 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the ring-opening of substituted cyclobutene to benzocyclobutene: analysis of π delocalization, hyperconjugation, and ring strain†‡
Paola
Nava§
and
Yannick
Carissan§
*
Aix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, Marseille, 13397, France. E-mail: yannick.carissan@univ-amu.fr; Fax: +33 4 9128 9179; Tel: +33 4 9128 9168
First published on 20th June 2014
Abstract
The influence of several substituents on the ring-opening elementary step of cyclobutene-like systems is analyzed computationally in detail. We focus on trans-1,2-disiloxycyclobutene-like molecules. Electronic effects (hyperconjugation and π delocalization) and geometrical constraints are decoupled and allow for an instructive analysis. It is found that the energy difference between closed and open forms is dictated mainly by the electronic structure of the open form, in which the rotation along the resulting simple C–C bond drives the electronic delocalization. Our calculations led us to quantify effects that determine the energy difference in the special case of disubstituted benzocyclobutene with respect to the disubstituted o-xylylene (aromaticity, π delocalization, ring strain). The relevant role of the siloxy-substituents is rationalized by an analysis of the molecular orbital interaction in an original manner. Finally, calculations are presented and show that the PBE0 functional must be preferred to the popular B3LYP functional for computations on substituted cyclobutene-like rings.
1 Introduction
Highly strained molecules are fascinating systems because of their enhanced reactivity.1–3 Among them, cyclobutene occupies historically a privileged position, since its thermal ring-opening led to the formulation of the famous Woodward and Hoffmann's rules, based on orbital symmetry conservation.4 Since then, the concerted conrotatory mechanism has been confirmed as the usual pathway for the thermal ring-opening of cyclobutene and benzocyclobutene, ref. 5 and 6 and references therein, even if it has been shown that it can be modified by mechanical forces.7–10Closed forms, i.e. the forms that contain the cyclobutene moiety, are in general less stable than open forms: the 1,3-butene is 11 kcal mol−1 (experimental value)7 and 9.9 kcal mol−1 (computed value for the s-cis conformation)5 more stable than the cyclobutene. This can be explained by considering the release in ring strain and the possible π electron delocalization in open forms. Quite interestingly, only in the case of the aromatic benzocyclobutenes, closed forms become more stable: the benzocyclobutene is about 13 kcal mol−1 more stable than the o-xylylene (experimental observations).11,12 Several theoretical studies have been conducted on the ring-opening of cyclobutene-like systems. Nevertheless, the comparison between open and closed forms was rarely the main issue,13–15 since many studies have been focusing on substitution effects on barrier heights or torquoselectivity.7,16,17 On the basis of Woodward and Hoffmann's work and of Longuet-Higgins and Abrahamson's study, Houk and coworkers have suggested a diagram that correlates the cyclobutene frontier molecular orbitals with those of the transition state for the ring-opening: the principal ingredients are the occupied π and σC–C orbitals, the latter accounts for the C1–C4 bond, and the corresponding virtual π* and σC–C* orbitals, Fig. 1. This picture helps understanding, for instance, the preference for the outward rotating structure of the 3-aminocyclobutene, which has been attributed to a stabilization due to the interaction between the lone pair and the σC–C* orbital in the transition state.16 This kind of stabilization takes place in general for allylic substituents that carry lone pairs, notably oxygen-based and siloxy.
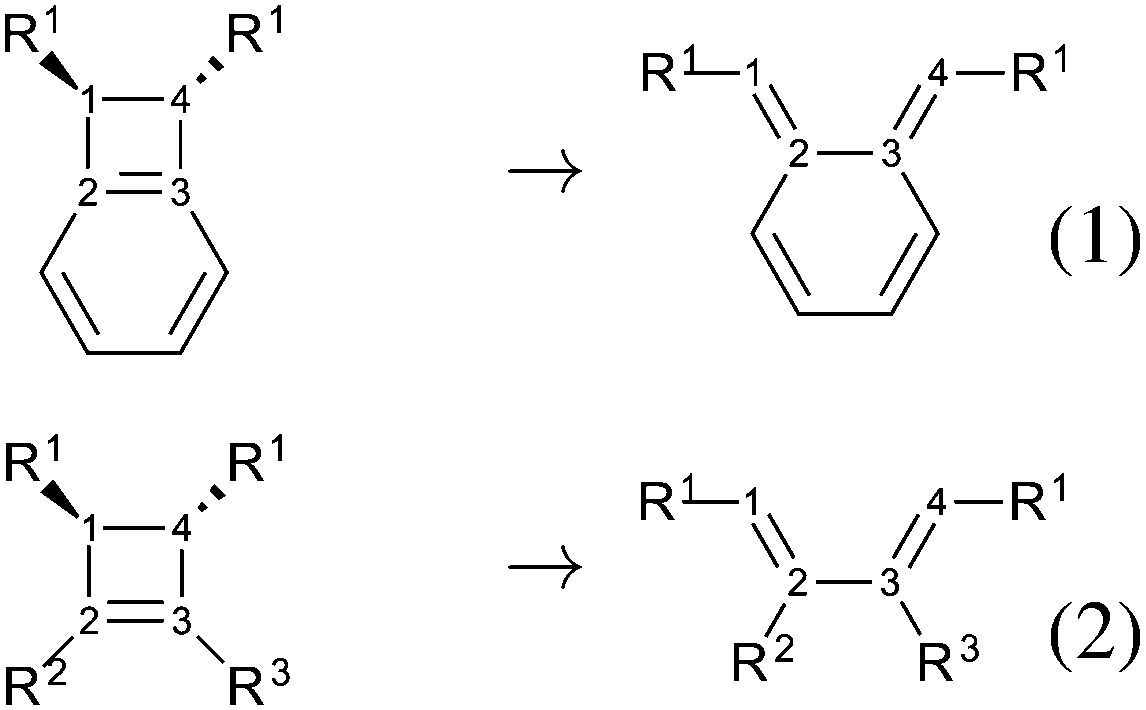 | ||
| Fig. 1 Ring opening reactions. If not specified otherwise in the text, R1 = OTMS. | ||
The present study is a computational work that aims at analyzing the effect of electronic delocalization and aromaticity on the relative stability between open and closed forms in a set of OTMS disubstituted molecules (OTMS = trimethylsilyloxy), as in reactions (1) and (2), Fig. 1. These reactions are elementary steps, the closed form is in the conformation that derives directly from the ring-opening of the cyclobutene moiety (s-cis conformation). Recently, it has been proven that the trans-disiloxybenzocyclobutene combines easily with dioxygen in its triplet state.18 This opens up the perspective of new, stable molecules capable of catching radical systems. In our previous work, it has been shown that the open form combines easily with dioxygen in its triplet state. The resulting triplet intermediate is crucial, since it undergoes spin orbit coupling, which allows the system to reach the singlet potential energy surface and to evolve in a barrierless process towards the product. In the overall reaction, the relevant parameter is the population of the triplet intermediate, which is directly driven by the amount of open form of the reactant. Thus, we focus on the thermodynamics of the opening reaction, since the reaction barrier of the opening process was crossed under experimental conditions.18 A spin-catalysis like mechanism could be at work here and lower the opening barrier.19
Our discussion is based on ΔHR values, defined as follows:
| ΔHR = HO − HC, | (3) |
| ΔH# = HTS − HC, | (4) |
Last, but not least, we shall present calculations that led us to choose the computational level. Those calculations concern ΔER and ΔE# values of benzocyclobutene (C8H6(R1)2, R2 = R3 = H) and cyclobutene (C4H4(R1)2, R2 = R3 = H), disubstituted on the allylic positions of the cyclobutene, R1 = H, NH2, OTMS, CH3, F, NO2. Throughout this work, calculations are performed using the PBE0 functional20 (within the frame of the Density Functional Theory methods) and the def2-TZVP basis set.21 Experimentally the substituents are OTBS, TBS = tert-butyldimethylsilyl; calculations were performed on OTMS analogous structures to reduce the computational cost, TMS = trimethylsilyl.
2 Results and discussion
Computed values of ΔHR and ΔG0R = ΔG298.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KR are reported in Table 1. As expected, those values follow the same trend, but ΔG0R values are lower than corresponding ΔHR values: entropy always favors open forms, because of the release of the cyclobutene ring constraint. This stabilization shifts down all reaction energies by 3 to 5 kcal mol−1. In the following, we shall analyze ΔHR, whose behavior is driven by the electronic structures of reactants and products.
KR are reported in Table 1. As expected, those values follow the same trend, but ΔG0R values are lower than corresponding ΔHR values: entropy always favors open forms, because of the release of the cyclobutene ring constraint. This stabilization shifts down all reaction energies by 3 to 5 kcal mol−1. In the following, we shall analyze ΔHR, whose behavior is driven by the electronic structures of reactants and products.
|
|
ΔHR | ΔG0R | τ(°) |
|---|---|---|---|
|
8.0 | 4.9 | 32.3 |
|
−1.8 | −6.6 | 57.2 |
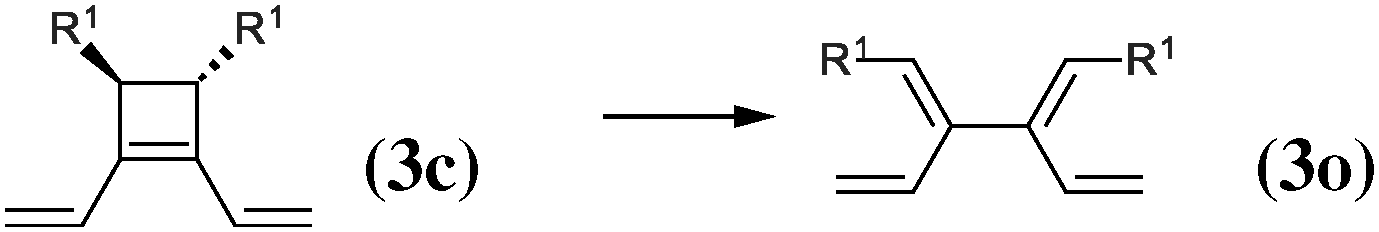
|
−2.4 | −4.6 | 57.1 |
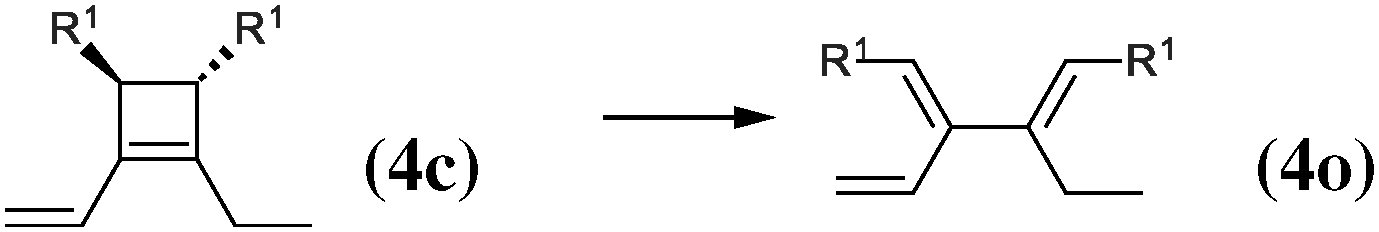
|
−3.3 | −7.2 | 49.8 |
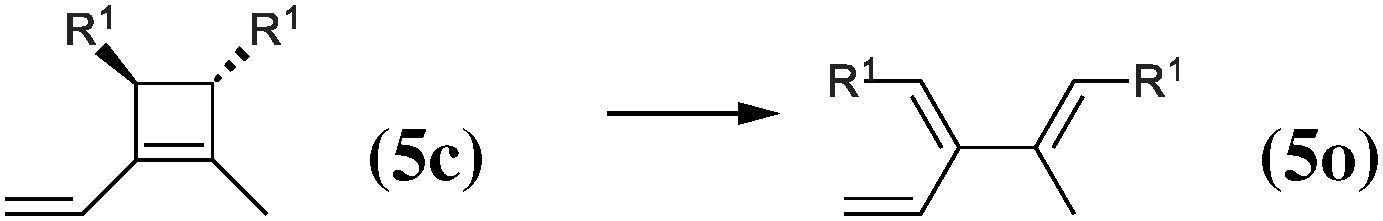
|
−4.1 | −6.0 | 57.6 |
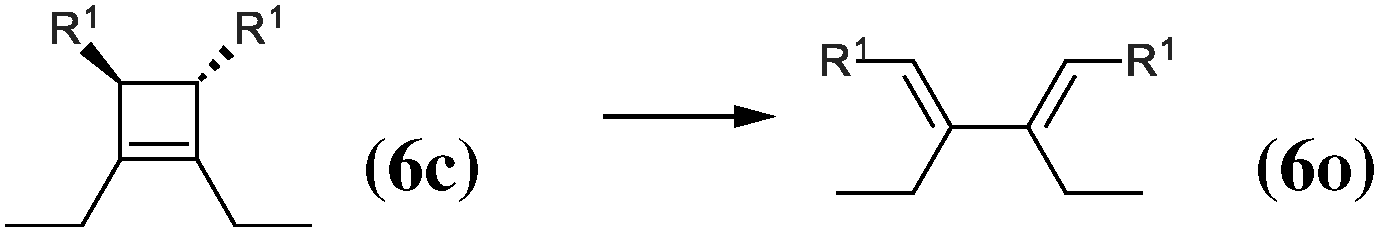
|
−4.1 | −7.2 | 45.9 |
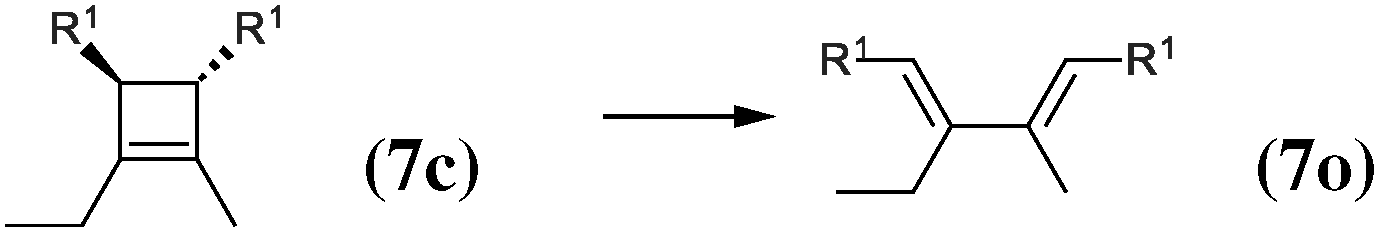
|
−6.4 | −7.9 | 49.2 |

|
−6.3 | −10.8 | 49.1 |

|
−10.2 | −12.0 | 44.7 |
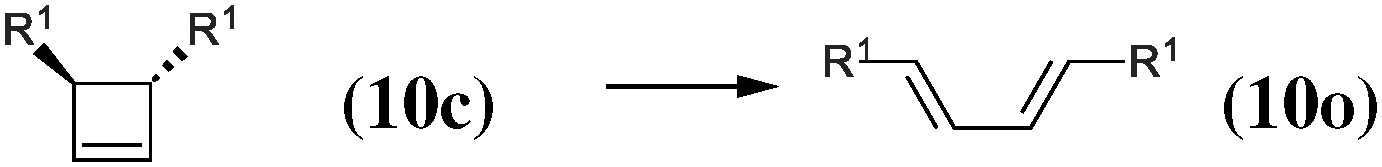
|
−12.5 | −15.3 | 40.2 |
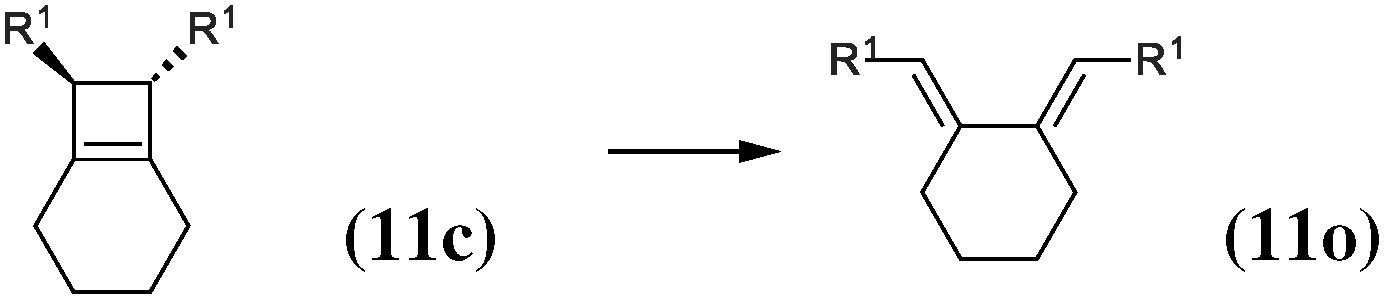
|
−7.7 | −12.1 | 45.6 |
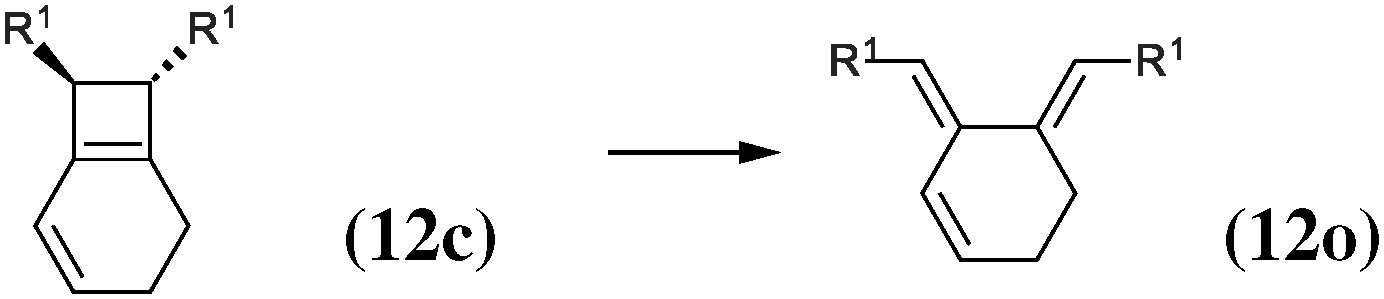
|
−17.2 | −21.6 | 38.6 |
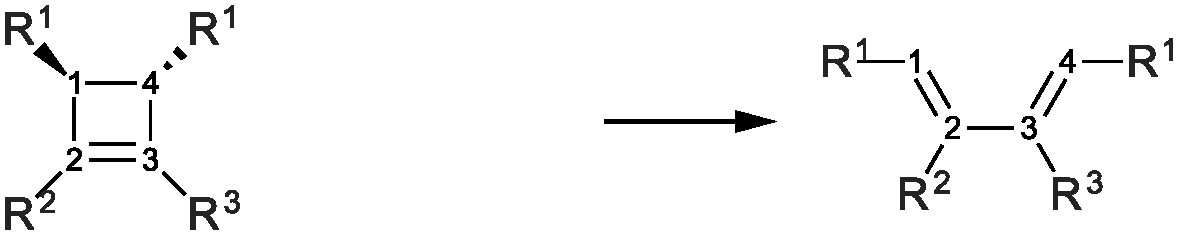
2.1 Influence of electronic delocalization
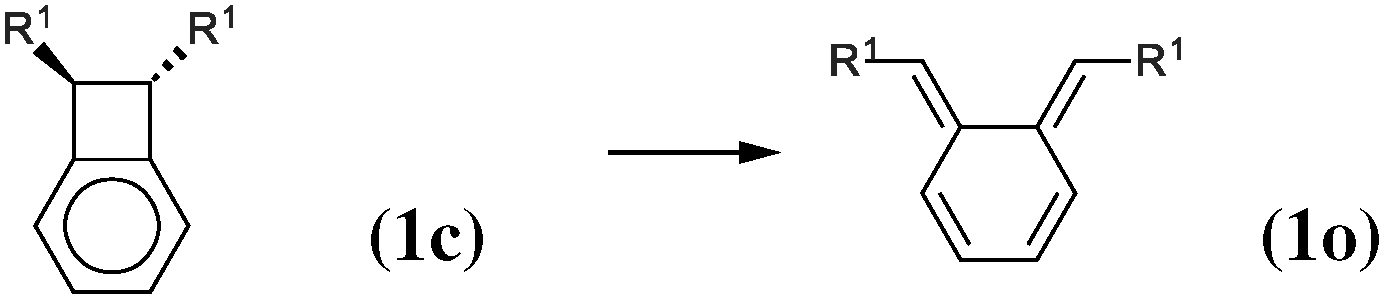
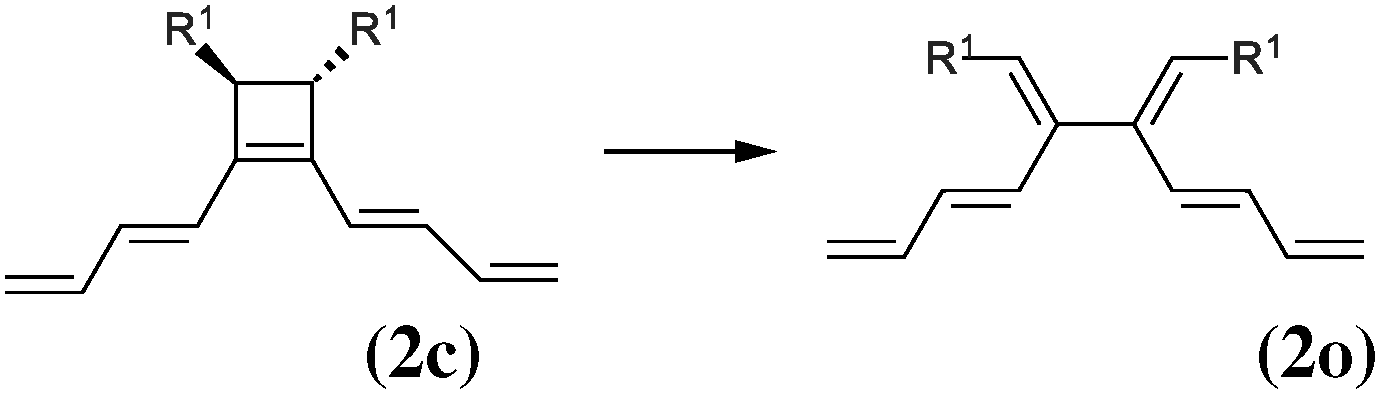
As our goal is to understand deeply the opening reaction of benzocyclobutene, from (1c) to (1o), we first study simpler molecules built in a systematic manner, Table 1. In order to analyze and quantify the effects that play a role in the relative stability between closed and open forms, we consider OTMS cyclobutene derivatives from (2) to (10), where R2 and R3 are non-cyclic alkylic substituents. Molecules (11) and (12) are then discussed. Finally, we shall focus on benzocyclobutene. For the cyclic systems, the rationalization of ΔHR values is supported by computations on hypothetical homodesmotic reactions, as defined by Houk and coworkers.22 Those reactions give access to energy differences that are not bond breaking and creation related, such as strain or aromaticity.In the closed form, because of the constraint due to the 4-membered ring, π delocalization can be efficient. In the open form, mesomery of π electrons along the C1–C2–C3–C4 backbone is favored when the torsion angle τ is low. The change from ethyl to vinyl (7) to (5) modifies again significantly the ΔHR values by 2.7 kcal mol−1. Now the effect is pure resonance due to the delocalization of the π electrons. It is worth keeping in mind that the open form has always two π electrons more than the closed form. The closed form (5c) is efficiently stabilized by mesomery: the p orbitals of the carbon atoms are orthogonal to the 4-membered ring, which leads to nice π delocalization of the four π electrons. In the open form (5o), there are six π electrons but τ is large (57.6°). This leads to two distinct π systems of 4 and 2 electrons, which repel each other because of electron pair repulsion. It is this repulsion that destabilizes the open form with respect to the closed one. Analysis of geometrical parameters corroborates the fact that π delocalization occurs better in the closed form than in the open one: the C2–C3 distance is equal to 148.0 pm in (5o), i.e. 4 pm longer than C2–R2. Thus, the π delocalization along the C2–C3 bond is weak. The same holds true from (5) to (2). The larger number of π electrons to delocalize in (2) stabilizes further the closed form with respect to the open form and ΔHR approaches zero.¶
In summary, hyperconjugation and π electronic delocalization are the key phenomena which explain the relative stability of the closed and open forms of the non-cyclic molecules (2) to (10). If R2 and R3 carry n2 and n3 π electrons, respectively, then the total number of π electrons is n2 + n3 + 2 in the closed form. As τ is constraint to values close to zero, the π system involves all these electrons through the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 bond and the stabilization is due to hyperconjugation and delocalization. In the open form, the breaking of C1–C4 releases the geometrical constraint on τ. The substituents R2 and R3 lead to two distinct π systems with n2 + 2 and n3 + 2 electrons, respectively, which possibly repel each other. The stability of the open form is due to a balance between π delocalization together with ring strain release and repulsion between π systems together with the breaking of the C1–C4 single bond.
C3 bond and the stabilization is due to hyperconjugation and delocalization. In the open form, the breaking of C1–C4 releases the geometrical constraint on τ. The substituents R2 and R3 lead to two distinct π systems with n2 + 2 and n3 + 2 electrons, respectively, which possibly repel each other. The stability of the open form is due to a balance between π delocalization together with ring strain release and repulsion between π systems together with the breaking of the C1–C4 single bond.
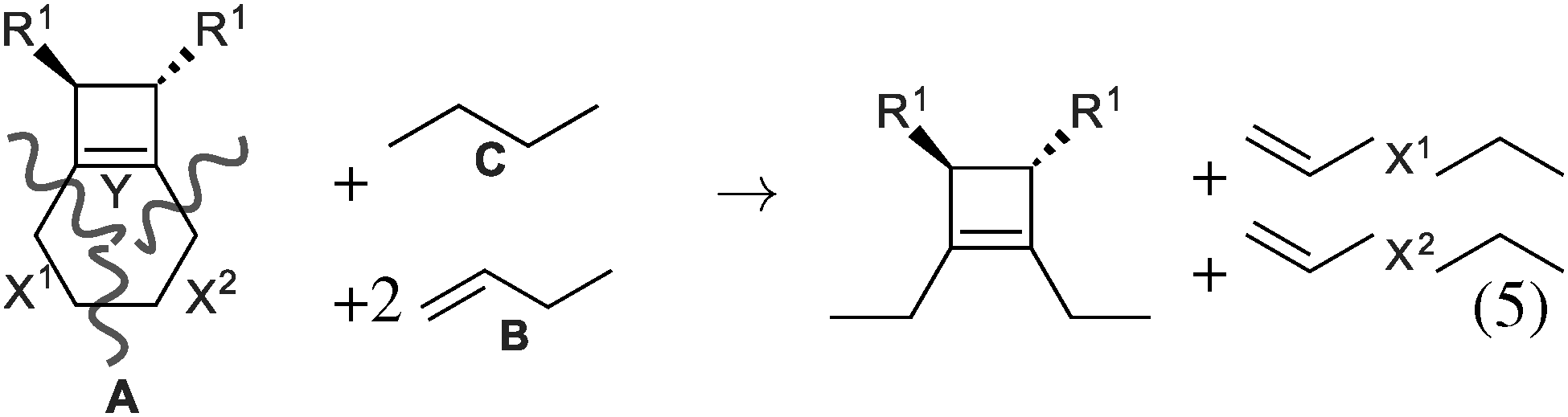 | ||
| Fig. 2 A homodesmotic reaction can be built as follows. The reactant A is cut into three parts (X1, X2 and Y). Fragment X1 is obtained by cleavage of one Csp3–Csp2 and one Csp3–Csp3 bond. It is thus inserted between the single and the double bond of B. The same procedure applies to X2 and Y and determines the nature of the other reactants. In the current case, one B is needed to insert X2 and one C to insert Y. This fully defines the left and right hand side of the arrow. This procedure ensures that the same number of each kind of bond is on each side of the reaction arrow. | ||
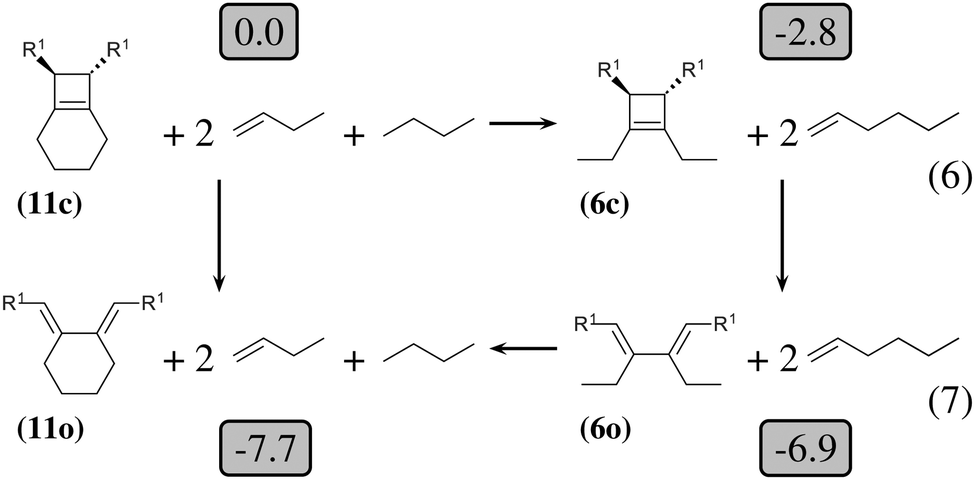 | ||
| Fig. 3 Thermodynamic cycle which decomposes the opening of (11c) into three steps. Relative energies in kcal mol−1 are indicated in gray boxes for R1 = OTMS. Reactions (6) and (7) are homodesmotic. | ||
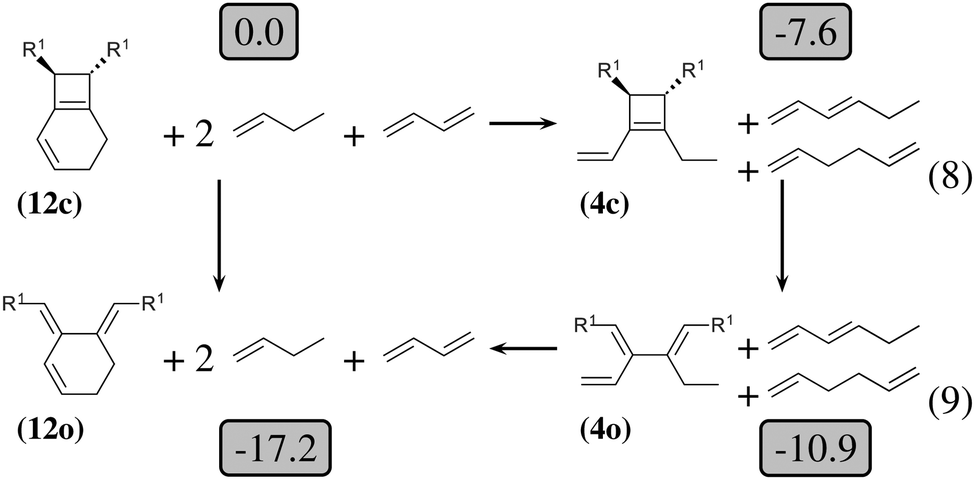 | ||
| Fig. 4 Thermodynamic cycle which decomposes the opening of (12c) into three steps. Relative energies in kcal mol−1 are indicated in gray boxes for R1 = OTMS. Reactions (8) and (9) are homodesmotic. | ||
The 4-membered ring-opening of (12c) is 9.5 kcal mol−1 more favorable than the same opening for (11c). To understand the reason for this difference, we decompose the transformation into three steps. The first step, reaction (8), releases 7.6 kcal mol−1, which is the ring strain energy of the 6-membered ring of (12c). It is larger than the corresponding strain energy of (11c) because of the lack of flexibility due to the sp2 carbon atoms. The opening of the cyclobutene moiety from (4c) to (4o) is 3.3 kcal mol−1 exothermic, which is similar to the opening of the cyclobutene moiety from (6c) to (6o) (4.1 kcal mol−1). Quite surprisingly, the closing of the 6-membered ring, reaction (9), is 6.3 kcal mol−1exothermic: the ring constraint in (12o) leads to a more stable molecule than (4o). Indeed, the cyclic constraint imposes a τ value of 38.6°: this is the lowest value in the set of molecules in Table 1, with the exception of the aromatic (1c). In (12o), the π electronic delocalization is favored because of this low τ value, thus the 17.2 kcal mol−1 exothermicity of the opening of the cyclobutene moiety in (12) comes from the stabilization energy due to π delocalization in (12o), induced by the 6-membered ring constraint.
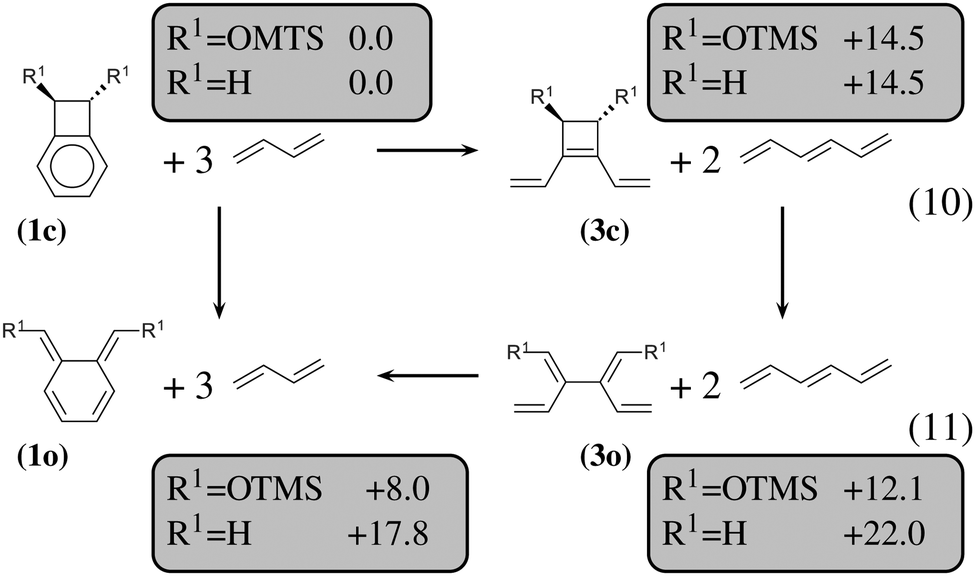 | ||
| Fig. 5 Thermodynamic cycle which decomposes the opening of (1c) into three steps. Relative energies in kcal mol−1 are indicated in gray boxes for R1 = OTMS and R1 = H. Reactions (10) and (11) are homodesmotic. | ||
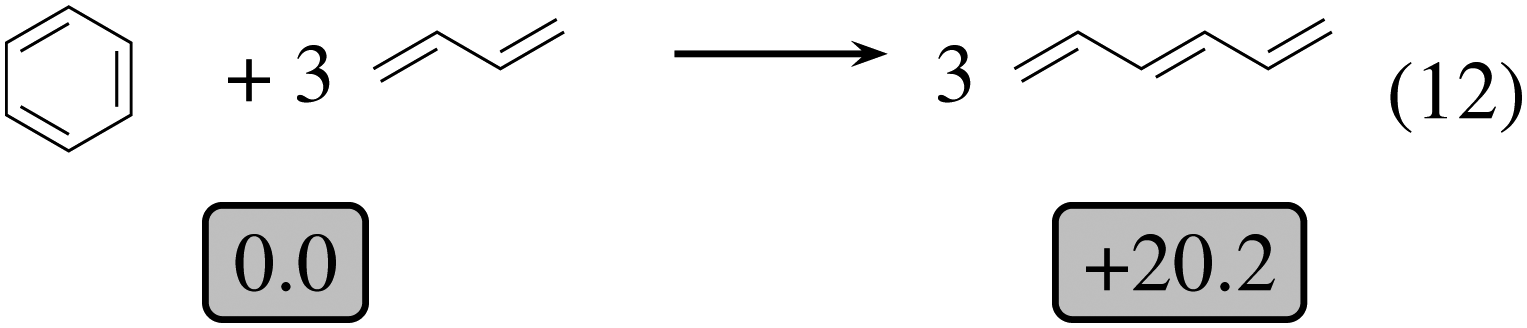 | ||
| Fig. 6 Homodesmotic opening reaction of benzene. Relative energies in kcal mol−1 are indicated in gray boxes. | ||
The most obvious result is that OTMS substituents do not have any influence on the energetics of the 6-membered ring-opening: the reaction energies of (10), which lead from (1c) to (3c), are of 14.5 kcal mol−1, and the reaction energies of (11), which lead from (3o) to (1o), are of about −4 kcal mol−1, both for R1 = H and R1 = OTMS. Nevertheless, the OTMS substituents have a large influence on the ring-opening of the cyclobutene moiety, of about 10 kcal mol−1. The effect is so strong that the relative stability of (3c) vs. (3o) is inverted for R1 = H (the closed form is the most stable) with respect to R1 = OTMS (the open form is the most stable).
We shall now analyze the energetic contributions of the reactions in the Hess cycle. According to the IUPAC definition,23 the reaction energy of (10) and (12) equals the aromaticity loss. For benzocyclobutene (substituted or not), this loss accounts for 14.5 kcal mol−1, while the opening of the benzene ring leads to an endothermicity of 20.2 kcal mol−1, reaction (12). The 5.7 kcal mol−1 difference corresponds to a partial loss of aromaticity in (1c), due to the geometrical constraint brought by the cyclobutene moiety (ring strain energy of the 6-membered ring in benzocyclobutene).
For reaction (11), from (3o) to (1o), an energy gain of about 4 kcal mol−1 is computed. There are eight π electrons in (3o) and in (1o). For (3o), the delocalization involves two π systems of four electrons each, with a large τ (57°, OTMS-disubstituted case) as described in the previous subsection. For (1o), the delocalization involves all the eight electrons, since it occurs mainly via the cycle rather than through the C2–C3 bond. It is, therefore, much more efficient.
In conclusion, the energetic decomposition of the ΔHR values is the following: in the OTMS-disubstituted case, ΔHR is of 8 kcal mol−1. The breaking of the aromaticity implies an energy loss of 14.5 kcal mol−1. The 6.5 kcal mol−1 that needs to be recovered are due to two contributions: the opening of the cyclobutene moiety releases 2.4 kcal mol−1; secondly, the efficient electron delocalization in (1o) accounts for the remaining 4.1 kcal mol−1. A similar picture can be drawn for the non-substituted case. Here, ΔHR is of 17.8 kcal mol−1 and the only difference with respect to the previous picture lies in the energetic demanding opening (7.5 kcal mol−1) of the cyclobutene moiety.
2.2 Substitution on cyclobutene allylic positions
In the previous section, we have pointed out that OTMS substitutes play an important role in the ring-opening of the cyclobutene moiety. As reminded in the Introduction, effects of substitution on allylic positions of the cyclobutene have been widely studied. Here we disclose a further aspect that affects ΔHR and ΔH# values and that has not been clearly reported so far, to the best of our knowledge.We consider disubstituted cyclobutenes and benzocyclobutenes, where R1 = H, OTMS, NH2, the substituents carry lone pairs. The ΔHR and ΔH# values are collected in Table 2. The SCS-MP227 reference values are also indicated. The discussion is based on the PBE0 values, but the SCS-MP2 values have been reported, because effects are slightly too pronounced at the PBE0 level, even if trends are preserved.
The destabilization of the closed forms with respect to the transition states and the open forms for substituted cases correlates well with the highest occupied molecular orbital pictures in Fig. 7. We recall that crucial orbitals in the closed form are those that correspond to bonds that need to be broken: the π and π* orbitals between C2 and C3, and the σC–C and σC–C* orbitals that can be ascribed to the C1–C4 bond.
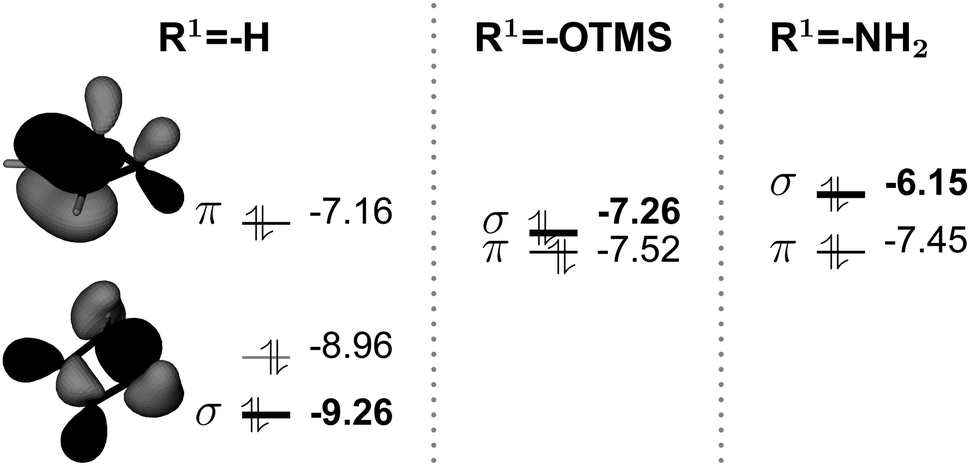 | ||
| Fig. 7 Highest occupied molecular orbitals for cyclobutene cases (energies in eV at the PBE0/def2-TZVP level, R1 = H, OTMS, NH2). σ stands for the molecular orbital that corresponds to the C1–C4 bond (in bold). Represented molecular orbital densities (contour ±0.05 a.u.) are for the cyclobutene π and the σ orbital. For cyclobutene, the σ orbital is the HOMO − 2, while for disubstituted cases the σ orbital is the HOMO. | ||
For R1 = H, the HOMO is the π orbital, but the σC–C orbital is the HOMO − 2 and relatively low in energy. For R1 = OTMS, NH2, the presence of the substituents does not significantly perturb the π system. Nevertheless their action is relevant to the σC–C orbital: its energy raises substantially and it becomes the new HOMO, with an energy close to that of the π orbital. This results in a weakening of the C1–C4 bond. As a consequence, the barrier height decreases and the closed form is destabilized with respect to the open form. The open form is further stabilized by the contribution of the substituents to the delocalized π system.
The raise in energy of the σC–C orbital finds its origin in the interaction with the two lone pairs on the substituents: this interaction leads to three molecular orbitals, as represented in Fig. 8 for cyclobutene and R1 = NH2. The most interesting outcome is that the most energetic orbital becomes the σC–C for the substituted cases.
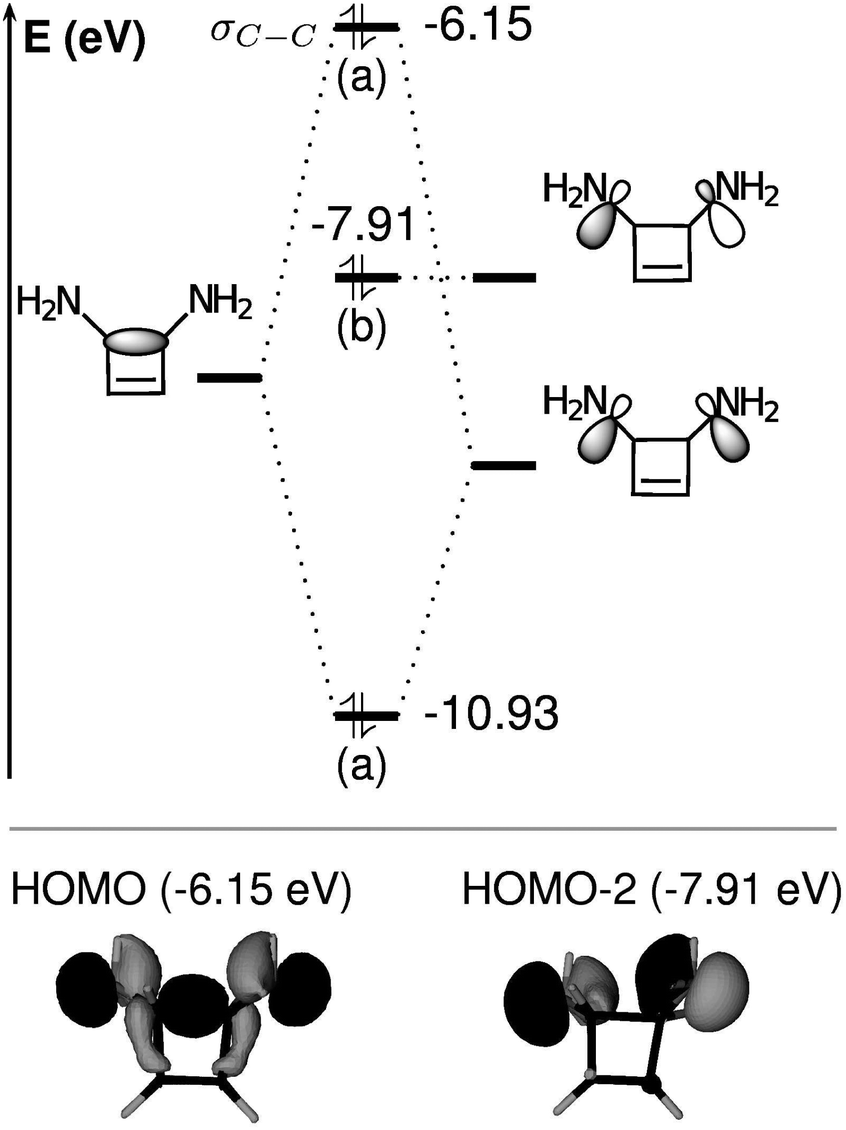 | ||
| Fig. 8 Schematic representation of the interaction between lone pairs and the σC–C orbital, in the disubstituted cyclobutene R1 = NH2. Symmetry labels are given in parentheses in the C2 point group of symmetry. The HOMO and HOMO − 2 orbitals are represented below the orbital interaction diagram. The HOMO (−6.15 eV) is the σC–C bond, the HOMO − 2 (−7.91 eV) is localized on the N atoms and the lowest energetic orbital is delocalized on the cycle. Energies are in eV (PBE0/def2-TZVP level). The energy of the not represented π orbital (the HOMO − 1) is of −7.45 eV. Energies before interaction are arbitrary. | ||
To summarize, several factors that intervene in the ring-opening of cyclobutene derivatives have been already discussed elsewhere,7,15–17 here we emphasize a further aspect, the weakening of the C1–C4 bond upon disubstitution on cyclobutene allylic positions by R1 = OTMS, NH2. This contributes to lower barrier heights and, in general, to destabilize closed forms.
2.3 Computational details
From a theoretical point of view, the functional B3LYP28–33 has been widely employed to study electrocyclic reactions of cyclobutene-like systems and its validity has been carefully tested with respect to hydrocarbon pericyclic reactions.34 Nevertheless, this type of benchmarking does not guarantee that a DFT functional maintains the same performances when substituents are introduced that perturb deeply the electronic structure of a molecule, such as OTMS. Thus, calculations on allylic substituted cyclobutene C8H6(R1)2 and benzocyclobutene C4H4(R1)2 have been employed to compare results from DFT methods to highly accurate CCSD(T) and SCS-MP2 values.Calculations at the CCSD(T) level were performed with the MOLPRO program package35 and give access to SCS-MP2 energies, as well. Otherwise, DFT (B3LYP, PBE,28,29,36,37 and PBE0) and additional SCS-MP2 calculations were performed with the program package TURBOMOLE.38 The def2-TZVP basis set was employed, unless specified. Structures were fully optimized at each DFT level. The ab initio calculations (SCS-MP2 and CCSD(T)) are obtained as single point energy calculations on PBE0 geometries. The Resolution of Identity approximation was employed.39–43 Comparison among methods is based on ΔER and ΔE# values, which do not contain zero-point energies. We do not have here the ambition to provide an extensive benchmark, but our aim is to investigate some specific cyclobutene derivatives, thus we have restricted our preliminary methodological analysis to a small set of similar molecules, R1 = H, NH2, OTMS, CH3, F, NO2. For the conciseness sake, we shall discuss results in terms of average errors (ae), mean absolute errors (mae), and maximum errors (max), details are reported in the ESI.‡ We employed the following scheme:
(i) the reliability of the CCSD(T) and SCS-MP2 methods has been checked with respect to the basis set choice. Thus, reference ΔER and ΔE# values of the C4H6 and C4H4F2 systems have been computed at the complete basis set (cbs) CCSD(T) level, the cbs extrapolation scheme uses cc-pVTZ, cc-pVQZ and cc-pV5Z basis sets.44–46 Results are reported in Table 3 and show that deviations for SCS-MP2 values are negligible (less than 1.5 kcal mol−1) and that ΔER and ΔE# have already converged with a def2-TZVP basis set (both for SCS-MP2 and CCSD(T) calculations).
| Method | Basis set | C4H6 | C4H4F2 | ||
|---|---|---|---|---|---|
| ΔER | ΔE# | ΔER | ΔE# | ||
| a Extrapolation from cc-pVTZ, cc-pVQZ, cc-pV5Z basis sets. b Experimental ΔH# = 32.5 ± 0.5 kcal mol−1.7 | |||||
| CCSD(T) | cbsa | −8.12 | 35.17 | −3.98 | 27.74 |
| def2-QZVP | −0.3 | −0.2 | −0.3 | −0.2 | |
| def2-TZVP | −0.4 | −0.4 | −0.6 | −0.5 | |
| SCS-MP2 | def2-QZVP | 0.5 | 1.2 | 0.4 | 0.7 |
| def2-TZVP | 0.4 | 1.1 | 0.2 | 0.6 | |
(ii) SCS-MP2 and DFT values of substituted cyclobutene C4H4(R1)2 have been compared to those at the CCSD(T) level, Table 4. Here, again, SCS-MP2 deviations from CCSD(T) results are within 1 kcal mol−1, which confirms that SCS-MP2 provides reliable reference data for those systems.
| R1 | PBE0 | PBE | B3LYP | SCS-MP2 |
|---|---|---|---|---|
| a R1 = H. b R1 = NH2. c R1 = OTMS. | ||||
| C4H4(R1)2 | ||||
| ΔER | ||||
| ae | 0.1 | −2.6 | −6.2 | 0.5 |
| mae | 1.7 | 3.2 | 6.2 | 0.5 |
| max | 3.7a | −5.6b | −8.3c | 0.9 |
| ΔE# | ||||
| ae | −0.5 | −6.0 | −3.8 | 0.6 |
| mae | 1.3 | 6.0 | 3.8 | 0.6 |
| max | −2.8 | −8.3 | −5.6 | 1.5 |
| C8H6(R1)2 | ||||
| ΔER | ||||
| ae | 0.4 | −4.5 | −6.0 | |
| mae | 1.5 | 4.6 | 6.0 | |
| max | 3.6a | −8.0b | −8.0b | |
| ΔE# | ||||
| ae | 0.6 | −6.7 | −3.7 | |
| mae | 1.3 | 6.7 | 3.7 | |
| max | 2.4 | −8.8 | −5.4 | |
(iii) DFT calculations for substituted benzocyclobutene C8H6(R1)2 have been consequently compared to SCS-MP2 only, Table 4.
Let us consider first DFT performances for ΔE# values: as expected, the PBE functional underestimates reaction barriers by about 6–7 kcal mol−1. Results for B3LYP and PBE0 are significantly better, the mean average errors are of 3.8 and 1.3 kcal mol−1, respectively.
Let us now consider ΔER values. The PBE0 functional behaves quite well, mean average errors are less than 2 kcal mol−1 and the largest error is for non-substituted cases (R = H: 3.7 kcal mol−1 for cyclobutene and 3.6 kcal mol−1 for benzocyclobutene, Table 2). PBE and B3LYP show larger deviations from our reference calculations. The B3LYP functional performs the worst and underestimates the ΔER values, thus suggesting open forms much too stable with respect to closed forms. Errors are not negligible, notably for disubstituted benzocyclobutene, R1 = OTMS, NH2. For those cases, PBE and B3LYP ΔER results are misleading, since they suggest that open and closed forms are almost degenerate, while the equilibrium is clearly displaced towards closed structures.
We conclude that, even if barrier heights are well reproduced, the validity of B3LYP is questionable for cyclobutene-like systems, when substitutions on allylic positions perturb their electronic structure. In the present work, since we largely focus on OTMS disubstituted systems, we have chosen to present PBE0 results that provide a better performance with respect to our reference calculations.
3 Conclusions
In this work we have suggested an analysis of key parameters that determine the relative stability between closed and open forms of OTMS disubstituted cyclobutene derivatives, where the open forms are in the conformations that derive directly from the cyclobutene ring-opening elementary step.The analysis of the relative stability of closed and open forms for a set of molecules shows that the nature of R2 and R3 plays a decisive role. The cyclobutene ring imposes a geometrical constraint that leads to a frustration (the ring strain), but allows for efficient hyperconjugation (systems (10c) → (6c)) and π delocalization around C2–C3 (systems (5c) → (2c)). The relative stability of the open form is determined by an equilibrium of several factors, notably the C4 strain release, the repulsion between R2 and R3, and the π delocalization along C2–C3 that is driven by the dihedral angle τ = τC1C2C3C4. Thus, electronic effects due to R2 and R3 differ in the open and closed form, which explains the evolution of ΔHR in the set of molecules studied.
When R2 and R3 are bound, leading to cyclic structures, strain energy plays a significant role in the relative stability of the open and closed forms. The balance between energy strain and π electronic delocalization leads to large variations in ΔHR. For benzocyclobutene, the closed form is more stable. This is related to the loss of the aromaticity, which accounts for 14 kcal mol−1. Even if this energetic loss is tempered by the constraint on the benzene due to the cyclobutene, the release of the strain of the cyclobutene and the efficient π delocalization through the C6 cycle in the o-xylylene are not sufficient to compensate it.
Finally, substitution on allylic positions of the cyclobutene by R = OTMS, NH2 impacts and weakens the C1–C4 bond in the closed form, lowering significantly ΔH# values with respect to R = H and destabilizing the closed form with respect to the open form. As mentioned in the Introduction, the trans-disiloxybenzocyclobutene is active towards radical species. Since in this kind of reaction, the rate limiting step is the ring-opening of the cyclobutene moiety, the amino-disubstituted benzocyclobutene is a potential candidate, which performs as good as or even better than the OTBS analogues.
Acknowledgements
We thank Pr. Maurice Santelli, Pr. Stéphane Humbel and Dr Laurent Commeiras for fruitful discussions. This work is dedicated to Guillaume Chailly, a dearest friend.References
- M. R. Wilson and R. E. Taylor, Angew. Chem., Int. Ed., 2013, 52, 4078–4087 CrossRef CAS PubMed.
- F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS PubMed.
- W. J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 CAS.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- S. Sakai, J. Phys. Chem. A, 2000, 104, 11615–11621 CrossRef CAS.
- D. C. Spellmeyer and K. N. Houk, J. Am. Chem. Soc., 1988, 110, 3412–3416 CrossRef CAS.
- P. Lee, S. Sakai, P. Horstermann, W. Roth, E. Kallel and K. Houk, J. Am. Chem. Soc., 2003, 125, 5839–5848 CrossRef CAS PubMed.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- J. Friedrichs, M. Luessmann and I. Frank, ChemPhysChem, 2010, 11, 3339–3342 CrossRef CAS PubMed.
- J. Ribas-Arino, M. Shiga and D. Marx, Chem. – Eur. J., 2009, 15, 13331–13335 CrossRef CAS PubMed.
- W. R. Roth, M. Biermann, H. Dekker, R. Jochems, C. Mosselmann and H. Hermann, Chem. Ber., 1978, 111, 3892–3903 CrossRef CAS.
- W. R. Roth and B. P. Scholz, Chem. Ber., 1981, 114, 3741–3750 CrossRef CAS.
- A. G. Csàszàr, J. Phys. Chem. A, 2004, 108, 2002–2007 CrossRef.
- L. Lo Presti, A. Ellern, R. Destro, R. Soave and B. Lunelli, J. Phys. Chem. A, 2011, 115, 12695–12707 CrossRef CAS PubMed.
- N. Mariet, H. Pellissier, J.-L. Parrain and M. Santelli, Tetrahedron, 2004, 60, 2829–2835 CrossRef CAS PubMed.
- P. Lee, X. Zhang and K. Houk, J. Am. Chem. Soc., 2003, 125, 5072–5079 CrossRef CAS PubMed.
- M. Murakami, M. Hasegawa and H. Igawa, J. Org. Chem., 2004, 69, 587–590 CrossRef CAS PubMed.
- J. Drujon, R. Rahmani, V. Héran, R. Blanc, Y. Carissan, B. Tuccio, L. Commeiras and J.-L. Parrain, Phys. Chem. Chem. Phys., 2014, 16, 7513–7520 RSC.
- O. Plachkevytch, B. Minaev and H. Ågren, J. Phys. Chem., 1996, 100, 8308–8315 CrossRef CAS.
- J. Perdew, M. Emzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. E. Wheeler, K. N. Houk, P. V. R. Schleyer and W. D. Allen, J. Am. Chem. Soc., 2009, 131, 2547–2560 CrossRef CAS PubMed.
- A. D. McNaught and A. Wilkinson, IUPAC. Compendium of Chemical Terminology, 2nd edn, (the “Gold Book”), XML on-line corrected version: http://goldbook.iupac.org (2006) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins., Blackwell Scientific Publications, Oxford, 1997.
- I. Fernàndez and G. Frenking, Chem. – Eur. J., 2006, 12, 3617 CrossRef PubMed.
- F. Liu, R. S. Paton, S. Kim, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 15642–15649 CrossRef CAS PubMed.
- A. Julg and P. François, Theor. Chim. Acta, 1967, 8, 249–259 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS PubMed.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, 123, 714–733 CrossRef CAS.
- J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS.
- S. Vosko, L. Wilk and M. Nussair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- V. Guner, K. Khuong, A. Leach, P. Lee, M. Bartberger and K. Houk, J. Phys. Chem. A, 2003, 107, 11445–11459 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2006.1, a package of ab initio programs, 2006 Search PubMed.
- J. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- TURBOMOLE V6.5 2013, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef PubMed.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2001, 116, 3175–3183 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
Footnotes |
| † This work is dedicated to Guillaume Chailly. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01695e |
| § These authors contributed equally to this work. |
| ¶ For (2), |ΔER| is smaller than the accuracy of the theoretical method used: the mean absolute error is found to be 1.7 kcal mol−1 (see Section 2.3). |
| This journal is © the Owner Societies 2014 |