
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally induced structural rearrangement of the Fe(II) coordination geometry in metallo-supramolecular polyelectrolytes
Wojciech
Szczerba
*a,
Marco
Schott
b,
Heinrich
Riesemeier
a,
Andreas F.
Thünemann
a and
Dirk G.
Kurth
b
aBAM Federal Institute for Materials Research and Testing, Unter den Eichen 87, 12205 Berlin, Germany. E-mail: wojciech.szczerba@bam.de
bJulius-Maximilians University Würzburg, Chemical Technology of Advanced Materials, Röntgenring 11, 97070 Würzburg, Germany
First published on 28th July 2014
Abstract
Rigid rod-type metallo-supramolecular coordination polyelectrolytes with Fe(II) centres (Fe-MEPEs) are produced via the self-assembly of the ditopic ligand 1,4-bis(2,2′:6′,2′′-terpyridine-4′-yl)benzene (tpy-ph-tpy) and Fe(II) acetate. Fe-MEPEs exhibit remarkable electrochromic properties; they change colour from blue to transparent when an electric potential is applied. This electrochemical process is generally reversible. The blue colour in the ground state is a result of a metal-to-ligand charge transfer at the Fe(II) centre ion in a quasi-octahedral geometry. When annealed at temperatures above 100 °C, the blue colour turns into green and the formerly reversible electrochromic properties are lost, even after cooling down to room temperature. The thermally induced changes in the Fe(II) coordination sphere are investigated in situ during annealing of a solid Fe-MEPE using X-ray absorption fine structure (XAFS) spectroscopy. The study reveals that the thermally induced transition is not accompanied by a redox process at the Fe(II) centre. From the detailed analysis of the XAFS spectra, the changes are attributed to structural changes in the coordination sphere of the Fe(II) site. In the low temperature state, the Fe(II) ion rests in a quasi-octahedral coordination environment surrounded by six nitrogen atoms of the pyridine rings. The axial Fe–N bond length is 1.94 Å, while the equatorial bond length amounts to 1.98 Å. In the high temperature state, the FeN6-site exhibits a distortion with the axial Fe–N bonds being shortened to 1.88 Å and the equatorial Fe–N bonds being elongated to 2.01 Å.
Introduction
The electrochromic effect is related to the well-known photochromic effect, where an impinging light triggers a colour change in photochromic materials. Such materials are broadly used, e.g., in sunshades with variable transparency. In the electrochromic effect, however, the application of an external electric potential triggers a change in the optical properties of the material. Such electrochromic materials are prospective candidates for applications in so-called smart windows, which can be shaded on demand for privacy, large scale displays or energy conservation in vehicles and buildings by minimizing air conditioning, heating and artificial lighting.The electrochromic effect was first discovered by Deb1 in WO3, which is colourless in its oxidized state and turns blue upon reduction. Although metal oxides such as WO3 are among the best studied electrochromic materials, their widespread commercial use is hindered by a slow switching speed, limited optical contrast and prohibitive costs. Thus, there is a high demand for alternative materials that can meet the stringent requirements needed for commercial use such as long-term reversible operation, low switching potential combined with short switching times and low capacities, high optical contrast, thermal stability, and low-cost mass production. Among these alternative materials are metallo-supramolecular coordination polyelectrolytes (MEPEs).
MEPEs form spontaneously as a result of the metal ion induced self-assembly of ditopic ligands, such as 1,4-bis(2,2′:6′,2′′-terpyridine-4′-yl)benzene (tpy-ph-tpy), and metal salts.2 The exchange of the counter ions with amphiphiles results in the corresponding polyelectrolyte–amphiphile complexes (PACs).3 While MEPEs are readily soluble in aqueous solutions, PACs are neutral and hydrophobic thus soluble in common organic solvents. MEPEs with Fe(II) as the central metal ion (Fe-MEPEs) exhibit a strong absorption band at around 590 nm, attributed to the metal-to-ligand charge transfer (MLCT) transition of the central Fe(II) ion in the quasi-octahedral coordination geometry to the coordinating terpyridine units, giving the Fe-MEPEs their deep blue colour.4,5 The corresponding Fe-PACs show an unusual temperature induced spin crossover.6,7 The modularity of self-assembly permits the introduction of different metal ions, ligands and counter ions, giving rise to a plethora of materials with a large range of properties.8,9 These macromolecular assemblies which utilize metal ion coordination, open a new dimension to the field of polymer materials. With different metal ions integrated in the polymer chain, several interesting functions become accessible, including magnetic, electrorheological, photophysical, electrochemical or electrochromic ones.10 The positive charge of the metal ion can be used for incorporating the MEPEs into different material architectures, including liquid crystals, nanostructures or thin films.11,12 Especially, rigid ditopic bis-terpyridine based MEPEs are attractive.13 The electrochemistry of polypyridines has been studied for many years.14,15 The metal ion complexes show metal ion and ligand based redox transitions with a total of up to five redox steps. Generally, a change in the redox state is associated with a change in the optical properties. MEPEs combine the versatile and generally reversible electrochemistry with the processing advantage of polymeric materials.16,17 MEPEs readily form thin films of high optical quality using various methods including layer-by-layer deposition18 or dip coating.19 Immobilized on transparent conducting electrodes, thin films of MEPE show the desired electrochromic properties, with high switching speeds and a low switching potential.19
The thermal stability, one of the key properties for a commercially successful electrochromic material, is in the scope of the present study. Studies on thin films of neat Fe-MEPEs showed that annealing above 100 °C leads to an irreversible colour change from blue to green and a loss of the electrochromic function. Hence, X-ray absorption fine structure (XAFS) experiments during in situ annealing of pure Fe-MEPE powder was set up. XAFS spectroscopy has the unique ability to track changes in the electronic structure and the local geometry of crystals and molecules making it the ideal method for this problem. In XAFS spectroscopy, X-ray photons are absorbed by excitation of the inner-core electrons of a specific element. The absorbing element is selected by choosing the appropriate energy range for the scan with monochromatic photons. If the energy of the impinging X-ray photon is sufficient to excite an electron, it will probe the unoccupied electronic states present in the absorbing atom. This part of the spectrum, called X-ray absorption near-edge structure (XANES), gives information on the oxidation state, hybridization, changes in the coordination chemistry, etc. Simply speaking XANES ‘feels’ the chemistry of the given element, and thus allows the identification of the chemical environment, in which the element under investigation is present.
With increasing photon energy, the excited photoelectrons have a surplus energy so that they can ‘leave’ the atom. Such quasi-free electrons are scattered over the electron densities of the neighbouring atoms. This phenomenon generates an interference pattern in the farther part of the XAFS spectrum called the extended X-ray absorption fine structure (EXAFS). The Fourier transformation gives a quasi-radial distribution of the electron densities, thus atoms, around the absorbing element.20,21 Using numerical analysis methods which employ computer simulations of model structures, it is possible to reconstruct the local geometry of the absorbing atom. There are several simulation codes available, in most cases following different theoretical approaches.22–24 In this study the FEFF code25 is used which is, in its newest version, capable of handling both parts of the XAFS spectrum.26
Experimental
Synthesis of Fe-MEPE
The ditopic ligand 1,4-bis(2,2′:6′,2′′-terpyridine-4′-yl)benzene (tpy-ph-tpy) was synthesized and characterized according to a literature procedure.27 The chemicals were purchased from Aldrich and used without further purification. The Fe-MEPE was synthesized with a metal ion to ligand molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.28 Iron powder was dissolved in refluxing glacial acetic acid under an inert gas atmosphere to get dispersed Fe(OAc)2. After cooling down to room temperature, the Fe(OAc)2 solution was added to 7.57 g (14 mmol) of tpy-ph-tpy dissolved in acetic acid (75%) and stirred for 2 h while refluxing. The deep blue solution was dried under vacuum and the obtained solid was dissolved in 100 mL ultrapure water and dried again. 10 g (14 mmol) of dark blue coloured solid Fe-MEPE was obtained.
:1.28 Iron powder was dissolved in refluxing glacial acetic acid under an inert gas atmosphere to get dispersed Fe(OAc)2. After cooling down to room temperature, the Fe(OAc)2 solution was added to 7.57 g (14 mmol) of tpy-ph-tpy dissolved in acetic acid (75%) and stirred for 2 h while refluxing. The deep blue solution was dried under vacuum and the obtained solid was dissolved in 100 mL ultrapure water and dried again. 10 g (14 mmol) of dark blue coloured solid Fe-MEPE was obtained.
X-ray absorption fine structure experiments
The XAFS experiments were carried out at the BAMline with a BESSY II synchrotron light source in Berlin, Germany.29 The XAFS spectra at the K-edge of iron (7112 eV) were recorded in transmission mode using two ionization chambers (Oxford Danfysik IC Plus 50). The incident beam intensity was monitored using an ionization chamber filled with air at ambient pressure giving a transmission rate of 0.92 at 7100 eV. The absorption signal was measured using a second ionization chamber of the same type filled with argon gas at ambient pressure with a transmission rate of approx. 0.25 in the energy range of interest. The energy was scanned using a Si(111) double crystal monochromator with a relative energy resolution of 2 × 10−4. The XAFS scans in the near edge region (XANES) were carried out in the range from 7032 eV (80 eV below the edge) to 7182 eV (70 eV above the edge) with a step of 1 eV. The extended absorption fine structure region (EXAFS) was scanned up to kmax = 15 Å−1 above the absorption edge (Emax = 7968 eV) with a constant step in the momentum space of 0.05 Å−1. The scans were recorded with an acquisition time of 4 s per point.The Fe-MEPE powder sample was placed in a quartz glass capillary into a PID controlled oven. The quartz glass capillary had 0.01 mm thick walls and a diameter of 2 mm. At each temperature point, four XAFS spectra were recorded, so that the last scan followed after a 2 h annealing time at the respective temperature. At each temperature, with the exception of T = 100 °C, the four spectra were identical, so that they could be merged into one spectrum. In the case of the 100 °C spectra, a time-dependent evolution was observed, see below, indicating a threshold temperature in this region, thus these spectra could not be merged into one.
The obtained XAFS spectra underwent a standard data treatment procedure using Ifeffit 1.2.11c.30,31 The k-range for the Fourier transformation was 1.7 Å−1 to 11.0 Å−1 using a Hannig-type window with a slope of 1.0 Å−1. Such parameters allowed for the use of the maximum width of the spectrum with a minimal influence from noise.
Results and discussion
The series of XANES spectra recorded while annealing the Fe-MEPE sample can be coarsely divided into two groups (Fig. 1), a low temperature state (LT) and a high temperature state (HT). The spectra of the LT state, below 100 °C, feature a double peak at the white line (principle maximum above the absorption edge step), whereas in the HT state, above 100 °C, only one peak is present at the white line. The XANES spectra measured at 100 °C exhibit a gradual evolution with time from the LT-like to the HT-like spectra. Above 150 °C, the XANES spectra do not change anymore, indicating that the transition temperature is somewhere in the range between 100 °C and 150 °C. Moreover, after cooling down to room temperature the XANES spectra retain their HT-like character. This means that the thermally induced transition is irreversible.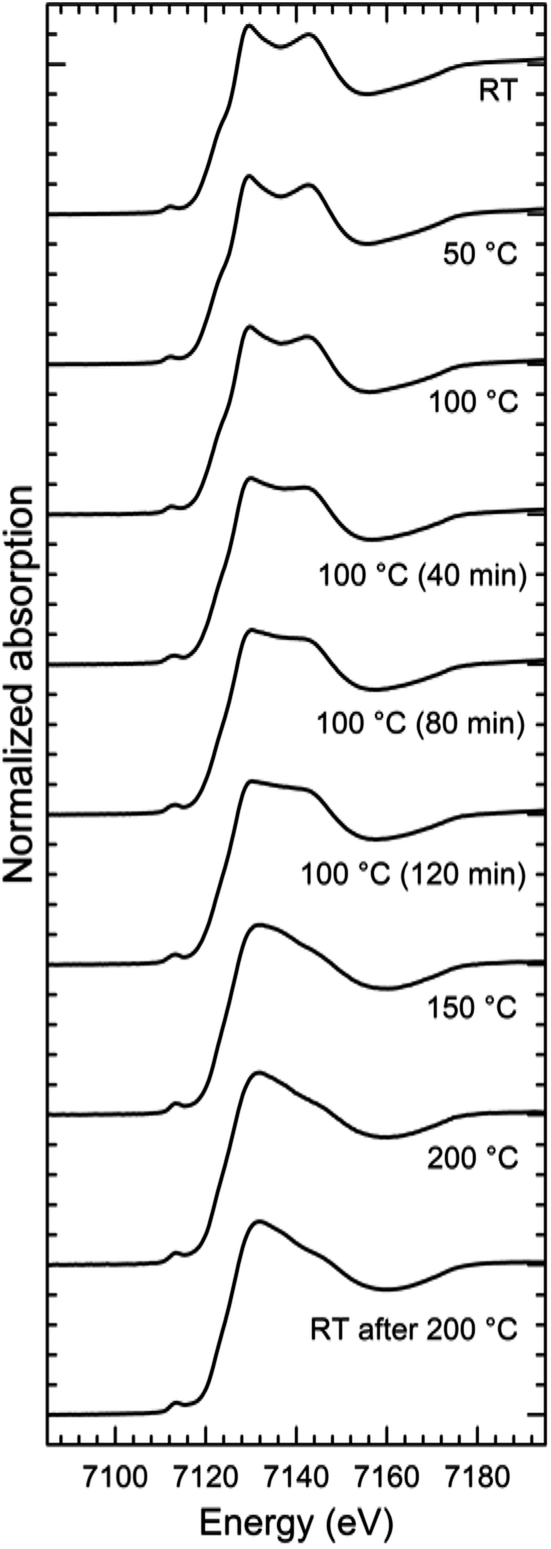 | ||
| Fig. 1 XANES Fe K-edge spectra of neat Fe-MEPE as a function of temperature. The spectra can be divided into two groups: the low temperature state (LT) below 100 °C featuring a double white-line peak and a shoulder at the absorption edge, and the high temperature state (HT) above 100 °C with a single white-line peak and a featureless absorption edge. The XANES spectra recorded at 100 °C slowly transit into the HT state. At all other temperatures the spectra do not change with time, thus the accumulated data is averaged into one spectrum. After cooling down to room temperature, the HT state structure is retained. Thus, the transition is irreversible. | ||
The XANES spectra of Fe-MEPE in the LT state are identical with the ground state spectra of Fe(tpy)2 (ref. 32) as well as with the Fe-PAC in the low spin state.6,7 However, the XANES spectra of Fe-MEPE in the HT state differ in details from those of the corresponding Fe-PAC in the high-spin state6,7 or the photo-excited Fe(tpy)2.32 Although in all the cases a strong first white-line peak is present with the second one reduced to a shoulder, the exact positions and intensities of the features are in each case slightly different.
A comparison of the XANES spectra measured at room temperature (RT) before and after annealing at 200 °C (Fig. 2) reveals that both spectra have the same absorption edge energy. No shift in the absorption edge energy means that no valency change at the Fe(II) centre is involved.33 Thus, no oxidation or reduction takes place in the Fe(II) complexes during the annealing process, and the differences in the XANES spectra observed between the two thermal states have to be attributed to changes in the geometry of the Fe(II) complexes.
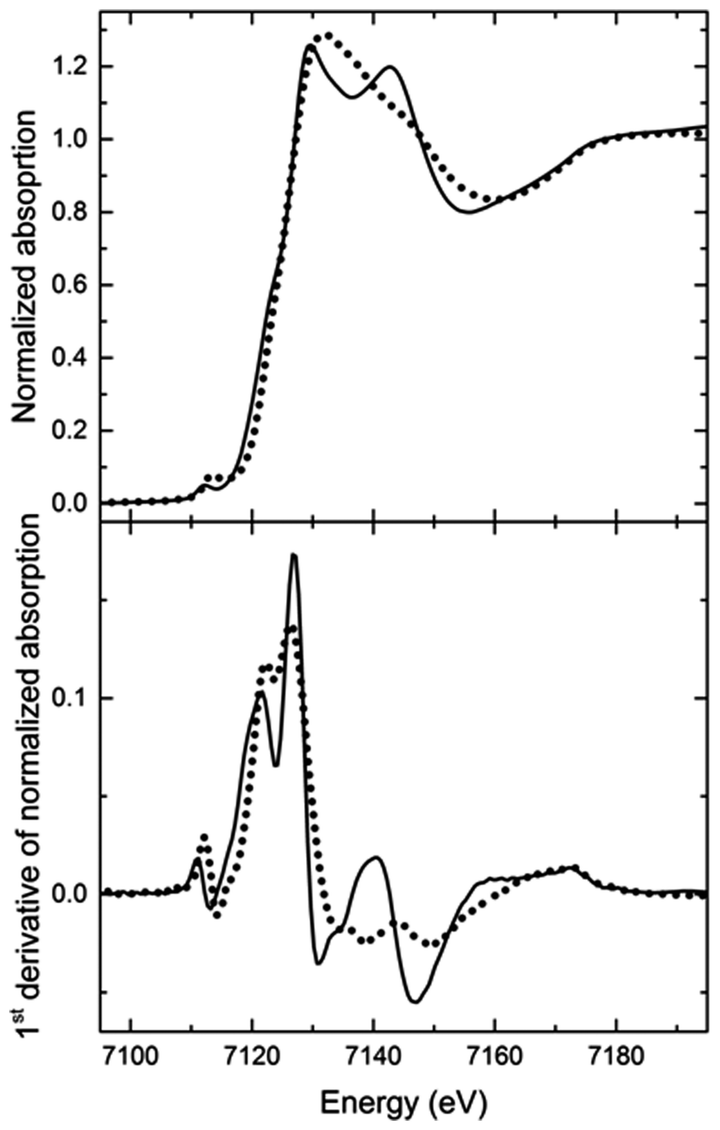 | ||
| Fig. 2 Normalized absorption (top) and first derivative (bottom) vs. energy plots of the Fe K-edge XANES spectra of the LT (solid line) and the HT state (dotted line) recorded at RT before and after heating to 200 °C, respectively. The most notable change is the transition of the double peak into a single peak and the disappearance of the shoulder in the absorption edge. The pre-peak below the absorption edge is slightly higher in the HT state. Altogether these features indicate a lowering of the local symmetry. The first derivative plot demonstrates clearly that no shift of the absorption edge energy occurs, thus no reduction or oxidation of the Fe(II) ions takes place. | ||
In order to track the changes in the geometry of the local symmetry around the Fe(II) centres, the Fourier transformed EXAFS spectra are analysed. The two FT-EXAFS patterns representative for the LT and the HT states exhibit significant differences (Fig. 3). In the case of the LT state, the modulus of the FT-EXAFS function shows clearly separated peaks, which can be attributed to the coordination shells. In general, these peaks consist of two or more sub-shell peaks. The intensity of the amplitudes decreases quite slowly; the decrease is proportional to 1/R2, as expected for a spherical or close to spherical symmetry. Additionally, the oscillations are well recognizable up to a distance of 6 Å. These features are strong evidence for a highly ordered local structure. In contrast, the FT-EXAFS signals of the HT state have a lower amplitude. The peaks of the first and second coordination shell are not separated and build up a congestion reaching 2.8 Å, which is followed by a significantly smaller peak at 3 Å. Beyond 3.5 Å, no significant oscillations are present in the FT-EXAFS patterns. These features indicate a lower symmetry environment of the Fe(II) coordination sphere in the HT state leading to less scattering events that contribute constructively to the interference pattern. In both cases, however, the first shell peak has the same width and position indicating that the average Fe–N bond length is approximately the same. The lower amplitude should then be a result of the aforementioned lowering of the local symmetry, e.g., by distortion of the coordination geometry. The first shell peak in both states features a shoulder at the low R slope. This is a hint that the first shell might consist of two Fe–N subshells, one of which with 2 nitrogen atoms closer to the Fe(II) centre.
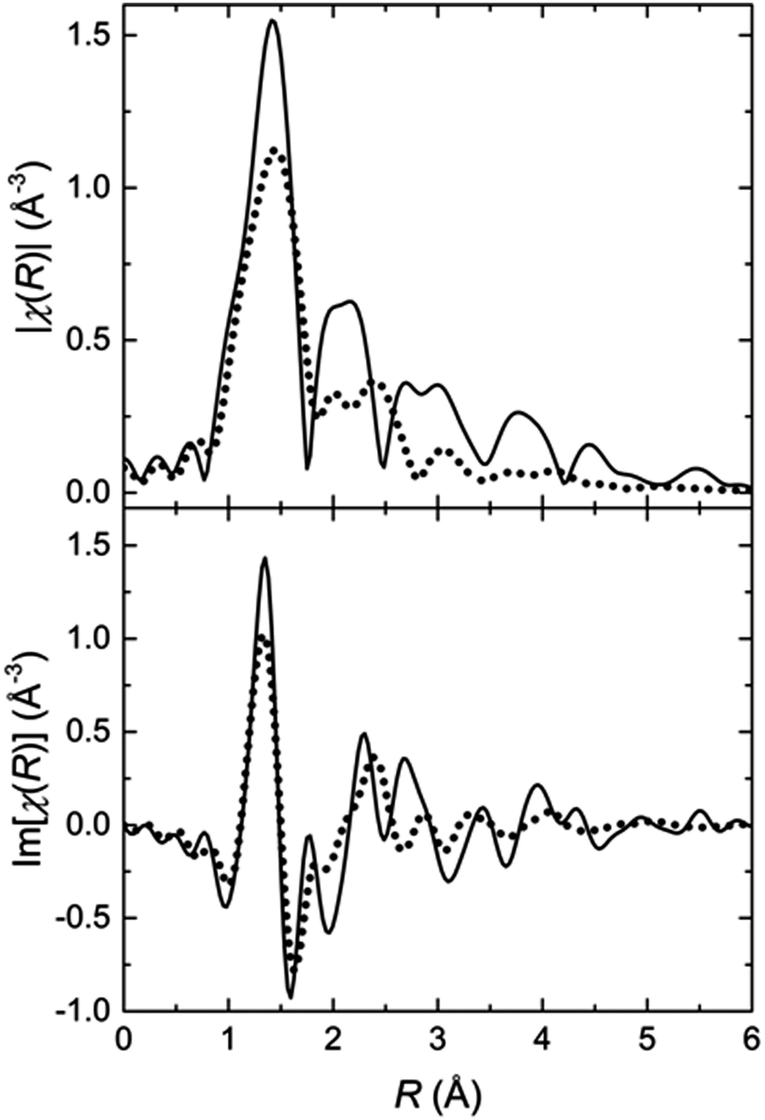 | ||
| Fig. 3 The Fe K-edge FT-EXAFS functions of the LT (solid line) and the HT (dotted line) states in direct comparison; (top) Module of the k2-weighted FT-EXAFS functions, (bottom) imaginary part of the k2 weighted FT-EXAFS functions. The decrease in the overall amplitude as well as the absence of distinctive peaks (oscillations) beyond 3 Å indicate a significant lowering of the local symmetry around the Fe(II) ions. | ||
The numerical analysis and interpretation of the EXAFS data follows a two-step approach. We begin with a first shell approximation including the central Fe(II) ion and the six next neighbouring nitrogen atoms. Then we use a structural model including the whole ligands (Scheme 1). The calculations of the model spectra are done using the FEFF 9 code.34
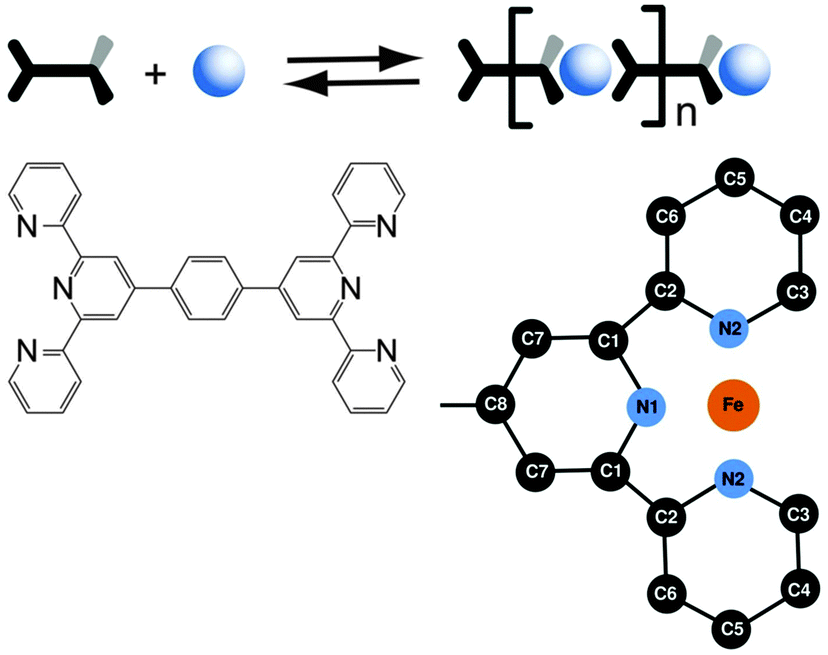 | ||
| Scheme 1 The self-assembly of 1,4-bis(2,2′:6′,2′′-terpyridine-4′-yl)benzene and FeOAc2 results in the formation of Fe-MEPE. The octahedral coordination geometry is indicated by the wedges and the numbering scheme used in the text. The Fe-MEPE polymer chain consists of tpy-ph-tpy ligands interconnected by Fe(II) ions. The zoom-in shows the numbering scheme used in the text for the Fe(tpy) unit. The second tpy-unit is perpendicular to the plane of the sketch with an overall octahedral D2d symmetry. For clarity the hydrogen atoms are omitted. | ||
The first shell model is a distorted octahedron including two subshells with the nearest neighbouring nitrogen atoms and the iron atom in the centre. The first subshell consists of two nitrogen atoms at a distance of 1.88 Å in the axial direction, whereas the second subshell is filled with four nitrogen atoms at a distance of 1.99 Å in the equatorial direction. This geometry is taken from the XAFS study of Fe-PACs.7 The calculated EXAFS curves are fitted to the series of EXAFS spectra using only two free parameters (Fig. 4) that is the distances from the central Fe(II) ion to the nitrogen atoms in the two subshells. The other three standard parameters present in the FEFF fit model by default, are set constant with values estimated in preliminary fits of the series. Hence, the amplitude reduction factor S02 = 0.5, the Debye–Waller factor σ2 = 0.025 Å2, and the inner core potential correction ΔE0 = 0. Employing this model gives the history of changes in the geometry of the FeN6 pseudo-octahedron that take place during the annealing process of the pure Fe-MEPE (Fig. 5).
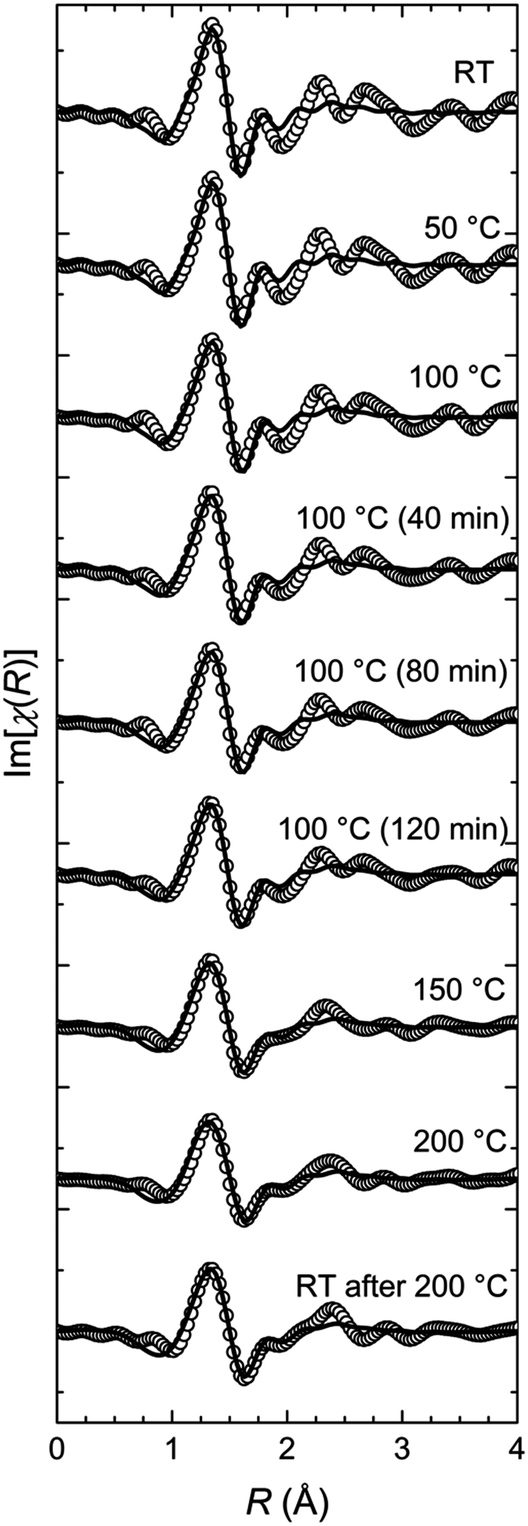 | ||
| Fig. 4 Experimental FT-EXAFS data (circles) and the respective fits of the first shell model of the FeN6 octahedron (solid line) plotted as the imaginary part of the FT. The structural model, consisting of two nitrogen sub-shells with a quasi-octahedral symmetry, is in very good agreement with the experimental data in the fitting region, 1.0 Å to 2.1 Å. | ||
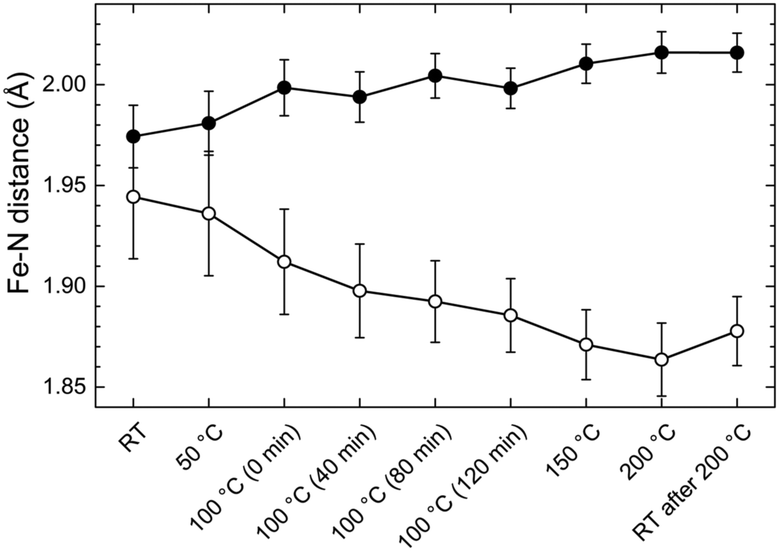 | ||
| Fig. 5 The Fe–N distances derived from the two sub-shell model during annealing of Fe-MEPE. The axial Fe–N bond length (hollow circles) decreases during annealing from 1.94 Å to 1.88 Å, whereas the equatorial Fe–N bonds (full circles) are slightly elongated from 1.98 Å to 2.02 Å. Thus, the approximately symmetric octahedron at room temperature evolves into a pseudo-octahedron that is compressed in the axial direction and slightly stretched in the equatorial direction. The distortion is irreversible, as the HT geometry is retained after cooling down to RT. | ||
In the initial LT state, the coordination geometry is quasi-octahedral. The Fe–N distance in the axial direction, along the main axis of the assembly, is 1.94 Å, whereas, the equatorial Fe–N bond length, perpendicular to the assembly axis, amounts to 1.98 Å. As the temperature is raised a gradual distortion of the FeN6 geometry is observed. The axial bonds undergo a steady contraction reaching a minimum value of 1.87 Å at 200 °C. The equatorial bonds expand continuously to reach a value of 2.02 Å at maximum temperature. Thus, in the HT state the FeN6 pseudo-octahedron exhibits a distortion, which may be related to a spin crossover in the Fe(II) complexes from low spin to high spin as reported previously for Fe-PAC6,7 and other Fe(II)-complexes.35
This distortion also affects the MLCT transition around 590 nm, causing a colour change from blue to green. The corresponding changes in the UV-Vis spectrum and the visible appearance of Fe-MEPE coated PET films are shown in Fig. 6. The transition is irreversible, as indicated by the XAFS and UV-Vis data.
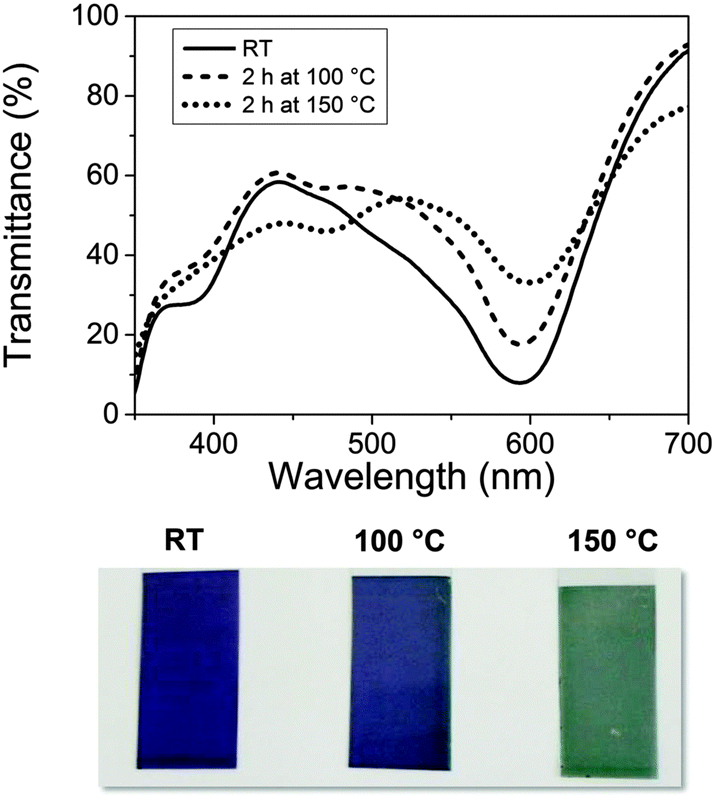 | ||
| Fig. 6 UV-Vis spectra and photographic images of dip-coated Fe-MEPE films on ITO coated PET foil, demonstrating the colour change of the material due to the thermal treatment. | ||
In the second step of the EXAFS analysis, the geometry of the Fe(II) complex in the initial and final states is explored using a structural model incorporating the central Fe(II) ion, the six nitrogen atoms at the nearest neighbouring positions, and the 20 carbon atoms in the farther coordination shells with the corresponding hydrogen atoms (Scheme 1). The structural analysis is applied to the RT spectra recorded before and after annealing representing the LT and HT state, respectively. The use of the RT spectrum after annealing instead of the 200 °C spectrum is preferred, because of the expected smaller values of the Debye–Waller factor at low temperatures. The Debye–Waller factor in the FEFF model and the XAFS theory is a measure for thermal disorder. Thus, the RT spectrum recorded after annealing provides less uncertainty.
The EXAFS spectra for the LT and HT states are calculated using models based on the literature.30,36 The theoretical spectra are fitted to the experimental ones in the range of 1.0 Å to 3.2 Å. This includes the two nearest coordination shells, i.e. six nitrogen atoms in the first shell and 12 carbon atoms, as well as hydrogen atoms attached to the C3 atoms. The model includes single-scattering paths to the neighbouring atoms and multiple-scattering paths of triangular trajectories, Tables 1 and 2. The multiple-scattering paths in this region have amplitudes that are comparable or even stronger than those of the single-scattering paths, and thus cannot be omitted. However, the single-scattering paths to the hydrogen atoms are weak enough to be excluded from the fitting model. The model is parameterized using the three default ‘spectral’ parameters S02, σ2 and ΔE0. Additionally, four ‘geometrical’ parameters are established. These allow for changes in the distances between the central Fe(II) ion and the atoms of the two shells. According to the designations introduced in Scheme 1, these are the distances to the nitrogen atoms, Fe–N1 and Fe–N2, and the distances to the nearest carbon atoms of the equatorial ring, Fe–C2 and Fe–C3. The distance Fe–C1 is not parametrized separately, since the C1 atom is part of the axial ring. Hence, when the N1 atom is pushed or pulled the C1 atom follows that motion. Therefore, its position is calculated from the change of distance of N1. From the molecular geometry it is possible to get all the remaining atomic positions of the structural model.
| # | Path degeneracy | R eff (Å) | Amplitude | Scattering path | |
|---|---|---|---|---|---|
| 1 | 2 | 1.951 | 100.00 | Fe–N1–Fe | * |
| 2 | 4 | 1.976 | 194.60 | Fe–N2–Fe | * |
| 3 | 4 | 2.871 | 76.40 | Fe–C2–Fe | * |
| 4 | 4 | 2.911 | 74.06 | Fe–C1–Fe | * |
| 5 | 4 | 2.990 | 69.70 | Fe–C3–Fe | * |
| 6 | 4 | 3.103 | 10.55 | Fe–H3–Fe | |
| 7 | 8 | 3.109 | 52.18 | Fe–C2–N2–Fe | * |
| 8 | 8 | 3.116 | 60.81 | Fe–C1–N1–Fe | * |
| 9 | 8 | 3.177 | 63.28 | Fe–C3–N2–Fe | * |
| 10 | 8 | 3.209 | 17.42 | Fe–N2–N1–Fe |
| # | Path degeneracy | R eff (Å) | Amplitude | Scattering path | |
|---|---|---|---|---|---|
| 1 | 2 | 1.875 | 100.00 | Fe–N1–Fe | * |
| 2 | 4 | 2.007 | 173.10 | Fe–N2–Fe | * |
| 3 | 4 | 2.842 | 71.91 | Fe–C1–Fe | * |
| 4 | 4 | 2.894 | 69.01 | Fe–C3–Fe | * |
| 5 | 4 | 2.912 | 11.25 | Fe–H3–Fe | |
| 6 | 4 | 3.020 | 62.63 | Fe–C2–Fe | * |
| 7 | 8 | 3.043 | 59.80 | Fe–C1–N1–Fe | * |
| 8 | 8 | 3.126 | 17.99 | Fe–N2–N1–Fe | |
| 9 | 8 | 3.145 | 43.46 | Fe–C3–N2–Fe | * |
| 10 | 8 | 3.192 | 60.22 | Fe–C2–N2–Fe | * |
The resulting fit curves are presented in Fig. 7 and the fit data are summarized in Table 3. The results confirm the findings on the coordination geometry of the first shell model discussed above. In the LT state the Fe–N1 bond length is 1.94 Å and the Fe–N2 bond length is 1.98 Å while in the HT state the respective lengths are 1.88 Å and 2.01 Å. Moreover, the averaged bond length in the LT state is 1.96 Å and in the HT state 1.97 Å.
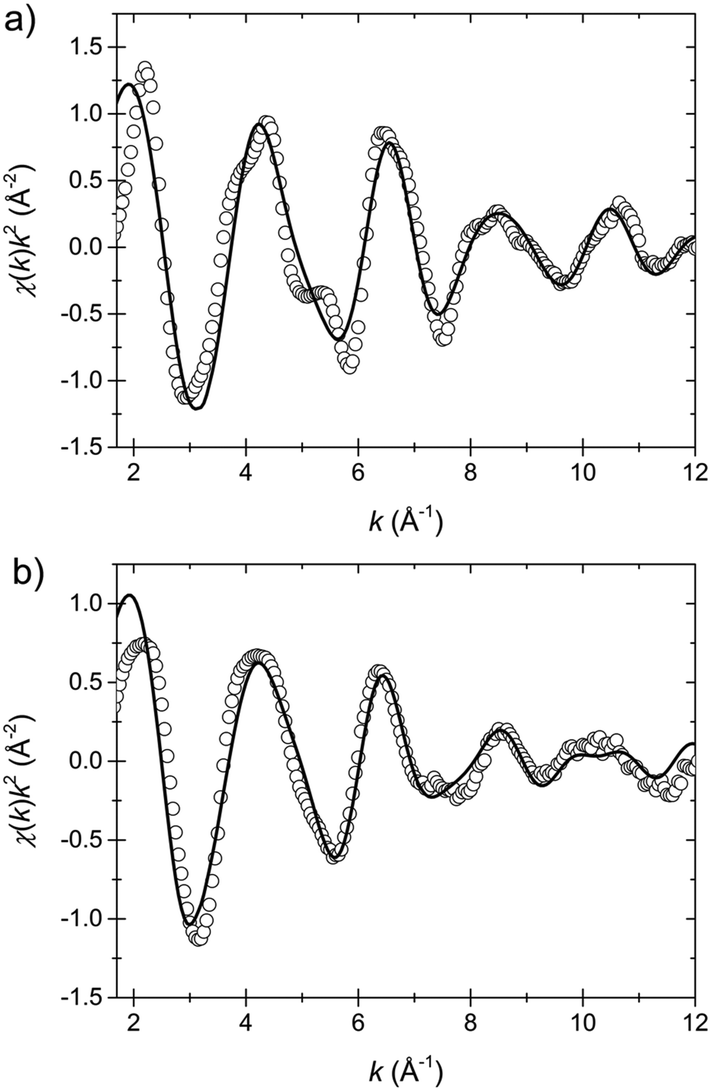 | ||
| Fig. 7 EXAFS data (circles) and fits (solid lines) in the k-space of the low temperature (a) and the high temperature state (b). The fit model includes 6 nitrogen atoms (first shell) and the 12 nearest carbon atoms (second shell), cf.Scheme 1. | ||
| State | S 0 2 | ΔE0 (eV) | σ 2 (Å2) | Δr (Å) | R | |||
|---|---|---|---|---|---|---|---|---|
| Fe–N1 | Fe–N2 | Fe–C2 | Fe–C3 | |||||
| LT | 0.61(6) | 8.1(9) | 0.0036(1) | −0.02(2) | 0.00(2) | −0.02(4) | 0.08(4) | 0.139 |
| HT | 0.55(5) | 7.7(9) | 0.0025(2) | 0.00(1) | 0.00(1) | 0.06(3) | 0.09(3) | 0.112 |
Overall, the equatorial nitrogen atoms are moved further away from the central Fe(II) ion. This causes a slight widening of the bite-angle of the terpyridine unit from 99.8° in the LT state to 115.4° in the HT state, see Scheme 2 and Table 4. The bite-angles found are smaller than the bite-angle of 120° of the uncoordinated tpy-ph-tpy ligand.36
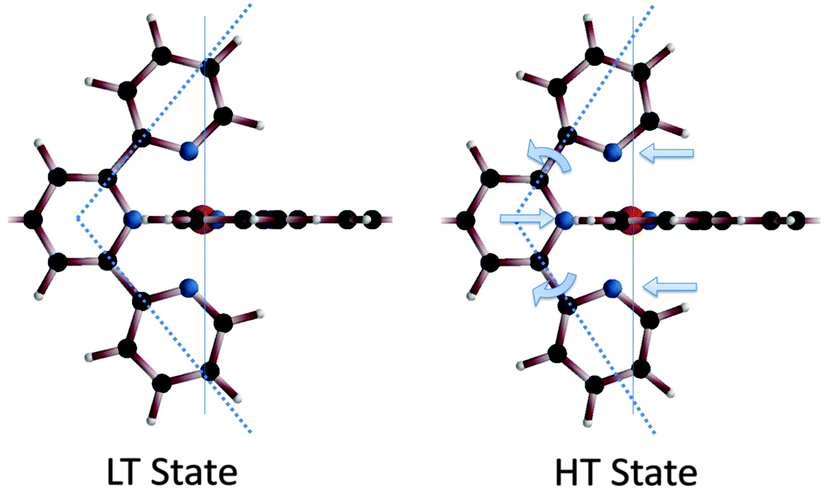 | ||
| Scheme 2 In the HT state the axial Fe–N bond length is shortened, whereas the equatorial bonds are elongated, opening slightly the bite-angle of the tpy unit, from 99.8° to 115.4° (dotted line). | ||
| LT | HT | |
|---|---|---|
| Distance Fe–N1 (Å) | 1.94 | 1.88 |
| Distance Fe–N2 (Å) | 1.98 | 2.01 |
| Angle N1–Fe–N1 | 180° | 180° |
| Angle N2–Fe–N2 | 157.5° | 150.4° |
| Angle Fe–N2–C3 | 124.5° | 115.7° |
| Angle Fe–N2–C2 | 117.1° | 125.8° |
| Angle N2–C2–C1 | 114.3° | 101.3° |
| Angle N1–C1–C2 | 108.4° | 116.2° |
| Bite-angle | 99.8° | 115.4° |
In the case of the photo-excited spin crossover of Fe(tpy)2 reported by Canton et al.,32 the excitation results in an elongation of all Fe–N bonds, axial and equatorial, causing a similar change in the angle between the C–C bonds connecting the central with the peripheral pyridine rings. However, the thermally induced transition of Fe-MEPE affects the coordination geometry in a different manner, causing the elongation of the equatorial Fe–N bonds while shortening the axial Fe–N bonds. The difference might arise from the connectivity of the coordination centres along the chain axis due to the rigid ligands.
Conclusions
Annealing neat Fe-MEPE above 150 °C causes a colour change from blue to green and a loss of the reversible electrochromic properties. An in situ XAFS study reveals that the cause for the colour change is a thermally induced distortion of the coordination geometry. Initially, the Fe(II) ions are surrounded by the tpy-units in a quasi-octahedral coordination environment in agreement with crystallographic data from analogous compounds.36 In the low temperature state, the axial Fe–N bonds are 1.94 Å, and the equatorial Fe–N bonds are 1.98 Å. Upon heating the geometry distorts to a pseudo-octahedral symmetry, where the axial Fe–N bonds are shortened and the equatorial bonds are elongated. The transition commences around 100 °C. Above 150 °C no further changes in the geometry of the complex are observed. In the high temperature state, the axial Fe–N bond length is 1.88 Å and the equatorial length is 2.01 Å. Within the accuracy of the experiment, the average Fe–N bond length is, however, the same in both states. The change in the coordination geometry affects the interaction between the central metal ion and the coordinating ligands, which is directly observable in the colour change. Finally, we have no evidence for a redox process at the Fe(II) centre. While the driving force behind the structural change is not clear, it might be associated with a temperature induced spin crossover. Due to the rigid nature of the ditopic ligand a relaxation upon cooling might be prohibited in the solid state, thus resulting in an irreversible process. Further research is needed to verify these points in detail.Acknowledgements
The authors wish to acknowledge the German Federal Ministry of Education and Research (BMBF) for financial support within the BMBF Joint Project ‘SmartWin MEPE’, Grant No. 13N11285.Notes and references
- S. K. Deb, Appl. Opt., Suppl., 1969, 3, 192 Search PubMed.
- G. Schwarz, Y. Bodenthin, T. Geue, J. Koetz and D. G. Kurth, Macromolecules, 2010, 43, 494 CrossRef CAS.
- Y. Yan and J. Huang, Coord. Chem. Rev., 2010, 254, 1072 CrossRef CAS PubMed.
- K. Sénéchal-David, J. P. Leonard, S. E. Plush and T. Gunnlaugsson, Org. Lett., 2006, 8, 2727 CrossRef PubMed.
- F. S. Han, M. Higuchi and D. G. Kurth, Adv. Mater., 2007, 19, 3928 CrossRef CAS PubMed.
- B. Arezki, G. Schwarz, Y. Bodenthin, D. Lutzenkirchen-Hecht, C. Markert, R. Wagner, R. Frahm, D. G. Kurth and U. Pietsch, ChemPhysChem, 2011, 12, 405 CrossRef CAS PubMed.
- Y. Bodenthin, U. Pietsch, H. Möhwald and D. G. Kurth, J. Am. Chem. Soc., 2005, 127, 3110 CrossRef CAS PubMed.
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553 CrossRef CAS PubMed.
- E. C. Constable and A. M. W. Cargill Thompson, J. Chem. Soc., Dalton Trans., 1995, 1615 Search PubMed.
- U. S. Schubert, A. Winter and G. R. Newkome, Terpyridine-based Materials, Wiley-VCH, 2012 Search PubMed.
- D. G. Kurth and M. Higuchi, Soft Matter, 2006, 2, 915 RSC.
- R. J. Mortimer, Annu. Rev. Mater. Res., 2011, 41, 241 CrossRef CAS.
- F. S. Han, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 2073 CrossRef CAS PubMed.
- P. Ceroni, A. Credi and M. Venturi, Electrochemistry of Functional Supramolecular Systems, Wiley, 2010 Search PubMed.
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge, 2007 Search PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- C. M. Amb, L. D. Aubrey and J. R. Reynolds, Chem. Mater., 2011, 23, 397 CrossRef CAS.
- M. Schütte, D. G. Kurth, M. R. Linford, H. Cölfen and H. Möhwald, Angew. Chem., 1998, 110, 3058 CrossRef.
- M. Schott, H. Lorrmann, W. Szczerba, M. Beck and D. G. Kurth, Sol. Energy Mater. Sol. Cells, 2014, 126, 68 CrossRef CAS PubMed.
- D. E. Sayers, E. A. Stern and F. W. Lythe, Phys. Rev. Lett., 1971, 27, 1204 CrossRef CAS.
- P. A. Lee and J. B. Pendry, Phys. Rev. B: Solid State, 1975, 11, 2795 CrossRef CAS.
- Y. Joly, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 125120 CrossRef.
- A. Filipponi, A. Di-Cicco and C. R. Natoli, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 15122 CrossRef CAS.
- M. Taillefumier, D. Cabaret, A.-M. Flank and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 195107 Search PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621 CrossRef CAS.
- J. J. Rehr and A. L. Ankudinov, Coord. Chem. Rev., 2005, 249, 131 CrossRef CAS PubMed.
- E. C. Constable and A. M. W. C. J. Thompson, J. Chem. Soc., Dalton Trans., 1992, 3467 RSC.
- J.-H. Li and M. Higuchi, J. Inorg. Organomet. Polym. Mater., 2010, 20, 10 CAS.
- H. Riesemeier, K. Ecker, B. R. Müller, W. Görner, M. Radtke and M. Krumrey, X-Ray Spectrom., 2005, 34, 160 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- S. E. Canton, X. Zhang, L. M. Lawson Daku, A. L. Smeigh, J. Zhang, Y. Liu, C.-J. Wallentin, K. Attenkofer, G. Jennings, C. A. Kurtz, D. Gosztola, K. Wärnmark, A. Hauser and V. Sundström, J. Phys. Chem. C, 2014, 118, 4536 CAS.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-Ray Absorption Fine Structure Spectroscopy, Cambridge University Press, 2010 Search PubMed.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503 RSC.
- T. G. Gopakumar, M. Bernien, H. Naggert, F. Matino, C. F. Hermanns, A. Bannwarth, S. Mühlenberend, A. Krüger, D. Krüger, F. Nickel, W. Walter, R. Berndt, W. Kuch and F. Tuczek, Chem. – Eur. J., 2013, 19, 15702 CrossRef CAS PubMed.
- E. C. Constable, J. Lewis, M. C. Liptrot and P. R. Raithby, Inorg. Chim. Acta, 1990, 178, 47 CrossRef CAS.
| This journal is © the Owner Societies 2014 |