 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnergy transfer in aggregates of bacteriochlorophyll c self-assembled with azulene derivatives†
Martina
Matěnová
a,
Viviana
Lorelei Horhoiu
bc,
Florian-Xuan
Dang
d,
Petr
Pospíšil
a,
Jan
Alster
a,
Jaroslav V.
Burda
a,
Teodor
Silviu Balaban
bcd and
Jakub
Pšenčík
*a
aFaculty of Mathematics and Physics, Charles University, Ke Karlovu 3, 121 16 Prague, Czech Republic. E-mail: psencik@karlov.mff.cuni.cz
bInstitute for Nanotechnology, Karlsruhe Institute for Technology (KIT), Postfach 3640, D-76021 Karlsruhe, Germany
cCenter for Functional Nanostructures, KIT, Postfach 3640, D-76021 Karlsruhe, Germany
dAix Marseille Université, Centrale Marseille, CNRS UMR 7313, Institut des Sciences Moléculaires de Marseille, Chirosciences, Marseille, France
First published on 23rd June 2014
Abstract
Bacteriochlorophyll (BChl) c is the main light-harvesting pigment of certain photosynthetic bacteria. It is found in the form of self-assembled aggregates in the so-called chlorosomes. Here we report the results of co-aggregation experiments of BChl c with azulene and its tailored derivatives. We have performed spectroscopic and quantum chemical characterization of the azulenes, followed by self-assembly experiments. The results show that only azulenes with sufficient hydrophobicity are able to induce aggregation of BChl c. Interestingly, only azulene derivatives possessing a conjugated phenyl ring were capable of efficient (∼50%) excitation energy transfer to BChl molecules. These aggregates represent an artificial light-harvesting complex with enhanced absorption between 220 and 350 nm compared to aggregates of pure BChl c. The results provide insight into the principles of self-assembly of BChl aggregates and suggest an important role of the π–π interactions in efficient energy transfer.
1. Introduction
Bacteriochlorophyll c (BChl c) is similar to BChls d and e, all three being found in chlorosomes, the light-harvesting antennae of photosynthetic bacteria of the phylum Chlorobi, some Chloroflexi, and the only one known Acidobacteria species.1,2 BChls c, d and e in chlorosomes are self-assembled into aggregates without any protein scaffold. The close proximity of BChl molecules in the aggregate leads to strong exciton coupling and consequently to a red shift of the Qy absorption band compared to the monomer. The aggregates form curved layers which are arranged in lamellar structures by a hydrophobic interaction between esterifying alcohols from neighboring layers.3–6 Tubular models also abound in the literature.1,6,7 Carotenoids and quinones are present in the chlorosomes as well, and located in the hydrophobic space between the lamellar layers.8 Such an arrangement ensures a close contact between these molecules and BChls without affecting excitonic coupling between BChls. Carotenoids serve for light-harvesting and photoprotective functions while quinones are involved in the protective excitation quenching in the presence of oxygen.1 The orderly arrangement of pigments in domains inside the chlorosomes allows for efficient light-harvesting and excitation energy transfer.6,9 In addition, aggregation increases the stability against photodegradation compared to monomeric BChls.10The ability of BChls c, d and e to self-assemble and their high light-harvesting efficiency make the BChl aggregates a promising material for artificial light-harvesting antennae.11,12 Both chlorosomal pigments and their synthetic analogs are intensively studied in this respect.12–20 BChl c forms aggregates in organic nonpolar solvents and also in aqueous environments. In the latter case, addition of a suitable nonpolar compound is necessary to induce the aggregation. Other constituents of the chlorosomes, e.g. lipids, carotenoids and quinones, were shown to induce such aggregation in vitro.21–23 Each of these additional components also affects the properties of the formed complexes in a distinctive way. This ability can be used to prepare artificial complexes with tailored properties. For instance, if the aggregation is induced by pigment molecules absorbing light at the wavelengths where the BChl c aggregates absorb poorly, it is possible to prepare artificial light-harvesting complexes with extended spectral coverage compared to pure BChl aggregates.24
In order to prepare an antenna combining BChls with another molecule, it is necessary to understand the nature of the involved interactions. The aggregation in the polar environment of the aqueous buffers is driven by a hydrophobic effect.22,25 In addition, it has been suggested that π–π or CH–π interactions play an important role in maintaining the close contact between carotenoids and BChls required for efficient energy transfer and photoprotection.26,27 To further investigate the role of the above-mentioned interactions in the co-aggregation with BChl c, we have studied the aggregation-inducing properties of several azulene derivatives which have different substituents, a conjugated phenyl ring and/or hydrophobic side chains.
Azulene (Fig. 1) is an aromatic 10 π conjugated molecule related to naphthalene. It consists of fused cyclopentadiene and cycloheptatriene rings. Its skeleton is naturally found in some mushrooms and plants.28–30 It is a textbook example of a compound violating Kasha's rule and exhibiting fluorescence from the S2 state.31 Azulene and its derivatives represent the first group of chromophores not found in chlorosomes, for which the ability to induce BChl c aggregation was investigated. Use of the series of derivatives differing in their side groups was instrumental in distinguishing between the role of the hydrophobicity, which is important for induction of BChl c aggregation, and the role of the π–π interactions, which seems to be critical for efficient transfer of excitation energy absorbed by azulenes to BChls. The derivatives containing a conjugated phenyl ring effectively extended the spectral coverage of the final assemblies into the near UV spectral region.
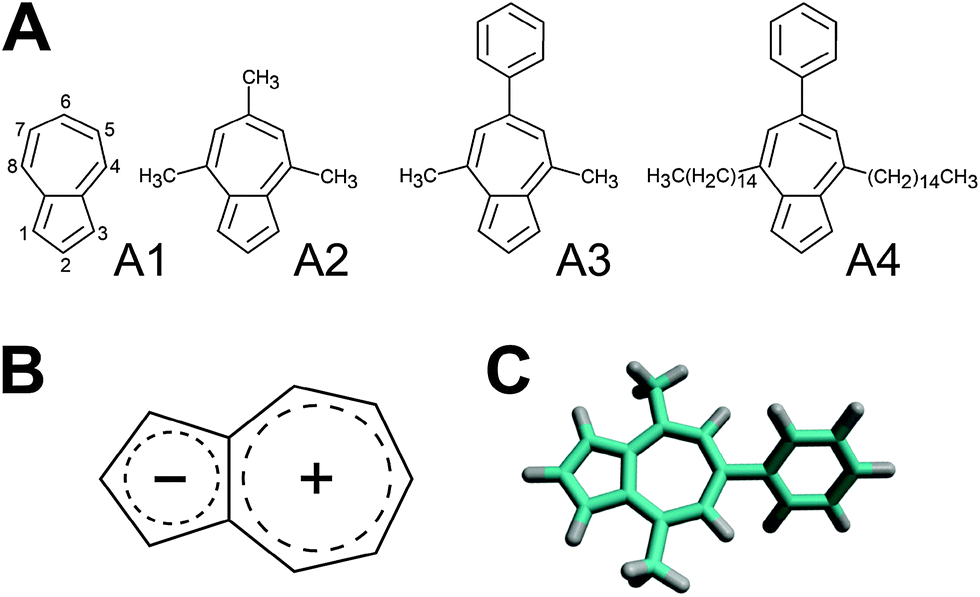 | ||
| Fig. 1 (A) Chemical structure of azulene (A1) and its three derivatives (A2–A4) used in this work. (B) Schematic representation of the charge distribution within the azulene skeleton. (C) The optimized geometry of azulene A3. | ||
2. Experimental methods
Azulene was purchased from Sigma-Aldrich (99%, A97203) and its three derivatives were synthesized by using Hafner's condensation of pyrylium salts32,33 with the sodium salt of cyclopentadiene (Scheme 1).34 Full experimental details are presented in the ESI.† For the numbering of pigments used within this work, see Fig. 1. | ||
| Scheme 1 A2 R = R′ = CH3; A3 R = CH3, R′ = C6H5; A4 R = −(CH2)14CH3, R′ = C6H5. | ||
The hydrophobicity of the azulenes was calculated as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P defined here as the partition coefficient between octanol and water using Molinspiration software (http://www.molinspiration.com).
P defined here as the partition coefficient between octanol and water using Molinspiration software (http://www.molinspiration.com).
The optimized geometry of the azulene molecules was determined by quantum chemical calculation using the B3PW91 functional and the 6-31G* basis set within the Gaussian 09 software.35 The optical spectra were calculated by several methods based on both molecular orbital (MO) formalism and the density functional theory (DFT) method (see ESI†). The most successful approach, the results of which are presented here, was the TDDFT/ω-B97-XD functional with aug-cc-pVDZ basis sets as implemented in a Gaussian programme package.35 Analysis of transition integrals between the corresponding molecular orbitals was performed using Molpro 2012.1 MATROP – matrix operation utility.36,37
Extinction coefficients of azulene and its derivatives were determined from absorption spectra. An amount of pigment between 1 and 5 mg was weighed and then dissolved in tetrahydrofuran (THF) in a 5 ml volumetric flask. This stock solution was diluted in a defined way and the absorbance at the absorption maximum in a 1 cm cuvette was determined using a Specord 250 spectrophotometer (Analytik Jena). Extinction coefficients were calculated from at least 10 samples according to the Lambert–Beer law.
BChl c was isolated from the whole cells of Chlorobaculum (Cba.) tepidum and purified by HPLC as described before22 with the following modifications: an Eclipse XDB-C18 column has been used and the mobile phase and the flow rate were, respectively, 100% methanol, 1.5 ml min−1 during the first 25 minutes and 80% methanol and 20% n-hexane, 2 ml min−1 for the next 15 minutes. All four well-resolved main BChl c homologues found in Cba. tepidum were then combined together to imitate the natural distribution of BChl c in chlorosomes.
Self assembly experiments were performed in the following way. First, stock solutions of chromophores were prepared. BChl c was dissolved in methanol and its concentration was determined using an extinction coefficient of 70 mM−1 cm−1 at the Qy maximum.38 Azulene was dissolved in THF and the extinction coefficient determined in this study was used to adjust the concentration. The stock solutions were mixed together to reach the required molar ratio and rapidly injected into 3 ml of stirred 50 mM Tris-HCl buffer, pH 8.0. The amount of injected organic solvents did not exceed 1% (v/v). The final concentration of BChl c was approximately 20 μM in all samples. Molar ratios of the azulene derivatives to BChl c ranged from 0.01:1 to 2:1. Aggregate formation was evaluated from the red shift of the BChl c Qy absorption band. Absorption spectra were measured immediately after preparation and after three days, when the aggregates were fully developed. It should be noted that the reported molar ratios were determined for initial mixtures. It is possible that a small fraction of the azulene molecules was not incorporated into the BChl c aggregates and precipitated in the buffer.
Energy transfer from azulenes to BChl c was examined by means of steady-state fluorescence spectroscopy. Fluorescence excitation spectra were recorded on a FluoroMax-2 fluorescence spectrometer (Jobin Yvon Spex). A 715 nm cut-off filter (Roper Scientific) was used to attenuate the stray light contribution and eliminate the emission of azulene as well as the transmission of the second order of the excitation light. The excitation spectra were corrected for the wavelength dependence of the excitation light intensity as described previously.39 Fluorescence spectra were recorded at least three days after the preparation of aggregates in order to have fully developed samples. The molar ratios of azulene to BChl c in aggregates for excitation spectra measurement ranged from 0.3:1 to 2:1, and absorbance of the sample was less than 0.2 in the BChl c Qy maximum to minimize the effect of re-absorption or self-quenching. Excitation energy transfer efficiency was determined from a comparison of absorption (1 − T) and fluorescence excitation spectra. Since aggregate formation generally increases light scattering, absorption spectra used for comparison with the excitation spectra were corrected for the scattering using the method of Latimer and Eubanks.40,41
3. Results
3.1. Extinction coefficient of azulenes
Fig. 2 shows the absorption spectra of the four azulenes used in this work. The spectra show a weak S0 → S1 transition between 500 and 650 nm, and a S0 → S2 transition at around 340 nm for azulene A1, at 350 nm for A2 and at approximately 370 nm for the A3 and A4 derivatives. The strongest S0 → S3 transition is at 276 nm for azulene A1, at 287 nm for A2 and at approximately 300 nm for derivatives A3 and A4. The absorption maxima of azulenes A3 and A4 which possess the 6-phenyl ring, are thus more red shifted. This is an unexpected observation, since the conjugated system of the phenyl ring is partially decoupled from the conjugated system of azulene: the geometry optimization of azulene A3 shows that the plane of the 6-phenyl ring forms a dihedral angle of ∼50° with the plane of the azulene moiety (Fig. 1C). The appendage of the optically transparent hydrocarbon chains (A3 → A4) leads to a much less pronounced red shift (from 300 nm to 304 nm at the main absorption band). The spectra of A3 and A4 exhibit a similar shape, which further indicates that the side chains do not affect the absorption properties of the molecules significantly. Upon increasing the number of carbon atoms in side chains the vibrational fine structure of the bands becomes less resolved, most probably due to conformational averaging (Fig. 2). Extinction coefficients of azulene and its derivatives in THF determined from absorption measurements are summarized in Table 1. | ||
| Fig. 2 Absorption spectra of azulene and its derivatives. (A) Absorption spectra of A1 (red) and A2 (blue). (B) Absorption spectra of A3 (red) and A4 (blue). Note the break in the X-axis and the change in scale. | ||
3.2. Calculations of the azulene spectra
The observed changes in extinction coefficients between the azulene derivatives are rather difficult to understand, considering that the conjugated backbone seems to be very similar in all explored molecules. To shed light on the quantum mechanical basis for these differences, electronic spectra were calculated using several methods based on both MO formalism and DFT.The order of the states and their oscillator strengths roughly agree with experiment only when calculated using “long range corrected density functionals” (in our case TDDFT/ω-B97-XD and CAM-B3LYP functional methods42,43) except that these methods include one additional state with a very small oscillator strength. To be consistent with the convention used for azulenes in the literature where S2 is used for the state from which fluorescence occurs31,44 and S3 for the state with the maximal oscillator strength,45 we denote the transition with a negligible oscillator strength obtained from calculations as S0 → Sx. More importantly, the obtained results quantitatively correctly describe the experimentally observed differences between the intensity of the strongest transition S0 → S3 (Table 2). Its oscillator strength is comparable for azulenes A1 and A3, but reduced approximately to one half for A2. The oscillator strength of this transition for azulene A4 is lower than that of A3. The difference between intensities of the S0 → S3 transition for azulenes A1 and A2 is even in a good quantitative agreement. In addition, the results obtained for azulene A1 agree well with previous studies46,47 and the minor differences are most probably caused by the different basis sets used. Because geometry optimization and electronic spectra calculation of the whole A4 molecule are computationally very demanding, the C15 aliphatic chains were replaced with ethyl, propyl, and butyl groups in order to examine the effect of the side chain length on the electronic spectra. The calculations revealed that the length of the side chain has almost no effect on the transition energies, but does have a small influence on the oscillator strengths. The calculated transition energies of A3 and A4 are thus very similar, in agreement with the experimental data (Table 2). Calculations where THF was included as an implicit solvent exhibit similar trends.
| Transition | A1 | A2 | ||
|---|---|---|---|---|
| Wavelength [nm] | f | Wavelength [nm] | f | |
| S0 → S1 | 517.9 | 0.008 | 412.3 | 0.027 |
| S0 → Sx | 332.5 | 0.003 | 313.5 | 0.000 |
| S0 → S2 | 260.8 | 0.073 | 261.4 | 0.300 |
| S0 → S3 | 247.6 | 1.121 | 243.6 | 0.678 |
| S0 → S4 | 236.7 | 0.000 | 232.7 | 0.000 |
| S0 → S5 | 220.1 | 0.001 | 214.5 | 0.000 |
| Transition | A3 | A4-Butyl | ||
|---|---|---|---|---|
| Wavelength [nm] | f | Wavelength [nm] | f | |
| S0 → S1 | 505.2 | 0.012 | 506.4 | 0.016 |
| S0 → Sx | 342.2 | 0.025 | 342.4 | 0.012 |
| S0 → S2 | 275.5 | 0.061 | 275.6 | 0.085 |
| S0 → S3 | 272.9 | 1.384 | 274.6 | 1.292 |
| S0 → S4 | 242.1 | 0.003 | 235.3 | 0.000 |
| S0 → S5 | 238.3 | 0.000 | 230.2 | 0.002 |
To gain a better understanding of the observed differences in extinction coefficients, the role of different molecular orbitals in the main electronic transitions was analyzed. Generally, the HOMO → LUMO excitation dominates in the S0 → S1 transition, however, its intensity is low. Next transition (between 310 and 350 nm) corresponds almost equally to HOMO − 1 → LUMO and HOMO → LUMO + 1 excitations and its intensity is close to a dark state. DFT calculations further predict that the third transition is predominately represented by the HOMO − 1 → LUMO + 1 excitation. Its energy was calculated to be around 260–270 nm which is distinctly higher than that of the S0 → S1 and S0 → Sx transitions. The most intense spectral line for all of the explored azulenes corresponds to the fourth excited state. Its energy is close to the third transition (within 5–20 nm), but the intensity is more than an order of magnitude higher in comparison with all previous transitions. To explain different intensities of the S0 → S3 transition for different azulene derivatives, two effects have to be taken into account: the composition of the excited state and the values of molecular transition integrals 〈Ψx|r|Ψy〉. First, the fourth transition is a combined excitation of HOMO − 1 → LUMO and HOMO → LUMO + 1, similarly to the second transition. However, detailed analysis of the transition integrals between the corresponding MOs revealed that the 〈HOMO − 1|r|LUMO〉 and 〈HOMO|r|LUMO + 1〉 integrals add up. This is in contrast to the second transition, in which the 〈HOMO − 1|r|LUMO〉 and 〈HOMO|r|LUMO + 1〉 integrals cancel each other. The second effect is connected with the fact that for A2 the S0 → S3 transition contains some additional non-negligible admixtures of the 〈HOMO − x|r|LUMO + y〉 type (where at least one of x, y > 1) with low integral values. Consequently the weights of the 〈HOMO − 1|r|LUMO〉 and 〈HOMO|r|LUMO + 1〉 integrals are decreased reducing the final transition intensity of this transition to approximately one-half of the value for A1.
3.3. Solubility of azulenes in water
It was proposed that the aggregation of BChl molecules in an aqueous environment is driven by the hydrophobic effect, and therefore the hydrophobicity of the aggregation-inducing molecule is expected to be an important parameter. The calculated hydrophobicities of azulenes A1–A4 are listed in Table 1. As can be expected already from the chemical structure of the molecules, the hydrophobicity increases from A1 through A2 and A3 to A4. This order was further confirmed by reverse phase HPLC chromatography and also by calculation of the solubility of azulenes in water (not shown). In agreement with all these estimates, we could observe that azulene A1 is partly soluble in water. Its solubility was previously determined to be 0.02 g l−1 (ref. 48), which corresponds to a 160 μM solution. The concentration of azulene A1 used for self-assembly experiments in this work was much lower. We have tested the solubility of all the azulenes in water. In all cases, the amount of azulene which corresponded to an intended final concentration of about 40 μM was injected into 3 ml of buffer. Absorption spectra were recorded after preparation and after three days to determine the degree of precipitation of azulene derivatives. The amount of azulenes in buffer decreased from A1 to A4. Azulene A1 remained dissolved in its original concentration, while azulenes A2, A3 and A4 precipitated to concentration of ∼20 μM, ∼10 μM and ∼3 μM, respectively. The concentration was determined using the same extinction coefficient as in THF.3.4. Self-assembly experiments
BChl c is partly soluble in water where it develops into a form whose absorption spectrum resembles that determined for an antiparallel dimer in dichlormethane.49 The Qy absorption maximum of this form is between 710 and 715 nm and we denote it as a dimer in this work. We use the term aggregate for assemblies in which the Qy band is distinctively red shifted above 715 nm.The ability of all azulenes to induce aggregation of BChl c was examined by measuring steady-state absorption spectra upon mixing with an increasing concentration of azulene or its derivatives. The concentrations of azulene derivatives necessary to induce a red shift of the BChl c Qy band above 715 nm were then compared. If the aggregation is driven by the hydrophobic effect, as previously proposed, the aggregation inducing ability should increase from A1 through A2 and A3 to A4. This is exactly what we have observed. The results are summarized in Fig. 3. Azulene A1 was used in concentrations at which it is fully soluble in water (<40 μM) and was not able to induce any aggregation even at the highest used molar ratio of A1 to BChl c (2:1). The maximum of the BChl c Qy band in the sample containing azulene A1 and BChl c in a molar ratio of 2:1 was found to be at ∼715 nm. The increase in the amount of azulene in the sample can be monitored by the intensity of the azulene absorption between 250 and 400 nm. The total absorption spectrum thus most probably represents just a sum of spectra of azulene A1 and BChl c dissolved in buffer. Azulene A2 induces aggregation only at the highest tested molar ratio of azulene to BChl c. For a molar ratio of azulene A2 to BChl c of 1:1 the Qy maximum was observed at ∼719 nm, while for the molar ratio of 2:1, the Qy band shifted to ∼730 nm. This shift has to be caused by the interaction of BChl c species with azulene A2.
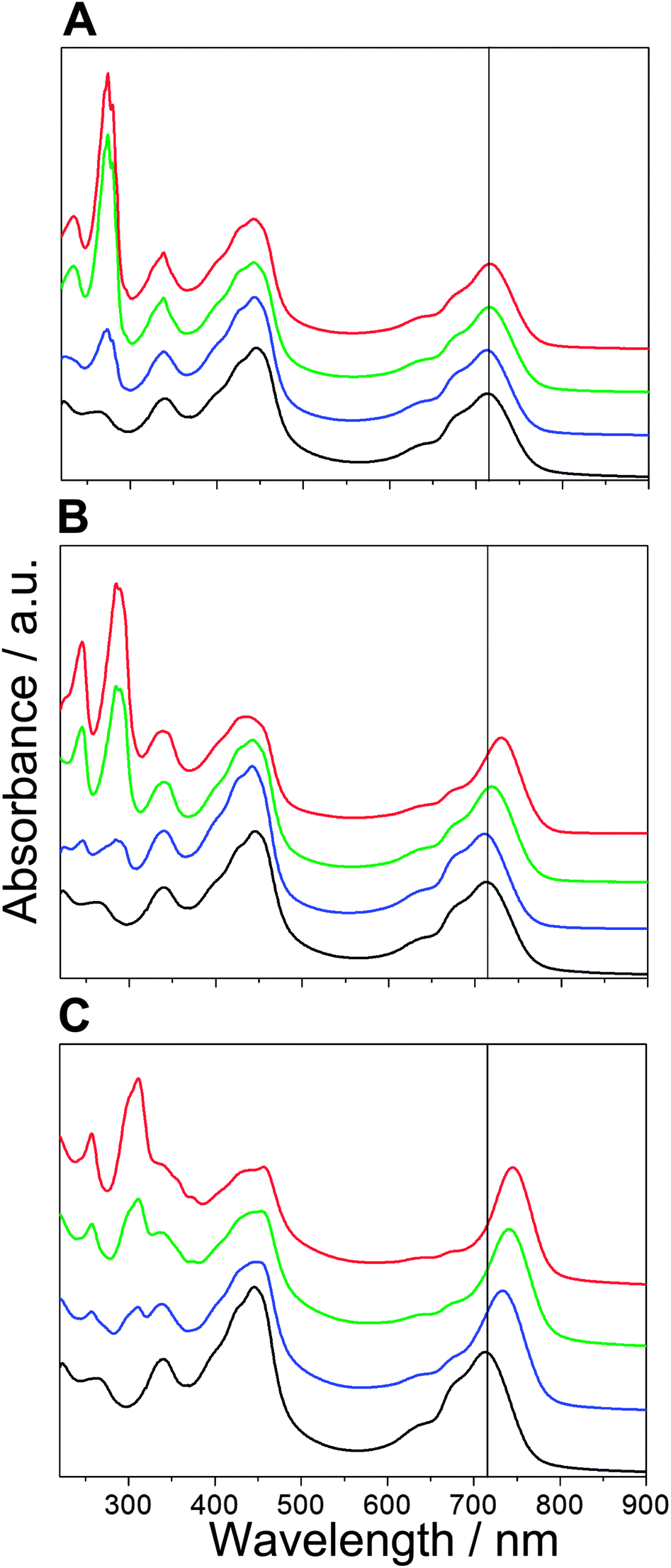 | ||
| Fig. 3 Absorption spectra of BChl c with A1 (A), A2 (B) and A4 (C) measured three days after preparation. The azulene to BChl c molar ratio is 0:1 (black), 0.3:1 (blue), 1:1 (green) and 2:1 (red). A vertical line at 715 nm is shown to assist the comparison of the BChl c Qy band positions. Spectra were normalized at the maximum of Qy band and offset along the absorbance axis for the sake of clarity. Results for A3 are very similar to those for A4. | ||
The aggregation-inducing ability, however, improves significantly upon appending the 6-phenyl ring. Very similar results were obtained from experiments with both A3 and A4. Fig. 3C shows the absorption of BChl c upon mixing with azulene A4. Addition of A4 to the sample leads to a significant red shift of the BChl c Qy band, indicating pronounced aggregation of BChl c. The red shift increases with an increase of azulene concentration in the sample, which can be again monitored by the intensity of the azulene bands between 250 and 400 nm. The Soret band of BChl c is also affected by the aggregation, but to a lesser degree. The molar ratio between azulene A4 and BChl c required to obtain the Qy band at a longer wavelength than 715 nm is about 0.1:1. The Qy band position of samples with the molar ratio of 0.1:1 was at ∼710 nm after preparation, but it shifted to ∼720 nm after aggregates fully developed within three days. The maximal red shift of ∼745 nm was observed for the highest molar ratio used (2:1). Azulene A3, which lacks the long hydrocarbon chains, was only slightly less efficient in inducing the aggregation of BChl c. The molar ratio of A3 to BChl c required to induce a red-shift up to 715 nm is 0.2:1. The maximal red shift of the BChl c Qy band was similar to azulene A4 at ∼745 nm and observed for the molar ratio of 2:1.
We can conclude that the absorption of azulene A1 observed in Fig. 3A after three days comes from the pigment dissolved in buffer. On the other hand, samples where azulenes A2–A4 induced aggregation of BChl c exhibit substantially higher intensity of azulene absorption than what corresponds to their solubility in water, and the azulenes do not precipitate. Quantitative estimation of the azulene concentration embedded in the aggregate structure is difficult due to the contribution of BChl c absorption and light-scattering at a given wavelength, and the necessity to use the extinction coefficient of azulene determined in THF. Nevertheless reasonable estimates were obtained. For instance, concentration of azulene A4 in the sample prepared from a mixture of this azulene and BChl c in a molar ratio of 2:1 was ∼20 μM, i.e. 6–7 times higher than its solubility. This fact certainly means that the hydrophobic interaction leading to the aggregate formation leads also to incorporation of the azulene molecules within the aggregate structure. In addition, the absorption spectra of the red shifted BChl–azulene assemblies exhibit substantially larger scattering contribution which indicates that the aggregation leads to formation of much larger particles than the dimers as can be seen in Fig. 3C for azulene A4. Similar results were also observed for azulene A3 (not shown). This is in contrast to what we observed for azulene A1 where any significant contribution of the scattering was absent (Fig. 3A).
3.5. Excitation energy transfer from azulenes to BChl c
Steady-state fluorescence excitation spectroscopy was used to check whether the excitation energy can be transferred from the incorporated azulenes to BChl c. The fluorescence quantum yield of BChl c aggregates is certainly lower than that of the monomers, and thus does not represent the aggregate induced fluorescence, a phenomenon described and recently explained by Tang and coworkers.50 However, the quantum yield was high enough to allow determination of the energy transfer efficiency with a sufficient accuracy. Excitation spectra were recorded for samples with an azulene-to-BChl c ratio of at least 0.3:1. For lower azulene-to-BChl c ratios, the contribution of azulenes to the absorption spectrum was too weak to allow an accurate determination of the efficiency. The excitation spectra were detected close to the maximum of BChl c emission (755 to 780 nm depending on the aggregation state of BChl c). The emission of azulene with a maximum at ∼420 nm (not shown) was eliminated using a cut-off filter. All measured excitation spectra exhibit spectral features of BChl c in the same proportions as in the absorption spectra which indicates that internal conversion and vibrational relaxation within BChl c occurs with close to 100% efficiency, as expected. In addition, the main absorption peak of azulene at ∼300 nm is discernible in the spectra from samples containing azulene A3 or A4, although its intensity does not correspond to that observed in the (1 − T) absorption spectra (Fig. 4). This means that only a part of the energy absorbed by azulene is transferred to BChl c. To determine the extent of the transferred energy, the excitation spectra were compared with absorption (1 − T) spectra of the sample as a reference for 100% efficiency.51,52 The efficiency of energy transfer was determined by comparing the intensity of 1 − T and fluorescence excitation spectra at ∼260 nm, where the absorption of azulene is negligible, and near ∼300 nm, where it is maximal. The estimated energy transfer efficiencies vary with the concentration of the azulene in the sample. For both azulenes A3 and A4 the highest efficiency was determined for aggregates with an azulene to BChl c molar ratio of around 1:1 (∼50% for A3 and ∼60% for A4). In aggregates with a higher molar ratio of azulene, the energy transfer efficiency approximately linearly decreases (∼40% efficiency for a molar ratio of azulene A4 to BChl c of 1.5:1; ∼30% efficiency for a molar ratio of 2:1). For a lower A3 or A4 to BChl c ratio (0.3–0.8:1) the energy transfer efficiency was estimated to be similar to that observed for 0.8–1.0:1 concentration.
 | ||
| Fig. 4 Comparison of the fluorescence excitation spectrum (green circles) and the 1 − T absorption spectrum (red) of A2–BChl c aggregates (molar ratio 2:1) (A) and A3–BChl c aggregates (molar ratio 1:1) (B). | ||
In contrast, the excitation spectra recorded for mixtures of BChl c with azulenes A1 and A2 did not reproduce any features of the azulene absorption. Most importantly, no sign of energy transfer was observed even for those samples where the concentration of azulene A2 was high enough to induce aggregation of BChl c (Fig. 4A). Thus, it can be safely concluded that these pigments do not transfer their excitation energy to BChl c molecules even if they are incorporated in the aggregate structure.
4. Discussion
Self-assembly of chlorosomal BChls in aqueous environments induced by other nonpolar molecules naturally found in chlorosomes (lipids, carotenoids, quinones) has been well studied before.12,21–23 These authors established that the hydrophobic interaction between the esterifying alcohols is not strong enough to drive the aggregation of BChl c molecules in aqueous environments. However, a nonpolar compound which is not soluble in water, but is soluble in a solvent miscible with water may induce aggregation. This is in contrast to the situation in nonpolar solvents, like hexane, where the hydrophilic interaction between the polar chlorin rings drives the interaction without the need for any additional molecule. To further characterize the properties of a molecule necessary to induce aggregation of BChl c in aqueous environments, we have chosen to investigate azulene and its three derivatives studied in this work. Neither azulene nor its derivatives are found in chlorosomes, however, their easy synthesis via the pyrylium route and versatility in modification of the structure and properties make them a valuable model system.The results show that an increase in hydrophobicity of the azulene molecule increases its ability to induce aggregation. Azulene A1 is partly soluble in water and is not able to induce aggregation even at the highest concentration used. On the other hand, both azulenes with a phenyl ring (A3 and A4) induce aggregation with a similar efficiency, with A4 being only slightly more efficient. This is interesting, because originally we assumed that the presence of long hydrophobic chains of A4 is required for aggregation induction. However, the hydrophobicity required to induce aggregation is clearly achieved already for azulene A3, and the effect of the additional hydrophobic side chains (A4vs.A3) is rather weak although still discernible. These results prove that a certain hydrophobicity of the co-aggregating molecule is sufficient to induce aggregation, regardless of whether it is reached by incorporation of nonpolar chains or not.
Importantly, the results also show that even if the hydrophobicity of the chromophore molecules is high enough to induce aggregation of BChls, it does not necessarily ensure that the energy absorbed by a molecule incorporated in the aggregate will be transferred efficiently to BChls. This was observed for azulene A2, which is able to induce aggregation if the molar ratio of A2 to BChl c is larger than 1:1, but even in this case we did not observe any energy transfer from azulene A2 to BChl c. In contrast, both A3 and A4 are able to transfer the absorbed excitation energy rather efficiently to BChl c molecules. Consequently, these two azulene derivatives extend the spectral coverage of the prepared assemblies into the near UV region (200–350 nm). The observed energy transfer, together with the aggregation inducing ability of these two azulene derivatives, is an indication of a strong interaction between azulene and BChl c molecules. This inference is further justified by the fact that azulenes A3 and A4 have very low solubility in water and precipitate in aqueous buffer, but not in assemblies with BChl c. The interaction between azulenes and BChls leading to BChl aggregate formation, and the persistence of azulenes in spectra, strongly indicate that azulenes A3 and A4 are incorporated in the aggregate structure.
The efficiency of ET is approximately constant up to a molar ratio of azulenes A3 and A4 to BChl c of ∼1:1, but then decreases with increasing azulene concentration. This is probably caused by excess of azulene molecules incorporated in the aggregates. Not all azulene molecules are then in close contact with the conjugated system of the BChl molecules and thus they are not able to transfer absorbed energy. Excitation energy transfer from azulene to BChl c occurs most probably from the S2 state of the azulene molecule to the Soret or Qy band of the BChl c, and not from the S1 state of azulene to the Qy state of BChl c (Fig. 5). Internal relaxation from S2 to S1 was suggested to be slow due to the small cross section between potential energy curves of extremely separated levels.31 Therefore energy transfer needs to compete only with a relatively slow fluorescence, which originates also from the S2 state.53
 | ||
| Fig. 5 Scheme of the energy levels and the most relevant transitions in the aggregates of BChl c with azulenes A3 and A4. Vertical arrows represent absorption (yellow) and internal relaxation (green), oblique arrows represent proposed energy transfer pathways (red) and wavy arrows (blue) represent fluorescence. Only the three lowest allowed states of azulene are shown for clarity. | ||
Our results imply an important role of the phenyl substituent in efficient energy transfer. The phenyl ring can be involved in π–π interactions between azulene and BChl molecules which may ensure the proximity of pigments required for sufficient interaction and efficient energy transfer. These results are in line with our recently proposed hypothesis concerning BChl–carotenoid interactions in chlorosomes.27 As there is no protein in the chlorosome interior which would maintain the optimal distance and orientation of the pigments, there must be some other way how self-assembly can ensure this. It was proposed that π–π (or CH–π) interactions between the end-group of a carotenoid and the conjugated system of a BChl molecule are responsible for strong interactions required for observed efficient energy transfer between carotenoids and BChls in chlorosomes.26,27 It seems that azulenes without phenyl rings are not capable of such π–π interactions with BChls. Both the cyclopentadiene and cycloheptatriene rings of the azulene molecule are components of the conjugated system that could potentially fit in the space available for interaction with the chlorin rings.27 However, the azulene A1 exhibits a permanent dipole moment of 1.08 D.54 To incorporate such a polar moiety is energetically unfavorable. In contrast, the phenyl group of azulenes A3 and A4 is well suited for π–π interactions with the conjugated system of the BChl molecule (Fig. 6). We propose that the interaction between azulene and BChl molecules does not alter the hydrogen bonding pattern between the BChl molecules forming the aggregates. Instead, the phenyl ring is interacting with the part of the conjugated system of the BChl molecule, which is accessible from the side of the BChl layers. The proposed arrangement ensures a close interaction between azulene and BChl molecules required to explain the observed energy transfer without disrupting the excitonic coupling between BChls. Azulene A2, which induces aggregation at higher concentration but does not transfer energy, is probably also located in the hydrophobic space formed by esterifying alcohols of BChls from the two neighboring chlorin layers, but without any close interaction with the chlorin ring. Further study including quantum mechanical calculation and molecular modeling is required to verify or disprove this hypothesis, and is now underway.
 | ||
| Fig. 6 Schematic representation of the proposed π–π interaction between the phenyl group (smaller blue squares) of azulene A3 or A4 and the conjugated system of the BChl molecules (green squares with tails). The larger blue squares represent the azulene moiety. | ||
In summary, this is the first study on incorporation of fully synthetic pigments with a spectral coverage different from the pure chlorosomal BChl. It proves the functional construct, where in certain cases efficient excitation energy transfer occurs, and paves the way for applications in biomimetic dye aggregate solar cells.55
5. Conclusions
Results of co-aggregation experiments of BChl c with azulene and three of its 4,6,8-trisubstituted derivatives provide further insight into the principles of self-assembly of BChl aggregates. They revealed that π–π interactions mediated via the phenyl ring are important for the efficient energy transfer to BChl c, and confirmed that a certain hydrophobicity of the molecule is required for the aggregation inducing ability of the admixed molecules, in this work azulenes. Once the aggregates are formed, the azulenes remain incorporated in the finally formed structures. Azulenes A3 and A4 are able to induce aggregation of BChl c molecules and transfer the excitation energy absorbed between 220 and 350 nm to BChl c. The formed complexes thus represent an artificial light-harvesting antenna with the spectral coverage extended into the near UV region compared to pure BChl c aggregates. This study may open the way to light-harvesting devices with tailored properties.Acknowledgements
This study was supported by Czech Science Agency, project No. P501/12/G055 and P208/12/0622. STB and VLH acknowledge financial support from the DFG through the Centre for Functional Nanostructures (CFN) within projects C3.2 and C3.6. The EU FP7 Future and Emerging Technologies (FET) Programme is thanked for the present generous funding of the Project PEPDIODE in Marseille.References
- N.-U. Frigaard and D. A. Bryant, in Complex Intracellular Structures in Prokaryotes, Microbiological Monographs, ed. J. Shively, Springer, Berlin, 2006, pp. 79–114 Search PubMed.
- D. A. Bryant, A. M. G. Costas, J. A. Maresca, A. G. M. Chew, C. G. Klatt, M. M. Bateson, L. J. Tallon, J. Hostetler, W. C. Nelson, J. F. Heidelberg and D. M. Ward, Science, 2007, 317, 523–526 CrossRef CAS PubMed.
- J. Pšenčík, T. P. Ikonen, P. Laurinmäki, M. C. Merckel, S. J. Butcher, R. E. Serimaa and R. Tuma, Biophys. J., 2004, 87, 1165–1172 CrossRef PubMed.
- G. T. Oostergetel, M. Reus, A. G. M. Chew, D. A. Bryant, E. J. Boekema and A. R. Holzwarth, FEBS Lett., 2007, 581, 5435–5439 CrossRef CAS PubMed.
- J. Pšenčík, M. Torkkeli, A. Župčanová, F. Vácha, R. E. Serimaa and R. Tuma, Photosynth. Res., 2010, 104, 211–219 CrossRef PubMed.
- G. T. Oostergetel, H. van Amerongen and E. J. Boekema, Photosynth. Res., 2010, 104, 245–255 CrossRef CAS PubMed.
- S. Ganapathy, G. T. Oostergetel, P. K. Wawrzyniak, M. Reus, G. M. A. Chew, F. Buda, E. J. Boekema, D. A. Bryant, A. R. Holzwarth and H. J. M. de Groot, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8525–8530 CrossRef CAS PubMed.
- J. Pšenčík, J. B. Arellano, T. P. Ikonen, C. M. Borrego, P. A. Laurinmäki, S. J. Butcher, R. E. Serimaa and R. Tuma, Biophys. J., 2006, 91, 1433–1440 CrossRef PubMed.
- J. Dostál, T. Mančal, R. Augulis, F. Vácha, J. Pšenčík and D. Zigmantas, J. Am. Chem. Soc., 2012, 134, 11611–11617 CrossRef PubMed.
- H. Kim, H. Li, J. A. Maresca, D. A. Bryant and S. Savikhin, Biophys. J., 2007, 93, 192–201 CrossRef CAS PubMed.
- T. S. Balaban, Acc. Chem. Res., 2005, 38, 612–623 CrossRef CAS PubMed.
- T. Miyatake and H. Tamiaki, Coord. Chem. Rev., 2010, 254, 2593–2602 CrossRef CAS PubMed.
- T. S. Balaban, in Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, ed. K. Kadish, K. Smith and R. Guilard, Singapore, 2010, vol. 1, pp. 221–306 Search PubMed.
- S. Shoji, T. Hashishin and H. Tamiaki, Chem. – Eur. J., 2012, 18, 13331–13341 CrossRef CAS PubMed.
- S. Patwardhan, S. Sengupta, L. D. A. Siebbeles, F. Würthner and F. C. Grozema, J. Am. Chem. Soc., 2012, 134, 16147–16150 CrossRef CAS PubMed.
- V. Huber, S. Sengupta and F. Würthner, Chem. – Eur. J., 2008, 14, 7791–7807 CrossRef CAS PubMed.
- S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. von Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378–6382 CrossRef CAS PubMed.
- S. Sengupta and F. Wurthner, Acc. Chem. Res., 2013, 46, 2498–2512 CrossRef CAS PubMed.
- N. Takahashi, H. Tamiaki and Y. Saga, Tetrahedron, 2013, 69, 3638–3645 CrossRef CAS PubMed.
- V. B. Shah and P. Biswas, ACS Nano, 2014, 8, 1429–1438 CrossRef CAS PubMed.
- M. Hirota, T. Moriyama, K. Shimada, M. Miller, J. M. Olson and K. Matsuura, Biochim. Biophys. Acta, Bioenerg., 1992, 1099, 271–274 CrossRef CAS.
- P. Klinger, J. B. Arellano, F. Vácha, J. Hála and J. Pšenčík, Photochem. Photobiol., 2004, 80, 572–578 CrossRef CAS.
- J. Alster, A. Župčanová, F. Vácha and J. Pšenčík, Photosynth. Res., 2008, 95, 183–189 CrossRef CAS PubMed.
- J. Alster, T. Polívka, J. Arellano, P. Chábera and J. Pšenčík, Chem. Phys., 2010, 373, 90–97 CrossRef CAS PubMed.
- A. Župčanová, J. B. Arellano, D. Bína, J. Kopecký, J. Pšenčík and F. Vácha, Photochem. Photobiol., 2008, 84, 1187–1194 CrossRef PubMed.
- M. Fuciman, P. Chábera, A. Župčanová, P. Hříbek, J. B. Arellano, F. Vácha, J. Pšenčík and T. Polívka, Phys. Chem. Chem. Phys., 2010, 12, 3112–3120 RSC.
- J. Pšenčík, J. B. Arellano, A. M. Collins, P. Laurinmäki, M. Torkkeli, B. Löflund, R. E. Serimaa, R. E. Blankenship, R. Tuma and S. J. Butcher, J. Bacteriol., 2013, 195, 1727–1734 CrossRef PubMed.
- S. Carret, A. Blanc, Y. Coquerel, M. Berthod, A. E. Greene and J.-P. Deprés, Angew. Chem., Int. Ed., 2005, 44, 5130–5133 CrossRef CAS PubMed.
- C. Zhang, H. Liang, G. Tu and Y. Zhao, Fitoterapia, 2010, 81, 849–851 CrossRef CAS PubMed.
- A. D. Harmon, K. H. Weisgraber and U. Weiss, Experientia, 1980, 36, 54–56 CrossRef CAS.
- M. Beer and H. C. Longuet-Higgins, J. Chem. Phys., 1955, 23, 1390–1391 CrossRef CAS PubMed.
- T. S. Balaban and A. T. Balaban, in Science of Synthesis - Houben-Weyl, Methods of Molecular Transformations, ed. E. J. Thomas, Thieme, Stuttgart, 2003, vol. 14, pp. 11–200 Search PubMed.
- T. S. Balaban and A. T. Balaban, in Science of Synthesis Knowledge Updates, ed. E. J. Thomas and A. P. Dobbs, Thieme, 2013, vol. 3, pp. 145–216 Search PubMed.
- K. Hafner and H. Kaiser, Justus Liebigs Ann. Chem., 1958, 618, 140–152 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, Molpro 2012.1, 2012 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- R. Y. Stanier and J. H. C. Smith, Biochim. Biophys. Acta, 1960, 41, 478–484 CrossRef CAS.
- J. Alster, T. Polívka, J. B. Arellano, P. Hříbek, F. Vácha, J. Hála and J. Pšenčík, Photosynth. Res., 2012, 111, 193–204 CrossRef CAS PubMed.
- K. R. Naqvi, M. N. Merzlyak and T. B. Melø, Photochem. Photobiol. Sci., 2004, 3, 132–137 CAS.
- P. Latimer and H. C. A. Eubanks, Arch. Biochem. Biophys., 1962, 98, 274–285 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- N. Tetreault, R. S. Muthyala, R. S. H. Liu and R. P. Steer, J. Phys. Chem. A, 1999, 103, 2524–2531 CrossRef CAS.
- M. Dierksen and S. Grimme, J. Chem. Phys., 2004, 120, 3544–3554 CrossRef CAS PubMed.
- D. P. Chong, Can. J. Chem., 2010, 88, 787–796 CrossRef CAS.
- L. I. Sweet and P. G. Meier, Bull. Environ. Contam. Toxicol., 1997, 58, 268–274 CrossRef CAS.
- M. Umetsu, R. Seki, T. Kadota, Z.-Y. Wang, T. Adschiri and T. Nozawa, J. Phys. Chem. B, 2003, 107, 9876–9882 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- R. Blankenship, Molecular Mechanism of Photosnythesis, Blackwell Science, 2002 Search PubMed.
- M. D. Yilmaz, O. A. Bozdemir and E. U. Akkaya, Org. Lett., 2006, 8, 2871–2873 CrossRef CAS PubMed.
- A. E. W. Knight and B. K. Selinger, Chem. Phys. Lett., 1971, 12, 419–421 CrossRef CAS.
- A. G. Anderson, Jr and B. M. Steckler, J. Am. Chem. Soc., 1959, 81, 4941–4946 CrossRef.
- P. L. Marek, H. Hahn and T. S. Balaban, Energy Environ. Sci., 2011, 4, 2366–2378 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of azulene derivatives A2–A4 and calculation of the azulene spectra. See DOI: 10.1039/c4cp01311e |
| This journal is © the Owner Societies 2014 |