 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The bioinspired construction of an ordered carbon nitride array for photocatalytic mediated enzymatic reduction†
Jian
Liu
*a,
Rémi
Cazelles
b,
Zu Peng
Chen
a,
Han
Zhou
a,
Anne
Galarneau
b and
Markus
Antonietti
a
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: Jian.Liu@mpikg.mpg.de; Fax: +49 331 567 9502; Tel: +49 331 567 9515
bInstitut Charles Gerhardt Montpellier, UMR 5253 CNRS/UM2/ENSCM/UM1, ENSCM, 8 rue de l'Ecole Normale, 34296 Montpellier Cedex 5, France
First published on 28th April 2014
Abstract
A carbon nitride array (CNA) material has been constructed using a sacrificial diatom template. A regular carbon nitride nanorod array could be replicated from the periodic and regular nanochannel array of the template. The directional charge transport properties and high light harvesting capability of the CNA gives much better performance in splitting water to give hydrogen than its bulk counterpart. Furthermore, by combining with a rhodium complex as a mediator, the nicotinamide adenine dinucleotide (NADH) cofactor of many enzymes could be photocatalytically regenerated by the CNA. The rate of the in situ NADH regeneration is high enough to reverse the biological pathway of the three dehydrogenase enzymes, which then leads to the sustainable conversion of formaldehyde to methanol and also the reduction of carbon dioxide into methanol.
Introduction
In recent years, substantial efforts, both from academia and industry, have been directed towards the development of high efficiency energy conversion systems for turning solar light into chemicals.1 Material and structure optimization are the key points determining the efficiency of light energy conversion in artificial systems.2,3 Biological materials are endless fountains of inspiration for the design and fabrication of such kinds of nanostructured materials.4–7 In natural photosynthesis, green plants and algae can transform carbon dioxide and water into sugars by utilizing light energy and release oxygen into the atmosphere. A wide range of nanomaterials have been designed and synthesized using biomaterials or bioinspired processes. The silica exoskeletons (frustules) of the single-celled algae called diatoms are one of the most spectacular examples of biologically evolved nanostructured materials.8,9 The diatom frustule features a unique architecture with hierarchical structures ranging from the micrometre to nanometre scale, which are thought to contribute to their high photosynthesis efficiency.10,11 Each frustule possesses a porous skeleton with a regular and periodic pattern, as illustrated in Scheme 1.12 Imitating the ultrastructure of the diatom frustule and reconstructing the frustules with photoactive materials could be promising for light energy conversion purposes.13,14 | ||
| Scheme 1 A schematic illustration of the inner shell and outer layer structures of a diatom. | ||
One dimensional semiconductor nanomaterial (nanorod, nanotube, nanowire) arrays have attracted enormous attention in the light energy conversion field due to potentially facile charge separation and their enhanced light absorbance properties.15–20 These merits have been demonstrated in TiO2 based materials but are yet to be explored in other visible-light-driven materials, such as graphitic carbon nitride. In recent years, graphitic carbon nitride was found to be a metal-free semiconductor material, and meanwhile has found enormous application in catalysis, photocatalysis, and bio-related applications due to its abundance, high thermal and chemical stability and visible-light-driven responsive properties.21–24 Various strategies have been proposed for further improving carbon nitride photocatalysis for practical purposes.24–32 However, there are few reports on constructing carbon nitride arrays derived from biological materials, which would be promising for light energy conversion processes including photocatalytic hydrogen evolution from water and nicotinamide adenine dinucleotide (NADH) regeneration (an essential coenzyme involved in many biological processes).
In this work, a carbon nitride array (CNA) material was prepared by replicating diatom frustules. Pt nanoparticle decorated CNA exhibited improved hydrogen evolution from water in the presence of triethanolamine compared with bulk carbon nitride. In addition, NADH, the biological form of hydrogen, could be photocatalytically regenerated from NAD+, which allows reversal of the biological pathway of dehydrogenase enzymes. In metabolism, NADH/NAD+ is involved in redox reactions, carrying electrons from one reaction to another. Maintaining a kinetically efficient cycle between NAD+ and NADH is extremely important for sustainable dehydrogenase enzymatic reactions.33–35 Photocatalytic NADH regeneration suggests great possibilities for using light to drive various enzymatic reactions. In situ regeneration of the NADH cofactor coupled with the yeast alcohol dehydrogenase enzyme (YADH) for the sustainable production of methanol from formaldehyde was realized, as an important step toward the enzymatic cascade conversion of CO2 to methanol.36,37 The work reported here, in our opinion, could provide a basic and general platform to construct artificial bio-inspired light energy conversion systems from sustainable biological materials.
Experimental
Materials
Formate dehydrogenase (FateDH), formaldehyde dehydrogenase (FaldDH), yeast alcohol dehydrogenase (YADH), β-nicotinamide adenine dinucleotide (β-NAD+), reduced nicotinamide adenine dinucleotide (NADH), Celpure p100 diatomite, cyanamide, triethanolamine (TEOA), rhodium(III) chloride hydrate, 2,2′-bipyridyl, and 1,2,3,4,5-pentamethylcyclopentadiene were purchased from Sigma-Aldrich.Sample fabrication and characterization
CNA was synthesized through a solid “impregnation incipient wetness” method. 0.5 g of cyanamide and 0.5 g of purified Celpure P100 diatomite was mixed evenly in the mortar, transferred into crucibles with lids, heated to 600 °C under N2 atmosphere (4 hours) and kept at that temperature for another 4 hours. The resultant yellow powder was treated with 4 M NH4HF2 solution under stirring for 48 hours (be cautious!). The dispersion was then filtered and the precipitate was rinsed with copious amounts of deionized water and ethanol. After the filtering procedure, the yellow powder was dried under vacuum at 60 °C overnight.[Cp*Rh(bpy)H2O]2+ (M) was synthesized as follows: RhCl3·H2O was refluxed in methanol with one equivalent of 1,2,3,4,5-pentamethylcyclopentadiene for 24 h. The resulting red precipitate was filtrated and suspended in methanol. On addition of two equivalents of 2,2-bipyridine, the suspension cleared up immediately and a yellowish solution was formed. [Cp*Rh(bpy)Cl]Cl was precipitated on the addition of diethyl ether to the obtained yellowish solution. Stock solutions (100 mM) were prepared in water and stored at room temperature avoiding direct light exposure ([Cp*Rh(bpy)Cl]Cl readily hydrolyzes to [Cp*Rh(bpy)(H2O)]2+).
XRD measurements were performed on a D8 Diffractometer from Bruker instruments (Cu Kα radiation, λ = 0.154 nm) equipped with a scintillation counter. N2 sorption experiments were done with a Quantachrome Autosorb-1 at liquid nitrogen temperature. TEM images were taken on Philips CM200 FEG (Field Emission Gun), operated at an acceleration voltage of 200 kV. SEM measurements were performed on a LEO 1550 Gemini instrument. The UV-Vis absorbance spectra were recorded on a T70 UV/Vis spectrophotometer.
Photocatalytic water splitting by CNA
In a typical experiment, 50 mg carbon nitride material was placed in the photoreactor (equipped with a magnetic stirrer), then the reactor was evacuated and refilled with argon five times. Then 38 mL of aqueous TEOA solution (10 vol% TEOA) containing the platinum precursor (Pt metal content: 3% weight of carbon nitride) was added under argon flow. The set-up was equipped with a thermostat and stirring plate. H2O and TEOA were pretreated before use. H2O was firstly degassed for 1 h under vacuum in an ultrasonic bath and secondly purged with argon for 1 h. TEOA was purged for 1 h with argon. After the reaction, a sample of the headspace was analyzed with GC for hydrogen content.The photocatalytic regeneration of NADH and enzymatic reactions
In a typical regeneration procedure, the reaction medium was composed of NAD+ (1 mM), TEOA (15 w/v%), phosphate buffer (100 mM) and the CNA (3 mg). The reaction system was placed into a quartz reactor equipped with a stirring bar and illuminated with a LED lamp (wavelength = 420 nm, OSA Opto Light GmbH). The distance between the reactor and LED lamp was fixed at 5.5 cm. During the illumination, the concentration of NADH was estimated by measuring the absorbance of the diluted reaction system at 340 nm. NAD+ has a peak absorption at a wavelength of 260 nm, with an extinction coefficient of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1. NADH has a peak absorption at 340 nm with an extinction coefficient of 6220 M−1 cm−1.
900 M−1 cm−1. NADH has a peak absorption at 340 nm with an extinction coefficient of 6220 M−1 cm−1.
The conversion of formaldehyde to methanol was performed in a quartz reactor. The reaction solution was composed of 3 mg CNA, NAD+ (1 mM), M (0.125 mM), formaldehyde solution (5 mM), and alcohol dehydrogenase (0.45 mg) in 3 mL 0.1 M PBS buffer (pH = 7.0). Prior to the addition of enzyme stock solution, the PBS buffer was firstly saturated with nitrogen. The amount of methanol was detected by GC.
The conversion of CO2 to methanol was performed in quartz reactor. The reaction solution was composed of 3 mg CNA, NADH (1 mM), KHCO3 (0.1 M), M (0.125 mM), formate dehydrogenase (0.02 mg), formaldehyde dehydrogenase (0.3 mg), and alcohol dehydrogenase (1.5 mg) in 2 ml 0.1 M PBS buffer (pH = 7.0). Prior to the addition of enzyme stock solution, the PBS buffer was firstly saturated with gaseous CO2. The amount of methanol was detected by GC.
Results and discussion
The CNA was synthesized by a simple sacrificial diatom template method.22,38 The commercially available diatom frustule was first purified by gravity sedimentation. During the repeated purification and washing process, the smallest fragments of diatom were removed due to their slower rate of sedimentation. For templating, only the largest fragments (>20 microns) were used, and the major components were large segments with ordered nanochannel arrays, corresponding to a final yield of 10 wt% (see Fig. S1 of the ESI†). The purified diatom template was dried under ambient conditions and mechanically mixed together with equal amounts of cyanamide in a mortar (see Fig. S2 of the ESI†). The resultant powder was put in a nitrogen oven at 600 °C for 4 hours for thermal condensation of the carbon nitride network (Fig. S3 of the ESI†). Finally, the resulting homogeneous yellow powder was treated with NH4HF2 to remove the diatom template, and hierarchical carbon nitride nanorod array structures were obtained (Fig. 1). Therefore, the whole process can be summarized as follows: (1) purification and drying of the diatom frustule; (2) mechanical mixing with cyanamide and high temperature thermal condensation; (3) removal of the diatom template.39 | ||
| Fig. 1 SEM images of CNA with a top (a) and lateral (b) view of the array of carbon nitride nanorods. (c) A TEM image of a single carbon nitride nanorod detached from the CNA. (d) A SEM image of the starting diatom frustule template featuring a periodic and regular porous pattern. | ||
Structural details revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are presented in Fig. 1. As shown in Fig. 1a, the carbon nitride nanorod array is maintained even after vigorous stirring during the NH4HF2 treatment process. A lateral view is presented in Fig. 1b to demonstrate the intact carbon nitride nanorod array. Additional views of the CNA are also presented in Fig. S4 of the ESI.† As can be seen from Scheme 1, the inner structure of the diatoms is a honeycomb structure of pores, which are connected to the external surface by smaller pores. The outer layer plays an important role in maintaining the dense structure of the regular nanorods of carbon nitride even after vigorous stirring during the template removal process, providing the mechanical robustness of the CNA. The nanorod array is attached to a plane coming from a replica of the outer surface of the diatoms, and is therefore different and easier to manipulate than discrete nanorods derived from anodic aluminum oxide or silica.27 A further look at a single and intact nanorod sheds some light on the formation mechanism. A TEM image of an intact carbon nitride nanorod is shown in Fig. 1c, revealing a nanoporous structure with a pore size of around 20 nm. It is also interesting to find that the vase-shaped nanorod features a small concave cavity on both ends. It can be speculated that its formation comes from non-confined condensation on the ends, compared with the nanoconfinement in the interior of the nanochannel, allowing minor shrinking. Both tips of the rod are narrower than the body size, corresponding to the original template. Practically no bulk carbon nitride particles are observed in the CNA sample, demonstrating the cyanamide is exclusively confined in the frustule pore channels. The cyanamide precursor in the evenly mixed solid phase with diatoms melts at elevated temperatures and is pulled by capillary forces into the interior of the channel, and undergoes thermal condensation into graphitic carbon nitride afterwards. TGA analysis (Fig. S5 of the ESI†) demonstrates that only around 1% of silica residue was found to remain in the final CNA samples, indicating that most of the silica template was successfully removed during the etching process.
The Fourier transform infrared spectroscopy (FTIR) and powder X-ray diffraction (XRD) characterizations depicted in Fig. 2 demonstrate that the obtained CNA is a typical polymeric graphitic carbon nitride material featuring novel hierarchical structures. The band at 810 cm−1 in the FTIR spectrum is ascribed to the s-triazine ring modes, while the bands at 1200–1600 cm−1 are characteristic of aromatic CN heterocycles (Fig. 2a). The XRD spectrum shows the two characteristic peaks of graphitic carbon nitride (Fig. 2b) at 13.0° and 27.4°, which are ascribed to the in-plane repeat period and stacking of the conjugated aromatic system, respectively.
 | ||
| Fig. 2 (a) The FTIR spectrum of the CNA showing typical C–N heterocycle stretches in the 1100–1600 cm−1 spectral range and the breathing mode of the tri-s-triazine units at 810 cm−1. (b) The XRD spectrum of the CNA with two peaks at 13.0° and 27.4° ascribed to the in-plane repeat period and stacking of the conjugated aromatic system, respectively. | ||
The optical properties of CNA were measured by UV-Vis diffuse reflection spectroscopy (Fig. 3a). The CNA exhibits typical semiconductor absorption in the blue region, similar to CNB (bulk carbon nitride). The photogenerated charge carrier separation and recombination were also investigated by photoluminescence spectroscopy under excitation at 380 nm. As shown in Fig. 3b, the samples exhibit a broad emission peak centered at 470 nm. Compared with CNB, the fluorescence emission of the CNA is greatly decreased, suggesting a suppression of photo-induced charge carrier recombination, which is beneficial for further heterogeneous photocatalysis.
 | ||
| Fig. 3 (a) The optical absorption and (b) photoluminescence spectra of the CNA and CNB, respectively. | ||
The CNA was firstly evaluated in a photocatalytic hydrogen evolution activity test by loading 3 wt% Pt as a cocatalyst and using TEOA as a hole scavenger. Under illumination, the electrons in the valence band are excited to the conduction band. The photoinduced electrons in the conduction band will further migrate to the Pt nanoparticles deposited on the surface of the carbon nitride by crossing the aligned Fermi energy levels. The electron vacancy in the valence band will be restored by the TEOA, producing glycolaldehyde and di(ethanol)amine as the oxidation products. The hydrogen evolution results are presented in Fig. 4. The performance in photocatalytic water splitting to give hydrogen is improved by a factor of 4 over that of CNB. The hydrogen evolution amount increases linearly and the hydrogen evolution capability did not show any obvious decay in a 10 hour run experiment. Considering the low surface area of the CNA (25 m2 g−1) compared to CNB (28 m2 g−1) (Fig. S6 of the ESI†), the higher activity can be attributed to the introduction of the well-ordered nanorod array structure. The directional charge transport properties of the carbon nitride nanorod array, together with the multiple light scattering of the segmented photocatalyst, contribute to the improvement of photocatalytic hydrogen evolution.40–42
 | ||
| Fig. 4 (a) A schematic diagram of hydrogen evolution by Pt nanoparticle decorated CNA, and (b) time plots of H2 evolution for the CNA and CNB during a 10 hour experiment. CB: conduction band; VB: valence band. | ||
The photocatalytic properties of the CNA were also investigated in the biocatalytic conversion of formaldehyde into methanol (selected as a test reaction) using YADH, for which the cofactor NADH needs to be regenerated for a sustainable production of methanol. YADHs are enjoying increasing interest as versatile and selective biocatalysts in both academic and industrial fields. The reduction of pro-chiral ketones is an area of application that is most attractive to organic chemists, while the reversible process of the oxidation of alcohols is less attractive due to the destruction of chirality. Usually, for the sustainable enzymatic synthesis of a chiral alcohol, another enzyme, such as formate dehydrogenase and phosphite dehydrogenase, is used to regenerate NADH. A natural photosystem such as plant chloroplasts could also be used for NADH regeneration, but presents very low stability over time. In order to increase the long term stability, and also reduce the complexity of the system, we have investigated light-driven NADH regeneration by the CNA, which is inherently sustainable. We investigated the regeneration of 1,4-NADH from NAD+ by CNA photocatalysis, in the presence of [Cp*Rh(bpy)H2O]2+ (abbreviated as M, see the HNMR in Fig. S7 of the ESI†) as an electron mediator and hydride transfer agent. The electrons are transferred to [Cp*Rh(bpy)H2O]2+ to form Cp*Rh(bpy), followed by coupling with one proton to generate hydriderhodium [Cp*Rh(bpy)H]+, which acts as a hydride transfer reagent towards NADH regeneration from NAD+. TEOA is employed as a sacrificial electron donor. The oxidized state of TEOA provides the protons during its decomposition path to glycolaldehyde and di(ethanol)amine. The NADH concentration gradually increases throughout illumination of the reaction medium, as evidenced from the absorbance peak intensity at a wavelength of 340 nm quantified by a UV-Vis spectrophotometer, shown in Fig. S8 of the ESI.† During the reduction of formaldehyde into methanol, NADH is oxidized into NAD+ and the light energy is used to reduce NAD+ back into NADH. Optimum enzymatic reaction conditions for YADH for the reduction of formaldehyde into methanol were previously determined and the highest activity was obtained for pH values between 5 and 7, in accordance with the literature for other reactions.43 The previously optimized conditions of the reaction were applied for the following photocatalytic regeneration of NADH with CNA.
The CNA based photocatalytic NADH regeneration system was successfully combined with YADH conversion, as illustrated in Fig. 5a. We proved that the in situ regenerated NADH is in fact useful as a hydride equivalent for enzymatic reduction. The rhodium complex mediated NADH regeneration by carbon nitride photocatalysis is obviously also rather efficient. According to the results shown, a complete and nearly linear conversion of formaldehyde to methanol was obtained in 3 hours. As a reference experiment, a dark enzymatic reaction gave no detectable methanol. The result for methanol production in solution is shown in Fig. 5b (0.21 mmolMeOH min−1 genzyme−1). It is thereby demonstrated that NADH regeneration by the carbon nitride photocatalysis plays an energetically driving role in the formaldehyde conversion process.
 | ||
| Fig. 5 (a) A schematic illustration of the conversion of formaldehyde into methanol by YADH, with NADH regenerated by CNA photocatalysis in the presence of M. (b) The production of methanol as a function of time obtained in the biophotocatalytic system. | ||
Furthermore, in view of the success of the preliminary transformation of formaldehyde to methanol, three consecutive enzymatic conversions of CO2 into methanol were consequently tested (Fig. 6a). Optimized enzymatic powder ratios between the three enzymes were used.43 CO2 was generated from NaHCO3 (0.1 M). The actual gaseous CO2 dissolved in the pH 7 buffer solution was 8 mM.44 We performed a continuous reduction of CO2 to methanol during 40 hours illumination; 1.8 μmol of methanol was produced, corresponding to a productivity of 0.07 mmolMeOH genzymes−1 in 3 hours (Fig. 6b). We can observe the approximately linear increase of methanol yield as the illumination time increases. Meanwhile, the dark enzymatic reaction does not produce any methanol. Therefore, NADH regeneration by the photosynthetic material is effective enough to reverse the natural reaction pathway of the three dehydrogenase enzymes. Enzymatic conversion of CO2 to methanol with photocatalytic NADH regeneration is therefore firstly proven feasible. The capability of the carbon nitride material for NADH regeneration has been compared to a biological system using the phosphite dehydrogenase enzyme. Some of the current authors obtained a productivity of 0.16 mmolMeOH genzymes−1 in 3 hours, but using a pressure of 5 bars of CO2, thus allowing the dissolution of 5 times more CO2 in aqueous media.43 The conversion efficiency of our current system can still be enhanced by using a suitable pressurized photocatalytic reactor.
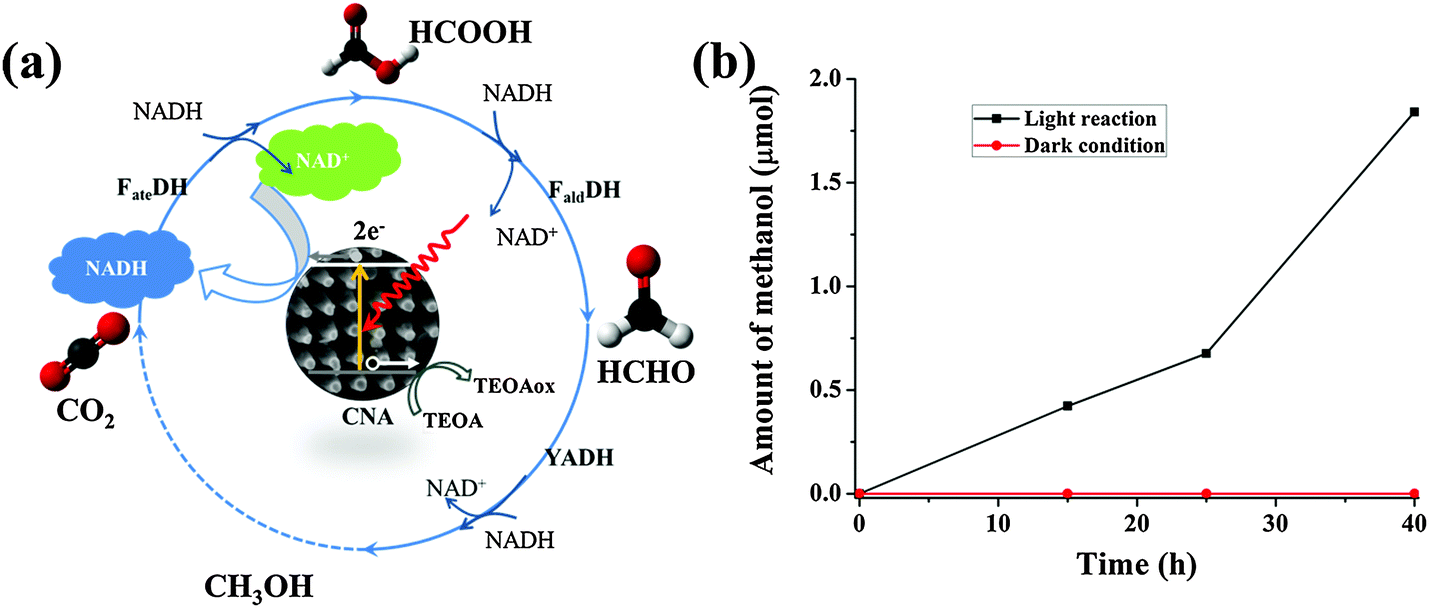 | ||
| Fig. 6 (a) A schematic illustration of three consecutive enzymatic reactions for converting CO2 into methanol with in situ regeneration of NADH by CNA photocatalysis; (b) the production of methanol as a function of time obtained in the three enzyme based biophotocatalytic system. | ||
Conclusions
In summary, diatom-inspired, hierarchical carbon nitride materials have been synthesized and used for the biophotocatalytic enzymatic conversion of formaldehyde to methanol as well as splitting water to give hydrogen. The work reported here, in our opinion, provides a general and basic platform to construct bio-inspired artificial light energy conversion systems.45 Future work might involve coupling the carbon nitride photocatalytic regeneration system with enzyme immobilization within the confinement of mesopores in the carbon nitride matrix. This would substantially improve the enzyme stability for long term use and also large scale applications.46 Furthermore, it is exciting to inquire into the diversity of diatom species to retro-analyze other structures for their efficiency in artificial photosynthesis. Finally, we proved that synthetic photosystems are suitable for the regeneration of the biologically active cofactor NADH in biophotosynthetic processes.Acknowledgements
J. Liu acknowledges the support of the Alexander von Humboldt Foundation and helpful assistance and discussion by Dr Hongqiang Wang of MPIKG. H. Zhou acknowledges the financial support of the National Natural Science Foundation of China (51102163).References
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- J. Huang, M. Antonietti and J. Liu, J. Mater. Chem. A, 2014, 2, 7686–7693 CAS.
- J. Liu, G. Liu, M. Li, W. Shen, Z. Liu, J. Wang, J. Zhao, L. Jiang and Y. Song, Energy Environ. Sci., 2010, 3, 1503–1506 CAS.
- A. R. Parker and H. E. Townley, Nat. Nanotechnol., 2007, 2, 347–353 CrossRef CAS PubMed.
- N. Nadine and L. Jacques, Chem. Soc. Rev., 2011, 40, 849–859 RSC.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2011, 112, 1555–1614 CrossRef PubMed.
- J. Liu, Q. Yang, W. Yang, M. Li and Y. Song, J. Mater. Chem. A, 2013, 1, 7760–7766 CAS.
- M. Sumper and E. Brunner, Adv. Funct. Mater., 2006, 16, 17–26 CrossRef CAS.
- H. Zhang, M.-A. Shahbazi, E. M. Mäkilä, T. H. da Silva, R. L. Reis, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 9210–9219 CrossRef CAS PubMed.
- D. Losic, J. G. Mitchell, R. Lal and N. H. Voelcker, Adv. Funct. Mater., 2007, 17, 2439–2446 CrossRef CAS.
- T. Fuhrmann, S. Landwehr, M. El Rharbi-Kucki and M. Sumper, Appl. Phys. B: Lasers Opt., 2004, 78, 257–260 CrossRef CAS PubMed.
- Z. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church and Z. Kang, Nature, 2007, 446, 172–175 CrossRef CAS PubMed.
- C. Jeffryes, J. Campbell, H. Li, J. Jiao and G. Rorrer, Energy Environ. Sci., 2011, 4, 3930–3941 CAS.
- H. Zhou, T. Fan, X. Li, J. Ding, D. Zhang, X. Li and Y. Gao, Eur. J. Inorg. Chem., 2009, 211–215 CrossRef CAS.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef CAS PubMed.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191–195 CrossRef CAS PubMed.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, J. Jin, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 10317–10324 CAS.
- J. Jiu, S. Isoda, F. Wang and M. Adachi, J. Phys. Chem. B, 2006, 110, 2087–2092 CrossRef CAS PubMed.
- K. Q. Peng and S. T. Lee, Adv. Mater., 2011, 23, 198–215 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76–80 CrossRef PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., 2012, 124, 3237–3241 CrossRef.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- X. Chen, Y.-S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed.
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 CAS.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- Y. Zhang, Z. Schnepp, J. Cao, S. Ouyang, Y. Li, J. Ye and S. Liu, Sci. Rep., 2013, 3, 2163 Search PubMed.
- Y. S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- S. Choudhury, J. O. Baeg, N. J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 124, 11792–11796 CrossRef.
- J. Ryu, S. H. Lee, D. H. Nam and C. B. Park, Adv. Mater., 2011, 23, 1883–1888 CrossRef CAS PubMed.
- P. Könst, H. Merkens, S. Kara, S. Kochius, A. Vogel, R. Zuhse, D. Holtmann, I. W. Arends and F. Hollmann, Angew. Chem., Int. Ed., 2012, 51, 9914–9917 CrossRef PubMed.
- R. Obert and B. C. Dave, J. Am. Chem. Soc., 1999, 121, 12192–12193 CrossRef CAS.
- S.-w. Xu, Y. Lu, J. Li, Z.-y. Jiang and H. Wu, Ind. Eng. Chem. Res., 2006, 45, 4567–4573 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- J. Liu, J. Huang, D. Dontosova and M. Antonietti, RSC Adv., 2013, 3, 22988–22993 RSC.
- F.-Q. Xiong, X. Wei, X. Zheng, D. Zhong, W. Zhang and C. Li, J. Mater. Chem. A, 2014, 2, 4510–4513 CAS.
- A. W. Maijenburg, E. J. Rodijk, M. G. Maas, M. Enculescu, D. H. Blank and J. E. ten Elshof, Small, 2011, 7, 2709–2713 CrossRef CAS PubMed.
- Y. Liu, H. Wang, Y. Wang, H. Xu, M. Li and H. Shen, Chem. Commun., 2011, 47, 3790–3792 RSC.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721–3730 RSC.
- Z. Duan and R. Sun, Chem. Geol., 2003, 193, 257–271 CrossRef CAS.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455–11461 CrossRef CAS PubMed.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01348d |
| This journal is © the Owner Societies 2014 |