 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays?
Matthias
Bauer
Department Chemie, Universität Paderborn, Warburger Str. 100, 33098 Paderborn, Germany. E-mail: matthias.bauer@upb.de; Tel: +49-5251-605614
First published on 6th June 2014
Abstract
In this perspective, the HERFD-XANES (high energy resolution fluorescence detected X-ray absorption near edge structure) and Kβ2,5- or V2C-XES (valence-to-core X-ray emission spectroscopy) methods are discussed as new and powerful tools for chemical research with hard X-rays. This includes a brief survey of the underlying physical processes and the introduction of experimental issues. The potential of both methods to overcome limitations of conventional XAS (X-ray absorption spectroscopy) and to push the limits for obtaining new information about the electronic and geometric structures of metal centers, in the solid state structure or heterogeneous catalysts, but also in metal complexes and homogeneous catalysts, is discussed by presenting a survey of representative references and recent own studies, rounded off by a conclusion and outlook.
 Matthias Bauer | Matthias Bauer is Professor of Inorganic Chemistry at the University of Paderborn (Germany). He studied chemistry in Stuttgart, where he also received his PhD in Physical Chemistry. After a Postdoc in Stuttgart and at the European Synchrotron Radiation Facility (Grenoble, France) he headed the group for Modern spectroscopic methods at the Karlsruhe Institute for Technology from 2010 to 2011. From 2011 to 2013 he was Assistant Professor of Analytics of catalytically active materials at the Technical University of Kaiserslautern (Germany). His work focuses on the development and investigation of sustainable catalytic reactions using synchrotron radiation methods. |
Introduction
Hard X-ray absorption spectroscopy (XAS) is undoubtedly a powerful tool to study chemical processes and structures, e.g. in catalytic reactions, using both XANES (X-ray absorption near edge structure) and EXAFS (extended X-ray absorption fine structure).1–13 In view of the topic of this perspective article these methods are referred to as ‘conventional’ X-ray absorption methods. While conventional XANES provides information on the qualitative or comparative oxidation states via fingerprinting, conventional EXAFS offers the chance to extract the local structure around an X-ray absorbing central catalyst metal, i.e. type, number and distances of coordinating atoms.14 Both XANES and EXAFS are valuable tools especially in chemical research, however they suffer from particular intrinsic limitations.Starting with the XANES region, a lot of structural and electronic information content is found in the pre-edge signal of transition metals, especially of the first transition metal row. Since such prepeaks are caused by a 1s → 3d transition,1,2,15 they basically contain information about the lowest unoccupied (LUMO) states of metal catalysts. These LUMO states unambiguously reflect the geometric (coordination symmetry) and electronic (oxidation state) structure. However, extracting LUMO information from conventional transition metal K-edge XANES spectra is complicated by the core–hole lifetime broadening of the 1s electron hole, which smears out features in the prepeak spectrum. Already in the mid-80s of the last century, Eisenberger et al.16 showed that the lifetime broadening in the XANES region could be reduced by detecting the X-ray absorption spectrum using the intensity of the emitted X-ray fluorescence in a narrow energy bandwidth. This technique was called high-energy-resolution fluorescence detected XANES (HERFD-XANES).17,18 In a pioneering study, this technique was applied by de Groot et al. to investigate iron zeolite catalysts.19 The benefit of recording high resolution XANES spectra becomes most obvious if prepeaks of the first transition metal row are considered, which are more often than not due to quadrupole transitions to unoccupied 3d states, and thus of low intensity.20 An example of a HERFD-XANES spectrum in comparison to a conventional XANES is given in Fig. 1, where the underlying transition is also shown for the example of ferrocene.21
 | ||
| Fig. 1 Top left: Kβ-detected HERFD-XANES spectrum (solid line) of ferrocene in comparison to the corresponding conventional XANES spectrum (dashed line), including the enlarged prepeak area. Bottom left: Kβ emission spectrum composed of the Kβ1,3 main line on the low energy side and the Kβ2,5 emission on the high energy side, which is also enlarged in the inset. Right: electronic transitions involved and studied in the prepeak signal of HERFD-XANES and Kβ2,5-XES for the example of ferrocene. | ||
Obviously, with HERFD-XANES, the LUMO structure at a central atom in catalysts can be probed, giving a precise measure of the symmetry dependent arrangement of molecular orbitals and their occupancy, i.e. a more physical substitute for the oxidation state formalism usually applied in chemistry. It thus presents an extension of the ‘fingerprinting approach’ of conventional XANES analysis concerning the oxidation state and coordination geometry.14 This approach relies on the comparison of the conventional XANES spectrum of an unknown sample with a set of structurally defined references. The coordination geometry is deduced from the prepeak intensity and the oxidation state from the energies of the prepeak and of the absorption edge. Although this approach is still of undoubted value especially in catalytic studies, the extraction of oxidation states can be tedious22 or bear the risk of misinterpretations, if for example the chemical nature of the atoms coordinating to the metal center is not identical to the unknown sample.23 Moreover, in order to make full use of the information provided by hard X-ray in chemistry, it would be desirable to extract more precise measures of the electronic structure.
On the other hand, standard EXAFS analysis suffers from a lack of sensitivity for light atom ligands coordinating a central metal atom. As a consequence, it is not possible to distinguish, for example, carbon, nitrogen and oxygen atoms in the proximity of the metal center. Especially in catalysis, this is a serious drawback: most heterogeneous systems are composed of oxidic materials or catalysts on an oxidic support that are used for the conversion of small molecules consisting mainly of C, H, N, like in Fischer–Tropsch synthesis24–28 or alkyne hydrogenation.29–32 Usually homogeneous catalysts are transition metal complexes composed of H, C, N, O containing ligands that undergo only slight changes in catalytic transformations, which are not detectable by EXAFS.1,2,33 Only in cases where strictly linear ligands like CO34 or [CN]− (ref. 35) are coordinating, strong multiple scattering signals could be used to discriminate such ligands in a multiple scattering analysis against other light ligand atoms.
Valence-to-core (V2C) or V2C X-ray emission spectroscopy (XES)18,36 could overcome these limitations while still using hard X-rays that are of undoubted benefit in many cases concerning the tolerance towards practically every experimental condition. In this technique, the 1s electron is non-resonantly excited into the continuum far off the threshold energy, and the following radiative HOMO → 1s relaxation of a valence electron is detected. These valence electrons originate from the highest occupied molecular orbitals (HOMOs). In a complex, these are formed by interaction of the metal and valence orbitals with ligand orbitals. Consequently, V2C-XES is highly sensitive to the ligands coordinating to a metal center, since the character of the valence orbitals changes the most for different chemical species. An example of a Kβ X-ray emission spectrum is given in Fig. 1, again for the case of ferrocene. Although the Kβ1,3 emission corresponding to the 3p → 1s relaxation channel is also of value for investigations into catalytic systems by providing a measure for the total spin at the metal center, its further discussion is outside the scope of this perspective. The comparatively low transition probability of the V2C emission in comparison to the Kβ1,3 line should nevertheless be noticed. It imposes a particular requirement on the used X-ray source, since a high X-ray flux is mandatory to obtain high quality V2C-XES spectra.
Both HERFD-XANES and V2C have the advantage that spectra can be recorded element specifically, which is beneficial for mixed metal systems as often found in catalysis37 and enzyme chemistry.38 Since they are hard X-ray based, there are few limitations on the sample environment, making high-pressure studies or studies of complexes in solution easily feasible. Such sensitive techniques, practically without restrictions on the sample environment, can find application in several important cases in which in situ changes need to be followed, such as monitoring ligand binding and other transformations in chemical reactions.
With this perspective, a personal account on the still rather rare applications of hard X-ray HERFD-XANES and V2C-XES in catalytic and chemical science will be given after a short introduction into experimental issues. The selected examples will shed light both on heterogeneous and homogeneous catalytic reactions – or in a broader sense on solid state and complex chemistry – rounded off by a glimpse of the theoretical description of HERFD and V2C spectra, to end up finally with an outlook on possible future applications. Since there already exist excellent reviews on the topics of X-ray absorption that also include HERFD and XES techniques mainly in catalysis,13,20,39,40 the focus of this perspective lies in the demonstration of the potential of HERFD-XANES and V2C-XES to overcome the limitations of conventional hard X-ray absorption spectroscopy and in the comparison of their individual sensitivity. Therefore the third important method of the group, core-to-core or Kβ1,3-XES, is not discussed here.19,41–43 The presented examples were selected to work out this particular aim, but in no sense present an exhaustive overview of all applications of HERFD-XANES and V2C-XES. Soft X-ray absorption spectroscopy, like at transition metal L-edges, and the corresponding emission spectroscopy44,45 are also beyond the scope of this perspective, but also excellent literature exists on this topic.42
Experimental
For both methods, HERFD-XANES and V2C-XES, the same type of spectrometer can be used. This can be either a scanning Johann46,47 or a dispersive von Hamos type.48,49 The data acquisition principle is shown in Fig. 2 for the example of a scanning Johann type spectrometer. This spectrometer type uses focusing analyzer crystals and a point detector. To record HERFD-XANES spectra the intensity of a single fluorescence channel, selected with the analyzer crystals, is monitored with a resolution smaller than the lifetime broadening, while sweeping the incident energy of the double crystal monochromator. Usually, the maximum of the Kα or Kβ main line (in Fig. 2 the Kβ line) is used for this purpose. In contrast, to record XES spectra the incident energy is fixed at a constant value off resonance beyond the edge position and the analyzer crystals are swept over the V2C emission energy range. Using a dispersive von Hamos spectrometer avoids any scanning movements by applying cylindrically bent crystals and a position sensitive detector.49 With such a spectrometer, the emission spectrum can be obtained in a single “shot”, which can reduce the acquisition time significantly, but at the price of lower signal intensities and reduced energy resolution. Although it is also possible to record HERFD spectra using a von Hamos spectrometer, the main advantage here is surely the change to increase the time resolution for V2C-XES and resonant inelastic scattering (RIXS), which is not further discussed here.50–52 The core parts of a Johann and von Hamos type spectrometer are shown schematically in Fig. 3. Since both types of spectrometers and spectroscopic methods require a high flux, they are usually carried out at undulator or wiggler beamlines, like ID26 (ESRF, Grenoble), P64 (PETRA III, Hamburg), BL6-2b (SSRL, Standford). However, so-called super bending magnet beamlines as found at SuperXAS (SLS, Villigen) provide sufficient flux. In the near future X-ray free electron lasers will probably provide the most exciting experimental stations to carry out HERFD or V2C-XES experiments.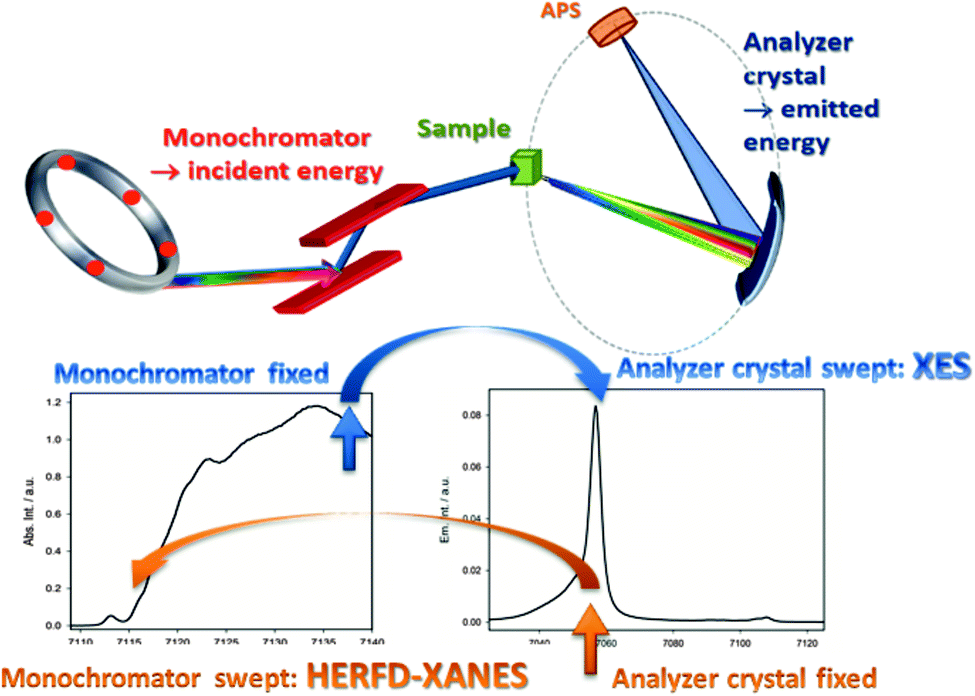 | ||
| Fig. 2 Experimental set-up using a Johann-type emission spectrometer. For HERFD-XANES spectra, the monochromator is swept over the absorption edge while the analyzer crystal is fixed at the maximum of an emission line (here Kβ1,3 main line). For Kβ2,5-XES spectra the monochromator is fixed and the analyzer crystal is swept over the emission energy range. | ||
 | ||
| Fig. 3 Schematic comparison of a Johann (left) and von Hamos type (right) spectrometer. | ||
Applications to solid state and heterogeneous catalytic systems
The V2C-XES spectra of ionic compounds as applied in heterogeneous catalysis show general spectral characteristics, which consist of a Kβ′′ signal at lower fluorescence energy and the V2C main signal immediately below the Fermi level.20 The spectral characteristics of metal particles and the investigation of such systems by HERFD-XANES and V2C-XES will not be discussed here.53,54Fig. 4 as an example shows the V2C spectrum of Fe2O3. The crossover Kβ′′ signal is assigned to ligand 2s to metal 1s transitions, i.e. it is caused by an oxygen 2s to iron 1s decay. Generally, different ligands show distinct Kβ′′ energies due to their characteristic 2s binding energies; therefore light atom ligands that are neighbors in the periodic table can also be distinguished by their Kβ′′ signal.55 Moreover, the intensity of this signal is correlated with the metal–ligand distance.20 | ||
| Fig. 4 Kβ2,5-XES spectrum of Fe2O3 with an indication for the Kβ2,5 main signal and the Kβ′′ cross-over signal. | ||
Safonov et al. studied the nature of the species in chromium deposits formed by electrodeposition using V2C-XES.56 They used the Kβ′′ signal mainly in a fingerprint approach by comparison with defined references to determine the presence of Cr–O, C–N, or Cr–C bonds.56 Confirmation of the signature assignment in the references is achieved by full multiple scattering calculations using FEFF 8.2.57 This study is one of the, if not the first, applications of XES to non-model systems to overcome the limitations of EXAFS concerning the discrimination of chemically similar coordinating neighbours at metal centers in solid state samples. The authors also point out the potential to interpret the spectra by application of a linear combination fit of references to unknown samples.
Although this work applies V2C-XES to ‘real’ systems, it is not yet applied to study a catalytic reaction. Safonova et al.58 applied V2C-XES in a similar fashion to ref. 56 for the analysis of heterogeneous vanadium catalysts for acrylonitrile production from propane. They investigated the local environment of the vanadium centers under operando conditions by V2C-XES after assignment of the spectral signatures using an exhaustive set of references in combination with full multiple scattering calculations. For example, they showed that the Kβ′′ line in VN corresponds to transitions originating from the N 2s states, while the V2C line is due to the band formed of mainly N 2p orbitals. They generally observed that the relative positions of Kβ′′ lines of the vanadium reference compounds depend more strongly on the nature of the ligands than on the formal oxidation state of vanadium and its local coordination. However, for oxidic vanadium compounds, the intensity of the Kβ′′ signal is related to the formal oxidation state of the vanadium center. Additionally, estimation of the effect of partial substitution of nitrogen through oxygen in the first coordination shell on the shape of V2C-XES spectra of vanadium oxides was tried. Even with one nitrogen substituting an oxygen in an octahedral coordination, an effect is visible in the V2C spectra by the appearance of shoulders on the right side of the Kβ′′ lines of oxides, close to the position of the Kβ′′ line of VN. Although this is an in silico result it demonstrates impressively the high potential of V2C-XES to extend the value of hard X-ray methods concerning the sensitivity towards the nearest neighbour environment.
In order to increase the practical value of V2C-XES, the step from a qualitative fingerprinting data analysis to a more quantitative description has to be made. Choosing appropriate structural models as inputs for calculations is a method that requires a certain kind of chemical knowledge. Gallo et al. simulated the V2C-XES of titania silicalite catalysts TS-1 by DFT instead of full multiple scattering methods.59 Such calculations are easy to obtain in the so-called one-electron approximation,60 which neglect multiplet structures,42 core–hole potential and multi-electron excitations. However, this simple method comes with the price of reduced accuracy in predicting spectral signatures, like the intensity of Kβ′′ signals. The authors show that a Z + 1 approach is valuable to account for core–hole effects. Although TS-1 represents a porous structure with long range order, a cluster model consisting of only one Ti center surrounded in a tetrahedral fashion as found in the TS-1 framework is sufficient to reproduce the V2C spectra, i.e. no necessity for larger clusters was observed (Fig. 5). Another key result of the study is that the result of V2C-XES calculations by DFT methods depends strongly on the used basis set and functional, as demonstrated in Fig. 5. For systems like TS-1 the best results were achieved using a meta-hybrid gradient corrected TPSSh functional, but also B3LYP yielded satisfactory results. Since first calculations on Co and Cu systems gave comparable results, the authors concluded that their findings are of general relevance for 3d transition metal systems.
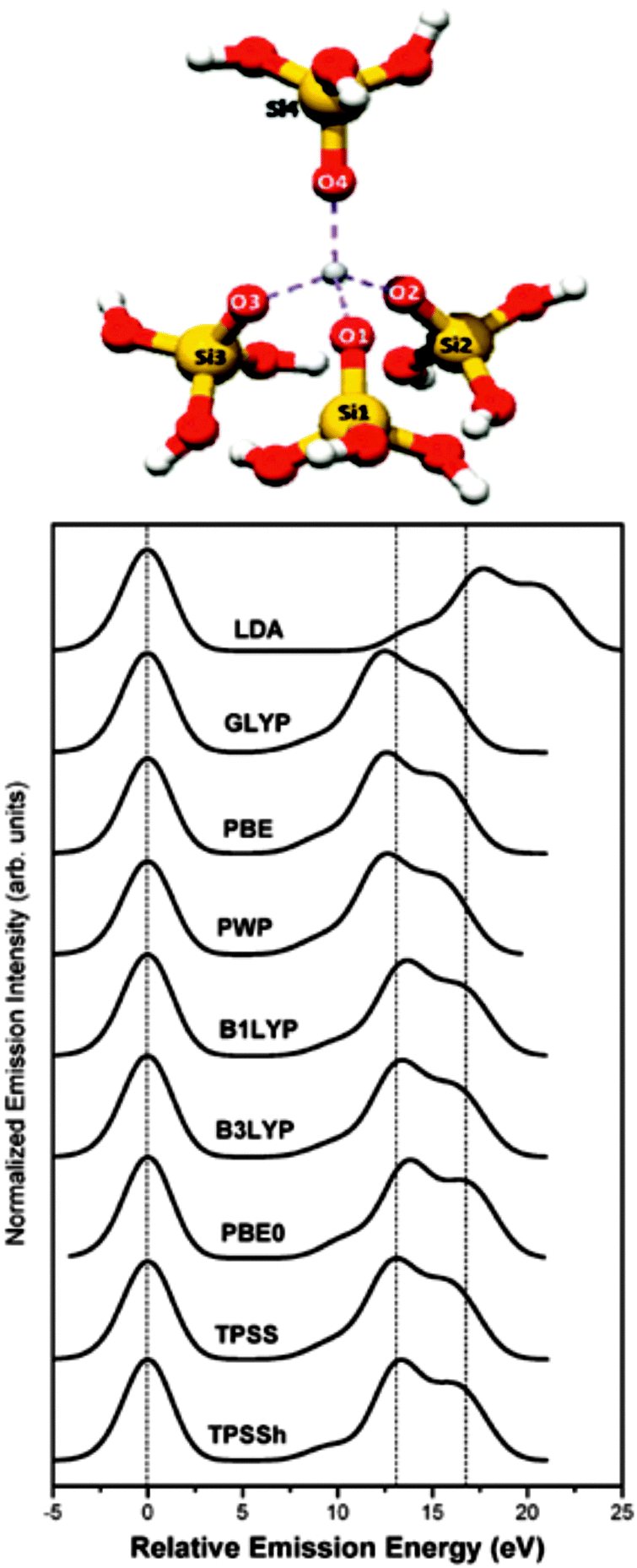 | ||
| Fig. 5 Structure used to study the valence electronic state of Ti silicalite-1 by Kβ2,5-XES (top), and density functional dependency of the simulated spectra (bottom). [Reproduced from ref. 59 with permission from the PCCP Owner Societies.] | ||
A succeeding study by Gallo et al.61 impressively showed that adsorption processes of water and ammonia can be studied by V2C-XES. The investigation of adsorption phenomena of CO on noble metal particles are possible using conventional XAS,62,63 but the determination of adsorbed light atom molecules on oxidic compounds is a major challenge. By V2C-XES it is even possible to define the number of adsorbed NH3 molecules on TS-1 by comparison with TD-DFT methods.
The first attempt to combine HERFD-XANES and V2C-XES in one study to gain deeper insights into the working principle of catalysts by using hard X-rays is provided by Swarbrick et al.64 They use the combination to study model compounds for homogeneous titania catalysts.65 Experimentally, they emphasize the risk and effects of radiation damages during high resolution X-ray spectroscopic experiments. Both methods are considered “flux hungry”, which requires high intensity and brilliant sources, with a large photon impact on the sample. Besides radiation damages as ultimate consequence of such a high X-ray intensity, which may hinder successful measurements by HERFD-XANES and – due to longer acquisition times – even more by V2C-XES, more subtle effects can occur, if immediate neighbours in the sample are excited at the same time. Contrary to the work discussed above, which employed full multiple scattering calculations, here TD-DFT methods using the ORCA program were applied.66 Such calculations have been proved to be applicable for the simulation of V2C-XES spectra by Smolentsev et al.67 For titania complexes that contain coordinated THF molecules, as relevant for Ziegler–Natta polymerization reactions, theoretically expected signals are not observed experimentally, which is explained through hybridization effects. The authors generally pointed out the different aspects of molecular orbital properties that can be probed with the applied combination of HERFD-XANES and V2C-XES.
Applications to metal complexes and homogeneous catalytic systems
Investigations applying HERFD-XAS and XES in solutions are still quite rare, and as such measurements require serious precautions to avoid radiation damage. As a matter of experience, liquid samples and transition metal complexes suffer much more from radiation damages and photoreduction due to covalent bond characteristics. Although in solid samples also radiation damage is observed, they can be more simply coped with by moving the solid sample in the beam. For liquid sample cells as used in studies on homogeneous reactions,1,2,68 only moving the cell in the beam is not enough due to the large energy impact on the liquid. Therefore a rather large volume of the solution sample has to be moved continuously. This can be done in a standard solution cell as developed in the authors group.68 Here the reaction solution is pumped from a multi-purpose reaction vessel – in which nearly every reaction condition can be achieved – through the cell.68 Alternatively, a capillary set-up can be used, in which the solution is injected in a silica capillary.67 Mixing of reactants is also possible with such a set-up, but the solution cannot be pumped in a cycle. The cutting edge technology to avoid radiation damage is certainly the so-called micro-jet technique.69,70 It makes use of glass nozzles through which the reaction solution is pumped with elevated pressures up to 40 bars.71–73 Consequently this approach allows a very fast exchange of sample volume in the X-ray beam. It has therefore two advantages: it can be used for pump–probe schemes to study ultra-fast processes with a high repetition rate, like structural responses to photoexcitation.70,74–77 Moreover, it avoids window materials, which can trigger or catalyze photoinduced processes on the sample. The jet-technique can therefore be considered as the ideal sample environment for high flux X-ray measurements, like HERFD-XAS and V2C-XES. With the advent of fourth generation sources, the jet-technique will be without alternative for time-dependent measurements in the liquid state. However, concerning a static structure determination, geometric or electronic, an individual compromise has to be found between the sample environment and the risk of radiation damages.In the first comprehensive study to extract structural information about transition metal complexes in solution beyond the possibilities of conventional XAS, Smolentsev et al. studied experimentally and theoretically the following complexes of manganese: [Mn(H2O)6]2+, [Mn(H2O)5(OH)]+ and [Mn(H2O)5(NH3)]2+.67 They showed that the ligands H2O, OH− and NH3 can be distinguished by V2C-XES, but not by standard XAS. This also means that N/O substitution and the protonation state of the ligands can be detected. As a fundamental difference, they also figured out the fact that interatomic distances influence the XANES shape, while the XES peak positions reflect the ligand type and the local symmetry. In a successive study, the conclusions of Smolentsev et al. were confirmed by studies on the protonation state in metal–organic manganese complexes with V2C-XES.78 Valence-to-core XES can thus be used to determine the chemical nature of the different ligands even if they are spatially at the same positions. With respect to manganese as the element of interest, these studies provide the basis to extend the insights already gained by conventional XAS into photosystem II.79,80 Since the so-called S states in PSII differ only by subtle changes of the coordination sphere around the manganese centers, the high resolution power of V2C-XES and HERFD-XANES can surely lead to refined conclusions. Although studies on PSII are of unbroken interest, the future impact of V2C-XES and HERFD-XANES measurements on manganese and related systems in the future will be of more general character, as they offer the possibility to understand and improve water oxidation catalysts for sustainable oxygen production.81,82
In order to examine an extension of the sensitivity of HERFD-XANES and V2C-XES towards ligand changes beyond the first coordination sphere, and to put current theory levels to the test with electronically more complex structures, we carried out combined experimental and theoretical studies with HERFD-XANES and V2C-XES on a carefully selected set of ferrocene model compounds.21,83 In Fig. 6, the investigated complexes and the corresponding HERFD-XANES spectra including the isolated prepeak areas are shown.
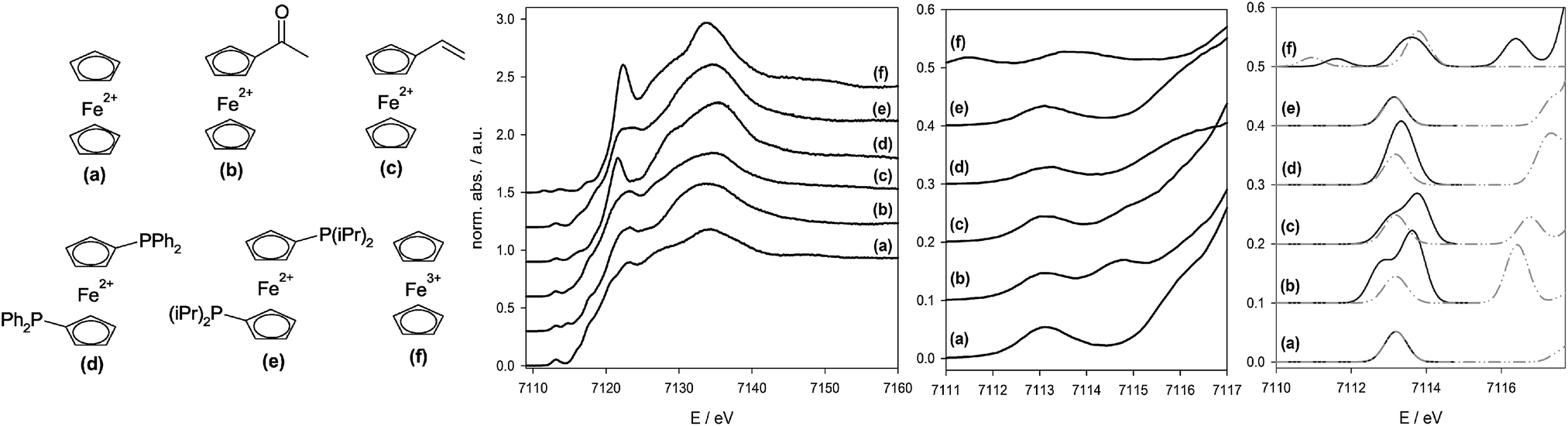 | ||
| Fig. 6 From left to right: ferrocene model compounds used for thorough investigation of the chemical sensitivity of HERFD-XANES and Kβ2,5-XES. Experimental HERFD-XANES spectra of these compounds. Enlarged prepeak area and simulated prepeak spectra (black solid line: B3LYP/def2-QZVPP, grey dotted dashed line: BP86/def2-QZVPP). | ||
In the HERFD-XANES spectra21 it is obvious that different substituents at the cyclopentadienyl ligand cause changes in the prepeak structure. TD-DFT methods are able to explain these differences by analysis of the unoccupied molecular orbitals. However, there is a strong influence of the functional used on the calculated HERFD-XANES spectra, for which the phosphine-substituted ferrocenes (d) and (e) (cf.Fig. 6) are a good example. If the phosphine-substituent does not contain a π-system (e), the prepeak intensity is predicted satisfactorily by both BP86 and B3LYP functionals. However, if a π-system is present (d), BP86 predicts a mixing of the π-system with the cyclopentadienyl ring, introducing a large dipole contribution to the otherwise dipole-forbidden 1s → LUMO transition. Theoretically this causes an increased prepeak intensity that is not found in reality. In contrast, ligands containing more immediate π-bonds, like in acetyl (b) or vinyl (c) ferrocene lead to a splitting of the final level, which is reflected in two prepeak signals. Although none of the two applied exchange correlation functionals BP86 and B3LYP are able to reproduce the experimental splitting correctly, a quantitative assignment to the involved MOs is possible that allows assigning and following the changes in the LUMO states during chemical reactions.
Considering applications to the investigation of catalytic systems, the usage of the same analyzer crystals for both the acquisition of HERFD-XANES and V2C-XES spectra is beneficial. In order to explore the potential of such an approach, we compared the V2C spectra of the same ferrocenes subject to HERFD-XANES measurements recorded using the same type of spherically bent Ge(662) analyzer crystals.83 These spectra are displayed in their background corrected form in Fig. 7. In contrast to the HERFD-XANES spectra, no significant differences can be observed in these spectra for exchanging the substituents at the Cp ring. This is also found by TD-DFT calculations, which are less (but still) sensitive to the exchange–correlation functional than in case of the HERFD-XANES spectra. The reason for this observation can be found in the particular transitions: as mentioned above, pre-edge peaks in HERFD-XANES probe dipole forbidden transitions to unoccupied d-orbitals, which mix to a certain extent with the substituent's p-orbitals, like in acetyl (b) and vinyl (c) ferrocene. In contrast, the V2C-XES spectra are dominated by dipole-allowed transitions originating from occupied ligand orbitals, with only small Fe p-orbital contributions. These ligand orbitals orbitals are less affected by substituents at the Cp ring. Although there are occupied orbitals which are split by substitution with an acetyl or a vinyl group, the corresponding transitions are not observable in the V2C-XES spectra as they are still significantly smaller than the dipole allowed transition corresponding to the unperturbed orbitals.
 | ||
| Fig. 7 Experimental Kβ2,5-XES spectra of the ferrocene model compounds given in Fig. 6. | ||
For resolving the differences in the occupied orbitals caused by substituents at the Cp ring, it would be necessary to increase the resolution of experimental V2C-XES spectra. For this purpose, analyzer crystals of larger Miller indices, i.e. providing higher resolution had to be used. Since this comes only with the price of a reduced flux at the detector diode, the applicability of higher order analyzers to studies in catalysis, especially under operando conditions is limited.41
Although the above mentioned results were obtained for the particular example of substituted ferrocenes, generalized statements can be deduced concerning the sensitivity of HERFD-XANES and V2C-XES. Usually, the pre-edge region in K-edge XAS spectra of transition metal complexes contains only the dipole-forbidden transitions to orbital levels of d-character. These will in general be very sensitive to changes in chemical reactions, both for directly coordinating atoms, but also beyond the first coordination shell. With the possibility to resolve such subtle changes with HERFD-XANES it is surely a very sensitive and powerful analytical tool. On the other hand, the V2C-XES spectra are dominated by dipole-allowed transitions originating from orbitals with contributions from metal p orbitals. Thus, changes in occupied metal d-orbitals will hardly be detectable since the corresponding transitions are much weaker. Nonetheless, this does not shorten the value of V2C-XES if changes appear at directly coordinating atoms to the metal center.
Although the higher sensitivity of XANES spectroscopy compared to X-ray emission spectroscopy was recently demonstrated for the nitrogen K-edge in peptides,84 our study in ref. 83 was the first systematic comparison on the chemical sensitivity of HERFD-XANES and V2C-XES using a combined experimental and theoretical approach. Beside the experimental developments achieved in the past, it is obvious that the increase of information content in X-ray spectra is equally dependent on the capability of theoretical methods to describe the obtained spectra. The investigation into the electronic structure of ferrocene complexes showed that for the calculation of XAS and especially prepeak intensities for hard X-rays the dipole approximation, in which the oscillator strengths are proportional to the square of the electric-dipole transition moments, is insufficient. This is due to the fact that the dipole approximation is based on the assumption that the wavelength of the electromagnetic radiation is large compared to the size of the core orbital, which is not the case in hard X-ray spectroscopy. This becomes even more important if the prepeaks have very low dipole intensity as they are dipole forbidden, i.e. in K-edge spectra of transition metal complexes. Here, the prepeak intensity can be due to effects that are not included in the dipole approximation.85,86 Different from currently used methods,87 Jacob et al. could show that contributions beyond the dipole approximation can be achieved in an origin-independent manner. In their approach all contributions to the oscillator strength, which are of the same order in the wave vector, are included.88
TD-DFT calculations of HERFD-XANES and V2C-XES spectra require a shift of 150–180 eV (depending on the used functional) of the theoretical spectrum to match the experiment.21,60,83,89 The major fraction of the shift in the absolute excitation energies is caused by relativistic effects that are usually neglected for the 1s core orbital. They can be considered by including scalar relativistic effects using the zeroth-order regular approximation (ZORA),90 which reduces the shifts to 20–50 eV. Although these absolute shifts do not affect relative excitation energies and intensities, their minimization would be beneficial to remove the remaining ambiguities with respect to the assignment of transitions. A promising approach is ΔSCF-DFT. Since ΔSCF-DFT methods are to date applied rarely to the interpretation of X-ray spectra, only a few applications to transition metal complexes exist in literature.91,92 This is even more surprising if the following is considered for the HERFD-XANES spectra: TD-DFT calculations are only able to yield the prepeak transitions below the absorption step with satisfactory accuracy. More ligand centered final states of quasi-continuous character, which can be found at energies close to the absorption step and beyond are not reproduced. Calculations on copper systems, which exhibit a broad variety of such quasi-continuum states are very promising to overcome this limitation.93
According to what has been discussed about the theoretical approaches to model HERFD-XANES and V2C-XES spectra in the preceding sections, it has to be concluded here that it is not yet possible to identify a priori a particular theoretical methodology or even a certain level of complexity within TD-DFT methods that work in a predictive way and that are independent of the particular specifications of the studied system. Therefore, equally important to the application of the new and powerful techniques HERFD-XANES and V2C-XES to open questions in chemistry and catalysis, is still the establishment of “error bars” within the determination of subtle effects by iterating and cross-checking theory and experiment against each other using well-defined references.
For transition metal carbonyls, a class of compounds, both used as catalyst in homogeneous catalytic reactions94 and relevant as intermediates in heterogeneous processes,26,95,96 we performed such an extensive V2C-XES benchmarking study.97 It includes different levels of V2C-XES spectra interpretation. The simplest one is a fingerprint approach, in which signals in the iron V2C emission are assigned to different kinds of ligands. For example, the intensity of the signal connected to the CO ligand can be correlated with the number of CO molecules per iron center as demonstrated in Fig. 8. Moreover, the signal of hydrocarbon molecules in iron carbonyl complexes is found at different energies than for the CO ligand, cf.Fig. 8. Since both hydrocarbons and CO coordinate with carbon atoms to the iron center, this is another example for the increased chemical sensitivity of V2C-XES with respect to the discrimination of light atom ligands. The information gained by such measurements can even be more refined and secured if TD-DFT calculations are employed. We performed such calculations for a row of model compounds like Fe(CO)5, Fe2(CO)9, Fe3(CO)12 and Fe2Cp2(CO)4. Again it turned out that in iron V2C-XES spectra Fe np contributions play a major role, which is in line with the results of the ferrocene studies.21,83 Both studies therefore prove the potential of not only eliminating limitations of conventional XAS, but also of opening completely new ways to follow changes in the electronic structure at catalyst centers during chemical reactions.
 | ||
| Fig. 8 Experimental Kβ2,5-XES spectra of the iron carbonyl compounds Fe(CO)5, Fe3(CO)12 and Fe2Cp2(CO)4 with a qualitative assignment of the emission signals to CO and hydrocarbon ligands. | ||
A first application of these results to homogeneous catalytic systems was carried out to explore the ground state of the broadly applicable catalyst [Fe(CO)3NO]−, known as the Hieber anion.98–100 The result of this study was a detailed and revised view on the electron density at the iron center, which can be used in the future to study the changes in the course of reactions catalysed by [Fe(CO)3NO]− on a molecular orbital level in situ.
Conclusion and outlook
With the presented examples it was intended to show the potential of both HERFD-XANES and V2C-XES spectroscopy to overcome the limitations of conventional X-ray absorption spectroscopy and to open up new views on solid state systems and transition metal complexes as for example applied in catalytic reactions. The two methods proved to be advantageous, since in contrast to conventional XAS, they provide insight into the electronic levels that determine the chemical and catalytic properties and that are subject to changes in the course of reactions. These are the HOMO and LUMO states if one talks about homogeneous catalytic systems, or the conduction and valence band in solid state systems. Since the information gained by HERFD-XANES and V2C-XES is constrained by the applying selection rules, the combination with other spectroscopic methods is always an advantage in order to get complementary information or to avoid misinterpretations. Simultaneous combinations can be frequently found for conventional XAS, e.g. with IR,101 Raman1,102 and UV/Vis1,68,103 spectroscopy.104 Although it is in principle possible to combine HERFD-XANES and V2C-XES with other methods, the geometric requirements are slightly different, and still such combinations are rare.105 Nonetheless, such combinations will be achieved, and new combinations will even provide more insights into the electronic structure at metal centers. In this direction, especially the combination of HERFD-XANES and V2C-XES with electron spin resonance is considered highly promising.Although heterogeneous reactions are surely of superior importance from an application point of view, they are certainly less suited to further establish the value and use of HERFD-XANES and V2C-XES. This is mainly due to the fact, that heterogeneous catalysts consist more often then not of a multitude of catalytically active species, like particles of different sizes or different oxide modifications. This indeed hinders a precise comparison to theoretical calculations for a particular catalyst structure. Another issue is the flux hunger, requiring high photon densities at the sample. Avoiding radiation damages is certainly the major experimental task for HERFD-XANES and V2C-XES measurements. Although the realization of a sample exchange system is possible for heterogeneous catalysts, it can be more easily realized for liquid samples with a liquid jet.
This issue became even more prominent with the advent of X-ray free electron lasers. The opportunities one can theoretically think of at such XFEL sources, like to obtain molecular movies, demand systems to translate them into a ‘catalytic reality’. For this purpose, homogeneous and solution reactions105 are without any alternative. It can therefore be assumed that two major fields will make use of HERFD-XANES and V2C-XES. One is bioinorganic106,107 and enzyme science38 and the other one chemical energy conversion, here especially photochemical water splitting.108–111 Photocatalytic reactions are particularly suited for experiments at XFELs, since they require the very high time resolution. Together with third generation synchrotron sources a bunch of methods and time scales are available to study thus the generation of hydrogen and oxygen from water, which is one of the promising approaches to overcome the shortage of fossil fuels.112
However, the prices to pay compared to conventional XAS spectroscopy are higher experimental efforts and less available beamlines. Therefore it has to be noticed, that the application of HERFD-XANES and V2C-XES should be the second step in an X-ray study on catalytic reactions. This is even more obvious if one considers that the theoretical methods do not yet work on a fully predictive general basis, especially for molecular complexes. In cases where conventional X-ray absorption cannot answer particular questions, like about coordinating or adsorbed reactants to the catalytic center, or about the molecular orbital levels involved in a catalytic reaction, both HERFD-XANES and V2C-XES are the methods of choice – if hard X-rays are required!
As a closing summary, both HERFD-XANES and V2C-XES spectroscopy are without any doubt a big step forward in terms of increased information compared to conventional XAS, and the question raised in the title can be answered by a clear YES. Nonetheless, to be able to push the limits fully, theory has to be further consolidated, the time resolution has to be further increased by allowing such measurements at XFELs, and finally the number of beamlines dedicated to catalytic studies that allow recording HERFD-XANES and V2C-XES spectra has to be increased. If these requirements are fulfilled, new and still unpredictable insights into chemical reactions and the working principle of catalysts can be expected.
Acknowledgements
The German Bundesministerium für Bildung und Forschung (BMBF) is acknowledged for funding within the two projects SusXES and SusChEmX aiming at building an infrastructure for time-resolved XES at PETRA III and MAX IV. The Forschergruppe 1405 of the German DFG is acknowledged for financial support. The ESRF is acknowledged for provision of beamtime to record data discussed here.References
- M. Bauer and C. Gastl, Phys. Chem. Chem. Phys., 2010, 12, 5575–5584 RSC.
- M. Bauer, T. Kauf, J. Christoffers and H. Bertagnolli, Phys. Chem. Chem. Phys., 2005, 7, 2664–2670 RSC.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- H. Bertagnolli and T. S. Ertel, Angew. Chem., Int. Ed., 1994, 33, 45–66 CrossRef.
- J. M. D. Cónsul, I. M. Baibich and M. C. M. Alves, Catal. Commun., 2011, 12, 1357–1360 CrossRef.
- R. J. Davis, S. M. Landry, J. A. Horsley and M. Boudart, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 10580–10583 CrossRef CAS.
- A. J. Dent, Top. Catal., 2002, 18, 27–35 CrossRef CAS.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- G. Guilera, M. A. Newton, C. Polli, S. Pascarelli, M. Guino and K. K. Hii, Chem. Commun., 2006, 4306–4308 RSC.
- A. S. K. Hashmi, C. Lothschütz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem. – Eur. J., 2010, 16, 8012–8019 CrossRef CAS PubMed.
- W. Hörner and H. Bertagnolli, J. Organomet. Chem., 2002, 649, 128–135 CrossRef.
- S. Opelt, V. Krug, J. Sonntag, M. Hunger and E. Klemm, Microporous Mesoporous Mater., 2012, 147, 327–333 CrossRef.
- P. Glatzel, T.-C. Weng, K. Kvashnina, J. Swarbrick, M. Sikora, E. Gallo, N. Smolentsev and R. A. Mori, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 17–25 CrossRef CAS.
- M. Bauer and H. Bertagnolli, Methods in Physical Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 231–269 Search PubMed.
- F. Farges, Y. Lefrère, S. Rossano, A. Berthereau, G. Calas and G. E. Brown Jr, J. Non-Cryst. Solids, 2004, 344, 176–188 CrossRef CAS.
- P. Eisenberger, P. M. Platzman and H. Winick, Phys. Rev. Lett., 1976, 36, 623–626 CrossRef CAS.
- O. V. Safonova, M. Tromp, J. A. van Bokhoven, F. M. F. de Groot, J. Evans and P. Glatzel, J. Phys. Chem. B, 2006, 110, 16162–16164 CrossRef CAS PubMed.
- P. Glatzel, M. Sikora, G. Smolentsev and M. Fernández-García, Catal. Today, 2009, 145, 294–299 CrossRef CAS.
- W. M. Heijboer, P. Glatzel, K. R. Sawant, R. F. Lobo, U. Bergmann, R. A. Barrea, D. C. Koningsberger, B. M. Weckhuysen and F. M. F. de Groot, J. Phys. Chem. B, 2004, 108, 10002–10011 CrossRef CAS.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- A. J. Atkins, C. R. Jacob and M. Bauer, Chem. – Eur. J., 2012, 18, 7021–7025 CrossRef CAS PubMed.
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, ACS Catal., 2012, 3, 95–102 CrossRef.
- J. Herrero-Martín, A. Mirone, J. Fernández-Rodríguez, P. Glatzel, J. García, J. Blasco and J. Geck, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 075112 CrossRef.
- P. Biloen, J. N. Helle and W. M. H. Sachtler, J. Catal., 1979, 58, 95–107 CrossRef CAS.
- B. H. Davis, Fuel Process. Technol., 2001, 71, 157–166 CrossRef CAS.
- B. H. Davis, Catal. Today, 2009, 141, 25–33 CrossRef CAS.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2002, 124, 11568–11569 CrossRef CAS PubMed.
- J. M. H. Lo and T. Ziegler, J. Phys. Chem. C, 2008, 112, 13681–13691 CAS.
- M. Bauer, R. Schoch, L. Shao, B. Zhang, A. Knop-Gericke, M. Willinger, R. Schlögl and D. Teschner, J. Phys. Chem. C, 2012, 116, 22375–22385 CAS.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, J. Catal., 2006, 242, 26–37 CrossRef CAS.
- M. W. Tew, M. Janousch, T. Huthwelker and J. A. van Bokhoven, J. Catal., 2011, 283, 45–54 CrossRef CAS.
- M. W. Tew, M. Nachtegaal, M. Janousch, T. Huthwelker and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2012, 14, 5761–5768 RSC.
- M. P. Feth, C. Bolm, J. P. Hildebrand, M. Köhler, O. Beckmann, M. Bauer, R. Ramamonjisoa and H. Bertagnolli, Chem. – Eur. J., 2003, 9, 1348–1359 CrossRef CAS PubMed.
- N. Binsted, J. Evans, G. N. Greaves and R. J. Price, Chem. Commun., 1987, 1330–1333 RSC.
- K. Hayakawa, K. Hatada, P. D'Angelo, S. Della Longa, C. R. Natoli and M. Benfatto, J. Am. Chem. Soc., 2004, 126, 15618–15623 CrossRef CAS PubMed.
- M. Rovezzi and P. Glatzel, Semicond. Sci. Technol., 2014, 29, 023002 CrossRef.
- J. Singh, C. Lamberti and J. A. van Bokhoven, Chem. Soc. Rev., 2010, 39, 4754–4766 RSC.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- J. Singh, C. Lamberti and J. A. van Bokhoven, Chem. Soc. Rev., 2010, 39, 4754–4766 RSC.
- G. D. Pirngruber, J.-D. Grunwaldt, J. A. van Bokhoven, A. Kalytta, A. Reller, O. V. Safonova and P. Glatzel, J. Phys. Chem. B, 2006, 110, 18104–18107 CrossRef CAS PubMed.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- A. Mijovilovich, H. Hayashi, N. Kawamura, H. Osawa, P. C. A. Bruijnincx, R. J. M. Klein Gebbink, F. M. F. de Groot and B. M. Weckhuysen, Eur. J. Inorg. Chem., 2012, 1589–1597 CrossRef CAS.
- E. Suljoti, R. Garcia-Diez, S. I. Bokarev, K. M. Lange, R. Schoch, B. Dierker, M. Dantz, K. Yamamoto, N. Engel, K. Atak, O. Kühn, M. Bauer, J.-E. Rubensson and E. F. Aziz, Angew. Chem., Int. Ed., 2013, 52, 9841–9844 CrossRef CAS PubMed.
- M. M. van Schooneveld, R. W. Gosselink, T. M. Eggenhuisen, M. Al Samarai, C. Monney, K. J. Zhou, T. Schmitt and F. M. F. de Groot, Angew. Chem., Int. Ed., 2013, 52, 1170–1174 CrossRef CAS PubMed.
- H. H. Johann, Z. Phys., 1931, 69, 185 CrossRef CAS.
- T. T. Johansson, Z. Phys., 1933, 82, 507 CrossRef CAS.
- L. v. Hamos, Naturwiss, 1932, 20, 705 CrossRef.
- J. Szlachetko, M. Nachtegaal, E. d. Boni, M. Willimann, O. Safonova, J. Sa, G. Smolentsev, M. Szlachetko, J. A. v. Bokhoven, J.-C. Dousse, J. Hoszowska, Y. Kayser, P. Jagodzinski, A. Bergamaschi, B. Schmitt, C. David and A. Lucke, Rev. Sci. Instrum., 2012, 83, 103105 CrossRef CAS PubMed.
- M. Zienkiewicz, A. Jablonska-Wawrzycka, J. Szlachetko, Y. Kayser, K. Stadnicka, W. Sawka-Dobrowolska, J. Jezierska, B. Barszcz and J. Sa, Dalton Trans., 2014, 53, 8599 RSC.
- J. Sa, Y. Kayser, C. J. Milne, D. L. Abreu Fernandes and J. Szlachetko, Phys. Chem. Chem. Phys., 2014, 16, 7692–7696 RSC.
- J. Szlachetko, J. Sá, O. V. Safonova, G. Smolentsev, M. Szlachetko, J. A. van Bokhoven and M. Nachtegaal, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 161–165 CrossRef CAS.
- A. I. Frenkel, M. W. Small, J. G. Smith, R. G. Nuzzo, K. O. Kvashnina and M. Tromp, J. Phys. Chem. B, 2013, 117, 23286–23294 CAS.
- P. Glatzel, J. Singh, K. O. Kvashnina and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 2555–2557 CrossRef CAS PubMed.
- U. Bergmann, C. R. Horne, T. J. Collins, J. M. Workman and S. P. Cramer, Chem. Phys. Lett., 1999, 302, 119–124 CrossRef CAS.
- V. Safonov, L. Vykhodtseva, Y. Polukarov, O. Safonova, G. Smolentsev, M. Sikora, S. Eeckhout and P. Glatzel, J. Phys. Chem. B, 2006, 110, 23192–23196 CrossRef CAS PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- O. V. Safonova, M. Florea, J. Bilde, P. Delichere and J. M. M. Millet, J. Catal., 2009, 268, 156–164 CrossRef CAS.
- E. Gallo, C. Lamberti and P. Glatzel, Phys. Chem. Chem. Phys., 2011, 13, 19409 RSC.
- N. Lee, T. Petrenko, U. Bergmann, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2010, 132, 9715–9727 CrossRef CAS PubMed.
- E. Gallo, F. Bonino, J. C. Swarbrick, T. Petrenko, A. Piovano, S. Bordiga, D. Gianolio, E. Groppo, F. Neese, C. Lamberti and P. Glatzel, ChemPhysChem, 2013, 14, 79–83 CrossRef CAS PubMed.
- M. K. Oudenhuijzen, J. A. van Bokhoven, J. T. Miller, D. E. Ramaker and D. C. Koningsberger, J. Am. Chem. Soc., 2005, 127, 1530–1540 CrossRef CAS PubMed.
- D. E. Ramaker, M. Teliska, Y. Zhang, A. Y. Stakheev and D. C. Koningsberger, Phys. Chem. Chem. Phys., 2003, 5, 4492–4501 RSC.
- J. C. Swarbrick, Y. Kvashnin, K. Schulte, K. Seenivasan, C. Lamberti and P. Glatzel, Inorg. Chem., 2010, 49, 8323–8332 CrossRef CAS PubMed.
- S. Bordiga, F. Bonino, A. Damin and C. Lamberti, Phys. Chem. Chem. Phys., 2007, 9, 4854–4878 RSC.
- F. Neese, 2.7.0 edn, 2009–2012.
- G. Smolentsev, A. V. Soldatov, J. Messinger, K. Merz, T. Weyhermüller, U. Bergmann, Y. Pushkar, J. Yano, V. K. Yachandra and P. Glatzel, J. Am. Chem. Soc., 2009, 131, 13161–13167 CrossRef CAS PubMed.
- M. Bauer, G. Heusel, S. Mangold and H. Bertagnolli, J. Synchrotron Radiat., 2010, 17, 273–279 CAS.
- E. Suljoti, R. Garcia-Diez, S. I. Bokarev, K. M. Lange, R. Schoch, B. Dierker, M. Dantz, K. Yamamoto, N. Engel, K. Atak, O. Kühn, M. Bauer, J.-E. Rubensson and E. F. Aziz, Angew. Chem., 2013, 125, 10024–10027 CrossRef.
- M. Bauer, T. Stalinski and E. F. Aziz, ChemPhysChem, 2011, 12, 2088–2091 CrossRef CAS PubMed.
- M. Faubel, K. R. Siefermann, Y. Liu and B. Abel, Acc. Chem. Res., 2011, 45, 120–130 CrossRef PubMed.
- D. P. DePonte, U. Weierstall, K. Schmidt, J. Warner, D. Starodub, J. C. H. Spence and R. B. Doak, J. Phys. D: Appl. Phys., 2008, 41, 195505 CrossRef.
- U. Weierstall, R. B. Doak, J. C. H. Spence, D. Starodub, D. Shapiro, P. Kennedy, J. Warner, G. G. Hembree, P. Fromme and H. N. Chapman, Exp. Fluids, 2008, 44, 675–689 CrossRef CAS.
- F. A. Lima, C. J. Milne, D. C. V. Amarasinghe, M. H. Rittmann-Frank, R. M. v. d. Veen, M. Reinhard, V.-T. Pham, S. Karlsson, S. L. Johnson, D. Grolimund, C. Borca, T. Huthwelker, M. Janousch, F. van Mourik, R. Abela and M. Chergui, Rev. Sci. Instrum., 2011, 82, 0631111 CrossRef PubMed.
- V.-T. Pham, T. J. Penfold, R. M. van der Veen, F. Lima, A. El Nahhas, S. L. Johnson, P. Beaud, R. Abela, C. Bressler, I. Tavernelli, C. J. Milne and M. Chergui, J. Am. Chem. Soc., 2011, 133, 12740–12748 CrossRef CAS PubMed.
- T. J. Penfold, C. J. Milne and M. Chergui, Advances in Chemical Physics, John Wiley & Sons, Inc., 2013, pp. 1–41 Search PubMed.
- G. Smolentsev, A. Guda, X. Zhang, K. Haldrup, E. S. Andreiadis, M. Chavarot-Kerlidou, S. E. Canton, M. Nachtegaal, V. Artero and V. Sundstrom, J. Phys. Chem. C, 2013, 117, 17367–17375 CAS.
- B. Lassalle-Kaiser, T. T. Boron, V. Krewald, J. Kern, M. A. Beckwith, M. U. Delgado-Jaime, H. Schroeder, R. Alonso-Mori, D. Nordlund, T.-C. Weng, D. Sokaras, F. Neese, U. Bergmann, V. K. Yachandra, S. DeBeer, V. L. Pecoraro and J. Yano, Inorg. Chem., 2013, 52, 12915–12922 CrossRef CAS PubMed.
- C. Glöckner, J. Kern, M. Broser, A. Zouni, V. Yachandra and J. Yano, J. Biol. Chem., 2013, 288, 22607 CrossRef PubMed.
- M. J. Latimer, V. J. DeRose, I. Mukerji, V. K. Yachandra, K. Sauer and M. P. Klein, Biochemistry, 1995, 34, 10898–10909 CrossRef CAS PubMed.
- F. Conrad, M. Bauer, D. Sheptyakov, S. Weyeneth, D. Jaeger, K. Hametner, P.-E. Car, J. Patscheider, D. Gunther and G. R. Patzke, RSC Adv., 2012, 2, 3076–3082 RSC.
- H. Junge, N. Marquet, A. Kammer, S. Denurra, M. Bauer, S. Wohlrab, F. Gärtner, M.-M. Pohl, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 12749–12758 CrossRef CAS PubMed.
- A. J. Atkins, M. Bauer and C. R. Jacob, Phys. Chem. Chem. Phys., 2013, 15, 8095–8105 RSC.
- W. Hua, Y.-J. Ai, B. Gao, H. Li, H. Agren and Y. Luo, Phys. Chem. Chem. Phys., 2012, 14, 9666–9675 RSC.
- G. Dräger, R. Frahm, G. Materlik and O. Brümmer, Phys. Status Solidi, 1988, 146, 287–294 CrossRef.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- S. DeBeer George, T. Petrenko and F. Neese, Inorg. Chim. Acta, 2008, 361, 965–972 CrossRef CAS.
- S. Bernadotte, A. J. Atkins and C. R. Jacob, J. Chem. Phys., 2012, 137, 204106 CrossRef PubMed.
- C. J. Pollock and S. DeBeer, J. Am. Chem. Soc., 2011, 133, 5594–5601 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- N. A. Besley, A. T. B. Gilbert and P. M. W. Gill, J. Chem. Phys., 2009, 130, 1243081–1243087 CrossRef PubMed.
- B. Brena, P. E. M. Siegbahn and H. Ågren, J. Am. Chem. Soc., 2012, 134, 17157–17167 CrossRef CAS PubMed.
- N. Vollmers, U. Gerstmann, W.-G. Schmidt and M. Bauer, HERFD-XANES analysis of biomimetic copper complexes, unpublished results.
- R. Jennerjahn, R. Jackstell, I. Piras, R. Franke, H. Jiao, M. Bauer and M. Beller, ChemSusChem, 2012, 5, 734–739 CrossRef CAS PubMed.
- H. P. Withers, K. F. Eliezer and J. W. Mitchell, Ind. Eng. Chem. Res., 1990, 29, 1807–1814 CrossRef CAS.
- M. Kollár, A. De Stefanis, H. E. Solt, M. R. Mihályi, J. Valyon and A. A. G. Tomlinson, J. Mol. Catal. A: Chem., 2010, 333, 37–45 CrossRef.
- M. U. Delgado-Jaime, S. DeBeer and M. Bauer, Chem. – Eur. J., 2013, 19, 15888–15897 CrossRef CAS PubMed.
- J. E. M. N. Klein, B. Miehlich, M. S. Holzwarth, M. Bauer, M. Milek, M. M. Khusniyarov, G. Knizia, H.-J. Werner and B. Plietker, Angew. Chem., 2014, 126, 1820–1824 CrossRef.
- A. P. Dieskau, M. S. Holzwarth and B. Plietker, Chem. – Eur. J., 2012, 18, 2423–2429 CrossRef CAS PubMed.
- M. Jegelka and B. Plietker, Chem. – Eur. J., 2011, 17, 10417–10430 CrossRef CAS PubMed.
- M. A. Newton, B. Jyoti, A. J. Dent, S. G. Fiddy and J. Evans, Chem. Commun., 2004, 2382 RSC.
- A. M. Beale, A. M. J. van der Eerden, K. Keryinen, M. A. Newton and B. M. Weckhuysen, Chem. Commun., 2005, 3015 RSC.
- M. Tromp, J. R. A. Sietsma, J. A. van Bokhoven, G. P. F. Strijdonck, R. J. van Haaren, A. M. J. van der Eerden, P. W. N. M. van Leeuven and D. C. Koningsberger, Chem. Commun., 2003, 584 Search PubMed.
- S. J. Tinnemans, J. G. Mesu, K. Kervinen, T. Visser, T. A. Nijhuis, A. M. Beale, D. E. Keller, A. M. J. van der Eerden and B. M. Weckhuysen, Catal. Today, 2006, 113, 3–15 CrossRef CAS.
- M. Makosch, C. Kartusch, J. Sá, R. B. Duarte, J. A. van Bokhoven, K. Kvashnina, P. Glatzel, D. L. A. Fernandes, M. Nachtegaal, E. Kleymenov, J. Szlachetko, B. Neuhold and K. Hungerbühler, Phys. Chem. Chem. Phys., 2012, 14, 2164–2170 RSC.
- A. Hoffmann, S. Binder, A. Jesser, R. Haase, U. Flörke, M. Gnida, M. Salomone Stagni, W. Meyer-Klaucke, B. Lebsanft, L. E. Grünig, S. Schneider, M. Hashemi, A. Goos, A. Wetzel, M. Rübhausen and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2014, 53, 299–304 CrossRef CAS PubMed.
- N. Leidel, P. Cherney, K. G. V. Havelius, S. Ezzaher, S. Ott and M. Haumann, Inorg. Chem., 2012, 51, 4546–4559 CrossRef CAS PubMed.
- F. Gärtner, B. Sundararaju, A.-E. Surkus, A. Boddien, B. Loges, H. Junge, P. H. Dixneuf and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9962–9965 CrossRef PubMed.
- F. Gärtner, A. Boddien, E. Barsch, K. Fumino, S. Losse, H. Junge, D. Hollmann, A. Brückner, R. Ludwig and M. Beller, Chem. – Eur. J., 2011, 17, 6425–6436 CrossRef PubMed.
- M. Kessler, S. Schüler, D. Hollmann, M. Klahn, T. Beweries, A. Spannenberg, A. Brückner and U. Rosenthal, Angew. Chem., 2012, 124, 6377–6380 CrossRef.
- Pioneering experiments on the investigation of photocatalytic water splitting by HERFD-XANES and Kβ2,5-XES were carried out by the author at ID26 of the ESRF.
- Two experimental stations for this purpose are currently built at PETRA III and MAX IV by the author, funded by the German ministry for Research and Education (BMBF).
| This journal is © the Owner Societies 2014 |