 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical predictions of isotope effects versus their experimental values for an example of uncatalyzed hydrolysis of atrazine†
Anna
Grzybkowska
,
Rafal
Kaminski
and
Agnieszka
Dybala-Defratyka
*
Institute of Applied Radiation Chemistry, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, Lodz, Poland. E-mail: agnieszka.dybala-defratyka@p.lodz.pl; Fax: +48 42 631 3199; Tel: +48 42 631 3198
First published on 2nd June 2014
Abstract
Kinetic isotope effects are one of the most powerful experimental techniques for establishing the nature of a chemical process. However their interpretation very often seeks support from electronic structure calculations in order to get detailed information regarding the transition state which is not experimentally available. For an example of atrazine hydrolysis we have shown how the match between experimentally and theoretically determined magnitudes of carbon, nitrogen and chlorine kinetic isotope effects can be used to discuss the mechanism under different reaction conditions. Two different density functionals combined with the explicit presence of solvent molecules and a continuum solvation model revealed that although the reaction proceeds via the same concerted mechanism regardless of the reaction conditions the transition state structure for an acid and base-catalyzed pathway is different.
Introduction
The elucidation of the detailed microscopic level mechanisms of chemical reactions is of fundamental importance in chemistry. Reaction dynamics studies provide this type of information as well as macroscopic kinetic data, i.e., thermal rate constants k(T).1 This information is available for reactants that differ only by their isotopic composition, providing in this manner a unique way of probing the nature of the chemical reactions' transition states. Hence, measurement of kinetic isotope effects (KIEs) is one of the most powerful experimental techniques for establishing the nature of a chemical process. However, very often in order to interpret them we seek support from theory. But since models we construct have certain and inevitable limitations it is advisable to test our selection of models, levels of theory and calculations against the available experimental data whenever possible. Unfortunately, there are no many systems described in literature for which we would have access to both experimental data on more than one element and its theoretical predictions. A few examples2–4 cover a small number of important reactions studied with the use of multiple isotope effects as a mechanistic tool. Such comparison and detailed analysis can be of particular interest in the era of using isotopic fingerprints in the description of environmental transformations,5 especially that concomitant analysis of more than one element is becoming more and more abundant in such studies these days.6Our previous, similar in its nature, study4 aimed at detailed exploration of IEs accompanying standard SN2 reaction between the cyanide anion and ethyl chloride using both experimental and theoretical approaches. The outcome from those studies clearly indicated the lack of the ability to reproduce all experimental KIEs determined in that work and called for further analysis. The follow-up study has been provided recently by Ruiz-Pernia and Williams7 and pointed to one of possible reasons of previous failure.
In this work we have made an attempt to test the performance of popular theoretical methods toward prediction of isotope effects for hydrolysis of atrazine (2-chloro-4-(N-ethylamino)-6-(N-isopropylamino)-1,3,5-s-triazine) – one of the s-triazine compounds, which were introduced into the environment more than half century ago as dyes, resins and herbicides.8–10 Especially herbicides based on the s-triazine ring have been used worldwide by direct application either to soils or plants. Their widespread presence and environmental accumulation leading to certain dysfunctions in vertebrate species or toxic conditions to other species present in the shared contaminated area have resulted in many laboratory studies aiming at their degradation.11 Atrazine degradation occurs via both chemical and biological processes, resulting in the formation of metabolites such as hydroxyatrazine, deethylatrazine and deisopropylatrazine.12 Hydroxyatrazine is considered to degrade much faster than atrazine so conversion of atrazine to this product alone seems to be sufficient to consider the herbicide detoxified.13 Uncatalyzed hydrolysis of atrazine has been postulated to occur via nucleophilic aromatic substitution reaction (Scheme 1). However the details of the mechanism have not been revealed to date. Although the first attempts toward their elucidation have been already undertaken using carbon and nitrogen isotopic analysis14 they did not answer key questions on this biomolecular reaction such as whether it proceeds as a single step reaction in a SN2-like manner or a stable intermediate so called a Meisenheimer complex is formed or whether any mechanistic scenario is influenced by reaction conditions. Despite providing useful information, the magnitudes of carbon and nitrogen kinetic isotope effects (KIEs) measured for the hydrolysis of atrazine did not allow for the satisfactory description of underlying mechanisms as by definition they were not able to capture directly the changes in the carbon–chlorine bond – one of the key interatomic distances in the studied reaction. Therefore, the further investigation of atrazine hydrolysis is crucial in order to propose the transition state structures of respective pathways. Such information at the molecular level is only accessible through electronic structure calculations. An insight provided this way may serve as a reference system for the enzymatic hydrolysis as based on the obtained data the understanding of catalytic potential of triazine chlorohydrolases will be possible.
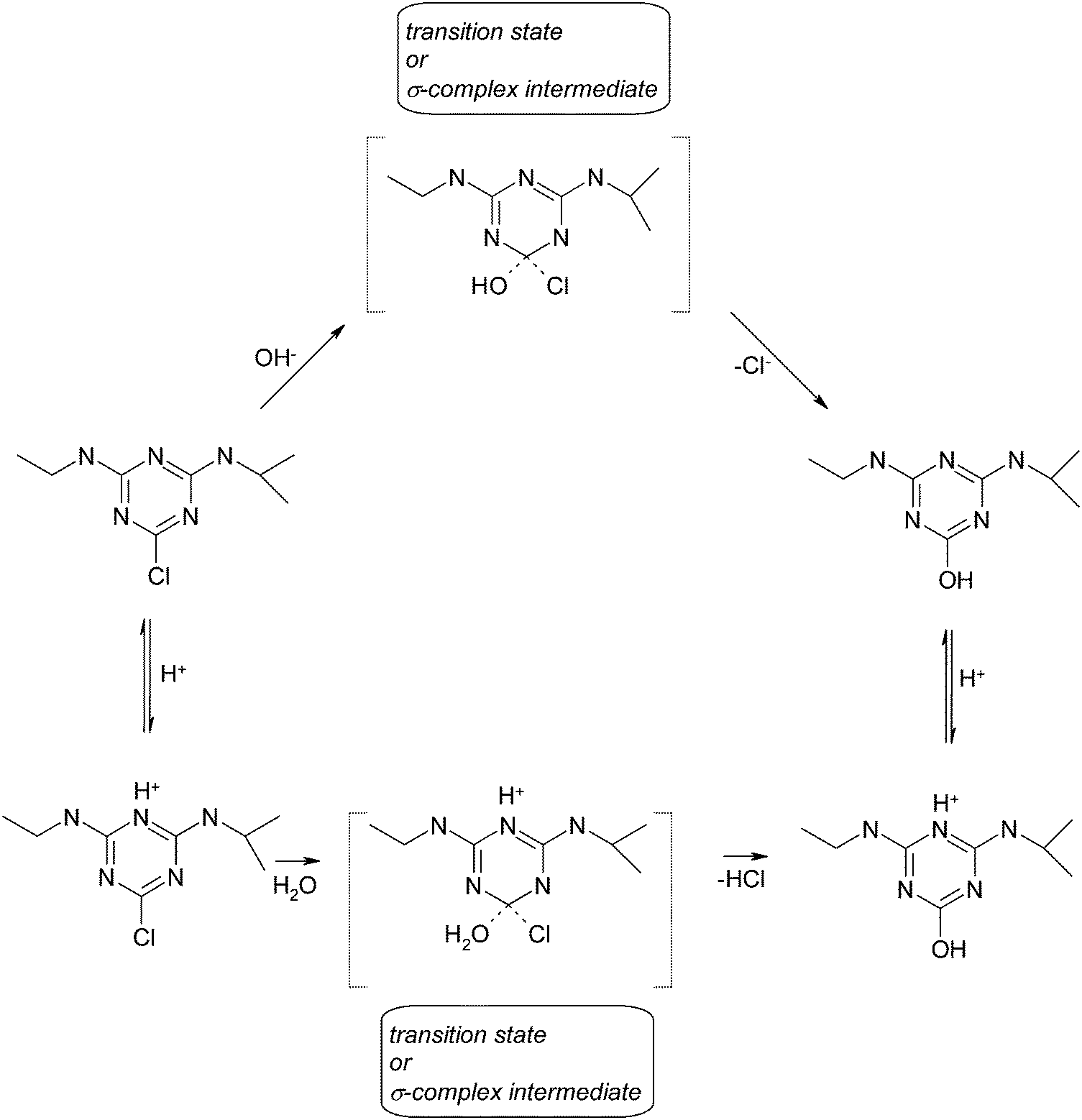 | ||
| Scheme 1 Postulated possible pathways of atrazine hydrolysis. | ||
To this end we determined chlorine KIEs using hybrid fast atom bombardment – isotope ratio (FAB-IR) mass spectrometry and modeled the reaction using the density functional theory methods and computational techniques. Chlorine KIEs have already proved to be very useful in elucidating the reaction mechanism in which negatively charged chloride is the leaving group.15 Although the magnitude of these effects is not large and therefore their range is not broad, the available precision with which these effects can be measured nowadays16–18 for more and more different reactions confirms their usefulness in mechanistic studies of chemical and biochemical reactions.16,19,20
Experimental methods
Materials and preparation of samples
Nitric acid (pure p.a.), silver nitrate (pure p.a.), ethanol (special pure), chloroform (pure p.a.), sodium dihydrogen phosphate monohydrate (pure p.a.), disodium hydrogen phosphate anhydrous from POCh Gliwice, mercuric(II) thiocyanate (pure p.a.), ion-exchange cation form DOWEX 50W X8 200–400 mesh from Fluka, ferric ammonium sulfate (L.P.P-H “OCh” Lublin. Poland), potassium hydroxide pellets GR for analysis (Merck), trisodium phosphate (pure p.a.), ammonia solution 25% (pure p.a.), n-hexane (pure p.a.) from Chempur, 1-hexanol (pure p.a., Sigma-Aldrich) were used without further purification.Atrazine (pure p.a., ZChO-Azot, Jaworzno, Poland) was purified by extracting into trichloromethane, washed four times with water, dried with magnesium sulfate, precipitated with n-hexane and dried in a desiccator. Due to very low solubility in water about 5 mg of atrazine was dissolved in 10 ml of 99.8% solution of ethanol and then this solution was used to prepare the final atrazine solution in 20 mM potassium phosphate buffer. The final concentration of atrazine was in the range of 110–121 μM. This difference resulted from different weights of atrazine used during preparation of its solution in ethanol. The solutions were prepared at pH 12.
Chlorine KIE measurements
In order to prepare samples for isotopic ratio determination 30 ml solutions at pH 12 were sealed in 10 ml glass ampoules and immersed in the water bath at 50 °C. The elevated temperature was used to decrease the reaction time.14 At the same time the reactions were also conducted in 2 ml glass ampoules which were used for monitoring the reaction progress. 2 ml ampoules were withdrawn from baths at various intervals of time estimated based on the available experimental rate constants.21 In order to determine the reaction progress removed ampoules were opened and the chloride ion concentration was measured using the colorimetric method in which the absorbance of the iron thiocyanate complex was measured.22 This complex is formed as a result of the reaction between chloride ions and mercury thiocyanate. Since the molar amount of forming chloride ions is equal to the molar amount of degrading atrazine, the atrazine concentration was determined using the chloride ion concentration from the following equation:| CAtr = C0 − CCl | (1) |
At the desired reaction progress (15–20% of conversion) the reaction was quenched by removing from the water bath and cooling down in a refrigerator. Then, the 10 ml ampoules were opened and all aliquots were mixed in one flask to obtain a sufficient amount of chlorine for further analysis.
In order to remove organic content, the resultant solution was extracted with 1-hexanol at pH around 12 which was obtained by adding 0.1 M KOH. The extraction was carried out four times. Next, the solution was concentrated using a vacuum dryer overnight. The concentrated solution (around 1.5 ml) was acidified to pH 1 with 65% HNO3 and AgCl was precipitated with 200 mM AgNO3 in the dark. After 12–24 hours the resultant precipitate was centrifuged (2000–3000 rpm) and washed twice with 1% HNO3. Then, AgCl was left to dry over NaOH in a desiccator in the dark.
The isotopic ratios were measured using CsCl solutions. For this purpose the AgCl precipitate was dissolved in 100 μl of 5% solution of NH4OH and the resultant solution was passed through a set of ion exchange columns. Each column was filled with DOWEX 50W X8 and then nitric acid (0.1 M) solution was passed through them. The first column with NH4+ resin was washed with ammonia solution (5%). In this column silver cations were exchanged for ammonium cations. The second column filled with H+ resin was washed with 0.1 M nitric acid. The third column with Cs+ resin was prepared by passing 0.1 M cesium nitrate solution. The final eluent was collected in a quartz crucible and subsequently evaporated to dryness in a vacuum dryer (50 °C, 130 mbar) overnight. The crystallized precipitate was dissolved in 40 μl of ultra pure water. Then, 1 μl of this solution was used to measure chloride ion concentration. The solution was transferred to an acid resistant steel plate and left to evaporate. Chlorine isotopic ratios, R, were measured using hybrid fast atom bombardment – isotope ratio (FAB-IR) mass spectrometer with a modified sample support that results in reduced sample requirement as described previously23 and kinetic isotope effects calculated from the equation:
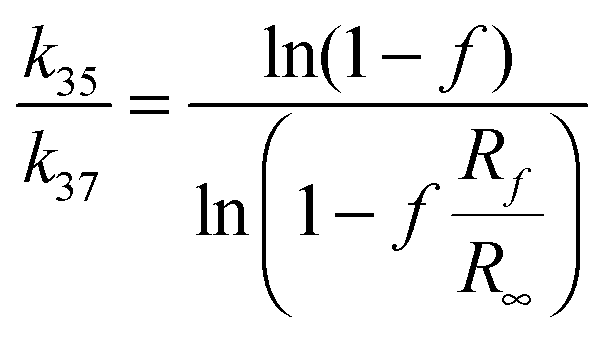 | (2) |
Computational methods
Models
In order to consider different reaction conditions we constructed two models in which either the hydroxide anion (alkaline hydrolysis) or the water molecule (acidic hydrolysis) played a role of the attacking nucleophile. Additionally, in the case of the latter we also explored protonation of the triazine ring. To account for any specific solute-solvent interactions two explicit solvent molecules were added symmetrically to stabilize the base in nucleophilic substitution reactions modeled in this study. A similar approach that allows for improving the match with experiment on one hand and does not require a proper sampling of water molecule positions on the other has been previously successfully used for elimination reactions promoted by OH− as a base.24,25 For the sake of comparison we also constructed a model that mimicked hydrolysis at neutral pH (no ring activation, water molecule attack).Methods
Free energy surfaces for the hydrolysis of atrazine were modeled using the MPW1K functional as the one shown to perform well for nonmetallic thermochemical data and thermochemistry,26 and the BP86 functional which was used by some of us previously in our initial studies on atrazine hydrolysis.27 They were combined with the split valence 6-31+G(d,p) basis set.28 This basis set is meant whenever we mention the level of calculation in this work, unless stated otherwise. All calculations were performed using the Gaussian 09 package.29 Tight convergence and optimization criteria were applied. For all geometry optimizations as well as single point energy calculations an ultrafine grid was used for the integration. Molecular geometries of all species were fully optimized and vibrational analyses were carried out to confirm the nature of the stationary points (3n − 6 real vibrations in the case of reactants and one imaginary frequency corresponding to the desired reaction coordinate in the case of transition states). IRC30 calculations were performed to validate transition states. In order to account for the bulk effect of water solvation we computed single point energies at the MPW1K/6-31+G(d,p) and the BP86/6-31+G(d,p) optimized geometries using the SMD31 solvation model along with the above functionals and the 6-311+G(2df,2p) basis set. Free energies in solution were calculated using zero-point energies and thermal contributions to the Gibbs free energy computed in vacuo using the 6-31+G(d,p) basis set and single point electronic energies calculated using the SMD model and the respective density functional. Harmonic and rigid-rotor approximations were used to obtain Hessians, zero point energies and thermal contributions to the Gibbs free energy. Wiberg bond indices32 were calculated using the NBO module33 implemented in the Gaussian09 package.Position-specific chlorine (k35/k37), carbon (k12/k13) and nitrogen (k14/k15) kinetic isotope effects as well as nitrogen equilibrium isotope effects have been calculated using the Bigeleisen equations34 and ISOEFF07 program.35 We neglected scaling frequencies as well as calculating tunneling correction as we found previously for heavy atom isotope effects prediction that the latter effect and anharmonicity have a tendency to cancel each other out.36
Results and discussion
Energetics of the atrazine hydrolysis
Experimentally determined values of activation energies for reaction under acidic and alkaline conditions are comparable.21,37 Sawunyama and Bailey in their computational studies on hydrolysis under acidic conditions have shown an important role of the solvation model.38 However, the activation energy obtained theoretically was in poor agreement with the available experimental values.We approached the free energy profiles for the hydrolysis of atrazine under different reaction conditions using two levels of calculations. In the case of alkaline hydrolysis both of them predicted one-step, SN2-like pathway for the attack of the hydroxyl anion on the acidic form of atrazine. No minima corresponding to σ-complexes were located on a 1-dimensional projection of the energy surfaces. This finding is consistent with other experimental studies indicating that anions reacting with heteroaromatics react via a single step nucleophilic aromatic substitution in aqueous solution.39 Gibbs free energies of activation and reaction were calculated two-fold. First, we estimated their values for in vacuo models. Then, in order to calculate free energies in aqueous solution we included the SMD correction to the electronic energy of stationary points. Finally, since we wanted to refer our findings to the existing experimental data we used the experimental value for free energy of solvation for water (−4.4 kcal mol−1) and also included the correction for 1 M standard state. The final values are presented in Table 1. In order to estimate the possible range of experimental Gibbs free energies of activation we adopted values of rate constants obtained for atrazine hydrolysis in aqueous alkaline solution of pH 11–13.21 The deduced range of values is 26.5–24.3 kcal mol−1. Also the value of free energy of activation calculated by Burkhard and Guth for the reaction in 0.1 M sodium hydroxide solution fits in this range (24.8 kcal mol−1).37 Our both in vacuo models predicted high energetic barriers, however the SMD and 6-311+G(2df,2p) correction to the gas phase electronic energy did decrease them significantly. The resultant Gibbs free energies of activation are slightly lower than the deduced experimental range (by 3.3 kcal mol−1). However, the estimation of experimental values was based on those determined by other rate constants and they were reported without accompanying errors or standard deviations.
In the case of acidic hydrolysis we explored the triazine ring protonation as it has been postulated to occur prior to nucleophilic attack based on the observed inverse nitrogen isotopic fractionation.14 Taking into consideration that hydrolysis may proceed either through the zwitterionic or acidic form of atrazine (Scheme 2) we included the protonation step in the overall reaction under acidic conditions. Therefore, we first calculated the Gibbs free energy of protonation. For this purpose, we used the fixed-concentration solvation free energy of the proton corrected for 1 mol per 1 L standard state of −264 kcal mol−1.40 The Gibbs free energies of protonation obtained for three possible protonation sites within the triazine ring (N1, N3 and N5) are shown in Table 2.
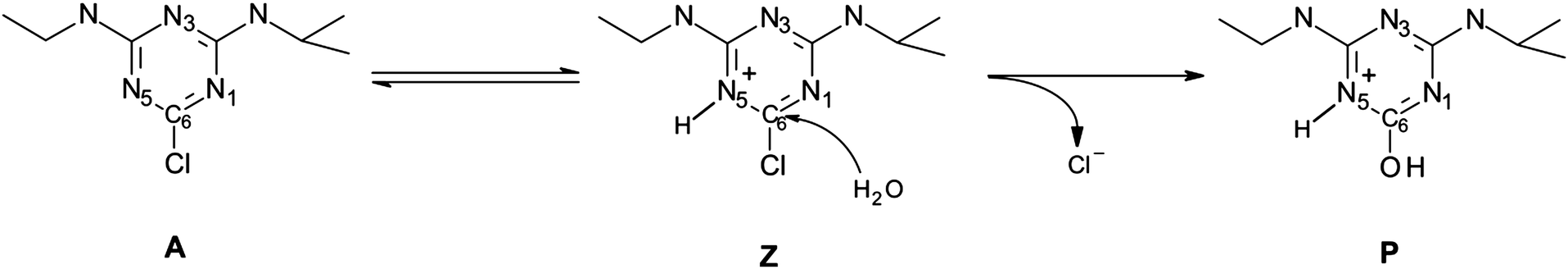 | ||
| Scheme 2 Hydrolysis of atrazine under acidic conditions on an example of the N5 protonation site. | ||
| SMD/MPW1K | SMD/BP86 | |
|---|---|---|
| N1 | 7.2 | 9.3 |
| N3 | 5.7 | 5.6 |
| N5 | 6.6 | 8.6 |
Based on the obtained values it is clear that the equilibrium is shifted toward the acid, A. This finding nicely corresponds to the inverse nitrogen isotope effect obtained for the acidic hydrolysis of atrazine by Meyer et al. which provides an additional proof that not all molecules under acidic conditions undergo immediate protonation in solution. Otherwise normal nitrogen fractionation would be observed. The differences in the Gibbs free energies of protonation observed among the possible protonation sites within the triazine ring clearly indicate a preference of the N3 position (Table 2). Therefore, we followed it in our subsequent modeling of the nucleophilic attack step.
In contrast to the hydrolysis under alkaline conditions, we found a Meisenheimer complex in acidic solution using the MPW1K level of calculations and its formation is the rate determining step of the overall reaction (Fig. 1). A very small energetic barrier (0.3 kcal mol−1) in the gas phase was predicted for the chloride ion elimination step. It disappeared when the energies of the stationary points were corrected for solvation (Fig. 1, black curve). Therefore it is very likely that it would be one transition state comprising both water molecule attachment and chloride elimination acts rather than a stable σ-complex separating two transition states representing these acts individually. Within the BP86 level no intermediate structure was found. Nevertheless the two studied functionals along with the correction for bulk solution predicted the Gibbs free energies of activation in very good agreement with the experimentally observed range21,41 (Table 1) suggesting that the inclusion of a solvent model and extra explicit solvent molecules is important for a correct prediction of energies of the system like the one under study. Both experiment and theory indicate that the hydrolysis of atrazine in alkaline solution proceeds slightly faster than the reaction in acidic solution (Table 1).
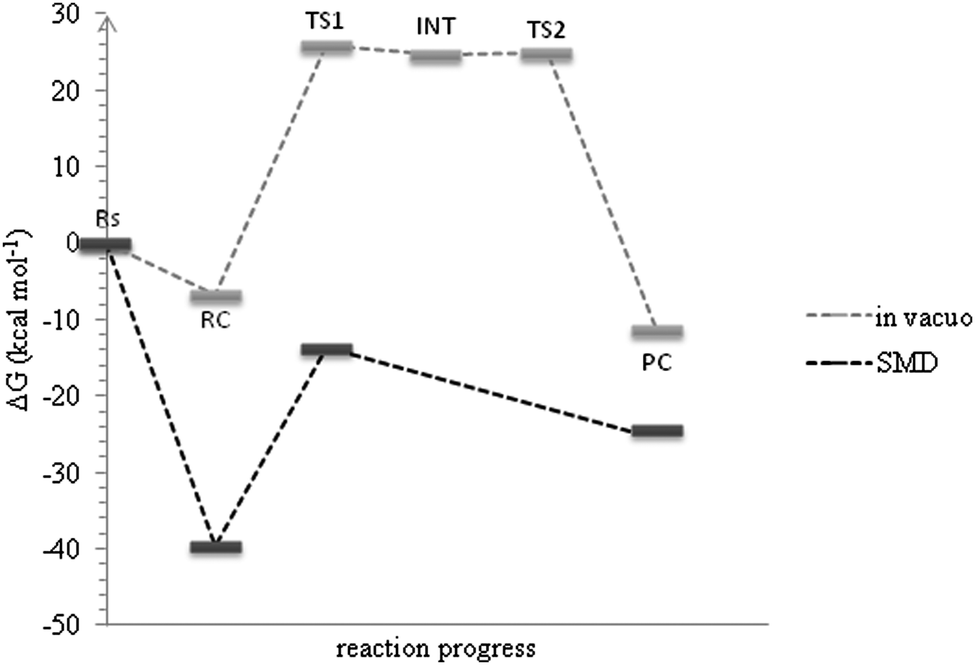 | ||
| Fig. 1 Free energy profiles for the uncatalyzed acidic hydrolysis of atrazine obtained at the MPW1K/6-31+G(d,p) (grey curve) and the SMDwater/MPW1K/6-311+G(2df,2p)//MPW1K/6-31+G(d,p) (black curve) levels of calculations. Rs, RC, TS, INT and PC denote separate reactants, reactants complex, transition state, intermediate and products complex, respectively. All energy values are related to the sum of energies of Rs. | ||
Kinetic isotope effects
Other so far undertaken studies aimed at characterizing the reaction of interest comprised measurements of nitrogen and carbon kinetics isotope effects.14 Carbon KIEs for uncatalyzed hydrolysis under both alkaline and acidic conditions were 1.031 ± 0.003 and 1.040 ± 0.003, respectively. The normal nitrogen kinetic isotope effect (N-KIE) was observed for alkaline hydrolysis which was interpreted as a tight SN2-like transition state. In contrast, inverse N-KIE was observed for acidic reaction which might suggest protonation at a nitrogen atom of the heterocyclic ring. Based on these observations it has been proposed that hydrolysis of atrazine can occur either by a direct substitution of the chlorine atom without increased bonding in the transition state (normal isotope effects) or the bonding involving the element of interest (nitrogen) becomes tighter in the transition state in comparison with the reactants (inverse nitrogen isotope effect).14 Similar magnitude of carbon KIEs obtained under two extremely different reaction conditions did not turn out to be a sufficient mechanistic indicator. Nitrogen isotope analysis might play this role, however, the values of N-KIEs indicate only whether the triazine ring undergoes protonation and if so set up the conditions for the chloride elimination step (increased polarization of the carbon–chloride bond) but do not provide the details of the transition state structures. Therefore, chlorine KIE determination seemed to us to be a complementary and necessary tool for the full description of underlying reaction mechanisms.27,38 The previously determined value of chlorine KIE for the hydrolysis of atrazine at pH 12 was equal to 1.0069.27 This value indicated significant carbon–chlorine bond elongation in the transition state and was also in agreement with the preliminary theoretically predicted value.27 It was however contradictory to the findings from N-KIEs that suggested a tight transition state.14 Hence, verification of this data both experimentally and theoretically and exploration of this discrepancy in more detail became necessary.Meyer et al.14 based on the obtained enrichment factors calculated apparent isotope effects for the transformation of atrazine into hydroxyatrazine under alkaline and acidic conditions at 60 °C. Since in the case of carbon we discuss the primary isotope effect as it is a reaction center (Scheme 2, C6 atom) we adopted the values of 1.031 ± 0.003 and 1.040 ± 0.003 from their work for alkaline and acidic hydrolysis, respectively. On the other hand, the effect on nitrogen is a secondary one so we decided to rely our comparison with the theoretically predicted values on the average nitrogen effect of 1.0010 ± 0.0002 and 0.9975 ± 0.0010 at pH = 12 and 3, respectively. The chlorine kinetic isotope effect measured in this study at 50 °C and pH = 12 is 1.0009 ± 0.0006. All isotope effects predicted for alkaline hydrolysis were calculated on the direct conversion of atrazine to hydroxyatrazine via the respective transition state. The calculated carbon (C6), chlorine and average nitrogen kinetic isotope effects of all five positions are presented in Table 3. Both tested levels of calculations listed in Table 3 gave the results within the experimental confidence interval for carbon14 and chlorine (this work). The derived average N-KIE however although reproduced the experimental direction (normal isotope effect) appeared to be slightly larger than the measured values. This resulted match with experiment is even more noticeable when we compare the magnitudes of isotope effects resulting from a model with none explicit water molecules (ESI,† p. S47).
| C-AKIE | N bulk | Cl-KIE | |
|---|---|---|---|
| a Values taken from ref. 14. | |||
| Level of calculations | |||
| Alkaline hydrolysis | |||
| BP86 | 1.0289 | 1.0021 | 1.0014 |
| MPW1K | 1.0314 | 1.0017 | 1.0003 |
| Experimental values | 1.031 ± 0.003a | 1.001 ± 0.000a | 1.0009 ± 0.0006 |
| Acidic hydrolysis | |||
| BP86 | 1.0322 | 0.9993 | 1.0011 |
| MPW1K | 1.0383 | 0.9982 | 1.0002 |
| Experimental values | 1.040 ± 0.003a | 0.9975 ± 0.0010a | n/d |
In the case of acidic hydrolysis, since the protonation of the triazine ring may be a source of isotopic fractionation we also included it in the prediction of an apparent KIE. Based on the earlier energetic analysis of acidic and zwitterionic forms of atrazine we took into account a transformation scenario in which the acidic form of the substrate is more stable and therefore the overall isotope effect can be calculated from this form.
Taking into account the experimentally determined uncertainty only the C-KIE magnitude predicted using the MPW1K functional lies within the range of measured values. The same observation can be made for nitrogen isotope fractionation. The agreement between the values obtained for the acidic form of atrazine and the experimental value would confirm the usefulness of the nitrogen isotope effect to describe in general the conditions of atrazine hydrolysis and to make prediction regarding the manner in which the chlorine substituent is removed from the atrazine molecule. Furthermore, it could also be an indication of the acidic form prevailing in the mechanism of atrazine hydrolysis in aqueous solution at pH = 2–3. In the case of Cl-KIE the predicted magnitudes are of 1.0010 at most. Based on all the above findings, the chlorine kinetic isotope effect for acidic hydrolysis of atrazine could be then in the range of 1.0000–1.0010. It would be rather a small leaving group KIE for SN2-like reaction40–44 and basically identical to the one predicted for the alkaline hydrolysis of atrazine. This observation could indicate that one should expect that the carbon–chlorine bond in the transition state might be elongated to a similar, almost negligible extent disregarding the reaction conditions.
Geometry of transition state structures
Since the interpretation of isotope effects is inextricably related to the elucidation of a transition state structure we also performed a detailed analysis of saddle points located on the modeled free energy surfaces. Their key properties are presented in Fig. 2. As the MPW1K level of calculations resulted in better agreement with the experimental values of KIEs we discussed the geometry of the transition state structures found using this method in this section. The results for BP86 are provided in the ESI.† In order to explore how different the reaction under neutral conditions would be we also modeled it using the MPW1K level of calculations. As expected we obtained much higher Gibbs free energy of activation of 38.6 kcal mol−1 and carbon, average nitrogen and chlorine kinetic isotope effects of 1.0362, 1.0024 and 1.0045, respectively. The calculated Wiberg bond indices for the carbon–chlorine bond (nC6–Cl) clearly indicate very early transition states in the case of alkaline (n = 1.03) and acidic (n = 1.03) conditions. It is also reflected in the predicted Cl-KIEs. The Wiberg bond index computed for neutral reaction is 0.87 and the carbon–chlorine bond is elongated to 1.87 Å. On the other hand, when analyzing the nucleophilic attack on the C6 atom the similarity of acidic and neutral hydrolysis shows up with respect to the oxygen–carbon distances in the transition state which is reflected in larger C-KIE compared with the alkaline conditions. The difference between acidic and alkaline conditions becomes evident when one looks at the nucleophile's attack site as the O–C6 bond under acidic conditions is almost halfway formed (nO–C6 = 0.45) whereas during alkaline hydrolysis in less than one third (nO–C6 = 0.24).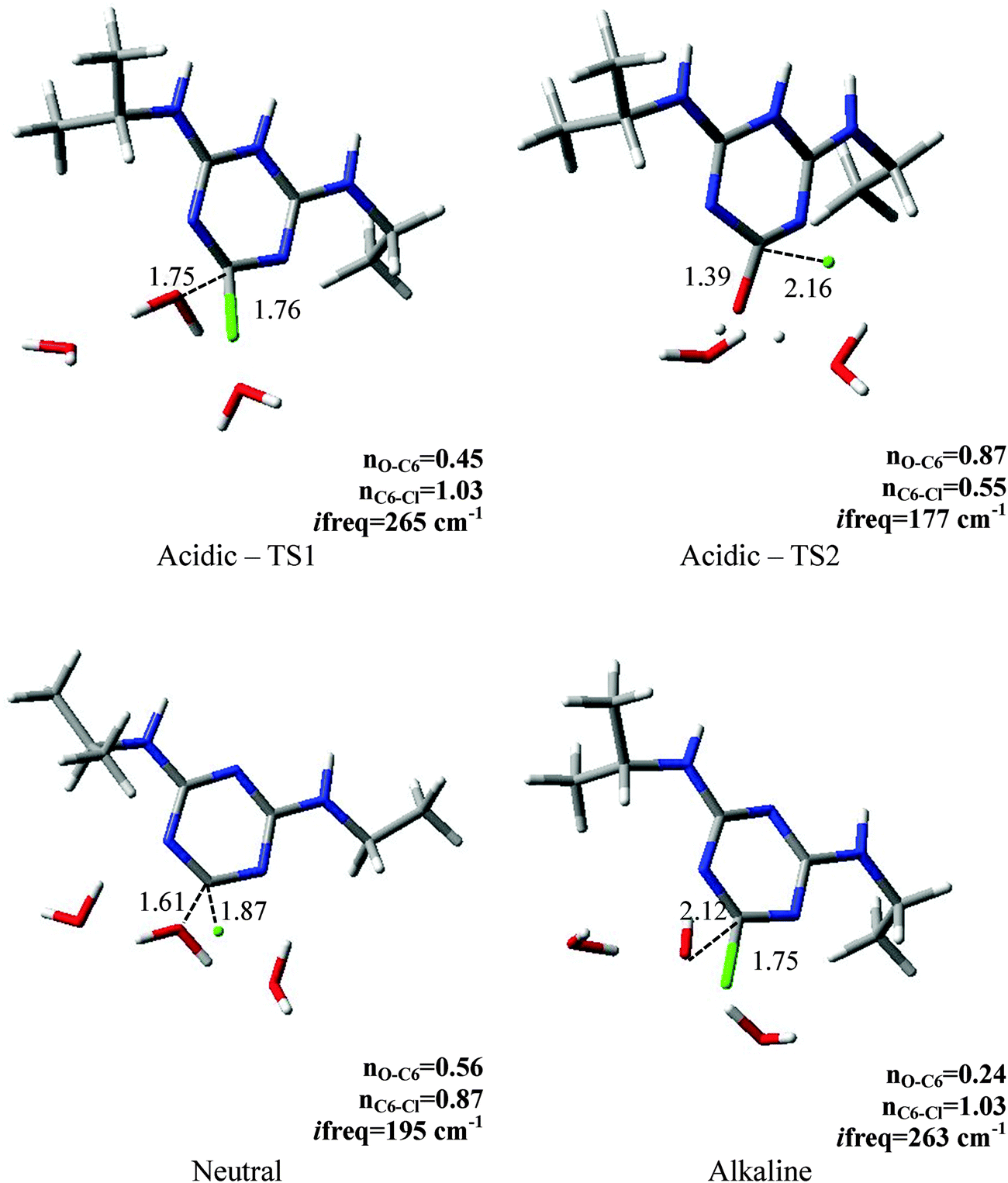 | ||
| Fig. 2 Key parameters for the transition state structures located for acidic, neutral and alkaline hydrolysis using the MPW1K level of calculations. The interatomic distances are given in Å. | ||
Conclusions
Density functionals tested in this study combined with microsolvation allowed for the correct prediction of kinetic isotope effects for three elements (carbon, nitrogen and chlorine). Both primary (carbon and chlorine) and secondary (nitrogen) effects are in qualitative and reasonable quantitative agreement with the same effects determined experimentally. Taking this into account we made an attempt to predict the magnitude of the chlorine kinetic isotope effect for acidic hydrolysis.The uncatalyzed hydrolysis of atrazine is a slow process, especially at neutral pH and at room temperature. Elevated temperatures and pH of solution much lower and greater than 7 can accelerate its rate which was also confirmed in the presented work by predicted Gibbs free energies of activation. Alkaline and acidic hydrolysis occur at similar rates, via the same concerted mechanism but the structure of the transition state is different and largely determined by the distance between the nucleophile oxygen atom and the attacked carbon center of the atrazine ring. This difference affects C-KIE which is slightly smaller for the alkaline hydrolysis, however not to the extent that would allow us to use it as a mechanistic descriptor of the reaction. As stated previously by Meyer et al. N-KIE could play this role but the additional information it provides is limited to the increased bonding to nitrogen atoms in the transition state upon the ring protonation. Therefore, in order to elucidate the full description of the transition state structures for different hydrolysis pathways information coming from electronic structure calculations and predicted chlorine isotopic fractionation was required. The magnitudes of the latter one however do not allow us to utilize it as a mechanistic indicator of the uncatalyzed hydrolysis of atrazine under different reaction conditions alone.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is a part of the project “The mechanism of hydrolysis of s-triazines catalyzed enzymatically”, which is funded by NCN (National Science Centre) Poland based on decision number DEC-2011/01/N/ST4/02461. Access to supercomputing facilities at the University of Minnesota (Minnesota Supercomputing Institute), and Cyfronet (Poland) is gratefully acknowledged.References
- A. Fernández-Ramos, J. A. Miller, S. Klippenstein and D. G. Truhlar, Modeling the kinetics of bimolecular reactions, Chem. Rev., 2006, 106, 4518–4584 CrossRef PubMed.
- M. Skarpeli-Liati, M. Jiskra, A. Turgeon, A. N. Garr, W. A. Arnold, C. J. Cramer, R. P. Schwarzenbach and T. B. Hofstetter, Using Nitrogen Isotope Fractionation to Assess the Oxidation of Substituted Anilines by Manganese Oxide, Environ. Sci. Technol., 2011, 45, 5596–5604 CrossRef CAS PubMed.
- E. S. Burgos, M. J. Vetticatt and V. L. Schramm, Recycling Nicotinamide. The Transition-State Structure of Human Nicotinamide Phosphoribosyltransferase, J. Am. Chem. Soc., 2013, 135, 3485–3493 CrossRef CAS PubMed.
- Y.-r. Fang, Y. Gao, P. Ryberg, J. Eriksson, M. Kolodziejska-Huben, A. Dybala-Defratyka, S. Madhavan, R. Danielsson, P. Paneth, O. Matsson and K. C. Westaway, Experimental and theoretical multiple kinetic isotope effects for an SN2 reaction. An attempt to determine transition-state structure and the ability of theoretical methods to predict experimental kinetic isotope effects, Chem. – Eur. J., 2003, 9, 2696–2709 CrossRef CAS PubMed.
- M. Elsner, M. A. Jochmann, T. B. Hofstetter, D. Hunkeler, A. Bernstein, T. C. Schmidt and A. Schimmelmann, Current challenges in compound-specific stable isotope analysis of environmental organic contaminants, Anal. Bioanal. Chem., 2012, 403, 2471–2491 CrossRef CAS PubMed.
- T. Kuder, B. M. van Breukelen, M. Vanderford and P. Philp, 3D-CSIA: Carbon, chlorine and hydrogen isotope fractionation in transformation of TCE to ethene by a Dehalococcoides culture, Environ. Sci. Technol., 2013, 47, 9668–9677 CrossRef CAS PubMed.
- J. J. Ruiz-Pernia and I. H. Williams, Ensemble-Averaged QM/MM Kinetic isotope effects for the SN2 reaction of cyanide anions with chloroethane in DMSO solution, Chem. – Eur. J., 2012, 18, 9405–9414 CrossRef CAS PubMed and references cited therein.
- E. L. Erickson and K. H. Lee, Degradation of atrazine and related s-triazines, Crit. Rev. Environ. Control, 1989, 19, 1–13 CrossRef.
- H. LeBaron, Herbicide resistance in plants, John Wiley & Sons, Inc., New York, N.Y., 1982, vol. 9–19, pp. 99–114 Search PubMed.
- J. M. E. Quirke, 1,3,5-Triazinesin Comprehensive heterocyclic chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, New York, N.Y., 1984, vol. 3, pp. 459–529 Search PubMed.
- J. W. Allran and W. H. Karasov, Effects of atrazine on embryos, larvae, and adults of Anuran amphibians, Environ. Toxicol. Chem., 2001, 20, 769–775 CrossRef CAS.
- M. R. Barrett, The environmental impact of pesticide degrades in groundwater, in Herbicide metabolites in surface water and groundwater, ed. M. T. Meyer and E. M. Thurman, ACS Symp Ser, American Chemical Society, Washington, DC, 1996, pp. 200–225 Search PubMed.
- L. C. Strong, H. McTavish, M. J. Sadowsky and L. P. Wackett, Field-scale of atrazine-contaminated soil using recombinant Escherichia coli expressing atrazine chlorohydrolase, Environ. Microbiol., 2000, 2(1), 91–98 CrossRef CAS.
- A. H. Meyer, H. Penning and M. Elsner, C and N Isotope Fractionation Suggests Similar Mechanisms of Microbial Atrazine Transformation Despite Involvement of Different Enzymes (AtzA and TrzN), Environ. Sci. Technol., 2009, 43, 8079–8085 CrossRef CAS PubMed.
- D. Sicinska, M. Rostkowski and P. Paneth, Chlorine Isotope Effects on Chemical Reactions, Curr. Org. Chem., 2005, 9, 75–88 CrossRef CAS.
- L. Szatkowski, M. K. Thompson, R. Kaminski, S. Franzen and A. Dybala-Defratyka, Oxidative dechlorination of halogenated phenols catalyzed by two distinct enzymes: horseradish peroxidase and dehaloperoxidase, Arch. Biochem. Biophys., 2011, 505, 22–32 CrossRef CAS PubMed.
- S. Cretnik, A. Bernstein, O. Shouakar-Stash, F. Loffler and M. Elsner, Chlorine Isotope Effects from Isotope Ratio Mass Spectrometry Suggest Intramolecular C–Cl Bond Competition in Trichloroethene (TCE) Reductive Dehalogenation, Molecules, 2014, 19, 6450–6473 CrossRef PubMed.
- A. Aeppli, H. Holmstrand, P. Andersson and O. Gustafsson, Direct Compound-Specific Stable Chlorine Isotope Analysis of Organic Compounds with Quadrupole GC/MS Using Standard Isotope Bracketing, Anal. Chem., 2010, 82, 420–426 CrossRef PubMed.
- P. Paneth, Chlorine Kinetic Isotope Effects on Biological Systems, in Isotope Effects in Chemistry and Biology, ed. A. Kohen and H. H. Limbach, CRC Press, Boca Raton, 2006, pp. 875–891 Search PubMed.
- C. Aeppli, D. Bastviken, P. S. Andersson and O. Gustafsson, Chlorine isotope effects and composition of naturally produced organochlorines from chloroperoxidases, flavin-dependent halogenases and in forest soil, Environ. Sci. Technol., 2013, 47, 6864–6871 CrossRef CAS PubMed.
- S. J. Plust, J. R. Loehe, F. J. Feher, J. H. Benedict and H. F. Herbrandson, Kinetics and mechanism of hydrolysis of chloro-1,3,5-triazines, Atrazine, J. Org. Chem., 1981, 46, 3661–3665 CrossRef CAS.
- I. Iwasaki, S. Utsumi, K. Hagino and T. Ozawa, A new spectrophotometric method for determination of small amounts of chloride using the mercuric thiocyanate method, Bull. Chem. Soc. Jpn., 1956, 29, 860–864 CrossRef CAS.
- K. C. Westaway, T. Koerner, Y.-R. Fang, J. Rudzinski and P. Paneth, A new method of determining chlorine kinetic isotope effects, Anal. Chem., 1998, 70, 3548–3552 CrossRef CAS PubMed.
- F. Duarte, S. Gronert and S. C. L. Kamerlin, Concerted or Stepwise: How Much Do Free-Energy Landscapes Tell Us about the Mechanism of Elimination Reactions?, J. Org. Chem., 2014, 79, 1280–1288 CrossRef CAS PubMed.
- Y. Kim, J. R. Mohrig and D. G. Truhlar, Free-Energy Surface for Liquid-Phase Reactions and Their Use to Study the Border between Concerted and Nonconcerted α,β-Elimination Reactions of Esters and Thioesters, J. Am. Chem. Soc., 2010, 132, 11071–11082 CrossRef CAS PubMed.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, Adiabatic Connection for kinetics, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- A. Dybala-Defratyka, L. Szatkowski, R. Kaminski, M. Wujec, A. Siwek and P. Paneth, Kinetic isotope effects on dehalogenations at aromatic carbon, Environ. Sci. Technol., 2008, 42, 7744–7750 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. J. S. Binkley, Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, J. W. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. P. Hratchian and H. B. Schlegel, Using Hessian Updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. C. Bochicchio, L. Lain and A. Torre, On the definition of bond orders at correlated level, Chem. Phys. Lett., 2003, 374, 567–571 CrossRef CAS.
- J. P. Foster and F. Weinhold, Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102(24), 7211–7218 CrossRef CAS.
- J. Bigeleisen, The relative Reaction Velocities of Isotopic Molecules, J. Chem. Phys., 1949, 17, 675–678 CrossRef CAS PubMed.
- V. Anisimov and P. Paneth, ISOEFF98. A program for studies of isotope effects using Hssian modifications, J. Math. Chem., 1999, 26, 75–86 CrossRef.
- P. Adamczyk, A. Dybala-Defratyka and P. Paneth, DFT study of trichloroethene reaction with permanganate in aqueous solution, Environ. Sci. Technol., 2011, 45, 3006–3011 CrossRef CAS PubMed.
- N. Burkhard and J. A. Guth, Chemical hydrolysis of 2-chloro-4,6-bis(alkylamino)-1,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption, Pestic. Sci., 1981, 12, 45–52 CrossRef CAS.
- P. Sawunyama and G. Bailey, Computational chemistry study of the environmentally important acid-catalyzed hydrolysis of atrazine and related 2-chloro-s-triazunes, Pest Manage. Sci., 2002, 58, 759–768 CrossRef CAS PubMed.
- A. H. M. Renfrew, et al., A single transition state in nucleophilic aromatic substitution: reaction of phenolate ions with 2-(4-nitrophenoxy)-4,6-dimethoxy-1,3,5-triazine in aqueous solution, J. Chem. Soc., Perkin Trans. 2, 1993, 1703–1704 RSC.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart and J. V. Coe, The Proton's Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef CAS.
- P. Paneth, Chlorine kinetics isotope effects on enzymatic dehalogenations, Acc. Chem. Res., 2003, 36, 120–126 CrossRef CAS PubMed.
- A. Dybala-Defratyka, M. Rostkowski, O. Matsson, K. C. Westaway and P. Paneth, A new interpretation of chlorine leaving group kinetic isotope effects; A theoretical approach, J. Org. Chem., 2004, 69, 4900–4905 CrossRef CAS PubMed.
- Y.-r. Fang, S. MacMillar, J. Eriksson, M. Kolodziejska-Huben, A. Dybala-Defratyka, P. Paneth, O. Matsson and K. C. Westaway, The Effect of Solvent on the Structure of the Transition State for The SN2 Reaction between Cyanide Ion and Ethyl Chloride in DMSO and THF Probed with Six Different Kinetic Isotope Effects, J. Org. Chem., 2006, 71, 4742–4747 CrossRef CAS PubMed.
- A. Pabis, P. Paluch, J. Szala and P. Panteh, A DFT Study of the Kinetic Isotope Effects on the Competing SN2 and E2 Reactions between Hypochlorite Anion and Ethyl Chloride, J. Chem. Theory Comput., 2009, 5, 33–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables containing Cartesian coordinates of all stationary points described in this work as well as key parameters for the other stationary point structures located for acidic hydrolysis using the MPW1K level of calculations and key parameters for the transition state structures located for acidic, alkaline and neutral hydrolysis using the BP86 level of calculations. See DOI: 10.1039/c4cp00914b |
| This journal is © the Owner Societies 2014 |