
A density functional theory study of oxygen reduction reaction on non-PGM Fe–Nx–C electrocatalysts†
Abstract
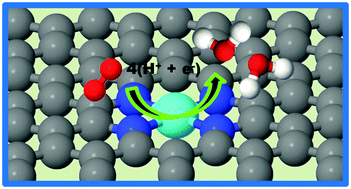
First-principles density functional theory (DFT) calculations were performed to explain the stability of catalytically active sites in Fe–Nx–C electrocatalysts, their ORR activity and ORR mechanism. The results show that the formation of graphitic in-plane Fe–N4 sites in a carbon matrix is energetically favorable over the formation of Fe–N2 sites. Chemisorption of ORR species O2, O, OH, OOH, and H2O and O–O bond breaking in peroxide occur on both Fe–N2 and Fe–N4 sites. In addition to the favorable interaction of ORR species, the computed free energy diagrams show that elementary ORR reaction steps on Fe–Nx sites are downhill. Thus, a complete ORR is predicted to occur via a single site 4e− mechanism on graphitic Fe–Nx (x = 2, 4) sites. Because of their higher stability and working potential for ORR, Fe–N4 sites are predicted to be prime candidate sites for ORR in pyrolyzed Fe–Nx–C electrocatalysts.
- This article is part of the themed collection: Electrocatalysis - Fundamental Insights for Sustainable Energy
 Please wait while we load your content...
Please wait while we load your content...

