 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe nature of chlorine-inhibition of photocatalytic degradation of dichloroacetic acid in a TiO2-based microreactor
M.
Krivec
ab,
R.
Dillert
c,
D. W.
Bahnemann
c,
A.
Mehle
a,
J.
Štrancar
a and
G.
Dražić
abd
aJožef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
bJožef Stefan International Postgraduate School, Jamova 39, SI-1000 Ljubljana, Slovenia
cInstitut für Technische Chemie, Gottfried Wilhelm Leibniz Universität Hannover, Callinstrasse 3, D-30167 Hannover, Germany
dNational Institute for Chemistry, Hajdrihova 19, SI-1000 Ljubljana, Slovenia
First published on 30th April 2014
Abstract
Photocatalytic degradation of dichloroacetic acid (DCA) was studied in a continuous-flow set-up using a titanium microreactor with an immobilized double-layered TiO2 nanoparticle/nanotube film. Chloride ions, formed during the degradation process, negatively affect the photocatalytic efficiency and at a certain concentration (approximately 0.5 mM) completely stop the reaction in the microreactor. Two proposed mechanisms of inhibition with chloride ions, competitive adsorption and photogenerated-hole scavenging, have been proposed and investigated by adsorption isotherms and electron paramagnetic resonance (EPR) measurements. The results show that chloride ions block the DCA adsorption sites on the titania surface and reduce the amount of adsorbed DCA molecules. The scavenging effect of chloride ions during photocatalysis through the formation of chlorine radicals was not detected.
Introduction
Photocatalytic degradation of polluted groundwater, hazardous chemical substances and toxic industrial wastes has become an important field of advanced oxidation processes (AOPs). In the majority of studies and applications, TiO2 in its nano-form is considered to be the best candidate for the efficient photocatalytic process due to its high efficiency, physical and chemical stability, low cost and low toxicity.1,2 The majority of photocatalytic reactions in aqueous media take place in slurry reactors with TiO2 catalyst in the form of dispersed particles. These systems have a uniform catalyst distribution throughout the whole reactor volume and high surface-to-volume ratio. On the other hand, suspended particles have to be separated from the products and recycled at the end of the process, which decreases the economic viability of this approach. Systems with immobilized titania permit a continuous use of the photocatalyst without the need for post-process particle recovery. TiO2 immobilized reactors have several drawbacks; the main one comes from the limited amount of exposed titania surface area and consequently small surface-to-volume ratio, which leads to mass-transfer limitations.3,4 In order to exploit photocatalysis with immobilized titania, new reactor designs have to be developed where this process could be intensified to an extent similar to slurry reactors.Microreactors have already proven to be highly effective tools when dealing with photochemical and photocatalytic reactions. Their characteristics with laminar flow, short molecular diffusion distances, high surface-to-volume ratios and good light penetration through the entire reactor give processes inside microreactors an advantage over conventional reactor systems. Moreover, microreactors can be optimized to their final version in a research laboratory while they do not need a scale-up: using several units in parallel (numbering-up) provide the possibility of working with larger volumes for the potential continuous industrial operations. Photocatalytic microreactors with immobilized TiO2 have high catalytic surface-to-volume ratios (in the order of 7000 to 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 m−3) which are in the same range as in the slurry reactors. Even more, the actual illuminated catalytic surface-to-volume ratios are considerably higher than those in the suspended titania systems due to the strong absorbing properties of TiO2 particles and consequently rapidly decreasing penetration depth of UV light in the slurry reactors.5
000 m2 m−3) which are in the same range as in the slurry reactors. Even more, the actual illuminated catalytic surface-to-volume ratios are considerably higher than those in the suspended titania systems due to the strong absorbing properties of TiO2 particles and consequently rapidly decreasing penetration depth of UV light in the slurry reactors.5
The effect of inorganic ions on the photocatalytic activity of TiO2 and photocatalytic degradation of organic pollutants has been discussed in many papers.6–15 In many of them an inhibiting effect of inorganic anions, such as Cl−, NO3−, PO43−, SO42−, was reported due to two possible effects. The first one is the competitive adsorption between the pollutant molecules and ions for the surface active sites of TiO2 which leads to reduced photocatalytic efficiency of the process. The second one is the potential scavenging function of anions: they can react with photogenerated holes and consequently reduce the number of oxidizing species for the degradation of pollutant molecules. Moreover, inorganic anions (e.g. Cl−) are often side products during photocatalytic degradation of a large number of organics (e.g. chlorinated organic molecules) which could inhibit the further degradation process. These findings were obtained using conventional slurry reactors with suspended particles and with the addition of different amounts of inorganic salts. According to Lindner et al., inorganic ions such as Cl− and NO3− could react with photogenerated holes and could be oxidized to a radical state while, on the other hand, PO43− and SO42− ions, due to the fact that their central atoms are already in their highest oxidation states, most likely block the surface sites for the adsorption of organic molecules.6 Chen et al. confirmed the competitive adsorption of various ions, including Cl−, with the adsorption of dichloroethane in aqueous titania suspensions: as the adsorption constants for dichloroethane decreased significantly when the system contained inorganic salts, a half-time smaller inhibiting effect was observed in the photocatalytic degradation.8 Calza et al. report that the inhibition of the photocatalytic degradation in the presence of halide ions is mostly due to the competition with organic molecules for the oxidative species (i.e. photogenerated holes) rather than the geometrical occupation of the adsorption sites.10 The true nature of inhibition with inorganic ions and its impact on the photocatalytic degradation has not yet been investigated on a micro-level with a limited amount of absolute photocatalytic surface area inside a microreactor set-up.
In this study, a highly efficient microreactor with an immobilized TiO2 nanoparticle/nanotube dual layer was used for the degradation of dichloroacetic acid (DCA). The concentrations of DCA in its ionic state (at pH 3) and chloride ions formed during the photocatalytic process were analysed with high-performance ionic chromatography (HPIC) while the mechanism of inhibition was determined using a combination of adsorption isotherms and electron paramagnetic resonance (EPR) measurements.
Results and discussion
Microreactor and photocatalyst characterization
Fig. 1 shows the design of the TiO2-based microreactor. A 390 mm-long and 0.5 mm-wide serpentine microchannel was immobilized with an approximately 10 μm-thick TiO2 nanoparticle/nanotube film. The SEM micrograph in Fig. 1 reveals the surface structure of the film, which consists of nanotubes with an average diameter of 100 nm that are surrounded and covered with an additional thin layer of small TiO2 nanoparticles. The X-ray diffraction pattern (not shown) shows the presence of single-phase anatase with some additional peaks that correspond to the titanium substrate. The prepared microreactor has a considerably smaller reactor volume (70 μL), a relatively high catalytic surface-to-volume ratio (6777 m2 m−3) and a high temperature stability (0.3 K h−1) under long-term operating conditions. | ||
| Fig. 1 A scheme of the microreactor with a top-view SEM micrograph of the TiO2 nanoparticle/nanotube dual layer. | ||
Photocatalytic degradation of DCA
The results of photodegradation of DCA at different hydrodynamic residence times, calculated from the equation τ = V/qV, where V is the reactor volume (70 μL) and qV is volumetric flow rate, are shown in Fig. 2a. The degradation of 1 mM DCA concentration is linear to a certain residence time depending on the intensity of the UV light: 2.5 min for low-intensity and 0.5 min for high-intensity UV source. When these residence times are achieved, no further degradation is observed. In both cases, the reactor degrades approximately 24% of the initial DCA concentration which suggests that either the remaining quantity of DCA molecules have diffusivity restrictions or that the reaction is somehow inhibited. The degradation of a higher initial quantity of DCA (1.5 mM) showed similar behaviour as in previous experiments: the degradation is linear until 16% of DCA is decomposed while further reaction is no longer effective. These results show that, independent of the DCA concentration and UV intensity, the microreactor degrades only approximately 0.24 mM quantity of DCA. The results in Fig. 2b show the concentration of chloride ions formed during DCA degradation at different hydrodynamic residence times. These results confirm previously reported findings as the chloride ions concentration increases linearly and according to the degradation stoichiometry to a certain point (roughly at around 0.5 mM) while after that the quantity does not change any more.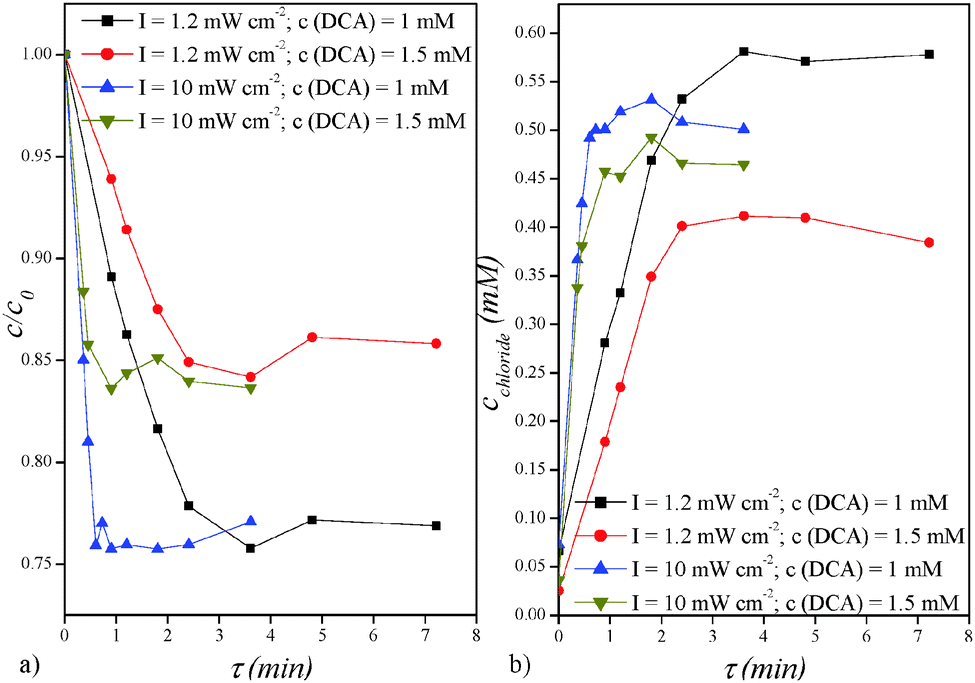 | ||
| Fig. 2 (a) Photocatalytic degradation of DCA and (b) the formation of chloride ions in the microreactor for two initial concentrations of DCA (1 and 1.5 mM) and UV intensities (1.2 and 10 mW cm−2). | ||
According to the reaction stoichiometry, the concentration of H+ ions increases from 1 mM (pH 3) to around 1.25 mM (pH 2.9) at the highest degradation of DCA. The small decrease in the pH has most probably no evident effect on the reaction kinetics and the complete deactivation of the photocatalytic process.
Several papers report the inhibiting effect of inorganic ions on the photocatalytic degradation of different pollutants.6,8,11 In order to evaluate the impact of the chloride ions which are formed during the degradation of DCA, experiments were conducted with two different initial concentrations of NaCl in the initial DCA solution. The results in Fig. 3 show the evident inhibition with chloride ions; in comparison to the DCA degradation experiment without NaCl where the reactor degraded a maximum of 24%, only around 10% of DCA with 0.3 mM concentration of NaCl decomposes and even less (5%) with 0.5 mM concentration of NaCl. Similarly, the formation of chloride ions ends at the same concentration (0.55 mM) in the cases of the samples with 0 and 0.3 mM NaCl while a slightly higher concentration (0.63 mM) is monitored with the highest amount of NaCl.
 | ||
| Fig. 3 (a) Photocatalytic degradation of DCA (1 mM) and (b) the formation of chloride ions in the microreactor in the presence of different concentrations of NaCl (0, 0.3 and 0.5 mM) at low-power UV intensity (1.2 mW cm−2). | ||
The same results were obtained using a high-power UV intensity (Fig. 4). Based on these results, we can confirm that when a certain (critical) concentration of chloride ions is present in our reaction system, the degradation of DCA cannot be initiated further. Moreover, results in Fig. 5 show the inhibiting effect of chloride ions on the initial reaction rates of DCA degradation (−r0) and chloride formation (r0). Initial reaction rates of DCA degradation decrease from 1.7 × 10−6 to 0.4 × 10−6 mol L−1 s−1 at low-power UV intensity and from 6.6 × 10−6 to 0.6 × 10−6 mol L−1 s−1 at high-power UV intensity after introducing 0.5 mM quantity of NaCl. The initial reaction rates of chloride ions formation are approximately 2 times higher than the rates of DCA degradation which is in agreement with the stoichiometry of the DCA degradation reaction.
 | ||
| Fig. 4 (a) Photocatalytic degradation of DCA (1 mM) and (b) the formation of chloride ions in the microreactor in the presence of different concentrations of NaCl (0, 0.3 and 0.5 mM) at high-power UV intensity (10 mW cm−2). | ||
 | ||
| Fig. 5 (a) Initial reaction rates (−r0) of DCA degradation (1 mM) and (b) initial reaction rates (r0) of chloride ions formation in the microreactor in the presence of different concentrations of NaCl (0, 0.3 and 0.5 mM) at two different UV intensities (1.2 and 10 mW cm−2). | ||
Inhibition mechanism of chloride ions
The influence of inorganic ions on the photocatalytic activity of systems containing TiO2 as the heterogeneous catalyst has been discussed in numerous papers.6,8,11,17 The majority of them are convinced that these ions, which are present in the solutions as salts or as degradation side products, inhibit the efficiency of the photocatalytic degradation of different organic molecules. They have proposed two possible inhibition mechanisms: the first one is a competitive adsorption of inorganic ions on the TiO2 active surface which blocks adsorption sites for the target molecules.8 The second is a possible scavenging effect of the adsorbed inorganic ions which consume photogenerated holes and, therefore, decrease the efficiency of the photocatalytic process.6,17 Moreover, according to Zalazar et al.,18 the direct attack of the photogenerated hole is the most significant oxidative step in the photocatalytic oxidation of DCA, which confirms the great importance of the adsorption ability of DCA and hole quantity for an efficient photocatalytic mineralization. In this study, both mechanisms were studied; competitive adsorption was evaluated with DCA adsorption isotherms while the scavenging effect of chloride ions was determined with EPR measurements.In our case, all experiments were conducted in an acidic environment (pH 3); therefore, based on the pH of the TiO2 isoelectric point (6.3) and the pKa value of the DCA (1.29), positively charged titania surface provides a good environment for the adsorption of DCA in its anionic form.6,10 Moreover, chloride anions can adsorb on the titania surface and sterically block the adsorption sites for DCA. In order to determine the effect of chloride anions on the DCA adsorption, adsorption isotherms were experimentally obtained following the adsorption of DCA molecules on the surface of commercial Degussa (Evonik) P25 powder without NaCl and in the presence of 0.5 mM concentration of NaCl, separately. Degussa P25 powder was used instead of the previously reported immobilized TiO2 nanoparticle/nanotube layer mainly because of its higher suspended surface area which gave us the ability to experimentally detect the adsorbed quantity of the DCA molecules on the titania surface. Moreover, our previous reports confirm that the adsorption coefficients of a different molecule (caffeine) are very similar in the reaction using a microreactor or slurry reactor with suspended Degussa P25 powder indicating a good correlation between the different set-ups and titania samples.16
The adsorption isotherms were described with a Langmuir adsorption model:8
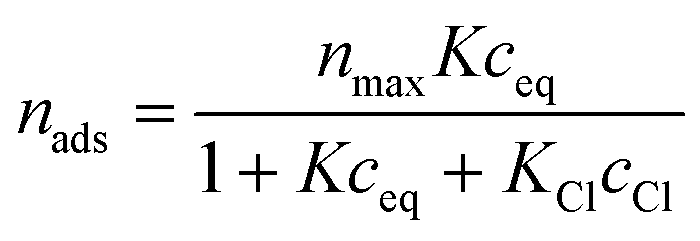 | (1) |
The linear transformation of the Langmuir adsorption model at constant concentration of chloride ions is described as:
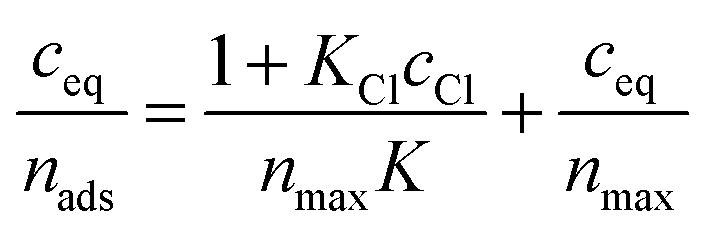 | (2) |
 versus ceq gives a linear plot with a slope equal to
versus ceq gives a linear plot with a slope equal to  and an intercept equal to
and an intercept equal to 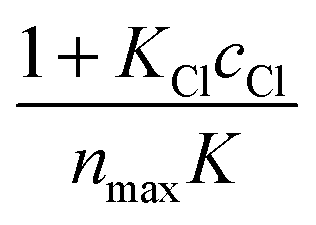 .
.
The adsorbed quantity of DCA molecules at individual equilibrium concentrations and Langmuir adsorption parameters are presented in Fig. 6 and Table 1. Maximal adsorption quantity of DCA per mass of the photocatalyst in the presence of NaCl (0.022 mmol g−1 TiO2) is nearly two times lower than in the case of samples without the presence of the salt (0.043 mmol g−1 TiO2). This confirms that DCA anions compete with chloride ions for the adsorption sites on the catalytic surface which results in a smaller amount of adsorbed DCA molecules. While chloride ions clearly reduce the number of DCA adsorption sites on TiO2, the adsorption constant for DCA stays the same (1.64 L mmol−1) and an adsorption constant for the chloride ion of 0.69 L mmol−1 was determined from the Langmuir adsorption model.
 | ||
| Fig. 6 Adsorbed quantity of DCA molecules (nads) on Degussa P25 particles at individual equilibrium concentrations of DCA (ceq). | ||
| C Cl (mM) | n max (mmol g−1 TiO2) | K (L mmol−1) | K Cl (L mmol−1) |
|---|---|---|---|
| 0 | 0.043 | 1.64 | — |
| 0.5 | 0.022 | 1.64 | 0.69 |
| hVB+ + Cl− → Cl˙ | (3) |
| Cl˙ + Cl− → Cl2˙− | (4) |
| Cl2˙− + eCB− → 2Cl− | (5) |
There have already been reports on the detection of chlorine radicals in non-aqueous media with EPR spectrometry.19–21 In our study, EPR measurements were conducted to detect the presence of chlorine radicals during the photocatalytic process by using the PBN spin-trap in chloroform solution. According to the literature, chlorine radicals react with the PBN spin-trap (I) and form a chlorinated PBN–Cl˙ radical (II) with hyperfine splitting constants: aN ≈ 12.7 G, aH ≈ 0.82 G and aCl ≈ 5.1–6.2 G (depending on the type of Cl isotope).19,21

For the detection of the scavenging mechanism with chloride ions, BTACl (the source of chloride ions) and PBN were dissolved in chloroform and illuminated on the photocatalytic TiO2 surface. The spectrum in Fig. 7b shows a spectrum superimposed of two weak double triplets corresponding to two spin adducts A and B with the weight of 83% and 17%, respectively, and hyperfine splitting constants of aN ≈ 14.1 G and aH ≈ 2.3 G, for species A, and aN ≈ 15.8 G and aH ≈ 4.5 G, for species B, respectively. These signals are attributed to the degradation products of the PBN spin-trap, while the characteristic hyperfine splitting constants of the PBN–Cl˙ radical were not identified. The latter (PBN–Cl˙ radical) should be identified with smaller hyperfine coupling at N and with characteristic larger spin values of the Cl nucleus which can be easily identified through additional splitting. Since the latter was not observed, this proves that the photocatalytic production of chlorine radicals from chloride ions in our system is not efficient enough for the detection of chlorine radicals with EPR spectrometry. In order to evaluate the sensitivity of the photocatalytic experiment on the TiO2 surface, a similar one was conducted using PBN in aqueous solution. Fig. 7d shows a characteristic EPR spectrum of PBN–OH˙ (III) radicals (aN ≈ 16.0 G, aH ≈ 3.2 G); therefore, the experimental set-up was appropriate and sensitive enough for the detection of OH˙ radicals during the photocatalysis. These results suggest that the chlorine radical formation is not the primary process of the photogenerated-hole consumption in the microreactor, although the existence of chlorine radical species during the photocatalytic process cannot be completely excluded.
 | ||
| Fig. 7 EPR spectra of samples prepared with 20 minute UVA illumination of (a) BTACl (0.1 M) and PBN (0.1 M) in chloroform solution; (b) BTACl (0.1 M) and PBN (0.1 M) in chloroform solution on TiO2 layer; (c) PBN (0.1 M) in deionized water solution and (d) PBN (0.1 M) in deionized water solution on the TiO2 layer. | ||
Experimental and methods
Chemicals
Titanium foil (Ti, 99.6 wt%, annealed, with the dimensions of 25 mm × 25 mm × 1 mm, Goodfellow Cambridge Ltd, Huntingdon, England), poly(methyl methacrylate) sheet (PMMA, Plexiglas grade, UV-transparent, with the dimensions of 25 mm × 25 mm × 2 mm, Acrytech d.o.o., Ljubljana, Slovenia), fluorinated ethylene propylene tubing (FEP, technical grade, of appropriate length, with an internal diameter of 0.5 mm, Vici AG International, Schenkon, Switzerland), titanium(IV) chloride (TiCl4, 99.9 wt%, Acros Organics, New Jersey, USA), ethylene glycol (EG, 99.5 wt%, Carlo Erba Reagents, Val de Reuil, France), ammonium fluoride (NH4F, reagent grade, Kemika d.d., Zagreb, Croatia) were used for the synthesis of the TiO2 film and the fabrication of the microreactor, described below.Dichloroacetic acid (DCA, >99 wt%, Sigma-Aldrich Chemie GmbH, Steinheim, Germany), sodium chloride (Fluka Chemie GmbH, Buchs, Switzerland), sodium carbonate (>99.8 wt%, Carl Roth GmbH + Co. KG, Karlsruhe, Germany) and sodium bicarbonate (>99.8 wt%, Fluka Chemie GmbH, Buchs, Switzerland) were used for the photocatalytic measurements and adsorption isotherms experiments without further purification. Deionized water was used to dissolve DCA prior the measurements while the pH 3 was adjusted using perchloric acid (70 wt%, Sigma-Aldrich Chemie GmbH, Steinheim, Germany) and sodium hydroxide (>99 wt%, Carl Roth GmbH + Co. KG, Karlsruhe, Germany).
N-tert-Butyl-α-phenylnitrone (PBN, ≥98%, Enzo Life Sciences, New York, USA), benzyltrimethylammonium chloride (BTACl, 95 wt%, Sigma-Aldrich Chemie GmbH, Steinheim, Germany), trichloromethane (>99.5 wt%, AppliChem GmbH, Gatersleben, Germany) and ethanol (absolute, Carlo Erba Reagents, Milano, Italy) were used for the electron paramagnetic resonance (EPR) measurements without further purification.
Construction and synthesis of TiO2-based microreactor
The detailed description of its construction and TiO2-immobilization was described in our previous work.16In brief, a serpentine micro-channel was engraved in a titanium foil using a high-precision CNC milling machine. The inner walls of the micro-channel were immobilized with a two-step synthesis. Firstly, the titanium-based flow microreactor was anodized in the ethylene glycol solution with a small portion of NH4F (0.3 wt%) at 60 V for 3 h. Secondly, the anodized microreactor was hydrothermally treated at 75 °C for 1 h in a Teflon-lined stainless-steel autoclave, filled with a water suspension of TiCl4 (0.2 M). Thirdly, the prepared microreactor was thermally treated at 400 °C for 3 h, covered with UV-transparent Plexiglas, and sealed with epoxy glue. The whole fabricated device was mounted in a stainless-steel housing with 4 integrated UV-LED diodes (Roithner Lasertechnik GmbH, Vienna, Austria) with the emission maximum at 365 nm.
The TiO2 film was investigated with a focused-ion-beam workstation (FIB, Helios NanoLab 650, FEI, Hillsboro, Oregon, USA).
Photocatalytic degradation experiments
The photocatalytic activity of the TiO2-based microreactor was characterized with the degradation of DCA. DCA has been chosen as a degradation molecule because of its relatively strong chloro-organic acidity and its presence in the industrial wastewaters. In aqueous solutions at pH > 2, DCA exists in an anion form and it photocatalytically degrades into two molecules of carbon dioxide, one proton and two chloride ions:22 | (6) |
Afterwards, the concentrations of DCA and chloride ions in the product solution were monitored by a high-performance ionic chromatography apparatus (HPIC, Dionex ICS-1000) equipped with conductivity detector (ICS-1000) and a solvent delivery pump. The isocratic elution at the constant flow rate of 0.3 mL min−1 was used in a separation column (Dionex 003461), using a water solution of sodium carbonate (8 mM) and sodium bicarbonate (1.5 mM) as a solvent.
Adsorption isotherms experiments
Different concentrations of DCA (from 0.05 to 2 mM) were prepared in a 50 mL plastic centrifuge flasks. 5 mL Aliquots were taken from the initial solutions to determine the initial concentration of DCA. Afterwards, 0.09 g of a commercial TiO2 powder (Degussa P25) was added into each solution, its pH was adjusted to 3 and the suspension was put under stirring at 250 rpm in the dark. 5 mL Aliquots of each suspension were taken out after 24 hours of stirring and filtered through cellulose acetate syringe filters. In order to analyse the impact of chloride ions on the DCA adsorption, parallel experiments were conducted with 0.5 mM sodium chloride in the initial DCA samples. DCA and chlorine concentrations were analysed by HPIC.EPR experiments
A piece of 0.5 cm × 0.5 cm titanium sheet with an immobilized TiO2 nanoparticle/nanotube layer was placed in the indent on the polyethylene plate. For the detection of chlorine radicals during a photocatalytic process, 3 μl of 1 M PBN solution and 27 μl of 0.1 M BTACl solution dissolved in chloroform were poured on top of the TiO2 surface. The polyethylene plate with the sample was placed inside a petri dish, partially filled with chloroform (to obtain a chloroform-saturated environment), and then irradiated with a UV-LED diode with a maximum at 360 nm for 20 min. For the detection of hydroxyl radicals during a photocatalytic process, similar experiments were conducted using 3 μl of the 1 M PBN solution and 27 μl of deionized H2O on the titania surface in the absence of chloroform saturation. Control samples were irradiated without the TiO2 surface and/or left in the dark for 20 min. Finally, the samples were suctioned into quartz glass capillaries. The electron paramagnetic resonance (EPR) spectra of the samples were analyzed using an EPR spectrometer (Bruker Elexsys E500). All measurements were recorded at room temperature using 1 G modulation amplitude, 100 kHz modulation frequency, 20 ms time constant, 15 scans with scan time 20 s, 20 mW microwave power and 150 G sweep width with center field positioned at 3320 G.Conclusions
Photocatalytic degradation of DCA was investigated in a continuous-flow regime inside a microreactor with an immobilized TiO2 nanoparticle/nanotube film. This system degrades a maximum of 0.24 mM DCA concentration independently from the initial concentrations, UV intensities and volumetric residence times. The reason for incomplete degradation of DCA molecules comes from the presence of chloride ions that are formed during the photocatalytic degradation; when chloride ions reach a certain limiting concentration at around 0.5 mM, the photocatalytic degradation inside the TiO2-based microreactor stops. Chloride ions compete with DCA molecules for the TiO2 adsorption sites: adsorbed ions sterically block the adsorption of DCA and inhibit the photocatalytic process. While these ions can theoretically react with photogenerated holes and form chlorine radicals, we did not detect any with EPR spectrometry. Therefore, the reduction of the DCA adsorption due to chloride ions is most probably the key inhibition mechanism in our system.Acknowledgements
This work was financially supported by the Slovenian Research Agency within the program P2-0084 and project J2-4309. The work is part of the PhD thesis of M. Krivec under Grant no. PR-03769. The financial support from the scholarship (Ad Futura) provided by the Slovene Human Resources Development and Scholarship Fund is gratefully acknowledged.Notes and references
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed.
- A. K. Ray and A. A. C. M. Beenackers, AIChE J., 1998, 44, 477–483 CrossRef CAS.
- H. de Lasa, B. Serrano and M. Salaices, Photocatalytic Reaction Engineering, Springer Science + Business Media, New York, 2005 Search PubMed.
- T. Van Gerven, G. Mul, J. Moulijn and A. Stankiewicz, Chem. Eng. Process., 2007, 46, 781–789 CrossRef CAS PubMed.
- M. Lindner, B. Hirthe, W. D. Griebler and D. W. Bahnemann, J. Sol. Energy Eng., 1997, 119, 120–125 CrossRef CAS.
- W. Zhang, T. An, M. Cui, G. Sheng and J. Fu, J. Chem. Technol. Biotechnol., 2005, 80, 223–229 CrossRef CAS.
- H. Y. Chen, O. Zahraa and M. Bouchy, J. Photochem. Photobiol., A, 1997, 108, 37–44 CrossRef CAS.
- N. Kashif and F. Ouyang, J. Environ. Sci., 2009, 21, 527–533 CrossRef CAS.
- P. Calza and E. Pelizzetti, Pure Appl. Chem., 2001, 73, 1839–1848 CrossRef CAS.
- C. Guillard, E. Puzenat, H. Lachheb, A. Houas and J.-M. Herrmann, Int. J. Photoenergy, 2005, 7, 1–9 CrossRef CAS.
- R. Yuan, S. Fan, H. Zhou, Z. Ding, S. Lin, Z. Li, Z. Zhang, C. Xu, L. Wu, X. Wang and X. Fu, Angew. Chem., Int. Ed., 2013, 52, 1035–1039 CrossRef CAS PubMed.
- R. Yuan, T. Chen, E. Fei, J. Lin, Z. Ding, J. Long, Z. Zhang, X. Fu, P. Liu, L. Wu and X. Wang, ACS Catal., 2011, 1, 200–206 CrossRef CAS.
- M. Lewandowski and D. F. Ollis, J. Catal., 2003, 217, 38–46 CAS.
- N. Petit, A. Bouzaza, D. Wolbert, P. Petit and J. Dussaud, Catal. Today, 2007, 124, 266–272 CrossRef CAS PubMed.
- M. Krivec, K. Žagar, L. Suhadolnik, M. Čeh and G. Dražić, ACS Appl. Mater. Interfaces, 2013, 5, 9088–9094 CAS.
- J. Moser and M. Grätzel, Helv. Chim. Acta, 1982, 65, 1436–1444 CrossRef CAS.
- C. S. Zalazar, C. A. Martin and A. E. Cassano, Chem. Eng. Sci., 2005, 60, 4311–4322 CrossRef CAS PubMed.
- E. G. Janzen, H. J. Stronks, D. E. Nutter, Jr., E. R. Davis, H. N. Blount, L. J. Poyer and P. B. McCay, Can. J. Chem., 1980, 58, 1596–1598 CrossRef CAS.
- D. Rehorek, E. G. Janzen and Y. Kotake, Can. J. Chem., 1991, 69, 1131–1133 CrossRef CAS.
- T. H. Walter, E. E. Bancroft, G. L. McIntire, E. R. Davis, L. M. Gierasch and H. N. Blount, Can. J. Chem., 1982, 60, 1621–1636 CrossRef CAS.
- D. W. Bahnemann, S. N. Kholuiskaya, R. Dillert, A. I. Kulak and A. I. Kokorin, Appl. Catal., B, 2002, 36, 161–169 CrossRef CAS.
| This journal is © the Owner Societies 2014 |