 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium-43 chemical shift and electric field gradient tensor interplay: a sensitive probe of structure, polymorphism, and hydration†
Cory M.
Widdifield
*ab,
Igor
Moudrakovski‡
ac and
David L.
Bryce
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie Pvt., Ottawa, Ontario, Canada. E-mail: dbryce@uottawa.ca; Fax: +1 613 562 5170; Tel: +1 613 562 5800 ext. 2018
bUniversité de Lyon, Institut des Sciences Analytiques (CNRS, ENS-Lyon, UCB Lyon 1), Centre de RMN à Très Hauts Champs, 5 rue de la Doua, 69100, Villeurbanne, France. E-mail: cory.widdifield@ens-lyon.fr; Fax: +33 4 78 89 67 61; Tel: +33 4 26 23 38 88
cSteacie Institute for Molecular Sciences, NRC, 100 Sussex Drive, Ottawa, K1A 0R6, Canada
First published on 15th May 2014
Abstract
Calcium is the 5th most abundant element on earth, and is found in numerous biological tissues, proteins, materials, and increasingly in catalysts. However, due to a number of unfavourable nuclear properties, such as a low magnetogyric ratio, very low natural abundance, and its nuclear electric quadrupole moment, development of solid-state 43Ca NMR has been constrained relative to similar nuclides. In this study, 12 commonly-available calcium compounds are analyzed via43Ca solid-state NMR and the information which may be obtained by the measurement of both the 43Ca electric field gradient (EFG) and chemical shift tensors (the latter of which are extremely rare with only a handful of literature examples) is discussed. Combined with density functional theory (DFT) computations, this ‘tensor interplay’ is, for the first time for 43Ca, illustrated to be diagnostic in distinguishing polymorphs (e.g., calcium formate), and the degree of hydration (e.g., CaCl2·2H2O and calcium tartrate tetrahydrate). For Ca(OH)2, we outline the first example of 1H to 43Ca cross-polarization on a sample at natural abundance in 43Ca. Using prior knowledge of the relationship between the isotropic calcium chemical shift and the calcium quadrupolar coupling constant (CQ) with coordination number, we postulate the coordination number in a sample of calcium levulinate dihydrate, which does not have a known crystal structure. Natural samples of CaCO3 (aragonite polymorph) are used to show that the synthetic structure is present in nature. Gauge-including projector augmented-wave (GIPAW) DFT computations using accepted crystal structures for many of these systems generally result in calculated NMR tensor parameters which are in very good agreement with the experimental observations. This combination of 43Ca NMR measurements with GIPAW DFT ultimately allows us to establish clear correlations between various solid-state 43Ca NMR observables and selected structural parameters, such as unit cell dimensions and average Ca–O bond distances.
1. Introduction
Calcium is present in various chemical compounds and biological systems. For example, intracellular calcium-binding proteins such as calbindin, calretinin, and calmodulin are important for calcium ion transport and for cellular regulation processes.1–4 Likewise, calcium occurs in a variety of minerals and glasses, and is present at a level of about 400 ppm in sea water.5 Calcium-containing materials find applications as high temperature superconductors,6–9 in catalysis,10–15 in hydrogen storage16,17 and in solid-state proton conductors.18 In light of this wide array of interesting applications, which in theory could be probed using calcium solid-state nuclear magnetic resonance (SSNMR) experiments, development of the spectroscopy of the only NMR-active nuclide of calcium (i.e., 43Ca) remains somewhat lethargic19–21 due to complications which arise when attempting to perform such experiments. To date, the vast majority of systems studied by 43Ca SSNMR are inorganic materials with high calcium content by mass.19,20 This is not to say that 43Ca NMR experiments have never been employed to study more calcium-dilute (by mass) systems, as a number of years ago, solution 43Ca NMR experiments using isotopically enriched samples were used to probe the active sites in a variety of calcium-binding proteins.22–24Thus far, it has been beyond question that if one wishes to perform 43Ca SSNMR experiments, the most debilitating factor is the very low natural abundance of the 43Ca nuclide (0.135(2)%),25 which unfortunately is the only stable calcium nuclide that is also NMR active. As alluded to above in the studies on calcium-binding proteins, isotopic enrichment is an option which may be pursued, but it is rather expensive, as sample costs for 60% isotopic enrichment may potentially run in excess of $400 per milligram.19,26 As well, unlike familiar spin-1/2 nuclei such as 1H and 13C, 43Ca possesses a quadrupolar nucleus (I(43Ca) = 7/2). The nuclear electric quadrupole moment (Q) associated with any quadrupolar nucleus will couple with the electric field gradient (EFG) at the nuclear site. This resulting quadrupole interaction (QI) has been demonstrated to have significant importance in the characterization of a diverse array of systems.27–31 At the same time, as SSNMR experiments are typically (and most conveniently) performed on powdered samples, the orientation-dependence of the QI-perturbed Zeeman interaction in a powdered sample results in NMR signal broadening. This creates a situation where the feeble 43Ca NMR signal becomes distributed over a range of frequencies and hence could be buried in the noise.
While 43Ca enrichment offers the most significant degree of 43Ca NMR signal enhancement, if one does not wish (or is unable) to pursue enrichment, it is worth noting that the magic-angle spinning (MAS) technique has been used on several occasions to detect 43Ca SSNMR signals at natural abundance. Likewise, at natural abundance there is often no other reasonable option than performing experiments within a very high external magnetic field (B0) to reduce second-order line shape broadening due to the QI and to boost the nuclear spin polarization.32 It is not surprising that coupling high-field data acquisition with MAS is probably the optimal strategy when performing experiments on non-enriched samples. In addition, when performing experiments at natural abundance in 43Ca, large amounts (i.e., several hundreds of milligrams to even grams) of sample are often required, but it appears that reduced mass samples may be evaluated at natural abundance if one uses a microcoil approach.33 For systems where one wishes to balance cost, experimental NMR sensitivity, and sample amount, it is likely preferable to modestly enrich the material in 43Ca (ca. 5–10%). Regardless, the sensitivity boost afforded by MAS experiments comes at the expense of the removal (or at least in the apparent reduction) of a number of anisotropic interactions, such as magnetic shielding anisotropy and the direct dipole–dipole coupling between NMR-active spins. Although not of significance in the systems studied here, one may envision cases where small J-coupling values (e.g., J(43Ca,17O)) could potentially be measured using samples enriched in both 43Ca and 17O, but this is expected to be very challenging. According to recent reviews of the 43Ca SSNMR literature,19–21 only a handful of attempts have been made to quantify calcium chemical shift anisotropy (CSA), which is the experimentally-observable form of magnetic shielding anisotropy. As knowledge of a given nuclide's CSA is of substantial utility in characterizing the local bonding environment (in addition, there is considerable evidence that the isotropic calcium chemical shift value (δiso) gives insight into both the coordination number and the average Ca–O bond distance in a variety of simple calcium-containing materials),20,34–37 we have undertaken the present study with the aim of characterizing calcium CSA in several common materials, with and without 43Ca enrichment. To enhance the potential of observing calcium CSA, and also to increase the experimental sensitivity, many of the measurements relied upon the use of a very large B0 of 21.1 T.
Herein, we present 43Ca SSNMR results for 12 common calcium-containing compounds: Ca(OH)2, α-calcium formate, CaCl2·2H2O, (±)-calcium tartrate tetrahydrate, calcium acetate monohydrate, calcium levulinate dihydrate, CaSO4·2H2O, CaCrO4, CaTiO3, Ca(NO3)2, CaH2, and two naturally-occurring samples of CaCO3 (both aragonite polymorph). Depending on the system, experiments have been performed either at natural abundance in 43Ca, or with moderate enrichment (ca. 7% in 43Ca). We establish links between the calcium CSA, as well as the values of the Euler angles which relate the EFG and CSA principal axis systems (PASs), with changes in the local calcium environment (i.e., ‘tensor interplay’). In cases where high-quality structural data were available, we performed gauge-including projector augmented-wave (GIPAW) density functional theory (DFT) calculations to complement the experimental SSNMR findings. While demonstrated upon 43Ca enriched materials on a few occasions,38,39 we also briefly consider the substantial signal enhancement that cross-polarization (CP) from the 1H spin network to the 43Ca spins at their natural abundance may afford.
2. Experimental
A. Sample preparation
Samples of Ca(OH)2, calcium levulinate dihydrate (Ca(C5H7O3)2·2H2O), CaCrO4, CaTiO3, and CaH2 were obtained from Sigma-Aldrich, while α-calcium formate (99%, α-Ca(CHO2)2) and CaCl2·2H2O were obtained from Fluka. To prepare racemic calcium tartrate tetrahydrate ((±)-Ca(C4H4O6)·4H2O), a literature procedure was followed with minor modifications.40 For details of the procedure, see the ESI,† additional experimental. The received CaTiO3 demonstrated poor crystallinity, so before NMR measurements it was calcined at 1450 °C for 48 h. For the synthesis of 43Ca enriched materials, enriched 43CaCO3 (57% in 43Ca; Trace Isotopes, Toronto) was used. Due to the small amount of available 43Ca enriched material (50 mg), it was diluted with a fine powder of natural calcite such that the 43Ca enrichment was about 7%. The carbonate was then calcined at 1200 °C in a platinum crucible. The obtained 43Ca-enriched lime was exposed to D2O vapor, with its conversion to 43Ca(OD)2 controlled gravimetrically. About one quarter of the obtained 43Ca(OD)2 was neutralized with diluted acetic acid, and the solvent was allowed to slowly evaporate at room temperature, resulting in a powder of 43Ca-enriched calcium acetate monohydrate. Anhydrous 43Ca(NO3)2 was isolated by drying the solution obtained from neutralizing 43Ca(OD)2 with 1 M HNO3 at room temperature, followed by gradually increasing the temperature to 200 °C and keeping the powder at this temperature until the measured mass was constant. Enriched 43Ca33SO4·2H2O was made by simple precipitation from a solution of 43Ca(NO3)2 and K233SO4 (49(1)% in 33S). The two natural samples of CaCO3 (aragonite polymorph) were obtained by powdering a single river clam pearl of about 5 mm in diameter and a small branch of less than 1 cm of a common Ivory Bush coral. The pearl was obtained from a craft store in Ottawa, while the coral was collected during a field trip to Cuba.While most of these materials are stable and available, or may be prepared with high purity, additional 13C SSNMR experiments were performed, where possible, to further confirm sample purity (see ESI† for 13C SSNMR results). In most cases, powder X-ray diffraction (pXRD) measurements were performed (see ESI† for pXRD experimental details and diffractograms). Before performing the 43Ca SSNMR experiments, all samples were gently ground and tightly packed into 7 mm o.d. Bruker MAS ZrO2 rotors, 10 mm glass tubes, or 14 mm rotors from Revolution NMR LLC (Fort Collins, CO).
B. Solid-state 43Ca NMR
Data were primarily acquired at the National Ultrahigh-field NMR Facility for Solids in Ottawa using a standard bore Bruker AVANCE II spectrometer operating at B0 = 21.1 T (ν0(1H) = 900.08 MHz; ν0(43Ca) = 60.58 MHz). Additional experiments were performed at the University of Ottawa using a wide bore Bruker AVANCE III spectrometer operating at B0 = 9.4 T (ν0(1H) = 400.17 MHz; ν0(43Ca) = 26.93 MHz) or a wide bore Bruker AVANCE spectrometer operating at 11.75 T (ν0(43Ca) = 33.66 MHz). Experiments were also performed at the National Research Council Canada (Montreal road campus in Ottawa, ON) using a wide bore Bruker AVANCE III spectrometer (B0 = 9.4 T) or a standard bore Bruker AVANCE III spectrometer (B0 = 11.75 T). At 21.1 T, 43Ca NMR experiments generally used 7 mm probes: either a Bruker low-γ X MAS probe or a home-built HX static probe. In select cases where 1H decoupling was not needed, a 10 mm home-built solenoid probe was used. At B0 = 11.75 T, experiments used either a 6 mm Varian T3 HX MAS probe with low-γ accessory, a 10 mm Bruker X static probe, or a 10 mm home-built HX static probe. At 9.4 T, experiments largely used 7 mm probes: either a Bruker HX MAS probe or a Bruker HX static probe, although in selected cases, MAS experiments used a re-built 14 mm Chemagnetics probe equipped with a stator from Revolution NMR LLC. Unless specified otherwise, 43Ca SSNMR spectra were referenced to 1 M or 2 M CaCl2(aq) (δ(43Ca) = 0.0 ppm), and non-selective π/2 pulse length calibrations were performed using a saturated aqueous solution of CaCl2. We checked for consistency between the 1 M and 2 M CaCl2(aq) reference solutions, and find no significant difference between chemical shifts measured against one standard solution versus the other. For further details, see the ESI,† Table S1. Unless stated otherwise, for 43Ca SSNMR experiments, the non-selective pulses were scaled by a factor of 1/(I + 1/2) = 1/4 to ensure that they were central-transition selective.41The 43Ca SSNMR data were often obtained using the simple Bloch decay experiment under either MAS or static (i.e., stationary sample) conditions. Typical parameters varied depending upon whether the sample was enriched in 43Ca or not. As such, selected acquisition details are provided in many of the figure captions, and in further detail in Table S1 of the ESI.† For certain experiments on Ca(OH)2, CP experiments from 1H nuclei were successful. Other experiments relied upon signal enhancement techniques using double frequency sweeps (DFS)42 or frequency-shifted Gaussian rotor-assisted population transfer (FSG-RAPT).43 Where relevant, 1H decoupling was applied during data acquisition under static conditions, although the sharpening of spectral features was modest. Calcium-43 spin–lattice relaxation time (T1) measurements were performed at 21.1 T for all available 43Ca-enriched materials using the saturation-recovery method with an MAS frequency of 5 kHz. The 43Ca T1 values were also estimated for several natural abundance samples using 3 to 5 Bloch decay experiments. A mixed Gaussian/Lorentzian apodization function was applied prior to Fourier transformation of the time-domain data. Subsequent analytical line shape modeling was carried out using WSolids1,44 while in selected cases, numerical simulations were performed with SIMPSON.45 For numerical simulation details, see the ESI,† additional experimental section.
C. Quantum chemical computations
Computations of EFG and nuclear magnetic shielding tensors were performed with the GIPAW DFT method, as implemented in CASTEP (v. 4.1 or 5.5).46–49 Input files were typically generated with Materials Studio (v. 3.2.0.0), and used ultrasoft pseudopotentials to describe the core electrons.48,50 For all calculations, the generalized gradient approximation (GGA) was used, with the exchange–correlation (XC) functional developed by Perdew, Burke, and Ernzerhof (PBE).51,52 Crystal structures were taken from a variety of literature sources, as indicated in Table 1 and also in the ESI,† Tables S3 and S4. For certain systems, optimization of the hydrogen positions was carried out prior to NMR property computations (see ESI,† Table S6 for optimized structural parameters), but the unit cell dimensions were not optimized. Additional pertinent computational details (atomic coordinates, k-points, basis set energy cutoff, pseudopotential details) can be found in the ESI,† Tables S3 and S4. To determine the computed Euler angles, EFGShield (v. 4.0 with GUI) was used.53 For calculations involving unit cell manipulations, these were carried out by a simple isotropic scaling of the unit cell lengths, without any additional modifications or optimizations.| Compound | Lattice system | Space group | Ca sites | Ca symmetry | Ca c.n.a |
![[r with combining overline]](https://www.rsc.org/images/entities/i_char_0072_0305.gif) Ca–O
/Å
Ca–O
/Å |
Structure reference |
|---|---|---|---|---|---|---|---|
| a Calcium first coordination sphere coordination number. b Average calcium–oxygen first coordination sphere distance for systems where only oxygen atoms are present in this coordination sphere. The subsequent number in brackets is the standard deviation when a distribution of distances is present. c Arrived at in the present study by conducting 13C and 43Ca solid-state NMR experiments. | |||||||
| Ca(OH)2 | Trigonal |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 m1 |
1 | m |
6 | 2.369 | 54 |
| Calcium acetate·H2O | Triclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2 | 1, 1 | 6, 7 | 2.393(80), 2.404(73) | 55 |
| α-Calcium formate | Orthorhombic | Pcab | 1 | 1 | 7 | 2.418(85) | 56 |
| (±)-Calcium tartrate·4H2O | Triclinic |
P |
1 | 1 | 8 | 2.448(41) | 57 |
| Calcium levulinate·2H2O | — | — | 1c | — | 8c | — | — |
| CaCl2·2H2O | Orthorhombic | Pbcn | 1 | 2 | 6 | n/a | 58 |
| CaCO3 (aragonite) | Orthorhombic | Pmcn | 1 | m | 9 | 2.527(83) | 59 |
| CaSO4·2H2O | Monoclinic | C2/c | 1 | 2 | 8 | 2.454(91) | 60 |
| CaCrO4 | Tetragonal | I41/amd | 1 | ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) m2 m2 |
8 | 2.440(64) | 61 |
| CaTiO3 | Orthorhombic | Pbnm | 1 | m | 6 | 2.46(11) | 62 |
| Ca(NO3)2 | Cubic |
Pa |
1 | |
6 | 2.502 | 63 |
| CaH2 | Orthorhombic | Pnma | 1 | m | 9 | n/a | 64 |
3. Results and discussion
Short discussions of the crystal structures and the local calcium environments for each compound studied are provided below (for an overview of selected structural parameters, see Table 1). To establish if clear links exist between these structural elements and measurable calcium SSNMR observables, we initially discuss systems with well-known structures. As the discussion progresses, further points will be made regarding the calcium environment in cases where the local and long-range structures are not known with such confidence. Experimentally measured 43Ca SSNMR parameters are summarized in Table 2. Where crystal structure data exist, the local Ca2+ environments are displayed in Fig. 1.| Compound | |CQ(43Ca)|b/MHz | η Q | δ iso /ppm | Ω/ppm | κ | α, β, γ/° | Notes |
|---|---|---|---|---|---|---|---|
| a Measurement errors are in parentheses with parameter definitions as follows: |V33| ≥ |V22| ≥ |V11|; CQ = eQV33/h; ηQ = (V11 − V22)/V33; δ11 ≥ δ22 ≥ δ33; δiso = (δ11 + δ22 + δ33)/3; Ω = δ11 − δ33; κ = 3(δ22 − δiso)/Ω. The Euler angles α, β, and γ define the relative orientation between the EFG tensor PAS and the CS tensor PAS. For further discussion, see ref. 69. b While CQ may take any real value, |CQ| is typically measured experimentally using solid-state NMR. c Calcium chemical shifts are relative to ca. 1 M or 2 M CaCl2 in H2O (δiso(43Ca) = 0.0 ppm). d Parameter does not influence NMR spectrum due to symmetry of the interaction tensors for this system. e EFG tensor parameters and isotropic chemical shifts were consistent between both natural samples. Euler angles included in static line shape models are from the computational results of ref. 67. f Value corrected for consistency with current chemical shift scale. | |||||||
| Ca(OH)2 | 2.56(0.03) | <0.05 | 70.4(0.6) | 36.5(2.0) | <−0.8 | —d, 90(4), 0(4) | This study |
| α-Calcium formate | 1.25(0.08) | 0.9(0.1) | 9.8(0.5) | 26(4) | 0.0(0.3) | 85(10), 82(10), 315(10) | This study, T1 ≈ 3.9 s |
| (±)-Calcium tartrate·4H2O | <1.2 | 0.6(0.1) | −0.5(0.5) | 23(3) | 0.1(0.2) | 175(10), 75(10), 112(10) | This study |
| Calcium acetate·H2O | 1.64(0.03) | 0.45(0.03) | −7.7(0.3) | 27(2) | −0.4(0.2) | 295(10), 86(5), 341(5) | This study, site 1, T1 = 8.4(0.4) s |
| 1.37(0.03) | 0.1(0.1) | 13.8(0.3) | 20(2) | −0.15(0.20) | 90(10), 90(10), 175(10) | This study, site 2, T1 = 5.5(0.3) s | |
| Calcium levulinate·2H2O | 1.3(0.1) | 0.55(0.2) | −4.5(1.0) | — | — | — | This study |
| Calcium p-aminosalicylate | 1.63(0.03) | 0.35(0.05) | −3.2(0.5) | 30(5) | −1.0(0.1) | 0, 0, 0 | Burgess et al.,65 site 1 |
| 2.60(0.05) | 0.60(0.03) | −5.5(0.5) | 65(5) | 0.6(0.1) | 210(20), 20(10), 110(10) | Burgess et al.,65 site 2 | |
| CaCl2 | 0.95(0.20) | 0.7(0.2) | 54.8(0.5) | 31(5) | −0.5(0.3) | 90(20), 90(5), 0(5) | Widdifield/Bryce66 |
| CaCl2·2H2O | 2.62(0.06) | 0.22(0.05) | 51.2(0.4) | 19(5) | 0.2(0.3) | 180, 82(3), 180 | This study |
| CaCO3, calcite | 1.39(0.10) | <0.05 | 19.3(0.4) | 8(2) | 0.9(0.1) | 0d, 0(5), 0d | Bryce67/Chan68 |
| CaCO3, aragonite (coral) | 0.25(0.05) | 0.4(0.1) | −27.0(0.4) | 49(2) | 0.39(0.05) | 0, 15, 90 | This studye, T1 ≈ 1.0 s (coral) |
| CaCO3, aragonite (pearl) | 48(2) | 0.44(0.05) | |||||
| CaCO3, aragonite | 0.256–0.263 | 0.35–0.5 | −27.2(0.1) | 49.6(0.5) | 0.44(0.01) | 0, 15, 90 | Bryce67/Chan68 |
| CaCO3, vaterite | 3.7(0.5) | 0.55(0.10) | 16(1)f | 70(20) | 0.0(0.5) | 0(20), 30(20), 120(20) | Bryce et al.67 |
| CaSO4·2H2O | 1.28(0.03) | 0.60(0.03) | −19.1(0.3) | 43(2) | −0.31(0.07) | 90(25), 0(10), 0(25) | This study, T1 = 64.4(3.3) s |
| CaCrO4 | 0.96(0.05) | <0.1 | −65.4(0.3) | 87(3) | −1 | —d, 45, 0 | This study, T1 ≈ 0.4 s |
| CaTiO3 | 2.09(0.05) | 0.75(0.05) | 21.0(0.3) | 53(3) | 0.8(0.1) | 90(20), 8(4), 0(20) | This study, T1 ≈ 2.7 s |
| Ca(NO3)2 | 1.57(0.01) | 0.0 | −50.9(0.2) | 11.4(1.0) | 1 | —d, 0(7), —d | This study, T1 = 6.3(0.4) s |
| CaH2 | 1.2(0.1) | 0.7(0.2) | 95.8(1.0) | 29(2) | 0.45(0.1) | 90, 11(5), 0 | This study |
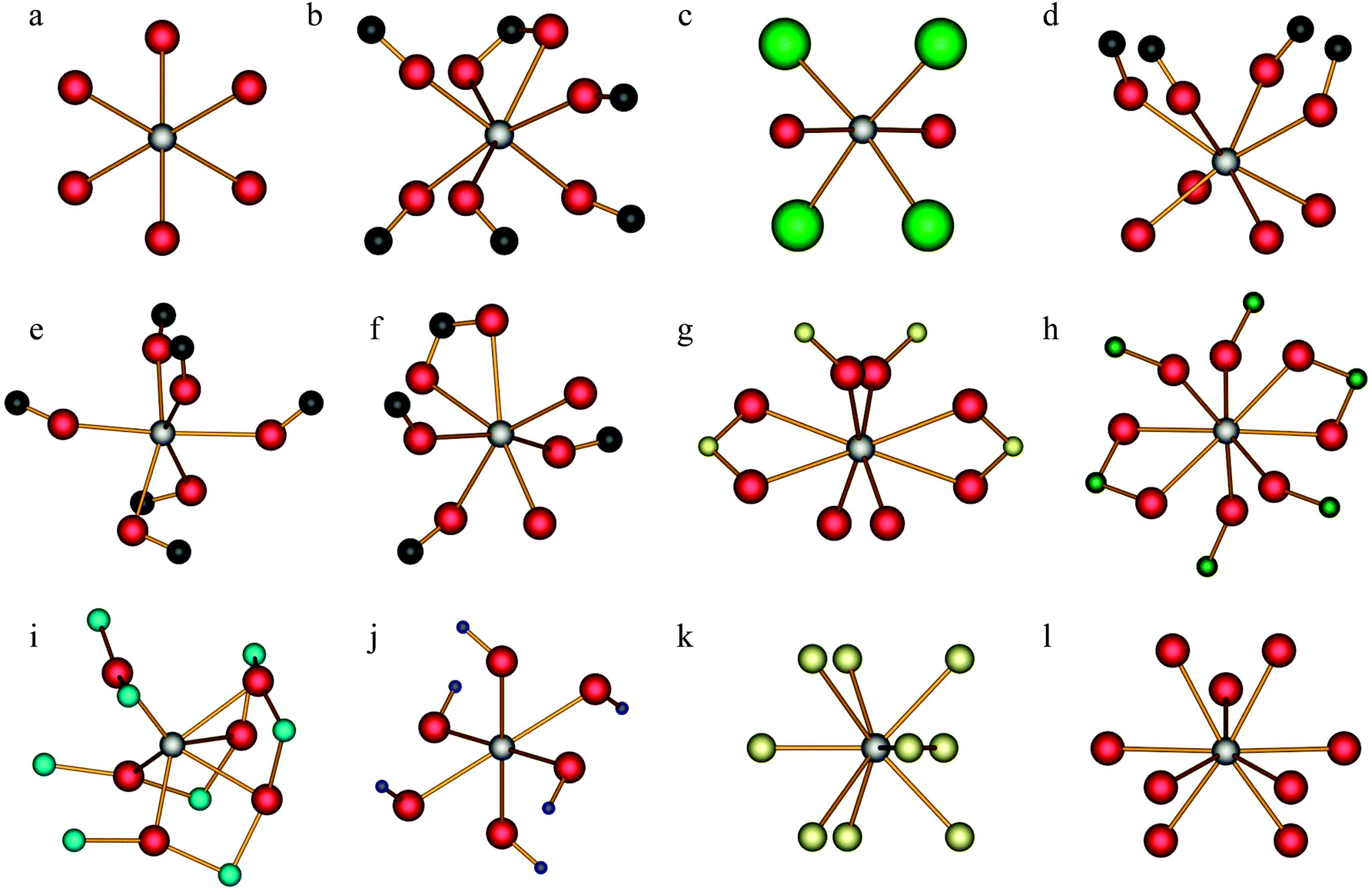 | ||
| Fig. 1 Local calcium environments for systems with known crystal structures. Ca(OH)2 (a); α-calcium formate (b); CaCl2·2H2O (c); (±)-calcium tartrate tetrahydrate (d); calcium acetate monohydrate, site 1 (e); calcium acetate monohydrate, site 2 (f); CaSO4·2H2O (g); CaCrO4 (h); CaTiO3 (i); Ca(NO3)2 (j); CaH2 (k); aragonite (l). Hydrogen atoms have generally been omitted (except CaH2) and only the most local structure about the calcium is shown. The atomic colour scheme is as follows: Ca (silver); O (red); C (black); Cl (light green); S (yellow); Cr (dark green); Ti (aqua); N (blue); H (gold). | ||
A. Calcium hydroxide, Ca(OH)2
The unit cell and heavy atom positions in Ca(OH)2 were established long ago by both X-ray and neutron diffraction techniques,70,71 while specification of the hydrogen atomic positions required additional studies.54,72–74 The stable ambient condition crystal structure for Ca(OH)2 is consistently observed to belong to the trigonal Pm1 space group, with the Ca atoms at m sites. Due to a C3 symmetry element passing through the calcium lattice positions (Fig. 1a), the 43Ca EFG tensor must possess axial symmetry (i.e., the asymmetry parameter of the EFG tensor, ηQ, must equal 0),75 which is interestingly in contrast with several earlier 43Ca NMR measurements that report a small, albeit consistently nonzero ηQ(43Ca) value.33,35,76,77 Oxygen-17 SSNMR results have also been presented,78,79 and while several measurements of the 43Ca EFG tensor in Ca(OH)2 have been conducted, we are not aware of any attempts to completely determine the calcium CSA tensor in this material, which also includes the measurement of the Euler angles relating the CSA and EFG tensor PASs.
A 43Ca MAS NMR experiment at B0 = 21.1 T facilitated the measurement of the relevant EFG tensor parameters, as well as the calcium δiso value (see ESI,† Fig. S1), and largely yielded values consistent with the majority of the literature reports: (CQ = 2.56(0.03) MHz; ηQ < 0.05; δiso = 70.4(0.6) ppm; NMR parameter definitions can be found in the footnotes of Table 2).33,35,76,77 For an illustration of Euler angle definitions, see Fig. S2 in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 69 Static 43Ca SSNMR experiments were also performed at multiple applied magnetic fields (Fig. 2 and Fig. S3, ESI†).
69 Static 43Ca SSNMR experiments were also performed at multiple applied magnetic fields (Fig. 2 and Fig. S3, ESI†).
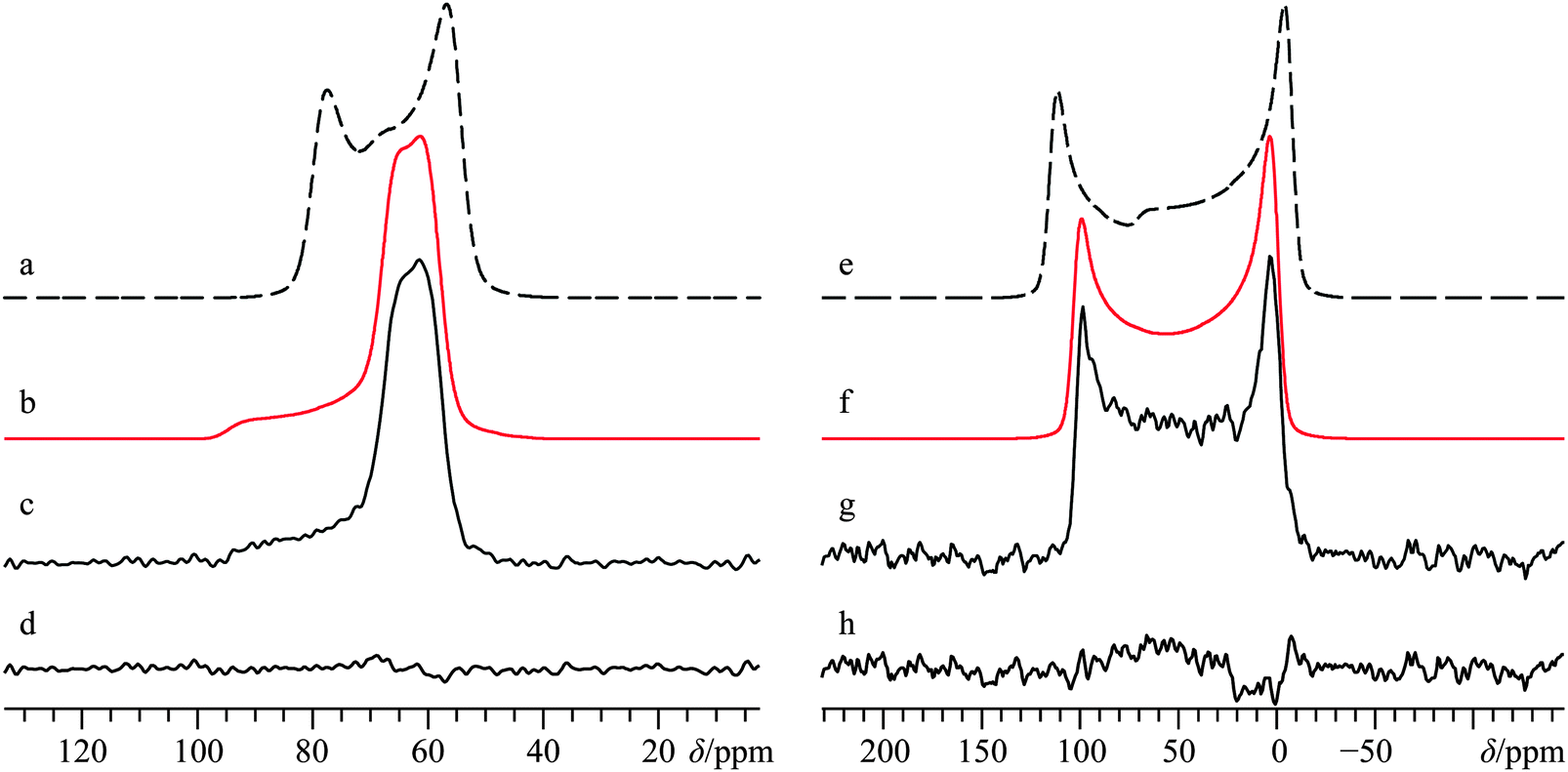 | ||
| Fig. 2 Analytical line shape simulations (a, b, e, f) and experimental (c, g) 43Ca{1H} CP/static SSNMR spectra of Ca(OH)2, acquired at B0 = 21.1 T (c) and B0 = 9.4 T (g). Traces (b) and (f) represent the best-fit simulations of the experimental spectra, while (a) and (e) ignore calcium CSA by setting Ω = 0 ppm. The difference spectrum between (b) and (c) is provided in (d), while the difference spectrum between (f) and (g) is provided in (h). The experimental spectrum in (c) results from the collection of 28200 transients with a recycle delay of 2.0 s (experiment time = 15.7 h), while (g) results from the collection of 273600 transients with a recycle delay of 0.7 s (experiment time = 53.2 h). | ||
Direct detection experiments at B0 = 11.75 T, even when using a single channel 10 mm solenoid probe and 4 days of acquisition time, resulted in a 43Ca SSNMR spectrum which did not have a satisfactory signal-to-noise (S/N) ratio (Fig. S3, ESI†), and which was difficult to quantitatively model. Due to the challenging nature of obtaining high quality 43Ca SSNMR spectra under these conditions (i.e., direct polarization at standard applied fields and at natural abundance in 43Ca), we carried out CP experiments using 1H nuclei as the polarization source. The CP technique has been applied several times before using samples isotopically enriched in 43Ca.33,38,39 Fortunately, the 1H spin–lattice relaxation time (i.e., T1) in Ca(OH)2 is rather short, and the CP experiment clearly afforded phenomenal gains in sensitivity relative to direct excitation of 43Ca (contrast Fig. 2g with Fig. S3, ESI†). The Hartmann–Hahn matching condition for half-integer quadrupolar nuclei described earlier (i.e., the nutation frequency of 1H = (I + 1/2) = 4 times the nutation frequency of the quadrupolar nucleus)41 was confirmed to be optimal for Ca(OH)2 at 21.1 T. Other matching conditions near the optimal value gave rise to significant polarization transfer (Fig. S4, ESI†), but were not as broad-banded (i.e., non-uniform enhancement across the powder pattern). We also performed optimizations of the CP contact time, finding that approximately 20 ms was optimal in this case (Fig. 3), which is roughly in accord with prior cases when CP has been used on 43Ca-enriched samples.38 The inclusion of phase-modulated 1H decoupling during the acquisition period (Fig. S5, ESI†) was explored, and while a slight variation in the observed signal was noted, the overall line shape distortion by 1H-43Ca through-space dipolar coupling is clearly minor, as postulated earlier,20 but never experimentally confirmed beforehand for Ca(OH)2.
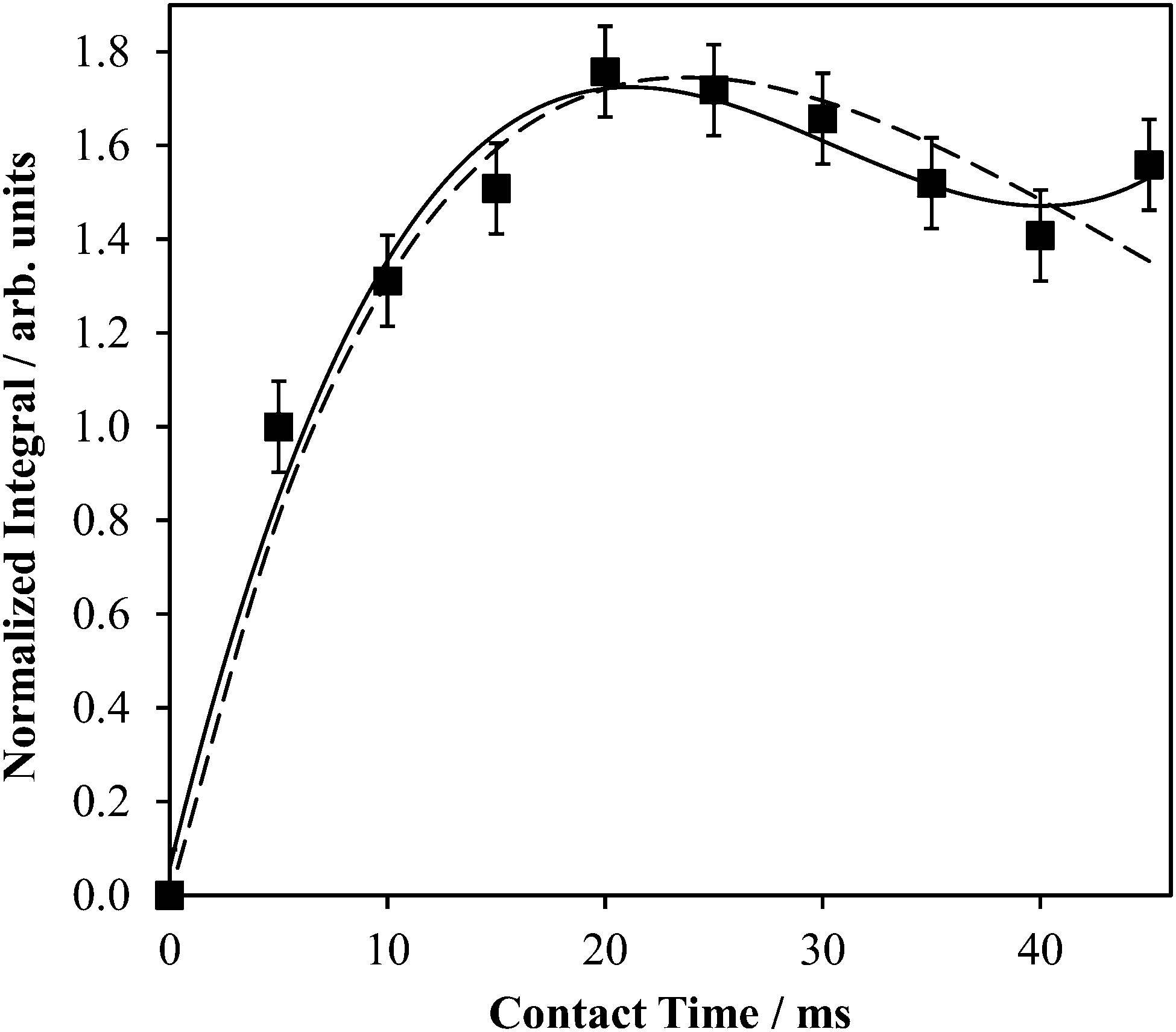 | ||
| Fig. 3 Experimental 1H → 43Ca CP build-up curve for powdered Ca(OH)2 at B0 = 9.4 T. The data were fit to a polynomial expression (solid line): S = 7.977 × 10−5(τCP)3 − 7.278 × 10−3(τCP)2 + 0.2001(τCP); R2 = 0.978, where S = the signal intensity (arbitrary units) and τCP is the CP contact time (ms), and a damped sinusoidal expression (dashed line): S = 80.98(sin(2.461 × 10−3τCP))exp(−0.04198τCP). | ||
The CP NMR spectrum in Fig. 2g was acquired at B0 = 9.4 T using a 7 mm solenoid coil (contrast with the 43Ca NMR spectrum acquired at B0 = 11.75 T using a 10 mm solenoid coil, Fig. S3, ESI†). To the best of our knowledge, the spectrum in Fig. 2g encompasses a couple of firsts: (i) it is the first CP 43Ca SSNMR spectrum acquired under static conditions and at natural abundance; and (ii) it allows for the first measurement of the full calcium chemical shift tensor at a standard applied magnetic field (i.e., B0 < 12 T). An additional CP/static 43Ca{1H} SSNMR spectrum was acquired at B0 = 21.1 T. Except for the rather poor quality direct-detect spectrum acquired at 11.75 T, by modeling all the above-mentioned spectra simultaneously, we precisely measured the calcium CSA in Ca(OH)2, including the Euler angle values (Ω = 36.5(2.0) ppm; κ < −0.8; β = 90(4)°; γ = 0(4)°, Table 2). It is rather interesting to see the 43Ca NMR spectrum of Ca(OH)2 acquired at 9.4 T is dominated by second-order quadrupolar broadening, while the 43Ca NMR spectrum of the same material acquired at 21.1 T is significantly affected by CSA. This dramatic example of the field dependencies of quadrupolar broadening and CSA nicely underscores the importance of, when possible, acquiring SSNMR data of half-integer quadrupolar nuclei in two very distinct applied fields. GIPAW DFT calculations of calcium-43 EFG and magnetic shielding tensors using the crystal structure of Ca(OH)2 (Table 3) established by Desgranges et al.54 produced values that are in very good agreement with those experimentally measured (Table 2), including the axial tensor symmetry for both the EFG and shielding tensors. As observed on several occasions for quadrupolar nuclei in the past, the eigenvectors that define the EFG and CS tensor PASs are seen to lie parallel to the unit cell axes (see ESI,† Fig. S6). It is also calculated that the direction of least magnetic shielding (i.e., σ11) is collinear with the direction of the largest EFG (i.e., V33).
| Compound | |CQ(43Ca)|/MHz | η Q | δ iso /ppm | Ω/ppm | κ | α, β, γ/° | Notes |
|---|---|---|---|---|---|---|---|
| a For a more complete disclosure of the computed EFG and magnetic shielding tensor eigenvalues, see Table S5 (ESI). b Generated using the mapping from magnetic shielding to chemical shifts denoted by Moudrakovski et al. in ref. 77. c The values here correspond to the polymorph provided in ref. 55. For details on the other polymorph, see the ESI, Table S5. d Calculations are similar to what appeared as supporting information in ref. 66, but differ as the present NMR property calculations employ a more modern calcium pseudopotential, which has been shown to be more accurate relative to the earlier pseudopotential.37 e Calculations employed an older calcium pseudopotential. f Parameter has vanishingly small effect on experimental NMR spectrum due to symmetry. | |||||||
| Ca(OH)2 | 2.28 | 0.000 | 71.9 | 38.5 | −0.998 | 30.1f, 90.0, 0.1 | This study |
| α-Calcium formate | 1.41 | 0.899 | 14.1 | 29.6 | −0.190 | 97.7, 74.9, 251.1 | This study |
| β-Calcium formate | 2.86 | 0.342 | −1.6 | 40.8 | 0.094 | 270.0, 65.3, 0.0 | This study |
| Calcium tartrate·3H2O | 1.73 | 0.499 | 25.5 | 36.5 | −0.072 | 174.9, 5.1, 290.9 | This study |
| (+)-Calcium tartrate·4H2O | 1.14 | 0.628 | 5.4 | 30.0 | −0.205 | 121.4, 75.2, 298.7 | This study |
| (±)-Calcium tartrate·4H2O | 0.99 | 0.713 | 4.4 | 17.0 | 0.497 | 245.8, 67.9, 62.6 | This study |
| Calcium acetate·H2O (1984)c | 1.92 | 0.366 | −7.0 | 38.7 | −0.666 | 280.1, 87.2, 345.2 | This study, site 1 |
| 1.35 | 0.080 | 17.7 | 22.7 | −0.367 | 99.5, 88.9, 184.4 | This study, site 2 | |
| CaCl2 | 0.98 | 0.541 | 67.7 | 52.7 | −0.24 | 90.0, 89.4, 0.0 | Widdifield/Bryce66 |
| CaCl2·2H2Od | 3.34 | 0.187 | 63.6 | 25.5 | 0.296 | 180.0, 84.6, 180.0 | This study |
| CaCl2·6H2O | 0.32 | 0.000 | −27.3 | 9.2 | −0.999 | 29.8, 90.0, 178.7 | Widdifield/Bryce66 |
| CaCO3, calcitee | 1.47 | 0.00 | 22.7 | 10.8 | 1.00 | 0.0f, 0.0, 0.0f | Bryce et al.67 |
| CaCO3, aragonitee | 0.32 | 0.58 | −27.1 | 52.2 | 0.49 | 0.0, 15.0, 90.0 | Bryce et al.67 |
| CaSO4·2H2O | 1.45 | 0.999 | −19.8 | 57.6 | −0.441 | 270.0, 89.7, 180.0 | This study |
| CaCrO4 | 1.35 | 0.000 | −62.7 | 85.8 | −1.000 | 90.0f, 90.0, 180.0 | This study |
| CaTiO3 | 1.97 | 0.683 | 19.3 | 69.6 | 0.420 | 90.0, 3.4, 0.0 | This study |
| Ca(NO3)2 (original) | 3.48 | 0.000 | −49.3 | 39.4 | 1.000 | 2.9f, 0.0, 90.1f | This study |
| Ca(NO3)2 (N, O optimized) | 2.72 | 0.000 | −61.0 | 27.6 | 1.000 | 1.8f, 0.0, 180.0f | This study |
| CaH2 | 1.14 | 0.849 | 120.1 | 35.3 | 0.449 | 270.0, 13.6, 180.0 | This study |
B. α-Calcium formate, α-Ca(CHO2)2
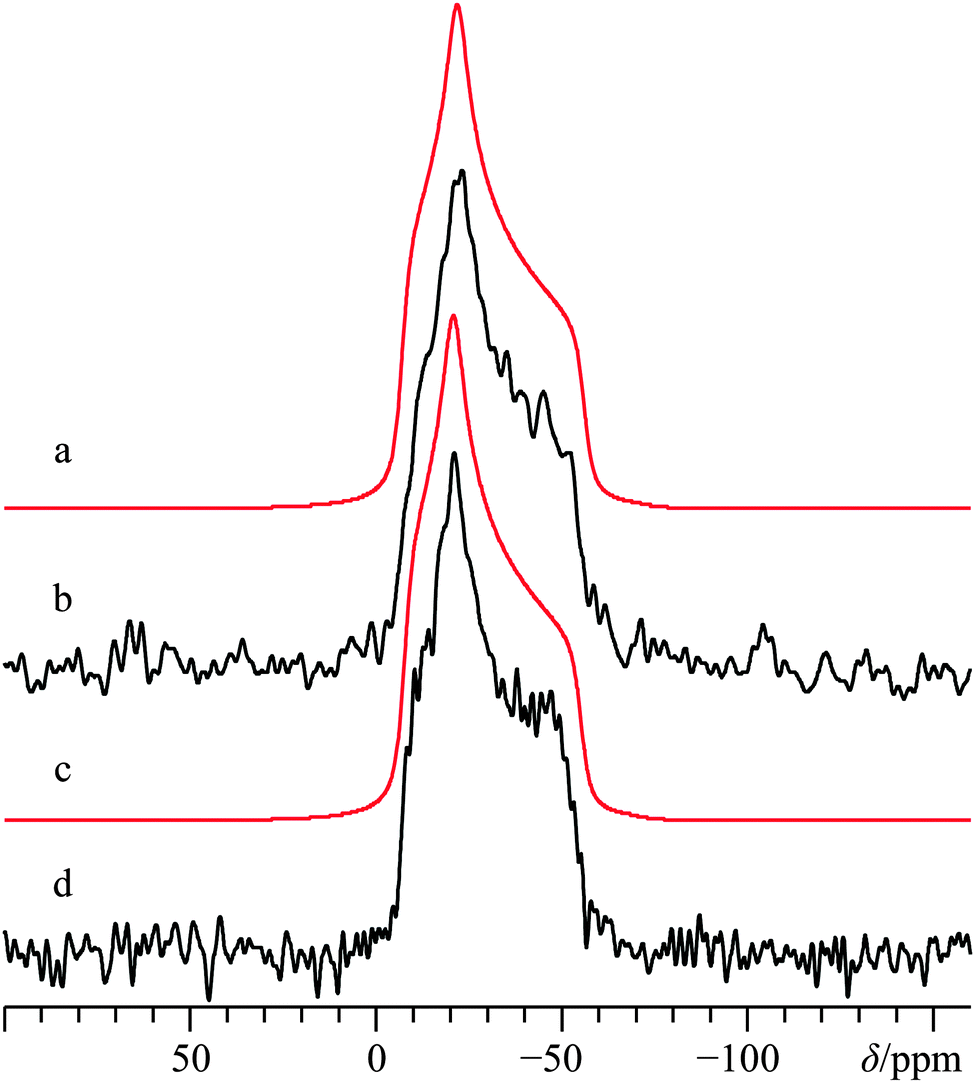
The crystal structure of α-calcium formate has been determined using both X-ray80 and neutron56 diffraction measurements: it belongs to the orthorhombic Pcab space group. In contrast with Ca(OH)2, the local Ca2+ coordination sphere is populated by seven oxygen atoms (Fig. 1b), and there is a distribution of Ca–O bond lengths. The α polymorph has been studied several times using solid-state 1H,812H,8213C,83–87 and 43Ca SSNMR.36 While the 43Ca EFG tensor parameters and the calcium δiso value are well-documented, we are not aware of measurements of the calcium CSA for this compound. Carbon-13 SSNMR experiments and 43Ca MAS NMR experiments reproduced prior literature values and confirmed the sample purity (13C CP/MAS and 43Ca MAS NMR spectra can be found in the ESI,† Fig. S7 and S8, respectively). While 1H → 43Ca CP experiments were attempted on this material, the relatively long proton T1 value (>10 s) made these types of experiments only moderately useful at high field. Static direct detection and CP experiments were attempted at standard applied fields of 9.4 and 11.75 T, but did not provide satisfactory results due to: (i) poor S/N even after several days of signal averaging for direct detection, or (ii) difficulties in optimizing the CP matching conditions due to the relatively long proton T1. The limited success of CP from 1H nuclei was also noted in an earlier study by Smith and co-workers when studying labeled hydroxyapatite.39 It appears that the amazing CP enhancement obtained for Ca(OH)2 may not be a general phenomenon for 43Ca under static conditions, unless other sample preparation precautions are undertaken (such as doping with paramagnetic species, as documented in 29Si SSNMR).88 As the T1(1H) values of the remaining samples in this study more closely resembled that of α-calcium formate rather than Ca(OH)2, additional CP experiments were not exhaustively pursued.Due to the precision with which the EFG tensor parameters have been determined previously, and confirmed here, the calcium CSA parameters may be extracted with high confidence from a fit of the static 43Ca SSNMR data acquired at B0 = 21.1 T. The significant impact of the CSA on the spectral line shape is underscored by including in Fig. 4 a fit which does not consider calcium CSA (Fig. 4c), and which is clearly inconsistent with the experimental spectrum.
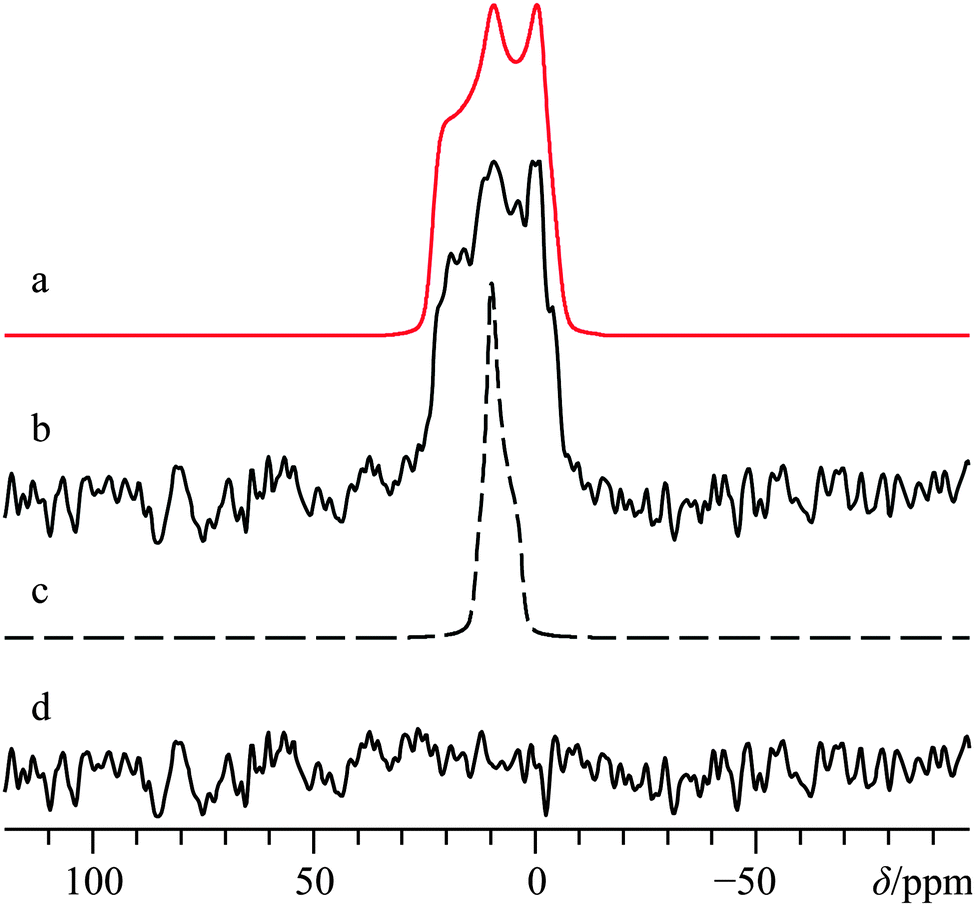 | ||
| Fig. 4 Analytical line shape simulations (a, c) and experimental (b) 43Ca{1H} static SSNMR spectrum of α-Ca(CHO2)2, acquired at B0 = 21.1 T. Trace (a) represents the best-fit spectrum, while (c) ignores calcium CSA by setting Ω = 0 ppm. The difference spectrum between (b) and (a) is provided in (d). The experimental spectrum in (b) results from the collection of 15952 transients with a recycle delay of 10.0 s (experiment time = 44.3 h). | ||
We were also able to experimentally measure the set of calcium Euler angles for this compound (Table 2), and found them to be in reasonably good agreement with the results of GIPAW DFT computations (Table 3), although there is a significant difference between experimental and computed γ values. Relative to Ca(OH)2, it is seen that the experimentally measured 43Ca EFG and chemical shift tensor magnitudes are reduced for α-Ca(CHO2)2, in accord with the GIPAW DFT computations. We note that prior GIPAW DFT calculations exist for α-calcium formate using a slightly different structure,67 and although there are subtle differences in the resulting calculated parameters, they are generally in good agreement. It also appears that GIPAW DFT computations of the 43Ca EFG tensor parameters for α-calcium formate are in better agreement with the experimental data than when compared to cluster-based methods.89
To comment upon the potential applicability of 43Ca NMR experiments in distinguishing polymorphic forms, we also performed GIPAW DFT computations on the accepted structures of the β polymorph of calcium formate.90 As we will highlight in this manuscript on several occasions, software using GIPAW DFT is able to accurately calculate both the calcium-43 EFG and magnetic shielding tensors for a variety of calcium-containing materials. The calculated CQ(43Ca) value for β-calcium formate is 2.86 MHz (Table 3), which is more than double the calculated (and experimentally observed) value for the α polymorph (these values were observed and computed to be 1.25(0.08) and 1.41 MHz). In addition, the calcium chemical shift value for the β polymorph is predicted to be roughly 15 ppm lower relative to the α polymorph. Both of these differences should be easily observable at natural abundance and add further support to the potential utility that 43Ca SSNMR experiments may have in distinguishing polymorphs.
C. Calcium chloride dihydrate, CaCl2·2H2O, and pseudopolymorphs thereof
As part of an earlier study,66 we focused on the utility of 35/37Cl SSNMR experiments in distinguishing anhydrous and hydrated calcium chloride pseudopolymorphs. It was found that 35/37Cl SSNMR parameters were able to differentiate between various stable calcium chloride hydrates, and it was predicted that if 43Ca SSNMR measurements could be performed on CaCl2·2H2O, they would produce calcium NMR tensor parameters that were unique compared to both CaCl2 and CaCl2·6H2O. To validate the computational prediction, we carried out 43Ca SSNMR experiments using the same sample employed in a prior chlorine NMR study of CaCl2·2H2O.91The crystal structure of CaCl2·2H2O determined using XRD methods58 belongs to the orthorhombic Pbcn space group, with the Ca2+ ions located on C2 sites (Fig. 1c). Prior 2H SSNMR experiments on various deuterated hydrates of CaCl2 have explored the various modes of water dynamics which occur in this class of systems.92 Presently, MAS experiments at B0 = 21.1 T clearly identify a single calcium environment whose 43Ca NMR line shape is broadened by the second-order QI (Fig. 5b). A satisfactory fit to the experimental signal is arrived at using CQ(43Ca) = 2.62(0.06) MHz, ηQ = 0.22(0.05), and δiso = 51.2(0.4) ppm. Subsequent static NMR experiments on this sample highlight a relatively modest calcium CSA (Ω = 19(5) ppm, Fig. 5f).
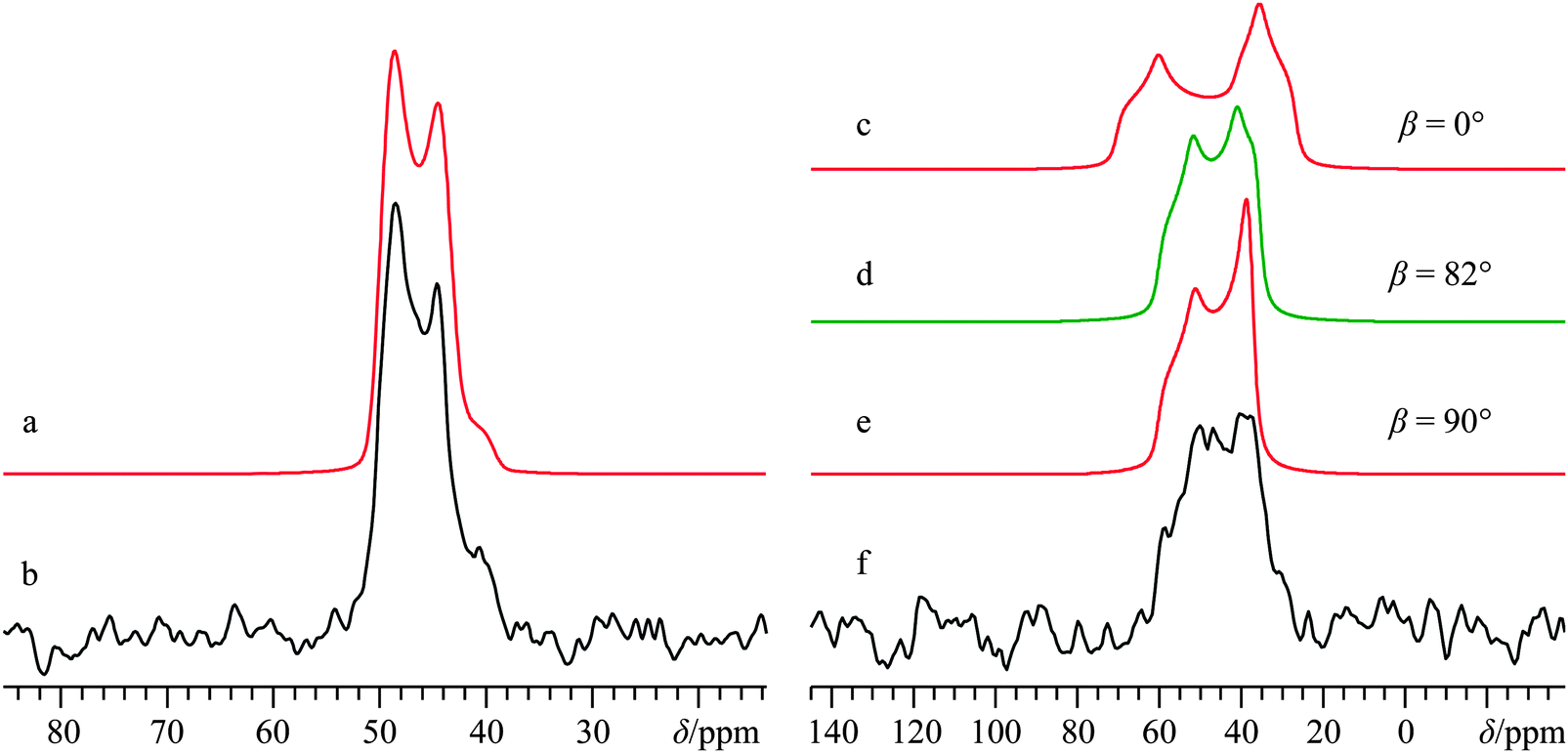 | ||
| Fig. 5 Analytical line shape simulations (a, c–e), experimental 43Ca SSNMR spectrum under MAS (νMAS = 4.5 kHz, b), and 43Ca{1H} SSNMR spectrum under static conditions (f) of CaCl2·2H2O, acquired at B0 = 21.1 T. The experimental spectrum in (b) results from the collection of 7920 transients with a recycle delay of 10.0 s (experiment time = 22.0 h), while the spectrum in (f) resulted from collecting 7220 transients with a recycle delay of 10.0 s (experiment time = 20.1 h). The pronounced sensitivity of the simulated static line shapes with respect to variation in β is highlighted in (c–e), with selected β values indicated to the right of the simulated line shapes. | ||
Based on the C2 site symmetry at the Ca, the parameter space for the α and γ Euler angles can be restricted significantly. This is supported by GIPAW DFT computations, and we establish that the largest components of the EFG and calcium shift tensors are non-coincident (i.e., β ≠ 0°). Importantly, we note the critical role that the β value plays in the observed 43Ca NMR signal for this system, as outlined in Fig. 5. We also note that high field NMR experiments were essential to acquire spectra with a reasonable S/N ratio for CaCl2·2H2O, as 43Ca NMR experiments at B0 = 11.75 T using a 10 mm coil gave barely any noticeable signal, even after nearly three days of signal averaging (Fig. S9, ESI†).
In full accord with the GIPAW DFT calculations presented in the earlier study,66 the observed 43Ca SSNMR spectra and corresponding EFG and CS tensor parameters for CaCl2·2H2O are clearly distinct from the anhydrous and hexahydrate forms of CaCl2. It is seen that the experimental |CQ(43Ca)| value for CaCl2·2H2O is somewhat less than the calculated value (Table 3), and postulate that the known fast (10 to 100 ps) motions of the water molecules92 are likely partially averaging the EFG at the calcium nuclei. Nevertheless, the present finding further highlights the potential utility of 43Ca SSNMR experiments when distinguishing pseudopolymorphs. We also note that GIPAW DFT calculations of the relevant NMR tensor parameters for the known (but unstable) polymorphs of CaCl2·4H2O (i.e., the α, β, and γ polymorphs, as in ref. 66) predict that 43Ca SSNMR experiments would very likely be able to distinguish these systems (i) from the three forms of CaCl2 described directly above, and (ii) from one another.
D. Racemic calcium tartrate tetrahydrate, (±)-Ca(C4H4O6)·4H2O
As commonly observed in the wine industry, calcium tartrate is naturally present as an enantiomerically pure tetrahydrate (i.e., (+)-Ca(L-C4H4O6)·4H2O),93 although other hydrated forms can be prepared.94 A number of prior XRD measurements95–97 have been carried out on the enantio-pure form, although recently a crystal structure for the racemic form of calcium tetrahydrate has been determined via Rietveld refinements.57 Due to the lack of point symmetry at the calcium (Fig. 1d), the corresponding EFG and CS tensor parameters can take any value, making data fitting more complicated in comparison to the three systems discussed above. Due to the reasonably high degree of correlation between the experimental and computed calcium NMR tensor parameters for the systems studied herein (vide infra), we relied on a complementary approach which uses both experimental NMR measurements and GIPAW DFT computed values to determine the calcium NMR tensor parameters.To the best of our knowledge, (±)-Ca(C4H4O6)·4H2O has never been studied via43Ca SSNMR. As such, we employed 13C SSNMR and pXRD measurements to verify sample purity (see Fig. S10 and S11 of the ESI†). No presence of the starting material (DL-tartaric acid) was observed, and small changes in the 13C chemical shifts of the tartrate anion were observed, which are consistent with product formation. The 43Ca MAS SSNMR signal recorded at B0 = 21.1 T (Fig. 6b) is rather narrow and quite shielded (δiso = −0.5(0.5) ppm).
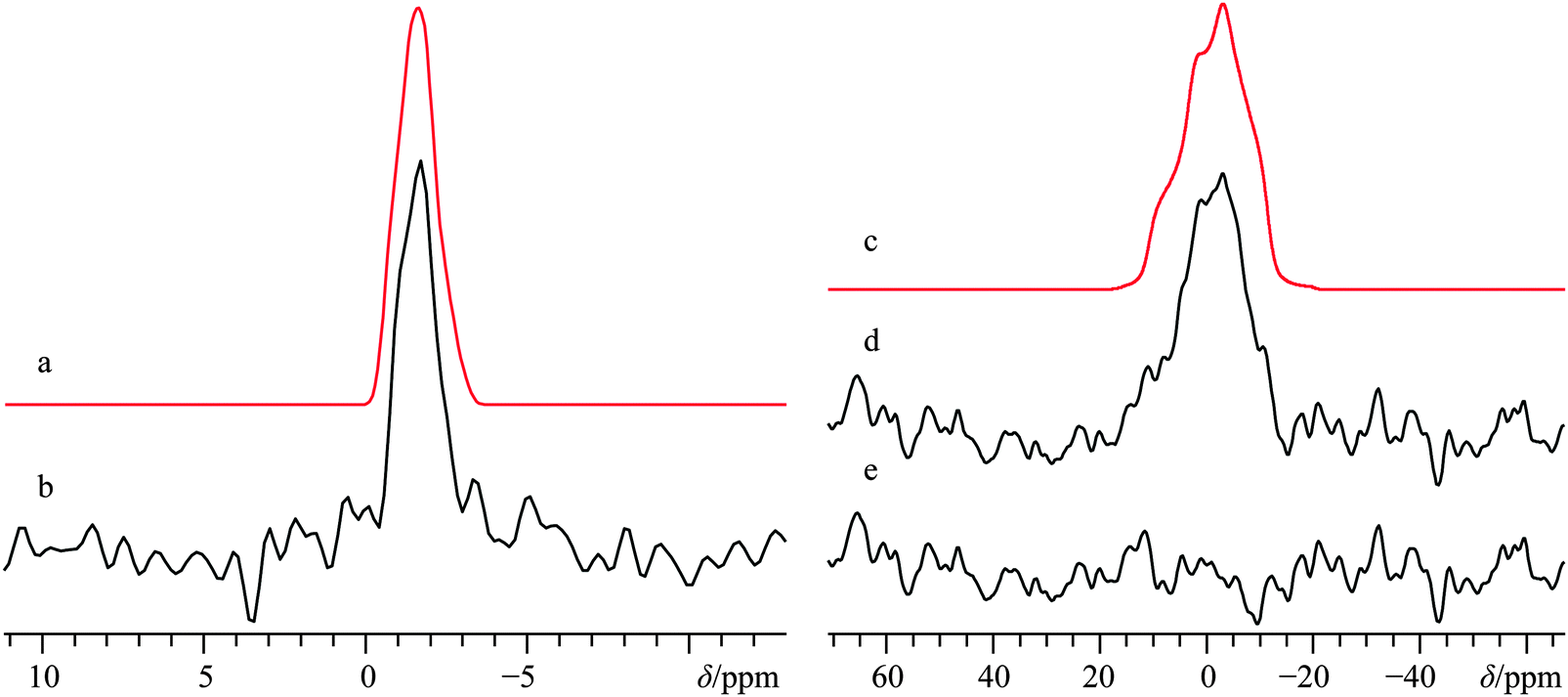 | ||
| Fig. 6 Analytical line shape simulations (a, c) and experimental 43Ca SSNMR spectrum of (±)-Ca(C4H4O6)·4H2O under MAS (νMAS = 5.0 kHz, b), and 43Ca{1H} SSNMR spectrum under static conditions (d), both acquired at B0 = 21.1 T. The difference spectrum between (c) and (d) is provided in (e). The experimental spectrum in (b) results from the collection of 4896 transients with a recycle delay of 7.0 s (experiment time = 9.5 h), while the spectrum in (d) resulted from collecting 24000 transients with a recycle delay of 10.0 s (experiment time = 66.7 h). | ||
According to the established relationship between δiso(43Ca) and coordination number,19,20 the observed calcium chemical shift is consistent with a calcium coordination number of 8, as observed in the known crystal structure. With such a high coordination number about the only unique calcium in the crystal structure, it is not surprising that |CQ| is rather small: CQ < 1.2 MHz; ηQ = 0.6(0.1).
With such a small CQ value, 43Ca NMR experiments conducted on a stationary sample at 21.1 T will give a signal that is broadened predominantly by CSA. Line shape modeling of the static 43Ca{1H} SSNMR spectrum presented in Fig. 6d (and detailed further in Fig. S12, ESI†), yields values of Ω = 23(3) ppm and κ = 0.1(0.2) and also establish that the EFG and magnetic shielding tensors are not coincident in this system. Tensor non-coincidence is consistent with the calcium site symmetry for this compound, and agrees with the GIPAW DFT computations for this material (Table 3). We also note that GIPAW DFT calculations predict that there is a substantial calcium chemical shift difference (ca. 20 ppm) between the tetrahydrate and trihydrate forms, based on the accepted structures of all forms.57,94,97 The CQ(43Ca) value for the trihydrate is also predicted to be significantly larger than either tetrahydrate (Table 3). This again highlights the potential of 43Ca SSNMR for distinguishing between hydrated forms of calcium-containing materials.
E. Enriched calcium acetate monohydrate, 43Ca(C2H3O2)2·H2O
A crystal structure of calcium acetate monohydrate is known for at least two polymorphs,55,98 where the form obtained can be controlled through the preparation method. A hemi-hydrated version may also be generated by performing the crystallization at temperatures greater than 60 °C.99,100 Spek and co-workers reported the structure of one polymorphic form of Ca(C2H3O2)2·H2O in 1984.55 They found the space group to be P with two inequivalent calcium atoms, each lying on a general lattice position (see Fig. 1e and f for the local calcium environments). A second polymorph of Ca(C2H3O2)2·H2O was presented by the same research group in 1987 and bears much in common with the 1984 polymorph (i.e., P, two non-equivalent Ca2+ ions on general positions).98 The most significant distinction between the two polymorphs from the perspective of the calcium is that both unique water molecules coordinate to the calcium in the 1984 polymorph, while only one coordinates in the 1987 polymorph. Prior 43Ca NMR measurements were carried out on this material, including the famous first example of 1H → 43Ca CP/MAS, which included the complete destruction of the NMR probe due to the very long and high-power contact times involved.38
Presently, 43Ca MAS SSNMR spectra at B0 = 9.4 T and 21.1 T were used to very precisely measure the isotropic calcium chemical shifts, as well as the 43Ca EFG tensor parameters for both sites (Fig. 7b, Fig. S13b, ESI,† and Table 2) using a sample which was isotopically enriched in 43Ca (ca. 7%). Although prior measurements of δiso(43Ca) exist for this system,36 we would suggest that the present values are more accurate due to data being collected at multiple applied fields. Interestingly, there is a rather large difference in the two observed calcium δiso values for this system of over 20 ppm. Additional 43Ca SSNMR experiments under static conditions at B0 = 9.4 and 21.1 T (Fig. 7d and Fig. S13d, ESI†) allow for the precise measurement of CSA for both overlapping sites, including Euler angle values.
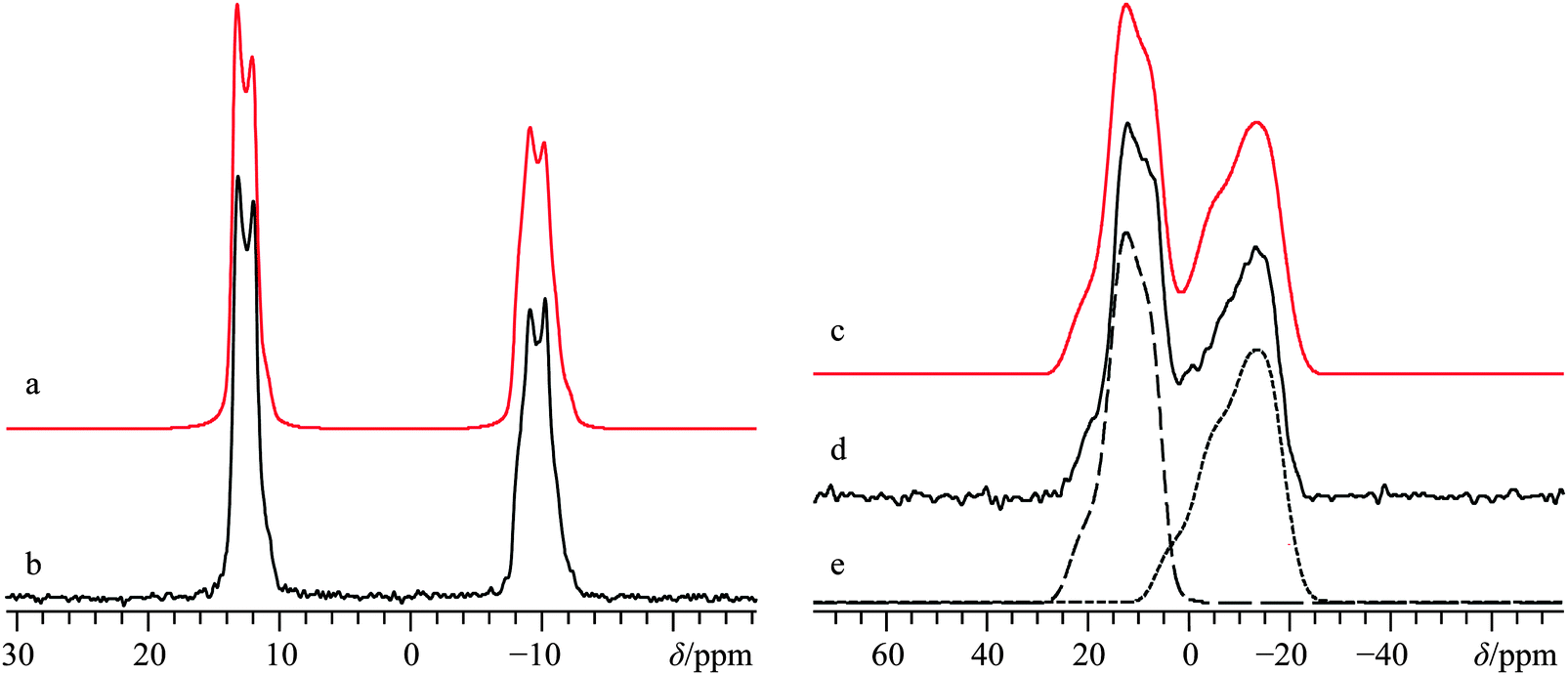 | ||
| Fig. 7 Analytical line shape simulations (a, c, e), experimental (b, d) 43Ca MAS (νMAS = 5.0 kHz, b) and static 43Ca{1H} (d) SSNMR spectra of 43Ca(C2H3O2)2·H2O, enriched to ca. 7% in 43Ca, and acquired at B0 = 21.1 T. The experimental spectra result from the collection of 64 transients with a recycle delay of 5.0 s (experiment time = 5.3 min) for (b), and from 256 transients with a recycle delay of 30.0 s (experiment time = 2.1 h) for (d). In (e), a deconvolution of the two calcium sites is provided. | ||
By coupling our 43Ca NMR results with complementary 13C SSNMR and pXRD results (see Fig. S14 and S15, ESI† for 13C SSNMR and pXRD results, respectively) we confirm that the sample used here corresponds to the monohydrated polymorph outlined by Spek and co-workers in 1984. It is immediately clear that the GIPAW DFT-computed NMR tensor parameters are in very good agreement with the experimentally measured values (Table 3), and the observed 43Ca SSNMR powder patterns may thus be confidently assigned to sites in the crystal lattice. GIPAW DFT calculations of the same NMR tensor parameters using the other known polymorph of calcium acetate monohydrate are in relatively poor agreement (Table S5). We note that the computed 13C chemical shift values for the acetate ligands are also in rather good agreement with the experimentally observed values.
F. Calcium levulinate dihydrate, Ca(C5H7O3)2·2H2O
While calcium levulinate dihydrate does not possess a known crystal structure, based upon prior studies using 43Ca SSNMR, as well as the results outlined in the systems directly above, it was thought that it would be possible to obtain important structural information by performing 43Ca SSNMR experiments on this compound. As with calcium tartrate tetrahydrate, 13C NMR experiments on calcium levulinate dihydrate confirmed sample purity via the anionic ligand. As such, the 13C NMR results are fully consistent with the presence of a single crystallographically distinct levulinate anion (Fig. S16, ESI†). Based on these combined NMR results (vide infra for 43Ca), it appears reasonably likely that Z′ = 1, a finding which may be of use in future attempts to determine the crystal structure of this compound.A 43Ca MAS NMR spectrum for this material is provided in Fig. 8, and at first glance appears rather similar to what was observed for the tartrate system. When considered in tandem, the small 43Ca CQ value of 1.3(0.1) MHz and the very shielded calcium chemical shift of −4.5(1.0) ppm are consistent with a high coordination number about the calcium cation, likely eight. Although the 43Ca SSNMR line width for calcium levulinate dihydrate is rather similar to the tartrate, due to the relatively bulkier levulinate anion (and hence clearly lower 43Ca content by mass), acquisition of the 43Ca MAS NMR spectrum was considerably more time consuming in the present case. While static 43Ca NMR experiments were attempted on this material at B0 = 21.1 T, we could not obtain data of high enough quality for data analysis which may have been able to extract calcium CSA.
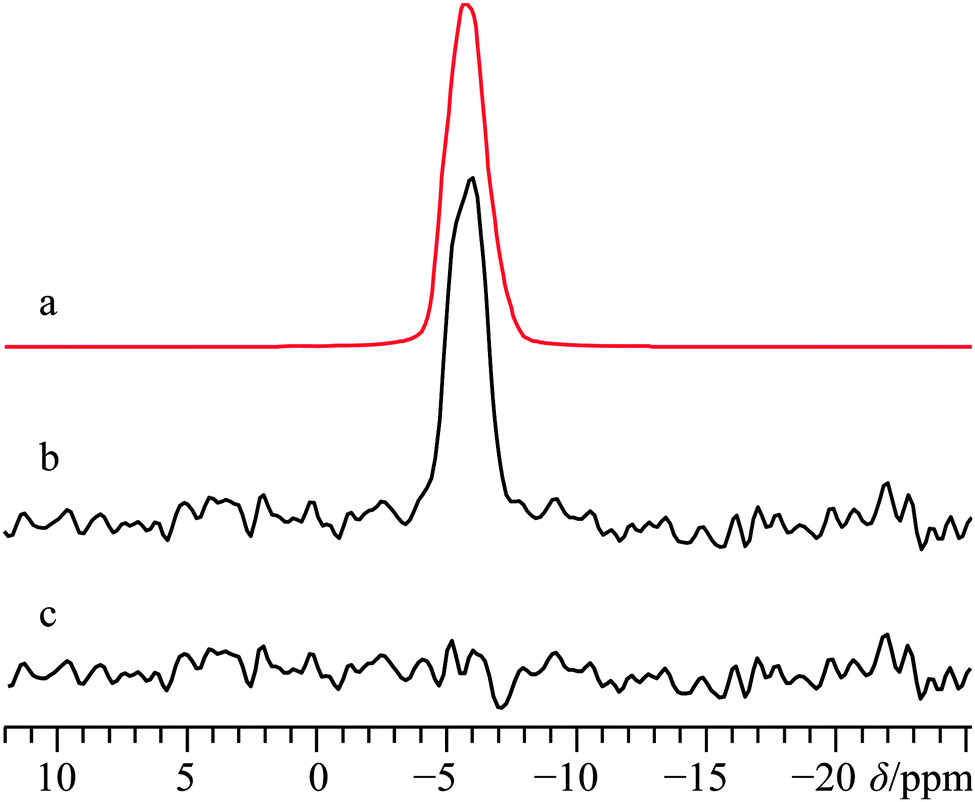 | ||
| Fig. 8 Analytical line shape simulation (a) and experimental 43Ca SSNMR spectrum of calcium levulinate dihydrate under MAS (νMAS = 4.5 kHz), (b), acquired at B0 = 21.1 T. The difference spectrum between (a) and (b) is provided in (c). The experimental spectrum in (b) results from the collection of 8495 transients with a recycle delay of 10.0 s (experiment time = 23.6 h). | ||
Thus, while calcium MAS NMR experiments on systems of this size are feasible, we clearly observed that even relatively small amounts of 43Ca dilution renders static 43Ca NMR at natural abundance a very substantial challenge. Signal enhancement techniques, such as RAPT,43,101 adiabatic DFS,42 and other interesting varieties,102,103 could be used for signal enhancement under MAS, but producing undistorted line shapes under static conditions can be somewhat challenging without the most modern spectrometers or extensive optimization. Likewise, due to the relatively narrow line shapes and unfavourable 43Ca T2 relaxation properties for several of the samples under study, we did not find much benefit in multiple-echo sensitivity enhancement techniques, such as the quadrupolar Carr-Purcell Meiboom-Gill (QCPMG) experiment.103 Due to these considerations, these signal enhancement routes were not explored further.
G. Enriched calcium sulfate dihydrate, 43Ca33SO4·2H2O
CaSO4 may be found in several distinct forms, including hydrates. Anhydrous β-CaSO4 can be found in nature as the mineral anhydrite104 while the anhydrous γ-CaSO4 form may be prepared by thermal decomposition of CaSO4 hydrates.105 Calcium sulfate hemihydrate, CaSO4·0.5H2O, also known as plaster of Paris, is occasionally found as the mineral Bassanite,106 and can be prepared on an industrial scale by the low temperature decomposition of CaSO4·2H2O at about 100 to 120 °C. This last form, CaSO4·2H2O, is perhaps the most well-known of all calcium sulfates, and is commonly found in mineral deposits as gypsum, and together with the plaster of Paris represents an important industrial and building material.107 The dihydrate was also one of the compounds used in Pake's seminal paper dealing with dipolar broadening of 1H NMR absorption lines in solids.108It appears that no 43Ca EFG tensor information exists for CaSO4·2H2O, as prior accounts34,67 report only experimental calcium peak positions. Several X-ray and neutron diffraction experiments have been performed to elucidate the crystal structure of this material,109–111 and we note that a recent X-ray crystal structure study places the calcium at lattice positions with 2-fold rotational symmetry in the space group C2/c.60 The space group assignment is consistent with the pXRD data acquired as part of this study (see ESI,† Fig. S17). The single unique Ca2+ is coordinated by eight oxygen atoms (Fig. 1g), which suggests a relatively shielded calcium environment, and which is borne out experimentally using a sample enriched to ca. 7% 43Ca (δiso = −19.1(0.3) ppm, Fig. 9b). The measured 43Ca EFG tensor parameters highlight a very symmetric electronic environment which does not possess axial symmetry and hence limits the rotational site symmetry to two-fold (CQ(43Ca) = 1.28(0.03); ηQ = 0.60(0.03)). The calcium CSA is significantly larger than observed for many of the organic calcium-containing systems (Ω = 43(2) ppm; κ = −0.31(0.07)). As expected by the local Ca site symmetry, the measured Euler angles depict non-coincident tensor frames, but indicate that they share some form of symmetry element. This is consistent with GIPAW DFT calculations on the known crystal structure for this material, (Table 3) although the actual values determined by GIPAW DFT using the structure of Comodi et al.60 differ from those experimentally observed. The discrepancy could be remedied by considering the possibility of sulfate librational motions, which can become pronounced in certain situations.112 Regardless, all NMR tensor parameters have been determined with high precision, and are fully consistent with MAS and static NMR measurements carried out at B0 = 9.4 T (Fig. S18 of the ESI†).
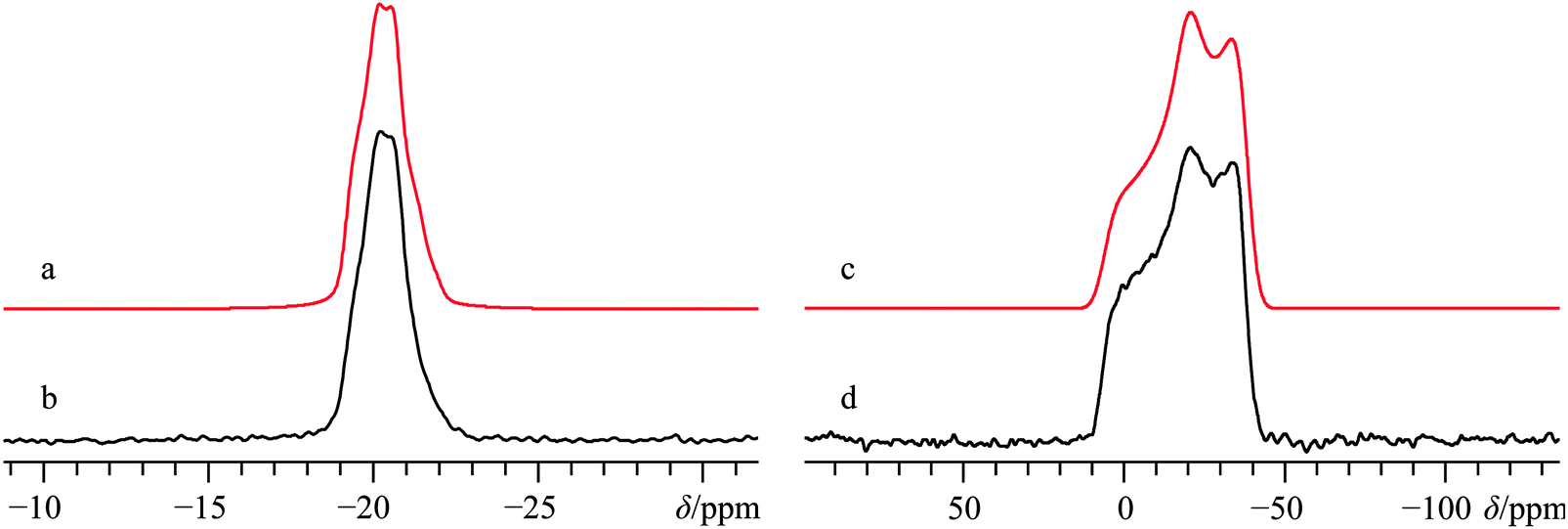 | ||
| Fig. 9 Analytical line shape simulations (a, c), experimental 43Ca MAS SSNMR spectrum (νMAS = 5.0 kHz, b), and experimental 43Ca{1H} static SSNMR spectrum (d) of 43Ca33SO4·2H2O (enriched to ca. 7% in 43Ca), acquired at B0 = 21.1 T. The experimental spectra result from the collection of 32 transients with a recycle delay of 200 s (experiment time = 1.8 h for b), and from 112 transients with a recycle delay of 200 s (experiment time = 6.2 h for d). | ||
H. Calcium chromate, CaCrO4
Preliminary structural studies of CaCrO4 using XRD were carried out nearly a century ago by Clouse,113,114 and relatively recently a slightly modified structure for this material was proposed.61 The common form of this material belongs to the tetragonal I41/amd space group with the calcium ions at very high symmetry locations (m2, Fig. 1h), which is expected to greatly constrain the resulting EFG and CS tensor parameters for this system. No prior 43Ca NMR information exists for this material, although 53Cr SSNMR studies have been performed by Forgeron and Wasylishen.115 In accord with the known crystal structure (see Fig. S19, ESI† for pXRD diffractogram), we conclude that the 43Ca NMR data acquired at B0 = 9.4 T (Fig. S20, ESI†) and 21.1 T (Fig. 10) are consistent with a high local site symmetry, as both tensors are, within measurement error, axially symmetric (ηQ < 0.1; κ = −1). The very small CQ value (0.96(0.05) MHz) highlights a highly symmetric environment about the calcium. This is very interesting when taken in concert with the very large CS tensor span (Ω = 87(3) ppm), which is the largest calcium Ω value measured to date. As part of the computational section below, we offer discussion of GIPAW DFT calculations which rationalize this observation. Likewise, the combination of a small CQ(43Ca) value with a substantial Ω value, while perhaps unexpected, is not unique for calcium-containing materials, as for example the CQ(43Ca) value for the aragonite polymorph of CaCO3 is about 260 kHz,67,68 while the span value is nearly 50 ppm.
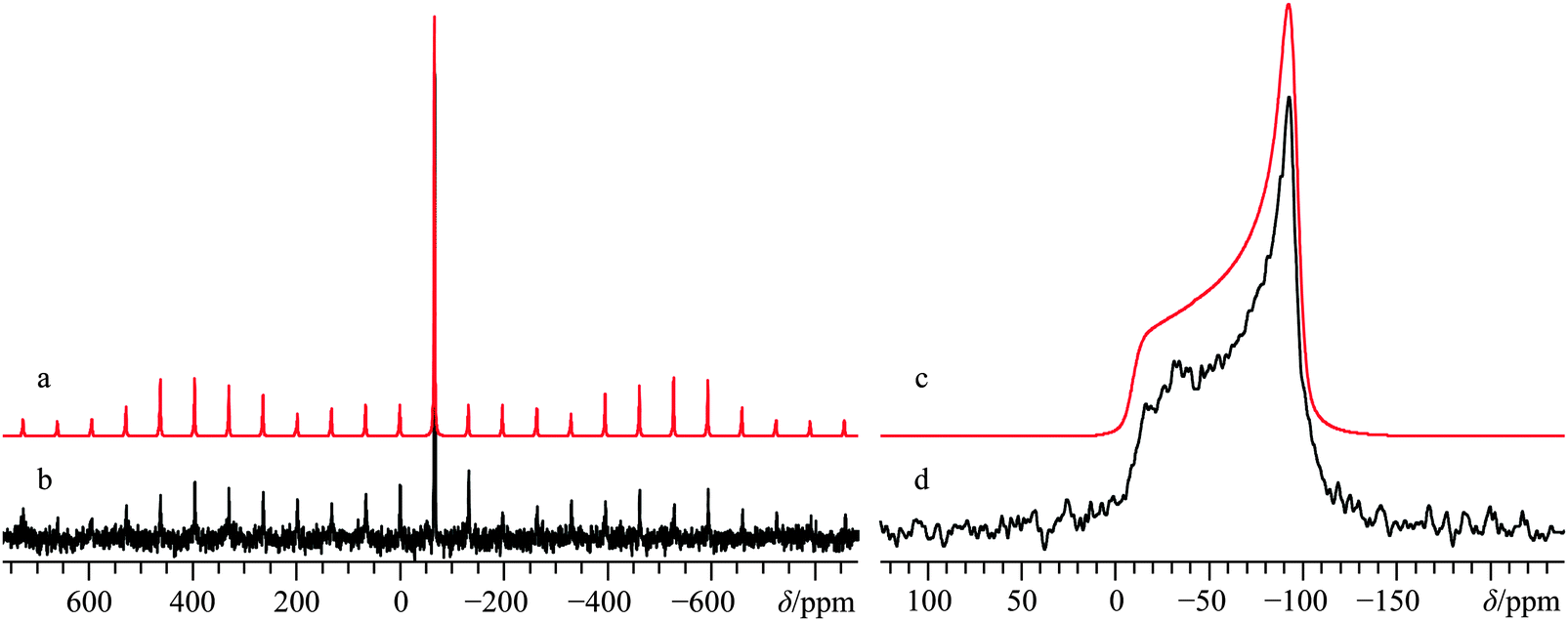 | ||
| Fig. 10 Numerical line shape simulation (a), analytical line shape simulation (c), experimental 43Ca MAS SSNMR spectrum (νMAS = 4.0 kHz, b), and experimental 43Ca static SSNMR spectrum (d) of CaCrO4, acquired at B0 = 21.1 T. The experimental spectra result from the collection of 22722 transients with a recycle delay of 3 s (experiment time = 18.9 h, b), and from 37485 transients with a recycle delay of 0.5 s (experiment time = 5.2 h, d). | ||
The highly shielded calcium environment (δiso = −65.4(0.3) ppm) is consistent with a high calcium coordination number and short calcium–oxygen bond distances, but is considerably shielded relative to the calcium systems which possess organic anions. This emphasizes that care must be taken when using prior data which correlates calcium δiso with Ca–O bond distances. While we further discuss the topic of the calcium CS tensor of CaCrO4 in the computational section, we note that GIPAW DFT computations using an accepted crystal structure61 for CaCrO4 provide calculated NMR tensor parameters that are in very good agreement with the experimentally observed values.
I. Calcium titanate, CaTiO3
The crystal structure of the ceramic material CaTiO3 belongs to the perovskite structure class, and even today its structure is the focus of active research, more specifically the evolution of the ambient condition structure (orthorhombic Pbnm space group116–119) to the high-temperature cubic structure via either one (or more) intermediate-temperature phases.62,120,121 The local site symmetry at the calcium is m, which will slightly restrict the potential values for some NMR parameters (see Fig. 1i for local Ca environment). Prior 17O,12243Ca,34,123 and 47/49Ti124 SSNMR data exist for CaTiO3, but no measurement of the 43Ca shift anisotropy has been reported. A 43Ca MAS SSNMR experiment at 21.1 T using a freshly calcined sample of CaTiO3 (see Fig. S21, ESI† for the pXRD diffractogram) yields a spectrum which is clearly broadened by the second-order QI and which is non-axially symmetric (CQ(43Ca) = 2.09(0.05) MHz; ηQ = 0.75(0.05), Fig. 11b). These present values are in excellent agreement with the prior literature values.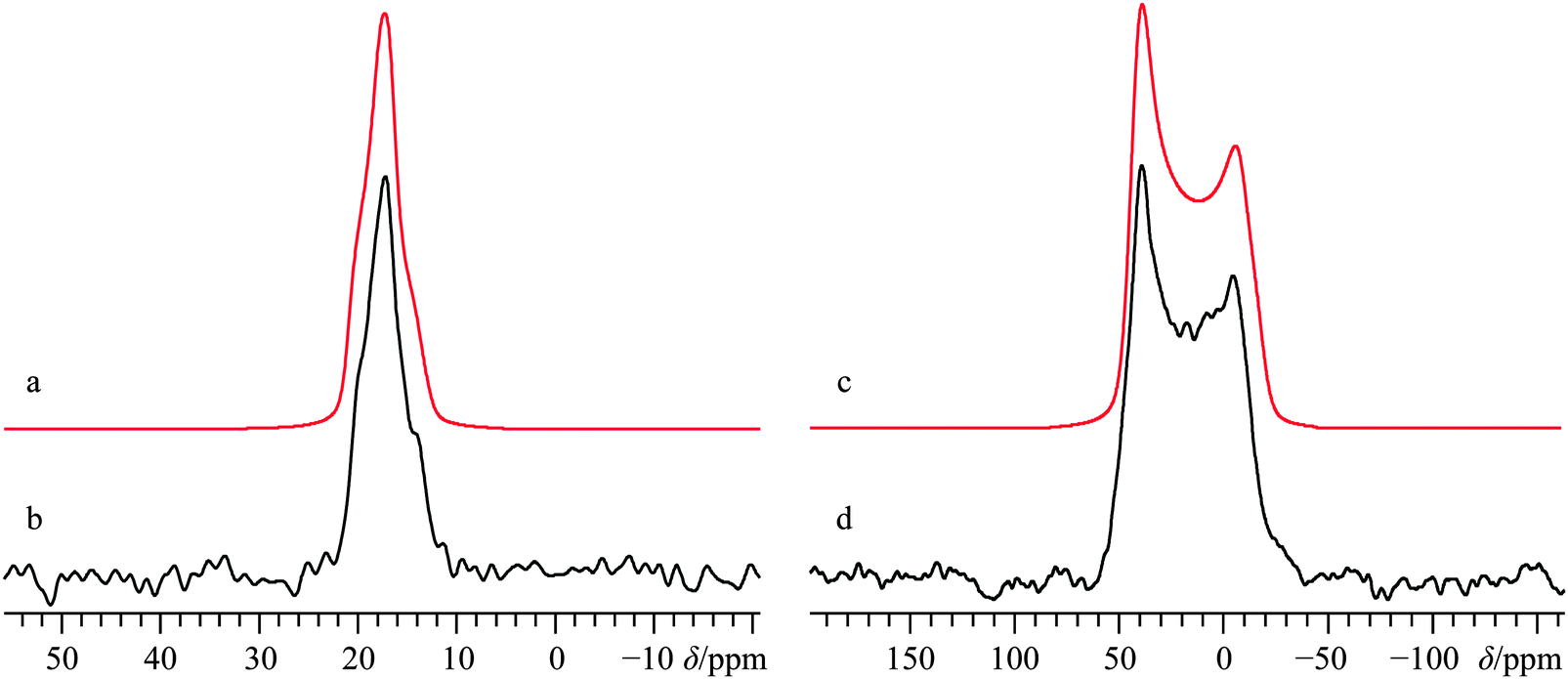 | ||
| Fig. 11 Analytical line shape simulations (a, c) and experimental (b, d) 43Ca MAS (νMAS = 5.0 kHz, b) and static (d) SSNMR spectra of CaTiO3, acquired at B0 = 21.1 T. The experimental spectra result from the collection of 13133 transients with a recycle delay of 5.0 s (experiment time = 18.2 h, b), and from 29071 transients with a recycle delay of 5.0 s (experiment time = 40.4 h, d). | ||
Static 43Ca SSNMR experiments at B0 = 21.1 and 11.75 T (Fig. 11d and Fig. S22, ESI†) establish that the calcium CS tensor for CaTiO3 is rather large (Ω = 53(3) ppm) and nearly axially symmetric (κ = 0.8(0.1)). GIPAW DFT calculations using the structure of Ali and Yashima62 result in EFG and CS tensor parameters which agree well with experiment, although the calculated Ω value overestimates the experimental value by about 20%.
J. Enriched calcium nitrate, 43Ca(NO3)2
Under normal atmospheric conditions, Ca(NO3)2 will freely absorb ambient water, with the most stable water solvate being the α-tetrahydrate,125,126 although many other hydrated and solvated forms of calcium nitrate may be prepared.127,128 The crystal structure for anhydrous calcium nitrate was determined approximately 90 years ago by XRD methods,129 and does not appear to have been updated since 1931.63 The crystal structure belongs to the cubic Pa space group, with calcium cations being positioned on high symmetry sites (Fig. 1j), which should confer axial symmetry on the CS and EFG tensors for calcium. Solid-state 43Ca NMR experiments have not been previously reported for the anhydrous form, but 43Ca NMR data exist for Ca(NO3)2·4H2O.35 Calcium-43 NMR experiments were carried out on this material at both B0 = 9.4 and 21.1 T, and generally yield “textbook” quality signals which are clearly broadened by the second-order QI, even at our highest B0 (CQ(43Ca) = 1.57(0.01) MHz; ηQ = 0.0; Fig. 12; for data acquired at 9.4 T, see Fig. S23 of the ESI†).
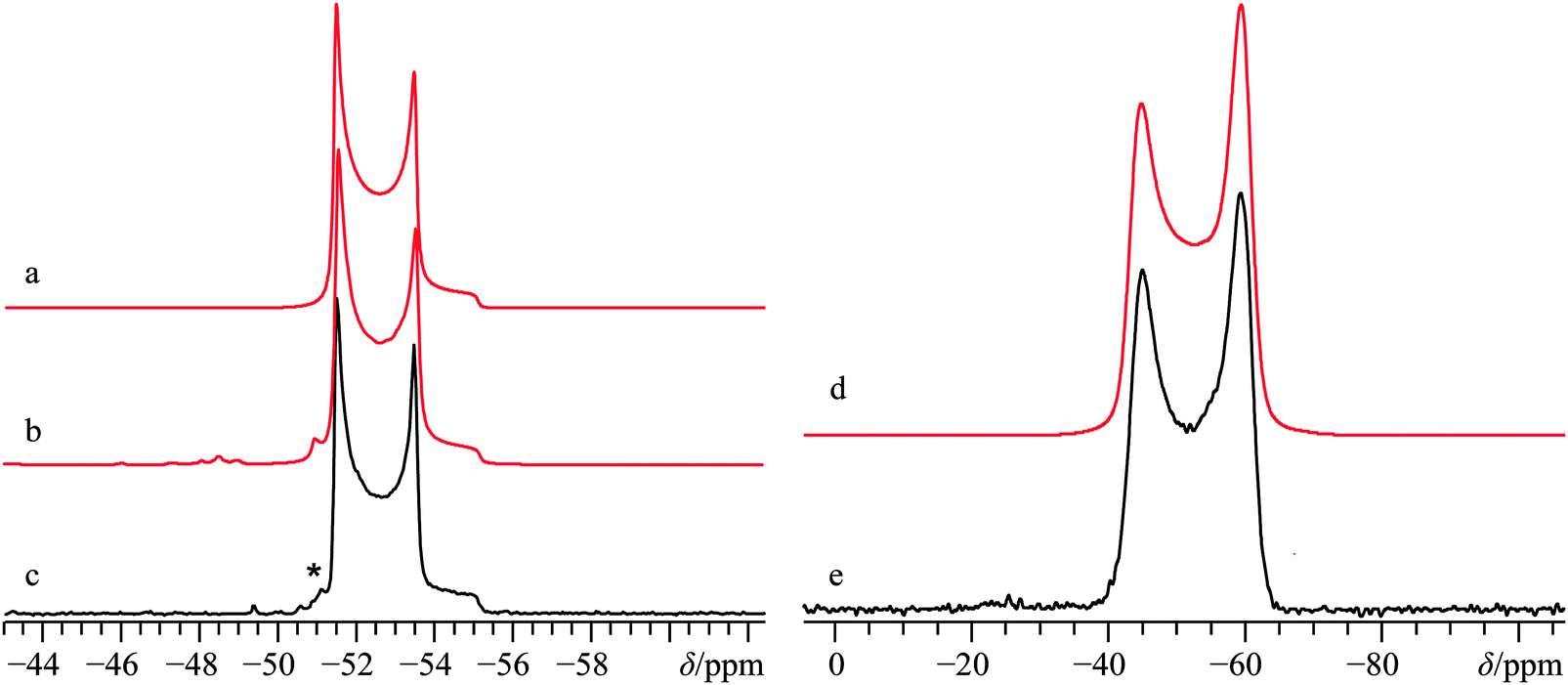 | ||
| Fig. 12 Analytical line shape simulations (a, d), numerical line shape simulation (b), and experimental (c, e) 43Ca MAS (νMAS = 5.0 kHz, c) and static SSNMR (e) spectra of 43Ca(NO3)2, (enriched to ca. 7% in 43Ca) acquired at B0 = 21.1 T. The experimental spectra result from the collection of 64 transients with a recycle delay of 7.0 s (experiment time = 7.5 min, c), and from 640 transients with a recycle delay of 10.0 s (experiment time = 1.8 h, e). The asterisk in (c) denotes a small amount of signal intensity from the calcium satellite transitions, which are reproduced in the numerical line shape simulations. | ||
The calcium CSA is rather small relative to other systems studied, (Ω = 11.4(1.0) ppm) and the data are consistent with an axially symmetric shift tensor (κ = 1). We did not find any enhancement in the agreement when allowing for non-zero Euler angles in our modelling; hence, the EFG and CS tensor frames are assumed to be coincident. While the precision of the measured NMR tensor parameters for Ca(NO3)2 is relatively high, it is rather noteworthy that for this system, GIPAW DFT calculations are not able to reproduce the calculated 43Ca EFG tensor magnitude using the available structure.63 At the same time, this finding is not terribly surprising given the relatively old source data for the crystal structure of Ca(NO3)2. Additionally, even 80 years ago, it was appreciated that the published crystal structure for Ca(NO3)2 was likely unsatisfactory,130 as it did not consider the very likely possibility of nitrate group rotations at ambient temperatures (see Fig. S24, ESI† for the pXRD diffractogram of the Ca(NO3)2 sample used in the NMR measurements). Based on the axial tensor symmetry exhibited at the Ca sites, we conclude that the local site symmetry is at least C3, which is consistent with the accepted structure. With these points in mind, we carried out an optimization of the crystal structure for this system (holding the calcium ions on the high symmetry positions), but note that the agreement between calculated and experimental 43Ca NMR tensor parameters remains rather poor, especially when considered relative to the very good agreement for the other systems in this study.
K. Calcium hydride, CaH2
As a simple binary compound of calcium, it is interesting to note that very recent efforts have been expended to determine the crystal structure for CaH2, likely due to its potential as a hydrogen storage material. Although consistently observed to belong to the Pnma space group,131,132 recent neutron diffraction experiments were used to comment on the non-identical nature of each hydrogen atom in the asymmetric unit, as well as the poor representation provided by approximating the constituent atoms as spheres.64 The 9-coordinate Ca2+ ions lie on mirror planes, which somewhat restricts the Euler angles between the CS and EFG tensor frames (Fig. 1k). Only the peak position in a featureless 43Ca MAS NMR spectrum obtained at one applied field was reported previously for this system,35 but more detailed 1H (CaH2) and 2H SSNMR (CaD2) measurements have been carried out.133 Our present NMR experiments on CaH2 were performed at natural abundance in 43Ca, and by careful measurements under MAS conditions at three magnetic fields (B0 = 9.4 T, 11.75 T, and 21.1 T), we were able to arrive at a set of EFG tensor parameters and a δiso(43Ca) value (Fig. 13b and Fig. S25, as well as Fig. S26, ESI† for pXRD results). The primary difficulty arises from the relatively featureless 43Ca NMR spectrum under MAS conditions, even at 9.4 T. This is indicative of a relatively small CQ(43Ca) value of 1.2(0.1) MHz. We also note that the line shape models are not very sensitive to the ηQ value, which leads to large errors associated with its measurement (ηQ = 0.7(0.2)). The high chemical shift value (δ(43Ca) = 95.8(1.0) ppm) could be anticipated due to the large amount of charge transfer believed to exist in this system, and consequently very low contributions to magnetic shielding at the calcium (i.e., positive chemical shifts).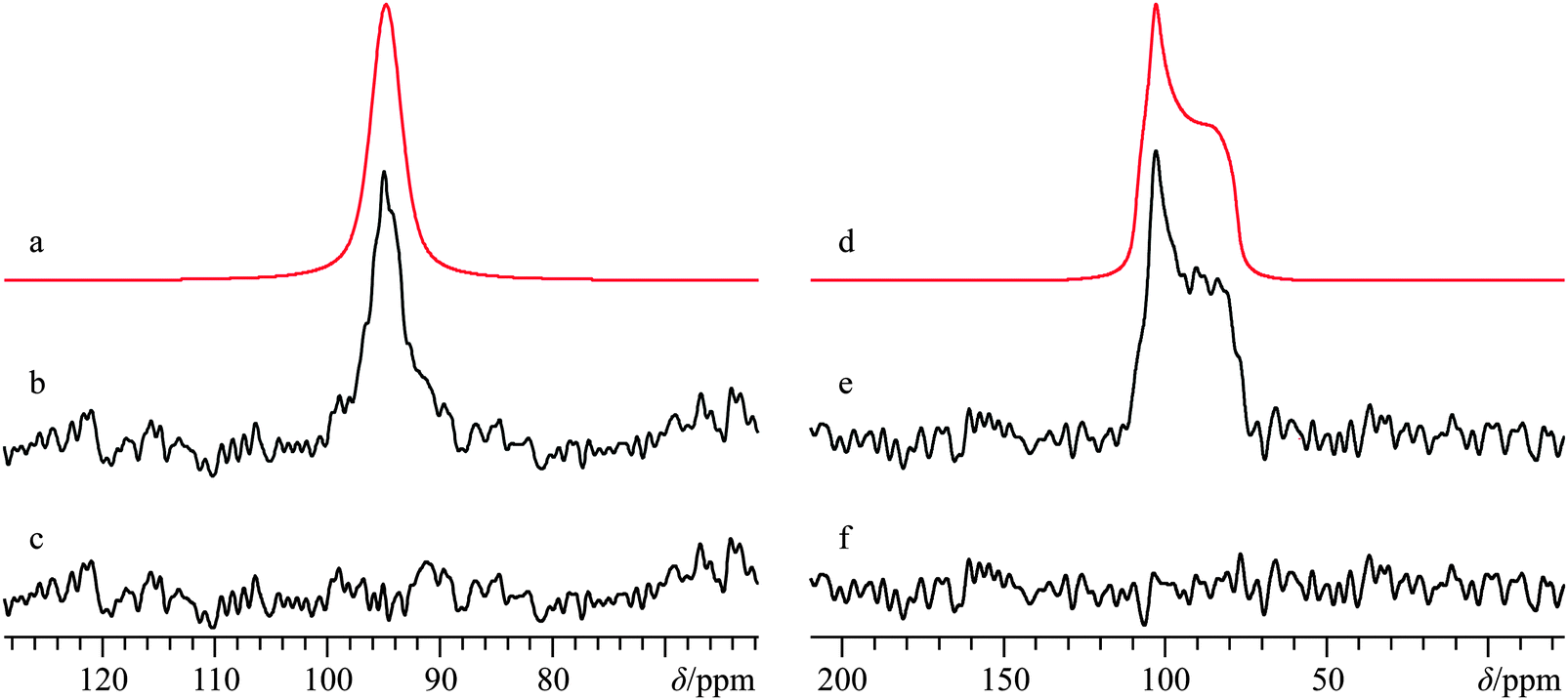 | ||
| Fig. 13 Analytical line shape simulations (a, d) experimental (b, e) 43Ca MAS (νMAS = 5.0 kHz, b) and static 43Ca{1H} (e) SSNMR spectra of CaH2, along with corresponding difference spectra in (c) and (f), acquired at B0 = 21.1 T. The experimental spectra result from the collection of 3072 transients with a recycle delay of 40.0 s (experiment time = 34.1 h) for (b), and from 5216 transients with a recycle delay of 40.0 s (experiment time = 58.0 h) for (e). | ||
Due to the very high quality of the neutron diffraction data, we find excellent agreement between the GIPAW DFT-calculated and experimental 43Ca NMR tensor parameters for this system, although the calculated δiso value is overestimated.
L. Natural samples of calcium carbonate, CaCO3
Much recent effort has been expended in characterizing the natural formation of CaCO3.134,135 While not the typical polymorph formed immediately during biomineralization, the calcite polymorph is considered to be the most stable form of CaCO3 under ambient conditions.136 Along with calcite, the aragonite polymorph (Fig. 1l) is typically present in natural samples, and while several accounts have led to a full characterization of the polymorphic forms of CaCO3 by 43Ca SSNMR,34,67,68 we are not aware of any attempts to characterize a naturally occurring sample of CaCO3. As such, we performed experiments on two natural samples of CaCO3, one from Cuban coral, and one from a river clam pearl. Casual inspection of the 43Ca MAS NMR spectra and pXRD spectra (Fig. S27 and S28, respectively, ESI†) made it clear that both samples of CaCO3 contained the aragonite polymorph exclusively. As noted earlier, the CQ(43Ca) value for this polymorph is very small, measured to be ca. 260 kHz by Chan and co-workers using samples which had been modestly enriched in 43Ca.68 To determine the CQ(43Ca) value for the natural samples, 43Ca MAS SSNMR experiments were carried out at B0 = 11.75 and 21.1 T. Experiments at the highest field were performed with relatively slow MAS (νMAS = 3000 Hz) and used non-CT-selective pulses to excite all single-quantum transitions as uniformly as possible. Line shape analysis using all the data yields: CQ = 250(50) kHz, ηQ = 0.4(0.1), and δiso = −27.0(0.4) ppm.With the EFG tensor parameters and δiso value accurately determined, static 43Ca SSNMR experiments were performed on both natural samples to see if there was any variation in the CS tensor parameters between: (i) natural samples and synthetic samples and (ii) between natural samples taken from different sources. Perhaps not surprisingly, we find that the variation in the CS tensor parameters between natural samples of aragonite is within experimental error (Fig. 14), which highlights that from the local perspective probed by 43Ca SSNMR, the structures are similar. When taken in tandem with pXRD measurements that probe long-range crystalline order (see ESI,† Fig. S28), we conclude that for these natural samples, there is a high level of similarity between the natural samples and between natural and synthetic samples of aragonite.
 | ||
| Fig. 14 Analytical line shape simulations (a, c), and experimental (b, d) static 43Ca SSNMR spectra of natural samples of the aragonite polymorph of CaCO3, as found in Cuban coral (b) and the pearl of a river clam (d), acquired at B0 = 21.1 T. The experimental spectra result from the collection of 20000 transients with a recycle delay of 1.5 s (experiment time = 8.3 h) for (b), and from 12000 transients with a recycle delay of 2.0 s (experiment time = 6.7 h) for (d). | ||
While there could be slight differences in the 43Ca NMR tensor parameters between natural samples (Table 2), the data are not of sufficient S/N to distinguish these subtle differences.
M. Quantum chemical calculations
As highlighted above, high-quality crystal structures exist for nearly all of the systems under study here. As such, we used GIPAW DFT to calculate the magnetic shielding and EFG tensor parameters, and compared these with the experimentally measured parameters, where possible. The vast majority of the calculated results can be found in Table 3, with additional calculations presented in the ESI,† Table S5.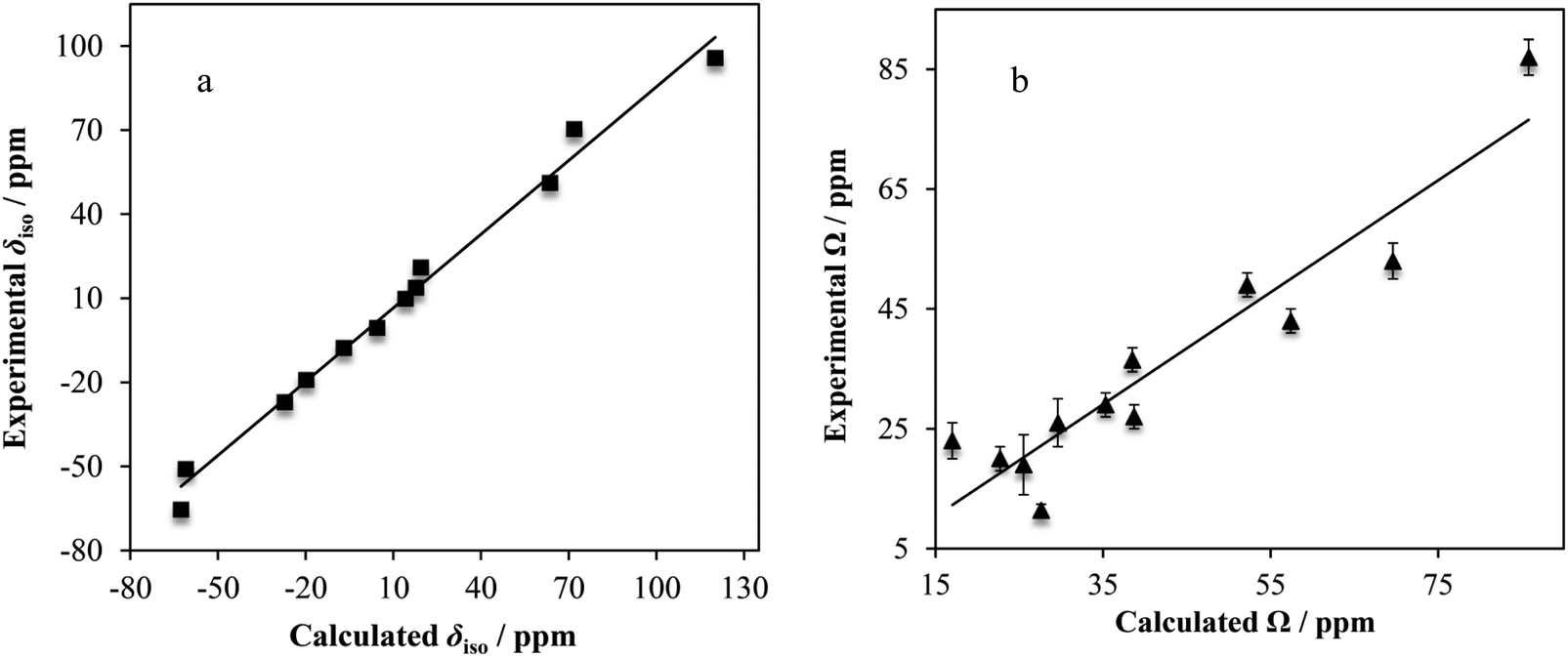 | ||
| Fig. 15 Experimental vs. GIPAW DFT-computed calcium chemical shift tensor parameters (δiso, a; Ω, b) for many of the systems under study. Lines of best-fit are displayed (in a, δiso(exp./ppm) = 0.877δiso(calc./ppm) − 2.14, R2 = 0.988; in b, Ω(exp./ppm) = 0.933Ω(calc./ppm) − 3.57, R2 = 0.885). Experimental error bars are actually present in both above plots, but lie within the data points of (a). | ||
Linear correlations with correlation coefficients near unity are observed when comparing the computed and experimental δiso (R2 = 0.988) and Ω (R2 = 0.885) values. For the calculated vs. experimental Ω values, we make the typical observation that the calculated values are nearly always greater than the experimental values (as observed with, for example, 13C).137 We rationalize this as being due to dynamics which may occur at room temperature, which will partially average the observed Ω. As isotropic values are not affected as strongly in this manner, we see a higher level of correlation between experimental and calculated δiso values.
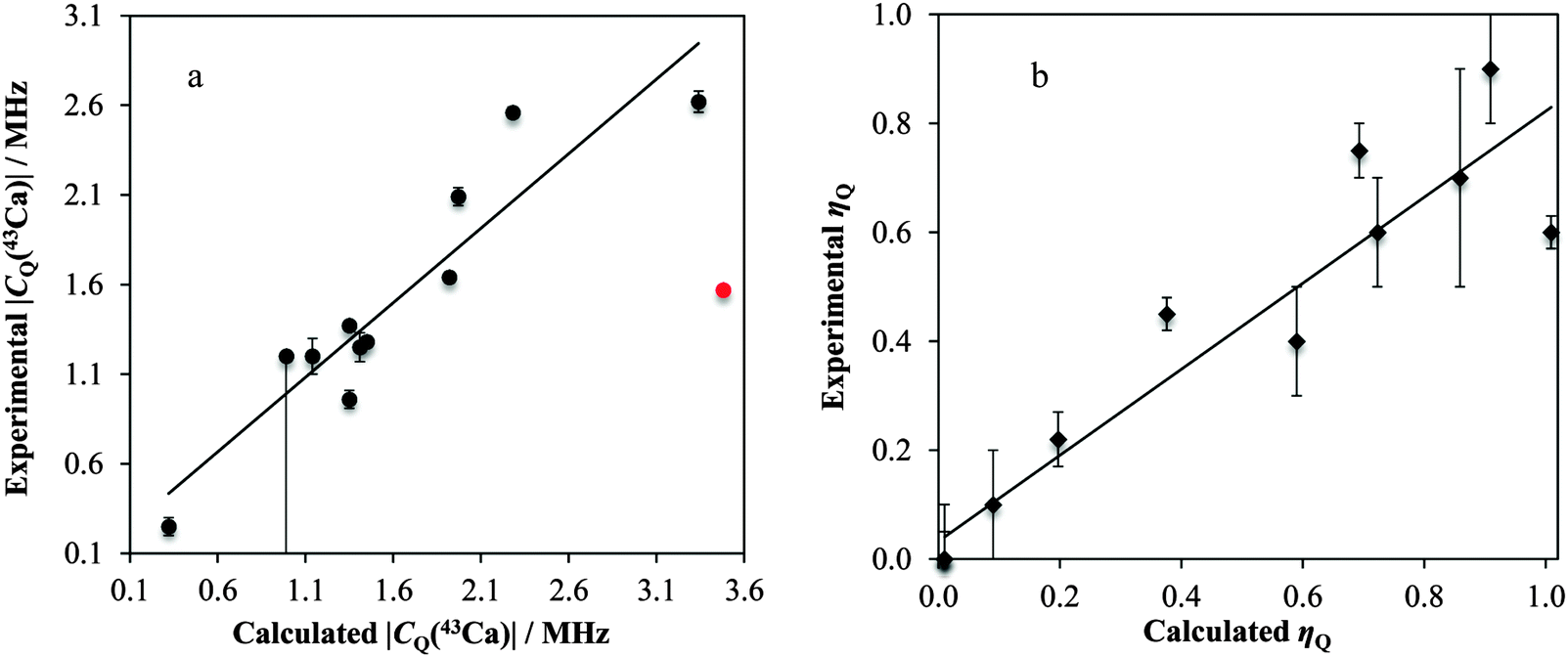 | ||
| Fig. 16 Experimentally observed vs. GIPAW DFT-computed EFG tensor parameters (|CQ(43Ca)|, a; ηQ, b) for many of the systems under study. The |CQ(43Ca)| datum for Ca(NO3)2 is not included in the fitting (red point). Lines of best-fit are displayed (in a, CQ(exp./MHz) = 0.832CQ(calc./MHz) + 0.168, R2 = 0.864; in b, ηQ(exp.) = 0.790ηQ(calc.) + 0.0408, R2 = 0.883). | ||
We postulate that the reason for the poor agreement for Ca(NO3)2 may stem from the age (and hence poor resolution) of the accepted crystal structure. As outlined below, very small alterations in the crystalline lattice should result in dramatic changes in the observable calcium NMR tensor parameters. Importantly, the GIPAW DFT and experimental measurements seem to consistently agree that the calcium atom is on a site of axial symmetry (ηQ equal to or approaching 0). Axial EFG tensor symmetry can be used in NMR crystallography approaches to restrict the local symmetry at the nuclear site.75
For Ca(OH)2, we observe significant changes in both the δiso and |CQ(43Ca)| values (Fig. 17), with the other tensor parameters remaining largely unchanged (for example, Ω, see ESI,† Fig. S29). The data suggest that alterations of the unit cell and calcium–oxygen first coordination sphere internuclear distances by as little as 1% could be detected through changes in CQ(43Ca) and δiso for Ca(OH)2. It is important to note that these NMR parameters can be simultaneously measured using primarily MAS experiments, which are much more sensitive for 43Ca compared to the static experiments. This finding offers hope that these parameters could be meaningfully used as structural restraints in crystal structure refinements, as demonstrated recently for other quadrupolar nuclei in the solid state.139,140
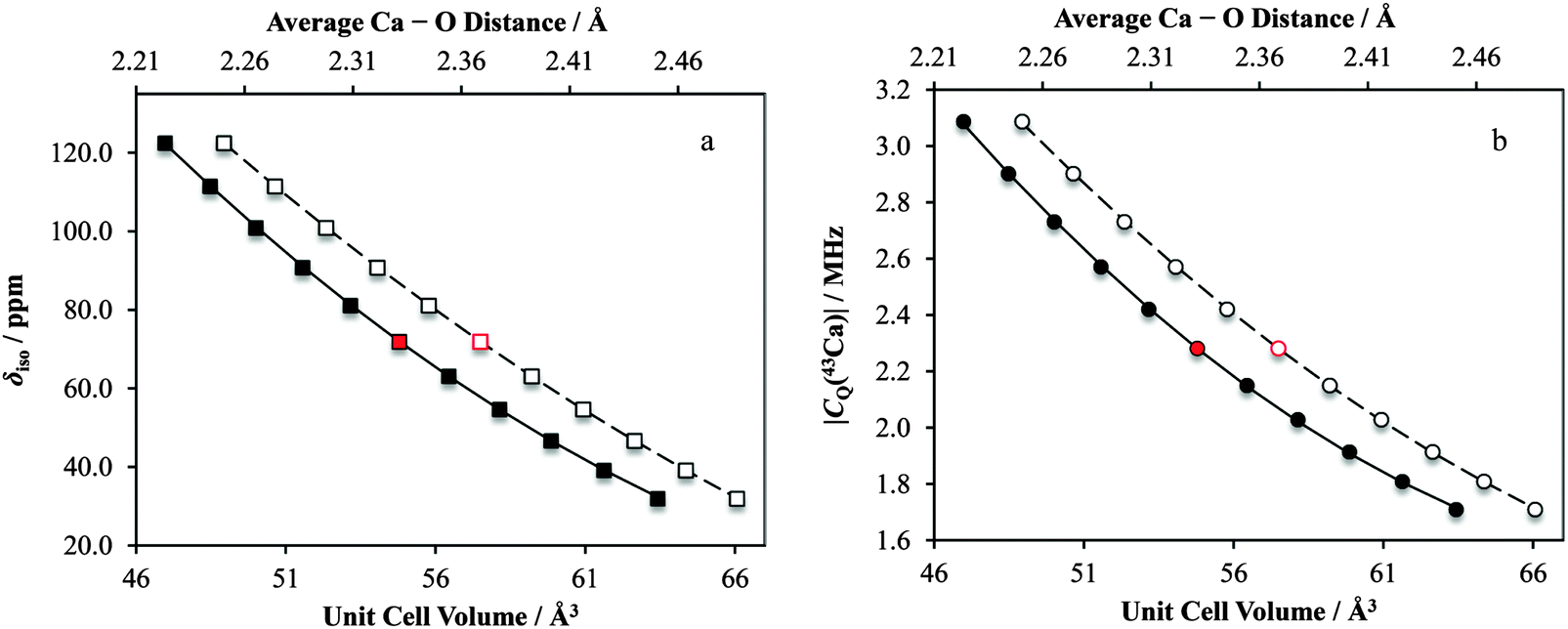 | ||
| Fig. 17 Results of GIPAW DFT computations which highlight the profound sensitivity of both the isotropic calcium chemical shift, as well as the |CQ(43Ca)| value to minor alterations in Ca(OH)2 structural parameters. The data points indicated in red correspond to the respective parameters calculated using the accepted crystal structure. Solid data points correspond to lower horizontal axis. Second-degree polynomial lines of best fit: in a, δiso = 0.112(Vcell)2 − 17.8(Vcell) + 713, R2 = 0.999; in b, |CQ(43Ca)| = 0.00223(Vcell)2 − 0.329(Vcell) + 13.6, R2 = 0.999, where Vcell is the unit cell volume in Å3. Open data points correspond to upper horizontal axis. Second-degree polynomial lines of best fit: in a, δiso = 382.7(rCa–O)2 − 2195(rCa–O) + 3124, R2 = 1.00; in b, |CQ(43Ca)| = 8.40(rCa–O)2 − 45.6(rCa–O) + 63.1, R2 = 1.00, where rCa–O is the average calcium–oxygen distance in Å. | ||
Likewise, for CaCrO4 we observe a similar sensitivity in |CQ(43Ca)| and δiso to small changes in the crystal lattice dimensions (and hence also calcium–oxygen first coordination sphere distances; ESI,† Fig. S30). Notably, for this system we also calculate that the CS tensor span (Ω) would serve as a very sensitive probe of small structural changes (Fig. 18). Of course, measurement of the CS tensor for calcium remains challenging, but we anticipate that our present findings will motivate future studies which involve the measurement of calcium CS tensors.
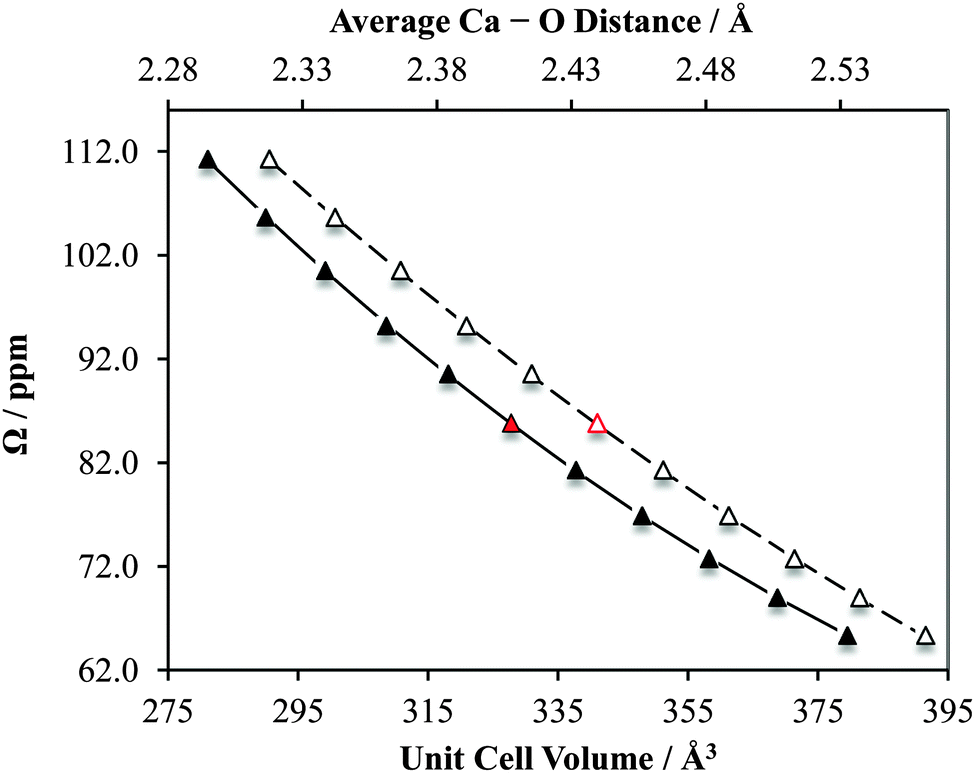 | ||
| Fig. 18 Results of GIPAW DFT computations which highlight the sensitivity of the calcium chemical shift tensor span (Ω) to alterations in CaCrO4 structural parameters. The data points indicated in red correspond to Ω calculated using the accepted crystal structure. Solid data points correspond to lower horizontal axis. Second-degree polynomial line of best fit: Ω = 0.00152(Vcell)2 − 1.47(Vcell) + 404; R2 = 0.999. Open data points correspond to upper horizontal axis. Second-degree polynomial line of best fit: Ω = 169.8(rCa–O)2 − 1017(rCa–O) + 1556; R2 = 1.00. | ||
As noted earlier, the observed calcium CS tensor Ω value for CaCrO4 is the largest measured to date. This could be reasonably anticipated due to the presence of a transition metal species near to the calcium. When considering the full calcium CS tensor, which includes the associated eigenvectors, it is seen that δ11 (the most de-shielded component) points exactly towards the most proximate chromium (rCa–Cr = 3.142 Å), as depicted in Fig. 19a. Due to the symmetry of the calcium CS tensor for CaCrO4 (κ = −1), the other two CS tensor eigenvectors (δ22 and δ33) define a plane of equivalent magnetic shielding which is oriented perpendicular to the Ca–Cr internuclear vector. The enhanced de-shielding in the direction of the Cr is consistent with larger contributions to the paramagnetic shielding mechanism,141–144 which is often seen in transition metals, and is due to the various d orbitals available.145 We also note that this directional dependence can explain the very pronounced changes in the calcium CS tensor as a function of the small unit cell changes that were shown in Fig. 18. While both the parallel and perpendicular components of the Ca CS tensor are sensitive to unit cell variation, GIPAW DFT computations make it clear that the component which lies in the direction of the Ca–Cr internuclear bond vector (i.e., δ11) changes more rapidly relative to the components which lie perpendicular to this direction (i.e., δ22/δ33; Fig. 19b).
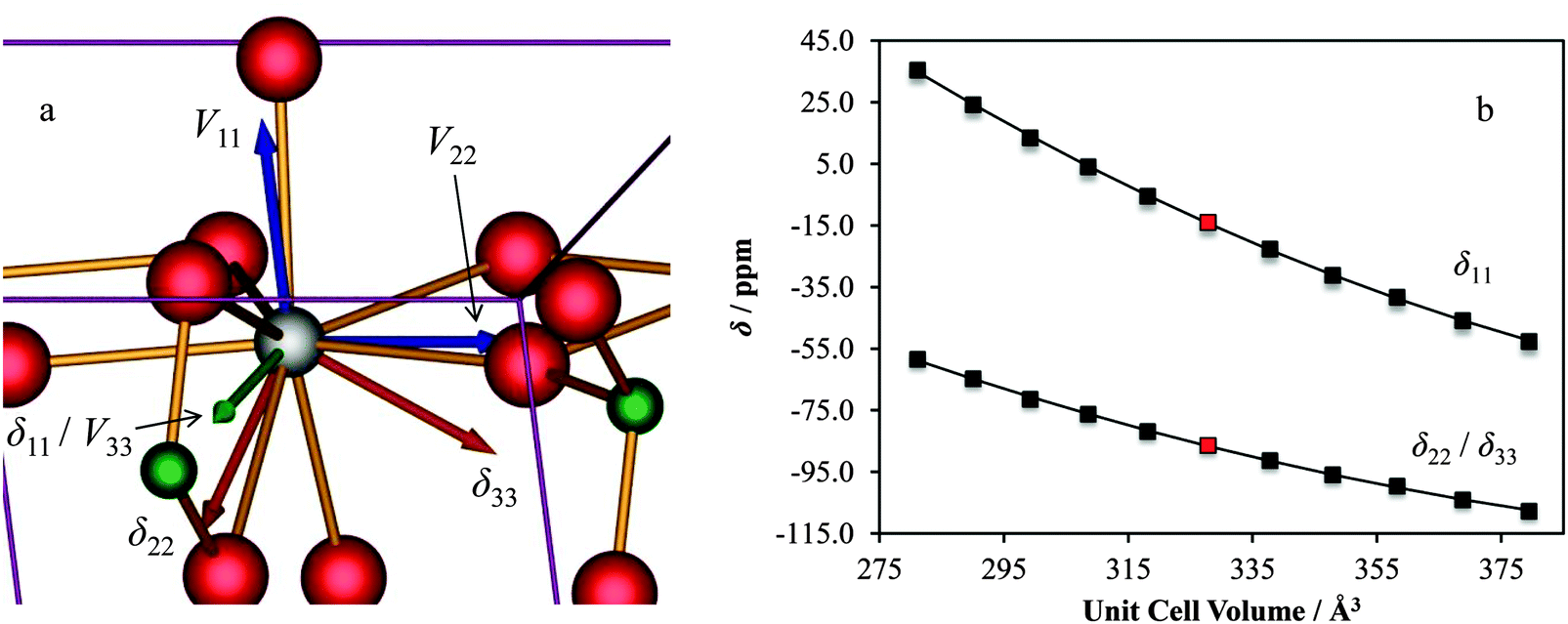 | ||
| Fig. 19 GIPAW DFT results for CaCrO4 which display (a) the computed EFG and CS tensor eigenvectors and (b) the sensitivity of the calcium chemical shift tensor eigenvalues to alterations in CaCrO4 structural parameters. For (a): POV-Ray rendering of the computed EFG tensor (Vii, i = 1, 2, 3, in blue) and chemical shift (δii, orange) eigenvectors in the unit cell reference frame. When the EFG and CS tensor eigenvectors overlap (as for V33 and δ11), they are displayed in green. For (b): red data points correspond to values calculated using the accepted crystal structure. Second-degree polynomial lines of best fit: δ11/ppm = 0.00321(Vcell)2 − 3.00(Vcell) + 626; R2 = 1.00; δ33/ppm = δ22/ppm = 0.00194(Vcell)2 − 1.77(Vcell) + 286; R2 = 0.999. | ||
4. Conclusions
Using a variety of modern experimental NMR spectroscopy techniques, including cross-polarization and population transfer, 43Ca solid state NMR data for a broad selection of common calcium-containing materials have been acquired and interpreted. Importantly, by acquiring data at both standard (9.4 and 11.75 T) and very high (21.1 T) applied magnetic fields, and by using MAS and static data acquisition, we were able to measure 13 calcium chemical shift tensors, which roughly triples the amount of data available in the literature. For a few select cases, modest isotopic enrichment in 43Ca (ca. 7%) was used to enable very high quality data acquisition. By coupling our experimental data with GIPAW DFT computations using accepted crystal structures, we were able to calculate, with reasonably high accuracy, the EFG and magnetic shielding tensors for nearly all of the samples considered. These results indicate that 43Ca solid-state NMR experiments would be valuable probes for distinguishing polymorphs (for example, α- vs. β-calcium formate) and hydrates (for example, CaCl2 and calcium tartrate hydrates). While demonstrated for nearly all of the samples considered here using only 1D NMR experiments, extracting full tensor information for systems with many calcium chemical sites will be exceptionally challenging due to resolution issues. It is also shown for two samples used herein, that the local calcium structure for natural samples of aragonite from different sources is retained. We also demonstrated, using Ca(OH)2 and CaCrO4 as representative examples, that both the calcium EFG tensor and the calcium chemical shift tensor are extremely sensitive probes of modest structural changes in the unit cell volume and the average calcium–oxygen first coordination sphere distance. As such, with further continued advances in solid-state NMR apparatus and signal enhancement techniques, these NMR observables could potentially find general application in NMR crystallographic structural refinement protocols, and potentially even structural determinations.Acknowledgements
C.M.W. and D.L.B. are grateful to the Natural Sciences and Engineering Research Council (NSERC) for funding. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh-Field NMR Facility for Solids (Ottawa, Canada), a national research facility funded by the Canada Foundation for Innovation, the Ontario Innovation Trust, Recherche Québec, the National Research Council Canada, and Bruker BioSpin and managed by the University of Ottawa (http://www.nmr900.ca). NSERC is acknowledged for a Major Resources Support grant. We are also grateful for the technical support provided by Dr V. Terskikh and Dr E. Ye at the high-field NMR facility and by Dr Tara Kell (uOttawa core X-ray facility) and Mr Kevin M. N. Burgess for recording the powder XRD spectra. We thank Mr Dominic Aebi for early work on this project, which resulted in the synthesis of the racemic calcium tartrate tetrahydrate sample.References
- Calcium: A Matter of Life or Death, ed. J. Krebs and M. Michalak, Elsevier, Amsterdam, 2007 Search PubMed.
- D. W. Rooklin, M. Lu and Y. Zhang, J. Am. Chem. Soc., 2012, 134, 15595–15603 CrossRef CAS PubMed.
- M. B. Johny, P. S. Yang, H. Bazzazi and D. T. Yue, Nat. Commun., 2013, 4, 1717 CrossRef PubMed.
- P. Yuan, M. D. Leonetti, Y. Hsiung and R. MacKinnon, Nature, 2012, 481, 94–97 CrossRef CAS PubMed.
- S. Forsén and B. Lindman, Annu. Rep. NMR Spectrosc., 1981, 11, 183–226 CrossRef.
- H. Wang, J. S. Tse, K. Tanaka, T. Iitaka and Y. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6463–6466 CrossRef CAS PubMed.
- W. Schnelle, A. Ormeci, A. Wosylus, K. Meier, Y. Grin and U. Schwarz, Inorg. Chem., 2012, 51, 5509–5511 CrossRef CAS PubMed.
- K. Kanetani, K. Sugawara, T. Sato, R. Shimizu, K. Iwaya, T. Hitosugi and T. Takahashi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19610–19613 CrossRef CAS PubMed.
- Y.-L. Li, W. Luo, Z. Zeng, H.-Q. Lin, H.-k. Mao and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9289–9294 CrossRef CAS PubMed.
- J. S. Wixey and B. D. Ward, Chem. Commun., 2011, 47, 5449–5451 RSC.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58–71 CrossRef CAS PubMed.
- B. Liu, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 13259–13264 CrossRef CAS PubMed.
- J.-M. Begouin and M. Niggemann, Chem. – Eur. J., 2013, 19, 8030–8041 CrossRef CAS PubMed.
- C. B. Minella, S. Garroni, D. Olid, F. Teixidor, C. Pistidda, I. Lindemann, O. Gutfleisch, M. D. Baró, R. Bormann, T. Klassen and M. Dornheim, J. Phys. Chem. C, 2011, 115, 18010–18014 Search PubMed.
- H. Chu, G. Wu, Y. Zhang, Z. Xiong, J. Guo, T. He and P. Chen, J. Phys. Chem. C, 2011, 115, 18035–18041 CAS.
- S. Horike, Y. Kamitsubo, M. Inukai, T. Fukushima, D. Umeyama, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 4612–4615 CrossRef CAS PubMed.
- D. L. Bryce, Dalton Trans., 2010, 39, 8593–8602 RSC.
- D. Laurencin and M. E. Smith, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 68, 1–40 CrossRef CAS PubMed.
- I. L. Moudrakovski, Annu. Rep. NMR Spectrosc., 2013, 79, 129–240 CrossRef CAS.
- T. Andersson, T. Drakenberg, S. Forsén, E. Thulin and M. Swärd, J. Am. Chem. Soc., 1982, 104, 576–580 CrossRef CAS.
- J. M. Aramini, T. Drakenberg, T. Hiraoki, Y. Ke, K. Nitta and H. J. Vogel, Biochemistry, 1992, 31, 6761–6768 CrossRef CAS.
- H. J. Vogel and S. Forsén, Biol. Magn. Reson., 1987, 7, 249–309 CAS.
- T. B. Coplen, J. K. Böhlke, P. De Bièvre, T. Ding, N. E. Holden, J. A. Hopple, H. R. Krouse, A. Lamberty, H. S. Peiser, K. Révész, S. E. Rieder, K. J. R. Rosman, E. Roth, P. D. P. Taylor, R. D. Vocke Jr. and Y. K. Xiao, Pure Appl. Chem., 2002, 74, 1987–2017 CrossRef CAS.
- G. M. Bowers and R. J. Kirkpatrick, J. Am. Ceram. Soc., 2009, 92, 545–548 CrossRef CAS PubMed.
- S. E. Ashbrook, Phys. Chem. Chem. Phys., 2009, 11, 6892–6905 RSC.
- J. Autschbach, S. Zheng and R. W. Schurko, Concepts Magn. Reson., Part A, 2010, 36, 84–126 Search PubMed.
- V. I. Bakhmutov, Chem. Rev., 2011, 111, 530–562 CrossRef CAS PubMed.
- C. Fernandez and M. Pruski, Top. Curr. Chem., 2012, 306, 119–188 CrossRef CAS.
- T. Bräuniger and M. Jansen, Z. Anorg. Allg. Chem., 2013, 639, 857–879 CrossRef.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, Amsterdam, 2002 Search PubMed.
- A. Wong, P. M. Aguiar, T. Charpentier and D. Sakellariou, Chem. Sci., 2011, 2, 815–818 RSC.
- R. Dupree, A. P. Howes and S. C. Kohn, Chem. Phys. Lett., 1997, 276, 399–404 CrossRef CAS.
- Z. Lin, M. E. Smith, F. E. Sowrey and R. J. Newport, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 224107 CrossRef.
- A. Wong, A. P. Howes, R. Dupree and M. E. Smith, Chem. Phys. Lett., 2006, 427, 201–205 CrossRef CAS PubMed.
- C. Gervais, D. Laurencin, A. Wong, F. Pourpoint, J. Labram, B. Woodward, A. P. Howes, K. J. Pike, R. Dupree, F. Mauri, C. Bonhomme and M. E. Smith, Chem. Phys. Lett., 2008, 464, 42–48 CrossRef CAS PubMed.
- R. G. Bryant, S. Ganapathy and S. D. Kennedy, J. Magn. Reson., 1987, 72, 376–378 CAS.
- D. Laurencin, A. Wong, J. V. Hanna, R. Dupree and M. E. Smith, J. Am. Chem. Soc., 2008, 130, 2412–2413 CrossRef CAS PubMed.
- A. F. Armington and J. J. O'Connor, in Inorganic Syntheses, ed. D. H. Busch, John Wiley & Sons, Toronto, 1980, vol. 20, pp. 1–8 Search PubMed.
- P. R. Bodart, J.-P. Amoureux, Y. Dumazy and R. Lefort, Mol. Phys., 2000, 98, 1545–1551 CrossRef CAS.
- A. P. M. Kentgens and R. Verhagen, Chem. Phys. Lett., 1999, 300, 435–443 CrossRef CAS.
- S. Prasad, H.-T. Kwak, T. Clark and P. J. Grandinetti, J. Am. Chem. Soc., 2002, 124, 4964–4965 CrossRef CAS PubMed.
- K. Eichele and R. E. Wasylishen, WSolids1, version 1.19.11, Universität Tübingen, Tübingen, Germany, 2009 Search PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- (a) M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed; (b) T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1–20 CrossRef CAS PubMed; (c) C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Adiga, D. Aebi and D. L. Bryce, Can. J. Chem., 2007, 85, 496–505 CrossRef CAS.
- L. Desgranges, D. Grebille, G. Calvarin, G. Chevrier, N. Floquet and J.-C. Niepce, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 812–817 Search PubMed.
- E. A. Klop, A. Schouten, P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 51–53 CrossRef.
- N. Burger, H. Fuess and S. A. Mason, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1968–1970 CrossRef.
- A. Le Bail, D. Bazin, M. Daudon, A. Brochot, V. Robbez-Masson and V. Maisonneuve, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 350–354 CAS.
- A. LeClaire and M. M. Borel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1608–1610 CrossRef.
- E. N. Caspi, B. Pokroy, P. L. Lee, J. P. Quintana and E. Zolotoyabko, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 129–132 CAS.
- P. Comodi, S. Nazzareni, P. F. Zanazzi and S. Speziale, Am. Mineral., 2008, 93, 1530–1537 CrossRef CAS.
- G. Weber and K. J. Range, Z. Naturforsch., B: Chem. Sci., 1996, 51, 751–753 CAS.
- R. Ali and M. Yashima, J. Solid State Chem., 2005, 178, 2867–2872 CrossRef CAS PubMed.
- L. Vegard and L. Bilberg, Skr. Nor. Vidensk.-Akad., [Kl.] 1: Mat.-Naturvidensk. Kl., 1931, 12, 1–22 Search PubMed.
- J. A. Alonso, M. Retuerto, J. Sánchez-Benítez and M. T. Fernández-Díaz, Z. Kristallogr., 2010, 225, 225–229 CrossRef CAS.
- K. M. N. Burgess, Y. Xu, M. C. Leclerc and D. L. Bryce, Inorg. Chem., 2014, 53, 552–561 CrossRef CAS PubMed.
- C. M. Widdifield and D. L. Bryce, Can. J. Chem., 2011, 89, 754–763 CrossRef PubMed.
- D. L. Bryce, E. B. Bultz and D. Aebi, J. Am. Chem. Soc., 2008, 130, 9282–9292 CrossRef CAS PubMed.
- Y.-C. Huang, Y. Mou, T. W.-T. Tsai, Y.-J. Wu, H.-K. Lee, S.-J. Huang and J. C. C. Chan, J. Phys. Chem. B, 2012, 116, 14295–14301 CrossRef CAS PubMed.
- D. L. Bryce, in NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons, West Sussex, United Kingdom, 2009, ch. 20, pp. 289–301 Search PubMed.
- E. A. Harrington, Am. J. Sci., 1927, 13, 467–479 CrossRef CAS.
- H. D. Megaw, Proc. R. Soc. London, Ser. A, 1933, 142, 198–214 CrossRef CAS.
- W. R. Busing and H. A. Levy, J. Chem. Phys., 1957, 26, 563–568 CrossRef CAS PubMed.
- H. E. Petch, Acta Crystallogr., 1961, 14, 950–957 CrossRef CAS.
- O. Chaix-Pluchery, J. Pannetier, J. Bouillot and J. C. Niepce, J. Solid State Chem., 1987, 67, 225–234 CrossRef CAS.
- O. H. Han and E. Oldfield, Inorg. Chem., 1990, 29, 3666–3669 CrossRef CAS.
- A. Trokiner, A. Bessière, R. Thouvenot, D. Hau, J. Marko, V. Nardello, C. Pierlot and J.-M. Aubry, Solid State Nucl. Magn. Reson., 2004, 25, 209–215 CrossRef CAS PubMed.
- I. L. Moudrakovski, R. Alizadeh and J. J. Beaudoin, Phys. Chem. Chem. Phys., 2010, 12, 6961–6969 RSC.
- G. Wu, D. Rovnyak, P. C. Huang and R. G. Griffin, Chem. Phys. Lett., 1997, 277, 79–83 CrossRef CAS.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- T. Watanabe and M. Matsui, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2731–2736 CrossRef.
- H. Post and U. Haeberlen, J. Magn. Reson., 1980, 40, 17–31 CAS.
- H. Schmitt, H. Zimmermann, O. Körner, M. Stumber, C. Meinel and U. Haeberlen, J. Magn. Reson., 2001, 151, 65–77 CrossRef CAS PubMed.
- J. L. Ackerman, J. Tegenfeldt and J. S. Waugh, J. Am. Chem. Soc., 1974, 96, 6843–6845 CrossRef CAS.
- M. E. Stoll, A. J. Vega and R. W. Vaughan, J. Chem. Phys., 1976, 65, 4093–4098 CrossRef CAS PubMed.
- R. Richarz and H. Sauter, J. Magn. Reson., 1983, 52, 308–309 CAS.
- T. Nakai, J. Ashida and T. Terao, J. Chem. Phys., 1988, 88, 6049–6058 CrossRef CAS PubMed.
- A. E. Bennett, C. M. Rienstra, M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef CAS PubMed.
- S. Inagaki, I. Kawamura, Y. Sasaki, K. Yoshida, Y. Kubota and A. Naito, Phys. Chem. Chem. Phys., 2013, 15, 13523–13531 RSC.
- A. Wong, D. Laurencin, G. Wu, R. Dupree and M. E. Smith, J. Phys. Chem. A, 2008, 112, 9807–9813 CrossRef CAS PubMed.
- M. Matsui, T. Watanabe, N. Kamijo, R. L. Lapp and R. A. Jacobson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1081–1086 CrossRef.
- D. L. Bryce and E. B. Bultz, Chem. – Eur. J., 2007, 13, 4786–4796 CrossRef CAS PubMed.
- D. I. Kolokolov, I. S. Glaznev, Y. I. Aristov, A. G. Stepanov and H. Jobic, J. Phys. Chem. C, 2008, 112, 12853–12860 CAS.
- R. Boese and O. Heinemann, Z. Kristallogr., 1993, 205, 348–349 CrossRef CAS PubMed.
- A. J. de Vries and J. Kroon, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1542–1544 CrossRef.
- R. C. Evans, Z. Kristallogr., 1935, 92, 154–155 CAS.
- G. K. Ambady, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 1548–1557 CrossRef CAS.
- F. C. Hawthorne, I. Borys and R. B. Ferguson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2461–2463 CrossRef.
- P. van der Sluis, A. Schouten and A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1922–1924 CrossRef.
- J. Panzer, J. Chem. Eng. Data, 1962, 7, 140–142 CrossRef CAS.
- C. Saury, R. Boistelle, F. Dalemat and J. Bruggeman, J. Chem. Eng. Data, 1993, 38, 56–59 CrossRef CAS.
- Z. Yao, H.-T. Kwak, D. Sakellariou, L. Emsley and P. J. Grandinetti, Chem. Phys. Lett., 2000, 327, 85–90 CrossRef CAS.
- R. Siegel, T. T. Nakashima and R. E. Wasylishen, Concepts Magn. Reson., Part A, 2005, 26, 47–61 Search PubMed.
- F. A. Perras, J. Viger-Gravel, K. M. N. Burgess and D. L. Bryce, Solid State Nucl. Magn. Reson., 2013, 51–52, 1–15 CrossRef CAS PubMed.
- F. C. Hawthorne and R. B. Ferguson, Can. Mineral., 1975, 13, 289–292 Search PubMed.
- C. Bezou, A. Nonat, J.-C. Mutin, A. N. Christensen and M. S. Lehmann, J. Solid State Chem., 1995, 117, 165–176 CrossRef CAS.
- P. Ballirano, A. Maras, S. Meloni and R. Caminiti, Eur. J. Mineral., 2001, 13, 985–993 CrossRef CAS.
- W. F. Cole and C. J. Lancucki, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 921–929 CrossRef.
- G. E. Pake, J. Chem. Phys., 1948, 16, 327–336 CrossRef CAS PubMed.
- W. A. Wooster, Z. Kristallogr., 1936, 94, 375–396 CAS.
- M. Atoji and R. E. Rundle, J. Chem. Phys., 1958, 29, 1306–1311 CrossRef CAS PubMed.
- B. F. Pedersen and D. Semmingsen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1074–1077 CrossRef.
- B. Ravikumar, S. Pandiarajan and S. Athimoolam, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o596 CAS.
- J. H. Clouse, Z. Kristallogr., 1930, 76, 285–286 CAS.
- J. H. Clouse, Z. Kristallogr., 1932, 83, 161–171 CAS.
- M. A. M. Forgeron and R. E. Wasylishen, Magn. Reson. Chem., 2008, 46, 206–214 CrossRef CAS PubMed.
- H. D. Megaw, Proc. Phys. Soc. London, 1946, 58, 133–152 CrossRef CAS.
- H. F. Kay and P. C. Bailey, Acta Crystallogr., 1957, 10, 219–226 CrossRef CAS.
- H. J. A. Koopmans, G. M. H. van de Velde and P. J. Gellings, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1323–1325 CrossRef.
- S. Sasaki, C. T. Prewitt, J. D. Bass and W. A. Schulze, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1668–1674 CrossRef.
- X. Liu and R. C. Liebermann, Phys. Chem. Miner., 1993, 20, 171–175 CrossRef CAS.
- B. J. Kennedy, C. J. Howard and B. C. Chakoumakos, J. Phys.: Condens. Matter, 1999, 11, 1479–1488 CrossRef CAS.
- T. J. Bastow, P. J. Dirken, M. E. Smith and H. J. Whitfield, J. Phys. Chem., 1996, 100, 18539–18545 CrossRef CAS.
- D. Padro, V. Jennings, M. E. Smith, R. Hoppe, P. A. Thomas and R. Dupree, J. Phys. Chem. B, 2002, 106, 13176–13185 CrossRef CAS.
- D. Padro, A. P. Howes, M. E. Smith and R. Dupree, Solid State Nucl. Magn. Reson., 2000, 15, 231–236 CrossRef CAS.
- B. Ribar and V. Divjaković, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1546–1548 CrossRef CAS.
- A. Leclaire and J.-C. Monier, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1861–1866 CrossRef.
- A. Leclaire, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 235–238 CrossRef.
- A. Leclaire, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1950–1953 CrossRef.
- L. Vegard, Z. Phys., 1922, 9, 395–410 CrossRef CAS.
- F. C. Kracek, S. B. Hendricks and E. Posnjak, Nature, 1931, 128, 410–411 CrossRef CAS.
- E. Zintl and A. Harder, Z. Elektrochem. Angew. Phys. Chem., 1935, 41, 33–52 CAS.
- J. Bergsma and B. O. Loopstra, Acta Crystallogr., 1962, 15, 92–93 CrossRef CAS.
- A. F. Andresen, A. J. Maeland and D. Slotfeldt-Ellingsen, J. Solid State Chem., 1977, 20, 93–101 CrossRef CAS.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Díaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed.
- A. F. Wallace, L. O. Hedges, A. Fernandez-Martinez, P. Raiteri, J. D. Gale, G. A. Waychunas, S. Whitelam, J. F. Banfield and J. J. de Yoreo, Science, 2013, 341, 885–889 CrossRef CAS PubMed.
- L. Kabalah-Amitai, B. Mayzel, Y. Kauffmann, A. N. Fitch, L. Bloch, P. U. P. A. Gilbert and B. Pokroy, Science, 2013, 340, 454–457 CrossRef CAS PubMed.
- J. C. Johnston, R. J. Iuliucci, J. C. Facelli, G. Fitzgerald and K. T. Mueller, J. Chem. Phys., 2009, 131, 144503 CrossRef PubMed.
- B. K. Sahoo, Phys. Rev. A: At., Mol., Opt. Phys., 2009, 80, 012515 CrossRef.
- C. M. Widdifield and D. L. Bryce, Phys. Chem. Chem. Phys., 2009, 11, 7120–7122 RSC.
- F. A. Perras and D. L. Bryce, J. Phys. Chem. C, 2012, 116, 19472–19482 CAS.
- N. F. Ramsey, Phys. Rev., 1950, 78, 699–703 CrossRef CAS.
- N. F. Ramsey, Phys. Rev., 1951, 83, 540–541 CrossRef CAS.
- N. F. Ramsey, Physica, 1951, 17, 303–307 CrossRef CAS.
- C. M. Widdifield and R. W. Schurko, Concepts Magn. Reson., Part A, 2009, 34, 91–123 Search PubMed.
- D. L. Bryce and R. E. Wasylishen, Phys. Chem. Chem. Phys., 2002, 4, 3591–3600 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01180e |
| ‡ Current address: Max-Planck-Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany. |
| This journal is © the Owner Societies 2014 |