 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1 J CH couplings in Group 14/IVA tetramethyls from the gas-phase NMR and DFT structural study: a search for the best computational protocol†
Ryszard B.
Nazarski‡
*a and
Włodzimierz
Makulski
b
aLaboratory of Molecular Spectroscopy, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: nazarski@uni.lodz.pl
bLaboratory of NMR Spectroscopy, Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 15th May 2014
Abstract
Four tetramethyl compounds EMe4 (E = C, Si, Ge, and Pb) were studied by high-resolution NMR spectroscopy in gaseous and liquid states at 300 K. Extrapolation of experimental vapor-phase C–H J-couplings to a zero-pressure limit permitted determining the 1J0,CHs in methyl groups of their nearly isolated molecules. Theoretical predictions of the latter NMR parameters were also performed in a locally dense basis sets/pseudopotential (Sn, Pb) approach, by applying a few DFT methods pre-selected in calculations of other gas-phase molecular properties of all these species and SnMe4 (bond lengths, C–H stretching IR vibrations). A very good agreement theory vs. experiment was achieved with some computational protocols for all five systems. The trends observed in their geometry and associated coupling constants (1JCHs, 2JHHs) are discussed and rationalized in terms of the substituent-induced rehybridization of the methyl group (treated as a ligand) carbon, by using Bent's rule and the newly proposed, theoretically derived values of the Mulliken electronegativity (χ) of related atoms and groups. All these χ data for the Group-14/IVA entities were under a lot of controversy for a very long time. As a result, the recommended χ values are semi-experimentally confirmed for the first time and only a small correction is suggested for χ(Ge) and χ(GeMe3).
1 Introduction
High-resolution nuclear magnetic resonance (NMR) spectroscopy is undoubtedly the most valuable technique for determining the structure and motion of molecules in all states of aggregation. Among various NMR parameters, magnetic shieldings σ (or related chemical shifts, δ) and scalar spin–spin coupling constants [hereafter referred to as J-couplings or JXYs] are the most important physical observables. Nowadays, first-principles calculations of the latter NMR data are available and published for many objects.1–4 But their performance is not as straightforward as predicting the σ data. As viewed from the classical (nonrelativistic) Ramsey theory of JXYs,5 there are four interactions between nuclear spins in molecules that can contribute to an indirect J-coupling, i.e., the Fermi contact (FC), spin–dipole (SD), paramagnetic spin–orbit (PSO), and diamagnetic spin–orbit (DSO) term. Although, in most cases, the FC contribution dominates the isotropic J value, none of these terms can be a priori excluded in any reliable JXY calculation.1–4 Moreover, a more advanced relativistic approach is needed for J-couplings embracing heavier elements of the 5th and higher rows of the periodic table.4,6 Fortunately, the impact of such atoms (e.g., beyond Kr) on JXYs that do not directly involve these nuclei is small unless a heavy nucleus is located directly in the coupling path.3,4 Currently, density-functional theory (DFT) computations seem to be the most promising in J evaluations for small and medium-sized molecules, such as, e.g., various organo-derivatives of tin7 and lead. It is obvious that all theoretical calculations may be verified only when accurate experimental data are accessible. The most valuable slices of such data come from the gas-phase studies, because results of this kind are not obscured by bulk effects arising from the surrounding medium and molecular effects coming from specific interactions.In this paper we present experimental results from the vapor-phase measurements of 1JCH couplings for a complete set of tetramethyl derivatives of the main Group-14/IVA elements, namely, 2,2-dimethylpropane (neopentane, CMe4), tetramethylsilane (TMS, SiMe4), tetramethylgermane (GeMe4), tetramethyltin (SnMe4), and tetramethyllead (PbMe4). Among all these isostructural systems EMe4, where the central element E = C to Pb, only the Sn derivative was studied in such a way very recently.8 All species were analyzed by means of 500 MHz 1H NMR spectra taken for their gaseous mixtures with krypton (Kr), xenon (Xe), sulfur hexafluoride (SF6), nitrous oxide (N2O), and carbon dioxide (CO2) used as buffer gases. Indirect 1JCH couplings, evaluated from the 13C satellite lines of related methyl group signals, were then extrapolated to zero-pressure limit.9 Such a procedure leads to determining the 1J0,CHs practically free from solvent effects. The latter experimental data were subsequently used for verification of related NMR JtheorCHs predicted with various locally dense basis sets (LDBS)10–13/effective core potential (ECP, for Sn and Pb) approaches, by using a few pre-selected DFT-level treatments. All these two-step protocols were a priori tested in computations of other gas-phase properties of these objects EMe4, such as bond lengths and C–H stretching infrared (IR) vibrations. A general methodology of quite accurate calculations of the geometries and spectroscopic properties of these organoelement compounds was established in this way. Related 1JCHs were also measured for neat (pure liquid) samples. A consideration of such NMR data brings up the magnitude of the gas-to-solution effects on the one-bond 13C–1H J-coupling. Finally, the impact of the change in a central E atom on JXYs in the attached methyl groups (treated as ligands) is discussed in terms of Bent's rule, new theoretical values of the Mulliken electronegativity of atoms (χa) or functional groups (χg), and distortion of a tetrahedral symmetry around the methyl carbons in all analyzed molecules. These latter efforts also targeted a semi-empirical verification of the newly determined χ values mentioned above, being under considerable controversy for a very long time.
Four crucial and inseparable issues were addressed in this work: (i) a highly reliable representation of a molecular geometry of all compounds EMe4, (ii) adequate calculations of 1JCHs and 2JHHs operative in these isostructural model systems, (iii) interpretation of the trends in JXY values observed for the species EMe4 in terms of changes in the molecular shape and net charge on their methyl carbons (reflected by the recent Mulliken electronegativities of attached groups), and (iv) experimental NMR data-based validation of these electronegativity data for all five Group 14/IVA elements and related EMe3 substituents. As far as we know, such a comprehensive internally consistent structure- and NMR-oriented investigation of the title systems has not yet been published.
2 Experimental details
2,2-Dimethylpropane (neopentane, Merck-Schuchardt, 98%) from a lecture bottle with a liquefied gas, tetramethylsilane (TMS, Aldrich, ≥99.9%), tetramethylgermane (Aldrich, 98%) and tetramethyllead (Aldrich, 65 wt% solution in xylene) from a glass container, were used without further purification. As reported in detail elsewhere,6,8,9b,14–16 gaseous samples were obtained by condensation of the studied compound and pure commercially available solvent gas (Kr, Xe, or SF6) transferred from the calibration part of our vacuum line. The solute gas, EMe4, was applied in a small concentration (∼0.005 mol L−1, pressure ∼100 Torr) and was mixed with different quantities of gaseous solvent (from ∼0.2 to ∼1.5 mol L−1).One-dimensional 1H NMR spectra were recorded at 300 K on a Varian INOVA FT-NMR spectrometer operating at the 500.6075 MHz proton Larmor frequency. The FID acquisition time was set to 2 s and a spectral width from 400 to 1000 Hz was applied. 1JCH couplings were directly evaluated in Hz as differences between the outside 13C satellites of methyl group signals. Liquid TMS was always used as an external NMR chemical-shift reference standard.
3 Computational details
All electronic structure ‘gas-phase’ calculations, i.e., for isolated vibration-free objects at 0 K in vacuum, were performed applying the Gaussian 09 suite of programs,17 by starting with the tetrahedral models of EMe4 systems. The hybrid B3LYP functional as implemented in the Gaussian code was employed,18 because its use has been found suitable for a large variety of previous computations, including Sn and Pb containing species.7,19–21 This exchange–correlation density functional has been one of the more successful tools for predicting different magnetic properties of organic systems.1,18,22–25 The threshold and convergence parameters were tightened in all geometry optimizations (carried out by taking advantage of the Td symmetry) and frequency calculations by specifying ‘Int = UltraFine’, ‘SCF = VeryTight’, and ‘Opt = VeryTight’ for numerical integration grids and convergence criteria in Gaussian;16 the former two keywords were also used in subsequent single-point NMR predictions. Moreover, the five pure d-functions have been mainly applied (5D option), see also Table S1 (ESI†). The corresponding vibrational wavenumbers, ωis, were computed analytically at the same theory level in a rigid rotor-harmonic oscillator approximation. These data were also applied to verify that all structures, localized as stationary points on pertinent Born–Oppenheimer energy hypersurfaces, represent real global minima (NImag = 0). Finally, a gauge-including atomic orbital (GIAO) formalism26 was employed to ensure the gauge independence of related calculated JXYs. Several pre-selected functional/basis set combinations were used as DFT protocols at two stages. A locally dense basis sets (LDBS) approach,10–13 was applied in the vast majority of calculations. In addition, simulations of the impact of nonpolar molecules of CCl4 and C6H6 on the geometry and NMR response properties of species EMe4 were performed in an improved equilibrium solvation protocol27 of the polarizable continuum model of solvation (IEF-PCM).28Thus, in the first step of a ground-state-geometry optimization, a centrally situated E atom was best represented by an economical def2-TZVPP29 (hereafter referred to as A) or def2-TZVPPD30 (B) basis set of triple-ζ valence quality, while the small double-ζ Gaussian-type orbitals (GTOs), that is, 6-31G(d,p) (C), 6-31+G(d,p) (D) or 6-31+G(2df,p) (E) bases,17 were applied to the C and H atoms in the methyl groups. The usage of basis sets C and D for analogous organometallics was found as a fully sufficient approach.19,20 The aug-cc-pVTZ (C,31 Si,32 Ge33) and aug-cc-pVTZ-PP (Sn and Pb)34 (F) basis sets were also a priori tested for the E atom, but with rather worse results. In these A, B, and F bases an all-electron description concerns C, Si, and Ge—however, the innermost orbitals of Sn and Pb are represented by small-core quasi-relativistic pseudopotentials in an effective core potential (ECP) approximation.7 In turn, five other basis sets were exploited for C and H in the second computational step, instead of the Pople style bases mentioned above, namely, IGLO-II (G), IGLO-III (H), aug-cc-pVTZ-J (I), pcJ-2 (J), and pcJ-3 (K). The former two Huzinaga-type atomic basis sets35 modified by Kutzelnigg and coworkers,36,37 also known as HII and HIII bases,38–41 have been widely used for calculations of NMR properties;36–45e.g., the compact basis set G of polarized triple-ζ valence quality consists of [9s5p1d]/(5s4p1d) C and [5s1p]/(3s1p) H functions in the [primitive]/(contracted GTOs) notation.41 The latter aug-cc-pVTZ-J46 and pcJ-n bases23 were specifically developed for J calculations. As for the E atoms, the following basis sets of quadruple-ζ valence quality were used for the JCH prediction: def2-QZVPP29 (L), def2-QZVPPD30 (M), (aug-)cc-pVQZ (C,31 Si,32 Ge33) and (aug-)cc-pVQZ-PP (Sn and Pb)34 [N and O (augmented)].
In addition, a few runs were conducted in order to verify the adopted methodology. In one series, the B3LYP functional was applied, at both computational steps, in conjunction with the all-electron basis sets G available for C–Sn. Moreover, owing to some reported22 disadvantages of B3LYP in 1JCH calculations, two other density functionals were used, i.e., PBE47 and PBE0,48 in a PBE/I(C,H),L(E)//PBE0/E(C,H),A(E) approach49 similar to that employed for hydrocarbons.22
On the whole, 15 different basis sets A–O were used in conjunction with related DFT functionals in the two computational steps; for their application in 14 different B3LYP/basis sets combinations tested to obtain the best results, see Table S1 (ESI†). All atomic orbitals inaccessible within Gaussian17 were downloaded from the EMSL base (ver. 1.2.2)50 and applied in their standard forms with optimized general contractions. The only exceptions were G orbitals for the Ge and Sn atoms, created by O. Malkina and V. G. Malkin, taken from an earlier release of the StoBe package.51 Such Sn basis has been used very recently.52 The only results from two most suitable procedures [i.e., B3LYP/G(C,H),L(E)//B3LYP/C(C,H),A(E) and, especially, B3LYP/G(C,H),M(E)//B3LYP/C(C,H),B(E) (henceforth called protocol I and II, respectively)] are discussed in depth, for the sake of brevity. An employment of a basis set J instead of G in both of the above protocols (referred to as approach I-pcJ-2 and II-pcJ-2, respectively) leads to similar or slightly worse results. Generally, the use of IGLO-II (G) in the second step was found the most reliable and efficient protocol of the DFT NMR calculations presented here. The relative computer time required for a single-point in vacuo B3LYP-prediction of J-couplings in Ge(Me)4 with the use of basis sets G, H, J, and K was determined as 1.0, 1.9, 4.7, and 34.4, respectively.
4 Results and discussion
In the gaseous phase, nuclear spin–spin coupling constants are modified by pairwise interactions between the coexisting molecules and by additional multiple interactions. At a constant temperature, the appropriate equation for a measured one-bond heteronuclear X, Y J-coupling value in a binary mixture of two gases, i.e., a solute gas (A) and buffer gas (B), is expressed as| JXY = J0,XY + JAA,XY × ρA + JAB,XY × ρB +… | (1) |
| JXY = J0,XY + JAB,XY × ρB | (2) |
In this work, one-bond JCH couplings between constituent atoms of the methyl groups (treated as ligands surrounding the centrally situated E atoms) in species EMe4 were measured with good accuracy. Also their determination was made with very high precision, because the methyl group 1H NMR signals of all these systems were flanked by sharp lines of more or less symmetrically disposed 13C satellites separated by JCHs. The J0,CH values obtained from the extrapolation of such experimental data points to a zero density of the buffer gas are summarized in Table 1, where intermolecular effects on such couplings are also listed. As can be easily seen in Fig. 1, JCHs are affected by different buffer gases in different ways. The greatest concentration dependence of 13C–1H J-couplings was found for PbMe4; JAB,CH > 200 Hz mL mol−1. Related JCH values measured for the neat samples of all tetramethyls are also given in Table 1, together with such data reported for liquids or solutions.53–55 Inspection of this table shows a small difference ΔJCH = JCH(neat) − J0,CH,av(gas) slowly growing from CMe4 to PbMe4, which may be recognized as a modulation of the J0,CH coupling—a solvent independent characteristic of the solute molecule—induced by the surrounding medium. Indeed, there is good, R2 = 0.9948, expected in view of ref. 24, quasi-linear correlation ΔJCH = f[2(ε − 1)/(2ε + 1)] between the J data and the values of the relative permittivity (formerly called dielectric constant, ε) of three normally liquid tetramethyls,56 accounted via the reaction field function (Fig. S1, ESI†); unfortunately, the value of ε for PbMe4 is not available in the literature. The above observation is also in line with the finding that the solvent induced change in the 1JCH coupling in a given solvent depends on the J value itself.24
| Solute gas (A) | Solvent gas (B) | J 0,CH [Hz] | J AB,CH [Hz mL mol−1] | J 0,CH,av [Hz] | J CH(neat)c [Hz] | ΔJCHc,d [Hz] | J CH for neat,e solution in C6H6f or CCl4g [Hz] |
|---|---|---|---|---|---|---|---|
| a Density dependence of J(CH); see eqn (2). b Weighted averages. c This work. d Differences [J(CH)neat − J0(CH)av]. e Ref. 53b. f Ref. 54. g Ref. 55. h Measured for a liquid at vapor pressure of ∼2 atm. i Ref. 8. j For 65 wt% solution in xylene. | |||||||
| CMe4 | Kr | 123.95(5) | 23(59) | 123.93(4) | 124.06(1)h | 0.13(5) | 124.5(1)e, 124.3f, 124.0(2)g |
| Xe | 123.92(6) | 163(63) | |||||
| SiMe4 | Kr | 117.88(5) | 52(67) | 117.88(4) | 118.15(2) | 0.27(6) | 118.2(1)e, 118.2f, 117.8(2)g |
| Xe | 117.88(7) | 99(83) | |||||
| GeMe4 | Kr | 124.05(2) | 76(30) | 124.05(1) | 124.40(1) | 0.35(2) | 124.0(4)e, 124.4f, 124.6(2)g |
| Xe | 124.05(1) | 53(21) | |||||
| SnMe4i | CO2 | 127.12(8) | 32(10) | 127.13(5) | 127.62(2) | 0.49(7) | 127.2(1)e, 127.7f, 127.8(2)g |
| N2O | 127.13(7) | 64(10) | |||||
| PbMe4 | SF6 | 133.28(4) | 314(42) | 133.29(3) | 134.07(2)j | 0.78(5)j | 134.0(2)e, 134.2f, 134.3(2)g |
| Xe | 133.31(5) | 205(52) | |||||
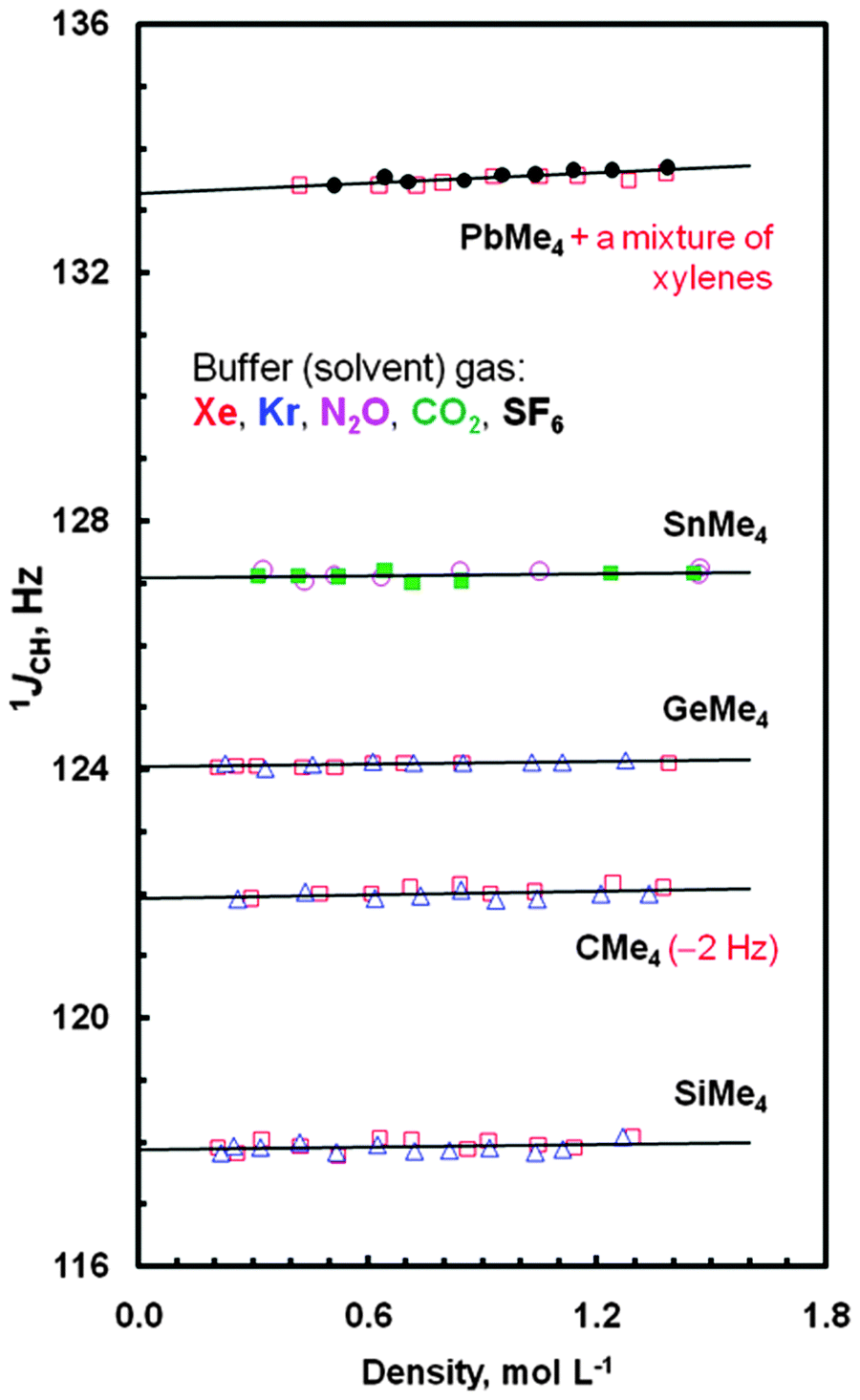 | ||
| Fig. 1 Dependence of the 1JCH data in compounds EMe4 on the density of solvent gases at 300 K (□ – Xe, △ – Kr, ○ – N2O, ■ – CO2, ● – SF6). The data points for CMe4 are shown with the (−2 Hz) vertical shift, for clarify. | ||
The foregoing vapor-phase J0,CHs and related liquid/solution data were subsequently compared with the high-quality computational results on JCHs in all five species EMe4 obtained for the first time. It is well-known that the Fermi-contact (FC) contribution to this type of J-couplings is a dominating term.1 Furthermore, this contribution is very sensitive to geometry and an applied calculational protocol (basis-set size and inclusion of the electron-correlation, in particular), while the SD, PSO, and DSO terms are less sensitive to these factors.1,38 Hence, special efforts were made in choosing a suitable DFT treatment. The use of experimental geometries often employed in J calculations on small molecules39,57 was discarded at the beginning, because our aim was to elaborate an accurate and cheap (from the viewpoint of calculation time) methodology available for other similar organic/organometallic species. Accordingly, only a few pre-selected computational approaches were used here.
4.1 Selection of a two-step computational protocol
In the first stage, reliable equilibrium ground-state geometries of tetramethyls of all members of the periodic table Group 14/IVA were modeled, while in the second estimations of their NMR spectroscopic parameters were made at a higher theory level, on the optimized geometries. The same DFT-level approach had to always be used for all objects, so that such computed JXYs are comparable and mutually consistent. The presence of heavy atoms in SnMe4 and PbMe4 forced the use of quasi-relativistic effective core potentials (ECPs) to describe their innermost orbitals. Recent results21 on several diatomics demonstrate that the B3LYP functional combined with a mixture of LANL08d58 (as an ECP) for Sn and Pb, and of a triple-ζ 6-311G(d,p) basis set for H and C atoms provides a sensible compromise between accuracy and computational cost. But an application of def2-TZVP29 instead of LANL08d was capable of predicting some molecular properties of these species slightly more accurately.21 As a consequence, only a few sufficiently flexible basis sets were tested and used in this work, for which the ECP approximation is fully consistent with an associated all-electron treatment available for the whole carbon group elements, i.e., atoms C through Pb.It is well known that NMR parameters depend critically on the electron density in the nuclear regions which may not be accurately modeled by the ECPs.1 However, we were mainly interested in the reliable evaluation of JXYs within the methyl groups around E atoms. Hence, our approach was fully compatible with the ECP-based studies on NMR properties of organic ligands linked to the metal atoms.59 Thus, all JCHs in species EMe4 were calculated with Gaussian 09 using the GIAO formalism coupled with B3LYP, as this DFT protocol was found suitable for many similar computations performed previously.1,18,24
Generally, a locally dense basis sets (LDBS) method10–13 was applied. However, in the geometry optimization step, this idea was employed in a manner opposite to that of the originally formulated one by Chesnut et al.10i.e., the E atom was described by the large (locally dense) basis set for best reproducing the E–C bond distances. Indeed, some problems with the choice of basis sets were reported for predicting the E–H bond lengths in Group-14/IVA tetrahydrides.60 The LDBS approach was successfully used in the calculations of many molecular properties including NMR chemical shifts10 and JXYs.12,13 Thus, fully balanced basis functions of the same level were employed here for the C and H atoms of methyl groups, that is, double-zeta split-valence Pople's bases and more extended Huzinaga–Kutzelnigg-type IGLO-II (or IGLO-III) atomic orbitals in our first and second step, respectively. The former compact basis set was widely adapted for calculating the NMR response of many different tin-containing species.38,42,52 It is also known that a subsequent use of IGLO sets at the geometries B3LYP-optimized in conjunction with double-ζ Pople's bases leads to adequate prediction of the experimental JCH couplings.41,43–45
Let us consider our results on some interatomic distances in five molecules of EMe4. All such geometric data obtained by a pre-selected DFT protocol II are collected in Table S2 (ESI†); for the plot, related statistics and Cartesian coordinates of all these species, see Fig. S2 and S8–S10 (ESI†), respectively. An inspection of Table S2 (ESI†) reveals an excellent agreement (R2 = 0.9998) with the experimental rg bond lengths from an electron diffraction analysis of vapor-phase samples,61–65 while perfect consistency (R2 = 0.9999) was found for the carefully examined CMe4.61 But such rg data65 for PbMe4 seem likely to be underestimated, especially for the Pb⋯H distance. Obviously, there is some dissimilarity in the physical meaning of these two quantities, i.e., thermally averaged rgs measured for vibrating species at room temperature vs. equilibrium res computed for motionless objects at 0 K. But their ratio is approximately constant over a wide distance range as these geometric parameters are related by the equation re ≅ rg − 3au2/2,66 where a and u are an associated Morse anharmonicity constant and a root-mean-square vibration amplitude for a related atom pair, respectively. A very good agreement (R2 = 0.9993) was also achieved between our C–E distances and those B3LYP-optimized within a relativistic elimination of small components (RESC) approach67 (ESI,† Table S2). Surprisingly, the greatest difference between such re data is found for SiMe4. Some shortening of the C–E bond lengths was, however, found in a relativistic DFT study on less sterically crowded molecules H–EMe3 possessing the post-d main-group central atom Ge–Pb.68
Very good conformity was also found (R2 = 0.9994 and 0.9998 for the protocols I and II, respectively) in comparison of in vacuo computed antisymmetric C–H IR stretchings, ωas(CH3), and their vapor-phase experimental counterparts, νas(CH3), determined by the same researchers,69 see Table S3 and Fig. S3 (ESI†). Unexpectedly, two such values were stated69 for CMe4 but only one compatible with the other independently reported70 values of this fundamental. The present calculations supported these one-value results. So, the problem of a relation between two types of molecular parameters recurs, which is commonly overcome with linear scaling theoretical data, using the equation ν = λ × ω71,72 or ν = a + b × ω.72 A better agreement obtained in the approach II also justifies a modification of the basis set A to B.30 The aforementioned findings confirmed our belief that the computed Td symmetric geometries of the title tetramethyls are sufficiently accurate, especially those found with protocols I and II. Hence, all subsequent predictions of their NMR spectroscopic properties based on such molecular structures should also be reliable.
4.2 DFT calculation results on 1JCHs
At this point, we present our gas- and solution-phase computational results on the total values of JCHs in five molecules EMe4 obtained with approaches I and II, including their partitioning into all four Ramsey contributions with dominant FC term participation as expected;1–4,6,25 see Table 2 and Fig. S4 (as its extended version, ESI†). The J values denoted as CCl4 or C6H6 are from the implicit solvent IEF-PCM27,28 simulations mimicking solvation of solutes EMe4 by molecules of carbon tetrachloride or benzene. A typical approach taking into account the scaling theoretical results according to the measured J data was used for assessing the JtheorXY = f(JexpXY) correlations, as in our previous works44,45,73 and in line with a recent benchmarking study.25 Generally, a fairly good agreement with an experiment was found (R2 = 0.9847, 0.9879, 0.9845, 0.9840, 0.9892, and 0.9930 for the approaches I, II, I-pcJ-2, II-pcJ-2, II-C6H6, and II-CCl4, respectively). The only one outlying data point for CMe4 was at first glance a considerable surprise in consideration of the relative simplicity of JCH-coupling predictions for hydrocarbons (vide infra). The best linear four-point plot JtheorCH = f(J0,CH) for gaseous samples is shown in Fig. 2. In either case, i.e., using IGLO-II vs. IGLO-III or pcJ-2 vs. pcJ-3, the better conformity in the second computational step was found with the basis set of smaller size. This suggests a fortuitous cancellation of calculational errors on the geometry and NMR spectroscopic properties occurring with the use of smaller basis sets.3 On the other hand, a little difference (1.1 Hz, protocol II) between the gas-phase experimental data for CMe4 and its predicted 1JCH value is considerably smaller than e.g., the 6–7 Hz discrepancy obtained at a similar theory level for some strained carbon atoms.41 An omission of the CMe4 point gives perfect relationships with R2 of 0.9999, 0.9999, 0.9997, 0.9997, 0.9995, and 0.9999, respectively. In this case, a slope of 1.036 and an intercept of −5.34 Hz [or only −0.34 Hz, after the +5Hz arbitral correction for a zero-point vibrational (ZPV) contribution to the C–H 1J-couplings22,25,40] was found for the best approach II, close to the ideal values of unity and zero. Hence, one can infer that J0,CHs [or JCH(CCl4)s] and pertinent JtheorCHs are perfectly correlated for the four remaining tetramethyls. The use of the recommended PBE/I(C,H),L(E)//PBE0/E(C,H),A(E) protocol22 afforded R2 of 0.9702 and 0.9998 after exclusion of CMe4. Even worse results were found in the all-electron B3LYP/G//B3LYP/G approach available for the E atoms from C to Sn; R2 of only 0.9456 and 0.9965 (without CMe4). In view of the latter finding, the employment of the LDBS approach is fully justified.| Compd | Medium | FC | SD | PSO | DSO | Total Jtheor |
|---|---|---|---|---|---|---|
| a Protocol II, and II-CCl4 was applied, respectively. | ||||||
| CMe4 | Gas | 119.70 | 0.18 | 0.92 | 0.73 | 121.52 |
| CCl4 | 119.67 | 0.18 | 0.92 | 0.73 | 121.50 | |
| SiMe4 | Gas | 114.25 | 0.29 | 1.46 | 0.68 | 116.68 |
| CCl4 | 114.24 | 0.29 | 1.46 | 0.68 | 116.67 | |
| GeMe4 | Gas | 120.70 | 0.33 | 1.13 | 0.98 | 123.13 |
| CCl4 | 120.67 | 0.33 | 1.13 | 0.98 | 123.10 | |
| SnMe4 | Gas | 124.15 | 0.39 | 1.21 | 0.63 | 126.38 |
| CCl4 | 124.08 | 0.39 | 1.22 | 0.63 | 126.31 | |
| PbMe4 | Gas | 130.51 | 0.44 | 1.10 | 0.57 | 132.62 |
| CCl4 | 130.41 | 0.44 | 1.11 | 0.57 | 132.53 | |
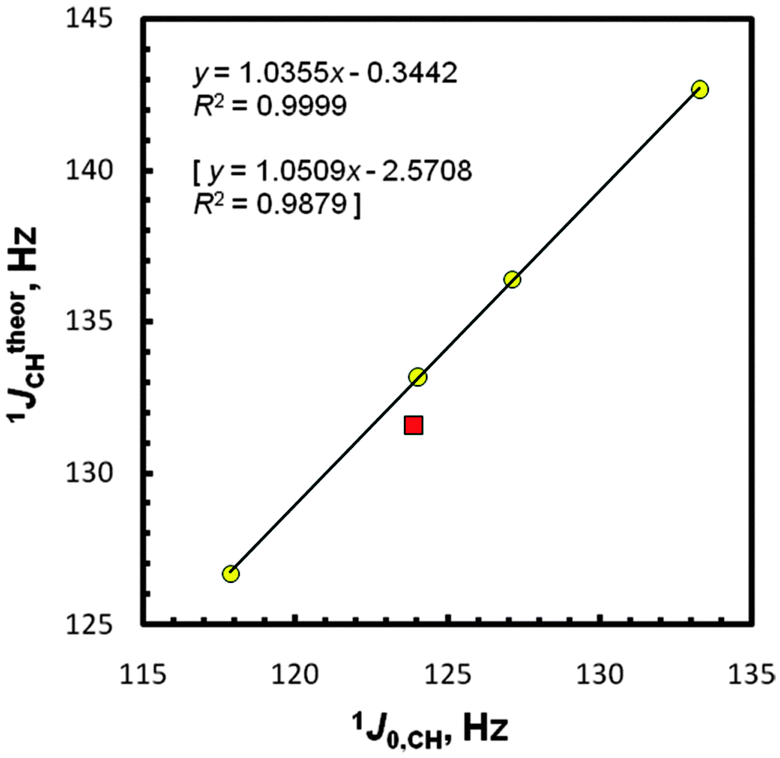 | ||
| Fig. 2 Correlation between (+5 Hz corrected) 1JCH computed (protocol II) and extrapolated 1J0,CH values.The statistics for the five-point plot with the CMe4 data (■) are in brackets. | ||
It is worth mentioning that our PCM calculations for CCl4 solutions failed to reproduce the observed trends, because they predict a slight decrease of JCHs vs. J0,CHs, from −0.009 Hz (Si) to −0.095 Hz (Pb) in the I-CCl4 data and from −0.012 Hz (Si) to −0.088 Hz (Pb) in II-CCl4 [Table S4 (ESI†)], in contradiction to a small increase accessed experimentally (Table 1); protocol II again turned out to be a better tool, however. A lot more remarkable is the ‘abnormal’ position of the CMe4 data point on the plot JtheorCH = f(J0,CH), see Fig. 2. This observation, apparently not fulfilling a ‘periodic correlation’, was made many years ago for liquid/solution samples of all five tetramethyls,53–55 but was only tentatively rationalized by (a) the effectiveness of an E–C orbital overlap53a or (b) the C–H bond rehybridization.54 The usage of the latter factor as an only criterion was criticized for molecules with the possible impact of electronegativity.74 The most probable explanation of this irregularity will be given afterwards (vide infra).
The next crucial data shown in Table 2 relates to spin–dipole (SD) contributions to the total values of JtheorCHs in five systems EMe4. In this instance, our data fulfilled the expected ‘periodic correlation’, at first sight. Thus, the increase of this generally small term was found as one goes down the periodic table from CMe4 (0.18 Hz) to PbMe4 (0.44 Hz), regardless of the computation level used. Such finding can be explained by a gradual change in the central E atom which modulates interactions of a nuclear magnetic moment with an electron spin within the methyl groups in question. An increase of the SD term with electronegativity is known for the directly coupled nuclei,38,57 including 1JEHs in tetrahydrides EH4 (E = C to Sn).38 To gain further information, various correlations were considered, using the newly proposed values of atomic Mulliken electronegativities (χas) and group electronegativities (χgs) of the EMe3 substituents attached to the methyl carbons in species EMe4versus the SD term magnitudes. All these χa and χg values (Table 3) were theoretically determined by Giju et al.68 with the inclusion of relativistic effects. In the cited work, an issue of the controversial electronegativity of the Group 14/IVA elements and, especially, related EMe3 substituents is discussed in details, but without experimental evidence of the correctness of these novel χ data. The NMR results considered in the present paper afforded such possibility for the first time (see also below). Thus, for the SD term values mentioned above the best least squares regression line (R2 = 0.997) was really found with the recent χgs, but again after omission of the CMe4 outlier. The plot with this bounding point is shown in Fig. 3. Obviously, any linear correlation embracing the SD terms of five species EMe4 does not exist.
| Compd | ∠ECH (gas) [°] | ∠HCH (gas) [°] | ∠HCH (CCl4) [°] | 2 J theorHH (CCl4) [Hz] | 2 J expHH (CCl4)b [Hz] | χ a for the E atomc,d,e [eV] | χ g for the EMe3 groupc,e [eV] |
|---|---|---|---|---|---|---|---|
| a Protocol II, and II-CCl4 was applied, respectively. b Ref. 55. c Ref. 68. d χ a for the H atom = 7.26 eV, ref. 68. e The proposed corrected value is in parenthesis, see the text. | |||||||
| CMe4 | 111.07 | 107.83 | 107.85 | −11.93 | −12.56 | 6.73 | 4.15 |
| SiMe4 | 111.46 | 107.41 | 107.46 | −13.47 | −14.05 | 4.96 | 3.80 |
| GeMe4 | 110.86 | 108.05 | 108.09 | −12.36 | −12.96 | 4.71 (∼4.54) | 3.85 (∼3.88) |
| SnMe4 | 110.51 | 108.41 | 108.47 | −12.12 | −12.37 | 4.31 | 3.95 |
| PbMe4 | 109.48 | 109.46 | 109.52 | −10.68 | −10.94 | 3.85 | 4.04 |
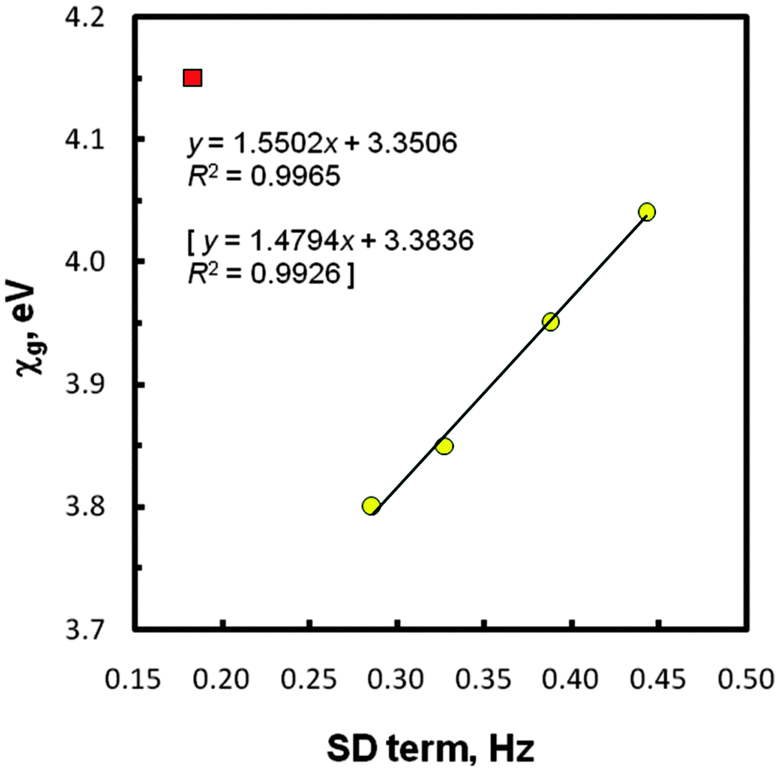 | ||
| Fig. 3 Group electronegativity vs. SD term (protocol II) values; the CMe4 point (■) was omitted. The statistics with χg corrected for GeMe4 (3.88 eV) are in brackets. | ||
4.3 Discussion of observed trends
As seen in Tables S2 and S3 (ESI†) as well as Fig. S2 and S3 (ESI†), some ‘periodic tendencies’ in the geometry and infrared properties of EMe4 are clearly shown. Indeed, related interatomic distances and, especially, C–H stretching IR frequencies change monotonically. It should be highlighted that differences in the computed tetrahedral-like geometry around the methyl carbon atoms in these species agree with Bent's model of isovalent hybridization.75–78 According to this concept, in a molecule with inequivalent R groups, smaller bond angles are formed between the electronegative R groups since a central atom tends to direct the spn-hybridized orbitals of greater p character towards its more electronegative R substituents. Based on the foregoing χg data of the groups attached to the methyl carbons (Table 3), it is clear that the fractional s character of a carbon 2s orbital forming the C–H bond in species EMe4 follows the order Si < Ge < Sn < Pb, exactly the same as is observed from the 1JCH data. Thus, an ‘abnormally large’ J-value found for CMe4 as an outlier (vide supra) is rationalized now to some degree, because such C–H couplings depend mainly on the FC interactions occurring for s electrons.54,74In order to discuss the gas-phase J0,CH-couplings in terms of χas and χg values,68 two series of these data were considered for the SD terms as previously; the point due to CMe4 was always found to be a strongly outlying one. In this case, a better four-point regression line plot was found for χas; R2 = 0.963 (Fig. 4) vs. R2 = 0.950 for χgs (Fig. S4, ESI†). It was also of interest to see how strong other linear relationships expected for compounds EMe4 are, i.e., 1J0,CH = f(2JHH), ∠HCH = f(2JHH), χa = f(2JHH), and χg = f(2JHH). The 2JHHs = 6.51 × 2JDHs (provided that there is no primary isotope effect on the J values) for species E(CH2D)4 in CCl4 solution55 and our PCM computed HCH bond angles were used accordingly (Table 3). The corresponding plots reflecting mutually consistent correlations (R2 = 0.994, 0.996, 0.973, and 0.957, respectively), again after omission of CMe4, are shown in Fig. S5–S8 (ESI†). Thus, the 2° opening of the HCH angle on going from Si to Pb causes an increase in the 2JHH value (from −14.05 to −10.94 Hz)55 and an increase in the s character of the C–H carbon spn-hybridized bonding orbitals, as expected. All the above data dealing with Mulliken electronegativities, based on a combination of experimental and DFT results, seem to prove the legitimacy of the newly reported χa and χg values.68 As far as we know, such χ data for this type of Group-14/IVA entities are confirmed for the first time. The analysis of the five plots [Fig. 3 and 4, Fig. S4, S7 and S8 (ESI†)] suggests, however, the need for a decrease of χa to ∼4.54 eV and increase of χg to ∼3.88 eV for Ge and –GeMe3, respectively, because there is 0.989 ≤ R2 ≤ 0.999 for the above correlations with the use of these corrected χ values.
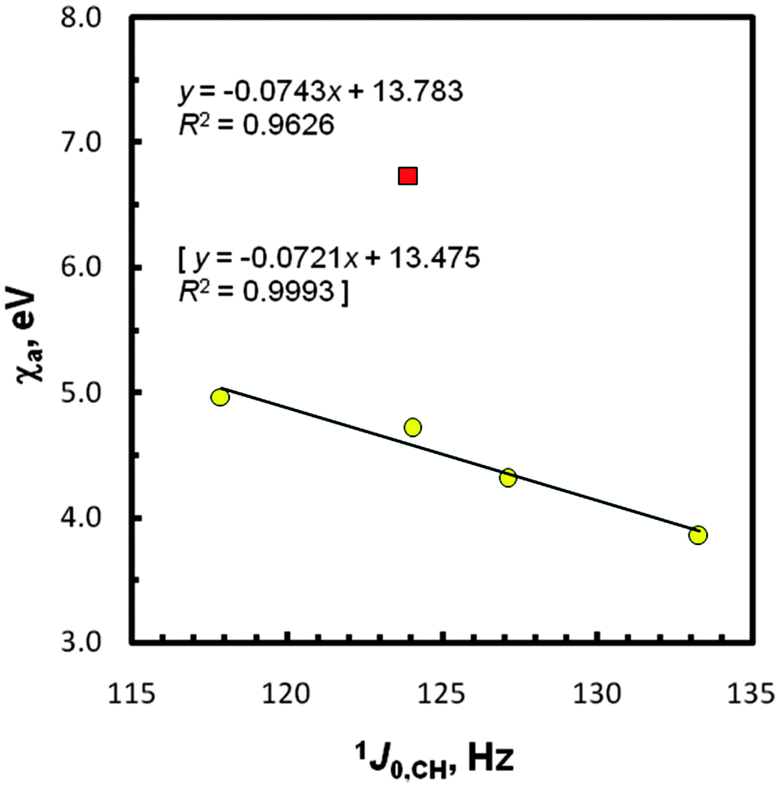 | ||
| Fig. 4 Correlation between the atom electronegativity and 1J0,CH data; the CMe4 point (■) was omitted. The statistics with χa corrected for Ge (4.54 eV) are in brackets. | ||
Finally, let us come back to some geometric parameters predicted for molecules EMe4 (Table 3). It is worth noting that all changes in their ECH and HCH bond angles also follow the trends anticipated from Bent's rule;75–78 the influence of the simulated ‘solvation’ by the molecules of CCl4 or C6H6 on the geometry is negligible. Indeed, such angles found for CMe4, being close in magnitude to those for GeMe4, reproduce the relation JCH(C) ≤ JCH(Ge) very well and so excellently explain the ‘abnormal’ position of the CMe4 data point in Fig. 2. Furthermore, the C–H distance in this object is in between those found for SiMe4 (the longest length, the greatest p character) and GeMe4. It seems that the computed geometry around all five methyl carbons in species EMe4 is mainly dependent on the C–E distance, which forces a large modification of the methyl group shape. Indeed, its strongly ‘stretched’ geometry, most distorted from the regular tetrahedron (∠ECH = 111.46°, ∠HCH = 107.41°) was found for SiMe4 with the shortest C–E bond length; Table 3 and Table S2 (ESI†). In contrast, a fully relaxed, practically idealized tetrahedral coordination was computed for the methyl carbons in PbMe4 possessing the longest C–E distance. Hence, the greatest J0,CH coupling in this compound is attributable in part to a relatively small difference in χgs of its carbon substituents (H, PbMe3) and to the greatest s character of the C–H bond among all tetramethyls under study.
In view of the foregoing facts, the ‘abnormal’ couplings 1JCH and 2JHH in CMe4, discussed here via related outlying data points, are attributable to the shortest C–E distance and greatest electronegativity of its E atom (or –EMe3 group) which characterize this molecule. On the contrary, there is a monotonic increase in the C–E distance for all four remaining tetramethyls (E = Si to Pb), associated with a diminished internal steric congestion around the E atom, and an increase in the pertinent values of χa and χg on going down a Group 14/IVA. As a result, their 1JCH couplings follow the observed Si < Ge < Sn < Pb sequence. The case of the system CMe4 is significantly different in this respect and that is the most probable origin of the aforesaid ‘anomalies’ in its molecular NMR response properties.
5 Conclusions
This investigation affords experimental one-bond C–H J-couplings in tetramethyl species EMe4 (E = C to Pb) in vapor and liquid states. Such gas-phase J0,CHs, obtained as J values extrapolated to a zero-pressure limit, are only slightly different from the JCHs measured for liquids. The former data show linear dependence on the density of the gaseous solvents used. The present study also demonstrates that current DFT protocols allow for a successful prediction of the experimental NMR J-couplings and other molecular properties as well as for their interpretation in terms of electronic and geometric structure. From the computational results on the title compounds it is concluded that: (1) Reliable geometries of all these molecules can be obtained applying the B3LYP functional in the LDBS approach which consists in the use of a def2-TZVPPD basis set for a central E atom (ECPs for Sn and Pb) and 6-31G(d,p) for the ligand (methyl group) atoms; this procedure can also be proposed for other structurally similar systems. (2) Subsequent LDBS calculations of JCHs, carried out with an IGLO-II basis sets for the C and H atoms and def2-QZVPPD for the E atoms (protocol II), afforded J values in good agreement with the experiment. (3) The use of pcJ-2 instead of IGLO-II gives a similar or worse result, but at the expense of computation time. (4) All four Ramsey contributions to JCHs in compounds EMe4 were computed with the Fermi-contact term being dominant participant. (5) Strong linear correlations were shown between the SD terms, J0,CHs, and JHHs vs. recently reported Mulliken electronegativities (χs) of the E atoms and EMe3 groups, excluding the CMe4 case. (6) These χ values were positively verified for the first time, however a small correction of χa (Ge) and χg for –GeMe3 is proposed. (7) Predicted alterations in the C–E distance and bond angles around the methyl carbons in species EMe4 in conjunction with χgs of their substituents and Bent's rule permitted an explanation of the trends observed in experimental 1JCHs and 2JHHs in terms of the gradual changes in the s character of carbon spn-hybridized orbitals forming the C–H bonds. (8) The ‘abnormal’ molecular NMR properties of CMe4 discussed above appear to result from the shortest C–E bond length and the greatest electronegativity of its E atom and EMe3 group among all tetramethyls under study.Note added in proof
Some growth of resemblance of the Mulliken electronegativity values to those determined for corresponding Sn(IV) entities,68 suggested here for the χ(Ge) and χ(GeMe3) data, is in line with the recent calculational results80 showing that electrophilicity and nucleophilicity values are quite similar for identically substituted germylenes and stannylenes, i.e., the divalent Ge(II) and Sn(II) analogues of carbenes.Acknowledgements
R.B.N. thanks the Academic Computer Centre CYFRONET (AGH – University of Science and Technology, Kraków, Poland) for access to the Gaussian 09 software (computational grant MNiSW/SGI3700/UŁódzki/057/2010).References
- T. Helgaker and M. Pecul, in Spin–Spin Coupling Constants with HF and DFT Methods in Calculation of NMR and EPR Parameters: Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004, pp. 101–121 Search PubMed.
- L. B. Krivdin and R. H. Contreras, Annu. Rep. NMR Spectrosc., 2007, 61, 133–245 CrossRef CAS.
- T. Helgaker, M. Jaszuński and M. Pecul, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 249–268 CrossRef CAS PubMed.
- Y. Yu. Rusakov and L. B. Krivdin, Russ. Chem. Rev. (Engl. Transl.), 2013, 82, 99–130 CrossRef PubMed.
- N. F. Ramsey, Phys. Rev., 1953, 91, 303–307 CrossRef CAS.
- A. Antušek, D. Kędziera, K. Jackowski, M. Jaszuński and W. Makulski, Chem. Phys., 2008, 352, 320–326 CrossRef PubMed.
- For a recent review see: S. R. Whittleton, R. J. Boyd and T. B. Grindley, in Computational Methods for Organotin Compounds in Tin Chemistry: Fundamentals, Frontiers, and Applications, ed. M. Gielen, A. Davies, K. Pannell and E. Tiekink, J. Wiley & Sons Ltd., Chichester, 2008, pp. 269–283 Search PubMed.
- W. Makulski, J. Mol. Struct., 2012, 1017, 45–50 CrossRef CAS PubMed.
- (a) K. Jackowski, Int. J. Mol. Sci., 2003, 4, 135–142 CrossRef CAS PubMed; (b) K. Jackowski and M. Jaszuński, Concept. Magn. Reson. A, 2007, 30, 246–260 CrossRef.
- D. B. Chesnut and K. D. Moore, J. Comput. Chem., 1989, 10, 648–659 CrossRef CAS.
- G. A. DiLabio, J. Phys. Chem. A, 1999, 103, 11414–11424 CrossRef CAS and references therein.
- M. Sanchez, P. F. Provasi, G. A. Aucar and S. P. A. Sauer, Adv. Quantum Chem., 2005, 48, 161–183 CrossRef.
- D. M. Reid, R. Kobayashi and M. A. Collins, J. Chem. Theory Comput., 2014, 10, 146–152 CrossRef CAS.
- M. Wilczek, W. Koźmiński and K. Jackowski, Chem. Phys. Lett., 2002, 358, 263–270 CrossRef CAS.
- W. Makulski, J. Mol. Struct., 2008, 872, 81–86 CrossRef CAS PubMed.
- W. Makulski, J. Mol. Struct., 2013, 1036, 168–173 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT 06492, USA, 2010, 23-Sep-2011 Search PubMed.
- J. R. Cheeseman, G. W. Truck, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS PubMed and references cited therein.
- S. R. Whittleton, R. J. Boyd and T. B. Grindley, J. Phys. Chem. A, 2006, 110, 5893–5896 CrossRef CAS PubMed.
- C. Gourlaouen, O. Parisel and J.-P. Piquemal, Chem. Phys. Lett., 2009, 469, 38–42 CrossRef CAS PubMed.
- P. Matczak, Comput. Theor. Chem., 2012, 983, 25–30 CrossRef CAS PubMed.
- T. W. Keal, T. Helgaker, P. Sałek and D. J. Tozer, Chem. Phys. Lett., 2006, 425, 163–166 CrossRef CAS PubMed.
- F. Jensen, Theor. Chem. Acc., 2010, 126, 371–382 CrossRef CAS.
- A. A. Shahkhatuni, A. B. Sahakyan, A. G. Shahkhatuni, S. S. Mamyan and H. A. Panosyan, Chem. Phys. Lett., 2012, 542, 56–61 CrossRef CAS PubMed.
- J. S. Fabián, J. M. García de la Vega, R. Suardíaz, M. Fernández-Oliva, C. Pérez, R. Crespo-Otero and R. H. Contreras, Magn. Reson. Chem., 2013, 51, 775–787 CrossRef PubMed.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS and references therein.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- J. Tomasi, B. Mennucci and E. Cancès, THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS PubMed.
- A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. Dunning, Jr., J. Chem. Phys., 1999, 110, 7667–7676 CrossRef CAS PubMed.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS PubMed.
- S. Huzinaga, Approximate Atomic Wave Approximate Atomic Functions. I, II. Technical Report, University of Alberta, Edmonton, AB, Canada, 1971 Search PubMed.
- M. Schindler and W. Kutzelnigg, J. Chem. Phys., 1982, 76, 1919–1933 CrossRef CAS PubMed.
- W. Kutzelnigg, U. Fleischer and M. Schindler, in The IGLO-Method: Ab-initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities in Deuterium and Shift Calculation NMR – Basic Principles and Progress, ed. P. Diehl, E. Fluck, H. Günther, R. Kosfeld and J. Seelig, Springer-Verlag, Berlin, 1991, vol. 23, pp. 165–262 Search PubMed.
- O. Malkina, D. R. Salahub and V. G. Malkin, J. Chem. Phys., 1996, 105, 8793–8800 CrossRef CAS PubMed.
- M. Pecul, M. Jaszuński and J. Sadlej, Chem. Phys. Lett., 1999, 305, 139–146 CrossRef CAS.
- T. A. Ruden, O. B. Lutnæs, T. Helgaker and K. Ruud, J. Chem. Phys., 2003, 118, 9572–9581 CrossRef CAS PubMed.
- H. Dodziuk, M. Jaszuński and W. Schilf, Magn. Reson. Chem., 2005, 43, 639–646 CrossRef CAS PubMed.
- R. Vivas-Reyes, F. De Proft, M. Biesemans, R. Willem and P. Geerlings, J. Phys. Chem. A, 2002, 106, 2753–2759 CrossRef CAS.
- A. Wu, D. Cremer, A. A. Auer and J. Gauss, J. Phys. Chem. A, 2002, 106, 657–667 CrossRef CAS.
- S. Leśniak, A. Chrostowska, D. Kuc, M. Maciejczyk, S. Khayar, R. B. Nazarski and Ł. Urbaniak, Tetrahedron, 2009, 65, 10581–10589 CrossRef PubMed.
- R. B. Nazarski, B. Pasternak and S. Leśniak, Tetrahedron, 2011, 67, 6901–6916 CrossRef CAS PubMed.
- P. F. Provasi, G. A. Aucar and S. P. A. Sauer, J. Chem. Phys., 2001, 115, 1324–1334 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS . Erratum: Phys. Rev. Lett., 1997, 78, 1396.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS PubMed.
- Throughout this work we follow the standard convention that w/x//y/z denotes a calculation with the method w and base x on a geometry optimized with the method y and base z, which was extended here for specifying the two basis-set partitions separated with comma.
- EMSL Basis Set Exchange Library. http://https://bse.pnl.gov/bse/portal.
- Orbital basis set, auxiliary functions and pseudopotentials for StoBe (Stockholm-Berlin version of deMon, a DFT molecule/cluster package). Dec. 2000. Database created by K. Hermann (The Fritz Haber Institute of the Max Planck Society, Berlin). Accessed on 27 June, 2006.
- P. Matczak, Comput. Theor. Chem., 2013, 1013, 7–14 CrossRef CAS PubMed.
- (a) R. S. Drago and N. A. Matwiyoff, J. Organomet. Chem., 1965, 3, 62–69 CrossRef CAS; (b) A. W. Douglas, J. Chem. Phys., 1966, 45, 3465 CrossRef CAS PubMed.
- F. J. Weigert, M. Winokur and J. D. Roberts, J. Am. Chem. Soc., 1968, 90, 1566–1569 CrossRef CAS and references therein.
- M. J. Lacey, C. G. Macdonald, A. Pross, J. S. Shannon and S. Sternhell, Aust. J. Chem., 1970, 23, 1421–1429 CrossRef CAS.
- S. Geer and R. A. Holroyd, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 5043–5046 CrossRef CAS.
- J. S. Fabián, E. Díez, J. M. García de la Vega and R. Suardíaz, J. Chem. Phys., 2008, 128, 084108 CrossRef PubMed.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS.
- M. Kaupp, V. G. Malkin and O. L. Malkina, in NMR of Transition Metal Compounds in Encyclopedia of Computational Chemistry, ed. P. v. R. Schleyer, J. Wiley & Sons Ltd., Chichester, 1998, vol. 3, pp. 1857–1866 Search PubMed.
- K. G. Dyall, P. R. Taylor, K. Faegri, Jr. and H. Partridge, J. Chem. Phys., 1991, 95, 2583–2594 CrossRef CAS PubMed.
- L. S. Bartell and W. F. Bradford, J. Mol. Struct., 1977, 37, 113–126 CrossRef CAS.
- A. R. Campanelli, F. Ramondo, A. Domenicano and I. Hargittai, Struct. Chem., 2000, 11, 155–160 CrossRef CAS.
- E. Csákvári, B. Rozsondai and I. Hargittai, J. Mol. Struct., 1991, 245, 349–355 CrossRef.
- M. Nagashima, H. Fuji and M. Kimura, Bull. Chem. Soc. Jpn., 1973, 46, 3708–3711 CrossRef CAS.
- T. Oyamada, T. Iijima and M. Kimura, Bull. Chem. Soc. Jpn., 1971, 44, 2638–2642 CrossRef CAS.
- B. Beagley, J. J. Monaghan and T. G. Hewitt, J. Mol. Struct., 1971, 8, 401–411 CrossRef CAS.
- W. Lie, D. G. Fedorov and K. Hirao, J. Phys. Chem. A, 2002, 106, 7057–7061 CrossRef CAS.
- K. T. Giju, F. De Proft and P. Geerlings, J. Phys. Chem. A, 2005, 109, 2925–2936 CrossRef CAS PubMed.
- H. Bürger and S. Biedermann, Spectrochim. Acta, Part A, 1972, 28, 2283–2286 CrossRef.
- K. Shimizu and H. Murata, Bull. Chem. Soc. Jpn., 1957, 30, 487–491 CrossRef CAS.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS.
- Z. A. Fekete, E. A. Hoffmann, T. Körtvélyesi and B. Penke, Mol. Phys., 2007, 105, 2597–2605 CrossRef CAS.
- R. B. Nazarski, Phosphorus, Sulfur, and Silicon, 2009, 184, 1036–1046 CrossRef CAS.
- D. M. Grant and W. M. Litchman, J. Am. Chem. Soc., 1965, 87, 3994–3995 CrossRef CAS.
- H. A. Bent, Chem. Rev., 1961, 61, 275–311 CrossRef CAS.
- J. E. Huheey, Inorg. Chem., 1981, 20, 4033–4035 CrossRef CAS.
- A. E. Reed and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 1434–1445 CrossRef CAS.
- Glossary of Terms Used in Theoretical Organic Chemistry (IUPAC Recommendations 1999) prepared for publication by V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919–1981 CrossRef CAS.
- E. A. Skorupska, R. B. Nazarski, M. Ciechańska, A. Jóźwiak and A. Kłys, Tetrahedron, 2013, 69, 8147–8154 CrossRef CAS PubMed.
- L. Broeckaert, P. Geerlings, A. Růžička, R. Willem and F. De Proft, Organometallics, 2012, 31, 1605–1617 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S9 and Fig. S1–S8. See DOI: 10.1039/c4cp01596g |
| ‡ Physical image vs. molecular structure relation, Part 18. For Part 17, see ref. 79. |
| This journal is © the Owner Societies 2014 |