB//S_extra_HB while all the other conformers exhibit major discrepancy. Although the smaller experimental VCD features in this region are somewhat noisy, the consistency they show in both CDCl3 and THF-d8 solvents gives one confidence in their reliability. In region II (shaded), the experimental VCD spectrum shows a beautiful sequence of +/−/+/−/+/− VCD signatures. This sequence is best matched with S_intra_HB//S_extra_HB, while all the other conformers exhibit markedly different patterns with the exception of perhaps R_intra_HB//S_extra_HB. The latter one demonstrates a somewhat similar pattern but with an additional negative band at the high wavenumber end. In region III, the most prominent experimental VCD feature at ∼1270 cm−1 is positive, while a negative band is predicted for all HB conformers except S_intra_HB//S_extra_HB, which exhibits a positive VCD band feature. From the above detailed comparison, it is clear that S_intra_HB//S_extra_HB is the only HB conformer which demonstrates consistent agreement with the experimental data in all three regions.
Since the previous ECD study indicated that the R_intra_NHB//R_extra_NHB conformer provides the best agreement between simulated and experimental ECD spectra, we also compare simulated VA and VCD spectra of all the relevant R_intra//R_extra HB and NHB conformers and S_intra//S_extra HB and NHB conformers in Fig. 5 for completion.
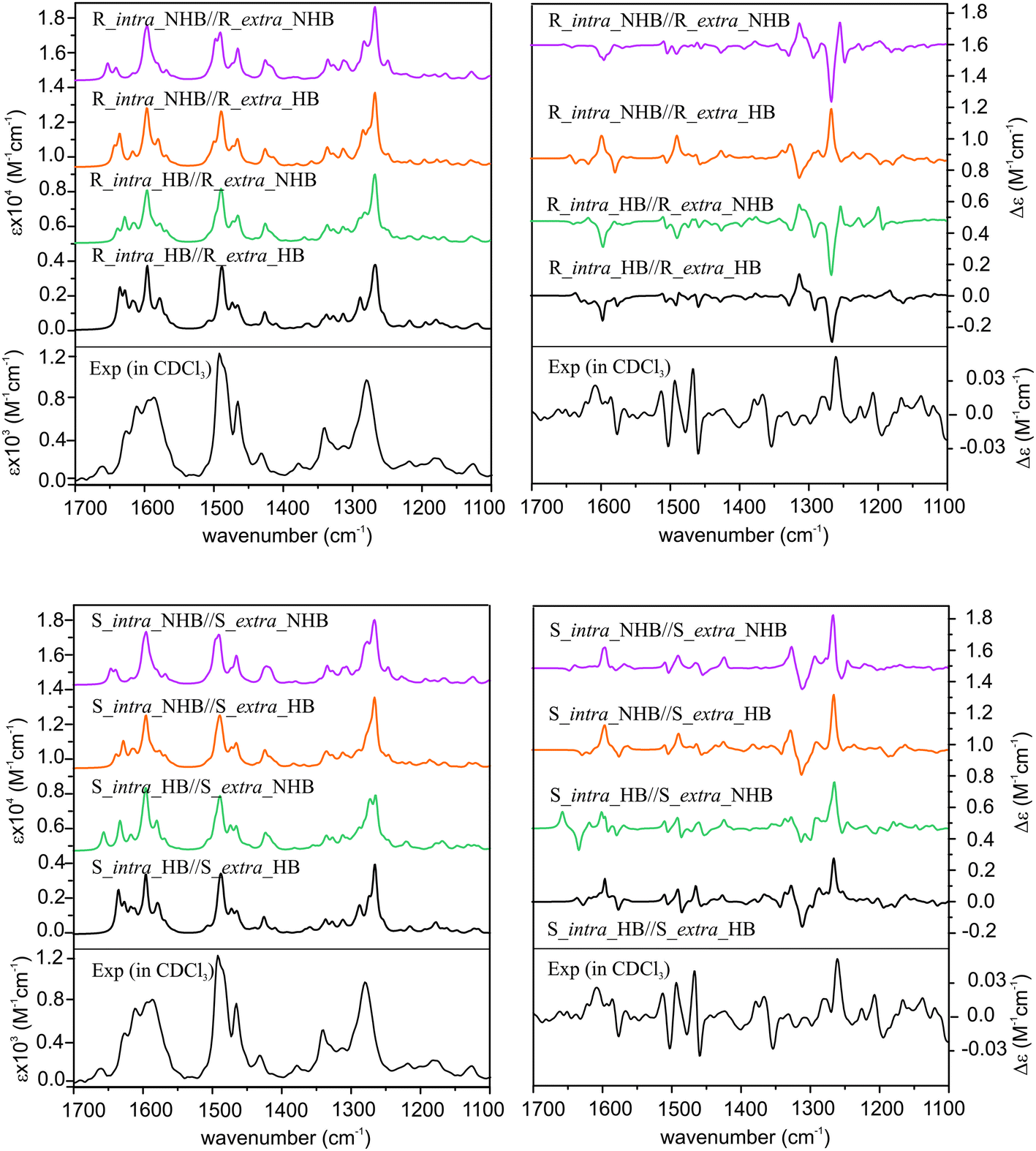 | ||
| Fig. 5 Calculated VA (left) and VCD (right) spectra of all the R_intra//R_extra HB or NHB conformers (top panel) and S_intra//S_extra HB or NHB conformers (bottom panel) of the AFX-155 molecule obtained at the PCM/B3LYP/6-31G(d) level compared with the experimental ones. | ||
From Fig. 5, it is immediately obvious that all R_intra//R_extra HB or NHB conformers show major discrepancies in the VCD features in the 1300–1200 cm−1 region. For the S_intra//S_extra conformers, the agreements between experiment and theory seem to be generally better than R_intra//R_extra at the first glance. Detailed comparison of VA and VCD spectra of S_intra_HB//S_extra_HB with the experimental data is summarized in Fig. 6. While the existence of other S_intra//S_extra NHB conformers in small amounts cannot be ruled out completely, S_intra_HB//S_extra_HB is evidently by far the dominant species. This preference for the HB species observed here is also consistent with the discussion on the relative energies above.
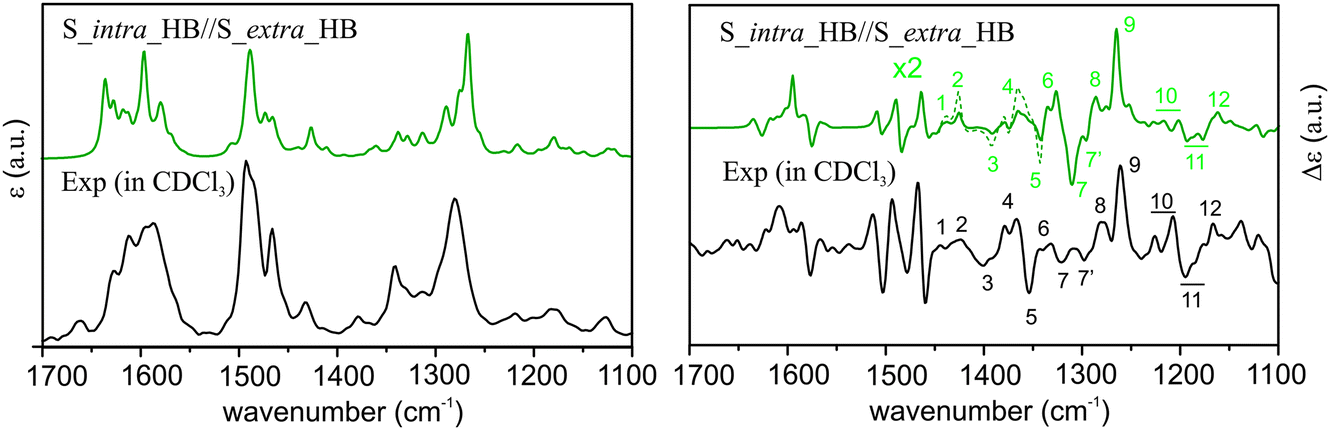 | ||
| Fig. 6 Comparison of the experimental VA and VCD spectra of AFX-155 in CDCl3 with the corresponding simulated spectra of S_intra_HB_S_extra_HB obtained at the PCM/B3LYP/6-31G(d) level of theory. The theoretical VCD band intensities in a small region between 1450 cm−1 and 1330 cm−1 are amplified by a factor of 2 and indicated with a dotted line for easier pattern recognition. The Arabic numerals are used to indicate the corresponding features in the experimental and theoretical data in the crowded region below 1450 cm−1. | ||
3.4 Concentration dependent diastereomeric preference
Based on the distinctive VCD spectral features described above, we can conclusively identify S_intra_HB//S_extra_HB as the dominant species in CDCl3. This conclusion, however, is in odd with the previous ECD and TPCD investigations of AFX-155 where R_intra//R_intra diastereomers were identified as the main species. This prompts us to ask a few interesting and important questions. Why do the previous ECD and the present VCD studies come to such different conclusions? Are there really different dominant species in CDCl3 in the present study and in THF-d8 in the previous studies? If yes, what are the plausible explanations for such strong preference of different diastereomers in solution?First, we examine if two different solvents, i.e. CDCl3 and THF-d8, are the cause for such noticeably different conclusions. For that, we already did the VA and VCD measurements in both solvents (see discussion before) and found no obvious difference in the VA and VCD spectral features of AFX-155 excluding absorption due to solvents themselves. Furthermore, both solvents have similar dielectric constants, 4.7113 and 7.4257, respectively. For completion, we also measured UV-Vis and ECD spectra in CDCl3 (Fig. 7). As one can see, these spectra are very much the same as those obtained in the previous study using THF-d8.15 Therefore, solvents themselves seem not to be the main issue here.
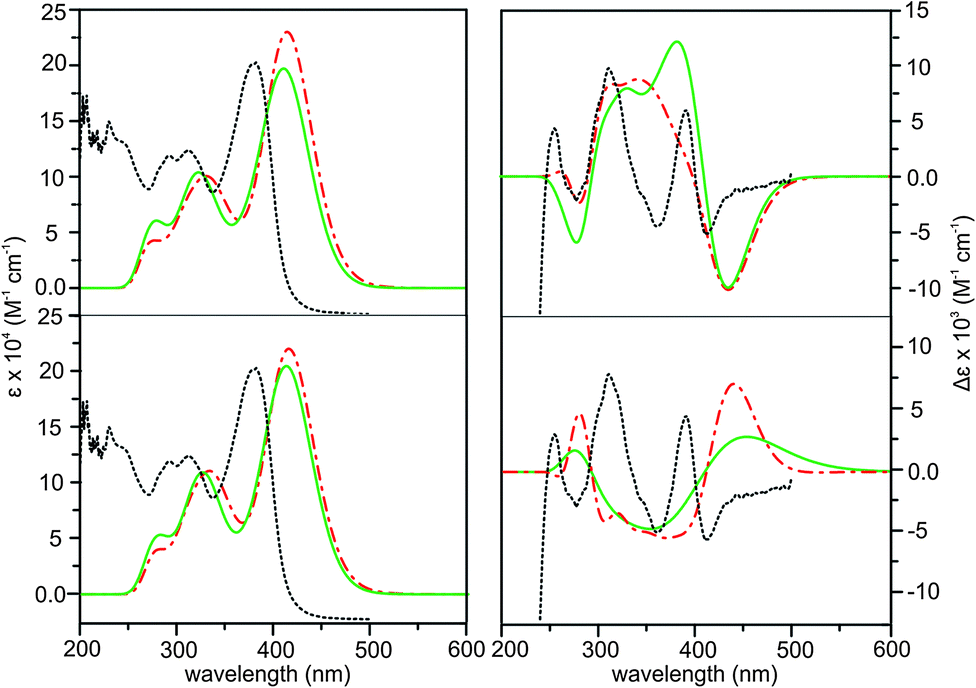 | ||
| Fig. 7 Comparison of the experimental ECD spectrum in CDCl3 (black dotted line) with the theoretical ECD spectra of R_intra//R_extra (upper row) and S_intra//S_extra (lower row) HB (red dashed–dotted line) and NHB (green solid line) conformers obtained at the PCM/B3LYP/6-31G(d) level of theory. | ||
Another noticeable difference is the concentrations used in ECD and VCD experiments. While ECD experiments typically use a highly diluted sample (≤1 × 10−3 M), a highly concentrated sample is needed for VCD measurements because VCD intensity is usually only 10−4 to 10−5 of VA bands. In the current experiment, a 70 fold higher concentration is used for VCD than ECD. For a more systematic comparison, we included also ORD measurements in CDCl3. Furthermore, we performed both ECD and ORD simulations for all conformers of interest using their full geometries, in contrast to the previous ECD study where some bulky groups –C6H13 were simplified with –CH3 groups. The results are summarized in Fig. 7 and 8 for ECD and ORD spectra, respectively. Generally, the simulated ECD spectra are similar to those reported before, suggesting that the truncation used before is justified. While the agreement between ECD experimental and simulated spectra is not as decisive as the corresponding VCD counterparts, it is nevertheless quite clear here that the ECD features of R_intra//R_extra conformers are in accord with the observed data while S_intra//S_extra conformers show severe discrepancies. The experimental ORD spectrum shows an upward trend going from short to long wavelengths. While R_intra_HB//R_extra_HB exhibits the same upward trend in agreement with the experimental observation, S_intra_HB//S_extra_HB shows a downward trend. It is noted that although the absolute ORD values are still challenging to be reproduced theoretically, the general variation trend with wavelength is typically better reproduced.29–31 Therefore the ORD study performed here also qualitatively supports the conclusion derived from both the current and previously reported ECD studies15,16 that R_intra//R_extra is the dominant species in the much diluted solution, rather than S_intra//S_extra.
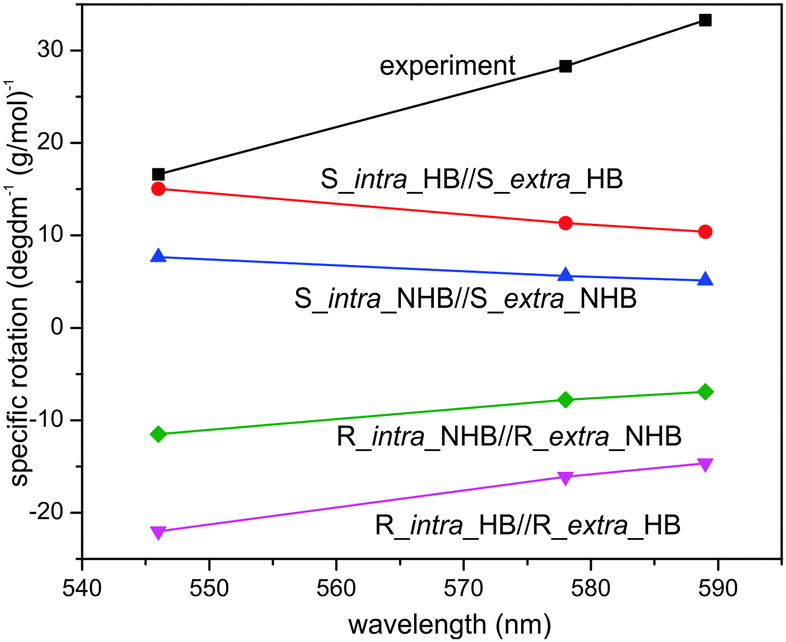 | ||
| Fig. 8 Comparison of the experimental ORD spectrum in CDCl3 with the theoretical ORD spectra of R_intra//R_extra HB and NHB conformers as well as S_intra//S_extra HB and NHB conformers obtained at the PCM/B3LYP/6-31G(d) level of theory. | ||
It therefore appears that concentration plays a crucial role in which diastereomer is preferred in solution. Although such observations appear not to have been extensively studied, there are a number of theoretical and experimental publications when similar concentration dependent conformational preferences were reported.32–34 For example, inversion of population distribution of felodipine conformations at increasing concentration in DMSO was observed by Khodov and co-workers in 2014.33 Using NMR spectroscopy, these authors concluded that conformational preference of felodipine in DMSO is concentration dependent: conformers that dominate in a dilute solution become the least abundant in the saturated one. Furthermore, they pointed out that conformers that dominate in the saturated solution are of the same type as revealed in the crystalline state by X-ray. The authors attribute the inversion of conformer distribution in the saturated solution to both increases in intermolecular felodipine–felodipine and felodipine–DMSO interactions. Ghorai applied molecular dynamics simulations to predict the concentration dependence of trans and gauche conformations of n-butane inside a confined medium such as zeolite NaY.34 He found that the percentage of gauche conformations inside zeolite increases with concentration and identified guest–guest interaction as the key factor for the enhancement of the gauche conformation.
To understand the observation, we performed a one-dimensional potential energy surface scan for the inter-conversion between R_intra_HB//R_extra_HB and S_intra_HB//S_extra_HB and found that the interconversion can occur through a planar TS with a barrier energy of 1.8 kcal mol−1 obtained at the B3LYP/3-21G level of theory. The interconversion between R_intra//R_extra and S_intra//S_extra conformers of AFX-155 is likely facilitated by solvent molecules. As indicated before, while R_intra//R_extra conformers exhibit a larger cavity, allowing easy access by solvent molecules, S_intra//S_extra conformers show a much smaller cavity (see Fig. S2, ESI†). One may expect that such differences in solvent–solute interactions may lead to preferentially stabilization of certain conformers, as recently reported in the case of a tris(diamine)nickel(II) complex.35 Further theoretical modelling will be required to understand this interesting and important phenomenon in greater details, but that is out-of-scope of the current study.
Conclusions
Axial chirality and conformations of a flexible multiple axial chiral molecule, AFX-155, have been extensively investigated using three chiroptical spectroscopies, i.e. VCD, ECD, and ORD, in conjugation with DFT calculations. Experimentally, both VCD and ECD spectra of AFX-155 in THF-d8 and CDCl3 look essentially the same, indicating that the polarity of solvent does not influence the axial chirality and conformational distribution of AFX-155 significantly. The combined VCD and DFT study clearly shows that the dominant species is R-binaphthyl, S_intra_HB//S_extra_HB (R-SS) in the concentrated solution. In the much dilute solution, the dominant species is determined to be R-binaphthyl, R_intra_HB//R_extra_HB (R-RR) based on the ECD and ORD investigations. The latter conclusion is consistent with the previously reported ECD study. This is an area of research which has not yet been fully explored, although some theoretical and experimental publications had reported similar concentration dependent behaviors. Further theoretical modeling will be highly desirable to explain the interesting observations reported here in detail.Acknowledgements
This research was funded by the University of Alberta and the Natural Sciences and Engineering Research Council of Canada. This research was enabled in part by support provided by WestGrid (http://www.westgrid.ca) and Compute Canada Calcul Canada (http://www.computecanada.ca). We gratefully acknowledge access to the computing facilities provided by the Shared Hierarchical Academic Research Computing Network (Sharcnet). The authors thank Dr Frazer for kindly providing the AFX-155 sample. YX holds a senior Canada Research Chair in Chirality and Chirality Recognition.References
- (a) B. Mennucci, C. Cappelli, R. Cammi and J. Tomasi, Chirality, 2011, 23, 717–729 CrossRef CAS; (b) P. Mukhopadhyay, G. Zuber, M. Goldsmith, P. Wipf and D. N. Beratan, ChemPhysChem, 2006, 7, 2483–2486 CrossRef CAS PubMed; (c) S. Coriani, A. Baranowska, L. Ferrighi, C. Forzato, D. Marchesan, P. Nitti, G. Pitacco, A. Rizzo and K. Ruud, Chirality, 2006, 18, 357–369 CrossRef CAS PubMed.
- G. Yang and Y. Xu, in Top. Curr. Chem., ed. R. Naaman, D. N. Beratan and D. H. Waldeck, Springer-Verlag, Berlin Heidelberg, 2011, vol. 298, pp. 189–236 Search PubMed.
- (a) P. L. Polavarapu, Chirality, 2012, 24, 909–920 CrossRef CAS PubMed; (b) M. R. Poopari, Z. Dezhahang, G. Yang and Y. Xu, ChemPhysChem, 2012, 13, 2310–2321 CrossRef CAS PubMed; (c) M. R. Poopari, P. Zhu, Z. Dezhahang and Y. Xu, J. Chem. Phys., 2012, 137, 194308 CrossRef PubMed; (d) P. Zhu, G. Yang, M. R. Poopari, Z. Bie and Y. Xu, ChemPhysChem, 2012, 13, 1272–1281 CrossRef CAS PubMed.
- Z. Dezhahang, M. R. Poopari and Y. Xu, Chem. – Asian J., 2013, 8, 1205–1212 CrossRef CAS PubMed; C. Merten and Y. Xu, Angew. Chem., 2013, 125, 2127–2130 ( Angew. Chem., Int. Ed. , 2013 , 52 , 2073–2076 ) CrossRef.
- M. R. Poopari, Z. Dezhahang and Y. Xu, Phys. Chem. Chem. Phys., 2013, 15, 1655–1665 RSC.
- R. W. Sinkeldam, M. H. C. J. van Houtem, K. Pieterse, J. A. J. M. Vekemans and E. W. Meijer, Chem. – Eur. J., 2006, 12, 6129–6137 CrossRef CAS PubMed.
- A. L. Hofacker and J. R. Parquette, Angew. Chem., 2005, 117, 1077–1081 ( Angew. Chem., Int. Ed. , 2005 , 44 , 1053–1057 ) CrossRef.
- (a) M. Losada, P. Nguyen and Y. Xu, J. Phys. Chem. A, 2008, 112, 5621–5627 CrossRef CAS PubMed; (b) M. Losada, H. Tran and Y. Xu, J. Chem. Phys., 2008, 128, 014508 CrossRef PubMed.
- (a) T. Taniguchi and K. Monde, J. Am. Chem. Soc., 2012, 134, 3695–3698 CrossRef CAS PubMed; (b) V. Andrushchenko, D. Tsankov, M. Krasteva, H. Wieser and P. Bour, J. Am. Chem. Soc., 2011, 133, 15055–15064 CrossRef CAS PubMed; (c) T. Brotin, D. Cavagnat, J. Dutasta and T. Buffeteau, J. Am. Chem. Soc., 2006, 128, 5533–5540 CrossRef CAS PubMed.
- S. Abbate, F. Lebon, G. Longhi, C. F. Morelli, D. Ubiali and G. Speranza, RSC Adv., 2012, 2, 10200–10208 RSC.
- P. Scafato, F. Caprioli, L. Pisani, D. Padula, F. Santoro, G. Mazzeo, S. Abbate, F. Lebon and G. Longhi, Tetrahedron, 2013, 69, 10752–10762 CrossRef CAS PubMed.
- S. Qiu, E. De Gussem, K. Abbaspour Tehran, S. Sergeyev, P. Bultinck and W. Herrebout, J. Med. Chem., 2013, 56(21), 8903–8914 CrossRef CAS PubMed.
- L. Andernach, L. P. Sandjo, J. C. Liermann, I. Buckel, E. Thines and T. Opatz, Eur. J. Org. Chem., 2013, 5946–5951 CrossRef CAS.
- P. L. Polavarapu, N. Jeirath, T. Kurtan, G. Pescitelli and K. Krohn, Chirality, 2009, 21, E202–E207 CrossRef CAS PubMed.
- C. Diaz, A. Fraser, A. Morales, K. D. Belfield, S. Ray and F. E. Hernández, J. Phys. Chem. A, 2012, 116, 2453–2465 CrossRef CAS PubMed.
- C. Díaz, L. Echevarria and F. E. Hernández, J. Phys. Chem. A, 2013, 117, 8416–8426 CrossRef PubMed.
- M. Losada and Y. Xu, Phys. Chem. Chem. Phys., 2007, 9, 3127–3135 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef; (b) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef; (c) J. K. Labanowski and J. W. Andzelm, Density Functional Methods in Chemistry, Springer-Verlag, New York, 1991 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (c) T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- M. R. Poopari, Z. Dezhahang and Y. Xu, Phys. Chem. Chem. Phys., 2013, 15, 1655–1665 RSC.
- Z. Dezhahang, C. Merten, M. R. Poopari and Y. Xu, Dalton Trans., 2012, 41(35), 10817–10824 RSC.
- V. P. Nicu, E. J. Baerends and P. L. Polavarapu, J. Phys. Chem. A, 2012, 116, 8366–8373 CrossRef CAS PubMed.
- P. Lahiri, K. B. Wiberg, P. H. Vaccaro, M. Caricato and T. D. Crawford, Angew. Chem., 2014, 126, 1410–1413 ( Angew. Chem., Int. Ed. , 2014 , 53 , 1386–1389 ) CrossRef.
- P. Mukhopadhyay, G. Zuber, M. Goldsmith, P. Wipf and D. N. Beratan, ChemPhysChem, 2006, 7, 2483–2486 CrossRef CAS PubMed.
- K. Ruud and R. Zanasi, Angew. Chem., 2005, 117, 3660–3662 ( Angew. Chem., Int. Ed. , 2005 , 44 , 3594–3596 ) CrossRef.
- R. Begum, T. Sagawa, S. Masatoki and H. Matsuura, J. Mol. Struct., 1988, 442, 243–250 CrossRef.
- I. A. Khodov, S. V. Efimov, M. Y. Nikiforov, V. V. Klochkov and N. Georgi, J. Pharm. Sci., 2014, 103, 392–394, DOI:10.1002/JPS.23833.
- P. Kr. Ghorai, J. Phys. Chem. B, 2010, 114, 6492–6499 CrossRef CAS PubMed.
- C. Merten, R. McDonald and Y. Xu, Inorg. Chem., 2014, 53, 3177–3182 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01704h |
| This journal is © the Owner Societies 2014 |